early initiation of prostacyclin in portopulmonary hypertension: 10 years of a transplant center’s...
TRANSCRIPT

PULMONARY HYPERTENSION
Early Initiation of Prostacyclin in Portopulmonary Hypertension:10 Years of a Transplant Center’s Experience
Rana L. A. Awdish • Hector R. Cajigas
Received: 14 February 2013 / Accepted: 5 August 2013 / Published online: 25 August 2013
� Springer Science+Business Media New York 2013
Abstract
Background Portopulmonary hypertension (PoPH) is a
subgroup of Group 1 pulmonary arterial hypertension
(PAH) with particularly poor prognosis. Delay in initiation
of parenteral therapy may be the reason for poor outcome.
Methods We conducted a prospective observational study
of all patients with Group 1 PoPH evaluated at Henry Ford
Hospital between January 2002 and July 2012. The cohort
of the REVEAL Registry patients with PoPH was used as
the comparator group. The patient survival rates at 5 years
after diagnosis and 2 years after enrollment, treatment
trends, and the freedom from all-cause hospitalization rates
at 2 years after enrollment were compared using v2
analysis.
Results Twenty-one patients were enrolled in the PH
Clinic with PoPH from January 2002 through July 2012.
Our patients were significantly more likely to be on pros-
tacyclin IV at 90 days as compared to REVEAL PoPH
patients (67 vs. 31 %; p = 0.002). Despite this, early out-
comes were not significantly different between the groups:
2-year survival from enrollment (70 and 67 %, respec-
tively; p = 0.77) and 2-year freedom from hospitalization
(35 vs. 49 %, respectively; p = 0.29). However, 5-year
survival from time of diagnosis was significantly higher in
our cohort of PoPH patients (71 and 40 %, respectively; p =
0.02).
Conclusions Early initiation of parenteral prostacyclin
therapy in PoPH patients at a single institution was asso-
ciated with improved 5-year patient survival from diag-
nosis as compared to the REVEAL Registry of PoPH
patients and allowed for clearance for transplant in 52 % of
patients within 1 year.
Keywords Model for end stage liver disease �Orthotopic liver transplant � Pulmonary arterial
hypertension � Portopulmonary hypertension �Transplant clearance � Prostacyclin
Abbreviations
ALT Alanine aminotransferase
AST Aspartate aminotransferase
6MWD Six minute walk distance
CO Cardiac output
ETRA Endothelin receptor antagonist
IPAH Idiopathic pulmonary arterial hypertension
mPAP Mean pulmonary arterial pressure
MELD Model for end stage liver disease
OLT Orthotopic liver transplant
PAH Pulmonary arterial hypertension
PASP Pulmonary artery systolic pressure
PCWP Pulmonary artery occlusion pressure
PDEI Phosphodiesterase inhibitor
PoPH Portopulmonary hypertension
PVR Pulmonary vascular resistance
NYHA New York Heart Association
REVEAL Registry to Evaluate Early and Long-term
PAH Disease Management
TPG Transpulmonary gradient
R. L. A. Awdish (&) � H. R. Cajigas
Pulmonary and Critical Care Medicine Division, Henry Ford
Hospital, 2799 West Grand Boulevard, K-17, Detroit, MI 48202,
USA
e-mail: [email protected]
123
Lung (2013) 191:593–600
DOI 10.1007/s00408-013-9501-5

Introduction
Portopulmonary hypertension (PoPH) is defined as pul-
monary arterial hypertension (PAH) associated with portal
hypertension, with or without liver disease [1]. PoPH is a
life-threatening disease characterized by elevation in pul-
monary vascular resistance (PVR), which leads to
increased pulmonary artery pressure, right ventricular
failure, and ultimately death [2]. Although the natural
history of the disease has been difficult to characterize,
multiple studies have demonstrated that despite what
would be considered ‘‘more favorable’’ hemodynamics
(higher cardiac index and lower PVR), mortality of these
patients is significantly higher than their idiopathic pul-
monary arterial hypertension (IPAH) counterparts [3, 4].
This fact, in addition to the increased perioperative mor-
bidity and mortality of this group during liver transplant
makes PoPH especially worthy of study [5].
Given the severity and poor prognosis of PoPH, early
and appropriate therapy is considered critical; however,
studies in this area are lacking. Treatment of PoPH has
focused on vasodilator therapy as continuous IV adminis-
tration of epoprostenol reduces PVR, mPAP, and improves
RV function [6]. Independent of transplant status, IV
prostacyclin impacts long-term survival in PoPH [5].
Recently, a report from the Registry to Evaluate Early and
Long-term PAH Disease Management (REVEAL) descri-
bed a large cohort of patients with PoPH, and reaffirmed
that survival from the time of enrollment was worse than
that observed for IPAH [3]. Because only 57 % of the
newly diagnosed patients were receiving PAH treatment,
the authors postulated that delayed initiation of PAH
treatment may be implicated in their lower survival [3]. As
a high-volume liver transplant center, with a structured
protocol for screening for PoPH that is in accordance with
the international guidelines, and early initiation of paren-
teral therapy strategy, our cohort of patients offer oppor-
tunity to expand the knowledge in this area. We describe
the characteristics and outcomes of a cohort of patients
with PoPH.
Methods and Materials
This prospective, observational study was approved by the
Institutional Review Board of Henry Ford Hospital. The
medical records of patients diagnosed with PoPH and
evaluated in the Pulmonary Hypertension Clinic at Henry
Ford Hospital between January 2002 and July 2012 were
reviewed. All patients met the UNOS criteria for liver
transplantation and were referred to our clinic when
screening 2D transthoracic echocardiography reported a
systolic pulmonary artery pressure (PASP) greater than
40 mmHg. Date of diagnosis was taken as the date of
cardiac catheterization confirming the diagnosis, regardless
of whether it was within our system. Date of enrollment
was documented as the date the patient was first seen by the
Pulmonary Hypertension Clinic at Henry Ford Hospital.
All patients underwent cardiac catheterization and were
diagnosed with PoPH when the following specific criteria
were met: portal hypertension and/or evidence of advanced
liver disease, mPAP C25 mmHg, PVR C240 dynes/s/cm5,
PCWP B15 mmHg, and no evidence of alternative car-
diopulmonary diseases associated with PAH [3]. As
advocated by expert opinion, and given the high prevalence
of volume overload and high-output heart failure in
advanced liver disease, patients with PCWP [15 mmHg
were not excluded if their transpulmonary gradient (TPG)
was [12 mmHg [5]. This is modeled as well on REVEAL
registry enrollment criteria [7].
The following demographic data were collected: patient
age at diagnosis, gender, and self-identified race. Hemo-
dynamic data were collected at the time of diagnosis and
following therapy. Surrogate markers, including New York
Heart Association Functional Class (NYHA), Model for
End-Stage Liver Disease (MELD) and Child-Turcotte-
Pugh scores, REVEAL Risk Scores, treatment, and six-
minute walk distance (6MWD), also were collected before
and after therapy. Modality of therapy and timing of ini-
tiation of therapy were documented.
The cohort of the REVEAL registry patients with PoPH
as described in Chest by Krowka et al. [3] was used as the
comparator group. Group comparisons for continuous
variables and categorical variables were made using t tests
and Chi square tests, respectively. For categorical data
involving expected frequency counts \5, the Fisher exact
test was used instead of the Chi square test. Group com-
parisons for ordered data were made using the Cochran–
Armitage trend test. Patient survival (during the first
5 years after diagnosis and the first 2 years after enroll-
ment) and freedom from all-cause hospitalization (during
the first 2 years after enrollment) were assessed using
Kaplan–Meier estimation. A true Kaplan–Meier analysis
was performed on the Henry Ford cohort. Because we did
not have the original raw patient data from the REVEAL
registry cohort, we were not able to produce a direct
Kaplan–Meier comparison of the two cohorts. However,
when the Chi square comparisons of the survival rates at
specific time points were performed, the patient censoring
information from the Henry Ford cohort was directly
accounted for using the raw patient data, and the patient
censoring information from the REVEAL registry cohort
was indirectly accounted for using number of patients still
at risk at various time intervals. Data analysis was per-
formed using version 9.2 of the SAS statistical package.
594 Lung (2013) 191:593–600
123
The patient survival rates at 5 years after diagnosis, the
patient survival rates at 2 years after enrollment, and the
freedom from all-cause hospitalization rates at 2 years after
enrollment were compared between our group and the
REVEAL registry group using Chi square analysis. Sta-
tistical significance was attained for p \ 0.05.
Results
The analysis included 21 patients with PoPH compared
with the REVEAL registry’s 174 patients (Table 1). At the
time of diagnosis and enrollment in the PH Clinic (our
surrogate for enrollment in the REVEAL registry), our
subgroup was similar in age, gender, race, and NYHA
functional class with the majority of patients in each group
in functional class II (24 and 35 % of patients respectively)
or III (52 and 49 % respectively). Our patients were pre-
dominately Child-Turcotte-Pugh B at diagnosis (n = 11;
52 %); five patients (24 %) were Class A, and five patients
(24 %) were Class C. Our patients had a pretreatment
MELD score mean of 12.5 (SD ± 5.1). REVEAL registry
data does not capture severity of liver disease.
Neither the Child-Turcotte-Pugh Class nor the MELD
score had statistically significant change when pretreatment
and posttreatment variables were compared. This also was
true of the REVEAL Risk Score. The pre-REVEAL score
had a mean of 8.6 with a standard deviation of 1.8, whereas
the post-REVEAL score had a mean of 8.7 with a standard
deviation of 2.4. The pre to post change in the REVEAL
score was not statistically significant (p = 0.71). Where
available, hemodynamic variables and 6MWD were com-
pared before and after therapy as represented in Table 2.
Although the timeframe was variable, posttherapy values
are generally within 6–12 months after initiation of ther-
apy. 6MWD increased an average of 30 meters
(p = 0.009). We observed a decrease in mPAP from a
mean of 45 to 33 mmHg (p \ 0.001) as well as a decrease
in PVR from 6.71 to 3.43 WU (p \ 0.001). There also was
an increase in CO from a pretreatment mean of 5.48 to 7.82
L/min (p = 0.005); CI increased from a mean of 2.73 to
4.01 L/min/m2 (p \ 0.001).
Compared with the REVEAL registry data, our PoPH
patients were significantly more likely to be on prostacy-
clin IV at 90 days (p = 0.002) with 67 % of patients begun
on prostacyclin by that time compared with 31 % of
REVEAL registry patients (Table 3). Our patients were
significantly less likely to be on phosphodiesterase inhibi-
tors (PDEI) at 90 days (p \ 0.001) with only 24 % of our
patients on PDEI versus 65 % of REVEAL registry
patients. In addition, 5 % were initiated on inhaled pros-
tacyclin therapy within 90 days of diagnosis and 5 % on an
endothelin receptor antagonist (ETRA). No patients were
untreated at 90 days, nor were any patients were on com-
bination therapy by 90 days. Two patients were on com-
bination therapy at 1 year.
Although our patients did have significantly higher
levels of PCWP at diagnosis, the REVEAL registry authors
do explain that they conducted a separate analysis,
including the 26 patients they had excluded with
PCWP [15 mmHg, which did not affect the results or
conclusions of their study. A separate analysis of our seven
patients with PCWP [15 mmHg demonstrated that they
benefited from treatment with a statistically significant
decrease in mPAP posttreatment (pre-mPAP 49 mmHg;
post-mPAP 31 mmHg; p = 0.03) and improvement in
PVR (pre-PVR 5.29; post-PVR 2.29; p = 0.01). Taken
together, these improvements in hemodynamics would
allow for transplant listing for these patients, having been
brought into the lower-risk category through treatment,
suggesting that it is appropriate to include them in our
analysis.
Survival was assessed both from the time of diagnosis as
well as at the time of enrollment in the Henry Ford
Table 1 Baseline demographic and clinical comparison results for
HFHS patients versus the REVEAL registry
Comparison variable HFHS
study
(N = 21)
REVEAL
registry
(N = 174)
Comparison
p value
Patient age, year
(mean ± SD)
55 ± 10 53 ± 10 0.39 (T)
Gender (female) 48 % 52 % 0.72 (C)
Race (white) 67 % 79 % 0.19 (C)
NYHA at enrollment 0.23 (C–A)
I 24 % 10 %
II 24 % 35 %
III 52 % 49 %
IV 0 % 6 %
AST at enrollment 0.15 (C–A)
Normal 43 % 45 %
B3 times ULN 29 % 42 %
[3 and B5 times ULN 14 % 12 %
[5 times ULN 14 % 2 %
ALT at enrollment 0.04 (C–A)*
Normal 57 % 71 %
B3 times ULN 19 % 20 %
[3 and B5 times ULN 14 % 8 %
[5 times ULN 10 % 1 %
Data are mean ± standard deviation
T two-sample t test, C Chi square test, C–A Cochran-Armitage trend
test, F Fisher exact test, K–M Kaplan–Meier estimation
* Statistically significant, p \ 0.05
Lung (2013) 191:593–600 595
123

Pulmonary Hypertension Clinic. As some patients were
diagnosed outside of the PH clinic and in some cases,
outside of Henry Ford Hospital, the estimate from the time
of enrollment is representative of a mixed population of
newly and previously diagnosed patients. Despite a sig-
nificantly higher proportion of our patients on IV prosta-
cyclin therapy, there were no statistically significant
differences in 2-year freedom from death (patient survival)
or freedom from all-cause hospitalization detected between
the two groups. However, there was a statistically signifi-
cant difference in 5-year patient survival from diagnosis in
our cohort of patients, who benefited from early therapy
with IV prostacyclin. The 2-year survival from enrollment
in Henry Ford PH clinic compared with patients from
REVEAL registry with PoPH is represented in Fig. 1 (2-
year survival estimates 70 and 67 %, respectively;
p = 0.86). The 5-year survival from diagnosis compared
with patients from REVEAL registry with PoPH is repre-
sented in Fig. 2 (5-year survival estimates 71 and 40 %,
respectively; p = 0.02). In subgroup analysis, patients who
did not receive a liver transplant had a probability of both
2-year survival from enrollment and 5-year survival from
diagnosis of 57 % (Figs. 1, 2).
As demonstrated in the REVEAL registry data, patients
with PoPH are prone to frequent hospitalization. Despite
what would be agreed was a more aggressive treatment
strategy, our patients demonstrated similar 2-year freedom
Table 2 Pre- to posttreatment changes
Variable N Mean SD p value
Pre-6MWD 20 330.75 82.77
Post-6MWD 20 360.65 92.56
Post- minus pre-6MWD 20 29.9 91.93 0.009 (W)*
Pre-mPAP 16 45.56 7.73
Post-mPAP 16 33.31 9.65
Post- minus pre-mPAP 16 -12.25 10.72 \0.001 (T)*
Pre-mRAP 16 9.94 8.9
Post-mRAP 16 5.44 3.16
Post- minus pre-mRAP 16 -4.5 9.4 0.108 (W)
Pre-PVR 16 6.71 2.59
Post-PVR 16 3.43 2.21
Post- minus pre-PVR 16 -3.28 2.25 \0.001 (T)*
Pre-PCWP 16 12.88 3.54
Post-PCWP 16 11.19 6.02
Post- minus pre-PCWP 16 -1.69 6.45 0.312 (T)
Pre-CO 16 5.48 2.30
Post-CO 16 7.82 3.05
Post- minus pre-CO 16 2.34 2.87 0.005 (T)*
Pre-CI 16 2.73 1.02
Post-CI 16 4.01 1.44
Post- minus pre-CI 16 1.27 1.39 0.002 (T)*
Pre-TPG 16 31.81 7.72
Post-TPG 16 21.75 8.59
Post- minus pre-TPG 16 -10.06 9.57 \0.001 (T)*
Pre RV systolic pressure 18 71.89 22.70
Post RV systolic pressure 18 55.11 29.96
Post- minus pre-RV systolic
pressure
18 -16.78 23.63 0.008 (T)*
Each pre to post analysis involves only patients with both pre- and
post-data
W Wilcoxon signed-rank test, T paired t test, SD standard deviation
* Statistically significant, p \ 0.05
Table 3 Treatment and outcome comparison results for the HFHS
study versus the REVEAL registry
Comparison variable HFHS
study
(N = 21)
REVEAL
Registry
(N = 174)
Comparison
p value
Prostacyclin IV at 90 days 67 % 31 % 0.002 (C)*
Prostacyclin inhaled at
90 days
5 % 16 % 0.32 (F)
PDEI at 90 days 24 % 65 % \0.001 (C)*
ETRA at 90 days 5 % 8 % 1.00 (F)
Prostacyclin IV at 365 days 55 % 35 % 0.09 (C)
Prostacyclin inhaled at
365 days
10 % 20 % 0.37 (F)
PDEI at 365 days 50 % 71 % 0.06 (C)
ETRA at 365 days 10 % 14 % 1.00 (F)
mPAP at diagnosis
(mean ± SD)
46 ± 9 49 ± 11 0.23 (T)
mRAP at diagnosis
(mean ± SD)
9 ± 8 9 ± 6 1.00 (T)
PVR at diagnosis
(mean ± SD)
7 ± 2 8 ± 4 0.26 (T)
PCWP at diagnosis
(mean ± SD)
13 ± 4 9 ± 3 \0.001 (T)*
Cardiac output at diagnosis
(mean ± SD)
6 ± 2 5 ± 2 0.20 (T)
Cardiac index at diagnosis
(mean ± SD)
2.8 ± 1.0 2.7 ± 0.8 0.60 (T)
TPG at diagnosis
(mean ± SD)
32 ± 8 40 ± 11 0.001 (T)*
6MWD at diagnosis
(mean ± SD)
334 ± 82 354 ± 125 0.48 (T)
2-year patient survival from
enrollment (K–M)
70 % 67 % 0.77 (C)
5-year patient survival from
diagnosis (K–M)
71 % 40 % 0.02 (C)*
2-year all-cause non-hosp.
from enrollment (K–M)
35 % 49 % 0.29 (C)
Data are mean ± standard deviation
T two-sample t test, C Chi square test, C–A Cochran–Armitage trend
test, F Fisher exact test, K–M Kaplan–Meier estimation
* Statistically significant, p \ 0.05
596 Lung (2013) 191:593–600
123

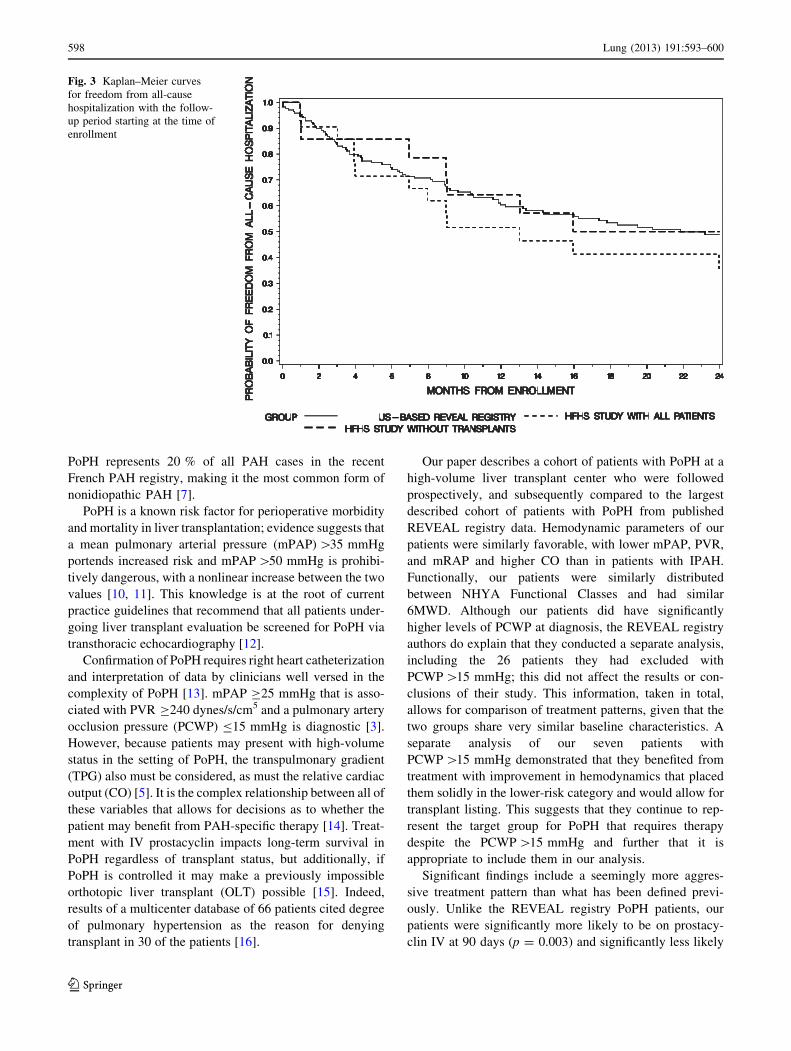
from hospitalization estimates, 35 versus 49 %, respec-
tively; p = 0.29). Figure 3 also plots subgroup analysis of
2-year freedom from hospitalization in patients who were
not transplanted; the resulting probability was 50 %.
Discussion
The amassed data on PoPH is limited by the fact that it is a
rare presentation of a still underrecognized disease (PAH).
PoPH is characterized by the 5th World Symposium on
Pulmonary Hypertension as being within the same group as
idiopathic PAH, which is a reflection of similar pathology
[8]. Due to concomitant cirrhosis, many of these patients
require liver transplantation, and the cohort patients who
receive both PAH-specific therapy and transplant have the
best survival [5]. The prevalence of PoPH reported in the
patients undergoing evaluation for liver transplant has been
reported as 5.3 and 8.5 % in two large series [9, 10]. As
awareness of the disorder increases, this may rise. In fact,
Fig. 1 Kaplan–Meier curves
for patient survival with the
follow-up period starting at the
time of enrollment
Fig. 2 Kaplan–Meier curves
for patient survival with the
follow-up period starting at the
time of diagnosis
Lung (2013) 191:593–600 597
123
PoPH represents 20 % of all PAH cases in the recent
French PAH registry, making it the most common form of
nonidiopathic PAH [7].
PoPH is a known risk factor for perioperative morbidity
and mortality in liver transplantation; evidence suggests that
a mean pulmonary arterial pressure (mPAP) [35 mmHg
portends increased risk and mPAP [50 mmHg is prohibi-
tively dangerous, with a nonlinear increase between the two
values [10, 11]. This knowledge is at the root of current
practice guidelines that recommend that all patients under-
going liver transplant evaluation be screened for PoPH via
transthoracic echocardiography [12].
Confirmation of PoPH requires right heart catheterization
and interpretation of data by clinicians well versed in the
complexity of PoPH [13]. mPAP C25 mmHg that is asso-
ciated with PVR C240 dynes/s/cm5 and a pulmonary artery
occlusion pressure (PCWP) B15 mmHg is diagnostic [3].
However, because patients may present with high-volume
status in the setting of PoPH, the transpulmonary gradient
(TPG) also must be considered, as must the relative cardiac
output (CO) [5]. It is the complex relationship between all of
these variables that allows for decisions as to whether the
patient may benefit from PAH-specific therapy [14]. Treat-
ment with IV prostacyclin impacts long-term survival in
PoPH regardless of transplant status, but additionally, if
PoPH is controlled it may make a previously impossible
orthotopic liver transplant (OLT) possible [15]. Indeed,
results of a multicenter database of 66 patients cited degree
of pulmonary hypertension as the reason for denying
transplant in 30 of the patients [16].
Our paper describes a cohort of patients with PoPH at a
high-volume liver transplant center who were followed
prospectively, and subsequently compared to the largest
described cohort of patients with PoPH from published
REVEAL registry data. Hemodynamic parameters of our
patients were similarly favorable, with lower mPAP, PVR,
and mRAP and higher CO than in patients with IPAH.
Functionally, our patients were similarly distributed
between NHYA Functional Classes and had similar
6MWD. Although our patients did have significantly
higher levels of PCWP at diagnosis, the REVEAL registry
authors do explain that they conducted a separate analysis,
including the 26 patients they had excluded with
PCWP [15 mmHg; this did not affect the results or con-
clusions of their study. This information, taken in total,
allows for comparison of treatment patterns, given that the
two groups share very similar baseline characteristics. A
separate analysis of our seven patients with
PCWP [15 mmHg demonstrated that they benefited from
treatment with improvement in hemodynamics that placed
them solidly in the lower-risk category and would allow for
transplant listing. This suggests that they continue to rep-
resent the target group for PoPH that requires therapy
despite the PCWP [15 mmHg and further that it is
appropriate to include them in our analysis.
Significant findings include a seemingly more aggres-
sive treatment pattern than what has been defined previ-
ously. Unlike the REVEAL registry PoPH patients, our
patients were significantly more likely to be on prostacy-
clin IV at 90 days (p = 0.003) and significantly less likely
Fig. 3 Kaplan–Meier curves
for freedom from all-cause
hospitalization with the follow-
up period starting at the time of
enrollment
598 Lung (2013) 191:593–600
123

to be on PDEI at 90 days (p \ 0.001). This treatment
pattern is influenced by the Mayo Clinic experience and
other published data clearly demonstrating the survival rate
for untreated PoPH patients is abysmally low and that
survival improves with vasodilator therapy regardless of
whether the patient receives liver transplant [5]. Although
the REVEAL registry data were not available to us at the
time of decision regarding initiation of therapy, it too
reinforces that patients with PoPH have significantly worse
survival compared with patients with IPAH and that the
simple diagnosis of PoPH compared with other PAH sub-
groups is an independent risk factor for increased mortality
[3]. Our treatment strategy is comparable to that used for
other types of PAH, with consideration of severity of dis-
ease, functional class, MELD score, and hemodynamics all
considered. Because of the poor prognosis of PoPH, how-
ever, all patients are considered for aggressive therapy with
a prostacyclin infusion. This is in contrast to PAH where
‘‘sicker’’ patients only are considered for more aggressive
therapy. In the setting of PoPH, what are traditionally
considered ‘‘favorable’’ hemodynamics for an IPAH
patient are relatively meaningless, as the prognosis even in
these patients is poor.
This construct is at the root of our treatment patterns,
and in fact, none of our patients with PoPH were untreated
at 90 days. In addition to the 65 % who were initiated on
IV prostacyclin, an additional 5 % were initiated on
inhaled prostacyclin therapy, 25 % on a PDEI, and 5 % on
an ETRA. Seven of our patients were transplanted suc-
cessfully; an additional four patients were cleared for
transplant (Table 4). Few of our patients were on combi-
nation therapy, however, at 365 days. This is perhaps
reflective of our reliance on prostacyclin to achieve
favorable hemodynamics. A majority of our patients were
treated with IV epoprostenol, with a mean dose of 20.8 ng/
kg/min (SD ± 13.9 ng/kg/min). Hemodynamic improve-
ment was noted with a significant decrease in mPAP, PVR,
and improvement in CO as well as CI (Table 2).
In the French series by Le Pavec et al. [17], patients with
PoPH demonstrated a 68 % 5-year survival from diagnosis.
This is despite only 15 % of their patients receiving IV
prostacyclin and is likely related to severity of liver disease
in a cohort where Child-Pugh class A predominated at
51 % of patients. In contrast, REVEAL registry data noted
a 40 % survival. The REVEAL registry researchers had
postulated that the severity of liver disease may have been
the variable influencing the difference in 5-year survival
between the two groups, and this is possible. However, our
cohort had a 5-year survival that surpassed the French
series at 71 %, despite our patients being predominately
Child-Pugh class B (55 %) and C (20 %) at the time of
diagnosis. We believe that the improved 5-year survival in
our patients, despite the severity of liver disease is a direct
reflection of an aggressive treatment strategy (Fig. 2). In
the French series, only 15 % of patients were on IV pros-
tacyclin therapy [17]. In the Mayo Clinic series, the
patients who received PAH therapy only, the 5-year sur-
vival was 45 % [5]. This may have reflected uncontrolled
and variable treatment patterns.
This improved 5-year survival was not apparent in
change in MELD score, Child-Pugh class, or REVEAL risk
score as there was not a statistically significant change
observed pre- and posttreatment in any of these variables.
This suggests to us that the improvement garnered with
therapy is not captured in a measured variable that is
included in these scores.
Small, prospective, cohort studies such as these provide
valuable information that can help broaden the under-
standing of this rare disease; nevertheless, there are many
limitations. Some patient censoring occurred before the end
of the 2- and 5-year follow-up periods. However, the Chi
square comparisons of the 2- and 5-year outcome propor-
tions were based on the underlying Kaplan–Meier esti-
mates and the denominators of those proportions were
adjusted accordingly and did not involve censored patients.
Given the small sample size, the ability to adjust our
Table 4 Patient outcomes
Patient
no.
Date of
death
Narrative outcome
1 Jul-11 Patient elected hospice enrollment due to
HCC
2 Alive Transplanted
3 Alive Transplanted
4 Alive Transplanted
5 Alive Developed HCC
6 Alive Transplanted
7 Alive Continues on medical therapy
8 May-08 Cleared for transplant; elected hospice
enrollment
9 Alive Continues on medical therapy
10 Jun-12 Elected hospice enrollment
11 Aug-11 Patient declined transplant
12 Alive Patient elected hospice enrollment due to
HCC
13 Alive Transplanted
14 Apr-08 Patient elected hospice enrollment
15 Aug-07 Patient elected hospice enrollment
16 Alive Continues on medical therapy
17 Alive Patient cleared for transplant
18 Jul-07 Patient cleared for transplant
19 Alive Not an OLT candidate due to ongoing
drinking
20 Alive Transplanted
21 Alive Transplanted
Lung (2013) 191:593–600 599
123

analysis for possible confounders is limited. Nevertheless,
we believe there is value in a single-center study of this
nature. The consistency and relatively uniform treatment
pattern, which is not true or possible in large registries,
allows insight into this complex and deadly disease.
Conclusions
Early initiation of IV prostacyclin therapy in PoPH patients
at a single institution was associated with improved 5-year
patient survival from diagnosis compared with the
REVEAL registry of PoPH patients and allowed for
clearance for transplant in 52 % of patients within 1 year.
Conflict of interest Dr. Cajigas has served as a consultant for
United Therapeutics Inc., Actelion and Gilead. He has served in the
speaker’s bureau for Actelion and United Therapeutics Inc, received
honoraria for service on Advisory boards from United Therapeutics,
Actelion and Gilead. Dr. Cajigas has received Grants and research
support from Pfizer, Actelion and United Therapeutics. Dr. Awdish
has received Grants and research support from Pfizer, Actelion and
United Therapeutics.
References
1. Rodriguez-Roisin R, Krowka MJ, Herve P et al (2004) Pulmon-
ary-hepatic vascular disorders (PHD). Eur Respir J 24:861–880
2. Krowka MJ, Edwards WD (2000) A spectrum of pulmonary
vascular pathology in portopulmonary hypertension. Liver
Transpl 6:241–242
3. Krowka MJ, Miller DP, Barst RJ et al (2012) Portopulmonary
hypertension: a report from the US-based REVEAL registry.
Chest 141:906–915
4. Kawut SM, Taichman DB, Ahya VN et al (2005) Hemodynamics
and survival of patients with portopulmonary hypertension. Liver
Transpl 11:1107–1111
5. Swanson KL, Wiesner RH, Nyberg SL et al (2008) Survival in
portopulmonary hypertension: Mayo Clinic experience catego-
rized by treatment subgroups. Am J Transplant 8:2445–2453
6. Fix OK, Bass NM, De Marco T et al (2007) Long-term follow-up
of portopulmonary hypertension: effect of treatment with epo-
prostenol. Liver Transpl 13:875–885
7. Frost AE, Badesch DB, Barst RJ et al (2011) The changing
picture of patients with pulmonary arterial hypertension in the
United States: how REVEAL differs from historic and non-US
Contemporary Registries. Chest 139:128–137
8. Simonneau G, Robbins IM, Beghetti M et al (2009) Updated
clinical classification of pulmonary hypertension. J Am Coll
Cardiol 54:S43–S54
9. Krowka MJ, Swanson KL, Frantz RP et al (2006) Portopulmo-
nary hypertension: results from a 10-year screening algorithm.
Hepatology 44:1502–1510
10. Ramsay MA, Simpson BR, Nguyen AT et al (1997) Severe
pulmonary hypertension in liver transplant candidates. Liver
Transpl Surg 3:494–500
11. Krowka MJ, Plevak DJ, Findlay JY et al (2000) Pulmonary
hemodynamics and perioperative cardiopulmonary-related mor-
tality in patients with portopulmonary hypertension undergoing
liver transplantation. Liver Transpl 6:443–450
12. Murray KF, Carithers RL Jr (2005) AASLD practice guidelines:
evaluation of the patient for liver transplantation. Hepatology
41:1407–1432
13. Sussman NL, Kochar R, Fallon MB (2011) Pulmonary compli-
cations in cirrhosis. Curr Opin Organ Transplant 16:281–288
14. McLaughlin VV, Archer SL, Badesch DB et al (2009) ACCF/
AHA 2009 expert consensus document on pulmonary hyperten-
sion a report of the American College of Cardiology Foundation
Task Force on Expert Consensus Documents and the American
Heart Association developed in collaboration with the American
College of Chest Physicians; American Thoracic Society, Inc.;
and the Pulmonary Hypertension Association. J Am Coll Cardiol
53:1573–1619
15. Hollatz TJ, Musat A, Westphal S et al (2012) Treatment with
sildenafil and treprostinil allows successful liver transplantation
of patients with moderate to severe portopulmonary hypertension.
Liver Transpl 18:686–695
16. Krowka MJ, Mandell MS, Ramsay MA et al (2004) Hepatopulmo-
nary syndrome and portopulmonary hypertension: a report of the
multicenter liver transplant database. Liver Transpl 10:174–182
17. Le Pavec J, Souza R, Herve P et al (2008) Portopulmonary
hypertension: survival and prognostic factors. Am J Respir Crit
Care Med 178:637–643
600 Lung (2013) 191:593–600
123