electron energy loss spectrum of graphane from first ...lu.ac.ir/usersfiles/698781312-3.pdf ·...
TRANSCRIPT

Ec
HD
a
ARRAA
KHCTECFD
1
iomaCoe
le(iaAooai2
(
h0
Micron 67 (2014) 30–36
Contents lists available at ScienceDirect
Micron
j our na l ho me page: www.elsev ier .com/ locate /micron
lectron energy loss spectrum of graphane from first-principlesalculations
. Nejati, M. Dadsetani ∗
epartment of Physics, University of Lorestan, Khorramabad, Iran
r t i c l e i n f o
rticle history:eceived 26 March 2014eceived in revised form 3 June 2014ccepted 11 June 2014vailable online 25 June 2014
eywords:
a b s t r a c t
In this study, the energy loss near edge structure (ELNES) of carbon atoms in chair and tricycle conformersof hydrogenated graphene, namely ‘graphane’, has been calculated in the density functional theory usingFP-LAPW method, and then, it has been compared with that of graphite and graphene. Using ELNES fromchair conformer, the carbon K-edge was found to have a few main features including electron transitionfrom 1s orbital of carbon atom to �*, �*, and a hybridization of these two states. The first feature intricycle conformer, however, has contributions of both �* and �* states. The comparison of ELNES andthe unoccupied density of states in each structure also justifies this. The energy difference between �*
ydrogenated graphenehairricycleLNESore-hole approximationP-LAPW
and �* features of graphane conformers was decreased relative to it in graphite and graphene. Since theinclusion of core-holes and super-cells is essential for accurate reproduction of features in graphite andgraphene, it may be essential as well for the ELNES spectra of graphane conformers.
© 2014 Elsevier Ltd. All rights reserved.
ensity functional theory
. Introduction
Graphene (Novoselov et al., 2004), a single sheet of graphite,s a two-dimensional carbon material consisting of a monolayerf carbon atoms in a honeycomb lattice. This material exhibitsany unusual properties in relevant areas of physics, chemistry
nd material science (Geim and Novoselov, 2007; Katsnelson, 2007;astro Neto et al., 2009) such as the high electrical conductivity, theptical transparency and the half quantized quantum Hall effect,tc.
Graphane, a fully hydrogenated graphene sheet, which is a cova-ently bonded hydrocarbon, was theoretically proposed by Sofot al. (2007), and it was synthesized in the laboratory by Elias et al.2009). Some various interesting properties of graphene includingnsulating wide band gap (Sofo et al., 2007; Boukhvalov et al., 2008)nd magnetization by partial dehydrogenation of the sheet (Neek-mal and Peeters, 2011) were reported. Experimentally, adsorptionf hydrogen on graphene was indeed observed to result in a gappening in the electron states (Elias et al., 2009). Therefore, the
dsorption of hydrogen turns the highly conductive graphene intonsulating graphane, as theoretical predictions show (Sofo et al.,007; Boukhvalov et al., 2008). However, the exact value of the band∗ Corresponding author. Tel.: +98 6616200192; fax: +98 6616200192.E-mail addresses: [email protected], dadsetani [email protected]
M. Dadsetani).
ttp://dx.doi.org/10.1016/j.micron.2014.06.003968-4328/© 2014 Elsevier Ltd. All rights reserved.
gap is still unknown. The thermal contraction effect and the heatcapacity of graphane are larger than those of graphene (Neek-Amaland Peeters, 2011). Six possible conformers of this hydrocarbonwere reported, known as ‘chair’, ‘boat-1’, ‘boat-2’, ‘stirrup’, ‘twist-boat’, and ‘tricycle’, for the connection of the hydrogen atoms to up-and down-side of carbon atoms in a honeycomb lattice (Sofo et al.,2007; Samarakoon and Wang, 2009; Flores et al., 2009; Chaoyuet al., 2012; Wen et al., 2011). The chair conformer is the moststable, and the tricycle conformer is the second one (Chaoyu et al.,2012). Other physical properties of graphane conformers are understudy.
Energy loss near edge structure (ELNES) spectroscopy, which isoften performed within a TEM (Egerton, 1996), is a routine tech-nique to detect and measure the electronic structure of materials.Since ELNES originates from the electron transition from a coreorbital to unoccupied bands, spectral features of the ELNES reflectthe partial density of states of unoccupied bands, and it can provideinformation on the atomic arrangements, electronic structures, andthe chemical bonding of an objective atom in the materials (Bottonet al., 1996; Mizoguchi et al., 2003, 2004). Despite the fact thatDFT is not intended for the calculation of electronically excitedstates, the calculation of ELNES and the low-loss spectra with theDFT works pretty well (Rez et al., 1995). Among a variety of codes
available for DFT calculations, in addition to the pseudopotentialCASTEP code (Segall et al., 2002), the FP-LAPW+lo code WIEN2k(Blaha et al., 2001) has been successfully applied to EELS calcula-tions of many different materials (Holec et al., 2008; Keast et al.,
i / Mic
2uaer(2caaLi(fc
gbsafLsataghubhE
mogsg
2
2
FtscobmhKG(KcSastTiew
H. Nejati, M. Dadsetan
003; McCullocha et al., 2012; Luitz et al., 2001). WIEN2k elegantlyses a number of approximations to find sensible results of ELNESlthough it explicitly calculates valence electronic states (Hébertt al., 2003). Using the WIEN2k code leads to highly accurateesults for ELNES, specially carbon K-edges in the carbon materialsDadsetani et al., 2010; Titantah et al., 2004; Titantah and Lamoen,005). It should be mentioned that another choice for excited statealculations is the Exciting-code (Anon., 2014) which recently hasttained considerable successes. While the Exciting-code is also
full-potential all-electron package implementing the families ofAPW methods, it is based on a different theory. Its particular focuss on excited states within both the many-body perturbation theoryMBPT) (Møller and Plesset, 1934) and the time-dependent densityunctional theory (TDDFT) (Runge and Gross, 1984). The core lossalculation has not been yet implemented in the Exciting-code.
Low-loss spectra of hydrogenated graphene for different hydro-en coverage and different corrugation angles have been calculatedy Bangert et al. (2010) using the WIEN2k-code. It has beenhown that hydrogenated graphene exhibits a �-plasmon peakt 4.7 eV. Bangert et al. showed the evolution of �-plasmon inully hydrogenated graphene with increasing corrugation angle.ike graphene, the flat hydrogenated graphene (angle 90◦) pos-esses a �-plasmon. The �-plasmon diminishes for a corrugationngle of 99◦, and it nearly disappears for corrugation angles largerhan 107.8◦. An additional peak at 6–7 eV emerges at the end of
pure �-plasmon, which establishes sp3 character in corrugatedraphene when all bonds are used. The core loss spectroscopy ofydrogenated graphene, to be sure, is still unknown. In this work,sing first-principles simulations, ELNES spectra of two more sta-le conformers of fully hydrogenated graphene, chair and tricycle,ave been calculated. In order to compare and check the results, theLNES spectra of graphite and graphene have also been calculated.
In this article, first we pointed to the ab initio calculationethod. Then, we presented theoretical estimations for the effect
f hydrogenation on the structural and electrical properties of theraphene layer. Afterwards, we calculated the energy loss near edgetructure of graphane and finally we compared ELNES of hydro-enated graphene to graphite and graphene.
. Computational details
.1. Calculation parameters
The calculations presented in this work are based on theP-LAPW method in which no shape approximation on poten-ial or the electronic charge density is made. The calculations oftructural and electronic properties have been done relativisti-ally. We used the WIEN2k (Blaha et al., 2001) implementationf the method, which allows inclusion of local orbitals in theasis, improving upon linearization and enabling consistent treat-ent of the semicore and valence states in an energy window,
ence ensuring proper orthogonality. In independent particleohn–Sham DFT, the exchange-correlation potential within theGA was calculated by means of the scheme of Perdew et al.
1996) for all spectra. The interstitial plane wave vector cut offMax was chosen in a way that converging RMTKMax of total energy,harge density and atomic forces equals 8 for the ground state.ince the use of super-cell increases the volume of calculations,nd given the presence of very light materials like hydrogen, amaller value of RMTKMax (5.0) for core-hole super-cell calcula-ions is sufficient (http://www.wien2k.at/reg user/faq/rmt.html).
he maximum l quantum number for the wavefunction expansionnside the atomic sphere was confined to lmax = 12. The Gmax param-ter was taken to be 18 Bohr−1. Brillouin zone (BZ) integrationsithin self-consistency cycles were performed via a tetrahedronron 67 (2014) 30–36 31
method containing 24 k-points in the irreducible BZ of all struc-tures. It corresponds to a mesh with dimensions 9 × 9 × 2 forgraphite, 14 × 14 × 1 for graphene and chair, and 1 × 5 × 15 for tri-cycle structures, respectively. Crystal lattices and atomic positionsof chair and tricycle conformers of graphane are fully optimizedup to the residual force on every atom less than 1 mRyd/a.u. Inorder to neglect any interlayer interactions, we set the distance15 A between hydrocarbon layers.
Although there are different studies on the energy gap of chair,there is no experimental report of it. Moreover, the lack of an exactcalculation of energy gap of tricycle reveals the need for knowledgeof energy gap values of graphane conformers through a precisemethod. Therefore, the many body G0W0 approximation (Hedin,1965) implemented in Exciting-code was used to calculate theexact values of chair and tricycle band gaps. It should be noted thatwhile G0W0 changes the band structures, these changes do not ingeneral improve the calculation of spectra and G0W0 data are nottypically used for XANES or ELNES modeling.
2.2. The spectra method
In a band structure, the ELNES is calculated within the first Bornapproximation with the assumption that the incoming and out-coming fast electrons are plane waves with wave vectors �ki and�kf . The double differential scattering cross section (DDSCS) for theinelastic electron scattering is given by (Bethe, 1930):
∂2�
∂˝∂E(E, q) =
[4�2
a20q4
]kf
ki
∑i,f
∣∣⟨f |ei�q.�r |i⟩∣∣2
ı(E − Ef + Ei) (1)
where �q = �ki − �kf is the momentum transfer, a0 is the Bohr radius,
E is the energy loss, and � =√
1 − ˇ2 is the relativistic factor. Thesummation is done over all occupied initial and unoccupied finalone-electron states. The initial state is the ground state of the tar-get electron and the final state is the state occupied by the targetelectron in the conduction band after interaction. Eq. (1) can berecast in terms of the unoccupied density of states, �(E). With someapproximation (Nelhiebel et al., 1999), we can write:
∂2�
∂˝∂E(E, q) =
∑lF
∣∣MlF (E, q)∣∣2
�lF (E) (2)
which is a sum of transitions to final states of lF-character (s, p, d,. . .). MlF (q, E) is a smooth and slowly decaying function of energyloss, representing the overall shape of the edge, in a way that vari-ations in DDSCS represent the energy dependence of the densitiesof states (DOS) above the Fermi level. To simulate the energy lossnear the edge structure, we used the TELNES3 program (Blaha et al.,2001), a modified version of TELNES (Nelhiebel et al., 1999), whichinvolves the fully relativistic effects, and which is a part of theWIEN2k program.
Since the core-hole left by the excited electron locally changesthe potential, and therefore, influencing the electron density, itserves as a perturbation, so the valence and conduction statescan be contracted and shifted. The projected density of states ischanged, and the overall shape of the spectrum can be stronglymodified, in a way that it may even lead to excitonic peaks inthe ELNES. The core-hole calculations have been carried out bythe reduction of the occupancy of the core level corresponding tothe observed edge. In order to avoid renormalization problems, themissing charge is added to the unit cell as a uniform backgroundcharge. In order to avoid the interaction among neighboring core-
holes, we used super-cell structures. This approach including theDFT, the super-cell, and the core-hole, namely, “Final State Rule”(FSR) (Jorissen, 2007), was used for all spectra. This rule states thatthe calculation is done via Fermi’s golden rule with final states being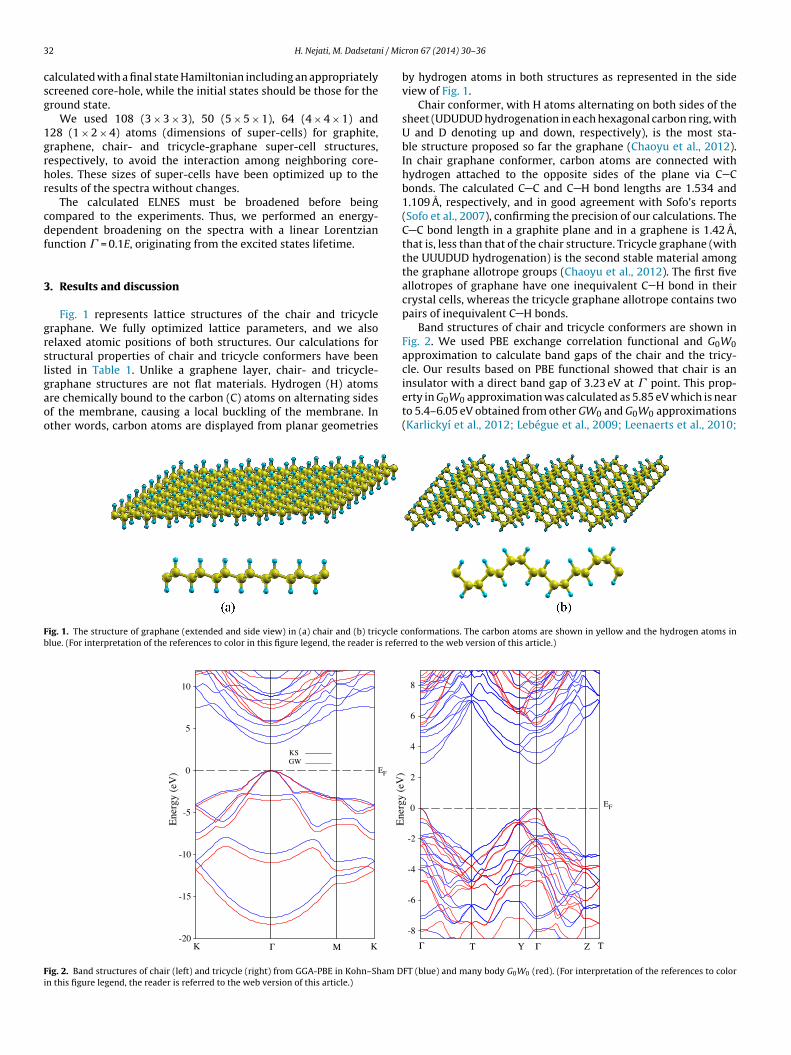
3 i / Mic
csg
1grhr
cdf
3
grslgaoo
Fb
Fi
2 H. Nejati, M. Dadsetan
alculated with a final state Hamiltonian including an appropriatelycreened core-hole, while the initial states should be those for theround state.
We used 108 (3 × 3 × 3), 50 (5 × 5 × 1), 64 (4 × 4 × 1) and28 (1 × 2 × 4) atoms (dimensions of super-cells) for graphite,raphene, chair- and tricycle-graphane super-cell structures,espectively, to avoid the interaction among neighboring core-oles. These sizes of super-cells have been optimized up to theesults of the spectra without changes.
The calculated ELNES must be broadened before beingompared to the experiments. Thus, we performed an energy-ependent broadening on the spectra with a linear Lorentzianunction � = 0.1E, originating from the excited states lifetime.
. Results and discussion
Fig. 1 represents lattice structures of the chair and tricycleraphane. We fully optimized lattice parameters, and we alsoelaxed atomic positions of both structures. Our calculations fortructural properties of chair and tricycle conformers have beenisted in Table 1. Unlike a graphene layer, chair- and tricycle-
raphane structures are not flat materials. Hydrogen (H) atomsre chemically bound to the carbon (C) atoms on alternating sidesf the membrane, causing a local buckling of the membrane. Inther words, carbon atoms are displayed from planar geometriesig. 1. The structure of graphane (extended and side view) in (a) chair and (b) tricycle clue. (For interpretation of the references to color in this figure legend, the reader is refer
Ene
rgy
(eV
)
-20
-15
-10
-5
0
5
10
Κ Γ Μ Κ
KSGW
EF
ig. 2. Band structures of chair (left) and tricycle (right) from GGA-PBE in Kohn–Sham Dn this figure legend, the reader is referred to the web version of this article.)
ron 67 (2014) 30–36
by hydrogen atoms in both structures as represented in the sideview of Fig. 1.
Chair conformer, with H atoms alternating on both sides of thesheet (UDUDUD hydrogenation in each hexagonal carbon ring, withU and D denoting up and down, respectively), is the most sta-ble structure proposed so far the graphane (Chaoyu et al., 2012).In chair graphane conformer, carbon atoms are connected withhydrogen attached to the opposite sides of the plane via C Cbonds. The calculated C C and C H bond lengths are 1.534 and1.109 A, respectively, and in good agreement with Sofo’s reports(Sofo et al., 2007), confirming the precision of our calculations. TheC C bond length in a graphite plane and in a graphene is 1.42 A,that is, less than that of the chair structure. Tricycle graphane (withthe UUUDUD hydrogenation) is the second stable material amongthe graphane allotrope groups (Chaoyu et al., 2012). The first fiveallotropes of graphane have one inequivalent C H bond in theircrystal cells, whereas the tricycle graphane allotrope contains twopairs of inequivalent C H bonds.
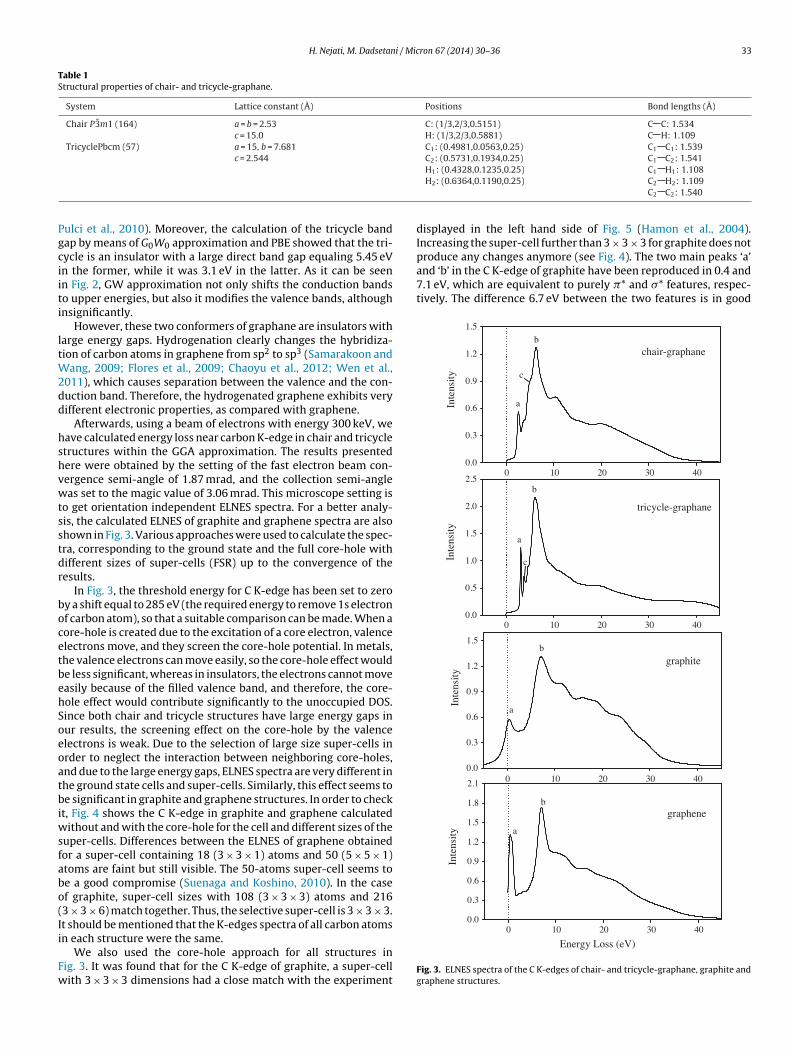
Band structures of chair and tricycle conformers are shown inFig. 2. We used PBE exchange correlation functional and G0W0approximation to calculate band gaps of the chair and the tricy-cle. Our results based on PBE functional showed that chair is an
insulator with a direct band gap of 3.23 eV at � point. This prop-erty in G0W0 approximation was calculated as 5.85 eV which is nearto 5.4–6.05 eV obtained from other GW0 and G0W0 approximations(Karlickyı et al., 2012; Lebégue et al., 2009; Leenaerts et al., 2010;onformations. The carbon atoms are shown in yellow and the hydrogen atoms inred to the web version of this article.)
Ene
rgy
(eV
)
-8
-6
-4
-2
0
2
4
6
8
Γ ZYT Γ T
EF
FT (blue) and many body G0W0 (red). (For interpretation of the references to color
H. Nejati, M. Dadsetani / Micron 67 (2014) 30–36 33
Table 1Structural properties of chair- and tricycle-graphane.
System Lattice constant (Å) Positions Bond lengths (Å)
Chair P3m1 (164) a = b = 2.53c = 15.0
C: (1/3,2/3,0.5151)H: (1/3,2/3,0.5881)
C C: 1.534C H: 1.109
TricyclePbcm (57) a = 15, b = 7.681c = 2.544
C1: (0.4981,0.0563,0.25)C2: (0.5731,0.1934,0.25)H : (0.4328,0.1235,0.25)
C1 C1: 1.539C1 C2: 1.541C H : 1.108
Pgciiti
ltW2dd
hshvwtsstdr
bocetbehSoeoatbiwsfabo(Ii
Fw
produce any changes anymore (see Fig. 4). The two main peaks ‘a’and ‘b’ in the C K-edge of graphite have been reproduced in 0.4 and7.1 eV, which are equivalent to purely �* and �* features, respec-tively. The difference 6.7 eV between the two features is in good
403020100
Inte
nsity
0.0
0.3
0.6
0.9
1.2
1.5
1.8
2.1
403020100
Inte
nsity
0.0
0.3
0.6
0.9
1.2
1.5
chair-graphane
403020100
Inte
nsity
0.0
0.3
0.6
0.9
1.2
1.5
graphite
graphene
a
b
a
b
b
a
403020100
Inte
nsity
0.0
0.5
1.0
1.5
2.0
2.5
tricycle-graphane
a
b
c
c
ulci et al., 2010). Moreover, the calculation of the tricycle bandap by means of G0W0 approximation and PBE showed that the tri-ycle is an insulator with a large direct band gap equaling 5.45 eVn the former, while it was 3.1 eV in the latter. As it can be seenn Fig. 2, GW approximation not only shifts the conduction bandso upper energies, but also it modifies the valence bands, althoughnsignificantly.
However, these two conformers of graphane are insulators witharge energy gaps. Hydrogenation clearly changes the hybridiza-ion of carbon atoms in graphene from sp2 to sp3 (Samarakoon and
ang, 2009; Flores et al., 2009; Chaoyu et al., 2012; Wen et al.,011), which causes separation between the valence and the con-uction band. Therefore, the hydrogenated graphene exhibits veryifferent electronic properties, as compared with graphene.
Afterwards, using a beam of electrons with energy 300 keV, weave calculated energy loss near carbon K-edge in chair and tricycletructures within the GGA approximation. The results presentedere were obtained by the setting of the fast electron beam con-ergence semi-angle of 1.87 mrad, and the collection semi-angleas set to the magic value of 3.06 mrad. This microscope setting is
o get orientation independent ELNES spectra. For a better analy-is, the calculated ELNES of graphite and graphene spectra are alsohown in Fig. 3. Various approaches were used to calculate the spec-ra, corresponding to the ground state and the full core-hole withifferent sizes of super-cells (FSR) up to the convergence of theesults.
In Fig. 3, the threshold energy for C K-edge has been set to zeroy a shift equal to 285 eV (the required energy to remove 1s electronf carbon atom), so that a suitable comparison can be made. When aore-hole is created due to the excitation of a core electron, valencelectrons move, and they screen the core-hole potential. In metals,he valence electrons can move easily, so the core-hole effect woulde less significant, whereas in insulators, the electrons cannot moveasily because of the filled valence band, and therefore, the core-ole effect would contribute significantly to the unoccupied DOS.ince both chair and tricycle structures have large energy gaps inur results, the screening effect on the core-hole by the valencelectrons is weak. Due to the selection of large size super-cells inrder to neglect the interaction between neighboring core-holes,nd due to the large energy gaps, ELNES spectra are very different inhe ground state cells and super-cells. Similarly, this effect seems toe significant in graphite and graphene structures. In order to check
t, Fig. 4 shows the C K-edge in graphite and graphene calculatedithout and with the core-hole for the cell and different sizes of the
uper-cells. Differences between the ELNES of graphene obtainedor a super-cell containing 18 (3 × 3 × 1) atoms and 50 (5 × 5 × 1)toms are faint but still visible. The 50-atoms super-cell seems toe a good compromise (Suenaga and Koshino, 2010). In the casef graphite, super-cell sizes with 108 (3 × 3 × 3) atoms and 2163 × 3 × 6) match together. Thus, the selective super-cell is 3 × 3 × 3.t should be mentioned that the K-edges spectra of all carbon atoms
n each structure were the same.We also used the core-hole approach for all structures inig. 3. It was found that for the C K-edge of graphite, a super-cellith 3 × 3 × 3 dimensions had a close match with the experiment
1
H2: (0.6364,0.1190,0.25)1 1
C2 H2: 1.109C2 C2: 1.540
displayed in the left hand side of Fig. 5 (Hamon et al., 2004).Increasing the super-cell further than 3 × 3 × 3 for graphite does not
Energy Loss (eV)
Fig. 3. ELNES spectra of the C K-edges of chair- and tricycle-graphane, graphite andgraphene structures.

34 H. Nejati, M. Dadsetani / Mic
403020100
Inte
nsity
0
1
2
3
Energy Loss (eV)
403020100
Inte
nsity
0
1
2
3
cellsupercell 2*2*1supercell 3*3*1
cellsupercell 2*2*1supercell 2*2*4
supercell 5*5*1
grap hene
graphite
supercell 3*3*3
without core- hole
with core -hole :
without core-h ole
wit h core-h ole :
superc ell 3*3*6
Fps
a(pfaKF
2Ts(ibhaigets
ig. 4. C K-edge ELNES of graphite (top) and graphene (bottom) structures: com-aring the calculations without and with core-hole for cell and different sizes of theuper-cells.
greement with previous theoretical and experimental reportsHamon et al., 2004; Titantah and Lamoen, 2004). The equivalenteaks exist in the graphene ELNES structure but with a 6.2 eV dif-erence between them. The reproduced features of graphene are ingreement with Suenaga et al.’s experimental work (Suenaga andoshino, 2010) as it has been presented in the right hand side ofig. 5.
The first feature in the ELNES of chair (tricycle), i.e. ‘a’, is at.55 (3.1) eV and the second feature, i.e. ‘b’, is at 6.25 (6.05) eV.herefore, the difference between two main peaks, �* and �*, con-iderably decreases in the hydrogenated graphene structure to 3.72.95) eV. This difference indicates that the bindings are changedn the presence of hydrogen atoms. In a graphene layer, each car-on atom is connected to others with sp2 hybridization. Whenydrogen atoms are added to a carbon layer, hybridization mustpparently be changed from sp2 to sp3. Moreover, C C lengthsn both graphane structures are larger than those in graphite and
raphene (see Table 1). It can justify the decreased energy differ-nce between �* and �* features of graphane conformers relativeo it in graphite and graphene. This effect has been observed in thetudies on the changes of ELNES with respect to the bond length inFig. 5. Experimental carbon K-edge ELNES spectra of graphite (left) and graph
ron 67 (2014) 30–36
carbon systems by Titantah and Lamoen (2005). They showed thatwhen the C C bond length is changed to larger values in graphiteand the amorphous carbon, the �* edge shifts to lower energy,while the �* edge is less sensitive to it. It is found that when car-bon is bonded to a foreign atom like hydrogen or nitrogen the shiftto lower energies can be drastic, resulting in an entirely differentenergy loss near edge structure, or near-edge X-ray absorption finestructure, at energies intermediate between the main �* and �*features. In all structures, the intensity of the first peak is less thanthe second one.
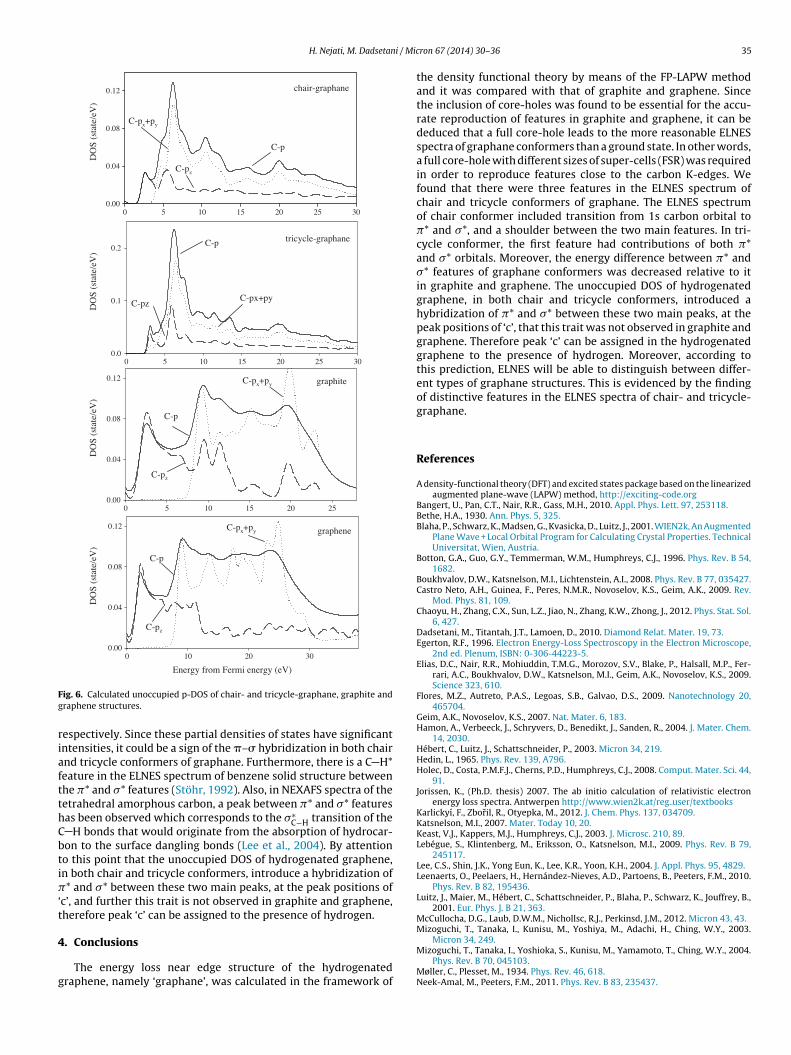
Fig. 6 shows the site and symmetry-projected DOS above theFermi energy of chair, tricycle, graphite and graphene structures,which directly interprets the ELNES of C K-edges. Since the matrixelements in Eq. (1) smoothly vary with respect to the energy, DDSCSis correlated with the unoccupied density of states of the excitedatom, projected on the proper symmetries allowed by the dipoleselection rules (Eq. (2)).
C p-DOS in chair graphane has a significant intensity at 2.55 eV,so peak ‘a’ observed in the same energy loss of C K-edge of chairgraphane could be considered as a transition to antibinding �* (pz)of the carbon atom. Similarly, peak ‘b’ in chair structure can be dueto transition to the antibinding �* (more px + py), because of themost intense peak in the partial DOS of 6.2 eV.
The symmetry-projected density of states in tricycle conformerhas a rather different behavior in comparison with the other threestructures, in a way that, in this structure, the first peak in C p-DOS at 3.1 eV is composed of not only �*(pz) orbitals, but also thecontribution of the �*(more px + py) is not zero. It has a significantDOS value, as compared to �*. Peak ‘a’ in the ELNES of tricycle con-former is due to the transition to both unoccupied �* and �* orbital,because of the common coincidence of ELNES and unoccupied DOS.Thus, peak ‘a’ in the tricycle structure can be attributed to a compo-sition of sp2 and sp3 hybridizations of bonds between each carbonatom with three carbons and one hydrogen atom in the nearestneighborhoods, whereas the first peak observed in the ELNES ofgraphite and graphene and chair-graphane, is the �*orbital becauseof sp2 hybridization of carbon atoms. Like the other three struc-tures, peak ‘b’ is composed of more �* and less �* orbitals.
When we look at Fig. 3, we can see that there is a feature at 4.95(3.8) eV between �* and �* peaks in the C K-edge of chair (tricy-cle) graphane, which was not observed in the C K-edge of graphiteand graphene. The origin of this feature as a rather broad shoul-der in chair (a small peak in tricycle) at the lower energy part of�*, that is ‘c’, is explained by a careful study of the unoccupied Cp-DOS of chair and tricycle graphane (see Fig. 6). The unoccupiedC p-DOS of chair (tricycle) shows a similar peak around 5 (3.8) eV,
corresponding to the position of feature ‘c’ in the C K-edge. At thisenergy in the unoccupied DOS of the chair (tricycle) conformer,the contribution of local DOS is composed of pz and px + py statesof carbon atoms equal to 0.027 (0.02) and 0.03 (0.016) states/eV,ene (right) structures (Suenaga and Koshino, 2010; Hamon et al., 2004).
H. Nejati, M. Dadsetani / Mic
3020100
DO
S (s
tate
/eV
)
0.00
0.04
0.08
0.12
C-p
graphene
2520151050
DO
S (s
tate
/eV
)
0.00
0.04
0.08
0.12
C-p
graphit e
Energy from Fermi energy (eV)
C-pz
C-px+py
C-pz
C-px+py
302520151050
DO
S (s
tate
/eV
)
0.00
0.04
0.08
0.12
C-px+py
C-pz
C-p
chair-gra pha ne
tricycle -graph ane
302520151050
DO
S (s
tate
/eV
)
0.0
0.1
0.2C-p
C-px+pyC-pz
Fg
riaftthCbti�‘t
4
g
ig. 6. Calculated unoccupied p-DOS of chair- and tricycle-graphane, graphite andraphene structures.
espectively. Since these partial densities of states have significantntensities, it could be a sign of the �–� hybridization in both chairnd tricycle conformers of graphane. Furthermore, there is a C H*eature in the ELNES spectrum of benzene solid structure betweenhe �* and �* features (Stöhr, 1992). Also, in NEXAFS spectra of theetrahedral amorphous carbon, a peak between �* and �* featuresas been observed which corresponds to the �∗
C−H transition of the H bonds that would originate from the absorption of hydrocar-on to the surface dangling bonds (Lee et al., 2004). By attentiono this point that the unoccupied DOS of hydrogenated graphene,n both chair and tricycle conformers, introduce a hybridization of* and �* between these two main peaks, at the peak positions of
c’, and further this trait is not observed in graphite and graphene,herefore peak ‘c’ can be assigned to the presence of hydrogen.
. Conclusions
The energy loss near edge structure of the hydrogenatedraphene, namely ‘graphane’, was calculated in the framework of
ron 67 (2014) 30–36 35
the density functional theory by means of the FP-LAPW methodand it was compared with that of graphite and graphene. Sincethe inclusion of core-holes was found to be essential for the accu-rate reproduction of features in graphite and graphene, it can bededuced that a full core-hole leads to the more reasonable ELNESspectra of graphane conformers than a ground state. In other words,a full core-hole with different sizes of super-cells (FSR) was requiredin order to reproduce features close to the carbon K-edges. Wefound that there were three features in the ELNES spectrum ofchair and tricycle conformers of graphane. The ELNES spectrumof chair conformer included transition from 1s carbon orbital to�* and �*, and a shoulder between the two main features. In tri-cycle conformer, the first feature had contributions of both �*and �* orbitals. Moreover, the energy difference between �* and�* features of graphane conformers was decreased relative to itin graphite and graphene. The unoccupied DOS of hydrogenatedgraphene, in both chair and tricycle conformers, introduced ahybridization of �* and �* between these two main peaks, at thepeak positions of ‘c’, that this trait was not observed in graphite andgraphene. Therefore peak ‘c’ can be assigned in the hydrogenatedgraphene to the presence of hydrogen. Moreover, according tothis prediction, ELNES will be able to distinguish between differ-ent types of graphane structures. This is evidenced by the findingof distinctive features in the ELNES spectra of chair- and tricycle-graphane.
References
A density-functional theory (DFT) and excited states package based on the linearizedaugmented plane-wave (LAPW) method, http://exciting-code.org
Bangert, U., Pan, C.T., Nair, R.R., Gass, M.H., 2010. Appl. Phys. Lett. 97, 253118.Bethe, H.A., 1930. Ann. Phys. 5, 325.Blaha, P., Schwarz, K., Madsen, G., Kvasicka, D., Luitz, J., 2001. WIEN2k, An Augmented
Plane Wave + Local Orbital Program for Calculating Crystal Properties. TechnicalUniversitat, Wien, Austria.
Botton, G.A., Guo, G.Y., Temmerman, W.M., Humphreys, C.J., 1996. Phys. Rev. B 54,1682.
Boukhvalov, D.W., Katsnelson, M.I., Lichtenstein, A.I., 2008. Phys. Rev. B 77, 035427.Castro Neto, A.H., Guinea, F., Peres, N.M.R., Novoselov, K.S., Geim, A.K., 2009. Rev.
Mod. Phys. 81, 109.Chaoyu, H., Zhang, C.X., Sun, L.Z., Jiao, N., Zhang, K.W., Zhong, J., 2012. Phys. Stat. Sol.
6, 427.Dadsetani, M., Titantah, J.T., Lamoen, D., 2010. Diamond Relat. Mater. 19, 73.Egerton, R.F., 1996. Electron Energy-Loss Spectroscopy in the Electron Microscope,
2nd ed. Plenum, ISBN: 0-306-44223-5.Elias, D.C., Nair, R.R., Mohiuddin, T.M.G., Morozov, S.V., Blake, P., Halsall, M.P., Fer-
rari, A.C., Boukhvalov, D.W., Katsnelson, M.I., Geim, A.K., Novoselov, K.S., 2009.Science 323, 610.
Flores, M.Z., Autreto, P.A.S., Legoas, S.B., Galvao, D.S., 2009. Nanotechnology 20,465704.
Geim, A.K., Novoselov, K.S., 2007. Nat. Mater. 6, 183.Hamon, A., Verbeeck, J., Schryvers, D., Benedikt, J., Sanden, R., 2004. J. Mater. Chem.
14, 2030.Hébert, C., Luitz, J., Schattschneider, P., 2003. Micron 34, 219.Hedin, L., 1965. Phys. Rev. 139, A796.Holec, D., Costa, P.M.F.J., Cherns, P.D., Humphreys, C.J., 2008. Comput. Mater. Sci. 44,
91.Jorissen, K., (Ph.D. thesis) 2007. The ab initio calculation of relativistic electron
energy loss spectra. Antwerpen http://www.wien2k.at/reg user/textbooksKarlickyı, F., Zboril, R., Otyepka, M., 2012. J. Chem. Phys. 137, 034709.Katsnelson, M.I., 2007. Mater. Today 10, 20.Keast, V.J., Kappers, M.J., Humphreys, C.J., 2003. J. Microsc. 210, 89.Lebégue, S., Klintenberg, M., Eriksson, O., Katsnelson, M.I., 2009. Phys. Rev. B 79,
245117.Lee, C.S., Shin, J.K., Yong Eun, K., Lee, K.R., Yoon, K.H., 2004. J. Appl. Phys. 95, 4829.Leenaerts, O., Peelaers, H., Hernández-Nieves, A.D., Partoens, B., Peeters, F.M., 2010.
Phys. Rev. B 82, 195436.Luitz, J., Maier, M., Hébert, C., Schattschneider, P., Blaha, P., Schwarz, K., Jouffrey, B.,
2001. Eur. Phys. J. B 21, 363.McCullocha, D.G., Laub, D.W.M., Nichollsc, R.J., Perkinsd, J.M., 2012. Micron 43, 43.Mizoguchi, T., Tanaka, I., Kunisu, M., Yoshiya, M., Adachi, H., Ching, W.Y., 2003.
Micron 34, 249.Mizoguchi, T., Tanaka, I., Yoshioka, S., Kunisu, M., Yamamoto, T., Ching, W.Y., 2004.
Phys. Rev. B 70, 045103.Møller, C., Plesset, M., 1934. Phys. Rev. 46, 618.Neek-Amal, M., Peeters, F.M., 2011. Phys. Rev. B 83, 235437.

3 i / Mic
N
N
PP
RRS
6 H. Nejati, M. Dadsetan
elhiebel, M., Louf, P.H., Schattschneider, P., Blaha, P., Schwarz, K., Jouffrey, B., 1999.Phys. Rev. B 59, 12807.
ovoselov, K.S., Geim, A.K., Morozov, S.V., Jiang, D., Zhang, Y., Dubonos, S.V., Gre-gorieva, I.V., Firsov, A.A., 2004. Science 306, 666.
erdew, J.P., Burke, K., Ernzerhof, M., 1996. Phys. Rev. Lett. 77, 3865.
ulci, O., Gori, P., Marsili, M., Garbuio, V., Seitsonen, A.P., Bechstedt, F., Cricenti, A.,Del Sole, R., 2010. Phys. Stat. Sol. A 207, 291.ez, P., Bruley, J., Brohan, P., Payne, M., Garvie, L.A.J., 1995. Ultramicroscopy 59, 159.unge, E., Gross, E.K.U., 1984. Phys. Rev. Lett. 52, 997.amarakoon, D.K., Wang, X., 2009. ACS Nano 3, 4017.
ron 67 (2014) 30–36
Segall, M.D., Lindan, P.J.D., Probert, M.J., Pickard, C.J., Hasnip, P.J., Clark, S.J., Payne,M.C., 2002. J. Phys. Condens. Matter 14, 2717.
Sofo, J.O., Chaudhari, A.S., Barber, G.D., 2007. Phys. Rev. B 75, 153401.Stöhr, J., 1992. NEXAFS Spectroscopy. Springer-Verlag, Berlin.Suenaga, K., Koshino, M., 2010. Nature 468, 1088.
Titantah, J.T., Lamoen, D., 2004. Phys. Rev. B 70, 075115.Titantah, J.T., Lamoen, D., 2005. Phys. Rev. B 72, 193104.Titantah, J.T., Jorissen, K., Lamoen, D., 2004. Phys. Rev. B 69, 125406.Wen, X.D., Hand, L., Labet, V., Yang, T., Hoffmann, R., Ashcroft, N.W., Oganov, A.,Lyakhov, A., 2011. Proc. Natl. Acad. Sci. U.S.A. 108, 6833.