
Novel Point Mutations, Deletions, and Polymorphisms in theCathepsin C Gene in Nine Families from Europe and NorthAfrica with Papillon±LefeÁvre Syndrome
Caroline LefeÁvre, Claudine Blanchet-Bardon,* Florence Jobard, Bakar Bouadjar,² Jean-FrancËois Stalder,³Susan Cure,§ Aude Hoffmann, Jean-FrancËois Prud'homme,¶ and Judith FischerCentre National de GeÂnotypage, Evry, France; *Department of Dermatology, Saint Louis Hospital, Paris, France; ²Department of Dermatology,
CHU of Bab-El-Oued, Algiers, Algeria; ³Department of Dermatology, CHU Nantes, France; §Genoscope, CNS, Evry, France; ¶GeÂneÂthon, Evry,
France
Papillon±LefeÁvre syndrome is an autosomal recessivedisorder characterized by palmoplantar keratoderma,periodontitis, and premature loss of dentition.Mutations in the CTSC gene that encodes cathepsinC have been described in families affected withPapillon±LefeÁvre syndrome. Cathepsin C is the leastunderstood of the lysosomal cysteine proteases; it hasbeen reported to participate in both intracellular andextracellular cleavage of proteins and activation ofserine proteases in immune and in¯ammatory cells.We report here eight new mutations in Papillon±LefeÁvre syndrome families: four deletions and fourpoint mutations, including a missense mutation in
the propeptide chain that could help elucidate struc-ture±function relationships in this protein. We alsofound that the 458C > T mutation, ®rst reported intwo families by Hart et al (2000c), was a neutral poly-morphism in our families, as suggested by Allendeet al (Cathepsin C gene: ®rst compound hetero-zygous patient with Papillon±LefeÁvre syndrome andnovel symptomless mutation. Hum Mutat 17:152±153,2001). Key words: cathepsin C mutations/chromosome11q14/palmoplantar keratoderma/Papillon±LefeÁvre syn-drome/periodontitis. J Invest Dermatol 117:1657±1661,2001
Inherited palmar and plantar keratodermas (PPK) are aheterogeneous group of skin diseases characterized byhyperkeratosis on the palms and soles and potentiallyectodermal or nonectodermal features, such as hair and nailanomalies, hearing loss, and mental retardation (Kelsell and
Stevens, 1999). Defects in various structural proteins such askeratins, connexins, and desmosomal proteins have been identi®edas the cause of diverse variants of PPK, most of which exhibitautosomal dominant transmission. Keratins, the major intermediate®laments of keratinocytes, and loricrin, the major precursor proteinof the corni®ed cell envelope, are mutated in a number of diverseepidermal disorders (Kelsell and Stevens, 1999). The desmosomalcell±cell junctions consist of membrane proteins such as desmoglein1 and desmoplakin, which are mutated in nonsyndromic striatePPK (Rickman et al, 1999) and syndromic PPK with cardiomyo-pathy and woolly hair (Norgett et al, 2000). Mutations in othermolecules involved in ®lament anchorage such as plakoglobin orplakophilin have been shown to underlie Naxos disease (McKoy etal, 2000) and ectodermal dysplasia/skin fragility syndrome(McGrath et al, 1997), respectively. Furthermore, defects of gap-junction proteins (connexins 26, 30, and 31) have been found to beinvolved in a spectrum of epidermal diseases (Kelsell and Stevens,1999). Recently, mutations in a secreted protein SLURP-1, which
is a member of the Ly-6/uPAR superfamily, were identi®ed in Malde Meleda (Fischer et al, 2001).
Papillon±LefeÁvre syndrome (PLS, MIM 245000) is a rareautosomal recessive genodermatosis with an estimated incidenceof 1±4 per million (Gorlin et al, 1964). It was ®rst described by twoFrench dermatologists (Papillon and LefeÁvre, 1924) who mistook itfor Mal de Meleda (MIM 248300). Both diseases are diffuse PPKwith disseminated keratotic lesions, in particular on the elbows andknees. PLS, however, is typically associated with premature loss ofdeciduous and permanent teeth and a severe periodontitis. Gingivalin¯ammation, pocket formation, and bleeding of the gums developshortly after tooth eruption, but disappear after exfoliation(Haneke, 1979).
The PLS gene was localized to chromosome 11q14 byhomozygosity mapping (Fischer et al, 1997; Laas et al, 1997). In1999, two different groups (Hart et al, 1999; Toomes et al, 1999)reported PLS to be due to mutations in the gene encodingcathepsin C, a lysosomal cysteine protease of the papain type alsoknown as dipeptidyl aminopeptidase (CTSC and DPP1, MIM602365). Shortly thereafter, Hart's group suggested that mutationsin this gene could also lead to Haim±Munk syndrome (HMS, MIM245010), which is characterized by typical PLS symptoms as well ashand deformities such as arachnodactyly, acroosteolysis, andonychogryposis, in descendants of a religious isolate fromCochin, India (Hart et al, 2000a). This group further reportedthat a CTSC mutation was associated with prepubertal periodon-titis in a consanguineous family from Jordan with no other PLS-type symptoms (Hart et al, 2000b). Hart has subsequently noted thatadditional symptoms could be due to homozygosity at other loci inthese consanguineous families, and might not be related to the
Manuscript received April 30, 2001; revised August 22, 2001; acceptedfor publication August 29, 2001.
Reprint requests to: Dr. Judith Fischer, 2 rue Gaston CreÂmieux, CP5721, 91057 Evry Cedex, France. Email: ®[email protected]
Abbreviations: PLS, Papillon±LefeÁvre syndrome; PPK, palmoplantarkeratoderma; CTSC, cathepsin C; HMS, Haim±Munk syndrome.
0022-202X/01/$15.00 ´ Copyright # 2001 by The Society for Investigative Dermatology, Inc.
1657
cathepsin C mutations (Hart et al, 2000c). We have analyzed ninefamilies from Algeria, France, Martinique, Morocco, and theNetherlands with PLS, and found eight novel mutations includingfour deletions predicted to cause frameshifts leading to prematuretermination codons. We also found that a mutation (458C > T)leading to T153I previously described in two families (Hart et al,2000c) was probably a neutral polymorphism, as suggested byAllende et al (2001).
MATERIALS AND METHODS
Subject We collected whole blood samples after obtaining writteninformed consent from 18 patients and 29 unaffected members of ninefamilies including six consanguineous kindreds. Five of these ninefamilies have been previously reported (Fischer et al, 1997). The familieswere from Algeria (two), France (four), Martinique (one), Morocco(one), and the Netherlands (one). All affected family members presentedsymptoms typical of PLS with no evidence of hand deformities or othersymptoms reported in HMS.
Mutation analysis We ®rst designed oligonucleotide primers ¯ankingexons 1 and 2 using the Primer3 program for an optimal annealingtemperature of 58°C from the sequence U79415, which was described asthe speci®c genomic sequence of CTSC (Rao et al, 1997). We de®ned
the forward primer 5¢-CAGGGGTAACATGCAAAG-3¢ and the reverseprimer 5¢-CCCTTTACAACTGATGCAG-3¢ for the ampli®cation ofexon ``2'' (which corresponds now to exon 7), according to the reportedsequence of CTSC with two exons (Rao et al, 1997). We foundmutations in two families in this exon, but attempts to amplify exon ``1''were repeatedly unsuccessful. As demonstrated by Toomes et al (1999),exon 1 is actually divided into six exons. We used the published primersof Toomes et al (1999) for ampli®cation of exons 2±6.
Based on the speci®c genomic sequence (AC011088) and by compar-ing the complete cDNA sequence (U79415) with the HTGS (HighThroughput Genomic Sequences) database using the BLAST program,we de®ned primers for the ampli®cation of exon 1 (forward primer 5¢-GTCGGCTTCCTGGTAATTCT-3¢ and reverse primer 5¢-CGAATC-CAGTCAAGATGCTC-3¢). The polymerase chain reaction (PCR) wasperformed in a 50 ml volume containing 100 ng of genomic DNA (in10 ml) using standard procedures (Vignal et al, 1993). After an initialdenaturation step at 96°C for 5 min, Taq polymerase was added at 94°C(hot start) and 35 cycles of ampli®cation were performed consisting of30 s at 94°C, 40 s at the optimal annealing temperature (between 55°Cand 61°C), and a 10 min terminal elongation step.
One to two microliters of puri®ed PCR products (corresponding to20±40 ng of template) were added to 1 ml of sense or antisense primer(10 mM) and 3 ml of BigDye terminator mix (PE Applied Biosystems).The linear ampli®cation consisted of an initial denaturation step at 96°C,25 cycles of 10 s of denaturation at 96°C, a 5 s annealing step (55°C±
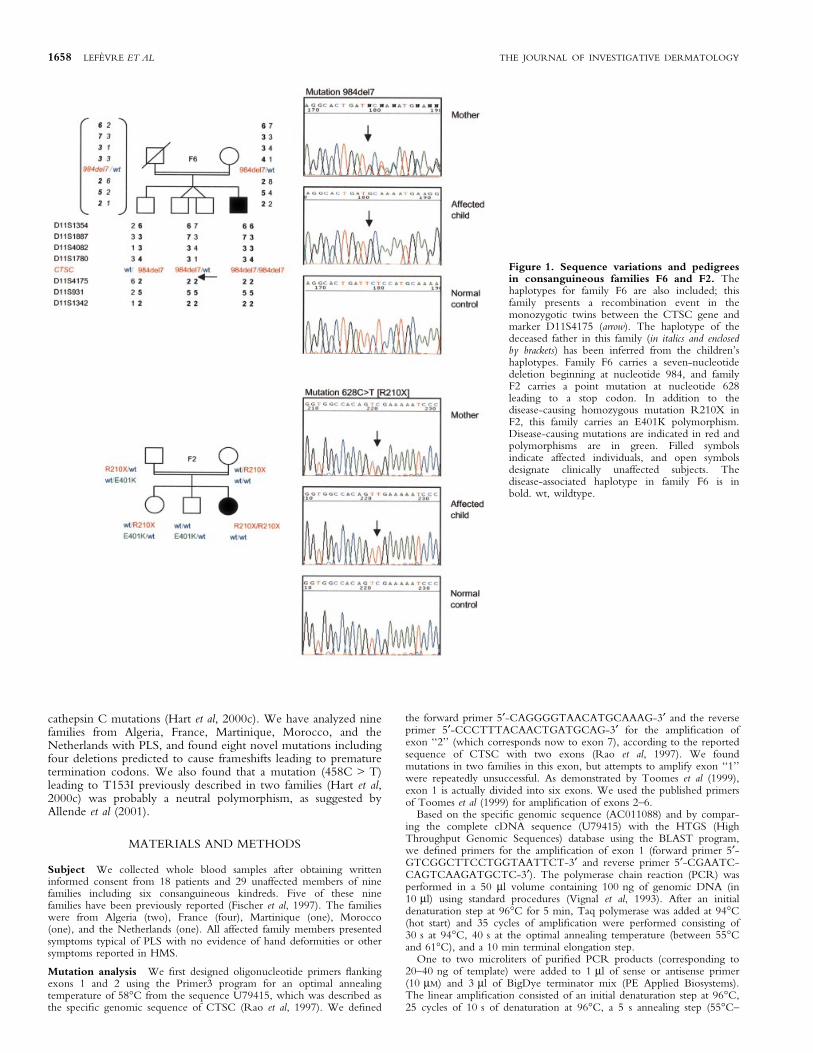
Figure 1. Sequence variations and pedigreesin consanguineous families F6 and F2. Thehaplotypes for family F6 are also included; thisfamily presents a recombination event in themonozygotic twins between the CTSC gene andmarker D11S4175 (arrow). The haplotype of thedeceased father in this family (in italics and enclosedby brackets) has been inferred from the children'shaplotypes. Family F6 carries a seven-nucleotidedeletion beginning at nucleotide 984, and familyF2 carries a point mutation at nucleotide 628leading to a stop codon. In addition to thedisease-causing homozygous mutation R210X inF2, this family carries an E401K polymorphism.Disease-causing mutations are indicated in red andpolymorphisms are in green. Filled symbolsindicate affected individuals, and open symbolsdesignate clinically unaffected subjects. Thedisease-associated haplotype in family F6 is inbold. wt, wildtype.
1658 LEFEÁVRE ET AL THE JOURNAL OF INVESTIGATIVE DERMATOLOGY

61°C), and a 4 min extension step at 60°C. The reaction products werepuri®ed and sequenced on an Applied Biosystems Sequencer 3700. Bothstrands from all patients, all family members, and controls weresequenced for the entire coding region and the intron±exon boundaries.
RESULTS
Genotyping results in all nine families showed linkage to chromo-some 11q14, con®rming the genetic homogeneity of this disorder.The maximum combined lod score was 6.53 at D11S931 at Q = 0.Haplotype analysis showed no evidence for a common haplotype inthese families, which have different geographic and ethnic origins.In family F6, which was from a small French island, a consanguinityof the seventh degree was reported. The haplotype in the affectedindividual includes a homozygous deletion (984del7) in the CTSCgene (Fig 1) and shows loss of homozygosity for markerscentromeric to CTSC. Interestingly, the same homozygoushaplotype for CTCS telomeric markers was also found in hisnonaffected monozygotic twin siblings. Haplotype analysis of thetwins showed that a recombination event in the maternalchromosome had occurred between CTSC and D11S4175,which re®ned the gene-containing interval to a 1 cM regionbetween D11S1780 and D11S4175. The new order of markers isbased on the physical map of the Golden Path database (http://genome.ucsc.edu) and differs from that originally reported (Fischeret al, 1997). Linkage analysis in this family for markers telomeric toCTSC produced negative lod scores. Sequencing of the PCR-ampli®ed exons 1±7 revealed 11 different mutations in our families:four point mutations (H127P, R272P, R339C, W429C), threenonsense mutations (C24X, Y32X, R210X), and four deletions(711del14, 984del7, 1056delT, 1141delC) (Table I). Cathepsin Cmutations were found in all the families, and were homozygous inthe six consanguineous families, whereas the nonconsanguineousfamilies all presented heterozygous mutations in this gene.Sequence variations in two of these families are presented inFig 1, and pedigrees and genotypes of the other affected familiesare shown in Fig 2. A total of 39 control alleles were screened formutations in the cathepsin C gene. The previously reportedR210X and the R272P mutations (Toomes et al, 1999) werepresent in an Algerian family and in two families from, respectively,France and the Netherlands. Another previously reported variant,R339C (Hart et al, 1999; Toomes et al, 1999), was found in onefamily from Martinique. One of the new missense mutations,W429C, occurs in the same codon as that in which both Toomes etal (1999) and Hart et al (1999) reported a mutation leading to a stopcodon, W429X.
We also found three different polymorphisms in three families(F2, F8, and F9) in exons 3 and 7, and in intron 4 (Fig 1,Table II). In a consanguineous Algerian family F2 with homo-zygous R210X mutations in affected individuals, a 1202G > Apolymorphism, which changes glutamic acid to lysine (E401K), was
present in unaffected individuals (one of the parents and twononaffected children). A 458C > T polymorphism, which changesthreonine to isoleucine (T153I), was present in one parent in eachof two French families (F8, F9), but not in all the affectedindividuals. Ninety-®ve control individuals were screened for thissequence variation (458C > T) in exon 3; half of them (47) werepopulation-matched controls from North Africa (Algeria andTunisia) and the other half (48) were from France. There were66 individuals homozygous for the C allele, two individualshomozygous for the T allele, and 27 subjects heterozygous forC/T. The allele frequencies are therefore 0.837 for the C and 0.163for the T. In family F8, both probands inherited a previouslyreported 815C > G mutation, R272P (Toomes et al, 1999; Hart etal, 2000c), from his father and a 1141delC mutation in exon 7 aswell as the 458C > T polymorphism probably from his mother,who was not available for analysis. In family F9, the proband washeterozygous for a deletion inherited from his father (1056delTleading to a frameshift and a premature stop codon at amino acid352) and a point mutation (W429C) inherited from his mother. Asthe mother was heterozygous for the 458C > T polymorphism(T153I, Fig 2), not transmitted to her affected son, and as she wasnot affected herself, one can conclude that the 458C > T changewas not disease-causing in this family.
DISCUSSION
Identi®cation of eight new mutations in families from Europe,Martinique, and North Africa brings the total number of reportedmutations to 36. Thirty-three of these mutations lead to PLSexclusively, one has been suggested to cause either PLS orprepubertal periodontitis (Y347C in exon 6), and two differentmutations in codon 286, also in exon 6, could lead either to HMS(Q286R) or PLS (Q286X) (Toomes et al, 1999; Hart et al, 1999,2000a, c; Allende et al, 2001; Nakano et al, 2001; Zhang et al,2001). The 458C > T polymorphism we found in two Frenchfamilies is the same as the mutation reported by Hart et al (2000c) asdisease-causing and by Allende et al (2001) as symptomless. Wedemonstrated the presence of this polymorphism in population-matched controls from France and North Africa in which we founda frequency of 16.3% for the T allele.
Cathepsin C is a lysosomal cysteine protease of the papain type(Paris et al, 1995). It is the least understood of the cysteine proteasesand differs from most other members of this family (cathepsin B, H,L, and S) in both structure and substrate preference. The protein isprocessed into a mature proteolytically active enzyme consisting ofa heavy chain, a light chain, and a propeptide that remainsassociated with the active enzyme (Wolters et al, 1998; Cigic et al,2000). Whereas the other cathepsins are monomers, CTSC is a200,000 kDa tetramer with four identical subunits, each composedof the three different polypeptide chains. All but three of themissense mutations were found in the heavy chain of the protein,
Table I. Mutations in cathepsin C in PLS families from Europe and North Africaa
Family Origin Consanguinity Site Mutation Effect
1 Morocco + Exon 1 72C > A C24X2 Algeria + Exon 4 628C > T R210X3 Algeria + Exon 5 711del14 Frameshift, premature stop codon at aa 2494 Holland + Exon 6 815G > C R272P5 Martinique + Exon 7 1015C > T R339C6 France + Exon 7 984del7bp Frameshift, premature stop codon at aa 330
Ile d'Yeu7 France ± Exon 1 96T > G Y32X
± Exon 3 380A > C H127P8 France ± Exon 6 815G > C R272P
± Exon 7 1141delC Frameshift, premature stop codon at aa 3949 France ± Exon 7 1056delT Frameshift, premature stop codon at 352
± Exon 7 1287G > C W429C
aNovel mutations are in boldface.
VOL. 117, NO. 6 DECEMBER 2001 NOVEL CATHEPSIN C MUTATIONS IN PLS 1659

which is the most conserved region of the protein across species andis suspected to be important for enzyme activity and speci®city(Wolters et al, 1998). Two missense mutations (Q252L andW429C) are in amino acids that are thought to be critical in theactive site of cysteine proteases in the heavy and light chains,respectively. Two other mutations, H127P and W39S, are in thepropeptide chain, con®rming the importance of this peptide inmaintaining the structural stability of the mature enzyme (Cigic etal, 2000). The enzyme can act as both an exopeptidase and anendopeptidase (Wolters et al, 1998). It has dipeptidyl aminopepti-dase activity with broad speci®city and a transferase activity atalkaline and neutral pH and is activated by chloride ion, whichseems to bind to the active site (Cigic and Pain, 1999; Wolters et al,2000). Cathepsin C is implicated in the intracellular andextracellular cleavage of proteins, and the activation of serineproteases such as leukocyte elastase, cathepsin G, and granzymes Aand B in immune and in¯ammatory cells. Cathepsin C can alsoactivate neuraminidase, factor XIII (a transglutaminase), and itcleaves endoproteolytically some proteins of the extracellularmatrix including collagen types I, III, and IV, and ®bronectin(Wolters et al, 1998, 2000).
The physiologic role of cathepsin C is still unclear. Expressed athigh levels in lung, kidney, and placenta (Rao et al, 1997), it is alsopresent in many other organs including the epithelial regionsaffected by PLS, the oral keratinized gingiva (Hart et al, 1999), and
cells involved in immune and in¯ammatory reactions such asneutrophils, alveolar macrophages, mast cells, and cytotoxiclymphocytes. De®ciency of cathepsin C in mutant mice had noobvious phenotypic effects, but their cytotoxic lymphocytes couldnot induce granzyme A and B mediated apoptosis in target cells dueto a defect in the processing of these granzymes (Pham et al, 1997).
Electronic database GenBank accession numbers: Completecds, U79415; CTSC mRNA NM_001814; AC011088, BACclone RP11±292E14, complete genomic sequence.
BLAST (http://www.ncbi.nlm.nih.gov/BLAST)Primer3 (http://www.genome.wi.mit.edu/cgi-bin/primer/
primer3.cgi)
Figure 2. Pedigrees and genotypes in familiesF1, F3, F4, F5, F7, F8, and F9. Symbols arethe same as in Fig 1. Double horizontal linesbetween parents indicate consanguineousmarriages. NA: not available.
Table II. Polymorphisms in the cathepsin C gene infamilies from France and Algeria
Family Origin Site Polymorphism Effect
2 Algeria Exon 7 1202G > A E401K8 France Exon 3 458C > T T153I9 France Exon 3 458C > T T153I8 France Intron 4 IVS5 + 38C > T9 France Intron 4 IVS5 + 38C > T
1660 LEFEÁVRE ET AL THE JOURNAL OF INVESTIGATIVE DERMATOLOGY

We would like to acknowledge the technical support of the GeÂneÂthon DNA bank.
We wish to thank Dr. Steijlen, the clinicians, and the families who participated.
This study was supported by the Center National de GeÂnotypage (CNG), the
Association FrancËaise contre les Myopathies (AFM), and GeÂneÂthon.
REFERENCES
Allende LM, Garcia-Perez MA, Moreno A, et al: Cathepsin C gene: ®rst compoundheterozygous patient with Papillon±LefeÁvre syndrome and novel symptomlessmutation. Hum Mutat 17:152±153, 2001
Cigic B, Pain RH: Location of the binding site for chloride ion activation ofcathepsin C. Eur J Biochem 264:944±951, 1999
Cigic B, Dahl SW, Pain RH: The residual pro-part of cathepsin C ful®lls the criteriarequired for an intramolecular chaperone in folding and stabilizing the humanproenzyme. Biochemistry 39:12382±12390, 2000
Fischer J, Blanchet-Bardon C, Prud'homme J-F, et al : Mapping of Papillon±LefeÁvresyndrome to the chromosome 11q14 region. Eur J Hum Genet 5:156±160, 1997
Fischer J, Bouadjar B, Heilig R, et al: Mutations in the gene encoding SLURP-1 inMal de Meleda. Hum Mol Genet 10:875±880, 2001
Gorlin RJ, Sedano H, Anderson VE: The syndrome of palmo-plantar hyperkeratosisand premature periodontal destruction of the teeth. A clinical and geneticanalysis of the Papillon±LefeÁvre syndrome. J Pediatr 65:896±908, 1964
Haneke E: The Papillon±LefeÁvre syndrome: keratosis palmoplantaris withperiodontopathy. Report of a case and review of the cases in the literature.Hum Genet 51:1±35, 1979
Hart TC, Hart PS, Bowden DW, et al: Mutations of the cathepsin C gene areresponsible for Papillon±LefeÁvre syndrome. J Med Genet 36:881±887, 1999
Hart TC, Hart PS, Michalec MD, et al: Haim±Munk syndrome and Papillon±LefeÁvresyndrome are allelic mutations in cathepsin C. J Med Genet 37:88±94, 2000a
Hart TC, Hart PS, Michalec MD, et al: Localisation of a gene for prepubertalperiodontitis to chromosome 11q14 and identi®cation of a cathepsin C genemutation. J Med Genet 37:95±101, 2000b
Hart PS, Zhang Y, Firatli E, et al: Identi®cation of cathepsin C mutations inethnically diverse Papillon±LefeÁvre syndrome patients. J Med Genet 37:927±932, 2000c
Kelsell DP, Stevens HP: The palmoplantar keratodermas: much more than palms andsoles. Mol Med Today 5:107±113, 1999
Laas MW, Hennies HC, Preis S, et al: Localisation of a gene for Papillon±LefeÁvresyndrome to chromosome 11q14-q21 by homozygosity mapping. Hum Genet101:376±382, 1997
McGrath JA, McMillan JR, Shemanko CS, et al: Mutations in the plakophilin 1 generesult in ectodermal dysplasia/skin fragility syndrome. Nat Genet 17:240±244,1997
McKoy G, Protonotarios N, Crosby A, et al: Identi®cation of a deletion inplakoglobin in arrhythmogenic right ventricular cardiomyopathy withpalmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355:2119±2124, 2000
Nakano A, Nomura K, Nakano O, et al: Mutation report: Papillon±LefeÁvresyndrome: mutations and polymorphisms in the cathepsin C gene. J InvestDermatol 116:339±343, 2001
Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al: Recessive mutation in desmoplakindisrupts desmoplakin±intermediate ®lament interactions and causes dilatedcardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 9:2761±2766,2000
Papillon MM, LefeÁvre P: Deux cas de keÂratodermie palmaire et plantaire symeÂtriquefamiliale (maladie de Meleda) chez le freÁre et la soeur: coexistence dans les deuxcas d'alteÂrations dentaires graves. Bull Soc FrancË Dermatol Syph 31:82±87, 1924
Paris A, Strukelj B, Pungercar J, Renko M, Dolenc I, Turk V: Molecular cloning andsequence analysis of human preprocathepsin C. FEBS Lett 369:326±330, 1995
Pham CTN, Armstrong RJ, Zimonjic DB, Popescu NC, Payan DG, Ley TJ:Molecular cloning, chromosomal localization, and expression of murinedipeptidyl peptidase I. J Biol Chem 272:10695±10703, 1997
Rao NV, Rao GV, Hoidal JR: Human dipeptidyl-peptidase I. Gene characterization,localization and expression. J Biol Chem 272:10260±10265, 1997
Rickman L, Simrak D, Stevens HP, et al: N-terminal deletion in a desmosomalcadherin causes the autosomal dominant skin disease striate palmoplantarkeratoderma. Hum Mol Genet 8:971±976, 1999
Toomes C, James J, Wood AJ, et al: Loss-of-function mutations in the cathepsin Cgene result in periodontal disease and palmoplantar keratosis. Nat Genet23:421±424, 1999
Vignal A, Gyapay G, Hazan J, et al: In: Adolph KW, ed. Gene and ChromosomeAnalysis ± Part A, Vol 1. San Diego: Academic Press, 1993:pp 211±221
Wolters PJ, Raymond WW, Blount JL, Caughey GH: Regulated expression,processing, and secretion of dog mast cell dipeptidyl peptidase I. J Biol Chem273:15514±15520, 1998
Wolters PJ, Laig-Webster M, Caughey GH: Dipeptidyl peptidase I cleaves matrix-associated proteins and is expressed mainly by mast cells in normal dog airways.Am J Respir Cell Mol Biol 22:183±190, 2000
Zhang Y, Lunddgren T, Renvert S, et al: Evidence of a founder effect for fourcathepsin C gene mutations in Papillon±LefeÁvre syndrome patients. J MedGenet 38:96±101, 2001
VOL. 117, NO. 6 DECEMBER 2001 NOVEL CATHEPSIN C MUTATIONS IN PLS 1661







