Download - Thalassemia By IPMS-KUM Peshawar

THALASSEMIAS Muhammad Asif Zeb

HEMOGLOBIN DISORDERS
Genetic disorders of hemoglobin are the most
common genetic disorders worldwide
Mutations in the globin genes are the most
prevalent monogenic disorders worldwide &
affect approx. 7% of the world population

Α AND Β THALASSEMIA
• The α gene is duplicated: 2 genes per chromosome, therefore 4 α genes in a normal adult. • The β gene is single therefore the normal adult will have two β genes
α and β globin gene clusters Chromosomes 11 and 16

NORMAL HUMAN HEMOGLOBIN
Embryonic Hemoglobin
• Hgb Gower 1 (ζ2,ε2)
• Hgh portland (ζ2, γ2)
• Hgb Gower 2 (α2, ε2)
Fetal Hemoglobin
• Hgb F (α2,γ2)
Adult Hemoglobin
• Hgb A (α2,β2) 98%
• Hgb A2 (α2,δ2) 1.5- 3.2%
• Hgb F (α2,γ2) <1%
Early embryogenesis Yolk sac erythroblasts
Major Hemoglobin of Intra-uterine life
Adult Hemoglobin


THALASSEMIA
Are characterized by a reduced rate of
production of normal hemoglobin due to
absent or decreased synthesis of one or
more types of globin polypeptide chain

TYPES OF THALASSEMIA
• Inherited anemia characterized by a decrease in synthesis one or more of the globin chains. The defect may affect the
α chains
β chains
γ chains
δ chains
• The clinical syndromes arise from a combination of
Inadequate production of hemoglobin
Unbalanced accumulation of globin chains


Α-THALASSEMIA SYNDROMES
• Group of disorders characterized by decreased
synthesis of α-chains usually due to gene
deletions
• The mutation is a functionally abnormal α
gene
• The clinical severity can be classified according
to the number of genes that are missing or
inactive

Α-THALASSEMIA SYNDROMES
Four possible α thalassemia syndromes:
1. α-thalassemia (silent carrier): One of the four α-
globin gene fails to function.
2. α-thalssemia-trait (α Thal trait minor): Two of the
four α-globin gene fails to functions.
3. Hgb H disease: Three of the four α-globin gene
fails to functions.
4. Bart’s Hydops Fetalis: Four of the four α-globin
gene fails to functions.

CLASSIFICATION


Syndrome # alpha genes
deleted
Newborn Hb
Barts (γ4) %
Clinical Picture
Silent Carrier 1 1-2 Silent
Alpha
thalassemia trait 2 3-10
Mild hypochromic,
microcytic anemia
Hb H Disease 3 25 Hb H (β4) mild
hemolytic anemia
Hydrops fetalis 4 80-100 Death in utero or
shortly after birth
α Thalassemia

Α-THALASSEMIA MAJOR
• --/--( α0 -thal) (Hydrops fetalis)
• Most severe form of α-thalassemia, involving the deletion of all 4 genes resulting the absence of α chains.
• In the absence of α chains, erythrocytes assemble hemoglobin using the δ,ϒ & β chains available.
• Therefore abnormal hemoglobin tetramer involving gamma chains (Hb Bart’s, ϒ4) & beta chains(HbH, β4 )is produced.

Α-THALASSEMIA MAJOR
Hb Bart’s has high affinity for oxygen so this
Hb can’t supply tissue with sufficient O2 to
sustain life & developing infant dies of
hypoxia & congestive heart failure.

CLINICAL FEATURES
• Infants that survive until birth exhibit significant physical changes upon routine exam
• The babies are
- Underweight, edematous with distend
abdomen,
- Hepatosplenomegaly due to extramedulary
hematopoiesis
- Massive bone marrow hyperplasia
• Hemolysis is severe, as there is extensive deposition of hemosiderin

LAB DIAGNOSIS
• There is severe anemia
• hemoglobin 3-10 g/dl
• Microcytic hypochromic RBCs
• marked anisocytosis & poikilocytosis
• Increased NRBCs
• Hb electrophoresis on cellulose acetate membrane at alkaline PH shows:
Hb Bart’s 80-90%
Hb Portland 10-20%
Hb H sometimes detectable
Hb A, HbA2, Hb F absent

HEMOGLOBIN H DISEASE
--/-α (HETEROZYGOUS)
Occurs when 3 of 4 α genes are deleted
This disorder usually results when 2
heterozygous parents, one with --/αα & the
other expressing –α/αα genotype, bear
children.

PATHOPHYSIOLOGY
• The reduction of α-chain synthesis results in the decrease in the assembly of HbA, HbA2, and HbF.
• A decrease in α-chain creates a relative excess
of beta chain which unite to form a tetrad of 4
beta chains called HbH.
• HbH is unstable & tend to
precipitate inside erythrocytes trigering chronic
hemolytic anemia.
• It has an O2 affinity 10 times that of
HbA, which reduces oxygen delivery to tissues
causes tissue hypoxia.

LAB DIAGNOSIS
• Hemoglobin level 8-10 g/dl
• Reticulocyte count moderately raised ranging
5-10%
• Microcytic hypochromic red cells
• Marked poikilocytosis
• Target cells
• Basophilic stippling
• NRBCs
Peripheral Blood Smear: stained With brilliant cresyl blue, showing Precipitated Hgb H

LAB DIAGNOSIS
• Hemoglobin electrophoresis of affected neonates shows
- Hb Bart’s 25% with decreased levels of
HbA, HbA2,HbF
• After birth β chains begin to replace gamma chains & Hb H replaces the Hb Bart’s.
- Hb H in Adults 2-40%
- HbA2 Decreased (1.5%)
- HbF Normal


DEMONSTRATION OF HB H INCLUSION BODIES
“In alpha thalssemia, red cell containing Hb H
are incubated with a solution of a redox
dye(brilliant cresyl blue), HbH which is relatively
unstable, precipitates & red cells are pitted by
numerous inclusions, an appearance likened to
the surface of a golf ball”


THALASSEMIA TRAIT
--/αα and -α/-α
Also called Thalassemia minor, occurs when
two of the four α gene, either on same or
opposite chromosomes, are missing
There is a measureable decrease in the
production of α- containing hemoglobins, the
unaffected hemoglobin genes are able to direct
synthesis of globin

THALASSEMIA TRAIT
chains faster than normal & compensate
for effected genes
• Patients with α-thalassemia trait are
asymptomatic with mild anemia and are
often diagnosed incidentally or when being
evaluated for family studies
• These patients have normal life span & do
not need medical intervention for their
thalassemia

LAB DIAGNOSIS
MCV 60-70 fl
Slight anemia
In peripheral blood film, red cells with
- Hypocromia & Microcytosis
- Poikilocytosis
- Target Cells
- Basophilic Stippling
In Hb electrophoresis, Hb Bart’s 5-6%

BETA THALASSEMIA
• occurs when one or both of the two genes needed for making the beta globin chain of hemoglobin are variant.
• The severity of illness depends on whether one or both genes are affected and the nature of the abnormality.
• If both genes are affected, anemia can range from moderate to severe.
• The severe form of beta thalassemia is also known as Cooley’s anemia.

THALASSEMIA
Greek letter used to designate globin chain:
+: Indicates diminished, but some production of globin chain by gene:
+
0 :Indicates no production of globin chain by gene:
0

CLASSIFICATION
Genotype Genetic
description Phenotype
βο/βο Homozygous Major
βο/β+ Heterozygous Major or
Intermedia
β+/β+ Homozygous Major or
Intermedia
βο/β Heterozygous Intermedia or
minor
β+/β Heterozygous Minor
β /β Homozygous Normal

Β-THALASSEMIA MAJOR
• βο/βο, βο/ β+, β+/ β+
• Is a severe, transfusion dependent, inherited
anemia
• Also referred to as COOLEY’S ANEMIA
• Caused by a homozygous or double
heterozygous inheritance of abnormal β gene
resulting in marked reduction or absence in β-
chain synthesis.
• Presents early in life

PATHOPHYSIOLOGY
• The molecular defects in - β thalassemia result in absent or reduced β-chain production. α-Chain synthesis is unaffected leading to an excess of α-chains
• Lack of beta chain production can be classified into four categories
– Reduced HbA
– Compensatory production of abnormal Hb
– Ineffective erythropoiesis
– Erythroid hyperplasia

PATHOPHYSIOLOGY

CLINICAL FEATURES
• Symptoms 1st observed in infants are;
- Irritability, pallor,
- Diarrhoea, fever, enlarged abdomen
- Growth retardation
- Brown pigmentation of skin
- Chronic hemolysis often produces
gallstones
- Gout & icterus

CLINICAL FEATURES
- Bone changes accompany the hyperplastic marrow, marrow cavities enlarge in every bone,expanding the bone & producing characteristic bossing of the skull
- Facial deformities & “hair-on-end” appearance of the skull on X-ray
- Massive splenomegaly
• Severe infections, septicemia,pericarditis
• Iron deposition leads to organ dysfunction, diabetes & cirrhosis

Marked bony changes from bone marrow proliferation

Bone marrow hyper expansion gives the classic “hair on end” appearance on X-ray of the skull

LAB DIAGNOSIS
On CBC TLC Normal
- Hb low (2-3 g/dl)
- MCV, MCH, low
PLT: Normal to Decrease
- Reticulocyte count increases to the degree
expected for the severity of anemia
because of the increase degree of
ineffective erythropoises

• On peripheral blood picture
- Marked anemia
- Marked microcytosis & hypochromia
- Anisocytosis & poikilocytosis
- Target cells, tear drop cells, fragmented RBC, polychromasia
- NRBCs
- Basophilic stippling
• Osmotic fragility test reveals increased resistance to hemolysis


• Bone Marrow:
- is not usually necessary for diagnosis
but, when performed, show marked
erythroid hyperplasia
• Bone Marrow iron stores increased

BIOCHEMISTRY
- Serum iron raised
- TIBC saturated
- Serum transferrin receptor variable
- Serum ferritin raised
- Serum unconjugated bilirubin raised
- Serum uric acid raised
- Urine dipyrrols (hB degradation products)
are present
serum & red cell folate reduced

HB ELECTROPHORESIS
In adults, the absence of HbA, 90% HbF, & low,
normal, or increase HbA2 is characteristic of
βο/βο thalassemia


Β-THALASSEMIA MINOR
βο/ β or β+/ β
Results from the heterozygous inheritance of
either a β+ or βο gene with one normal β gene
The normal β gene directs synthesis of
sufficient amount β-chains to synthesize
enough HbA for near normal oxygen delivrey &
erythrocyte survival

Β-THALASSEMIA MINOR
In case of a heterozygous β+ patient, the
thalassemic gene can also contribute to β-
chain production
They are asymptomatic except in periods of
stress as can occur during pregnancy,
infections & folic acid deficiency
Under these conditions,a moderate microcytic
anemia develops

LAB DIAGNOSIS
Erythrocyte count is doubled (>5×10/L) for what is expected at the given Hb conc.
MCV 55-70fl
MCH less than 22pg
MCHC 29-33 g/dl
Mild anemia with hemoglobin ranges 9-14 g/dl
Reticulocyte count mild elevated
LAB DIAGNOSIS
• Peripheral blood picture shows mild anemia, Anisocytosis & poikilocytosis with
- Target cells
- Basophilic stippling
- NRBCs are not usually found
• On Hb electrophoresis
- HbF normal
- HbA > 3.5%


Β-THALASSEMIA INTERMEDIA
• βο/ β+, β+/ β+ or βο/ β+
• All three patterns of inheritance, homozygous, heterozygous, double heterozygous can produce β-Thalassemia intermedia
• The homozygous & double heterozygous forms represent a mutation in both β alleles resulting in a moderate degree of β chain synthesis
• Symptoms intense during pregnancy & infection

LAB DIAGNOSIS
CBC reflects a moderate microcytic
hypochromic anemia with Hb value 6-10 g/dl
Erythrocyte count raised
Target cells are prominent poikilocytes
observed, basophilic stippling & NRBCs are
present
HbA2 5-10%
HbF 30-75%

SECOND LINE IDENTIFICATION OF THALASSEMIA
• Second line identification tests based on results of Hb electrophoresis on cellulose acetate membrane are;
- Estimation of HbA2
- Estimation HbF by
– Alkali denaturation by modified betke method
– Acid elution method

OTHER TESTS
PCR for identification of mutation
Hb Electrophoresis


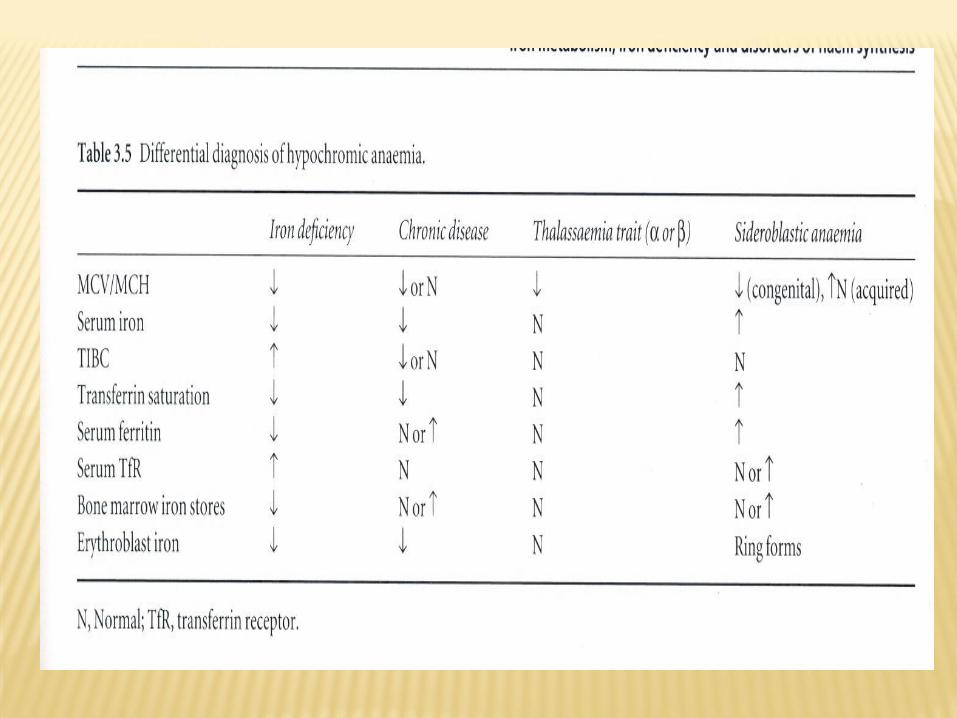
DIFFERENTIAL DIAGNOSIS
• Iron deficiency anemia
• Anemia of chronic disease
• Sideroblastic anemia

THANX







