drug-antibody-platelet interaction in quinine- and...
TRANSCRIPT

Drug-Antibody-Platelet Interaction in Quinine- andQuinidine-induced Thrombocytopenia
DOUGLASJ. CHRISTIE and RICHARD H. ASTER, Blood Center of SoutheasternWisconsin, Milwaukee, Wisconsin 53233; Departments of Medicine andPathology, Medical College of Wisconsin, Milwaukee, Wisconsin 53226
A B S T R A C T Binding of quinine- and quinidine-de-pendent antibodies to platelets was studied using anelectroimmunoassay to measure platelet-bound IgG.Antibodies from four patients with drug-inducedthrombocytopenia differed significantly in their inter-action with platelets: association constants for bindingto platelets at high drug concentrations ranged from0.29 to 2.6 X 107 M-1, the maximum number of an-tibody molecules bound ranged from 36,000 to161,000/platelet, the amount of drug necessary toachieve half-maximum binding of antibodies to plate-lets ranged from 2 to 60 ,tM, and only one of theantibodies cross-reacted with the stereoisomer of thedrug to which the patient was sensitized. Binding ofthe antibodies to platelets was enhanced at the highestachievable molar ratio of drug:antibody, 10,000:1,rather than being inhibited, as would be expected ina conventional, hapten-dependent reaction. The drug-antibody-platelet reaction was unaffected by FactorVIII/von Willebrand protein, nonspecifically aggre-gated IgG, or heat-labile complement components.After pretreatment with tritiated quinine, plateletsretained several hundred thousand molecules of drugeach, but failed to bind detectable amounts of anti-body. However, platelets treated simultaneously withquinine-dependent antibody and tritiated quinine re-tained significantly more drug after repeated washesthan platelets treated with drug and normal serum.
These findings support the proposition that in qui-nine- and quinidine-induced thrombocytopenia, drugand antibody combine first in the soluble phase to forma complex, which then binds with high affinity to areceptor on the platelet surface (innocent bystanderreaction), and demonstrate that these antibodies are
A preliminary report was presented at the VlIIth Inter-national Congress on Thrombosis and Haemostasis, Toronto,Canada, 1981. Thromb. Haemostasis. 46: 296.
Received for publication 13 January 1982 and in revisedform 30 June 1982.
heterogeneous in respect to the amount of drug re-quired to promote their binding to platelets, the num-ber of platelet receptors they recognize, and theirbinding affinities.
INTRODUCTION
More than 50 different medications are capable of trig-gering immunologic destruction of platelets in man(1). Of these various medications the stereoisomersquinine and quinidine appear to provoke this disordermost commonly. Antibodies associated with this phe-nomenon generally bind to platelets only in the pres-ence of drug, but the molecular basis of the drug-an-tibody-platelet interaction (DAP)l has not been fullycharacterized. Two mechanisms have been suggestedto explain the etiology of drug-induced immunologicthrombocytopenia. Ackroyd (2) proposed that drug,acting as a hapten, modifies some platelet constituentto produce an antigen that stimulates antibody pro-duction. Subsequent challenge of a sensitized individ-ual by the same drug would lead to drug-coated plate-lets that bind antibody and are destroyed. An alternatehypothesis developed independently by Miescher andMiescher (3) and Shulman (4) is that the antigen isformed by drug modification of some body constituentunrelated to platelets and that rechallenge with druginduces antibodies that combine first with drug to formsoluble immune complexes and then to platelets by amechanism not yet understood. The latter hypothesishas been termed "innocent bystander" (5) because theplatelet itself is not involved in formation of the pri-mary antigen. Shulman (4, 6) provided experimentalsupport for the latter proposal by showing that quin-
' Abbreviations used in this paper: DAP, drug-antibody-platelet interaction or complex; ddAb, drug-dependent an-tibody; [3H]Qn, tritiated quinine; PAIgG, platelet-associatedIgG; TX-100, Triton X-100; VIIIR:Ag, Factor VIII-relatedantigen; vWd, von Willebrand's disease.
J. Clin. Invest. © The American Society for Clinical Investigation, Inc. * 0021-9738/82/11/0989/10 $1.00Volume 70 November 1982 989-998
989

idine binds only weakly to the platelet surface and istherefore unlikely to form an antigenic hapten-carriercomplex and that high concentrations of quinidine failto inhibit the binding of antibody to platelets, as wouldbe expected in a classic hapten-antibody reaction.
Recently, Hosseinzadeh et al. (7) provided new ev-idence to support Ackroyd's hypothesis (2) by showingthat lymphocytes from patients who had experiencedquinine- or quinidine-induced thrombocytopenia couldbe stimulated by platelets previously exposed to drugin the presence of plasma and then washed, but notby drug or platelets alone. They suggested that thestimulating antigen was formed by drug complexedto the platelet membrane with the aid of a plasmacofactor. Further complexity has been introduced bythe finding of Pfueller et al. (8) that quinine- and quin-idine-dependent antibodies fail to induce platelet im-munoinjury in plasma lacking Factor VIII-related an-tigen (VIIIR:Ag), which suggests that the Factor VIIIcomplex is involved in DAP formation.
Hundreds of different medications appear to be ca-pable of inducing tissue injury by immunologic means(9-11) and it seems possible that better characteriza-tion of the mechanism(s) of drug-induced immuno-logic thrombocytopenia may provide clues to thepathogenesis of a broader spectrum of disease. Wehave therefore studied the interaction of platelet, drug,and drug-dependent antibody (ddAb), using tritium-labeled drug and a direct method for the determina-tion of platelet-associated IgG (PAIgG).
METHODSChemicals. Quinidine hydrochloride and bovine serum
albumin (Fraction V) were from Sigma Chemical Co. (St.Louis, MO), and quinine hydrochloride was from AldrichChemical Co. (Milwaukee, WI). Sodium 51Cr with specificactivity of 200-500 Ci/g (for platelet labeling) was fromAmersham-Searle Corp. (Arlington Heights, IL). DEAE-Se-phadex A50 was from Pharmacia Fine Chemicals (Piscata-way, NJ). Agarose (Standard Low-Mr), Triton X-100 (TX-100), and Coomassie Blue R were from Bio-Rad Laboratories(Richmond, CA). Gel supports were made from Gel-Bond(Marine Colloids, Inc., Rockland, ME). Complete CountingCocktail 3a20 was from Research Products InternationalCorp. (Elk Grove, IL). All other chemicals were standardanalytical grade reagents.
Patients. Four patients fulfilling the clinical and labo-ratory diagnostic criteria of drug-induced immunologicthrombocytopenia were studied (1). Each developed pro-found thrombocytopenia after courses of therapy rangingfrom 1 to 4 wk and recovered within 3 to 7 d after theoffending drug was discontinued. Two possessed quinine-dependent antibodies (I.L. and R.S.) and two quinidine-de-pendent antibodies (D.D. and M.S.). Each antibody wasreadily detectable by complement-mediated lysis of 51Cr-tagged platelets (12) and indirect immunofluorescence usinganti-IgG (13) and reacted with platelets only in the presenceof drug.
Platelet preparation. Blood drawn from normal healthy
volunteers and patients was anticoagulated with eitherEDTA or sodium citrate at final concentrations of 8 and 30mM, respectively. Platelet-rich plasma was prepared by dif-ferential centrifugation, as previously described (12). Insome experiments, von Willebrand's disease (vWd) and nor-mal platelets frozen in dimethylsulfoxide were used (14).Platelets were isolated from blood of a patient with the Ber-nard-Soulier syndrome (kindly supplied by Dr. MargaretJohnson, Wilmington, DE), as previously described (15, 16).Platelet concentration was determined by phase microscopy.Protein concentrations of platelets solubilized in TX-100were determined by the method of Markwell et al. (17).Unless otherwise stated, experiments were performed withplatelets pooled from at least three normal group 0 donors(PAIgG levels 4.0±1.1 fg/platelet) out of a group of sevenindividuals. EDTAwas present at 7.6 mMin all experiments.
Electroimmunoassay for PAIgG. Serum or plasma wasincubated with 1.5-3.0 X 108 buffer-washed or unwashedplatelets in the presence of the appropriate drug or saline.In preliminary experiments it was determined that maxi-mumbinding of antibody to platelets occurred in <20 minat 22°C with gentle agitation. It was also found that plateletswashed several times in buffer bound - 50-60% more ddAbthan unwashed platelets. Except where otherwise noted, un-washed platelets were used in the experimental studies. Afterthe incubation, platelets were washed twice at 22°C inEDTA, phosphate-buffered saline (EDTA-PBS) (7.6 mM),pH 7.3, containing a concentration of drug equal to thatused in the initial incubation, except where otherwise noted.Two washes were found to be sufficient to remove all looselybound IgG and PAIgG values were unchanged by up to fouradditional washes. Except where otherwise indicated, twowashes were used before determining all PAIgG values.
Determination of PAIgG was accomplished by an elec-troimmunoassay method described elsewhere by Kunickiand Aster (18). Platelets previously incubated with ddAb andthen washed twice were solubilized by agitation in 0.5% TX-100 in 0.1 M sodium acetate, pH 5.0, at 4°C for 30 min.Solubilized samples of 10-45 Al were loaded into wells of a1% agarose gel containing a predetermined amount of car-bamoylated rabbit IgG specific for the gamma chains ofhuman IgG. Carbamoylation lowered the pl of the rabbitIgG from -7 to 5, preventing its migration during electro-phoresis (19). Gels were electrophoresed at 2 V/cm for 18h at 15°C in 0.1 Msodium acetate, pH 5.0, washed in saline,dried, and stained with 0.5% (wt/vol) Coomassie Blue R in45% ethanol/10% acetic acid. Destaining was carried out in45% ethanol/10% acetic acid. The resulting peak heights ofthe immunoprecipitates were directly proportional to thetotal amount of PAIgG present in each sample. A linearstandard curve was obtained using known quantities of pu-rified human IgG in 10% (wt/vol) bovine serum albumin.PAIgG of platelets incubated in seven different normalplasma samples in the presence or absence of drug or in thefour ddAb-containing plasmas in the absence of drug rangedfrom 10,000 to 20,000 molecules (2.5-5.0 fg)/platelet, withinthe normal range reported by Kunicki and Aster (18) andothers (20, 21). The term "ddAb", as used in Figs. 1, 2, 3and Table I refers to the net PAIgG determined by sub-tracting control PAIgG (measured in the absence of drug)from total PAIgG (measured in the presence of drug). Theterm "bound IgG", as used in Figs. 4 and 5, refers to totalPAIgG.
Measurement of VIIIR:Ag. VIIIR:Ag in platelets andplasma was determined with a modification of the quanti-tative immunodiffusion assay reported by Glode et al. (22),in which plasma and platelets (solubilized in 0.1% TX-100)
990 D. J. Christie and R. H. Aster

are electrophoresed into 0.9% agarose gels containing 1251_rabbit antihuman VIIIR:Ag antibody. The resulting rocketswere detected by autoradiography.
Preparation of ddAb free of VIIIR:Ag. Plasma sampleswere made essentially free of VIIIR:Ag by a modificationof the technique of Zimmerman et al. (23). Briefly, sampleswere treated with DEAE-Sephadex A50 beads in 0.005 MKH2PO4, 0.005 M K2HPO4, pH 8.0. Beads packed by cen-trifugation at 12,800g for 2 min in an Eppendorf MicroCentrifuge (Brinkmann Instruments, Inc., Westbury, NY),after removal of buffer, were mixed with plasma containingddAb at a ratio of 1 ml beads (unpacked volume) to 0.5 mlplasma and rotated gently for 15 min at ambient tempera-ture. The mixture was centrifuged at 12,800g for 2 min.The supernatant contained levels of VIIIR:Ag ranging from0.1 to 0.0001% (limit of detection) of that present in thestarting plasma. Levels of IgA and IgM were <20% of thosein starting plasma, as measured by radial immunodiffusion(Helena Laboratories, Beaumont, TX), whereas IgG levelswere not significantly reduced.
Preparation and use of tritiated quinine ([3H]Qn). Qui-nine hydrochloride was submitted to New England Nuclear(Boston, MA) for catalytic hydrogen exchange with carrier-free tritium oxide. The tritiated product was purified bypreparative thin-layer chromatography on silica gel 60 F254S(MCB Reagents, E. Merck, Darmstadt, Cincinnati, OH) witha solvent system of chloroform:triethylamine:ethanol (90:5:5)(24). The tritiated drug, localized by fluorescence, was elutedfrom the gel in absolute ethanol, evaporated to dryness usinga Buchi Rotavapor-R (Brinkmann Instruments) and recon-stituted in distilled water. Material prepared in this way wasstored at 4°C for up to several weeks. Periodic analysis forracemization by analytical thin-layer chromatography al-ways showed tritium comigrating only with nonradiolabeledquinine and distinct from quinidine. The solvent system usedseparates all of the common sterioisomers of quinine (24).
The molar absorptivity of quinine hydrochloride was mea-sured in water and found to be 4,700 M-1 cm-' at a wave-
go'0_
.04
a
0
E
.0
49
m
length of 330 nm. Concentrations of [3H]Qn (optical spec-trum identical to quinine hydrochloride) were determinedspectrophotometrically using this value. The specific activityof purified [3H]Qn was 10 mCi/mmol.
Tritium bound to platelets was assayed after solubilizationin 3a20:TX-100 (2:1) in an Isocap/300 6872 liquid scintil-lation system (Searle Radiographics, Des Plaines, IL).
RESULTS
Binding of antibody to platelets as a function ofdrug concentration. Platelets were incubated withddAb in the presence of quinine or quinidine to de-termine the relationship between drug concentrationand antibody binding. As shown in Fig. 1 and TableI, the four antibodies required different amounts ofdrug to promote attachment of IgG to platelets. Underconditions described in the legend to Fig. 1, theamount of drug required to produce one-half maxi-mumddAb binding per platelet ranged from 2 (M.S.plus quinidine) to 60 AM (I.L. plus quinine).
Maximum number of platelet receptors for drug-dependent antibody. Using drug concentrations de-termined to be sufficient for maximum antibody bind-ing (Fig. 1), we incubated platelets with increasingamounts of plasma containing ddAb to determine themaximum amount of antibody each was capable ofattaching to platelets. As shown in Fig. 2 and TableI, the number of antibody binding sites measured inthis way ranged from 36,000 (M.S. plus quinidine) to161,000/platelet (I.L. plus quinine). To determinewhether variability in the amount of IgG bound to
00-F
80l
601
401
201
0.01 0J
[Drug] (mm)
FIGURE 1 Binding of ddAb to platelets at various drug concentrations. Plasma from patientsI.L., 1.0 ml (-); R.S., 1.0 ml (U); D.D., 1.0 ml (-); and M.S., 2.0 ml (X) were incubated with3 X 108 platelets in the presence of quinine ( ) or quinidine (--- -). After incubation for20 min at 22°C, the platelets were washed twice in EDTA-PBS containing 0.5 mMdrug.Platelet-bound ddAb was calculated as described in Methods. Data points are the average ofduplicate determinations for a single experiment. Results shown are representative of two
separate experiments performed with each antibody, each of which yielded essentially identicalresults.
Quinine- and Quinidine-induced Thrombocytopenia
I
991
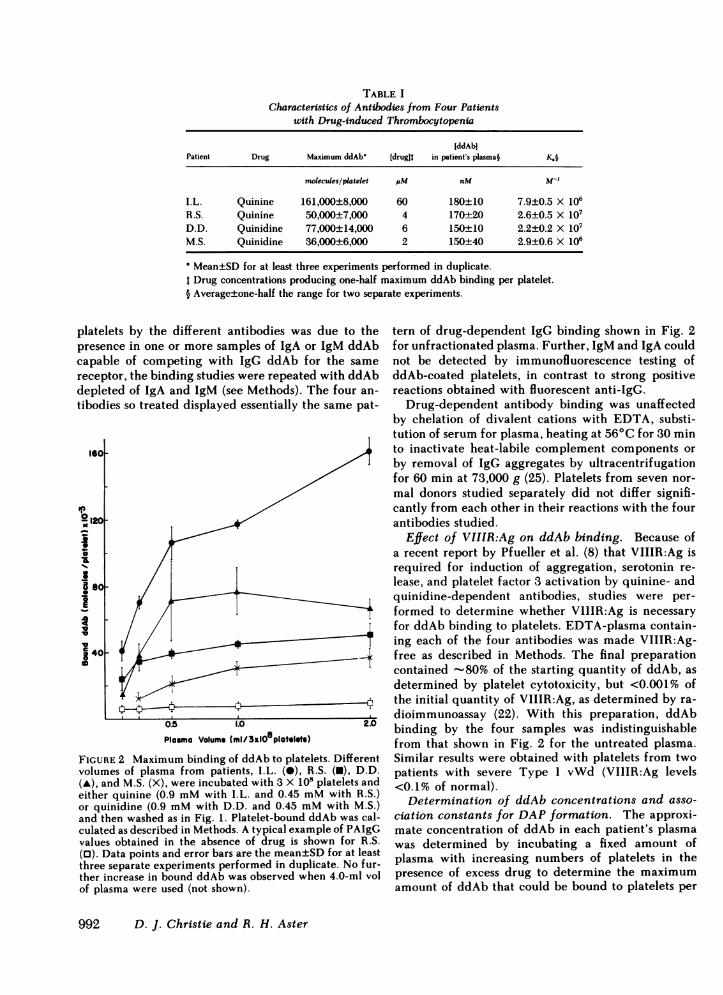
TABLE ICharacteristics of Antibodies from Four Patients
with Drug-induced Thrombocytopenia
IddAbiPatient Drug Maximum ddAb' [drugit in patient's plasma§ K.§
molecules/platelet piM nM M-l
I.L. Quinine 161,000±8,000 60 180±10 7.9±0.5 X 106R.S. Quinine 50,000±7,000 4 170±20 2.6±0.5 X 107D.D. Quinidine 77,000±14,000 6 150±10 2.2±0.2 X 107M.S. Quinidine 36,000±6,000 2 150±40 2.9±0.6 X 106
Mean±SD for at least three experiments performed in duplicate.Drug concentrations producing one-half maximum ddAb binding per platelet.
§ Average±one-half the range for two separate experiments.
platelets by the diff erent antibodies was due to thepresence in one or more samples of IgA or IgM ddAbcapable of competing with IgG ddAb for the samereceptor, the binding studies were repeated with ddAbdepleted of IgA and IgM (see Methods). The four an-tibodies so treated displayed essentially the same pat-
1601
ob 120
ID06-120K
la
la
j 400
O5 1.0 YUPlasma Volume (ml/3xlO8platelets)
FIGURE 2 Maximum binding of ddAb to platelets. Differentvolumes of plasma from patients, I.L. (0), R.S. (U), D.D.(A), and M.S. (X), were incubated with 3 X 108 platelets andeither quinine (0.9 mMwith I.L. and 0.45 mMwith R.S.)or quinidine (0.9 mMwith D.D. and 0.45 mMwith M.S.)and then washed as in Fig. 1. Platelet-bound ddAb was cal-culated as described in Methods. A typical example of PAIgGvalues obtained in the absence of drug is shown for R.S.(0). Data points and error bars are the mean±SD for at leastthree separate experiments performed in duplicate. No fur-ther increase in bound ddAb was observed when 4.0-ml volof plasma were used (not shown).
tern of drug-dependent IgG binding shown in Fig. 2for unfractionated plasma. Further, IgM and IgA couldnot be detected by immunofluorescence testing ofddAb-coated platelets, in contrast to strong positivereactions obtained with fluorescent anti-IgG.
Drug-dependent antibody binding was unaffectedby chelation of divalent cations with EDTA, substi-tution of serum for plasma, heating at 56°C for 30 minto inactivate heat-labile complement components orby removal of IgG aggregates by ultracentrifugationfor 60 min at 73,000 g (25). Platelets from seven nor-mal donors studied separately did not differ signifi-cantly from each other in their reactions with the fourantibodies studied.
Effect of VIIIR:Ag on ddAb binding. Because ofa recent report by Pfueller et al. (8) that VIIIR:Ag isrequired for induction of aggregation, serotonin re-lease, and platelet factor 3 activation by quinine- andquinidine-dependent antibodies, studies were per-formed to determine whether VIIIR:Ag is necessaryfor ddAb binding to platelets. EDTA-plasma contain-ing each of the four antibodies was made VIIIR:Ag-free as described in Methods. The final preparationcontained -80% of the starting quantity of ddAb, asdetermined by platelet cytotoxicity, but <0.001% ofthe initial quantity of VIIIR:Ag, as determined by ra-dioimmunoassay (22). With this preparation, ddAbbinding by the four samples was indistinguishablefrom that shown in Fig. 2 for the untreated plasma.Similar results were obtained with platelets from twopatients with severe Type I vWd (VIIIR:Ag levels<0.1% of normal).
Determination of ddAb concentrations and asso-ciation constants for DAPformation. The approxi-mate concentration of ddAb in each patient's plasmawas determined by incubating a fixed amount ofplasma with increasing numbers of platelets in thepresence of excess drug to determine the maximumamount of ddAb that could be bound to platelets per
992 D. J. Christie and R. H. Aster

milliliter of plasma as shown for antibody R.S. in Fig.3. Little difference in ddAb concentration was foundin the plasma of the four patients studied (Table I).
The Ka for DAP formation was calculated from astandard equilibrium expression for antibody bindingto a cell receptor, on the assumption that the equilib-rium between free antibody and antibody bound toplatelets is not significantly influenced by excess drug.This assumption is reasonable because a study by Shul-man (6) and our own findings described below indicatethat excess drug promotes, rather than inhibits, anti-body binding. On the assumption that the stoichiom-etry of binding between antibody and platelet receptoris 1:1, at the point where half of the antibody is bound(see arrow, Fig. 3)/the reciprocal of the molar con-centration of free platelet receptor sites provides anestimate of the Ka for DAP formation. The four an-tibodies studied differed significantly in their Ka values(Table I): the Ka for R.S. and D.D. were an order ofmagnitude greater than the Ka for M.S., and approx-imately three times higher than the Ka for I.L. Thehigh end of our Ka.range of 0.29-2.6 X 107 M` is closeto the value of 4 Xi07 M-' estimated by Shulman (4)for a single quinidine-dependent antibody using a dif-ferent experimental approach.
Cross-reactivity with quinine and quinidine. Thequinine-dependent antibody R.S. differed from theothers studied in that quinidine also promoted bindingof IgG to platelets and release of 5"Cr from taggedplatelets. No IgG binding or 51Cr release was observedwith the other three antibodies using the stereoisomerof the compound to which the patients were sensitized.
Effect of washing in the presence and absence ofdrug on the binding of antibody to platelets. Plate-
lets were incubated with either quinine or quinidinein the presence of quinine- and quinidine-dependentantibodies, respectively, and then washed in eitherEDTA-PBS or EDTA-PBS containing the same drugat a concentration of 1 mM, a 3,000-fold molar excessof drug:antibody. As shown in Fig. 4 for antibody I.L.,significantly more IgG was retained by platelets whenthey were washed in the presence of drug than in itsabsence. After four washes, 72,800±9,900 moleculesof IgG were bound per platelet when drug was in-cluded in wash solutions, but only 28,900±7,400 whendrug was absent (P < 0.001). It is apparent that excessdrug, rather than promoting dissociation of antibodyfrom platelets as would be expected in a classic hapten-antibody reaction (26), increases the affinity of anti-body for its receptor. Association of normal IgG withplatelets was not affected by the presence or absenceof drug in wash solutions (not shown).
Binding of quinine to platelets. To determinewhether platelets pretreated with drug retain signifi-cant quantities of drug after washing, 3 X 10' un-washed platelets were incubated with 0.4 mM[3H]Qnin 1.0 ml fresh normal plasma for 20 min at 37°Cunder the same conditions as shown in Fig. 5. Aftertwo washes, the platelet mixture retained 1.8±0.2X 10" (mean±SD, n = 3) drug molecules. This is >300times the amount expected on the basis of simple di-lution (5.4 X 10") calculated by using 3 IL as the totalvolume of the platelet pellet and 2.0 ml as the volumeper wash and represents a retention of 5.9±0.6 X 105drug molecules by each platelet. When platelets wereincubated with 0.4 mM[3H]Qn in' the absence ofplasma, 4.9±0.3 X 105 drug molecules were bound perplatelet.
80001
0-f
' 6000 _
4 _la 4000-la I-
2000 _
Q2 0.4 as 2.0 4.0
Platelets x 109/0.25ml plasma
FIGURE 3 Determination of total ddAb. Increasing numbers of platelets were incubated with0.25 ml of plasma from R.S. in 0.45 mMquinine, then washed in EDTA-PBS containing 0.45mMquinine. Bound ddAb was determined as described in Methods. Results shown are theaverage of two experiments. The arrow indicates the point of half-saturation used to determinethe Ka for complex formation of drug, antibody, and platelet receptor site (see text).
Quinine- and Quinidine-induced Thrombocytopenia 993

0
100 I
0
x
S
S
aa
S
o
S
E0
op
CL
20
a
80 1
601-
40 F
20F
2 3
Washes
FIGURE 4 Effect of washing platelets in the presence andabsence of drug on antibody binding. Plasma aliquots of 0.5ml from patient I.L., containing a quinine-dependent anti-body, were incubated with 1.5 X 108 platelets and 0.9 mMquinine for 20 min at 22°C. Samples were then washed two,three, or four times in either EDTA-PBS containing 1.0 mMquinine (@) or in EDTA-PBSalone (0). Data shown are fromtwo separate experiments where the lines connect the av-erages (two and three washes) and means (four washes) ofthe various sets of points for each wash. Similar results wereobtained with antibodies from R.S. and M.S.
Incubation of antibody with platelets pretreatedwith drug. Hosseinzadeh et al. (7) have reported thatplatelets preincubated with quinine and quinidine inthe presence of plasma and then washed are capableof inducing proliferation of lymphocytes from personswith quinine- and quinidine-induced thrombocyto-penia. The following studies were performed to de-termine whether platelets pretreated with drugs arecapable of binding ddAb. Platelets were pretreatedwith saline or quinine at various concentrations in thepresence and absence of autologous plasma. The drug-and saline-treated platelets were then washed threetimes in EDTA-PBSand tested for their ability to bindddAb. As shown in Fig. 5, platelets preincubated withquinine in plasma failed to bind ddAb unless addi-tional soluble drug was added. The same results (notshown) were obtained when autologous plasma wasomitted or when only two washes rather than threewere performed after the primary incubation. Resultssimilar to those shown in Fig. 5 for I.L. were also
obtained with plasma from R.S. Thus, platelets coatedin plasma with several hundred thousand quinine mol-ecules each (see above) by preincubation with the drug
* do not express receptors suitable for the attachmentof ddAb unless additional drug is added.
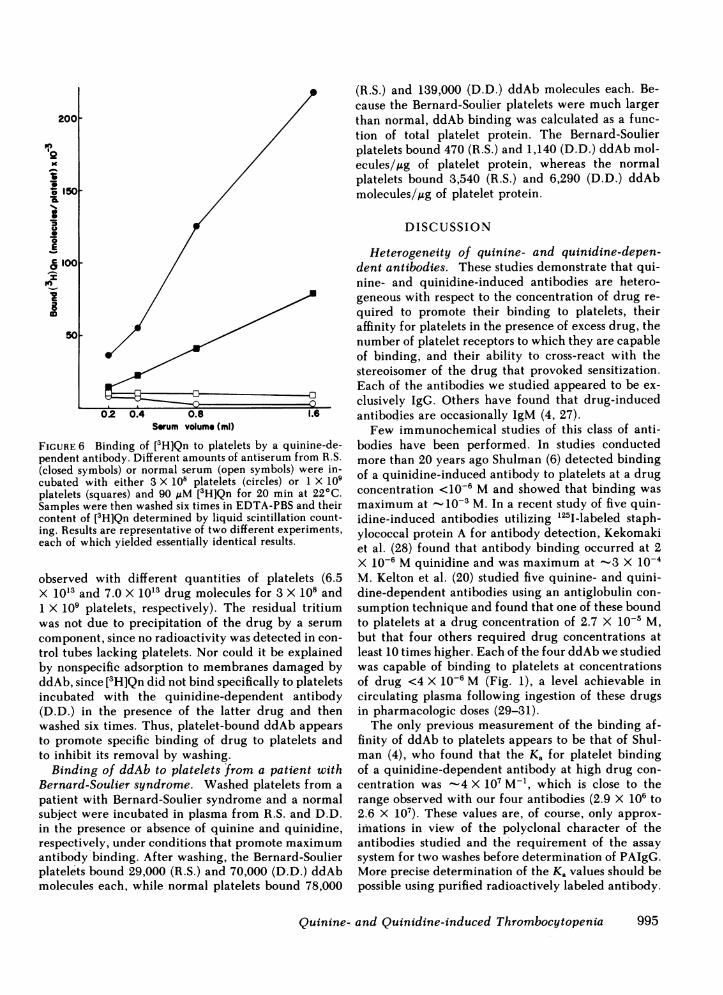
Antibody-dependent binding of quinine to plate-* lets. Whenantibody R.S. was incubated with platelets
>1 in the presence of 90 ,uM [3H]Qn and the antibody-coated platelets were then washed six times with
* buffer, significantly more quinine was retained by theplatelets than in controls using normal serum (Fig. 6).Because the six washes were performed in the absenceof added drug, we can estimate from Fig. 4 that at
0 least 75% of the platelet-bound ddAb was removed.00 On this assumption, the final estimated molar ratios
of platelet-bound drug:antibody would be 8:1 and00 17:1, respectively, for 1 X 109 and 3>X 108 platelets at
a serum volume of 1.6 ml. Binding of more drug percell with 3 X 108 than with 1 X 109 platelets presum-ably reflects the fact that antibody was no longer inexcess with the larger number of platelets. Nonspecifictrapping of quinine by aggregates of platelets is an
4 unlikely explanation for the greater binding of tritiumin the presence than in the absence of antibody becausedetectable clumping of the resuspended platelets didnot occur and the same degree of drug retention was
0
x~0Ke
6
6
aUe00
Ea
0
S
0
so
40
30
20
10
F F Fsaline 0.1
I0.3 1.0 6.0
Quinine concentration inprimary incubation (mM)
FIGURE 5 Binding of antibody to platelets pretreated withdrug. 3 X 10' unwashed normal platelets in 0.45 ml of au-tologous plasma were preincubated with varying amountsof quinine, as indicated along the abscissa, for 20 min at37°C with stirring. Platelets were then washed three timesin EDTA-PBS and incubated with 0.5 ml of plasma fromI.L. either in the presence (hatched bars) or absence (openbars) of 0.45 mMquinine as described in Fig. 1. All sampleswere then washed twice in EDTA-PBS. Data shown are rep-resentative of three experiments.
994 D. J. Christie and R. H. Aster
_2
o150-
...
8100-
50-
02 0.4 0.8 1.6Serum volume (mI)
FIGURE 6 Binding of [3H]Qn to platelets by a quinine-de-pendent antibody. Different amounts of antiserum from R.S.(closed symbols) or normal serum (open symbols) were in-cubated with either 3 X 108 platelets (circles) or 1 X 109platelets (squares) and 90 zIM [3H]Qn for 20 min at 22°C.Samples were then washed six times in EDTA-PBSand theircontent of [3H]Qn determined by liquid scintillation count-ing. Results are representative of two different experiments,each of which yielded essentially identical results.
observed with different quantities of platelets (6.5X 10'3 and 7.0 X 10'3 drug molecules for 3 X 10' and1 X 109 platelets, respectively). The residual tritiumwas not due to precipitation of the drug by a serumcomponent, since no radioactivity was detected in con-trol tubes lacking platelets. Nor could it be explainedby nonspecific adsorption to membranes damaged byddAb, since [3H]Qn did not bind specifically to plateletsincubated with the quinidine-dependent antibody(D.D.) in the presence of the latter drug and thenwashed six times. Thus, platelet-bound ddAb appearsto promote specific binding of drug to platelets andto inhibit its removal by washing.
Binding of ddAb to platelets from a patient withBernard-Soulier syndrome. Washed platelets from apatient with Bernard-Soulier syndrome and a normalsubject were incubated in plasma from R.S. and D.D.in the presence or absence of quinine and quinidine,respectively, under conditions that promote maximumantibody binding. After washing, the Bernard-Soulierplatelets bound 29,000 (R.S.) and 70,000 (D.D.) ddAbmolecules each, while normal platelets bound 78,000
(R.S.) and 139,000 (D.D.) ddAb molecules each. Be-cause the Bernard-Soulier platelets were much largerthan normal, ddAb binding was calculated as a func-tion of total platelet protein. The Bernard-Soulierplatelets bound 470 (R.S.) and 1,140 (D.D.) ddAb mol-ecules/,ug of platelet protein, whereas the normalplatelets bound 3,540 (R.S.) and 6,290 (D.D.) ddAbmolecules/,ug of platelet protein.
DISCUSSION
Heterogeneity of quinine- and quinidine-depen-dent antibodies. These studies demonstrate that qui-nine- and quinidine-induced antibodies are hetero-geneous with respect to the concentration of drug re-quired to promote their binding to platelets, theiraffinity for platelets in the presence of excess drug, thenumber of platelet receptors to which they are capableof binding, and their ability to cross-react with thestereoisomer of the drug that provoked sensitization.Each of the antibodies we studied appeared to be ex-clusively IgG. Others have found that drug-inducedantibodies are occasionally IgM (4, 27).
Few immunochemical studies of this class of anti-bodies have been performed. In studies conductedmore than 20 years ago Shulman (6) detected bindingof a quinidine-induced antibody to platelets at a drugconcentration <10-6 M and showed that binding wasmaximum at '10-1 M. In a recent study of five quin-idine-induced antibodies utilizing '25I-labeled staph-ylococcal protein A for antibody detection, Kekomakiet al. (28) found that antibody binding occurred at 2X 10-6 Mquinidine and was maximum at -3 X 10-4M. Kelton et al. (20) studied five quinine- and quini-dine-dependent antibodies using an antiglobulin con-sumption technique and found that one of these boundto platelets at a drug concentration of 2.7 X 10-' M,but that four others required drug concentrations atleast 10 times higher. Each of the four ddAb we studiedwas capable of binding to platelets at concentrationsof drug <4 X 10-6 M (Fig. 1), a level achievable incirculating plasma following ingestion of these drugsin pharmacologic doses (29-31).
The only previous measurement of the binding af-finity of ddAb to platelets appears to be that of Shul-man (4), who found that the Ka for platelet bindingof a quinidine-dependent antibody at high drug con-centration was -4 X 107 M-', which is close to therange observed with our four antibodies (2.9 X 106 to2;6 X 107). These values are, of course, only approx-imations in view of the polyclonal character of theantibodies studied and the requirement of the assaysystem for two washes before determination of PAIgG.More precise determination of the Ka values should bepossible using purified radioactively labeled antibody.
Quinine- and Quinidine-induced Thrombocytopenia 995

The variability in the number of platelet receptorsfor ddAb, ranging from 36,000 (M.S.) to 161,000(I.L.)/platelet, was unexpected in the light of our pre-vious finding that the primary receptor for quinine-and quinidine-induced antibodies appears to be situ-ated on glycoprotein Ib, which is not known to varysignificantly in freshly isolated platelets of normal sub-jects (16). On the basis of total platelet protein, anti-bodies R.S. and D.D. bound only 13 and 18% as well,respectively, to Bernard-Soulier platelets as to normalplatelets in the presence of drug. This is consistent withour previous finding that Bernard-Soulier platelets lackor have reduced levels of the receptor for quinine- andquinidine-dependent antibodies (16). In a recent re-port, Kelton et al. (20) found that three quinidine-de-pendent antibodies bound between 40 and 69 fg ofIgG/platelet at a quinidine concentration of 10-3 M,and that one quinine-dependent antibody inducedbinding of 15 fg of IgG/platelet at 10-3 Mquinine. Ifone assumes a molecular weight for IgG of 150,000and subtracts the mnaximum value of 10 fg obtainedby Kelton et al. (20) for normal PAIgG, this translatesinto a range of 20,000 to 236,000 ddAb binding sites/platelet. Although it is not clear whether antibody wasin excess in Kelton's study, this range for antibodybinding is comparable to the one we observed (36,000-161,000) (Table I). Our studies of platelets from dif-ferent normal donors showed that this variability is aproperty of the antibodies themselves and not theplatelets. Low values of IgG attached to platelets byM.S. may well have been related to its relatively lowKa. However, antibody I.L. bound more than twiceas much IgG per platelet as R.S. and D.D., despitehaving a lower Ka than the latter two antibodies andapproximately the same concentration. Thus, our find-ings suggest that these antibodies differ in their spec-ificities for receptor(s) on the platelet surface. Hos-seinzadeh et al. (32) also described differences in thebinding of quinine- and quinidine-dependent anti-bodies to platelet membrane proteins isolated by gelelectrophoresis in a recent preliminary report. Thebasis for this variability is a subject of current inves-tigation.
One of our antibodies (R.S.) also bound to plateletsin the presence of the stereoisomer (quinidine) towhich the patient was originally sensitized (quinine).This cross-reactivity appeared to be a property of asingle antibody, since all reactivity with quinidinecould be adsorbed with platelets in the presence ofquinine. Kelton et al. (20), studying eight quinine- andquinidine-dependent antibodies, found that one qui-nine-dependent antibody reacted equally well withquinidine, whereas in their study of five quinidine-induced antibodies Kekomaki et al. (28) failed to de-tect any cross-reactivity with quinine. Shulman found
that 20% of an unspecified number of quinine- andquinidine-induced antibodies cross-reacted with theappropriate stereoisomer (33).
Role of the Factor VIII complex in binding of ddAbto platelets. As noted previously, Pfueller et al. (8),in studies of seven quinine- and quinidine-inducedantibodies, found that antibody activity as measuredby tests involving release of platelet factor 3, induc-tion of platelet aggregation, and serotonin releasecould not be detected with citrated platelet-richplasma from patients with severe vWd. On this basis,they suggested that the Factor VIII complex, knownto be lacking in plasma of patients with vWd, is re-quired for the interaction of ddAb with platelets. Ourstudies demonstrate that ddAb binds normally to plate-lets from normal subjects and from patients with vWdat levels of VIIIR:Ag <0.001% of normal and at un-detectable levels of VIII coagulant activity. Thesefindings, together with those of Pfueller et al. (8), sug-gest the interesting possibility that the Factor VIIIcomplex or one of its constituent parts is required forthe mediation of immunoinjury after antibody hasbeen affixed to the platelet membrane but not for thebinding of antibody to platelets per se. The additionalfinding by Pfueller et al. (8) that VIIIR:Ag is requiredfor drug-treated platelets to induce proliferation oflymphocytes from patients with drug-induced throm-bocytopenia is not necessarily in conflict with our ob-servations, since drug loosely bound to platelets afterthe two washes used by Pfueller might have directlystimulated T lymphocytes in their preparation. Clearly,additional studies are needed to elucidate the possiblerole of VIIIR:Ag in quinine- and quinidine-inducedthrombocytopenia.
Mechanism of ddAb-platelet binding. Shulman (6),in an elegant series of studies of the interaction of aquinidine-dependent antibody with platelets, and aidedby a theoretical analysis involving methods of statis-tical mechanics (34), concluded that his data did notpermit a distinction to be made between two alter-native models for DAP formation: one in which quin-idine attaches first to antibody and a second in whichquinidine attaches first to platelets. His study was lim-ited by the need to use quantitative complement fix-ation as an index of antibody levels. However, it waspostulated on the basis of the relatively weak bindingof quinidine alone. to platelets and the failure of highconcentrations of the drug to inhibit antibody bindingthat a drug-antibody complex was formed first andthat this complex subsequently became bound to aplatelet receptor (4). Our findings that concentrationsof drug -10,000 times greater than antibody on amolar basis promote, rather than inhibit, binding ofddAb to platelets (Fig. 1, Table I) and that reversalof ddAb binding by repeated washes is inhibited by
996 D. J. Christie and R. H. Aster

excess drug (Fig. 4) support this viewpoint. The ob-servation that platelets preincubated with drug at var-ious concentrations and then washed fail to bind de-tectable amounts of ddAb (Fig. 5) even under con-ditions where several hundred thousand drug moleculesremain attached to each platelet is also consistent withthis concept. The finding that platelets coated withddAb in the presence of tritiated quinine and thenwashed repeatedly retain significantly greater amountsof drug than platelets exposed to the drug in normalserum (Fig. 6), suggests the possibility that each mol-ecule of ddAb promotes retention of several drug mol-ecules on the platelet surface, perhaps where its Fabtermini, occupied by drug, bind to the putative plateletreceptor. An alternative explanation for DAP forma-tion not altogether ruled out by the available data isthat drug binds first to a site on the platelet to forman unstable neoantigen that is stabilized by the at-tachment of antibody. Additional studies are neededto distinguish between this possibility and binding viathe innocent bystander mechanism.
In vivo correlates. In a series of in vivo studies,Shulman (35) showed in a patient with a potent quin-idine-dependent antibody that platelet destructionsufficient to reduce platelet levels from 300,000 to77,000/,ul occurred at a plasma concentration of drugthat did not exceed 4 X 10-8 M. In a second patientwith a weaker antibody, infusion of a much largeramount of quinidine resulted only in a slight reductionof platelets to -200,000/,ul. Quinine and quinidineare rapidly adsorbed from the gastrointestinal tractand plasma levels of patients on conventional therapyreach 20 (31) and 14 ,M (29, 30), respectively. If oneassumes that antibody levels in our four patients at theonset of thrombocytopenia were approximately thesame as when samples were obtained for study a fewdays later, the number of IgG molecules bound toplatelets in vivo at 20 uM drug and at a circulatingplatelet concentration of 300,000 (I.L., R.S., D.D.) and150,000 (M.S.)/,ul can be estimated from Fig. 1: I.L.,28,000; R.S., 46,000; D.D., 61,000; and M.S., 31,000.These quantities of platelets would adsorb 10-35% ofthe antibody in the four patients studied. After theinitial destruction of platelets and reduction in plateletlevels, even greater numbers of IgG molecules mightbind to the remaining platelets, accelerating the rateof platelet clearance. On the basis of a single study,Shulman suggested that as few as 400 quinidine-de-pendent antibody molecules might suffice to induceplatelet destruction (4). Using serum from patient I.L.,we found that 5,000 IgG molecules became bound tonormal platelets at a quinine level of 2.7 X 10-8 M, afigure well below the therapeutic range. The relativelylarge number of quinine- and quinidine-induced ddAbmolecules that become bound to platelets under con-
ditions likely to exist in vivo in a sensitized patientwho ingests these drugs may explain the dramatic,sometimes catastrophic, bleeding symptoms mani-fested by such patients.
In view of the heterogeneity in binding affinity, levelof drug required to promote binding, and the numberof antibody binding sites per platelet displayed by theantibodies we studied, it seems permissable to specu-late that patients who develop profound thrombocy-topenia with hemorrhagic symptoms upon taking qui-nine or quinidine may constitute a minority of a muchlarger group of sensitized patients whose drug-inducedantibodies either have a low Ka or bind to plateletswell only at very high drug concentrations. Furtherstudies to test these possibilities seem warranted.
ACKNOWLEDGMENTS
Wethank Dr. Thomas Kunicki for his many helpful discus-sions throughout the course of this work and Dr. RobertMontgomery for his assistance in obtaining severe von Wil-lebrand's platelets and for performing the Factor VIII-re-lated antigen measurements. Wealso thank Kathleen Reedfor her excellent technical assistance and the word processingdepartment for the typing of this manuscript.
This work was supported by National Heart, Lung, and BloodInstitute grant HL-13629 and Fellowship HL06255-01.
REFERENCES
1. Aster, R. H. 1977. Thrombocytopenia due to enhancedplatelet destruction. In Hematology. W. J. Williams, E.Beutler, A. J. Erslev, and R. W. Rundles, editors.McGraw-Hill, Inc., New York. 2nd edition. 1326-1359.
2. Ackroyd, J. F. 1953. Allergic purpura, including purpuradue to foods, drugs and infections. Am. J. Med. 14: 605-632.
3. Miescher, P., and R. Miescher. 1952. Drie Sedormid-An-aphlaxie. Schweiz. Med. Wochenschr. 82: 1279-1282.
4. Shulman, N. R. 1963. Mechanism of blood cell damageby absorption of antigen-antibody complex. In Immu-nopathology, Third International Symposium. P. Grabarand P. A. Miescher, editors. Schwabe and Co., Basel.338-352.
5. Croft, J. D., Jr., and R. I. Weed. 1968. Coombs'-testpositive induced by drugs: mechanisms of immunologicreactions, and red cell destruction. Ann. Intern. Med.68: 176-187.
6. Shulman, N. R. 1958. Immunoreactions involving plate-lets. I. A steric and kinetic model for formation of acomplex from a human antibody, quinidine as a haptene,and platelets; and for fixation of complement by thecomplex. J. Exp. Med. 107: 665-690.
7. Hosseinzadeh, P. K., B. G. Firkin, and S. L. Pfueller.1980. Study of the factors that cause specific transfor-mation in cultures of lymphocytes from patients withquinine- and quinidine-induced immune thrombocyto-penia. J. Clin. Invest. 66: 638-645.
8. Pfueller, S. L., P. K. Hosseinzadeh, and B. G. Firkin.1981. Quinine- and quinidine-dependent antiplateletantibodies. Requirement of Factor VIII-related antigenfor platelet damage and for in vitro transformation of
Quinine- and Quinidine-induced Thrombocytopenia 997

lymphocytes from patients with drug-induced throm-bocytopenia. J. Clin. Invest. 67: 907-910.
9. Petz, L. D., and H. H. Fudenberg. 1975. Immunologicmechanisms in drug-induced cytopenia. Prog. Hematol.9: 185-206.
10. Petz, L. D. 1980. Drug-induced immune haemolyticanaemia. Clin. Haematol. 9: 455-482.
11. Young, G. A. R., and P. C. Vincent. 1980. Drug-inducedagranulocytosis. Clin. Haematol. 9: 483-504.
12. Aster, R. H., and S. E. Enright. 1969. A platelet andgranulocyte membrane defect in paroxysmal nocturnalhemoglobinuria. Usefulness for the detection of plateletantibodies. J. Clin. Invest. 48: 1190-1210.
13. Lalezari, P., and S. C. Pryce. 1980. Detection of neu-trophil and platelet antibodies in immunologically-in-duced neutropenia and thrombocytopenia. In Manualof Clinical Immunology. N. R. Rose and H. Friedman,editors. American Society for Microbiology, Washing-ton. 2nd edition. 744-748.
14. Lizak, G. E., and F. C. Grumet. 1979. Storage of reagentplatelets for antiplatelet antibody testing in the 51Crplatelet lysis assay. Clin. Pathol. 32: 191-192.
15. Nurden, A. T., D. Dupuis, T. J. Kunicki, and J. P. Caen.1981. Analysis of the glycoprotein and protein compo-sition of Bernard-Soulier platelets by single and two-di-mensional sodium dodecyl sulfate-polyacrylamide gelelectrophoresis. J. Clin. Invest. 67: 1431-1440.
16. Kunicki, T. J., M. M. Johnson, and R. H. Aster. 1978.Absence of the platelet receptor for drug-dependent an-tibodies in the Bernard-Soulier syndrome. J. Clin. In-vest. 62: 716-719.
17. Markwell, M. A. K., S. M. Haas, L. L. Bieber, andN. E. Tolbert. 1978. A modification of the Lowry pro-cedure to simplify protein determination in membraneand lipoprotein samples. Anal. Biochem. 87: 206-210.
18. Kunicki, T. J., and R. H. Aster. 1982. Direct quantitationof total platelet-associated IgG by electroimmunoassay.Blood. 60: 54-58.
19. Bjerrum, 0. J., A. Ingild, H. Lowenstein, and B. Weeke.1973. Carbamylated antibodies used for quantitation ofhuman IgG. A routine method. Scand. J. Immunol.2(Suppl. 1): 145-148.
20. Kelton, J. G., D. Meltzer, J. Moore, A. R. Giles, W. E.Wilson, J. Hirsh, P. B. Neame, P. J. Powers, I. Walker,F. Bianchi, and C. J. Carter. 1981. Drug-induced throm-bocytopenia is associated with increased binding of IgGto platelets both in vivo and in vitro. Blood. 58: 524-529.
21. Pfueller, S. L., L. Cosgrove, B. G. Firkin, and D. Tew.1981. Relationship of raised platelet IgG in thrombo-cytopenia to total platelet protein content. Br. J. Hae-matol. 49: 293-302.
22. Glode, L. M., R. R. Montgomery, C. G. Smith, and
D. B. Link. 1982. Use of monoclonal antibody to increasesensitivity and specificity in quantitative immunodif-fusion assays. J. Immunol. Methods 48: 13-22.
23. Zimmerman, T. S., J. Roberts, and T. S. Edgington. 1975.Factor VIII-related antigen: multiple molecular formsin human plasma. Proc. Natl. Acad. Sci. USA. 72: 5121-5125.
24. Uskokovic, M. R., and J. Gutszwiller. 1973. Reinvesti-gation of the classical synthesis of cinchona alkaloids. II.The synthesis of quinine and its naturally occurring dias-tereomers from quinotoxine. Helv. Chim. Acta. 56:1494-1503.
25. Sugiura, K., M. Steiner, and M. Baldini. 1981. Physio-logical effects of nonimmune platelet associated im-munoglobulin G. Thromb. Haemostasis. 45: 27-33.
26. Kabat, E. A. 1976. Antigenic determinants and the sizeof the antibody combining site: determinants of cell-mediated immunity. In Structural Concepts in Immu-nology and Immunochemistry. Holt, Reinhart and Win-ston, New York. 2nd edition. 119-166.
27. Eisner, E. V., and B. C. Korbitz. 1972. Quinine-inducedthrombocytopenic purpura due to an IgM and an IgGantibody. Transfusion (Phila.). 12: 317-321.
28. Kekomaki, R., A. Rajamaki, and G. Myllyla. 1980. De-tection of quinidine-specific antibodies with platelet 125I_labeled staphylococcal protein A test. Vox Sang. 38: 12-18.
29. Kessler, K. M., D. T. Lowenthal, H. Warner, T. Gibson,W. Briggs, and M. M. Reidenberg. 1974. Quinidine elim-ination in patients with congestive heart failure or poorrenal function. N. Engi. J. Med. 290: 706-709.
30. Hirschfeld, D. S., C. T. Ueda, M. Rowland, and M. M.Scheinman. 1977. Clinical and electrophysiological ef-fects of intravenous quinidine in man. Br. Heart J. 39:309-316.
31. Rollo, I. M. 1980. Drugs used in the chemotherapy ofmalaria. In The Pharmacological Basis of Therapeutics.L. S. Goodman and A. Gilman, editors. MacMillan Pub-lishing Co., Inc., New York. 1038-1060.
32. Hosseinzadeh, P., S. Pfueller, and B. G. Firkin. 1981.The recognition of two platelet antigens by quinine/quinidine antibodies. Clin. Res. 29: 335A.
33. Shulman, N. R. 1972. Immunologic reactions to drugs.N. Engl. J. Med. 287: 408-409.
34. Hill, T. L. 1958. Immunoreactions involving platelets.II. Theoretical analysis of the model. J. Exp. Med. 107:691-695.
35. Shulman, N. R. 1958. Immunoreactions involving plate-lets. IV. Studies on the pathogenesis of thrombocyto-penia in drug purpura using test doses of quinidine insensitized individuals. Their implications in idiopathicthrombocytopenia purpura. J. Exp. Med. 107: 711-729.
998 D. J. Christie and R. H. Aster