dual-promoter lentiviral system allows inducible expression of noxious proteins in macrophages
TRANSCRIPT

Journal of Immunological Methods 329 (2008) 31–44www.elsevier.com/locate/jim
Research paper
Dual-promoter lentiviral system allows inducible expression ofnoxious proteins in macrophages
Hui Pan a, Gustavo Mostoslavsky b, Evgeny Eruslanov a,1,Darrell N. Kotton b,2, Igor Kramnik a,⁎
a Department of Immunology and Infectious Diseases, Harvard School of Public Health, 667 Huntington Avenue, Boston, MA 02115, USAb Department of Genetics, Harvard Medical School, 4 Blackfan Circle-Harvard Institute of Medicine-Room 405, Boston, MA 02115, USA
Received 10 July 2007; received in revised form 14 September 2007; accepted 19 September 2007Available online 18 October 2007
Abstract
In-depth studies of innate immunity require efficient genetic manipulation of macrophages, which is especially difficult in primarymacrophages. We have developed a lentiviral system for inducible gene expression both in macrophage cell lines and in primarymacrophages. A transgenic mouse strain C3H.TgN(SRA–rtTA) that expresses reverse tetracycline transactivator (rtTA) under thecontrol of macrophage-specific promoter, a modified human Scavenger Receptor A (SRA) promoter was generated. For genedelivery, we constructed a dual-promoter lentiviral vector, in which expression of a “gene-of-interest” is driven by a doxycycline-inducible promoter and the expression of a selectable surface marker is driven by an independent constitutive promoter UBC. Thisvector is used for transduction of bone marrow-derived macrophage precursors. The transduced cells can be enriched to 95–99%purity usingmarker-specific monoclonal antibodies, expanded and differentiated into mature macrophages or myeloid dendritic cells.We also successfully used this approach for inducible protein expression in hard to transfect macrophage cell lines.
Because many proteins, which are expressed by activated or infected macrophages, possess cytotoxic, anti-proliferative or pro-apoptotic activities, generation of stablemacrophage cell lines that constitutively express those proteins is impossible. Ourmethodwill beespecially useful to study immunity-relatedmacrophage proteins in their physiological context duringmacrophage activation or infection.© 2007 Elsevier B.V. All rights reserved.
Keywords: Lentiviral vectors; Macrophage; Tetracycline-inducible expression; Ipr1 gene; Interferon-activation; Protein–protein interaction
1. Introduction
Macrophages are versatile cells that play diverse rolesin host defenses, tissue homeostasis and remodeling
⁎ Corresponding author.E-mail address: [email protected] (I. Kramnik).
1 Current address: Department of Urology, University of FloridaCollege of Medicine, 1600 SW Archer Rd., Gainesville, FL 32610-0247, USA.2 Current address: The Pulmonary Center, Boston University School
of Medicine, 715 Albany St, R-304, Boston, MA 02118, USA.
0022-1759/$ - see front matter © 2007 Elsevier B.V. All rights reserved.doi:10.1016/j.jim.2007.09.009
under normal and pathologic conditions such as theonset, progression and resolution of various inflamma-tory and infectious diseases, as well as in the control oftumor progression (Ross and Auger, 2002; Sasmono andHume, 2004; Henson and Hume, 2006). Macrophagesperform both regulatory and effector functions byproducing secreted pro-inflammatory and pro-angiogenicmediators, growth factors, antibacterial peptides, reactiveoxygen and nitrogen species as well as via direct contactwith target cells. Macrophages also ingest particles andlive pathogens. They can either destroy the ingested

32 H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
pathogens or serve as reservoirs for their propagationpromoting survival and dissemination of infectiousagents, such as HIV and Mycobacterium tuberculosisamong others. The ability to understand genetic control ofmacrophage function will certainly allow development ofrational interventions to enhance beneficial and preventdisease-promoting macrophage activities.
Modern approaches to dissecting cellular functionsbroadly utilize expression ofmodified genes and reporterconstructs in target cells. However, the delivery ofrecombinant genetic constructs into macrophages isdifficult, which limits utilization of powerful molecularapproaches to the studies of the macrophage biology. It iswell known that the widely used transfection methodsusing synthetic carriers such as liposomes, lipoplexes ordiethylaminoethyl (DEAE)-dextran, to deliver nakedplasmid DNA, are inefficient in primary macrophages(Heider et al., 2000). Only a few monocyte ormacrophage cell lines, such as THP-1 or RAW264.7,can be efficiently transfected by plasmid DNA. Elec-troporation has been used to transfer plasmid DNA intomonocytic cell lines or macrophages with high efficien-cy (Weir and Meltzer, 1993; Hume et al., 2001). Usually,electroporation causes significant cell death and releaseof intracellular contents, which is known to inducemacrophage responses (Krysko et al., 2006). Moreover,both transfection and electroporation with plasmid DNAcontaining unmethylated CpG (cytosine followed byguanine) dinucleotides may activate macrophagesthrough Toll-like receptor (TLR)-9 to produce pro-inflammatory cytokines (Stacey et al., 1996; Sester et al.,1999; Jiang et al., 2006).Thus, the existing methods ofplasmid delivery perturb the normal physiological statusof the macrophage population and complicate interpre-tation of the experimental data. Recombinant vectorsbased on adenovirus (De et al., 1998; Foxwell et al.,1998; Heider et al., 2000) and lentivirus (Naldini et al.,1996; Corbeau et al., 1998; Schroers et al., 2000) havebeen used to deliver genetic constructs into macrophagesmuch more efficiently. This includes attempts to expresstherapeutic genes in macrophages, and use of thetransduced macrophages as a vehicle for adoptiveimmunotherapy (Burke et al., 2002).
The efficiency of transduction of primary macro-phages is much lower as compared to standard cell lines,which requires selection of the transduced cells. A popularstrategy for identification and enrichment of the virallytransduced cells is based on bicistronic constructs, inwhich two genes, a gene-of-interest and a selectablemarker, are encoded by the same transcript, and therefore,are expressed simultaneously. Thus, the cells expressingthe gene-of-interest may be identified and positively
selected. However, many genes related to innate immu-nity are expressed in activated or infected macrophagesonly in an inducible manner. These genes often possesscytotoxic, anti-proliferative or pro-apoptotic activities,which are directly related to their role in immunity topathogens. This precludes stable long-term expression ofthose immune-related macrophage genes in dividingcells, such as hematopoietic stem cells, and makes geneticcomplementation tests in a context of experimental infec-tion in vivo impossible.
Macrophages are the primary host cells of M.tuberculosis (MTB) as well as many other intracellularbacteria. Previously, we have mapped the sst1 locus onmouse chromosome 1 (supersusceptibility to tubercu-losis, 49–52 cM) that mediates host resistance totuberculosis (Kramnik et al., 2000). A strong candidategene Ipr1 (intracellular pathogen resistance 1) hasbeen identified within the sst1 locus using a positionalcloning approach. This is an inducible protein which isexpressed in interferon-activated and/or MTB-infectedmacrophages of the sst1-resistant (C57BL/6J), but notsst1-susceptible (C3HeB/FeJ) mice (Pan et al., 2005).To perform complementation tests and to study themolecular basis of Ipr1-mediated macrophage function(s), we attempted expression of the Ipr1 gene inhematopoietic stem cells and bone marrow-derivedmacrophages of the Ipr1-negative C3HeB/FeJ miceusing a standard bicistronic lentiviral vector, and failed,because overexpression of Ipr1 blocked cell divisionand increased apoptosis. Therefore, we have developeda system for efficient genetic manipulation of bothmacrophage cell lines and primary mature macrophagesbased on a set of dual-promoter lentiviral vectors. Theseenabled us to study the Ipr1 protein function in aphysiologically-relevant context of activated andMTB-infected macrophages.
2. Materials and methods
2.1. Mice
C3HeB/FeJ mice were obtained from the JacksonLaboratory (Bar Harbor, ME, USA). To make atransgene construct we used a pBlueScript KS (Strata-gene, Cedar Creek, TX, USA) derived plasmid contain-ing the reverse tetracycline transactivator (rtTA) and theSV40 polyadenylation signal (SV40pA). The XhoI–EcoRI DNA fragment of a modified human ScavengerReceptor A (SRA) promoter was derived from plasmidpAL1 (a generous gift of Dr Christopher Glass (Horvaiet al., 1995)). This promoter fragment was inserted intothe pBlueScript KS upstream of the rtTA using the same

33H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
restriction sites. To generate the transgenic mouse strainC3H.TgN(SRA–rtTA), the above plasmid was digestedwith XhoI and NotI to isolate the 6777-bp SRA–rtTA–SV40pA transgene. The fragment was purified fromagarose gel and introduced into fertilized C3HeB/FeJoocytes by pronuclear injection. Offspring was geno-typed by PCR with primers rtTA-4F (5′-CGC TAGACG ATT TCG ATC TGG AC-3′) and rtTA-4R (5′-TTC CAA GGG CAT CGG TAA ACA-3′). Transgene-bearing founder mice were backcrossed to the C3HeB/FeJ mice. The transcription of the transgene wasconfirmed by RT-PCR. Homozygote transgenic micewere generated by intercrossing transgene-positiveanimals and selection for the transgene homozygotesusing quantitative PCR. The sequence of the SRA–rtTA–SV40pA transgene is available upon request.Mice were bred and maintained under specific-patho-gen-free conditions in animal facility at the HarvardMedical School and given autoclaved chow and water adlibitum. All experiments were performed with the fullknowledge and approval of the Standing Committee onAnimals at Harvard Medical School.
2.2. Cell lines and BMDMs culture
Human renal epithelial cell line 293T, mouse fibro-blast cell line NIH/3T3, mouse macrophage cell linesRAW264.7 and J774A.1 were cultured in DMEM/F12(Mediatech, Inc., Herndon, VA, USA) containing 7.5%Tet-system approved fetal bovine serum (FBS, Clon-tech, Mountain View, CA, USA) and 10 mM Hepesbuffer (Mediatech, Inc.). Isolation of mouse bonemarrow and culture of BMDMs were describedpreviously (Pan et al., 2005). Briefly, mouse femursand tibias were homogenized in DMEM (Mediatech,Inc.) containing 2% FBS. Bone marrow cells werefiltered through a 70-μm strainer (BD Biosciences, SanJose, CA, USA) and further purified on a gradient ofNycoPrep A-1.077 (Axis-Shield Plc, Dundee, UK).Purified bone marrow cells were cultured in DMEM/F12 containing 10% FBS, 1 ng/mL recombinant mouseinterleukin-3 (rmIL-3, R&D Systems, Minneapolis,MN, USA), and 10% L-929 cell conditioned mediumas a source of colony stimulation factor-1 (CSF-1) for3 days. Non-adherent cells were collected and expandedin the same medium in the ultra-low cluster plates(Corning, Acton, MA, USA) for additional 6–20 dayswith medium changed every 2 days. Appropriate numberof cells were plated in tissue culture plastic ware inDMEM/F12 containing 10% FBS and 20% L-929conditioned medium (w/o IL-3) to form a monolayer ofmacrophages.
2.3. Construction of dual-promoter lentiviral vectors
The pHAGE backbone lentiviral vector used in theexperiments was an optimized self-inactivating non-replicative vector derived from the pHR'CMV-lacZvector (Naldini et al., 1996). The original pHAGE vectorwill be described in detail elsewhere (Balazs et al., inpreparation). Briefly, the original pHR'CMV-lacZ fromNaldini et al. was modified to create the pHRST vectorby adding a polypurine tract between the 3′ end of theenv sequence and the 5′ end of the CMV promoter. Inaddition, the LacZ gene was changed to eGFP and aWoodchuck Hepatitis virus post-transcriptional regula-tory element (WPRE) was cloned downstream of eGFPat the KpnI site. In order to remove extra sequences andalleviate cloning strategies the pHRST vector was furthermodified to create the pHAGE vector, by first movingthe viral backbone from 5′-LTR to 3′ flanking region intoa minimal pUC backbone containing an SV40 origin ofreplication. Subsequent cloning re-created the centralpolypurine tract and added a unique SpeI cloning site tothe 5′end and NotI site at the 3′ end of the CMVpromoter region to simplify swapping of the internalpromoter, and removed a large amount of the exogenous3′ flanking sequence that remained from the originalviral integrant. This pHAGE vector served as the basisfor the creation of all other pHAGE derivatives. Forinducible gene expression, the CMV promoter wasreplaced by the TRE promoter that contains seven copiesof the 42-bp Tet operator sequence and the minimalCMV promoter and was obtained from pLP-RevTREvector (Clontech) by digestion with XhoI and EcoRI.The truncated human cell surface selection markersLNGFR and CD4 were cloned from plasmid pMACS-LNGFR and pMACS4.1 (Miltenyi Biotec, Auburn, CA,USA) by PCR using primers LNGFR-NdeI 5′-TTT CATATG GGG GCA GGT GCC ACC GGC CGC GCC AT-3′, LNGFR-ClaI 5′-AAA ATC GAT CTA TCA CCTCTT GAA GGC TAT GTAGGC CAC AAG ACC CACAACCACAGCA-3′, and CD4-NdeI 5′-TTT CATATGAAC CGG GGA GTC CCT TTTAGG CAC TTG CTTC-3′, CD4-ClaI 5′-AAA ATC GAT CTA TCA GTGCCG GCA CCT GAC ACA GAA GAA GAT G-3′respectively, followed by digestion with NdeI and ClaI,and inserted in the same restriction sites downstream ofthe UBC promoter. The rtTA was cloned from plasmidpTet-ON (Clontech) by PCR using primers rtTA-NotI 5′-TTT GCG GCC GCC ATG TCT AGATTA GATAAAAGTAAAGTGATT-3′ and rtTA-BamHI 5′-AAG GATCCT TAC TAC CCA CCG TAC TCG TCA ATT CCAAGG GCA TCG GTA AAC-3′. The Ipr1 cDNA wascloned by PCR from lung of C3H.B6-sst1 mice as

34 H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
described previously (Pan et al., 2005). Sequences of thepHAGE constructs are available upon request.
2.4. Lentivirus production
Recombinant lentiviruses were produced by a five-plasmid transfection procedure (Mostoslavsky et al.,2005). Briefly, 293T cells were co-transfected usingTrans IT-293 liposome reagent (Mirus, Madison, WI,USA) and the pHAGE or pHRST backbone lentiviralvector together with four expression vectors encodingthe packaging proteins Gag-Pol, Rev, Tat and the G-protein of the vesicular stomatitis virus (VSVG). Thevirus supernatants were collected 24, 36, 48 and 60 hafter transfection, pooled and filtered through 0.45-μmfilters. If necessary, the viral supernatants were concen-trated to about 1/100 volume by ultracentrifugation at15,000 ×g for 3 h. Viral titers were determined by thepercentage of LNGFR+ or CD4+ 293T cells transducedwith serial dilutions of lentivirus supernatants.
2.5. Lentiviral transduction
Transduction of cell lines was performed in 6-wellplate in 4 mL volume per well. Lentivirus supernatantsand 10μg/mLPolybrenewere added to 1×106 target cellsat m.o.i. of 10 (or 5 for each virus in a co-transduction) inDMEM/F12 medium containing 2% FBS, 10 mM Hepesbuffer. Transduction of BMDMs was performed in theultra-low cluster 6-well plate (Corning) at m.o.i. of 10 andinDMEM/F12medium containing 10%FBS, 10%L-929conditioned medium, and 1 ng/mL rmIL-3. To achievehigher efficiency of transduction, plates were spun at1000 ×g for 1 h at room temperature (Kotani et al., 1994).Cells were then immediately washed twice with mediumand cultured for at least 24 h before FACS analysis ormagnetic selection.
2.6. Magnetic selection and FACS analysis
Transduced cells were enriched using the MACSelectsystems (Miltenyi Biotec) according to manufacturer'sinstruction. Briefly, adherent cells were collected usingcell scrapers after being incubated in PBS buffer(Mediatech, Inc.) containing 1% FBS and 5 mM EDTAfor 20 min. 4×107 cells were resuspended in 4 mL PBSbuffer containing 0.5% bovine serum albumin (BSA,Sigma-Aldrich, St. Louis, MO, USA) and 5 mM EDTA.Cells were incubated with 4 μg low-endotoxin rat anti-mouse CD16/CD32 (FcγRIII/II) (AbD Serotec, Raleigh,USA) on ice for 15min, and thenwith 80μL anti-LNGFRor anti-CD4 Microbeads (Miltenyi Biotec) for additional
15min on ice. Cells were passed through a pre-separationfilter to remove clumps and loaded on a pre-equilibratedMASC LS column in a magnetic holder, followed bywashing 4 times with 3 mL PBS buffer containing 0.5%BSA and 5 mMEDTA. The columns were removed fromthe magnetic holder and the antibody-bound cells wereflashed out with 5mL cell medium. Either immediately or2 days culturing after magnetic selection, cells were pre-blockedwith rat anti-mouseCD16/CD32 and then labeledwith Allophycocyanin (APC)-conjugated anti-LNGFR orPE-conjugated anti-CD4 (Miltenyi Biotec) and analyzedusing a FACScan flow cytometry (Becton-Dickinson, SanJose, CA, USA).
2.7. RNA isolation and RT-PCR
Total RNA was isolated by using TRIzol (Invitrogen,Carlsbad, CA, USA) and further cleaned up by usingRNeasy Mini kit (Qiagen, Valencia, CA, USA) withDNase I digestion. 2 μg total RNA was reversetranscripted by using oligo-dT primers and SuperScriptII (Invitrogen). One twenty-fifth of each product wasamplified by 3-primer PCRwith primers Ifi75-9F 5′-AGACATTAAGACATCTGGAGCAGAAAG-3′, Ifi75-9R5′-GCA CATATC AGG TCA GGA GTT CAT C-3′, andUBC-1R 5′-CGG GCG GAA GGATCA GGA-3′.
2.8. Cell cloning by limiting dilution
Transduced J774A.1 cells were seeded in 96-wellplates at a density of approximately 0.5 cell per well, sothat 20–30% of the wells had clones grown up. About400 clones were picked and transferred into duplicateplates. One plate was left untreated, while doxycyclinewas added to another plate. Cells were examined underthe fluorescent microscope for the nuclear eGFPexpression 24 and 48 h later. Clones with undetectablebasal level of the eGFP–Ipr1 fusion protein expressionand doxycycline-induced eGFP–Ipr1 expression wereselected and further tested by flow cytometry.
2.9. Cell lysis, nuclear extraction, antibodies andimmunoblotting
2×107 cells were washed twice with PBS, and scrapedin 1.2 mL RIPA buffer (50 mM TrisCl pH7.4, 150 mMNaCl, 1% Nonidet P-40, 0.25% Sodium deoxycholate,0.1% SDS) to prepare whole cell lysate. Alternatively,cells were scraped in 1.2 mL hypotonic buffer (10 mMHEPES pH7.9, 10 mM KCl, 1.5 mM MgCl2, 0.2%Nonidet P-40). Cytosolic lysate and nuclei were separatedby centrifugation at 15,000 ×g for 3 min. Nuclei were

Fig. 1. Schematic diagrams of the pHRST bicistronic and pHAGE dual-promoter lentiviral vectors used in this study. LTR, long terminal repeat;ΔLTR, U3 region-deleted LTR; Ψ, Psi packaging signal; RRE, Rev-responsive element; P, central polypurine tract, W, woodchuck hepatitisvirus post-transcriptional regulatory element; MAR, matrix attachmentregion; PCMV, cytomegalovirus immediate early promoter; PUBC, humanubiquitin C promoter; TRE, tetracycline responsible element; eGFP,enhanced green fluorescent protein; Ipr1, intracellular pathogen resistantgene 1; Flag, FLAG-tag; dLNGFR, truncated human low-affinity nervegrowth factor receptor; dCD4, truncated human CD4.
35H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
washed with 0.6 mL hypotonic buffer and then extractedwith 200 μL nuclear extraction buffer (20 mM HEPESpH7.9, 420 mMNaCl, 1.5 mMMgCl2, 25%Glycerol) ona rotary mixer at 4 °C for 2 h. All buffers weresupplemented with 1% protease inhibitor cocktail andphosphatase inhibitor cocktails I and II (Sigma-Aldrich).Protein concentration was measured by using BCAprotein assay kit (Pierce Biotechnology, Rockford, IL,USA). 20 μg protein was purified by using PAGEprep kit(Pierce Biotechnology) and separated by 10% SDS-PAGE. Proteins were transferred to PVDF membranes(Bio-Rad, Hercules, CA, USA) and immunoblotted. Allmembranes were developed with SuperSignal chemilu-minescent substrate (Pierce Biotechnology). The mouseanti-FLAGM2monoclonal antibodywas purchased fromSigma-Aldrich, and the rat anti-Hsc70 monoclonalantibody was from Assay Designs, Inc. (Ann Arbor,MI, USA). The Ipr1-specific rabbit anti-serum wasgenerated by using the service of Covance ResearchProducts, Inc. (Denver, CO, USA).
2.10. Immunoprecipitation and Nano-LC tandem massspectrometry
Immunoprecipitation was performed by usingμMACS GFP-tagged protein isolation kit (MiltenyiBiotec). Briefly, 200 μg nuclear extract was diluted with9 volumes of IP dilution buffer (50 mM TrisCl pH7.4,150 mM NaCl, 1% Triton X-100) and immunoprecipi-tated with 50 μL anti-GFP Microbeads according tomanufacturer's manual. The eluate were separated by10% SDS-PAGE and analyzed by either immunoblottingor Coomassie Blue R-250 staining. The Coomassiestained bands were excised from the gels and digestedwith trypsin. Protein digests were desalted on a C18 solidphase extraction (“trapping”) column, eluted and separat-ed on a Nano-LC revered-phase self-packed fused silicacolumn (75 μm i.d.×15 mm; HCPF) with linear gradientof eluent. The eluent was introduced into the LCQ DecaXP Plus mass spectrometer (MS) by nanoelectrospray.TheMSoperation andMSdata processingwas performedby investigators at the Harvard School of Public HealthCore Proteomics Facility (HCPF).
3. Results
3.1. Failure to constitutively express the Ipr1 gene inmouse bone marrow-derived macrophages using astandard bicistronic lentiviral vector
Initially, to express the Ipr1 gene in bone marrow-derived macrophages (BMDMs) of the Ipr1-negative
C3HeB/FeJ mice, we used the bicistronic lentiviralvector, in which the co-expression of the Ipr1 gene andselectable marker eGFP was driven by a constitutiveCMV promoter (pHRST-CMV.Ipr1-IRES.eGFP.W.MAR, see Fig. 1) (Mostoslavsky et al., 2005). Wefound that less than 10% of the cells could betransduced. Attempts to expand the Ipr1 expressingBMDMs after enrichment using FACS sorting wereunsuccessful because GFP-positive (Ipr1-expressing)cells rapidly declined after sorting (data not shown).Taken together, our failure to constitutively express thefunctional Ipr1 protein in macrophages suggested that itmight have deleterious effects on proliferation orsurvival of these cells both in vivo and in vitro. This isconsistent with the previous report that overexpressionof the human homologue of Ipr1, SP110b/IFI41, was“toxic” for a cell line (Kadereit et al., 1993). Therefore,

36 H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
we wanted to develop a gene delivery system, whichwould overcome difficulties associated with lowtransducibility of macrophages, avoid effects of foreignDNA and viral particles on macrophage activation statusduring experiments and allow inducible expression ofpotentially “toxic” products. Ideally, this system shouldbe applicable to studies of macrophage function in vitroand in vivo.
3.2. Dual-promoter lentiviral vectors do allow separatecontrol of the expression of both the gene-of-interest anda selectable marker in macrophages using constitutiveand inducible promoters
The backbone lentiviral vectors pHRST and pHAGEused in our experiments are derivatives of the self-inactivating non-replicative vector pHR′CMV-lacZ.They both have an intact HIV-1-derived 5′-longterminal repeat (5′-LTR) and a 3′-LTR deleted of U3region (Naldini et al., 1996). Both backbone vectors alsocontain several cis-acting elements, which enhance thetransduction efficiency or gene expression (Fig. 1). TheRev-responsive element (RRE) is the binding site of theHIV-1 Rev protein, which helps exporting the viralRNA genomes to the cytoplasm of virus-producing cellsand thus increases the production of large-size recom-binant lentiviruses (Pollard and Malim, 1998). Thecentral polypurine tract (cPPT) is a cis-acting determi-nant for the nuclear import of HIV pre-integrationcomplex, which is necessary for transduction of non-dividing cells (Follenzi et al., 2000; Zennou et al.,2000). The woodchuck hepatitis virus post-transcrip-tional regulatory element (WPRE) enhances geneexpression by facilitating the nuclear export of RNAtranscripts (Zufferey et al., 1999). In addition, thepHRST vector contains 7 copies of the humaninterferon-β gene matrix attachment region (MAR)which has been shown to increase gene expression(Murray et al., 2000).
3.2.1. Constitutive expressionTo achieve separate control of expression of a
selectable marker and a gene-of-interest, we developeddual-promoter lentiviral vectors (Fig. 1). The upstreampromoters, either a constitutive CMV promoter or aninducible promoter composed of tetracycline responseelement (TRE) and a minimal promoter, were used tocontrol the expression of a gene-of-interest. A constitutiveubiquitin C (UBC) promoter was placed downstream andused to control expression of one of two selectable surfacemarkers, either the human low-affinity nerve growthfactor receptor (LNGFR) or human CD4. The cytoplas-
mic domains of both selectable markers were deleted toeliminate signaling through these molecules. Expressionof the selectable markers on the cell surface allowedidentification of the transduced cells by FACS usingspecific antibodies as well as their enrichment usingmagnetic cell sorting (MACS) (Gaines and Wojchowski,1999) (Figs. 2D and 4B).
To determine whether the dual-promoter lentiviralvectors can simultaneously co-express the gene-of-interestand a selectable marker, we transduced the mousefibroblast NIH/3T3 cell line with a lentiviral vectorencoding the Ipr1 or FLAG-tagged Ipr1 (FLAG-Ipr1)under the control of the CMV promoter and the cellsurface marker LNGFR under the control of the UBCpromoter (pHAGE-CMV.FLAG-Ipr1.UBC.dLNGFR.Wor pHAGE-CMV.FLAG-Ipr1.UBC.dLNGFR.W, seeFig. 1). Seven days post-transduction, about 90% of thecells expressed the cell surface marker LNGFR (Fig. 2B).Meanwhile, strong expression of the Ipr1 or FLAG-Ipr1was detected in the nuclear extracts using immunoblotwith either FLAG- or Ipr1-specific antibodies (Fig. 2A).No expression of the endogenous Ipr1 was detectable inthe control sample (mock transduction). Next, we used thesame lentiviral construct to transduce a mouse macro-phage cell line J774A.1. However, in this macrophage cellline, the expression level of the FLAG-Ipr1 protein rapidlydeclined as compared to that in NIH/3T3 fibroblasts.Meanwhile, the transcript level of the vector-encodedFLAG-Ipr1 (Fig. 2C), as well as the proportion of theLNGFR-expressing (transduced) cells, which representedabout 97% of the population after magnetic sorting(Fig. 2D) remained stable. Similar results were observedusing another mouse macrophage cell line RAW264.7(data not shown).
When the eGFP was substituted for the FLAG-Ipr1in these constructs, we could achieve high-levelexpression of both the eGFP and LNGFR proteins inJ774A.1 macrophage cell line (data not shown), thus,indicating that there was no interference between thepromoters of the dual-promoter lentiviral vectors neitherin fibroblast nor in macrophage cell lines. Perhaps, post-transcriptional regulation was responsible for thedramatic decrease of the Ipr1 protein expression overtime specifically in macrophage, but not fibroblast celllines. Thus, to study the Ipr1 protein in macrophages, wedeveloped an inducible system.
3.2.2. Inducible expression of eGFPTwo lentiviral vectors were constructed for inducible
gene expression in cell lines. The first vector containedthe reverse tetracycline transactivator (rtTA) driven bythe constitutive CMV promoter and the CD4 under the
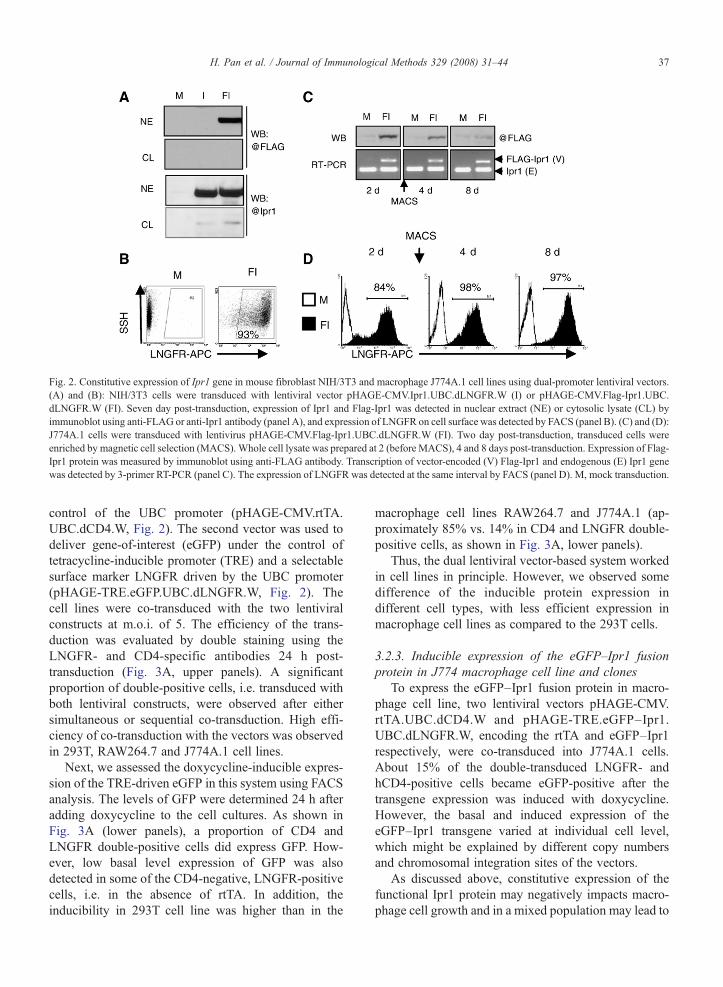
Fig. 2. Constitutive expression of Ipr1 gene in mouse fibroblast NIH/3T3 and macrophage J774A.1 cell lines using dual-promoter lentiviral vectors.(A) and (B): NIH/3T3 cells were transduced with lentiviral vector pHAGE-CMV.Ipr1.UBC.dLNGFR.W (I) or pHAGE-CMV.Flag-Ipr1.UBC.dLNGFR.W (FI). Seven day post-transduction, expression of Ipr1 and Flag-Ipr1 was detected in nuclear extract (NE) or cytosolic lysate (CL) byimmunoblot using anti-FLAG or anti-Ipr1 antibody (panel A), and expression of LNGFR on cell surface was detected by FACS (panel B). (C) and (D):J774A.1 cells were transduced with lentivirus pHAGE-CMV.Flag-Ipr1.UBC.dLNGFR.W (FI). Two day post-transduction, transduced cells wereenriched by magnetic cell selection (MACS). Whole cell lysate was prepared at 2 (before MACS), 4 and 8 days post-transduction. Expression of Flag-Ipr1 protein was measured by immunoblot using anti-FLAG antibody. Transcription of vector-encoded (V) Flag-Ipr1 and endogenous (E) Ipr1 genewas detected by 3-primer RT-PCR (panel C). The expression of LNGFR was detected at the same interval by FACS (panel D). M, mock transduction.
37H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
control of the UBC promoter (pHAGE-CMV.rtTA.UBC.dCD4.W, Fig. 2). The second vector was used todeliver gene-of-interest (eGFP) under the control oftetracycline-inducible promoter (TRE) and a selectablesurface marker LNGFR driven by the UBC promoter(pHAGE-TRE.eGFP.UBC.dLNGFR.W, Fig. 2). Thecell lines were co-transduced with the two lentiviralconstructs at m.o.i. of 5. The efficiency of the trans-duction was evaluated by double staining using theLNGFR- and CD4-specific antibodies 24 h post-transduction (Fig. 3A, upper panels). A significantproportion of double-positive cells, i.e. transduced withboth lentiviral constructs, were observed after eithersimultaneous or sequential co-transduction. High effi-ciency of co-transduction with the vectors was observedin 293T, RAW264.7 and J774A.1 cell lines.
Next, we assessed the doxycycline-inducible expres-sion of the TRE-driven eGFP in this system using FACSanalysis. The levels of GFP were determined 24 h afteradding doxycycline to the cell cultures. As shown inFig. 3A (lower panels), a proportion of CD4 andLNGFR double-positive cells did express GFP. How-ever, low basal level expression of GFP was alsodetected in some of the CD4-negative, LNGFR-positivecells, i.e. in the absence of rtTA. In addition, theinducibility in 293T cell line was higher than in the
macrophage cell lines RAW264.7 and J774A.1 (ap-proximately 85% vs. 14% in CD4 and LNGFR double-positive cells, as shown in Fig. 3A, lower panels).
Thus, the dual lentiviral vector-based system workedin cell lines in principle. However, we observed somedifference of the inducible protein expression indifferent cell types, with less efficient expression inmacrophage cell lines as compared to the 293T cells.
3.2.3. Inducible expression of the eGFP–Ipr1 fusionprotein in J774 macrophage cell line and clones
To express the eGFP–Ipr1 fusion protein in macro-phage cell line, two lentiviral vectors pHAGE-CMV.rtTA.UBC.dCD4.W and pHAGE-TRE.eGFP–Ipr1.UBC.dLNGFR.W, encoding the rtTA and eGFP–Ipr1respectively, were co-transduced into J774A.1 cells.About 15% of the double-transduced LNGFR- andhCD4-positive cells became eGFP-positive after thetransgene expression was induced with doxycycline.However, the basal and induced expression of theeGFP–Ipr1 transgene varied at individual cell level,which might be explained by different copy numbersand chromosomal integration sites of the vectors.
As discussed above, constitutive expression of thefunctional Ipr1 protein may negatively impacts macro-phage cell growth and in a mixed population may lead to

Fig. 3. Lentiviral system for inducible gene expression in macrophagecell lines. (A) 293T, RAW264.7 and J774A.1 cells were co-transducedwith two lentiviral vectors pHAGE-CMV.rtTA.UBC.dCD4.W andpHAGE-TRE.eGFP.UBC.dLNGFR.W. One day post-transduction,cells were cultured in medium containing 1 μg/mL doxycycline foradditional 1 day. The expressions of cells surface markers LNGFR andCD4 (upper panels), as well as eGFP in LNGFR-positive cells (lowerpanels) were measured by FACS. (B) J774A.1 cells were co-transduced with two lentiviral vectors pHAGE-CMV.rtTA.UBC.dCD4.W and pHAGE-TRE.eGFP–Ipr1.UBC.dLNGFR.W. Individualclones were isolated and tested for induction of eGFP–Ipr1 in mediumcontaining 1 μg/mL doxycycline for additional 1, 2, and 4 days byimmunoblot using Ipr1-specific antibody, (C) The green fluorescenceof eGFP–Ipr1 in a typical clone were also measured by FACS.
38 H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
positive selection of cells that express aberrant non-functional forms of the Ipr1 protein. Therefore, to obtaincells in which the eGFP–Ipr1 protein expression istightly regulated, we cloned the co-transduced J774A.1cells by limiting dilution. Approximately 400 resultingclones were obtained, split and tested for the eGFP–Ipr1expression in the presence and absence of doxycyclineby microscopy. About 25 clones were selected forfurther testing using FACS analysis and titration of
doxycycline, from which a final set of clones wasselected based on undetectable GFP–Ipr1 levels withoutinduction and high levels of inducible expression with alow dose of doxycycline. A typical clone is shown inFig. 3C.
We observed that even in those selected clones thelevel of eGFP–Ipr1 expression decreased within 4 daysas detected both by FACS (Fig. 3C) and immunoblotusing the Ipr1-specific antibodies (Fig. 3B). Thisdecreasing of eGFP–Ipr1 expression was not due tothe deactivation of doxycycline since we refreshed thedoxycycline-containing medium everyday. Also, weobserved no decrease of the eGFP protein expressionusing the same vector encoding eGFP alone as a control.These results indicated that the prolonged expression ofthe Ipr1 protein in macrophage cell lines was inhibited,most likely, at post-transcriptional level, although themechanism of this inhibition remains unknown. Never-theless, the inducible system provided a windowbetween the induction with doxycycline and theIpr1protein degradation that allowed us to study thisprotein's function in macrophage cell lines within2 days post-induction.
3.2.4. Inducible expression of the eGFP–Ipr1 fusiongene in primary bone marrow-derived macrophages
For inducible expression of genes in non-transformedprimary mouse macrophages, we generated a transgenicmouse strain C3H.TgN (SRA–rtTA) that expressed thertTA protein under the control of a macrophage-specificmodified human Scavenger Receptor A (SRA) promoter.The SRA promoter controlled transgene expression inCSF-1 differentiated macrophages, but not in bonemarrow progenitors (Horvai et al., 1995). The transgenewas introduced directly in C3HeB/FeJmice, which do notexpress the Ipr1 gene (Pan et al., 2005). To express theeGFP–Ipr1 fusion protein we have developed thefollowing procedure: first, the bone marrow-derivedmacrophage progenitors were enriched and transducedwith lentiviral constructs, next, the transduced cells werepositively selected using magnetic cell sorting, expandedand differentiated intomacrophages and then, treated withdoxycycline to induce the eGFP–Ipr1 gene expression.
To enrich for macrophage progenitors, the C3H.TgN(SRA–rtTA) bone marrow cells were cultured inmedium containing recombinant mouse interleukin-3(rmIL-3) and colony stimulating factor-1 (CSF-1) forthree days and depleted of more mature adherent cells.The non-adherent cells were transduced with thelentiviral vectors pHAGE-TRE.eGFP–Ipr1.UBC.dLNGFR.W or pHAGE-TRE.eGFP.UBC.dLNGFR.W,which encoded the GFP–Ipr1 fusion protein or eGFP
39H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
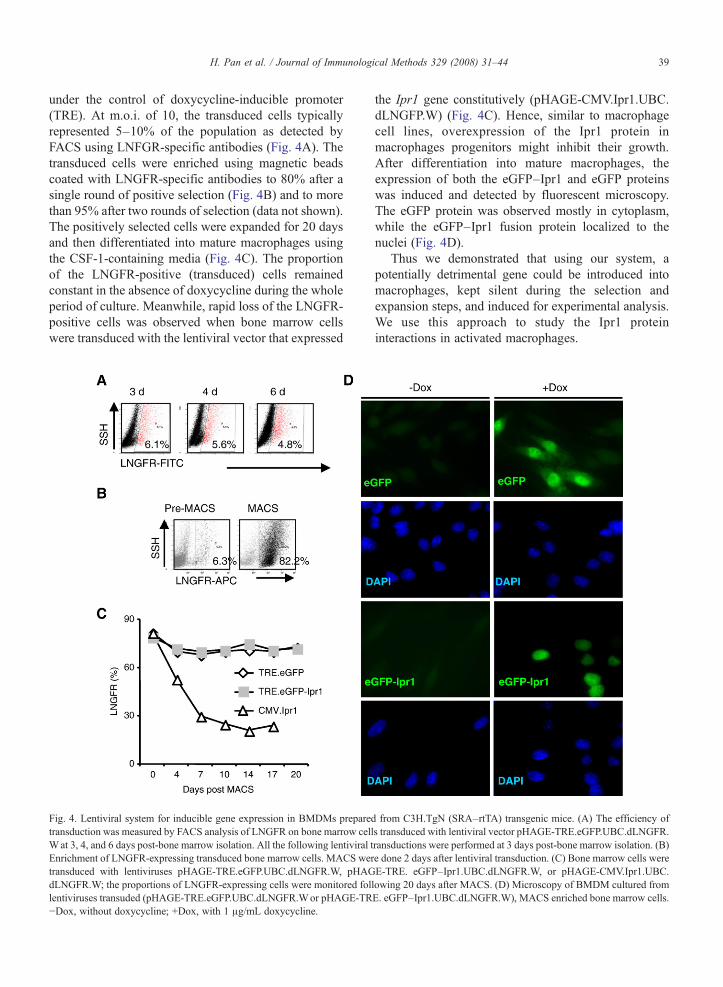
under the control of doxycycline-inducible promoter(TRE). At m.o.i. of 10, the transduced cells typicallyrepresented 5–10% of the population as detected byFACS using LNFGR-specific antibodies (Fig. 4A). Thetransduced cells were enriched using magnetic beadscoated with LNGFR-specific antibodies to 80% after asingle round of positive selection (Fig. 4B) and to morethan 95% after two rounds of selection (data not shown).The positively selected cells were expanded for 20 daysand then differentiated into mature macrophages usingthe CSF-1-containing media (Fig. 4C). The proportionof the LNGFR-positive (transduced) cells remainedconstant in the absence of doxycycline during the wholeperiod of culture. Meanwhile, rapid loss of the LNGFR-positive cells was observed when bone marrow cellswere transduced with the lentiviral vector that expressed
Fig. 4. Lentiviral system for inducible gene expression in BMDMs preparedtransduction was measured by FACS analysis of LNGFR on bone marrow cellWat 3, 4, and 6 days post-bone marrow isolation. All the following lentiviral tEnrichment of LNGFR-expressing transduced bone marrow cells. MACS wertransduced with lentiviruses pHAGE-TRE.eGFP.UBC.dLNGFR.W, pHAGdLNGFR.W; the proportions of LNGFR-expressing cells were monitored follentiviruses transuded (pHAGE-TRE.eGFP.UBC.dLNGFR.Wor pHAGE-TR−Dox, without doxycycline; +Dox, with 1 μg/mL doxycycline.
the Ipr1 gene constitutively (pHAGE-CMV.Ipr1.UBC.dLNGFP.W) (Fig. 4C). Hence, similar to macrophagecell lines, overexpression of the Ipr1 protein inmacrophages progenitors might inhibit their growth.After differentiation into mature macrophages, theexpression of both the eGFP–Ipr1 and eGFP proteinswas induced and detected by fluorescent microscopy.The eGFP protein was observed mostly in cytoplasm,while the eGFP–Ipr1 fusion protein localized to thenuclei (Fig. 4D).
Thus we demonstrated that using our system, apotentially detrimental gene could be introduced intomacrophages, kept silent during the selection andexpansion steps, and induced for experimental analysis.We use this approach to study the Ipr1 proteininteractions in activated macrophages.
from C3H.TgN (SRA–rtTA) transgenic mice. (A) The efficiency ofs transduced with lentiviral vector pHAGE-TRE.eGFP.UBC.dLNGFR.ransductions were performed at 3 days post-bone marrow isolation. (B)e done 2 days after lentiviral transduction. (C) Bone marrow cells wereE-TRE. eGFP–Ipr1.UBC.dLNGFR.W, or pHAGE-CMV.Ipr1.UBC.lowing 20 days after MACS. (D) Microscopy of BMDM cultured fromE. eGFP–Ipr1.UBC.dLNGFR.W), MACS enriched bone marrow cells.

40 H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
3.3. Identification of proteins that interact with Ipr1 inactivated macrophages
To identify nuclear proteins that interact with Ipr1 inactivated macrophages, we used a clone of J774A.1cells (clone 21), which was obtained after transductionwith lentiviral vectors pHAGE-CMV.rtTA.UBC.dCD4.W and pHAGE-TRE.eGFP–Ipr1.UBC.dLNGFR.W, asdescribed above (Fig. 3). The expression of the GFP–Ipr1 fusion protein was induced with doxycycline (1 μg/mL) for 24 h. To induce macrophage activation rIFN-γwas added for the last 16 h of culture to a finalconcentration of 100 U/mL. Using Western blot analysiswe observed that both the endogenous Ipr1 and theGFP–Ipr1 fusion proteins localize to the nucleus ofinterferon-activated and non-activated macrophages.Therefore, nuclear extracts were isolated from thosecells and immunoprecipitated with GFP-specific anti-
Fig. 5. Characterization of Ipr1-containing protein complexes in macrophageIpr1in doxycycline-inducible manner, were cultured in medium containiImmunoprecipitation of nuclear extracts was performed using GFP-specific a10% SDS-PAGE and visualized using Coomassie Brilliant Blue R-250 strespectively, using Nano-LC tandemmass spectrometry. (B) Western blot of Ipwere prepared as in (A), immunoprecipitation (IP) was performed using eithamount of nuclear extract (input) were immunoblotted using Hsc70-specific aIFN-γ activated clone 21 J774A.1 cells was demonstrated by immunoblexpression in naïve and IFN-γ-activated clone 21 cells. MFI, mean fluoresc
bodies coupled to magnetic beads. The precipitatedproteins were separated on SDS-PAGE gel andvisualized by staining with Coomassie Brilliant Blue.Individual bands were cut from the gel, digested withtrypsin and analyzed using mass spectrometry (seeMethods for details). Interestingly more Ipr1-interactingproteins were detected after macrophage activation withIFN-γ, as compared to naïve macrophages (Fig. 5A).
One of the most prominent proteins that interactedwith Ipr1 was identified by mass spectroscopy as heatshock cognate protein 70 (Hsc70) (Fig. 5A, band IV).Using co-immunoprecipitation with GFP-specific anti-bodies and immunoblot analysis with Hsc70-specificantibody, we confirmed the GFP–Ipr1 and Hsc70protein interactions and demonstrated that this interac-tion occurred specifically in the nuclei of interferon-activated, but not naïve macrophages (Fig. 5B, lowerpanel). Using immunoblot of nuclear extracts with Ipr1
nuclei. (A) Cells of clone 21 of J774A.1 cells, which express eGFP–ng 1 μg/mL doxycycline with or without rmIFN-γ (100 U/mL).ntibodies. Proteins co-precipitated with eGFP–Ipr1 were separated onaining. Bands III and IV were identified as eGFP–Ipr1 and Hsc70,r1-interacting proteins with Hsc70-specific antibodies: nuclear extractser anti-GFP or IgG isotype-matched control; eluate from IP and 1:10ntibodies. (C) Increased amount of eGFP–Ipr1 proteins in the nuclei ofot using Ipr1-specific antibodies. (D) FACS analysis of eGFP–Ipr1ence intensity.

41H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
specific antibody we detected accumulation of theGFP–Ipr1 fusion protein (Fig. 5C), as well asendogenous Ipr1 (not shown), in the macrophage nucleiupon interferon treatment. Accumulation of the eGFP–Ipr1 fusion protein in clone 21 cells treated withdoxycycline and IFN-γ was also demonstrated usingFACS analysis: the IFN-γ activated macrophagesshowed higher percentage of GFP-positive cells and,notably, 7-fold higher main fluorescence intensity ascompared to doxycycline-treated, but not activatedmacrophage cells (Fig. 5D). Meanwhile, the amount ofHsc70 proteins in the nuclei of the interferon-activatedand naïve macrophages did not change (Fig. 5B, toppanel). Thus the interferon-inducible interaction of theIpr1 protein with the Hsc70 chaperon may be due tomodifications of one or both proteins specific formacrophage activation leading to stabilization of theIpr1-containing protein complexes.
4. Discussion
Macrophages are among the most versatile andimportant animal cells (Ross and Auger, 2002; Sasmonoand Hume, 2004). However, currently our ability tostudy these cells using modern molecular approaches isgreatly limited as compared to other cell types, becausemacrophages resist genetic manipulation. Direct plas-mid DNA transfection or electroporation is onlysuccessful with a few monocyte or macrophage celllines such as THP-1 or RAW264.7 (Weir and Meltzer,1993; Hume et al., 2001), which is not suitable for othermacrophage cell lines (e.g. J744A.1) and primarymacrophages (Heider et al., 2000). Perhaps, this isrelated to their natural function in tissue homeostasisand host defense, such as recognition and elimination offoreign materials. There was evidence that certainmacrophage cell lines (e. g. RAW264.7) had specificdefects in inflammatory response to foreign materialsi.e. prostaglandin-dependent autoregulation of tumornecrosis factor-alpha secretion upon lipopolysaccharidestimulation (Rouzer et al., 2005). Thus it is beneficial tobe able to use different macrophage cell lines andprimary cells from different sources.
Here, we presented an improved strategy enablinggenetic manipulation of macrophages more efficiently.The major features of our approach include: 1) dual-promoter lentiviral vectors for the inducible expressionof the gene-of-interest and constitutive expression of theselectable surface marker; 2) the C3H.TgN (SRA–rtTA)transgenic mouse that enables doxycycline-inducibleexpression of lentivirally delivered genes-of-interest inprimary macrophages. This system is particularly useful
when the efficiency of transduction is low and a gene-of-interest is either unstable or exerts adverse effect ontarget cells. As discussed below, both conditions applyto studies of immunity-related genes in macrophages.
It is important to have independent controls ofexpression of a selectable surface marker and a gene-of-interest. Therefore, we used dual-promoter lentiviralvectors. Although there were several studies reportingthat dual-promoter vectors based on oncoretrovirusbackbones showed severe reduction of transgeneexpression due to promoter interference (Emerman andTemin, 1984; Overell et al., 1988), a similar strategy wassuccessful when using lentiviral vectors (Yu et al., 2003).Thus, Yu et al. have achieved efficient and consistent co-expression of two genes in cord-blood CD34+ HSCs orprimary endothelial cells (Yu et al., 2003). Our datademonstrate that the dual-promoter vectors may be usedin macrophage cell lines and primary BMDMs as well.
To obtain high purity of transduced primary macro-phages, we have developed a procedure, in whichmacrophage progenitors are transduced by lentiviralvectors containing a selectable surface marker expressedunder the control of a constitutive UBC promoter andthe transduced cells, are enriched using magnetic cellsorting, expanded and differentiated into macrophages.Positive selection using surface markers in case ofmacrophages has an advantage as compared to drugselection. Since macrophages are phagocytic cells, theyingest dead cells during the drug selection procedure,which affects their growth and behavior. Positiveselection using magnetic beads is rapid and avoids theadverse effects associated with drug selection. We havedemonstrated that the selectable surface markers used inour vectors are expressed at relatively constant levelsduring the whole period of observation (up to one monthin our studies) and therefore the selection proceduremight be repeated several times. Using this strategy wetypically obtain 108 transduced primary bone marrow-derived macrophages. Because the gene-of-interest iscontrolled separately by a doxycycline-inducible pro-moter, it is kept silent during the selection process anddoes not interfere with macrophage growth.
Hundreds of genes are expressed in activatedmacrophages in an inducible manner. Their productsare involved in host defense displaying pro-inflamma-tory, immunoregulatory and anti-microbial activities(Ehrt et al., 2001; Schroder et al., 2004; Kota et al.,2006). Some of them exert anti-proliferative or pro-apoptotic effects on macrophages. Thus, macrophagesthat are genetically manipulated to constitutivelyexpress such genes may be under negative selection,i.e. either selectively eliminated or outgrown by the cells

42 H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
that express non-functional genes inactivated by muta-tions, for example. We have identified the Ipr1 gene bypositional cloning as a candidate gene that controls amacrophage-mediated mechanism of host resistance tointracellular pathogens, MTB and Listeria monocyto-genesis. This gene encodes an interferon-inducibleprotein. We observed that similar to some otherinterferon-inducible proteins, the Ipr1 exerts an anti-proliferative effect on macrophages: cells that expressedfunctional Ipr1 under the control of a constitutive CMVpromoter were rapidly lost during cell expansion.Actually, we have observed cell-cycle arrest at the G2/M transition in macrophages overexpressing eGFP–Ipr1proteins. We also observed partial deletions in Ipr1 thatinactivated this protein in cell lines that were con-structed to express Ipr1 constitutively (data not shown).
To address these problems we utilized an induciblesystem to express the gene-of-interest in macrophagesonly when desired. We use two methods to express thedoxycycline-regulated reverse tetracycline transactiva-tor (rtTA) in target cells. For the macrophage cell lineswe use a lentiviral vector that constitutively expressesthe rtTA under control of CMV promoter. It can beeither used to establish stable cell lines that express thertTA or simply co-transduced with a lentiviral vectorencoding a gene-of-interest. Although utilization oftransformed macrophage cell lines in vitro is conve-nient, the analysis of gene function ideally has to beextended to primary macrophages. Therefore, we havedeveloped a transgenic mouse strain C3H.TgN (SRA–rtTA) that enabled us to use the lentiviral system for theinducible gene expression in primary BMDMs. TheC3HeB/FeJ mice also serve as a susceptible partner inour genetic analysis of host resistance to tuberculosis, inwhich four host resistance loci were mapped in additionto the sst1 (Yan et al., 2006a). Therefore the C3H.TgN(SRA–rtTA) transgenic mice will be useful for theanalysis of the Ipr1 as well as other candidate hostresistance genes in primary macrophages in vitro andpossibly in vivo.
Recently, Yan et al. have published a macrophage-specific tetracycline-inducible system for in vivoexpression in mice, which utilizes the c-fms (CSF-1receptor) promoter/intron regulatory element (Yan et al.,2006b). For inducible expression these mice have to bebred with another transgenic mouse, in which rtTAinducible promoter drives expression of “gene-of-interest”. The c-fms gene promoter is active inmacrophages and bone marrow progenitors (Sasmonoet al., 2003). Our system is distinct, because the SRApromoter expresses a transgene only in mature macro-phages (Horvai et al., 1995) and in bone marrow-
derived dendritic cells (Pan, unpublished observations).Therefore, it is suitable for in vivo expression of geneswith anti-proliferative effects.
We applied the tetracycline-inducible system, tocharacterize the Ipr1-interacting proteins in macro-phages and observed that macrophage activation withIFN-γ significantly enhanced Ipr1 interactions withnuclear proteins. In additional to transcriptional upregu-lation of Ipr1 upon macrophage activation with inter-ferons, we also observed accumulation of this protein inthe macrophage nuclei and inducible interactions of Ipr1with molecular chaperon Hsc70. It is possible thatinterferon signaling enables the Ipr1 and/or Hsc70protein interactions via phosphorylation of one or bothproteins. In addition, interferon might induce theexpression of a “bridge” molecule, which connects theIpr1 and Hsc70 proteins. In either case the observedinteractions are dependent on macrophage activationstatus and might not be detectable in other cell types,although they might be more convenient for co-transfection. These findings highlight the importanceof studying host defense-related proteins in a specificcellular environment that is related to their biologicalfunction, such as macrophage interactions with patho-genic intracellular bacteria in the case of the Ipr1 protein.Our method permits further analysis of the dynamic Ipr1-containing multiprotein complexes during the course ofmacrophage activation and infection with virulent strainsof M. tuberculosis in order to elucidate post-transcrip-tional modifications, traffic, turnover of the Ipr1 proteinin macrophages, as well as its role in innate immunity.
The lentiviral system that we have developedcontains a set of components that may allow forcomprehensive analysis of macrophage genes both invitro and in vivo. Initially, the lentiviral vector expres-sing a gene-of-interest in inducible manner can be testedusing a macrophage cell line, such as J774A.1, as wellas in primary BMDM isolated from the C3H.TgN(SRA–rtTA) transgenic mice. Next, the same constructcan be used to transduce hematopoietic stem cellsisolated from the C3H.TgN (SRA–rtTA) transgenicmice and generate bone marrow chimeras that wouldexpress a gene-of-interest in mature macrophages and,possibly, myeloid dendritic cells, after stimulation withdoxycycline. Finally, the same lentiviral vector may beused to generate transgenic mice on the C3H.TgN(SRA–rtTA) genetic background (Pfeifer et al., 2002)(Szulc et al., 2006) that would express the gene-of-interest in macrophages after induction with doxycy-cline. In the future, it may be used in combination withnovel systems for tetracycline-inducible expression oftransgene and/or RNAi (Szulc et al., 2006) and

43H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
macrophage progenitor expansion (Odegaard et al.,2007; Wang et al., 2006) to make genetic and functionalanalysis of macrophages more efficient.
Acknowledgement
The authors are grateful to Dr. Lester Kobzik forhelpful discussions and critical reading of the manu-script and Dr. Alex Ivanov for expert advice and massspectrometric analysis of the protein samples. This workwas supported by grants AI49421 and P01 AI056296from the National Institutes of Health.
References
Burke, B., Sumner, S., Maitland, N., Lewis, C.E., 2002. Macrophages ingene therapy: cellular delivery vehicles and in vivo targets. J. Leukoc.Biol. 72, 417.
Corbeau, P., Kraus, G., Wong-Staal, F., 1998. Transduction of humanmacrophages using a stable HIV-1/HIV-2-derived gene deliverysystem. Gene Ther. 5, 99.
De, S.K., Venkateshan, C.N., Seth, P., Gajdusek, D.C., Gibbs, C.J.,1998. Adenovirus-mediated human immunodeficiency virus-1 Nefexpression in human monocytes/macrophages and effect of Nef ondownmodulation of Fcgamma receptors and expression of mono-kines. Blood 91, 2108.
Ehrt, S., Schnappinger, D., Bekiranov, S., Drenkow, J., Shi, S.,Gingeras, T.R., Gaasterland, T., Schoolnik, G., Nathan, C., 2001.Reprogramming of the macrophage transcriptome in response tointerferon-gamma and Mycobacterium tuberculosis: signalingroles of nitric oxide synthase-2 and phagocyte oxidase. J. Exp.Med. 194, 1123.
Emerman, M., Temin, H.M., 1984. Genes with promoters in retrovirusvectors can be independently suppressed by an epigeneticmechanism. Cell 39, 449.
Follenzi, A., Ailles, L.E., Bakovic, S., Geuna, M., Naldini, L., 2000.Gene transfer by lentiviral vectors is limited by nucleartranslocation and rescued by HIV-1 pol sequences. Nat. Genet.25, 217.
Foxwell, B., Browne, K., Bondeson, J., Clarke, C., de Martin, R.,Brennan, F., Feldmann, M., 1998. Efficient adenoviral infectionwith IkappaB alpha reveals that macrophage tumor necrosis factoralpha production in rheumatoid arthritis is NF-kappaB dependent.Proc. Natl. Acad. Sci. U. S. A. 95, 8211.
Gaines, P., Wojchowski, D.M., 1999. pIRES-CD4t, a dicistronicexpression vector forMACS- or FACS-based selection of transfectedcells. Biotechniques 26, 683.
Heider, H., Verca, S.B., Rusconi, S., Asmis, R., 2000. Comparison oflipid-mediated and adenoviral gene transfer in human monocyte-derived macrophages and COS-7 cells. Biotechniques 28 (260-5),268.
Henson, P.M., Hume, D.A., 2006. Apoptotic cell removal indevelopment and tissue homeostasis. Trends Immunol. 27, 244.
Horvai, A., Palinski,W.,Wu, H., Moulton, K.S., Kalla, K., Glass, C.K.,1995. Scavenger receptor A gene regulatory elements target geneexpression to macrophages and to foam cells of atheroscleroticlesions. Proc. Natl. Acad. Sci. U. S. A. 92, 5391.
Hume, D.A., Underhill, D.M., Sweet, M.J., Ozinsky, A.O., Liew, F.Y.,Aderem, A., 2001. Macrophages exposed continuously to lipopoly-
saccharide and other agonists that act via toll-like receptors exhibit asustained and additive activation state. BMC Immunol. 2, 11.
Jiang, W., Reich, C.F., Pisetsky, D.S., 2006. In vitro assay ofimmunostimulatory activities of plasmid vectors. Methods Mol.Med. 127, 55.
Kadereit, S., Gewert, D.R., Galabru, J., Hovanessian, A.G., Meurs, E.F.,1993. Molecular cloning of two new interferon-induced, highlyrelated nuclear phosphoproteins. J. Biol. Chem. 268, 24432.
Kota, R.S., Rutledge, J.C., Gohil, K., Kumar, A., Enelow, R.I.,Ramana, C.V., 2006. Regulation of gene expression in RAW 264.7macrophage cell line by interferon-gamma. Biochem. Biophys.Res. Commun. 342, 1137.
Kotani, H., Newton III, P.B., Zhang, S., Chiang, Y.L., Otto, E.,Weaver, L., Blaese, R.M., Anderson, W.F., McGarrity, G.J., 1994.Improved methods of retroviral vector transduction and productionfor gene therapy. Hum. Gene Ther. 5, 19.
Kramnik, I., Dietrich, W.F., Demant, P., Bloom, B.R., 2000. Geneticcontrol of resistance to experimental infection with virulent My-cobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 97, 8560.
Krysko, D.V., D'Herde, K., Vandenabeele, P., 2006. Clearance ofapoptotic and necrotic cells and its immunological consequences.Apoptosis 11, 1709.
Mostoslavsky, G., Kotton, D.N., Fabian, A.J., Gray, J.T., Lee, J.S.,Mulligan, R.C., 2005. Efficiency of transduction of highly purifiedmurine hematopoietic stem cells by lentiviral and oncoretroviralvectors under conditions of minimal in vitro manipulation. Mol.Ther. 11, 932.
Murray, L., Travis, M., Luens-Abitorabi, K., Olsson, K., Plavec, I.,Forestell, S., Hanania, E.G., Hill, B., 2000. Addition of the humaninterferon beta scaffold attachment region to retroviral vectorbackbones increases the level of in vivo transgene expressionamong progeny of engrafted human hematopoietic stem cells.Hum. Gene Ther. 11, 2039.
Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F.H.,Verma, I.M., Trono, D., 1996. In vivo gene delivery and stabletransduction of nondividing cells by a lentiviral vector. Science272, 263.
Odegaard, J.I., Vats, D., Zhang, L., Ricardo-Gonzalez, R., Smith, K.L.,Sykes, D.B., Kamps, M.P., Chawla, A., 2007. Quantitativeexpansion of ES cell-derived myeloid progenitors capable ofdifferentiating into macrophages. J. Leukoc. Biol. 81, 711.
Overell, R.W., Weisser, K.E., Cosman, D., 1988. Stably transmittedtriple-promoter retroviral vectors and their use in transformation ofprimary mammalian cells. Mol. Cell. Biol. 8, 1803.
Pan, H., Yan, B.S., Rojas, M., Shebzukhov, Y.V., Zhou, H., Kobzik, L.,Higgins, D.E., Daly, M.J., Bloom, B.R., Kramnik, I., 2005. Ipr1gene mediates innate immunity to tuberculosis. Nature 434, 767.
Pfeifer, A., Ikawa, M., Dayn, Y., Verma, I.M., 2002. Transgenesis bylentiviral vectors: lack of gene silencing inmammalian embryonic stemcells and preimplantation embryos. Proc. Natl. Acad. Sci. U. S. A. 99,2140.
Pollard, V.W., Malim, M.H., 1998. The HIV-1 Rev protein. Annu. Rev.Microbiol. 52, 491.
Ross, J.A., Auger, M.J., 2002. The biology of macrophage. In: Burke,B., Lewis, C.E. (Eds.), The Macrophage. Oxford University Press,UK, p. 1.
Rouzer, C.A., Jacobs, A.T., Nirodi, C.S., Kingsley, P.J., Morrow, J.D.,Marnett, L.J., 2005. RAW264.7 cells lack prostaglandin-dependentautoregulation of tumor necrosis factor-alpha secretion. J. LipidRes. 46, 1027.
Sasmono, R.T., Hume, D.A., 2004. The biology of macrophages. In:Kaufmann, S.H.E., Medzhitov, R., Gordon, S. (Eds.), The Innate

44 H. Pan et al. / Journal of Immunological Methods 329 (2008) 31–44
Immune Response to Infection. Am. Society for Microbiology Press,p. 71.
Sasmono, R.T., Oceandy, D., Pollard, J.W., Tong, W., Pavli, P.,Wainwright, B.J., Ostrowski, M.C., Himes, S.R., Hume, D.A.,2003. A macrophage colony-stimulating factor receptor-greenfluorescent protein transgene is expressed throughout the mono-nuclear phagocyte system of the mouse. Blood 101, 1155.
Schroder, K., Hertzog, P.J., Ravasi, T., Hume, D.A., 2004. Interferon-gamma: an overviewof signals,mechanisms and functions. J. Leukoc.Biol. 75, 163.
Schroers, R., Sinha, I., Segall, H., Schmidt-Wolf, I.G., Rooney, C.M.,Brenner, M.K., Sutton, R.E., Chen, S.Y., 2000. Transduction ofhuman PBMC-derived dendritic cells and macrophages by an HIV-1-based lentiviral vector system. Mol. Ther. 1, 171.
Sester, D.P., Stacey, K.J., Sweet, M.J., Beasley, S.J., Cronau, S.L.,Hume, D.A., 1999. The actions of bacterial DNA on murinemacrophages. J. Leukoc. Biol. 66, 542.
Stacey, K.J., Sweet, M.J., Hume, D.A., 1996. Macrophages ingest andare activated by bacterial DNA. J. Immunol. 157, 2116.
Szulc, J., Wiznerowicz, M., Sauvain, M.O., Trono, D., Aebischer, P.,2006. A versatile tool for conditional gene expression andknockdown. Nat. Methods 3, 109.
Wang, G.G., Calvo, K.R., Pasillas, M.P., Sykes, D.B., Hacker, H.,Kamps, M.P., 2006. Quantitative production of macrophages orneutrophils ex vivo using conditional Hoxb8. Nat. Methods 3, 287.
Weir, J.P., Meltzer, M.S., 1993. Transfection of human immunodefi-ciency virus type 1 proviral DNA into primary human monocytes.Cell. Immunol. 148, 157.
Yan, B.S., Kirby, A., Shebzukhov, Y.V., Daly, M.J., Kramnik, I.,2006a. Genetic architecture of tuberculosis resistance in a mousemodel of infection. Genes Immun. 7, 201.
Yan, C., Lian, X., Li, Y., Dai, Y.,White, A., Qin, Y., Li, H., Hume, D.A.,Du, H., 2006b. Macrophage-specific expression of humanlysosomal acid lipase corrects inflammation and pathogenicphenotypes in lal−/−mice. Am. J. Pathol. 169, 916.
Yu, X., Zhan, X., D'Costa, J., Tanavde, V.M., Ye, Z., Peng, T.,Malehorn, M.T., Yang, X., Civin, C.I., Cheng, L., 2003. Lentiviralvectors with two independent internal promoters transfer high-level expression of multiple transgenes to human hematopoieticstem-progenitor cells. Mol. Ther. 7, 827.
Zennou, V., Petit, C., Guetard, D., Nerhbass, U., Montagnier, L.,Charneau, P., 2000. HIV-1 genome nuclear import is mediated by acentral DNA flap. Cell 101, 173.
Zufferey, R., Donello, J.E., Trono, D., Hope, T.J., 1999. Woodchuckhepatitis virus posttranscriptional regulatory element enhancesexpression of transgenes delivered by retroviral vectors. J. Virol.73, 2886.