drug interactions - 2017 course materials · ddi drug-drug interactions gi gastrointestinal ......
TRANSCRIPT

12/9/2015
1
Drug Interactions Sarah Robertson, Pharm.D. Director, Department of Clinical Pharmacology Vertex Pharmaceuticals Inc. Boston, MA, USA December 10, 2015
1

12/9/2015
2
Overview
• Epidemiology and Categories of Drug Interactions
• Mechanisms Affecting Drug Absorption
• Alteration in Drug Distribution
• Drug Interactions by Alteration in Drug Metabolism
• Modulation of Transport Proteins
• Alteration in Renal Elimination
• Enzyme/Transporter Interplay and Complex Drug Interactions
• Clinical Interpretation of Drug Interactions
• Drug Interaction Information in Product Labeling
• Resources
2

12/9/2015
3
Abbreviations AUC Area under the concentration vs. time curve
BCRP Breast Cancer Resistance Protein
CAR Constitutive Androstane Receptor
Cmax Maximum Observed Concentration
CNS Central Nervous System
CYP450 Cytochrome P450
DDI Drug-drug interactions
GI Gastrointestinal
IC50 Half-maximal inhibition concentration
NDA New Drug Application
NSCLC Non-Small Cell Lung Cancer
OAT Organic anion transporter
OCT Organic cation transporter
OATP Organic anion transporting polypeptide
P-gp P-glycoprotein
PPI Proton-pump inhibitor
PXR Pregnane X receptor
TB Tuberculosis
UGT UDP glucuronosyltransferase
Vd Volume of distribution
3

12/9/2015
4
Epidemiology of Drug Interactions
• True incidence not easily quantifiable
• Review of Medicaid records for 8860 patients from 2005-2009
found 16.6% had ≥1 clinically significant DDI1
• ↑ risk among elderly, patients with comorbidities or polypharmacy
• An FDA review of NDAs approved in 2013:2
• All compounds were metabolized by at least one CYP450 enzyme (77% by CYP3A)
• 77% showed possible inhibition or induction of ≥ 1 metabolizing enzyme in vitro; 85% showed a possible interaction with ≥ 1 transport protein in vitro
• Overall, 45% had a metabolism-based DDI that resulted in a change in exposure of clinical significance 4
1Nelson SD et al, J Pharm Pract. Aug 2014 Epub; 2Yu J et al, Drug Metab Dispos. Sept 2014 Epub

12/9/2015
5
Types of Drug Interactions
• Pharmacodynamic
• Related to drug’s effect on target (either safety or efficacy)
• One drug modulates that of another (additive, synergistic, or antagonistic)
• Most frequently identified in recent review by Nelson et al (mainly among drugs used to treat psychiatric/seizure/sleep disorders and pain)
• Pharmacokinetic
• Impact how a drug is absorbed, distributed, metabolized, or excreted (i.e. impact the concentration of drug at the site of activity or at a site of toxicity)
• Most commonly the result of inhibition or induction of CYP450 enzymes
5

12/9/2015
6
Pharmacodynamic Interactions
• Additive combinations
• Pharmacologic effect = sum of the 2 drugs
• Beneficial: ibuprofen + acetaminophen
• Harmful: neutropenia with zidovudine + ganciclovir
• Synergistic combinations
• Pharmacologic effect > sum of the 2 drugs
• Beneficial: aminoglycosides + penicillin
• Harmful: barbiturates + alcohol
• Antagonistic combinations
• Pharmacologic effect < either drug alone
• Beneficial: naloxone for opiate overdose
• Harmful: zidovudine + stavudine in treatment of HIV
6

12/9/2015
7
Pharmacokinetic Interactions
• Absorption: Gastrointestinal (GI) motility, pH, chelate formation, GI transport proteins
• Distribution: Transport proteins, plasma protein binding
• Metabolism: Phase I (CYP450 enzymes) +/- transport proteins, Phase II (conjugation)
• Elimination: Renal excretion (glomerular filtration; tubular secretion), biliary secretion
7

12/9/2015
8
Altered Absorption: pH Affects
• Many drugs are dependent on pH for optimal solubility
• Increasing pH in the gut with H2-antagonists (e.g. ranitidine) or PPIs (e.g. omeprazole) can ↓ or ↑ drug absorption
• Examples: • Atazanavir and omeprazole: Atazanavir AUC ↓ by 94%
• Raltegravir and omeprazole: Raltegravir AUC ↑ 3-fold
• Erlotinib and omeprazole: Erlotinib AUC ↓ ~50%
Retrospective review of patient records found concurrent treatment with acid suppressors and erlotinib was sig. associated with shorter overall survival after accounting for other factors (12.9 vs. 16.8 months)1
8
1Chu MP et al, Clin Lung Cancer. Aug2014 Epub.
12/9/2015
9
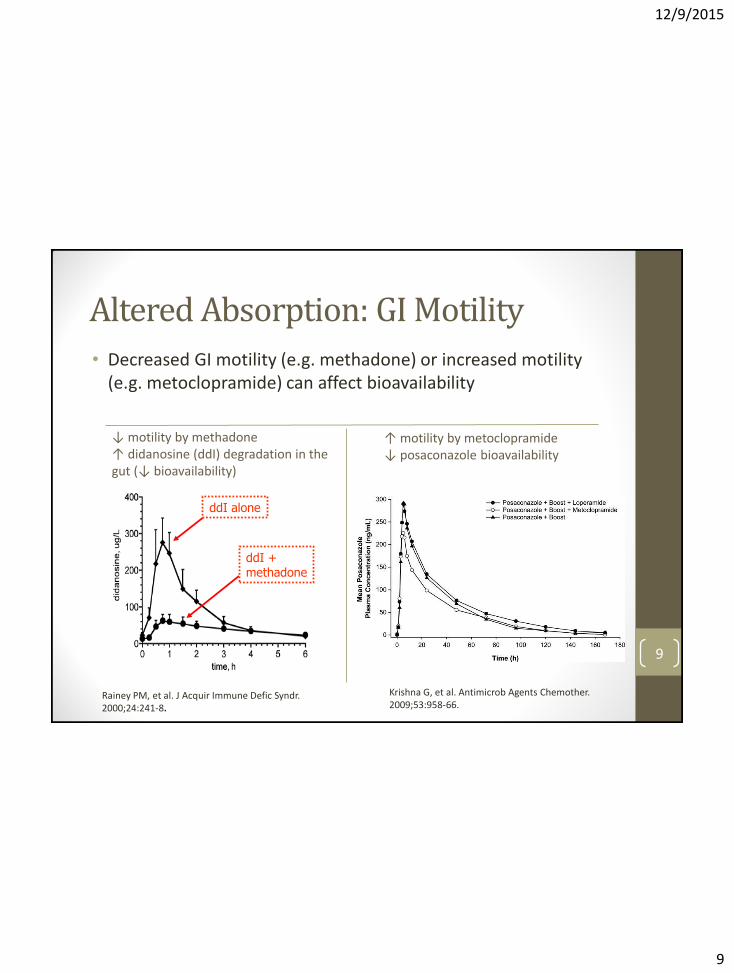
Altered Absorption: GI Motility
• Decreased GI motility (e.g. methadone) or increased motility (e.g. metoclopramide) can affect bioavailability
9
ddI alone
ddI + methadone
↓ motility by methadone ↑ didanosine (ddI) degradation in the gut (↓ bioavailability)
↑ motility by metoclopramide ↓ posaconazole bioavailability
Krishna G, et al. Antimicrob Agents Chemother. 2009;53:958-66.
Rainey PM, et al. J Acquir Immune Defic Syndr. 2000;24:241-8.

12/9/2015
10
Altered Absorption: Chelation
• Irreversible binding of drug in the GI tract
• Usually can be avoided by separation of administration
• E.g. Tetracyclines, quinolone antibiotics with ferrous sulfate (Fe+2), antacids (Al+3, Mg+2), dairy products (Ca+2)
10
Trovafloxacin Alone / Maalox ® 2 hrs after Trovafloxacin / Trovafloxacin + omeprazole
Maalox® 30 min before Trovafloxacin
12/9/2015
11
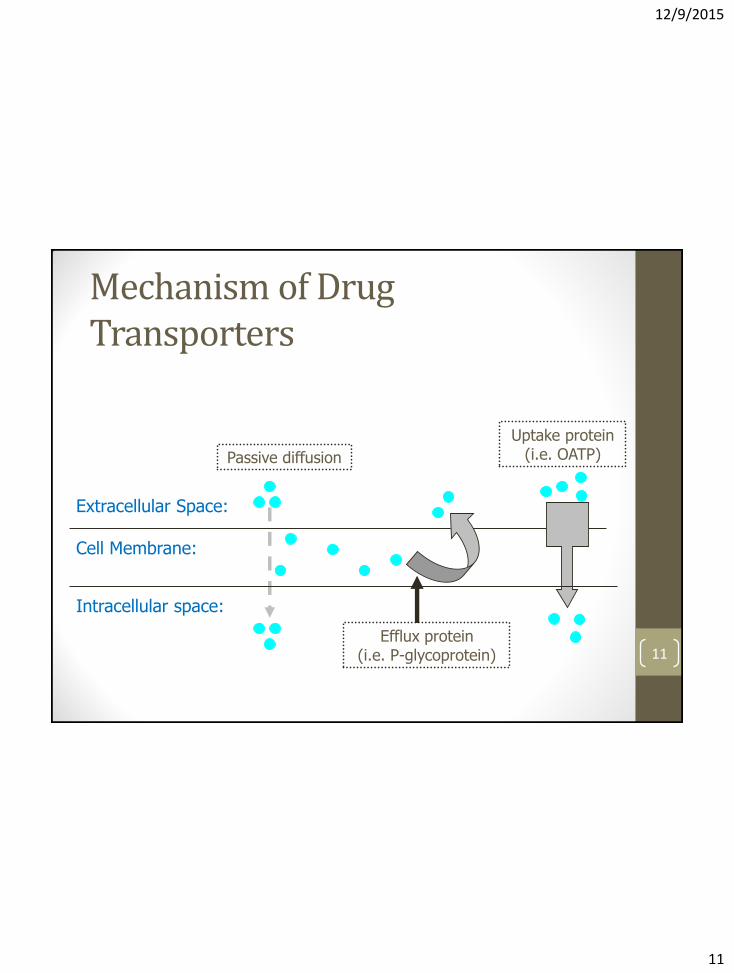
Mechanism of Drug Transporters
11
Cell Membrane:
Intracellular space:
Efflux protein (i.e. P-glycoprotein)
Passive diffusion
Uptake protein (i.e. OATP)
Extracellular Space:
12/9/2015
12
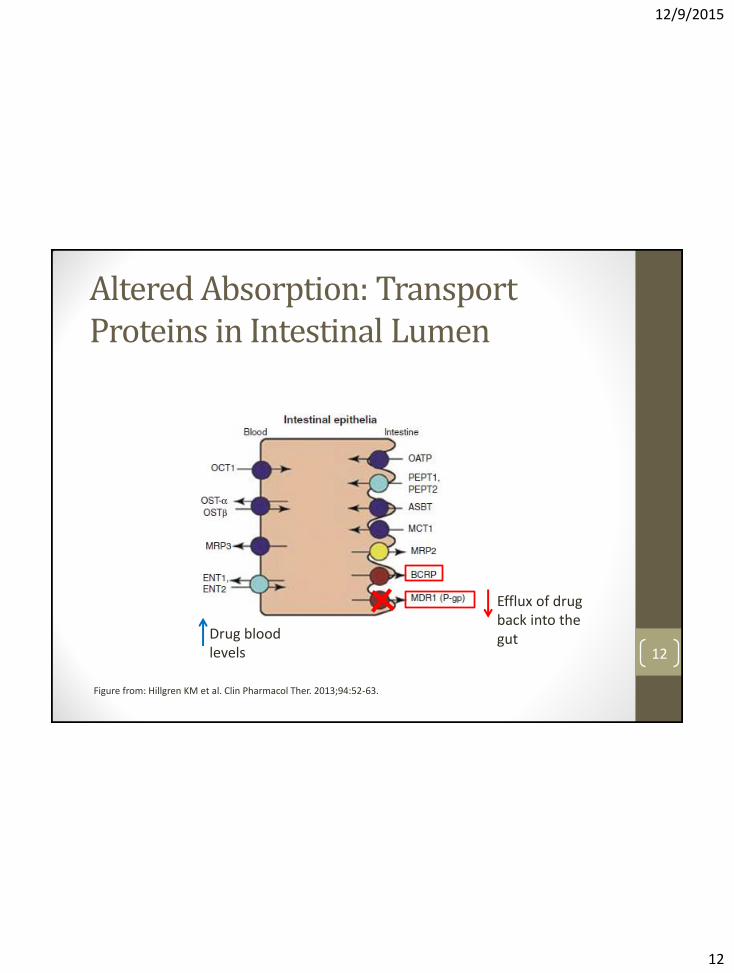
Altered Absorption: Transport Proteins in Intestinal Lumen
12
Figure from: Hillgren KM et al. Clin Pharmacol Ther. 2013;94:52-63.
Drug blood levels
Efflux of drug back into the gut

12/9/2015
13
Altered Absorption: Transport Proteins in Intestinal Lumen • Uptake and efflux transporters in the gut can be inhibited
or induced by drugs, which may ↑ or ↓ bioavailability of drugs that rely on the transporters (i.e. substrates)
• Reliance on uptake/efflux transporters for absorption depends largely on compound permeability The lower the permeability of a compound, the more its absorption is affected by membrane transporters.
• Of the many gut transporters, only 2 are generally associated with clinical DDIs: • P-glycoprotein (P-gp, MDR1, ABCB1)
• Breast Cancer Resistance Protein (BCRP, ABCG2) 13

12/9/2015
14
Altered Absorption: Transport Proteins in Intestinal Lumen
• E.g. DDI between quinidine (P-gp inhibitor) and digoxin (sensitive substrate) is well documented. In a perfusion catheter study, quinidine caused a 2.5-fold increase in the amount of digoxin absorbed1
• Digoxin DDI studies are often conducted as part of development for new drugs found to be potential P-gp inhibitors in vitro
• Clinical P-gp inhibitors include: cyclosporine, erythromycin, verapamil, itraconazole, and others
14
1Igel S, et al. Clin Pharmacokinet. 2007;46:777-85.

12/9/2015
15
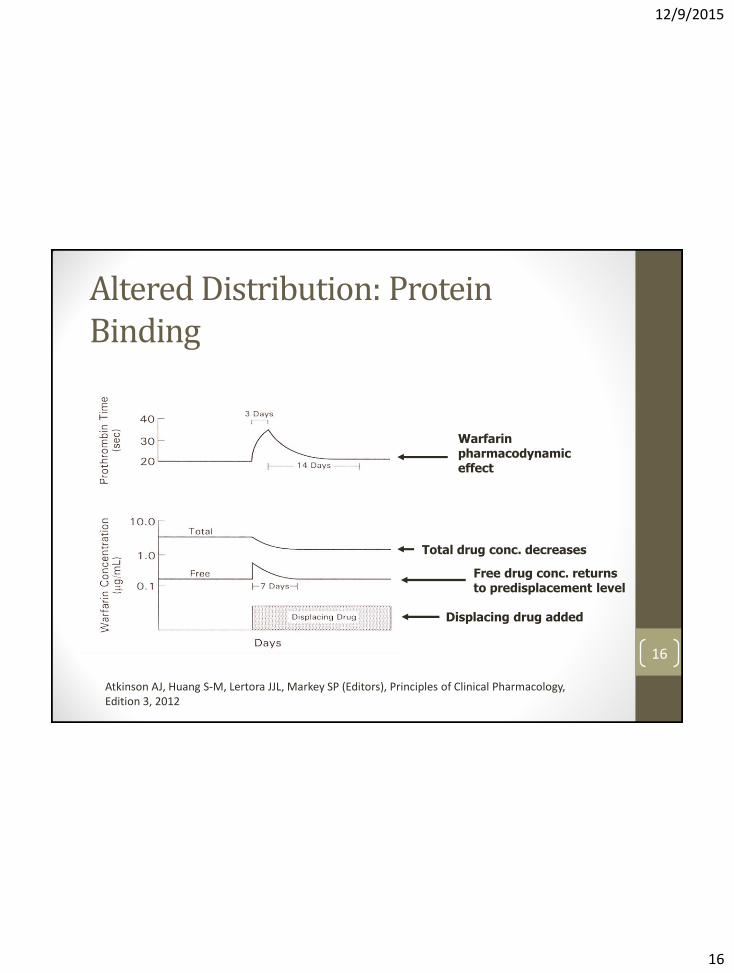
Altered Distribution: Protein Binding
• Theoretical mechanism of DDI for restrictively cleared drugs (i.e. small fraction of drug is extracted during passage through eliminating organ)
• Only unbound drug in plasma is cleared. Thus, ↑ fu leads to an increase in total drug CL (and ↓ in plasma concentrations)
• However, unbound plasma concentrations typically return to pre-displacement values after a transient increase
• Only likely to be clinically significant for drugs with high protein binding, small Vd, and narrow therapeutic index (e.g. warfarin)
• “…the overall clinical importance of plasma protein binding displacement interactions continues to be overstated…”
15 Sansom LN & Evans AM. Drug Safety 1995;12:227-233. Rolan PE. Br J Clin Pharmacol 1994;37:125-128.
12/9/2015
16
Altered Distribution: Protein Binding
16
Displacing drug added
Free drug conc. returns to predisplacement level
Total drug conc. decreases
Warfarin pharmacodynamic effect
Atkinson AJ, Huang S-M, Lertora JJL, Markey SP (Editors), Principles of Clinical Pharmacology, Edition 3, 2012
12/9/2015
17
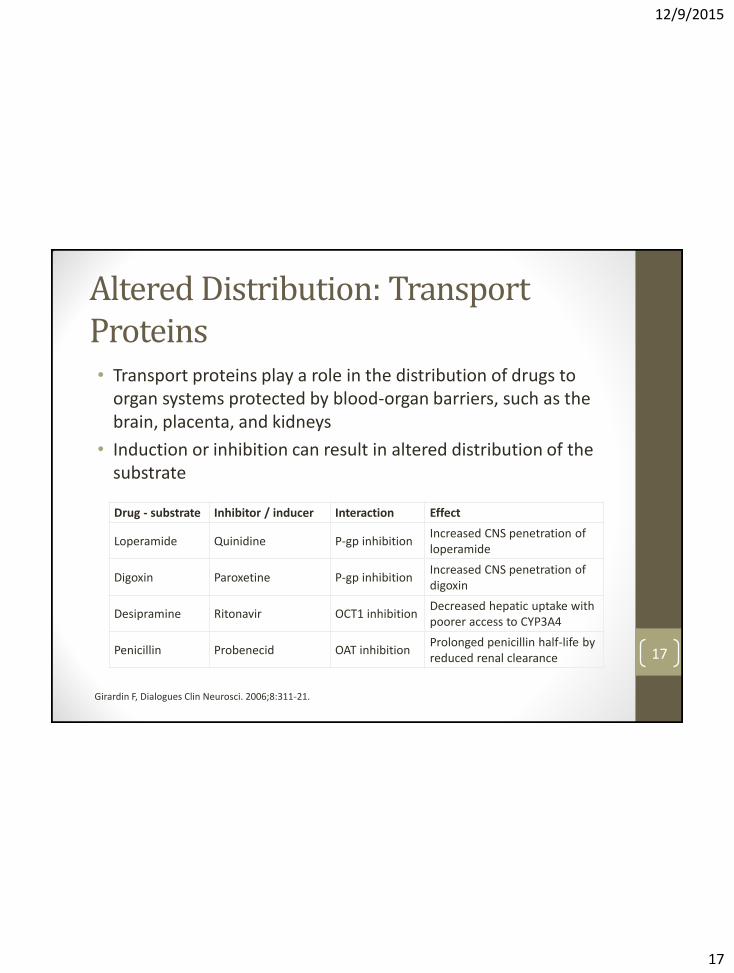
Altered Distribution: Transport Proteins • Transport proteins play a role in the distribution of drugs to
organ systems protected by blood-organ barriers, such as the brain, placenta, and kidneys
• Induction or inhibition can result in altered distribution of the substrate
17
Drug - substrate Inhibitor / inducer Interaction Effect
Loperamide Quinidine P-gp inhibition Increased CNS penetration of loperamide
Digoxin Paroxetine P-gp inhibition Increased CNS penetration of digoxin
Desipramine Ritonavir OCT1 inhibition Decreased hepatic uptake with poorer access to CYP3A4
Penicillin Probenecid OAT inhibition Prolonged penicillin half-life by reduced renal clearance
Girardin F, Dialogues Clin Neurosci. 2006;8:311-21.

12/9/2015
18
Metabolism Overview Phase 1: CYP450 enzymes, present in gut and liver
• Primary source of adverse drug interactions
• Proportion of drugs metabolized by CYP450 enzymes:1
Phase 2: Conjugation enzymes; most prevalent in liver
• UGTs, Methyltransferase, sulfotransferases, N-acetyltransferase, etc.
• Modulation of Phase 2 enzymes is rarely associated with clinically significant DDIs
18
CYP3A4/5
CYP2D6
CYP1A/2
CYP2C19
CYP2C8/9
CYP2E1
CYP2B6
1 Kashuba and Bertino. In Drug Interactions in Infectious Diseases. Humana Press. 2001

12/9/2015
19
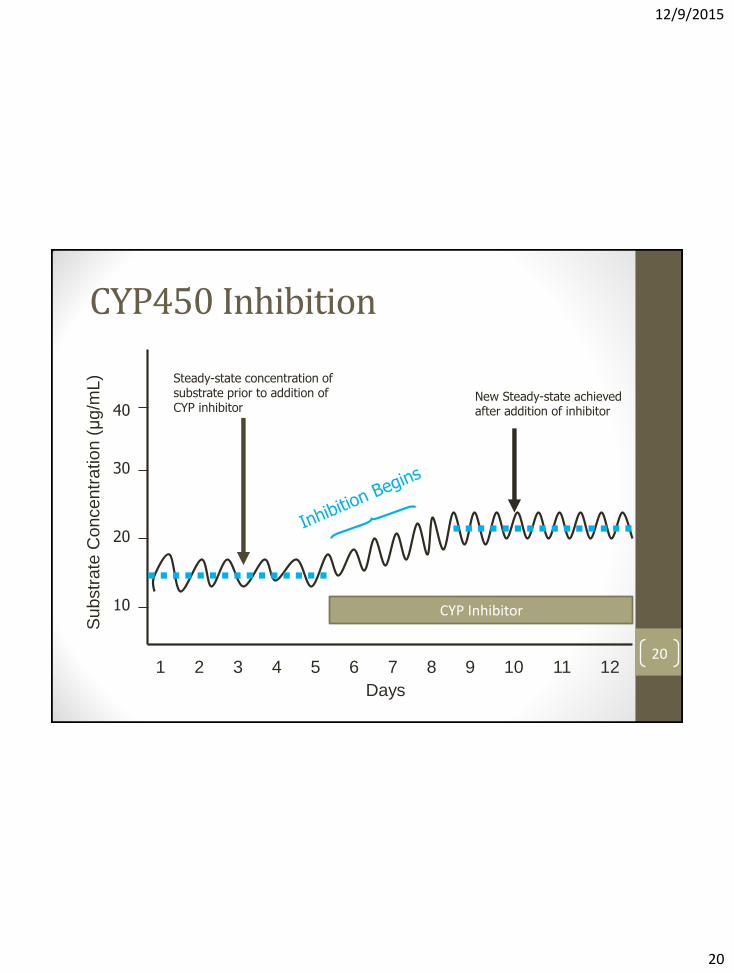
Altered Metabolism: Inhibition of CYP450 enzymes
• May occur in the liver and/or GI tract
• Results in ↑substrate potential for toxicity
• Usually by competitive binding to enzyme site
• Onset and offset of effect generally occurs quickly (depends on the time to steady-state of the inhibitor)
• Time to maximum interaction effect dependent on time required for substrate drug to reach new steady-state 19
12/9/2015
20
CYP450 Inhibition
20
Substr
ate
Concentr
ation (μ
g/m
L)
1 2 3 4 5 6 7 8 9 10 11 12
Steady-state concentration of substrate prior to addition of CYP inhibitor
New Steady-state achieved after addition of inhibitor
10
20
30
40
Days
CYP Inhibitor

12/9/2015
21
Altered Metabolism: Inhibition of CYP45 enzymes
• Effect of inhibition on substrate exposure is greater if the substrate relies on the inhibited enzyme as its sole route of metabolism (e.g. Midazolam and CYP3A4); drugs with >1 route of metabolism are less sensitive to inhibition of 1 route (e.g. voriconazole: CYP2C9, 2C19, 3A4)
• Mechanism-based enzyme inactivation • Formation of reactive metabolites which bind covalently to
enzyme or form a metabolic inhibitory complex (MIC)
• Results in irreversible or quasi-irreversible inactivation of CYP
• More profound and prolonged inhibitory effect
• Examples include macrolides, grapefruit juice
• Duration of inhibition depends on time to restore active enzyme
21
12/9/2015
22
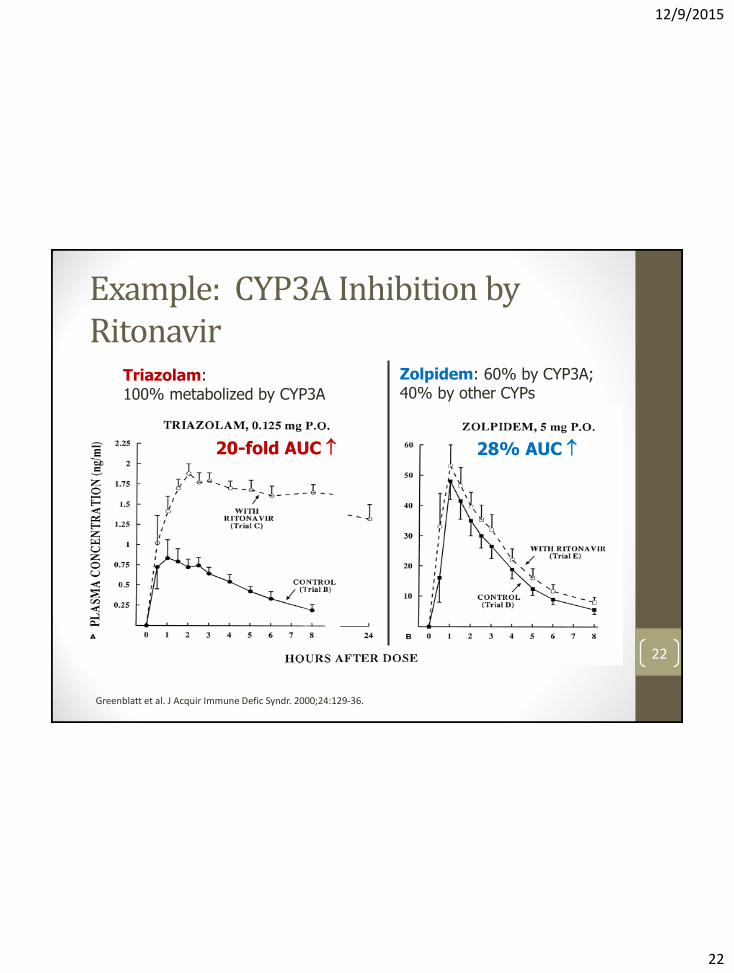
Example: CYP3A Inhibition by Ritonavir
22
Triazolam: 100% metabolized by CYP3A
Zolpidem: 60% by CYP3A; 40% by other CYPs
20-fold AUC 28% AUC
Greenblatt et al. J Acquir Immune Defic Syndr. 2000;24:129-36.

12/9/2015
23
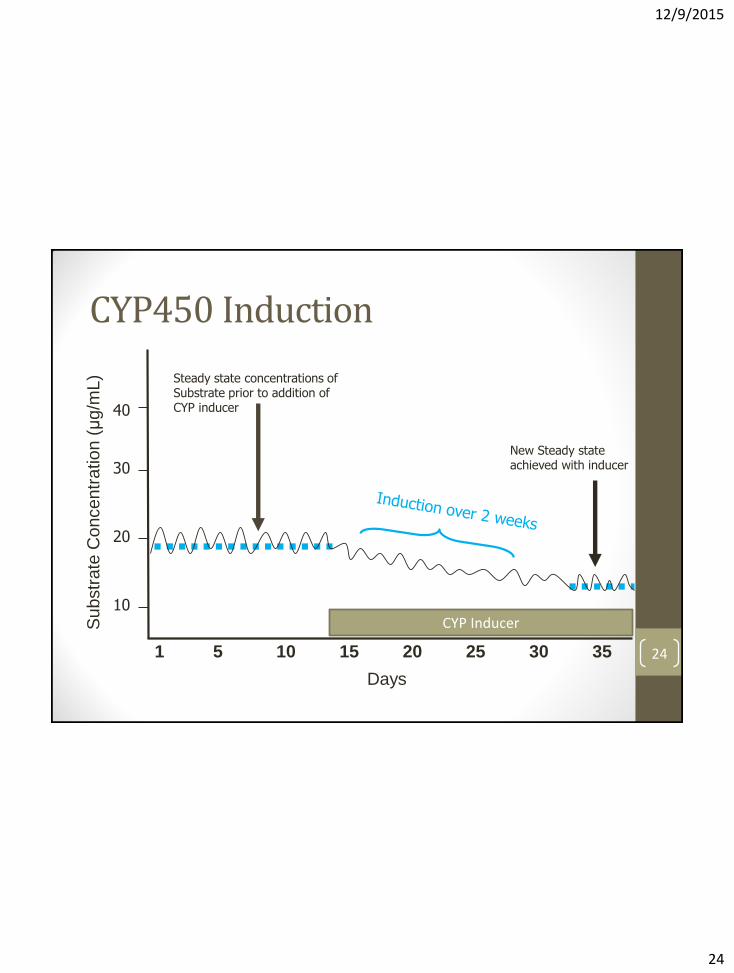
• Involves increased DNA transcription via nuclear receptor activation (e.g. PXR, CAR) synthesis of new CYP enzymes
• Slower onset and offset relative to inhibition; depends on half-life of inducer, time to make new CYP proteins, and rate of degradation of CYP proteins
• Results in ↓substrate potential for reduced activity, or formation of toxic metabolites
• Removal of inducer without a dose adjustment of substrate may lead to toxic concentrations of substrate
• Unlike inhibition, induction can be significant even when the particular CYP enzyme being induced is a minor pathway for the substrate
23
Altered Metabolism: Induction
12/9/2015
24
CYP450 Induction
24
Substr
ate
Concentr
ation (μ
g/m
L) Steady state concentrations of
Substrate prior to addition of CYP inducer
New Steady state achieved with inducer
10
20
30
40
Days
1 5 10 15 20 25 30 35
CYP Inducer
12/9/2015
25
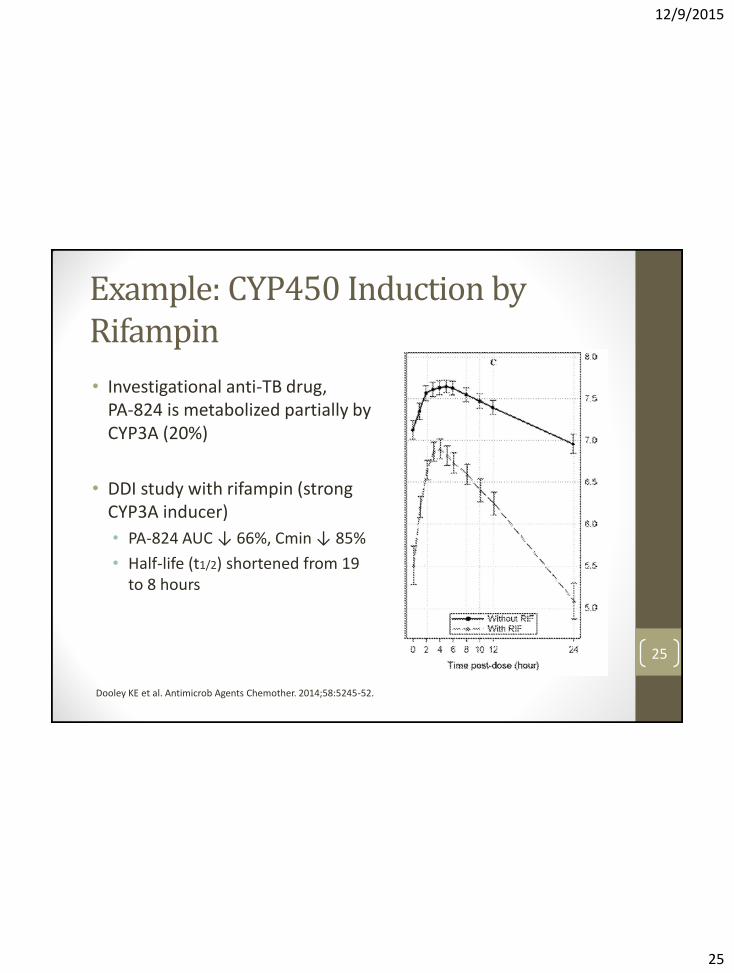
Example: CYP450 Induction by Rifampin
• Investigational anti-TB drug, PA-824 is metabolized partially by CYP3A (20%)
• DDI study with rifampin (strong CYP3A inducer)
• PA-824 AUC ↓ 66%, Cmin ↓ 85%
• Half-life (t1/2) shortened from 19 to 8 hours
25
Dooley KE et al. Antimicrob Agents Chemother. 2014;58:5245-52.

12/9/2015
26
Classification of Common CYP450 Inhibitors/Inducers
Inhibitors Inducers
Strong Moderate Weak Strong Moderate Weak
CYP3A Clarithromycin Itraconazole Posaconazole Ritonavir Telithromycin Voriconazole
Aprepitant Diltiazem Erythromycin Fluconazole Grapefruit juice Verapamil
Alprazolam Atorvastatin Cimetidine Cyclosporine Fluoxetine Isoniazid
Avasimibe Carbamazepine
Phenobarbital Phenytoin Rifampin St. John’s wort
Bosentan Efavirenz Etravirine Modafinil Nafcillin
Aprepitant Armodafinil Pioglitazone Prednisone
CYP2D6 Bupropion Fluoxetine Paroxetine
Duloxetine Terbinafine
Celecoxib Diltiazem Sertraline
None None None
CYP1A2 Ciprofloxacin Fluvoxamine
Mexiletine Zileuton
Acyclovir Allopurinol Famotidine Verapamil
None Montelukast Phenytoin Cigarette smoking
Moricizine Omeprazole Phenobarbital
CYP2C19 Fluconazole Fluvoxamine Ticlopidine
Esomeprazole Fluoxetine Omeprazole Voriconazole
Carbamazepine Cimetidine Ethinyl Estradiol Etravirine
None Rifampin Artemisinin
CYP2C8 Gemfibrozil None
Fluvoxamine Trimethoprim
None Rifampin None
CYP2C9 None
Amiodarone Fluconazole
Cotrimoxazole Fluvastatin
None Carbamazepine
Rifampin Aprepitant Bosentan
26

12/9/2015
27
Altered Hepatic or Biliary Elimination: Transport Proteins • Inhibition of uptake transporters (e.g. OATP1B1) decreased
hepatic update less hepatic metabolism, higher systemic exposure
• Inhibition of efflux transporters (e.g. BCRP) decreased biliary excretion higher systemic exposure (or increased hepatic metabolism, if drug is metabolized)
27
Figure from: Hillgren KM et al. Clin Pharmacol Ther. 2013;94:52-63.

12/9/2015
28
Example: OATP2 Inhibition by Gemfibrozil
28
Rosuvastatin AUC ↑ 88%
Schneck DW et al. Clin Pharmacol Ther. 2004;75:455-63.

12/9/2015
29
Transporter/CYP interplay
Interacting Drug Effect on Atorvastatin AUC
Cyclosporine - P-gp inhibitor - OATP1B1 inhibitor - Weak CYP3A inhibitor
↑ 8.7-fold
Clarithromycin - P-gp inhibitor - Strong CYP3A inhibitor
↑ 4.4-fold
Diltiazem - P-gp inhibitor - Moderate CYP3A inhibitor
↑ 51%
Gemfibrozil - OATP1B1 inhibitor
↑ 35%
29
Example: Atorvastatin Substrate for CYP3A, P-gp, OATP1B1, OATP2B1

12/9/2015
30
Altered Elimination: Renal
30

12/9/2015
31
Altered Elimination: Renal
31
• Inhibition of uptake transporters (e.g. OAT1, OAT3) decreased renal elimination increased systemic exposure
• Inhibition of efflux transporters (e.g. P-gp) decreased secretion into urine increased systemic exposure
• E.g. Cimetidine (OCT inhibitor): ↑ metformin and pramipexole exposure
• E.g. Probenecid (OAT1/3 inhibitor): ↑ cidofovir, furosemide, acyclovir exposure
Figure from: Hillgren KM et al. Clin Pharmacol Ther. 2013;94:52-63.

12/9/2015
32
Complex Drug Interactions
• Concurrent inhibition and induction of one enzyme (e.g. ritonavir and CYP3A) unpredictable and time-dependent effect on substrates
• Concurrent inhibition or induction of an enzyme and transporter potentially additive effect on substrate
• Combination of 2 inhibitors of different enzymatic pathways used by 1 substrate (e.g. CYP3A and CYP2C9) greater increase in substrate exposure than either drug alone (effect may be synergistic, not additive)
• Inhibition of the alternative enzyme pathway in a population of poor metabolizers (PMs) of the primary enzymatic pathway greater effect on substrate
• Enzyme/transporter inhibitors in patients with altered renal or hepatic elimination due kidney or liver disease
32

12/9/2015
33
Predicting Clinical DDIs
• In early drug development, clinically relevant DDIs are predicted with in vitro experiments (e.g. recombinant CYP enzymes, liver microsomes, hepatocytes, liver slices, etc.)
• Elucidate method of elimination – Describes study drug potential as a “victim”
• If metabolized, which enzyme is responsible?
• If renally eliminated, is active transport involved?
• If excreted in bile, are efflux transporters involved?
• Determine if study drug causes inhibition or induction of enzymes or transporters? – Describes study drug potential as a “perpetrator”
33

12/9/2015
34
Predicting Clinical DDIs, cont.
• If inhibition/induction of enzymes or transporters is observed in vitro, the probability of an in vivo effect is determined:
• Concentration at which inhibition or induction is observed in vitro (e.g. IC50) is compared to in vivo concentrations (e.g. Cmax or [I])
• If inhibition/induction is possible (e.g. [I]/IC50 > 0.1), clinical DDI studies must be considered, or mechanistic modeling may be performed to evaluate potential for DDI in vivo
34

12/9/2015
35
Assessing Clinical DDIs: Phase 1 studies • Initially probe DDI studies are performed:
• For a substrate, assess effect of a strong inhibitor (e.g. Itraconazole for CYP3A) and strong inducer (e.g. Rifampin for CYP3A) on the study drug
• For a potential perpetrator, assess effect on a sensitive substrate (e.g. single dose oral midazolam for CYP3A; digoxin for P-gp; rosuvastatin for OATP1B1)
• Depending on results of these initial studies, addt’l DDI studies should be considered:
• Likely coadministered drugs
• Moderate inhibitors or inducers
• Drugs with mixed enzyme/transporter effects
• Proton-pump inhibitor DDI for pH-dependent solubility drugs
• Etc.
35

12/9/2015
36
Evaluating Risk in the Clinical Setting • Consider the therapeutic index of the “victim” drug
• E.g. 50% increase in atorvastatin not likely clinically significant; 50% increase in tacrolimus may be clinically significant
• Are other potential “perpetrators” involved?
• What is the likely time course of the interaction?
• Consider both the addition and withdrawal of potential “perpetrators” and the implications to the substrate
• Is the DDI a class effect? Or are there other options?
• E.g. rosuvastatin vs. simvastatin – different susceptibility to DDIs and different therapeutic indices
• Are there other confounders that may magnify the DDI? (e.g. organ impairment, older age)
36

12/9/2015
37
DDI Information in U.S. Product Labeling
1. Indications and Usage
2. Dosage and Administration
3. Dosage Forms and Strengths
4. Contraindications
5. Warnings and Precautions
6. Adverse Reactions
7. Drug Interactions
8. Use in Specific Populations
9. Drug Use and Dependence
10. Overdosage
11. Description
12.Clinical Pharmacology
13. Nonclinical Toxicology
14. Clinical Studies
37

12/9/2015
38
Section 7: Drug Interactions Describes Clinical Interpretation of DDI Study Results or In Vitro Findings
Example: Kalydeco®(ivacaftor)
Potential for other drugs to affect ivacaftor
7.1 Inhibitors of CYP3A
Ivacaftor is a sensitive CYP3A substrate. Co-administration with ketoconazole, a strong CYP3A inhibitor, significantly increased ivacaftor exposure [measured as area under the curve (AUC)] by 8.5-fold. Based on simulations of these results, a reduction of the KALYDECO dose to 150 mg twice a week is recommended for co-administration with strong CYP3A inhibitors, such as ketoconazole, itraconazole, posaconazole, voriconazole, telithromycin, and clarithromycin.
Co-administration with fluconazole, a moderate inhibitor of CYP3A, increased ivacaftor exposure by 3-fold. Therefore, a reduction of the KALYDECO dose to 150 mg once daily is recommended for patients taking concomitant moderate CYP3A inhibitors, such as fluconazole and erythromycin.
Co-administration of KALYDECO with grapefruit juice, which contains one or more components that moderately inhibit CYP3A, may increase exposure of ivacaftor. Therefore, food containing grapefruit or Seville oranges should be avoided during treatment with KALYDECO [see Clinical Pharmacology (12.3)].
7.2 Inducers of CYP3A
Co-administration with rifampin, a strong CYP3A inducer, significantly decreased ivacaftor exposure (AUC) by approximately 9-fold. Therefore, co-administration with strong CYP3A inducers, such as rifampin, rifabutin, phenobarbital, carbamazepine, phenytoin, and St. John’s Wort is not recommended [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)]. 38
Kalydeco (ivacaftor) USPI, June 2014. Vertex Pharmaceuticals Inc., Boston, MA.

12/9/2015
39
Section 12: Clinical Pharmacology Provides DDI Study Results in Table or Forest Plot
39
Figure 2: Impact of Other Drugs on KALYDECO
Note: The data obtained for KALYDECO without co-administration of inducers or inhibitors are used as reference.
The vertical lines are at 0.8, 1.0 and 1.25, respectively.
Kalydeco (ivacaftor) USPI, June 2014. Vertex Pharmaceuticals Inc., Boston, MA.

12/9/2015
40
Resources and Tools
40
Site Web Address
Martindale1 https://www.medicinescomplete.com/mc/martindale/current/
Micromedex1 www.micromedex.com
UCSF2 http://hivinsite.ucsf.edu/arvdb?page=ar-00-02&post=7
Indiana University3 http://medicine.iupui.edu/clinpharm/ddis
Natural Products Database4 http://www.naturaldatabase.com
Lexi-Comp Lexi-Interact1 www.lexi-comp.com
University of Washington Drug Interaction Database5
http://www.druginteractioninfo.org/
1Includes all drugs; paid subscription required
2Focus on HIV meds; free 3Exhaustive tables of CYP substrates, inhibitors, and inducers; free 4Focuses on natural products; paid subscription required 5Comprehensive and thoroughly referenced database of in vitro and in vivo data related to DDIs, including transporter-mediated DDIs; database serves as a reference for FDA guidance and decision trees

12/9/2015
41
Questions
41