enzyme-mediated cross-linking of pluronic copolymer micelles for injectable and in situ forming...
TRANSCRIPT

Acta Biomaterialia 7 (2011) 1468–1476
Contents lists available at ScienceDirect
Acta Biomaterialia
journal homepage: www.elsevier .com/locate /ac tabiomat
Enzyme-mediated cross-linking of Pluronic copolymer micelles for injectableand in situ forming hydrogels
Soo Hyeon Lee a, Yuhan Lee a, Sang-Woo Lee a, Ha-Yeun Ji a, Ji-Hee Lee a, Doo Sung Lee b, Tae Gwan Park a,⇑a Department of Biological Sciences and Graduate School of Nanoscience and Technology, Korea Advanced Institute of Science and Technology, Daejeon 305-701, Republic of Koreab Theranostic Macromolecules Research Center, Department of Polymer Science and Engineering, Sungkyunkwan University, Suwon 440-746, Republic of Korea
a r t i c l e i n f o
Article history:Received 6 September 2010Received in revised form 30 October 2010Accepted 22 November 2010Available online 25 November 2010
Keywords:Pluronic copolymersEnzyme-mediated cross-linkingInjectable hydrogelSol–gel transitionTissue-adhesiveness
1742-7061/$ - see front matter � 2010 Acta Materialdoi:10.1016/j.actbio.2010.11.029
⇑ Corresponding author. Tel.: +82 42 350 2621; faxE-mail address: [email protected] (T.G. Park).
a b s t r a c t
A new class of injectable and erodible hydrogels exhibiting highly robust gel strength at body tempera-ture was fabricated by enzyme-mediated cross-linking between Pluronic copolymer micelles. Tyramine-conjugated Pluronic F-127 tri-block copolymers at two terminal ends of polyethylene oxide (PEO) sidechains were synthesized and utilized to form self-assembled micelles in aqueous solution. Tyrosinasewas employed to convert tyramine-conjugated micelles to highly reactive catechol conjugated micellesthat could further cross-link individual Pluronic copolymer micelles to form a highly stable gel structure.The enzyme cross-linked Pluronic hydrogels showed far lower critical gelation concentration, concomi-tantly showing enhanced gel strength compared to unmodified Pluronic copolymer hydrogels, suitablefor sustained delivery of bioactive agents. Rheological studies demonstrated that the enzyme cross-linkedhydrogels exhibited a fast and reversible sol–gel transition in response to temperature while maintainingsufficient mechanical strength at the gel state. In situ formed hydrogels were eroded gradually, releasingFITC-labeled dextran in an erosion-controlled manner. Moreover, they showed tissue-adhesive propertiesdue to the presence of unreacted catechol groups in the gel structure. Enzyme cross-linked Pluronichydrogels could be potentially used for delivery applications of drugs and cells.
� 2010 Acta Materialia Inc. Published by Elsevier Ltd. All rights reserved.
1. Introduction
Stimuli-sensitive and injectable sol–gel transition hydrogelsthat can be administered into the body via a minimally invasiveroute have attracted much attention for potential drug deliveryand tissue engineering applications [1–5]. Most of the injectablehydrogels are based on self-assembled nano-sized micelles andfibers that are composed of amphiphilic block copolymers andpeptides. When they are injected using a syringe, sudden changesof surrounding environments such as temperature, pH, and otherbiochemical signals alter their conformation and/or inter- and in-tra-molecular noncovalent interactions, resulting in a sol-to-gelphase transition at critical conditions. In particular, temperature-responsive polymer hydrogels showing a lower critical solutiontemperature (LCST) behavior just below the body temperaturehave been extensively studied, because a polymer solution canbe readily mixed with therapeutic agents and cells at roomtemperature, but rapidly solidified into a gel state at the injectionsite [6,7]. Various copolymers and their derivatives such aspoly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide)(PEO–PPO–PEO) tri-block copolymers, poly(D,L-lactide-co-glyco-
ia Inc. Published by Elsevier Ltd. A
: +82 42 350 2610.
lide)–poly(ethylene oxide)–poly(D,L-lactide-co-glycolide) (PLGA–PEO–PLGA) tri-block copolymers, rationally designed amphiphilicpolypeptides, and polyorganophosphazenes are typical thermo-sensitive polymers showing such injectable sol–gel transitionbehaviors [8–11].
Among them, PEO–PPO–PEO tri-block copolymers, also knownas Pluronic copolymers, are the most extensively studied materialsto achieve in situ forming depots for sustained delivery of proteindrugs. It is widely accepted that the sol–gel transition behavior ofPluronic copolymers is mainly ascribed to the self-association ofthe amphiphilic copolymers into spherical micelles. Above the tran-sition temperature, the spherical micelles are closely packed to-gether, resulting in the physically cross-linked hydrogels [12,13].However, their biomedical applications have been severely limitedbecause of several shortcomings including low mechanicalstrength, fast dissolution, and rapid drug release under physiologi-cal conditions, primarily caused by the immediate dilution of poly-mer concentration with the body fluid below a critical gelationconcentration [8,12–14]. To reinforce their poor mechanicalproperties, a number of approaches were attempted to enhancechemical and physical interactions between self-assembled andclose-packed individual Pluronic micelles. They included the useof multi-block Pluronic copolymers linked by chain extenders andcross-linkers, photo-induced inter-micelle cross-linking after thegel formation, and stereocomplexed Pluronic copolymer micelles
ll rights reserved.

S.H. Lee et al. / Acta Biomaterialia 7 (2011) 1468–1476 1469
comprising a pair of enantiomeric oligo(lactide)s [15–18]. However,chemically modified PEO–PPO–PEO hydrogels can provoke severalside-effects due to the use of toxic cross-linking agents (e.g., diiso-cyanates, hydrogen peroxide, and UV) [19,20]. Thus, it is desirableto develop a more biocompatible cross-linking method for fabricat-ing high-strength injectable Pluronic hydrogels for controlled drugdelivery.
Herein, we demonstrate a bio-inspired cross-linking method forstabilizing Pluronic hydrogels by employing Pluronic copolymersterminally conjugated with tyramine. The tyramine moietiesexposed on the shell layer of micelles could be oxidized to catecholgroups by an enzyme, tyrosinase (also called tyrosine hydroxylase)that is widely found in animals and plants. Tyrosinase is responsi-ble for the production of various types of melanin in animal skinand blackening of fruits [21]. The key mechanism of the oxidativetyrosinase reaction is the conversion of phenol into catechol byadding an additional hydroxyl group on the aromatic ring, and fur-ther oxidation of catechol generates reactive catecholquinone. Inmelanogenesis (a process of melanin generation), the resultantreactive catecholquinone readily forms a covalent bond with vari-ous functional groups including amines, thiols, and other catechol-quinones to yield dark-colored polymerized products [21–24]. Inthis study, tyramines were attached at both terminal ends ofPEO–PPO–PEO tri-block copolymer as tyrosinase substrates to in-duce the enzyme-mediated cross-linking of Pluronic micelles forfabricating high-strength injectable hydrogels. It was demon-strated that the enzyme-mediated cross-linking reaction producedmultimeric PEO–PPO–PEO copolymers, exhibiting temperature-responsive sol–gel transition behavior with greatly enhancedmechanical strength under physiological conditions (Fig. 1A). Rhe-ological studies were performed to determine the change in gelstrength after enzymatic cross-linking and to characterize ther-mo-sensitive and reversible sol–gel transition behaviors. A modelmacromolecular drug, fluorescein isothiocyanate-dextran (FITC-dextran, molecular weight (MW) 20,000), was incorporated withinthe in situ formed hydrogel to investigate sustained release pat-terns with gel erosion profiles. Lastly, possible in vivo applicationsfor injectable, bio-adhesive, and erodible drug-releasing hydrogelmatrices were also explored with an expectation of catechol-in-duced interactions with mucosal tissues.
2. Materials and methods
2.1. Materials
Pluronic F-127 (MW 12,600 Da, PEO content: 70 wt.%), tyramine,p-nitrophenyl chloroformate (p-NPC), mushroom tyrosinase, tetra-hydrofuran (THF), anhydrous dimethylsulfoxide-d6 (DMSO-d6),and FITC-dextran (MW 20,000) were purchased from Sigma–Aldrich(Minnesota, USA). Methylene chloride and N,N-dimethylformamide(DMF) were purchased from Daejung Chemical (Daejeon, Korea) andJunsei chemical (Tokyo, Japan), respectively. Methylene chloridewas distilled before use.
2.2. Synthesis of tyramine-conjugated Pluronic F-127 (Plu-Tyr)
Plu-Tyr conjugates were synthesized via a two-step conjugationprocess using p-NPC as an amine-reactive coupling reagent. Amine-reactive Pluronic F-127 was synthesized as reported in previousstudies (Fig. 1B) [25]. Briefly, 30 g of Pluronic F-127 was dissolvedin methylene chloride (40.0 ml) and 2.71 g of p-NPC was added(the molar ratio of Pluronic F-127 to p-NPC was 1:6). The reactionwas carried out overnight at room temperature. After the reaction,the p-NPC-activated Pluronic F-127 was precipitated in excessamount of cold (�20 �C) diethyl ether, and dried in vacuum. The sub-
stitution degree of p-NPC on Pluronic F-127 was around 100% asconfirmed from 1H NMR (Bruker Avance, 400 MHz). Twenty gramsof p-NPC-activated Pluronic F-127 and 1.23 g of tyramine were dis-solved in DMF (18.9 ml) and reacted for 12 h at room temperature(the molar ratio of Pluronic F-127 to tyramine was 1:6). The solutionwas dissolved in methanol and the residual tyramine was removedby ultra-filtration (MWCO 0.45 lm). The final product was dialyzedusing MWCO 3500 membrane in deionized water for 5 days at 4 �C.The degree of tyramine substitution was calculated using 1H NMRand UV–vis spectrophotometer (UV-1601, Shimadzu, Japan) (absor-bance at 280 nm): 96.5% (1H NMR) and 105.1 ± 1.6% (UV–vis).
2.3. Cross-linking of Plu-Tyr conjugates by tyrosinase (Enz-Plu-Tyr)
To fabricate Enz-Plu-Tyr hydrogels, Plu-Tyr conjugate was dis-solved in pH 7.4 phosphate buffered saline (PBS) solution at 4 �C,mixed with 250 U ml�1 tyrosinase, and reacted for 48 h at roomtemperature. The color of the reaction solution slowly turned darkbrown after adding the enzyme. After terminating the enzymaticreaction, samples were cooled at 4 �C, far below critical gelationtemperature, to produce sol state materials [26–28]. The sol statehydrogels could be easily handled with pre-chilled tips and tubesfor further injectable experiments. To evaluate the effect of enzymeconcentration and reaction time on hydrogel properties, the con-centration of tyrosinase and reaction time were varied.
2.4. UV–vis analysis for measuring intermolecular cross-linking of Plu-Tyr hydrogels
Kinetic rates of oxidative coupling were measured by a spectro-metric method using a UV–vis spectrophotometer (NanoDrop™,Thermo Scientific, Waltham, MA). Briefly, 10 wt.% of Plu-Tyr inpH 7.4 PBS solution was mixed with 250 U ml�1 of tyrosinase atroom temperature. The UV–vis spectra of the solution at reactiontimes of 0, 2, 4, and 48 h were recorded. In addition, 10 wt.% ofPlu-Tyr in PBS solution was cross-linked using 50, 125, and250 U ml�1 of tyrosinase for 48 h to determine the influence of en-zyme concentration on the hydrogel strength.
2.5. Gel permeation chromatography (GPC) analysis
To confirm that Plu-Tyr conjugates cross-linked by tyrosinaseproduced oligomeric Pluronic F-127 copolymers, GPC analysiswas carried out using a Shodex GPC LF-804 column at 37 �C. Fiveweight percentage solution of Plu-Tyr was reacted with 0, 25,and 250 U ml�1 of tyrosinase for 24 h. The excess amount of colddeionized water was added to the Enz-Plu-Tyr hydrogel, anddialyzed overnight in 4 �C water to remove salts, and thenfreeze-dried. The resultant polymer powder was dissolved in THFand filtered using a 0.45 lm filter. Molecular weight was analyzedby GPC (Waters 600 S pump, Waters, USA) using THF as an eluant,employing GPC LF-804 (Shodex, Japan, MW detection range:100–2,000,000 Da) as a column, and using a refractive index detec-tor (Shimadzu RID-10A refractometer). Polystyrene standards withvarying molecular weights from 13,000 to 114,000 were used toobtain a calibration curve.
2.6. Sol–gel transition phase diagram
The sol–gel transition curves of Enz-Plu-Tyr hydrogels weredetermined by a vial tilting method as explained in our previousstudies, using Pluronic F-127 as a control [8,9]. Briefly, each 1 mlof Enz-Plu-Tyr hydrogel with the concentration ranging from 4 to30 wt.% was prepared in a 2 ml test tube. The hydrogels wereincubated with increasing temperature from 4 to 80 �C with anincrement of 2 �C. A gel state was determined by inverting the vial

HO
OO
OHn nm
p-NPCin Methylene Chloride
OO
OO
n m
N+ O
ClO
-O
O
N+
O
O
-O O
N+
O
O
O-O
n
Tyraminein DMF
OHH2N
OO
OO
n m
O
O
n OH
HN
HONH
B
A
C
OH
NHPlu
HO
OH
NHPlu
O
O
NHPlu
HO OHHO OH
HN NHPlu Plu
KM=1.5 mM KM=0.5 mM
Plu-Tyr280 nm
Plu-DOPA272 nm
Plu-DOPA (quinone)395 nm
Crosslinked Plu-DOPA
Tyrosinase –mediated crosslinking of Plu-Tyr conjugates
thermo-responsive sol-gel transition
Temp.>LCSTTemp.<LCST
Fig. 1. (A) Schematic illustration of Plu-Tyr hydrogel formation by enzyme mediated cross-linking and its thermo-responsive sol–gel transition. (B) Synthesis of Pluronic–tyramine conjugates and (C) the oxidative conversion of tyramine to catechol or catecholquinone, and cross-linking by tyrosinase, and wavelength values at UV–vis spectrumpeak of each reaction intermediates.
1470 S.H. Lee et al. / Acta Biomaterialia 7 (2011) 1468–1476
when no fluidity was visually observed in 1 min in an isothermalcondition.
2.7. Rheological analysis of Enz-Plu-Tyr hydrogels
All the rheological analyses were carried out with a rheometer(Gemini Nano, Bohlin Instruments, Germany) using a parallel plate(plate diameter: 2 cm, gap size: 150 lm) in an oscillatory mode. Toevaluate the effect of enzyme concentration on mechanical proper-ties of hydrogels, elastic (G0) and viscous (G00) modulus values of10 wt.% Enz-Plu-Tyr hydrogels reacted with 50 or 250 U ml�1 oftyrosinase were measured. To determine kinetic changes in thehydrogel mechanical strength during the enzymatic reaction, G0
and G00 values of 10 wt.% Plu-Tyr dissolved in PBS solution were mea-sured every hour after mixing 250 U ml�1 tyrosinase. A frequency
sweep on the hydrogels was performed from 0.1 to 10 Hz at stressvalue of 100 Pa in an isothermal condition at 37 �C. In addition, toevaluate the reversible sol–gel transition behavior of Enz-Plu-Tyrhydrogels, G0 and G00 values of 10 wt.% Enz-Plu-Tyr hydrogelwere monitored with repetitive thermal cycles between 4 and37 �C.
2.8. Mass erosion behavior of Enz-Plu-Tyr hydrogels
A mass erosion test was performed for 13 days at 37 �C. Briefly,1 ml of 20 wt.% Enz-Plu-Tyr hydrogels was prepared in 2 ml testtubes. Twenty weight percentage of Pluronic F-127 and Plu-Tyrhydrogels were used as controls. Onto each hydrogel mass,0.5 ml of pH 7.4 PBS solution was added and kept at 37 �C withgentle shaking. After predetermined time intervals, the

A
B
Elution Time (min)6 7 8 9 10
Inte
nsity
(a.u
.)
-20
0
20
40
60
80
100
120
140
160
180
w/ 0 units/mlw/ 25 units/mlw/ 250 units/ml
C
Reaction time
Wavelength (nm)250 300 350 400 450 500
Abs
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
48 hr4 hr2 hrw/o enzyme
Enzyme amount
Wavelength (nm)250 300 350 400 450 500
Abs
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
250 units/ml125 units/ml50 units/mlw/o enzyme
Fig. 2. UV-vis spectra of Enz-Plu-Tyr hydrogels with (A) different reaction times and (B) enzyme concentrations. (C) GPC analysis of Enz-Plu-Tyr conjugates with differentenzyme concentrations.
S.H. Lee et al. / Acta Biomaterialia 7 (2011) 1468–1476 1471

1472 S.H. Lee et al. / Acta Biomaterialia 7 (2011) 1468–1476
supernatant was collected, and replaced with 0.5 ml of fresh PBSsolution. The collected solution was freeze-dried, and dry weightwas measured. At day 13, the remnant of the gel was freeze-driedand remaining mass was also determined.
2.9. Release kinetics of model macromolecular drug from Enz-Plu-Tyrhydrogels
Release rates of FITC-dextran, a model drug, from Enz-Plu-Tyrhydrogels were determined. Briefly, 1 ml of 20 wt.% Enz-Plu-Tyrhydrogels was prepared in 2 ml test tubes and 0.1% (w/v) ofFITC-dextran was added into the hydrogel. Twenty weight percent-age Pluronic F-127 hydrogels and 20 wt.% Plu-Tyr hydrogels withthe same concentration of FITC-dextran were used as controls.Onto each hydrogel mass, 0.5 ml of pH 7.4 PBS solution was addedand kept at 37 �C with gentle shaking. After predetermined timeintervals, the supernatant was collected. The fluorescence of thecollected solution was measured using a spectrofluorophotometer(SLM-AMINCO 8100, SLM Instruments Inc., Rochester, NY) at anexcitation wavelength of 490 nm and an emission wavelength of520 nm.
2.10. Evaluation of mucoadhesive properties of Enz-Plu-Tyr hydrogels
To determine mucoadhesive properties of Enz-Plu-Tyr hydro-gels, porcine stomach mucin was blended with the hydrogel, andtheir interaction was analyzed by measuring a G0 value of the mix-ture at 37 �C. Two hundred microliters of 10 wt.% mucin dissolvedin PBS solution were added to 200 ll of 10 wt.% Enz-Plu-Tyr hydro-gel prepared as mentioned above. Five weight percentage mucinsolution and the mixture of 200 ll of 10 wt.% Enz-Plu-Tyr hydro-gels and 200 ll of PBS solution were used as controls. All thesamples were in triplicate.
3. Results and discussion
3.1. Inter-micellar cross-linking of Plu-Tyr hydrogels by enzymaticreaction
When highly concentrated Plu-Tyr solution (10 wt.%) was trea-ted with tyrosinase, the apparent color of Enz-Plu-Tyr hydrogels
Concentratio0 5 10 15
Tem
pera
ture
(o C)
0
20
40
60
80
100
PluronicPlu-Tyr (w/o enzyme)Enz-Plu-Tyr
Sol
Fig. 3. Sol–gel transition phase diagrams of Pluronic hydrogels (d), Plu-Tyr hydrogel wichange. Captured images represent the Enz-Plu-Tyr hydrogels in a sol (left image) or ge
was slowly changed from transparent colorlessness to dark browndue to the enzymatic conversion of phenol groups to catecholgroups with subsequent oxidation and self-cross-linking betweenneighboring Pluronic micelles. In this manner, individually packedPluronic micelles are expected to be covalently interlocked, result-ing in a more stable three-dimensional network structure. Whencatechol groups were formed and covalently cross-linked, a typicalUV absorption peak at 280 nm for a phenol group of tyramine res-idue significantly red-shifted to a higher wavelength over 310 nm(Fig. 1C) [21]. To evaluate the oxidative cross-linking reaction as afunction of time and enzyme concentration, UV–vis absorptionspectra of Enz-Plu-Tyr hydrogels was monitored. As shown inFig. 2A, Plu-Tyr solution shows a narrow UV absorption peak ofphenol groups at 280 nm. As the enzymatic cross-linking reactionproceeded, a broad peak in the range of 310–370 nm appearedand its absorbance value was gradually increased until the incre-ment was saturated at the time point of 48 h. The UV–vis absorptionspectra as a function of enzyme concentration also showed similarpatterns at the saturation enzyme concentration of 250 U ml�1 inthe hydrogel (Fig. 2B). This implies that, by the enzymatic oxidationreaction, tyramine molecules exposed on the closely packedPluronic micelles were reacted each other to produce cross-linkedPluronic copolymers. It should be mentioned that a residual peakaround 280 nm was also observed in the UV–vis spectrum of fullyreacted Enz-Plu-Tyr hydrogels, indicating that a portion of initialphenol groups in tyramine remained un-cross-linked. Thus, it canbe deduced that Enz-Plu-Tyr hydrogels were mostly composed ofa mixture of monomeric and dimeric Pluronic copolymers. Thecross-linked Pluronic copolymers could bridge between the packedPluronic micelles, thereby mechanically stabilizing the resultanthydrogels through inter-micellar interactions.
3.2. Quantitative analysis of cross-linking degree by using GPC
The polymer composition in Enz-Plu-Tyr hydrogel was evalu-ated by GPC analysis (Fig. 2C). As the enzyme concentration wasincreased up to 250 U ml�1, the peak intensity at an elution timeof 8.3 min (MW: �24,000 Da) was gradually enhanced while thatat 8.9 min (MW: �12,900 Da, Plu-Tyr peak) was diminished corre-spondingly. The intensity ratio of the peak at 8.3 min (I8.3/Itotal) wasincreased up to 56.3% after the enzyme reaction with 250 U ml�1 of
n (%, w/v)20 25 30 35
Gel
thout enzymatic reaction (s), and Enz-Plu-Tyr (.) with respect to the temperaturel state (right image), respectively.

S.H. Lee et al. / Acta Biomaterialia 7 (2011) 1468–1476 1473
tyrosinase. The result suggests that the cross-linking reaction in-duced by tyrosinase produced mostly Pluronic F-127 dimers. Itseems that the formation of dimers might be attributed to the factthat the two tyramine groups terminally conjugated to a Pluroniccopolymer backbone had a little chance to be oxidized and cross-linked simultaneously, although many tyramine moieties wereclustered in the shell of Pluronic micelles. In addition, the physicalentrapment of tyrosinases in the highly concentrated Pluronichydrogel might impede their spatial movement, limiting furtheroxidative reactions.
3.3. Sol–gel phase transition diagram
As shown in Fig. 3, the dimer formation of Pluronic copolymerssignificantly alters the sol–gel transition phase diagram of Enz-Plu-Tyr hydrogels. For the Plu-Tyr hydrogel, the critical gelation tem-perature (CGT, same as LCST) and critical gelation concentration
B
A
Frequ0.1
Mod
ulus
(Pa)
0.1
1
10
100
1000
10000
Freq0.1
Mod
ulus
(Pa)
0.1
1
10
100
1000
10000
Fig. 4. Enhancement of mechanical properties using tyrosinase-mediated cross-linkingreaction with 50 (s) and 250 U ml�1 of tyrosinase (d) for 48 h. Ten weight percentage Pmodulus (G0) of 10 wt.% Plu-Tyr solution at 37 �C after reaction with 250 U ml�1 of enzymenzyme reaction was used as a control (4).
(CGC) were only slightly changed from those for Pluronic F-127hydrogel, possibly due to the minor change in the micelle packingstructure (Fig. 3). The CGC values of Pluronic and Plu-Tyr hydrogelwithout enzyme treatment were around 15% and 19%, respectively.After the tyrosinase reaction for 48 h, the CGC value of Enz-Plu-Tyrhydrogels was significantly lowered to 4 wt.%, and the gel-to-soltransition curve was eliminated. At a concentration lower than4 wt.%, we did not observe any significant sol–gel transitionproperties. This suggests that the enzymatic dimerization ofPluronic F-127 copolymers generated loops and tails on the shelllayer of packed Pluronic micelles, providing physically and chemi-cally interconnected and tightly bound junctions between them. Inour previous study, a similar class of stable stereocomplexedPluronic hydrogels was reported by employing a blend mixtureof Pluronic copolymers terminally conjugated with enantiomericoligo(D-lactic acid) and oligo(L-lactic acid) [29]. It should be notedthat the apparent mechanical strength of Enz-Plu-Tyr hydrogels
Enzyme amount
ency (Hz)1 10
50 units/ml (G')250 units/ml (G')Plu-Tyr w/o Enz (G')
uency (Hz)1 10
1 hr (G')4 hr (G')24 hr (G')Plu-Tyr w/o Enz (G')
Reaction time
of Pluronic. (A) The elastic modulus (G0) of 10 wt.% Plu-Tyr solution at 37 �C afterlu-Tyr solution without enzyme reaction was used as a control (.). (B) The elastice for 1 h (d), 4 h (s), and 24 h (.). Ten weight percentage Plu-Tyr solution without

1474 S.H. Lee et al. / Acta Biomaterialia 7 (2011) 1468–1476
was dramatically enhanced to an extent that was comparable tothose of multimeric Pluronic copolymers with higher degrees ofpolymerization [30]. Thus, this clearly indicates that the partial for-mation of dimers found in this system is sufficient for the fabrica-tion of high-strength injectable hydrogels.
3.4. Rheological properties
The enhancement of mechanical properties of Enz-Plu-Tyrhydrogels was also examined by rheological analysis (Fig. 4A andB). Rheological properties of 10 wt.% Enz-Plu-Tyr hydrogels treatedwith 50 or 250 U ml�1 of tyrosinase were measured at 37 �C by afrequency sweep from 0.1 to 10 Hz. As shown in Fig. 4A, by increas-ing the enzyme concentration from 50 to 250 U ml�1, the value ofthe elastic modulus (G0) was significantly increased by 14.5 timesat a frequency of 1 Hz. Their mechanical properties as a functionof reaction time were also evaluated using 10 wt.% Enz-Plu-Tyrhydrogels treated with 250 U ml�1 tyrosinase for 1, 4, and 24 h atroom temperature (Fig. 4B). As the reaction proceeded, the G0 valuewas gradually increased. It is notable that the G0 value of the hydro-gel showed around 100-fold increase after enzymatic cross-linkingreaction in a short period of time at a frequency of 1 Hz, and the G0
value was rapidly saturated, showing no significant increase evenafter the additional incubation for 24 h. This is consistent withthe previous UV–vis analysis. It is possible to conclude that the for-mation of initial inter-micellar cross-linking by dimeric Pluroniccopolymers played an important role in stabilizing the gelstructure.
The reversible sol–gel transition of cross-linked Plu-Tyr hydro-gels was also analyzed by rheological studies. The repeated shift ofelastic and viscous modulus values was observed through analternating temperature cycle between 4 and 37 �C (Fig. 5). Bothsol-to-gel and gel-to-sol phase transitions were rapid with a tem-
Tim0 100 200 30
G',
G'' (
Pa)
1e-3
1e-2
1e-1
1e+0
1e+1
1e+2
1e+3
1e+4
1e+5
4
37
200Tem
pera
ture
()
Ti
Sol
GelGel
Sol
Fig. 5. The elastic (G0) and viscous (G00) modulus changes of Enz-Plu-Tyr hydrogel with aldetermined as sol–gel transition point.
perature change, taking less than 5 s. This rapid temperature-responsive behavior is necessary for injectable therapeutic applica-tions, because the polymer/drug mixture in a sol state must be eas-ily prepared at room temperature, but it should be formedinstantly into a gel state at 37 �C without diffusion from the injec-tion site.
3.5. In vitro assessment of mass erosion and release pattern ofmacromolecular drug
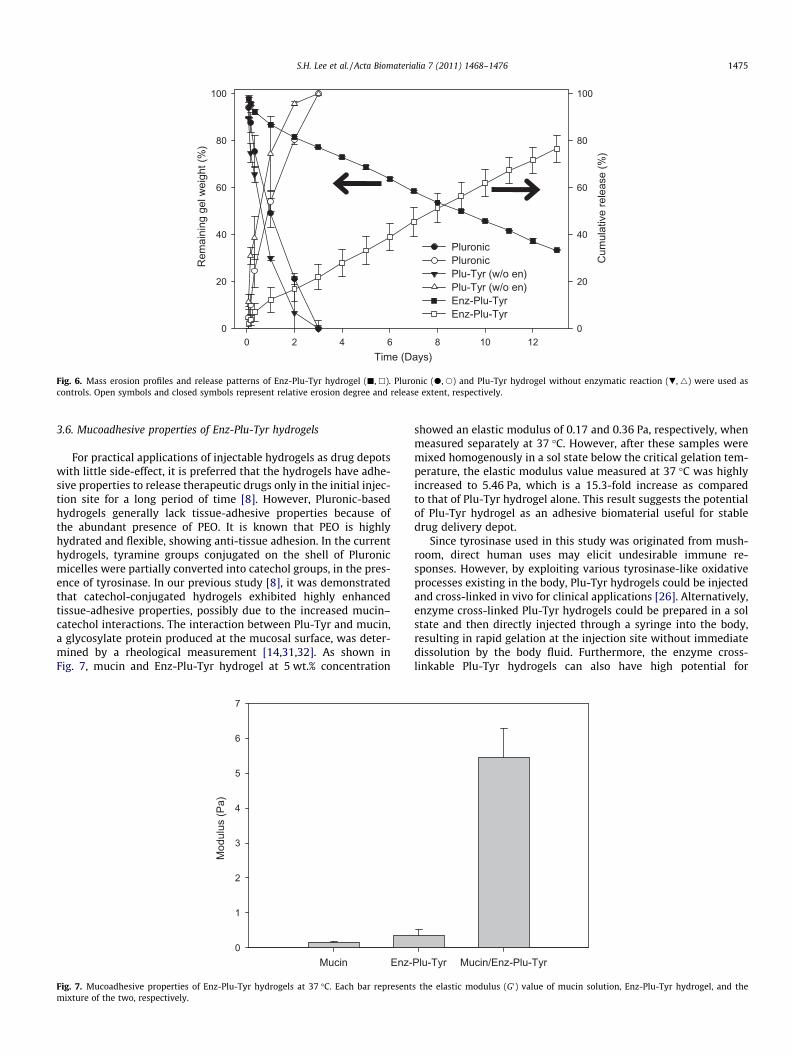
An erosion test was conducted to compare the physical durabil-ity of Enz-Plu-Tyr hydrogels in an aqueous environment. Twentyweight percentage of Pluronic F-127 and Plu-Tyr conjugatestreated with or without tyrosinase were incubated in PBS solutionat 37 �C, and daily mass loss was measured to examine the erosionprocess. After 3 days, Pluronic F-127 and Plu-Tyr hydrogels with-out enzyme treatment, control groups, were completely degraded,while more than 30% of Plu-Tyr hydrogel with the enzyme treat-ment remained even after 13 days (Fig. 6, left y-axis). This erosionpattern demonstrated that the Enz-Plu-Tyr hydrogels exhibitedhighly sustained degradation as compared to that of the othertwo types of hydrogels. The release test was simultaneouslyconducted with the erosion test under the same conditions. Thesupernatant collected in the erosion test contained FITC-labeleddextran (FITC-dextran), and the release amount of FITC-dexranwas measured by spectrofluorophotometer for 30 days. As shownin Fig. 6 (right y-axis), Pluronic F-127 and Plu-Tyr hydrogelswithout the enzyme treatment released out most of FITC-dextranwithin 3 days due to the rapid mass erosion of the hydrogels. Incontrast, Enz-Plu-Tyr hydrogels showed highly sustained releaseof FITC-dextran for 13 days. Taken together, the current Enz-Plu-Tyr hydrogels could serve as a macromolecular drug depotfor sustained release.
e (s) 0 400 500 600
G'G''
400 600 800mes (s)
Gel Gel
Sol Sol Sol
ternating temperature cycles of 4 �C and 37 �C. The cross-over point of G0 and G00 was
Time (Days)0 2 4 6 8 10 12
Rem
aini
ng g
el w
eigh
t (%
)
0
20
40
60
80
100
0
20
40
60
80
100
PluronicPluronicPlu-Tyr (w/o en)Plu-Tyr (w/o en)Enz-Plu-Tyr Enz-Plu-Tyr
Cum
ulat
ive
rele
ase
(%)
Fig. 6. Mass erosion profiles and release patterns of Enz-Plu-Tyr hydrogel (j, h). Pluronic (d, s) and Plu-Tyr hydrogel without enzymatic reaction (.,4) were used ascontrols. Open symbols and closed symbols represent relative erosion degree and release extent, respectively.
S.H. Lee et al. / Acta Biomaterialia 7 (2011) 1468–1476 1475
3.6. Mucoadhesive properties of Enz-Plu-Tyr hydrogels
For practical applications of injectable hydrogels as drug depotswith little side-effect, it is preferred that the hydrogels have adhe-sive properties to release therapeutic drugs only in the initial injec-tion site for a long period of time [8]. However, Pluronic-basedhydrogels generally lack tissue-adhesive properties because ofthe abundant presence of PEO. It is known that PEO is highlyhydrated and flexible, showing anti-tissue adhesion. In the currenthydrogels, tyramine groups conjugated on the shell of Pluronicmicelles were partially converted into catechol groups, in the pres-ence of tyrosinase. In our previous study [8], it was demonstratedthat catechol-conjugated hydrogels exhibited highly enhancedtissue-adhesive properties, possibly due to the increased mucin–catechol interactions. The interaction between Plu-Tyr and mucin,a glycosylate protein produced at the mucosal surface, was deter-mined by a rheological measurement [14,31,32]. As shown inFig. 7, mucin and Enz-Plu-Tyr hydrogel at 5 wt.% concentration
Mucin Enz-
Mod
ulus
(Pa)
0
1
2
3
4
5
6
7
Fig. 7. Mucoadhesive properties of Enz-Plu-Tyr hydrogels at 37 �C. Each bar representmixture of the two, respectively.
showed an elastic modulus of 0.17 and 0.36 Pa, respectively, whenmeasured separately at 37 �C. However, after these samples weremixed homogenously in a sol state below the critical gelation tem-perature, the elastic modulus value measured at 37 �C was highlyincreased to 5.46 Pa, which is a 15.3-fold increase as comparedto that of Plu-Tyr hydrogel alone. This result suggests the potentialof Plu-Tyr hydrogel as an adhesive biomaterial useful for stabledrug delivery depot.
Since tyrosinase used in this study was originated from mush-room, direct human uses may elicit undesirable immune re-sponses. However, by exploiting various tyrosinase-like oxidativeprocesses existing in the body, Plu-Tyr hydrogels could be injectedand cross-linked in vivo for clinical applications [26]. Alternatively,enzyme cross-linked Plu-Tyr hydrogels could be prepared in a solstate and then directly injected through a syringe into the body,resulting in rapid gelation at the injection site without immediatedissolution by the body fluid. Furthermore, the enzyme cross-linkable Plu-Tyr hydrogels can also have high potential for
Plu-Tyr Mucin/Enz-Plu-Tyr
s the elastic modulus (G0) value of mucin solution, Enz-Plu-Tyr hydrogel, and the

1476 S.H. Lee et al. / Acta Biomaterialia 7 (2011) 1468–1476
mucoadhesive drug delivery depot as an in situ forming adhesivebiomaterial for sustained and local drug release at the desired site[8]. Furthermore, Enz-Plu-Tyr hydrogels could also be applied forcell delivery to regenerate various tissues after optimizing hydro-gel properties for cell encapsulation and survival [33].
4. Conclusion
Pluronic F-127 copolymers were dimerized in a closely packedhydrogel state by introducing tyramine moieties at both terminalends of Pluronic F-127 copolymers using tyrosinase. The enzymaticreaction that coverts phenol groups to catechol groups, resulting incross-linked Pluronic micelles, ultimately enhanced physicalstrength of the hydrogel dramatically. The enzyme-mediatedcross-linking of Pluronic hydrogels showed controlled erosion withsustained release of a model macromolecular drug. They alsoshowed bio-adhesive, thermo-sensitive, and injectable properties,ideal for in situ depot formation in the tissue. Taken together,robust Pluronic hydrogels reinforced by an enzymatic treatmentcan possibly be utilized for sustained delivery of therapeuticproteins, genes, and chemical drugs.
Acknowledgements
This work was supported by Basic Science Research Program(2010-0027955) and Converging Research Center Program fromthe National Research Foundation (NRF) of Korea, the WCUProgram funded by the Korea government (MEST), and the URPProgram from KAIST.
Appendix A. Figures with essential colour discrimination
Certain figures in this article, particularly Figures 1 and 3, are dif-ficult to interpret in black and white. The full colour images can befound in the on-line version, at doi:10.1016/j.actbio.2010.11.029.
References
[1] Hoffman AS. Hydrogels for biomedical applications. Adv Drug Del Rev2002;43:3–12.
[2] Jeong B, Bae YH, Lee DS, Kim SW. Biodegradable block copolymers as injectabledrug-delivery systems. Nature 1997;388:860–2.
[3] Chun KW, Lee JB, Kim SH, Park TG. Controlled release of plasmid DNA fromphoto-cross-linked pluronic hydrogels. Biomaterials 2005;26:3319–26.
[4] Elisseeff J, Anseth K, Sims D, Mcintosh W, Randolphand M, Langer R.Transdermal Photopolymerization for Minimally Invasive Implantation. ProcNatl Acad Sci USA 1999;96:3104–7.
[5] Ta HT, Dass CR, Dunstan DE. Injectable chitosan hydrogels for localised cancertherapy. J Control Release 2008;126:205–16.
[6] Ruel-Gariepy E, Leroux JC. In situ forming hydrogels – review of temperature-sensitive systems. Eur J Pharm Biopharm 2004;58:409–26.
[7] Jeong B, Kim SW, Bae YH. Thermosensitive sol–gel reversible hydrogels. AdvDrug Deliv Rev 2002;54:37–51.
[8] Lee Y, Chung HJ, Yeo SH, Ahn CH, Lee HS, Messersmith PB, et al. Thermo-sensitive, injectable, and tissue adhesive sol–gel transition hyaluronic acid/Pluronic composite hydrogels prepared from bio-inspired catechol-thiolreaction. Soft Matter 2010;6:977–83.
[9] Jeong B, Bae YH, Kim SW. Drug release from biodegradable injectablethermosensitive hydrogel of PEG–PLGA–PEG triblock copolymers. J ControlRelease 2000;63:155–63.
[10] Hiemstra C, Zhong Z, van Tomme SR, van Steenbergen MJ, Jacobs JJL, Otter WD,et al. In vitro and in vivo protein delivery from in situ forming poly(ethyleneglycol)–poly(lactide) hydrogels. J Control Release 2007;119:320–32.
[11] Kang GD, Cheon SH, Khang G, Song S. Thermosensitive poly(organophosphazene) hydrogels for a controlled drug delivery. Eur J PharmBiopharm 2006;63:340–6.
[12] Fusco S, Borzacchiello A, Netti PA. Perspectives on: PEO–PPO–PEO triblockcopolymers and their biomedical applications. J Bioact Comp Polym2006;21:149–64.
[13] Cohn D, Lando G, Sosnik A, Garty S, Levi A. PEO–PPO–PEO-based poly(etherester urethane)s as degradable reverse thermo-responsive multiblockcopolymers. Biomaterials 2006;27:1718–27.
[14] Huang K, Lee BP, Ingram DR, Messersmith PB. Synthesis and characterizationof self-assembling block copolymers containing bioadhesive end groups.Biomacromolecules 2002;3:397–406.
[15] Hennink WE, van Nostrum CF. Novel crosslinking methods to designhydrogels. Adv Drug Del Rev 2002;54:13–36.
[16] Park SY, Lee YH, Bae KH, Ahn CH, Park TG. Temperature/pH-sensitive hydrogelsprepared from Pluronic copolymers end-capped with carboxylic acid groupsvia an oligolactide spacer. Biomaterials 2006;27:1718–27.
[17] Yoo HS. Photo-cross-linkable and thermo-responsive hydrogels containingchitosan and Pluronic for sustained release of human growth hormone (hGH). JBiomater Sci Polym Ed 2007;18:1429–41.
[18] Lee YH, Park SY, Park TG. Novel stereocomplexed sol–gel transition hydrogelsprepared from physical packing of self-assembled PEO–PPO–PEO and PPO–PEO–PPO copolymer nanoscale micelles. J Nanosci Nanotechnol2008;8:5236–41.
[19] van Tomme SR, Storm G, Hennink WE. In situ gelling hydrogels forpharmaceutical and biomedical applications. Int J of Pharm 2008;355:1–18.
[20] Kurisawa M, Chung JE, Yang YY, Gao SJ, Uyama H. Injectable biodegradablehydrogels composed of hyaluronic acid–tyramine conjugates for drug deliveryand tissue engineering. Chem Comm 2005;34:4312–4.
[21] Lee BP, Dalsin JL, Messersmith PB. Synthesis and gelation of DOPA-modifiedpoly(ethylene glycol) hydrogels. Biomacromolecules 2002;3:1038–47.
[22] Mason HS. The chemistry of melanin: III. Mechanism of the oxidation ofdihydroxyphenylalanine by tyrosinase. J Biol Chem 1948;172:83–99.
[23] Braunschweig AB, Elnathan R, Willner I. Monitoring the activity of tyrosinaseon a tyramine/dopamine-functionalized surface by force microscopy. NanoLett 2007;7:2030–6.
[24] Baron R, Willner B, Willner I. Biomolecule–nanoparticle hybrids as functionalunits for nanobiotechnology. Chem Comm 2007;4:323–32.
[25] Choi SH, Lee SH, Park TG. Temperature-sensitive pluronic/poly(ethylenimine)nanocapsules for thermally triggered disruption of intracellular endosomalcompartment. Biomacromolecules 2006;7:1864–70.
[26] Gong CY, Shi S, Dong PW, Zheng XL, Fu SZ, Guo G, et al. In vitro drug releasebehavior from a novel thermosensitive composite hydrogel based on Pluronicf127 and poly(ethylene glycol)-poly(epsilon-caprolactone)-poly(ethyleneglycol) copolymer. BMC Biotechnol 2009;9:8.
[27] Chung HJ, Lee Y, Park TG. Thermo-sensitive and biodegradable hydrogels basedon stereocomplexed Pluronic multi-block copolymers for controlled proteindelivery. J Control Release 2008;27:22–30.
[28] Terada S, Yoshimoto H, Fuchs JR, Sato M, Pomerantseva I, Selig MK, et al.Hydrogel optimization for cultured elastic chondrocytes seeded onto apolyglycolic acid scaffold. J Biomed Mater Res A 2005;75:907–16.
[29] Park SY, Chung HJ, Lee Y, Park TG. Injectable and sustained delivery of humangrowth hormone using chemically modified Pluronic copolymer hydrogels.Biotechnol J 2008;3:669–75.
[30] Jiang J, Malal R, Li C, Lin MY, Colby RH, Gersappe D, et al. Rheology ofthermoreversible hydrogels from multiblock associating copolymers.Macromolecules 2008;41:3646–52.
[31] Catron ND, Lee H, Messersmith PB. Enhancement of poly(ethylene glycol)mucoadsorption by biomimetic end group functionalization. Biointerphases2006;1:134–41.
[32] Burke SA, Ritter-Jones M, Lee BP, Messersmith PB. Thermal gelation and tissueadhesion of biomimetic hydrogels. Biomed Mater 2007;2:203–10.
[33] Park KM, Lee SY, Joung YK, Na JS, Lee MC, Park KD. Thermosensitive chitosan-Pluronic hydrogel as an injectable cell delivery carrier for cartilageregeneration. Acta Biomater 2009;5:1956–65.