ema update on qbd and pat - welcome to ifpacnet agency of the european union ifpac, 23 january 2012...
TRANSCRIPT

An agency of the European Union
IFPAC, 23 January 2012
EMA Update on QbD and PAT
Evdokia Korakianiti, PhD
Section Head Chemicals, Quality of Medicines SectorEuropean Medicines Agency

2
Presentation Overview
EU perspective on:
• QbD and the patient
• Where we are
• What we have done so far
• What are the challenges
• What needs to be done further

3
Quality by Design (QbD) and the patient
BenefitsRisks
QbD:
“Build quality into the product”
= reducing uncertainties
Benefit to the patient
??
• Systems approach• Leverages information/ knowledge • Smart study designs, tools • Better decision-making (risk assessment and management)

4
Where we are? QbD submissions in Centralised Procedure (CP)
•No of submissions received so far (NCEs)
• Initial MAAs:20 (6 incl. RTRT)• Post-authorisation: 6• Sci. Advice requests: 6
Initial MAAs Post-authorisation: 6 Sci. Advice requests
•Biologicals? Initial MAAs: 2 (1 withdrawn on safety grounds), some at pre-submission level
•Generics? None so far

5
Where we are? QbD submissions at EMA • Are we there yet? Not yet, we are far from seeing dynamic
processes adjusted real time with feedforward or feedback controls as the norm in submissions, but progress is being made
• The numbers of applications with QbD and / or PAT elements received are steadily increasing over the past 5 years
• It seems that the companies that have received approval for QbD approach in one product are then implementing QbD across several of their products
• The content of the QbD submissions is now more comprehensive includes defining CQAs, developing design space, in some cases RTRT or CPV
• First contacts on continuous processes

6
What have we seen in QbD dossiersSystematic use of the following tools:
•Risk assessment –E.g. Ishikawa diagrams, FMEA, CFD to determine criticality
•Statistical models – Design Space definition, E.g. DOEs
– Surrogates for end product testing e.g. multivariate model to predict dissolution
• Process Analytical Tools for: –on-line/ in-line measurements e.g. moisture measurement in a fluid bed dryer, blend
uniformity, drying step and particle size for active substances
–At-line measurement e.g. NIR for assay and content uniformity or identity testing
•Elimination of some end product testing in the specifications
–E.g. replacement of dissolution by disintegration, content uniformity replaced by upstream blend uniformity

7
Where we are? ICH Regulatory toolbox• ICH Q8, Q9 and Q10
• ICC- IWG Questions and Answers
– Knowledge Management, Design Space, Real Time Release, Control Strategy and Pharmaceutical Quality System
• Points to consider on :
– Criticality of Quality Attributes and Process Parameters
– Control Strategy
– Level of Documentation R/A and DoE
– Design space, modelling, process validation/continuous process verification
*http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html

8
What have we done? Implementation of ICH vision in EU
Build knowledgeIdentify knowledge gaps
Share Knowledge
Interactions with Industry Evaluation of applications
Workshops with Industry Expert meetings
Guidance developmentTraining
EU PAT team and WPs

9
What have we done? EU PAT Team
Aim: Prepare a harmonised approach within EU on assessment and inspection of QbD submissions
Activities:
• Informal scientific discussions with applicants at pre-submission level
• Advice during evaluation upon request from Rapporteurs
• Training of assessors – 3 trainings for Assessors and Inspectors, ICH-WG training, regular
presentations to Working parties
• Workshops with Industry
– EMEA-EFPIA Workshops on Design Space (2006) and ‘Quality by Design' (2009)
– Mock Inspections /Mock submissions

10
What have we done? Guidelines revision by Quality Working Party (QWP)
•Use of Near Infrared Spectroscopy (NIRS) by the Pharmaceutical Industry and the Data Requirements for New Submissions and Variations (EMEA/CHMP/CVMP/WP/17760/2009 Rev 1)* (2nd draft to be released for public consultation within 1Q 2012)
•Real Time release Testing (EMA/CHMP/QWP/811210/2009)* (finalised)
•Process validation (EMA/CHMP/CVMP/QWP/809114/2009)* (concept paper released, draft Guideline to be released for consultation within 1Q 2012)
*http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content
_000081.jsp&murl=menus/regulations/regulations.jsp&mid=WC0b01ac0580027546

11
What are the challenges?
• Common misconceptions
• Presentation of information in dossiers
• Clarification of regulatory requirements
• Enable post-approval regulatory flexibility
• Harmonised implementation

12
What are the challenges? Common misconceptions
•QbD = more scrutiny, more questions, delays in approvals
•QbD = pre-approval inspections, delays in approvals
– Product specific like with traditional applications
– When RTRT is proposed and the site in question has not been authorised for RTRT before. Depends also on complexity of the proposed strategy
•Release based on compliance with DS (no need for specifications)
•Design Space presentation and Process descriptions (to be discussed later)
• Criticality vs risk: If a parameter is controlled it stops being critical (common misconception)
•Proven Acceptable Ranges (PAR) means the same as Design Space

13
What are the challenges? Presentation of information in dossiers
•Current approach: The less that is included the better, nice graphs are enough…•BUT the basic regulatory requirements remain, e.g. for specifications, process descriptions•Often only conclusions of studies are presented with no explanation about how they have been reached•DS boundaries not clearly described (which parameters consist the DS and their ranges)•Ranges investigated in lab scale not corresponding with the DS boundaries with no further explanations•No information on statistical validity of models / DoEs• QbD dossiers need to be better structured, the aim of the studies should be clear, what is applied for should be clear

14
What are the challenges? Presentation of information in dossiers
Summary of risk assessment (R/A) outputs (extract from an application file): what is often missing
1515
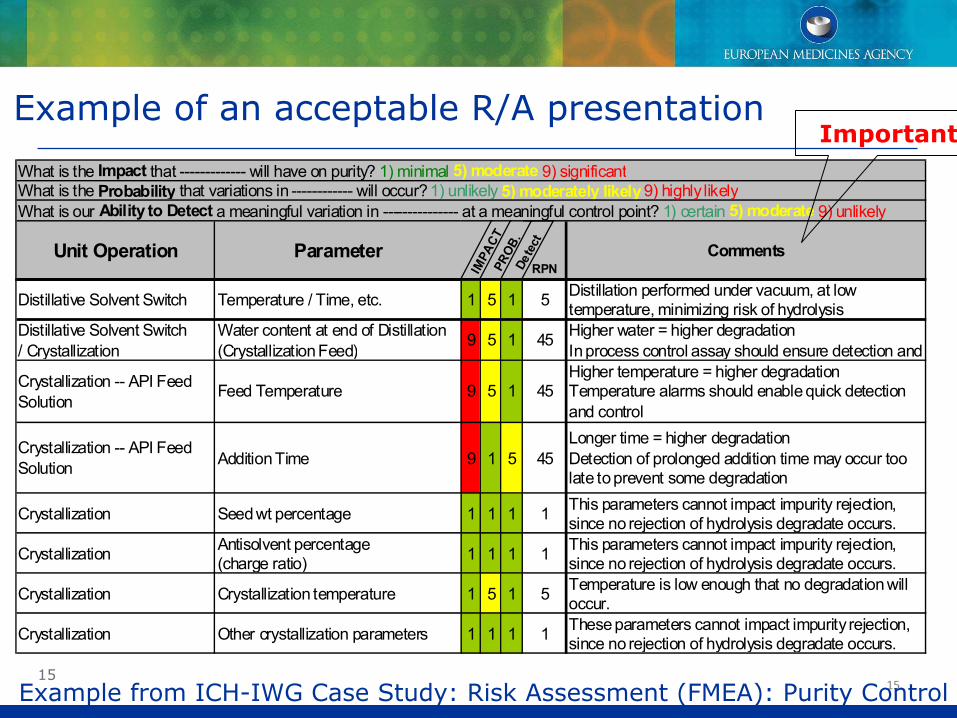
Example of an acceptable R/A presentation
What is the Impact that ------------- will have on purity? 1) minimal 5) moderate 9) significantWhat is the Probability that variations in ------------ will occur? 1) unlikely 5) moderately likely9) highly likelyWhat is our Ability to Detect a meaningful variation in --------------- at a meaningful control point? 1) certain 5) moderate 9) unlikely
Unit Operation Parameter
IMPA
CTPR
OB.
Dete
ct
RPNComments
Distillative Solvent Switch Temperature / Time, etc. 1 5 1 5 Distillation performed under vacuum, at low temperature, minimizing risk of hydrolysis
Distillative Solvent Switch/ Crystallization
Water content at end of Distillation (Crystallization Feed) 9 5 1 45 Higher water = higher degradation
In process control assay should ensure detection and
Crystallization -- API Feed Solution Feed Temperature 9 5 1 45
Higher temperature = higher degradationTemperature alarms should enable quick detection and control
Crystallization -- API Feed Solution Addition Time 9 1 5 45
Longer time = higher degradationDetection of prolonged addition time may occur too late to prevent some degradation
Crystallization Seed wt percentage 1 1 1 1 This parameters cannot impact impurity rejection, since no rejection of hydrolysis degradate occurs.
Crystallization Antisolvent percentage (charge ratio) 1 1 1 1 This parameters cannot impact impurity rejection,
since no rejection of hydrolysis degradate occurs.
Crystallization Crystallization temperature 1 5 1 5 Temperature is low enough that no degradation will occur.
Crystallization Other crystallization parameters 1 1 1 1 These parameters cannot impact impurity rejection, since no rejection of hydrolysis degradate occurs.
Example from ICH-IWG Case Study: Risk Assessment (FMEA): Purity Control
Important

Presentation in dossiers: Process descriptions
• Need for adequate well described processes to enable the assessor to make an informed decision
• This includes all CPPs and ranges with target set point for all non CPPs
• How this affects post-approval flexibility?
• To be discussed later but having not adequately described processes in the dossier only raises questions
delays in approval
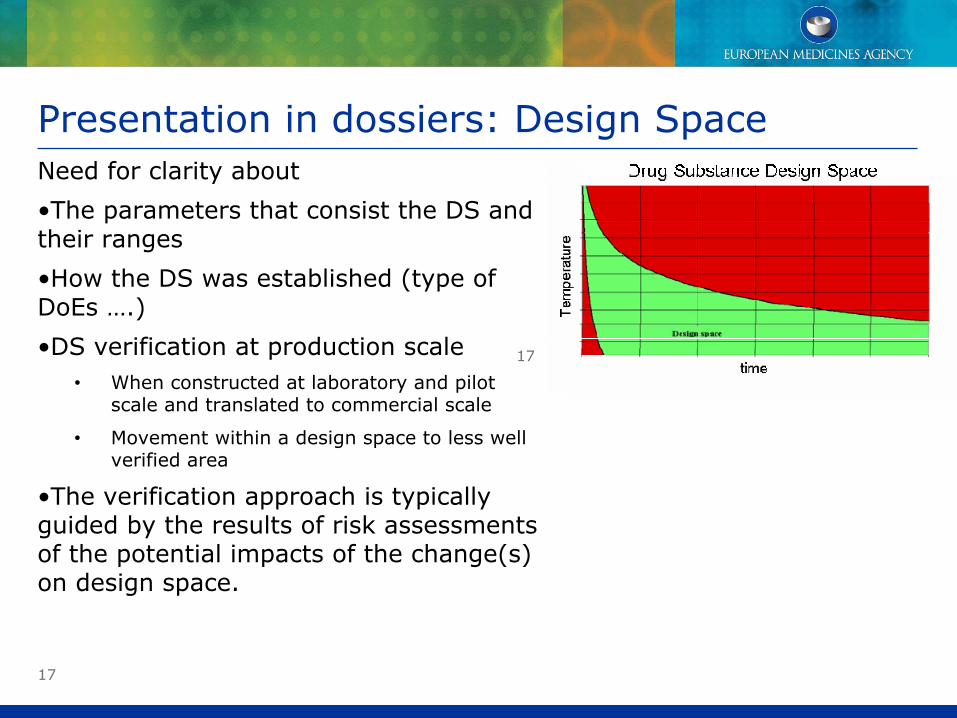
Presentation in dossiers: Design SpaceNeed for clarity about
•The parameters that consist the DS and their ranges
•How the DS was established (type of DoEs ….)
•DS verification at production scale• When constructed at laboratory and pilot
scale and translated to commercial scale
• Movement within a design space to less well verified area
•The verification approach is typically guided by the results of risk assessments of the potential impacts of the change(s) on design space.
17
17

Level of info in the dossier
• Adequate to allow the assessor to make an informed decision
• Consider impact for finished product quality.
• Consider available Guidance on level of detail e.g. ICH PtC for Models
Level of detail in a regulatory submission
Low-Impact Models High -Impact ModelsMedium-Impact Models
.
Impact

19
Design space (DS)•DS limits vs statistical risk
•DS verification vs scale and equipment
•Movement to less well verified areas within a DS
•DS for individual unit operations, what is the interaction amongst all the unit operations in the manufacturing process?
Modelling•When is a model appropriate for RTRT and CPV?
•How will model validity be verified through the product lifecycle?
•Model vs risk to quality
Quality systems and QbDWhat change constitutes a variation and what a change that can be handled under the Quality System of the company?
QbD and established regulatory standardse.g. Acceptance criteria for content Uniformity when using large sample sizes (ongoing work from EDQM)
Manufacturing process descriptionsvs post approval changes
What are the challenges?Clarification of regulatory requirements

20
What are the challenges? Post-approval changes
•Should better understanding of process risks mean less regulatory commitments and how?
• Risk based framework that enables handling of changes post approval
•Risk based protocols
-Draft Q&A on EMA website*
One size fits all!
*http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000478.jsp&murl=menus/regulations/regulations.jsp&mid=WC0b01ac0580251bd9
From J. Berridge, PQRI March 2005

21
Post-approval Change Management Protocols
+
Strategy•Planned studies
•Acceptance criteria
•Methods
Results +
Strategy•Planned studies
•Acceptance criteria
•Methods
Results
CurrentlyEvaluation of a
proposed variation as a ‘whole’ (Strategy +
Results)
Early Step 1:Submission of
a Protocol
Quick Step 2:Implementation of
the change
Major Variation (Type II) or in
initial MAA
Minor Variation (Type IA or IB)

Changes to non-CPPs
• The variations Classification Guideline is under revision
• The current thinking is that changes to non CPPs could be handled under the PQS and notified to the Competent Authorities post implementation in the form of annual reporting in order to keep the dossier updated
22

23
What are the challenges? Harmonised implementation in EU
• Assessment scrutiny Risk to finished product quality
•Adequate expertise at a time of limited resources at national and European level.
•Harmonisation– Training, Guidance, Peer review
•Assessor/Inspector interaction-Standardise the communication between assessor and inspector
-Inspectors focus on system related issues-Joint inspections

24
What are the challenges? Harmonised implementation in ICH regions
EMA-FDA pilot for parallel assessment of QbD applications(NCE products only). Japan participating as observer.
Objective : Increase assessors awareness using actual applications and toensure consistent implementation in the evaluation process of ICH Q8-10 concepts between EU and US
Outcome• Same LoQ and LoOI for the parts of the application subject to the pilot •Guidance development
For more details: http://www.ema.europa.eu/ema/index.jsp?curl=pages/partners_and_networks/document_listing/document_listing_000228.jsp&murl=menus/partners_and_networks/partners_and_networks.jsp&mid=WC0b01ac058003176e

Status of the QbD pilot
• One application ongoing
• Several TCs between EU and FDA, very useful interactions
• Aim to exchange views and harmonise where possible
• However in some cases regional regulatory requirements need to be respected
• WPs informed at key milestones
• Lessons learnt to be published at the end of the evaluation process
25

26
What needs to be done•Guidance to address the current challenges and future trends
– Multivariate Statistical Process Control (MSPC) based models proposed as surrogate for traditional release tests
– Setting clinically meaningful specifications
– QbD and analytical development
– Implementation of the QbD paradigm for continuous manufacturing
•Share knowledge with the network of Assessors and Inspectors in EU
– Training, Webinars, Guidance, Monitor consistency in the evaluation more closely
•Harmonised implementation across ICH regions

27
Summary• We haven’t reached the desired state yet, but progress is made• Assessors are requested to evaluate new types of data –Need
for appropriate expertise• Guidance documents are being drafted/revised• Need to continue dialogue with Industry to understand better
the concerns• Regulatory flexibility is not achieved by including minimal
information in dossiers. – Consider available Guidance on level of detail e.g. ICH Q&A and PtC
• Regulators try to ensure a harmonised approach in the implementation , e.g Parallel assessment pilot with FDA
• Advise may be sought in EU in the following ways:– Contact the PAT Team ([email protected])
– Seek formal scientific advice ([email protected])

28
Final remark
QbD Better process and product understanding
Higher assurance of product quality and better management
of risks
Benefit to the patientBenefitsRisks
??

29
Thank you for your attention!
Questions?
Acknowledgements:
EU PAT Team: J-L Robert, K.Pugh, L.Ertle, M.Diller, R.Cejka, K.Ho, M.Welin, M.Wierer, M.Catibusic, G.Lorenti, D.Makohon