www.sciencemag.org/cgi/content/full/315/5810/513/DC1
Supporting Online Material for
A Virus in a Fungus in a Plant: Three-Way Symbiosis and Thermal
Tolerance Luis M. Márquez, Regina S. Redman, Russell J. Rodriguez, Marilyn J. Roossinck*
*To whom correspondence should be addressed. E-mail: [email protected]
Published 26 January 2007, Science 315, 513 (2007)
DOI: 10.1126/science.1136237
This PDF file includes
Materials and Methods Figs. S1 to S6 Table S1 References

Report 1136237 Márquez et al. SOM 2
Materials and Methods
Double stranded RNA extraction: Isolates of the fungus, Curvularia protuberata, were
cultured from the panic grass, Dichanthelium lanuginosum, as described elsewhere (1).
Fungal isolates were grown in 500 ml of liquid 1 X (24g/l) Potato Dextrose (PD) medium
supplemented with ampicillin, streptomycin and tetracycline at 50 µg/ml each, with
agitation (100-150 rpm) for one week at 25ºC. Mycelium mats were filtered through
Whatman paper, frozen at -80ºC for 30 min and lyophilized over night. Two hundred mg
of lyophilized mycelium per isolate were ground in liquid nitrogen until completely
pulverized. Enrichment of double-stranded RNA was done by a modification of the
method of Dodds (2). Nucleic acids were extracted in 10 ml of extraction buffer (0.1 M
NaCl, 50 mM Tris pH 8, 1 mM EDTA pH 8, 1% SDS, 0.1% 2-Mercaptoethanol) and 10
ml of phenol:chloroform (1:1, w:w). The aqueous phase was removed and adjusted to
16.5% ethanol and passed through a column packed with approximately 100 mg of CF11
cellulose using a slow speed (∼200 rpm) centrifuge. Columns were washed with three
column volumes of application buffer (0.1 M NaCl, 50 mM Tris pH 8, 0.5 mM EDTA pH
8, 16.5 % ethanol). The dsRNA was eluted in 4.5 ml of elution buffer (0.1 M NaCl, 50
mM Tris pH 8, 0.5 mM EDTA pH 8) brought to 0.3 M Sodium Acetate (pH 6) and
precipitated at -20°C overnight with 10 ml of ice cold absolute ethanol (100%). After
centrifugation, pellets were resuspended in 50µl of 0.1 mM EDTA pH 8. A 10 µl aliquot
of the sample was separated on a 1.2% agarose gel in TBE and dsRNA bands were
visualized by staining with ethidium bromide using standard procedures.
Double stranded RNA cloning: Approximately 2 µg of dsRNA were mixed with 800
ng of primer (5'CCTGAATTCGGATCCTCCNNNNNN3') and sterile water for a final
volume of 11 µl, and boiled for 2 min. The sample was quenched on ice, and 9 µl of
Reverse Transcription (RT) mix (Superscipt, Gibco/BRL, and buffers and reagents
recommended by the manufacturer) were added, and the sample was incubated at 50°C
for 2 hours. One µl of the RT reaction was used as template for a 15 µl thermal cycling
reaction with TaKaRa Ex Taq (Takara Shuzo Co. Ltd.), buffers and dNTPs supplied by
the manufacturer, 1 µM concentration of the primer (5'CCTGAATTCGGATCCTCC3'),
and 0.5 µg of RNase A (Sigma). The reaction was completed in a Idaho Technologies

Report 1136237 Márquez et al. SOM 3
Rapid Cycler with a slope setting of 5, using the following cycles: 1 cycle of 94°C, 1
min.; 40 cycles of 94°C, 0 sec., 45°C, 0 sec., and 72°C 15 sec.; 1 cycle of 72°C, 5 min.; 1
cycle of 37°C 5 min. The product was cloned into the vector pGemTEasy (Promega)
according to the manufacturers instructions. The sequence was determined using an ABI
3700 sequencer for 60 clones, and the data was assembled into contigs using software in
the GCG sequence analysis package. Approximately 10 fold coverage was obtained for
the sequences of the two RNAs with no ambiguities. The terminal sequences were
determined by rapid amplification of cDNA ends (3).
Sequence analysis: The 5' and 3' ends were defined arbitrarily, using the orientation that
gave +sense matches for the open reading frames (ORFs). The ORFs and polyprotein
sequences related to them were identified respectively using the ORF finder and
BLASTP 2.2.13 (4) programs accessible online at the NCBI website.
RNA probe synthesis: To make a labeled probe for each viral RNA, cDNA clones were
selected that were representative of the two RNAs. One µg (4 µl) of the appropriate
plasmid, linearized with SphI (Amersham) for the RNA 1 probe and Aat II (New England
Biolabs Inc.) for the RNA 2 probe, 4 µl of 5X Ribomax buffer (400 mM HEPES-KOH
pH7.5, 60 mM MgCl2, 10 mM Spermidine, 200 mM DTT), 2 µl of a mixture of ATP,
CTP, GTP (30 mM) and UTP (1 mM), 5 µl of 20 µCi/ µl of 32-P UTP, 1 µl (40 U) of
RNAsin (Promega), 1 µl (0.9 U) of inorganic pyrophosphatase (Sigma), 1 µl (20 U) of
SP6 RNA polymerase (New England Biolabs) and 2 µl of H2O were mixed and incubated
at 37 ºC for 1.5 hours (5). The remaining plasmid DNA was digested by adding 3 µl of
RQ1 DNAse 10 X Buffer, 3 µl (1 U/µl) of RQ1 DNAse (Promega) and 4 µl of H2O to the
mix and incubating for 15 additional min. This was followed by extraction with
Phenol:Chloroform (1:1, w:w), and the aqueous phase was removed and brought to 2.5 M
ammonium acetate and precipitated with ethanol at -20°C over night. The probe was
pelleted, the pellet washed with 70% ethanol and 30% 2.5 M ammonium acetate and
resuspended in 100 µl of 0.1 mM EDTA.

Report 1136237 Márquez et al. SOM 4
Northern blot: Approximately 200 ng of each dsRNA sample were denatured by
incubating at 65 ºC in 12.5 µl of de-ionized Formamide, 2.5 µl of 10 X MOPS buffer and
4 µl of 37% Formaldehyde. The samples were then chilled on ice and 2.5 µl of loading
buffer (50% v/v glycerol containing 0.1 mg/ml of bromophenol blue) were added. The
samples were electrophoresed in 1.2 % agarose gels in MOPS buffer and incubated for 20
min in 20 X SSC prior to transfer. Denatured dsRNA was transferred by capillary action
overnight from the gels to nylon membranes (Hybond N+ Amersham). The membranes
were baked for 3 hours at 80ºC in a vacuum oven. Hybridizations and washings were
carried out at 65 ºC, using the method of Church and Gilbert (6).
RT-PCR: RT-PCR reactions for viral RNA detection were essentially the same as
described for cloning, except that the primer used for the RT was
5'GACGCCTCCGAGATCGAG3'. For the thermal cycling reaction we used this same
primer in conjunction 5'ACCCCAATTAACCGGATGCC3'. Thermal cycling reaction
conditions were as follows: slope setting of 7, 1 cycle of 94°C, 1 min.; 40 cycles of
94°C, 1 sec., 68°C, 1 sec., and 72°C 40 sec.; 1 cycle of 72°C, 5 min.; 1 cycle of 37°C 5
min.
Sequencing of rDNA ITS1-5.8S-ITS2 region: Approximately 10ng of fungal DNA was
used as template for a 10 µl thermal cycling reaction with TaKaRa Ex Taq (Takara Shuzo
Co. Ltd.), buffers and dNTPs supplied by the manufacturer, and 1 µM each of primers
ITS1F (5'TTGGTCATTTAGAGGAAGTAA3') and ITS4
(5'TCCTCCGCTTATTGATATGC3'). Thermal cycling reaction were done in an Idaho
Technologies Rapid Cycler using the following conditions: slope setting of 0.5ºC/second,
1 cycle of 94ºC, 1 min.; 40 cycles of 94°C, 1 sec., 60°C, 1 sec., and 72°C 40 sec.; 1 cycle
of 72°C, 5 min.; 1 cycle of 37°C 5 min. Amplified products were directly sequenced
using an ABI 3700 sequencer.
Simple Sequence Repeat (SSR) Analysis: Polymerase chain reactions were performed
in 20 µl volumes containing 10 mM Tris-HCl (pH 9.0), 50 mM KCl, 2.5 mM MgCl2,
0.2% Triton X-100, 200 µM each of dATP, dCTP, dGTP, dTTP, 0.2 units Taq DNA

Report 1136237 Márquez et al. SOM 5
polymerase, 500 ng of each oligonucleotide primer and 20 ng of fungal DNA.
Amplification reactions were carried out in MJ research thermocyclers programmed as
follows: initial denaturation at 93ºC for 2 min; denaturation at 93ºC for 15 seconds,
primer annealing (60°C) for 1.5 min following a 0.2°C/sec ramp from 93ºC, and
synthesis at 72ºC for 1.5 min following a 0.2°C/sec ramp from annealing temperatures.
The products were electrophoresed in 1.2% agarose gels in TBE and bands were
visualized by staining with ethidium bromide using standard procedures.
Virus particle isolation: Mycelium grown for about 1 month in 500 ml of 1 X PD
medium was filtered and used for virus particle isolation using the method described for
Cucumber mosaic virus (7). For negative staining, the virus preparation was diluted four
fold in de-ionized water and mixed with the same volume of 1% uranyl acetate for 1 min
at room temperature and then allowed to adhere to 200 mesh copper grids with 0.25%
formvar. The grids were air-dried and examined at 75kV with a transmission electron
microscope (Hitachi 7100, Electron Microscope Facility, University of Montana).
Curing the fungus: Mycelium of the isolate obtained from sectoring of the wild type of
C. protuberata was grown for about one week on 500 ml of 1 X PD medium. The
mycelium mat was filtered through filter paper, frozen at -80ºC for half an hour and
lyophilized over night. Small pieces of about 10 mg of this lyophilized mycelium were
used to inoculate new 500 ml liquid cultures. One virus free isolate, out of four, was
obtained in this way.
Restriction enzyme-mediated integration (REMI): Transformation of Curvularia
protuberata was conducted using standard protocols as previously described (8) with the
following exceptions: 10 µg of pCT74 vector (9) was linearized with restriction enzyme
XhoI (Promega) and 40 units of the same restriction enzyme was added to carry out the
REMI protoplast transformation reactions. Genomic DNA was extracted and
transformation of fungal isolates verified using standard molecular techniques as
previously described, with XhoI linearized pCT74 vector as the probe (8).

Report 1136237 Márquez et al. SOM 6
Anastomosis: Mycelium inocula of the donor wild type isolate and the recipient
hygromycin resistant virus free isolate of C. protuberata were picked from pure single-
conidiospore cultures and placed adjacent to one another at one end of a Petri dish
containing 1 X PD and 2 X agar (30g/l) with antibiotics (ampicillin, streptomycin and
tetracycline at 50 µg/ml each). Mycelia plugs from the zone of contact between the two
colonies were transferred to PD media plates containing 50 µg/ml of hygromycin.
Isolates exhibiting hygromycin resistance were transferred to 1/10 (2.4g/l) PD media with
2 X agar Petri dishes to induce sporulation. Single conidiospore isolates were then used
to inoculate 500 ml 1 X PD liquid cultures for dsRNA extraction as described above.
Heat stress experiments: Experiments using tropical panic grass, Dichanthelium
lanuginosum, were performed as follows: Seed coats of D. lanuginosum were removed
and naked seeds were surface sterilized by washing with agitation in 2% hypochlorite for
10-15 minutes and rinsed thoroughly with 7-10 volumes of de-ionized water to generate
endophyte-free plants. These sterilized seeds were planted in sterile Magenta boxes (the
"Petri dish" of plant tissue culture) containing autoclaved sand. After 1 month, seedlings
were inoculated with C. protuberata of either the wild type (Wt), the virus-free (VF) or
the hygromycin-resistant isolate newly infected with the virus through hyphal
anastomosis (An), by pipetting 50-100 spores (suspended in a 0.035% agar solution)
between the crown and first leaf of each plant. Non-symbiotic (NS) controls were not
inoculated with fungal spores. To assess the percentage of plants successfully infected
with the fungal isolates, approximately 40 plants from each treatment (Wt, VF, An and
NS) were washed and surface-sterilized by agitation in 70% ethanol for 5 min, 2%
sodium hypochlorite for 20 min, 70% ethanol for 5 min, then rinsed in sterile water,
dissected into sections and plated on 0.1 X PD Agar medium. This confirmed that non-
symbiotic plants were truly endophyte-free, while 100% of the plants inoculated with
each of the three different C. protuberata isolates were colonized. The time between
inoculation and application of heat stress was 2-3 weeks. Root zones were heated using
thermal soil simulators constructed as described in Redman et al. (1). Rhizosphere
temperature was maintained at 65°C for 10 hours and 37ºC for 14 hours/day for 14 days
under green-house conditions.

Report 1136237 Márquez et al. SOM 7
For the heat stress experiments using tomato (Solanum lycopersicon, var Rutgers), seeds
were surface sterilized with hypochlorite as described above. One week old seedlings
were inoculated with one of the three isolates of C. protuberata: the wild type (Wt), the
virus-free (VF) or the hygromycin-resistant isolate newly infected with CThTV by
anastomosis (An), by incubating the roots and lower 1/3 portion of the stems
hydroponically in a solution of 0.035% agar containing 105 conidiospores per ml. Non-
symbiotic (NS) controls were incubated in 0.035% agar alone, without fungal spores.
After three days of incubation, the spore solution was discarded and replaced with de-
ionized H2O. The endophyte colonization was continued for 3 more days before assessing
the inoculation success and transferring the seedlings to the thermal soil simulators
described in Redman et al. (1). The endophyte-free status of the NS plants as well as the
infection success of Wt, VF and An plants was assessed as described above by plating
sections of approximately 40 surface sterilized plants per treatment on 0.1 X PDA. Non-
symbiotic plants were truly endophyte-free, while 87-94% of the plants inoculated with
each of the three different C. protuberata isolates were colonized. The endophyte was
recovered from colonized plants from all parts of the plants: roots, stems and leaves. The
seedlings were allowed to acclimate for 2-10 days in the thermal soil simulators before
applying heat stress. The time between inoculation and application of heat stress was 2-3
weeks. The variation in time was due to differences in growth rates among the different
batches of seedlings; plants were used at the same developmental stage rather than the
same age. Rhizosphere temperature was maintained at 65°C for 10 hours and ambient
temperature for 14 hours/day for 14 days under green-house conditions.
Osmolyte concentrations: One half gram of fresh plant tissue was cut into pieces of
uniform size on a glass platform suspended over ice to keep the tissue cold. Plant tissue
was transferred to a microfuge tube containing 1 ml ice cold sterile deionized water.
Tissue was ground using a small hand pestle, boiled for 30 minutes and cooled on ice for
10 minutes. Prior to determining osmolyte content, samples were brought to room
temperature, vortexed quickly and plant debris removed by centrifugation for 30sec at
7000 rpm in a benchtop microfuge. Osmotic content was determined on the supernatant

Report 1136237 Márquez et al. SOM 8
using an Advanced Micro Osmometer model 3300 (Advanced Instruments, Norwood,
MA).
Test for reactive oxygen species (ROS) generation: Panic grass and tomato plants were
non-symbiotic (NS) or symbiotically colonized with C. protuberata fungal isolates:
virus-containing wild type (Wt) or virus-free (VF) as indicated above for the heat stress
experiments. Plants were maintained at 22°C (- Stress) or exposed to heat stress using the
thermal soil simulators as indicated above for 12 hours at 50 - 65°C and 12 hours at 22°C
(+ Stress) for 10-14 days. Three to five mm wide plant leaf disks per plant were exposed
to 1µM of the herbicide Paraquat (N, N'-Dimethyl-4, 4'-bipyridinium dichloride)
(Syngenta,Basel). In the presence of light, Paraquat is reduced to its radical ion by
electron transfer from plant photosystem-I; the ion is subsequently oxidized by molecular
oxygen, resulting in the generation of superoxide ions and photobleaching (10). Exposure
to Paraquat was maintained for 24-48 hours in the presence of 24 hours of light. Three
plants per treatment were used in each assay and the assays were repeated 3 times for a
total of 9 plants per treatment. Bleaching of plant tissue indicated oxidation due to the
presence of ROS. A lack of ROS indicates successful scavenging of the ROS generated
by the Paraquat treatment.
Supplementary References
1. R. S. Redman, K.B. Sheehan, R. G. Stout, R. J. Rodriguez, J. M. Henson. Science
298, 1581 (2002).
2. J. A. Dodds, T. J. Morris, R. L. Jordan, Ann. Rev. Phytopathol. 22, 151 (1984).
3. M. A. Frohman, M. K. Dush, G. R. Martin, Proc. Natl. Acad. Sci. U.S.A. 85, 8998
(1988).
4. S. F. Altschul et al., Nucleic Acids Res. 25, 3389 (1997).
5. G. S. Beckler, Promega Notes 39, 12 (1992).
6. G. M. Church, W. Gilbert, Proc. Natl. Acad. Sci. U.S.A. 81, 1991 (1984).
7. M. J. Roossinck, P. S. White, in Plant Virology Protocols, G. D. Foster, S. C. Taylor,
Eds. (Humana Press, Totowa, NJ, 1998), pp. 189–196.
8. R.S. Redman et al., Mol. Plant-Microbe Interact. 11, 969 (1999).

Report 1136237 Márquez et al. SOM 9
9. J. M. Lorang et al., Appl. Environ. Microbiol. 67, 1987 (2001).
10. K. C. Vaughn, S. O. Duke, Plant Cell Environ. 6, 13 (1983).

Report 1136237 Márquez et al. SOM 10
Figure captions
Fig. S1. Double stranded RNA extractions of field samples of Curvularia protuberata,
isolated from individual plants of the panic grass, Dichanthelium lanuginosum, growing
in geothermic soils in Yellowstone National Park. Four out of the 10 isolates containing
the virus also show a subgenomic RNA band <1 kb.
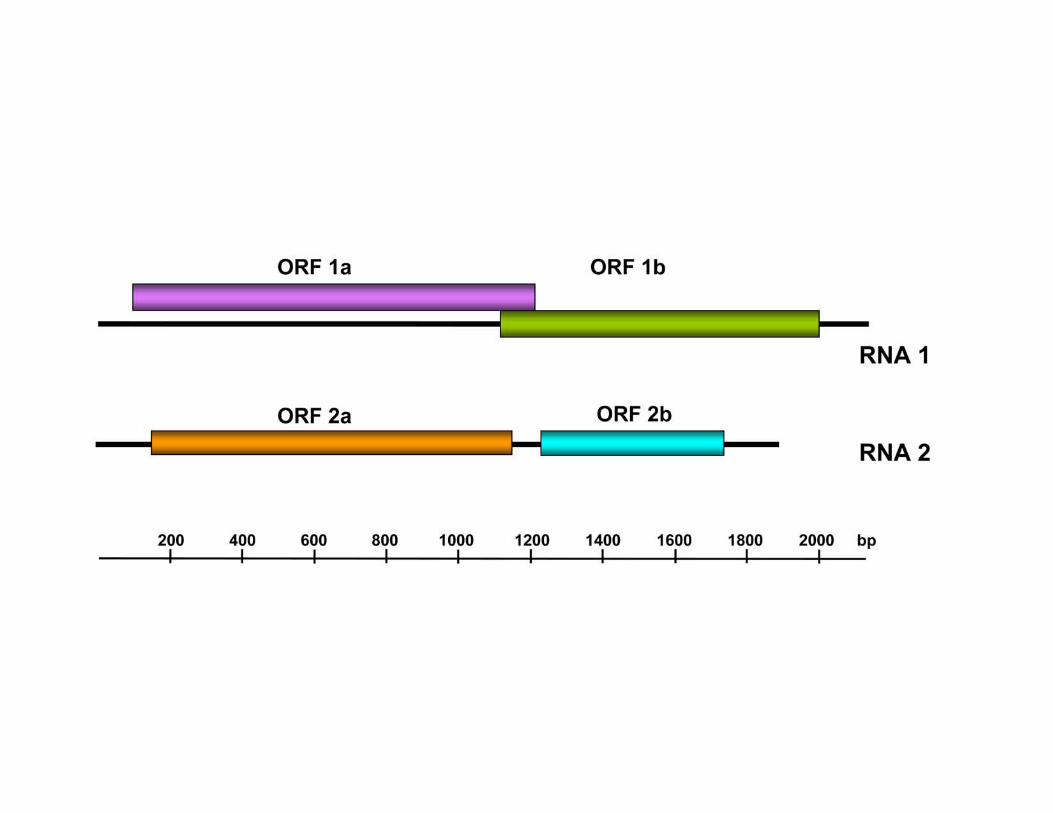
Fig. S2. Genome organization of Curvularia thermal tolerance virus (CThTV). The first
open reading frame, ORF 1b, in RNA 1 corresponds to the putative RNA dependent RNA
polymerase.
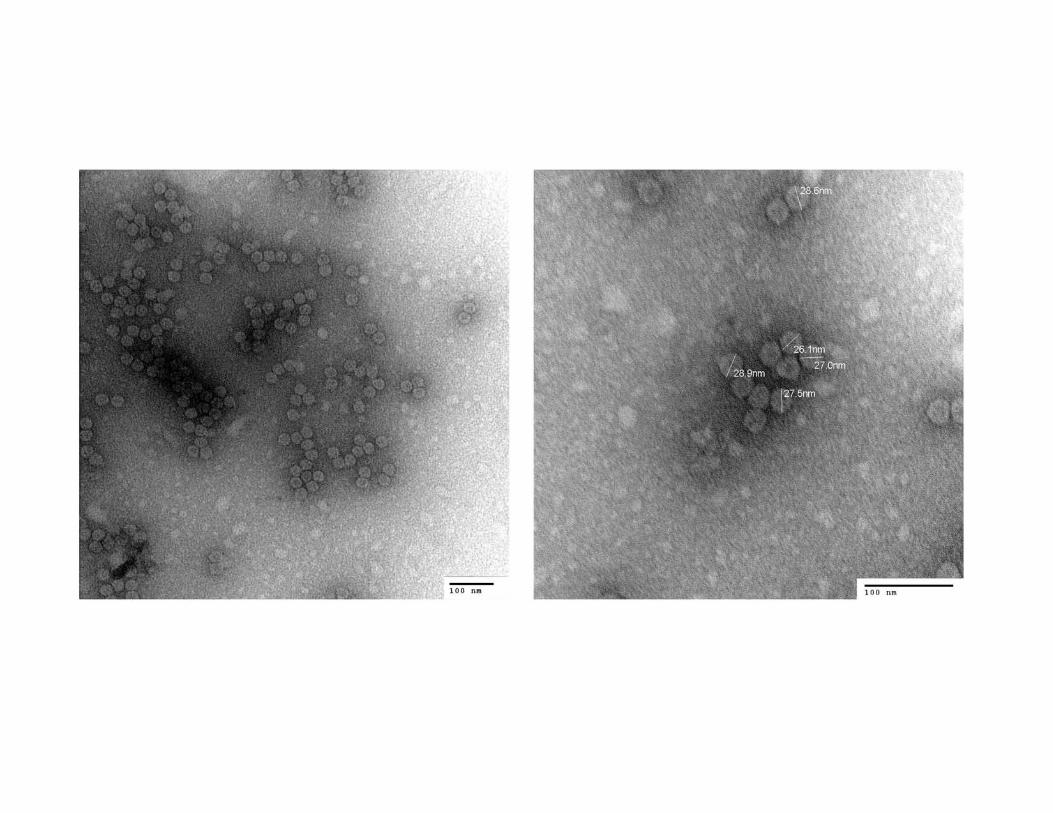
Fig. S3. Transmission electron micrographs of the Curvularia thermal tolerance virus
(CThTV). Direct magnifications were 100,000 X (A) and 200,000 X (B) at 75kV. Virus
particles are spherical and approximately 27 nm in diameter, similar to those of other
fungal viruses.
Fig. S4. Alignment of the rDNA ITS1-5.8S-ITS2 of the CThTV infected wild type (Wt)
and the virus-free (VF) isolates of the fungal endophyte C. protuberata.
Fig. S5. PCR analysis of Curvularia protuberata and C. ineaqualis with simple sequence
repeat (SSR) primers. Lanes 1 and 2 are C. protuberata virus-containing wild type (Wt)
and virus-free (VF) isolates, lanes 3 and 4 are C. ineaqualis isolate 5851, and negative
control (no DNA) amplified with the primer 5'-AGGAGGAGGAGGAGG-3',
respectively. Lanes 5-8 represent C. protuberata Wt, C. protuberata VF, C. ineaqualis
isolate 5851, and negative control (no DNA) amplified with the primer 5'-
GTCGTCGTCGTCGTC-3'. Lanes M are 100bp molecular weight standards.
Fig. S6. Effect of colonization with virus-free and CThTV infected isolates of C.
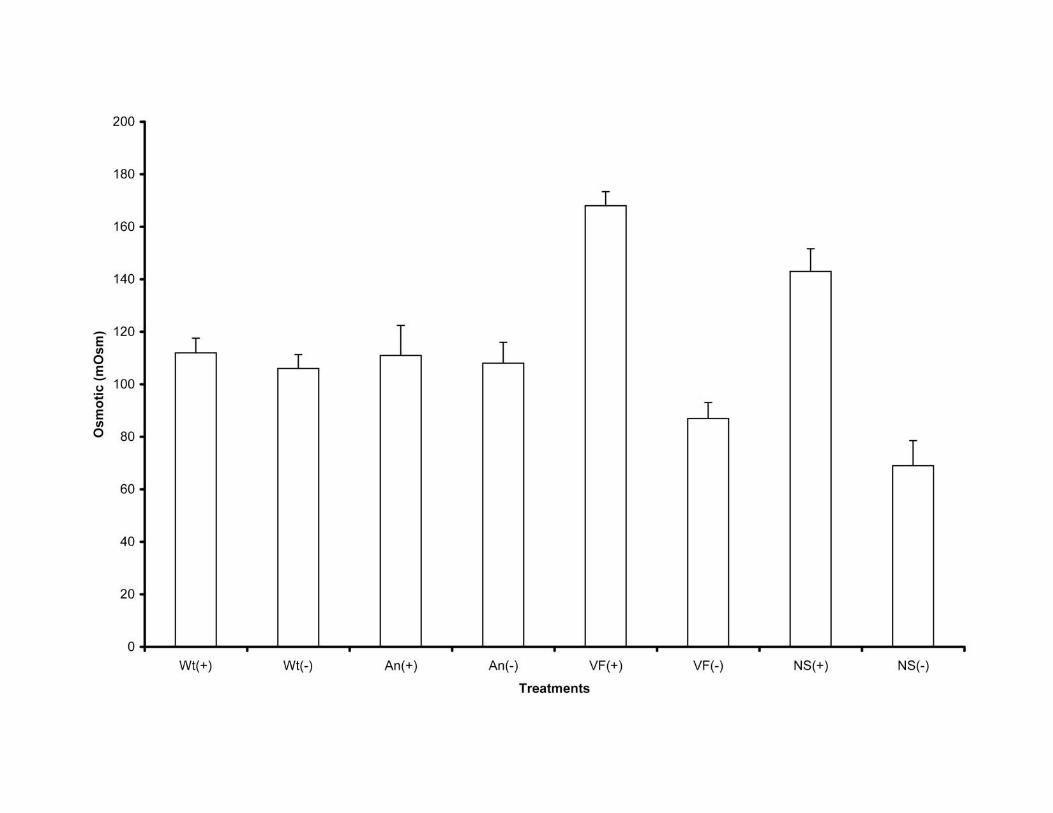
protuberata on osmolyte concentration (mOsm) in D. lanuginosum plants in the presence
and absence of heat stress. Error bars represent standard deviation values that ranged
from 5.3 to 11.4. ANOVA single factor analysis indicated that there were no significant

Report 1136237 Márquez et al. SOM 11
differences in osmotic concentration before and after heat induction in plants colonized
with the CThTV infected wild type isolate of the fungus (Wt) (P= 0.10) and isolate newly
infected with CThTV by hyphal anastomosis (An) (P= 0.66). There were significant
differences before and after heat induction in nonsymbiotic (NS) (P= 4.2e-05) and plants
colonized with the virus-free isolate of the fungus (VF) (P= 1.0e-07). Assays were
repeated a minimum of three times.

Report 1136237 Márquez et al. SOM 13
Table S1. Test for reactive oxygen species (ROS) generation in tropical panic grass,
Dichanthelium lanuginosum, and tomato (Solanum lycopersicon, var Rutgers) plants in
the presence and absence of temperature stress (see text for experiment details). The
numbers indicate how many plants out of the total (N=9) showed at least one bleached
leaf after exposure to Paraquat. Panic grass and tomato plants were non-symbiotic (NS)
or symbiotically colonized with Curvularia protuberata fungal isolates: virus-containing
wild type (Wt) or virus-free (VF) (see text for description of isolates).
Treatment Panic grass Tomato
- Stress + Stress - Stress + Stress
NS 0/9 8/9 0/9 8/9
Wt 0/9 0/9 0/9 1/9
VF 0/9 1/9 1/9 2/9


Report 1136237 Márquez et al. SOM 12
Figure S4
---------+---------+---------+---------+---------+---------+---------+---------+
Wt TTGGTCATTTAGAGGAAGTAAAAGTCGTAACAAGGTCTCCGTAGGTGAACCTGCGGAGGGATCATTACACAATAACATAT
VF TTGGTCATTTAGAGGAAGTAAAAGTCGTAACAAGGTCTCCGTAGGTGAACCTGCGGAGGGATCATTACACAATAACATAT
---------+---------+---------+---------+---------+---------+---------+---------+
Wt GAAGGCTGTACGCCGCTGCGCCCCCCGGGCCAGTTGGCTGAGGCTGGATTATTTATTACCCTTGTCTTTTGCGCACTTGT
VF GAAGGCTGTACGCCGCTGCGCCCCCCGGGCCAGTTGGCTGAGGCTGGATTATTTATTACCCTTGTCTTTTGCGCACTTGT
---------+---------+---------+---------+---------+---------+---------+---------+
Wt TGTTTCCTGGGCGGGTTCGCCCGCCTCCAGGACCACACCATAAACCTTTTTTATGCAGTTGCAATCAGCGTCAGTACAAC
VF TGTTTCCTGGGCGGGTTCGCCCGCCTCCAGGACCACACCATAAACCTTTTTTATGCAGTTGCAATCAGCGTCAGTACAAC
---------+---------+---------+---------+---------+---------+---------+---------+
Wt AAATGTAAATCATTTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATACGT
VF AAATGTAAATCATTTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATACGT
---------+---------+---------+---------+---------+---------+---------+---------+
Wt AGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCAAAGGGCATGCCTG
VF AGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCAAAGGGCATGCCTG
---------+---------+---------+---------+---------+---------+---------+---------+
Wt TTCGAGCGTCATTTGTACCCTCAAGCTTTGCTTGGTGTTGGGCGTTTTTTGTCTTTGGTTTGCCAAAGACTCGCCTTAAA
VF TTCGAGCGTCATTTGTACCCTCAAGCTTTGCTTGGTGTTGGGCGTTTTTTGTCTTTGGTTTGCCAAAGACTCGCCTTAAA
---------+---------+---------+---------+---------+---------+---------+---------+
Wt ACGATTGGCAGCCGGCCTCCTGGTTACGCAGCGCAGCACATTTTTGCGCTTGCAATCAGCAAGAGGGCGGCACTCCATCA
VF ACGATTGGCAGCCGGCCTCCTGGTTACGCAGCGCAGCACATTTTTGCGCTTGCAATCAGCAAGAGGGCGGCACTCCATCA
---------+---------+---------+---------+---------+---------+---------+---------+
Wt AGACTCCTTCTCACGTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAA
VF AGACTCCTTCTCACGTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAA