
Theoretische Physik IV:Thermodynamik und Statistik
Skript zur gleichnamigen VorlesungWintersemester 2006/2007 an der Universitat Wurzburg
Priv.-Doz. Dr. Michael Potthoff

2

Inhaltsverzeichnis
1 Grundbegriffe der Thermodynamik 111.1 System und Zustand . . . . . . . . . . . . . . . . . . . . . . 111.2 Temperatur . . . . . . . . . . . . . . . . . . . . . . . . . . . . 131.3 Einfache thermodynamische Systeme . . . . . . . . . . . . . 131.4 Thermodynamische Prozesse . . . . . . . . . . . . . . . . . . 15
2 Erster Hauptsatz 212.1 Arbeit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 212.2 Energieerhaltung . . . . . . . . . . . . . . . . . . . . . . . . . 222.3 Warmekapazitaten . . . . . . . . . . . . . . . . . . . . . . . . 252.4 Adiabaten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 272.5 Carnot-Prozess . . . . . . . . . . . . . . . . . . . . . . . . . . 28
3 Zweiter Hauptsatz 313.1 perpetuum mobile zweiter Art . . . . . . . . . . . . . . . . . 313.2 Irreversibilitat . . . . . . . . . . . . . . . . . . . . . . . . . . . 323.3 Thermodynamische Temperaturskala . . . . . . . . . . . . . 343.4 Entropie als Zustandsgroße . . . . . . . . . . . . . . . . . . . 393.5 Grundgleichung der Thermodynamik . . . . . . . . . . . . . 413.6 Entropie und Irreversibilitat . . . . . . . . . . . . . . . . . . 423.7 Grundrelation der Thermodynamik . . . . . . . . . . . . . . 47
4 Thermodynamische Potenziale 514.1 Potenziale fur Gase . . . . . . . . . . . . . . . . . . . . . . . 514.2 Stabilitatsbedingungen . . . . . . . . . . . . . . . . . . . . . 554.3 Gibbs-Duhem-Relation . . . . . . . . . . . . . . . . . . . . . 594.4 Freie Energie des klassischen idealen Gases . . . . . . . . . 614.5 Dritter Hauptsatz . . . . . . . . . . . . . . . . . . . . . . . . 654.6 Phasen und Komponenten . . . . . . . . . . . . . . . . . . . 674.7 Gibbssches Paradoxon . . . . . . . . . . . . . . . . . . . . . . 70
5 Elementare Statistik 835.1 Wahrscheinlichkeit . . . . . . . . . . . . . . . . . . . . . . . . 845.2 Shannon-Information . . . . . . . . . . . . . . . . . . . . . . 89
3

5.3 Zufallsvariable . . . . . . . . . . . . . . . . . . . . . . . . . . 945.4 Zentraler Grenzwertsatz . . . . . . . . . . . . . . . . . . . . 985.5 Binomialverteilung . . . . . . . . . . . . . . . . . . . . . . . . 1005.6 Stirling-Formel . . . . . . . . . . . . . . . . . . . . . . . . . . 102
6 Zufallsgroßen im Zustandsraum 1056.1 Klassische Mechanik, Phasenraum . . . . . . . . . . . . . . 1056.2 Gemischter Zustand, statistische Gesamtheit . . . . . . . . 1076.3 Liouville-Gleichung . . . . . . . . . . . . . . . . . . . . . . . . 1106.4 Quantenmechanik, Hilbert-Raum . . . . . . . . . . . . . . . 1136.5 Gemischter Zustand, Dichteoperator . . . . . . . . . . . . . 1156.6 Systeme identischer Teilchen . . . . . . . . . . . . . . . . . . 121
7 Thermodynamische Zustande und Prozesse 1317.1 Mikrokanonische Gesamtheit . . . . . . . . . . . . . . . . . . 1337.2 Mikrokanonische Gesamtheit mit festem E, V, N . . . . . . 1387.3 Quasistatische Parameteranderung . . . . . . . . . . . . . . 1427.4 Warme und erster Hauptsatz . . . . . . . . . . . . . . . . . 1467.5 Temperatur und Nullter Hauptsatz . . . . . . . . . . . . . . 1487.6 Entropie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
8 Einfache Anwendungen 1578.1 Gleichverteilungssatz . . . . . . . . . . . . . . . . . . . . . . 1578.2 Maxwellsche Geschwindigkeitsverteilung . . . . . . . . . . . 1608.3 Zustandsdichte eines idealen Gases . . . . . . . . . . . . . . 1638.4 Entropie eines klassischen idealen Gases . . . . . . . . . . . 1678.5 Sattelpunktsmethode . . . . . . . . . . . . . . . . . . . . . . 1698.6 Quantenmechanisches System harmonischer Oszillatoren . 171
9 Allgemeiner Aufbau der statistischen Mechanik 1819.1 Allgemeiner Kontakt . . . . . . . . . . . . . . . . . . . . . . 1819.2 Bestimmung der a priori-Wahrscheinlichkeiten . . . . . . . 1829.3 Zustandssumme . . . . . . . . . . . . . . . . . . . . . . . . . 1839.4 Thermodynamische Potenziale . . . . . . . . . . . . . . . . . 1869.5 Entropie und Information . . . . . . . . . . . . . . . . . . . . 1889.6 Quasistatische Prozesse . . . . . . . . . . . . . . . . . . . . . 1909.7 Fluktuationen . . . . . . . . . . . . . . . . . . . . . . . . . . . 1929.8 Allgemeiner Kontakt zwischen Systemen . . . . . . . . . . . 1959.9 Prinzip der Maximierung der Entropie . . . . . . . . . . . . 200
10 Kanonische und großkanonische Gesamtheit 20510.1 Kanonische Gesamtheit . . . . . . . . . . . . . . . . . . . . . 20510.2 Großkanonische Gesamtheit . . . . . . . . . . . . . . . . . . 20710.3 Klassisches ideales Gas . . . . . . . . . . . . . . . . . . . . . 209
4

10.4 Quantenmechanisches System harmonischer Oszillatoren . 21010.5 Dritter Hauptsatz . . . . . . . . . . . . . . . . . . . . . . . . 21210.6 Eindimensionales Ising-Modell, Transfermatrixmethode . . 21410.7 Heisenberg-Modell, Molekularfeldtheorie . . . . . . . . . . . 222
11 Ideale Quantengase 22911.1 Fermi- und Bose-Gas . . . . . . . . . . . . . . . . . . . . . . 22911.2 Fermi- und Bose-Verteilungsfunktion . . . . . . . . . . . . . 23211.3 Thermodynamik des idealen Fermi-Gases . . . . . . . . . . 23511.4 Klassischer Grenzfall . . . . . . . . . . . . . . . . . . . . . . . 23811.5 Entartetes Fermi-Gas . . . . . . . . . . . . . . . . . . . . . . 23911.6 Quasiteilchen . . . . . . . . . . . . . . . . . . . . . . . . . . . 24411.7 Phononen- und Photonengase . . . . . . . . . . . . . . . . . 24711.8 Phononen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25111.9 Massives Bose-Gas . . . . . . . . . . . . . . . . . . . . . . . 253
5

Theoretische Physik IV:
Thermodynamik und Statistik
• Thermodynamik
– Grundbegriffe der Thermodynamik
– Erster Hauptsatz
– Zweiter Hauptsatz
– Thermodynamische Potenziale
• Statistik
– Elementare Statistik
– Zufallsgroßen im Zustandsraum
– Thermodynamisches Gleichgewicht
– Einfache Anwendungen
– Statistische Gesamtheiten
– Entropie und Information
– Ideale Quantengase
– Approximative Methoden

Literaturauswahl
• E. Fermi: Thermodynamics (Dover)
• R. Becker: Theorie der Warme (Springer)
• W. Nolting: Grundkurs Theoretische Physik, Band 4 (Springer)
• W. Nolting: Grundkurs Theoretische Physik, Band 6 (Springer)
• W. Kinzel: Statistische Mechanik und Thermodynamik (Skript)
• F. Schwabl: Statistische Mechanik (Springer)
• C. Kittel: Physik der Warme (Wiley)
• B. Diu, C. Guthmann, D. Lederer, B. Roulet: Grundlagen der Stati-
stischen Physik (de Gruyter)
• W. Weidlich: Thermodynamik und statistische Mechanik (Akad.Verlagsges.)
• J. Honerkamp: Statistical Physics (Springer)
• S.-K. Ma: Statistical Mechanics (World Scientific)

klassische Mechanik Quantenmechanik
InformationstheorieWahrscheinlichkeitstheorie
PlasmaphysikAstrophysik
(Anwendung)
(Reduktion)
(Reduktion)
statistische Physik
Thermodynamikphänomenologische
mathematische Statistik
physikalische Chemie
Festkörperphysik
Hochenergiephysik
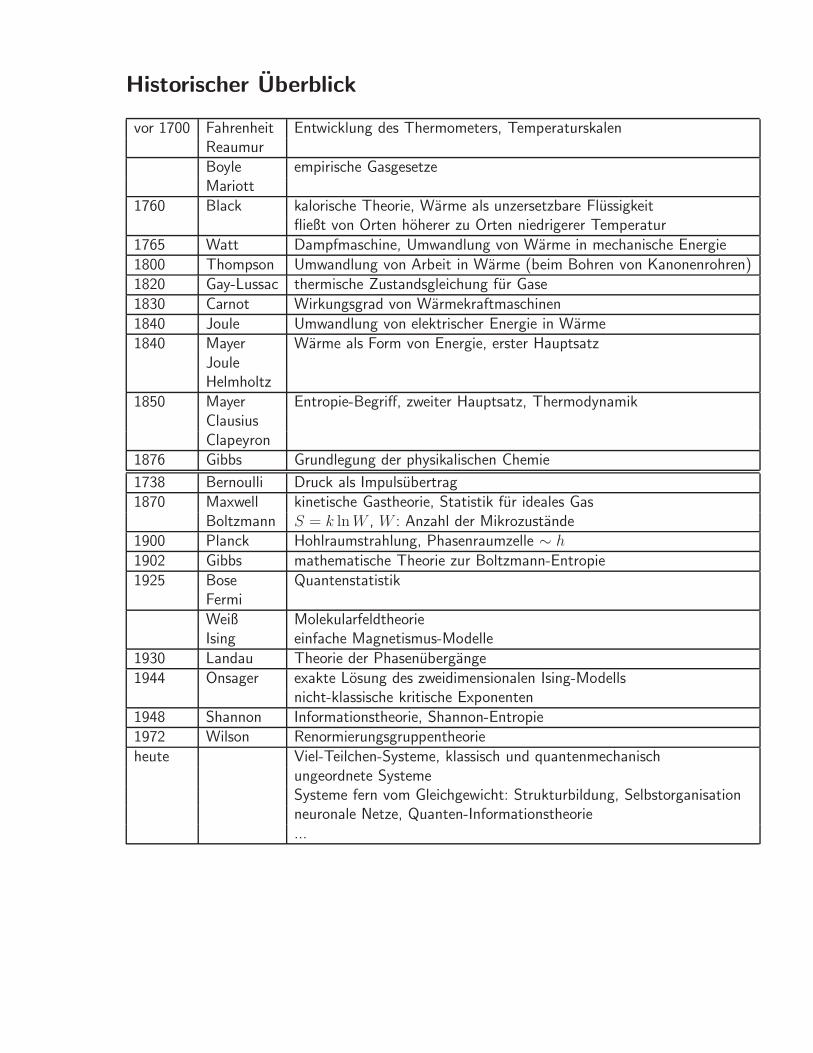
Historischer Uberblick
vor 1700 Fahrenheit Entwicklung des Thermometers, TemperaturskalenReaumurBoyle empirische GasgesetzeMariott
1760 Black kalorische Theorie, Warme als unzersetzbare Flussigkeitfließt von Orten hoherer zu Orten niedrigerer Temperatur
1765 Watt Dampfmaschine, Umwandlung von Warme in mechanische Energie1800 Thompson Umwandlung von Arbeit in Warme (beim Bohren von Kanonenrohren)1820 Gay-Lussac thermische Zustandsgleichung fur Gase1830 Carnot Wirkungsgrad von Warmekraftmaschinen1840 Joule Umwandlung von elektrischer Energie in Warme1840 Mayer Warme als Form von Energie, erster Hauptsatz
JouleHelmholtz
1850 Mayer Entropie-Begriff, zweiter Hauptsatz, ThermodynamikClausiusClapeyron
1876 Gibbs Grundlegung der physikalischen Chemie
1738 Bernoulli Druck als Impulsubertrag1870 Maxwell kinetische Gastheorie, Statistik fur ideales Gas
Boltzmann S = k ln W , W : Anzahl der Mikrozustande1900 Planck Hohlraumstrahlung, Phasenraumzelle ∼ h1902 Gibbs mathematische Theorie zur Boltzmann-Entropie1925 Bose Quantenstatistik
FermiWeiß MolekularfeldtheorieIsing einfache Magnetismus-Modelle
1930 Landau Theorie der Phasenubergange1944 Onsager exakte Losung des zweidimensionalen Ising-Modells
nicht-klassische kritische Exponenten1948 Shannon Informationstheorie, Shannon-Entropie1972 Wilson Renormierungsgruppentheorieheute Viel-Teilchen-Systeme, klassisch und quantenmechanisch
ungeordnete SystemeSysteme fern vom Gleichgewicht: Strukturbildung, Selbstorganisationneuronale Netze, Quanten-Informationstheorie...

10

Kapitel 1
Grundbegriffe derThermodynamik
1.1 System und Zustand
(Def.) thermodynamisches Systemjedes makroskopische System, d.h. N groß
(Def.) thermodynamischer Zustand, GleichgewichtszustandZustand des Systems, der sich (makroskopisch) nach langer Zeit bei zeit-lich festen außeren Bedingungen von selbst einstellt
Zeit spielt in der TD keine Rolle !
empirische Tatsache:
die makroskopischen Eigenschaften eines TD Systems im Gleichgewicht konnendurch wenige Großen vollstandig bestimmt werden
X1, ..., Xf vollstandiger Satz von Zustandsgroßen
X = X(X1, ..., Xf) fur jede weitere Zustandsgroße
X = X(X1, ..., Xf): Zustandsgleichung
X1, ..., Xf spannen Raum der (Gleichgewichts-)Zustande auf, Zustandsraum
f : Dimension
—————————————————————————————————
11

typische Zustandsgroßen:
Gas: p, V, N, T, U, S, µ
Gasgemisch: N1, N2, ..., µ1, µ2,...
Magnet: M, B
—————————————————————————————————
Zustand ⇔ X1, ..., Xf ⇔ Y1, ..., Yf (weiterer vollst. Satz)
Xi = Xi(Y1, ..., Yf) und Yj = Yj(X1, ..., Xf)
(eindeutige Auflosbarkeit wird stets angenommen)
weitere Zustandsgroßen:∂Xi
∂Yj=
∂Xi
∂Yj(Y1, ..., Yf)
Achtung:∂Xi
∂Yj
mehrdeutig !
Bsp: f = 2, und X, Y, Z, A Zustandsgroßen
A = A(X, Y ) aber auch A = A(X, Z)
und
∂A(X, Y )
∂X6= ∂A(X, Z)
∂X
daher Schreibweise:
∂A(X, Y )
∂X=
(∂A
∂X
)
Y
=∂A
∂X
∣∣∣∣∣∣Y
es gilt:(
∂A
∂X
)
Y
=
(∂A
∂X
)
Z
+
(∂A
∂Z
)
X
(∂Z
∂X
)
Y
—————————————————————————————————
Systeme sind homogen oder bestehen aus mehreren homogenen Phasen
Zustandsgleichungen sind charakteristisch fur jeweilige Phase
intensive Zustandsgroßen: raumlich konstant innerhalb einer Phase
extensive Zustandsgroßen: proportional zum von der Phase eingenommenen Vo-lumen
intensiv: p, T, µ, B
extensiv: V, N, U, S, M
12

1.2 Temperatur
empirische Tatsache:
zwei Systeme in thermischem Kontakt haben im Gleichgewicht (t → ∞)mindestens eine makroskopische messbare Eigenschaft gemein: Temperatur T
Nullter HS
Hauptsatze: empirische Tatsachen bzw. Postulate, ableitbar (?) aus der statisti-schen Physik
(Def.) ideales Gasreales Gas in starker Verdunnung, V/N groß gegen “Teilchenvolumen”
(Def.) absolute Temperatur, Kelvin-SkalaT = pV
kNmit k = 1, 3805 · 10−23 J/K
p, V, N : ideales GasEinheit: K
Boyle-Marriott-Gesetz:
pV
N= const, nur T -abhangig
V, N fest: p ∝ T Gasthermometer
universell (gilt fur alle idealen Gase)
Probleme:T → 0 (Kondensation)T → ∞ (Wande schmelzen)ideales Gas strenggenommen nicht existent
spater: substanzunabhangige Definition von T
1.3 Einfache thermodynamische Systeme
(klassisches) ideales Gas
thermische Zustandsgleichung
pV = NkT
oder pV = nRT mit
13

n = N/NA Stoffmenge
NA = 6, 023 · 1023 Avogadro-Konstante
R = Nk/n = kNA allgemeine Gaskonstante, R = 8, 3166 J/mol K
—————————————————————————————————
van der Waals-Gas
thermische Zustandsgleichung(p + a
n2
V 2
)(V − nb) = nRT
van der Waals-Gleichung, a, b: Materialkonstanten
Motivation:
p =nRT
V − nb− an2
V 2
1) Eigenvolumen der Molekule, bn p → ∞ fur V → nb
2) schwach attraktive vdW-WW p → p − ∆p, ∆p ∝ N2/V 2
—————————————————————————————————
idealer Paramagnet
• Atome/Molekule mit permanenten magnetischen Momenten mi, m = |mi|
• “ideal”: keine WW
• Gesamtmoment M =∑N
i=1 mi extensiv, Magnetisierung: M/V intensiv
• zufallige Richtungen M = 0
• Ausrichtung in außerem Feld B 6= 0
• Zustandsgleichung:
M = M0L(mB/kT ) (idealer klassischer Paramagnet)
L(x) = coth x − 1
xLangevin-Funktion
mit M0 = Nm und L(x) = coth(x) − 1/x Langevin-Funktion
• T → 0 (B > 0 fest): M → M0 = Nm = max.
• T → ∞ (B > 0 fest): M = CB/T (Curie-Gesetz)
mit C = Nm2/3k Curie-Konstante
14

—————————————————————————————————
thermische Zustandsgleichung (N = const.)
Gas: f(−p, V, T ) = 0 Magnet: f(B, M, T ) = 0
Antwortkoeffizienten
κT = − 1
V
(∂V
∂p
)
T
χT =1
V
(∂M
∂B
)
T
isotherme Kompressibilitat isotherme Suszeptibilitat
αp =1
V
(∂V
∂T
)
p
µB =1
V
(∂M
∂T
)
B
Ausdehnungskoeffizient
βV =1
p
(∂p
∂T
)
V
ϕM =
(∂B
∂T
)
M
Spannungskoeffizient
beachte: (−p, V, T ) ↔ (B, M, T )
alle Koeffizienten positiv außer µB
Kompressionsmodul KT = κ−1T = −V
(∂p
∂V
)
T
Zusammenhang: αp = pκT βV
1.4 Thermodynamische Prozesse
(Def.) thermodynamischer Prozessjeder Ubergang zwischen zwei thermodynamischen Zustanden Z1 und Z2
(also zwischen Gleichgewichtszustanden)
Z1
Z2
Zustandsraum
(Bsp.) freie Expansion eines Gases ins Vakuum
15

zunachst Hahn geschlossen,Z1: rechts: Gas T1, V1, p1, links: Vakuum(Gleichgewicht! Beschreibung durch wenige Zustandsgroßen)
nach Offnen des Hahns und langerem Warten:Z2: Gas T2, V2, p2
(Gleichgewicht!)
1
Z2
Z
?
Z1 und Z2 sind Punkte im Zustandsraum
zwischenzeitliche Nichtgleichgewichtszustande dagegen nicht !
(Def.) quasistatischer ProzessSystem bleibt stets im Gleichgewicht
quasistatischer Prozess: Kurve im Zustandsraum
Z1
Z2
Zustandsraum
Z
(Bsp.) langsames Verschieben einer Trennwandpinnen = paußen + ∆p∆p → 0, quasistatisch, Prozeß dauert unendlich lange
16

innenp paußen
(Def.) Kreisprozessthermodynamischer Prozess mit Z1 = Z2
Z1
—————————————————————————————————
Anderung von Zustandsgroßen bei TD Prozessen
sei f = 2 und A, X, Y Zustandsgroßen, also z.B. A = A(X, Y )
bei infinitesimalem quasistatischem Prozess
X → X + dX
Y → Y + dY
gilt:
A → A + dA mit
dA =
(∂A
∂X
)
Y
dX +
(∂A
∂Y
)
X
dY = F (X, Y )dX + G(X, Y )dY
F und G sind Zustandsgroßen
dA ist ein vollstandiges (totales) Differenzial
es gilt:(
∂F
∂Y
)
X
=
(∂G
∂X
)
Y
Maxwell-Relation
Anderung von A bei endlichem Prozess:
∆A =∫ 2
1(F (X, Y )dX + G(X, Y )dY ) =
∫ 2
1dA = A2 − A1
das Integral ist wegunabhangig (prozessunabhangig)
17

—————————————————————————————————
andere Situation:
seien F = F (X, Y ) und G = G(X, Y ) beliebige Zustandgroßen
definiere fur infinitesimalen TD Prozess
X → X + dX
Y → Y + dY
die Große
δA = F (X, Y )dX + G(X, Y )dY
bzw. fur endlichen Prozess X1 → X2, Y1 → Y2
∆A =∫
CδA =
∫
C(F (X, Y )dX + G(X, Y )dY )
δA, ∆A sind i.allg. prozessabhangig !
δA, ∆A Prozessgroße
Schreibweise: beliebiges Differenzial: δAtotales Differenzial: dA
—————————————————————————————————
(∂F
∂Y
)
X
=
(∂G
∂X
)
Y
Integrabilitatsbedingung
⇔∆A =
∫
C(FdX + GdY ) ist prozessunabhangig,
nur abhangig von Anfangs- und Endzustand
⇔δA = FdX + GdY ist ein totales Differenzial, δA = dA
⇔
∃ Zustandsgroße A(X, Y ) mit F =
(∂A
∂X
)
Y
und G =
(∂A
∂Y
)
X
—————————————————————————————————
Definition von Zustandsgroßen durch Angabe der Anderung bei TD Prozessen
sei δA = FdX + GdY mit gegebenen F (X, Y ), G(X, Y ) und sei(
∂F
∂Y
)
X
=
(∂G
∂X
)
Y
18

dann definiert
A(X, Y ) =∫ (X,Y )
(X0,Y0)(FdX + GdY ) + A(X0, Y0)
eine Zustandsgroße bis auf eine additive Konstante
19

20

Kapitel 2
Erster Hauptsatz
2.1 Arbeit
an einem Gas verrichtete Arbeit δW bei infinitesimaler quasistatischer
Kompression dV < 0
Expansion dV > 0:
dss
extF F
Ap
dV = A ds < 0
δW = −Fds = −pAds = −pdV
mechanische Arbeit
δW = −pdV
magnetische Arbeit
δW = BdM
chemische Arbeit
δW = µdN (Def µ s.u.)
mehrere Teilchensorten: δW =∑
k µkdNk
allgemeine Form:
21

δW =∑
i
Kidqi
Ki: verallgemeinerte Krafte, intensiv, −p, B, µ
qi: verallgemeinerte Koordinaten, extensiv, V, M, N
—————————————————————————————————
typische Zustandsgleichung Ki = Ki(T, q1, q2, ...)
(T, q1, q2, ...) vollstandiger Satz
δW =∑
i
Kidqi + 0dT
Integrabilitatsbedingung (i.allg.) nicht erfullt:(
∂Ki
∂T
)
q1,q2,...
6=(
∂0
∂qi
)
T,qj ,...
= 0
Arbeit ist eine Prozessgroße
δW kein totales Differenzial
2.2 Energieerhaltung
KM, QM: Gesamtenergie erhaltene Zustandsgroße
TD: nur innere Energie U
(kinetische und potenzielle Energie des makroskopischen Korpers bleiben i.allg.unberucksichtigt)
—————————————————————————————————
Gay-Lussac-Veruch: freie Expansion eines idealen Gases V → V ′ > V , isoliert
Beobachtung: T = T ′
isoliertes System: U = U ′, N = const.
U(T, V ) = U(T, V ′)
U = U(T )
U extensiv U = Nu(T )
empirisch:
U =3
2NkT einatomige ideale Gase (Bsp: Ar)
22

U =5
2NkT zweiatomige ideale Gase (Bsp: O2)
U = 3NkT raumliche Gasmolekule (Bsp: CO2)
allgemein: U = U(T, V ) = U(T, qi) kalorische Zustandsgleichung
—————————————————————————————————
KM, QM: Energieerhaltung
∆U = U2 − U1 =∫ 2
1dW =
∫ 2
1
∑
i
F exti dri
dU = dW , dW totales Differenzial (F konservativ)
TD: δW nur auf makroskopischer Beschreibungsebene definiert, δW = −pdV
(makroskopisch nicht kontrollierbare mechanische Arbeit nicht in δW enthalten,z.B. Energieubertrag auf einzelne Molekule durch eine ruhende Wand hindurch)
δW kein totales Differenzial
Konsequenz:
da U Zustandsgroße, kann Energieerhaltung in der Form dU = δW nicht gelten
—————————————————————————————————
Erster Hauptsatz
Fur jedes thermodynamische System gibt es eine Zustandsgroße Umit dU = dW fur Prozesse ohne WarmeaustauschFur beliebige Prozesse sei δQ := dU − δWWarme δQ ist eine Prozessgroße
kurz:
dU = δW + δQ
statistische Physik: Def. der Warme aus mikroskopischer Theorie (KM, QM)
wichtig:
U : Zustandsgroße (Mengengroße)
δW , δQ: Prozessgroßen (Fluss)
23

Wδ
Qδ
dU
U
—————————————————————————————————
Vorzeichenkonvention:
Arbeit:
δW > 0 Arbeit wird am System verrichtet
δW < 0 Arbeit wird frei, Arbeit wird vom System verrichtet
Warme:
δQ > 0 Warme wird dem System zugefuhrt
δQ < 0 Warme wird dem System entzogen
—————————————————————————————————
chemisches Potenzial:
dU = δW + δQ = −pdV + µdN + δQ
µ =
(∂U
∂N
)
V,δQ=0
µ = Anderung der inneren Energie bei Hinzufugen eines Teilchens ohne Warme-austausch und Verrichten von mechanischer Arbeit
—————————————————————————————————
TD Begriffe:
quasistatischer Prozess mit:
δQ = 0 adiabatisch System thermisch isoliertdT = 0 isotherm System im WarmebadδW = δQ = 0 System isoliert
speziell fur Gase:
dV = 0 isochor System dynamisch isoliertdp = 0 isobar System im Volumenbaddµ = 0 System im Teilchenbad
24

2.3 Warmekapazitaten
(Def.) Warmekapazitatnotwendige zuzufuhrende Warme δQ, um bei konstantem x die Tempe-raturerhohung dT zu erreichen:
Cx =
(δQ
dT
)
x
Cx ist eine Zustandsgroße
typischerweise:
Gas: CV , Cp
Magnet: CM , CB
spezifische Warmekapazitat (“spezifische Warme”): Cx/M mit Gesamtmasse M
molare Warmekapazitat (“Molwarme”) Cx/n mit Stoffmenge n
—————————————————————————————————
Berechnung von CV =
(δQ
dT
)
V
fur Gase:
mit 1.HS und N = const. ist:
δQ = dU − δW = dU + pdV =
(∂U
∂T
)
V
dT +
(∂U
∂V
)
T
dV + pdV
Warmezufuhr bei konstantem V , dV = 0, also:
CV =
(∂U
∂T
)
V
(Zustandsgroße)
erfordert kalorische Zustandsgleichung
z.B. einatomiges ideales Gas: U =3
2NkT CV =
3
2Nk
—————————————————————————————————
Berechnung von Cp =
(δQ
dT
)
p
fur Gase:
δQ = dU + pdV
=
(∂U
∂T
)
p
dT +
(∂U
∂p
)
T
dp + p
(∂V
∂T
)
p
dT + p
(∂V
∂p
)
T
dp
25

=
(
∂U
∂T
)
p
+ p
(∂V
∂T
)
p
dT + (· · · )dp
Warmezufuhr bei konstantem p, dp = 0, also:
Cp =
(∂U
∂T
)
p
+ p
(∂V
∂T
)
p
(Zustandsgroße)
erfordert kalorische und thermische Zustandsgleichung
z.B. einatomiges ideales Gas: U =3
2NkT , pV = NkT
Cp =3
2Nk + Nk =
5
2Nk
offensichtlich ist
Cp − CV = Nk fur alle idealen Gase
—————————————————————————————————
allg. Zusammenhang zwischen CV und Cp?
mit U(T, p) = U(T, V ) = U(T, V (T, p)) folgt:(
∂U
∂T
)
p
=
(∂U
∂T
)
V
+
(∂U
∂V
)
T
(∂V
∂T
)
p
= CV +
(∂U
∂V
)
T
(∂V
∂T
)
p
Cp = CV +
((∂U
∂V
)
T
+ p
)(∂V
∂T
)
p
aus 2.HS (Vorgriff) folgt:(
∂U
∂V
)
T
+ p = T
(∂p
∂T
)
V
damit:
Cp = CV + T
(∂p
∂T
)
V
(∂V
∂T
)
p
= T (pβV )(V αp) = TV pβV αp
mit αp = pκT βV ist pβV = αp/κT und
Cp = CV + TVα2
p
κT
26

2.4 Adiabaten
Prozesse mit δQ = 0 (adiabatische Prozesse eines thermisch isolierten Systems)
1.HS
(∂T
∂V
)
δQ=0
= −Cp − CV
CV
(∂T
∂V
)
p
fur ideales Gas gilt:(
∂T
∂V
)
δQ=0
=T
V
damit folgt:
dT
T= −(γ − 1)
dV
V
mit:
γ =Cp
CVAdiabatenexponent
γ =CV + Nk
CV
einatomig: γ = 5/3
zweiatomig: γ = 7/5
raumlich: γ = 4/3
Integration liefert Adiabatengleichungen:
TV γ−1 = const. pV γ = const. T γp1−γ = const.
Isotherme: pV = const
Adiabate: pV γ = const, wegen γ =Cp
CV> 1 steiler im p-V -Diagramm
γpV = constAdiabate
V
p pV = constIsotherme
27

2.5 Carnot-Prozess
Carnot-Prozess:
quasistatischer Kreisprozess bestehend aus 2 Adiabaten und 2 Isothermen mitidealam Gas als Arbeitssubstanz
∆U =∮
dU = 0 (Kreisprozess, U Zustandsgroße)
∆W =∮
δW 6= 0
∆Q =∮
δQ 6= 0
δQ=0
pV =0γ
δQ=0
pV =0γT1
T2
T1 T2>
A
B
C
D
pV=const, dT=0
pV=const, dT=0
p
V
idealesGas
Carnot-Prozess kann in beiden Richtungen durchlaufen werden
betrachte A → B → C → D → A
—————————————————————————————————
A → B adiabatische Kompression
∆T = T1 − T2 > 0
∆Q = 0 ∆W = ∆U =r
2Nk(T1 − T2)
pAV γA = pBV γ
B
—————————————————————————————————
B → C isotherme Expansion
T = T1 = const.
28

∆U = 0 ∆Q = −∆W
∆W = −∫ C
BpdV = −NkT1
∫ VC
VB
dV
V= −NkT1 ln
VC
VB< 0
pBVB = pCVC
—————————————————————————————————
C → D adiabatische Expansion
∆T = T2 − T1 < 0
∆Q = 0 ∆W = ∆U =r
2Nk(T2 − T1)
pCV γC = pDV γ
D
—————————————————————————————————
B → C isotherme Kompression
T = T2 = const.
∆U = 0 ∆Q = −∆W
∆W = −NkT2 lnVA
VD> 0
pDVD = pAVA
—————————————————————————————————
Bilanz:
∆U = 0
∆W = −NkT1 lnVC
VB− NkT2 ln
VA
VD
∆Q = −∆W
es ist:
VAVC
VBVD= 1
lnVC
VB= − ln
VA
VD
∆W = −Nk(T1 − T2) lnVC
VB< 0
∆Q > 0
29

Warmekraftmaschine
∆Q >01
T1
T2
T1 T2
∆Q <0
∆
2
W<0>
1. HS: ∆W + ∆Q1 + ∆Q2 = 0
Definition Wirkungsgrad η:
η =Nutzen
Aufwand=
frei gewordene Arbeit
zugefuhrte Warme=
−∆W
∆Q1
∆Q2: “Abwarme”
fur Carnot-Maschine:
ηc =−∆W
∆Q1=
Nk ln(VC/VB)(T1 − T2)
Nk ln(VC/VB)T1
ηc = 1 − T2
T1
(T1 > T2)
—————————————————————————————————
Carnot-Prozess reversibel
Warmepumpe
∆W > 0
∆Q < 0
T1
T2
∆Q <01
∆
∆
2Q >0
W>0
30

Kapitel 3
Zweiter Hauptsatz
3.1 perpetuum mobile zweiter Art
Definition:
Ein ppm2 ist eine periodisch arbeitende Maschine, die weiter nichts bewirktals Verrichten von Arbeit und Warmeentnahme aus einem Bad
Kreisprozess, Umgebung ebenfalls unverandert, außer: ∆W < 0, ∆Q > 0
1.HS: ∆W + ∆Q = 0
vergleiche: Carnot-Maschine: 2 Bader
ppm2 Carnot ppm1
T∆Q>0
∆W<0
∆Q >01
T1
T2
T1 T2
∆Q <0
∆
2
W<0>
∆W<0
1. Hauptsatz: ein ppm1 ist unmoglich
2. Hauptsatz: ein ppm2 ist unmoglich
Konsequenzen:
31

• Irreversibilitat bestimmter Prozesse
• Thermodynamische Temperaturskala
• Zustandsgroße Entropie
3.2 Irreversibilitat
(Def.) Irreversibler ProzessEin Prozess, der auf keine einzige Weise vollstandig ruckgangig ge-macht werden kann, heißt irreversibel (sonst: reversibel)
System und Umgebung wieder im Ausgangszustand !
—————————————————————————————————
Warmeerzeugung durch Reibung ist irreversibel
Beweis:
Reibung:
T∆Q<0
∆W>0
Annahme: Prozess reversibel
dann existiert:
T∆Q>0
∆W<0
dies ist ein ppm2, also unmoglich
Annahme falsch
—————————————————————————————————
Warmeleitung ist irreversibel
Beweis:
32

Warmeleitung:
T1
T2
T1 T2
∆Q<0
∆Q>0
>
−
Annahme: Prozess reversibel
dann existiert:
T1
T2
T1 T2
∆Q<0
∆
>
Q>0
−
somit existiert auch:
T1
T2
T1 T2
∆Q >01∆Q <02
∆Q >02 ∆Q <0
∆
2
W<0>
−
equivalent mit:
T
∆W<0
∆Q 1+∆Q >02
dies ist ein ppm2, also unmoglich
Annahme falsch
—————————————————————————————————
die freie Expansion eines Gases ist irreversibel
Beweis:
freie Expansion: V’V
Annahme: Prozess reversibel
dann existiert: V’ V
an diesen Prozess anschließend: adiabatische Expansion mit ∆Q = 0, ∆W < 0
33

bis V ′ wieder erreicht, dabei ∆T < 0:V
anschließend: Warmezufuhr ∆Q > 0 bis T ′ wieder erreicht:
(∆W = 0) V’T’
Bilanz: System und Umgebung unverandert, bis auf ∆W < 0, ∆Q > 0
dies ist ein ppm2, also unmoglich
Annahme falsch
—————————————————————————————————
quasistatische Prozesse sind reversibel
Beweis:
quasistatischer Prozess gefuhrt durch Kontrollparameter λ:
λ = p, T, µ fur Arbeits-, Warme-, Teilchenaustausch (δW, δQ, dN 6= 0)
quasistatisch, falls λSystem = λUmgebung + ∆λ mit ∆λ → 0
Annahme: Prozess irreversibel
aber Prozess mit ∆λ → −∆λ stets moglich und fur ∆λ → 0 identisch mitRuckprozess
Ruckprozess existiert (mit beliebig kleinen Anderungen in der Umgebung)
Annahme falsch
3.3 Thermodynamische Temperaturskala
M : beliebige periodische zwischen zwei Warmebadern mit T1 > T2
arbeitende Warmekraftmaschine
nicht notwendig reversibel, beliebiges Arbeitsmedium
34

∆Q >01
T1
T2
T1 T2
∆Q <0
∆
2
W<0> M
Wirkungsgrad: (∆W + ∆Q1 + ∆Q2 = ∆U = 0)
η =−∆W
∆Q1
=∆Q1 + ∆Q2
∆Q1
= 1 +∆Q2
∆Q1
Satz:
η ≤ ηc = 1 − T2
T1
und
η = ηc genau dann, wenn M reversibel
ηc (Carnot) ist der maximal mogliche Wirkungsgrad fur WK-Maschinen
ηc wird von allen WK-Maschinen erreicht, die reversibel arbeiten(unabhangig vom Arbeitsmedium!)
Beweis:
fuge reversible Warmepumpe M ′rev hinzu:
∆Q >01
T1
T2
∆W<0
∆Q <02 ∆Q >02
∆Q <01
∆W >0M revM’
’
’
’
und wahle ∆Q′1 = −∆Q1
Bilanz:
• Warmebad bei T1 unverandert
• M , M ′rev unverandert (nach 1 vollen Zyklus)
• Warmebad bei T2: Warme ∆Q2,tot = ∆Q2+∆Q′2 wird M+M ′
rev zugefuhrt
35

• Arbeit ∆Wtot = ∆W + ∆W ′ wird am System verrichtet
• 1. HS: ∆Wtot + ∆Q2,tot = 0
effektiv:
T2
revM’
∆W tot
∆Q 2,tot
M+
unterscheide 3 Moglichkeiten:
• ∆Wtot < 0 ∆Q2,tot > 0
also: ppm2, unmoglich
• ∆Wtot = 0 ∆Q2,tot = 0
auch Bad bei T2 unverandert
M ′rev hat Effekte von M vollstandig ruckgangig gemacht
M ist reversibel
mit 0 = ∆Q2,tot = ∆Q2 + ∆Q′2 gilt:
η = 1 +∆Q2
∆Q1= 1 − ∆Q2
∆Q′1
= 1 +∆Q′
2
∆Q′1
wahle speziell M ′rev als Carnot-Warmepumpe, dann ist
η = ηc = 1 − T2
T1
• ∆Wtot > 0 ∆Q2,tot < 0
M irreversibel
mit 0 > ∆Q2,tot = ∆Q2 + ∆Q′2 (und somit ∆Q2 < −∆Q′
2) gilt:
η = 1 +∆Q2
∆Q1= 1 +
∆Q2
−∆Q′1
< 1 +∆Q′
2
∆Q′1
= η′
(denn: fur WK-Maschine M ist ∆Q1 > 0, somit −∆Q′1 > 0)
η < η′ = ηc
beachte: fur M , als Warmepumpe betrieben, gilt mit obigem Beweis:
η ≥ ηc = 1 − T2
T1
36

und
η = ηc genau dann, wenn M reversibel
—————————————————————————————————
der Beweis zeigt ferner:
fur zwei beliebige reversible WK-Maschinen M und M ′ gilt
−∆Q2
∆Q1
= −∆Q′2
∆Q′1
unabhangig vom Arbeitsmedium
−∆Q2
∆Q1= f(T1, T2)
mit einer vom Arbeitsmedium unabhangigen (“universellen”) Funktion f
es gilt sogar (s.u.) f(T1, T2) =g(T2)
g(T1)mit einer universellen Funktion g und somit
−∆Q2
∆Q1=
g(T2)
g(T1)
wahlt man einen festen Bezugspunkt (z.B. Gefrierpunkt von Wasser bei Nor-malbedingungen), und setzt dessen Temperatur willkurlich auf θ1 = g(T1) =273, 15K fest, dann kann man durch Betreiben einer reversiblen Maschine zwi-schen dem Bezugsbad und einem weiteren beliebigen Bad dessen Temperaturdefinieren durch:
θ = − ∆Q
∆Q1θ1
dies ist die (substanzunabhangige!) thermodynamische Temperaturskala
weiter gilt (siehe Beweis oben):
θ
θ1= − ∆Q
∆Q1=
T
T1
thermodynamische Temperaturskala = Kelvin-Skala
(bei der angegeben Wahl des Bezugspunkts)
—————————————————————————————————
noch zu zeigen:
f(T1, T2) =g(T2)
g(T1)
Kopplung zweier beliebiger aber reversibler WK-Maschinen mit ∆Q′2 = −∆Q2
37

∆Q >01
T1
T1 T2
T2 T3
T2
T3
∆Q’ >02
T1
T3
∆W’<0
∆W’<0∆W+
∆ 3Q’ <0 ∆ 3Q’ <0
∆Q >01
∆Q <0
∆
2
W<0>
>
M
M’
M + M’
es gilt:
−∆Q2
∆Q1
= f(T1, T2)
−∆Q′3
∆Q′2
= f(T2, T3)
−∆Q′3
∆Q1= f(T1, T3)
also:
f(T1, T2)f(T2, T3) =∆Q2
∆Q1
∆Q′3
∆Q′2
= −∆Q′3
∆Q1= f(T1, T3)
Funktionalgleichung
f(T1, T2)f(T2, T3) = f(T1, T3)
ln f(T1, T2) + ln f(T2, T3) = ln f(T1, T3)
∂
∂T1ln f(T1, T2) =
∂
∂T1ln f(T1, T3)
unabhangig vom zweiten Argument,
∂
∂T1
ln f(T1, T2) = a′(T1)
Integrieren:
ln f(T1, T2) = a(T1) + b(T2)
mit von T2 abhangiger Integrationskonstante
38

f(T1, T2) = A(T1)B(T2)
einsetzen in Funktionalgleichung:
A(T1)B(T2)A(T2)B(T3) = A(T1)B(T3)
A(T ) =1
B(T )
f(T1, T2) =B(T2)
B(T1)
3.4 Entropie als Zustandsgroße
Satz:
Fur jeden quasistatischen Kreisprozess P gilt:
∮
P
δQ
T= 0
Beweis:
zunachst:P Prozess zwischen 2 Badern bestehend aus 2 Adiabaten und 2 Isothermen
T1
T2
δQ=0
δQ=0
dT=0
dT=0
P quasistatisch P reversibel
also:
η = ηc
39

d.h.
1 +∆Q2
∆Q1= 1 − T2
T1
∆Q1
T1+
∆Q2
T2= 0 bzw.
∮
P
δQ
T= 0
jetzt beliebiger Kreisprozess in zweidimensionalem Zustandsraum
Approximation durch quasistatische Kreisprozesse mit 2 Adiabaten und 2 Iso-thermen:
δQ=0
dT=0
Gesamtprozess P = limK→∞
(P1 + P2 + ... + PK)
(Prozesse im Inneren des von P umschlossenen Gebiets neutralisieren sich)
fur Pi ist (s.o.):∮
Pi
δQ
T= 0
∮
P1+...+PK
δQ
T= 0
∮
P
δQ
T= 0
analoge Verallgemeinerung auf beliebigen f -dimensionalen Zustandsraum moglich
40

P approximieren durch Prozesse Pi auf f−1-dimensionalen Hyperflachen δQ = 0und dT = 0
—————————————————————————————————
Konsequenz:∫
P
δQ
Tist prozessunabhangig
δQ
Tist ein totales Differenzial
es existiert eine Zustandsgroße S mit
dS =δQ
TEntropie nach Clausius
S = S(Z) =∫ Z
Z0
δQ
T+ S(Z0)
definiert bis auf additive Konstante (Z0: Referenzzustand)
3.5 Grundgleichung der Thermodynamik
1. HS: dU = δW + δQ =∑
i
Kidqi + δQ
2. HS: dS =δQ
T
TdS = dU −∑
i
Kidqi Grundgleichung der Thermodynamik
beachte: nur Zustandsgroßen bzw. totale Differenziale
Anwendung fur Gas: δW = −pdV , N = const.
dS totales Differenzial Maxwell-Relationen, z.B.
dS =1
TdU +
p
TdV
∂
∂V
(1
T
)
U=
∂
∂U
(p
T
)
V
besser: T und V als unabhangige Variable
dS =1
TdU +
p
TdV
41

=1
T
(∂U
∂T
)
V
dT +1
T
(∂U
∂V
)
T
dV +p
TdV
=1
T
(∂U
∂T
)
V
dT +
[1
T
(∂U
∂V
)
T
+p
T
]dV
Maxwell-Relation:
∂
∂V
[1
T
(∂U
∂T
)
V
]=
∂
∂T
[1
T
(∂U
∂V
)
T
+p
T
]
1
T
∂2U
∂V ∂T=
1
T
∂2U
∂T∂V− 1
T 2
(∂U
∂V
)
T
+1
T
(∂p
∂T
)
V
− 1
T 2p
(
∂U
∂V
)
T
= T
(∂p
∂T
)
V
− p (s.o.)
Bsp: ideales Gas(
∂p
∂T
)
V
=Nk
VT
(∂p
∂T
)
V
− p =NkT
V− p = 0
(∂U
∂V
)
T
= 0 U(T, V ) = U(T )
(Ergebnis des Gay-Lussac-Versuchs als notwendige Konsequenz der thermischenZustandsgleichung idealer Gase und der Hauptsatze !)
3.6 Entropie und Irreversibilitat
Prinzip der Maximierung der Entropie:
In einem isolierten System verlauft jeder Prozess derat, dass die Entropienicht abnimmt (sofern S definiert ist)
dS = 0 ⇔ Prozess reversibel
dS > 0 ⇔ Prozess irreversibel
(System isoliert)
Problem: Definition von S (und T , p, µ, etc.) fur Nichtgleichgewichtszustande ?
Definition unmoglich:
42

Definition moglich:
System besteht aus k Subsystemen, jedes Subsystem fur sich im TD GG-Zustand,vollstandig beschrieben durch f Zustandsgroßen
TD Zustand des Gesamtsystems: ((S1, T1, ...), ..., (Sk, Tk, ...))
Dimension des Zustandsraums: k · fDefinitionen fur extensive Großen:
S =k∑
i=1
Si Gesamtentropie
V =k∑
i=1
Vi Gesamtvolumen
N =k∑
i=1
Vi Gesamtteilchenzahl
U =k∑
i=1
Ui gesamte innere Energie
—————————————————————————————————
Prinzip der Maximierung der Gesamtentropie fur isoliertes Gesamtsystem:
dS = 0 ⇔ Prozess reversibel dS > 0 ⇔ Prozess irreversibel
Beweis:
betrachte beliebigen Prozess P im isolierten System:
43

((S1, T1, ...), ..., (Sk, Tk, ...)) → ((S ′1, T
′1, ...), ..., (S
′k, T
′k, ...))
und quasistatische/reversible Ruckprozesse R1, ..., Rk, so dass
((S ′1, T
′1, ...), ..., (S
′k, T
′k, ...)) → ((S1, T1, ...), ..., (Sk, Tk, ...))
beachte: bei R1, ..., Rk Kontakt mit Umgebung
P + R1 + ... + Rk ist ein Kreisprozess
Ruckprozess R1 :
• (S ′1, T
′1, ...)1 → (S ′
1, T′2, ...)1 adiabatisch
(quasistatische Expansion/Kompression)
Ziel: gleiche Temperaturen von Subsystem 1 und 2
(ggfs. vorab adiabatische Wiederherstellung X ′ → X der Werte der ubrigenZustandsgroßen)
• (S ′1, T
′2, ...)1, (S
′2, T
′2, ...)2 → (S1, T
′′2 , ...)1, (S
′′2 , T ′′
2 , ...)2 adiabatisch
(Subsysteme 1 und 2 in thermischem Kontakt, Warmeaustausch ∆Q1 =−∆Q2, Expansion/Kompression)
Ziel: S ′1 → S1, aber mittels eines fur das Gesamtsystem adiabatischen
Prozesses (daher thermischer Kontakt mit anderem Subsystem notwendigund somit, um quasistatisch zu arbeiten, gleiche Temperaturen notwendig)
• (S1, T′′2 , ...)1 → (S1, T1, ...)1 adiabatisch
(quasistatische Expansion/Kompression)
Ziel: Subsystem 1 mit gleicher Temperatur wie vor Prozess P
Ruckprozess R2 :
• analog zu R1, aber mit Subsystemen 2 und 3
...
Ruckprozess Rk−1 :
• analog zu R1, aber mit Subsystemen k-1 und k
nach R1, ..., Rk sind Subsysteme 1, ..., k-1 im Ausgangszustand:
((S1, T1, ...), ..., (Sk−1, Tk−1, ...), (S′′k , T
′′k , ...))
Ruckprozess Rk :
44

• (S ′′k , T ′′
k , ...)k → (S ′′k , Tk, ...)k adiabatisch
(quasistatische Expansion/Kompression)
Ziel: gleiche Temperatur wie im Ausgangszustand
• (S ′′k , Tk, ...)k → (Sk, Tk, ...)k isotherm
(isotherme Expansion/Kompression, ∆Q = Tk∆Sk = Tk(Sk − S ′′k))
Ziel: Wiederherstellung des Ausgangszustands auch fur Subsystem k
—————————————————————————————————
Bilanz:
kR
’ ’
k k
isoliert
adiabatisch
adiabatisch
adiabatisch
isotherm
R1
R2
R k−1
P ...
1’ ’
2 2’’
S T , ... , S T S T , ... , S T , S T
S T , ... , S T k’ ’’S T , S T , ..., S T
1 1 k−1 k−1’’
1 1 k
k’
1 k
’’k
1 1 k
System unverandert (Kreisprozess) ∆UP+R = 0
1. HS ∆WP+R = −∆QP+R
P : Prozess im isolierten System ∆QP = 0
R = R1 + ... + Rk: adiabatisch bis auf letzten Teilprozess ∆QR = Tk∆Sk = Tk(Sk − S ′′
k)
∆QP+R = Tk(Sk − S ′′k )
effektiv:
∆QT
∆
k
W
P+R
P+R
weiter ist: Stot−S ′tot = ∆SR = ∆SR1 +· · ·+∆SRk−1
+∆SRk= ∆SRk
= Sk−S ′′k
Tk(Stot − S ′tot) = ∆QP+R
45

• Stot > S ′tot:
∆QP+R = −∆WP+R > 0 ppm2
P unmoglich
• Stot = S ′tot:
∆QP+R = ∆WP+R = 0System und Umgebung unverandert
R hat P vollstandig ruckgangig gemacht
P reversibel
• Stot < S ′tot:
∆QP+R = −∆WP+R < 0
Umkehrung von P+R ware ein ppm2, R war reversibel
P irreversibel
—————————————————————————————————
Diskussion von Prozessen in isolierten Systemen:
• ein Nicht-GG-Zustand entwickelt sich zeitlich gemaß
dS > 0 irreversible Zeitentwicklung
(sofern definiert)
• Entropie wachst bis
dS = 0 Gleichgewichtsbedingung
• im GG sind Zustandsgroßen zeitlich konstant
nur noch quasistatische/reversible Prozesse sind moglich mit dS = 0
• fur virtuelle (phys. unmogliche) Prozesse, die aus dem GG herausfuhren,gilt
dS < 0 Stabilitatsbedingung
• in isolierten System ist das TD GG charakterisiert durch
S = max. Entropieprinzip
Probleme:
• Gesamtentropie des Universums (isoliert?) wachst bis dS = 0 (“Warme-tod”, keine lebenserhaltenden irreversiblen Prozesse mehr)
46

• irreversible Zeitentwicklung mit dS > 0 bedeutet Asymmetrie zwischenVergangenheit und Zukunft, 2.HS “definiert den Zeitpfeil”
(gegeben Zustande Z1, Z2: t1 > t2 ⇔ S1 > S2)
aber: mikroskopische Bewegungsgleichungen zeitumkehrinvariant !
Widerspruch?
3.7 Grundrelation der Thermodynamik
dS ≥ 0 fur isoliertes System
Verallgemeinerung auf beliebige Kontakte mit Umgebung ?
Nicht-GG-Zustand eines Gases: ((U1, V1, N1), ..., (Uk, Vk, Nk))
S = S((U1, V1, N1), ..., (Uk, Vk, Nk)) =k∑
i=1
Si(Ui, Vi, Ni)
—————————————————————————————————
S = max. fur isoliertes System
⇔S = max. unter den Nebenbedingungen
U =∑
i
Ui = const.
V =∑
i
Vi = const.
N =∑
i
Ni = const.
⇔S − λ1
∑
i
Ui − λ2
∑
i
Vi − λ3
∑
i
Ni = max. (ohne Nebenbedingungen)
(λ1, λ2, λ3 Lagrange-Parameter)
d.h. Ui, Vi, Ni frei variierbar, beliebige Prozesse
—————————————————————————————————
es folgen die GG-Bedingungen:
∂
∂Uj
[S(..., Ui, Vi, Ni, ...) − λ1
∑
i
Ui − λ2
∑
i
Vi − λ3
∑
i
Ni
]= 0
47

∂
∂Vj
[...] = 0
∂
∂Nj
[...] = 0
also:
∂S
∂Uj−λ1 = 0
∂S
∂Vj−λ2 = 0
∂S
∂Nj−λ3 = 0
das j-te Subsystem ist fur sich im GG, also:
∂S(..., Ui, Vi, Ni, ...)
∂Uj=
(∂Sj
∂Uj
)
VjNj
=1
Tj
denn: S =∑
j
Sj und:
TjdSj = dUj + pjdVj − µjdNj
analog:
∂S
∂Vj=
pj
Tj
∂S
∂Nj= −µj
Tj
GG-Bedingungen:
1
Tj= λ1 =
1
Tunabhangig von j
pj
Tj= λ2 =
p
T
−µj
Tj= λ3 = −µ
T
also:
im TD GG haben alle Subsysteme eines Systems gleiches T , p und µ
—————————————————————————————————
es folgt:
S − 1
TU − p
TV +
µ
TN = max.
bzw.
dS − 1
TdU − p
TdV +
µ
TdN ≥ 0 fur reale Prozesse
beachte:
S, U, V, N : Funktionen des Nicht-GG-Zustands ((U1, V1, N1), ..., (Uk, Vk, Nk))
48

1
T,p
T,−µ
T: Lagrange-Parameter
(phys. Bedeutung: GG-Werte fur Temperatur, Druck, chem. Potenzial)
kurz:
TdS ≥ dU − δW reale reversible (=), irreversible (>) Prozesse
thermodynamische Grundrelation
49

50

Kapitel 4
Thermodynamische Potenziale
TD Potenzial:
Zustandsgroße P (X, Y, Z) mit einfachen partiellen Ableitungen,extremal im GG
X, Y, Z: naturliche Variablen des Potenzials P ,beschreiben Art des Kontakts mit Umgebung
Dimension: Energie (Ausnahme: Entropie)
4.1 Potenziale fur Gase
Ausgangspunkt: TD Grundgleichung
TdS = dU + pdV − µdN
a) Entropie
dS =1
TdU +
p
TdV − µ
TdN
S = S(U, V, N)
(∂S
∂U
)
V,N
=1
Tetc.
Entropie-Prinzip dS ≥ 0 falls U, V, N = const.
isoliertes System: S maximal
—————————————————————————————————
b) innere Energie
dU = TdS − pdV + µdN
51

U = U(S, V, N)
(∂U
∂S
)
V,N
= T etc.
TD Grundrelation: TdS ≥ dU + pdV − µdN
dU ≤ 0 falls S, V, N = const.
U minimal, genauer:
U = U(..., Si, Vi, Ni, ...) = min. fur∑
i
Si = S,∑
i
Vi = V ,∑
i
Ni = N
(S = const. schwer zu kontrollieren, z.B. fur irreversible Prozesse bei Warme-austausch mit Umgebung)
—————————————————————————————————
weitere TD Potenziale durch Legendre-Transformationen
Ausgangspunkt
Potenzial: F = F (X) = F (X, ...)
totales Differenzial: dF = Y dX + ... mit Y =∂F
∂X
F extremal: F = min. fur X = X1 + X2 + ... = const.
dF ≤ 0 fur X = const.
Legendre-Transformation:
G = F − ∂F
∂XX = F − Y X
naturliche Variable von G ist Y , also G = G(Y ) = G(Y, ...), denn
dG = d(F − Y X) = Y dX − Y dX − XdY
dG = −XdY∂G
∂Y= −X
G extremal, denn aquivalent sind:
dF ≤ 0 fur X = const.
F = min. fur X = const.
F − Y X = min. Y Lagrange-Parameter
dF − Y dX ≤ 0
d(G + Y X) − Y dX ≤ 0
dG + XdY ≤ 0
G + XY = min. X Lagrange-Parameter
52

G = min. fur Y = const.
dG ≤ 0 fur Y = const.
beachte: X extensiv Y intensiv
—————————————————————————————————
innere Energie freie Energie großkanonisches Potenzial
U = U(S, V, N) F = F (T, V, N) Ω = Ω(T, V, µ)
Enthalpie freie Enthalpie/Gibbssches Potenzial ?
H = H(S, p, N) G = G(T, p, N) ?
S U V
H F
−p G T
(N = const.)
SUV H(ilft) F(ysikern) p(ei) G(uten) T(aten)
Bsp:
(∂U
∂S
)
V
= T
(∂G
∂(−p)
)
T
= −V
—————————————————————————————————
c) freie Energie
F = U − TS = F (T, V, N)
dF = dU − TdS − SdT = TdS − pdV + µdN − TdS − SdT
dF = −SdT − pdV + µdN
dF ≤ 0 F = min. fur T, V, N = const. (V =∑
i
Vi, N =∑
i
Ni)
53

System im Warmebad
V,NT
fur isotherme Prozesse ist dF = δW , d.h. am System isotherm verrichtete Arbeitfuhrt zur Erhohung der freien Energie
—————————————————————————————————
d) Enthalpie
H = U + pV = H(S, p, N)
dH = dU + pdV + V dp = TdS − pdV + µdN + pdV + V dp
dH = TdS + V dp + µdN
dH ≤ 0 H = min. fur S, p, N = const. (S =∑
i
Si, N =∑
i
Ni)
Realisierung der Nebenbedingungen? (dS > 0 fur irreversible Prozesse im Systemdurch Warmeentzug und damit dS < 0 ausgleichen, schwer zu handhaben)
e) freie Enthalpie
G = F + pV = H − TS = U − TS + pV = G(T, p, N)
dG = d(H − TS) = TdS + V dp + µdN − TdS − SdT
dG = −SdT + V dp + µdN
dG ≤ 0 G = min. fur T, p, N = const. (N =∑
i
Ni)
System im Warme- und Volumenbad
kg
Tp,N
f) großkanonisches Potenzial
Ω = F − µN = U − TS − µN = Ω(T, V, µ)
dΩ = d(F − µN) = −SdT − pdV + µdN − µdN − Ndµ
dΩ = −SdT − pdV − Ndµ
54
dΩ ≤ 0 Ω = min. fur T, V, µ = const. (V =∑
i
Vi)
System im Warme- und Teilchenbad (z.B. metallischer Kontakt)
—————————————————————————————————
Diskussion:
• Potenziale liefern weitere Maxwell-Relationen
• extremal (minimal außer S) bei festen naturlichen Variablen
• liefern (bei Kenntnis der funktionalen Abhangigkeiten) vollstandige In-formation uber Zusammenhange zwischen Zustandsgroßen (Zustandsglei-chungen)
• implizieren Ungleichungen fur Antwortkoeffizienten (s.u.)
• Bestimmung eines Potenzials Hauptaufgabeder TD (aus empirisch bestimmten Zustandsgleichungen)und der stat.M. (aus Hamilton-Funktion, -Operator)
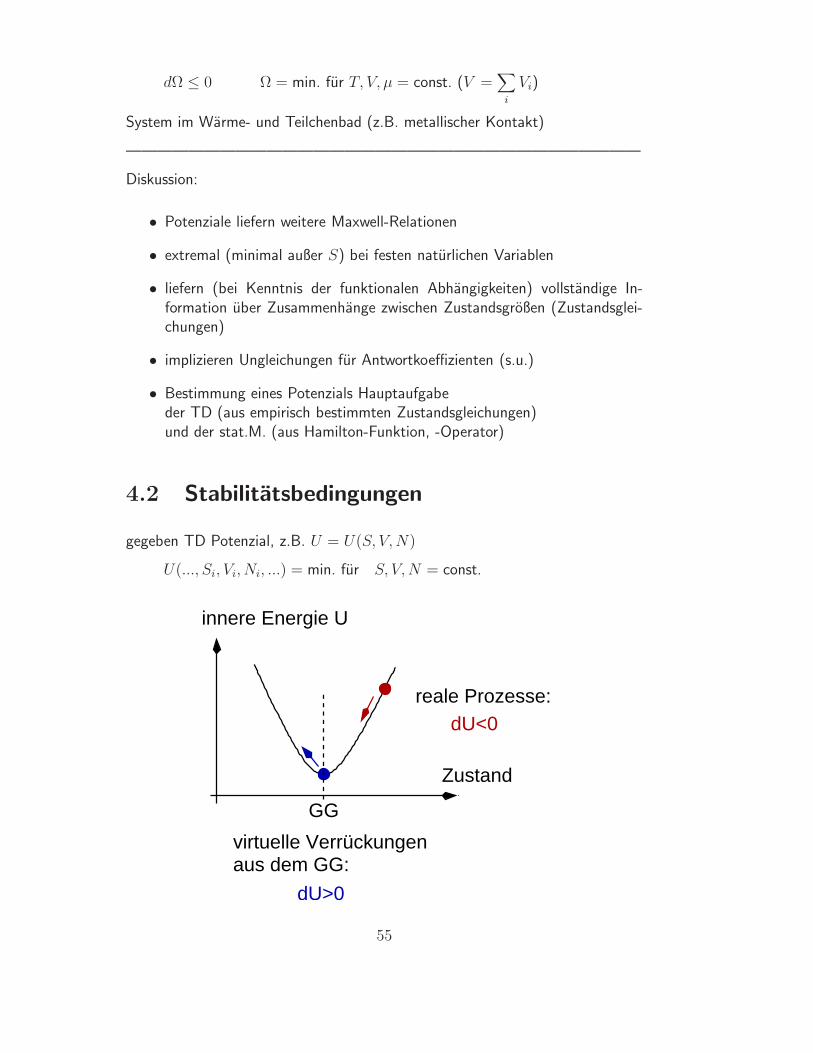
4.2 Stabilitatsbedingungen
gegeben TD Potenzial, z.B. U = U(S, V, N)
U(..., Si, Vi, Ni, ...) = min. fur S, V, N = const.
innere Energie U
Zustand
GG
virtuelle Verrückungenaus dem GG:
reale Prozesse:dU<0
dU>0
55

Stabilitatsbedingung: dU > 0
Lagrange-Parameter: T, p, µ
U − TS + pV − µN = min.
Entwickeln um das GG in “virtuellen Verruckungen” dSi, dVi, dNi
—————————————————————————————————
erste Ordnung:
∂
∂Si(U−TS+pV −µN) = 0
∂U
∂Si−T = Ti−T = 0
(s.o.) analog:
pi − p = 0 µi − µ = 0
(bei k Subsystemen:) 3k−3 Gleichungen und 3 Lagrange-Parameter bestimmenGG-Werte der 3k Unbekannten (..., Si, Vi, Ni, ...)
—————————————————————————————————
zweite Ordnung:
1
2
∑
kl
∂2(U − TS + pV − µN)
∂Xk∂Xl
dXkdXl > 0
fur beliebige dXk, dXl mit X = (..., Si, Vi, Ni, ...)
S, V, N linear in Si, Vi, Ni
0 <1
2
∑
kl
∂2U
∂Xk∂XldXkdXl
Subsysteme indiziert durch i:
0 <1
2
∑
ij
(∂2U
∂Si∂Sj
dSidSj + 2∂2U
∂Si∂Vj
dSidVj + 2∂2U
∂Si∂Nj
dSidNj + · · ·)
Terme fur i = j verschwinden, denn U =∑
i Ui(Si, Vi, Ni)
0 <1
2
∑
i
(∂2Ui
∂Si∂SidSidSi + 2
∂2Ui
∂Si∂VidSidVi + · · ·
)
fur beliebige dSi, dVi, dNi
(· · · ) = 0 fur jedes Subsystem i
jetzt gilt: Ui(Si, Vi, Ni) = U(Si, Vi, Ni), denn jedes Subsystem i ist fur sich imGG, wird also durch die gleiche Zustandsgleichung beschrieben
56

0 <1
2
(∂2U
∂S2dS2 + 2
∂2U
∂S∂VdSdV +
∂2U
∂V 2dV 2 + · · ·
)
mit∑
rs
xrArsxs = xT Ax > 0 fur beliebige x = (..., xr, ...) ⇔ A positiv definit
folgt: (N = const.)(
∂2U∂S2
∂2U∂S∂V
∂2U∂V ∂S
∂2U∂V 2
)positiv definit
bzw. in den Variablen S, V, N :
Hesse U(S, V, N) positiv definit
allgemein ist:
Hesse(TD Potenzial(ext.nat.Var.)) positiv definit
(Entropie: negativ definit)
—————————————————————————————————
Auswertung mit Satz von Hurwitz:
A positiv definit ⇔ det Ak > 0 ∀k
mit Ak: obere, linke k × k Untermatrix von A
—————————————————————————————————
Beispiel: fur U(S, V, N) folgt (N = const.)
(1)∂2U
∂S2> 0
(2)∂2U
∂V 2> 0
(3)∂2U
∂S2
∂2U
∂V 2−(
∂2U
∂V ∂S
)2
> 0
—————————————————————————————————
(1), es ist (dU = TdS − pdV )(
∂U
∂S
)
V
= T
also:(
∂T
∂S
)
V
> 0
57

bzw. nach Invertieren(
∂S
∂T
)
V
> 0
also:
CV =
(δQ
dT
)
V
= T
(∂S
∂T
)
V
> 0 CV > 0
—————————————————————————————————
(2), weiter ist:(
∂U
∂V
)
S
= −p
also:
−(
∂p
∂V
)
S
> 0
bzw. nach Invertieren(
∂V
∂p
)
S
< 0
definiere:
adiabatische Kompressibilitat
κS = − 1
V
(∂V
∂p
)
S
adiabatischer Ausdehnungskoeffizient
αS =1
V
(∂V
∂T
)
S
adiabatischer Spannungskoeffizient
βS =1
p
(∂p
∂T
)
S
es ist somit
κS > 0
—————————————————————————————————
(3) schließlich:
0 <∂2U
∂S2
∂2U
∂V 2−(
∂2U
∂V ∂S
)2
58

=
(∂T
∂S
)
V
(∂(−p)
∂V
)
S
−(
∂T
∂V
)2
S
=T
CV
1
V κS
− 1
V 2α2S
also:
TV α2S > CV κS
—————————————————————————————————
• van der Waals-Isothermen: V -Bereich mit CV < 0 System nicht im TDGG in diesem Bereich
• beachte: positiv definite Hesse-Matrix nur in extensiven (naturlichen) Va-riablen
∂2F (T, V, N)
∂T 2= −
(∂S
∂T
)
V,N
= − 1
TCV < 0 !
4.3 Gibbs-Duhem-Relation
freie Enthalpie G = G(T, p, N)
G, N : extensiv, T, p: intensiv
Ableitung einer Homogenitatsrelationdurch Vergroßerung des Systems um Faktor α
G → αG N → αN T, p = const.
also:
αG = G(T, p, αN)
mit ∂/∂α ergibt sich:
G =
(∂G
∂N
)
T,p
N
somit:
G = µN
liefert Interpretation von µ: µ ist die freie Enthalpie pro Teilchen
mit G = U − TS + pV = µN folgt:
U − TS + pV − µN = 0 Gibbs-Duhem-Relation
59

allgemein: U − TS −∑
i
Kiqi = 0
mit Ω = U − TS − µN ist:
Ω = −pV
beachte: p, V (bzw. µ, N) sind nicht die naturlichen Variablen von Ω (bzw. G)
—————————————————————————————————
beachte: U = min. fur S, V, N = const., also:
U − TS + pV − µN = min mit T, p, µ Lagrange-Parameter
Gibbs-Duhem: min. = 0
U − TS + pV − µN = 0 im TD GG
U − TS + pV − µN > 0 fur Nicht-GG-Zustande
(U =∑
Ui etc., T : GG-Temperatur aller Subsysteme etc.)
—————————————————————————————————
mit der TD Grundgleichung (Kombination von 1. und 2. HS)
dU − TdS + pdV − µdN = 0
folgt aus:
0 = d(U − TS + pV − µN)
= dU − TdS + pdV − µdN − SdT + V dp − Ndµ
die Relation:
−SdT + V dp − Ndµ = 0
Interpretation:
U(S, V, N) ↔ H(S, p, N)
F (T, V, N) ↔ G(T, p, N)
Ω(T, V, µ) ↔ P (T, p, µ) ?
es ist:
dP = −SdT + V dp − Ndµ
da T, p, µ intensiv und P extensiv (Legendre-Transformierte), gilt weiter:
αP (T, p, µ) = P (T, p, µ)
somit
60

P (T, p, µ) = 0
der Wert des TD Potenzials P ware stets durch 0 gegeben, aber:
Problem: der Satz (T, p, µ) ist nicht vollstandig
(und mit Hinzunahme einer weiteren extensiven Große ubervollstandig)
denn: (T, p, µ) ↔ (T, V, N) 6= (T, αV, αN) ↔ (T, p, µ)
4.4 Freie Energie des klassischen idealen Gases
Hauptaufgabe der TD: Bestimmung eines TD Potenzials (in den naturlichenVariablen) ausgehend von (empirisch bestimmten) Zustandsgleichungen
Bsp.: ideales Gas
gegeben:
pV = NkT CV N =s
2kT
gesucht:
F (T, V, N)
(weitere Potenziale aus Legendre-Transformation)
—————————————————————————————————
es ist dF = −SdT − pdV + µdN
F bestimmen durch Integration von(
∂F
∂T
)
V N
= −S
benotigt: S(T, V, N), es gilt:
CV =
(δQ
dT
)
V N
= T
(∂S
∂T
)
V N
= C(T, V, N)
also:
S(T, V, N) =∫ T
T0
dT ′C(T ′, V, N)
T ′ + S(T0, V, N)
=s
2Nk
∫ T
T0
dT ′ 1
T ′ + S(T0, V, N)
=s
2Nk ln
T
T0+ S(T0, V, N)
61

—————————————————————————————————
damit:
F (T, V, N) = −∫ T
T0
dT ′S(T ′, V, N) + F (T0, V, N)
= −s
2Nk
∫ T
T0
dT ′ lnT ′
T0− (T − T0)S(T0, V, N) + F (T0, V, N)
= −s
2NkT0
∫ T/T0
1d(T ′/T0) ln
T ′
T0− (T − T0)S(T0, V, N) + F (T0, V, N)
= −s
2NkT0[x ln x − x]
T/T0
1 − (T − T0)S(T0, V, N) + F (T0, V, N)
= −s
2NkT ln
T
T0+
s
2NkT − s
2NkT0− (T −T0)S(T0, V, N)+F (T0, V, N)
also:
F (T, V, N) = −s
2NkT ln
T
T0+
s
2Nk(T −T0)−(T −T0)S(T0, V, N)
+F (T0, V, N)
—————————————————————————————————
Problem: F (T0, V, N), S(T0, V, N) = ?
dazu:(
∂F
∂V
)
TN
= −p = −NkT
V
F (T, V, N) = −NkT∫ V
V0
dV ′ 1
V ′ + F (T, V0, N)
mit der Definition: V0 =NkT0
p0
F (T, V, N) = −NkT lnV
V0+ F (T, V0, N)
also:
F (T0, V, N) = −NkT0 lnV
V0+ F (T0, V0, N)
und mit S(T, V, N) = −(
∂F
∂T
)
V,N
= Nk lnV
V0+ S(T, V0, N) ist:
62

S(T0, V, N) = Nk lnV
V0+ S(T0, V0, N)
—————————————————————————————————
jetzt noch F (T0, V0, N), S(T0, V0, N) unbekannt Homogenitatsrelation ausnutzen:
F (T, V, N) = Nf(T, V, N) = Nf(T, V/N) = Nf(T, v) v =V
N
damit ist:(
∂f
∂v
)
T,N
=
(∂F/N
∂V/N
)
T,N
=
(∂F
∂V
)
T,N
= −p = −NkT
V= −kT
v
integrieren:
f(T, v) = −kT lnv
v0
+ f(T, v0) mit v0 =V0
N0
Nf(T, v) = −NkT lnv
v0
+ Nf(T, v0)
F (T, V, N) = −NkT lnV N0
NV0+
N
N0F (T, V0, N0)
F (T0, V, N) = −NkT0 lnV N0
NV0+
N
N0F (T0, V0, N0)
jetzt ist F (T0, V, N) bis auf eine echte Konstante bestimmt
analog:
S(T0, V, N) = Nk lnV N0
NV0+
N
N0S(T0, V0, N0)
—————————————————————————————————
einsetzen: (F0 = F (T0, V0, N0), S0 = S(T0, V0, N0)
F (T, V, N) = −s
2NkT ln
T
T0+
s
2Nk(T − T0)
−(T − T0)(Nk ln
V N0
NV0+
N
N0S0
)− NkT0 ln
V N0
NV0+
N
N0F0
also:
F = −s
2NkT ln
T
T0+ Nk(T − T0)
(s
2− S0
N0k
)− NkT ln
V N0
NV0+
N
N0F0
63

TD Potenzial in naturlichen Variablen F (T, V, N)
—————————————————————————————————
Entropie des klassischen idealen Gases:
−S =
(∂F
∂T
)
V,N
= −s
2Nk ln
T
T0
− s
2Nk +Nk
(s
2− S0
N0k
)−Nk ln
V N0
NV0
S = Nk(
s
2ln
T
T0+
S0
N0k+ ln
V N0
NV0
)
S = Nk(
s
2ln T + lnV − ln N − s
2ln T0 − lnV0 + ln N0 +
S0
N0k
)
S = Nk(
s
2ln T + lnV − ln N + const.
)
mit nur vom Referenzzustand abhangiger Konstante
S nicht in naturlichen Variablen gegeben! S = S(U, V, N)
U =s
2NkT T =
2
sk
U
N
ln T = ln U − ln N + const.
S = Nk(
s
2ln U + lnV −
(s
2+ 1
)ln N + const.
)
const. =S
Nk− s
2ln U − ln V −
(s
2+ 1
)ln N
const. =S0
N0k− s
2ln U0 − ln V0 −
(s
2+ 1
)ln N0
einsetzen:
S = Nk(
s
2ln
U
U0+ ln
V
V0−(
s
2+ 1
)ln
N
N0+
S0
N0k
)
Bsp.:
1
T=
(∂S
∂U
)
V,N
= Nks
2
1
U
Zustandsgleichung mit µ:
−µ
T=
(∂S
∂N
)
U,V
=S
N− k
(s
2+ 1
)
µ = k(
s
2+ 1
)T − TS
N
64

4.5 Dritter Hauptsatz
TD Potenziale bis auf additive Konstante definiert
Satz von Nernst, 3. HS
S(T, V, N), S(T, p, N) → 0 fur T → 0
empirisch bestatigt, Ableitung: statistische Mechanik
Konsequenzen?
Annahme:
Cx =
(δQ
dT
)
x
= T
(∂S
∂T
)
x
→ C0 > 0 fur T → 0
(
∂S
∂T
)
x
→ C0
Tfur T → 0
S(T, x) → C0 ln T + S0 fur T → 0
also S divergent im Widerspruch zum 3. HS
es folgt somit:
Cp, CV → 0 fur T → 0
—————————————————————————————————
ideales Gas, kalorische Zustandsgleichung U =s
2NkT CV =
s
2Nk
kalorische Zustandsgleichung des idealen Gases inkorrekt fur T → 0
—————————————————————————————————
Differenz der Warmekapazitaten unahangig von kalorischer Zustandsgleichung,denn:
Cp − CV = T
(∂p
∂T
)
V
(∂V
∂T
)
p
(s.o.)
ausdrucken als Ableitungen der Entropie:
mit dF = −SdT − pdV ist:(
∂p
∂T
)
V
= −∂2F (T, V )
∂T∂V=
(∂S
∂V
)
T
und mit dG = −SdT + V dp:
65

(∂V
∂T
)
p
=∂2G(T, p)
∂T∂p= −
(∂S
∂p
)
T
also:
Cp − CV = −T
(∂S
∂V
)
T
(∂S
∂p
)
T
S(T, x) → 0 fur beliebige x ∂S
∂x→ 0
also:
Cp − CV
T→ 0 fur T → 0
(Differenz der Warmekapazitaten schneller als linear gegen Null)
—————————————————————————————————
ideales Gas: Cp − CV = Nk
thermische Zustandsgleichung des idealen Gases inkorrekt fur T → 0
—————————————————————————————————
direkter: Antwortkoeffizienten
Ausdehnungskoeffizient:
αp =1
V
(∂V
∂T
)
p
= − 1
V
(∂S
∂p
)
T
→ 0
Spannungskoeffizient
βV =1
p
(∂p
∂T
)
V
=1
p
(∂S
∂V
)
T
→ 0
αp, βV → 0
—————————————————————————————————
ideales Gas:
αp =1
V
(∂(NkT/p)
∂T
)
p
=1
V
Nk
p=
1
T→ ∞
βV =1
p
(∂(NkT/V )
∂T
)
V
=1
p
Nk
V=
1
T→ ∞
thermische Zustandsgleichung des idealen Gases inkorrekt fur T → 0
66

4.6 Phasen und Komponenten
Phase: (physikalisch und chemisch) homogener Bereich
Zwei-Phasen-Systeme:
– Wasser/Wasserdampf
Drei-Phasen-Systeme:
– Wasser/Wasserdampf/Eis (“Tripelpunkt”)
Frage: Dimension des Zustandsraums?
—————————————————————————————————
isoliertes (einkomponentiges) P -Phasen-System:
jede Phase i = 1, ..., P vollstandig durch (Ui, Vi, Ni) charakterisiert
Gesamtentropie S =∑
i
Si =∑
i
Si(Ui, Vi, Ni) = max.
beachte: Si(.., .., ..) 6= Sj(.., .., ..), verschiedenen Phasen haben unterschiedlichephysikalische/chemische Eigenschaften, unterschiedliche Zustandsgleichungen
Anzahl der Freiheitsgrade kleiner als 3 P wegen GG-Bedinungen:
T1 = T2 = ... = TP
p1 = p2 = ... = pP
µ1 = µ2 = ... = µP
3(P − 1) Bedingungen, z.B. T1(U1, V1, N1) = T2(U2, V2, N2) etc.
also: f = 3P − 3(P − 1) = 3 (wie fur homogenes (Ein-Phasen-) System!)
ein trivialer Freiheitsgrad pro Phase: Skalierung der Phase um Faktor α:
(Ui, Vi, Ni) → (αUi, αVi, αNi)
Anzahl “intensiver Freiheitsgrade”:
fint = 3 − P Gibbssche Phasenregel
—————————————————————————————————
P -Phasen-System im Warmebad:
jede Phase i = 1, ..., P vollstandig durch (T, Vi, Ni) charakterisiert
gesamte freie Energie F =∑
i
Fi =∑
i
Fi(T, Vi, Ni) = min.
GG-Bedinungen:
67

p1 = p2 = ... = pP
µ1 = µ2 = ... = µP
2(P − 1) Bedingungen
also: f = 1 + 2P − 2(P − 1) = 3
fint = 3 − P
—————————————————————————————————
P -Phasen-System im Warme- und Volumenbad:
Phase i: (T, p, Ni)
gesamte freie Enthalpie G =∑
i
Gi =∑
i
Gi(T, p, Ni) = min.
GG-Bedinungen:
µ1 = µ2 = ... = µP
P − 1 Bedingungen
also: f = 2 + P − (P − 1) = 3
fint = 3 − P
Beispiel: Wasser, Wasser/Wasserdampf, Wasser/Wasserdampf,Eis
P = 1 fint = 2 intensive Freiheitsgrade p und T
P = 2 fint = 1 intensiver Freiheitsgrad p = p(T ), Dampfdruckkurve
P = 3 fint = 0 Tripelpunkt
Phasendiagramm mit Phasenubergangen
p
T
gasförmigp=p(T)fest
flüssig
auf der Dampfdruckkurve: µgasf.(T, p) = µfl.(T, p) sonst µgasf.(T, p) 6= µfl.(T, p)
µ stetig am Phasenubergang
differenzierbar? kontinuierliche, diskontinuierliche Phasenubergange
Tripelpunkt von Wasser: T = 273.16000 als Festlegung des Bezugspunkts der
68

Kelvin-Skala
—————————————————————————————————
Komponenten: die verschiedenen chemischen Substanzen, aus denen ein Sy-stem besteht
Bsp: Gasgemisch H2, CO2, N2
System mit k = 1, ..., K Komponenten
Teilchenzahlen: Nk, N =∑
k
Nk
definiere:
Konzentration:
xk =Nk
N(∑
k
Nk = N)
Partialdruck:
pk = xkp (∑
k
pk = p)
—————————————————————————————————
Bsp: Gemisch idealer Gase
wiederum ideales Gas pV = NkT
pkV = xkpV = xkNkT = NkkT
pkV = NkkT
pk =NkkT
V
Partialdruck pk:Druck des nur aus Komponente k bestehenden Gases bei gleichem T, V
—————————————————————————————————
K-komponentiges System mit P Phasen im Warme- und Volumenbad:
i = 1, ..., P Phasen, k = 1, ..., K Komponenten
freie Enthalpie:
G = G(T, p, Nki)GG-Bedingungen:
∂G
∂Nk1=
∂G
∂Nk2= ... =
∂G
∂NkPfur k = 1, ..., K
69

K(P − 1) Bedingungen
f = 2 + KP − K(P − 1) = 2 + K
Skalierung jeder Phase moglich
fint = 2 + K − P Gibbssche Phasenregel
Bsp:
K = 1: s.o.
K = 2, P = 1: homogenes Gasgemisch, fint = 3 (intensive Variable: T, p, N1/N2)
K = 2, P = 2: Dampfdruck einer zweikomponentigen Flussigkeit, fint = 2(intensive Variable: T, p)
4.7 Gibbssches Paradoxon
grundlegende Diskussion der Dimension des Zustandsraums f !
P = 1, K = 2: Durchmischung zweier verschiedener idealer Gase (einatomig)
f = 4 GG-Zustand: (p, T, N1, N2)
—————————————————————————————————
Prozess P :
prapariere einen Nicht-GG-Zustand durch Herausziehen einer Trennwand:
Zustand A
1 N1 V 2 N2
p T
V
das System ist isoliert
nach einiger Zeit: TD GG
70

V=V +V1 2N=N +N1 2
p T
Zustand B
System isoliert, also Erwartung:
SB − SA > 0 (d.h. Prozess irreversibel)
—————————————————————————————————
Berechnung von ∆S = SB − SA durch
∆S = −∆SR = −∫ A
BdS
fur quasistatischen, reversiblen Ruckprozess R = R1 + R2: B → C → A
setze nur fur Teilchensorte 1 bzw. 2 durchlassige Wande ein:
(Bsp: Teilchen der Sorte 1 zu groß, um durch porose Wand 1 durchzudiffundieren,Teilchen der Sorte 2 dagegen klein genug; Teilchen der Sorte 1 ungeladen unddurchlaufen Wand 2, Teilchen der Sorte 2 erfahren dagegen elektronstatischeAbstoßung an Wand 2)
V=V +V1 2N=N +N1 2
Zustand B
durchlässig nur für durchlässig nur für
R1 fuhrt B nach C: (quasistatisch, reversibel)
fur jede Teilchensorte: lediglich langsames Verschieben des jeweiligen Behalters
Zwischenzustand
beachte: fur beide Behalter gilt: gleiche Krafte auf die Wande links und rechts
∆WR = 0
71

V N1 V N2
Zustand C
die Behalter sind thermisch isolierend
∆WR = 0, ∆QR = 0, ∆UR = 0
T ebenfalls unverandert, da die inneren Energien konstant bleiben
die Drucke sinken:
Druck links: p1 =N1kT
V
Druck rechts: p2 =N2kT
V
entsprechen Partialdrucken im Zustand B, p1 +p2 =(N1 + N2)kT
V=
NkT
V= p
—————————————————————————————————
R2 fuhrt C zuruck nach A, isotherm: (quasistatisch, reversibel)
(isotherme Kompression, separat fur jedes Teilsystem)
Zustand A
1 N1 V 2 N2
p T
V
hierbei ist:
∆WR = −∫ V1
V
N1kT
V ′ dV ′ −∫ V2
V
N2kT
V ′ dV ′
= −N1kT lnV1
V− N2kT ln
V2
V
im Zustand A ist:
pV1 = N1kT pV2 = N2kT
im Zustand B bzw. C ist:
pV = (p1 + p2)V = N1kT + N2kT = NkT
also:
72

V1
V=
p
NkT
N1kT
p=
N1
N
V2
V=
N2
N
und somit:
∆WR =∫ A
BδW = −N1kT ln
N1
N− N2kT ln
N2
N
∆WR > 0, am System wird Arbeit verrichtet
∆UR = 0, Prozess isotherm
∆QR = −∆W , 1. HS
somit folgt
∆S = −∆SR = −∫
RdS = −
∫
R
δQ
T= − 1
T∆QR =
1
T∆WR
∆S = SB − SA = N1k lnN
N1+ N2k ln
N
N2
wie erwartet: ∆S > 0, Prozess P irreversibel
ist speziell N1 = N2 =N
2, dann gilt:
∆S = SB − SA = Nk ln 2
—————————————————————————————————
Entropie des idealen Gases:
S = S(T, V, N) = Nk(
s
2ln T + ln V − ln N + σ0
)
mit nur vom Referenzzustand abhangiger Konstante σ0
Zustand A: (beide Gase mit gleichem s, z.B. H2, O2)
SA = S(T, V1, N1) + S(T, V2, N2)
= N1k(
s
2ln T + ln V1 − ln N1 + σ0
)+N2k
(s
2ln T + ln V2 − ln N2 + σ0
)
= (N1 + N2)ks
2ln T + N1k ln
V1
N1
+ N2k lnV2
N2
+ (N1 + N2)kσ0
mit V1/N1 = V2/N2 = V/N ist:
SA = Nk(
s
2ln T + ln
V
N+ σ0
)
SA = S(T, V, N1 + N2)
im “geordneten Zustand”: N Teilchen im Volumen V bei Temperatur T
73

Zustand B:
SB = SA + ∆S
= Nk(
s
2ln T + ln
V
N+ σ0
)+ N1k ln
N
N1
+ N2k lnN
N2
= Nk(
s
2ln T + ln V + σ0
)+ N1k ln
1
N1+ N2k ln
1
N2
SB = S(T, V, N1) + S(T, V, N2)
im “durchmischten Zustand”: Ni Teilchen im Volumen V bei Temperatur T
∆S = S(T, V, N1) + S(T, V, N2) − S(T, V, N1 + N2)
—————————————————————————————————
Gibbssches Paradoxon
– verschiedene Gase: ∆S = Nk ln 2
– gleiche Gase: offensichtlich ist ∆S = 0 (A und B sind gleiche TD Zustande)
Forderungen:
• Man sollte ignorieren konnen, dass die Gase verschieden sind!
(welchen Effekt sollte es z.B. haben, wenn N1 rote und N2 blaue (alsoverschiedene) Molekule vorliegen, die “Farbe” aber gar keinen Einfluss aufderen Dynamik hat?)
• Es kann nicht sein, dass ∆S = Nk ln 2, falls die Gase verschieden sind,dies uns aber nicht bekannt ist!
(historisches Beispiel: Entdeckung der Isotope)
• Die Anzahl f der (unabhangigen) Zustandsgroßen sollte also nicht festsondern wahlbar sein!
Unterschiedliche Beschreibungsebenen sollten zulassig sein!
(p, T, N1, N2) vs. (p, T, N)
Beschreibung des Mischungsexperiments auf der Ebene (p, T, N), also so, als obdie Gase gleich waren:
A → B:
keine Zustandsanderung (statt irreversibler Durchmischung mit ∆S > 0)
C → A:
jeweils isotherme Kompression mit ∆WR = −∆QR > 0 (unverandert)
74

B → C:
1) Einsatz von semipermeablen Wanden:
Zwischenzustand
• wie vorher ist ∆WR = ∆QR = 0
• also: A → B → C → A ist ein reversibler Kreisprozess mit ∆W = −∆Qbei Warmeaustausch mit einem Bad der Temperatur T
• −(P + R) ist ein ppm2!
Fazit:
• Makrovariablen, die in einem TD Prozess manipuliert werden, mussenzur Zustandscharakterisierung mit einbezogen werden (Konsistenz der Be-schreibungsebene)
2) kein Einsatz von semipermeablen Wanden:
p1 und p2 konnen nicht separat manipuliert werden
• B → C nur uber isotherme Expansion moglich
dabei: ∆WBC = ∆QBC < 0
und: ∆WBC = ∆WCA
• also: A → B → C → A ist ein reversibler Kreisprozess mit ∆W, ∆Q = 0
Fazit:
• konsistente Beschreibung mit weniger Zustandsvariablen moglich, “Parado-xien” (z.B. ppm2) entstehen nur bei Vermischung verschiedener Beschrei-bungsebenen
• eine real beobachtete Verletzung des 2.HS zeigt an, dass die Beschreibungdes TD GG nicht vollstanig ist
• die Großen Entropie, Warme, Arbeit sind keine absoluten Großen sondernabhangig vom gewahlten Satz von Zustandsgroßen, d.h. von der Beschrei-bungsebene
75

Mischung von Gasen: paradox?
Paradoxon
• ∆S = Nk ln 2 ist unabhangig von der Art der Gase
• gleiche Gase durchmischen sich auch, was klar wird, wenn man die einzelnenGasmolkule betrachtet
• warum sollte also ∆S = 0 fur gleiche Gase gelten ?
Auflosung
• Mischung produziert Entropie heißt:
man kann die Gase wieder entmischen und das System reversibel in denAusgangszustand zuruckfuhren, wobei eine Anderung in der Umgebungzuruckbleibt
• Ruckkehr in den Ausgangszustand aber meint Ruckkehr in den TD Aus-gangszustand (Makrozustand nicht Mikrozustand)
• dazu ist aber bei gleichen Gasen gar kein Prozess notwendig, Anfangs- undEndzustand sind gleich, also ∆S = 0
Fazit
• es ist wichtig, zwischen Mikro- und Makrozustand zu unterscheiden
• die Entropie bezieht sich auf den Makrozustand
76

Paradoxon
• man nehme an, dass die zwei Gase aus (hypothetischen) Teilchen bestehen,die sich nur in einem Parameter (z.B. Masse m) unterschieden
• dieser Parameter sei kontinuierlich varrierbar
• fur ∆m → 0 kontinuierlich ist auch ∆S → 0, aber diskontinuierlich !
Auflosung
• der Mikrozustand der Gase andert sich durchaus kontinuierlich mit m
• die TD bezieht sich aber nicht auf den Mikro- sondern auf den Makro-zustand (es wird keineswegs der ursprungliche Mikrozustand wiederherge-stellt)
• was also diskontinuierlich ist, ist die Bedeutung von “TD Zustand wieder-hestellen konnen” und “reversibel”
• entweder man hat die Fahigkeit (auch bei kleinster Massendifferenz), se-mipermeable Wande zu bauen, oder nicht
• wenn nicht, macht es fur die TD Beschreibung keinen Sinn, die Massezur Unterscheidung von Zustandsvariablen heranzuziehen (z.B. N1 und N2
oder Partialdrucke)
Fazit
• die Definition der Entropie hangt davon ab, welche Variablen zur Beschrei-bung des Makrozustands herangezogen werden, in dieser Wahl liegt dieDiskontinuietat
77

Paradoxon
• man nehme an, die zwei Gase bestunden aus Teilchen, die sich nur in einerEigenschaft unterscheiden, die fur den Durchmischungsprozess dynamischirrelevant und nur in anderen Experimenten wichtig ist
• dies Gase sind unterschiedlich, also ∆S = Nk ln 2
• der Prozess ist aber komplett bis ins kleinste mikroskopische Detail iden-tisch mit einem Prozess, wo die Gase gerade nicht unterschiedlich sind,und hier ist ∆S = 0 !
Auflosung
• der TD Zustand ist charakterisiert durch wenige (typisch f = 2 − 4) Zu-standsgroßen X1, .., Xf und es ist S = S(X1, ..., Xf)
• Zustandsgroßen konnen auch einen Mikrozustand charakterisieren, mussen
aber nicht (Beispiel U vs. p, p nicht definiert wahrend freier Expansioneines Gases)
• ware S durch den Mikrozustand bestimmt, ware in der Tat ∆S = 0 plausi-bel, aber das ist falsch (Vorgriff auf SM: S hat zu tun mit dem “Volumen”einer Klasse von Mikrozustanden)
• ∆S kann also durchaus verschieden sein kann, auch wenn der Prozessmikroskopisch vollig identisch ablauft
• wichtig ist, dass zwischen den Gasen in anderen Experimenten diskrimi-niert werden kann, denn das bedeutet, dass ein reversibler Ruckprozesskonstruiert werden kann, also ∆S = Nk ln 2
Fazit
• Paradoxien entstehen nur, wenn man irrtumlich annimmt, dass die Entropieeine Funktion des Mikrozustands ist
78

Paradoxon
• man nehme an, man konnte die Gasmolekule “anmalen” (links rot, rechtsblau) und dies hatte keinerlei Einfluss auf deren Einzeltrajektorien - auchnicht in anderen Experimenten
• die Molekule und also die Gase sind dann verschieden also ∆S = NK ln 2
• der Mischungsprozess ware aber absolut bis ins kleinste Detail identischmit einem Prozess “gleichfarbiger” Molekule, fur den ∆S = 0
Auflosung
• Mischung produziert Entropie heißt: Man kann die Gase wieder entmischenund das System reversibel in den Ausgangszustand zuruckfuhren, wobeieine Anderung in der Umgebung zuruckbleibt
• dies ist hier nicht moglich, entsprechende semipermeable Wande lassensich nach Voraussetzung nicht konstruieren, der Ausgangszustand kannalso nicht wiederhergestellt werden
• die Variable “Farbe” ist also thermodynamisch sinnlos, TD Zustande konnennicht mit ihr unterschieden werden, es ist sinnlos, den “durchmischten” undden “geordneten” Zustand als verschieden zu betrachten, ∆S = 0
• naturlich ist es denkbar, jede einzelne Trajektorie zu verfolgen und zu be-einflussen, aber eine Beeinflussung des Systems mittels MakrovariablenX1, X2, ... ist nicht moglich
Fazit
• die Entropie ist ein Konzept zur Beschreibung von Makrozustanden undihrer Manipulation mittels Makrovariablen
79

Paradoxon
• ∆S = 0 fur gleiche Gase, aber was ware, wenn uns die Gase nur als gleicherschienen (ein Gas G), in Wirklichkeit aber verschieden sind (zwei Gase G1und G2)? (historisch ahnlich gelagerter Fall: die Entdeckung von Isotopen)
• was, wenn im Jahr 2100 die Substanzen Whifnium und Whafnium gefundenwerden, so dass das eine Gas (G1) durch Whifnium diffundieren kann (G2aber nicht) und umgekehrt Whafnium nur fur G2 durchlassig ist
• nach heutiger Kenntnis: ∆S = 0, zukunftig ∆S = Nk ln 2?
Auflosung
• im Jahr 2100 kann man die Gase trennen und somit das Durchmischenreversibel ruckgangig machen
• die Beschreibung durch ∆S = Nk ln 2 ist korrekt bei Verwendung einesentsprechenden Satzes von TD Zustandsgroßen (N1, N2, ...)
• die Definition von S hangt vom Satz der Zustandsgroßen ab und auchheute konnte man N1 und N2 wahlen, was aber sinnlos ware, wie durch“Farbe” markierte Molekule (s.o.), da man keine entsprechende Kontrolleuber die Makrovariablen hat
• der Wechsel der Beschreibung von heute zu derjenigen in 2100 reprasen-tiert verbesserte Fahigkeiten, und ∆S = Nk ln 2 hat im Jahr 2100 realeKonsequenzen:
• ∆S 6= 0 bedeutet, dass man das System (messbare!) Arbeit verrichtenlassen kann: Statt die irreversible Durchmischung zuzulassen, ist dazu le-diglich der reversible Ersatzprozess zu schalten (s. Rechnung)
• auch nach der Entdeckung der Substanzen Whifnium und Whafnium konn-te man aber auf einer Beschreibung durch die alten Makrovariablen beste-hen (dann: ∆S = 0)
Fazit
• die Entropie hangt von der Wahl des Satzes der Makrovariablen ab
• der Entropiebegriff ist also abhangig von der Beschreibungsebene
• der Entropiebegriff ist deswegen nicht “subjektiv”, denn Nk ln 2 kann prin-zipiell zur Verrichtung von Arbeit ausgenutzt werden (wenn man G1 undG2 handhaben kann)
80

• “Die Zahl der Fische, die man fangen kann, ist eine objektive Tatsache,hangt aber von der subjektiven Kenntnis uber das Verhalten von Fischenab” – S ist sozusagen die Zahl der Fische, die man fangen kann
81

Paradoxon
• man betrachte zwei Beobachter (Experte und Ignorant), die um die Exi-stenz von Whifnium und Whafnium und G1 und G2 wissen bzw. nichtwissen
• Experiment 1: spontane Durchmischung von G1 und G2 (Zustand Zini →Zfin):
fur beide Beobachter ist ∆W , ∆Q, ∆U = 0
fur den Ignoranten ist auch Zini = Zfin
• Experiment 2: mit semipermeablen Wanden werden G1 und G2 reversibelgemischt (Zustand Zini → Zfin quasistatisch)
jetzt wird die Arbeit ∆W < 0 frei, ∆Q > 0 wird also zugefuhrt (s.Rechnung)
fur beide Beobachter bleibt jetzt eine Anderung in der Umgebung bestehen(∆Q), die klar messbar ist
fur den Ignoranten gilt: ∆Q wird vollstandig in ∆W umgewandelt, sonstpassiert nichts, das ist ein perpetuum mobile 2. Art !
fur den Experten gilt: ∆Q wird zwar vollstandig in ∆W umgewandelt,aber das System ist nicht gleich geblieben (Zini 6= Zfin), seine Entropie ist∆S = Nk ln 2 gewachsen
• beide Beobachter konnten jetzt wiederum durch einen dritten ausgetrickstwerden, der weiss, dass G2 aus G2a und G2b besteht und der um dieExistenz von Whoofnium weiß etc.
Auflosung
• so ist es
Fazit
• der 2. Hauptsatz (Nichtexistenz eines ppm2) gilt nur, solange man sichfur einen Satz von Makrovariablen entschieden hat und auch bei dieserEntscheidung in der Erklarung von Phanomenen bleibt
• eine beobachtete Verletzung des 2. Hauptsatzes sollte also Anlass geben,nach der nicht in die Beschreibung einbezogenen Variable zu suchen
82

Kapitel 5
Elementare Statistik
Aufgaben der statistischen Mechanik
• mikroskopische Charakterisierung des thermodynamischen Gleichgewichts
• Nullter Hauptsatz: mikroskopische Definition der Temperatur
• Ableitung der Zustandsgleichung des klassischen idealen Gases
• Zustandsgleichung des idealen Paramagneten
• Ableitung der kalorischen Zustandsgleichung idealer Gase
• Definition der Warme
• Begrundung des ersten Hauptsatzes
• Begrundung des zweiten Hauptsatzes
• Anwachsen der Entropie inkonsistent mit Zeitinversionssymmetrie der dy-namischen Grundgleichungen der KM, QM?
• Definition der Entropie fur beliebige Nichtgleichgewichtszustande?
• Bestimmung thermodynamischer Potenziale aus der Hamilton-Funktion(aus dem Hamilton-Operator)
• dritter Hauptsatz
• Zustandsgleichungen des idealen Gases fur T → 0?
83

5.1 Wahrscheinlichkeit
mathematisch-axiomatische Einfuhrung des WK-Begriffs:
WK werden Ereignissen zugeordnet
Struktur des Ereignisraums fur konsistente WK-Definition? Kolmogorov (1933)
Menge Ω
ω ∈ Ω Ausgang eines Versuchs
A ⊂ Ω Ereignis
A = ω elementares Ereignis
E ⊂ A|A ⊂ Ω Ereignisraum
Def: E heißt Borel-Raum, falls
– Ω ∈ E– A ∈ E ⇒ A = Ω \ A ∈ E– A1, A2 ∈ E ⇒ A1 ∪ A2 ∈ Eeinfache Folgerungen:
– A1, A2 ∈ E ⇒ A1 ∩ A2 ∈ E– ∅ ∈ EDef: zwei Ereignisse A1, A2 heißen exklusiv falls
– A1 ∩ A2 = ∅Def: A1, ..., An ∈ E heißen Partition, falls
– Ai ∩ Aj = ∅ fur i 6= j
– A1 ∪ ... ∪ An = Ω
—————————————————————————————————
Beispiel: Wurfel
Ω = 1, 2, 3, 4, 5, 6 Versuchsausgange: i
elementares Ereignis: iein stets moglicher (trivialer) Borel-(Ereignis-)Raum:
E = A|A ⊂ Ωz.B. 1, ∅, Ω, 1, 3, 5 ∈ E
84

nicht-trivialer Borel-Raum:
seien 1, 2, 3, 4 ∈ E
∅, Ω ∈ E (immer)
3, 4, 5, 6 ∈ E 1, 2, 5, 6 ∈ E (Komplemente)
1, 2, 3, 4 ∈ E (Vereinigung)
5, 6 ∈ E (Komplement)
E = 1, 2, 3, 4, 5, 6, 1, 2, 3, 4, 1, 2, 5, 6, 3, 4, 5, 6, ∅, Ω ist einBorel-Raum
—————————————————————————————————
(mathematische) Wahrscheinlichkeit:
jedem Ereignis A ∈ E wird eine Zahl P (A) zugeordnet
P : E → R A → P (A)
wobei P bestimmte Eigenschaften hat (Regeln der WK-Rechnung):
– P (A) ≥ 0 fur alle A ∈ E– P (Ω) = 1
– fur A1, ..., An paarweise exklusiv: P (A1 ∪ ... ∪ An) = P (A1) + ... + P (An)
Summenregel
notwendige und hinreichende (!) Axiome fur komplette WK-Theorie
(auch fur n → ∞, abzahlbar unendlich)
weitere (ableitbare) WK-Regeln:
– P (A) ≤ 1
– P (∅) = 0
– A ⊂ B ⇒ P (A) ≤ P (B)
– P (A) = 1 − P (A)
– P (A ∪ B) + P (A ∩ B) = P (A) + P (B)
– fur Partition A1, ..., An gilt:n∑
i=1
P (Ai) = P (A1 ∪ ... ∪ An) = P (Ω) = 1
– fur Partition A1, ..., An gilt: P (B) =n∑
i=1
P (Ai ∩ B)
Marginalisierungsregel
85

Def: A, B ∈ E heißen unabhangig, falls
P (A ∩ B) = P (A)P (B)
Def: bedingte Wahrscheinlichkeit
P (A|B) =P (A ∩ B)
P (B)
alle WK-Regeln gelten auch fur bedingte WK
z.B. A1 ⊂ A2 ⇒ P (A1|B) ≤ P (A2|B)
bedingte WK sind WK
weiter ist:
P (A ∩ B) = P (A|B)P (B) = P (B|A)P (A) Produktregel
und somit:
P (A|B) =P (B|A)P (A)
P (B)Bayessches Theorem
fur Partition A1, ..., An:
P (B) =n∑
i=1
P (Ai ∩ B) =n∑
i=1
P (B|Ai)P (Ai)
Marginalisierungsregel
P (Ai|B) =P (B|Ai)P (Ai)∑n
i=1 P (B|Ai)P (Ai)
Bayessches Theorem
A, B unabhangig, falls
P (A|B) = P (A)
—————————————————————————————————
Probleme des mathematischen (axiomatischen) WK-Begriffs:
Bsp: gezinkte Munze, A = Kopf, A = Zahl
falls P (A) = 1−P (A) gegeben, kann WK berechnet werden, in n Wurfen k-malA zu finden, etc.
WK-Regeln proliferieren WKen
aber:
a priori WKen (z.B. P (A) im Bsp) beim axiomatischen Zugang offen
Problem der a priori WK
86

—————————————————————————————————
naive Definition der WK: Pascal, Fermat
P (A) =NA
N=
Anzahl gunstiger Falle
Anzahl moglicher Falle
Probleme des naiven Definition der WK:
Bsp: WK, 7 Augen bei zweimaligem Wurfeln zu bekommen:
N = 11: 2,3,...,12, NA = 1: 7
P (A) =1
11?? (korrekt ist P (A) = 1/6)
P (A) =NA
Nnur gultig, falls alle Falle gleichwahrscheinlich sind
was ist bei nicht gleichwahrscheinlichen Fallen?
noch schlimmer:
zyklische Definition!
—————————————————————————————————
statistische Definition der WK:
WK als empirisch bestimmbare Große
tritt Ereignis A in N wiederholten, gleichen Versuchen NA-mal auf, dann ist
NA
Ndie relative Haufigkeit
definiere:
P (A) = limN→∞
NA
N
naive sowie statistische Def. der WK liefern die obigen WK-Regeln !
Probleme der statistischen Definition der WK
– Messvorschrift mit limN→∞
nie durchfuhrbar, nur relative Haufigkeiten messbar
– N ist oft klein (teure Experimente)
– unwiederholbare “Versuche”: Astrophysik, also P undefiniert??
– “50% Regen-WK fur morgen”: Realisierung gleicher Versuchsbedingungen?
starke Einschrankungen
Vorgriff auf Statistische Physik: Festlegung der WK von mikroskopischen Zustandenso nicht moglich
87

—————————————————————————————————
umfassenderer WK-Begriff (Jaynes):
jede WK ist eine bedingte WK: P (A|Bedingungskomplex B)
Interpretation der bedingten WK als Implikationsmaß:
P (A|B) = 1 falls A sicher (zwingend ableitbar aus B)
P (A|B) = 0 falls A unmoglich (vor dem Hintergrund B)
P (A|B) < 1 falls A plausibel (P : Plausibilitatsgrad)
WK fur Aussagen (statt “Ereignisse”)
Aussagen konnen sicher (wahr), unmoglich (falsch) oder plausibel (wahr-scheinlich) sein
—————————————————————————————————
A1, A2 Aussagen A1 ∪ A2 Aussage
A1 ∪ A2 ⇔ A1 → A2 (wenn A1 dann A2)
Bsp: sei A1 ∈ B und A1 → A2 ∈ B, dann folgt A2
P (A2|B) = 1
gewohnliches logisches Schließen kann durch WK ausgedruckt werden
—————————————————————————————————
Bsp: A1: “Es hat geregnet”, A2: “Die Straße ist nass”
es ist: P (A2|A1) = 1 (weitere Bedingungen B (Alltagswissen) nicht explizitangegeben)
P (A1|A2) =P (A2|A1)P (A1)
P (A2)Bayessches Theorem
also:
P (A1|A2) =P (A1)
P (A2)
und somit:
P (A2) > P (A1)
nasse Straße plausibler als Regen! (Straßenreinigung etc.)
Implikationsmass als Erweiterung der Logik
—————————————————————————————————
Bsp: gezinkte Munze, A = Kopf, A = Zahl
88

B gebe keinerlei Bevorzugung von A oder A
P (A|B) = P (A|B) = 1/2 Laplacesches Indifferenzprinzip
beachte:
• keine Interpretation von P (A|B) als messbare Eigenschaft der Munze
• reine Symmetrieuberlegung
• nach vielen Wurfen (z.B. mit Ausgang “Kopf” bei allen) andert sich B →B′ (und somit P (A|B′) ≈ 1)
Symmetrie von Aussagen (vor gegebenem Bedingungskomplex) erlaubt Fest-legung von a priori WKen
Vorgriff auf Statistische Physik: gleichwahrscheinliche Mikrozustande bei prak-tisch vollstandig fehlender Information
Formalisierung dieser Idee:
5.2 Shannon-Information
Versuch mit endlich vielen Ausgangen k = 1, ..., M (z.B. Wurfel)
Ω = 1, 2, ..., MElementareignisse:
Ak = kEreignisraum
E = A|A ⊂ ΩWK fur Elementarereignis Ak:
P (Ak) = pk (i.allg. verschieden)
Elementarereignisse bilden eine Partition:
Ai ∩ Aj = ∅ A1 ∪ ... ∪ AM = Ω
—————————————————————————————————
Def: I(k), Informationswert von Ak: Anzahl der notwendigen, vorab festgeleg-ten binaren Fragen zu dessen vollstandiger Festlegung × ln 2
Bsp: M = 2r, pk = p =1
M
89

r binare Fragen, also
I(k) = r ln 2 = log2 M · ln 2 = ln M = − ln pk
gleichwahrscheinliche Ereignisse Ak:
I(k) = − ln pk
—————————————————————————————————
allgemeinerer Algorithmus zur Bestimmung von I(k):
(1) setze Ω′ = Ω
(2) partitioniere Ω = Ω1 ∪ Ω2, Ω1 ∩ Ω2 = ∅, so dass
P (Ω1) = P (Ω2) =1
2
(3) setze Ω′ = Ω1 falls k ∈ Ω1, sonst Ω′ = Ω2
(4) pk → 2pk fur alle k ∈ Ω′
(5) gehe zu (2) und iteriere bis pk = 1
jetzt ist:
I(k) = Anzahl der Iterationen × ln 2 = r ln 2
(denn: r = Anzahl der Fragen)
weiter gilt:
pk → 2pk → · · · → 2rpk = 1
also:
pk =1
2rr = − log2 pk
und
I(k) = r ln 2 = − log2 pk · ln 2 = − ln pk
Algorithmus setzt oben benutzte Partitionierbarkeit voraus, kein Problem furgroße M
definiere jetzt allgemein:
I(k) = − ln pk Informationswert von Ak
“Wert der Information, die man erhalt, wenn einem das sichere Vorliegen vonEreignis Ak mitgeteilt wird”, Neuigkeitswert
—————————————————————————————————
90

es ist:
• I(k) ≥ 0, denn pk ≤ 1
• I(k) = 0 ⇔ pk = 1, kein Informationsgewinn bei Mitteilung eines sowiesoschon sicheren Ereignisses
• I(k) → ∞ fur pk → 0, je unwahrscheinlicher das Ereignis desto großer derInformationswert
der mittlere Informationswert ist gegeben durch
σ =∑
k
pkI(k) = −∑
k
pk ln pk Shannon-Information
sind die pk bekannt, ist σ = σ(p1, ..., pM) ein Maß fur die Unkenntnis uber denVersuchsausgang
—————————————————————————————————
Eigenschaften der Shannon-Information:
• σ(p1, ..., pM) ≥ 0
• σ(p1, ..., pM) = 0 ⇔ pk = δkk0 fur ein k0
(minimale Unkenntnis)
• Beitrag von k zu σ klein fur
pk → 1 (kaum neue Info)
oder
pk → 0 (wegen x ln x → 0 unwichtig fur mittlere Info)
• fur pk =1
M= const. (Gleichverteilung) ist
σ(p1, ..., pM) = −M∑
k=1
1
Mln
1
M= lnM
• Monotonie
σM ≡ σ(1
M, ...,
1
M) monoton wachsend
(großere Unkenntnis bei wachsender Anzahl gleichwahrscheinlicher Moglich-keiten)
• Stetigkeit
σ(p1, ..., pM) stetig in allen Argumenten
91

• Additivitat
σ(p1, ..., pM) = σ(p, p3, ..., pM) + pσ(p1
p,p2
p)
fur p = p1 + p2
(folgt durch direktes Nachrechnen)
Unkenntnis unabhangig davon, ob EE in Klassen zusammengefasst werden
Monotonie, Stetigkeit und Additivitat bestimmen σ eindeutig bis auf eine Kon-stante (Shannon)
—————————————————————————————————
Bedeutung (und Verallgemeinerung) der Additivitat:
fasse EE A1, ..., AM in Klassen B1, ..., BN zusammen (N ≥ M):
B1 = A1 ∪ ... ∪ Ar
B2 = Ar+1 ∪ ... ∪ Ar+s
...
es ist:
P (B1) = p1 + ... + pr ≡ w1
P (B2) = pr+1 + ... + pr+s ≡ w2
...
Unkenntnis uber A1, ..., AM gegeben durch Unkenntis uber Klassen B1, ..., BN
plus, gegeben dass B1 (WK w1), Unkenntnis uber A1, ..., Ar plus ...
(1 Gewinnlos in einer von M Schachteln ⇔ Gewinnlos in einer von N Boxen mitjeweils r, s, ... Schachteln)
σ(p1, ..., pM) = σ(w1, ..., wN)
+w1σ(
p1
w1, ...,
pr
w1
)
+w2σ(
pr+1
w2, ...,
pr+s
w2
)
...
denn (z.B.) bedingte WK fur A1, gegeben dass B1, ist (mit Bayesschem Theo-rem)
P (A1|B1) = P (A1|A1 ∪ ... ∪ Ar)
=P (A1 ∪ ... ∪ Ar|A1)P (A1)∑ri=1 P (A1 ∪ ... ∪ Ar|Ai)P (Ai)
92

=P (A1)∑ri=1 P (Ai)
=p1
p1 + ... + pr=
p1
w1
—————————————————————————————————
Beweis(skizze) des Shannonschen Theorems:
betrachte p1 = ... = pM = 1/M als Spezialfall
σ(1/M, ..., 1/M) = σ(w1, ..., wN)+w1σ(1/Mw1, ..., 1/Mw1)+w2...
also:
σM = σ(w1, ..., wN) +N∑
l=1
wlσ(1/Mwl, ..., 1/Mwl)
und somit
σ(w1, ..., wN) = σM −N∑
l=1
wlσMwl
fur w1 = ... = wN = 1/N ist speziell:
σN = σM − σM/N
Funktionalgleichung mit Losung
(hier strenggenommen Approximation reeller durch rationale Zahlen notwendig Stetigkeit)
σN = σ0 lnN
Konstante σ0 > 0 (Monotonie)
einsetzen:
σ(w1, ..., wN) = σ0 ln M −N∑
l=1
wlσ0 ln(Mwl) = −σ0
N∑
l=1
wl lnwl
—————————————————————————————————
Prinzip der Maximierung der Shannon-Information:
a priori WK konnen durch Forderung nach maximaler Unkenntnis (evtl.unter Nebenbedingungen) festgelegt werden
einzig rationale Moglichkeit
andere Festlegungen der a priori WK sind nicht “unvoreingenommen”
—————————————————————————————————
Beispiel:
gezinkte Munze, WK p1, p2 mit p1 + p2 = 1 (Nebenbedingung)
93

σ(p1, p2) = −p1 ln p1 − p2 ln p2
σ(p1, p2) − λ(p1 + p2) = max.
∂
∂pi(σ(p1, p2) − λ(p1 + p2)) = 0
− ln pi − 1 − λ = 0
pi = e−1−λ = const.
wahle Lagrange-Parameter λ so, dass NB erfullt, also
p1 = p2 =1
2Laplacesches Indifferenzprinzip
—————————————————————————————————
(unfairer) “Wurfel” mit N Seiten, Seite i mit Xi = i Augen, WK: pi
gegeben: mittlere Augenzahl 〈X〉 ≡N∑
i=1
piXi = X0
Maximierung der Shannon-Information unter 2 NB:
0 =∂
∂pi
−
N∑
j=1
pj ln pj − µ∑
j
pj − λ∑
j
pjXj
0 = − ln pi − 1 − µ − λXi
also:
pi = exp(−1 − µ − λXi) = const. exp(−λXi)
µ: Normierungsbedingung
pi =e−λXi
∑Ni=1 e−λXi
λ: Festlegung des Erwartungswerts
X0 =
∑i Xie
−λXi
∑i e
−λXi= X0(λ) λ(X0)
z.B. X0 =∑
i
Xi/N λ = 0
5.3 Zufallsvariable
betrachte Ereignisse der Form
Ai = X hat den Wert xi (xi reell)
94

– X heißt Zufallsvariable
– Wertebereich x1, x2, ... (diskret)
– p : x1, x2, ... → R mit p(xi) = P (Ai) WK-Funktion (WK-Verteilung)
A1, A2, ... bilden eine Partition ∑
i
p(xi) = 1
—————————————————————————————————
X Zufallsvariable Xn Zufallsvariable
Def: n-tes Moment von X
〈Xn〉 =∑
i
xni p(xi)
grobe Charakterisierung der WK-Funktion
Erwartungswert von X (Mittelwert)
〈X〉 =∑
i
xip(xi)
Varianz von X (mittlere quadratische Schwankung)
σ2 = 〈(X − 〈X〉)2〉 = 〈X2 − 2〈X〉X + 〈X〉2〉 = 〈X2〉 − 〈X〉2
Standardabweichung
σ =√〈X2〉 − 〈X〉2
—————————————————————————————————
Bsp: der Informationswert I(k) ist eine Zufallsvariable
Erwartungswert: 〈I〉 =∑
k
I(k)pk = σ
(hier ist speziell I(k) = − ln pk eine Funktion der pk)
—————————————————————————————————
kontinuierlicher Wertebereich: kontinuierliche Zufallsvariable
ρ(x) WK-Dichte
ρ(x)dx WK, dass X ∈ [x, x + dx]
Normierung:∫ ∞
−∞dxρ(x) = 1
Momente:
95

〈Xn〉 =∫ ∞
−∞dxxnρ(x)
—————————————————————————————————
WK-Dichte fur diskrete Zufallsvariable:
ρ(x) =∑
i
p(xi)δ(x − xi) (normiert!)
Bsp:
〈X〉 =∫
dx xρ(x) =∑
i
p(xi)∫
dx xδ(x − xi) =∑
i
p(xi)xi
WK-Dichte allgemeiner als WK-Funktion
—————————————————————————————————
charakteristische Funktion:
ϕ(k) =∫
dxeikxρ(x)
(Fourier-Transformation der WK-Dichte)
Taylor-Entwicklung:
ϕ(k) =∫
dx∞∑
n=0
(ikx)n
n!ρ(x)
=∞∑
n=0
(ik)n
n!
∫dxxnρ(x) =
∞∑
n=0
(ik)n
n!〈Xn〉
ϕ(k) = 〈eikX〉
charakteristische Funktion erzeugt Momente / Kumulanten
Momente:
〈Xn〉 =1
indn
dknϕ(k)
∣∣∣∣k=0
Kumulanten:
Cn(X) =1
indn
dknln ϕ(k)
∣∣∣∣k=0
speziell:
C0(X) = 0
C1(X) = 〈X〉C2(X) = 〈X2〉 − 〈X〉2 (“zentriert”)
C3(X) = 〈X3〉 − 3〈X2〉〈X〉 + 2〈X3〉
96

...
—————————————————————————————————
X Zufallsvariable Y = F (X) Zufallsvariable
ρ(x) gegeben, ρ(y) gesucht (ρ(x) kurz fur ρX(x))
angeben lasst sich zunachst:
bedingte WK dafur, dass Y in dy bei y, falls X = x:
ρ(y|x)dy = δ(y − F (x))dy (beachte:∫
dyρ(y|x) = 1)
Marginalisierungsregel: P (B) =∑
i
P (Ai)P (B|Ai), kontinuierlich:
ρ(y)dy =∫
dx ρ(x) ρ(y|x)dy
also:
ρ(y) =∫
dx ρ(x) δ(y − F (x))
ρ(y) = 〈δ(y − F (X))〉
sind xi(y) die Losungen von y = F (x), dann ist
δ(y − F (x)) =∑
i
1
|F ′(xi(y))|δ(x − xi(y))
also:
ρ(y) =∑
i
1
|F ′(xi(y))| ρ(xi(y))
—————————————————————————————————
k Zufallsvariablen X1, ..., Xk
gemeinsame WK-Funktion/WK-Dichte: ρ(x1, ..., xk)
– ρ(x1, ..., xk) ≥ 0
–∫
dx1 · · · dxkρ(x1, ..., xk) = 1 (kontinuierlich)
–∑
x1
· · ·∑
xk
p(x1, ..., xk) = 1 (diskret)
– 〈Xni 〉 =
∫dx1 · · ·dxk xn
i ρ(x1, ..., xk) n-tes Moment von Xi
– 〈Xni 〉 =
∑
x1
· · ·∑
xk
xni p(x1, ..., xk)
– Erwartungswert, Varianz von Xi
97

Def: Kovarianz von Xi und Xj:
cov(Xi, Xj) = 〈(Xi − 〈Xi〉)(Xj − 〈Xj〉)〉 = 〈XiXj〉 − 〈Xi〉〈Xj〉Def: Korrelation von Xi und Xj:
cor(Xi, Xj) =cov(Xi, Xj)
σXiσXj
es gilt:
cov(Xi, Xi) = 〈X2i 〉 − 〈Xi〉2 = σ2
Xi
cor(Xi, Xi) = 1
|cor(Xi, Xj)| ≤ 1
Def: X1, ..., Xk heißen unabhangig, falls
ρ(x1, ..., xk) = ρ(x1) · · · ρ(xk)
sind X und Y unabhangig, dann ist
〈XY 〉 = 〈X〉〈Y 〉und somit auch cov(X, Y ) = cor(X, Y ) = 0 (dies ist aber nicht hinreichend!)
weiter gilt: σ2X+Y = σ2
X + σ2Y
die Varianzen unabhangiger Zufallsvariablen sind additiv
5.4 Zentraler Grenzwertsatz
seien X1, ..., XN unabhangige Zufallsvariable und
Y =1
N
N∑
i=1
Xi
im Grenzfall N → ∞ gilt:
Y ist normalverteilt
mit
〈Y 〉 =1
N
N∑
i=1
〈Xi〉
σ2Y =
1
N2
N∑
i=1
σ2Xi
(Xi muss endliche Momente besitzen)
98

—————————————————————————————————
falls 〈Xi〉 = 〈X〉 und σXi= σX , ist
〈Y 〉 = 〈X〉 σ2Y =
1
Nσ2
X
Diskussion:
• Z =N∑
i=1
Xi typische Form extensiver Zustandsgroßen
Xi Mikrovariable eines Teilchens i
• Z/N normalverteilt (falls Xi unabhangig)
Varianz: σ2Z/N = O(1/N) falls σ2
X = O(1)
Standardabweichung: σZ/N = O(1/√
N)
Standardabweichung: σZ = O(√
N)
relative Standardabweichung: σZ/〈Z〉 = O(1/√
N) → 0
• sichere Aussagen trotz statistischer Beschreibung im Limes N → ∞thermodynamischer Limes
—————————————————————————————————
Beweis:
ρ(y) = 〈δ(y − N−1∑
i
Xi)〉
charakteristische Funktion:
ϕ(k) =∫
dy eikyρ(y) =∫
dy eiky〈δ(y − N−1∑
i
Xi)〉
= 〈ei kN
∑iXi〉 = 〈
∏
i
ei kN
Xi〉 =∏
i
〈ei kN
Xi〉 (Unabhangigkeit !)
also:
ϕY (k) =∏
i
ϕXi(k/N)
Taylor-Entwicklung:
ln ϕXi(k/N) =
∞∑
n=0
(ik/N)n
n!Cn(Xi)
= C0(Xi) + ik
NC1(Xi) −
1
2
k2
N2C2(Xi) + O(k3/N3)
99

mit C0 = 0, C1 = 〈Xi〉, C2 = σ2Xi
folgt:
ϕXi(k/N) = exp
(ik
N〈Xi〉 −
1
2
k2
N2σ2
Xi+ O(k3/N3)
)
somit:
ϕY (k) = exp
(ik
N
∑
i
〈Xi〉 −1
2
k2
N2
∑
i
σ2Xi
+ NO(k3/N3)
)
fur N → ∞ und |k| = O(N2/3) ist
Term 3 = O(1) ≪ Term 2 = O(N1/3) ≪ Term 1 = O(N2/3)
(fur |k| ≤ O(N2/3) dann sicher auch)
ρ(y) =1
2π
∫dke−ikyϕY (k) =
1
2π
∫dke−iky exp
(ik
N
∑
i
〈Xi〉 −1
2
k2
N2
∑
i
σ2Xi
)
definiere
〈Y 〉 ≡ 1
N
N∑
i=1
〈Xi〉 σ2Y ≡ 1
N2
N∑
i=1
σ2Xi
dann ist:
ρ(y) =1
2π
∫dke−iky exp
(ik〈Y 〉 − 1
2k2σ2
Y
)=
1√2πσ2
Y
exp
(−(y − 〈Y 〉)2
2σ2Y
)
(Gauß-Integral)
also Y normalverteilt und 〈Y 〉, σ2Y Mittelwert bzw. Varianz von Y
beachte: signifikante Beitrage zum Integral nur fur k mit
|k| ≤ O(1/σY ) = O(N1/2) ≤ O(N2/3)
Anwendung auf:
5.5 Binomialverteilung
Xi fur i = 1, ..., N unabhangige Zufallsvariable mit Wertebereich 1, 0WK-Funktion:
p(1) = p p(0) = q (fur alle i gleich)
N -maliges Werfen einer (gezinkten) Munze
100

X =N∑
i=1
Xi
Wertebereich:
0, 1, .., NWK-Funktion:
pN(n) n = 0, 1, ..., N
Erwartungswert und Varianz von X fur N → ∞ ?
—————————————————————————————————
Anwendung des ZGWS:
Y ≡ 1
N
N∑
i=1
Xi =X
N
ist normalverteilt (Gauß) mit
〈Y 〉 =1
N
N∑
i=1
〈Xi〉 = 〈Xi〉
σ2Y =
1
N2
N∑
i=1
σ2Xi
=1
Nσ2
Xi
es ist
〈Xi〉 = p · 1 + q · 0 = p
σ2Xi
= 〈X2i 〉 − 〈Xi〉2 = p · 12 + q · 02 − p2 = p(1 − p)
und somit
〈X〉 = N〈Y 〉 = pN
σ2X = N2σ2
Y = N2 1
Np(1 − p) = p(1 − p)N
wiederum:
σX
〈X〉 =
√Np(1 − p)
Np=
√1 − p
p
1√N
= O(N−1/2)
—————————————————————————————————
direkte Rechnung (N beliebig)
WK fur n-mal 1 bei N Wurfen in ganz bestimmter Reihenfolge:
pnqN−n (Produktregel, Unabhangigkeit)
Anzahl der Moglichkeiten n-mal 1 zu finden:
101

(N
n
)=
N !
n!(N − n)!
WK fur n-mal 1 bei N Wurfen: (Summenregel, Exklusivitat)
pN (n) =(
N
n
)pnqN−n Binomialverteilung
es gilt:
N∑
n=0
pN(n) =N∑
n=0
(N
n
)pnqN−n = (p + q)N = 1
Erwartungswert:
〈X〉 =N∑
n=0
npN (n) =N∑
n=0
p∂
∂ppN (n)
∣∣∣∣q=1−p
= p∂
∂p
N∑
n=0
pN(n)
∣∣∣∣q=1−p
= p∂
∂p(p + q)N
∣∣∣∣q=1−p
= pN(p + q)N−1∣∣∣∣q=1−p
= pN
also:
〈X〉 = pN
〈X2〉 =N∑
n=0
n2pN (n) =N∑
n=0
(p
∂
∂p
)2
pN(n)∣∣∣∣q=1−p
=
(p
∂
∂p
)2
(p + q)N∣∣∣∣q=1−p
= p∂
∂ppN(p + q)N−1
∣∣∣∣q=1−p
= pN(p + q)N−1∣∣∣∣q=1−p
+ p2N(N − 1)(p + q)N−2∣∣∣∣q=1−p
= pN + p2N(N − 1) = (pN)2 + (p − p2)N
Varianz:
σ2X = 〈X2〉 − 〈X〉2 = (p − p2)N
also:
σ2X = p(1 − p)N
in Ubereinstimmung mit ZGWS
5.6 Stirling-Formel
Approximation der Fakultat:
102

n! =√
2πn nne−n(1 + O(1/n))
ln n! = n lnn − n +1
2ln n + ln
√2π + O(1/n)
Approximation von n! mit verschwindendem relativen Fehler:
n! −√
2πn nne−n
n!= O(1/n)
Approximation von ln n! mit verschwindendem absoluten Fehler:
ln n! − (n ln n − n +1
2lnn + ln
√2π) = O(1/n)
Approximation von ln n! mit verschwindendem relativen Fehler:
ln n! − (n ln n − n)
ln n!= O(1/n)
ln n! − n ln n
lnn!= O(1/ lnn)
Beweis mit Hilfe der Sattelpunktsmethode (s.u.)
—————————————————————————————————
Beispiel Binomialverteilung:
pN(n) =N !
n!(N − n)!pn(1 − p)N−n
fur N, n, N − n groß ist (Vernachlassigung log. Korrekturen)
ln pN(n) = lnN !
n!(N − n)!pn(1 − p)N−n
= ln N ! − ln n! − ln(N − n)! + ln pn + ln(1 − p)N−n
≈ N ln N −N − n ln n + n− (N − n) ln(N − n) + N − n + n ln p
+(N − n) ln(1 − p)
= N ln N −n ln n− (N −n) ln(N −n)+n ln p+(N −n) ln(1−p)
Maximum von pN(n):
0 =∂
∂nln pN(n) ≈ − ln n − 1 + ln(N − n) + 1 + ln p − ln(1 − p)
= ln
(N − n
n
p
1 − p
)
also:
nmax ≈ pN = 〈X〉
103

wahrscheinlichster Wert ≈ Mittelwert bei scharfer Verteilung
104

Kapitel 6
Zufallsgroßen im Zustandsraum
mikroskopische Theorie Thermodynamik ?
Situation der KM / QM:
• dynamische Grundgleichungen bekannt
• N ∼ NA ∼ 1023
• Anfangsbedingungen nicht kontrollierbar
• (bei weitem) unzureichende Information uber Mikrozustand
• statistische Bescheibung: Mikrozutand k liegt mit WK pk vor
• Bestimmung der pk uber Maximierung der Shannon-Information
6.1 Klassische Mechanik, Phasenraum
N -Teilchen System, j = 1, ..., N
f = 3N Freiheitsgrade, i = 1, ..., f
(Mikro-)Zustand:
π = (q, p) = (q1, ..., qf , p1, ..., pf) “Phase”
qi: verallgemeinerte Koordinaten, pi: verallgemeinerte Impulse
Zustandsraumm:
π = q, p Phasenraum
zeitliche Entwicklung des Zustands:
105

π = π(t) = (q(t), p(t) Phasentrajektorie
Anfangsbedingung:
π(t0) = π0
Dynamik: Hamilton-Funktion (konservatives Hamiltonsches System)
H = H(π) = H(q, p) = H(q1, ..., qf , p1, ..., pf) = T + V
Bewegungsgleichungen:
qi =∂H(q, p)
∂pi
pi = −∂H(q, p)
∂qi
(Hamilton-Gleichungen)
—————————————————————————————————
Beispiele:
H =N∑
j=1
p2j
2m+ V (r1, ..., rN)
ideales Gas im Volumen V :
V (r1, ..., rN) = VWand(r1) + · · ·+ VWand(rN)
mit
V (r) = 0 fur r ∈ V
V (r) = ∞ fur r /∈ V
Gitterschwingungen:
V (r1, ..., rN) =f∑
i=1
1
2mω2x2
i
—————————————————————————————————
Observable: reelle Zustandsfunktionen
A = A(q, p)
Bsp.: H = H(q, p)
zeitliche Entwicklung:
A(t) = A(q(t), p(t))
Bewegungsgleichung:
dA(t)
dt=∑
i
(∂A
∂qi
qi +∂A
∂pi
pi
)
106

=∑
i
(∂A
∂qi
∂H
∂pi− ∂A
∂pi
∂H
∂qi
)
definiere Poisson-Klammer:
A, B =∑
i
(∂A
∂qi
∂B(q, p)
∂pi− ∂A
∂pi
∂B(q, p)
∂qi
)
damit:
dA(t)
dt= A, H
speziell:
qi = qi, H pi = pi, HErhaltungsgroßen:
A, H = 0 dA
dt= 0 A = const.
Energieerhaltung:
H, H = 0
die Phasentrajektorie liegt in der (6N-1)-dim. Hyperflache H(q, p) = E = const.
6.2 Gemischter Zustand, statistische Gesamtheit
Mikrozustand:
π reiner Zustand
bei unvollstandiger Information:
πk mit WK p(πk) = pk gemischter Zustand
gemischter Zustand = Satz von reinen Zustanden mit zugehorigen WK
unvollstandig praparierte Systeme (unbekannte Anfangsbedingungen) wer-den durch einen gemischten Zustand beschrieben, d.h. durch eine WK-Funktion
Zustand πk ist eine Zufallsgroße
Observable A = A(πk) = Ak ist eine Zufallsgroße
statistische statt deterministische Beschreibung
107

q
p
π
reiner Zustand
πk kp
q
pgemischter Zustand
Punkte: System in verschienen moglichen reinen Zustanden
—————————————————————————————————
Beschreibung durch WK-Dichte:
ρ(π) =∑
k
pkδ(π − πk)
Dichte im 6N -dimensionalen Phasenraum!
ρ(π) = ρ(q, p) = ρ(q1 · · · qf , p1 · · · pf)
Normierung:∫
dπρ(π) =∫
dπ∑
k
pkδ(π − πk) =∑
k
pk = 1
ρ(q, p)dqdp = WK, System bei (q, p) in Phasenvolumen dqdp zu finden
gemischter Zustand durch ρ(q, p) vollstandig charakterisiert
Erwartungswert einer Observablen A = A(q, p):
〈A〉 =∫
dqdp ρ(q, p)A(q, p)
oder:
〈A〉 =∫
dπ∑
k
pkδ(π − πk)A(π) =∑
k
pkA(πk)∫
dπ∑
k
δ(π − πk)
〈A〉 =∑
k
pkA(πk)
Varianz: analog
—————————————————————————————————
Ubergang kontinuierlich diskretisiert
sei ρ(q, p) = ρ(π) gegeben
Einteilung des Phasenraums in Zellen k, in jeder Zelle reiner Zustand πk
108

∆ q
∆p
q
p
πk
Zelle k
Phasenvolumen der k-ten Zelle: ∆v = ∆q∆p
definiere:
pk =∫
Zelle kdqdp ρ(q, p) (es ist:
∑k pk = 1)
fur kleine ∆q, ∆p aquivalente Bescheibung desselben gemischten Zustands:
ρ(π) ≈∑
Zelle k
pkδ(π − πk)
—————————————————————————————————
Shannon-Information:
σ = σ(p1, ..., pk, ...) = −∑
k
pk ln pk
= − lim∆v→0
∑
k
ρ(πk)∆v ln(ρ(πk)∆v)
= − lim∆v→0
∫dπρ(π) ln(ρ(π)∆v)
= −∫
dπρ(π) ln ρ(π) − lim∆v→0
∫dπρ(π) ln∆v
= −∫
dπρ(π) ln ρ(π) − lim∆v→0
ln∆v → ∞
divergent! (ρ(π) 6= 0 uberall)
Shannon-Information nur fur diskrete Darstellung
physikalische Dimension des Zellenvolumens: (Wirkung)3N
∆v = ∆q∆p =3N∏
i=1
∆qi∆pi
sinnvolle Wahl z.B. ∆v = h3N
QM: ∆qi∆pi ≥~
2—————————————————————————————————
reiner Zustand: pk = δkk0
σ(pk) = −∑
k
pk ln pk = 0 (minimal)
109

minimale Unkenntnis
gemischter Zustand maximaler Unkenntnis?
0 =∂
∂pkσ(pk) − λ
∑
k
pk = − ln pk − 1 − λ
pk = const.
jeder Mikrozustand (reiner Zustand) gleichwahrscheinlich
—————————————————————————————————
Interpretation der pk als relative Haufigkeiten (statistische Def. der WK):
pk =Mk
M
M identische Kopien des Systems, davon Mk in Zelle k
statistisches Ensemble / statistische Gesamtheit
• M → ∞ bzw. sehr viele mehr hypothetische Systeme als Zellen notwendig
• tatsachliches Messen der WK’s in Praxis unmoglich, zeigt wiederum dieSchwierigkeit, WK als messbare Große aufzufassen
6.3 Liouville-Gleichung
zeitliche Entwicklung eines gemischten Zustands?
System z.Zt. t0:
πk(t0) mit WK pk
reiner Zustand πk = πk(t) gemaß Hamilton-Gleichungen
System z.Zt. t:
πk(t) mit WK pk
WK-Dichte z.Zt. t0:
ρ(π, t0) = ρ0(π)
in diskretisierter Darstellung:
ρ(π, t0) =∑
k
pkδ(π − πk(t0))
WK-Dichte z.Zt. t:
110

ρ(π, t) =∑
k
pkδ(π − πk(t))
Bewegungsgleichung ?
beachte: ρ(π, t) 6= ρ(π(t)), Bewegungsgleichung fur Phasenfunktion ρ(t) ≡ρ(π(t)): ρ(t) = ρ, Hstatt dessen: (beachte: πk = (qk1, ..., qkf , pk1, ..., pkf))
∂
∂tρ(π, t) =
∑
k
pk∂
∂tδ(2f)(π − πk(t)) (f = 3N)
(Kettenregel!)
= −∑
k
pk
f∑
i=1
[qki(t)
∂
∂qiδ(2f)(q − qk(t), p − pk(t)) + pki(t)
∂
∂piδ(2f)(q − qk(t), p − pk(t))
]
= −∑
k
pk
f∑
i=1
[∂H
∂pi
(qk(t), pk(t))∂
∂qi
δ(2f)(q − qk(t), p − pk(t))
−∂H
∂qi(q(t), p(t))
∂
∂piδ(2f)(q − qk(t), p − pk(t))
]
= −∑
k
pk
f∑
i=1
[∂H
∂pi(q, p)
∂
∂qi− ∂H
∂qi(q, p)
∂
∂pi
]δ(2f)(q − qk(t), p − pk(t))
= −f∑
i=1
[∂H
∂pi(q, p)
∂
∂qi− ∂H
∂qi(q, p)
∂
∂pi
]∑
k
pkδ(π − πk(t))
= −f∑
i=1
[∂H(π)
∂pi
∂ρ(π, t)
∂qi− ∂H(π)
∂qi
∂ρ(π, t)
∂pi
]
=f∑
i=1
[∂H(π)
∂qi
∂ρ(π, t)
∂pi
− ∂H(π)
∂pi
∂ρ(π, t)
∂qi
]
also:
∂
∂tρ(π, t) = H, ρ(π, t) Liouville-Gleichung
—————————————————————————————————
es gilt:
∂
∂tρ(π, t) = H, ρ(π, t) =
f∑
i=1
[∂H
∂qi
∂ρ
∂pi− ∂ρ
∂qi
∂H
∂pi
]
=f∑
i=1
[∂
∂pi
(∂H
∂qiρ
)− ∂
∂qi
(∂H
∂piρ
)]
111

= −f∑
i=1
[∂
∂pi(piρ) +
∂
∂qi(qiρ)
]
= −2f∑
j=1
∂
∂πj
(ρvj)
mit:
vj = qj fur j = 1, .., f
vj = pj−f fur j = f + 1, .., 2f
also:
v = π (2f -dimensionale Vektoren)
damit:
∂
∂tρ(π, t) + ∇(ρ(π, t)v(π, t)) = 0 Kontinuitatsgleichung
definiere Stromdichte (Wahrscheinlichkeitsstrom):
j(π, t) = ρ(π, t)v(π, t)
∂
∂tρ + divj = 0
fur endlichen Bereich im Phasenraum B:
d
dt
∫
Bdπρ(π, t) = −
∫
∂Bj(π, t)dA
Wahrscheinlichkeitserhaltung
—————————————————————————————————
weiter gilt:
divv =f∑
i=1
(∂
∂qiqi +
∂
∂pipi
)=
f∑
i=1
(∂2H
∂qi∂pi− ∂2H
∂pi∂qi
)= 0
Liouvillscher Satz
Bedeutung von divv = 0 in der Hydrodynamik: inkompressible Flussigkeit
sei ρ(π, t) 6= 0 innerhalb B und ρ(π, t′) 6= 0 innerhalb B′
Phasenraumvolumina von B, B′ gleich!
—————————————————————————————————
Observable A = A(π) = A(π(t)) = A(t)
112

zeitliche Anderung von Mittelwerten?
d
dt〈A(t)〉 =
d
dt
∑
k
pkA(πk(t)) =∑
k
pkA, H(πk(t)) = 〈A, H(t)〉
oder:
d
dt〈A(t)〉 =
d
dt
∫dπρ(π, t)A(π) =
∫dπ
∂ρ(π, t)
∂tA(π) =
∫dπH, ρ(π, t)A(π)
Thermodynamisches Gleichgewicht: keine zeitliche Anderung von Makrovariablen
〈A(t)〉 = const.
fur ρ(π) = ρ(H(π)) gilt:
H, ρ(H) = 0
also (mit Liouville-Gleichung):
ρ(π, t) = ρ(π) = const.
Erwartung: das TD GG ist durch einen gemischten Zustand ρ = ρ(H) gegeben
6.4 Quantenmechanik, Hilbert-Raum
Zustand
|Ψ〉 ∈ H mit 〈Ψ|Ψ〉 = 1
bzw.
|Ψ〉〈Ψ| (Projektor)
Zustandsraum:
|Ψ〉 = H Hilbert-Raum
Observable: hermitesche Operatoren auf H:
A = A†
mogliche Messwerte: Eigenwerte ar von A
A|ar, γ〉 = ar|ar, γ〉 (γ = 1, ..., gr, gr: Entartung von ar)
|ar, γ〉 Eigenzustand von A zum Eigenwert ar
QM:
• keine sichere Vorhersage einzelner Messungen
113

• prinzipielle Unvorhersagbarkeit, auch bei vollstandiger Praparation ei-nes reinen Zustands |Ψ〉
• objektive WK
• Quantenstatistik: gemischte Zustande mit weiteren (“subjektiven”) WK
Messung von A am System im Zustand |Ψ〉 liefert ar mit WK
P (ar) = |〈ar|Ψ〉|2 = 〈Ψ|ar〉〈ar|Ψ〉 = 〈Π(ar)〉ΨΠ(ar): Projektor auf den Unterraum zum Eigenwert ar
bei Entartung von ar:
P (ar) =∑
γ
|〈ar, γ|Ψ〉|2 = 〈Ψ|gr∑
γ=1
|ar, γ〉〈ar, γ|Ψ〉 = 〈Π(ar)〉Ψ
Systemzustand nach der Messung (mit Resultat ar):
Π(ar)|Ψ〉 “Kollaps”, akausale Entwicklung
Praparation eines reinen Zustands |Ψ〉.Messung eines vollstandigen Satzes kommutierender Observabler
|ar, bs, ...〉 = Π(ar)Π(bs) · · · |Ψ〉Erwartungswert von A bei Messung im Zustand Ψ:
〈A〉Ψ =∑
r
P (ar)ar =∑
r,γ
|〈ar, γ|Ψ〉|2ar
=∑
r,γ
〈Ψ|ar, γ〉ar〈ar, γ|Ψ〉 = 〈Ψ|A|Ψ〉
—————————————————————————————————
zeitliche Entwicklung des Zustands:
|Ψ〉 = |Ψ(t)〉Anfangsbedingung:
|Ψ(t0)〉 = |Ψ0〉Dynamik: Hamilton-Operator
H = H(q, p) klassische Hamilton-Fkt. aber [q, p] = i~
Bewegungsgleichung
i~d
dt|Ψ(t)〉 = H|Ψ(t)〉 Schrodinger-Gleichung
formale Losung:
114

|Ψ(t)〉 = exp(− i
~H(t − t0)
)|Ψ(t0)〉 = U(t − t0)|Ψ(t0)〉
mit Zeitentwicklungsoperator U (H nicht explizit zeitabhangig)
Zeitabhangigkeit eines Erwartungswerts:
〈A〉t = 〈Ψ(t)|A|Ψ(t)〉 Schrodinger-Bild
= 〈Ψ(0)|e i~HtAe−
i~Ht|Ψ(0)〉
= 〈Ψ(0)|A(t)|Ψ(0)〉 Heisenberg-Bild
mit: A(t) ≡ ei~HtAe−
i~Ht
Schrodinger-Bild:
– A = A(q, p) Operatoren zeitunabhangig
– |Ψ(t)〉 Zustande zeitabhangig Schrodinger-Gleichung
Heisenberg-Bild
– Operatoren zeitabhangig Heisenberg-Gleichung
– Zustande |Ψ〉 zeitunabhangig
Heisenberg-Gleichung (A nicht explizit zeitabhangig):
i~d
dtA(t) = i~
d
dt
(e
i~HtAe−
i~Ht)
= [A, H ](t)
Erhaltungsgroßen:
[A, H ] = 0 A = const.
Energieerhaltung:
[H, H ] = 0 H = const.
6.5 Gemischter Zustand, Dichteoperator
Mikrozustand:
|Ψ〉 reiner Zustand
bei unvollstandiger Information:
|Ψk〉 mit WK p(|Ψk〉) = pk gemischter Zustand
gemischter Zustand = Satz von reinen Zustanden mit zugehorigen WK
Zustand |Ψk〉 ist eine Zufallsgroße
115

zusatzliche statistische Beschreibung
KM: WK-Dichte:
ρ(π) =∑
k
pkδ(π − πk)
δ(π − πk) projiziert auf reinen Zustand πk !
QM: definiere Dichteoperator
ρ =∑
k
pk|Ψk〉〈Ψk|
Operator im Hilbert-Raum (aber nicht als Observable aufzufassen)
—————————————————————————————————
beachte: die Aussagen Ak: “System im Zustand |Ψk〉” sind paarweise exklusivund vollstandig (Partition), falls |Ψk〉 ONB
denn: |Ψk〉 konnen als Zustande nach einer Messung einer Observablen (odereinen Satzes) B mit paarweise exklusiven und vollstandigen Messresultaten (Ei-genwerten) bk aufgefasst werden
—————————————————————————————————
Eigenschaften:
Normierung: (|Ψk〉 ONB)
Sp ρ =∑
k
〈Ψk|ρ|Ψk〉 =∑
k
pk = 1
Sp ρ = 1
Positive Definitheit: sei |Φ〉 beliebig:
〈Φ|ρ|Φ〉 = 〈Φ|∑
k
pk|Ψk〉〈Ψk||Φ〉 =∑
k
pk|〈Φ|Ψk〉|2 ≥ 0
ρ ≥ 0
Hermitizitat:
ρ† =
(∑
k
pk|Ψk〉〈Ψk|)†
=∑
k
pk|Ψk〉〈Ψk| = ρ
ρ = ρ†
umgekehrt:
ρ = ρ†, ρ ≥ 0, Sp ρ = 1 Eigenwerte reell, nicht negativ, Summe: 1
Eigenwerte als WK interpretierbar
116

Operator mit diesen Eigenschaten charakterisiert (eindeutig!) einen gemisch-ten Zustand |Ψk〉, pk—————————————————————————————————
reiner Zustand als Spezialfall:
pk = δkk0 ρ =∑
k
pk|Ψk〉〈Ψk| = |Ψk0〉〈Ψk0|
weiter gilt:
Sp (ρ2) =∑
k,l
〈Ψk|ρ|Ψl〉〈Ψl|ρ|Ψk〉 =∑
k
p2k 〈Ψk|Ψk〉 =
∑
k
p2k ≤ 1
Sp (ρ2) = 1 ⇔ ρ = |Ψ〉〈Ψ| fur ein |Ψ〉 ⇔ ρ reiner Zustand
—————————————————————————————————
ρ =∑
k
pk|Ψk〉〈Ψk|
messbare Großen: direkt durch ρ darstellbar?
System mit WK pk im Zustand |Ψk〉. Messung von A. WK ar zu messen:
P (ar) =∑
k
P (ar| System im Zustand |Ψk〉) · pk
(Marginalisierungsregel)
beachte: “System im Zustand |Ψk〉” bilden eine Partition
(evtl nach Erganzung zu einer ONB)
P (ar) =∑
k
〈Π(ar)〉Ψkpk mit Π(ar) =
gr∑
γ=1
|ar, γ〉|ar, γ〉
P (ar) =∑
k
∑
n
〈Ψk|n〉〈n|Π(ar)|Ψk〉 pk
=∑
n
〈n|Π(ar)∑
k
|Ψk〉pk〈Ψk|n〉 = Sp (Π(ar)ρ) = Sp (ρΠ(ar))
nur uber Dichteoperator von |Ψk〉 und pk abhangig
Erwartungswert einer Observablen A:
〈A〉 =∑
r
arP (ar) =∑
r
ar Sp (ρΠ(ar))
= Sp
(ρ∑
r
arΠ(ar)
)= Sp (ρA)
nur uber Dichteoperator von |Ψk〉 und pk abhangig
117

somit:
〈A〉ρ = Sp (ρA)
und
P (ar) = 〈Π(ar〉ρ—————————————————————————————————
speziell fur einen reinen Zustand ρ = |Ψ〉〈Ψ|:〈A〉Ψ = Sp(|Ψ〉〈Ψ|A) =
∑
n
〈n|Ψ〉〈Ψ|A|n〉 =∑
n
〈Ψ|A|n〉〈n|Ψ〉 = 〈Ψ|A|Ψ〉
und:
P (ar) = 〈Π(ar)〉Ψ = ... = 〈Ψ|∑
γ
|ar, γ〉〈ar, γ|Ψ〉 =∑
γ
|〈ar, γ|Ψ〉|2
—————————————————————————————————
Beispiel: 2-dimensionaler Hilbert-Raum
Basis:
| ↑〉 =
(10
)| ↓〉 =
(01
)
normierter reiner Zustand:
|Ψ〉 =
(αβ
)= α| ↑〉 + β| ↓〉 (|α|2 + |β|2 = 1)
“koharente” Superposition reiner Zustande
normierter gemischter Zustand:
ρ = |α|2| ↑〉〈↑ | + |β|2| ↓〉〈↓ | (|α|2 + |β|2 = 1 Sp ρ = 1)
“inkoharente” Mischung reiner Zustande
ρ = |α|2(
10
)(1, 0) + |β|2
(01
)(0, 1) =
(|α|2 00 |β|2
)
beachte: Sp ρ2 = |α|4 + |β|4 < 1 fur α 6= 0, α 6= 1
unterschiedliche Messresultate, z.B. Observable Sx =~
2
(0 11 0
)=
~
2σx
〈σx〉ρ = Sp (ρσx) = Sp
((|α|2 00 |β|2
)(0 11 0
))
= Sp
(0 |α|2
|β|2 0
)= 0
118

dagegen:
〈σx〉Ψ = (α∗, β∗)
(0 11 0
)(αβ
)= (α∗, β∗)
(βα
)= α∗β + β∗α
(Interferenz von WK-Amplituden)
Dichteoperator des reinen Zustands ρΨ:
ρΨ = |Ψ〉〈Ψ| =
(αβ
)(α∗, β∗) =
(|α|2 βα∗
αβ∗ |β|2)
Interferenzterme fehlen auf der Nebendiagonalen von ρ
—————————————————————————————————
Shannon-Information eines gemischten Zustands ρ =∑
k
pk|Ψk〉〈Ψk|:
σ = σ(pk) = −∑
k
pk ln pk
pk Eigenwerte von ρ, |Ψk〉 Eigenzustande
|Ψk〉 Eigenzustande von ρ ln ρ, Eigenwerte:
ρ ln ρ|Ψk〉 = pk ln pk|Ψk〉
pk ln pk = 〈Ψk|ρ ln ρ|Ψk〉und somit:
σ = −∑
k
〈Ψk|ρ ln ρ|Ψk〉
σ = − Sp (ρ ln ρ)
Beispiel von oben:
σ = σ(|α|2, |β|2) = −|α|2 ln |α|2 − |β|2 ln |β|2
maximale Unkenntnis fur σ =max., also fur |α|2 = |β|2 = 1/2
gemischter Zustand maximaler Unkenntnis:
ρ =
(1/2 00 1/2
)=
1
2
(10
)(1, 0) +
1
2
(01
)(0, 1)
oder:
ρ =
(1/2 00 1/2
)=
1
2
1√2
(11
)1√2(1, 1) +
1
2
1√2
(1−1
)1√2(1,−1)
System mit WK 1/2 in Zustand mit Spin in ±x-Richtung
119

—————————————————————————————————
zeitliche Entwicklung eines gemischten Zustands?
System z.Zt. t0:
|Ψk(t0)〉 mit WK pk
reiner Zustand |Ψk〉 = |Ψk(t)〉 gemaß Schrodinger-Gleichung
System z.Zt. t:
|Ψk(t)〉 mit WK pk
WK-Dichte z.Zt. t:
ρ(t) =∑
k
pk|Ψk(t)〉〈Ψk(t)|
Bewegungsgleichung ?
i~d
dtρ(t) = i~
d
dt
∑
k
pk|Ψk(t)〉〈Ψk(t)|
=∑
k
pk
[i~
d|Ψk(t)〉dt
〈Ψk(t)| + |Ψk(t)〉i~d〈Ψk(t)|
dt
]
=∑
k
pk (H|Ψk(t)〉〈Ψk(t)| − |Ψk(t)〉〈Ψk(t)|H)
= Hρ(t) − ρ(t)H
also:
i~d
dtρ(t) = [H, ρ(t)] von Neumann-Gleichung
vergleiche: Liouville-Gleichung:∂
∂tρ(π, t) = H, ρ(π, t)
vergleiche: Heisenberg-Gleichung: i~d
dtA(t) = [A, H ](t)
—————————————————————————————————
zeitliche Entwicklung der Shannon-Information:
d
dtσ(t) = − d
dtSp(ρ(t) ln ρ(t)) = − Sp
(dρ(t)
dtln ρ(t)
)− Sp
(ρ(t)
d ln ρ(t)
dt
)
= − 1
i~Sp ([H, ρ(t)] ln ρ(t)) − Sp
(ρ(t)ρ(t)−1 dρ(t)
dt
)
WK-Erhaltung: Sp ρ(t) = 1 = const., also
120

= − 1
i~Sp ([H, ρ(t)] ln ρ(t))
= − 1
i~Sp (Hρ(t) ln ρ(t) − ρ(t)H ln ρ(t)) = 0
zeitliche Erhaltung der Shannon-Information
—————————————————————————————————
zeitliche Anderung von Erwartungswerten?
Heisenberg-Bild:
Observable A = A(t), Zustand ρ = const.
i~d
dt〈A(t)〉 = i~
d
dtSp (ρA(t)) = Sp (ρ[A, H ](t)) = 〈[A, H ](t)〉
oder Schrodinger-Bild:
Observable A = const., Zustand ρ = ρ(t)
i~d
dt〈A(t)〉 = i~
d
dtSp (ρ(t)A) = Sp ([H, ρ]A)
Thermodynamisches Gleichgewicht: keine zeitliche Anderung von Makrovariablen
〈A(t)〉 = const.
fur ρ = ρ(H) gilt:
[H, ρ(H)] = 0
also (mit von Neumann-Gleichung):
ρ(t) = ρ = const.
Erwartung: das TD GG ist durch einen gemischten Zustand ρ = ρ(H) gegeben
6.6 Systeme identischer Teilchen
Ein-Teilchen-Hilbert-Raum: H1
ONB von H1:
|φα〉〈φα|φβ〉 = δαβ (Orthonormierung)∑
α
|φα〉〈φα| = 1 (Vollstandigkeit)
—————————————————————————————————
121

N -Teilchen-Hilbert-Raum:
HN = H(1)1 ⊗H(2)
1 ⊗ · · · ⊗ H(N)1 (Tensorprodukt)
H(i)1 : Hilbert-Raum des i-ten Teilchens
ONB von HN :
|φ(1)α1〉|φ(2)
α2〉 · · · |φ(N)
αN〉
Index oben: Teilchen
Index unten: vollstandiger Satz von Quantenzahlen
es folgt: dimHN =dimHN1 (falls H1 endlichdimensional)
Orthogonalitat:
〈φ(1)α1|〈φ(2)
α2| · · · 〈φ(N)
αN| · |φ(1)
β1〉|φ(2)
β2〉 · · · |φ(N)
βN〉 = 〈φ(1)
α1|φ(1)
β1〉 · · · 〈φ(N)
αN|φ(N)
βN〉 = δα1β1 · · · δαN βN
Vollstandigkeit:∑
α1,...,αN
|φ(1)α1〉 · · · |φ(N)
αN〉〈φ(1)
α1| · · · 〈φ(N)
αN| = 1
—————————————————————————————————
Bsp: Ortsdarstellung (spinlose Teilchen, |r〉 ist ONB)
ONB von H1:
φα(r) mit φα(r) = 〈r|φα〉
ONB von H(i)1 :
φα(ri)
ONB von HN = H(1)1 ⊗H(2)
1 ⊗ · · · ⊗ H(N)1 :
φα1(r1)φα2(r2) · · ·φαN(rN )
fur beliebiges Φ(r1, ..., rN) ∈ HN ist
Φ(r1, ..., rN ) =∑
α1,...,αN
cα1,...,αNφα1(r1) · · ·φαN
(rN)
mit Koeffizienten
cα1,...,αN=∫
d3r1 · · ·∫
d3rN φ∗α1
(r1) · · ·φ∗αN
(rN) · Φ(r1, ..., rN)
—————————————————————————————————
122
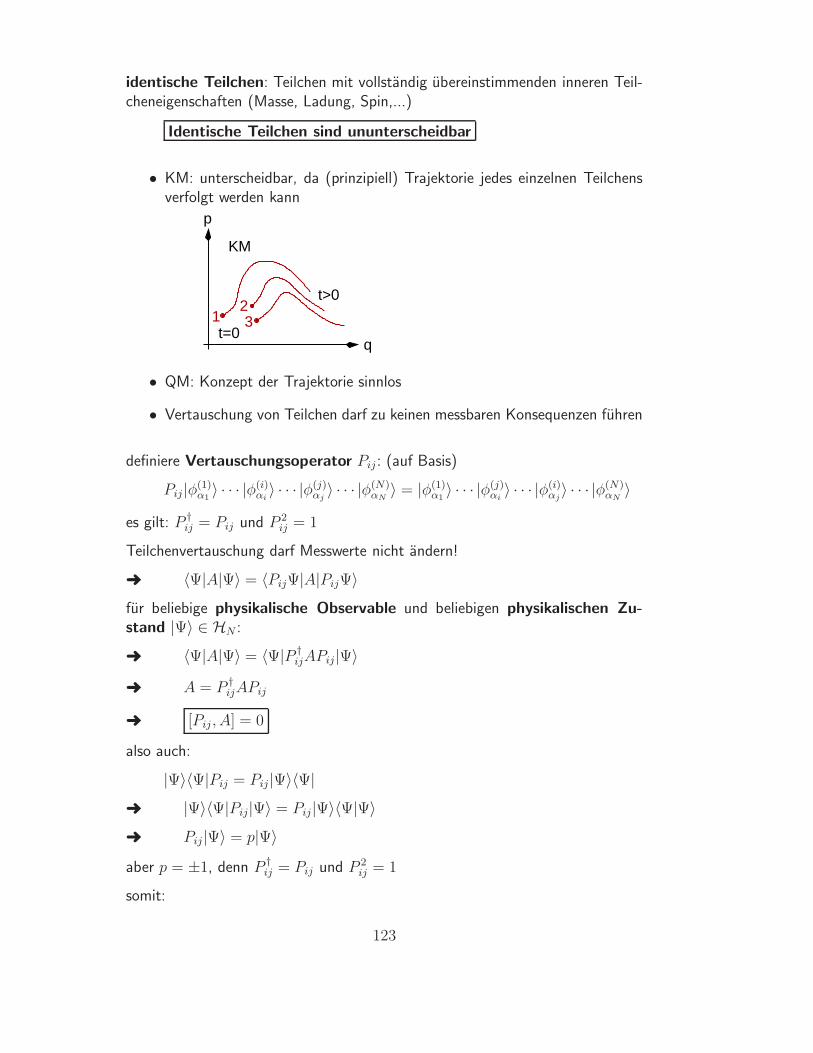
identische Teilchen: Teilchen mit vollstandig ubereinstimmenden inneren Teil-cheneigenschaften (Masse, Ladung, Spin,...)
Identische Teilchen sind ununterscheidbar
• KM: unterscheidbar, da (prinzipiell) Trajektorie jedes einzelnen Teilchensverfolgt werden kann
q
p
32
1t=0
t>0
KM
• QM: Konzept der Trajektorie sinnlos
• Vertauschung von Teilchen darf zu keinen messbaren Konsequenzen fuhren
definiere Vertauschungsoperator Pij : (auf Basis)
Pij |φ(1)α1〉 · · · |φ(i)
αi〉 · · · |φ(j)
αj〉 · · · |φ(N)
αN〉 = |φ(1)
α1〉 · · · |φ(j)
αi〉 · · · |φ(i)
αj〉 · · · |φ(N)
αN〉
es gilt: P †ij = Pij und P 2
ij = 1
Teilchenvertauschung darf Messwerte nicht andern!
〈Ψ|A|Ψ〉 = 〈PijΨ|A|PijΨ〉fur beliebige physikalische Observable und beliebigen physikalischen Zu-stand |Ψ〉 ∈ HN :
〈Ψ|A|Ψ〉 = 〈Ψ|P †ijAPij|Ψ〉
A = P †ijAPij
[Pij , A] = 0
also auch:
|Ψ〉〈Ψ|Pij = Pij |Ψ〉〈Ψ| |Ψ〉〈Ψ|Pij|Ψ〉 = Pij|Ψ〉〈Ψ|Ψ〉 Pij|Ψ〉 = p|Ψ〉aber p = ±1, denn P †
ij = Pij und P 2ij = 1
somit:
123

Pij |Ψ〉 = ±|Ψ〉—————————————————————————————————
Bsp.:
Pij
(· · · |φ(i)
αi〉 · · · |φ(j)
αj〉 · · ·
)=(· · · |φ(j)
αi〉 · · · |φ(i)
αj〉 · · ·
)6= ±
(· · · |φ(i)
αi〉 · · · |φ(j)
αj〉 · · ·
)
Produktzustande nicht physikalisch
Bsp.:
r(i) Ortsoperator des i-ten Teilchens
P †ijr
(i)Pij = r(j) 6= r(i)
r(i) nicht physikalisch
Bsp.:
H =N∑
i=1
p(i)2
2m+
1
2
∑
i6=j
α
|r(i) − r(j)|
P †ijHPij = H
H physikalisch
—————————————————————————————————
Fermionen BosonenPij|Ψ〉 = −|Ψ〉 Pij|Ψ〉 = +|Ψ〉antisymmetrische symmetrische Zustande
Hilbert-Raum H(−)N Hilbert-Raum H(+)
N
halbzahliger Spin ganzzahliger Spin (Spin-Statistik-Zusammenhang QFT)
(Observable stets symmetrisch)
H(±)N ⊂ HN Hilbert-Raum der physikalisch erlaubten Zustande
—————————————————————————————————
Def.: Permutationsoperator P:
P|φ(1)α1〉 · · · |φ(N)
αN〉 = |φ(i1)
α1〉 · · · |φ(iN )
αN〉
wobei (i1, ..., iN) Permutation P der Zahlen 1, ..., N
P Produkt von p Vertauschungsoperatoren
P ist unitar, P−1 = P†
Def.: (Anti-)Symmetrisierungsoperator S(±)N :
S(±)N =
1
N !
∑
P(±1)pP
124

Eigenschaften:
• PijS(±)N = ±S
(±)N
also: S(±)N |Ψ〉 (anti-)symmetrisch
• (S(±)N )2 = S
(±)N (Idempotenz)
• (S(±)N )† = S
(±)N (Hermitizitat)
also: S(±)N Projektoren
es ist:
H(±)N = S
(±)N HN
—————————————————————————————————
Def.: (anti-)symmetrisierter Produktzustand:
|n1, n2, ..., nd〉 = K± S(±)N
(|φ(1)
α1〉|φ(2)
α2〉 · · · |φ(N)
αN〉)
(d = dimH1)
mit Normierungskonstante K± (〈n1, n2, ..., nd|n1, n2, ..., nd〉 = 1) und:
ni = Anzahl der Teilchen im Zustand φi Besetzungszahl
ni = Anzahl Indizes mit αj = i
es gilt: (ohne Beweis)
|n1, n2, ..., nd〉 ist ONB von H(±)N (NB:
∑α nα = N)
Orthonormalitat:
〈n1, n2, · · · , nα, · · · |n′1, n
′2, · · · , n′
α, · · ·〉 = δn1n′1δn2n′
2· · ·
Vollstandigkeit:
∑
n1
∑α
nα=N∑
n2
· · ·∑
nα
· · · |n1, n2, · · · , nα, · · ·〉〈n1, n2, · · · , nα, · · · | = 1H(±)N
Produktzustand:
φ1φ4 φ5 φ8φ8 φ4 φ8Teilchen
1 ...32 N
(anti-)symmetrisierter Produktzustand in Besetzungszahldarstellung:
125

φ3φ1φ2 φd Zustände...
Ein−Teilchen−
(Teilchen ohne Identitat)
—————————————————————————————————
Bsp.: N = 2 Teilchen
Produktzustand:
|φ(1)α 〉|φ(2)
β 〉 ∈ HN
(Anti-)symmetrisierter Produktzustand:
S(±)N |φ(1)
α 〉|φ(2)β 〉 =
1
2!
(|φ(1)
α 〉|φ(2)β 〉 ± |φ(2)
α 〉|φ(1)β 〉
)= ±S
(±)N |φ(2)
α 〉|φ(1)β 〉
= K−1± |0 · · ·nα = 1 · · ·nβ = 1 · · ·0〉 ∈ H(±)
N
Normierung: (K± reell)
K−2± =
∣∣∣S(±)N |φ(1)
α 〉|φ(2)β 〉
∣∣∣2
=1
4
(〈φ(1)
α |〈φ(2)β | ± 〈φ(2)
α |〈φ(1)β |) (
|φ(1)α 〉|φ(2)
β 〉 ± |φ(2)α 〉|φ(1)
β 〉)
=1
4
(〈φ(1)
α |〈φ(2)β | · |φ(1)
α 〉|φ(2)β 〉 + 〈φ(2)
α |〈φ(1)β | · |φ(2)
α 〉|φ(1)β 〉
±〈φ(1)α |〈φ(2)
β | · |φ(2)α 〉|φ(1)
β 〉 ± 〈φ(2)α |〈φ(1)
β | · |φ(1)α 〉|φ(2)
β 〉)
=1
4(1 + 1 ± δαβ ± δαβ) =
1
2(1 ± δαβ)
K± =
√2
1 ± δαβ
allgemein:
K± =
√N !
∏α nα!
K− =√
N !
—————————————————————————————————
Fermionen:
S(±)N
(|φ(1)
α1〉|φ(2)
α2〉 · · · |φ(N)
αN〉)
=1
N !
∑
P(−1)pP|φ(1)
α1〉 · · · |φ(N)
αN〉
126

=1
N !det
|φ(1)α1〉 · · · |φ(N)
α1〉
··
|φ(1)αN
〉 · · · |φ(N)αN
〉
(Entwicklungssatz von Leibniz)
“Slater-Determinante”
nicht-verschwindend nur fur αi 6= αj falls i 6= j
Pauli-Prinzip
Fermionen: nα = 0, 1
Bosonen: nα = 0, 1, 2, ...
φ3φ1φ2 φd Zustände...
QM, Fermionen
Ein−Teilchen−
φ3φ1φ2 φd Zustände...
Ein−Teilchen−
QM, Bosonen
—————————————————————————————————
Vergleich mit KM:
reiner Zustand in KM: Punkt im 6N -dimensionalen Phasenraum:
1p=(p ,..., p )3N
1q=(q ,..., q )3N
KM
oder: N Punkte im 6-dimensionalen Ein-Teilchen-Phasenraum (hier: N = 7)
zum Abzahlen der Ein-Teilchen-Zustande: diskretisierter Ein-Teilchen-Phasenraum
127

p=(p ,p ,p )x y z
x y zq=(q ,q ,q )
KM
2
6
1
74
3
5
p=(p ,p ,p )x y z
x y zq=(q ,q ,q )
KM
2
6
1
74
3
5
Darstellung eines reinen N -Teilchen-Zustands:
Ein−Teilchen−Zustände2 3 d1 ...
3 1 2 6 54 7
KM
durch Teilchenvertauschung (z.B.1 ↔ 5) entstehen (in KM) neue Zustande:
Ein−Teilchen−Zustände2 3 d1 ...
3 2 64 7
KM
15
Anzahl der Zustande:
M = dN (klassisch)
—————————————————————————————————
Vergleich mit QM:
φ1φ4 φ5 φ8φ8 φ4 φ8Teilchen
1 ...32 N
QM
M = dN QM, unterscheidbare Teilchen
analog zur KM, M = Dimension des Hilbert-Raums HN
—————————————————————————————————
φ3φ1φ2 φd Zustände...
QM, Fermionen
Ein−Teilchen−
M =
(dN
)QM, identische Fermionen
H = Dimension des Hilbert-Raums H(−)N ⊂ HN
128

—————————————————————————————————
φ3φ1φ2 φd Zustände...
Ein−Teilchen−
QM, Bosonen
M =
(d + N − 1
N
)QM, identische Bosonen
M = Dimension des Hilbert-Raums H(+)N ⊂ HN
Bilder beziehen sich auf:
• reine N -Teilchen-Zustande!
• Basis-Zustande! (allg. Zustand in der QM: Linearkombination)
korrektes Zahlen der Mikrozustande entscheidend in der statistischen Mechanik!
129

130

Kapitel 7
Thermodynamische Zustande undProzesse
Situation:
• N = O(1023) Teilchen
• diskreter Satz von Mikrozustanden k = 1, ..., M , M ≫ N
KM: diskretisiert, QM: M = dimH(±)N
• f kontrollierbare Makrovariable z = (z0, ..., zs)
mit f = s + 1, z0: Gesamtenergie
• (bei weitem) unvollstandige Information f ≪ M
• statistische Beschreibung mittels gemischtem Zustand:
Mikrozustand k mit WK pk
KM: WK-Dichte ρ(π), QM: Dichteoperator ρ
statistische Theorie des TD GG:
Festlegung der pk durch Maximierung der Shannon-Information unter NBen
σ(p1, ..., pM) = max f NBen: zj fest fur j = 0, ..., s
damit:
pk = pk(z0, z1, ..., zs)
Beschreibung des TD GG durch gemischten Zustand mit
ρ = ρz0,z1,...,zs(π) (KM)
131

ρ = ρz0,z1,...,zs (QM)
sei A = A(π) (KM), A = A† (QM) beliebige extensive Observable:
〈A〉 =∫
dπρz0,z1,...,zs(π)A(π) = A(z0, ..., zs) (KM)
〈A〉 = Sp (ρz0,z1,...,zsA) = A(z0, ..., zs) (QM)
Werte der Makrovariable A konnen um 〈A〉 fluktuieren, aber:
σA
〈A〉 = O(1/√
N)
(A extensiv, somit makroskopische Summe unabhangiger Zufallsvariabler, ZGWS)
makroskopische Abweichungen von TD GG-Werten extrem unwahrscheinlich
—————————————————————————————————
Bsp: nach ZGWS ist
ρ(x) =1√
2πσ2exp
(−(x − 〈x〉)2
2σ2
)
WK fur große Fluktuation (setze σ = 1):
P (x > 〈x〉 + a) =∫ ∞
〈x〉+aρ(x)dx =
1√2π
∫ ∞
adx exp
(−x2
2
)
a = 0 P = 0.5a = 1 P = 0.1587a = 2 P = 0.0228a = 3 P = 0.00135a = 5 P = 3 · 10−7
—————————————————————————————————
f -dimensionaler TD Zustandsraum der TD (f = s + 1):
A = 〈A〉 = A(z0, ..., zs) (Zustandsgleichungen)
Makrozustand:
(z0, ..., zs)
f gegeben durch die Anzahl der kontrollierbaren Makrovariablen
(beachte: Gibbs’sches Paradoxon)
TD GG ↔ nur f Makrovariable kontrolliert, sonst maximale Unkenntnis
—————————————————————————————————
Bsp: Box mit Gas, z.Zt. t = 0 Trennwand rausnehmen
132

t>0
(partielle) Information uber Mikrozustand vorhanden
Beschreibung mit pk aus maximierter Shannon-Information nicht korrekt
kein TD GG fur kleine t > 0
—————————————————————————————————
Bsp: prapariere Mikrozustand:
keine Stoße der Teilchen, exakt reflektierende Wande
Mikrozustand fur alle t berechenbar
Beschreibung mit pk aus maximierter Shannon-Information nicht korrekt
kein TD GG
aber: bei leicht nichtidealen Wanden nach kurzer Zeit (bis aufMakrovariablen zj) vollstandig unkontrollierter Mikrozustand
TD GG
7.1 Mikrokanonische Gesamtheit
z0, z1, ..., zs fest System isoliert
betrachte zunachst nur z0 = H = H(π) (KM), z0 = H = H† (QM)
es ist:
E − ∆E < H(π) = H(q, p) < E (KM)
E − ∆E < En < E mit H|En, γ〉 = En|En, γ〉 (QM)
E kontrollierbar bis auf (infinitesimale) Unscharfe
∆E ≪ E
∆E Hilfsgroße, “Messungenauigkeit”, am Ende ∆E → 0,
133

GG TD von ∆E unabhangig
Mikrozustande (mit Sicherheit) in Energieschale E, ∆E
H(q,p)=Ep
q
E
QMKM
nEigenenergien E
H(q,p)=E− E∆E− E∆
weitere kontrollierbare Makrovariable V, N, ... s.u.
—————————————————————————————————
Shannon-Information:
σ(p1, ..., pM) − λ∑
k
pk = max
NB:
pk = 0 fur Mikrozustand k außerhalb Energieschale E, ∆E
pk = const. innerhalb Energieschale
—————————————————————————————————
KM, kontinuierliche WK-Dichte:
ρE(q, p) = const. fur E − ∆E < H(q, p) < E
WK-Dichte der mikrokanonischen Gesamtheit
mit Normierung:
ρE(q, p) =1
Σ(E)∆Efur E − ∆E < H(q, p) < E, ρE(q, p) = 0 sonst
mit dem Phasenvolumen der Energieschale:
Σ(E)∆E =∫
E−∆E<H(q,p)<Edqdp
damit gilt:∫
dqdp ρE(q, p) = 1
134

—————————————————————————————————
Bsp: harmonischer Oszillator
H(q, p) =p2
2m+
1
2mωq2
H(q, p) = E: Ellipse im Phasenraum
—————————————————————————————————
Schreibweise mit δ-Funktion (∆E → 0):
δ(E−H(q, p))∆E =Θ(E − H(q, p)) − Θ(E − ∆E − H(q, p))
∆E∆E
= Θ(E − H(q, p)) − Θ(E − ∆E − H(q, p))
= 1 falls E − ∆E < H(q, p) < E
= 0 sonst
damit lautet die NB:
δ(E − H(q, p))∆E = 1 (∆E → 0)
also:
ρE(q, p) =1
Σ(E)∆Eδ(E − H(q, p))∆E
ρE(q, p) =1
Σ(E)δ(E − H(q, p))
und
Σ(E) =∫
dqdp δ(E − H(q, p)) Zustandsdichte
bzw.
Σ(E) =dΓ(E)
dEΓ(E) =
∫dqdp Θ(E − H(q, p)) =
∫
H(q,p)<Edqdp
Γ(E): das von der Hyperflache H(q, p) = E eingeschlossene Phasenvolumen
135

H(q,p)=Ep
q
KM
(E)Γ
wichtig fur die TD: E-Abhangigkeit von ρE Σ(E), Γ(E)
—————————————————————————————————
hochdimensionaler Phasenraum:
gesamtes Phasenvolumen Γ(E) in beliebig dunner Energieschale konzentriert:
Γ(E) − Σ(E)∆E
Γ(E)→ 1 Γ(E) ∼ Σ(E)∆E
im Limes hoher Dimensionen
—————————————————————————————————
beachte:
ρE(q, p) = ρE(H(q, p))
also:
ρE(q, p, t) = ρE(q, p) = const.
—————————————————————————————————
QM, diskreter Zustandsraum, ONB von H: |En, γ〉, γ = 1, ..., gn
pE(En, γ) = const. fur E − ∆E < En < E
WK-Funktion der mikrokanonischen Gesamtheit
mit Normierung:
pE(En, γ) =1
Σ(E)∆Efur E − ∆E < En < E, pE(En, γ) = 0 sonst
mit der Anzahl der Zustande im Energieintervall [E − ∆E, E]:
Σ(E)∆E =E−∆E<En<E∑
n,γ
1 =E−∆E<En<E∑
n
gn Zustandssumme
136

damit gilt:∑
n,γ
pE(En, γ) = 1
—————————————————————————————————
mikrokanonischer Dichteoperator:
ρE =∑
n,γ
pE(En, γ)|En, γ〉〈En, γ| =1
Σ(E)∆E
E−∆E<En<E∑
n,γ
|En, γ〉〈En, γ|
mit Sp ρE = 1
—————————————————————————————————
NB E − ∆E < En < E durch δ(E − En)∆E = 1 ausdrucken:
ρE =1
Σ(E)∆E
∑
n,γ
|En, γ〉〈En, γ|δ(E − En)∆E
=1
Σ(E)
∑
n,γ
δ(E − En)|En, γ〉〈En, γ|
=1
Σ(E)
∑
n,γ
δ(E − H)|En, γ〉〈En, γ|
=1
Σ(E)δ(E − H)
also:
ρE =1
Σ(E)δ(E − H)
Σ(E) = Sp δ(E − H) Zustandsdichte
bzw.
Σ(E) =dΓ(E)
dE
mit
Γ(E) = Sp Θ(E − H) =∑
n,γ
Θ(E − En) =En<E∑
n,γ
1 =En<E∑
n
gn
—————————————————————————————————
analog zur KM ist auch hier:
Γ(E) ∼ Σ(E)∆E
und
137

ρE = ρE(H)
also:
ρE(t) = ρE = const.
—————————————————————————————————
innere Energie U
U = 〈H〉 = Sp (ρEH) =1
Σ(E)Sp (δ(E − H)H)
=1
Σ(E)
∑
n
gnδ(E − En)En = E1
Σ(E)
∑
n
gnδ(E − En) = E
〈H2〉 =1
Σ(E)Sp (δ(E − H)H2) = E2
also:
U = 〈H〉 = E σH = 0
KM: analog
H ist keine “echte” Zufallsvariable in MK Gesamtheit
7.2 Mikrokanonische Gesamtheit mit festem E, V, N
V und N als weitere kontrollierbare Makrovariable
MK WK-Dichte:
ρE,V,N(q, p) =1
Σ(E, V, N)∆E∆V ∆Nfur
E − ∆E < H(q, p) < E
V − ∆V < V (q, p) < V
N − ∆N < N(q, p) < N
(sonst: 0)
NB durch δ-Funktion ausdrucken (∆E, ∆V, ∆N → 0), z.B.
δ(V −V (q, p))∆V =Θ(V − V (q, p)) − Θ(V − ∆V − V (q, p))
∆V∆V
= Θ(V − V (q, p)) − Θ(V − ∆V − V (q, p))
= 1 falls V − ∆V < V (q, p) < V
138

= 0 sonst
also:
ρE,V,N(q, p) =δ(E − H(q, p))∆E δ(V − V (q, p))∆V δ(N − N(q, p))∆N
Σ(E, V, N)∆E∆V ∆N
WK-Dichte:
ρE,V,N(q, p) =1
Σ(E, V, N)δ(E − H(q, p))δ(V − V (q, p))δ(N − N(q, p))
Normierung∫
dqdp ρE,V,N(q, p) = 1
Zustandsdichte:
Σ(E, V, N) =∫
dqdp δ(E − H(q, p))δ(V − V (q, p))δ(N − N(q, p))
Phasenvolumen der unter den NBen moglichen Zustande:
Σ(E, V, N)∆E∆V ∆N
—————————————————————————————————
V = V (q, p) als Observable auffassen:
KM: betrachte kleines Volumen v mit
V
N≪ v ≪ V
definiere (bis auf irrelvanten Fehler):
V (q, p) = V (q) = V (q1, ..., q3N)
= das von den Teilchen bei q mit umgebendem v uberdeckte Volumen
vV
Definition fur beliebigen Mikrozustand
QM: V = V (q, p) mit [qi, pj] = i~δij
es ist:
[H, V ] ≈ 0
139

(Makrovariablen gleichzeitig scharf messbar, Kommutator makroskopisch klein)
—————————————————————————————————
N = N(q, p) als Observable auffassen:
sinnvoll bei verallgemeinertem Zustandsraum:
(q1, p1), (q1, q2, p1, p2), ...Phasenraum eines Systems mit variabler Teilchenzahl
definiere fur (q, p) = (q1, ..., p1, ...):
N(q, p) = N(q1, ..., p1, ...) ≡ N falls (q, p) = (q1, ..., q3N , p1, ..., p3N)
QM:
definiere Fock-Raum HH = ⊕∞
N=0HN (orthogonale Summe)
ONB:
|n1, n2, ..., nd〉 (ohne NB:∑
α nα = N)
definiere (auf ONB) Teilchenzahloperator:
N |n1, n2, ..., nd〉 ≡∑
α
nα|n1, n2, ..., nd〉
es gilt:
[H, V ], [H, N ], [V , N ] ≈ 0
—————————————————————————————————
QM, sei |En, Vm, Np, γ〉 ONB gemeinsamer Eigenzustande von H, V , N
pE,V,N(En, Vm, Np, γ) = const.
falls δ(E − En)∆E = δ(V − Vm)∆V = δ(N − Np)∆N = 1
Anzahl der unter den NBen moglichen Zustande:
Σ(E, V, N)∆E∆V ∆N
normierte WKen:
pE,V,N(En, Vm, Np, γ) =1
Σ(E, V, N)δ(E − En)δ(V − Vm)δ(N − Np)
Eigenwerte von
140

ρE,V,N =1
Σ(E, V, N)δ(E − H)δ(V − V )δ(N − N)
MK Dichteoperator
Zustandsdichte:
Σ(E, V, N) =∑
n,m,p,γ
δ(E − En)δ(V − Vm)δ(N − Np)
oder:
Σ(E, V, N) = Sp (δ(E − H)δ(V − V )δ(N − N))
—————————————————————————————————
Realisierung der NBen: V, N als Parameter in der Hamilton-Funktion:
H(q, p) = HV,N(q, p)
H erzwingt NB: N fest (teilchenundurchlassige Wande), V fest (starre Wande)
also:
HV,N(q, p) = ∞bei Verletzung der NBen
δ(V − V (q, p))∆V = 1 δ(N − N(q, p))∆N = 1
fur die MK WK-Dichte
ρE,V,N(q, p) =1
Σ(E, V, N)δ(E − H(q, p))δ(V − V (q, p))δ(N − N(q, p))
gilt daher auch:
ρE,V,N(q, p) =1
Σ(E, V, N) ∆V ∆Nδ(E − HV,N(q, p))
und aus der Normierungsbedingung folgt:
Σ(E, V, N) =1
∆V ∆N
∫dqdp δ(E − HV,N(q, p))
beachte: das Phasenvolumen
Σ(E, V, N)∆E∆V ∆N
hat die Dimension (Wirkung)3N
—————————————————————————————————
QM: analoge Uberlegungen
141

Hamilton-Operator von V und N abhangig (Wande), H = HV,N mit
EV,N ;n = ∞ falls NBen verletzt
also:
ρE,V,N =1
Σ(E, V, N)∆V ∆Nδ(E − HV,N)
und aus der Normierungsbedingung folgt:
Σ(E, V, N) =1
∆V ∆NSp (δ(E − HV,N))
7.3 Quasistatische Parameteranderung
bislang: TD Zustand, GG Zustand, jetzt: TD Prozesse
betrachte Anderung der inneren Energie U = E durch außere (quasistatische)Anderung der Kontrollparameter z
quasistatisch gemischter Zustand ρE,z(q, p) fur alle z GG-Zustand:
ρE,z(q, p) =1
Σ(E, z)∆zδ(E − Hz(q, p))
innere Energie:
〈Hz〉 =∫
dqdp1
Σ(E, z)∆zδ(E − Hz(q, p)) Hz(q, p) = E
somit:
〈Hz〉 =∫
dqdp1
Σ(〈Hz〉, z)∆zδ(〈Hz〉 − Hz(q, p)) Hz(q, p)
und:
∂〈Hz〉∂z
=∫
dqdp1
Σ(〈Hz〉, z)∆zδ(〈Hz〉 − Hz(q, p))
∂Hz(q, p)
∂z
+∫
dqdp Hz(q, p)∂
∂z
(1
Σ(〈Hz〉, z)∆zδ(〈Hz〉 − Hz(q, p))
)
=
⟨∂Hz
∂z
⟩+ 〈Hz〉
∂
∂z
∫dqdp
1
Σ(〈Hz〉, z)∆zδ(〈Hz〉 − Hz(q, p))
2.Term = 0 wegen Normierungsbedingung, also
142

∂〈Hz〉∂z
=
⟨∂Hz
∂z
⟩quasistatische Kontrollparameteranderung
—————————————————————————————————
QM:
ρE,z =1
Σ(E, z)∆zδ(E − Hz)
also:
〈Hz〉 = Sp
(1
Σ(〈Hz〉, z)∆zδ(〈Hz〉 − Hz) Hz
)
Auswertung in ONB von Energieeigenzustanden: |En(z), γ〉 mit
Hz|En(z), γ〉 = En(z)|En(z), γ〉liefert:
〈Hz〉 =∑
n,γ
(1
Σ(〈Hz〉, z)∆zδ(〈Hz〉 − En(z)) En(z)
)
und damit:
∂〈Hz〉∂z
=∑
n,γ
1
Σ(〈Hz〉, z)∆zδ(〈Hz〉 − En(z))
∂En(z)
∂z
wegen Normierungsbedingung kein weiterer Beitrag
mit dem Hellmann-Feynman-Theorem
∂En(z)
∂z= 〈En(z), γ| ∂Hz
∂z|En(z), γ〉
folgt:
∂〈Hz〉∂z
=∑
n,γ
1
Σ(〈Hz〉, z)∆zδ(〈Hz〉 − En(z)) 〈En(z), γ| ∂Hz
∂z|En(z), γ〉
=∑
n,γ
〈En(z), γ|(ρ〈Hz〉,z
∂Hz
∂z
)|En(z), γ〉
= Sp
(ρ〈Hz〉,z
∂Hz
∂z
)=
⟨∂Hz
∂z
⟩
wie in KM-Rechnung:
∂〈Hz〉∂z
=
⟨∂Hz
∂z
⟩quasistatische Kontrollparameteranderung
143

—————————————————————————————————
Beweis des Hellmann-Feynman-Theorems (ohne Entartung, mit: analog)
es gilt:
0 =∂
∂z1 =
∂
∂z〈En(z)|En(z)〉
=∂〈En(z)|
∂z|En(z)〉 + 〈En(z)|∂|En(z)〉
∂z
also:
∂En(z)
∂z=
∂
∂z〈En(z)|Hz|En(z)〉
=∂〈En(z)|
∂zHz|En(z)〉 + 〈En(z)|Hz
∂|En(z)〉∂z
+ 〈En(z)| ∂Hz
∂z|En(z)〉
= En(z)
(∂〈En(z)|
∂z|En(z)〉 + 〈En(z)|∂|En(z)〉
∂z
)+ 〈En(z)| ∂Hz
∂z|En(z)〉
= 〈En(z)| ∂Hz
∂z|En(z)〉
—————————————————————————————————
Verallgemeinerung: z = (z1, ..., zs)
∂〈Hz〉∂zj
=
⟨∂Hz
∂zj
⟩
definiere
Kj =
⟨∂Hz
∂zj
⟩verallgemeinerte Krafte
dann ist:
dU =s∑
j=1
⟨∂Hz
∂zj
⟩dzj =
s∑
j=1
Kjdzj
Energieanderung durch Arbeit außerer Krafte
—————————————————————————————————
speziell:
statistische Definition des mechanischen Drucks:
144

−p =
⟨∂HV,N
∂V
⟩
statistische Definition des chemischen Potentzials:
µ =
⟨∂HV,N
∂N
⟩
damit z.B. dU = −pdV (N fest) Ubereinstimmung mit TD Definition
—————————————————————————————————
Bsp: mechanischer Druck
HV (q, p) =3N∑
i=1
p2i
2m+ V (q1, ..., q3N ) +
3N∑
i=1
WV (qi)
W : Potenzial der Wand
betrachte Volumenanderung durch Verschieben eines Flachenstucks A bei x0
bei x0 ist W = w(xi − x0) = αΘ(xi − x0) mit α → ∞
x0
A
x
V
es ist:
∂HV (q, p)
∂V=
1
A
∂HV (q, p)
∂x0=
1
A
3N∑
i=1
∂Wx0(xi)
∂x0=
1
A
∑
i
∂w(xi − x0)
∂x0
= − 1
A
∑
i
∂Wx0(xi)
∂xi=
1
A
∑
i
F(ext)i (xi) = −F (q1, ..., q3N)
A
F(ext)i : vom Wandelement auf i-tes Teilchen ausgeubte Kraft
F : von allen Teilchen auf das Wandelement ausgeubte Kraft
F : Zufallsgroße mit Fluktuationen
makroskopisch: Druck = mittlere Kraft (pro Flache)
p = −⟨
∂HV
∂V
⟩
145

7.4 Warme und erster Hauptsatz
Phasenvolumen (das von Hz(q, p) = E eingeschlossene Volumen):
Γ(z0, z1, ...zs) = Γ(E, z) (z0 = E, z = (z1, ..., zs))
=1
∆z
∫
Hz(q,p)<Edqdp (∆z = ∆z1 · · ·∆zs)
somit:
dΓ =∂Γ(E, z)
∂EdE +
s∑
j=1
∂Γ(E, z)
∂zjdzj
es ist:
∂Γ(E, z)
∂E=
∂
∂E
1
∆z
∫dqdp Θ(E − Hz(q, p))
=1
∆z
∫dqdp δ(E − Hz(q, p)) = Σ(E, z)
und:
∂Γ(E, z)
∂zj
=∂
∂zj
1
∆z
∫dqdp Θ(E − Hz(q, p))
= − 1
∆z
∫dqdp δ(E − Hz(q, p))
∂Hz(q, p)
∂zj
= −Σ(E, z)∫
dqdp ρz(q, p)∂Hz(q, p)
∂zj
= −Σ(E, z)
⟨∂Hz
∂zj
⟩
also:
dΓ = Σ(E, z) dE −s∑
j=1
Σ(E, z)
⟨∂Hz
∂zj
⟩dzj
= Σ(E, z)
dE −
s∑
j=1
⟨∂Hz
∂zj
⟩dzj
und:
dE =1
Σ(E, z)dΓ +
s∑
j=1
⟨∂Hz
∂zj
⟩dzj erster Hauptsatz
quasistatische TD Prozesse durch Kontakt mit Umgebung:
1) (langsame) Anderung eines Kontrollparameters zj :
146

• kontrollierte Anderung des GG-Zustands ρz(q, p)
• Anderung der inneren Energie: dU = δW
• Verrichten von Arbeit δW =s∑
j=1
Kjdzj
• dΓ = 0 adiabatische Invarianz des Phasenvolumens
2) (langsame) Anderung bei festen Kontrollparametern
• unkontrollierte Anderung des GG-Zustands ρz(q, p)
• Anderung der inneren Energie: dU = δQ
• Zufuhr von Warme δQ
• δQ =dΓ
Σ(E, z)statistische Definition der Warme
Zufuhr von Warme: Anwachsen des Phasenvolumens
zusammen: dU = δW + δQ
δW , δQ uber Prozesse definiert keine totalen Differenziale
Verrichten von Arbeit Zufuhr von Warme
V V
—————————————————————————————————
Bsp. mit N = 1, Pendel: Hl(q, p), Erhohung der inneren Energie, dE, durch
Verrichten von Arbeit Zufuhr von Warme
l → l + dl, Γ = const. Γ → Γ + dΓ, l = const.
δW = 〈∂Hl/∂l〉dl δQ = dΓ/Σ(E, l)
kontrollierte Parameteranderung unkontrollierter Eingriff
147

Stoßbeachte:
• fur N = 1 konnte auch der “unkontrollierte” Stoß als kontrollierbarbetrachtet werden
• Unterscheidung zwischen Arbeit und Warme erst fur N → ∞ sinnvoll
—————————————————————————————————
QM: Hz|En(z), γ〉 = En(z)|En(z), γ〉
Γ(E, z) =1
∆z
En(z)<E∑
n,γ
=1
∆z
∑
n,γ
Θ(E−En(z)) =1
∆zSpΘ(E−Hz)
es ist:
∂Γ(E, z)
∂E=
1
∆zSp δ(E − Hz) = Σ(E, z)
und:
∂Γ(E, z)
∂zj= − 1
∆zSp
(δ(E − Hz)
∂Hz
∂zj
)
= Σ(E, z) Sp
(ρz
∂Hz
∂zj
)= Σ(E, z)
⟨∂Hz
∂zj
⟩
also:
dΓ = Σ(E, z)
dE −
s∑
j=1
⟨∂Hz
∂zj
⟩dzj
und:
dE =1
Σ(E, z)dΓ +
s∑
j=1
⟨∂Hz
∂zj
⟩dzj erster Hauptsatz
7.5 Temperatur und Nullter Hauptsatz
zwei beliebige Systeme in thermischem Kontakt:
148

V V1 2
Austausch von Warme(unkontrollierter Energieaustausch auf mikroskopischer Ebene)
V1, V2, N1, N2 fest
Hamilton-Funktion/-Operator:
H(q, p) = H1(q1, p1) + H2(q2, p2) + H12 H = H1 + H2 + H12
H12: Oberflachenterm, vernachlassigbar gegen H1, H2
H = E fest, H1, H2 Zufallsgroßen
es ist: (∆z = ∆V1∆N1∆V2∆N2 = ∆z1∆z2)
Σ(E, z) =1
∆z
∫dqdpδ(E − Hz(q, p))
=1
∆z1∆z2
∫dq1dp1
∫dq2dp2 δ(E − Hz1(q1, p1) − Hz2(q2, p2))
=1
∆z1∆z2
∫dq1dp1
∫dq2dp2
∫dE1δ(E1−Hz1(q1, p1))δ(E−E1−Hz2(q2, p2))
=∫
dE11
∆z1
∫dq1dp1δ(E1−Hz1(q1, p1))
1
∆z2
∫dq2dp2δ(E−E1−Hz2(q2, p2))
=∫
dE1Σ1(E1, z1)Σ2(E − E1, z2)
bzw. (QM)
Σ(E, z) =1
∆zSp δ(E −Hz) =
1
∆z1∆z2
Sp 1 Sp 2δ(E −Hz1 −Hz2)
=1
∆z1∆z2Sp 1 Sp 2
∫dE1δ(E1 − Hz1)δ(E − E1 − Hz2)
=∫
dE11
∆z1Sp 1δ(E1 − Hz1)
1
∆z2Sp 2δ(E − E1 − Hz2)
=∫
dE1Σ1(E1, z1)Σ2(E − E1, z2)
—————————————————————————————————
Bemerkung:
Sp (A1A2) = Sp 1A1 Sp 2A2
149

gilt fur Operatoren des Systems 1 bzw. 2 nur bei Verwendung der ONB vonnicht-(anti-)symmetrisierten Produktzustanden, aber:
Sp HNA = Sp H(±)
N
A
falls A physikalische Obersable ([A,P] = 0, [A, S(±)N ] = 0)
Beweis(skizze):
SpH(±)N
A =∑
n
〈n|A|n〉 =∑
m
〈m|S(±)N
†AS
(±)N |m〉 =
∑
m
〈m|A|m〉 = SpHNA
mit |n〉: ONB von H(±)N (Besetzungszahldarstellung)
und |m〉: ONB von HN (Produktzustande)
und |n〉 = K±S(±)N |m〉
—————————————————————————————————
Analyse der Zustandsdichte:
Σ(E, z) =∫
dE1Σ1(E1, z1)Σ2(E − E1, z2)
Σ1(E1, z1) wachst extrem schnell mit E1
Σ2(E − E1, z2) fallt extrem schnell mit E1
Integral als Riemann-Summe schreiben, dE → ∆E
E1(max)
E1
Σ1 1(E )Σ2(E−E )1
Approximation der Summe durch maximalen Summanden liefert:∑
E1
∆E Σ1(E1, z1)Σ2(E −E1, z2) = Σ1(E(max)1 , z1)Σ2(E −E
(max)1 , z2)∆E
und somit:
150

Σ(E, z)∆E = Σ1(E1, z1)∆E Σ2(E2, z2)∆E
oder:
ln Γ(E, z) = ln Γ1(E1, z1) + ln Γ2(E2, z2)
ln Phasenvolumen additiv
fur E1 = E(max)1 und E2 = E
(max)2 ≡ E − E
(max)1
Bestimmung des Maximums:
0 =∂
∂E1(Σ1(E1, z1)Σ2(E − E1, z2))
∣∣∣∣∣∣E1=E
(max)1
0 =∂Σ1(E1, z1)
∂E1Σ2(E − E1, z2) − Σ1(E1, z1)
∂Σ2(E − E1, z2)
∂E2
∣∣∣∣∣∣E1=E
(max)1
mit E(max)2 ≡ E − E
(max)1 ist also:
1
Σ1(E(max)1 , z1)
∂Σ1(E(max)1 , z1)
∂E1=
1
Σ2(E(max)2 , z2)
∂Σ2(E(max)2 , z2)
∂E2
also:
zwei beliebige Systeme in thermischem Kontakt haben im GG denselben Wert
der Große1
Σ(E, z)
∂Σ(E, z)
∂E, Dimension: 1/Energie
also:
T1 = T2 Nullter Hauptsatz
gilt, falls
1
kT=
1
Σ(E, z)
∂Σ(E, z)
∂Estatistische Definition der Temperatur
(fur jedes System gibt es also die) thermische Zustandsgleichung:
T = T (E, z) bzw. E = E(T, z)
Ableitung der thermischen Zustandsgleichung eines Systems:
• H(q, p) / H = H† aufstellen
• Σ(E, z)∆z =∫
dqdp δ(E − H(q, p) / Σ(E, z)∆z = Sp δ(E − H(q, p)
berechnen
151

• 1/kT = (∂Σ(E, z)/∂E)/Σ(E, z) nach E auflosen
7.6 Entropie
betrachte quasistatischen Prozess, es gilt:
dE =1
Σ(E, z)dΓ +
s∑
j=1
⟨∂Hz
∂zj
⟩dzj
zugefuhrte Warme:
δQ =1
Σ(E, z)dΓ =
dΓ∂Γ(E,z)
∂E
Konzentration des Phasenvolumens in dunner Schicht unter Flache H(q, p) = E:
Γ(E, z) = Σ(E, z)∆E (N → ∞)
also:
δQ =dΣ∆E
∂(Σ(E,z)∆E)∂E
=dΣ
∂Σ(E,z)∂E
=Σ
∂Σ(E,z)∂E
· dΣ
Σ(E, z)= kT
dΣ
Σ(E, z)
Entropie nach Clausius: dS = δQ/T , also
dS = kdΣ
Σ(E, z)
dS ist ein totales Differenzial, denn
dS = k d ln(αΣ(E, z)∆E∆z)
beachte: αΣ(E, z)∆E∆z dimensionslos, mit:
α = 1 (QM)
α =1
h3NN !(KM)
Bemerkungen:
• α wird so gewahlt, dass SQM → SKM im klassischen Limes (s.u.)
• Diskretisierung des Phasenraums in Zellen mit Volumen ∆v = h3N
αΣ(E, z)∆E∆z: Anzahl der Zellen (Zustanden im diskretisierten Pha-senraum), die mit NBen (E, z fest) vertraglich sind
• Clausius-Formel dS = δQ/T nur fur Prozesse mit N = const. abgeleitet
α darf N -abhangig sein
152

damit motiviert:
S(E, z) = k ln(αΣ(E, z)∆E∆z)
statistische Definition der Entropie nach Boltzmann
“S = k log W” Inschrift auf Grabstein von Boltzmann in Wien !
Integrationskonstante = 0 gesetzt (nur damit ist 3. HS erfullt, s.u.)
jetzt gilt unmittelbar:
TdS = δQ = dE − δW Grundgleichung der TD
und (da ln Phasenvolumen additive Große):
S ist extensiv
—————————————————————————————————
S(E, z) = k ln(αΣ(E, z)∆E∆z) = k ln(αΣ(E, V, N)∆E∆V ∆N)
Phasenvolumen wachst extrem schnell mit E, V, N (s.u. ideales Gas)
Entropie als (logarithmisches) Maß fur erlaubtes Phasenvolumen
—————————————————————————————————
Zusammenhang mit Shannon-Information:
σ(p1, ..., pM) = −M∑
k=1
pk ln pk
mikrokanonische WK: (QM: k = (En, Vm, Np, γ))
pk = p(En, Vm, Np, γ) =1
Σ(E, V, N)δ(E−En)δ(V −Vm)δ(N−Np)
=1
Σ(E, V, N)∆E∆V ∆N
falls NB (δ(E − En)∆E = 1, etc.) erfullt
pk = 0 sonst
damit:
σ = −NBen∑
En,Vm,Np,γ
1
Σ(E, V, N)∆E∆V ∆Nln
(1
Σ(E, V, N)∆E∆V ∆N
)
mit der Normierungsbedingung folgt:
= − ln
(1
Σ(E, V, N)∆E∆V ∆N
)
153

= ln (Σ(E, V, N)∆E∆V ∆N) =1
kS(E, V, N)
Entropie ist k× Maximalwert (unter NBen) der Shannon-Information:
S(E, z) = k maxE,z fest
σ(p1, ..., pM)
• Shannon-Information: Funktion des (gemischten) Mikrozustands
• Entropie: Funktion des Makrozustands, abhangig von den kontrollierbarenMakrovariablen (vergl: Gibbs-Paradox)
—————————————————————————————————
S(E, V, N) = k ln(αΣ(E, V, N)∆E∆V ∆N)
Konsistenzchecks:(
∂S
∂E
)
V,N
=∂
∂Ek ln(αΣ(E, V, N)∆E∆V ∆N)
= k∂Σ(E, V, N)/∂E
Σ(E, V, N)= k
1
kT=
1
T
statistische Definition der Temperatur(
∂S
∂V
)
E,N
=∂
∂Vk ln(αΣ(E, V, N)∆E∆V ∆N)
= k∂Σ(E, V, N)/∂V
Σ(E, V, N)=
1
T
Σ(E, V, N)
∂Σ(E, V, N)/∂E
∂Σ(E, V, N)/∂V
Σ(E, V, N)
=1
T
∂Σ(E, V, N)/∂V
∂Σ(E, V, N)/∂E= − 1
T
(∂E
∂V
)
Σ,N
= − 1
T
(∂E
∂V
)
S,N
= − 1
T
⟨∂H
∂V
⟩
S,N=
p
T
(quasistatische Kontrollparameteranderung (adiabatisch))
statistische Definition des Drucks(
∂S
∂N
)
E,V
=∂
∂Nk ln(αΣ(E, V, N)∆E∆V ∆N)
= k∂Σ(E, V, N)/∂N
Σ(E, V, N)=
1
T
Σ(E, V, N)
∂Σ(E, V, N)/∂E
∂Σ(E, V, N)/∂N
Σ(E, V, N)
=1
T
∂Σ(E, V, N)/∂N
∂Σ(E, V, N)/∂E= − 1
T
(∂E
∂N
)
Σ,V
= − 1
T
(∂E
∂N
)
S,V
154

= − 1
T
⟨∂H
∂N
⟩
S,V= −µ
T
quasistatische Kontrollparameteranderung (adiabatisch)
statistische Definition des chemischen Potenzials
beachte hier:(
∂X
∂Y
)
Z
(∂Y
∂Z
)
X
(∂Z
∂X
)
Y
= −1
155

156

Kapitel 8
Einfache Anwendungen
8.1 Gleichverteilungssatz
Jeder “Freiheitsgrad” (qi oder pi), der quadratisch in die Hamilton-Funktioneingeht, liefert einen Beitrag
1
2kT zur inneren Energie U
bzw.
k
2zur Warmekapazitat CV N =
(∂U
∂T
)
V,N
beachte: Gleichverteilungssatz gilt nur im Rahmen der KM
—————————————————————————————————
Bsp: klassisches einatomiges ideales Gas
HV,N(q, p) =3N∑
i=1
p2i
2m+ VWand(q1, ..., q3N )
3N Freiheitsgrade, die quadratisch in H eingehen (pi)
3N Freiheitsgrade, die keinen Beitrag zu U liefern (qi)
also:
U = 3N1
2kT kalorische Zustandsgleichung
—————————————————————————————————
Beweis:
mit πi = qi oder πi = pi ist
157

⟨πi
∂HV,N (π)
∂πi
⟩=∫
dπ ρE,V,N(π)πi∂HV,N (π)
∂πi
=∫
dπ ρE,V,N(π)πi∂HV,N (π)
∂πi
mit
ρE,V,N(π) =1
Σ(E, V, N) ∆V ∆Nδ(E − HV,N(π))
folgt:⟨πi
∂HV,N (π)
∂πi
⟩=
1
Σ(E, V, N) ∆V ∆N
∫dπ δ(E−HV,N(π))πi
∂HV,N (π)
∂πi
=1
Σ(E, V, N) ∆V ∆N
∂
∂E
∫dπ Θ(E − HV,N(π))πi
∂HV,N (π)
∂πi
es ist:
πi∂HV,N(π)
∂πi=
∂
∂πi(πi(HV,N(π) − E)) − (HV,N(π) − E)
1.Term eingesetzt:∫
dπ Θ(E−HV,N (π))∂(πi(HV,N(π) − E))
∂πi
=∫
H<Edπ
∂Fi(π)
∂πi
= 0
denn Fi(π) ≡ (πi(HV,N(π) − E)) = 0 auf HV,N(q, p) = E
bleibt Beitrag des 2. Terms:⟨πi
∂HV,N (π)
∂πi
⟩
=1
Σ(E, V, N) ∆V ∆N
∂
∂E
∫dπ Θ(E − HV,N(π))(E − HV,N(π))
=1
Σ(E, V, N) ∆V ∆N
∫dπ Θ(E − HV,N(π))
=Γ(E, V, N)
Σ(E, V, N)=
Γ(E, V, N)∂Γ(E,V,N)
∂E
=Σ(E, V, N)
∂Σ(E,V,N)∂E
mit Γ = Σ∆E fur N → ∞, und deshalb:⟨πi
∂HV,N (π)
∂πi
⟩=
1∂ ln(αΣ(E,V,N)∆E∆V ∆N
∂E
=k
∂S(E,V,N)∂E
= kT
—————————————————————————————————
es ist also:
158

⟨πi
∂HV,N (π)
∂πi
⟩= kT unabhangig von i
sei
H =p2
i
2m+ cq2
i + H ′ mit H ′ unabhangig von qi, pi, c = const.
dann ist:⟨
p2i
2m
⟩=⟨pi
mqi
2m
⟩=
1
2
⟨piqi
⟩=
1
2
⟨pi
∂H
∂pi
⟩=
1
2kT
und:⟨cq2
i
⟩=
1
2
⟨qi2cqi
⟩=
1
2
⟨qi
∂H
∂qi
⟩=
1
2kT
—————————————————————————————————
Anwendung auf Festkorper:
N Kristallatome im Gitter: harmonische Schwingungen
hohe T kl.Statistik gultig
3N Impulse, 3N Koordinaten quadratisch in H
also:
CV,N =
(∂U
∂T
)
V,N
=1
2k × 6N = 3Nk
Dulong-Petitsches Gesetz
– empirisch gut bestatigt, solange kT ≫ ~ω (ω: Schwingungsfrequenz)
– kT ∼ ~ω QM Rechnung (s.u.):
CV,N = 3Nk
( β~ω2
sinh β~ω2
)2
(β = 1/kT )
T → ∞, β → 0: CV,N → 3Nk, denn sinh x = x + O(x3), klassischer Limes
T → 0, β → ∞: CV,N → 3Nk(β~ωe−β~ω/2)2 → 0 (konsistent mit 3.HS !)
CV,N durch Quanteneffekte fur T → 0 exponentiell unterdruckt
159

3
kT/ h ω
klassischer LimesV,NC /Nk
—————————————————————————————————
Anwendung auf Gase bei hohen T :
einatomig, Bsp: Ar,Xe
3N Freiheitsgrade (pi) quadratisch in H
CV,N =3
2Nk einatomig
zweiatomig, mehratomig, Bsp: O2, N2, H2O
2 (oder mehr) zusatzliche Schwingungsfreiheitsgrade
bei Raumtemperatur aber meist kTRaum ≪ ~ωSchwingungen
durch Quanteneffekte unterdruckt, Schwingungsfreiheitsgrade “ausgefroren”
2 bzw. 3 Rotationsfreiheitsgrade (zwei-/mehratomige Gase)
~ω ∼ ~2
2ΘΘ: Tragheitsmoment
zweiatomig: Θ bzgl. Molekulachse klein kTRaum ≪ ~ω
also: fur Raumtemperatur typischerweise:
CV,N =5
2Nk zweiatomig
CV,N = 3Nk mehratomig
8.2 Maxwellsche Geschwindigkeitsverteilung
klassisches (nicht ideales, wechselwirkendes) Gas
v1 = p1/m: Geschwindigkeit des Teilchens “1” ist eine Zufallsvariable
160

WK, dass Teilchen 1 die Geschwindigkeit v1 in d2v1 besitzt:
ρ(v1)d3v1 =?
es ist:
ρ(mv1) =∫
d3p2 · · ·∫
d3pN
∫dq3
1 · · ·∫
d3qN ρE,V,N(q, p)
=∫
d3p1 · · ·∫
d3pN
∫dq3
1 · · ·∫
d3qN ρE,V,N(q, p)δ(p1 − mv1)
= 〈δ(p1 − mv1)〉 =1
m〈δ(p1/m − v1)〉 =
1
mρ(v1)
MK WK-Dichte:
ρE,V,N(q, p) =1
Σ(E, V, N)∆V ∆Nδ(E − HV,N(q, p))
also (nur die v1-Abhangigkeit ist hier von Interesse):
ρ(v1) ∝∫
d3p1 · · ·∫
d3pN
∫d3Nq δ(E −HV,N (q, p))δ(p1/m−v1)
= m∫
d3p2 · · ·∫
d3pN
∫d3Nq δ(E − HV,N(q, p)|p1=mv1)
Hamilton-Funktion:
HV,N(q, p) =N∑
j=1
p2j
2m+ V (q1, ..., qN)
HV,N(q, p)|p1=mv1 −1
2mv2
1 = HV,N(0, p2, ..., pN , q) ≡ HV,N(q, p)
damit:
ρ(v1) ∝∫
d3p2 · · ·∫
d3pN
∫d3Nq δ(E − 1
2mv2
1 − HV,N(q, p))
∝ Σ(E − 1
2mv2
1, V, N) ≈ Σ(E − 1
2mv2
1, V, N)
in sehr guter Naherung !(die v1-Abhangigkeit andert sich quasi nicht, falls N → N − 1, etc.)
jetzt ist1
2mv2
1 ≪ E, also:
ln Σ(E − 1
2mv2
1, V, N) ≈ ln Σ(E, V, N) − ∂ ln Σ(E, V, N)
∂E· 1
2mv2
1
(Entwicklung des ln wegen der extrem starken E-Abhangigkeit von Σ sinnvoller)
und mit 1/kT = (∂/∂E) ln Σ(E, V, N):
ρ(v1) ∝ exp(lnΣ(E − 1
2mv2
1, V, N))
161

= exp
(ln Σ(E, V, N) − ∂ ln Σ(E, V, N)
∂E· 1
2mv2
1
)
∝ exp(− 1
kT
1
2mv2
1
)
Normierung (Index 1 weglassen):∫
d3v ρ(v) = 1
legt Konstante fest (einfaches Gaußintegral):
ρ(v) =(
m
2πkT
)3/2
exp
(−mv2
2kT
)
Maxwellsche Geschwindigkeitsverteilung
– gilt fur klassische System mit beliebigem WW-Potenzial !
– (trotz Gaußverteilung) keine Relation zum ZGWS (v keine extensive Variable)
es ist ρ(v) = ρ(vx)ρ(vy)ρ(vz) und
ρ(vi) =(
m
2πkT
)1/2
exp
(−mv2
i
2kT
)
symmetrische Verteilung
〈vi〉 = 0
Varianz:
〈v2i 〉 =
∫dvi v2
i
(m
2πkT
)1/2
exp
(−mv2
i
2kT
)
= −2kT
m
∂
∂x
∫dvi
(m
2πkT
)1/2
exp
(−x
mv2i
2kT
) ∣∣∣∣∣∣x=1
= −2kT
m
∂
∂xx−1/2
∣∣∣∣∣∣x=1
=kT
m
oder einfacher mit GVS:
〈v2i 〉 =
2
m
⟨p2
i
2m
⟩=
2
m
1
2kT =
kT
m
es folgt:
kT = m〈v2i 〉
Temperatur: mittlere quadratische Geschwindigkeitsschwankung
(allgemeine, mikroskopische Veranschaulichung des Temperaturbegriffs !)
162

8.3 Zustandsdichte eines idealen Gases
ideales (einatomiges) Gas:
HV,N(q, p) =3N∑
i=1
p2i
2m+ WWand,V(q1, ..., q3N)
Zustandsdichte: Σ(E, V, N)
klassisch:
Σ(E, V, N)∆V ∆N =∫
dqdp δ(E − HV,N(q, p))
= V N∫
dp1 · · · dp3N δ
(E −
3N∑
i=1
p2i
2m
)
δ(...) = 0, falls qi /∈ V , und W (...) = 0, falls qi ∈ V
= V N ∂
∂E
∫dp1 · · · dp3N Θ
(E −
3N∑
i=1
p2i
2m
)
= V N ∂
∂EV3N (
√2mE)
VD(R): Volumen einer D-dimensionalen Kugel vom Radius R
VD(R) =∫
dx1 · · · dxDΘ(R2 −∑
i
x2i ) = RDVD(1)
VD(1) =∫
dΩ∫ 1
0dr rD−1 = SD(1)
1
DrD
∣∣∣∣1
0=
1
DSD(1)
SD(1): Oberflache der D-dimensionalen Einheitskugel
SD(1) = 2πD/2
(D/2 − 1)!
damit folgt:
Σ(E, V, N)∆V ∆N = V N ∂
∂E
(√2mE
3N 1
3N2
π3N/2
(3N/2 − 1)!
)
= V N 3N
2(2mE)(3N/2)−12m
1
3N2
π3N/2
(3N/2 − 1)!
Σ(E, V, N)∆V ∆N = V N 1
E(2mE)3N/2 π3N/2
(3N/2 − 1)!
—————————————————————————————————
gleiche Rechnung auf Basis der QM fur hohe T
163

Zustandsdichte:
Σ(E, V, N) =1
∆V ∆NSp (δ(E − HV,N))
zur Auswertung der Spur: Energieeigenzustande
Hamilton-Operator:
H =N∑
j=1
H(j)1 mit H1 =
p2
2m+ W1,Wand,V (r)
Ein-Teilchen-Problem:
H1|φ〉 = ε|φ〉Ortsdarstellung:
− ~2
2m∆φ(r) = εφ(r)
Wandpotenzial: zyklische Randbedingungen auf Wurfel mit V = L3:
φ(r + Leµ) = φ(r)
eµ: Einheitsvektor in µ = x, y, z-Richtung
(Form der RB und des Volumens unwichtig, Ergebnisse nur von V abhangig)
Losungsansatz:
φk(r) = const. eikr
Losung, falls:
~2k2
2m≡ ε(k) = ε
Normierung:∫
Vd3r |φk(r)|2 = 1
also:
φk(r) =1√V
eikr Ein-Teilchen-Energieeigenzustande
RB:
eik(r+Leµ) = eikr ⇒ eiLkeµ = eiLkµ = 1
164

2 /Lπ
k
also:
kµ =2π
Lnµ nµ ganze Zahl erlaubte k-Vektoren
Rastervolumen im k-Raum:
∆3k =(2π)3
V—————————————————————————————————
N-Teilchen-Problem:
H|Ψ〉 = EΨ
wegen H =∑
j
H(j)1 sind Produktzustande Eigenzustande:
H|φ(1)k1〉 · · · |φ(N)
kN〉 =
N∑
j=1
ε(kj)
|φ(1)
k1〉 · · · |φ(N)
kN〉
physikalischer Hilbert-Raum: (anti-)symmetrisierte Produktzustande
HS(±)N |φ(1)
k1〉 · · · |φ(N)
kN〉 = S
(±)N H|φ(1)
k1〉 · · · |φ(N)
kN〉
=
N∑
j=1
ε(kj)
S
(±)N |φ(1)
k1〉 · · · |φ(N)
kN〉
ebenfalls Eigenzustande
Eigenenergien:
E = E(k1, ..., kN) =N∑
j=1
ε(kj)
—————————————————————————————————
Zustandsdichte:
Σ(E, V, N)∆V ∆N = Sp(δ(E−HV,N)) ≈′∑
k1
· · ·′∑
kN
δ(E−E(k1, ..., kN))
Summation nur uber (k1, ..., kN), die nicht durch Permutation auseinander her-
165

vorgehen (Ununterscheidbarkeitsprinzip !)
also:
Σ(E, V, N) ≈ 1
∆V ∆N
1
N !
∑
k1
· · ·∑
kN
δ(E − E(k1, ..., kN))
jetzt mit nicht eingeschrankter Summation
—————————————————————————————————
beachte: nur gultig, falls Mehrfachbesetzungen selten klassischer Limes
Bosonen: fur nk ≥ 2 liefern Permutationen von Teilchen mit gleichem k keinenneuen Zustande, und durfen schon in
∑′ nicht mehrfach gezahlt werden
Fermionen: Zustande mit nk ≥ 2 durfen in∑′ gar nicht gezahlt werden
nk ≤ 1, falls N ≪ d, (d: Anzahl zuganglicher Ein-Teilchen-Zustande)
klassischer Limes fur hohe T oder geringe Dichte v = V/N → ∞—————————————————————————————————
Abschatzung der korrekten Anzahl von Zustanden fur N ≪ dund d = dimH1 endlich:
Terme in∑′: M =
1
N !dN
ln M ≈ N ln d − N ln N
Bosonen: M =
(d + N − 1
N
)
ln M ≈ (d + N − 1) ln(d + N − 1) − N ln N − (d − 1) ln(d − 1)
≈ d ln d + N ln d − N ln N − d ln d = N ln d − N ln N
Fermionen: M =
(dN
)
ln M ≈ d ln d − N ln N − (d − N) ln(d − N)
≈ d ln d − N ln N − d ln d + N ln d = N ln d − N ln N
—————————————————————————————————
fur L → ∞ (V → ∞) ist
∑
k
=V
(2π)3
∑
k
∆3k → V
(2π)3
∫d3k
somit:
166

Σ(E, V, N) =1
∆V ∆N
V N
(2π)3N
1
N !
∫d3k1 · · ·
∫d3kNδ
E −
N∑
j=1
~2k2
j
2m
mit Substitution pj = ~kj:
Σ(E, V, N) =1
∆V ∆N
V N
(2π~)3N
1
N !
∫d3p1 · · ·
∫d3pNδ
E −
N∑
j=1
p2j
2m
Σ(E, V, N) =1
∆V ∆N
1
(2π~)3N
1
N !
∫dqdp δ (E − HV,N(q, p))
also (fur hohe T ):
ΣQM(E, V, N) =1
(2π~)3N
1
N !ΣKM(E, V, N) = αΣKM(E, V, N)
– erklart Wahl von α
–1
N !: “korrekte Boltzmann-Abzahlung”
–1
(2π~)3N: Diskretisierung des Phasenraums in Zellen mit Phasenvolumen (2π~)3N
System mit verschiedenen Teilchensorten (Komponenten):
α =1
∏α Nα!(2π~)3Nα
(Permutation unterscheidbarer Teilchen liefert neue Zustande)
8.4 Entropie eines klassischen idealen Gases
Entropie:
S(E, V, N) = k ln (αΣ(E, V, N)∆E∆V ∆N)
mit: α = 1/(h3NN !)
Zustandsdichte (klassisch):
Σ(E, V, N)∆V ∆N = V N 1
E(2mE)3N/2 π3N/2
(3N/2 − 1)!
damit:
S(E, V, N) = k ln
[(V
h3
)N 1
N !
∆E
E(2mEπ)3N/2 1
(3N/2 − 1)!
]
mit N ! ≈ NNe−N , 3N/2 − 1 → 3N/2 ist:
167

S(E, V, N) = k ln
(
V e
h3N
)N ∆E
E
(2mEπe
3N/2
)3N/2
ln ∆E/E kann gegen (extensive) Terme ∝ N vernachlassigt werden:
S(E, V, N) = Nk ln
(
V e
h3N
)(2mEπe
3N/2
)3/2
= Nk ln
[V
N
(E
N
)3/2 (4mπ
3h2
)3/2
e5/2
]
S(E, V, N) = Nk[ln
V
N+
3
2ln
E
N+
3
2ln
4πm
3h2+
5
2
]
Sackur-Tetrode-Gleichung
– gleiches Resultat wie in TD
– zusatzlich: Entropie-Konstante σ0 =3
2ln
4πm
3h2+
5
2
– S extensiv
—————————————————————————————————
Zustandsgleichungen des idealen Gases:
1
T=
(∂S
∂E
)
V,N
=3
2Nk
1
E(kalorische Zustandsgleichung)
p
T=
(∂S
∂V
)
E,N
= Nk1
V(thermische Zustandsgleichung)
—————————————————————————————————
Gultigkeitsbereich?
mit E = 3NkT/2 ist:
S = S(T, V, N) = Nk(ln
V
N+
3
2ln
3
2kT + σ0
)
= Nk
(ln
[V
N
(3
2kT)3/2 (4πm
3h2
)3/2]
+5
2
)
also:
S(T, V, N) = Nk(ln
v
λ3+
5
2
)
mit v = V/N und
168

λ =h√
2πmkTthermische de Broglie-Wellenlange
Interpretation:
– Teilchen mit thermischer Energie ε ∼ kT (Gleichverteilungssatz!)
– freies Teilchen p =√
2mε
– de Broglie-Wellenlange λ = h/p
also: λ ∼ h/√
2mkT
λ3 ≪ v = V/N Volumen pro Teilchen groß gegen typisches Volumen, beidem QM Effekte wichtig werden
klassische Approximation korrekt fur λ3 ≪ v = V/N (große E, hohe T )
8.5 Sattelpunktsmethode
mathematischer Einschub:Sattelpunktsmethode wird fur die Auswertung der QM Zustandsdiche benotigt
—————————————————————————————————
f(z) analytisch mit f ′(z0) = 0, C: beliebiger Weg
∫
Cdz eNf(z) =
√2π
−Nf ′′(z0)eNf(z0) fur N → ∞
also:
ln∫
Cdz eNf(z) = Nf(z0) + O(lnN)
—————————————————————————————————
schreibe f = u + iv, z = x + iy
fur z = z0 gilt:
df
dz= 0
∂f
∂x=
∂u
∂x+ i
∂v
∂x= 0
∂u
∂x= 0,
∂v
∂x= 0
analog (fur z = z0):
∂u
∂y= 0,
∂v
∂y= 0
mit Cauchy-Riemann-DGLs:
∂u
∂x=
∂v
∂y
∂u
∂y= −∂v
∂x
169

folgt weiter:
∂2u
∂x2=
∂
∂x
∂v
∂y=
∂
∂y
∂v
∂x= −∂2u
∂y2
analog:∂2v
∂x2= −∂2v
∂y2
u(x, y), v(x, y) haben in z0 einen Sattelpunkt
wahle C ′ durch z0 mit gleichem Anfangs- und Endpunkt, es ist∫
Cdz... =
∫
C′
dz...
Im zz0
C
C’
Re z
Entwicklung von f um z0, Polardarstellung:
f(z) = f(z0 + reiϕ) ≈ f(z0) +1
2f ′′(z0)r
2e2iϕ
(Terme dritter und hoherer Ordnung vernachlassigt)
schreibe f ′′(z0) = ReiΦ, damit ist:
f(z) = f(z0) +1
2ReiΦr2e2iϕ = f(z0) +
1
2Rr2ei(Φ+2ϕ)
wahle C ′ so, dass (bei z0):
ϕ = −1
2Φ +
π
2
dann ist:
f(z) = f(z0) −1
2Rr2
Realteil
u(z) = u(z0) −1
2Rr2
hat in z0 entlang ϕ ein Maximum
(Imaginarteil v(z) konstant bei z0 entlang C ′)
zweiter Faktor des Integranden nahe bei z0:
170

eNf(z) = eiNv(z0)eNu(z)
hat bei z0 entlang C ′ ein extrem scharfes Maximum (N → ∞), also:∫
Cdz eNf(z) = eNf(z0)
∫
C′
dz eN 12f ′′(z0)(z−z0)2 (N → ∞)
mit dz = eiϕdr (ϕ = −Φ/2 + π/2 fest) folgt fur N → ∞:
= eNf(z0)∫ r0
−r0
dr eiϕ e−12NRr2
= eNf(z0)eiϕ∫ ∞
−∞dr e−
12NRr2
mit y = r√
NR/2 ist:
= eNf(z0)eiϕ
√√√√ 112NR
∫ ∞
−∞dy e−y2
= eNf(z0)eiϕ
√π
12NR
= eNf(z0)
√π
12NRe−2iϕ
= eNf(z0)
√π
12NRei(Φ−π)
= eNf(z0)
√2π
−Nf ′′(z0)
Probleme bei mehreren Sattelpunkten
8.6 Quantenmechanisches System harmonischer
Oszillatoren
N Teilchen, Koordinaten qi, Impulse pi (i = 1, ..., N), eindimensional
[qi, pj] = i~δij (i, j = 1, ..., N)
Hamilton-Operator des i-ten Teilchens: harmonischer Oszillator
H(i)1 =
p2i
2m+
1
2mω2
i q2i
Gesamtsystem:
HN =N∑
i=1
H(i)1
Realisierung:
– Atome auf Platzen eines (D = 1) Gitters
– qi: Auslenkung relativ zum Gitterpunkt i
– vereinfachend: ungekoppelte Oszillatoren (sonst: Hauptachsentransformation)
171

– kleine Auslenkungen harmonisches Potenzial
—————————————————————————————————
Strategie:
1. Eigenwertproblem zu H1
2. Eigenzustande und -energien zu HN
3. Kontrollparameter: E und N
Zustandsdichte Σ(E, N)∆N = Sp δ(E − HN)
Auswertung der Spur: ONB aus Energieeigenzustanden
4. Anwendung der Sattelpunktsmethode
5. Entropie: S(E, N) = k ln(Σ(E, N)∆E∆N)
Ableitung von Zustandsgleichungen und Antwortkoeffizienten
—————————————————————————————————
1) Transformation auf Erzeuger/Vernichter:
qi =
√~
2mωi
(ci + c†i ) pi = −i
√m~ωi
2(ci − c†i)
es ist:
[qi, pj] = i~δij [ci, c†j] = δij
und:
H(i)1 = ~ωi
(ni +
1
2
)
Besetzungszahloperator:
ni = c†ici
Eigenwertgleichung:
ni|ni〉 = ni|ni〉mit ni = 0, 1, 2, ... und gni
= 1 (ni nicht entartet)
es ist: H(i)1 |ni〉 = εi(ni)|ni〉 mit
εi(ni) = ~ωi
(ni +
1
2
)ni = 0, 1, 2, ...
—————————————————————————————————
2) Viel-Teilchen-Problem
172

kleine Auslenkungen:
kein Uberlapp der Wellenfunktionen zweier Teilchen
niemals zwei Teilchen im gleichen Oszillator
jedes Teilchen hat seinen eigenen Ein-Teilchen-Hilbert-Raum
Teilchen unterscheidbar
HN = ⊗Ni=1H(i)
1
ONB von HN :
|n1〉|n2〉 · · · |nN〉 (Produktzustande)
mit ONB von H(i)1 :
|ni〉—————————————————————————————————
M = dimHN , d = dimH1 (endlich)
ununterscheidbare Teilchen:
φ3φ1φ2 φd Zustände...
Ein−Teilchen−
QM, Bosonen
φ3φ1φ2 φd Zustände...
QM, Fermionen
Ein−Teilchen−
M =
(d + N − 1
N
)M =
(dN
)
(durch raumliche Trennung) unterscheidbare Teilchen:
ZuständeEin−Teilchen−
φ3φ1φ2 φd... φ3φ1φ2 φd... φ3φ1φ2 φd...
QM, Teilchen in separaten Subsystemen
M = dN
d: Dimension desjenigen Unterraums des gesamten Ein-Teilchen-Hilbert-Raums,in dem sich ein Teilchen aufhalten kann
—————————————————————————————————
(fur Puristen:) Einschub zur Begrundung der Unterscheidbarkeit
beachte: prinzipiell sind auch identische Teilchen, die raumlich getrennt sind(kein Uberlapp von Wellenfunktionen), ununterscheidbar und somit als Bose-/Fermi-System zu behandeln, aber die Behandlung als unterscheidbare Teilchenliefert dieselben Ergebnisse:
173

betrachte Messung einer “lokalen” Observablen A, d.h.
A =N∑
i=1
A(i)
Erwartungswert in (anti-)symmetrisiertem Produktzustand:
〈A〉 = K2±〈φ(1)
α1| · · · 〈φ(N)
αN|S(±)
N
N∑
i=1
A(i)S(±)N |φ(1)
α1〉 · · · |φ(N)
αN〉
die Teilchen befinden sich in paarweise verschiedenen Ein-Teilchen-Zustanden(dimH1 = Nd bei Beschreibung mit ununterscheidbaren Teilchen!)
Besetzungszahlen nα = 0, 1 K2± = N !
〈A〉 = N !N∑
i=1
〈φ(1)α1| · · · 〈φ(N)
αN|A(i)S
(±)N |φ(1)
α1〉 · · · |φ(N)
αN〉
Permutationsoperator P in S(±)N =
∑
P(±1)pP/N ! wirkt auf Teilchenindizes oder
(nach Umsortieren des Produkts) auf Quantenzahlen αi:
〈A〉 =N∑
i=1
∑
P(±1)p〈φ(1)
α1| · · · 〈φ(N)
αN|A(i)|φ(1)
αp(1)〉 · · · |φ(N)
αp(N)〉
wegen nα = 0, 1 liefert nur P = 1 einen nichtverschwindenden Beitrag:
〈A〉 =N∑
i=1
〈φ(i)αi|A(i)|φ(i)
αi〉
es ist also:
〈A〉 =N∑
i=1
〈φ(1)α1| · · · 〈φ(N)
αN|A(i)|φ(1)
α1〉 · · · |φ(N)
αN〉
〈A〉 = 〈φ(1)α1| · · · 〈φ(N)
αN|A|φ(1)
α1〉 · · · |φ(N)
αN〉
Erwartungswert in Produktzustand (unterscheidbare Teilchen)
mit analoger Rechnung fur 〈Ψ1 + cΨ2|A|Ψ1 + cΨ2〉 mit c = 1, i und N -Teilchen-Zustanden |Ψ1,2〉 sind auch allgemeine Matrixelemente lokaler hermitescher Ope-ratoren mit (nicht (anti-)symmetrisierten) Produktzustanden ausdruckbar
beachte weiter:
• fur ein Bose-/Fermi-System ergibt sich nα = 0, 1 auch im klassischenLimes, da fur T → ∞ oder v = V/N → ∞ Doppelbesetzungen unwahr-scheinlich werden; im klassischen Limes kann also mit unterscheidbarenTeilchen gerechnet werden
174

• um die richtige Anzahl von Zustanden beizubehalten (z.B. bei der Auswer-tung der Spur, siehe ideales Quantengas im klassischen Limes oder Systemharmonischer Oszillatoren hier) ist zu berucksichtigen, dass (falls nα =0, 1) die N ! Produktzustande P|φ(1)
α1〉 · · · |φ(N)
αN〉 (fur die N ! Permutationen)
zum gleichen (anti-)symmetrisierten Produktzustand S(±)N |φ(1)
α1〉 · · · |φ(N)
αN〉
korrespondieren und denselben physikalischen Zustand beschreiben
• daher ist der Faktor 1/N ! zu erganzen (korrekte Boltzmann-Abzahlung)oder der Zustandsraum auf den 1/N !-ten Teil zu begrenzen, was dadurcherreicht werden kann, dass Teilchen “1” sich nur im ersten Teil des Ein-Hilbert-Teilchen-Raums, Teilchen “2” nur im zweiten Teil etc. (Letzteresist Grundlage der folgenden Rechnung, siehe Formel fur die Zustandsdichteunten)
—————————————————————————————————
Gesamtenergie:
H|n1〉 · · · |nN〉 =N∑
i=1
H(i)1 |n1〉 · · · |nN〉 =
N∑
i=1
εi(ni)|n1〉 · · · |nN〉
E = E(n1, ..., nN ) =N∑
i=1
εi(ni) = ~ωN∑
i=1
(ni +
1
2
)ni = 0, 1, 2, ...
vereinfachend: Oszillatoren gleicher Frequenz ωi = ω
E i.allg. stark entartet, GZE: E0 =1
2~ωN (nicht entartet)
—————————————————————————————————
3) Zustandsdichte:
Σ(E, N)∆N = Sp δ(E − HN)
=∞∑
n1=0
· · ·∞∑
nN=0
δ(E − E(n1, ..., nN ))
mit δ(E) =1
2π
∫ ∞
−∞ds eiEs ist:
Σ(E, N)∆N =∞∑
n1=0
· · ·∞∑
nN=0
1
2π
∫ ∞
−∞ds exp (i(E − E(n1, ..., nN))s)
=1
2π
∫ ∞
−∞ds
∞∑
n1=0
· · ·∞∑
nN=0
exp(iEs) exp
−is
N∑
j=1
~ω(nj + 1/2)
=1
2π
∫ ∞
−∞ds ei(E−E0)s
∞∑
n1=0
· · ·∞∑
nN=0
N∏
j=1
e−is~ωnj
175

=1
2π
∫ ∞
−∞ds ei(E−E0)s
N∏
j=1
∞∑
nj=0
(e−is~ω
)nj
es ist:∞∑
n=0
(e−is~ω
)n=
1
1 − e−is~ω=
e12is~ω
e12is~ω − e−
12is~ω
= e12is~ωe− ln(2i sin( 1
2s~ω))
—————————————————————————————————
Zwischenbemerkung:∞∑
n=0
(e−is~ω
)nist eine divergente Reihe!
Ausweg: ersetze∫ ∞
−∞dsF (s) durch lim
ε→0
∫ ∞
−∞dsF (s − iε)
damit wird z ≡ e−is~ω → z = e−is~ωe−ε~ω mit |z| < 1 und∑
n
zn = 1/(1 − z)
—————————————————————————————————
mit E0 = N1
2~ω gilt:
Σ(E, N)∆N =1
2π
∫ ∞
−∞ds ei(E−E0)s
(e
12is~ωe− ln(2i sin( 1
2s~ω))
)N
=1
2π
∫ ∞
−∞ds exp
[N(is
E
N− ln(2i sin(
1
2s~ω))
)]
Σ(E, N)∆N =1
2π
∫ ∞
−∞ds exp [Nf(s)]
mit
f(s) = isE
N− ln(2i sin(
1
2s~ω))
—————————————————————————————————
4) fur N → ∞:
Σ(E, N)∆N = O(N−1/2) exp [Nf(s0)] mit f ′(s0) = 0
(damit ln Σ(E, N) = Nf(s0) + O(ln N))
Bestimmung von s0:
0 = f ′(s) = iE
N−
cos(
12s~ω
)
sin(
12s~ω
) 1
2~ω
176

cot(
1
2s~ω
)= 2i
E
N
1
~ω= i
E
E0
1
2s~ω = arccot
(iE
E0
)= −iarccoth
(E
E0
)
=1
i
1
2ln
(E/E0 + 1
E/E0 − 1
)=
1
2iln(
E + E0
E − E0
)
s0 = −i1
~ωln(
E + E0
E − E0
)
einsetzen: f(s0) =?
sin(
1
2s0~ω
)= sin
(1
2iln
E + E0
E − E0
)=
1
isinh
(1
2ln
E + E0
E − E0
)
=1
isinh
(ln(
E + E0
E − E0
)1/2)
=1
2i
(eln... − e− ln...
)
=1
2i
((E + E0
E − E0
)1/2
−(
E − E0
E + E0
)1/2)
=1
2i
(E + E0
E − E0
)1/2 (1 − E − E0
E + E0
)
=1
2i
(E + E0
E − E0
)1/2 2E0
E + E0=
1
2i
2E0
(E + E0)1/2(E − E0)1/2
damit:
f(s0) = is0E
N− ln(2i sin(
1
2s0~ω))
=1
~ω
E
Nln
E + E0
E − E0− ln
2E0
(E + E0)1/2(E − E0)1/2
=E
2E0ln
E + E0
E − E0− ln
2E0
(E + E0)1/2(E − E0)1/2
=(
E
2E0+
1
2
)ln(E + E0) −
(E
2E0− 1
2
)ln(E − E0) − ln(2E0)
f(s0) =E + E0
2E0
ln(
E + E0
2E0
)− E − E0
2E0
ln(
E − E0
2E0
)
also:
Σ(E, N)∆N = O(N−1/2) exp [Nf(s0)]
—————————————————————————————————
5) Entropie
177

S(E, N) = k ln(Σ(E, N)∆N∆E)
= Nkf(s0) + kO(ln N)
S(E, N) = Nk(
E + E0
2E0ln(
E + E0
2E0
)− E − E0
2E0ln(
E − E0
2E0
))
kalorische Zustandsgleichung:
1
T=
(∂S
∂E
)
N
= Nk1
2E0ln
E + E0
E − E0
lnE + E0
E − E0=
2E0
NkT
e2E0NkT − 1 =
E + E0
E − E0− 1 =
2E0
E − E0
E = E0 + 2E01
e2E0NkT − 1
also mit E0 = N~ω/2:
E = N~ω
1
exp(
~ωkT
)− 1
+1
2
= E(T, N) innere Energie
T → 0 E → E0
klassischer Limes:
T → ∞ exp
(~ω
kT
)≈ 1 +
~ω
kT E → N~ω
kT
~ω= NkT
(GVS: Beitrag kT/2 zu E, 2N Freiheitsgrade quadratisch in H)
—————————————————————————————————
es ist:
E = 〈H〉 =N∑
i=1
~ω(〈ni〉 +
1
2
)= N~ω
(n +
1
2
)
mittlere Besetzungszahl eines Oszillators n ≡ 〈ni〉:
n =1
exp(
~ωkT
)− 1
es gilt (mit x = ~ω/kT ):
178

E − E0
2E0=
1
ex − 1= n
E + E0
2E0=
ex
ex − 1=
1
1 − e−x= 1 + n
und somit:
S(T, N) = Nk[(1 + n) ln(1 + n) − n ln n]
fur T → 0 ist n → 0, also S → 0 3.HS erfullt
—————————————————————————————————
Warmekapazitat:
CN =
(δQ
dT
)
N
=
(∂E
∂T
)
N
= T
(∂S
∂T
)
N
klassischer Limes: E = NkT
CN = Nk fur T → ∞ (Dulong-Petit)
tiefe Temperaturen Quanteneffekte (x = ~ω/kT ):
CN = N~ωex
(ex − 1)2
~ω
k
1
T 2
= Nkx2ex
ex(ex/2 − e−x/2)2= Nk
(x
ex/2 − e−x/2
)2
= Nk
(x/2
sinh(x/2)
)2
also (β = 1/kT ):
CN = Nk
(β~ω/2
sinh(β~ω/2)
)2
(vergl. Diskussion der Warmekapazitat und des GVS oben)
179

180

Kapitel 9
Allgemeiner Aufbau derstatistischen Mechanik
9.1 Allgemeiner Kontakt
Mikrozustande k = 1, ..., M , M ≫ N
f kontrollierbare Makrovariable, u.a. H
—————————————————————————————————
mikrokanonische Gesamtheit:
H und s weitere extensive Großen z = (z1, ..., zs) fest:
E − ∆E < H(q, p) < E
zj − ∆zj < zj(q, p) < zj fur j = 1, ..., s
f = s + 1, System isoliert
—————————————————————————————————
kanonische Gesamtheit:
z = (z1, ..., zs) fest
zj − ∆zj < zj(q, p) < zj fur j = 1, ..., s
x0 = H fluktuierende Zufallsvariable, Erwartungswert 〈H〉 kontrolliert
f = s + 1, System im Warmebad
—————————————————————————————————
allgemeinkanonische Gesamtheit:
181

z = (z1, ..., zs) fest
H und x = (x1, ..., xr) Zufallsvariable, Erwartungswerte 〈H〉, 〈xi〉 kontrolliert
f = s + r + 1, System im Warme- und xj-Bad
KM: H(q, p), xi(q, p), zj(q, p) (i = 1, ..., r, j = 1, ..., s)
QM: H , xi, zj
—————————————————————————————————
Bsp: H, V, N als kontrollierte Makrovariable
Gesamtheit: MK K GK Gibbs allgemeinKontakt: System thermisch thermisch mit thermisch mit
isoliert (Warmebad) Teilchenaust. Arbeitsaust.ausgetauschteGroßen: - H H, N H, V H, xfesteGroßen: H, V, N V, N V N z
9.2 Bestimmung der a priori-Wahrscheinlichkeiten
TD GG:
Festlegung der pk durch Maximierung der Shannon-Information unter NBen
σ(p1, ..., pM) = max
r + 1 NBen: 〈H〉, 〈xi〉 fest fur i = 1, ..., r
Lagrange-Parameter λ0, λi
s NBen: zj fest fur j = 1, ..., s
pk 6= 0 nur fur δ(zj − zj(q, p))∆zj = 1
damit:
pk = pk(λ0, λ1, ..., λr, z1, ..., zs) = pk(λ0, λ, z)
und
ρ = ρλ0,λ,z(π) (KM)
ρ = ρλ0,λ,z (QM)
—————————————————————————————————
182

sei A = A(π) (KM), A = A† (QM) beliebige extensive Observable:
〈A〉 =∫
dπρλ0,λ,z(π)A(π) = A(λ0, λ, z) (KM)
〈A〉 = Sp (ρλ0,λ,zA) = A(λ0, λ, z) (QM)
Werte der Makrovariable A konnen um 〈A〉 fluktuieren, aber:
σA
〈A〉 = O(1/√
N)
(A extensiv, somit makroskopische Summe unabhangiger Zufallsvariabler, ZGWS)
makroskopische Abweichungen von TD GG-Werten extrem unwahrscheinlich
—————————————————————————————————
Maximierung von σ:
0 =∂
∂pk
[−∑
k
pk ln pk − α∑
k
pk − λ0
∑
k
pkH(k) −s∑
i=1
λi
∑
k
pkxi(k)
]
KM: xi(k) = xi(q, p) (diskretisierter Phasenraum)
QM: xi|Ψk〉 = xi(k)|Ψk〉 mit ONB |Ψk〉(als Makrovariablen kommutieren H, xi paarweise bis auf mikroskopischen Fehler)
Lagrange-Parameter: α, λ0, λi
es ist:
0 = − ln pk − 1 − α − λ0H(k) −s∑
i=1
λixi(k)
also:
pk ∝ e−λ0H(k)−∑
iλixi(k)
s∏
j=1
(δ(zj − zj(k))∆zj)
allgemeinkanonische WK
λ0, λ1, ..., λr, z1, ..., zs bzw. λ0, λ, z
naturliche Variablen der AK Gesamtheit
9.3 Zustandssumme
KM:
WK-Dichte:
183

ρλ0,λ,z(q, p) =α
Ψ(λ0, λ, z)e−λ0H(q,p)−
∑iλixi(q,p)
s∏
j=1
δ(zj − zj(q, p))
Normierung:∫
dqdp ρ(q, p) = 1
Ψ(λ0, λ, z) =∫
dqdp α e−λ0H(q,p)−∑
iλixi(q,p)
s∏
j=1
δ(zj − zj(q, p))
AK Zustandsintegral
Ψ(λ0, λ, z)∆z (∆z ≡ ∏j ∆zj) dimensionslos mit
α =1
h3NN !—————————————————————————————————
beachte:
• Faktor h−3N entspricht Diskretisierung des klassischen Phasenraums
der Wert des Rastervolumens im Phasenraum (h3N ) ist so gewahlt, dassQM Resultate im klassischen Limes reproduziert werden
notwendig (s.o.) fur Anwendung des Prinzips der Maximierung der Shannon-Information!
•∫
dqdp lauft uber verallgemeinerten Phasenraum (beliebige Teilchenzahl)
α steht unter dem Phasenraum-Integral
• Einfugen des Faktors α, und zwar des 1/N !-Terms (wobei ja N = N(q, p)),ist eine ad hoc Abweichung vom Prinzip maximaler Shannon-Information
• Motivation fur den 1/N !-Faktor: Anschluss an die QM im klassischen Limes
Zustande π und π′, die durch Teilchenpermutation auseinander hervorge-hen, durfen nicht als physikalisch verschieden betrachtet werden (klassischeVersion des QM Ununterscheidbarkeitsprinzips, das die QM Resultate imklassischen Limes reproduziert)
klassische physikalische Zustande (in diesem Sinn) nehmen den Anteil 1/N !des gesamten Phasenraums ein
bei Anwendung des Prinzips der Maximierung der Shannon-Informationsollen nur physikalische Zustande (im diesem Sinn) benutzt werden
oder: (und so wird’s in der Praxis gemacht)
es werden wieder alle (mathematischen) Zustande in Betracht gezogen,aber mit um den Faktor 1/N ! verringerter WK fur jeden einzelnen Zustand
184

—————————————————————————————————
QM:
Dichteoperator:
ρλ0,λ,z ∝∑
k
pk(λ0, λ, z)|Ψk〉〈Ψk|
=∑
k
e−λ0H(k)−∑
iλixi(k)
s∏
j=1
δ(zj − zj(k))|Ψk〉〈Ψk|
=∑
k
e−λ0H−∑
iλixi
s∏
j=1
δ(zj − zj)|Ψk〉〈Ψk|
denn Makrovariablen kommutieren paarweise, |Ψk〉 gemeinsamer Eigenzustand
ρλ0,λ,z =1
Ψ(λ0, λ, z)e−λ0H−
∑iλixi
s∏
j=1
δ(zj − zj)
Normierung: Sp ρ = 1
Ψ(λ0, λ, z) = Sp
e−λ0H−
∑iλixi
s∏
j=1
δ(zj − zj)
AK Zustandssumme
(Ψ(λ0, λ, z)∆z dimensionslos)
—————————————————————————————————
H → Hz1,...,zs = Hz erzwingt die NB bzgl. z
(es ist Hz = ∞ und somit e−λ0Hz = 0 (λ0 > 0), falls δ(z − z(q, p))∆z 6= 1, undHz = H sonst)
KM:
ρλ0,λ,z(q, p) =α
Ψ(λ0, λ, z)∆ze−λ0Hz(q,p)−
∑iλixi(q,p)
Ψ(λ0, λ, z) =1
∆z
∫dqdp α e−λ0Hz(q,p)−
∑iλixi(q,p)
QM:
ρλ0,λ,z =1
Ψ(λ0, λ, z)∆ze−λ0Hz−
∑iλixi
Ψ(λ0, λ, z) =1
∆zSp
(e−λ0Hz−
∑iλixi
)
185

9.4 Thermodynamische Potenziale
es ist (x = (..., xi, ...)):
Ψ(λ0, λ, z) =∫
dEdx1
∆z
∫dqdp αδ(E−Hz(q, p))δ(x−x(q, p))e−λ0Hz(q,p)−λx(q,p)
=∫
dEdx e−λ0E−λx 1
∆z
∫dqdp α δ(E − Hz(q, p))δ(x − x(q, p))
=∫
dEdx e−λ0E−λxα1
∆z∆x
∫dqdp δ(E − Hx,z(q, p))
Ψ(λ0, λ, z) =∫
dEdx e−λ0E−λxα Σ(E, x, z)
r + 1-fache Laplace-Transformation
Umkehrung?
—————————————————————————————————
eindimensionale Laplace-Transformation:
Ψ(λ) =∫ ∞
0dx Σ(x)e−λx (λ > 0)
betrachte:
1
2πi
∫ i∞
−i∞dλ Ψ(λ)eλx (x > 0)
=1
2πi
∫ i∞
−i∞dλ
∫ ∞
0dx′ Σ(x′)e−λx′
eλx
=∫ ∞
0dx′ Σ(x′)
1
2πi
∫ i∞
−i∞dλ eλ(x−x′)
substituiere: λ = iλ′:
=∫ ∞
0dx′ Σ(x′)
1
2π
∫ ∞
−∞dλ′ eiλ′(x−x′)
=∫ ∞
0dx′ Σ(x′)δ(x − x′)
Umkehrung der Laplace-Transformation:
Σ(x) =1
2πi
∫ i∞
−i∞dλ Ψ(λ)eλx
—————————————————————————————————
weiter gilt:
ln Ψ(λ) = ln∫ ∞
0dx Σ(x)e−λx
186

= ln∫ ∞
0dx eln Σ(x)−λx
(beachte: ln Σ(x) − λx = O(N)) mit der Sattelpunktsmethode
= ln Σ(x) − λx
hier ist x bestimmt durch:
0 =∂
∂x(ln Σ(x) − λx)
zusammen ist also:
Ψ(λ) =∫ ∞
0dx Σ(x)e−λx
Laplace-Transformation: Ψ(λ) ⇔ Σ(x)
ln Ψ(λ) = ln Σ(x) − λx λ =∂
∂xln Σ(x)
Legendre-Transformation: ln Ψ(λ) ⇔ ln Σ(x)
—————————————————————————————————
aus der AK Zustandssumme
Ψ(λ0, λ, z) =∫
dEdx e−λ0E−λxα Σ(E, x, z)
folgt also:
λ0 =∂
∂Eln(α Σ(E, x, z)) =
1
k
∂
∂ES(E, x, z)
(S(E, x, z) = k ln αΣ(E, x, z)∆E∆x∆z)
physikalische Bedeutung des Lagrange-Parameters λ0:
λ0 =1
kT
analog (fur i = 1, ..., r):
λi =∂
∂xiln(α Σ(E, x, z)) =
1
k
∂
∂xiS(E, x, z)
mit TdS = dU − Kdx ist
λi = −Ki
kT
—————————————————————————————————
definiere:
A(λ0, λ, z) = −kT ln(Ψ(λ0, λ, z)∆z)
187

bzw.
A(T, K, z) = −kT ln(Ψ(T, K, z)∆z) AK Potenzial
es ist:
A(T, K, z) = −kT
(ln(αΣ(E, x, z)∆z) − λ0E −
∑
i
λixi
)
= −TS(E, x, z) + E −∑
i
Kixi
mit E = E(T, K, z) und x = x(T, K, z)
physikalische Bedeutung des AK Potenzials:
A = −TS + E −∑
i
Kixi
Legendre-Transformation bzgl. E und x
TD Potenzial des durch die natulichen Variablen T, K, z beschriebenen Kontakts
—————————————————————————————————
Bsp: x = V , z = N
A(T, p, N) = −TS + E + pV = G(T, p, N)
freie Enthalpie, Gibbs’sches Potenzial
G(T, p, N) = −kT ln(Ψ(T, p, N)∆N)
= −kT ln(
1
∆N
∫dqdp α e−λ0Hz(q,p)−
∑iλixi(q,p)∆N
)
= −kT ln(∫
d3Nqd3Np α e−(HN (q,p)+pV (q,p))/kT)
QM:
G(T, p, N) = −kT ln(
Sp N e−(H+pV )/kT)
9.5 Entropie und Information
Zusammenhang mit Shannon-Information:
zunachst QM:
σ = − Sp (ρ ln ρ)∣∣∣∣max
ausgewertet fur GG-Zustand
188

ρT,K,z =1
Ψ(T, K, z)∆ze−(Hz−
∑iKixi)/kT
liefert:
σ = − Sp
e−(Hz−
∑iKixi)/kT
Ψ(T, K, z)∆zln
e−(Hz−∑
iKixi)/kT
Ψ(T, K, z)∆z
= ln Ψ(T, K, z)∆z− Sp
e−(Hz−
∑iKixi)/kT
Ψ(T, K, z)∆zln e−(Hz−
∑iKixi)/kT
= − 1
kTA(T, K, z) +
(〈Hz〉 −
∑
i
Ki〈xi〉)
/kT
=1
kT
(−A + E −
∑
i
Kixi
)=
1
kTTS
also:
max σ[ρ] =1
kS
—————————————————————————————————
KM:
σ(pk) = −∑
k
pk ln pk
ausgewertet fur GG-Zustand
pk =∫
Zelle kdqdp
α
Ψ(λ0, λ, z)∆ze−λ0Hz(q,p)−
∑iλixi(q,p)
=1
Ψ(T, K, z)∆zh3N αe−(Hz(qk,pk)−
∑iKixi(qk,pk))/kBT
liefert:
σ(pk)
= − 1
Ψ(T, K, z)∆z
∑
k
h3Nαe(··· ) ln
(1
Ψ(T, K, z)∆zαe(··· )
)
= ln(Ψ(T, K, z)∆z) − 1
Ψ(T, K, z)∆z
∑
k
h3Nαe(··· ) ln(h3Nαe(··· )
)
= −A(T, K, z)
kBT−∑
k
pk((· · · ) + ln(h3Nα))
= −A(T, K, z)
kBT− 〈(· · · )〉 − 〈ln(1/N !)〉
189

=1
kBT
(−A(T, K, z) + 〈H〉 −
∑
i
Ki〈xi〉)
+ ln N !
=1
kBT
(−A + E −
∑
i
Kixi
)+ N ln N =
1
kBTTS − N ln N
=1
kBS + N lnN
also:
σ(pk)∣∣∣∣GG
=1
kBS + N ln N
—————————————————————————————————
Ausgangssituation, um an QM anzuschließen:
– WK pk fur physikalische Zustande (phys.∑
k
pk = 1)
in Praxis:
– WK pk = pk/N ! fur gesamten Phasenraum (∑
k
pk = 1)
aquivalent bezuglich Berechnung von Erwartungswerten (z.B. Entropie S)
aber σ = −〈ln p〉 = −〈ln(p/N !)〉 = −〈ln p〉 + ln N ! = σ + N ln N
mehrere gleichwahrscheinliche Alternativen vergroßern die Shannon-Information
9.6 Quasistatische Prozesse
quasistatische TD Prozesse gefuhrt durch T, K, z
dA(T, K, z) =∂A
∂TdT +
r∑
i=1
∂A
∂Ki
dKi +s∑
j=1
∂A
∂zj
dzj
—————————————————————————————————
es ist:
∂A(T, K, z)
∂zj
= − ∂
∂zj
kT ln(Ψ(T, K, z)∆z)
= −kT∂
∂zj
ln∫
dqdp α e−(Hz(q,p)−∑
iKixi(q,p))/kT
=
∫dqdp α e−(Hz(q,p)−
∑iKixi(q,p))/kT ∂Hz(q,p)
∂zj
∫dqdp α e−(Hz(q,p)−
∑iKixi(q,p))/kT
190

also:
∂A(T, K, z)
∂zj
=
⟨∂Hz
∂zj
⟩= K ′
j
(intensive) verallgemeinerte Krafte treten auf als
– Lagrange-Parameter λi bzw. Ki
– zu zj konjugierte Großen (und damit Erwartungswerte) K ′j
je nach Art des Kontaktes mit der Umgebung
—————————————————————————————————
weiter ist:
∂A(T, K, z)
∂Ki= − ∂
∂KikT ln(Ψ(T, K, z)∆z)
= −kT∂
∂Kiln∫
dqdp α e−(Hz(q,p)−∑
iKixi(q,p))/kT
also:
∂A(T, K, z)
∂Ki
= −〈xi〉
(extensive) verallgemeinerte Koordinaten treten auf als
– Kontroll-Parameter zj
– zu Ki konjugierte Großen (Erwartungswerte)
je nach Art des Kontaktes mit der Umgebung
—————————————————————————————————
und:
∂A(T, K, z)
∂T= − ∂
∂T(kT ln(Ψ(T, K, z)∆z))
= −k ln(Ψ(T, K, z)∆z)) − kT∂
∂Tln∫
dqdp α e−(Hz(q,p)−∑
iKixi(q,p))/kT
= A(T, K, z)/T − kT
(〈Hz〉 −
∑
i
Ki〈xi〉)
1
kT 2
=1
T
(A − E +
∑
i
Kixi
)
also:
191

∂A(T, K, z)
∂T= −S
—————————————————————————————————
insgesamt:
dA(T, K, z) =1
T
(A(T, K, z) − 〈Hz〉 +
∑
i
Ki〈xi〉)
dT −s∑
i=1
〈xi〉dKi +s∑
j=1
⟨∂Hz
∂zj
⟩dzj
dA(T, K, z) = −SdT −s∑
i=1
xidKi +s∑
j=1
K ′jdzj
beachte: Erwartungswerte 〈Hz〉, 〈xi〉 der AK Gesamtheit konnen mit festen Kon-trollparametern E, xi der MK Gesamtheit idendifiziert werden
(ZGWS: relative Standardabweichung ∼ N−1/2 → 0)
9.7 Fluktuationen
zweite Ableitungen des AK Potenzials A(T, K, z)
es ist
∂A(T, K, Z)
∂Ki= −〈xi〉
und
−∂2A(T, K, Z)
∂Ki∂Kj=
∂
∂Kj〈xi〉
=∂
∂Kj
1
Ψ(T, K, z)∆zSp z
(xie
−(Hz−∑
iKixi)/kT
)
=−1
Ψ(T, K, z)2∆z2
∂
∂KjSpz
(e−(Hz−
∑iKixi)/kT
)Spz
(xie
−(Hz−∑
iKixi)/kT
)
+1
Ψ(T, K, z)∆z
∂
∂Kj
Sp z
(xie
−(Hz−∑
iKixi)/kT
)
=−1
Ψ(T, K, z)2∆z2
1
kTSpz
(xje
−(Hz−∑
iKixi)/kT
)Spz
(xie
−(Hz−∑
iKixi)/kT
)
+1
Ψ(T, K, z)∆z
1
kTSp z
(xixje
−(Hz−∑
iKixi)/kT
)
=1
kT(〈xixj〉 − 〈xi〉〈xj〉)
192

∂2A(T, K, Z)
∂Ki∂Kj
= − ∂Kj
∂〈xi〉= − ∂Ki
∂〈xj〉= − 1
kT(〈xixj〉 − 〈xi〉〈xj〉)
—————————————————————————————————
fur beliebige α1, ..., αr ist
αT · (HesseKA) · α
=r∑
i=1
r∑
j=1
αi∂2A(T, K, Z)
∂Ki∂Kjαj
= − 1
kT
r∑
i=1
r∑
j=1
αi(〈xixj〉 − 〈xi〉〈xj〉)αj
= − 1
kT(〈
r∑
i=1
αixi
r∑
j=1
αjxj〉 − 〈r∑
i=1
αixi〉〈r∑
j=1
αjxj〉)
= − 1
kT(〈X2〉 − 〈X〉2) = − 1
kT〈(X − 〈X〉)2〉 < 0
also:
HesseKA negativ definit
—————————————————————————————————
Bsp:
0 >∂2Ω(T, V, µ)
∂µ2= −∂N
∂µ
d.h.∂N
∂µ> 0
—————————————————————————————————
mit B ≡ Hz −∑
i Kixi ist
∂2
∂β2ln Sp e−βB
= − ∂
∂β
1
Ψ(T, K, z)∆zSp
(Be−βB
)
=1
Ψ(T, K, z)2∆z2
∂
∂βSp(e−βB
)Sp(Be−βB
)+
1
Ψ(T, K, z)∆zSp(B2e−βB
)
= −〈B〉2 + 〈B2〉also (A = −kT ln(Ψ∆z)):
193

∂2
∂β2(βA) = −〈(Hz −
∑
i
Kixi)2〉 + 〈Hz −
∑
i
Kixi〉2 < 0
—————————————————————————————————
Bsp:
0 >∂2(βF (T, V, N))
∂β2=
∂
∂β
(F + β
∂F
∂β
)= 2
∂F
∂β+ β
∂2F
∂β2
∂F
∂β=
∂T
∂β
∂F
∂T= −kT 2(−S) = kT 2S
∂2F
∂β2= −kT 2 ∂
∂T(kT 2S) = −2k2T 3S − k2T 4 ∂S
∂T
also:
0 > 2kT 2S − 2kT 2S − kT 3 ∂S
∂T= −kT 2CV,N
d.h. CV,N > 0
—————————————————————————————————
insgesamt:
Hesseλ0,λ lnΨ(λ0, λ, z) positiv definit
(λ0 = 1/kT = β, λi = −Ki/kT )
—————————————————————————————————
speziell:
∂〈H〉∂T
=∂λ0
∂T
∂
∂λ0Sp
(1
Ψ(λ0, λ, z)∆zHe−λ0Hz−
∑iλixi
)
= − 1
kT 2
∂
∂λ0
1
Ψ(λ0, λ, z)Sp z
(He−λ0H−
∑iλixi
)
=1
kT 2
1
Ψ(λ0, λ, z)2
∂Ψ(λ0, λ, z)
∂λ0
Sp z
(He−λ0H−
∑iλixi
)
+1
kT 2
1
Ψ(λ0, λ, z)Sp z
(H2e−λ0H−
∑iλixi
)
= − 1
kT 2
1
Ψ(λ0, λ, z)2Spz
(He−λ0H−
∑iλixi
)Spz
(He−λ0H−
∑iλixi
)
+1
kT 2
1
Ψ(λ0, λ, z)Sp z
(H2e−λ0H−
∑iλixi
)
also:
194

∂〈H〉∂T
=1
kT 2
(〈H2〉 − 〈H〉2
)
—————————————————————————————————
aus TD bekannt (Zerlegung von Systemen):
HessezA positiv definit
verifizierbar?
∂2A(T, K, z)
∂zi∂zj
= −kT∂
∂zi
∂ ln(Ψ(T, K, z)∆z)
∂zj
=∂
∂zi
[1
Ψ(T, K, z)∆zSp
(∂Hz
∂zj
e−(Hz−∑
iKixi)/kT
)]
=1
Ψ(T, K, z)∆zSp
(∂2Hz
∂zi∂zje−(Hz−
∑iKixi)/kT
)
− 1
kT
1
Ψ(T, K, z)∆zSp
(∂Hz
∂zj
∂Hz
∂zi
e−(Hz−∑
iKixi)/kT
)
− 1
Ψ(T, K, z)2∆z2Sp
(∂Hz
∂zje−(Hz−
∑iKixi)/kT
)∂
∂ziSp(e−(Hz−
∑iKixi)/kT
)
∂2A(T, K, z)
∂zi∂zj=
⟨∂2Hz
∂zi∂zj
⟩− 1
kT
⟨∂Hz
∂zi
∂Hz
∂zj
⟩+
1
kT
⟨∂Hz
∂zi
⟩⟨∂Hz
∂zj
⟩
beachte:
∂2A(T, K, z)
∂zi∂zj= O(1/N)
rechte Seite:
1.Term = O(1/N), indefinit, ohne anschauliche Bedeutung
2.Term, 3.Term = O(1)
also: 2.+3.Term = O(1/N), negativ definit
9.8 Allgemeiner Kontakt zwischen Systemen
betrachte: Gesamtsystem = System 1 + System2
Gesamtsystem isoliert
System i (i = 1, 2): zi fest, Hi, xi ausgetauscht
195

beachte: gleichzeitiger Austausch aller makroskopisch kontrollierter Großen nichtsinnvoll, da Subsysteme nicht makroskopisch definierbar
Additivitat (keine Oberflachenbeitrage):
H = H1 + H2 x = x1 + x2 (H und x sind fest)
z = z1 + z2 (z und z1, z2 sind fest)
z1 z2
1H=H +H 2
1x=x +x 2z1 z2
H1x1
H2x2
System 1 System 2
fest:
,
Zustandsdichte des Gesamtsystems: (KM)
Σ(E, x, z1, z2) =1
∆x∆z1∆z2
∫dqdp δ(E − Hx,z1,z2(q, p))
mit ∫dE1dx1 δ(E1 − H1(q1, p1)) δ(x1 − x1(q1, p1)) = 1
ist:
Σ(E, x, z1, z2) =
=∫
dqdp δ(E −H(q, p))δ(x− x(q, p))δ(z1 − z1(q1, p1))δ(z2 − z2(q2, p2))
=∫
dqdpδ(E−H1(q1, p1)−H2(q2, p2))δ(x−x1(q1, p1)−x2(q2, p2))
×δ(z1 − z(q1, p1))δ(z2 − z(q2, p2))
=∫
dq1dp1dq2dp2
∫dE1dx1 δ(E1 − H1(q1, p1)) δ(x1 − x1(q1, p1))
×δ(E − E1 − H2(q2, p2))δ(x − x1 − x2(q2, p2))
×δ(z1 − z(q1, p1))δ(z2 − z(q2, p2))
=∫
dE1dx1
∫dq1dp1δ(E1−H1(q1, p1))δ(x1−x1(q1, p1))δ(z1−z(q1, p1))
196

×∫
dq2dp2 δ(E −E1 −H2(q2, p2))δ(x− x1 − x2(q2, p2))δ(z2 − z(q2, p2))
=∫
dE1dx11
∆x1∆z1
∫dq1dp1 δ(E1 − H1,x1,z1(q1, p1))
× 1
∆x2∆z2
∫dq2dp2 δ(E − E1 − H2,x−x1,z−z1(q2, p2))
=∫
dE1dx1 Σ1(E1, x1, z1) Σ2(E − E1, x − x1, z2)
also:
Σ(E, x, z1, z2)?=∫
dE1dx1 Σ1(E1, x1, z1) Σ2(E − E1, x − x1, z2)
—————————————————————————————————
zusatzliche Komplikation: Faktor α =1
h3NN !
Gesamtsystem: N fest
Zustandsdichte des Gesamtsystems ist von der Form:∫
d3π1 · · ·∫
d3πN f(π1, ..., πN)
N ! Zustande, die jeweils durch Teilchenpermutation auseinanger hervorgehen,werden getrennt gezahlt (klassische Unterscheidbarkeit identischer Teilchen)
Beispiel fur N = 3, N ! = 6 Zustande
12 3
21 3
13 2
31 2
23 1
32 1
eine mogliche Zerlegung des 6N -dimensionalen Integrals in 6N1- und 6(N−N1)-dimensionale Integrationen:
∫d3πi1 · · ·
∫d3πiN1
∫d3πj1 · · ·
∫d3πjN−N1
f(π1, ..., πN)
mit i1, ..., iN1 ⊂ 1, ..., N paarweise verschieden
197

und j1, ..., jN−N1 = 1, ..., N\i1, ..., iN1die Integration
∫d3πi1 · · ·
∫d3πiN1
zahlt die N1! Zustande getrennt, die durch Permutation der N1 Teilchen in Sy-stem 1 entstehen, analog die zweite Integration
das sind N1!(N − N1)! Zustande
Beispiel: N = 3, N1 = 2, gezahlt werden 2! · 1! = 2 Zustande
12 3
21 3
bei der Zerlegung des Integrals sind aber auch die restlichen Permutationen zuzahlen, also:
∫d3π1 · · ·
∫d3πN f(π1, ..., πN) =
=N∑
N1=0
∑
i1,...,iN1⊂1,...,N
∫d3πi1 · · ·
∫d3πiN1
∫d3πj1 · · ·
∫d3πjN−N1
f(π1, ..., πN)
fur gegebenes N1 (fur einen Summanden) liefern dieN !
N1!(N − N1)!Terme der
Summe∑
i1,...,iN1⊂1,...,N
den gleichen Beitrag (der Wert von f ist fur Systeme
identischer Teilchen invariant unter Teilchenpermutationen), also:∫
d3π1 · · ·∫
d3πN f(π1, ..., πN) =
=N∑
N1=0
N !
N1!(N − N1)!
∫d3πi1 · · ·
∫d3πiN1
∫d3πj1 · · ·
∫d3πjN−N1
f(π1, ..., πN )
Beispiel: N = 3, N1 = 2,
die Summe∑
i1,...,iN1
zahltN !
N1!(N − N1)!=
3!
2! 1!= 3 weitere Zustande
198

12 3
13 2
23 1
mit h3N = h3N1h3N2 und αN =1
h3NN !folgt also:
αNΣ(E, x, z1, z2) =∫
dE1dx1 αN1Σ1(E1, x1, z1) αN−N1Σ2(E − E1, x − x1, z2)
—————————————————————————————————
beachte: dies gilt auch, falls kein Teilchenaustauschkontakt vorliegt!
falls z1 = N1, z2 = N2 erzwingt die Form von f(π1, ..., πN), dass nur ein
N1-Term in der SummmeN∑
N1=0
einen Beitrag liefert
(allerdings gilt meist, dass die Resultate (bei N1, N2 fest) nicht von der Mitnahmeder α-Faktoren abhangen, denn die N1-Abhangigkeit ist dann meist uninteres-sant; so ist auch die (etwas vorschnelle) Rechnung oben zum Nullten Hauptsatzzu rechtfertigen)
—————————————————————————————————
QM: es ist:
Zustandsdichte des Gesamtsystems:
Σ(E, x, z1, z2) =1
∆x∆z1∆z2Sp δ(E − Hx,z1,z2)
mit∫
dE1dx1 δ(E1 − H1) δ(x1 − x1) = 1
ist:
Σ(E, x, z1, z2) = Sp[δ(E−H)δ(x− x)δ(z1− z1)δ(z2− z2(q2, p2))
]
= Sp[δ(E − H1 − H2)δ(x − x1 − x2)δ(z1 − z1)δ(z2 − z2)
]
199
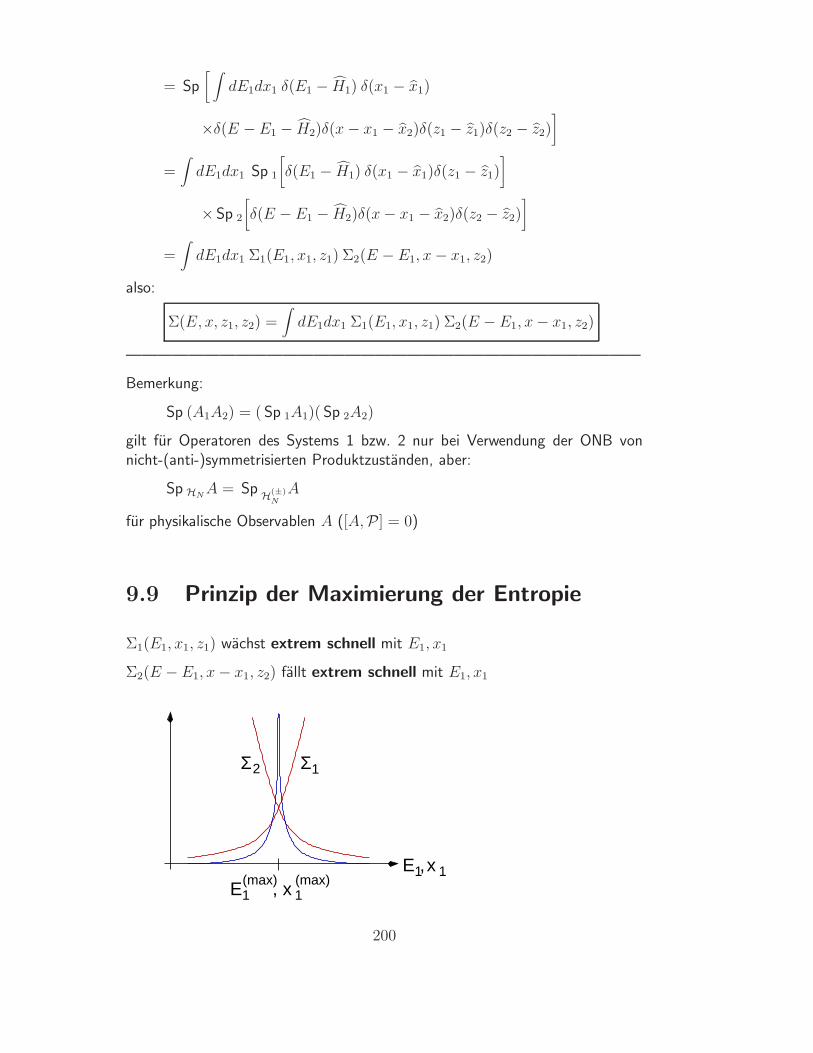
= Sp[ ∫
dE1dx1 δ(E1 − H1) δ(x1 − x1)
×δ(E − E1 − H2)δ(x − x1 − x2)δ(z1 − z1)δ(z2 − z2)]
=∫
dE1dx1 Sp 1
[δ(E1 − H1) δ(x1 − x1)δ(z1 − z1)
]
× Sp 2
[δ(E − E1 − H2)δ(x − x1 − x2)δ(z2 − z2)
]
=∫
dE1dx1 Σ1(E1, x1, z1) Σ2(E − E1, x − x1, z2)
also:
Σ(E, x, z1, z2) =∫
dE1dx1 Σ1(E1, x1, z1) Σ2(E − E1, x − x1, z2)
—————————————————————————————————
Bemerkung:
Sp (A1A2) = ( Sp 1A1)( Sp 2A2)
gilt fur Operatoren des Systems 1 bzw. 2 nur bei Verwendung der ONB vonnicht-(anti-)symmetrisierten Produktzustanden, aber:
Sp HNA = Sp H(±)
N
A
fur physikalische Observablen A ([A,P] = 0)
9.9 Prinzip der Maximierung der Entropie
Σ1(E1, x1, z1) wachst extrem schnell mit E1, x1
Σ2(E − E1, x − x1, z2) fallt extrem schnell mit E1, x1
Σ2 Σ1
x 1E ,1E ,1
(max)x 1
(max)
200

Integral als Riemann-Summe schreiben, dE1 → ∆E, dx1 → ∆x
Approximation der Summe durch maximalen Summanden liefert:∑
E1
∆E∑
x1
∆x Σ1(E1, x1, z1)Σ2(E − E1, x − x1, z2)
= Σ1(E(max)1 , x
(max)1 , z1)Σ2(E −E
(max)1 , x−x
(max)1 , z2)∆E∆x
und somit (wahle ∆z1 = ∆z2):
Σ(E, x, z1, z2)∆E∆x∆2z = Σ1(E1, x1, z1)∆E∆x∆z Σ2(E2, z2)∆E∆x∆z
oder mit Γ(E, x, z) = Σ(E, x, z)∆E∆x∆z
ln Γ(E, x, z1, z2) = ln Γ1(E1, x1, z1) + ln Γ2(E2, x2, z2)
ln Phasenvolumen additiv
fur E1 = E(max)1 und E2 = E
(max)2 ≡ E − E
(max)1
—————————————————————————————————
die Uberlegungen gelten auch auf Basis der KM, mit Σ → αΣ
es folgt:
S(E, x, z1, z2) = S1(E1, x1, z1) + S2(E2, x2, z2)
—————————————————————————————————
Bestimmung des Maximums:
0 =∂
∂E1(Σ1(E1, x1, z1)Σ2(E − E1, x − x1, z2))
=∂
∂E1exp
(1
kS1(E1, x1, z1) +
1
kS2(E − E1, x − x1, z2)
)
= exp(· · · ) 1
k
(∂S1(E1, x1, z1)
∂E1+
∂S2(E − E1, x − x1, z2)
∂E1
)
also:
T1 = T2
analog:
0 =∂
∂x1
(Σ1(E1, x1, z1)Σ2(E − E1, x − x1, z2))
= exp(· · · ) 1
k
(∂S1(E1, x1, z1)
∂x1
+∂S2(E − E1, x − x1, z2)
∂x1
)
also:
201

K1 = K2
Zwei Systeme in E, x-Kontakt haben im TD GG gleiche Temperaturen undKrafte
—————————————————————————————————
betrachte Gesamtsystem mit:
E ′1, x
′1, z1, E
′2, x
′2, z2 fest
d.h. vor Ermoglichung von E, x-Ausgleichsprozessen
dagegen nur noch
E1 + E2, x1 + x2, z1, z2 fest
nach E, x-Ausgleich
—————————————————————————————————
allgemein: x = (x1, ..., xr), z = (z1, ..., zs), hier o.B.d.A.: r = 1, s = 1
Entropie vor dem Ausgleich
S ′(E ′1, x
′1, z1, E
′2, x
′2, z2) = k ln(# mit 6 NB vertragliche Zustande)
Entropie nach dem Ausgleich
S(E1+E2, x1+x2, z1, z2) = k ln(# mit 4 NB vertragliche Zustande)
weniger NB, also
# mit 6 NB vertragliche Zustande ≪ # mit 4 NB vertragliche Zustande
und nach Logarithmieren:
S ′(E ′1, x
′1, z1, E
′2, x
′2, z2) < S(E1 + E2, x1 + x2, z1, z2)
—————————————————————————————————
vor dem Ausgleich: Nicht-GG-Zustand, nach dem Ausgleich: TD GG, also
SNicht−GG ≤ SGG
in einem isolierten System kann die Entropie (sofern definiert) nicht abnehmen
die Entropie ist im TD GG maximal
Beweis des 2.HS (des MaxEnt-Prinzips) statt Postulat!
• S nur definiert fur Gesamtsystem, das aus im TD GG befindlichen (ma-kroskopischen) Subsystemen besteht (zwischen den Subsystemen aber keinGG)
202

• Nicht-GG-Zustande konnen nur durch Kontrolle vieler Makrovariablen prape-riert werden
• Zulassen von Ausgleichsprozessen / Herstellen von Kontakten bedeutetWegfall von Kontrollparametern
• somit werden Ausgleichsprozesse durch Anwachsen der Unkenntnis (Infor-mationsgewinn) charakterisiert
(der Informationszuwachs bei Aufklarung des Mikrozustands ist großer,wenn weniger uber das System bekannt ist)
• Ausgleichsprozesse implizieren Anwachsen der Entropie
• beachte: im Gegensatz zur Shannon-Information ist die Entropie keineFunktion des Mikrozustands!
und: die Shannon-Information als dynamische Große (Observable) ist zeit-lich konstant!
• das MaxEnt-Prinzip sagt nichts uber die zeitliche Dynamik eines Systemsaus, sondern nur uber die (fast triviale) Relation zwischen Kontrollparame-tern und Unkenntnis des Mikrozustands
• als nicht-dynamisches Prinzip ist das MaxEnt-Prinzip daher nicht im Kon-flikt mit der Zeitumkehrsymmetrie der mikroskopischen Bewegungsglei-chungen
• also konsequenterweise: der 2. HS definiert keinen Zeitpfeil
203

204

Kapitel 10
Kanonische und großkanonischeGesamtheit
10.1 Kanonische Gesamtheit
thermischer Kontakt, naturliche Variablen: T, V, N
fest (z): V, N , ausgetauscht (λ0): H
—————————————————————————————————
KM:
WK-Dichte:
ρ(q, p) = ρT,V,N(q, p) =α
Z(T, V, N)e−βHV,N (q,p)
Zustandsintegral:
Z(T, V, N) = α∫
dqdp e−βHV,N (q,p)
beachte: Z(T, V, N) entspricht Ψ(λ0, z)∆z
TD Potenzial: freie Energie
F (T, V, N) = −kT ln Z(T, V, N)
—————————————————————————————————
QM:
Dichteoperator:
ρT,V,N =1
Z(T, V, N)e−βHV,N
205

Zustandssumme:
Z(T, V, N) = Sp e−βHV,N = e−βF (T,V,N)
—————————————————————————————————
MK Gesamtheit: H = E = const., K Gesamtheit: H Zufallsgroße, aber:
σH
〈H〉 ∝ 1√N
(denn: H extensive Große, ZGWS)
kanonisch
ρ (H)ρ
∆E N −1/2
E <H>H H
mikrokanonisch
(H)
Aquivalenz der MK und der K Gesamtheit
〈H〉 = E = U
direkte Berechnung der Varianz von H (QM, KM analog):
σ2H = 〈(H − 〈H〉)2〉
=1
ZSp (H2e−βH) − 1
Z2
(Sp He−βH
)2
= − 1
Z
∂
∂βSp (He−βH) − 1
Z2
(Sp He−βH
)2
= − ∂
∂β
(1
ZSp (He−βH)
)+
(∂
∂β
1
Z
)Sp (He−βH) − 1
Z2
(Sp He−βH
)2
= − ∂
∂β〈H〉 +
1
Z2Sp (He−βH) Sp (He−βH) − 1
Z2
(Sp He−βH
)2
= − ∂
∂β〈H〉 = −∂T
∂β
∂
∂T〈H〉 =
1
k
1
β2
∂
∂T〈H〉
also:
σ2H = kT 2∂〈H〉
∂T∝ N
206

und:
σ2H = kT 2CV,N (Energieschwankung ∼ Warmekapazitat)
—————————————————————————————————
Rechnungen in K Gesamtheit meist einfacher als in MK Gesamtheit wegen
Zerlegungssatz:
sei (QM):
H = H1 + H2
mit
H1 : H1 → H1 H2 : H2 → H2
(also: [H1, H2] = 0), und
H = H1 ⊗H2
dann ist:
Z = Sp e−βH = Sp 1 Sp 2e−βH1e−βH2
also:
Z = Z1Z2 F = F1 + F2
KM:
Z
α=∫
dqdpe−βH(q,p) =∫
dq1dp1
∫dq2dp2e
−βH1(q1,p1)e−βH2(q2,p2)
Z
α=
Z1
α1
Z2
α2
10.2 Großkanonische Gesamtheit
thermischer und Teilchenaustauschkontakt, naturliche Variablen: T, V, µ
fest (z): V , ausgetauscht (λ0, λ): H, N
—————————————————————————————————
KM:
WK-Dichte:
ρ(q, p) = ρT,V,µ(q, p) =α
Ξ(T, V, µ)e−β(HV (q,p)−µN(q,p))
großkanonisches Zustandsintegral:
207

Ξ(T, V, µ) =∫
dqdp αe−β(HV (q,p)−µN(q,p))
=∞∑
N=0
∫dq1 · · · dq3Ndp1 · · · dp3N α e−β(HV (q,p)−µN)
beachte: α = α(N) und:Ξ(T, V, µ) entspricht Ψ(λ0, λ, z)∆z
TD Potenzial: GK Potenzial
Ω(T, V, µ) = −kT ln Ξ(T, V, µ)
—————————————————————————————————
QM:
Dichteoperator:
ρT,V,µ =1
Ξ(T, V, µ)e−β(HV −µN)
GK Zustandssumme:
Ξ(T, V, µ) = Sp e−β(HV −µN) =∞∑
N=0
Sp N e−β(HV −µN) = e−βΩ(T,V,µ)
—————————————————————————————————
definiere:
z = eβµ Fugazitat
dann ist (KM, QM analog):
Ξ(T, V, µ) =∞∑
N=0
∫dq1 · · · dq3Ndp1 · · · dp3N α e−β(HV (q,p)−µN)
=∞∑
N=0
α eβµN∫
dq1 · · ·dq3Ndp1 · · ·dp3N e−βHV (q,p)
=∞∑
N=0
(eβµ
)Nα∫
dqdp e−βHV,N (q,p)
also:
Ξ(T, V, µ) =∞∑
N=0
zNZ(T, V, N)
es gilt (QM, KM analog):
〈N〉 =1
Ξ(T, V, µ)Sp
(Ne−β(HV −µN)
)
208

=1
Ξ(T, V, µ)
∞∑
N=0
NeβµN Sp Ne−βHV
=1
Ξ(T, V, µ)
∞∑
N=0
NzNZ(T, V, N)
also:
〈N〉 =
∑N NzNZ(T, V, N)∑
N zNZ(T, V, N)
d.h. ρ(N) ∝ zNZ(T, V, N)
—————————————————————————————————
Varianz von N ?(
∂〈N〉∂µ
)
T,V
=∂
∂µ
∑N NzNZ(T, V, N)∑
N zNZ(T, V, N)
=∂z
∂µ
∂
∂z
∑N NzNZ(T, V, N)∑
N zNZ(T, V, N)
= βz
(1
Ξ
∑
N
N2zN−1Z(T, V, N) − 1
Ξ2
∑
N
NzN−1Z(T, V, N)∑
N
NzNZ(T, V, N)
)
= β
1
Ξ
∑
N
N2zNZ(T, V, N) − 1
Ξ2
(∑
N
NzNZ(T, V, N)
)2
definiere Antwortkoeffizient (“Ladungssuszeptibilitat”):
κT,V =1
N
(∂〈N〉∂µ
)
T,V
dann ist:
σ2N = N kTκT,V
und auch
σN
N∝ 1√
N
10.3 Klassisches ideales Gas
Rechnung in kanonischer Gesamtheit
HV,N(q, p) =3N∑
i=1
p2i
2m+ WWand,V(q1, ..., q3N)
209

Zustandsintegral:
Z(T, V, N) = α∫
dqdp e−βHV,N (q,p)
=1
h3NN !
∫d3Np e−β
∑ip2
i /2m∫
d3Nq e−βWWand,V
=1
h3NN !
(∫dp e−βp2/2m
)3N
V N
(W = 0, exp(−βW ) = 1 fur r ∈ V ; W = ∞, exp(−βW ) = 0 fur r /∈ V )∫
dp e−βp2/2m =√
2m/β∫
ds e−s2
=√
2mkT√
π =h
λ
λ: thermische de Broglie-Wellenlange
also:
Z(T, V, N) =1
h3NN !
(h
λ
)3N
V N
bzw.
Z(T, V, N) =1
N !
(V
λ3
)N
freie Energie:
F (T, V, N) = −kT ln Z(T, V, N) ≈ NkT (ln N + 3 ln λ − ln V )
thermische Zustandsgleichung:
−p =
(∂F
∂V
)
T,N
= NkT−1
V
kalorische Zustandsgleichung:
E = 〈H〉 =1
Zα∫
dqdp He−βH = − ∂
∂βln(α
∫dqdp e−βH = − ∂
∂βln Z
= − ∂
∂βln(const.β−3N/2) = − 1
β−3N/2
(−3N
2
)β−3N/2−1 =
3
2NkT
10.4 Quantenmechanisches System harmonischer
Oszillatoren
Rechnung in kanonischer Gesamtheit
210

H =N∑
i=1
~ωi
(ni +
1
2
)
beachte: beliebige Frequenzen!
Eigenzustande:
|n1〉|n2〉 · · · |nN〉Eigenenergien:
E =N∑
i=1
~ωi
(ni +
1
2
)
mit ni = 0, 1, 2, ...
Zustandssumme:
Z = Z(T, N) = Sp e−βH =∞∑
n1=0
· · ·∞∑
nN=0
e−β∑
i~ωi(ni+1/2)
=∞∑
n1=0
· · ·∞∑
nN=0
N∏
i=1
(e−β~ωinie−β~ωi/2
)
=N∏
i=1
∞∑
n=0
(e−β~ωine−β~ωi/2
)
=N∏
i=1
Zi(T, N)
mit
Zi(T, N) = e−β~ωi/2∞∑
n=0
(e−β~ωi
)n
= e−β~ωi/2 1
1 − e−β~ωi
=1
eβ~ωi/2 − e−β~ωi/2
=1
2 sinh(β~ωi/2)
also:
Z(T, N) =N∏
i=1
1
2 sinh(β~ωi/2)
freie Energie:
211

F (T, N) = −kT ln Z(T, N) = kTN∑
i=1
ln(2 sinh(β~ωi/2))
mittlere Besetzungszahl des i-ten Oszillators?
∂
∂ωilnZ =
1
Z
∂
∂ωiSp e−β
∑i~ωi(ni+1/2)
=1
ZSp
(e−β
∑i~ωi(ni+1/2)(−β~(ni + 1/2))
)
= −β~(〈ni〉 + 1/2)
andererseits:
∂
∂ωilnZ = − ∂
∂ωi
∑
i
ln(2 sinh(β~ωi/2))
= − 1
2 sinh(β~ωi/2)2 cosh(β~ωi/2)β~/2
= −β~/21
tanh(β~ωi/2)
also:
〈ni〉 + 1/2 =1
2 tanh(β~ωi/2)
mit x = β~ωi/2:
〈ni〉 =1
2
(1
tanh(x)− 1
)=
1
2
(ex + e−x
ex − e−x− 1
)=
1
2
e−x
ex − e−x
=1
e2x − 1
also:
〈ni〉 =1
e~ωi/kT − 1
Verallgemeinerung des Resultats fur ωi = ω
10.5 Dritter Hauptsatz
QM Rechnung!
Dichteoperator der kanonischen Gesamtheit:
ρT,V,N =1
Z(T, V, N)e−βHV,N
212

Energie-Eigenwertgleichung:
HV,N |En, γ〉 = En|En, γ〉 mit γ = 1, .., gn
damit:
ρT,V,N =1
Z(T, V, N)
∑
En,γ
|En, γ〉〈En, γ|e−βHV,N
=
∑En,γ e−βEn |En, γ〉〈En, γ|
∑En,γ e−βEn
=
∑En,γ e−β(En−E0)|En, γ〉〈En, γ|∑
En,γ e−β(En−E0)
mit GZE E0; fur β → ∞ ist:
e−β(En−E0) → δEn,E0
also:
ρT,V,N →∑
En,γ δEn,E0|En, γ〉〈En, γ|∑En,γ δEn,E0
=
∑γ |E0, γ〉〈E0, γ|∑
γ 1
g0: GZ-Entartung, P0 =∑
γ |E0, γ〉〈E0, γ|: Projektor auf Eigenraum zur GZE
ρT,V,N → 1
g0P0 (T → 0)
fur T = 0 befindet sich ein Quantensystem im GG in dem gemischten Zustand,bei dem alle Grundzustande die gleiche WK haben (K wie auch MK!)
Entropie:
S(T, V, N) = kσ[ρT,V,N ] = −k Sp (ρ ln ρ) = −k〈ln ρ〉= −k〈ln(P0/g0)〉 = k ln g0 − k〈ln P0〉
mit
〈ln P0〉 =1
g0
∑
γ
〈E0, γ| lnP0|E0, γ〉 =1
g0
∑
γ
〈E0, γ| ln 1|E0, γ〉 = 0
folgt:
S(T, V, N) = k ln g0 (T → 0)
3. HS gilt bei fur ln g0 < O(N) also fur g0 < O(eN )
213

10.6 Eindimensionales Ising-Modell, Transferma-
trixmethode
Ising-Modell:
– Bechreibung magnetischer Ordnung
– Demonstrationsmodell der statistischen Physik
– Hamilton-Operator:
H = −JN∑
i=1
SiSi−1 − BN∑
i=1
Si (J, B > 0)
– Si kurz fur Siz, lokale Spins an Platzen eines Gitters: i = 1, ..., N
– alle Observablen kommutieren “klassisches” (aber diskretes) Modell
– Darstellung eines Spins: Si =
(1 00 −1
)(ohne Faktor ~/2!)
– mogliche Werte (Eigenwerte): si = ±1
– hier eindimensionales Modell
D = 1: losbar (Ising, Lenz ∼ 1920)
D = 2: losbar fur B = 0 (Onsager 1944)
D = 3: nicht analytisch losbar, numerische Losung problemlos
D = ∞: exakt losbar (Weiß’sche Molekularfeldtheorie, s.u.)
– periodische Randbedingung: i = 0 ⇔ i = N
– Energie eines Spins im Feld B:
−B, falls si = +1 (Spin parallel zum Feld, B > 0)
B, falls si = −1 (Spin antiparallel zum Feld, B > 0)
– Wechselwirkung zwischen nachsten Nachbarn
WW-Energie −J falls SiSi−1 = +1 (bevorzugt parallele Spins, J > 0)
i=N
B
+J +J +J
+J
−J −J −J −Ji=1 i=2
– GZE: E0 = −N(J + B), GZ: s1 = ... = sN = +1
214

– magnetisches Gesamtmoment: M =∑
i
〈Si〉 (allg.: M = −~−1gµB
∑
i
〈Si〉)
—————————————————————————————————
Kontrollparameter: H, M, N
kanonische Gesamtheit: T, B, N
– B als Lagrange-Parameter zur NB M(s1, ..., sN) = M
Zustandssumme: β = 1/kT
Z(T, B, N) = Sp e−βHN (B)
ONB:
|s1〉 · · · |sN〉also:
Z =∑
s1=±1
· · ·∑
sN=±1
exp
(βJ
∑
i
siss−1 + βB∑
i
si
)
=∑
s1=±1
· · ·∑
sN=±1
∏
i
exp (βJsisi−1 + βBsi)
(entspricht klassischer Zustandssumme)
—————————————————————————————————
Transfermatrixmethode∏
i
exp (βJsisi−1 + βBsi) =∏
i
exp(βJsisi−1 +
1
2βB(si + si−1)
)=∏
i
Qsi,si−1
mit Transfermatrix
Q =
(eβJ+βB e−βJ
e−βJ eβJ−βB
)
definiere Vektoren (Eigenvektoren von Si zu si = ±):
| + 1〉 =
(10
)| − 1〉 =
(01
)
dann ist:
〈si|Q|si−1〉 = Qsi,si−1
und:
Z =∑
s1=±1
· · ·∑
sN=±1
N∏
i=1
Qsi,si−1
215

=∑
s1=±1
· · ·∑
sN=±1
〈sN |Q|sN−1〉 · · · 〈s2|Q|s1〉〈s1|Q|sN〉
=∑
sN=±1
〈sN |QN |sN〉
Z = Sp QN
Q reell und symmetrisch
Q = UqUT
mit U reell und orthogonal, UUT = 1, q diagonal
Z = Sp QN = Sp(UqUT · · ·UqUT
)= Sp qN
= Sp
(λ1 00 λ2
)N
= Sp
(λN
1 00 λN
2
)= λN
1 + λN2
λi: Eigenwerte von Q
fur N → ∞ ist also:
Z = λN1 + λN
2 = λNmax
(1 +
λNmin
λNmax
)→ λN
max
Z = λNmax
beachte λmax nicht entartet!
—————————————————————————————————
Theorem von Perron und Frobenius:
sei A eine reelle M × M-Matrix mit Aij > 0
– es gibt einen nichtentarteten reellen Eigenwert λ > 0
– mit λ > |λ′| fur alle Eigenwerte λ′ 6= λ
– ∃ x = (x1, ..., xM) Eigenvektor zu λ mit xi > 0
—————————————————————————————————
statt Beweis: direkte Rechnung:
0 = det
(eβJ+βB − λ e−βJ
e−βJ eβJ−βB − λ
)
= λ2 − λ(eβJ+βB + eβJ−βB
)+ e2βJ − e−2βJ
also:
λ = eβJ 1
2
(eβB + e−βB
)±√
1
4e2βJ (eβB + e−βB)2 − e2βJ + e−2βJ
216

= eβJ(cosh(βB) ±
√cosh2(βB) − 1 + e−4βJ
)
(konsistent mit P.-F.-Theorem)
somit:
Z(T, B, N) = eNβJ(cosh(βB) +
√cosh2(βB) − 1 + e−4βJ
)N
Ableitung der Thermodynamik, z.B.
—————————————————————————————————
magnetisches Gesamtmoment
M =N∑
i=1
〈Si〉 =1
ZSp
N∑
i=1
〈Si〉e−βH =1
β
∂
∂Bln Z
=N
β
1
[· · · ]∂
∂B
[eβJ
(cosh(βB) +
√cosh2(βB) − 1 + e−4βJ
)]
=N
β
eβJ
[· · · ]
β sinh(βB) +
2 cosh(βB) sinh(βB)β
2√
cosh2(βB) − 1 + e−4βJ
=N
β
1
cosh(βB) +√· · ·
β√· · ·(sinh(βB)
√· · · + cosh(βB) sinh(βB)
)
= Ncosh(βB) −√· · ·
cosh2(βB) − cosh2(βB) + 1 − e−4βJ
sinh(βB)√· · · + cosh(βB) sinh(βB)√· · ·
= Ncosh2(βB) sinh(βB) − sinh(βB)
√· · ·2(1 − e−4βJ )
√· · ·
= N− sinh(βB)(e−4βJ − 1)
(1 − e−4βJ)√· · ·
also:
M = Nsinh(βB)√
cosh2(βB) − 1 + e−4βJthermische Zustandsgleichung
Diskussion:
– B = 0 M = 0
– B → ∞ M → N tanh(βB) → N = M0
– M(T, B, N) = −M(T,−B, N)
– T → 0, β → ∞ M → M0
217

– T → ∞, β → 0 M → 0
M0
T>0B
M
0
0
J=0J>0
0−M
fur T > 0 ist
limB→0
M(T, B, N) = 0
keine spontane magnetische Ordnung, Paramagnet
beachte:
0 = limT→0
limB→0
M(T, B, N) 6= limB→0
limT→0
M(T, B, N) = ±M0
—————————————————————————————————
idealer Paramagnet: J = 0 (nicht-w.w. Momente)
M(T, B, N) = N tanh(βB)
isotherme Suszeptibilitat des idealen Paramagneten:
χ =1
N
(∂M
∂B
)
T,N
∣∣∣∣∣∣B=0
=cosh2(βB) − sinh2(βB)
cosh2(βB)β
∣∣∣∣∣∣B=0
= β =1
kT
Curie-Gesetz
—————————————————————————————————
warum keine magnetische Ordnung? betrachte Korrelationen
mit
H = −JN∑
i=1
SiSi−1 −N∑
i=1
BiSi (J, Bi > 0)
ist
Z = Z(T, Bi, N) F = F (T, Bi, N)
und:
218
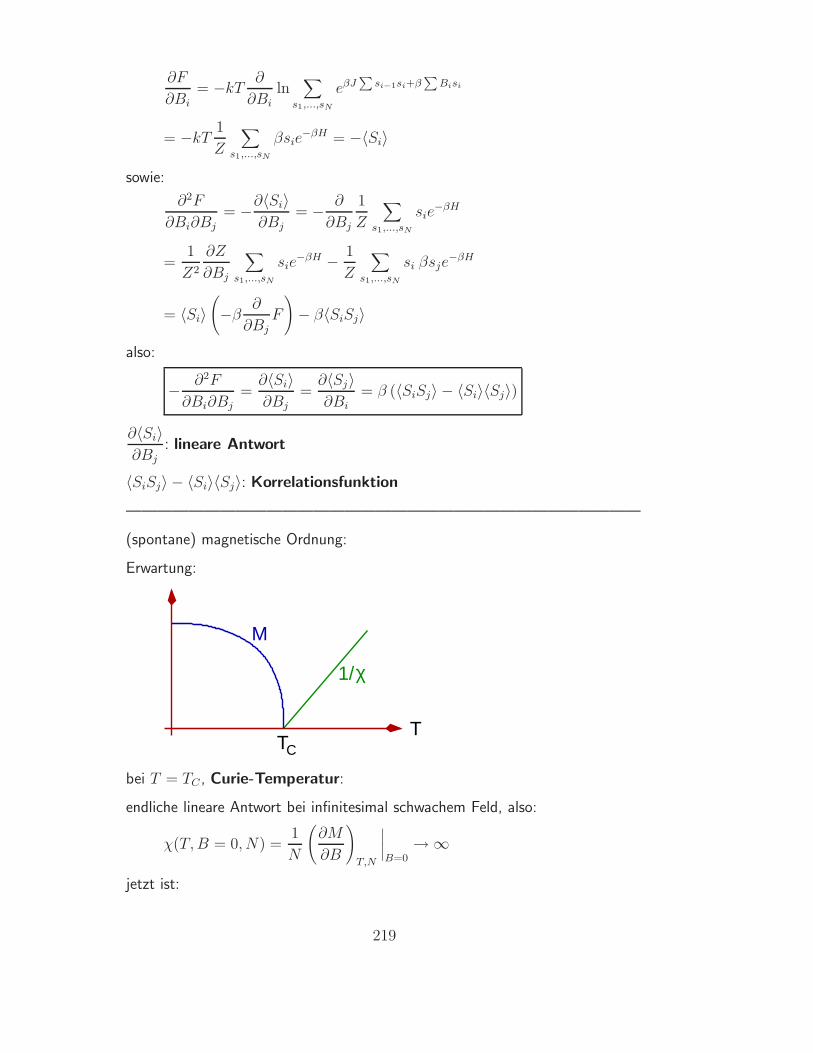
∂F
∂Bi
= −kT∂
∂Bi
ln∑
s1,...,sN
eβJ∑
si−1si+β∑
Bisi
= −kT1
Z
∑
s1,...,sN
βsie−βH = −〈Si〉
sowie:
∂2F
∂Bi∂Bj= −∂〈Si〉
∂Bj= − ∂
∂Bj
1
Z
∑
s1,...,sN
sie−βH
=1
Z2
∂Z
∂Bj
∑
s1,...,sN
sie−βH − 1
Z
∑
s1,...,sN
si βsje−βH
= 〈Si〉(−β
∂
∂BjF
)− β〈SiSj〉
also:
− ∂2F
∂Bi∂Bj=
∂〈Si〉∂Bj
=∂〈Sj〉∂Bi
= β (〈SiSj〉 − 〈Si〉〈Sj〉)
∂〈Si〉∂Bj
: lineare Antwort
〈SiSj〉 − 〈Si〉〈Sj〉: Korrelationsfunktion
—————————————————————————————————
(spontane) magnetische Ordnung:
Erwartung:
χ
TTC
M
1/
bei T = TC , Curie-Temperatur:
endliche lineare Antwort bei infinitesimal schwachem Feld, also:
χ(T, B = 0, N) =1
N
(∂M
∂B
)
T,N
∣∣∣∣B=0
→ ∞
jetzt ist:
219

χ =1
N
∂
∂BM =
1
N
∑
i
∂
∂Bi
∑
j
〈Sj〉 =1
N
∑
ij
∂〈Sj〉∂Bi
=1
Nβ∑
ij
(〈SiSj〉 − 〈Si〉〈Sj〉)
Translationsinvarianz ausnutzen: 〈SiSj〉 = 〈Si−jS0〉 = 〈SdS0〉, Abstand d = i−j
χ =1
NβN
∑
d
(〈SdS0〉 − 〈S0〉〈S0〉) = β∑
d
(〈SdS0〉 − 〈S0〉〈S0〉)
definiere Korrelationslange ξ:
〈SdS0〉 − 〈S0〉〈S0〉 ∝ e−d/ξ fur d → ∞Erwartung: exponentieller Zerfall der Korrelationsfunktion fur große Abstande
(Sd und S0 unabhangig bei n.N. bzw. kurzreichweitiger WW)
offensichtlich gilt, dass
χ → ∞ ⇒ ξ → ∞Korrelationslange divergiert bei TC!
—————————————————————————————————
Berechnung von ξ
Z(T, Bi, N nicht bekannt, direkte Berechnung der Korrelationsfunktion furB = 0:
fur B = 0 ist: 〈Si〉 = 〈Sj〉 = 0, bleibt also:
〈SiSj〉 =1
Z
∑
s1,...,sN
sisjeβJ∑
isisi−1
=1
Z
∑
s1
· · ·∑
sN
sisj
N∏
i=1
〈si|Q|si−1〉 Q =
(eβJ e−βJ
e−βJ eβJ
)
=1
Z
∑
si
∑
sj
∑
sN
〈sN |QN−i|si〉si〈si|Qi−j |sj〉sj〈sj|Qj |sN〉
falls i > j; mit
σ ≡∑
si
|si〉si〈si| =
(1 00 −1
)
(z-Pauli-Matrix) ist jetzt
〈SiSj〉 =1
Z
∑
sN
〈sN |QN−iσQi−jσQj|sN〉
=1
ZSp
(QN−iσQi−jσQj
)
220

=1
ZSp
(σQi−jσQN−i+j
)
Diagonalisierung der Transfermatrix:
Q = UqUT UUT = 1
mit
q =
(2 cosh(βJ) 0
0 2 sinh(βJ)
)U = UT =
1√2
(1 11 −1
)
es folgt:
〈SiSj〉 =1
ZSp
(σUqi−jUT σUqN−i+jUT
)
und mit
σ′ = UT σU =1√2
(1 11 −1
)(1 00 −1
)1√2
(1 11 −1
)=
(0 11 0
)
ist:
〈SiSj〉 =1
ZSp
(σ′qi−jσ′qN−i+j
)
=1
(2N cosh(βJ)NSp
(2 sinh(βJ) 0
0 2 cosh(βJ)
)i−j (2 cosh(βJ) 0
0 2 sinh(βJ)
)N−(i−j)
=1
(cosh(βJ)N
(sinhi−j(βJ) coshN−(i−j)(βJ) + coshi−j(βJ) sinhN−(i−j)(βJ)
)
= tanhi−j(βJ) + tanhN−(i−j)(βJ)
da tanh x < 1 fur x 6= 0, ist im Limes N → ∞:
〈SiSj〉 = tanh(i−j)(βJ)
Korrelationsfunktion nur vom Abstand abhangig
〈SiSj〉 = 〈SdS0〉 = tanhd(βJ)
Bestimmung der Korrelationslange (x = tanh(βJ)):
〈SiSj〉 = xd = eln(xd) = e− ln(1/xd) = e−d ln(1/x)
mit
ξ−1 ≡ ln(1/x) = ln coth(βJ)
ist dann
〈SiSj〉 = e−d/ξ
221

Divergenz von ξ nur fur T → 0, β → ∞es ist:
χ =1
Nβ∑
ij
(〈SiSj〉 − 〈Si〉〈Sj〉)
=1
Nβ∑
i
〈S2i 〉 + 2
1
Nβ∑
i>j
〈SiSj〉
= β + 21
Nβ∑
i
∞∑
d=1
tanhd(βJ)
= β
(1 + 2
∞∑
d=1
tanhd(βJ)
)
= β
(1 − 2 + 2
1
1 − tanh(βJ)
)
χ = β
(2
1 − tanh(βJ)− 1
)
• J = 0 (idealer Paramagnet, s.o.): χ = 1/kTdivergent bei T = 0 wegen GZ-Entartung g = 2N
• T → ∞, β → 0: χ = β
(2
1 − βJ + O(β3)− 1
)= β + 2Jβ2 + O(β3J2)
Curie-Gesetz: χ ∝ 1/T fur hohe T
• T → 0, β → ∞: tanh(βJ) → 1, χ → ∞ exponentiell
• Phasenubergang nur bei TC = 0
Q: Phasenubergange bei endlichen T ?
10.7 Heisenberg-Modell, Molekularfeldtheorie
Heisenberg-Modell:
H = −N∑
i=1
N∑
j=1
JijSiSj − BN∑
i=1
Si
mit lokalen Spins Si (hier: Spinquantenzahl S = 1/2),
[Six, Sjy] = i~Siz (und zyklische Permutationen)
222

auf Gitter mit i = 1, ..., N Platzen
Modell-Varianten:
H = −N∑
i,j=1
Jij [α(SixSjx + SiySjy) + βSizSjz]
α = β: Heisenberg-Modell
α = 0, β = 1: Ising-Modell
α = 1, β = 0: XY-Modell
—————————————————————————————————
approximative Losung des Heisenberg-Modells in Molekularfeld-Naherung
es gilt:
SiSj = Si〈Sj〉 + Sj〈Si〉 − 〈Si〉〈Sj〉 + F
mit Fluktuationsterm
F = (Si − 〈Si〉)(Sj − 〈Sj〉)Molekularfeld-Naherung: F = 0 fur i 6= j
(es sei Jii ≡ 0)
damit H → HMF:
HMF = −∑
ij
2Jij〈Sj〉Si +∑
ij
Jij〈Sj〉〈Si〉 − B∑
i
Si
= −∑
i
∑
j
2Jij〈Sj〉 + B
Si +
∑
ij
Jij〈Sj〉〈Si〉
= −∑
i
(B
(A)i + B
)Si +
∑
ij
Jij〈Sj〉〈Si〉
mit Austauschfeld
B(A)i ≡
∑
j
2Jij〈Sj〉
—————————————————————————————————
vereinfachende Annahmen:
Feld in z-Richtung:
B = (0, 0, B)
kollineare homogene Ordnung (Ferromagnetismus, Jij > 0):
〈Si〉 = miez
223

Translationsinvarianz:
mi = m ∀i
nachst-Nachbar-WW:
Jij = J > 0 nur fur n.N. i, j
Anzahl n.N.: q (Koordinationszahl)
damit ist:
HMF = − (BA + B)∑
i
Siz + NqJm2
und
BA = 2qJm
Diskussion:
• H : Si spurt fluktuierendes lokales “Feld”∑
j JijSj
• HMF: homogenes Austauschfeld, “Molekularfeld” BA
• MFT: Gitter-Modell atomares Modell
• Selbstkonsistenz: m BA HMF FMF m
—————————————————————————————————
Auswertung der MFT:
FMF = −kT ln Sp exp
(β(BA + B)
∑
i
Siz − βNqJm2
)
m = − 1
N
∂FMF
∂B
Berechnung von FMF:
sei |µi〉 mit µi = ±1/2 Eigenzustand von Siz, dann ist:
ZMF = Sp e−βHMF =∑
µ1µ2···〈µ1µ2 · · · |eβ
∑i(BA+B)Siz |µ1µ2 · · ·〉e−βNqJm2
= e−βNqJm2 ∑
µ1µ2···
∏
i
eβ(BA+B)µi
= e−βNqJm2 ∏
i
∑
µi=±1/2
eβ(BA+B)µi
= e−βNqJm2
(eβ(BA+B)/2 + e−β(BA+B)/2)N
224

es folgt:
FMF = −kT ln ZMF
= −NkT ln(eβ(BA+B)/2 + e−β(BA+B)/2
)+ NqJm2
also:
m = − 1
N
∂FMF
∂B= kT (β/2)
eβ(BA+B)/2 − e−β(BA+B)/2
eβ(BA+B)/2 + e−β(BA+B)/2
=1
2tanh
(1
2β (2qJm + B)
)
Mean-Field-Gleichung:
m =1
2tanh (β (qJm + B/2))
Selbstkonsistenz-Gleichung fur m
—————————————————————————————————
T → 0, β → ∞ : m → ±1
2Sattigungsmoment
T → ∞, β → 0 : m → 0 paramagnetische Losung
—————————————————————————————————
B = 0:
m = 0 ist Losung, paramagnetische Phase
m 6= 0: Ferromagnetismus ?
m → 0, tanhx = x − 1
3x3 + O(x5)
m =1
2βCqJm
TC =1
2qJ (Curie-Temperatur)
typisches MF-Resultat: TC ∝ J, q
m =1
2βqJm − 1
2
1
3(βqJm)3
m2 =3
4
T 2
T 2C
TC − T
TC
T → TC : m ∝√
TC − T
TCkritischer Exponent: β = 1/2
225

charakteristisch fur MF!
(m = tβ , t = (TC − T )/TC , β = 0.3646 3D Heisenberg)
—————————————————————————————————
B > 0:
T > TC , B → 0 m → 0 x klein in tanh x:
m =1
2βqJm +
1
2β
B
2
m =β
4B
1
1 − 12βqJ
=B
4
1
kT − kTC
∂m
∂B∝ 1
T − TC
Curie-Weiß-Verhalten der Suszeptibilitat
kritischer Exponent γ = 1 (χ = (−t)−γ, γ = 1.3866 3D Heisenberg)
—————————————————————————————————
alternative Herleitung der Molekularfeldtheorie mittels Variationsprinzip fur F :
definiere freie Energie als Funktional beliebiger Dichteoperatoren ρ:
F [ρ] ≡ Sp (ρ(H + kT ln ρ))
fur ρ = ρK = e−βH/Z (Dichteoperator der K Gesamtheit) ist:
F [ρK] = 〈H〉 + kT Sp (ρK ln ρK) = 〈H〉 − TS = F (T, N)
und
− 1
kTF [ρ] = − Sp(ρ ln ρ)− 1
kTSp(ρH) = σ[ρ]−λ0 Sp(ρH) = max
fur Dichteoperatoren ρ mit Sp ρ = 1
(Maximierung der Shannon-Information unter den NB Sp ρ = 1 und 〈H〉 = E,Lagrange-Parameter: λ0 = 1/kT )
also:
F [ρ] = min fur ρ = ρK , F [ρK] = F (T, N)
Verallgemeinerung des Ritzschen Variationsprinzips (klassisch: Gibbs)
—————————————————————————————————
Variationsmethode:
• wahle Ansatz fur ρ = ρ(s) mit Sp ρ = 1 und Parametern s = (s1, s2, ...)
226

• berechne F (ρ(s))
• bestimme s0 aus∂
∂sF (ρ(s0)) = 0
• approximiere F = F (ρ(s0))
—————————————————————————————————
Weißsche Molekularfeldtheorie:
Ansatz:
ρ = ρ(BA) =e−βHMF
Sp e−βHMF
mit:
HMF = −(BA + B)N∑
i=1
Siz
langere Rechnung ergibt:
BA = 2qJm
227

228

Kapitel 11
Ideale Quantengase
N wechselwirkungsfreie, identische Teilchen
Motivation:
• klassisch: Zustandsgleichungen in Widerspruch zum 3.HS
• quantenmechanische Modifikation der Zustandsgleichungen?
• Unterschiede zwischen Fermionen oder Bosonen?
• neue (Quanten-)Phanomene?
Beispiele:
1) System von Elektronen, z.B. Valenzelektonen im Festkorper (Fermionen),naherungsweise ww-frei
2) He-Gas/Flussigkeit (Bosonen)
3) unterscheidbare Teilchen (s.u.)
11.1 Fermi- und Bose-Gas
Ein-Teilchen-Hilbert-Raum: H1
ONB von H1:
|φα〉
H(i)1 : Hilbert-Raum des i-ten Teilchens, ONB: |φ(i)
α 〉—————————————————————————————————
229

Hilbert-Raum N identischer Teilchen (Fermionen (−) oder Bosonen (+)):
H(±)N = S
(±)N HN
mit
HN = H(1)1 ⊗H(2)
1 ⊗ · · · ⊗ H(N)1 (Tensorprodukt)
und
S(±)N =
1
N !
∑
P(±1)pP
ONB von H(±)N :
|n1, n2, ..., nd〉 (NB:∑
α nα = N)
(anti-)symmetrisierte Produktzustande
|n1, n2, ..., nd〉 = K± S(±)N
(|φ(1)
α1〉|φ(2)
α2〉 · · · |φ(N)
αN〉)
(d = dimH1)
Besetzungszahl:
nα = Anzahl der Teilchen im Zustand φα
Fermionen: nα = 0, 1 Bosonen: nα = 0, 1, 2, ...
—————————————————————————————————
Fermionen H(−)N Bosonen H(+)
N
Pij|Ψ〉 = −|Ψ〉 Pij |Ψ〉 = +|Ψ〉antisymmetrische symmetrische Zustandehalbzahliger Spin ganzzahliger Spin
M = Dimension des Hilbert-Raums
identische Fermionen: M =
(dN
)
φ3φ1φ2 φd Zustände...
Ein−Teilchen−
identische Bosonen: M =
(d + N − 1
N
)
φ3φ1φ2 φd Zustände...
Ein−Teilchen−
—————————————————————————————————
230

Hamilton-Operator (“ideal”: keine WW):
HN =N∑
i=1
H(i)N
mit:
H1 =p2
2m+ WV (r)
WV (r): Wandpotenzial
Ein-Teilchen-Zustande:
H1|φα〉 = εα|φα〉Teilchen mit Spin S, Spinprojektionsquantenzahl:
σ = −S, ..., (S − 1), S
also Sz|σ〉 = ~σ|σ〉System im Volumen V mit periodischen RB: α ≡ (k, σ) mit
kµ =2π
Lnµ nµ ganze Zahl, µ = x, y, z
Ein-Teilchen-Zustand zum Wellenvektor k:
|φα〉 = |φk〉|σ〉 = φk(r)|σ〉 =1√V
exp(ikr)|σ〉
Ein-Teilchen-Energie:
ε(k) =~
2k2
2m
N -Teilchen-Energie:
E =N∑
i=1
ε(ki) bzw. E =∑
k
∑
σ
nk,σε(k) =∑
α
nαεα
N -Teilchen-Zustand (Energieeigenzustand zu E):
|n1, ..., nα, ..., nd〉 mit∑
α
nα = N , d = dimH1 = ∞
Besetzungszahloperator nα:
nα|n1, ..., nα, ..., nd〉 = nα|n1, ..., nα, ..., nd〉es ist N =
∑
α
nα
231

11.2 Fermi- und Bose-Verteilungsfunktion
GK Rechnung:
Ξ(T, V, µ) = Sp e−β(HV −µN) =∞∑
N=0
Sp Ne−β(HV −µN)
=∞∑
N=0
′∑
n1
′∑
n2
· · ·′∑
nd
exp
(−β(
∑
α
nαεα − µ∑
α
nα)
)
=∞∑
N=0
zNZ(T, V, N)
′∑· · ·
′∑Summation unter der NB
∑
α
nα = N , beachte: d = ∞
K Rechnung fur Z(T, V, N) wegen NB schwierig
GK Rechnung NB wird aufgehoben:
Ξ(T, V, µ) =∑
n1
∑
n2
· · ·∑
nd
exp
(−β(
∑
α
nαεα − µ∑
α
nα)
)
Summation ohne Einschrankungen! also:
Ξ(T, V, µ) =∑
n1
∑
n2
· · ·∑
nd
∏
α
exp (−βnα(εα − µ)) =∏
α
∑
n
exp (−βn(εα − µ))
Fermionen: nα = 0, 1
Ξ(T, V, µ) =∏
α
(1 + e−β(εα−µ)
)
Ω(T, V, µ) = −kT∑
α
ln(1 + e−β(εα−µ)
)
Ω(T, V, µ) = −kT (2S + 1)∑
k
ln(1 + e−β(ε(k)−µ)
)
Bosonen: nα = 0, 1, 2, ...
Ξ(T, V, µ) =∏
α
1
1 − e−β(εα−µ)
Ω(T, V, µ) = kT∑
α
ln(1 − e−β(εα−µ)
)
Ω(T, V, µ) = kT (2S + 1)∑
k
ln(1 − e−β(ε(k)−µ)
)
fur µ muss gelten (Konvergenzradius der geometrischen Reihe):
232

µ < ε0 = ε(k = 0) = 0
ε0: kleinste Ein-Teilchen-Energie
—————————————————————————————————
mittlere Besetzungszahl:
〈nα〉 =1
Ξ(T, V, µ)
∑
n1
∑
n2
· · ·∑
nd
nα exp
(−β(
∑
α
nα(εα − µ))
)
also:
〈nα〉 = − 1
β
∂
∂εαln Ξ(T, V, µ)
Fermionen:
〈nα〉 = − 1
β
1
1 + e−β(εα−µ)e−β(εα−µ)(−β)
〈nα〉 =1
eβ(εα−µ) + 1= f(εα − µ)
mit f(x) = 1/(eβx + 1) Fermi-Dirac-Verteilungsfunktion
Bosonen:
〈nα〉 = − 1
β(−1)
1
1 − e−β(εα−µ)(−e−β(εα−µ))(−β)
〈nα〉 =1
eβ(εα−µ) − 1= b(εα − µ)
mit b(x) = 1/(eβx − 1) Bose-Einstein-Verteilungsfunktion
—————————————————————————————————
Diskussion
Fermionen:
ε −µαx=
f(x)
0
14kT
• f(0) = 1/2, f(x) + f(−x) = 1
233

• x → −∞ (εα ≪ µ) f(x) → 1
• x → ∞ (εα ≫ µ) f(x) → 0
• f ′(0) =−βeβx
(eβx + 1)2
∣∣∣∣x=0
= −β
4= − 1
4kT
1 ∼ |∆f | ≈ |f ′(0)|∆x =1
4kT∆x
Aufweichzone: ∆x = 4kT ; T = 300K 4kT ≈ 0.1 eV
• T = 0 f(x) = Θ(−x)
Def: εF ≡ µ(T = 0) Fermi-Energie
Interpretation von µ fur T = 0:
εF trennt zwischen besetzten (εα < εF ) und unbesetzten (εα > εF ) Ein-Teilchen-Zustanden |φα〉
Bosonen:
ε −µαx=−1
b(x)
0
• b(x) hat Pol erster Ordnung bei x = 0
• x → ∞ (εα ≫ µ) b(x) → 0
• x → −∞ (εα ≪ µ) b(x) → −1
• T → 0: b(x) → 0 fur x > 0
• x ≤ 0, εα ≤ µ: Konvergenzradius der geometrischen Reihe uberschritten
unphysikalisch, Ξ, Ω divergent
(schon fur d = 1: Ξ =∑∞
n=0(e−β(ε−µ))n = ∞)
234

11.3 Thermodynamik des idealen Fermi-Gases
Ω(T, V, µ) = −kT∑
α
ln(1 + e−β(εα−µ)
)
Teilchenzahl:
〈N〉 = −∂Ω
∂µ=∑
α
1
eβ(εα−µ) + 1=∑
α
〈nα〉
Innere Energie:
U = 〈H〉 = − ∂
∂βln Ξ + µ〈N〉 =
∑
α
εα1
eβ(εα−µ) + 1=∑
α
εα〈nα〉
Entropie:
S = −(
∂Ω
∂T
)
V,µ
= k∑
α
ln(1 + e−β(εα−µ)) +1
T
∑
α
(εα − µ)1
eβ(εα−µ) + 1
3.HS ? T → 0, β → ∞
• εα − µ > 0 S → k∑
α ln 1 + 0 = 0
• εα − µ < 0 S → k∑
α
ln e−β(εα−µ) +1
T
∑
α
(εα − µ) = 0
alternative Darstellung fur S (direktes Umformen):
S = −k∑
α
[(1 − 〈nα〉) ln(1 − 〈nα〉) + 〈nα〉 ln〈nα〉]
S → 0 fur T → 0
—————————————————————————————————
V -Abhangigkeit?
Ω(T, V, µ) = −kT (2S + 1)∑
k
ln(1 + e−β(ε(k)−µ)
)
mit∑
k
→ V
(2π)3
∫d3k
ist:
Ω(T, V, µ) = −kT (2S + 1)V
(2π)3
∫d3k ln
(1 + e−β( ~
2k2
2m−µ))
= −kT (2S + 1)V
(2π)34π∫ ∞
0dkk2 ln
(1 + e−β( ~
2k2
2m−µ))
235

Substitution: x = ~k√
β/2m
Ω(T, V, µ) = −kT (2S+1)V
(2π)34π
(√2m
β
1
~
)3 ∫ ∞
0dxx2 ln
(1 + eβµe−x2
)
Fugazitat z = eβµ, λ = h/√
2πmkT
Ω(T, V, z) = −kT (2S + 1)V
λ3f5/2(z)
—————————————————————————————————
mit:
f5/2(z) =4√π
∫ ∞
0dxx2 ln
(1 + ze−x2
)
= − 4
3√
π
∫ ∞
0dxx3−2xze−x2
1 + ze−x2
=8
3√
π
∞∑
n=0
∫ ∞
0dxx4ze−x2 · (−1)nzne−nx2
=8
3√
π
∞∑
n=1
(−1)n+1zn∫ ∞
0dxx4e−nx2
substituiere: y = nx2, dy = 2nxdx, dx =dy
2nx=
dy
2√
ny:
f5/2(z) =8
3√
π
∞∑
n=1
(−1)n+1zn∫ ∞
0dy
1
2√
ny
y2
n2e−y
=4
3√
π
∞∑
n=1
(−1)n+1zn 1
n5/2
∫ ∞
0dyy3/2e−y
=4
3√
π
∞∑
n=1
(−1)n+1 zn
n5/2Γ(5/2)
mit Γ(5/2) = (3/2)Γ(3/2) = (3/2)(1/2)Γ(1/2) = (3/4)√
π ist:
f5/2(z) =∞∑
n=1
(−1)n+1 zn
n5/2
—————————————————————————————————
thermische Zustandsgleichung:
p = −(
∂Ω
∂V
)
T,z
= (2S + 1)kT
λ3f5/2(z) = −Ω
V
(mit Gibbs-Duhem-Relation)
236

µ eleminieren! dazu: Ξ = Sp e−β(H−µN) = Sp (zNe−βH) und:
〈N〉 = z∂
∂zln Sp
(zNe−βH
)
= z∂
∂zln Ξ
= z∂
∂z(−βΩ) (Ω = −pV )
= βV z∂
∂zp∣∣∣∣T,V
= βV z∂
∂z(2S + 1)
kT
λ3f5/2(z)
also:
〈N〉 = (2S + 1)V
λ3f3/2(z)
mit
f3/2(z) = z∂
∂zf5/2(z) =
∞∑
n=1
(−1)n+1 zn
n3/2=
4√π
∫ ∞
0dxx2 1
z−1ex2 + 1
d.h. 〈N〉 = 〈N〉(T, V, z), Umkehrung: z = z(T, V, 〈N〉), und damit:
p = (2S + 1)kT
λ3f5/2(z(T, V, 〈N〉)) thermische Zustandsgleichung
—————————————————————————————————
kalorische Zustandsgleichung:
U = − ∂
∂βln Sp e−β(H−µN) + µ〈N〉 = − ∂
∂βln Ξ + µ〈N〉
es ist:
− ∂
∂βln Ξ = − ∂
∂β(−βΩ)
= − ∂
∂β(2S + 1)
V
λ3f5/2(z)
=3
2
1
β(2S + 1)
V
λ3f5/2(z) − (2S + 1)
V
λ3
∂
∂zf5/2(z)
∂z
∂β
mit∂
∂β
1
λ3= −3
2
1
β
1
λ3
= −3
2Ω − (2S + 1)
V
λ3z
∂
∂zf5/2(z)µ mit
∂z
∂β= µz
237

=3
2pV − µ〈N〉
also:
U =3
2pV kalorische Zustandsgleichung
gilt so auch klassisch!
oder:
U =3
2(2S + 1)
kT
λ3f5/2(z(T, V, 〈N〉)) V
11.4 Klassischer Grenzfall
Auflosen der Gleichung
〈N〉 = (2S + 1)V
λ3f3/2(z)
fur hohe Temperaturen T , geringe Dichten 〈N〉/V
λ3 ≪ v =V
〈N〉 klassischer Limes
es ist:
(2S + 1)f3/2(z) =λ3
v≪ 1
und
f3/2(z) = z + O(z2)
einfachste Approximation somit:
(2S + 1)z + O(z2) =λ3
v
ein Schritt weiter:
f3/2(z) = z − z2
23/2+ O(z3)
damit:
z − z2
23/2+ O(z3) =
λ3
(2S + 1)v≡ z0
z(1 − 1
23/2z0
)+ O(z3) = z0
238

z(1 +
1
23/2z0
)−1
+ O(z3) = z0
z + O(z3) = z0
(1 +
1
23/2z0
)
einsetzen in:
p = (2S + 1)kT
λ3f5/2(z) = (2S + 1)
kT
λ3
(z − z2
25/2+ O(z3)
)
= (2S + 1)kT
λ3
(z0
(1 +
1
23/2z0
)− z2
0
25/2+ O(z3)
)
= (2S + 1)kT
λ3z0
(1 +
1
23/2z0 −
z0
25/2+ O(z2)
)
= kT1
v
(1 +
1
25/2z0 + O(z2)
)
also:
pV = NkT
(1 +
1
25/2
λ3
(2S + 1)v+ O(z2)
)
fur festes V und T hoherer Druck
effektive Abstoßung der Fermionen durch Pauli-Prinzip
11.5 Entartetes Fermi-Gas
entgegengesetzter Limes: tiefe Temperaturen T , hohe Dichten 1/v
λ3 ≫ v =V
〈N〉 entartetes Fermi-Gas
definiere Ein-Teilchen-Zustandsdichte
D(ε)dε = Anzahl der |φkσ〉 mit Energien in [ε, ε − dε]
=∑
k
∑
σ
(Θ(ε − ε(k)) − Θ(ε − dε − ε(k)))
= (2S + 1)∑
k
δ(ε − ε(k))dε
also:
D(ε) = (2S + 1)∑
k
δ(ε − ε(k))
239

D(ε) = (2S + 1)V
(2π)34π∫ ∞
0dk k2δ(ε − ~
2k2
2m)
= (2S + 1)V
(2π)34π
∂
∂ε
∫ ∞
0dk k2Θ(ε − ~
2k2
2m)
= (2S + 1)V
(2π)34π
∂
∂ε
1
3k3∣∣∣∣k=
√2mε/~
= (2S + 1)V
(2π)34π
1
3
∂
∂ε
(√2mε
~
)3
= (2S + 1)V
(2π)34π
1
3
(√2m
~
)33
2ε1/2
D(ε) = (2S + 1)(2m)3/2V
4π2~3ε1/2
—————————————————————————————————
damit gilt:
Ω(T, V, µ) = −kT (2S + 1)∑
k
ln(1 + e−β(ε(k)−µ)
)
Ω(T, V, µ) = −kT∫ ∞
0dεD(ε) ln
(1 + e−β(ε−µ)
)
und
〈N〉 = (2S + 1)∑
k
1
eβ(ε(k)−µ) + 1
〈N〉 =∫ ∞
0dεD(ε)f(ε− µ)
sowie
U = (2S + 1)∑
k
ε(k)1
eβ(ε(k)−µ) + 1
U =∫ ∞
0dεD(ε) ε f(ε − µ)
und:
S = −k(2S + 1)∑
k
[(1 − 〈nkσ〉) ln(1 − 〈nkσ〉) + 〈nkσ〉 ln〈nkσ〉]
S = −k∫ ∞
0dεD(ε) (f(µ − ε) ln f(µ − ε) + f(ε − µ) ln f(ε − µ))
—————————————————————————————————
240

Teilchenzahl und innere Energie von der Form
I(T ) =∫ ∞
−∞dε F (ε)f(ε− µ)
D(ε) = 0 fur ε < 0, F (ε) T -unabhangig
entartetes Fermi-Gas bei fester Dichte: T → 0
Taylor-Entwicklung:
I(T ) =∫ µ
−∞dε F (ε) +
π2
6(kT )2F ′(µ) + O(T 4)
—————————————————————————————————
allgemein gilt:
I(T ) = I(0) + 2∞∑
n=1
(1 − 21−2n)ζ(2n)(kT )2nF (2n−1)(µ)
Sommerfeld-Entwicklung
mit
ζ(n) =∞∑
p=1
1
pnRiemannsche ζ-Funktion, ζ(2) =
π2
6
—————————————————————————————————
T -Abhangigkeit von µ fur festes N = 〈N〉:
N =∫
dεD(ε)f(ε− µ)
=∫ µ
−∞dεD(ε) +
π2
6(kT )2D′(µ) + O(T 4)
=∫ εF
−∞dεD(ε) +
∫ µ
εF
dεD(ε) +π2
6(kT )2D′(µ) + O(T 4)
erster Term: N(T = 0) = N (fest), also:
0 =∫ µ
εF
dεD(ε) +π2
6(kT )2D′(µ) + O(T 4)
0 = D(εF )(µ − εF ) + O(µ − εF )2 +π2
6(kT )2D′(µ) + O(T 4)
µ − εF = −π2
6(kT )2 D′(µ)
D(εF )+ O(µ − εF )2 + O(T 4)
µ − εF = −π2
6(kT )2D′(εF )
D(εF )+ O(T 4)
241

mit D(ε) = const.ε1/2 istD′(εF )
D(εF )=
1
2εF
wegen εF ∼eV (Valenzelektronen eines Metalls) und kT ≈ 1/40 eV fur T = 300K, ist µ − εF ∼ 0.001 eV
—————————————————————————————————
T -Abhangigkeit von U :
U =∫
dεD(ε) ε f(ε − µ)
=∫ µ
−∞dε D(ε) ε +
π2
6(kT )2(εD′(ε) + D(ε))
∣∣∣∣ε=µ
+ O(T 4)
=∫ εF
−∞dε D(ε) ε + (µ − εF )D(εF ) εF
+π2
6(kT )2(εF D′(εF ) + D(εF )) + O(T 4)
=∫ εF
−∞dε D(ε) ε − π2
6(kT )2D′(εF )εF (s.o.)
+π2
6(kT )2(εF D′(εF ) + D(εF )) + O(T 4)
= U(T = 0) +π2
6(kT )2D(εF ) + O(T 4)
also:
U = U(0) +π2
6(kT )2D(εF )
Warmekapazitat bei tiefen Temperaturen:
CV =
(∂U
∂T
)
V,N
=π2
3k2D(εF )T
also:
CV = γT mit Konstante γ
vergleiche: C(klassisch)V =
3
2Nk = const. (Dulong-Petit, GVS)
Metalle: lineares CV experimentell bestatigt!
242

εFkT
fur tiefe T konnen nur wenige Fermionen die thermische Energie kT aufnehmen(Pauli-Prinzip) CV → 0
—————————————————————————————————
fur T = 0 ist
N =∫ εF
0dε D(ε) =
∫ εF
0dε const.ε1/2 = const.
2
3ε3/2F
mit der Konstanten const. = (2S + 1)(2m)3/2V
4π2~3
innere Energie:
U(T = 0) =∫ εF
−∞dε D(ε) ε = const.
∫ εF
0dε ε3/2 = const.
2
5ε5/2F
weiter gilt:
D(εF ) = const.ε1/2F
also:
U = const.2
5ε5/2F +
π2
6(kT )2const.ε
1/2F = const.
2
5ε5/2F
1 +
5π2
12
(kT
εF
)2
=2
5
3
2εFN
1 +
5π2
12
(kT
εF
)2
mit U =3
2pV gilt also:
p =2
5εF
N
V
1 +
5π2
12
(kT
εF
)2
T = 0: Nullpunktsdruck p0 =2
5εF
N
V
klassisch: p = 0 fur T = 0 fur feste Dichte N/V
243

11.6 Quasiteilchen
betrachte ideales Quantengas aus N unterscheidbaren Teilchen
Hamilton-Operator:
HN =N∑
i=1
H(i)1
Hilbert-Raum:
HN = H(1)1 ⊗H(2)
1 ⊗ · · · ⊗ H(N)1 (Tensorprodukt)
ONB:
|φ(1)α1〉|φ(2)
α2〉 · · · |φ(N)
αN〉
—————————————————————————————————
Beispiele:
1) paarweise verschiedene Teilchen
– H-Atom: 1 Elektron, 1 Proton
– 4 verschiedene Atome: H, He, Li, Be
φ1φ4 φ5 φ8φ8 φ4 φ8Teilchen
1 ...32 N
M = dN
keine relevanten Beispiel-Systeme
—————————————————————————————————
2) identische Teilchen, aber raumlich getrennt
– qubits, Zweiniveausysteme, lokale Spins mit S = 1/2
– System harmonischer Oszillatoren (atomare Schwingungen)
ZuständeEin−Teilchen−
φ3φ1φ2 φd... φ3φ1φ2 φd... φ3φ1φ2 φd...
M = dN
physikalisch wichtige Beispiele
—————————————————————————————————
System von N qubits:
Hamilton-Operator eines qubits (dimH1 = 2):
244

H1 = ε1|φ1〉〈φ1| + ε2|φ2〉〈φ2| (|φ1〉, |φ2〉 normiert, 〈φ1|φ2〉 = 0)
o.B.d.A. (Wahl des Energienullpunkts, Umnumerierung):
ε1 = 0 ε2 = ε
damit:
|φα〉 = |n〉 mit α = 1 ↔ n = 0 und α = 2 ↔ n = 1
also:
H1 = ε∑
n=0,1
n |n〉〈n|
—————————————————————————————————
System von N harmonischen Oszillatoren:
Hamilton-Operator eines Oszillators (dimH1 = ∞):
H1 =p2
2m+
1
2mω2q2
wahle Energienullpunkt als GZE, dann gilt fur die
Eigenenergien:
εn = ε n
mit ε = ~ω und n = 0, 1, 2, ..., d = ∞Eigenzustande nicht entartet, also:
H1 = ε∞∑
n=0
n |n〉〈n|
—————————————————————————————————
N qubits/Oszillatoren mit charakteristischen Ein-Teilchen-Energie εi (i = 1, ..., N)
Produktzustande:
|n1〉|n2〉 · · · |nN〉 mit ni = 0, 1 bzw. ni = 0, 1, 2, ...
bilden ONB aus Energie-Eigenzustanden:
HN |n1〉|n2〉 · · · |nN〉 = E|n1〉|n2〉 · · · |nN〉mit
E =N∑
i=1
niεi
—————————————————————————————————
vergleiche mit Gas von M identischen Fermionen/Bosonen wobei
245

HM =M∑
m=1
H(m)1
wobei
H1 =d∑
i=1
εi|i〉〈i| (dim H1 = d)
ONB von H(±)M aus Energie-Eigenzustanden:
|n1〉|n2〉 · · · |nd〉 mit ni = 0, 1 bzw. ni = 0, 1, 2, ...
Eigenenergie:
E =d∑
i=1
niεi
—————————————————————————————————
Aquivalenz fur
d = N und M =N∑
i=1
ni
—————————————————————————————————
N qubits/harmonische Oszillatoren mit Ein-Teilchen-Energien niεi
(ni = 0, 1/ni = 0, 1, 2, ...) fur qubit/Oszillator i
aquivalent zu
M =N∑
i=1
ni freie Fermionen/Bosonen mit Ein-Teilchen-Energien εi
fur i = 1, ..., d und d = N
—————————————————————————————————
beachte: Fermi-/Bose-Gas mit nicht kontrollierbarer Teilchenzahl M
Quasiteilchen
GK Beschreibung: Ω = Ω(T, V )Lagrangeparameter µ wird nicht benotigt, Quasiteilchenzahl M nicht kontrolliert
(kein Lagrangeparameter µ = 0)
K (und MK) Beschreibung: F = F (T, V )M ist kein Kontrollparameter
(F unabhangig von N
(∂F
∂M
)
T,V
= 0)
246

11.7 Phononen- und Photonengase
Anwendung des Quasiteilchenkonzepts:
1) Schwingungen von Atomen um Gitterpositionen
N (entkoppelte) Eigenschwingungen mit Wellenvektor k
kleine Auslenkungen Ein-Teilchen-Energien nkεk
Dispersion εk ≈ ~c|k| (fur k → 0), c: Schallgeschwindigkeit
Phononengas (Bosonen)
2) elektromagnetisches Feld
N stehende Wellen (Eigenmoden) mit Wellenvektor k
Wellengleichung Ein-Teilchen-Energien nkεk
Dispersion εk = ~ωk = ~c|k|, c: Lichtgeschwindigkeit
Photonengas (Bosonen)
Bose-Gas
H =M∑
m=1
H(m)1
aus Quasiteilchen
H1 =N∑
k=1
εk|k〉〈k| (N = d = ∞)
mit linearer Dispersion
εk = ~ωk = ~c|k|periodische RB auf Volumen V = L3:
kµ =2π
Lnµ nµ ganze Zahl, µ = x, y, z
Rastervolumen im k-Raum:
∆3k =(2π)3
V
V : Festkorpervolumen bzw. Hohlraum/schwarzer Korper
(beachte: Rechnung fur System von harmonischen Oszillatoren oben mit εk =εi = ε = const., d.h. dispersionslose Phononen: Einstein-Modell)
—————————————————————————————————
GK Potenzial:
247

Ω = Ω(T, V ) = −kT ln Z(T, V )
= −kT ln∑
n1
∑
n2
· · ·∑
nk
· · · exp
(−β
∑
k
~c|k|nk
)
= −kT ln∏
k
∞∑
n=0
exp (−β~c|k|)n
= kT∑
k
ln(1 − exp(−β~c|k|))
= kTV
(2π)34π∫ ∞
0dk k2 ln(1 − exp(−β~ck))
= −kTV
(2π)34π∫ ∞
0dk
1
3k3 1
1 − exp(−β~ck)exp(−β~ck)β~c
= −1
3~c
V
(2π)34π∫ ∞
0dk k3 1
exp(β~ck) − 1
= −1
3~c
V
(2π)34π
1
(β~c)4
∫ ∞
0dx x3 1
ex − 1
Wert des Integrals: π4/15
= − 1
45~c
V
(2π)34π
π4
(β~c)4
also:
Ω(T, V ) = − π2V
90(~c)3(kT )4
—————————————————————————————————
Photonen: 2 Polarisationsrichtungen
Ω(T, V ) = −2π2V
90(~c)3(kT )4
bzw.
Ω(T, V ) = −1
3αV T 4
mit
α =π2k2
15(~c)3≈ 7.578 · 10−16 J
m3K4 Stefan-Boltzmann-Konstante
Druck (mit Ω = −pV ):
p =1
3αT 4
248

Entropie:
S = −(
∂Ω
∂T
)
V
=4
3αV T 3
(3.HS erfullt)
wegen µ = 0 ist Ω = F = U − TS, also
U = Ω + TS = −1
3αV T 4 + T
4
3αV T 3
U = αV T 4 Stefan-Boltzmann-Gesetz
es folgt
CV =1
4αV T 3
—————————————————————————————————
fur Phononen ist also ebenfalls
CV ∝ T 3 Debyesches T 3-Gesetz
solange die Dispersion als linear angenommen werden darf, d.h. fur k → 0(langwelliger Limes), εk → 0, und somit fur T → 0
vergleiche:
CV ∝(
β~ω/2
sinh(β~ω/2)
)2
fur εk = ~ω = const. (Einstein-Modell)
und
CV = const. (Dulong-Petitsches Gesetz)
—————————————————————————————————
Zustandsraum 2-dimensional (Ω = Ω(T, V )) Bosonenzahl M = M(T, V )M kein unabhangiger Kontrollparameter
GK Gesamtheit: M fluktuiert fur festes T, V
mittlere Gesamtphotonenzahl:
〈M〉 = −∂Ω(T, V, µ)
∂µ
∣∣∣∣∣∣µ=0
somit (Faktor 2 fur die beiden Polarisationsrichtungen):
249

〈M〉 = 2∑
k
〈nk〉
mittlere Besetzungszahl der Mode k:
〈nk〉 = b(εα − µ)
∣∣∣∣µ=0
=1
eβεk − 1=
1
eβ~ck − 1
d.h.
〈M〉 = 2∑
k
1
eβ~ck − 1
= 2V
(2π)34π∫ ∞
0dk k2 1
eβ~ck − 1
= 2V
(2π)34π
1
(β~c)3
∫ ∞
0dx x2 1
ex − 1
mit∫
dk(· · · ) = 2!ζ(3) und ζ(3) =∞∑
p=1
1
p3≈ 1.202
〈M〉 = 2ζ(3)V
π2(~c)3(kT )3
beachte: 〈M〉 → 0 fur T → 0, denn fur T → 0 GZ des elektromagnetischenFelds, also nk = 0
—————————————————————————————————
es gilt weiter:
U = 〈H〉 = 2∑
k
εk〈nk〉
= 2V
(2π)34π∫ ∞
0dk k2 ~ck
eβ~ck − 1
Energiedichte:
u =U
V= 2
1
(2π)34π∫ ∞
0dk k2 ~ck
eβ~ck − 1
= 21
(2π)34π
1
c3
∫ ∞
0dω
~ω3
eβ~ω − 1
also:
u =∫ ∞
0dω u(ω, T ) u(ω, T ): spektrale Energiedichte
mit
250
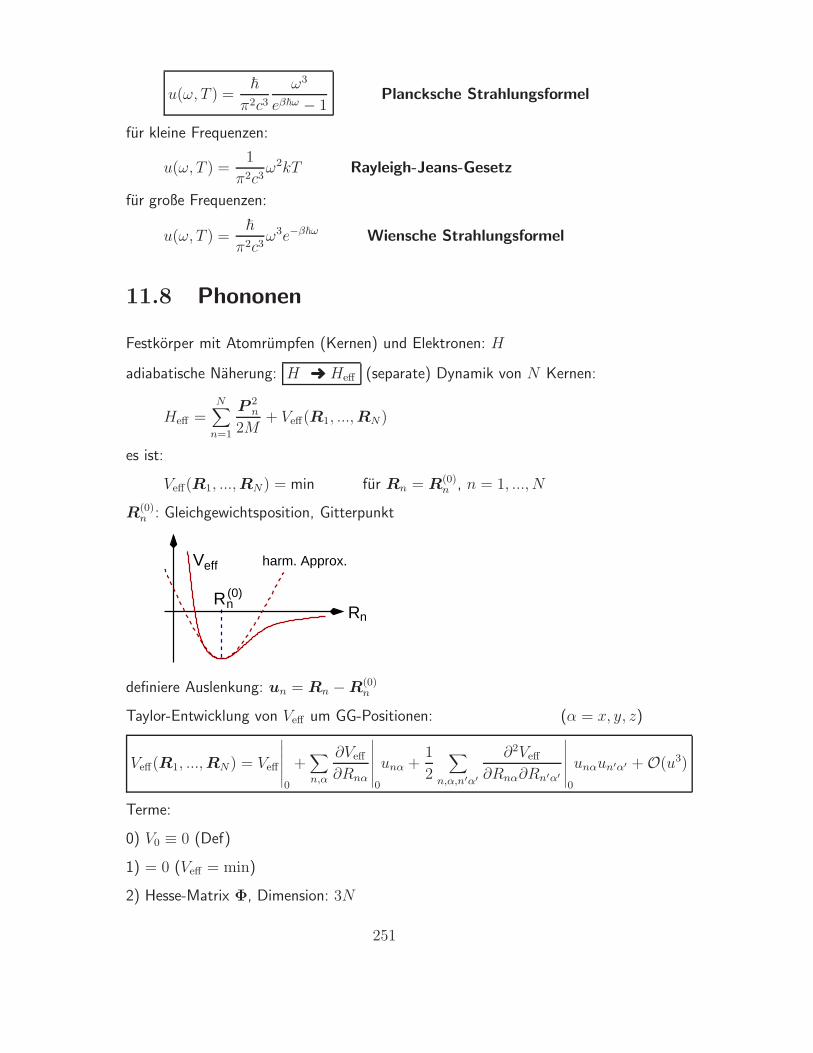
u(ω, T ) =~
π2c3
ω3
eβ~ω − 1Plancksche Strahlungsformel
fur kleine Frequenzen:
u(ω, T ) =1
π2c3ω2kT Rayleigh-Jeans-Gesetz
fur große Frequenzen:
u(ω, T ) =~
π2c3ω3e−β~ω Wiensche Strahlungsformel
11.8 Phononen
Festkorper mit Atomrumpfen (Kernen) und Elektronen: H
adiabatische Naherung: H Heff (separate) Dynamik von N Kernen:
Heff =N∑
n=1
P 2n
2M+ Veff(R1, ..., RN)
es ist:
Veff(R1, ..., RN) = min fur Rn = R(0)n , n = 1, ..., N
R(0)n : Gleichgewichtsposition, Gitterpunkt
n
V harm. Approx.
R n(0)
R
eff
definiere Auslenkung: un = Rn − R(0)n
Taylor-Entwicklung von Veff um GG-Positionen: (α = x, y, z)
Veff(R1, ..., RN) = Veff
∣∣∣∣∣∣0
+∑
n,α
∂Veff
∂Rnα
∣∣∣∣∣∣0
unα +1
2
∑
n,α,n′α′
∂2Veff
∂Rnα∂Rn′α′
∣∣∣∣∣∣0
unαun′α′ + O(u3)
Terme:
0) V0 ≡ 0 (Def)
1) = 0 (Veff = min)
2) Hesse-Matrix Φ, Dimension: 3N
251

3) vernachlassigbar fur kleine |un|, T → 0 harmonische Naherung
—————————————————————————————————
Matrixschreibweise:
Veff =1
2uTΦu
mit uT = (u1x, u1y, u1z, u2x, ..., uNz) (3N Komponenten)
Kraftkonstanten:
Φnα,n′α′ =∂2Veff
∂Rnα∂Rn′α′
∣∣∣∣∣∣0
Kraft in α-Richtung auf Atom n bei (Einheits-)Auslenkung eines Atoms bei n′ inα′-Richtung: (unα = δnn′δαα′)
Fnα = −∂Veff
∂unα
= (Φu)nα =∑
n′′α′′
Φnα,n′′α′′un′′α′′ = Φnα,n′α′
—————————————————————————————————
fur T → 0, geringer mittlere kinetische Energie, kleine Auslenkungen:
Heff ≈ Hharm. =1
2MP T P +
1
2uT Φu
P T = (P1x, ..., PNz)
Ortsdarstellung:
Pnα = −i~∂
∂Rnα= −i~
∂
∂unα
somit:
[unα, Pn′α′ ] = i~δnn′δαα′
—————————————————————————————————
Φ reell, symmetrisch, positiv definit Hauptachsentransformation:
Φ = OT DO mit OT O = 1
und
D = M
ω21 0 0 0
0 ω22 0 0
0 · · ·0 ω2
3N
also:
Hharm. =1
2MP T OT OP +
1
2uT OT DOu
252

und mit p = OP und q = Ou:
Hharm. =1
2MpT p +
1
2qT Dq =
3N∑
k=1
(p2
k
2M+
1
2Mω2
kq2k
)
—————————————————————————————————
q, p kanonisch konjugiert: (m = (n, α))
[qk, pk′] = [∑
m
Okmum,∑
m′
Ok′m′pm′ ] =∑
mm′
OkmOk′m′ i~δmm′
= i~∑
m
OkmOTm′k′ = i~δkk′
3N unabhangige harmonische Oszillatoren
Hharm. =3N∑
k=1
~ωk
(nk +
1
2
)
11.9 Massives Bose-Gas
fur T = Tc: Bose-Einstein-Kondensation
qualitative Ursache: effektive attraktive “WW” zwischen Bosonen
MK Modell-Rechung (E, N fest) mit Ein-Teilchen-Energie
εα = αε α = 0, 1, 2, ...
Beispiel:
253

4ε32
εεε0
4ε32
εεε0
N=3 E=3 ε
Bosonen
4ε32
εεε0
1 11 1
1 1
1
1 1
22
2
22
2
2
2 2
33
3
33
3
3
3 3
321
klassisch Ferm. bosonische Quasiteilchen(N unkontrolliert)
33%WK für Einfachbesetzung von Zuständen
60% 100%
Bosonen “ziehen sich an”
Rechung fur E = 10ε
Bosonen klassischN # Zustande mit nα ≤ 1 WK WK # Zustande mit nα ≤ 11 1 1 1.0 1.0 1 12 6 5 0.83 0.91 11 103 14 8 0.57 0.73 66 484 23 5 0.22 0.42 286 1205 30 1 0.03 0.12 1001 1206 35 0 0. 0. 3003 0N # Zustande mit nα ≤ 2 WK WK # Zustande mit nα ≤ 21 1 1 1.0 1.0 1 12 6 6 1.0 1.0 11 113 14 14 1.0 1.0 66 664 23 19 0.83 0.94 286 2705 30 16 0.53 0.78 1001 7806 35 8 0.23 0.48 3003 1440
GK Rechnung, N = 〈N〉 kontrolliert, 3-dimensionaler Zustandsraum:
Ω(T, V, µ) = kT (2S + 1)∑
k
ln(1 − e−β(ε(k)−µ)
)
254

im Folgenden: S = 0
N = −∂Ω(T, V, µ)
∂µ=∑
k
1
eβ(ε(k)−µ) − 1=∑
k
1
z−1eβε(k) − 1
Fugazitat z = eβµ
Konvergenz der geometrischen Reihe (s.o.) erfordert:
z < 1 (denn µ < 0 = ε(k) = 0)
—————————————————————————————————
mit∑
k
→ V
(2π)3
∫d3k
ist:
Ω(T, V, µ) = kTV
(2π)34π∫
dkk2 ln(1 − ze−β~2k2/2m
)
x Substitution: x = ~k√
β/2m
Ω(T, V, µ) = kTV
(2π)34π
(√2m
β
1
~
)3 ∫ ∞
0dxx2 ln
(1 − ze−x2
)
λ = h/√
2πmkT
Ω(T, V, z) = −kTV
λ3g5/2(z)
—————————————————————————————————
mit:
g5/2(z) = − 4√π
∫ ∞
0dxx2 ln
(1 − ze−x2
)
=4
3√
π
∫ ∞
0dxx3 2xze−x2
1 − ze−x2
=8
3√
π
∞∑
n=1
zn∫ ∞
0dxx4e−nx2
=4
3√
π
∞∑
n=1
zn 1
n5/2
∫ ∞
0dyy3/2e−y
=4
3√
π
∞∑
n=1
zn
n5/2Γ(5/2)
255

=∞∑
n=1
zn
n5/2
vergleiche: ideales Fermi-Gas (Spin S)
Ω(T, V, z) = −kT (2S + 1)V
λ3f5/2(z)
f5/2(z) =∞∑
n=1
(−1)n+1 zn
n5/2
—————————————————————————————————
Teilchenzahl:
N = −∂Ω
∂µ= kT
V
λ3
∂g5/2(z)
∂z
∂z
∂µ
also:
N =V
λ3g3/2(z)
mit:
g3/2(z) = z∂
∂zg5/2(z) =
∞∑
n=1
zn
n3/2
3/2g (z)
z1
2.612
g3/2(z) monoton wachsend
—————————————————————————————————
es seien jetzt V, N (und damit v = V/N) fest, es ist
g3/2(z) =λ3
v= const.T−3/2
mit fallendem T wachst g3/2(z) und damit z bis zum Maximalwert g(z = 1) ≈2.612, dies definiert ein Tc:
g3/2(1) =λ3(Tc)
v
beachte: λ3 ∼ v markiert die Temperatur, bei der Quanteneffekte wichtig werden
256

—————————————————————————————————
was passiert fur T < Tc?
Modifikation vonN
V=
g3/2(z)
λ3∝ T 3/2g3/2(z) notwendig, denn:
N/V fest, T weiter fallend, g3/2(z) maximal fur z = 1
k = 0-Term separat behandeln:
N =∑
k
1
z−1eβε(k) − 1=
1
z−1 − 1+∑
k6=0
1
z−1eβε(k) − 1
fur z = 1 −O(1/N) wird N0 = nk=0 = O(N) und
N =1
z−1 − 1+∑
k6=0
1
eβε(k) − 1= N0 +
V
(2π)3
∫d3k
1
eβε(k) − 1
denn im TD Limes (N → ∞) ist
1
eβε(k) − 1
fur k 6= 0 stetig und integrierbar
N0
k=0k
n(k)
fur z = 1 −O(1) ist N0 ≪ N ′ (dies reproduziert Resultate fur T > Tc oben)
—————————————————————————————————
also folgt fur T < Tc:
N = N0(T ) +V
λ3(T )g3/2(z = 1) (N, V fest)
mit der Definition von Tc ist dann:
N = N0(T ) +V
λ3(T )
λ3(Tc)
v= N0(T ) + N
(T
Tc
)3/2
also:
257

N0(T ) = N
[1 −
(T
Tc
)3/2]
Tc
N0
N
T
T < Tc: N0 = O(N) = N − N(
T
Tc
)3/2
T > Tc: N0 = O(1) bzw. N0/N = 0 + O(1/N)
Bose-Einstein-Kondensation:
– makroskopische Besetzung des Ein-Teilchen-Grundzustands unterhalb Tc
– Phasenubergang in ww-freiem System
—————————————————————————————————
makroskopische Besetzung auch fur andere Zustande?
kµ =2π
Lnµ nµ ganze Zahl, µ = x, y, z
betrachte z.B. k1 = (1, 0, 0):
ε(k1) =~
2k21
2m=
~2(2π)
2mL
damit ist:
N1 = nk=k1 =1
z−1eβε(k1) − 1≤ 1
eβε(k1) − 1
=1
1 − β ~2(2π)2mL
+ O(L−2) − 1= O(L) ≪ O(L3) = O(N) = N0
—————————————————————————————————
weitere TD Eigenschaften:
Ω(T, V, µ) = kT∑
k
ln(1 − e−β(ε(k)−µ)
)
separate Behandlung des k = 0-Terms?
258

Ω(T, V, z) = kT ln(1 − z) − kTV
λ3g5/2(z)
aber fur z = 1 −O(1/N) ist
kT ln(1 − z) = lnO(N−1) = O(ln N) ≪ O(N) = kTV
λ3g5/2(z)
also:
Ω(T, V, z) = −kTV
λ3g5/2(z)
Gibbs-Duhem-Relation Ω = −pV thermische Zustandsgleichung:
p =kT
λ(T )3g5/2(z) T > Tc
p =kT
λ(T )3g5/2(1) T < Tc (g5/2(1) = ζ(5/2) ≈ 1.342)
mit z = z(T, V, µ) als Umkehrung von N = N(T, V, z)
—————————————————————————————————
aus Ω = Ω(T, V, µ) folgt S(T, V, µ) = − ∂∂T
Ω(T, V, µ) und:
CV,N = Nk15
4
v
λ3g5/2(z) − Nk
9
4
g3/2(z)
g1/2(z)(T > Tc)
CV,N = Nk15
4
g5/2(1)
g3/2(1)
(T
Tc
)3/2
(T < Tc)
T
CV
Tc
3Nk/2
– Verlauf ahnlich der sog. λ-Anomalie von He4 (Tc = 2.18K)
– Theorie: Tc = 3.14K fur spezifisches Volumen von He
aber: He4 ist (stark) ww Bose-Gas!
259

• BEC: Vorhersage 1924
• experimentell realisiert: 1995 (Wieman, Cornell, Ketterle, Nobelpreis 2001)
(ca. 2000 spin-polarisierte 87Rb-Atome, Tc ≈ 170 · 10−9K)
260







