electrochemical characterization of optically passive cevo4 counterelectrodes
TRANSCRIPT

Electrochemical characterization of optically passive CeVO4
counterelectrodes
G. Picardia,*, F. Varsanoa, F. Deckera, U. Opara-Krasovecb, A. Surcab,B. Orelb
aChemistry Department, University of Roma ``La Sapienza'', I-00185 Roma, ItalybNational Chemistry Institute, Hajdrihova 19, SI-1001 Ljubljana, Slovenia
Received 7 September 1998; received in revised form 23 November 1998
Abstract
Ceria±vanadia stoichiometric oxides represent a new class of optically passive counterelectrodes with favorableproperties for the use as counterelectrodes in an electrochromic window. They are very transparent both in thepristine state and in the lithium-intercalated condition and have a good charge capacity (around 20 mC cmÿ2). Inthe present paper we report on the electrochemical properties of Ceria±Vanadia stoichiometric oxides, in liquid PC±
LiClO4 anhydrous electrolytes. The cyclic voltammograms of such electrodes show reversible reduction/oxidationpeaks in two distinct regions, around 1.2±2.2 V vs. Li and 2.9±3.9 V vs. Li. GITT experiments have beenperformed, with the following results: Li di�usion coe�cient goes from 10ÿ10 for pristine samples down to 10ÿ13
cm2 sÿ1 for fully intercalated samples. The impedance of Ceria±vanadia is that of a ceramic thin ®lm, going from aslightly p-type semiconducting state to a more insulating sample with resistivity of 0.1 GO cm when fully Li-intercalated.
The optical properties of the samples are that of a colorless, very transparent electrode (T>88% in thewavelength range between 450 and 740 nm). Extremely weak cathodic and anodic electrochromism can be detectedin the red and in the violet part of the visible spectrum, respectively. This extremely low absorption is of no harm tothe visible appearance of the samples. # 1999 Elsevier Science Ltd. All rights reserved.
1. Introduction
Ceria±vanadia stoichiometric oxides represent a new
class of optically passive counterelectrodes with favor-
able properties for use as counterelectrodes in an elec-
trochromic (EC) window [1]. In fact, they show not
only a very transparent state both in the pristine and
in the lithium-intercalated condition, but have also a
good charge capacity (around 20 mC cmÿ2). The idea
behind the synthesis of such mixed oxides has been to
combine the excellent stability and transparency of Ce-
oxides with the high ion-storage capacity typical of the
V-oxides, thus balancing the ion-charge capacity
required by WO3, which is the coloring electrode in an
EC device. Other ceria-based ®lms exhibit a character-
istic high transmittance (above 90%), associated with a
reasonable ion-storage capacity and optical passiveness
with respect to charging/discharging reactions, like
CeO2/TiO2 ®lms [2,3]. Comparing Ti-oxide with V-
oxide ®lms as ion-storage electrodes, the last one has
the advantage of very large charge capacities (30±40
mC cmÿ2) and the disadvantage of a strong residual
yellow coloration and of cathodic/anodic electrochro-
Electrochimica Acta 44 (1999) 3157±3164
0013-4686/99/$ - see front matter # 1999 Elsevier Science Ltd. All rights reserved.
PII: S0013-4686(99 )00033-X
* Corresponding author.
E-mail addresses: [email protected] (F. Decker),
[email protected] (B. Orel)

mism [4]. Since mixing of ceria with vanadia can leadto the quenching of the yellow color, typical of thislast oxide, a Ce/V-oxide with the appropriate electro-
chemical insertion properties can result in an excellention-storage transparent electrode.Among various Ce/V-oxide compounds, the in
nature occurring CeVO4 mineral wake®eldite belongsto the rare-earth orthovanadates [5] crystallizing in zir-con type (I41/amd) structure which consists of VO4 tet-
rahedra sharing corners and edges with CeO8
dodecahedra. The chain of CeO8, interrupted by dis-torted VO4 units, extends along the c-crystal axis. This
structure is capable, in principle, of Li+ insertionbecause the channels extending along its c-crystal axishave a diameter close to the ionic radius of the Li+
ion (i.e. 0.6 AÊ ). Moreover, CeVO4 is potentially an
intercalation electrode due to a certain degree of elec-tronic conductivity by valence-band holes. Although apure and stoichiometric form of this compound should
be a ceramic insulator, p-type conductivity has beenmeasured by Rao et al. [6], who attributed this conduc-tivity to an intervalence, thermally activated transition
between Ce4+ and Ce3+ states from atoms in equival-ent lattice sites. X-ray absorption spectra [7] have con-®rmed that nearly all the cerium ions are in the
trivalent state. The Ce4+ acceptor states would there-fore induce the electronic conductivity, which weexpect to be largely independent of the valence changeof the vanadium 5+ ions, which is usually associated
with the Li-intercalation process in the V-oxides.CeVO4 and other rare-earth vandates are also lumi-
nescent materials [8] and potential host lattices for anoptical maser due to their high thermal stability. No
further characterization of their optical and electricalproperties, however, has been reported so far in the lit-erature. To make such studies easier, an appropriate
solid-state reaction synthesis of CeVO4 has beenalready developed by Orel et al. [1]. This synthesis con-sisted of a sol±gel reaction, taking advantage of the
control of the Ce/V molar ratio determined by therelative concentration of the precursors. The sol±gelroute combined with the dip-coating deposition tech-
nique allowed the deposition of uniform and homo-geneous thin ®lms with a speci®c Ce/V ratio and a wellde®ned crystal structure. The structural and spectro-electrochemical properties of such ®lms have been
reported as well, suggesting that CeVO4 is indicated aspassive counter electrode in Li-ion intercalating ECdevices due to its small (0.1±0.5 cm2 Cÿ1) coloration
e�ciency. A representation of the crystal structure oftetragonal CeVO4±W, along the c-crystal axis, is givenin Fig. 1.
A further electrochemical study about Li+ intercala-tion and de-intercalation reactions is the aim of thepresent paper. In the ®eld of the EC devices, in fact,
the lack of reliable, optically passive and transparentcounter-electrodes is still of concern [9]. In order toyield an e�cient counter-electrode for an EC device,ion charge capacity and optical passivity and transpar-
ency are only two among several, stringent require-ments. Vanadia ®lms studied so far, such as mixedNb/V-oxide [10] and Ti/V-oxide [11] are almost opti-
cally passive, good ion-storage electrode, but theiroptical transmittance in the bleached state is too low(75±80%). The ®rst objective of our work, therefore, is
to demonstrate the feasibility of an ion-storage elec-trode with very high transmittance in the bleachedstate. Special attention needs to be paid to the stabilityand to the electrochemical kinetics in order to assess
the durability and the applicability of the counter-elec-trode. The exact Li-ion content of the thin ®lm andthe resulting electrode properties as a consequence of
the intercalation reactions are of preeminent import-ance for future, practical applications. Such issues,about the CeVO4 ®lm electrode material, will be
addressed in this work.
2. Experimental
CeVO4 ®lms were prepared by dip-coating on ITO-glass using an in-house built dipping unit with a vari-
able pulling speed from 1 to 10 cm/min. The dippingbath consisted in V-oxoisopropoxide (Fluka) added toceria sols made by an inorganic precursor
CeCl3 � 7H2O (Fluka) mixed with citric acid in theproportion 1:2 and dissolved in EtOH. The stability of
Fig. 1. Pictorial representation of the crystalline structure of
CeVO4±W (I41/amd), along the c-axis, showing the channels
for intercalation of guest ions.
G. Picardi et al. / Electrochimica Acta 44 (1999) 3157±31643158

such sols is limited to about 48 h, depending on the
vanadium concentration. Gelling time can be extended
by appropriate cooling of the sols down to 58C. After
dipping, the ®lms were dried at 4008C for di�erenttimes. Film thickness was measured with an Alpha
Step 200 pro®lometer. Electrochemical measurements
were performed in an Argon-®lled dry box, or in cells
hermetically sealed under Ar atmosphere, using EG&G
PAR 273 and PAR 362 potentiostat/galvanostat
coupled to a Macintosh CI computer for data record-ing and elaboration. The electrochemical cell consisted
of the working electrode, counter and reference electro-
des of Li metal, immersed in 1 M LiClO4/propylenecarbonate (water free). A HP 8452A diode-array
Spectrophotometer was used for transmittancemeasurements, taking a spectro±electrochemical cell®lled with the above electrolyte as the background. ASolartron Mod. 1250 Frequency Response Analyzer,
coupled with a Solartron Mod. 1286 ElectrochemicalInterface, was used to measure the complex electrodeimpedance, taking a AC modulation amplitude of 10
mV with a frequency range 10 mHz±65 kHz.
3. Results
3.1. Cyclic voltammetry
Typical results of cyclic voltammetries (CV) in thepotential window between 1.2 V and 4.0 V vs. Li metalelectrode (in PC/LiClO4 electrolyte) are reported in
Fig. 2, where the scan rate is the parameter. CV resultsin the potential windows between 1.2 and 2.4 V andbetween 2.4 and 4.0 V, are shown in Fig. 3 and Fig. 4,
respectively. Two electrochemically active regionsappear, one at higher potentials, associated with theinsertion of roughly 0.3 equivalent of Li-ions per oxidemole and a second one at lower potentials, where
further Li intercalation occurs. Three reduction andthree corresponding oxidation peaks have been ident-i®ed and denoted by the letters AA ', BB ' and CC ' inFig. 2. The oxidation peak at 3.0 V seems to be relatedto the occurrence of the reduction reaction appearingas a negative peak at 1.7 V: in fact this oxidation
vanishes unless a potential limit lower than 1.8 V isexceeded in the CV. Such pair BB ' of voltammetricpeaks (1.7 and 3.0 V) tends to grow up linearly, with
Fig. 2. Cyclic voltammograms (in the steady state) of CeVO4
in 1 M LiClO4/PC anhydrous electrolyte, with a Li±metal
reference electrode. Potential window: from 4.0 to 1.2 V.
Lower curve: scan rate 1 mV sÿ1. Higher curve: scan rate 10
mV sÿ1.
Fig. 3. Cyclic voltammograms of CeVO4 in 1 M LiClO4/PC
anhydrous electrolyte, with a Li-metal reference electrode
(scan rate 10 mV sÿ1). Potential window: solid line, from 4.0
to 1.2 V; dashed line, from 2.4 to 1.2 V.
Fig. 4. Cyclic voltammograms of CeVO4 in 1 M LiClO4/PC
anhydrous electrolyte, with a Li-metal reference electrode
(scan rate 10 mV sÿ1). Potential window: solid line, from 4.0
to 1.2 V; dashed line, from 4.0 to 2.4 V.
G. Picardi et al. / Electrochimica Acta 44 (1999) 3157±3164 3159
increasing scan rate, whereas all the remaining peaks
in the CVs have a lower power-law dependence on thescan rate. This di�erence in scan rate dependence altersthe shape of the CVs, making such pair of peaks pre-
dominant in the voltammetries taken with the higherpotential sweep velocity.
3.2. Transmittance variation
Transmittance spectra have been taken at di�erentelectrode potentials in the same window as in the CVs
(from 4.0 to 1.2 V and vice versa), with intervals of 0.2V (Fig. 5). From such spectra a moderate cathodicelectrochromic e�ect appears (vanishes) in the potentialregion corresponding to peak C ' (C), consisting in a
weak absorption a�ecting all wavelengths. Conversely,
some anodic electrochromic e�ect is associated with
peak A, disappearing with the corresponding reductionin A '. The process labelled with the pair of peaks BB 'in the CV, seems to have no e�ect on the transmit-
tance spectra. All the optical changes reported aboveare reversible and reproducible after several polariz-
ation cycles. The maximum transmittance variations in
the visible range (400±650 nm) are of the order of 3%.CeVO4 ®lms can therefore be described as optically
transparent and passive counter electrodes with respectto the electrochemical insertion of Li+. From the
above transmittance spectra, an indirect bandgap
energy of 3.36 eV has been derived by linear ®tting ofthe square root of the ®lm absorbance versus the pho-
ton energy, at the open circuit potential 3.2 V (Fig. 6).
The extinction coe�cient k of the material (not
shown here) was calculated out of transmittance andre¯ectance spectra performed ex situ. On the basis of
this calculation it is evident that the extinction coe�-
cient is not equal to zero in the range 300±900 nm.Besides the fundamental absorption (the one associated
with the band gap) there is a further optical absorption
centered at 2.4 eV, approximately 50 times weaker.This very broad and weak absorption makes uncertain
the determination of the band gap out of the ®tting of
the absorbance as given in Fig. 6.
Nevertheless a reversible shift of the absorbancespectra is always seen associated with the negative cur-
rent peak C ' (Fig. 6). This shift is well known [4] andis characteristic also for other vanadium containing
mixed oxide ®lms (V/Ti, V/Fe, V/Cr). The band gap
Fig. 5. Transmittance spectra vs. wavelength, measured in situ
during Li insertion performed in 1 M LiClO4/PC anhydrous
electrolyte by potential step experiments.
Fig. 6. (ahv )1/2 versus photon energy for a CeVO4 ®lm, from
transmittance spectra in Fig. 5. Linear extrapolation yields
the bandgap energy resulting from indirect transition.
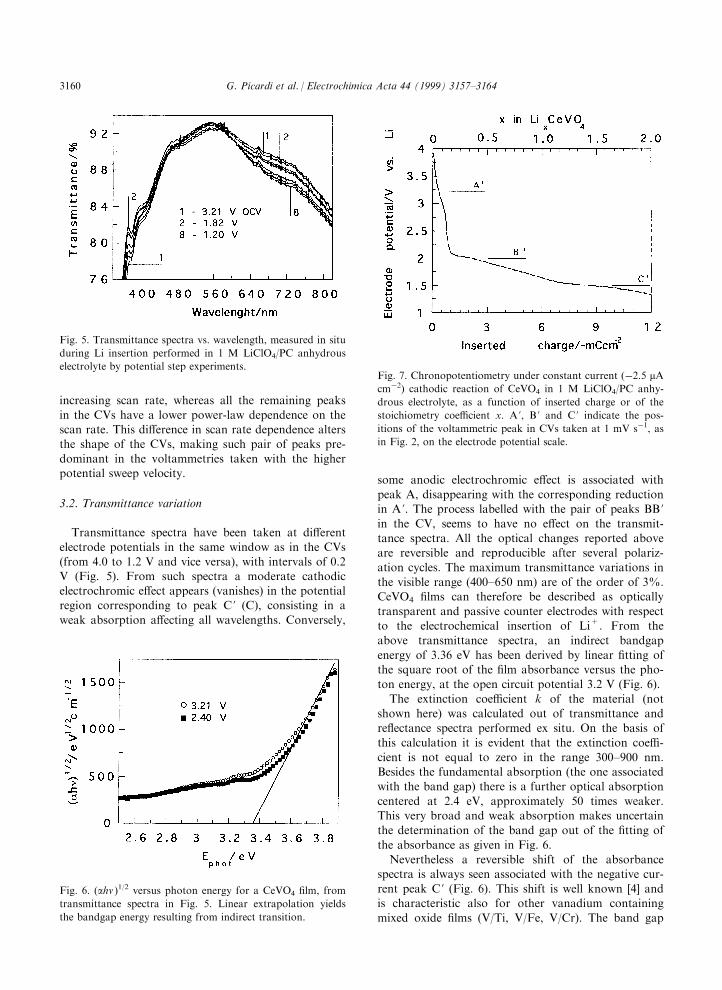
Fig. 7. Chronopotentiometry under constant current (ÿ2.5 mAcmÿ2) cathodic reaction of CeVO4 in 1 M LiClO4/PC anhy-
drous electrolyte, as a function of inserted charge or of the
stoichiometry coe�cient x. A ', B ' and C ' indicate the pos-
itions of the voltammetric peak in CVs taken at 1 mV sÿ1, asin Fig. 2, on the electrode potential scale.
G. Picardi et al. / Electrochimica Acta 44 (1999) 3157±31643160

widening was explained in terms of valence theoryonly for V2O5. No data are available about the elec-tronic structure of other mixed V-containing oxides.
3.3. Chronopotentiometry and GITT
The electrode potential has been measured as a func-
tion of time during constant current insertion at a veryslow rate, such as the chronopotentiometry experimentreported in Fig. 7 (current density 2.5 mA cmÿ2). Theplateaux in such plot (portions of the curve with the
shallower slope) appear at electrode potentials corre-
sponding to the potentials of the peaks A ', B ' and C 'in the CVs taken with the lower scan rate. The steeper
slope in the curve of Fig. 7 occurs after insertion of1.5 mC cmÿ2 (at around 2.5 V). From a simple calcu-lation of the Li+ charge inserted (supposing all the
current to be used for the faradic reaction) and of the®lm electrode mass (taking 4.76 g/cm3 as the density of
the oxide) it follows that the composition of the com-pound is: (i) Li0.4CeVO4 after the higher potential
region during insertion; (ii) Li1CeVO4 after reaction B 'during insertion; (iii) Li2CeVO4 at the end point of thegalvanostatic insertion.
The galvanostatic intermittent titration technique(GITT) [12] was performed with 300 mA current pulses
of the duration of 3 s, followed by relaxation intervalsof the duration of 7200 s, on ®lm electrodes that have
been already charged/discharged several times in theLi+-containing liquid electrolyte. Li di�usion coe�-
cient D in the CeVO4 ®lm was calculated for a ®lm110 nm thick using the same equations as in Ref. [12].
The calculated D ranges from an higher value of 10ÿ10
cm2 sÿ1, when the insertion reaction begins, to a lowervalue of 10ÿ13 cm2 sÿ1, at the end point of the inser-
tion reaction (see Fig. 8).The thermodynamic curve where the equilibrium cell
potential is reported as a function of the insertedcharge shows the same features as the galvanostatic
discharge curve taken at low current density, i.e. theslopes and the three plateaux observed in Fig. 7.
In another experiment with the same sample, butunder current pulses of 100 mA, the equilibrium poten-
tials after relaxation and the calculated di�usion coe�-cients are almost the same as in the ®rst GITT
experiment reported above.
Fig. 8. GITT results from experiments performed in 1 M
LiClO4/PC anhydrous electrolyte. Current steps from 0 to
ÿ300 mA cmÿ2, of 3 s duration, each followed by a potential
relaxation of 120 min. Di�usion coe�cients calculated as indi-
cated in Ref. [12].
Fig. 9. (a) Imaginary vs. real component of impedance (Nyquist plots); CeVO4 in 1 M LiClO4/PC anhydrous electrolyte at three
di�erent potentials, after equilibrium is reached. (b) magni®cation of the part of the same Nyquist plot, for 100<Z '<300 Ohms;
0<ÿZ0<200 O.
G. Picardi et al. / Electrochimica Acta 44 (1999) 3157±3164 3161

3.4. Film resistance and impedance
The LixCeVO4 resistance between the ITO substrateand an In disklet (geometric area 1 mm2) pressed onthe outer ®lm surface has been measured in a glove
box by means of an high internal resistance ohmmeter.The ®lm was cleaned and dried after the equilibrium atvarious inserted charges was reached. Typical resist-
ance values of 8 MO (which would correspond to anoxide resistivity of 1010 O cm) were measured, exceptfor the region of lower potentials (peaks A and A ' inthe CVs), where the resistance was found to increaseto 14 MO. An increase of the oxide resistance withlithium (an electron donor) concentration is in agree-ment with a mechanism of charge compensation within
the p-type semiconductor behavior of the Ce/V-oxide,due to insertion. Such a ®lm resistivity as the above,however, seems unreasonably high for this oxide and
would be detrimental to the EC device operationbecause of the associated IR drop, especially whenlarge currents are required. To check the above resis-
tivity values, AC electrochemical impedance spectrawere measured in the frequency range from 5 mHz to65 kHz, with a ®lm 30 nm thick with 1 cm2 of geo-
metric area. Impedance spectroscopy is widely used toextract the series resistance of the cell and the chemicaldi�usion coe�cient for the Li+-electron couple in theelectrode where insertion takes place [13]. The results
are shown in the form of Nyquist plots in Fig. 9. The®lm resistance can be taken from the high frequencylimit of such spectra and ranges from a minimum of
120 O to a maximum of 180 O depending of the degreeof Li+ insertion. These values correspond to oxide res-istivity values from 107 O cm to 108 O cm, a reason-
able value for such a high bandgap semiconductor,besides showing the expected trend with the charge ofthe Li+ inserted. The discrepancy between the lowerresistivity obtained with the electrolyte contact and the
higher one measured with the solid indium contact canbe explained in terms of the real areas of the twodi�erent contacts. The ratio of the two contact sur-
faces would be of 500 in favor of the liquid contact,after normalizing for the same geometrical area.Although a high crystallinity of the ®lm (grain dimen-
sions 0280 AÊ ) is not in favor of porosity such a largedi�erence in surface area between solid and liquid con-tact is not surprising.
The low frequency part of the impedance plots inFig. 9 are straight lines with a slope slightly largerthan one. This indicates a semi-in®nite mass-transportcontrol (due to Li+ di�usion in the ®lm) and a certain
e�ect of the ®lm ®nite capacity. The capacitive beha-vior associated with the ®nite dimension of the ®lm didnot appear clearly in the impedance spectra, because
of the low frequency limit of the apparatus and of apossible Li+ insertion in the underlying ITO current
collector. Further measurements, however, are in pro-gress to study such e�ects in more detail.
3.5. X-ray structural analysis of powders and ®lms
CeVO4 mineral Wake®eldite (CeVO4±W) [1,5]belongs to the class of rare-earth orthovanadates.These, with the exception of LaVO4 [14], crystallize in
a zircon type tetragonal crystal system with a spacegroup of I41/amd. The XRD spectra of Ce/V (1:1)oxide powder heat-treated for 1 h reveal the simul-
taneous formation of the CeVO4±H and CeVO4±Wphases. The latter phase prevails and, to expectation,the extinction of the monoclinic CeVO4±H phaseoccurs in powders heat-treated for 4 h. The appearance
of a single CeVO4±W phase is accompanied by anincrease in grain size from 300 to 500 AÊ . The XRDspectra of CeVO4 ®lms con®rm the results obtained
from powders indicating that the formation of thephases in the ®lms and powders is similar, with theexception that the tetragonal CeVO4±W phase in the
®lms develops faster; after 30 min of heat-treatment awell crystalline CeVO4±W phase could be observed.The tetragonal CeVO4±W phase prevails in the ®lms,
with the amount of monoclinic CeVO4±H phase beingmuch smaller relative to that noted in the XRD spec-tra of powders.
4. Discussion and conclusions
The Li ion-storage capacity of the CeVO4 ®lms hasbeen proven by CV, galvanostatic discharge, GITTand potentiostatic experiments. For a 30 nm-thick
®lm, this charge capacity is typically of 10 mC cmÿ2
and increases with ®lm thickness. According to thesol±gel preparation technique described elsewhere [1],the ion-storage capacity can be increased proportion-
ally with the ®lm thickness. provided only one or twodipping cycles are used for ®lm deposition. CeVO4 ®lmcounterelectrodes, however, can store enough Li charge
to drive the clectrochromic reaction of a WO3 ®lm,with comparable thickness and area, between bleachedand fully colored states. The maximum guest ion con-
centration, from our experiments, is 2 Li equivalentsper mole of the stoichiometric CeVO4±W oxide, pro-vided slow charge/discharge processes are allowed.
Di�usion times of the order of 10 s or more areexpected, in fact, to equilibrate concentration di�er-ences across a 100 nm CeVO4 thick ®lm, consideringthat the di�usion coe�cient is only 10ÿ12 cm2 sÿ1.More work on the ®lm preparation procedures will beneeded, therefore, in order to improve the ionic mobi-lity in the oxide by means of an appropriate ®lm mor-
phology and crystallinity.The spectral coloration e�ciencies calculated for the
G. Picardi et al. / Electrochimica Acta 44 (1999) 3157±31643162

CeVO4 ®lms are as low as few cm2 Cÿ1 in all the vis-
ible range. Only in the UV region (l<450 nm), largerabsorbance changes (about ÿ40 cm2 Cÿ1) areobserved, due mainly to energy bandgap shifts upon
charge insertion. The optical transmittance of the ®lmsin the visible region (450 nm< l<650 nm) is between92% and 88%. Such optical data rank the CeVO4 as
one of the most promising transparent counter-elec-trode materials with an optically passive response
reported so far.As far as the redox processes associated with Li+
insertion in CeVO4 are concerned, we can separate (at
least) two di�erent steps in the reaction mechanism, inagreement with the high- and the low-potential regions
appearing in the CVs. X-ray absorption measurements[7] and IR spectroscopy on tetragonal CeVO4±W haverevealed that only Ce3+ and not Ce4+ are present in
the structure of the pristine oxide. The peak C 'observed at higher potentials in the CVs, consequently,can be attributed to a V5+/V4+ redox process invol-
ving only part of the vanadium ions. The cyclic vol-tammograms, indeed, show a striking similarity with
the CV response of crystalline vanadium pentoxide®lms [15]. Further Li-ion insertion and di�usion is as-sociated with the group of the voltammetric peaks tak-
ing place at lower potentials. Several mechanisms canoccur here, i.e. the reduction of V4+ to V3+ and of the
remaining V5+ to V4+ and to V3+. Nevertheless, theabove sequence is not straightforward and a phasetransition with the participation of cerium ions to
allow further Li-insertion cannot be excluded a priori.The insertion of Li+ in Ce-oxide has been associatedwith the appearance of a voltammetric peak at 1.5 V
vs. Li [16], similar to that present in our CVs (Fig. 2).In fact, the availability of a high density of Ce 4f states
in the middle of the band gap of the Ce-oxide shouldgive a peak in the CV [17] and in the derivative takenfrom the composition±potential curve (ÿdx/dE ). A
sharp maximum in such derivative has been calculatedfrom our chronopotentiometry experiments in Fig. 7,occurring for x>1. At such level of insertion, however,
domains with a lower occupancy of Li ions are in equi-librium with domains with a higher occupancy, like in
a multi-phase system. In a crystalline oxide with theMO2 structure inserted with Li+ to a level correspond-ing to a Li/M ratio >0.5, othorhombic distortion of
the starting tetragonal structure is likely to occur [18].Such distortion has been associated with the fact that
Li ions start occupying the tetrahedral sites, after hav-ing ®lled all the octahedral sites available for insertionand the energetics of the inserted electrons is changed.
The coexistence of two di�erent phases, one with theoriginal tetragonal structure and a second one, withhigher Li content and already distorted, is the most
reasonable explanation for the levelling of the electrodepotential around 1.5 V in chronopotentiometry (Fig. 7)
[19]. This explanation of the electrochemical behavior,in terms of structural phase changes of the material,
does not account for the exact oxidation state of thedi�erent metal ions in the oxide material. Although wecannot account for the exact reduction process, the
®nal oxidation states of the metal ions should be Ce3+
and V3+ in the fully intercalated oxide.Considering de-insertion reactions, the removal of
structural distortions is a possible reason for the widepotential di�erence between peak B ' and its corre-sponding B counterpart in CV (Fig. 2), which is a sig-
nature for electrochemical irreversibility and a premisefor decrease in the ion storage capacity of the material.The structural changes during insertion/de-insertion inour samples have not been experimentally investigated
so far, but it has been observed that excessively largecrystalline grains or too high Li charges lead to irre-versible changes in electrochemical properties.
However, visual inspection of the ®lms has shown nooptical defects in the ®lms and their adherenceremained una�ected after several hundred cycles. Also
CVs show little variation in shape after cycling, withsharp peaks denoting the maintenance of an orderedcrystal structure of the ®lm. The charge capacity
decreases upon cycling, however, by about 10% after300 cycles. It is not yet clear if such degradation, prob-ably associated with changes in oxide ®lm structure,can be prevented with an appropriate heat-treatment
following sol±gel deposition.In conclusion, crystalline CeVO4 ®lms obtained by
the sol±gel method have been shown to behave as
transparent, optically passive counterelectrodes for ECdevices, with a ion-storage capacity exceeding 20 mCcmÿ2 (300 mC nmÿ1 cmÿ2). The ®lm behaves as a cer-
amic, p-type semiconductor with high resistivity (107 Ocm to 108 O cm), showing a Li-ion di�usivity in therange 10ÿ11 cm2 sÿ1 to 10ÿ13 cm2 sÿ1. Further work isin progress in order to improve the insertion kinetics
and the long term durability of the electrode. In par-ticular, the incorporation of structural Li in the oxidestructure, with the addition of appropriate precursors
in the sol±gel bath composition, has been recentlytested, seeming to improve the long term electrochemi-cal reversibility, without altering the other electrode
properties.
Acknowledgements
This work was supported by The Ministry ofScience and Technology of Slovenia and The
Ministries of Foreign A�airs of Italy and Slovenia.Part of the work was completed in the framework ofthe COST Action 518 from the EU.
G. Picardi et al. / Electrochimica Acta 44 (1999) 3157±3164 3163

References
[1] U. Opara-Krasovec, B. Orel, R. Reisfeld, Electrochem.
Solid-State Lett. 1 (1998) 104.
[2] D. Ke omany, J.-P. Petit, D. Deroo, Proc. SPIE 2255
(1994) 363.
[3] M. Stromme Mattsson, A. Azens, G.A. Niklasson, C.G.
Granqvist, J. Appl. Phys. 81 (1997) 6432.
[4] A. Talledo, C.G. Granqvist, J. Phys. D.: Appl. Phys. 27
(1994) 2445.
[5] C. Baudracco-Gritti, S. Quartieri, G. Vezzalini, F.
Permingeat, F. Pillard, R. Rinaldi, Bull. Mine ral 110
(1987) 657.
[6] N. Suresh Rao, O.G. Planna, Bull. Mater. Sci. 18 (1995)
593.
[7] R.F. Reidy, K.E. Swider, J. Am. Ceram. Soc. 78 (1995)
1121.
[8] L.H. Brixner, E. Abramson, J. Electrochem. Soc. 112
(1965) 70.
[9] C.G. Granqvist, Handbook of Inorganic Electrochromic
Materials, Elsevier, Amsterdam, 1995.
[10] S.F. Cogan, R.D. Rauch, N.M. Nguye, T.D. Plante,
J.D. Westwood, J. Electrochem. Soc. 140 (1993) 112.
[11] H. Hiroshima, S. Kamimura, Mater. Res. Soc. Symp.
Proc. 121 (1988) 779.
[12] W. Weppner, R.A. Huggins, J. Electrochem. Soc. 124
(1977) 1569.
[13] W. Weppner, in: P.G. Bruce (Ed.), Solid State
Electrochemistry, Cambridge Univ. Press, Cambridge,
1993, Chap. 8.
[14] V.S. Stubican, R. Roy, Z. Kristalographie 119 (1963)
90.
[15] J. Livage, Solid State Ionics 86±88 (1995) 935.
[16] S.-Y. Zhang, A.M. Andersson, B. Stjerna, C.G.
Granqvist, Appl. Opt. 32 (1993) 6303.
[17] P. Baudry, A.C.M. Rodriguez, M.A. Aegerter, L.O.
BulhoÄ es, J. Non-Cryst. Solids 121 (1990) 319.
[18] B. Zachau-Christiansen, K. West, S. Skaarup, Solid
State Ionics 53±56 (1992) 364.
[19] K. West, B. Zachau-Christiansen, T. Jacobsen, S.
Skaarup, in: G. Nazri, J. Tarasco, M. Armand (Eds.),
Solid State Ionics, III, Materials Research Soc,
Pittsburg, PA, 1993, pp. 9±47.
G. Picardi et al. / Electrochimica Acta 44 (1999) 3157±31643164