egfr signals downregulate tumor suppressors mir-143 and ... › content › molcanres › 9 › 7...
TRANSCRIPT

Signaling and Regulation
EGFR Signals Downregulate Tumor Suppressors miR-143 andmiR-145 in Western Diet–Promoted Murine Colon Cancer:Role of G1 Regulators
Hongyan Zhu1, Urszula Dougherty1, Victoria Robinson2, Reba Mustafi1, Joel Pekow1, Sonia Kupfer1,Yan-Chun Li1, John Hart3, Kathleen Goss2, Alessandro Fichera2, Loren Joseph3, and Marc Bissonnette1
AbstractEpidermal growth factor receptors (EGFR) contribute to colonic tumorigenesis in experimental models
of colon cancer. We previously showed that EGFR was also required for colonic tumor promotion byWestern diet. The goal of this study was to identify EGFR-regulated microRNAs that contribute to diet-promoted colonic tumorigenesis. Murine colonic tumors from Egfrwt and hypomorphic Egfrwa2 mice werescreened using micro RNA (miRNA) arrays and miR-143 and miR-145 changes confirmed by Northern,real-time PCR, and in situ analysis. Rodent and human sporadic and ulcerative colitis (UC)-associated coloncancers were examined for miR-143 and miR-145. Effects of EGFR on miR-143 and miR-145 expressionwere assessed in murine and human colonic cells and their putative targets examined in vitro and in vivo.miR-143 and miR-145 were readily detected in normal colonocytes and comparable in Egfrwt and Egfrwa2
mice. These miRNAs were downregulated in azoxymethane and inflammation-associated colonic tumorsfrom Egfrwt mice but upregulated in Egfrwa2 tumors. They were also reduced in human sporadic andUC colon cancers. EGFR signals suppressed miR-143 and miR-145 in human and murine colonic cells.Transfected miR-143 and miR-145 inhibited HCT116 cell growth in vitro and in vivo and downregu-lated G1 regulators, K-Ras, MYC, CCND2, cdk6, and E2F3, putative or established targets of thesemiRNAs. miRNA targets Ras and MYC were increased in colonic tumors from Egfrwt but not Egfrwa2 micefed a Western diet. EGFR suppresses miR-143 and miR-145 in murine models of colon cancer.Furthermore, Western diet unmasks the tumor suppressor roles of these EGFR-regulated miRNAs. MolCancer Res; 9(7); 960–75. �2011 AACR.
Introduction
Colon cancer is a leading cause of cancer-related deaths inthe Western world (1). Tumors arise from colonic epithelialcells transformed by growth-promoting mutations (2). Inprior studies, we showed that epidermal growth factorreceptor (EGFR) signals were required for efficient colonictumorigenesis and tumor progression in the azoxymethane(AOM) and azoxymethane/dextran sulfate sodium (AOM/DSS) models of sporadic and inflammation-associated
colon cancer, respectively (3–5). In the AOM/DSS study,we examined the role of EGFR and diet in colonic tumor-igenesis, comparing Egfr wild-type (Egfrwt) mice to micehomozygous for hypomorphic waved-2 Egfr mutations(Egfrwa2) that abrogate more than 90% of receptor kinaseactivity in vitro (6). In this model, we showed that tumorpromotion by Western diet required EGFR signals (4). Inthe presence of wild-type EGFR, Western diet upregulatedproto-oncogenes MYC and K-Ras (4, 7). These proto-oncogenes control several G1 cell-cycle regulators and theirdysregulations play critical roles in colonic tumor develop-ment in humans and experimental animals (8, 9).Micro RNAs (miRNA) are small, noncoding RNAs that
regulate gene expression by base pairing with mRNAs,leading to mRNA destabilization or inhibition of mRNAtranslation (10). In normal cells, miRNAs control numer-ous processes including stem cell fate, proliferation, anddifferentiation (11). Because in general, miRNAs targetmultiple mRNAs, individual miRNA could potentiallyalter complex cellular processes such as cell growth andapoptosis. Aberrant miRNA levels occur in many tumorsincluding colon cancer (12). Some miRNAs that are lost
Authors' Affiliations: Departments of 1Medicine, 2Surgery, and 3Patho-logy, University of Chicago, Chicago, Illinois
Note: Supplementary data for this article are available at Molecular CancerResearch Online (http://mcr.aacrjournals.org/).
H. Zhu and U. Dougherty contributed equally to the work.
Corresponding Author: Marc Bissonnette, Department of Medicine,University of Chicago Hospitals and Clinics, 900 East 57th Street, Chi-cago, IL 60637. Phone: (773) 702-8597; Fax: (773) 702-2281; E-mail:[email protected]
doi: 10.1158/1541-7786.MCR-10-0531
�2011 American Association for Cancer Research.
MolecularCancer
Research
Mol Cancer Res; 9(7) July 2011960
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

during tumorigenesis seem to function as tumor suppres-sors, whereas other miRNAs are upregulated and mightmediate proto-oncogenic signals. EGFR was recently shownto suppress several miRNAs that target proto-oncogenictranscription factors (13). MYC and Ras that are down-stream effectors of EGFR in colonic tumorigenesis havebeen shown to regulate miRNA expression (14, 15). Therole of miRNAs in mediating more proximal EGFR onco-genic effects, including tumor-promoting effects of Westerndiet, however, has not been examined in colonic tumor-igenesis. The availability of sporadic and inflammation-associated colonic tumors from Egfrwt and Egfrwa2 micefed standard or Western diet allowed us to ask whetherEGFR and diet might regulate miRNAs in colonic tumor-igenesis. For these studies, we examined tumors induced byAOM or AOM/DSS and also examined adenomas from Apcmutant Min mice, a genetic model of colon cancer. Inaddition, we investigated the expression levels of severalorthologues of EGFR-regulated miRNAs identified in ourmouse models in human sporadic and inflammation-asso-ciated colon cancers.We identified miR-143 and miR-145 as negatively regu-
lated EGFR targets by comparing their expression patternsin AOM and AOM/DSS-induced colonic tumors fromEgfrwt and Egfrwa2mice. ThesemiRNAs form a polycistroniccluster located on human chromosome 5 and mouse chro-mosome 18. Prior investigations reported that these miR-NAs were downregulated in human colon cancers (15, 16).Recent studies show that these miRNAs regulate embryonicand smooth muscle stem cell fates (17, 18). Furthermore, invitro studies showed that transfected miR-143 or miR-145inhibited the growth of colon cancer cells, consistent withtheir putative roles as tumor suppressors (19). These miR-NAs were later noted to be decreased in other tumorsincluding breast, lung, bladder, and prostate cancers (20–23). The availability of tumors from our experimentalmodels provided an opportunity to investigate EGFR andWestern diet regulation of demonstrated or in silico predictedtargets of these miRNAs in colonic malignant transforma-tion. We identified several putative targets of these miRNAsas G1 regulators controlled by EGFR, thus linking EGFRregulation of miRNAs to cell-cycle control in colonic tumor-igenesis. Our findings also suggest that tumor suppressoreffects of miR-143 and miR-145 are uncovered in thepresence of a Western diet that promotes colonic tumor-igenesis and upregulates targets of these miRNAs that are nolonger restrained in the absence of these miRNAs.
Materials and Methods
MaterialsAOM was obtained from Midwest Research, the
National Cancer Institute Chemical Carcinogen ReferenceStandard Repository. Standard chow diet and Western dietwere prepared as described (4). FokI restriction enzyme forwaved-2 genotyping was purchased from New EnglandBiolabs. EGF was purchased from Calbiochem. C225anti-EGFR neutralizing antibodies were obtained from
ImClone. Gefitinib was provided by AstraZeneca. Polyclo-nal antibodies to extracellular signal–regulated kinase(ERK5; #3372) were obtained from Cell Signaling. Anti-bodies to K-Ras, (SC-522), MYC (SC-40, clone 9E10),CCND2 (SC-181), cdk6 (SC-177), and E2F3 (SC-879)were obtained from Santa Cruz Biotechnology. Rabbitpolyclonal (#160106) and monoclonal (160112) anti-PTGS2 antibodies were purchased from Cayman Chemi-cals. MAP/ERK kinase (MEK2) antibodies were purchasedfrom Epitomics. Monoclonal antibodies against Ki67 (cloneSP1) were obtained from Neomarkers. Monoclonal ACTB(b-actin) antibodies were purchased from Sigma-Aldrich.Mimics of mature miR-143 and miR-145 were purchasedfrom Ambion. Custom PCR primers were obtained fromIntegrated DNA Technologies, Inc. Other PCR reagents,including Moloney murine leukemia virus reverse transcrip-tase and random hexamers, were purchased from AppliedBiosystems.
MethodsRodent tissue. We examined tissue obtained from
different studies to address the influence of EGFR onmiRNAs in both sporadic and inflammation-associatedcolonic tumors. These models included AOM/DSS mouse,AOM (mouse and rat), and Min mouse model.AOM/DSS study. Flash-frozen normal mouse colonic
mucosa and AOM/DSS-induced colonic tumors from micefed standard rodent chow or Western diet were availablefrom a prior study (5).AOM study. To examine the effects of Egfr genotype on
miRNAs in a sporadic model of colon cancer, C57BL6/JEgfrwt/wa2 mice were interbred with A/J Egfrwt/wa2 mice togenerate F1 hybrid C57BL6/J � A/J mice. Mice weregenotyped to identify experimental groups Egfrwt andhomozygous Egfrwa2 mice. We induced tumors by weeklyinjections with AOM [12.5 mg intraperitoneally (i.p.)/kgbody weight] � 6 weeks. We used this AOM dose, as thehybrid mice were relatively resistant to AOM.Mice were fedstandard or Western chow (20% fat) diets as described (4).Animals were sacrificed 40 weeks later and tumors wereharvested.AOM rat study. Normal rat colonic mucosa and AOM-
induced colonic tumors from rats fed standard chow orchow supplemented with gefitinib were available from aprevious study (5).Min mouse study. We used frozen Apc mutant Min
adenomas and adjacent normal appearing small bowelmucosa to assess miRNA in the Min model.Human tissue. Human sporadic and ulcerative colitis
(UC)-associated colonic adenocarcinomas and adjacentcolonic mucosa, as well as normal colonic mucosa fromcolons resected for diverticular disease, were obtained fromthe Department of Surgical Pathology at the University ofChicago under an Institutional Review Board-approvedprotocol. Resected tissues were placed in an ice bath andtransported promptly to the Department of Surgical Patho-logy. Representative tumor sections and adjacent normalappearing colonic mucosal sections were dissected free of
EGFR Controls miR-143 and miR-145 in Colonic Tumorigenesis
www.aacrjournals.org Mol Cancer Res; 9(7) July 2011 961
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

underlying muscle and flash frozen in liquid nitrogen. Carewas taken to avoid areas with visible necrosis.miRNA arrays. RNA was extracted using RNeasy Li-
pid Tissue Mini Kit (Qiagen). RNA from murine AOM/DSS tumors was labeled with Hy3 and RNA from controlmucosa was labeled with Hy5 using miRCURY Hy3/Hy5power labeling kit (Exiqon). A separate array was used foreach Hy3-labeled tumor. Hy5-labeled control RNA fromanimals matched for Egfr genotype was spotted on eacharray (n ¼ 4 for each Egfr genotype). Samples were hybri-dized on the miRCURY LNA Array (v.9.2) using a hybri-dization station. The quantified signals (backgroundsubtraction) were normalized using the global lowess(LOcally WEighted Scatterplot Smoothing) regression algo-rithm that utilizes within-slide normalization to minimizedye-dependent differences in intensity. Exiqon carried outthe miRNA labeling, hybridization, and preliminary arrayanalyses.Northern analysis of miRNAs. RNA was transferred to
nylon membranes using a full-immersion system. Oligo-nucleotides were synthesized with locked nucleic acids(LNA; Exiqon) and miRNA antisense probes generatedby oligonucleotide end labeling with [g32P] ATP usingT4 polynucleotide kinase. Filter hybridization was conduc-ted overnight in hybridization solution containing radiola-beled probes (106 cpm/mL), followed by several washeswith 2� SSC/0.1% SDS. Membranes were stripped withseveral washes in 0.1%SDS at 95�C and re-probed with anLNA probe specific for U6 small nuclear RNA (snRNA) as aloading control.Real-time PCR. For mRNA, 1 mg total RNA was
reverse transcribed into cDNA using SuperScript III Plati-num Two-Step qRT-PCR kit in 20 mL total volume. Theresulting first-strand cDNA was used as template for quan-titative PCR in triplicate using SYBR Green QPCR MasterMix kit. Primers were designed using Primer3 and sequencesare available on request (24). Where possible, one of theprimers was designed to span intron–exon junctions. Reversetranscribed cDNA (1 mL of 1:8 dilution) and primerswere mixed with SYBR Green dye I master mixture in finalvolume of 25 mL. Negative controls (reactions lacking eitherreverse transcriptase or template) yielded no PCR products.PrimersandTaqManprobes for themature formsofmiR-143and miR-145 were obtained from ABI and conditions forreverse transcription and PCR followed the manufacturer'srecommendations.Reverse transcriptase reactionswereruninduplicate andPCR reactions in triplicate.Datawere analyzedusing the comparative 2expð�DDCtÞ method (25, 26).mRNA levels were normalized to b-actin and miRNA levelsnormalized toRNU48 (human tissue) or snoRNA202 (mur-ine tissue) and expressed as fold-control (26).In situ hybridization. We carried out in situ staining
for miR-143 and miR-145 in human and mouse tissue fromthe AOM/DSS study using Exiqon miRCURY LNA oli-gonucleotide probes labeled 50 and 30 with digoxigenin(DIG) as described (27, 28). We used an Exiqon probewith a scrambled sequence as a negative control that gave nospecific staining. Briefly, 10 mm cryostat sections were
collected on Superfrost Plus slides and dried for 3 minutesat room temperature (RT) and stored at �80�C untilstaining. Sections were fixed in 4% (w/v) paraformaldehydefor 10 minutes and washed twice in diethyl pyrocarbonate(DEPC)-PBS for 3 minutes. Sections were incubated for 5minutes in acetylation buffer followed by DEPC-PBS rin-ses. Sections were incubated for 60 minutes with biotiny-lated LNA probes (0.025 mmol/L) in hybridization buffer attemperature 25�C below melting temperature (Tm) forLNA probe. Sections were washed 3 times for 10 minutesin 0.1� SSC at 4�C to 8�C above hybridization tempera-ture and then once for 5 minutes in 2� SSC at RT withagitation. Slides were treated for 20 minutes with 3% (v/v)H2O2 at RT and then for 30 minutes at RT in blockingbuffer. Sections were incubated with primary antibodies(100 mL, 1:4,000; anti-DIG/HRP in blocking buffer) for30minutes at RT followed by 3washes inTNTbuffer at RT.For detection, slides were incubated with 1:50 dilutionfluorescein isothiocyanate (FITC)-tyramide (100 mL) inamplification buffer for 10 minutes at RT in the dark. Then,25 mL of ProLong gold antifade reagent was added andsections were secured with glass coverslips. Sections wereimaged using an epifluorescence microscope equipped withcharge-coupled device camera and image analysis software.Cell culture. HCT116 colon cancer cells and CCD-
18Co human colonic fibroblasts were obtained from Amer-ican Type Culture Collection (ATCC). HCA-7 colorectalcancer cells were obtained from Susan Kirkland (ImperialCancer Research Fund, London, UK). Cells were main-tained at 37�C in a humidified atmosphere of 5% CO2–95% air as recommended by ATCC. HCT116 and HCA-7cells were cultured in McCoy's 5A modified medium con-taining 10% serum and CCD-18Co cells were cultured inEagle's minimum essential medium with 15% serum.Young adult mouse colonocytes (YAMC) are a condition-ally immortalized murine colonic epithelial cell line isolatedfrom the H-2Kb-tsA58 mouse expressing a heat-labile SV40large T antigen (29). YAMCs were provided by the Diges-tive Diseases Research Core Center at the University ofChicago and used between passages 25 and 32. Cells weregrown on culture dishes in RPMI 1640 medium underpermissive conditions at 33�C in a humidified atmospherewith 5% CO2 until confluent.Cell treatment. To modulate EGFR signals, cells were
stimulated with EGF (10 ng/mL) or EGFR was blockedwith C225 neutralizing anti-EGFR antibodies (20 mg/mL)or EGFR kinase inhibitor gefitinib (60 nmol/L) for theindicated times. Control cells were treated with PBS. Toexamine the effects of miR-143 and miR-145 on cell growthand to assess putative targets of these miRNAs, cells weretransfected with mimics of mature miR-143 or miR-145(Ambion) at the indicated concentrations. Cell proliferationwas measured using WST-1 assay (Roche) according to themanufacturer's recommendations. DNA synthesis was mea-sured by incorporation of 5-bromo-20-deoxyuridine (BrdU)into proliferating cell DNA using the BrdU ProliferationAssay Kit (EMD Biosciences). For BrdU assays, cells wereplated in sera in 96-well plates (1 � 105 cells per well)
Zhu et al.
Mol Cancer Res; 9(7) July 2011 Molecular Cancer Research962
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

overnight and then serum starved and incubated withindicated reagents for 24 hours. Six hours prior to harvest,BrdU (1:2,000 final dilution) was added to cells. Cells werethen fixed and incubated with anti-BrdU antibodies fol-lowed by horseradish peroxidase (HRP) substrate. Opticaldensities were read at 450 and 540 nm using the SynergyHT Microplate Reader (Bio-Tek). Absorbance differences(450 � 540 nm) were calculated as means � SD andexpressed as percentage of EGF treated.Luciferase 30UTR assays. Cdk6 is a putative miR-145
target, as its 30 untranslated region (UTR)containsa conservedseed match for miR-145 at positions 3,884 to 3,892 (N-M_001259.6).A440-bp fragment flanking this position frombase3,638 to4,078wasPCRamplified fromcDNAgeneratedby reverse-transcriptase PCR using total RNA extracted fromHCT116 cells. For PCR, the following primers were used:(forward) 50-CCCTCGAGGGTCCACAGCATTCAAG-30and (reverse) 50-GCGGCCGCTTCAGAG-AGGCTGA-GAT-30. The PCR product was subcloned into psiCHECK2dual luciferase reporter vector (Promega). Amutant constructthat deleted the 8-bp sequence complementary to the miR-145 seed sequence was generated by 2-step PCRwith forwardprimer 50-AATGCAGCTGTTCTGTTTTTCAGCATT-CTTTAG-30 and reverse primer 50-CTAAAGAATGC-TGAAAAACAGAACAGCTGCATT-30. The constructswere confirmed by DNA sequencing. HCT116 cells werecotransfected with psiCHECK2 (EV) or psiCHECK2 con-taining bases coding for a portion of wild-type or mutant30UTR of cdk6, together with a scrambled oligonucleotide(control) or mimic of mature miR-145 (Ambion). Renillaluciferase activity was normalized to firefly luciferase activity.HCT116 tumor xenografts expressing pre-miR-143
and pre-miR-145. DNA oligonucleotides coding forpre-miR-143 or pre-miR-145 stem loops were subclonedinto pCDH-CMV-MCS-EF1-copGFP (pCDH EV) lenti-viral vector for constitutive expression. Lentivirus was pre-pared in 293T cells by cotransfecting pCDH-miR-143,pCDH-miR-145, or pCDH-EV with vectors expressinggag, rev, and VSV-G. HCT116 cells were transduced withlentivirus expressing pre-miRNA or EV. TransducedHCT116 (5 � 106 cells) were implanted subcutaneouslyinto flanks of nu/numice (5 mice per group). Tumor growthwas monitored and tumors excised 3 week after implanta-tion. Tissue was fixed in 10% buffered formalin or snapfrozen in liquid nitrogen.Western blotting. Proteins were extracted in SDS-con-
taining Laemmli buffer, quantified by RC-DCprotein assay,and subjected to Western blotting as described (30). Briefly,proteins were separated by SDS-PAGE on 4% to 20%resolving PAGE gradient gels and electroblotted to polyvi-nylidene difluoride (PVDF) membranes. Prestained mole-cular markers were included on each gel. Blots wereincubated overnight at 4�C with specific primary antibodiesfollowed by 1-hour incubation with appropriate peroxidase-coupled secondary antibodies that were detected by enha-nced chemiluminescence using X-OMAT film. Xerogramswere digitized using an Epson scanner and band intensityquantified using UN-SCAN-IT gel software (V 5.3, Silk
Scientific). Protein levels in tumors were normalized tob-actin levels and expressed as fold of control colonicmucosa(means � SD), matched for Egfr genotype. Protein lysatesfrom tumors and control colonic mucosa with equal proteinabundance as assessed by RC-DC assays showed comparableb-actin levels by Western blotting. Tumors of comparablestage were used for Western blotting comparisons.Immunostaining. Sections (5 mm) were mounted on
Vectabond-coated Superfrost Plus slides. Sections wereheated to 60�C for 1 hour, deparaffinized by 3 washes �5 minutes in xylene, hydrated in a graded series of ethanolwashes, and rinsed in distilled water. Epitope retrieval forKi67 was achieved by microwave heating for 15 minutes inTris-EDTA buffer, pH 9, followed by 3 washes � 2minutes in TBS with 0.1% Tween-20 (TBST). Endogen-ous peroxidase activity was quenched with methanol/H2O2solution (0.5%). Sections were washed 3 times in TBST �2 minutes and blocked in protein block for 20 minutes.Sections were incubated with 1:300 dilution of anti-Ki67antibodies for 1 hour at RT. After 3 TBST washes, slideswere incubated at RT with 1:200 dilution of biotinylatedsecondary antibodies for 30 minutes. Antigen–antibodycomplexes were detected using an HRP-labeled DAKOEnVisionþ System and 3,30-diaminobenzidine as substrate.For negative controls, sections were incubated with isotype-matched nonimmune antibodies. After washing in distilledwater, slides were stained with Gill's III hematoxylin, rinsedwith water, dehydrated in ethanol, and cleared with xylene.It should be noted that tumors of comparable histology wereused for immunostaining comparisons between Egfrwt andEgfrwa2 mice.Immunostaining quantitation. We detected Ki67 by
nuclear staining quantified by an automated cellular imagingsystem (ACIS; Clarient San Juan). Color-specific thresholdswere used to determine brown (positive) and blue (negative)nuclei within the outlined regions of interest to calculate thefraction of positively stained nuclei. Proliferation was expres-sed as the percentage of nuclei positive for Ki67. At least 5fields per tumor and 3 tumors per group (�50,000 cells percondition) were scanned for quantitation.Statistical methods. Continuous data were expressed as
means � SD and compared using Student's t test. Real-time PCR samples were run in triplicate and Ct valuesaveraged. Untransformed Ct values were compared using anonparametric Wilcoxon test (26). Levels of relative abun-dance, expressed as 2ð�DDCtÞ, were calculated by exponen-tiating Ct differences between individual groups. Values ofP < 0.05 were considered statistically significant. Unlessotherwise indicated, data are representative of at least 3independent experiments.
Results
EGFR is required for miR-143/miR-145downregulation in murine colonic tumorsAOM/DSS model. To identify potential EGFR-regu-
lated miRNAs, we screened AOM/DSS-induced tumorsfrom Egfrwt and Egfrwa2 mice by miRNA arrays. These
EGFR Controls miR-143 and miR-145 in Colonic Tumorigenesis
www.aacrjournals.org Mol Cancer Res; 9(7) July 2011 963
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

tumors were available from a prior study (4). We initiallyfocused on mice fed standard chow to assess EGFR effectswithout Western diet influences (4). Shown in Figure 1Ais a heat map of miRNAs that were altered in tumorscompared with control mucosa. Relative miRNA expres-sion levels in tumors in the Egfrwt and Egfrwa2 mice werenormalized to their Egfr genotype–matched respectivecontrols. We observed that miR-143 and miR-145were downregulated in tumors from Egfrwt mice, whereasthese miRNAs were upregulated in tumors from Egfrwa2
mice. We quantified differences in expression levels ofthese miRNAs by real-time PCR. As shown in Figure 1Band as compared with control colonic mucosa matched forEgfr genotype (control expression levels normalized to 1),mature miR-143 and miR-145 were downregulated morethan 60% in tumors from Egfrwt mice, whereas thesemiRNAs were increased by more than 4-fold in tumorsfrom Egfrwa2 mice.We confirmed our array findings by Northern blot
analysis that showed reduced mature miR-145 (�20bp) in the Egfrwt tumor, whereas mature miR-145 wasincreased in the Egfrwa2 tumor (Fig. 1C). In contrast to thereduction in mature miR-145, in the Egfrwt tumor, theprecursor form (�100 bp) appeared to be increased(Fig. 1C). This could reflect deregulated posttranscrip-tional processing of miRNAs that occurs in cancer (31).Interestingly, mature levels of miR-145 appeared compar-able in mucosa from control Egfrwt and Egfrwa2 mice
(Fig. 1C), suggesting that basal levels of these miRNAsare not controlled by EGFR in the absence of neoplastictransformation. These observations, however, will requirefurther study.
miR-143 and miR-145 are expressed in colonicepithelial cellsTo determine the cell type in the colon expressing miR-
143 and miR-145, we assessed their expression by in situhybridization. As shown in Figure 2, miR-143 and miR-145 were predominantly expressed in colonic epithelial cellsespecially on the colonic surface and maturation zone of thecrypts in both Egfrwt and Egfrwa2 mice (see insets in Fig. 2B,C, H, and I). These miRNAs were downregulated in tumorsfrom Egfrwt mice (Fig. 2E and F) but preserved in tumorsfrom Egfrwa2 mice (Fig. 2K and L). Consistent with priorreports of miR-143 and miR-145 expression in muscle (17,32), these miRNAs were also expressed in colonicmuscularismucosa and muscularis propria, albeit at lower levels (e.g.,miR-143 in the muscle layers in Fig. 2B, white arrows).Although expression levels of miR-143 and miR-145 in
Egfrwt and Egfrwa2 tumors were significantly different, thesemiRNA differences did not translate into differences intumor multiplicity or progression in animals on standardchow (4). Rather, the effects of EGFR on tumorigenesiswere uncovered in the presence of a Western diet (4). Wenext examined these miRNAs in tumors from animals onWestern diet. As in the case of tumors from mice on
Figure 1. EGFR controls miR-143 and miR-145 expression in murine colonic tumors. A, miRNA heat map. Unsupervised clustering of miRNAs in colonictumors induced by AOM/DSS in Egfrwt and Egfrwa2 mice on standard chow. RNA was extracted from tumors and control mucosa. Normal colonic mucosalRNA was labeled with Hy5 and tumor RNAwith Hy3. Tumor and control RNA (matched for Egfr genotype) were hybridized to miRCURY LNA arrays. Quantifiedsignals were normalized using the global lowess regression algorithm. Scale of relative intensity (log2) is shown below heat map. Green indicates expressionlevels below control mucosa and red indicates expression levels greater than control mucosa. Note that miR-143 and miR-145 shown in bold weredownregulated in tumors from Egfrwtmice and upregulated in tumors from Egfrwa2mice. B, real-time PCR analysis of miR-143 and miR-145. Mature miRNA intumors from Egfrwt and Egfrwa2 mice induced by AOM/DSS were quantified by real-time PCR as described in Materials and Methods. Black bars are tumorsfrom Egfrwt mice and gray bars are tumors from Egfrwa2 mice (n ¼ 4 tumors per group, with each tumor from a separate mouse; * and†, P < 0.05compared with normal mucosa). Note miRNA expression levels in tumors were normalized to control mucosa indicated by the dotted horizontal line (Control).C, Northern blot analysis of miR-145. RNA was extracted from Egfrwt and Egfrwa2 tumors (T) and Egfr genotype–matched normal colonic mucosa (N). RNA (2.5mg) was separated on 15% denaturing PAGE and transferred to nylon membranes. LNA oligonucleotide probes were end labeled with [g32P] ATP usingT4 polynucleotide kinase. Filter hybridization was conducted overnight in solution containing 106 cpm/mL probe, followed by washes in 2� SSC/0.1% SDS.Specific miRNA probes were stripped from membranes in 0.1% SDS at 95�C and membranes reprobed with LNA specific for U6 snRNA as a loading control.
Zhu et al.
Mol Cancer Res; 9(7) July 2011 Molecular Cancer Research964
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

standard chow, miR-143 (Fig. 3A) and miR-145 (Fig. 3B)were downregulated in tumors from Egfrwt mice and upre-gulated in tumors from Egfrwa2mice.Western diet modestlyreduced the upregulation of these miRNAs in Egfrwa2
tumors compared with standard diet (Fig. 3). Taken
together, Egfr genotype and to a lesser extent, diet modu-lated expression of these miRNAs in tumors.AOM mouse model. We next assessed changes in
miR-143 and miR-145 in the AOM model of sporadiccolon cancer to dissect the role of inflammation in the
Figure 2. miR-143 and miR-145 are expressed in colonic epithelial cells and downregulated in tumors from Egfrwt but not Egfrwa2 mice. Cryosectionsfrom normal colon from Egfrwt (A) and Egfrwa2 (G) mice were stained with H&E. Tumors from Egfrwt (D) and Egfrwa2 mice (J) were also stained with H&E.Tumor regions are indicated with T and normal mucosa with N. Adjacent cryosections were stained for miR-143 (B, E, H, and K) and miR-145 (C, F, I, and L) asdescribed in Materials and Methods. miRNA staining from digoxigenin-labeled probes is shown in green and 40,6-diamidino-2-phenylindole (DAPI)-stainednuclei are blue. Micrographs are at 10� and insets are 63�. Scrambled control showed no specific staining. Note loss of miR-143 and miR-145 expression intumor from Egfrwt mouse (E and F) compared with tumor from Egfrwa2 mouse (K and L). In Egfrwt tumor, miR-143 and miR-145 are present in adjacentmucosa (see insets for Fig. 2E and F). White arrows in 2B indicate muscularis mucosa (top arrow) and muscularis propria (bottom arrow). Note lower miR-143expression in muscle compared with epithelial cells.
EGFR Controls miR-143 and miR-145 in Colonic Tumorigenesis
www.aacrjournals.org Mol Cancer Res; 9(7) July 2011 965
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

downregulation of these miRNAs. As in the case of theAOM/DSS model, a Western diet promoted tumordevelopment in Egfrwt but not in Egfrwa2 mice (Fig. 4-A). Similar to our findings in the AOM/DSS model,these miRNAs were downregulated in AOM-inducedtumors from Egfrwt mice and upregulated in AOM tu-mors from Egfrwa2 mice on standard chow (Fig. 4B).These results indicate that downregulation of thesemiRNAs was not dependent on inflammation inducedby DSS but mediated by increased EGFR signals inmalignant transformation.AOM rat model and Min mouse model. In addition to
AOM and AOM/DSS mouse models, we examined theAOM rat model and Min mouse model of colon cancerto assess the generality of our findings regarding changes inthese miRNAs. Using tumors obtained from a prior study(5), we found that these miRNAs were also decreasedin colonic tumors from AOM-treated rats. Furthermore,gefitinib, an EGFR kinase inhibitor partially preserved thesemiRNAs in rat tumors at a dose that concomitantly inhibitedtumorigenesis (5). ThesemiRNAs also appeared to be down-regulated in the Apc mutant Min mouse model of inheritedcolon cancer (Fig. 4D). In this regard, EGFR signals havebeen shown to be upregulated in Min adenomas (33).
miR-143 and miR-145 are downregulated in humansporadic and UC-associated colon cancersIn agreement with others (16, 34–36), we showed by
Northern blot analysis that miR-143 and miR-145 weredownregulated in sporadic human colon cancers comparedwith adjacent normal-appearing colonicmucosa (Fig. 5A andSupplementary Fig. S1). We also assessed miR-145 expres-sion levels by in situ hybridization in normal human colonand sporadic colon cancers. Hematoxylin and eosin (H&E)-
stained sections are shown in Figure 5B (i) and (iii). As in themouse, miR-145 was abundant in colonic epithelial cells inthe normal human colonic mucosa [Fig. 5B (iii)] but down-regulated in colonic tumors [Fig. 5B (iv)]. Because AOM/DSS is a murine model of inflammation-associated coloncancer, we examined these miRNAs in human colon cancersassociated with UC. As shown in Figure 5C, miR-143 andmiR-145were significantly downregulated inUC-associatedcancers. Thus, in agreement with our findings in the mouseAOM and AOM/DSS models, miR-143 and miR-145 weredownregulated in both sporadic and colitis-associated humancolon cancers. Interestingly, mucosa adjacent to UC tumorsalso appeared to have reductions in thesemiRNAs, suggestinga tumor-related field effect, or an effect of UC on thesemiRNAs as tumors arose in a field of quiescent UC.We havesince verified in a larger study that these miRNAs are down-regulated inUC compared with normal colonic mucosa (37).In this regard, UC is a premalignant condition associatedwith increased colon cancer risk.
EGFR signals suppress miR-143 and miR-145expression in colonic cellsAs Egfr genotype controlled miR-143 and miR-145
expression in colonic tumors, we asked whether EGFRsignals could regulate these miRNAs in vitro in cell culture.As shown in Figure 6, EGF treatment of HCT116 cells didnot significantly suppress levels of miR-143 or miR-145.These miRNAs were already significantly downregulated inHCT116 cells compared with normal colonic tissue (38).Because EGFR is constitutively activated by autocrinesignals in HCT116 cells, we next blocked these upregulatedsignals with C225 anti-EGFR neutralizing antibodies.EGFR blockade significantly increased miR-143 andmiR-145 compared with vehicle-treated HCT116 cells.
Figure 3. EGFR negatively regulates miR-143 and miR-145 in AOM/DSS tumors from animals fed standard or Western diet. RNA was extracted fromtumors from a prior AOM/DSS study (4). miR-143 and miR-145 were measured by real-time PCR as described in Materials and Methods. A, miR-143.Values were normalized to fold of control mucosa from saline-treated mice matched for Egfr genotype and diet (n ¼ 4 tumors and 4 matched controls ineach group). B, miR-145. * and †, P < 0.05, compared with Egfr genotype–matched control mucosa; z, P < 0.05, compared with tumors from Egfrwa2 mice onstandard diet. Ctl, control.
Zhu et al.
Mol Cancer Res; 9(7) July 2011 Molecular Cancer Research966
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

In contrast to HCT116 cells, basal EGFR activation is lowin human CCD-18Co colonic fibroblasts. We, therefore,activated this receptor with EGF and showed that EGFRsignals significantly suppressed these miRNAs in colonicfibroblasts (Fig. 6). We also examined YAMCs, an immor-talized murine colonic epithelial cell line with low basalEGFR signals (29). EGF treatment suppressed thesemiRNAs in this nontumorigenic cell (Fig. 6). Takentogether, our studies indicate that EGFR signals down-regulate these miRNAs in colonic cells.
Transfected miR-143/miR-145 inhibit EGF-inducedproliferation and DNA synthesisTo assess the growth consequences that accompany loss of
miR-143 and miR-145 in colonic tumorigenesis, we trans-fected mimics of mature miR-143 or miR-145 intoHCT116 colon cancer cells. These transfected miRNAsdid not alter basal proliferation but inhibited EGF-induced
cell proliferation (Fig. 7A). Presumably, targets of thesemiRNAs do not regulate basal proliferation. Alternatively,compensating mechanisms might be activated in the pre-sence of transfected miRNAs. These transfected miRNAsalso inhibited EGF-induced DNA synthesis (Fig. 7B), sug-gesting that EGFR-induced proliferation is mediated in partby downregulating these miRNAs in colonic tumorigenesis.Identification of G1 regulators as miR-143/145 targets
in vitro. We searched miRNA target databases and priorreports for potential targets of miR-143 and miR-145 thatmight mediate the antiproliferative effects of these miRNAs.We focused on G1 cell-cycle regulators implicated as in silicotargets, as miR-143 and miR-145 strongly inhibited DNAsynthesis (Fig. 7B). We identified K-Ras (miR-143) andMYC, CCND2, cdk6, and E2F3 (miR-145) as putativetargets, as they possessed 30UTR sequences that comple-mented seed sequences for these miRNAs. K-Ras and MYCwere previously identified as targets of miR-143 and
Figure 4. EGFR controls diet-related tumor promotion and downregulates miR-143 and miR-145 in the AOM model. A, tumor incidence. C57BL6/J � A/Jhybrid mice with Egfrwt or Egfrwa2 genotype were treated with AOM (12.5 mg/kg body weight) or saline (AOM vehicle) weekly � 6 week. Animals werefed standard or Western diet beginning 2 week after AOM treatment and sacrificed 40 week later. Diets were described in a prior study (4). Shown aretumor incidences for adenomas and cancers. (N ¼ 56 Egfrwt/þ and N ¼ 15 Egfrwa2 mice; * and †, P < 0.05 compared with Egfrwt mice on standard diet).B, miR-143 and miR-145 in AOM tumors. RNA was extracted from AOM-induced tumors from mice on standard chow. Indicated mature miRNA werequantified by real-time PCR. Black bars are tumors from Egfrwt mice and gray bars are tumors from Egfrwa2 mice (n¼ 4 tumors in each group; * and †, P < 0.05compared with normal mucosa). Note miRNA expression levels in tumors were normalized to control mucosa indicated by the dotted horizontal line(Control). C, miR-143 and miR-145 in AOM rat tumors. Values were normalized to fold of control mucosa from saline-treated rats matched for gefitinibsupplemented or unsupplemented diets (n¼ 4 tumors and matched controls in each group. *, P < 0.05, compared with control mucosa; †, P < 0.05, comparedwith AOM alone). D, miR-143 and miR-145 in Min adenomas. Values were averages of 2 tumors expressed as fold of adjacent small bowel mucosa.
EGFR Controls miR-143 and miR-145 in Colonic Tumorigenesis
www.aacrjournals.org Mol Cancer Res; 9(7) July 2011 967
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

miR-145 (39, 40), respectively but not studied in an in vivomodel of colonic tumorigenesis. As shown in Figure 7C(left), transfected miR-145 inhibited protein expression ofMYC, cdk6, CCND2, and E2F3 cell-cycle regulators inHCT116 cells. To assess the potential direct regulation ofcdk6 by miR-145, we cloned a portion of cdk6 30UTR thatcontained the target sequence complementing the miR-145seed sequence, downstream of a luciferase reporter(Fig. 7D). We also mutated the 30UTR to abrogate thepredicted miR-145-cdk6 30UTR interaction. As shown inFigure 7E, miR-145 significantly inhibited the expression ofRenilla luciferase regulated by wild-type cdk6-30UTR butnot mutant cdk6-30UTR. We examined the effect of EGFon transcripts of these targets and found that EGF signifi-cantly induced cdk6 and E2F3 mRNA in HCT116 cells,whereas gefitinib blocked these inductions (SupplementaryData and Fig. S2). As noted earlier, however, EGF did notfurther suppress miR-143 andmiR-145 in these cells, whichwere already significantly downregulated compared with
normal colonocytes. Based on these results, we speculatethat miR-143 and miR-145 downregulations contribute totumorigenesis but are not sufficient for induction of theseG1 regulators in transformed colon cancer cells.We also investigated the effects of miR-143 transfection
on K-Ras, MEK2, ERK5, and PTGS2, as these proteinsare demonstrated or predicted targets of this miRNA (39,41–43). For this purpose, we selected HCA-7 cells, whichexpress PTGS2 protein. (HCT116 cells express onlyPTGS2 transcripts). In agreement with these predictions,transfected miR-143 downregulated K-Ras, MEK2, ERK5,and PTGS2 in HCA-7 cells compared with a controloligonucleotide (with irrelevant sequence; Fig. 7C, right).
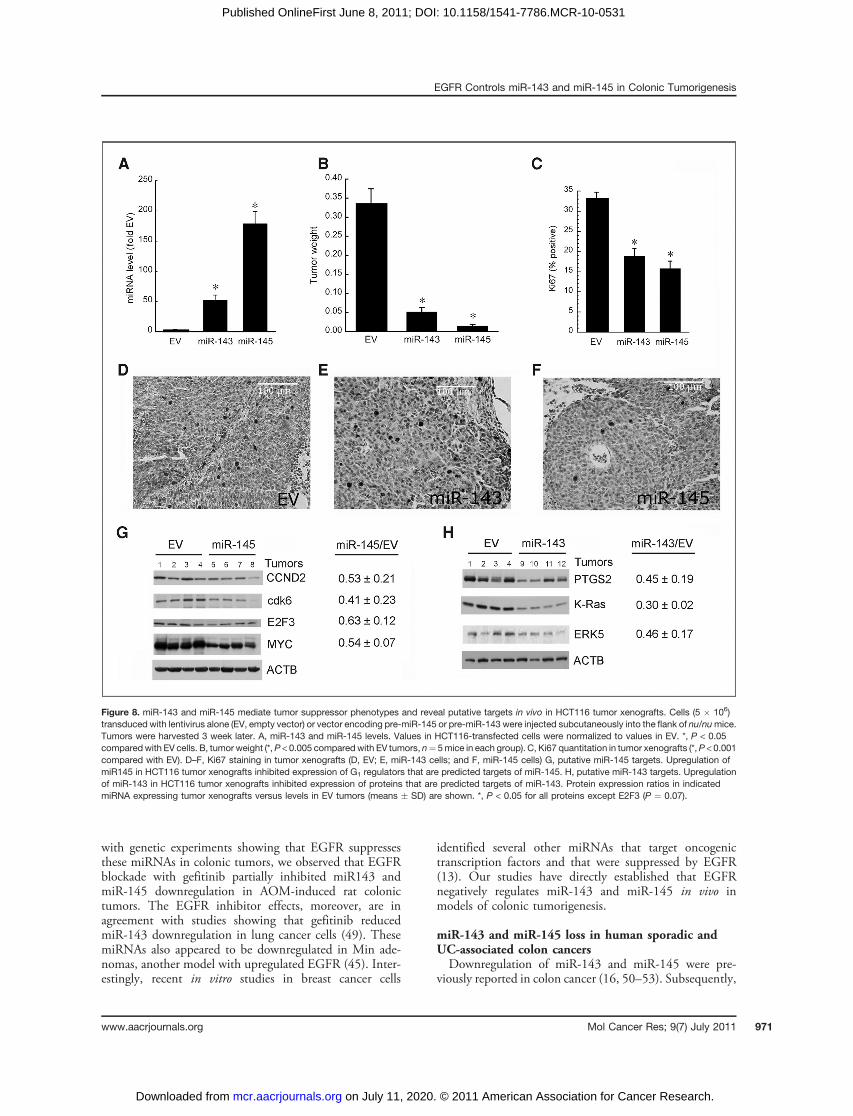
miR-143 and miR-145 are tumor suppressors in coloncancer cell xenograftsOur cell culture experiments suggested that miR-143 and
miR-145 drive growth-inhibiting signals mediated at leastin part by suppressing G1 cell-cycle regulators. Our animal
A
B
C
Figure 5. miR-145 is downregulated in sporadic and inflammation-associated human colon cancer. A, miR-145 Northern blot analysis. RNA wasextracted from colon cancers (T) and adjacent mucosa (N). RNA (7.5 mg) was separated on 15% denaturing PAGE and transferred to nylon membranes.LNA oligonucleotide probes were end labeled with [g32P] ATP using T4 polynucleotide kinase and Northern blots prepared as described inMaterials and Methods. B, miR-145 in situ expression. Frozen optimum cutting temperature (OCT)-embedded colonic tumors and adjacent normalappearing mucosa were fixed in 4% paraformaldehyde for H&E staining and for miRNA detection by in situ staining as described in Materialsand Methods. i, H&E-stained normal colon. ii, H&E-stained colon cancer. iii, miR-145 in normal colon. iv, miR-145 in colon cancer. Digoxigenin-labeledmiRNA is shown in green (white arrow) and DAPI-stained nuclei in blue. C, miR-143 and miR-145 are downregulated in UC-associated colon cancers.RNA was extracted from flash-frozen UC-associated tumors and adjacent mucosa as well as normal control colons (resected for diverticulosis)and miR-143 and miR-145 were measured by real-time PCR. n ¼ 3 control colons and n ¼ 3 UC-associated colonic cancers and adjacent mucosa.*, P < 0.05 compared with normal colonic mucosa.
Zhu et al.
Mol Cancer Res; 9(7) July 2011 Molecular Cancer Research968
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

studies suggested that miR-143 and miR-145 may be tumorsuppressors under Western diet conditions. To directlyassess the tumor suppressor phenotype of these miRNAsin vivo, we transduced HCT116 cells with lentivirus alone(EV) or lentivirus-expressing pre-miR-143 or pre-miR-145.We confirmed that mature miR-143 and miR-145 weresignificantly upregulated in pre-miRNA–transduced cells,compared with EV-transduced HCT116 cells (Fig. 8A).Upregulation of these miRNAs decreased tumor xenograftweight supporting their postulated tumor suppressor phe-notype (Fig. 8B). Consistent with cell-cycle effects of thesemiRNAs in cell culture, cells transfected with pre-miRNAsformed tumors with significantly reduced Ki67 immunos-taining (Fig. 8C). Representative tumors stained for Ki67are shown in Figure 8D–F.
Transfected miR-143/miR-145 downregulate putativetargets in tumor xenograftsTo identify possible proteins mediating the growth-
suppressive effects of these miRNAs in vivo, we examinedexpression levels of putative targets in tumor xenografts.As shown in Figure 8G, protein levels of CCND2, cdk6,E2F3, and MYC were downregulated in tumor xenograftsfrom miR-145 expressing cells compared with EV-trans-duced HCT116 cells. These G1 regulators were notsignificantly altered in tumor xenografts from miR-143–transduced cells (�80%–90% of EV tumors, P >0.3; Supplementary Data and Fig. S3), supporting thespecificity of miR-145 to downregulate these proteins. Inaddition, we observed downregulation of PTGS2, K-Ras,and ERK5 in tumors with upregulated miR-143
(Fig. 8H). The latter are putative or established targetsof miR-143 (39, 41, 42).
Putative miR-143 and miR-145 targets are upregulatedin tumors from Egfrwt miceExperiments in vitro in colon cancer cells and in vivo
in tumor xenografts using cells transfected with miR-145suggested that this miRNA negatively controls G1 reg-ulators cdk6, CCND2, and E2F3. We next examinedthese G1 regulators in colonic tumors from Egfrwt andEgfrwa2 mice to assess their regulation by EGFR, andtheir potential control by miR-145 in colonic tumori-genesis. As shown in representative tumors in Supple-mentary Figure S4A and quantified in SupplementaryFigure S4B, CCND2 and cdk6 were significantlyincreased in tumors from Egfrwt mice compared withEgfrwa2 mice, consistent with loss of miR-145 in Egfrwt
tumors. Increases in E2F3 in Egfrwt tumors comparedwith Egfrwa2 tumors did not reach statistical significance(P ¼ 0.3). Because tumor promotion occurred withWestern diet only in Egfrwt mice, we examined theexpression of K-Ras and MYC, targets of these miRNAsin tumors from mice on Western diet. As shown inFigure 9A and B, K-Ras and MYC were significantlyincreased in tumors from Egfrwt but not Egfrwa2 mice. Inprior studies, we also showed that the MYC increase wassignificantly higher in tumors from Egfrwt mice onWestern diet compared with standard diet. Tumors fromEgfrwt mice also had significantly higher proliferation asassessed by Ki67 staining and shown in Figure 9C (compareFig. 9E with G). In contrast, colonic epithelial cell prolifera-tion was comparable in control Egfrwt and Egfrwa2 mice(Fig. 9D and F).
Discussion
EGFR regulates expression of miR-143 and miR-145 inmurine colonic tumorsEGFR is increased in many tumors including colon
cancer (44). Furthermore, studies in experimental modelsof colon cancer have shown that this receptor plays a keyrole in colonic tumorigenesis (3–5, 45). Beginning with amiRNA array discovery approach, in the current report, weshowed for the first time that miR-143 and miR-145 werenegatively regulated by EGFR signals in murine colonictumors. Specifically, miR-143 and miR-145 were decreasedin AOM/DSS-induced tumors in Egfrwt mice, whereas theywere significantly increased in colonic tumors from Egfrwa2
mice on either standard or Western diet. We confirmedthese miRNA array results by real-time PCR and Northernblot analysis (miR-145). We also showed that miR-143 andmiR-145 were more highly expressed in colonic epithelialcells than in stromal cells in normal mouse colon. Wespeculate that EGFR mediators required for tumor promo-tion by Western diet include targets of miR-143 or miR-145. This provides an example of epigenetic gene regulationby environmental cues (Western diet). In support of thishypothesis in the current study, we showed that Ras and
Figure 6. EGFR signals suppress miR-143 and miR-145 in coloniccells. Indicated cells were plated in 6-well multiwell plates in 10% sera.When 60% confluent, cells were treated with 10 ng/mL EGF, or C225anti-EGFR (20 mg/mL) antibodies or PBS. Twenty-four hours later, cellswere lysed and RNA was isolated. miR-143 (gray bars) and miR-145 (blackbars) were measured by real-time PCR. Values were expressed asfold-control PBS-treated cells in each case. Note expression levels in cellstreated with PBS are indicated by the dotted line (Control). * and †, P < 0.05compared with vehicle-treated cells; n ¼ 2 independent platings intriplicate wells.
EGFR Controls miR-143 and miR-145 in Colonic Tumorigenesis
www.aacrjournals.org Mol Cancer Res; 9(7) July 2011 969
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

MYC were upregulated by Western diet in colonic tumorsfrom Egfrwt but not Egfrwa2 mice and that these proto-oncogenes are targets of miR-143 and miR-145, respec-tively in tumor xenografts. Prior studies have shown gene–dose thresholds for these proto-oncogenes in controllingtumorigenesis (46, 47). A gene–dose effect of cdk6 con-trolling proliferation has also been reported (48). Thus,upregulation of miR-143 and miR-145 in Egfrwa2 micewould be predicted to restrain Western diet–inducedincreases in these proto-oncogenes and inhibit tumor pro-motion. This hypothesis is also supported by our findingsthat miR-143 and miR-145 decreased MYC and K-Ras and
potently suppressed colon cancer cell growth in tumorxenografts.As in the case of AOM/DSS-induced tumors, we
observed that miR-143 and miR-145 were downregulatedin tumors induced by AOM alone in Egfrwt mice, whereasthey were upregulated in AOM-induced tumors in Egfrwa2
mice. Thus, expression levels of these miRNAs were nega-tively controlled by EGFR in murine models of bothsporadic and inflammatory colon cancer. EGFR was alsorequired for tumor promotion byWestern diet in the AOMmodel. Targets of these miRNAs likely also block diet-related AOM tumor promotion in Egfrwa2 mice. Consistent
Figure 7. Transfected miR-143 and miR-145 inhibit EGF-stimulated colon cancer cell proliferation and DNA synthesis and suppress expression of G1
targets. HCT116 cells were plated overnight in 10% sera on 6-well (WST-1 assay) or 24-well plates (BrdU assay). After serum removal, cells were transfectedwith miR-143 or miR-145 (20 nmol/L) or scrambled (Scr) control oligonucleotides. Forty-eight hours later, cells were treated with vehicle or EGF (10 ng/mL).Cell proliferation was assessed by WST-1 and DNA synthesis by BrdU incorporation 48 hours after EGF addition. A, WST-1 assay. *, P < 0.05 compared withcells treated with vehicle; †, P < 0.05 compared with cells treated with EGFþ scrambled miRNA. B, BrdU incorporation. *, P < 0.05 compared with cells treatedwith vehicle; z, P < 0.05 compared with cells treated with EGF þ scrambled miRNA. C, transfected miR-143 and miR-145 downregulate G1 cell-cycleregulators. HCT116 or HCA-7 colorectal cancer cells were transfected with 20 nmol/L scrambled oligonucleotide or indicated microRNA and 48 hourslater, cell lysates probed for putative target proteins. ACTB is shown as loading control. Blots are representative of experiments repeated 3 times. D, cdk6-30UTR sequence complementing miR-145 seed sequence. Shown in marked region is the complementation sequence at positions 3,884 to 3,892(NM_001259.6). E, miR-145 targets cdk6 via 30UTR region of cdk6. HCT116 cells were transfected with 100 ng luciferase (control EV) or luciferase containing afragment of cdk6 with wild-type 30UTR or 30UTR mutated in bases complementing miR-145 seed sequence as described in Materials and Methods. Cellswere cotransfected with control oligonucleotide or miR-145 (10 nmol/L). Forty-eight hours later, cells were lysed and luciferase measured. Renilla luciferasewas normalized to firefly luciferase. Experiments were repeated twice with comparable results. Values are means of 6 replicates.
Zhu et al.
Mol Cancer Res; 9(7) July 2011 Molecular Cancer Research970
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531
with genetic experiments showing that EGFR suppressesthese miRNAs in colonic tumors, we observed that EGFRblockade with gefitinib partially inhibited miR143 andmiR-145 downregulation in AOM-induced rat colonictumors. The EGFR inhibitor effects, moreover, are inagreement with studies showing that gefitinib reducedmiR-143 downregulation in lung cancer cells (49). ThesemiRNAs also appeared to be downregulated in Min ade-nomas, another model with upregulated EGFR (45). Inter-estingly, recent in vitro studies in breast cancer cells
identified several other miRNAs that target oncogenictranscription factors and that were suppressed by EGFR(13). Our studies have directly established that EGFRnegatively regulates miR-143 and miR-145 in vivo inmodels of colonic tumorigenesis.
miR-143 and miR-145 loss in human sporadic andUC-associated colon cancersDownregulation of miR-143 and miR-145 were pre-
viously reported in colon cancer (16, 50–53). Subsequently,
Figure 8. miR-143 and miR-145 mediate tumor suppressor phenotypes and reveal putative targets in vivo in HCT116 tumor xenografts. Cells (5 � 106)transduced with lentivirus alone (EV, empty vector) or vector encoding pre-miR-145 or pre-miR-143 were injected subcutaneously into the flank of nu/numice.Tumors were harvested 3 week later. A, miR-143 and miR-145 levels. Values in HCT116-transfected cells were normalized to values in EV. *, P < 0.05compared with EV cells. B, tumor weight (*,P < 0.005 compared with EV tumors, n¼ 5mice in each group). C, Ki67 quantitation in tumor xenografts (*,P < 0.001compared with EV). D–F, Ki67 staining in tumor xenografts (D, EV; E, miR-143 cells; and F, miR-145 cells) G, putative miR-145 targets. Upregulation ofmiR145 in HCT116 tumor xenografts inhibited expression of G1 regulators that are predicted targets of miR-145. H, putative miR-143 targets. Upregulationof miR-143 in HCT116 tumor xenografts inhibited expression of proteins that are predicted targets of miR-143. Protein expression ratios in indicatedmiRNA expressing tumor xenografts versus levels in EV tumors (means � SD) are shown. *, P < 0.05 for all proteins except E2F3 (P ¼ 0.07).
EGFR Controls miR-143 and miR-145 in Colonic Tumorigenesis
www.aacrjournals.org Mol Cancer Res; 9(7) July 2011 971
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

other tumor types were noted to have reduced levels of thesemiRNAs (20–23). In this study, we confirmed this findingand showed by in situ hybridization that miR-145 miRNAwas predominantly expressed in epithelial cells in normalhuman colon and downregulated in sporadic human coloncancers. We also showed for the first time that miR-143 andmiR-145 were downregulated in UC-associated colon can-cers. These results extend our recent study showing thatthese miRNAs were downregulated in chronic UC (37).Further investigation will be required to determine whethermechanisms mediating suppression of these miRNAs insporadic versus UC-associated colon cancer are different.
EGFR suppresses miR-143 and miR-145 in colonic cellsTo assess whether EGFR suppression of these miRNAs
could occur in a cell autonomous manner, we examinedcells in culture. Consistent with findings in tumors, EGFstimulation of CCD-18Co colonic fibroblasts and murinecolonocytes decreased these miRNAs. Conversely, EGFRblockade in HCT116 colon cancer cells increased miR-143and miR-145. These findings are in agreement with a recentstudy showing that miR-145 was inhibited by cellularproliferation (54). The failure of EGF to further reducethese miRNAs in HCT116 cells likely reflects their down-
regulated state under basal conditions. It should be notedthat these cells possess autocrine-activated EGFR signals. Inthe current study, we also showed that these miRNAssuppressed EGFR-induced colon cancer cell growth. Thus,while EGF-induced growth in HCT116 cells did notrequire further suppression of miR-143 and miR-145,increases in these miRNAs suppressed EGF-induced growthin vitro and in tumor xenograft growth in vivo. A growth-promoting circuit regulated by EGFR and involving miR-145 suppression was recently described in lung cancer cells(22). Taken together, we postulate that EGFR signalsdriving colonic tumorigenesis are mediated in part byEGFR suppression of these miRNAs. The recent demon-stration that oncogenic Ras suppresses these miRNAs inpancreatic cancer cells suggests that Ras might mediate theirdownregulation by EGFR in colon cancers (15). The role ofRas in EGFR suppression of these miRNAs, however, willrequire further study.
G1 regulators—targets of miR-145 and miR-145We showed that transfected miR-143 and miR-145
inhibited the expression of K-Ras, MYC, cdk6, CCND2,and E2F3 in colon cancer cells growing in vitro or in tumorxenografts in vivo. Furthermore, transfection of miR-143 or
Figure 9. K-Ras and MYC are upregulated in tumors from Egfrwt but not Egfrwa2 mice on Western diet. A, representative Western blots of K-Ras andMYC in control mucosa and tumors from Egfrwt and Egfrwa2 mice (n ¼ 4 tumors per group). B, quantitative densitometry of K-Ras and MYC expression intumors normalized to Egfr genotype–matched control colonic mucosa (ctl mucosa ¼ 1). *, P < 0.05 compared with control mucosa; †, P < 0.05 comparedwith tumors from Egfrwa2 mice. C, quantitation of Ki67 expression in Egfrwt and Egfrwa2 tumors (n ¼ 3 tumors in each group). *, P < 0.05 compared withEgfrwa2 tumors. D, Ki67 in normal colonic mucosa from Egfrwa2 mouse. E, Ki67 in Egfrwa2 tumor. F, Ki67 in normal colonic mucosa from Egfrwt mouse.G, Ki67 in Egfrwt tumor.
Zhu et al.
Mol Cancer Res; 9(7) July 2011 Molecular Cancer Research972
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

miR-145 inhibited proliferation and DNA synthesis in cellculture and reduced Ki67 staining in tumor xenografts.These results suggest an important role for these miRNAs incell-cycle regulation. cdk6, a major cyclin D–dependentkinase, is increased in human colon cancers (55). Weconfirmed that cdk6 is a direct target of miR-145 byshowing that this miRNA significantly downregulated aluciferase reporter linked to wild-type but not mutant cdk630UTR containing a target sequence for miR-145. CCND2is another putative target of miR-145 and a member of thecyclin D family that can activate cdk6. CCND2 was linkedto early progression in human colonic carcinogenesis (56,57). CCND2–cdk6 complex can phosphorylate the retino-blastoma protein, Rb causing the release of E2F3 (58).E2F3 in turn drives cell-cycle progression by upregulatinggenes controlling G1 to S-phase transition (59). Pathwaysinvolving E2F3 are active in many tumors including humancolon cancer (60–62). Our in vitro and in vivo studiessuggest that K-Ras, MYC, cdk6, CCND2, and E2F3 aretargets of miR-143 or miR-145 that link these miRNAs toEGFR and cell-cycle control. Experiments using luciferasereporters linked to 30UTR for CCND2 and E2F3 are inprogress to further characterize their miR-145 target statusin colon cancer cells.To further assess several of these G1 regulators as in vivo
targets of miR-145 in colonic tumorigenesis, we comparedtheir expression levels in colonic tumors arising in Egfrwt andEgfrwa2mice. CCND2 and cdk6were upregulated in tumorsfrom Egfrwt compared with Egfrwa2 mice. These results areconsistent with the differential effects of mutant and wild-type Egfr onmiR-145 expression in colonic tumors and within silico predictions of these G1 regulators as targets of miR-145. Clinical differences in tumor multiplicity and tumorstage in Egfrwt and Egfrwa2 mice, however, required aWestern diet that enhanced additional EGFR mediators,including K-Ras and MYC that are also targets of miR-143and miR-145, respectively.While miR-143 and miR-145 appear to drive tumor
suppressor phenotypes based on our tumor xenograft stu-dies, development of tumors in Egfrwa2 mice suggests thatalternative mechanisms allow tumor escape in these mutantmice. Such tumors likely exploit EGFR-independent onco-genic pathways. Increases in ErbB2, ErbB3, and c-Met wereimplicated as escape mechanisms in other tumors (63–65).Taken together with earlier reports, our results indicate
that miR-143 and miR-145 are tumor suppressors in coloncancer and play important restraining roles on cell-cycleprogression (40, 66). Loss of these miRNAs may be espe-cially critical for tumor development under stress conditionsof a Western diet. Our results have uncovered a potentiallyimportant role of miRNAs in EGFR regulation of the cell
cycle. The findings that EGFR signals coordinately controlmultiple G1 regulators via miR-143 (K-Ras) and miR-145(MYC, cdk6, CCND2, and E2F3) are consistent withstudies showing that EGFR regulates other orchestratedevents via coordinate miRNA changes (13). We wouldpredict that by targeting multiple G1 regulators, thesecotranscribed tumor suppressor miRNAs would play animportant restraining role on cell-cycle progression. Futurestudies restoring levels of individual targets could assesswhether loss of a specific target plays a dominant role in cell-cycle arrest induced by miR-143 or miR-145 as suggestedfor other targets (67).In summary, we have shown that EGFR signals nega-
tively regulate expression of miR-143 and miR-145 incolonic tumorigenesis. Because these miRNAs drive tumorsuppressor phenotypes in tumor xenograft models, theirdownregulation by EGFR uncovers an important miRNA-dependent mechanism mediating oncogenic effects of thisreceptor. The tumor suppressor role of these miRNAsappears to be conditional in the AOM and AOM/DSSmodels and revealed by Western diet that activates EGFRsignals and promotes tumorigenesis. A schema summariz-ing this pathway is shown in Supplementary Figure S5.This mechanism involves increased G1 cell-cycle regulatorscdk6, CCND2, and E2F3, as well as enhanced K-Ras andMYC that promote proliferation in colonic tumorigenesis.Approaches that prevent downregulation of these miRNAscould provide new strategies for colon cancer prevention.The recent demonstration that these miRNAs can beupregulated by small molecules, moreover, offers an excit-ing potential strategy for cancer prevention in high-riskindividuals (68).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. John Kwon (University of Chicago) for critical reading andhelpful suggestions.
Grant Support
These studies were funded in part by the following grants: P30DK42086(Digestive Diseases Research Core Center), and CA036745 (M. Bissonnette), aswell as the Samuel Freedman Research Laboratories for Gastrointestinal CancerResearch. The University of Chicago Department of Pathology Research Coreprovided additional funding.
Received November 22, 2010; revised May 16, 2011; accepted May 31, 2011;published OnlineFirst June 8, 2011.
References1. Rim SH, Seeff L, Ahmed F, King JB, Coughlin SS. Colorectal cancer
incidence in theUnitedStates, 1999–2004: an updated analysis of datafrom the National Program of Cancer Registries and the Surveillance,Epidemiology, and End Results Program. Cancer 2009;115:1967–76.
2. Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends inGenetics 1993;9:138–41.
3. Fichera A, Little N, Jagadeeswaran S, Dougherty U, Sehdev A, MustafiR, et al. EGFR signaling is required for microadenoma formation in the
EGFR Controls miR-143 and miR-145 in Colonic Tumorigenesis
www.aacrjournals.org Mol Cancer Res; 9(7) July 2011 973
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

mouse azoxymethane model of colonic carcinogenesis. Cancer Res2007;67:827–35.
4. Dougherty U, Cerasi D, Taylor I, Kocherginsky M, Tekin U, Badal S,et al. Epidermal growth factor receptor is required for colonic tumorpromotion by dietary fat in the azoxymethane/dextran sulfate sodiummodel: roles of transforming growth factor-alpha and PTGS2. ClinCancer Res 2009;15:6780–9.
5. Dougherty U, Sehdev A, Cerda S, Mustafi R, Little N, Yuan W, et al.Epidermal growth factor receptor controls flat dysplastic aberrantcrypt foci development and colon cancer progression in the ratazoxymethane model. Clin Cancer Res 2008;14:2253–62.
6. Luetteke NC, Phillips HK, Qiu TH, Copeland NG, Earp HS, JenkinsNA, et al. The mouse waved-2 phenotype results from a pointmutation in the EGF receptor tyrosine kinase. Genes Dev 1994;8:399–413.
7. Singh J, Hamid R, Reddy BS. Dietary fat and colon cancer: modulatingeffect of types and amount of dietary fat on ras-p21 function duringpromotion and progression stages of colon cancer. Cancer Research1997;57:253–8.
8. Adhikary S, Eilers M. Transcriptional regulation and transformation byMyc proteins. Nat Rev Mol Cell Biol 2005;6:635–45.
9. Calcagno SR, Li S, Colon M, Kreinest PA, Thompson EA, Fields AP,et al. Oncogenic K-ras promotes early carcinogenesis in the mouseproximal colon. Int J Cancer 2008;122:2462–70.
10. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in generegulation. Nat Rev Genet 2004;5:522–31.
11. Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. MicroRNAexpression and function in cancer. Trends Mol Med 2006;12:580–7.
12. Calin GA, Croce CM.MicroRNA signatures in human cancers. Nat RevCancer 2006;6:857–66.
13. Avraham R, Sas-Chen A, Manor O, Steinfeld I, Shalgi R, Tarcic G, et al.EGF decreases the abundance of microRNAs that restrain oncogenictranscription factors. Sci Signal 2010;3:ra43.
14. Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, et al.Widespread microRNA repression by Myc contributes to tumorigen-esis. Nat Genet 2008;40:43–50.
15. Kent OA, Chivukula RR, Mullendore M, Wentzel EA, Feldmann G, LeeKH, et al. Repression of the miR-143/145 cluster by oncogenic Rasinitiates a tumor-promoting feed-forward pathway. Genes Dev 2010;24:2754–9.
16. Michael MZ, SM OC, van Holst, Pellekaan NG, Young GP, James RJ.Reduced accumulation of specific microRNAs in colorectal neoplasia.Mol Cancer Res 2003;1:882–91.
17. Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN,et al. miR-145 and miR-143 regulate smooth muscle cell fate andplasticity. Nature 2009;460:705–10.
18. Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. Micro-RNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripo-tency in human embryonic stem cells. Cell 2009;137:647–58.
19. Akao Y, Nakagawa Y, Naoe T. MicroRNAs 143 and 145 are possiblecommon onco-microRNAs in human cancers. Oncol Rep 2006;16:845–50.
20. Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al.MicroRNA gene expression deregulation in human breast cancer.Cancer Res 2005;65:7065–70.
21. Ostenfeld MS, Bramsen JB, Lamy P, Villadsen SB, Fristrup N,Sørensen KD, et al. miR-145 induces caspase-dependent and -inde-pendent cell death in urothelial cancer cell lines with targeting of anexpression signature present in Ta bladder tumors. Oncogene2010;29:1073–84.
22. Cho WC, Chow AS, Au JS. Restoration of tumour suppressor hsa-miR-145 inhibits cancer cell growth in lung adenocarcinoma patientswith epidermal growth factor receptor mutation. Eur J Cancer2009;45:2197–206.
23. Ozen M, Creighton CJ, Ozdemir M, Ittmann M. Widespread dereg-ulation of microRNA expression in human prostate cancer. Oncogene2008;27:1788–93.
24. Rozen S, Skaletsky H. Primer3 on the WWW for general users and forbiologist programmers. In:Krawetz S, Misener S, editors. Bioinfor-
matics methods and protocols. Methods in molecular biology.Totowa, NJ: Humana Press; 2000. p. 365–86.
25. Livak KJ, Schmittgen TD. Analysis of relative gene expression datausing real-time quantitative PCR and the 2(-Delta Delta C(T)) Method.Methods 2001;25:402–8.
26. Yuan JS, Reed A, Chen F, Stewart CN Jr. Statistical analysis of real-time PCR data. BMC Bioinformatics 2006;7:85–97.
27. Obernosterer G, Martinez J, Alenius M. Locked nucleic acid-based insitu detection of microRNAs in mouse tissue sections. Nat Protoc2007;2:1508–14.
28. Silahtaroglu AN, Nolting D, Dyrskjot L, Berezikov E, Møller M,Tommerup N, et al. Detection of microRNAs in frozen tissue sec-tions by fluorescence in situ hybridization using locked nucleic acidprobes and tyramide signal amplification. Nat Protoc 2007;2:2520–8.
29. Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, Jat PS. Estab-lishment of conditionally immortalized epithelial cell lines from bothcolon and small intestine of adult H-2Kb-tsA58 transgenic mice. ProcNatl Acad Sci U S A 1993;90:587–91.
30. Bissonnette M, Khare S, von Lintig FC, Wali RK, Nguyen L, Zhang Y,et al. Mutational and nonmutational activation of p21ras in rat colonicazoxymethane-induced tumors: effects on mitogen-activated proteinkinase, cyclooxygenase-2, and cyclin D1. Cancer Res 2000;60:4602–9.
31. Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNAprocessing enhances cellular transformation and tumorigenesis. NatGenet 2007;39:673–7.
32. Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, et al.MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamicsand responsiveness of smooth muscle cells to injury. Genes Dev2009;23:2166–78.
33. Moran AE, Hunt DH, Javid SH, Redston M, Carothers AM, BertagnolliMM. Apc deficiency is associated with increased Egfr activity in theintestinal enterocytes and adenomas of C57BL/6J-Min/þmice. J BiolChem 2004;279:43261–72.
34. Arndt GM, Dossey L, Cullen LM, Lai A, Druker R, Eisbacher M, et al.Characterization of global microRNA expression reveals oncogenicpotential of miR-145 in metastatic colorectal cancer. BMC Cancer2009;9:374.
35. Wang CJ, Zhou ZG, Wang L, Yang L, Zhou B, Gu J, et al. Clinico-pathological significance of microRNA-31, -143 and -145 expressionin colorectal cancer. Dis Markers 2009;26:27–34.
36. Borralho PM, Kren BT, Castro RE, da Silva IB, Steer CJ, RodriguesCM, et al. MicroRNA-143 reduces viability and increases sensitivity to5-fluorouracil in HCT116 human colorectal cancer cells. The FEBSjournal 2009;276:6689–700.
37. Pekow J, Dougherty U, Mustafi R, Zhu H, Kocherginsky M, Rubin DT,et al. miR-143 and miR-145 are down-regulated in ulcerative colitis:putative regulators of inflammation and protooncogenes. InflammBowel Dis 2011 May 6. [Epub ahead of print].
38. Lee EJ, Baek M, Gusev Y, Brackett DJ, Nuovo GJ, Schmittgen TD.Systematic evaluation of microRNA processing patterns in tissues,cell lines, and tumors. RNA 2008;14:35–42.
39. Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y, et al. Role of miR-143targeting KRAS in colorectal tumorigenesis. Oncogene 2009;28:1385–92.
40. Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, et al. p53represses c-Myc through induction of the tumor suppressor miR-145.Proc Natl Acad Sci U S A 2009;106:3207–12.
41. Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, RavichandranLV, et al. MicroRNA-143 regulates adipocyte differentiation. J BiolChem 2004;279:52361–5.
42. Griffiths-Jones S, Saini HK, van Dongen S. Enright AJ. miRBase: toolsfor microRNA genomics. Nucleic Acids Res 2008;36:D154–8.
43. Yang Y, Chaerkady R, Kandasamy K, Huang TC, Selvan LD, DwivediSB, et al. Identifying targets of miR-143 using a SILAC-based pro-teomic approach. Mol Biosyst 2010;6:1873–82.
44. Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signalingnetwork: receptor heterodimerization in development and cancer.EMBO J 2000;19:3159–67.
Zhu et al.
Mol Cancer Res; 9(7) July 2011 Molecular Cancer Research974
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

45. Roberts RB, Min L, Washington MK, Olsen SJ, Settle SH, Coffey RJ,et al. Importance of epidermal growth factor receptor signalingin establishment of adenomas and maintenance of carcinomasduring intestinal tumorigenesis. Proc Natl Acad Sci U S A 2002;99:1521–6.
46. Shachaf CM, Gentles AJ, Elchuri S, Sahoo D, Soen Y, Sharpe O, et al.Genomic and proteomic analysis reveals a threshold level of MYCrequired for tumor maintenance. Cancer Res 2008;68:5132–42.
47. Radner H, el-Shabrawi Y, Eibl RH, Br€ustle O, Kenner L, Kleihues P,et al. Tumor induction by ras andmyc oncogenes in fetal and neonatalbrain: modulating effects of developmental stage and retroviral dose.Acta Neuropathol 1993;86:456–65.
48. Zhang F, Taipale M, Heiskanen A, Laiho M. Ectopic expression ofCdk6 circumvents transforming growth factor-beta mediated growthinhibition. Oncogene 2001;20:5888–96.
49. Wang G, Mao W, Zheng S, Ye J. Epidermal growth factor receptor-regulated miR-125a-5p–a metastatic inhibitor of lung cancer. FEBS J2009;276:5571–8.
50. Bandres E, Cubedo E, Agirre X, Malumbres R, Z�arate R, Ramirez N,et al. Identification by real-time PCR of 13 mature microRNAs differ-entially expressed in colorectal cancer and non-tumoral tissues. MolCancer 2006;5:29.
51. Akao Y, Nakagawa Y, Naoe T. MicroRNA-143 and -145 in coloncancer. DNA Cell Biol 2007;26:311–20.
52. Schepeler T, Reinert JT, Ostenfeld MS, Christensen LL, SilahtarogluAN, Dyrskjøt L, et al. Diagnostic and prognostic microRNAs in stage IIcolon cancer. Cancer Res 2008;68:6416–24.
53. Slaby O, Svoboda M, Fabian P, Smerdova T, Knoflickova D, Bed-narikova M, et al. Altered expression of miR-21, miR-31, miR-143 andmiR-145 is related to clinicopathologic features of colorectal cancer.Oncology 2007;72:397–402.
54. La Rocca G, Shi B, Audia A, Ferrari-Amorotti G, Mellert HS, CalabrettaB, et al. Regulation of microRNA-145 by growth arrest and differentia-tion. Exp Cell Res 2011;317:488–95.
55. Salh B, Bergman D, Marotta A, Pelech SL. Differential cyclin-depen-dent kinase expression and activation in human colon cancer. Antic-ancer Res 1999;19:741–8.
56. Bartkova J, Thullberg M, Slezak P, Jaramillo E, Rubio C, ThomassenLH, et al. Aberrant expression of g1-phase cell cycle regulators in flatand exophytic adenomas of the human colon. Gastroenterology2001;120:1680–8.
57. Mermelshtein A, Gerson A, Walfisch S, Delgado B, Shechter-Maor G,Delgado J, et al. Expression of D-type cyclins in colon cancer and incell lines from colon carcinomas. Br J Cancer 2005;93:338–45.
58. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell1995;81:323–30.
59. Leone G, DeGregori J, Yan Z, Jakoi L, Ishida S, Williams RS, et al.E2F3 activity is regulated during the cell cycle and is required for theinduction of S phase. Genes Dev 1998;12:2120–30.
60. Cooper CS, Nicholson AG, Foster C, Dodson A, Edwards S, FletcherA, et al. Nuclear overexpression of the E2F3 transcription factor inhuman lung cancer. Lung Cancer 2006;54:155–62.
61. Olsson AY, Feber A, Edwards S, Te Poele R, Giddings I, Merson S,et al. Role of E2F3 expression in modulating cellular proliferation ratein human bladder and prostate cancer cells. Oncogene 2007;26:1028–37.
62. Feng XD, Huang SG, Shou JY, Liao BR, Yingling JM, Ye X, et al.Analysis of pathway activity in primary tumors and NCI60 cell linesusing gene expression profiling data. Genomics Proteomics Bioinfor-matics 2007;5:15–24.
63. Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al.Escape from HER-family tyrosine kinase inhibitor therapy by thekinase-inactive HER3. Nature 2007;445:437–41.
64. Rajput A, Koterba AP, Kreisberg JI, Foster JM, Willson JK, BrattainMG, et al. A novel mechanism of resistance to epidermal growth factorreceptor antagonism in vivo. Cancer Res 2007;67:665–73.
65. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. METamplification occurs with or without T790Mmutations in EGFRmutantlung tumors with acquired resistance to gefitinib or erlotinib. Proc NatlAcad Sci U S A 2007;104:20932–7.
66. Akao Y, Nakagawa Y, Hirata I, Iio A, Itoh T, Kojima K, et al. Role of anti-oncomirs miR-143 and -145 in human colorectal tumors. CancerGene Ther 2010;17:398–408.
67. Shi B, Sepp-Lorenzino L, Prisco M, Linsley P, deAngelis T, BasergaR. Micro RNA 145 targets the insulin receptor substrate-1 andinhibits the growth of colon cancer cells. J Biol Chem 2007;282:32582–90.
68. Melo S, Villanueva A, Moutinho C, Davalos V, Spizzo R, Ivan C, et al.Small molecule enoxacin is a cancer-specific growth inhibitorthat acts by enhancing TAR RNA-binding protein 2-mediatedmicroRNA processing. Proc Natl Acad Sci U S A 2011;108:4394–9.
EGFR Controls miR-143 and miR-145 in Colonic Tumorigenesis
www.aacrjournals.org Mol Cancer Res; 9(7) July 2011 975
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531

2011;9:960-975. Published OnlineFirst June 8, 2011.Mol Cancer Res Hongyan Zhu, Urszula Dougherty, Victoria Robinson, et al.
Regulators1Promoted Murine Colon Cancer: Role of G−miR-145 in Western Diet
EGFR Signals Downregulate Tumor Suppressors miR-143 and
Updated version
10.1158/1541-7786.MCR-10-0531doi:
Access the most recent version of this article at:
Material
Supplementary
http://mcr.aacrjournals.org/content/suppl/2011/06/08/1541-7786.MCR-10-0531.DC1
Access the most recent supplemental material at:
Cited articles
http://mcr.aacrjournals.org/content/9/7/960.full#ref-list-1
This article cites 66 articles, 25 of which you can access for free at:
Citing articles
http://mcr.aacrjournals.org/content/9/7/960.full#related-urls
This article has been cited by 6 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mcr.aacrjournals.org/content/9/7/960To request permission to re-use all or part of this article, use this link
on July 11, 2020. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst June 8, 2011; DOI: 10.1158/1541-7786.MCR-10-0531