duocarmycins - natures prodrugs?
TRANSCRIPT

Current Pharmaceutical Design, 2002, 8, 1375-1389 1375
Duocarmycins – Natures Prodrugs?
Mark Searcey*
Dept. of Pharmaceutical and Biological Chemistry, University of London School of Pharmacy, 29/39 BrunswickSquare, London WC1N 1AX, UK
Abstract. The duocarmycins and (+)-CC-1065 are amongst the most potent antitumour antibioticsdiscovered to date and yet have not progressed into the clinic. The natural products are extremely stableto nucleophilic attack until bound to their DNA target and are not substrates for any other biologicalnucleophile. The mechanism for this target activation of the duocarmycins is discussed with relation toboth an acid-catalyzed activation and a binding-induced conformational change leading to ground statedestabilization. It is suggested that targeting of the duocarmycins to their site of action in a tumour maybe more important than introducing systemically-activated prodrugs as the natural product itself can beconsidered to be a type of prodrug, activated only on binding to its targets. Methods that have been usedto target CC-1065 and the duocarmycins are reviewed as well as efforts towards systemically activatedprodrugs. A simple analysis of the approaches that could be taken to vary the structure for targeting issuggested.
INTRODUCTION STRUCTURE OF THE DUOCARMYCINS AND CC-1065
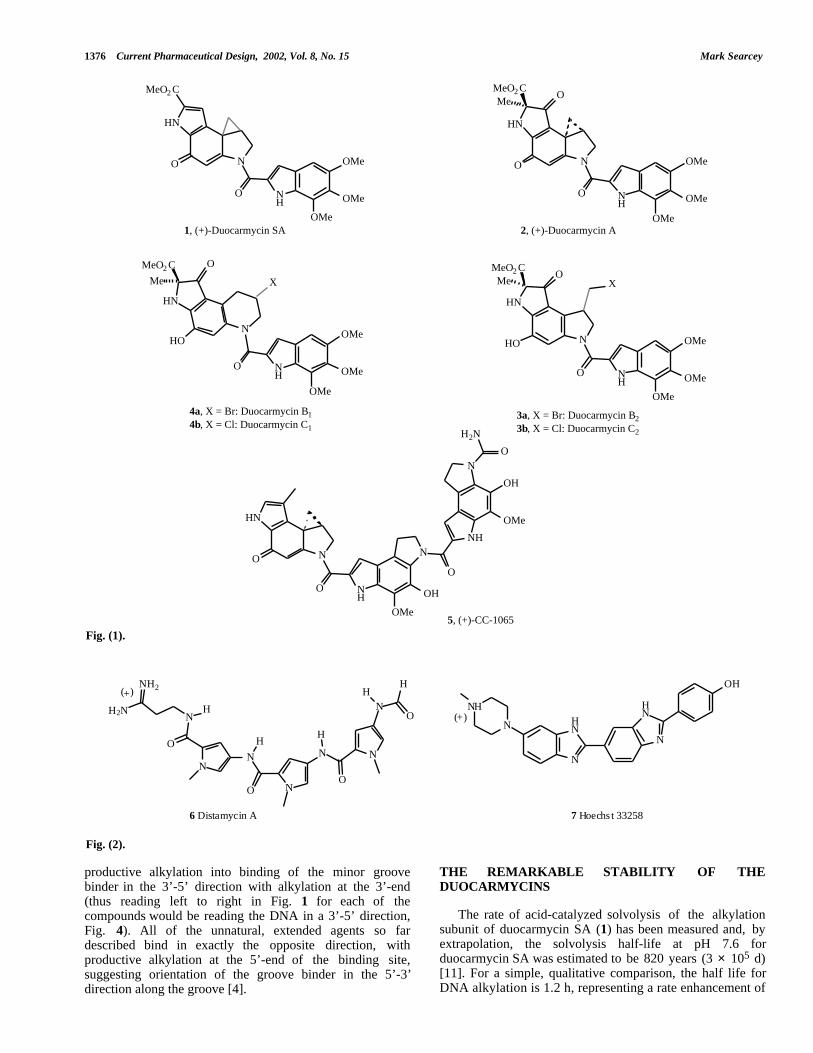
The duocarmycins 1 – 4 , isolated from a culture broth ofstreptomyces species, are parent members of a family ofantitumour antibiotics that also includes CC-1065 (5) [1].These extremely potent cytotoxic agents derive theirbiological activity from an ability to sequence-selectivelyalkylate DNA at the N3 of adenine in the minor groove,which initiates a cascade of effects that terminates in anapoptotic cell death mechanism [2]. Studies of both thesynthetic [3] and mechanistic [4] aspects of these compoundshave been extensively reviewed. One of the extraordinaryfeatures of the duocarmycins is their remarkable stability insolution at neutral or near neutral pH. Alkylation of DNA,however, takes place rapidly at room temperature (or lower)to give covalently bound adenine adducts. This suggests thatthe innate “ground-state” stability of the unboundduocarmycin is removed upon binding to its target. Twodiffering explanations of this enzyme-like activation of CC-1065 and the duocarmycins upon binding to DNA have beenput forward [5,6]. In this review, we describe the evidenceproduced to date that supports a binding-induced activationmechanism for the duocarmycins. We will also look at thevarious methods disclosed to design prodrugs of theduocarmycins and ask the question as to whether thebinding-induced activation of the duocarmycins is, in itself,a form of prodrug release, and whether efforts to selectivelytarget the compounds to their site of action, i.e. tumours,would be a more effective than a simple, systemicallyactivated prodrug approach.
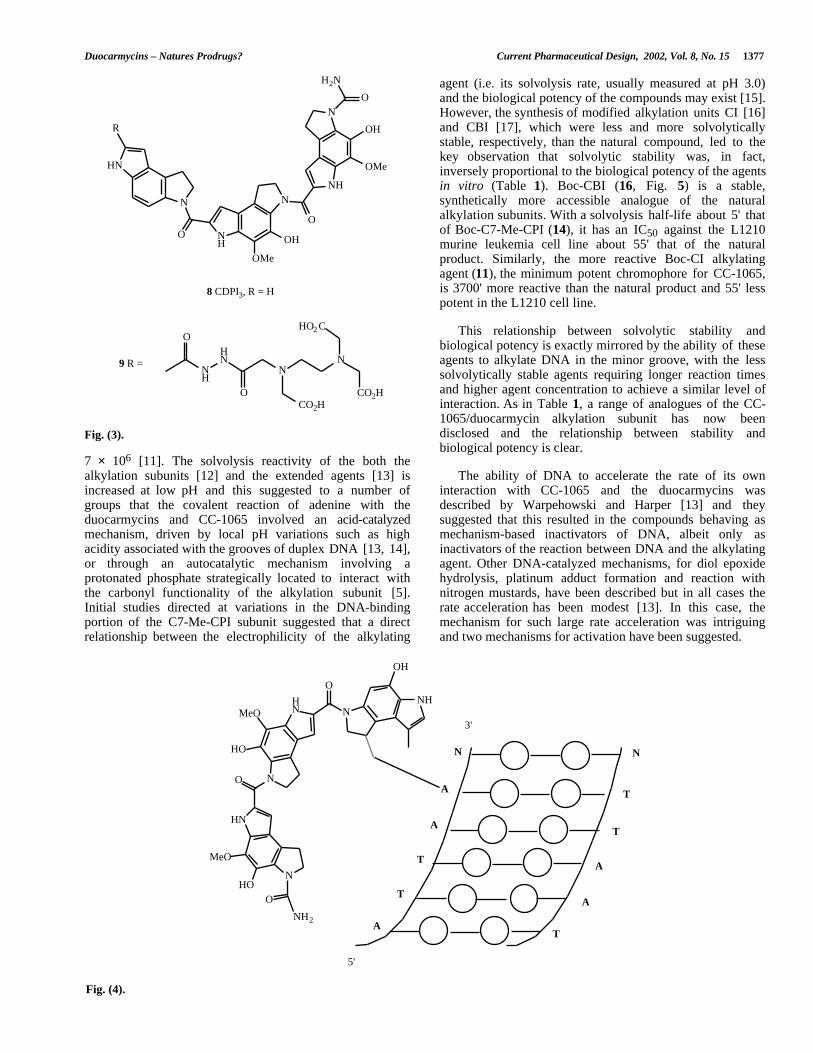
Inspection of the structures of both the duocarmycins andCC-1065 reveals planar curved shapes that are stronglyreminiscent of minor groove binders such as the antibioticdistamycin A 6 and the synthetic dye Hoechst 33258 7 (Fig.2) [7]. Synthesis of an analogue of CC-1065 (CDPI3, 8)lacking the cyclopropane ring revealed that non-alkylatinganalogues had a high affinity for poly[dA]-poly[dT] in asimilar fashion to the classical minor groove binders andsuggested that these compounds are minor groove binderswith an additional alkylating functionality [8]. A welldescribed method for the study of the sequence selectivity ofminor groove binders is the design of analogues carryingaffinity cleavage groups based upon Fe(III)-EDTA [9]. ACDPI3 derivative 9 (Fig. 3) with such an appendage wasshown to cleave DNA in a manner that suggested binding inAT rich sequences [10]. It also revealed that the CC-1065minor groove binding structure could bind in eitherorientation in an AT sequence, confirming that thealkylation sequence selectivity in the natural products isdependant upon the stereochemistry of the cyclopropane ringand the presence of an adenine in the required proximity tothe ring upon binding of the groove binder.
The alkylating group of the natural products isstereochemically defined and has the (+)-(8aR, 7bS)-orientation (using the DSA/C7-Me-CPI∗ numbering). All ofthe natural products isolated to date have the samestereochemical organization. This configuration directs
*Address for correspondence to that author at the Dept. of Pharmaceuticaland Biological Chemistry, University of London School of Pharmacy,29/39 Brunswick Square, London WC1N 1AX, UK; e-mail:[email protected]
∗ The alkylation subunit of CC-1065 was originally given the acronym CPI butthe recent description of the parent, C-7 unsubstituted analogue (see Boger et al,2000) suggests that the CC-1065 subunit should be denoted C-7 MeCPI.
1381-6128/02 $35.00+.00 © 2002 Bentham Science Publishers Ltd.
1376 Current Pharmaceutical Design, 2002, Vol. 8, No. 15 Mark Searcey
HN
O N
O NH
NNH
NO
H2N
OMe
OH
OMe
OH
O
HN
MeO2 C
O N
O NH
OMe
OMe
OMe
HN
O N
O NH
OMe
OMe
OMe
OMe
MeO2 C
HN
HO N
O NH
OMe
OMe
OMe
OMe
MeO2 C
HN
HO
O NH
OMe
OMe
OMe
O
Me
MeO2 C
X
N
X
5, (+)-CC-1065
2, (+)-Duocarmycin A1, (+)-Duocarmycin SA
3a, X = Br: Duocarmycin B23b, X = Cl: Duocarmycin C2
4a, X = Br: Duocarmycin B14b, X = Cl: Duocarmycin C1
Fig. (1).
N
NN
O
H
O
N
NH
O
H
H
N
O
NHH2N
NH2
N
HN
N
HN
N
OH
NH
(+)
6 Distamycin A
(+)
7 Hoechs t 33258
Fig. (2).
productive alkylation into binding of the minor groovebinder in the 3’-5’ direction with alkylation at the 3’-end(thus reading left to right in Fig. 1 for each of thecompounds would be reading the DNA in a 3’-5’ direction,Fig. 4). All of the unnatural, extended agents so fardescribed bind in exactly the opposite direction, withproductive alkylation at the 5’-end of the binding site,
THE REMARKABLE STABILITY OF THEDUOCARMYCINS
The rate of acid-catalyzed solvolysis of the alkylationsubunit of duocarmycin SA (1) has been measured and, byextrapolation, the solvolysis half-life at pH 7.6 forduocarmycin SA was estimated to be 820 years (3 × 105 d)[11]. For a simple, qualitative comparison, the half life forDNA alkylation is 1.2 h, representing a rate enhancement of
suggesting orientation of the groove binder in the 5’-3’direction along the groove [4].
Duocarmycins – Natures Prodrugs? Current Pharmaceutical Design, 2002, Vol. 8, No. 15 1377
HN
N
O NH
NNH
NO
H2N
OMe
OH
OMe
OH
O
R
NH
OHN
O
N
CO2H
N
CO2H
HO2 C
8 CDPI3, R = H
9 R =
Fig. (3).
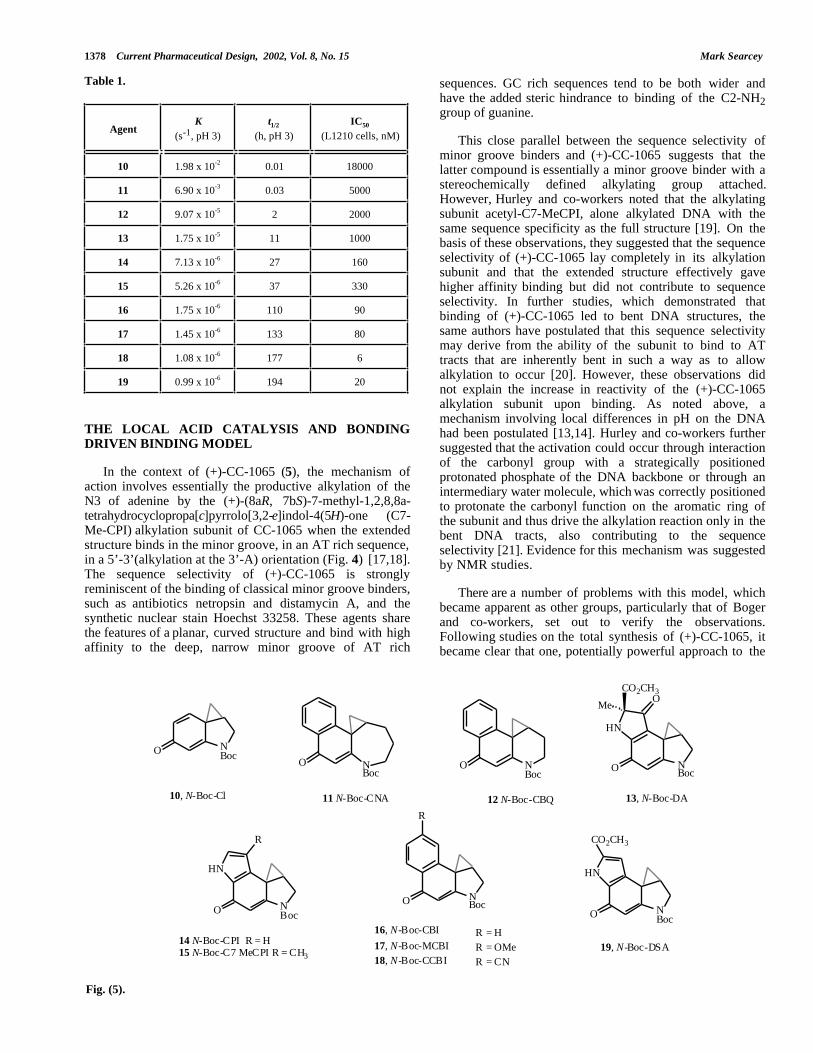
agent (i.e. its solvolysis rate, usually measured at pH 3.0)and the biological potency of the compounds may exist [15].However, the synthesis of modified alkylation units CI [16]and CBI [17], which were less and more solvolyticallystable, respectively, than the natural compound, led to thekey observation that solvolytic stability was, in fact,inversely proportional to the biological potency of the agentsin vitro (Table 1). Boc-CBI (16, Fig. 5) is a stable,synthetically more accessible analogue of the naturalalkylation subunits. With a solvolysis half-life about 5' thatof Boc-C7-Me-CPI (14), it has an IC50 against the L1210murine leukemia cell line about 55' that of the naturalproduct. Similarly, the more reactive Boc-CI alkylatingagent (11), the minimum potent chromophore for CC-1065,is 3700' more reactive than the natural product and 55' lesspotent in the L1210 cell line.
This relationship between solvolytic stability andbiological potency is exactly mirrored by the ability of theseagents to alkylate DNA in the minor groove, with the lesssolvolytically stable agents requiring longer reaction timesand higher agent concentration to achieve a similar level ofinteraction. As in Table 1, a range of analogues of the CC-1065/duocarmycin alkylation subunit has now beendisclosed and the relationship between stability andbiological potency is clear.
7 × 106 [11]. The solvolysis reactivity of the both thealkylation subunits [12] and the extended agents [13] isincreased at low pH and this suggested to a number ofgroups that the covalent reaction of adenine with theduocarmycins and CC-1065 involved an acid-catalyzedmechanism, driven by local pH variations such as highacidity associated with the grooves of duplex DNA [13, 14],or through an autocatalytic mechanism involving aprotonated phosphate strategically located to interact withthe carbonyl functionality of the alkylation subunit [5].Initial studies directed at variations in the DNA-bindingportion of the C7-Me-CPI subunit suggested that a directrelationship between the electrophilicity of the alkylating
The ability of DNA to accelerate the rate of its owninteraction with CC-1065 and the duocarmycins wasdescribed by Warpehowski and Harper [13] and theysuggested that this resulted in the compounds behaving asmechanism-based inactivators of DNA, albeit only asinactivators of the reaction between DNA and the alkylatingagent. Other DNA-catalyzed mechanisms, for diol epoxidehydrolysis, platinum adduct formation and reaction withnitrogen mustards, have been described but in all cases therate acceleration has been modest [13]. In this case, themechanism for such large rate acceleration was intriguingand two mechanisms for activation have been suggested.
A
T
A
A
N
O
NH2
MeO
HO
MeO
HO
O
NH
OH
N
O
HN
N
HN
N
T
N
A
T
A
T
T
5'
3'
Fig. (4).
1378 Current Pharmaceutical Design, 2002, Vol. 8, No. 15 Mark Searcey
Table 1.
AgentK
(s-1, pH 3)t1/2
(h, pH 3)IC50
(L1210 cells, nM)
10 1.98 x 10-2 0.01 18000
11 6.90 x 10-3 0.03 5000
12 9.07 x 10-5 2 2000
13 1.75 x 10-5 11 1000
14 7.13 x 10-6 27 160
15 5.26 x 10-6 37 330
16 1.75 x 10-6 110 90
17 1.45 x 10-6 133 80
18 1.08 x 10-6 177 6
19 0.99 x 10-6 194 20
sequences. GC rich sequences tend to be both wider andhave the added steric hindrance to binding of the C2-NH2group of guanine.
This close parallel between the sequence selectivity ofminor groove binders and (+)-CC-1065 suggests that thelatter compound is essentially a minor groove binder with astereochemically defined alkylating group attached.However, Hurley and co-workers noted that the alkylatingsubunit acetyl-C7-MeCPI, alone alkylated DNA with thesame sequence specificity as the full structure [19]. On thebasis of these observations, they suggested that the sequenceselectivity of (+)-CC-1065 lay completely in its alkylationsubunit and that the extended structure effectively gavehigher affinity binding but did not contribute to sequenceselectivity. In further studies, which demonstrated thatbinding of (+)-CC-1065 led to bent DNA structures, thesame authors have postulated that this sequence selectivitymay derive from the ability of the subunit to bind to ATtracts that are inherently bent in such a way as to allowalkylation to occur [20]. However, these observations didnot explain the increase in reactivity of the (+)-CC-1065alkylation subunit upon binding. As noted above, amechanism involving local differences in pH on the DNAhad been postulated [13,14]. Hurley and co-workers furthersuggested that the activation could occur through interactionof the carbonyl group with a strategically positionedprotonated phosphate of the DNA backbone or through anintermediary water molecule, which was correctly positionedto protonate the carbonyl function on the aromatic ring ofthe subunit and thus drive the alkylation reaction only in thebent DNA tracts, also contributing to the sequenceselectivity [21]. Evidence for this mechanism was suggestedby NMR studies.
THE LOCAL ACID CATALYSIS AND BONDINGDRIVEN BINDING MODEL
In the context of (+)-CC-1065 (5), the mechanism ofaction involves essentially the productive alkylation of theN3 of adenine by the (+)-(8aR, 7bS)-7-methyl-1,2,8,8a-tetrahydrocyclopropa[c]pyrrolo[3,2-e]indol-4(5H)-one (C7-Me-CPI) alkylation subunit of CC-1065 when the extendedstructure binds in the minor groove, in an AT rich sequence,in a 5’-3’(alkylation at the 3’-A) orientation (Fig. 4) [17,18].The sequence selectivity of (+)-CC-1065 is stronglyreminiscent of the binding of classical minor groove binders,such as antibiotics netropsin and distamycin A, and thesynthetic nuclear stain Hoechst 33258. These agents sharethe features of a planar, curved structure and bind with highaffinity to the deep, narrow minor groove of AT rich
There are a number of problems with this model, whichbecame apparent as other groups, particularly that of Bogerand co-workers, set out to verify the observations.Following studies on the total synthesis of (+)-CC-1065, itbecame clear that one, potentially powerful approach to the
HN
O NBoc
CO2CH3O
Me
HN
O NBoc
CO2CH3
HN
O NBoc
R
O NBoc
R
O NBoc
19, N-Boc-DSA
13, N-Boc-DA
16, N-Boc-CBI
17, N-Boc-MCBI18, N-Boc-CCBI
10, N-Boc-Cl
O NBoc
11 N-Boc-CNA
O NBoc
12 N-Boc-CBQ
R = H R = OMe R = CN
14 N-Boc-CPI R = H15 N-Boc-C7 MeCPI R = CH3
Fig. (5).

Duocarmycins – Natures Prodrugs? Current Pharmaceutical Design, 2002, Vol. 8, No. 15 1379
NH
N
O
O
N
NN
N
NH2
R
H+
DNA binding unit
Required for affini ty but not sequence selectivity or catalysis
Acid catalysed either by a protonated phosphate or through an intermediary water molecule
Sequence selectivity derives from the correct posit ioning of the phosphates and water to catalyse the reaction and this is embodied in the DNA so that the alkylation subunit
has the same sequence selectivity as the ful l agent
Fig. (6). The bonding-driven binding model.
study of the mechanism of action of natural products ingeneral and of CC-1065 and the duocarmycins in particular,was the design and synthesis of analogues of the naturalproduct that allowed the introduction of subtle, structuralchanges. These changes should then be reflected in thebiological activity of the resulting compounds and, throughcomparison with the natural product, conclusions about themechanism of action could be drawn [4]. These studiesinitially demonstrated that the opposite enantiomer of (+)-CC-1065 (with the (-)-8aR, 7bS stereochemistry) alkylatedDNA in a manner that suggested productive binding in theopposite orientation to the natural product – in itself strongsupport for the argument that sequence selectivity is derivedmainly from the minor groove binding of the planar curvedstructure [22]. Hurley and co-workers described similarobservations with (-)-CC-1065 [23]. However, they alsofound that the alkylation subunit alone had similaralkylation sequence selectivity to the natural compound, butat substantially higher concentrations, and that other (-)-analogues did not alkylate DNA. They held this to supporttheir hypothesis that the reactivity for the natural agents isinherent in the alkylation sites rather than in the ligand andthat (+)-CC-1065 is a special case, whereas the unnaturalisomer is, as expected, a minor groove binder carrying analkylating function (Fig. 7).*
These findings were quickly followed by the disclosurethat the minimum potent pharmacophore CI, which had anexactly similar alkylation pattern to the natural product CC-1065, also alkylated with the same sequence selectivity,albeit with minimal potency, even when the aromaticcarbonyl was alkylated, or removed completely. Thissuggests that the carbonyl group is required for potency (i.e.for ring closure to the cyclopropyl moiety), but does notcontribute to the sequence selectivity of the agent, arequirement for the phosphate/water protonation mechanismof the other model [25]. This was further supported recentlyby the synthesis of an analogue (termed iso-CBI) in whichthe carbonyl carbon must point into the groove uponbinding, is not at all accessible to the DNA backbone andyet maintains full biological activity of the natural product[26]. The discovery of the duocarmycins and the synthesis ofduocarmycin SA and its alkylation subunit, have allowedfurther investigations of the sequence selectivity of theseagents that strongly suggest that the alkylation specificity ofthis family of compounds derives from an alkylation eventbased upon a mechanism of non-covalent binding. While itcan be argued that (+)-CC-1065 may be a special case, inwhich nature has selected a compound for its ability to bondto certain sequences and that the non-covalent interactionmerely potentiates this reactivity (as has been suggested inthe comparison of (+) and ent(-)-CC-1065 [23]), the growingevidence suggests that this is unlikely.In the same study that demonstrated the reverse binding
of (-)-CC-1065, a key second observation was that both (+)-Boc-C7-MeCPI 10 and (+)-acetyl-C7-MeCPI alkylated DNAwith a different sequence selectivity than the parent (+)-CC-1065 – essentially with the sequence selectivity of atruncated minor groove binder [22]. While (+)-CC-1065required an AT sequence of 4 and more often 5 bases length,the truncated units alkylated any 3 base pair sequence ofadenines and thymines in which the A was correctlypositioned for alkylation. This effect has now been observedfor all the natural and synthetic alkylation subunits incomparison with the extended compounds described sincethese early studies and is a strong argument against thebonding-driven binding model.
THE BINDING-DRIVEN BONDING MODEL
These observations, and others as reviewed in [5], ledBoger and co-workers to suggest an alternative model forbinding which ultimately led to a new model for theactivation of the duocarmycins upon binding to DNA.Essentially, the binding-driven bonding model (Fig. 8)suggests that the duocarmycins and CC-1065 are planarcurved molecules which, in a similar fashion to otherclassical minor groove binders, bind preferentially to thenarrow, deep, minor groove of AT sequences. Upon bindingin sequences in which an adenine is correctly positioned tointeract with the cyclopropane ring, then alkylation canoccur. This model, as it stands, fails to address the questionof the increased reactivity of the cyclopropane ring uponbinding. However, studies to confirm the binding driven
* This similarity in alkylation for the (+) and (-)-alkylation subunits has also beenobserved for duocarmycin SA and and can be explained, in the context of thebinding driven bonding model by the orientation of the agents within a short 5’-AAA sequence which leads to alkylation at the same base [24].

1380 Current Pharmaceutical Design, 2002, Vol. 8, No. 15 Mark Searcey
HN
O N
O NH
NNH
NO
H2N
OMe
OH
OMe
OH
O
HN
O N
O NH
NNH
NO
H2N
OMe
OH
OMe
OH
O
HN
O
N
O
OHN
O
N
O
O
(+)-CC1065 Consensus sequence 5'-A/T A/T A/T A/T A Pu>Py-3'
(-)-CC1065 Consensus sequence 5'-A/T A A/T A/T A/T N
(+)-Boc-C7-MeCPI Consensus sequence 5'-A/T A Pu>Py-3'
(-)-Boc-C7-MeCPI Consensus sequence 5'-A/T A Pu>Py-3'
Fig. (7).
bonding model led to further observations with regard to thecatalysis of the alkylation reaction.
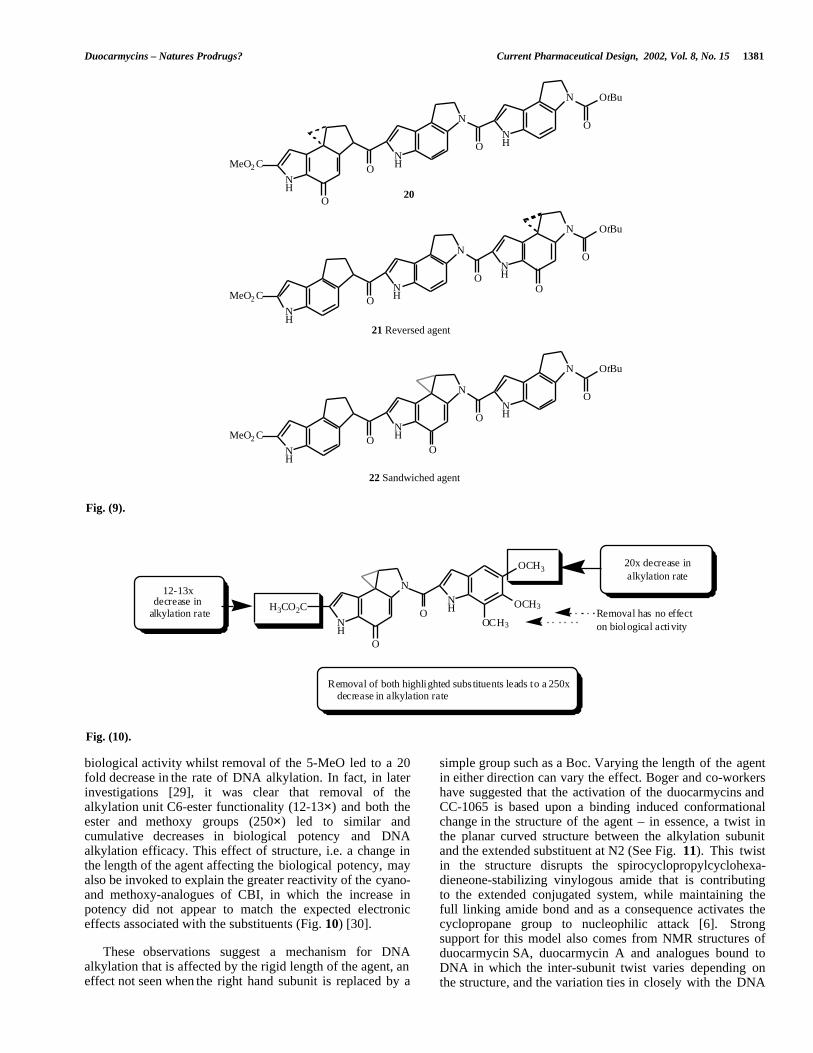
driven binding model, as the sequence dependence foralkylation resides only in the alkylation subunit, alkylationshould always be on the same adenine). While the resultsstrongly supported the binding driven bonding model, asurprising observation was that the reversed agents 21, whilehaving the expected sequence selectivity, lost their ability toalkylate the DNA and as a result suffered a concomitantdecrease in biological potency. The sandwiched agents 22,however, maintained both. This result suggested that thestructure to the right of the alkylating functionality (the“DNA-binding part”) had a crucial effect on the DNAalkylation reaction even though studies had shown that ithad little effect on the solvolytic stability [13,15].
SHAPE DEPENDENT CATALYSIS
While designing compounds to demonstrate the bindingdriven bonding model, Boger and co-workers synthesised anumber of reversed and sandwiched analogues ofduocarmycin SA and CC-1065 in which the DNA bindingunit was maintained, but the alkylation subunit was placedeither in the middle (sandwiched) or to the right (reversed) ofthe trimeric structure (See Fig. 9) [27]. In the binding drivenbonding model for these agents, the reversed agents shouldalkylate the same adenine as the unnatural isomer of thenormal agent, whereas the sandwiched agent should alkylatethe central portion of a run of As, on a different adenine thaneither the normal or reversed compounds (in the bonding
A second key observation was the requirement for the 5-MeO substituent of the right hand indole to be maintainedfor biological activity of duocarmycin SA [28]. Removal ofthe 6- and 7-MeO groups led to no discernible decrease in
NH
N
O
O
N
NN
N
NH2
R
H+
DNA binding unit
Required for affinity, for sequence selectivity and for catalysis (see later)
Nucleophilic attack is a simple, uncatalysed SN2 reaction with a
cyclopropane activated for nucleophilic attack through a
binding induced conformational change
Fig. (8). The binding-driven bonding model.
Duocarmycins – Natures Prodrugs? Current Pharmaceutical Design, 2002, Vol. 8, No. 15 1381
NH
N
O
ONH
N
NH
OtBu
O
MeO2 C
O
NH
N
O
ONH
N
NH
OtBu
O
MeO2 CO
NH
N
O
ONH
N
NH
OtBu
O
MeO2 CO
20
21 Reversed agent
22 Sandwiched agent
Fig. (9).
NH
H3CO2C
N
O
NHO
OCH3
OCH3
OCH3
20x decrease inalkylation rate
12-13x
alkylation rate Removal has no effecton biological activity
Removal of both highlighted subs tituents leads to a 250x decrease in alkylation rate
decrease in
Fig. (10).
biological activity whilst removal of the 5-MeO led to a 20fold decrease in the rate of DNA alkylation. In fact, in laterinvestigations [29], it was clear that removal of thealkylation unit C6-ester functionality (12-13×) and both theester and methoxy groups (250×) led to similar andcumulative decreases in biological potency and DNAalkylation efficacy. This effect of structure, i.e. a change inthe length of the agent affecting the biological potency, mayalso be invoked to explain the greater reactivity of the cyano-and methoxy-analogues of CBI, in which the increase inpotency did not appear to match the expected electroniceffects associated with the substituents (Fig. 10) [30].
simple group such as a Boc. Varying the length of the agentin either direction can vary the effect. Boger and co-workershave suggested that the activation of the duocarmycins andCC-1065 is based upon a binding induced conformationalchange in the structure of the agent – in essence, a twist inthe planar curved structure between the alkylation subunitand the extended substituent at N2 (See Fig. 11). This twistin the structure disrupts the spirocyclopropylcyclohexa-dieneone-stabilizing vinylogous amide that is contributingto the extended conjugated system, while maintaining thefull linking amide bond and as a consequence activates thecyclopropane group to nucleophilic attack [6]. Strongsupport for this model also comes from NMR structures ofduocarmycin SA, duocarmycin A and analogues bound toDNA in which the inter-subunit twist varies depending onthe structure, and the variation ties in closely with the DNA
These observations suggest a mechanism for DNAalkylation that is affected by the rigid length of the agent, aneffect not seen when the right hand subunit is replaced by a

1382 Current Pharmaceutical Design, 2002, Vol. 8, No. 15 Mark Searcey
NH
H3CO2 C
N
O
NHO
OCH3
OCH3
OCH3
Binding induced twist between the subunits
After binding the linking amide is still fully engaged
The vinylogous amide is disrupted, which destabilizes the spirocyclopropylcyclohexadieneone system
Fig. (11).
alkylation and biological data for the same compounds [31-33].
property unique to the tumour must be identified andexploited. A number of reviews in this issue will detail thevarious approaches currently under investigation. Here, wewill focus on efforts disclosed to date to target theduocarmycins, either to tumours specifically or to particularnucleic acid sequences, an approach that may eventually havesome utility either in antisense or antigene therapy.
DESIGN OF PRODRUGS BASED UPON THE TWOMODEL MECHANISMS
The binding driven bonding model for activation of theduocarmycins and CC-1065 has profound implications forthe design of prodrugs based upon these natural products.Using the acid-catalysis mechanism as a starting point, itwould seem reasonable that simply blocking the phenol ofring-opened duocarmycin precursors would be sufficient toblock the activity of the agents, preventing protonation.However, if duocarmycin SA is inactive to nucleophilicattack until it binds to DNA, and there is evidence in thecase of CC-1065 that suggests that it does not react withother biological nucleophiles [34], then it is, in effect, anatural prodrug that is only activated at its target. If thegroup blocking the phenol is removed systemically, it willbe of little benefit in preventing side effects associated withDNA alkylation and, if anything, will only delay any toxiceffects seen with the drug itself. A pre-requisite of drugdesign in the development of duocarmycins as drugs orantitumour agents would appear to be a methodology for thetargeting of the duocarmycins to their site of action. In thenext sections, we will review efforts to generate prodrugsand targeted versions of these compounds.
Amongst the methods for selective targeting of tumours,the use of antibodies directed against tumour-specificantigens has probably met with the most success, with anumber of antibody-based therapies having finally reachedthe clinic, or in very late stage development. Theconjugation of seco-(+)-CBI-indole (23, Fig. 12) to anantibody to the tumour antigen CD19 led to a potentimmunoconjugate with high specificity for CD19-expressingtumour cells, both in vitro and in vivo [35]. Conjugation ofthe synthetic analogue was achieved through disulfide bondformation from the left side of the extended structure, via alinker, to the antibody, giving a conjugate carrying 4-5duocarmycin analogues. The resulting conjugates, both inmurine and humanized form, had potent in vitrocytotoxicities against cells expressing CD19, with IC50values in the low pM range (8pM for murine,15 pM forhumanized). More importantly for this approach, the authorsalso demonstrated that the biological activity of the agentswas selective for cells expressing the particular tumourantigen, with a selectivity against a non-CD19 expressingcell line of >260-500. A second conjugate against a differentantigen gave similar results for cytotoxicity and selectivity.In vivo, the immunoconjugates gave a median increase in lifespan of SCID mice containing a human lymphoma xenograftof 170%. This particularly aggressive tumour, when treatedwith conventional agents gave an increased life span of only22-91%.
TARGETING OF DUOCARMYCINS
The design of prodrugs that are systemically hydrolyzed,as above, relies on the gradual release of potent antitumouragents at doses that are lower than would be achieved bydirect application of the naked agent. It is hoped that the lowdoses maintained will avoid toxicity to other dividing cells,whilst maintaining cytotoxicity in the target tumour. Thereis evidence for the production of general proteases in thevicinity of a developing tumour, perhaps to aid invascularization and migration, and this may give an elementof targeting to the tumour for these compounds; howeverthis is more of a serendipitous occurrence than a designedadvantage. In order to selectively target tumour tissues, a
Wang and co-workers have also described the synthesisof a series of duocarmycin and prodrug analogues and theirconjugation to antibodies [36]. Specifically, they conjugateda seco-CBI-indole derivative to a tumour specific antibodyvia a disulfide linkage that they hoped would be cleaved invivo after targeting to the tumour. The IC50 value for theconjugate in vitro is similar to that for the unconjugateddrug, suggesting that the drug is internalized in the cell andreaches its site of action.
Duocarmycins – Natures Prodrugs? Current Pharmaceutical Design, 2002, Vol. 8, No. 15 1383
N
OH
NHO
NH2
N
OH
NHO
HN
O
O
N
S+HN
OO
S
O-
CO2H
R
O
OOAc
OAc
AcO
CO2H
O
Cl
Cl
23 CBI-5-aminoindole
25 R =
24 R =
Fig. (12).
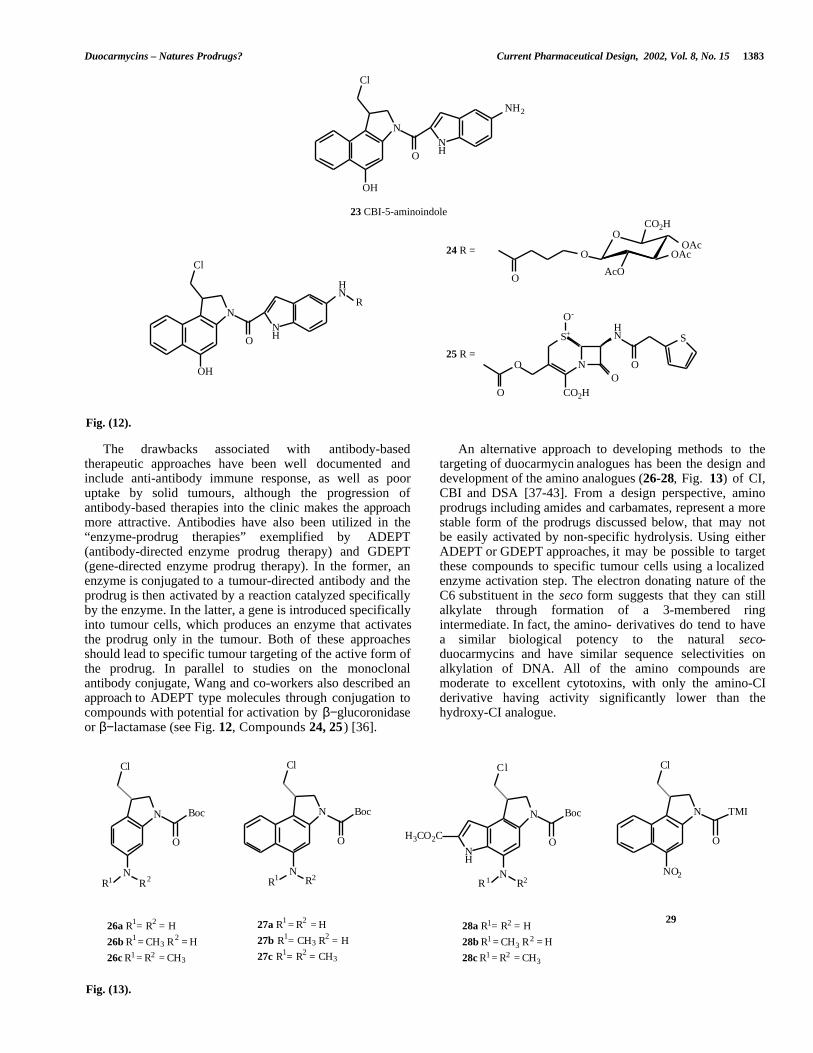
The drawbacks associated with antibody-basedtherapeutic approaches have been well documented andinclude anti-antibody immune response, as well as pooruptake by solid tumours, although the progression ofantibody-based therapies into the clinic makes the approachmore attractive. Antibodies have also been utilized in the“enzyme-prodrug therapies” exemplified by ADEPT(antibody-directed enzyme prodrug therapy) and GDEPT(gene-directed enzyme prodrug therapy). In the former, anenzyme is conjugated to a tumour-directed antibody and theprodrug is then activated by a reaction catalyzed specificallyby the enzyme. In the latter, a gene is introduced specificallyinto tumour cells, which produces an enzyme that activatesthe prodrug only in the tumour. Both of these approachesshould lead to specific tumour targeting of the active form ofthe prodrug. In parallel to studies on the monoclonalantibody conjugate, Wang and co-workers also described anapproach to ADEPT type molecules through conjugation tocompounds with potential for activation by β−glucoronidaseor β−lactamase (see Fig. 12, Compounds 24, 25 ) [36].
An alternative approach to developing methods to thetargeting of duocarmycin analogues has been the design anddevelopment of the amino analogues (26-28, Fig. 13) of CI,CBI and DSA [37-43]. From a design perspective, aminoprodrugs including amides and carbamates, represent a morestable form of the prodrugs discussed below, that may notbe easily activated by non-specific hydrolysis. Using eitherADEPT or GDEPT approaches, it may be possible to targetthese compounds to specific tumour cells using a localizedenzyme activation step. The electron donating nature of theC6 substituent in the seco form suggests that they can stillalkylate through formation of a 3-membered ringintermediate. In fact, the amino- derivatives do tend to havea similar biological potency to the natural seco-duocarmycins and have similar sequence selectivities onalkylation of DNA. All of the amino compounds aremoderate to excellent cytotoxins, with only the amino-CIderivative having activity significantly lower than thehydroxy-CI analogue.
N
N
O
Boc
Cl
R1 R2
N
N
O
Boc
Cl
R1 R2
N
N
O
Boc
Cl
NH
H3CO2C
R1 R2
N
NO2
O
TMI
Cl
26a R1= R2 = H
26b R1 = CH3 R2 = H
26c R1 = R2 = CH3
27a R1 = R2 = H
27b R1= CH3 R2 = H
27c R1= R2 = CH3
28a R1= R2 = H
28b R1 = CH3 R2 = H
28c R1 = R2 = CH3
29
Fig. (13).

1384 Current Pharmaceutical Design, 2002, Vol. 8, No. 15 Mark Searcey
A further advantage of the amino-derivatives could be theability to tune the reactivity of the prodrug linkage throughthe use of either the primary or secondary amine at the C6position. The secondary amine 27b in the amino-CBI-TMIseries was consistently more active in 3 tumour cell linesthan the unsubstituted compound 27a, perhaps reflecting theincrease in electrophilicity of the substituted amine [41]. Aprodrug derived from the secondary amine would beparticularly resistant to non-specific enzyme hydrolysis.
[e.g 44-46]. A selection are shown in Fig. 14. While manyof these compounds have interesting characteristics, it seemsunlikely that they will lead to clinically relevant entities.
TARGETING DUOCARMYCINS USING OLIGO-NUCLEOTIDES
The targeting of oligodeoxynucleotides to either single-or double-stranded DNA remains an attractive goal in thedevelopment of gene-specific agents, although it continuesto suffer from a number of drawbacks. For instance, bindingof an oligodeoxynucleotide (ODN) to single stranded DNAdoes not induce the degradation of the duplex in the sameway that antisense initiates RNaseH degradation of the targetmessage. Targeting of double-stranded DNA through thetriplex approach continues to suffer from a lack of completecode to bind all sequences. Stimulated by the concept of“hybridization-triggered” agents, in which a single-strandedODN bears a moiety that is only active upon forming aduplex, Lukhtanov and co-workers designed a series of C7-MeCPI-tethered ODN targeted both to single-strand [47] anddouble-stranded [48] DNA. In the single-strand targetedagents, the C7-MeCPI was, as expected, inactive until theformation of the duplex with the target DNA. Interestingly,this study reiterated that high binding affinity is not asufficient requirement to turn the simple moiety into aneffective DNA alkylating agent. The ODN carrying a simpleC7-MeCPI analogue 30 was an order of magnitude lesseffective in alkylating DNA than an extended agent 31, inspite of the high affinity of the tethered ODN for its targetsequence (Fig. 15). The more effective nature of the extendedagent is further, indirect support for the model of activationfor alkylation by a binding induced effect that is onlypresent in the extended agents. A similar effect was noted inthe study of triplex forming ODN-C7-MeCPI conjugatestargeted to duplex DNA [48]. No traces of “non-specific”alkylation, i.e. alkylation by the minor groove alkylatingmoiety alone, were seen at low concentrations. However, athigher concentration, in the µM range, non-specific
The amine-based duocarmycin derivatives also provide anentry to a further mechanism of targeting to tumours – thatof bioreductively-activated prodrugs. As a solid tumourdevelops, it begins to outstrip it's vascular network, whichwill eventually lead to the development of three distinctregions: the well-oxygenated region close to the oxygensupply; a necrotic region where cells have succumbed to lowoxygen and a hypoxic region, lying between the dead andwell oxygenated cells, where the tumour is surviving underanaerobic conditions. These cells have a number of uniquefeatures, including their refractory nature to radiation, thatfor many years has made them the subject of extensivestudies to determine methods for their control and drugs thatspecifically target hypoxic fractions in tumours are currentlyundergoing clinical trial and are discussed elsewhere withinthis issue. More recently, the use of these fractions togenerate a bystander effect and activate prodrugs throughtheir unique reducing environment, which then allows thedrug to diffuse into oxygenated tissue and exert its effect,has also been suggested and in this context the amino-duocarmycins may have some potential. C6-nitro-CBI-TMI29 is about 4000' less cytotoxic than the primary amine,10000' less than the secondary amine and could, given thecorrect reduction potential, be an effective bioreductivelyactivated prodrug for the amino derivatives [41]. To date, noinformation regarding the efficacy of the nitro compounds asprodrugs of this type has been disclosed.
Small molecules that are known to have characteristicsthat generate relatively selective tumour uptake have alsobeen conjugated to generate various duocarmycin analogues
N
OH
O
(CH2)n
Cl
N
NH
NCH3
N
OH
O
ClHN H
O
NH
O
H3 C
H3CO2 C
N
O
O
NCH3
HN
O
X
NCH3
HN CH3
O
n = 1-3
X = CH2 , N
Fig. (14).

Duocarmycins – Natures Prodrugs? Current Pharmaceutical Design, 2002, Vol. 8, No. 15 1385
alkylation became more prevalent, although at such highconcentrations, it is not clear whether this effect is caused bycontaminating, unlinked alkylating agent (in other studies,similar untethered agents show characteristic cleavage in the10-100 nM range). A further interesting design was of triplehelix forming ODNs that could crosslink DNA, with a C7-MeCPI at both the 3’- and 5’-ends [48]. It should be notedin the triplex forming case that a major groove targetedcompound is directing an agent into the minor groove.
activity both in vitro and in vivo. A similar approach hasbeen adopted by researchers at the Kyowa Hakko Kogyo Co.in Japan in their search for prodrug analogues ofduocarmycin A and its synthetic rearrangement product DU-86. A series of analogues of the ring-opened duocarmycin B2were synthesized, either esters or carbamates, in whichvarious solubilizing or hydrophobic blocking groups wereappended [50]. Careful study of the stability of the agents inaqueous solution, in culture medium or in fetal calf serumrevealed that a dialkyl carbamate was inactive when in cellculture but had potent antitumour activity in vivo.Although these compounds are of some interest, it seems
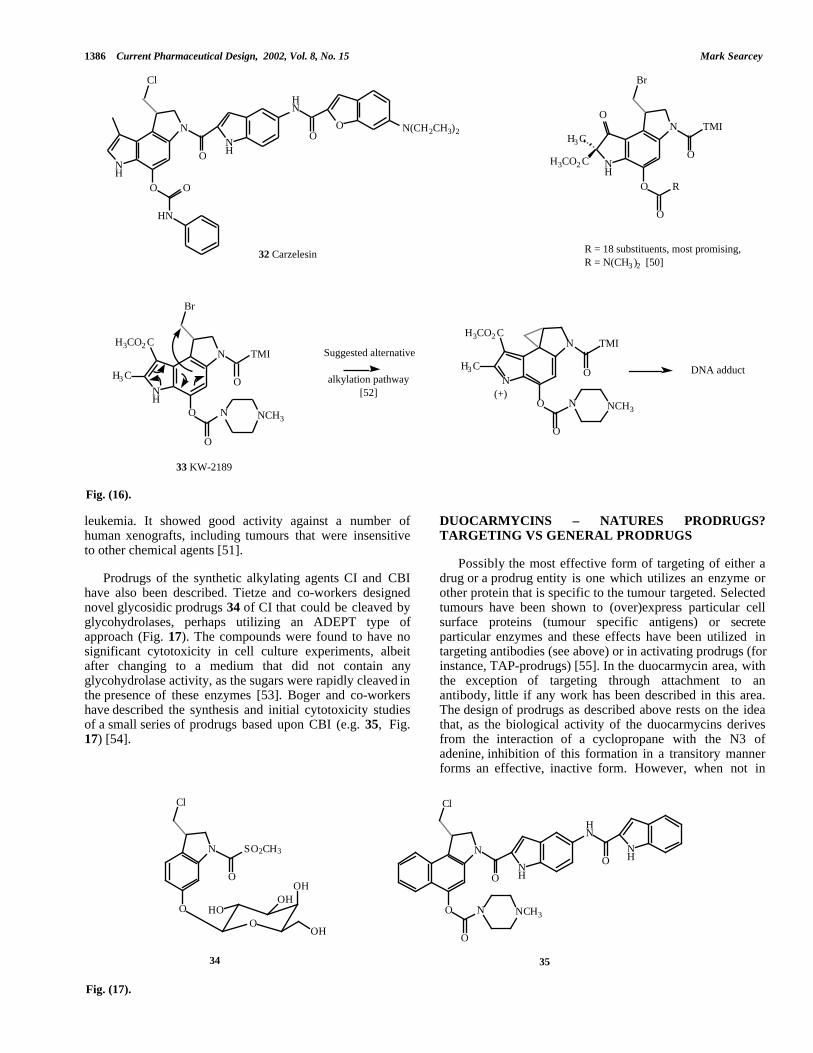
unlikely that oligonucleotides of this type will progresssuccessfully into the clinic. KW-2189 33 is a water-soluble synthetic analogue of
DU-86 that has progressed into clinical trials [51]. Althoughdesigned as a prodrug that is activated by enzymatichydrolysis to release DU-86 as the active metabolite, it wassuggested that KW-2186 could, in fact, alkylate DNAthrough an alternative pathway (see Fig. 16). In order to testthis hypothesis, which suggests that alkylation may not beblocked by O-5 substitution, a series of analogues of KW-2189 were synthesized containing various ether, ester,carbonate, carbamate, sulphonate groups at the O-5 as wellas one compound unsubstituted at this position [52]. Theresults disclosed clearly confirmed that release of DU-86through removal of substitution at the phenolic oxygen wasa requirement for biological activity. KW-2189 inhibited thegrowth of solid tumours in vivo and was also active against
GENERAL PRODRUGS BASED UPON CC-1065 ANDTHE DUOCARMYCINS
Initial studies on the potential clinical utility of (+)-CC-1065 focused on the design of active agents that did notdisplay the delayed lethal toxicity of the natural product[49]. From these studies carzelesin (32, Fig. 16), a prodruganalogue was identified, which carries a phenyl carbamate onthe OH of the seco-agent, to block spirocyclization and adiethylamine moiety on the right hand DNA bindingsubunit to aid solubility. Carzelesin acts by general serumbased hydrolysis of the carbamate and has antitumour
N
O
ONH
NH(CH2)6O-Oligo
O
N
O
ONH
NH(CH2 )6O-Oligo
O
NH
N
O
T T
T T
5'-CTTTGTCT CCTCTTT CCTCT CCTCTCT CCTTCTC TTCCAGGCTTAAGCCTGTAG
3'-GAAACAGAGGAGAAAGGAGAGGAGAGAGGAAGAGAAGGTCCGAATTCGGACATC
30
31
G CC GG CA TA TA TA TA TA TCGTGTTGTACCC5'
GCACAACATGGG
30 or 31
5'-AGAAAGGAGAGGAGAGAGGAAGAG-3'30 or 31
Triplex targeting
Single strand targeting
Fig. (15).
1386 Current Pharmaceutical Design, 2002, Vol. 8, No. 15 Mark Searcey
N
O
ONH
NH
HN
OO N(CH2CH3)2
Cl
HN
O
NH
O
H3 C
H3CO2 C
N
Br
TMI
O
O
O
R
NH
H3CO2 CN
Br
TMI
O
O
O
N NCH3
H3 C N
H3CO2 CN TMI
O
O
O
N NCH3
H3 C
32 Carzelesin R = 18 substituents, most promising, R = N(CH3 )2 [50]
33 KW-2189
Suggested alternative
alkylation pathway[52] (+)
DNA adduct
Fig. (16).
leukemia. It showed good activity against a number ofhuman xenografts, including tumours that were insensitiveto other chemical agents [51].
DUOCARMYCINS – NATURES PRODRUGS?TARGETING VS GENERAL PRODRUGS
Possibly the most effective form of targeting of either adrug or a prodrug entity is one which utilizes an enzyme orother protein that is specific to the tumour targeted. Selectedtumours have been shown to (over)express particular cellsurface proteins (tumour specific antigens) or secreteparticular enzymes and these effects have been utilized intargeting antibodies (see above) or in activating prodrugs (forinstance, TAP-prodrugs) [55]. In the duocarmycin area, withthe exception of targeting through attachment to anantibody, little if any work has been described in this area.The design of prodrugs as described above rests on the ideathat, as the biological activity of the duocarmycins derivesfrom the interaction of a cyclopropane with the N3 ofadenine, inhibition of this formation in a transitory mannerforms an effective, inactive form. However, when not in
Prodrugs of the synthetic alkylating agents CI and CBIhave also been described. Tietze and co-workers designednovel glycosidic prodrugs 34 of CI that could be cleaved byglycohydrolases, perhaps utilizing an ADEPT type ofapproach (Fig. 17). The compounds were found to have nosignificant cytotoxicity in cell culture experiments, albeitafter changing to a medium that did not contain anyglycohydrolase activity, as the sugars were rapidly cleaved inthe presence of these enzymes [53]. Boger and co-workershave described the synthesis and initial cytotoxicity studiesof a small series of prodrugs based upon CBI (e.g. 35, Fig.17) [54].
N
O
O
Cl
N
O
O
O
N NCH3
SO2CH3
OHO
OHOH
OH
NH
HN
ONH
Cl
34 35
Fig. (17).

Duocarmycins – Natures Prodrugs? Current Pharmaceutical Design, 2002, Vol. 8, No. 15 1387
N
ONH
A1
Cl
A
B
C OX
B1
C1
N
ONH
A1
A
B
C O
B1
C1
X - substitution blocks ring closure. For effective prodrug it must also have some element of targeting - amino- group? ADEPT or GDEPT activated. Systemic activation not sufficientA, A1 - Substituents may affect DNA binding - steric effects or effects on DNA binding induced conformational change.B, B1, C, C1 - substituents point out of the groove and have little effect on binding. Targeted to tumour by e.g, receptor binding? Care with B and C as may have electronic effects on the reactivity of the alkylating subunit.
Fig. (18).
contact with DNA, the alkylation subunit of theduocarmycins has been shown to be exceptionally stable tohydrolysis. In early studies, it was shown that CC-1065 didnot appear to interact with any other biological nucleophiles.In fact, reaction with nucleophiles (the N3 of adenine) onlyappears to significantly occur upon binding to DNA. Asevidence increases for a ground state destabilizationmechanism through a binding induced twist in the extendedstructure of the antibiotic, it becomes clear that nature hasdeveloped an antibiotic prodrug that is unreactive tobiological targets other than the one to which it is targeted –DNA. As nature did not design the compounds asantitumour agents, it remains to the medicinal chemist toget the compounds to the site of action that we desire –specifically to the tumour. If we consider the structuralfeatures required for effective biological activity, there are 3possible methods for targeting to tumours (Fig. 18). Onlyone of these involves substitution at C5, the preferredmethod for prodrug design as noted above. This onlybecomes useful if the seco- agent prodrug can be specificallytargeted to the tumour, for instance through conjugation to aspecific receptor binding protein [56]. Targeting of the“active” cyclopropyl-containing agent can be addressedthrough substitution of the fused pyrrole (in the naturalproduct) or benzene ring (in the synthetic analogue CBI),although care is required in drug design on both counts.Firstly, substitution at the C6 and C7 positions in bothstructures and also at the C8 in CBI may lead to decreasedDNA binding and thus a decrease in biological activity.Secondly, substitution in the fused ring can have an effecton the reactivity of the alkylating moiety, either of anelectronic nature or more likely through effects on thebinding induced twist in the extended structure.
N1-position proton point out of the minor groove,interacting minimally with DNA and not contributing to theextended structure that induces the conformational changeand activates the agent for DNA alkylation. These positionsare clearly available for substitution and with theidentification of tumour specific ligands may allow thedevelopment of tumour selective targeted duocarmycins.Boger and co-workers have recently published an extensivestudy of the effects of substitution in the DNA binding righthand subunits. [57]
CONCLUSIONS
The duocarmycins and CC-1065 are amongst the mostwell studied and most potent antitumour antibiotics so farisolated from natural sources. The design and synthesis ofcarefully designed synthetic analogues of the naturalproducts has allowed the study of the mechanism of actionof these agents in a more detailed fashion than would havebeen possible with the natural products alone and has helpedto define their mechanism of action at a molecular level.This level of detailed knowledge has helped to define themechanism by which the extremely stable duocarmycinsbind to and alkylate duplex DNA. This mechanism, whichinvolves a binding induced conformational change thatactivates the agents for DNA alkylation, suggests that theduocarmycins are, in themselves, a prodrug evolvednaturally to only interact with its target. In order to designantitumour agents based upon the duocarmycins that areactive in the clinic, it is likely that tumour specific targetingof the agents, rather than prodrugs designed to “inactivate”the alkylating core, would be of benefit.
Possibly more profitable is an approach utilizingconjugation to tumour targeting moieties throughsubstitution in the DNA binding/non-alkylating portions ofthe structure. In the duocarmycin series, it has been shownthat the 5-methoxyindole analogue of duocarmycin SAretains essentially all of the DNA binding ability andbiological activity of the full natural product structure. NMRanalysis clearly shows that the 6, 7 methoxy groups and the
ACKNOWLEDGEMENTS
Work in the authors laboratory is supported by theSchool of Pharmacy, the European Union and theEngineering and Physical Sciences Research Council.

1388 Current Pharmaceutical Design, 2002, Vol. 8, No. 15 Mark Searcey
REFERENCES [16] Boger, D. L.; Wysocki, R. J.; Ishizaki, T. J. Am. Chem. Soc.1990, 112, 5230
[1] Ichimura, M.; Ogawa, T.; Takahashi, K.; Kobayashi, E.;Kawamoto, I.; Yasuzawa, T.; Takahashi, I.; Nakano, H. J.Antibiot. 1990, 43 , 1037. Ichimura, M.; Ogawa, T.;Katsumata, S.; Takahashi, K.; Takahashi, I.; Nakano, H. J.Antibiot. 1991, 44 , 1045. Ohba, K.; Watabe, H.; Sasaki,T.; Takeuchi, Y.; Kodama, Y.; Nakazawa, T.; Yamamoto,H.; Shomura, T.; Sezaki, M.; Kondo, S. J. Antibiot. 1988,41 , 1515. Takahashi, I.; Takahashi, K.; Ichimura, M.;Morimoto, M.; Asano, K.; Kawamoto, I.; Tomita, F.;Nakano, H. J. Antibiot. 1988, 41 , 1915. Yasuzawa, T.;Iida, T.; Muroi, K.; Ichimura, M.; Takahashi, K.; Sano, H.Chem. Pharm. Bull. 1988, 36 , 3728. Ichimura, M.; Muroi,K.; Asano, K.; Kawamoto, I.; Tomita, F.; Morimoto, M.;Nakano, H. J. Antibiot. 1988, 41 , 1285. Ishii, S.;Nagasawa, M.; Kariya, Y.; Yamamoto, H.; Inouye, S.;Kondo, S. J. Antibiot. 1989, 42 , 1713. Hanka, L. J.; Dietz,A.; Gerpheide, S. A.; Kuentzel, S. L.; Martin, D. G. J.Antibiot. 1978, 31 , 1211. Chidester, C. G.; Krueger, W.C.; Mizsak, S. A.; Duchamp, D. J.; Martin, D. G. J. Am.Chem. Soc. 1981, 103, 7629.
[17] Boger, D. L.; Ishizaki, T.; Kitos, P. A.; Suntornwat, O. J.Org. Chem. 1990, 55 , 5823.
[18] Hurley, L. H.; Reynolds, V. L.; Swenson, D. H.; Petzold,G. L.; Scahill, T. A. Science 1984, 226, 843. Reynolds, V.L.; Molineux, I. J.; Kaplan, D. J.; Swenson, D. H.; Hurley,L. H. Biochemistry 1985, 24 , 6228.
[19] Hurley, L. H.; Lee, C.-S.; McGovren, J. P.; Warpehowski,M. A.; Mitchell, M. A.; Kelly, R. C.; Aristoff, P. A.Biochemistry 1988, 27 , 3886.
[20] Sun, D.; Lin, C. H.; Hurley, L. H. Biochemistry 1993, 32 ,4487.
[21] Hurley, L. H.; Draves, P. H. in Molecular Aspects ofDrug-DNA interactions, Vol 1 Eds. S. Neidle, M. J.Waring. CRC, Boca Raton, FL. 1993, pp 89-133
[22] Boger, D. L.; Coleman, R. S.; Invergo, B. J.; Sakya, S. M.;Ishizaki, T.; Munk, S. A.; Zarrinmayeh, H.; Kitos, P. A.;Collins Thompson, S. J. Am. Chem. Soc. 1990, 112, 4623[2] Boger, D. L.; Johnson, D. S.; Wrasidlo, W. Bioorg. Med.
Chem. Lett. 1994, 4, 631-636.[23] Hurley, L. H.; Warpehowski, M. A.; Lee, C.-S.; McGovren,
J. P.; Scahill, T. A.; Kelly, R. C.; Mitchell, M. A.;Wicnienski, N. A.; Gebhard, I.; Johnson, P. D.; Bradford,V. S. J. Am. Chem. Soc. 1990, 112, 4633.
[3] Boger D. L.; Boyce, C. W.; Garbaccio, R. M.; Goldberg, J.A. Chem. Rev. 1997, 97 , 787.
[4] Boger D. L.; Johnson, D. S. Angew. Chem. Int. Ed. Engl.1996, 35 , 1438. [24] Boger, D. L.; Johnson, D. S.; Yun, W. J. Am. Chem. Soc.
1990, 116, 1635.[5] Warpehowski, M. A.; Hurley, L. H. Chem. Res. Toxicol.1988, 1, 315. [25] Boger, D. L.; Zarrinmayeh, H.; Munk, S. A.; Kitos, P. A.;
Suntornwat, O. Proc. Natl. Acad. Sci. USA 1991, 88 ,1431. Boger D. L.; Munk, S. A.; Zarrinmeyah, H. J. Am.Chem. Soc. 1991, 113, 3980.
[6] Boger, D. L.; Garbaccio, R. M. Acc. Chem. Res. 1999, 32 ,1043. Boger, D. L.; Garbaccio, R. M. Bioorg. Med. Chem.1997, 5, 263.
[26] Boger, D. L.; Garbaccio, R. M.; Jin, Q. J. Org. Chem. 1997,62 , 8875.[7] Zimmer, C.; Wähnert, U. Prog. Biophys. Mol. Biol. 1986,
47 , 31.
[27] Boger, D. L.; Bollinger, B.; Hertzog, D. L.; Johnson, D. S.;Cai, H.; Goldberg, J.; Mésini, P.; Garbaccio, R. M.; Jin,Q.; Kitos, P. A. J. Am. Chem. Soc. 1997, 119, 4987.
[8] Boger, D. L.; Invergo, B. J.; Coleman, R. S.; Zarrinmayeh,H.; Kitos, P. A.; Thompson, S. C.; Leong, T.; McLaughlin,L. W. Chem-Biol. Interact. 1990, 73 , 29.
[28] Boger, D. L.; Bollinger, B.; Johnson, D. S. Bioorg. Med.Chem. Lett. 1996, 6, 2207.[9] Schultz, P. G.; Taylor, J. S.; Dervan, P. B. J. Am. Chem.
Soc. 1982, 104, 6861.
[29] Boger, D. L.; Santillán, A.; Searcey, M.; Brunette, S. R.;Wolkenberg, S. E.; Hedrick, M. P.; Jin, Q. J. Org. Chem.2000, 65 , 4101-4111.
[10] Boger, D. L.; Zhou, J.; Cai. H. Bioorg. Med. Chem. 1996,4, 859
[11] Boger, D. L.; Boyce, C. W.; Johnson, D. S. Bioorg. Med.Chem. Lett. 1997, 7, 233 [30] Boger, D. L.; McKie, J. A.; Han, N.; Tarby, C. M.; Riggs,
H. W.; Kitos, P. A. Bioorg. Med. Chem. Lett. 1996, 6, 659.
[12] See Table 1. Data taken from Boger, D. L.; Santillán, A.;Searcey, M.; Brunette, S. R.; Wolkenberg, S. E.; Hedrick,M. P.; Jin Q. J. Org. Chem. 2000, 65 , 4101-4111.
[31] Lin, C. H.; Patel, D. J. J. Mol. Biol. 1995, 248, 162.
[32] Eis, P.S.; Smith, J. A.; Rydzewski, J. M.; Case, D. A.;Boger, D. L.; Chazin, W. J. J. Mol. Biol. 1997, 272, 237.[13] Warpehowski, M. A.; Harper, D. E. J. Am. Chem. Soc.
1994, 116, 7573. [33] Schnell, J. R.; Ketchem, R. R.; Boger, D. L.; Chazin, W. A.J. Am. Chem. Soc. 1999, 121I, 5645.[14] Lamm, G.; Pack, G. R. Proc. Natl. Acad. Sci. USA 1990,
87 , 9033. [34] Li, L. H.; Swenson, D. H.; Schpok, S. L. F.; Kuentzel, S. L.;Dayton, B. D.; Krueger, W. C. Cancer Res. 1982, 42 , 999.[15] Warpehowski, M. A.; Gebhard, I.; Kelly, R. C.; Krueger,
W. C.; Li, L. H.; McGovren, J. P.; Prairie, M. D.;Wicnienski, N.; Wierenga, W. J. Med. Chem. 1988, 31 ,590.
[35] Chari, R. V. J.; Jackel, K. A.; Bourret, L. A.; Derr, S. M.;Tadayoni, B. M.; Mattocks, K. M.; Shah, S. A.; Liu, C.;Blättler, W. A.; Goldmacher, V. S. Cancer Res. 1995, 55 ,4079.

Duocarmycins – Natures Prodrugs? Current Pharmaceutical Design, 2002, Vol. 8, No. 15 1389
[36] Wang, Y.; Wright, S. C.; Larrick, J. L. US Patent5,843,937 1998.
[48] Lukhtanov, E. A.; Mills, A. G.; Kutyavin, I. V.; Gorn, V.V.; Reed, M. W.; Meyer, R. B. Nucleic Acids Res. 1997,25 , 5077.
[37] Tercel, M.; Denny, W. A.; Wilson, W. R. Bioorg. Med.Chem. Lett. 1996, 6, 2735 [49] Li, L. H.; DeKoning, T. F.; Kelly, R. C.; Krueger, W. C.;
McGovren, J. P.; Padbury, G. E.; Petzold, G. L.; Wallace,T. L.; Ouding, R. J.; Prairie, M. D.; Gebhard, I. CancerRes. 1992, 52 , 4904
[38] Atwell, G. J.; Wilson, W. R.; Denny, W. A. Bioorg. Med.Chem. Lett. 1997, 7, 1493
[39] Tercel, M.; Denny, W. A. J. Chem. Soc. Perkin Trans. 11998, 509
[50] Nagamura, S.; Kanda, Y.; Kobayashi, E.; Gomi, K.; Saito,H. Chem. Pharm. Bull. 1995, 43 , 1530
[40] Giesig, M. A.; Matejovic, J.; Denny, W. A. Anti-CancerDrug Des. 1999, 14 , 77.
[51] Kobayashi, E.; Okamoto, A.; Asada, M.; Okabe, M.;Nagamura, S.; Asai, A.; Saito, H.; Gomi, K.; Hirata, T.Cancer Res. 1994, 54 , 2404.
[41] Atwell, G. J.; Tercel, M.; Boyd, M.; Wilson, W. R.; Denny,W. A. J. Org. Chem. 1998, 63 , 9414 [52] Nagamura, S.; Kobayashi, E.; Gomi, K.; Saito, H. Bioorg.
Med. Chem. 1996, 4, 1379.[42] Tercel, M.; Giesig, M. A.; Denny, W. A.; Wilson, W. R. J.
Org. Chem. 1999, 64 , 5946. [53] Tietze, L. F.; Hanneman, R.; Buhr, W.; Löger, M.;Menningen, P.; Lieb, M.; Starck, D.; Grote, T.; Döring, A.;Schuberth, I. Angew. Chem. Int. Ed. Engl. 1996, 35 , 2674[43] Milbank, J. B. J.; Tercel, M.; Atwell, G. J.; Wilson, W. R.;
Hogg, A.; Denny, W. A. J. Med. Chem. 1999, 42 , 649[54] Boger D. L.; Boyce, C. W.; Garbaccio, R. M.; Searcey, M.;
Jin, Q. Synthesis 1999, 1505-1509.[44] Fan, J.-Y.; Tercel, M.; Denny, W. A. Anti-Cancer DrugDes. 1997, 12 , 277
[55] Denny, W. A. Eur. J. Med. Chem. 2001, 36 , 577.[45] Shishido, K.; Haruna, S.; Yamamura, C.; Iitsuka, H.;
Nemoto, H.; Shinohara, Y.; Shibuya, M. Bioorg. Med.Chem. Lett. 1997, 7, 2167
[56] Sebestyén, M. G.; Ludtke, J. J.; Bassik, M. C.; Zhang, G.;Budker, V.; Lukhtanov, E. A.; Hagstrom, J. E.; Wolff, J. A.Nature Biotech. 1998, 16 , 80.
[46] Tao, Z.-F.; Fujiwara, T.; Saito, I.; Sugiyama, H. Angew.Chem. Int. Ed. Engl. 1999, 38 , 650. [57] Boger, D. L.; Stauffer, F.; Hedrick, M. P. Bioorg. Med.
Chem. Lett. 2001, 9, 2021.[47] Lukhtanov, E. A.; Kutyavin, I. V.; Gorn, V. V.; Reed, M.
W.; Adams, A. D.; Lucas, D. D.; Meyer, R. B J. Am. Chem.Soc. 1997, 119, 6214.
RELATED ARTICLES RECENTLY PUBLISHED INCURRENT PHARMCEUTICAL DESIGN
Hill, B.T. Vinflunine, a Second Generation Novel VincaAlkaloid with a Distinctive Pharmacological Profile,Now in Clinical Development and Prospects forFuture Mitotic Blockers. Curr. Pharm. Des., 2001,7(13), 1199-212.
Nakajima, S.; Graham, D.Y.; Hattori, T. and Bamba, T.Strategy for Treatment of Helicobacter pyloriInfection in Adults. II. Practical Policy in 2000.Curr. Pharm. Des., 2000, 6(15), 1515-29.
Ünak, T. Potential use of Radiolabeled GlucuronideProdrugs with Auger and/or Alpha Emitters inCombined Chemo- and Radio-therapy of Cancer.Curr. Pharm. Des., 2000, 6(11), 1127-42.