dystrophin-deficient cardiomyopathy · advances in management, namely corticosteroid treatments,...
TRANSCRIPT

Listen to this manuscript’s
audio summary by
JACC Editor-in-Chief
Dr. Valentin Fuster.
J O U R N A L O F T H E AM E R I C A N C O L L E G E O F C A R D I O L O G Y V O L . 6 7 , N O . 2 1 , 2 0 1 6
ª 2 0 1 6 B Y T H E AM E R I C A N C O L L E G E O F C A R D I O L O G Y F O UN DA T I O N I S S N 0 7 3 5 - 1 0 9 7 / $ 3 6 . 0 0
P U B L I S H E D B Y E L S E V I E R h t t p : / / d x . d o i . o r g / 1 0 . 1 0 1 6 / j . j a c c . 2 0 1 6 . 0 2 . 0 8 1
REVIEW TOPIC OF THE WEEK
Dystrophin-Deficient Cardiomyopathy
Forum Kamdar, MD, Daniel J. Garry, MD, PHDABSTRACT
Fro
rep
Ma
Dystrophinopathies are a group of distinct neuromuscular diseases that result from mutations in the structural
cytoskeletal Dystrophin gene. Dystrophinopathies include Duchenne muscular dystrophy (DMD) and Becker muscular
dystrophy (BMD), X-linked dilated cardiomyopathy, as well as DMD and BMD female carriers. The primary presenting
symptom in most dystrophinopathies is skeletal muscle weakness. However, cardiac muscle is also a subtype of striated
muscle and is similarly affected in many of the muscular dystrophies. Cardiomyopathies associated with dystrophin-
opathies are an increasingly recognized manifestation of these neuromuscular disorders and contribute significantly to
their morbidity and mortality. Recent studies suggest that these patient populations would benefit from cardiovascular
therapies, annual cardiovascular imaging studies, and close follow-up with cardiovascular specialists. Moreover, patients
with DMD and BMD who develop end-stage heart failure may benefit from the use of advanced therapies. This review
focuses on the pathophysiology, cardiac involvement, and treatment of cardiomyopathy in the dystrophic patient.
(J Am Coll Cardiol 2016;67:2533–46) © 2016 by the American College of Cardiology Foundation.
DYSTROPHINOPATHIES:
A HISTORICAL PERSPECTIVE
The earliest detailed descriptions of muscular dys-trophies were by Meryon (1) in England in 1852 and byDuchenne (2) in France in 1868, identifying a diseasethat affected young boys with severe muscularweakness and an abnormal increase in calf size.Duchenne (2) observed fatty-fibrous replacement ofskeletal muscle in muscle biopsies obtained fromthese boys; for this contribution, the disease wasnamed after him. In 1886, Gowers (3) described how achild affected with muscular dystrophy used his armsto rise from the ground. Although Duchennemuscular dystrophy (DMD) had been known as aclinical entity since the 1860s, its cause was notelucidated until over 100 years later. In a landmarkdiscovery, Louis Kunkel’s laboratory (1986) identifiedthe DMD gene responsible for causing DMD and, ayear later, demonstrated that mutations resultedin the absence of the 427-kDa rod-like protein dys-trophin (4,5). These major advancements allowed for
m the Cardiovascular Division, Lillehei Heart Institute, University of Min
orted that they have no relationships relevant to the contents of this pap
nuscript received December 14, 2015; revised manuscript received Febru
the development of diagnostic and genetic testing, aswell as the establishment of disease models that haveenhanced our understanding of DMD. Although anumber of questions remain regarding the musculardystrophies, the rich history of discovery has signifi-cantly accelerated our knowledge of DMD in the last30-year period.
DYSTROPHINOPATHIES: INHERITANCE
DMD is the most common form of muscular dys-trophy, affecting 1 in every 5,000 boys born in theUnited States (Table 1) (6–8). DMD is a result ofan inherited or spontaneous mutation of the DMDgene. DMD is a 2.5-Mb gene located on chromosomeXp21.1 (5,9), and is the largest known gene,harboring 79 exons and encoding a 14-kb transcript(Figure 1). The full-length dystrophin gene has 3promoters: the M promoter produces the Dp427misoform, expressed in skeletal and cardiac muscle;the B promoter produces Dp427c, expressed inthe brain; and the P promoter produces Dp427p,
nesota, Minneapolis, Minnesota. Both authors have
er to disclose.
ary 16, 2016, accepted February 23, 2016.

TABLE 1 DMD Vers
Dystrophin protein
Incidence
Mean age at onset, y
Mean age of becominnonambulatory, y
Mean life expectancy
Onset of cardiomyopa
BMD ¼ Becker muscular
ABBR EV I A T I ON S
AND ACRONYMS
ACEI = angiotensin-converting
enzyme inhibitor
BMD = Becker muscular
dystrophy
CMR = cardiac magnetic
resonance imaging
CRISPR = clustered regularly-
interspaced short palindromic
repeat
DGC = dystroglycan complex
DMD = Duchenne muscular
dystrophy
ECG = electrocardiogram
ICD = implantable
cardioverter-defibrillator
LVAD = left ventricular
assist device
LVEF = left ventricular ejection
fraction
Kamdar and Garry J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6
DMD and BMD Cardiomyopathy M A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6
2534
expressed in the Purkinje cells in the brain(Figure 1) (10).
An out-of-frame mutation of 1 or several ofthe 79 exons in the full-length Dystrophingene results in an absence of a functionaldystrophin protein, causing the DMD pheno-type. DMD is inherited in an X-linked reces-sive manner, where a female carrier with 1 Xchromosome carrying the DMD mutation hasa 50% chance of passing on the mutated Xchromosome to her son. Female carriers canalso have symptoms on the basis of theirX-inactivation. Although the majority of DMDmutations are inherited, spontaneous muta-tions account for 30% of DMD cases (11).
DYSTROPHIN AND DYSTROGLYCAN
COMPLEX ORGANIZATION
Dystrophin, a rod-shaped cytoplasmic pro-tein, connects the dystroglycan complex
(DGC) to the intracellular contractile apparatus andextracellular matrix (ECM) of the cell (Figure 2) (12).Dystrophin is similar to spectrin and consists of 2ends separated by long, flexible, rod-like regions. TheN-terminus binds actin and the C-terminus binds toglycoproteins in the sarcolemma. The role of dystro-phin is to stabilize the plasma membrane by trans-mitting forces generated by the sarcomericcontraction to the ECM (Figure 2).
The DGC is a multimeric complex composed ofglycated integral membrane proteins and peripheralproteins that form a structural link between thefilamentous (F)-actin cytoskeleton and the ECM inboth cardiac and skeletal muscle (13). The DGCis comprised of cytoskeletal proteins, dystrophin,syntrophins, dystroglycans, sarcoglycans, DGC-associated proteins, neuronal nitric oxide synthase,and dystrobrevin (14). The fully nucleated DGC pro-vides mechanical support to the skeletal or cardiacplasma membrane during contraction, and loss of
us BMD
DMD BMD
Absent Partially functional
1:5,000 male births 1:19,000
rs 3–5 12
grs
w12 w27
, yrs Mid to late 20s 40s
thy, yrs 16–18 Variable; cardiomyopathymay precede skeletalsymptoms
dystrophy; DMD ¼ Duchenne muscular dystrophy.
function or absence of 1 or several of these DGC pro-teins leads to plasma membrane fragility. DMD andBecker muscular dystrophy (BMD) arise from muta-tions in dystrophin, and other forms of muscular dys-trophies arise from mutations in DGC components. Inskeletal muscle, the DGC is located at regular intervalsin structures known as costameres, whereas in cardiacmuscle, the DGC is not located in discrete costameresand may have a unique composition (15,16).
PATHOGENESIS AND PHYSICAL FINDINGS
Skeletal muscle and heart lacking functional dystro-phin are mechanically weak, and contraction of thecell (skeletal myocytes and cardiac myocytes) leadsto membrane damage (17–19) (Central Illustration).Loss of membrane integrity results in a cascade ofincreased calcium influx into the cell and eventualcell death. Clinically, dystrophin loss manifests asprogressive muscle weakness (6). Symptoms are firstnoted in early childhood and include calf pseudohy-pertrophy, an inability to stand without using thearms for assistance (Gower maneuver), toe walking,and difficulty keeping up with peers. As the diseaseprogresses, patients’ skeletal muscle becomesincreasingly weak, causing atrophy and contractures,which subsequently results in the loss of ambulationin their teens. Due to ongoing muscle damage, pa-tients with DMD have markedly elevated serum levelsof the muscle protein creatine kinase (CK), which maybe 10� to over 100� the normal limit, and an elevatedCK level is a diagnostic sign (20). The diagnosis ofDMD can be confirmed by muscle biopsy demon-strating the absence of dystrophin (Figure 3) andusing genetic testing for dystrophin mutations.
SURVIVAL
Historically, this progressive muscle weakness resul-ted in loss of ambulation by 10 to 12 years of age anddeath during the second decade of life, primarily dueto respiratory failure. However, with the advent ofnocturnal ventilation, spinal stabilization surgery,and steroid treatment, the life expectancy of boyswith DMD has increased to the late twenties to earlythirties (21) (Figure 4). Although respiratory failurewas previously the major cause of death, cardiomy-opathy has now emerged as the leading cause ofdeath in patients with DMD (22).
NONCARDIAC TREATMENTS
Advances in management, namely corticosteroidtreatments, spinal stabilization, and improved pul-monary support, have significantly improved the life

FIGURE 1 Dystrophin Gene and Protein
A
B
B/Dp427
M/Dp427
P/Dp427500 kb 1000 kb
R/Dp260 B,K/Dp140 S/Dp116 G/Dp71
1500 kb 2500kb
COOH
Dystrobrevin binding siteSyntrophin binding site
Dystroglycan
Syntrophindystrobrevin
DystroglycanBinding site
nNOSbinding
Rod Domain
Actinbindingdomain
1 2 10
H1
H2
H3
H4
241
20
30
40
45
50
56
60
63
70
79
Gene
Exons
Structural NH2
N terminus
(A) The 79 exons of the full-length dystrophin gene with promoters for the alternatively spliced isoforms indicated by red arrows and boxes
specific for each promoter. Full-length dystrophin promoters include: M, muscle promoter; B, brain promoter; and P, Purkinje promoter. Other,
shorter dystrophin isoforms include: Dp260, expressed in the retina (R); Dp140, expressed in kidney (K) and brain (B); Dp116, expressed in
Schwann cells (S); and Dp71, which is ubiquitously expressed. (B) Schematic of the protein structure of dystrophin, including the actin-binding
site at the N-terminus and the DGC protein-binding site at the C-terminus. The rod domain contains 24 spectrin-like repeats with 4 hinge
regions. DGC ¼ dystroglycan complex.
J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6 Kamdar and GarryM A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6 DMD and BMD Cardiomyopathy
2535
expectancy of boys/men with DMD to the latetwenties and early thirties (21). Glucocorticoids areone of the mainstays of treatment for DMD and havebeen shown to improve muscle strength, function,and pulmonary function (23). Glucocorticoids arecurrently recommended in patients with DMD whoare 5 years of age or older who are not gaining or havea decline in motor skills (24). Current glucocorticoidprotocols that have been tested in randomized clin-ical trials include: daily prednisone 0.75 mg/kg/day;intermittent prednisone 0.75 mg/kg/day, 10 days onand 10 days off; and daily deflazacort 0.9 mg/kg/day(25). Deflazacort is another steroid that is used in theDMD patient population and may have a more limitedside effect profile compared with prednisone (26).
Scoliosis and kyphosis, common, progressivesequelae of DMD, subsequently result in decreasedpulmonary function and discomfort in these patients;however, implementing spinal stabilization surgeryusing Harrington rods has improved survival, com-fort, and pulmonary function (27). Respiratory com-plications occur frequently in patients with DMD aslosing respiratory muscle strength leading todecreased ventilation, and they are at increased riskfor pneumonia, atelectasis, and respiratory failure(28). The majority of deaths in end-stage DMD pre-viously occurred as a result of respiratory failure andinfections; however, the introduction of noninvasivenocturnal mechanical ventilation has improved sur-vival in patients with DMD and may improvecardiac function through afterload reduction (29,30).
Additionally, nutrition, exercise therapy, and psy-chosocial support are critical in the interdisciplinarycare of patients with DMD (27).
CARDIAC INVOLVEMENT IN DMD
The reduction of respiratory-related deaths due tonocturnal ventilation and spinal stabilization hascontributed to the increase of DMD cardiomyopathydue to the increased survival and advanced age ofpatients with DMD (21,31). The incidence of cardio-myopathy in DMD increases with age. Although it isestimated that 25% of boys have cardiomyopathy at6 years of age and 59% by 10 years of age, cardiacinvolvement is nearly ubiquitous in older patientswith DMD, as more than 90% of young men over18 years of age demonstrate evidence of cardiacdysfunction (22). Dilated cardiomyopathy typicallyhas an onset in the midteen years and progressivelyremodels and contributes to the demise of patientswith DMD (22,32) (Figure 5). Distinct dystrophinmutations have been correlated to an increasedincidence of cardiomyopathy and possible responseto treatment (33). Recognition of heart failuresymptoms in patients with DMD can be challengingdue to physical inactivity and other respiratorycomplaints that can obscure the diagnosis (34).Currently, clinical guidelines recommend the initialcardiac screening at the time of diagnosis of DMD,every 2 years until 10 years of age, and then yearlythereafter (24).

FIGURE 2 Schematic Diagram of the DGC
ECMLaminin
Sarcoglycans
Dystrophin
Actin cytoskeleton
Extracellular
Intracellular
Syntrophin
nNOS
Sarcolemma
αα-dystroglycan
β-dystroglycan
Dystrobrevin
COOH
NH3
The DGC spans the sarcolemma and links the cytoplasmic actin cytoskeleton to the ECM via dystrophin. Transmembrane components of the DGC
include sarcoglycans, beta-dystroglycan, and extracellular alpha-dystroglycan, which binds laminin. Cytoplasmic components include dystrophin
(or utrophin), which binds dystrobrevin, syntrophin, and nitric oxide synthase. DGC ¼ dystroglycan complex; ECM ¼ extracellular matrix.
Kamdar and Garry J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6
DMD and BMD Cardiomyopathy M A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6
2536
Most patients with DMD will have abnormal elec-trocardiographic tracings. The classical pattern dem-onstrates tall R waves and increased R/S amplitude inlead V1, Q waves in the left precordial leads, right axisdeviation, or complete right bundle branch block(Figure 6) (35–37). These electrocardiographic (ECG)findings correlate with the pathological studies thathave demonstrated a predilection for fibrosis in thebasal posterior wall of the heart in patients withmuscular dystrophies and may reflect a reduction inelectrical activity in the inferobasal ventricular wall(38). ECG findings are believed to precede echocar-diographic findings of cardiomyopathy; however, nocorrelation between ECG findings and the presence ofcardiomyopathy has yet been established (35,39). Theuse of serum biomarkers, such as cardiac troponin orB-type natriuretic peptides (BNPs), has not been wellestablished for DMD screening and will need furtherexamination in the future.
Arrhythmias are a common cardiac involvement inpatients with muscular dystrophies. Sinus tachy-cardia is a common finding in patients with DMD(40–42). Dystrophic patients have elevated heartrates, even when compared with patients with othermuscular dystrophies or deconditioned patients (42).Pathological examination of the heart in dystrophicpatients has demonstrated fibrosis of the conductionsystem, in addition to the myocardium (43), whichmay, in part, explain the autonomic dysfunctionin the DMD population (44,45). Additionally, sinustachycardia may correlate with cardiac dysfunction in
patients with DMD (46). Dalmaz et al. (47) demon-strated that urinary catecholamines increased inpatients with DMD around 10 years of age, whichcorresponded temporally to heart rate elevation andcardiac involvement. In patients with DMD cardio-myopathy, atrial arrhythmias, including atrial fibril-lation, atrial flutter, and atrial tachycardias, can alsooccur. Ventricular tachycardia, premature ventricularcomplexes, and other conduction abnormalities havebeen noted, and patients with DMD with left ven-tricular ejection fraction (LVEF) <35% have a signifi-cantly higher burden of ventricular tachycardia(48,49). In addition to low LVEF, increased QTdispersion has been identified as a risk factor forventricular arrhythmias in DMD; however, there wasno prognostic value of signal-averaged ECG (49,50).
The role of implantable cardioverter-defibrillators(ICDs) is well-established for primary prevention ofsudden cardiac death in patients with an LVEF <35%;however, the use of ICDs has not been well estab-lished in patients with DMD cardiomyopathy (51). Theguidelines discourage the use of ICDs for a patientwith a life expectancy <1 year, and ICDs may not havebeen utilized in the past due to the previous highmortality in patients with DMD. The recent use ofneurohormonal therapy may also improve LVEF toreduce the risk of sudden cardiac death. We wouldrecommend ICD implantation candidacy for patientswith DMD with LVEF <35%; however, an individual-ized discussion with patients and families is neces-sary, given the course of disease progression.

CENTRAL ILLUSTRATION Duchenne Muscular Dystrophy: Pathogenesis and Therapies
DUCHENNE MUSCULAR DYSTROPHY PATHOGENESIS THERAPIES
Disease typically affects boys(1 in every 5,000 boys born in the U.S.)
Inherited or spontaneous mutation in the DMD gene
Possible learning difficulties
Scoliosis
Weak respiratory
muscles
Muscle loss and weakness
leading toloss of
ambulation
Absence of dystrophin—the anchor that connects the extracellular matrixand membrane proteinsto the cell cytoskeleton
Contraction of muscle cell leads to cell membrane damage
Increased calcium influx into damaged cell leads to cell death
Progressive muscle weakness
Cardiomyopathy
Current cardiac treatments
• Steroid treatment (glucocorticoids)• Neurohormonal therapy: Angiotensin
converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs)
Current non-cardiac treatments
• Noninvasive nocturnal ventilation• Nutrition, exercise therapy,
and psychological support• Spinal stabilization
Emerging treatments
• Implantable cardioverter defibrillation • Beta-blockers• Aldosterone inhibitors• Exon skipping• Dystrophin gene replacement• Gene editing mediated with CRISPR/Cas9• Advanced heart failure therapies
Dystrophin
Kamdar, F. et al. J Am Coll Cardiol. 2016;67(21):2533–46.
ACEI ¼ angiotensin-converting enzyme inhibitor; ARB ¼ angiotensin receptor blocker; CRISPR/Cas9 ¼ clustered regularly interspaced short palindromic
repeats/CRISPR-associated systems; DMD ¼ Duchenne muscular dystrophy.
J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6 Kamdar and GarryM A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6 DMD and BMD Cardiomyopathy
2537
CARDIAC IMAGING IN DMD
Cardiac imaging can be challenging in patients withend-stage DMD due to scoliosis, ventilation abnor-malities, and contractures. Current, commonly usedimaging modalities include echocardiography andcardiac magnetic resonance (CMR). Echocardiographyin DMD cardiomyopathy has demonstrated regionalwall motion abnormalities in the posterior basal wall,left ventricular dilation, and overall reduced systolicfunction (52). Current guidelines recommend anechocardiogram at the time of diagnosis or by 6 yearsof age, with repeat echocardiograms every 1 to 2 yearsuntil 10 years of age. After 10 years of age, it is rec-ommended that patients have an annual echocar-diogram to assess left ventricular function (27).Although echocardiography is easily accessible, rela-tively quick, and a cost-effective imaging modalityto utilize, it can be technically challenging inpatients with DMD due to chest wall deformities,scoliosis, and respiratory dysfunction, thus limitingthe diagnostic yield (53). CMR has been used to imagepatients with DMD and can provide one of the mostaccurate assessments of left ventricular size and
function (54–56). Silva et al. (54) performed gadolin-ium contrast-enhanced CMR on 10 patients withdystrophinopathies (8 patients with DMD and 2 withBMD) and were the first to demonstrate late gado-linium enhancement (LGE) by CMR in the dystrophicheart. They further demonstrated that LGE was pre-sent by echocardiography, even with normal leftventricular function (54). Puchalski et al. (57) subse-quently performed a study of 74 patients with DMD,where the majority of patients had LGE in the post-erobasal region of the left ventricle in a subepicardialdistribution (Figure 7). This pattern of LGE in thebasal inferior and inferolateral walls is consistentwith the pathological findings of fibrosis in the infe-rior basal wall (38,58) (Figure 8). Myocardial fibrosis,as assessed by LGE in DMD, has been demonstrated toincrease with age and correlates with a decline inLVEF (59,60). Thus, CMR may provide earlier detec-tion of cardiovascular involvement in DMD, allow foraccurate and reproducible quantification of left ven-tricular function and size, and promote initiation ofearlier cardioprotective treatment. CMR has manyimaging benefits for patients with DMD; however, itcan also be challenging in the pediatric population
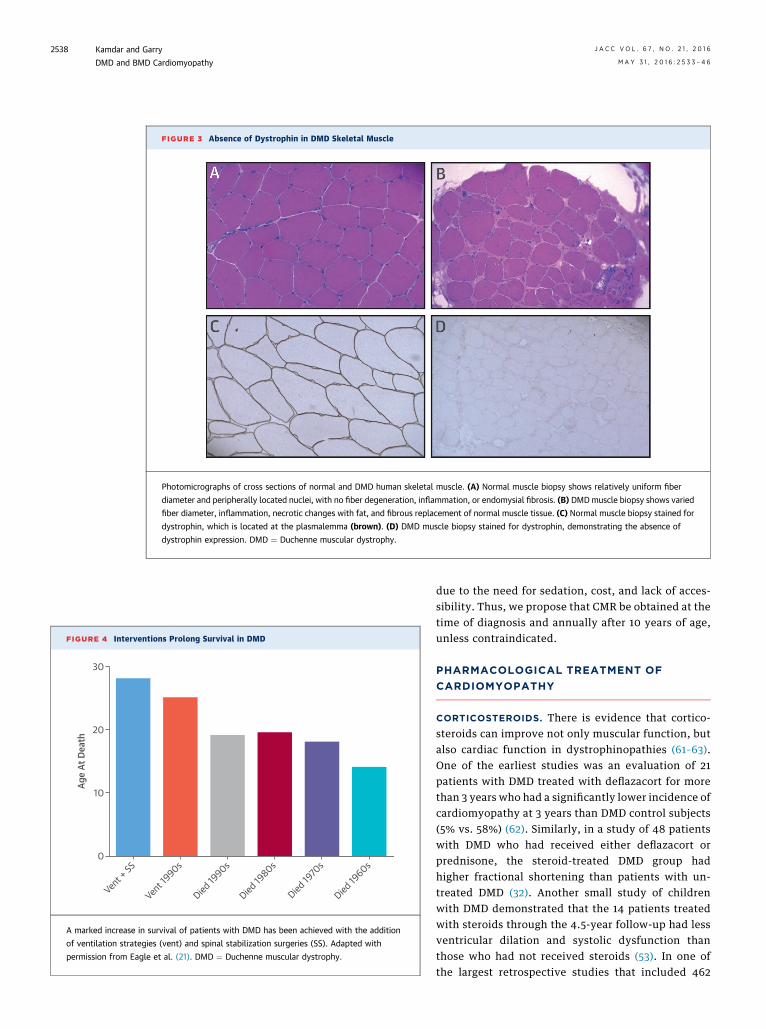
FIGURE 4 Interventions Prolong Survival in DMD
30
20
10
Age
At D
eath
0
Vent +
SS
Vent 1
990s
Died 19
90sDied
1980s
Died 19
70s
Died 19
60s
A marked increase in survival of patients with DMD has been achieved with the addition
of ventilation strategies (vent) and spinal stabilization surgeries (SS). Adapted with
permission from Eagle et al. (21). DMD ¼ Duchenne muscular dystrophy.
FIGURE 3 Absence of Dystrophin in DMD Skeletal Muscle
Photomicrographs of cross sections of normal and DMD human skeletal muscle. (A) Normal muscle biopsy shows relatively uniform fiber
diameter and peripherally located nuclei, with no fiber degeneration, inflammation, or endomysial fibrosis. (B) DMDmuscle biopsy shows varied
fiber diameter, inflammation, necrotic changes with fat, and fibrous replacement of normal muscle tissue. (C) Normal muscle biopsy stained for
dystrophin, which is located at the plasmalemma (brown). (D) DMD muscle biopsy stained for dystrophin, demonstrating the absence of
dystrophin expression. DMD ¼ Duchenne muscular dystrophy.
Kamdar and Garry J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6
DMD and BMD Cardiomyopathy M A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6
2538
due to the need for sedation, cost, and lack of acces-sibility. Thus, we propose that CMR be obtained at thetime of diagnosis and annually after 10 years of age,unless contraindicated.
PHARMACOLOGICAL TREATMENT OF
CARDIOMYOPATHY
CORTICOSTEROIDS. There is evidence that cortico-steroids can improve not only muscular function, butalso cardiac function in dystrophinopathies (61–63).One of the earliest studies was an evaluation of 21patients with DMD treated with deflazacort for morethan 3 years who had a significantly lower incidence ofcardiomyopathy at 3 years than DMD control subjects(5% vs. 58%) (62). Similarly, in a study of 48 patientswith DMD who had received either deflazacort orprednisone, the steroid-treated DMD group hadhigher fractional shortening than patients with un-treated DMD (32). Another small study of childrenwith DMD demonstrated that the 14 patients treatedwith steroids through the 4.5-year follow-up had lessventricular dilation and systolic dysfunction thanthose who had not received steroids (53). In one ofthe largest retrospective studies that included 462

FIGURE 5 DMD Cardiac Disease Progression
Schematic outlining DMD cardiomyopathy disease progression. Initially, patients with DMD have structurally normal hearts. Subsequently, patients with
DMD develop fibrosis of the inferobasal wall as the earliest sign of myocardial involvement. Over time, this leads to progressive fibrosis, left ventricular (LV)
dysfunction, and dilation leading to end-stage heart failure. DMD ¼ Duchenne muscular dystrophy.
J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6 Kamdar and GarryM A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6 DMD and BMD Cardiomyopathy
2539
patients with DMD, those treated with steroids had adelay in the onset of cardiomyopathy (64). Addition-ally, another retrospective study that included 86patients with DMD concluded that patients with DMD
FIGURE 6 ECG Changes in DMD
Electrocardiogram (ECG) from a patient with DMD. The ECG demonstrat
V1 and V2. DMD ¼ Duchenne muscular dystrophy.
who received steroids had significantly lower cardio-vascular mortality and incidence of new cardiomy-opathy than control subjects, which was largelydriven by a reduction in heart failure–related
es sinus tachycardia, Q waves in leads I, aVL, and V4 to V6, and large R waves in

FIGURE 7 CMR Demonstrating Dilated Cardiomyopathy and
Fibrosis in a Patient With DMD
CMR delayed enhancement image in the basal short-axis view,
performed on a 1.5-T MRI. This image demonstrates near trans-
mural enhancement (gray) in the left ventricular (LV) basal-
midanterolateral, inferolateral, and lateral wall (area denoted by
dashed white line) consistent with myocardial scar or fibrosis. This
is in an 18-year-old male patient with DMD and exon 24 deletion
with associated cardiomyopathy (LV ejection fraction 33%). CMR ¼cardiac magnetic resonance; DMD¼ Duchennemuscular dystrophy;
MRI ¼ magnetic resonance imaging; RV ¼ right ventricle.
Kamdar and Garry J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6
DMD and BMD Cardiomyopathy M A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6
2540
deaths (65). However, a confounding variable in thisstudy is that the patients with DMD treated with ste-roids also began taking angiotensin-convertingenzyme inhibitors (ACEIs) approximately 3 yearsearlier than the control subjects, which may have alsobeen cardioprotective and had an effect on the results.
Although several studies have demonstrated adelay in onset of cardiomyopathy in patients withDMD, these have all been retrospective, observationalstudies with inherent limitations. Additionally, thevariability of steroid dosing and duration poses achallenge to interpretation of the data and broadapplication of the results. To rigorously assess theeffect of corticosteroid use on ventricular function inpatients with DMD, a prospective randomizedcontrolled trial will be necessary.
ACEIs AND ANGIOTENSIN RECEPTOR BLOCKERS.
ACEIs and angiotensin receptor blockers (ARBs) are acornerstone of neurohormonal modulation in heartfailure and were shown to be effective in the reduc-tion of cardiovascular mortality in patients with heartfailure (51). Duboc et al. (66) assessed the effect ofACEIs in patients with DMD with preserved left
ventricular function. They randomized 57 childrenwith DMD (mean age 10.7 years) to perindopril (2 to 4mg/day) or placebo. At 3 years of follow-up, there wasno significant difference in LV function between thechildren treated with perindopril or placebo. How-ever, 3 years following enrollment, all patients wereswitched to perindopril treatment and followed for anadditional 2 years. After crossing over to perindoprilat 2 years, there was no difference in mean LV func-tion between those patients treated with perindoprilinitially versus those initially treated with placebo.However, in the initial placebo group, 8 of 29 patientshad LVEF <45%, whereas only 1 of 27 patients had LVdysfunction in the perindopril group (p ¼ 0.02),supporting the notion that early treatment with per-indopril was effective in preventing progression toleft ventricular dysfunction in DMD. After a 10-yearfollow-up period, only 65% of patients in the initialplacebo group were alive versus 92.9% in the initialperindopril group (p ¼ 0.013), which emphasized thatearly initiation of an ACEI reduced mortality in pa-tients with DMD (67). Current guidelines for DMDrecommend initiation of ACEI in patients with DMDonly after LV dysfunction has developed (27); how-ever, on the basis of studies by Duboc et al. (66,67),we would recommend initiation of an ACEI (by 10years of age) before the development of left ventric-ular dysfunction in patients with DMD, as they are athigh risk for developing left ventricular dysfunction(ACC heart failure stages A and B). Additionally, forthose patients who are intolerant to ACEIs, ARBs canalso be used, as they were demonstrated to be aseffective as ACEIs in DMD (68).BETA-ADRENERGIC RECEPTOR BLOCKERS. Althoughthe benefit of ACEIs in DMD cardiomyopathy has beendefinitively established, the efficacy of beta-blockersin DMD cardiomyopathy has been less clear. The useof carvedilol has been assessed in pediatric patientswith DMDwho have elevated atrial natriuretic peptide(ANP) or BNP and a low ejection fraction (EF <40%) byechocardiography with no significant difference incarvedilol-treated patients with respect to symptomsor left ventricular dysfunction (69). However, in astudy by Rhodes et al. (70), carvedilol was shown to beefficacious in patients with DMD cardiomyopathy.When superimposed on background therapy of ACEIs,the use of carvedilol in this patient population re-mains unclear. In an analysis of 13 patients with DMDwho were treated with ACEI versus ACEI and carve-dilol, echocardiography revealed a beneficial effect ofbeta-blocker therapy in increasing left ventricularshortening and decreasing left ventricularend-diastolic dimensions (71). In contrast, a recentstudy by Viollet et al. (72) tested ACEI alone versus

TABLE 2 DMD Cardiomyopathy Treatment
Level of Evidence
Corticosteroids þþACE inhibitors þþþBeta-blockers þMineralocorticoid receptor antagonists þ
ACE ¼ angiotensin-converting enzyme; DMD ¼ Duchenne muscular dystrophy.
FIGURE 8 Cardiac Fibrosis in DMD
Photomicrograph of the basal posterior wall of the left ventricle in a patient with DMD
demonstrating subepicardial fibrotic replacement (blue) of the myocardium using Masson
trichrome stain. DMD ¼ Duchenne muscular dystrophy.
J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6 Kamdar and GarryM A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6 DMD and BMD Cardiomyopathy
2541
ACEI and metoprolol; in this study, a low-dosebeta-blocker was added only for heart rates above100 beats/min or if arrhythmias occurred. The resultsshowed an improvement from pre-treatment LVEFin both groups, but no difference between them.Further research with larger groups of patients andmore robust trial designs are needed to definitivelyaddress the use of beta-blockers in DMD; however, onthe basis of current guidelines, we would recommendthat beta-blockers be initiated in patients with DMDwho have left ventricular dysfunction (51).
MINERALOCORTICOID RECEPTOR ANTAGONISTS.
Aldosterone inhibitors, such as spironolactone oreplerenone, are standard heart failure therapy forpatients with LVEF <35% and New York Heart Asso-ciation functional class II to VI symptoms (51). In arecently completed randomized double-blinded clin-ical trial, eplerenone, an aldosterone antagonist, orplacebo was added to background therapy of ACEIs orARBs in patients with DMD who had normal LVfunction to assess the efficacy of eplerenone in pre-venting cardiomyopathy in DMD. Twenty patientswere randomized to eplerenone and 20 to placebo,and they were followed for 6 and 12 months withCMR. The primary endpoint was change in left ven-tricular circumferential strain, which is a markerof contractility, at 12 months. The investigatorsobserved that the decline in LV circumferential strainwas lower in the group treated with eplerenone thanin the placebo group, although there was no overallchange in left ventricular function in either group(73). This small study demonstrated attenuation ofprogressive left ventricular dysfunction that occurs inDMD; although this study is positive, further studiesto assess the effect of eplerenone on DMD survival arewarranted. Table 2 summarizes cardiac pharmaco-logical treatments for DMD cardiomyopathy.
CARDIAC TRANSPLANTATION AND
LEFT VENTRICULAR ASSIST DEVICES
The only curative therapy for end-stage heart failureremains cardiac transplantation. Heart failure withmultisystem organ involvement and inability torehabilitate after cardiac transplantation has been arelative contraindication for orthotopic heart trans-plantation, and thus has limited the broad use of thistherapy in the DMD/BMD population. Rees et al. (74)were the first to describe heart transplantationin patients with muscular dystrophies in a singleGerman center. Of 582 transplants performed, 3patients with DMD and 1 patient with BMD underwentcardiac transplantation with a mean duration offollow-up of 40 months. These patients tolerated
immunosuppression, had no difference in post-operative intubation, and were able to be rehabili-tated (74). Ruiz-Cano et al. (75) described a Spanishsingle-center experience with heart transplantation in3 patients with BMD who underwent cardiac trans-plantation with a mean follow-up duration of 57months; this study also demonstrated that patientswith BMD had an intraoperative and post-operativecourse comparable to nonmuscular dystrophypatients undergoing heart transplantation. Patanèet al. (76) also described a single case of successfultransplantation in a patient with cardiomyopathysecondary to BMD. We performed a more recentmulticenter registry analysis of cardiac trans-plantation using the Cardiac Transplant ResearchDatabase and identified 29 patients with musculardystrophies, of whom 15 had BMD and 3 had DMD, whounderwent cardiac transplantation between 1995 and2005 and compared them with 275 nonmuscular dys-trophy, nonischemic patients matched for age, body

Kamdar and Garry J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6
DMD and BMD Cardiomyopathy M A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6
2542
mass index, sex, and race (77). We demonstrated thatthere was no significant difference in 1- or 5-year sur-vival, transplant rejection, infection, or allograft vas-culopathy between the patients with and withoutmuscular dystrophy. These studies described compa-rable outcomes of cardiac transplantation in a smalland select group of patients with DMD and BMD withend-stage cardiomyopathy; however, the functionalstatus of these patients prior to transplantation wasnot documented, and these studies may have aselection bias. We propose that orthotopic hearttransplantation should be considered for the patientwith DMD or BMD who has end-stage heart failure,provided that other comorbidities do not limit survival(i.e., respiratory failure). Further research regardingcardiac transplantation in patients with DMD and BMDwho have end-stage cardiomyopathy is warranted.
Given the scarcity of organs for heart trans-plantation, the use of left ventricular assist devices(LVADs), demonstrated to be to be effective in treat-ing patients with end-stage or advanced heart failure,is applicable to a larger population, including thosewith muscular dystrophies, as LVADs can be used asdestination therapy without the need for trans-plantation (78,79). Two groups recently reportedcases of successful implantation of LVADs as desti-nation therapy in patients with DMD (80,81). Amodeoet al. (80) were the first to describe LVAD implanta-tion in 2 pediatric patients with DMD. These in-vestigators implanted the Jarvik 2000 LVAD (JarvikHeart Inc., New York, New York) in a 15-year-old boywith DMD who had inotrope-refractory heart failureand in a 14-year-old boy with DMD who was bridgedfrom extracorporeal membrane oxygenation to a Jar-vik 2000 LVAD. These patients were discharged 3 and6 months after LVAD implantation, respectively.Ryan et al. (81) subsequently described implantationof the HeartMate II LVAD (Thoratec, Pleasanton,California) in a 29-year-old male patient with DMDand end-stage heart failure and of the HeartWareLVAD (HeartWare, Framingham, Massachusetts) in a23-year-old female symptomatic DMD carrier withend-stage heart failure.
The LVAD as destination therapy is a potentiallypromising therapy to address end-stage heart failurein patients with dystrophin-deficient heart failure;however, post-operative complications, includingrespiratory failure, rehabilitation, bleeding, stroke,and arrhythmias, will need to be evaluated further inthis population. Extensive pre- and post-operativemanagement in an experienced center would benecessary for LVAD implantation in the DMD popu-lation, and larger studies will be needed to evaluatethe efficacy and outcomes in this population.
BMD AND CARDIOMYOPATHY
In 1955, German physicians Becker and Kiener (82)described an X-linked muscular dystrophy with amilder clinical course than DMD, now known asBecker muscular dystrophy (83). In 1984, it wasdemonstrated that the gene responsible for BMD islocated on the X chromosome (84) and, subsequently,that mutations in the DMD gene resulted in BMD (85).BMD has an incidence of 1:19,000 and is an X-linkedrecessive disorder resulting from a mutation of thedystrophin gene (86). However, in comparison withpatients with DMD who have a complete absence ofdystrophin, dystrophin mutations in BMD tend to bein-frame and result in misfolded or abnormal and lessfunctional protein (87). Patients with BMD have atypically later age of onset and less severe clinicalinvolvement compared with patients with DMD(Table 1) (88). Although the muscular symptoms maybe less severe, over 70% of patients with BMD alsodevelop cardiomyopathy (89), and it is the leadingcause of death in patients with BMD (27). Cardiomy-opathy may be more severe in patients with BMD thanin patients with DMD (90,91). The onset of cardio-myopathy is variable in BMD and is not correlated toskeletal muscle involvement (89,92). CMR studieshave demonstrated an inferobasal fibrotic pattern inpatients with BMD similar to that seen in DMD (93).Patients with BMD cardiomyopathy should receivestandard medical heart failure therapy (51). As pa-tients with BMD have a relatively milder skeletalmuscle phenotype, selected patients with BMD havereceived LVADs and heart transplantation for severecardiomyopathy with good outcomes (74–77).
CARRIER STATUS AND CARDIOMYOPATHY
As DMD and BMD affect males, given that it isinherited in an X-linked recessive manner, femalescan be carriers of DMD and BMD. The majority of DMDand BMD is inherited, and thus there are a significantnumber of female carriers of DMD and BMD. Themajority of female carriers of DMD and BMD areasymptomatic; however, female carriers can becomesymptomatic or become manifesting carriers. Mani-festing carriers can have symptoms, such as mildmuscle weakness, elevated serum creatinine kinase,and cardiomyopathy (94,95). Hoogerwaard et al.(96,97) evaluated 90 women who were carriers ofdystrophin mutations and identified 22% with symp-toms, including muscle weakness, and 18% with evi-dence of dilated left ventricles. The age of onset ofcarriers is variable, and the proportion of symptomaticcarriers has ranged from 2.5% of 22% in prior studies

J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6 Kamdar and GarryM A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6 DMD and BMD Cardiomyopathy
2543
(96,97). X-inactivation is themechanismwhere 1 of the2 X chromosomes in female cells randomly becomestranscriptionally inactive. It is postulated that carrierscan become symptomatic on the basis of the extent ofrandom X-inactivation of the normal X chromosomeversus the dystrophic X chromosome. Although DMDand BMD carriers have been identified as having car-diomyopathy, the prevalence and severity of diseaseare incompletely defined (96,98). We recommend thatall female dystrophinopathy carriers be screened forcardiomyopathy, including CMR, at the time of diag-nosis of their children (99,100).
X-LINKED DILATED CARDIOMYOPATHY
X-linked dilated cardiomyopathy (XLDCM) was firstdescribed in 1987 and is an extremely rare cardiomy-opathy resulting from mutations in the DMD gene thataffects teenage males and their mothers (101,102).Patients with XLDCMhave normal dystrophin levels inskeletal muscle, but a complete loss of dystrophin incardiac muscle, thus making it a distinct phenotype(i.e., cardiomyopathy without overt skeletal muscledisease) from DMD or BMD (103). The rapidly pro-gressive cardiomyopathy associated with XLDCMresults in death between 10 and 20 years of age.
EMERGING THERAPIES
A major challenge for treatment of DMD is therapiesthat address dystrophin deficiency and target bothcardiac and skeletal muscle (Central Illustration). Anumber of promising therapies are emerging,including exon skipping, dystrophin gene replace-ment, and gene-editing strategies.EXON SKIPPING. DMD mutations are heterogeneous,but the majority of mutations arise from anout-of-frame deletion or duplication within the DMDgene’s 79 exons. These mutations affect the normaltranscription of dystrophin messenger ribonucleicacid open reading frame and subsequently preventthe full-length dystrophin protein from being tran-scribed. A novel therapy for the treatment of DMDinvolves exon skipping within the central roddomain, which can restore the normal messengerribonucleic acid reading frame and convert anout-of-frame mutation to a less severe in-framemutation, comparable to those seen in BMD. Thiscan be achieved through the use of antisense oligo-nucleotides (AONs), which are short, syntheticnucleic acid fragments that regulate ribonucleic acidsplicing (104). Antisense oligonucleotides for exonskipping that target exon 51 of the DMD gene, whichaccounts for w13% of DMD mutations, including dri-sapersen (GSK-2402698, GlaxoSmithKline, Brentford,
United Kingdom; Prosensa, Leiden, the Netherlands)and eteplirsen (AVI 4658, Sarepta Therapeutics,Cambridge, Massachusetts), are currently beingexamined in clinical trials. Initial phase I and II trialsfor both eteplirsen and drisapersen were promisingin restoring dystrophin expression, but did notdemonstrate a significant change in the primaryendpoint, the 6-min walk test. Major side effectsincluded renal toxicity and thrombocytopenia(105,106). These drugs are being further evaluated inphase III clinical trials.
DYSTROPHIN GENE AND CELL THERAPY. As dystro-phin mutations lead to complete absence of thefunctional dystrophin protein in DMD, 1 of the stra-tegies to mitigate the disease sequelae is genereplacement therapy using recombinant adenoviralvirus vectors (rAAV). In comparison to the exonskipping strategy, which currently is available onlyfor exon 51 skipping, gene replacement strategies canbe beneficial for all patients with DMD, regardless oftheir specific DMD mutation. A limitation of rAAVtechnology is the inability to deliver the large,full-length dystrophin gene due to packaging limita-tions (107). However, the observation of mildlyaffected patients with BMD who had very large dys-trophin gene deletions led to the understanding thattruncated dystrophins could be functional (85,108).This led to the development of micro- or mini-dystrophins, which contain only the essential, func-tional regions of the gene and can be packagedwithin the rAAV (109). Dystrophin gene replacementtherapy has been effective in mouse models; howev-er, in large animal models, efficacy has been limiteddue to the host immune response (110,111). In a hu-man pre-clinical trial of mini-dystrophin gene trans-fer, 6 patients with DMD received injections ofmini-dystrophin, but none demonstrated significantincreases in dystrophin levels and 1 patient had animmune response (112,113). In addition, cell therapyinitiatives for DMD cardiomyopathy usingcardiosphere-derived cells and/or induced pluripo-tent stem cell derivatives have shown improvementin both cardiac function and exercise capacity of themdx (dystrophin knockout) mouse model. Genereplacement and cell therapies, although limited byimmune side effects, may be overcome by immuno-suppressant regimens and may represent promisingtherapies for DMD cardiomyopathy.
DYSTROPHIN GENE EDITING. Gene editing is anemerging technology for DMD. Gene editing can bemediated by clustered regularly interspaced shortpalindromic repeats (CRISPR)/CRISPR-associatedsystems (Cas) to alter or edit the genome (114,115).

Kamdar and Garry J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6
DMD and BMD Cardiomyopathy M A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6
2544
The CRISPR/Cas9 system binds to the target gene andgenerates a double-stranded deoxyribonucleic acidbreak, which then can be replaced by the correctedgene sequence. This system precisely removes themutated gene of interest and replaces it with a func-tional copy of the gene. Gene editing using aCRISPR/Cas9 strategy was utilized in vivo to correctthe DMD gene mutation in the germline of mdx mice(116). In mosaic mdx mice with only 17% correction ofdystrophin by gene editing, a normal muscle pheno-type was observed. This nascent technology haspossible therapeutic benefits in patients with DMD,and further animal studies evaluating cardiac func-tion are needed. Recently, adult mdx mice were alsosuccessfully treated using a gene-editing strategy(117,118).
CONCLUSIONS
Cardiomyopathy is a leading cause of death in theDMD population. Early detection and intervention
with medical and device therapies are warranted.Given the significant cardiomyopathic phenotype inthe dystrophinopathy population, we have estab-lished a neurocardiomyopathy clinic where thesepatients have a comprehensive evaluation with heartfailure cardiologists and neuromuscular neurologists.We believe that increasing attention by heart failure–trained subspecialists and the establishment ofdedicated neurocardiomyopathy clinics will enhanceour understanding of the mechanisms of DMD car-diomyopathy and ultimately have an effect on themorbidity and mortality of this disease.
ACKNOWLEDGMENT The authors are grateful forartwork assistance by Ms. Cynthia DeKay.
REPRINT REQUESTS AND CORRESPONDENCE: Dr.Daniel J. Garry, Cardiovascular Division, LilleheiHeart Institute, University of Minnesota, 2231 6thStreet Southeast, 4-146 CCRB, Minneapolis, Minne-sota 55455. E-mail: [email protected].
RE F E RENCE S
1. Meryon E. On granular and fatty degenerationof the voluntary muscles. Medico-Chirugical Trans1852;35:73–4.
2. Duchenne GBA. Recherches sur la paralysiemusculaire pseudo-hypertrophique ou paralysiemyo-sclerosique. Arch Gen Med 1868;11:5–25.
3. Gowers WR. A manual of disease of the nervoussystem1. London, UK: Churchill, 1886:391–4.
4. Monaco AP, Neve RL, Colletti-Feener C, et al.Isolation of candidate cDNAs for portions of theDuchenne muscular dystrophy gene. Nature 1986;323:646–50.
5. Hoffman EP, Brown RH Jr., Kunkel LM. Dys-trophin: the protein product of the Duchennemuscular dystrophy locus. Cell 1987;51:919–28.
6. Emery AE. The muscular dystrophies. Lancet2002;359:687–95.
7. Cox GF, Kunkel LM. Dystrophies and heart dis-ease. Curr Opin Cardiol 1997;12:329–43.
8. Mendell JR, Shilling C, Leslie ND, et al.Evidence-based path to newborn screening forDuchenne muscular dystrophy. Ann Neurol 2012;71:304–13.
9. Koenig M, Hoffman EP, Bertelson CJ, et al.Complete cloning of the Duchenne muscular dys-trophy (DMD) cDNA and preliminary genomic or-ganization of the DMD gene in normal andaffected individuals. Cell 1987;50:509–17.
10. Ahn AH, Kunkel LM. The structural and func-tional diversity of dystrophin. Nat Genet 1993;3:283–91.
11. Dent KM, Dunn DM, von Niederhausern AC,et al. Improved molecular diagnosis of dystrophi-nopathies in an unselected clinical cohort. Am JMed Genet A 2005;134:295–8.
12. Rybakova IN, Patel JR, Ervasti JM. The dys-trophin complex forms a mechanically strong linkbetween the sarcolemma and costameric actin.J Cell Biol 2000;150:1209–14.
13. Ervasti JM, Campbell KP. A role for thedystrophin-glycoprotein complex as a trans-membrane linker between laminin and actin. J CellBiol 1993;122:809–23.
14. Cohn RD, Campbell KP. Molecular basis ofmuscular dystrophies. Muscle Nerve 2000;23:1456–71.
15. Klietsch R, Ervasti JM, Arnold W, et al.Dystrophin-glycoprotein complex and laminincolocalize to the sarcolemma and transverse tubulesof cardiac muscle. Circ Res 1993;72:349–60.
16. Johnson EK, Zhang L, Adams ME, et al.Proteomic analysis reveals new cardiac-specificdystrophin-associated proteins. PloS One 2012;7:e43515.
17. Cox GA, Cole NM, Matsumura K, et al. Over-expression of dystrophin in transgenic mdx miceeliminates dystrophic symptoms without toxicity.Nature 1993;364:725–9.
18. Petrof BJ, Shrager JB, Stedman HH, et al.Dystrophin protects the sarcolemma from stressesdeveloped during muscle contraction. Proc NatlAcad Sci U S A 1993;90:3710–4.
19. Yasuda S, Townsend D, Michele DE, et al.Dystrophic heart failure blocked by membranesealant poloxamer. Nature 2005;436:1025–9.
20. Konagaya M, Takayanagi T. Regularity in thechange of serum creatine kinase level in Duchennemuscular dystrophy. A study with long-termfollow-up cases. Jpn J Med 1986;25:2–8.
21. Eagle M, Baudouin SV, Chandler C, et al.Survival in Duchenne muscular dystrophy:
improvements in life expectancy since 1967 andthe impact of home nocturnal ventilation. Neuro-muscul Disord 2002;12:926–9.
22. Nigro G, Comi LI, Politano L, et al. The inci-dence and evolution of cardiomyopathy inDuchenne muscular dystrophy. Int J Cardiol 1990;26:271–7.
23. Angelini C. The role of corticosteroids inmuscular dystrophy: a critical appraisal. MuscleNerve 2007;36:424–35.
24. Bushby K, Finkel R, Birnkrant DJ, et al., for theDMD Care Considerations Working Group. Diag-nosis and management of Duchenne musculardystrophy, part 1: diagnosis, and pharmacologicaland psychosocial management. Lancet Neurol2010;9:77–93.
25. Manzur AY, Kuntzer T, Pike M, et al. Gluco-corticoid corticosteroids for Duchenne musculardystrophy. Cochrane Database Syst Rev 2008:CD003725.
26. Bonifati MD, Ruzza G, Bonometto P, et al.A multicenter, double-blind, randomized trial ofdeflazacort versus prednisone in Duchennemuscular dystrophy. Muscle Nerve 2000;23:1344–7.
27. Bushby K, Finkel R, Birnkrant DJ, et al., for theDMD Care Considerations Working Group. Diag-nosis and management of Duchenne musculardystrophy, part 2: implementation of multidisci-plinary care. Lancet Neurol 2010;9:177–89.
28. Gozal D. Pulmonary manifestations of neuro-muscular disease with special reference toDuchenne muscular dystrophy and spinal muscularatrophy. Pediatr Pulmonol 2000;29:141–50.
29. Eagle M, Bourke J, Bullock R, et al. ManagingDuchenne muscular dystrophy—the additive effectof spinal surgery and home nocturnal ventilation

J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6 Kamdar and GarryM A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6 DMD and BMD Cardiomyopathy
2545
in improving survival. Neuromuscul Disord 2007;17:470–5.
30. Finder JD, Birnkrant D, Carl J, et al. Respira-tory care of the patient with Duchenne musculardystrophy: ATS consensus statement. Am J RespirCrit Care Med 2004;170:456–65.
31. American Academy of Pediatrics Section onCardiology and Cardiac Surgery. Cardiovascularhealth supervision for individuals affected byDuchenne or Becker muscular dystrophy. Pediat-rics 2005;116:1569–73.
32. Markham LW, Spicer RL, Khoury PR, et al.Steroid therapy and cardiac function in Duchennemuscular dystrophy. Pediatr Cardiol 2005;26:768–71.
33. Jefferies JL, Eidem BW, Belmont JW, et al.Genetic predictors and remodeling of dilated car-diomyopathy in muscular dystrophy. Circulation2005;112:2799–804.
34. Romfh A, McNally EM. Cardiac assessment inDuchenne and Becker muscular dystrophies. CurrHeart Fail Rep 2010;7:212–8.
35. Thrush PT, Allen HD, Viollet L, et al. Re-examination of the electrocardiogram in boys withDuchenne muscular dystrophy and correlationwith its dilated cardiomyopathy. Am J Cardiol2009;103:262–5.
36. Perloff JK, Roberts WC, de Leon AC Jr., et al. Thedistinctive electrocardiogram of Duchenne’s progres-sive muscular dystrophy. An electrocardiographic-pathologic correlative study. Am J Med 1967;42:179–88.
37. Sanyal SK, Johnson WW, Thapar MK, et al. Anultrastructural basis for electrocardiographicalterations associated with Duchenne’s progres-sive muscular dystrophy. Circulation 1978;57:1122–9.
38. Frankel KA, Rosser RJ. The pathology of theheart in progressive muscular dystrophy: epi-myocardial fibrosis. Human Pathol 1976;7:375–86.
39. Markham LW, Michelfelder EC, Border WL,et al. Abnormalities of diastolic function precededilated cardiomyopathy associated with Duchennemuscular dystrophy. J Am Soc Echocardiogr 2006;19:865–71.
40. Boas EP, Lowenburg H. The heart rate inprogressive muscular dystrophy. Arch Intern Med1931;47:376–83.
41. Fitch CW, Ainger LE. The Frank vectorcardio-gram and the electrocardiogram in Duchenneprogressive muscular dystrophy. Circulation 1967;35:1124–40.
42. Kovick RB, Fogelman AM, Abbasi AD, et al.Echocardiographic evaluation of posterior leftventricular wall motion in muscular dystrophy.Circulation 1975;52:447–54.
43. Nomura H, Hizawa K. Histopathological studyof the conduction system of the heart in Duchenneprogressive muscular dystrophy. Acta Pathol Jpn1982;32:1027–33.
44. Yotsukura M, Sasaki K, Kachi E, et al. Circadianrhythm and variability of heart rate in Duchenne-type progressive muscular dystrophy. Am J Car-diol 1995;76:947–51.
45. Yotsukura M, Fujii K, Katayama A, et al. Nine-year follow-up study of heart rate variability inpatients with Duchenne-type progressive musculardystrophy. Am Heart J 1998;136:289–96.
46. Thomas TO, Morgan TM, Burnette WB, et al.Correlation of heart rate and cardiac dysfunction inDuchenne muscular dystrophy. Pediatr Cardiol2012;33:1175–9.
47. Dalmaz Y, Peyrin L, Mamelle JC, et al. Thepattern of urinary catecholamines and their me-tabolites in Duchenne myopathy, in relation todisease evolution. J Neural Transm 1979;46:17–34.
48. Villa CR, Czosek RJ, Ahmed H, et al. Ambula-tory monitoring and arrhythmic outcomes inpediatric and adolescent patients with Duchennemuscular dystrophy. J Am Heart Assoc 2015;5:e002620.
49. Corrado G, Lissoni A, Beretta S, et al. Prog-nostic value of electrocardiograms, ventricularlate potentials, ventricular arrhythmias, and leftventricular systolic dysfunction in patients withDuchenne muscular dystrophy. Am J Cardiol2002;89:838–41.
50. Yotsukura M, Yamamoto A, Kajiwara T, et al.QT dispersion in patients with Duchenne-typeprogressive muscular dystrophy. Am Heart J1999;137:672–7.
51. Yancy CW, Jessup M, Bozkurt B, et al. 2013ACCF/AHA guideline for the management of heartfailure: a report of the American College of Car-diology Foundation/American Heart AssociationTask Force on Practice Guidelines. J Am Coll Car-diol 2013;62:e147–239.
52. Rapezzi C, Leone O, Biagini E, et al. Echocar-diographic clues to diagnosis of dystrophin relateddilated cardiomyopathy. Heart 2007;93:10.
53. Markham LW, Kinnett K, Wong BL, et al.Corticosteroid treatment retards development ofventricular dysfunction in Duchenne musculardystrophy. Neuromuscul Disord 2008;18:365–70.
54. Silva MC, Meira ZM, Gurgel Giannetti J, et al.Myocardial delayed enhancement by magneticresonance imaging in patients with muscular dys-trophy. J Am Coll Cardiol 2007;49:1874–9.
55. Hagenbuch SC, Gottliebson WM, Wansapura J,et al. Detection of progressive cardiac dysfunctionby serial evaluation of circumferential strain inpatients with Duchenne muscular dystrophy. Am JCardiol 2010;105:1451–5.
56. Hor KN, Wansapura J, Markham LW, et al.Circumferential strain analysis identifies strata ofcardiomyopathy in Duchenne muscular dystrophy:a cardiac magnetic resonance tagging study. J AmColl Cardiol 2009;53:1204–10.
57. Puchalski MD, Williams RV, Askovich B, et al.Late gadolinium enhancement: precursor to car-diomyopathy in Duchenne muscular dystrophy? IntJ Cardiovasc Imaging 2009;25:57–63.
58. Verhaert D, Richards K, Rafael-Fortney JA,et al. Cardiac involvement in patients withmuscular dystrophies: magnetic resonance imag-ing phenotype and genotypic considerations. CircCardiovasc Imaging 2011;4:67–76.
59. Hor KN, Taylor MD, Al-Khalidi HR, et al.Prevalence and distribution of late gadoliniumenhancement in a large population of patients
with Duchenne muscular dystrophy: effect of ageand left ventricular systolic function. J CardiovascMagn Reson 2013;15:107.
60. Tandon A, Villa CR, Hor KN, et al. Myocardialfibrosis burden predicts left ventricular ejectionfraction and is associated with age and steroidtreatment duration in Duchenne muscular dystro-phy. J Am Heart Assoc 2015;4:e001338.
61. Dec GW. Steroid therapy effectively delaysDuchenne’s cardiomyopathy. J Am Coll Cardiol2013;61:955–6.
62. Silversides CK, Webb GD, Harris VA, et al. Ef-fects of deflazacort on left ventricular function inpatients with Duchenne muscular dystrophy. Am JCardiol 2003;91:769–72.
63. Mavrogeni S, Papavasiliou A, Douskou M, et al.Effect of deflazacort on cardiac and sternocleido-mastoid muscles in Duchenne muscular dystrophy:a magnetic resonance imaging study. Eur J Pae-diatr Neurol 2009;13:34–40.
64. Barber BJ, Andrews JG, Lu Z, et al. Oral cor-ticosteroids and onset of cardiomyopathy inDuchenne muscular dystrophy. J Pediatr 2013;163:1080–4.e1.
65. Schram G, Fournier A, Leduc H, et al. All-causemortality and cardiovascular outcomes with pro-phylactic steroid therapy in Duchenne musculardystrophy. J Am Coll Cardiol 2013;61:948–54.
66. Duboc D, Meune C, Lerebours G, et al. Effectof perindopril on the onset and progression of leftventricular dysfunction in Duchenne musculardystrophy. J Am Coll Cardiol 2005;45:855–7.
67. Duboc D, Meune C, Pierre B, et al. Perindoprilpreventive treatment on mortality in Duchennemuscular dystrophy: 10 years’ follow-up. AmHeart J 2007;154:596–602.
68. Allen HD, Flanigan KM, Thrush PT, et al.A randomized, double-blind trial of lisinopril andlosartan for the treatment of cardiomyopathy inDuchennemuscular dystrophy [published correctionappears in PLoS Curr 2015;7]. PLoS Curr 2013;5.
69. Saito T, Matsumura T, Miyai I, Nozaki S,Shinno S. [Carvedilol effectiveness for leftventricular-insufficient patients with Duchennemuscular dystrophy]. Rinsho Shinkeigaku 2001;41:691–4.
70. Rhodes J, Margossian R, Darras BT, et al.Safety and efficacy of carvedilol therapy for pa-tients with dilated cardiomyopathy secondary tomuscular dystrophy. Pediatr Cardiol 2008;29:343–51.
71. Kajimoto H, Ishigaki K, Okumura K, et al. Beta-blocker therapy for cardiac dysfunction in patientswith muscular dystrophy. Circ J 2006;70:991–4.
72. Viollet L, Thrush PT, Flanigan KM, et al. Effectsof angiotensin-converting enzyme inhibitorsand/or beta blockers on the cardiomyopathy inDuchenne muscular dystrophy. Am J Cardiol 2012;110:98–102.
73. Raman SV, Hor KN, Mazur W, et al. Eplerenonefor early cardiomyopathy in Duchenne musculardystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2015;14:153–61.
74. Rees W, Schüler S, Hummel M, et al.Heart transplantation in patients with muscular

Kamdar and Garry J A C C V O L . 6 7 , N O . 2 1 , 2 0 1 6
DMD and BMD Cardiomyopathy M A Y 3 1 , 2 0 1 6 : 2 5 3 3 – 4 6
2546
dystrophy associated with end-stage cardiomy-opathy. J Heart Lung Transplant 1993;12:804–7.
75. Ruiz-Cano MJ, Delgado JF, Jiménez C, et al.Successful heart transplantation in patients withinherited myopathies associated with end-stagecardiomyopathy. Transplant Proc 2003;35:1513–5.
76. Patanè F, Zingarelli E, Attisani M, et al. Suc-cessful heart transplantation in Becker’s musculardystrophy. Eur J Cardiothorac Surg 2006;29:250.
77. Wu RS, Gupta S, Brown RN, et al. Clinicaloutcomes after cardiac transplantation in musculardystrophy patients. J Heart Lung Transplant 2010;29:432–8.
78. Rose EA, Gelijns AC, Moskowitz AJ, et al., forthe Randomized Evaluation of Mechanical Assis-tance for the Treatment of Congestive HeartFailure (REMATCH) Study Group. Long-term useof a left ventricular assist device for end-stageheart failure. N Engl J Med 2001;345:1435–43.
79. Slaughter MS, Rogers JG, Milano CA, et al., forthe HeartMate II Investigators. Advanced heartfailure treated with continuous-flow left ventric-ular assist device. N Engl J Med 2009;361:2241–51.
80. Amodeo A, Adorisio R. Left ventricular assistdevice in Duchenne cardiomyopathy: can wechange the natural history of cardiac disease? Int JCardiol 2012;161:e43.
81. Ryan TD, Jefferies JL, Sawnani H, et al. Im-plantation of the HeartMate II and HeartWare leftventricular assist devices in patients withDuchenne muscular dystrophy: lessons learnedfrom the first applications. ASAIO J 2014;60:246–8.
82. Becker PE, Kiener F. [A new x-chromosomalmuscular dystrophy]. Archiv Psychiatr Nervenkr ZGesamte Neurol Psychiatr 1955;193:427–48.
83. Becker PE. Two families of benign sex-linkedrecessive muscular dystrophy. Rev Can Biol 1962;21:551–66.
84. Kingston HM, Sarfarazi M, Thomas NS, et al.Localisation of the Becker muscular dystrophygene on the short arm of the X chromosome bylinkage to cloned DNA sequences. Hum Genet1984;67:6–17.
85. Koenig M, Beggs AH, Moyer M, et al. Themolecular basis for Duchenne versus Beckermuscular dystrophy: correlation of severity withtype of deletion. Am J Hum Genet 1989;45:498–506.
86. Worton R. Muscular dystrophies: diseases ofthe dystrophin-glycoprotein complex. Science1995;270:755–6.
87. Monaco AP, Bertelson CJ, Liechti-Gallati S,et al. An explanation for the phenotypic differ-ences between patients bearing partial deletionsof the DMD locus. Genomics 1988;2:90–5.
88. Bradley WG, Jones MZ, Mussini JM,Fawcett PR. Becker-type muscular dystrophy.Muscle Nerve 1978;1:111–32.
89. Melacini P, Fanin M, Danieli GA, et al.Myocardial involvement is very frequent among
patients affected with subclinical Becker’smuscular dystrophy. Circulation 1996;94:3168–75.
90. Finsterer J, Stöllberger C. Cardiac involvementin Becker muscular dystrophy. Can J Cardiol 2008;24:786–92.
91. Hoogerwaard EM, de Voogt WG, Wilde AA,et al. Evolution of cardiac abnormalities in Beckermuscular dystrophy over a 13-year period.J Neurol 1997;244:657–63.
92. Nigro G, Politano L, Nigro V, et al. Mutation ofdystrophin gene and cardiomyopathy. NeuromuscDisord 1994;4:371–9.
93. Yilmaz A, Gdynia HJ, Baccouche H, et al. Car-diac involvement in patients with Becker musculardystrophy: new diagnostic and pathophysiologicalinsights by a CMR approach. J Cardiovasc MagnReson 2008;10:50.
94. Norman A, Harper P. A survey of manifestingcarriers of Duchenne and Becker muscular dys-trophy in Wales. Clin Genet 1989;36:31–7.
95. Comi LI, Nigro G, Politano L, et al. The car-diomyopathy of Duchenne/Becker consultands. IntJ Cardiol 1992;34:297–305.
96. Hoogerwaard EM, van der Wouw PA,Wilde AA, et al. Cardiac involvement in carriers ofDuchenne and Becker muscular dystrophy. Neu-romusc Disord 1999;9:347–51.
97. Hoogerwaard EM, Bakker E, Ippel PF, et al.Signs and symptoms of Duchenne muscular dys-trophy and Becker muscular dystrophy amongcarriers in The Netherlands: a cohort study. Lancet1999;353:2116–9.
98. Schade van Westrum SM, Hoogerwaard EM,Dekker L, et al. Cardiac abnormalities in a follow-up study on carriers of Duchenne and Beckermuscular dystrophy. Neurology 2011;77:62–6.
99. Schelhorn J, Schoenecker A, Neudorf U, et al.Cardiac pathologies in female carriers of Duchennemuscular dystrophy assessed by cardiovascularmagnetic resonance imaging. Eur Radiol 2015;25:3066–72.
100. Lang SM, Shugh S, Mazur W, et al. Myocardialfibrosis and left ventricular dysfunction inDuchenne muscular dystrophy carriers using car-diac magnetic resonance imaging. Pediatr Cardiol2015;36:1495–501.
101. Berko BA, Swift M. X-linked dilated cardio-myopathy. N Engl J Med 1987;316:1186–91.
102. Muntoni F, Cau M, Ganau A, et al. Briefreport: deletion of the dystrophin muscle-promoter region associated with X-linked dilatedcardiomyopathy. N Engl J Med 1993;329:921–5.
103. Towbin JA, Hejtmancik JF, Brink P, et al.X-linked dilated cardiomyopathy. Moleculargenetic evidence of linkage to the Duchennemuscular dystrophy (dystrophin) gene at the Xp21locus. Circulation 1993;87:1854–65.
104. Aartsma-Rus A, Fokkema I, Verschuuren J,et al. Theoretic applicability of antisense-mediatedexon skipping for Duchenne muscular dystrophymutations. Hum Mutat 2009;30:293–9.
105. Goemans NM, Tulinius M, van den Akker JT,et al. Systemic administration of PRO051 inDuchenne’s muscular dystrophy. N Engl J Med2011;364:1513–22.
106. Cirak S, Arechavala-Gomeza V, Guglieri M,et al. Exon skipping and dystrophin restoration inpatients with Duchenne muscular dystrophy aftersystemic phosphorodiamidate morpholino olig-omer treatment: an open-label, phase 2, dose-escalation study. Lancet 2011;378:595–605.
107. Pichavant C, Aartsma-Rus A, Clemens PR,et al. Current status of pharmaceutical and genetictherapeutic approaches to treat DMD. Mol Ther2011;19:830–40.
108. England SB, Nicholson LV, Johnson MA, et al.Very mild muscular dystrophy associated with thedeletion of 46% of dystrophin. Nature 1990;343:180–2.
109. Harper SQ, Hauser MA, DelloRusso C, et al.Modular flexibility of dystrophin: implications forgene therapy of Duchenne muscular dystrophy.Nat Med 2002;8:253–61.
110. Gregorevic P, Allen JM, Minami E, et al.rAAV6-microdystrophin preserves muscle functionand extends lifespan in severely dystrophic mice.Nat Med 2006;12:787–9.
111. Banks GB, Chamberlain JS, Froehner SC.Truncated dystrophins can influence neuromus-cular synapse structure. Mol Cell Neurosci 2009;40:433–41.
112. Mendell JR, Campbell K, Rodino-Klapac L,et al. Dystrophin immunity in Duchenne’s musculardystrophy. N Engl J Med 2010;363:1429–37.
113. Bowles DE, McPhee SW, Li C, et al. Phase 1gene therapy for Duchenne muscular dystrophyusing a translational optimized AAV vector. MolTher 2012;20:443–55.
114. Jinek M, Chylinski K, Fonfara I, et al.A programmable dual-RNA-guided DNA endonu-clease in adaptive bacterial immunity. Science2012;337:816–21.
115. Cong L, Ran FA, Cox D, et al. Multiplexgenome engineering using CRISPR/Cas systems.Science 2013;339:819–23.
116. Long C, McAnally JR, Shelton JM, et al. Pre-vention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science2014;345:1184–8.
117. Long C, Amoasii L, Mireault AA, et al. Post-natal genome editing partially restores dystrophinexpression in a mouse model of muscular dystro-phy. Science 2016;351:400–3.
118. Nelson CE, Hakim CH, Ousterout DG, et al.In vivo genome editing improves muscle functionin a mouse model of Duchenne muscular dystro-phy. Science 2016;351:403–7.
KEY WORDS Becker muscular dystrophycardiomyopathy, Duchenne musculardystrophy cardiomyopathy, musculardystrophy cardiomyopathy