
UNIVERSITÀ DEGLI STUDI DI TRIESTE
Sede Amministrativa del Dottorato di Ricerca Laboratorio Nazionale Centro Interuniversitario per le Biotecnologie
XX CICLO DEL
DOTTORATO DI RICERCA IN MEDICINA MOLECOLARE
MODULATION OF p53 ACTIVITIES BY THE PROLYL-ISOMERASE PIN1 AND THE
BROMODOMAIN PROTEIN BRD7
Settore scientifico-disciplinare BIO/13
DOTTORANDA COORDINATORE DEL COLLEGIO DEI DOCENTI FRANCESCA TOCCO CHIAR.MO PROF. GIANNINO DEL SAL, Università degli Studi di Trieste
SUPERVISORE E RELATORE
CHIAR.MO PROF. GIANNINO DEL SAL,
Università degli Studi di Trieste
CORRELATORE
DOTT.SSA FIAMMA MANTOVANI,
Università degli Studi di Trieste


INDEX
INDEX
INTRODUCTION……………..……………………………………………………..…..3
1. The tumor suppressor p53………….…………………………...………………….....6
2. The p53family...…...……………………………………………………………….... 7
3. p53 isoforms...…………………………… ………………………………………....10
4. Structure of the p53 protein…………………………………………………….........12
4.1. Transactivation Domain…………………………………………….…………..12
4.2. Proline Rich Domain……………………………………………………..….….13
4.3. DNA-binding Domain………………………………………………………......14
4.4. Oligomerization Domain……………………………………………………......16
4.5. C-terminal Domain………………………………………………………..….....16
4.6. Nuclear localization and nuclear export signals…………………………..….…17
5. Upstream events engaging the p53 pathway……………………………….……..…..18
5.1. DNA damage………………………………………………………….…….…..19
5.2. Oncogenic signaling………………………………………………….…….…..21
5.3. Other kinds of stimuli………………………………………………….…….….22
6. Outcomes of p53 activation…………………………………………………..….…...23
6.1. Cell cycle arrest…………………………………………………………..….…. 24
6.2. DNA-damage repair………………………………………………………...…...25
6.3. Senescence……………………………………………………………….……...29
6.4. Apoptosis…………………………………………………………………...…...28
6.5. Pro-survival and antioxidant functions……………………………………...…..33
7. Regulation of p53……………………………………………………………………. 34
7.1. Regulation of translation………………………………………………………...34
7.2. Regulation by the Mdm2/MdmX proteins………………………….…………...35
7.3. Regulation by other ubiquitin ligases………………………………….…...…...42
7.4. Localization at the Nuclear Bodies (NBs)……………………...….……..……..44
7.5. Post-translational modifications………………………………………….…..….45
7.5.1. Phosphorylation………………………………………….………….….45
7.5.2. Prolyl-isomerization……………………………………………..….…..50
7.5.3. Acetylation/Deacetylation……………………………………...……….51
1

INDEX
2
7.5.4. Other modifications affecting Lysine residues………...………..…….. 55
7.6. Regulation by co-factors………………………………………….……....56
7.7. Cooperation with other transcription factors………………..……...…… 64
8. Bromodomain proteins in transcriptional regulation……………….………………...66
9. The Bromodomain-containing protein Brd7………...……………………..….……. .68
AIM of the THESIS…..…………………………………...………………………...…… 71
RESULTS Part1…………..……………………………………………......………….…..72
Pin1 is required for p53-dependent apoptosis in response to DNA-damage in………….
human cells…………………………………………………………………………......73
Pin1 is required for dissociation of the iASPP-p53 complex upon DNA damage.…......74
Pin1 directly mediates dissociation of p53 from iASPP……...………………..………..75
Ser46 is required for Pin1-mediated regulation of p53-iASPP interaction…..……..…..78
RESULTS Part2…………………….………………………………………………………81
Analysis of Brd7 expression levels and half-life………………………………………..81
Brd7 acetylation and oligomerization….………………………………………………..83
Characterization of the interaction between Brd7 and the p53 family proteins.………..86
Validation of the binding between Brd7 and the p53 family proteins in vitro……....86
Mapping of the binding between Brd7 and the p53 family members……………….87
Analysis of the interaction between Brd7 and the p53 family members in human…….
cells…………………………………………………………………………………..91
Analysis of Brd7 biological functions………………………………………………......93
DISCUSSION………………………………………………………………………………99
APPENDIX ……………………………………………….………………………………109
MATHERIALS and METHODS….……………………….……………………………110
REFERENCES……………………………………………………………………………117
ACKNOWLEDGEMENTS………………………………………………………………148

INTRODUCTION
INTRODUCTION Cancer is a complex and frequently lethal disease. Due to the great variety and complexity
of tumors, for a long time cancer treatment has been approached only with blunt therapies
aimed at simply blocking the proliferation or inducing apoptosis of rapidly dividing cells,
thus frequently causing as much damage to the patient as to the tumour. Yet, rapid
advances in cancer research have been achieved and the mechanisms of the tumorigenic
process have been increasingly unveiled. This great knowledge has been exploited to
design targeted therapies that block the growth of cancer cells by interfering with specific
mechanisms and molecules needed for carcinogenesis and tumor growth and leave normal
cells unaffected.
According to classical cancer genetics, the cancer-causing mutations occur mostly against
two major categories of genes, oncogenes and tumor suppressors. Oncogenes like Ras,
myc, erB-2, Bcl-2, hTERT, derive from normal cellular genes (proto-oncogenes) that exert
different functions in promoting cell growth and survival. Thus, mutations enhancing their
expression or activity can cause a cell to divide in an unregulated manner. Conversely,
tumor suppressor genes like p53, Rb or PTEN that limit cellular growth, proliferation and
survival processes, are often inactivated in cancer so leading to a loss of inhibitory growth
control. The actual picture is furthermore complicated by the fact that malignant tumors
often display multiple mutations, harbour epigenetic abnormalities, and contain
chromosomal aberrations that include aneuploidy and loss of heterozygosity at numerous
loci.
Molecular dissection of the signalling networks that drive and maintain tumours, although
confirming their complexity and subtlety, has shown that tumour cells harbour the means
of their own potential destruction. It has been reported that many tumors, even when full
established, rely for their maintenance on the persistent activation of certain cancer-
promoting genes. This dependence, described by Weinstein as “oncogene addiction”
(Weinstein and Joe, 2006) has been demonstrated in various mouse models by generating
transgenic mice that over express an oncogene in a specific target tissue that can be
conditionally switched on or off. By this means it was observed that activation of an
3

INTRODUCTION
oncogene lead to the development of tumors, however, when this gene was subsequently
switched off the tumor cells stopped dividing and displayed differentiation and apoptosis
(Felsher and Bishop, 1999); (Huettner et al., 2000); (Fujita et al., 1999); (Jackson et al.,
2001). This dependency that renders tumors vulnerable to the inhibition of a cancer-
promoting agent, although seeming counterintuitive in the beginning, has been largely
exploited in drug design leading to clinical application of molecular targeted therapeutic
agents.
Indeed, several such inhibitors have already proven to be effective in cancer treatment.
Examples include imatinib mesylate (Gleevec®) , which targets the oncogenic BCR/ABL
protein in chronic myeloid leukaemia (Druker et al., 1996) and the EGFR-targeted drugs
gefitinib (Iressa) and erlotinib (Tarceva) in non-small-cell lung carcinoma (NSCLC),
pancreatic cancer, and glioblastoma (Grunwald and Hidalgo, 2002).
Although the concept of oncogene addiction has proven to be true in several tumors, it is
apparent from clinical experience with molecular targeted agents that cancers can ‘escape’
from this state of dependence probably due to their genomic instability.
Combination therapy directed against multiple targets within the tumour cells, has proven
to be more effective than the use of a single molecular targeted agent and has rendered
possible to achieve long lasting remissions in many human cancers. Synergistic or additive
effects on tumor growth inhibition might be obtained by combining therapies that exploit
oncogene addiction to others aimed at restoring the loss of a tumor suppressor gene.
Among the tumor suppressor genes candidate for targeted therapy, p53 is one of the most
studied. Indeed, p53 constitutes a central node in a complex signalling pathway evolved to
sense a great variety of cytotoxic and genotoxic stresses which may compromise genomic
stability and promote neoplastic transformation. Once activated by a stress and depending
on the cellular context, p53 may mediate a series of cellular outcomes that vary from cell-
cycle arrest to DNA-repair, senescence and apoptosis.
The key role played by p53 in tumor suppression is furthermore highlighted by the
observation that direct inactivation of this gene is the most common mutation in human
cancer, occurring in more than 50% of malignancies. Moreover, individuals affected by Li
Fraumeni syndrome, in which one mutant allele of p53 is germ-line inherited, show 25-fold
increase in the chance of developing early-onset cancers, compared with the general
population (Evans and Lozano, 1997). In addition, other components of the p53 pathway
4

INTRODUCTION
are frequently altered in tumors bearing wild type p53. It is thus possible to maintain that
the p53 pathway is compromised to some degree in all human cancers.
Three new studies published in the last year clearly demonstrated that restoration of p53 is
an effective means to induce tumor regression in vivo. Like in “oncogene addiction”, it was
in fact demonstrated that tumors remain addicted to the loss of p53 function thus assessing
the key role of p53 in inhibiting not only tumor development but also tumor maintenance.
With three distinct genetic approaches the three groups obtained engineered mice in which
lack of p53 function gene could be conditionally reverted by treating the animals with a
particular chemical.
Despite the different technical approaches and tumour types examined, the reinstatement of
p53 expression led universally to a prompt and impressive regression of established, in situ
tumours. How p53 carries out its anticancer function seems to differ according to the tumor
type and its context. For example, restoring p53 function in p53-deficient lymphomas
(blood cancers) rapidly induces apoptosis (Martins et al., 2006; Ventura et al., 2007). By
contrast, p53 reactivation in solid soft tissue sarcoma and hepatocellular carcinoma induces
a potent growth arrest characterized by the hallmarks of cellular senescence (Ventura et al.,
2007; Xue et al., 2007). It is not clear which features of a cancer determine whether its
response to p53 activation is apoptosis or senescence but both outcomes are associated with
tumour regression. Most importantly p53 activation occurred only in cancer cells and not in
normal ones that probably lack the specific environment to activate the p53 pathway
(Ventura et al., 2007). This furthermore suggests that p53 restoration would be effective in
tumor therapy to develop interventions that selectively kill tumor cells relative to normal
cells.
Indeed, the last years, several approaches to reactivate the p53 pathway in human cancers
have been proposed. p53 signalling could be reactivated in tumours by Nutlins, small
molecules that highly increase p53 stability (Thompson et al., 2004). DNA-methyl
transferases inhibitors such as azacytidin have been exploited to reverse epigenetic
silencing of genes positively affecting the p53 pathway (Fang et al., 2004). Moreover,
restoration of wild type properties in mutant p53 has been achieved by means of small
molecules like PRIMA-1 and CP-31398 (Bykov et al., 2002; Demma et al., 2004; Rippin et
al., 2002; Wang et al., 2006) .
5

INTRODUCTION
1. The tumor suppressor p53
p53 was first identified in 1979 in association with simian virus 40 (SV40) large T-antigen.
Throughout nearly 30 years of intensive studies a great knowledge has been achieved on
the p53 pathway and a great extent of complexity has been unveiled.
Due to its potentially lethal effect, in normal conditions p53 is maintained at low levels by
continuous ubiquitylation and subsequent degradation by the 26S proteasome (Haupt et al.,
1997; Kubbutat et al., 1997) . Yet, upon stimulation by stress signals such as DNA damage,
hypoxia, unscheduled oncogene expression, viral infection and ribonucleotide depletion,
p53 is rapidly stabilized and exerts its role of "guardian of the genome" and acts as a
transcription factor coordinating a program that eventually leads to cell cycle arrest,
senescence or apoptosis (Vousden and Lu, 2002). Obviously this is a very simplified view
of p53 activity in fact it has been recently demonstrated to exert also transcription-
independent functions within the apoptotic response and to be involved in other cellular
processes as metabolism, autophagy, DNA repair or antioxidant response to increased
ROS levels (Crighton et al., 2006; Sablina et al., 2005; Zhou et al., 2001).
p53 activity is tightly regulated and, depending on the stimulus, the protein undergoes a
series of post-translational modifications at specific residues that affect its stability, sub-
cellular localization and its ability to interact with different co-factors and to bind to its
target promoters (Gostissa et al., 2003; Vousden and Lu, 2002).
Despite the huge knowledge achieved on p53 and its pathway in the last years, many issues
about its functions and regulation remain partly unsolved. What are the stimuli leading to
the activation of p53 tumor suppressive activity, which modifications and interactions are
essential for p53 functions, what are the mechanisms by which p53 specifically selects
among different subsets of target genes leading to distinct cellular outcomes, are some out
of many interesting topics on which research needs to focus in order to achieve greater
insights on p53 biology.
6

INTRODUCTION
2. The p53 family
p53, together with its homologues p63 and p73 belongs to a family of related transcription
factors (Kaghad et al., 1997; Yang et al., 1998).
p53 family members have high structural similarity (Fig. 1). All the three contain a central
DNA binding domain (DBD), an N-terminal transactivation domain (TA) and an
oligomerization domain (OD). Since the recent identification of p53 isoforms (described in
detail below) it was believed that only p63 and p73 could be expressed in different
variants. p63 and p73 are in fact transcribed from two major promoters thus can be
expressed with (TA forms) or without (ΔN forms) the amino terminal TA domain (Stiewe
et al., 2002). Moreover, p63 and p73 splicing variants, designated by greek letters, differ in
the regions at the carboxy terminal end. Here also resides the most obvious structural
difference between p53 and its siblings consisting in the fact that the α-forms of p63 and
p73 contain a sterile α motif (SAM) that is absent in p53 (Kim and Bowie, 2003). The
homology shown by p53, p63 and p73 has at first suggested that the products of this gene
family share similar or even redundant functions. However, difference in regulation and
activity among the p53 members emerged quite rapidly. Many reports highlighted that the
three members of p53 family might be critical regulator of different processes. In
particular, p53 is mainly involved in DNA-damage response while p73 and p63 play key
roles in development. This is strikingly highlighted by the phenotypes of mice with
targeted deletions for each of the three family members. While p53-null mice are tumor-
prone, highlighting the key function of p53 in tumor suppression, they lack a clear
developmental phenotype that is instead evident upon disruption of p63 and p73.
The phenotype of p63-null mice demonstrates that p63 expression is absolutely essential
for limb formation and epidermal morphogenesis. p63-null animals show in fact absence of
limbs or severe limb truncations, absence of skin and craniofacial malformations. They
also fail to develop skin and most epithelial tissues (e.g., prostate and mammary glands).
Clearly, the animals do not survive beyond a few days postnatally (Yang et al., 1999).
Notably, heterozygous germ line point mutations of p63 in humans cause six rare
autosomal dominant developmental disorders with alterations reminescent of the knock-
out phenotype in mice (Brunner et al., 2002). Importantly, basal cells of normal human
epithelium including the epidermis strongly express p63 proteins but lose them as soon as
these cells withdraw from the stem cell compartment. Consistent with this notion,
keratinocyte differentiation is associated with the disappearance of ΔNp63α, a variant that
7

INTRODUCTION
Figure 1. (A) Homology between the functional domains in the p53 family proteins: % denotes percent identity . AD, activation domain; PRD, proline-rich domain; DBD, DNA-binding domain; NLS, nuclear localization signal; TD, tetramerization domain; BD, basic domain; SAM, sterile-a-motif domain. (B) p63 protein isoforms: TAp63 proteins encoded from promoter P1 contain the conserved N-terminal transactivation domain (TA). ΔNp63proteins encoded from promoter P2 are amino-truncated proteins containing an N-terminal domain different from TAp63 proteins. Aternative splicing originates the α, β and γ isoforms, numbers indicate the exons encoding p63 protein isoforms. (B) p73 protein isoforms: TAp73 proteins encoded from promoter P1 contain the conserved N-terminal transactivation domain (TA). Ex2p73 proteins are due to alternative splicing of exon 2. They have lost the conserved N-terminal TA domain, but still contain part of the transactivation domain (Exon-3). Ex2/3p73 proteins are due to alternative splicing of exons 2 and 3. They have entirely lost the TA domain and are initiated from exon 4. The protein encoded by ΔN’p73 mRNA has not been described. ΔN’p73 variant is often overexpressed at the mRNA level in tumours. ΔN’p73 is due to alternative splicing of exon 30 contained in intron 3. Theoretically, ΔN’p73 mRNA would encode either for a short p73 protein or p73 protein isoforms identical to ΔNp73. DN’p73 mRNA contains the normal initiation site of translation in exon 2 (ATG in perfect kozak sequence) and a stop codon in exon-30. Therefore, it could encode for a short p73 protein composed only of the transactivation domain. It is possible that translation of ΔN’p73 mRNA is initiated from the third ATG available present in exon 30and leading to p73 protein identical to ΔNp73 protein isoforms. ΔNp73 proteins encoded from promoter P2 are amino-truncated proteins containing an N-terminal domain different from TAp73 proteins. Alternative splicing generates 7different isoforms denoted by greek letters, numbers indicate the exons encoding p73 protein isoforms.
8

INTRODUCTION
shows a negative role in p21 regulation (Pellegrini et al., 2001). Together, these data
clearly establish a fundamental role of p63 in epithelial stem cell biology and in skin
development.p73 also has distinct developmental roles. Its expression is required for
neurogenesis of specific neural structures, for pheromonal signalling, and for normal fluid
dynamics of cerebrospinal fluid. Indeed, p73-null mice exhibit hippocampal dysgenesis
and failure in cortex organization, have severe malformations of the limbic telencephalon
and suffer from hydrocephalus. Moreover, they show hyper-inflammatory response of the
respiratory mucosa likely due to mucus hyper secretion. Alterations in the neuroepithelium
of the vomero-nasal organ causes defects in pheromone detection and is responsible of
altered social and reproductive behaviour in p73-null mice (Yang et al., 2000). Contrary to
p63, no human genetic disorders have been associated yet with germ line mutation of the
p73 gene.
Interestingly, phylogenetic analysis demonstrated that the primordial ancestor of all three
genes is actually much more related to p63 and p73, while p53 appears as the most
recently evolved member of its family (Yang et al., 2002). It might be hypothesized that
p53 lost some of its ancestral functions that are instead carried out by its homologues, yet it
has gained the ability to protect cells from tumorigenesis. This seems also to be confirmed
by the evidence that p53 is mutated in almost 50% of all human malignancies, while
mutations in the p63 and p73 genes are rare (Irwin and Kaelin, 2001; Nomoto et al., 1998).
However, there are reports showing that also p63 and p73 have tumor suppressive activities
in human tumors. On the one hand they contribute to p53 tumor suppression, as it has been
shown that mice heterozygous for mutations in both p53 and p63 or p53 and p73, lead to a
more aggressive tumor phenotype (Flores et al., 2005) and that p53 requires at least one of
its homologues to function properly as an inducer of pro-apoptotic genes upon DNA
damage (Flores et al., 2002). One the other hand they might have a specific role in tumor
suppression as their loss or down-expression leads to tumorigenesis at specific tissues
(Ahomadegbe et al., 2000; Park et al., 2000; Park et al., 2004; Puig et al., 2003; Urist et al.,
2002) with a tumor spectrum that differs from that of p53 and that reflects their pattern of
expression (mostly epithelial tissues).
9

INTRODUCTION
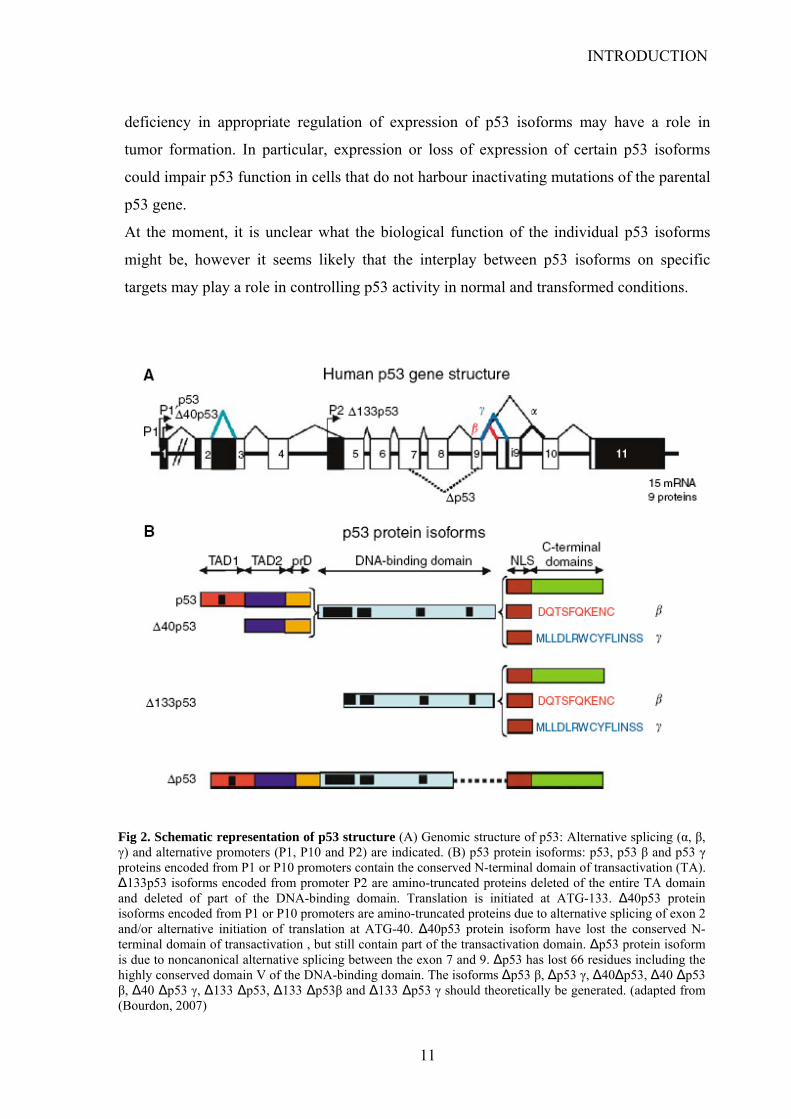
3. p53 isoforms
While the p53 gene was cloned almost thirty years ago, the existence of splice variants has
been ignored for long time. Only recently it has been established that the human TPp53
gene has a gene structure similar to p73 and p63 genes (Fig.2) (Bourdon et al., 2005). p53
gene transcription can be initiated from two distinct sites upstream of exon1 and from an
internal promoter located in intron 4. The alternative promoter leads to the expression of an
N-terminally truncated p53 protein initiated at codon 133 (Δ133p53) and lacking the entire
transactivation domain (TAD1and TAD2) and proline rich domain (PRD) and part of the
DNA-binding domain (DBD). The intron 9 can be alternatively spliced to produce three
isoforms: p53, p53β and p53γ, where the p53β and p53γ isoforms lack the oligomerization
domain (OD). Therefore, the human p53 gene can encode at least nine different p53 protein
isoforms which are named accordingly to p63/p73 nomenclature p53, p53β, p53γ,
Δ133p53, Δ133p53β and Δ133p53γ due to alternative splicing of the intron 9 and usage of
the alternative promoter in intron 4, and also Δ40p53, Δ40p53β, Δ40p53γ deleted of part of
the transactivation domain (TAD1) due to alternative splicing of the intron 9 and
alternative initiation of translation or alternative splicing of the intron 2 (Ghosh et al.,
2004). Interestingly, an additional p53 isoform (Δp53) has been described, which bears a
deletion of 66 amino acids within the core domain (corresponding to aa 257-322).
p53 mRNA variants are expressed in several normal human tissues in a tissue-dependent
manner, indicating that the internal promoter and the alternative splicing of p53 can be
regulated (Bourdon et al., 2005). Moreover, p53 isoforms can have distinct biological
activities. p53β was shown to hetero-dimerize with wt p53 and to preferentially bind to the
promoters of the Bax and p21 genes but not to Mdm2. p53β appeared to act in concert with
wt p53 on the Bax-gene promoter, particularly under conditions of stress.
However, this interaction seemed not to correlate with apoptosis induction, as co
expression of wt p53 together with p53β did not lead to increased apoptosis, when
compared with cells over expressing wt p53 alone. Conversely, a different isoform of p53,
Δ133p53 clearly acted as a potent dominant negative for wt p53 in apoptosis induction
(Bourdon et al., 2005) Finally, the recently described Δp53 appears to induce only p53-
target genes involved in cell-cycle arrest, thereby participating in a specific intra-S phase
checkpoint (Rohaly et al., 2005).
p53, p53β, p53γ, and Δ133p53 were found to be differentially expressed in normal versus
tumor breast as well as in different breast tumors (Bourdon et al., 2005). Possibly,
10
INTRODUCTION
deficiency in appropriate regulation of expression of p53 isoforms may have a role in
tumor formation. In particular, expression or loss of expression of certain p53 isoforms
could impair p53 function in cells that do not harbour inactivating mutations of the parental
p53 gene.
At the moment, it is unclear what the biological function of the individual p53 isoforms
might be, however it seems likely that the interplay between p53 isoforms on specific
targets may play a role in controlling p53 activity in normal and transformed conditions.
Fig 2. Schematic representation of p53 structure (A) Genomic structure of p53: Alternative splicing (α, β, γ) and alternative promoters (P1, P10 and P2) are indicated. (B) p53 protein isoforms: p53, p53 β and p53 γ proteins encoded from P1 or P10 promoters contain the conserved N-terminal domain of transactivation (TA). Δ133p53 isoforms encoded from promoter P2 are amino-truncated proteins deleted of the entire TA domain and deleted of part of the DNA-binding domain. Translation is initiated at ATG-133. Δ40p53 protein isoforms encoded from P1 or P10 promoters are amino-truncated proteins due to alternative splicing of exon 2 and/or alternative initiation of translation at ATG-40. Δ40p53 protein isoform have lost the conserved N-terminal domain of transactivation , but still contain part of the transactivation domain. Δp53 protein isoform is due to noncanonical alternative splicing between the exon 7 and 9. Δp53 has lost 66 residues including the highly conserved domain V of the DNA-binding domain. The isoforms Δp53 β, Δp53 γ, Δ40Δp53, Δ40 Δp53 β, Δ40 Δp53 γ, Δ133 Δp53, Δ133 Δp53β and Δ133 Δp53 γ should theoretically be generated. (adapted from (Bourdon, 2007)
11

INTRODUCTION
4. Structure of the p53 protein The p53 protein, like many other transcription factors, has a modular structure
characterized by the presence of evolutionarily conserved functional domains (Fig.3). This
organization is common to the other p53 family members p63and p73 (Kaghad et al., 1997;
Yang et al., 1998).
Fig. 3. Schematic rapresentation of p53 functional domains. TAD1and 2, transactivation domain 1 and 2; PRD, proline-rich domain; DBD, DNA-binding domain; OD, tetramerization domain; CT, C-terminal domain; NES, Nuclear export signal; NLS, nuclear localization signal
4.1. Transactivation Domain.
Early experiments already demonstrated that the N-terminal acidic domain of p53 is
responsible for transcriptional activity (Fields and Jang, 1990). This portion contains two
transactivation domains (TA1 aa 1-42 ad TA2 aa 43-62) that are independently sufficient to
activate transcription when fused to a heterologous DNA-binding domain (DBD) (Unger et
al., 1992) and that interact with components of the basal transcriptional machinery as the
TATA-binding protein (TBP) and TBP associated proteins (TAFs) as well as with
p300/CBP (Liu et al., 1993). These two domains are both necessary for p53 full
transcriptional activity but with different functions, the TA1 is in fact required for inducing
p21 transcription and G1 arrest while the TA2, together with the proline rich domain
(PRD), is essential for apoptotic response (Sabbatini et al., 1995; Zhu et al., 2000).
The transcriptional activation domain is finely regulated both by post-translational
modifications and by interaction with protein partners such as p300/CBP, Mdm2 and Pin1
(see below). Mdm2, the most critical negative regulator of p53, interacts with residues 17–
27 of TA1 and mediates the ubiquitination of p53 C-terminal lysine residues. Moreover
12

INTRODUCTION
Mdm2, together with MdmX, prevents the binding of transcriptional co-activators such as
p300 and interferes with the transactivation of TA1-dependent target genes.
4.2. Proline Rich Domain
Adjacent to the transactivation domain there is a proline-rich domain (PRD, aa 61-94)
containing five repeats of the amino acid motif PXXP (where P designates proline and X
any amino acid) in humans while only two in mice (Walker and Levine, 1996). This region
was thought to be significant for p53 regulation since PXXP motifs create binding sites for
Src homology 3 (SH3) domain-containing proteins and can modulate signal transduction
(Kay et al., 2000). Indeed, the p53 PXXP motifs may contribute to interactions with the
transcription co activator p300 (Dornan et al., 2003a) thus influencing p53 acetylation.
Furthermore, the prolyl isomerase Pin1 binds to Thr81-Pro82 site upon Thr81
Phosphorylation and induces a conformational change on Pro82. This may reduce Mdm2
binding thus influencing p53 stability (Wulf et al., 2002; Zacchi et al., 2002), consistently
with other evidence indicating that the PRD modulates Mdm2 binding (Berger et al., 2001;
Dumaz et al., 2001). Furthermore, the functional importance of the PRD is suggested by
studies showing that this domain is dispensable for cell cycle arrest but is essential for
apoptosis (Sakamuro et al., 1997; Venot et al., 1998) being required both for transcriptional
activation of pro-apoptotic genes (Bergamaschi et al., 2006; Venot et al., 1999; Zhu et al.,
2003) and for direct apoptotic function of p53 at mitochondria (Chipuk et al., 2004). Yet, a
recent study (Toledo et al., 2007) conducted in mice indicated that, although the PRD may
play some role in the regulation of p53, as mouse p53∆P (that bear p53 lacking the PRD)
displays increased Mdm2-mediated degradation and decreased transactivation capacity
(Toledo et al., 2007), the PXXP motifs are not essential for p53 tumor suppressor
functions, as their depletion does not significantly affect p53 accumulation and
transactivation, and exhibits only little effect on cell cycle control or apoptosis.
A common polymorphism is located within p53’s PRD at codon 72, encoding either
proline or arginine. This polymorphism is unique to humans while in the majority of
vertebrates proline is encoded in the corresponding residue. In early studies, the differential
electrophoretic gel mobility of the two isoforms raised the possibility that these variant
could differ structurally and therefore also functionally (Matlashewski et al., 1987). Indeed,
many reports have shown that the Arg72 variant is more efficient than Pro72 at inducing
apoptosis (Bonafe et al., 2004; Dumont et al., 2003; Sullivan et al., 2004; Thomas et al.,
13

INTRODUCTION
1999). The mechanistic basis appears to involve, on one hand, the enhanced localization of
p53 Arg72 to the mitochondria as compared to p54 Pro72 (Dumont et al., 2003) and on the
other hand its lower affinity for the iASPP protein, which binds preferentially to the PRD
and selectively blocks access of p53 to the promoters of apoptosis-related genes such as
Bax and PIG3 (Bergamaschi et al., 2006).
Extensive studies have been carried out to investigate the link between the expression of
p53 polymorphic variants at codon 72 (p53Pro72 and p53Arg72) and cancer susceptibility.
The results are controversial, reflecting a lack of understanding of how p53Pro72 and
p53Arg72 function in vivo (de Oliveira et al., 2004; Granja et al., 2004; Matakidou et al.,
2003). Homozygosity for the highly apoptotic wt p53-Arg72 variant is associated with
greater sensitivity of tumor cells to anticancer drugs, and is predictive of a more favourable
clinical response to chemotherapy in squamous carcinomas of the head and neck (Sullivan
et al., 2004). Yet Homozygosity for wtPro72 variant has shown to increase longevity
suggesting that enhanced lifespan associated with the lesser active Pro72 allele may
outweigh the deleterious effects of cancer susceptibility
4.3. DNA-binding Domain.
The central third of p53 (aa 100-300) contains the sequence-specific DNA binding domain
(DBD) required for p53 to function as a transcriptional activator (el-Deiry et al., 1992). The
canonical p53-responsive element contains two decamers or half sites,
PuPuPuC(A/T)(A/T)GPyPyPy, which are separated by a spacer of 0–13 bp. Several studies
indicate that a p53 monomer binds the pentameric sequence and that a tetramer binds the
full consensus site (Cho et al., 1994; Ma et al., 2005). Notably, variation within the
individual p53 responsive elements (REs) resulted in up to 1000-fold difference in
transactivation when tested in a yeast model system (Inga et al., 2002; Resnick and Inga,
2003). Besides, p53 may also recognize DNA sequences that differ from its canonical
consensus. For instance, a gene that is weakly induced by p53 is AQP3, in which, the RE is
made up of three pairs of pentamers (Zheng and Chen, 2001). Another example is the
repressive p53 RE located within the promoter of the MDR1 gene that shows orientation of
pentamers “head-to-tail” instead of classical “head-to-head”(Johnson et al., 2001).
Moreover, the microsatellite region within the PIG3 gene that was shown to function even
more effectively as a p53 RE than a more canonical binding site within the same promoter
(Contente et al., 2002). Also DNA topology is an important determinant of sequence-
14

INTRODUCTION
specific DNA binding by p53 (Gohler et al., 2002). Indeed, many p53REs deviating from
the consensus display an internal twofold symmetry that allows for intra-strand pairing and
formation of stem-loop structures. When presented in linear conformation, these sites are
poorly recognized by p53, while in stem-loop conformation, they are bound with high
affinity. It has been shown that p53 specifically binds to CTG_CAG trinucleotide repeats
that undergo topological alterations (Walter et al., 2005) or to cruciform-forming
sequences (Jett et al., 2000) that have no resemblance to the p53RE consensus.
The crystal structure of the p53-DBD bound to DNA has revealed that the conserved
regions are crucial for the p53–DNA interaction (Cho et al., 1994). The larger part of the
DBD forms an antiparallel β-sandwich. This β-sandwich serves as a scaffold that supports
the structures important for the interaction with DNA, specifically two large loops and a
loop-sheet-helix motif.
The two large loops are held together by a zinc atom which is coordinated by three
cysteines and one histidine (Cys176, His179, Cys238, and Cys242). The loop-sheet-helix
motif contains residues that contact the DNA phosphate backbone (Arg273, Ala276,
Arg283) as well as the major groove (Cys277 and Arg280). One additional residue,
Lys120, is important for the interaction with the major groove and the DNA phosphate
backbone. Importantly, all of the residues that are essential for the interaction with DNA
are conserved in p63 and p73 except R283, which is a Lysine in p63 and p73. However, the
proteins also possess unique functions and regulate distinct downstream target genes.
Subtle differences in the DBD are likely to contribute to the distinct functions of the
transcription factors (Harms and Chen, 2006).
Although the DBD is less involved than the N-term and C-terminal domains in protein
interactions, some proteins have been found to interact with it, among them 53BP1, Hzf,
ASPP1 and ASPP2 positively affect p53 activity. 53BP1 was demonstrated to enhance p53
transcriptional activation (Iwabuchi et al., 1998). Moreover it localizes at sites of DNA
damage (Anderson et al., 2001) where it participates in DNA damage signalling (Rappold
et al., 2001; Schultz et al., 2000) and repair (Iwabuchi et al., 2003). The roles of ASPP1/2
and Hzf will be treated in detail below. The p53-DBD also interacts with proteins that
negatively regulate p53. The ubiquitin ligase Mdm2 has been shown to interact with
residues in the DBD(Shimizu et al., 2002) besides its well known interaction with the N-
terminus of p53 (Oliner et al., 1993). In line with this observation, it was shown that the
binding of Mdm2 to full-length p53 is 10-fold stronger than binding to the N-terminal
15

INTRODUCTION
domain alone (Dawson et al., 2003). The SV40 oncoprotein large T antigen impairs p53
transcriptional functions by binding to its DBD (Jiang et al., 1993; Tan et al., 1986).
The importance of sequence-specific DNA binding for p53 to function as a tumor
suppressor is highlighted by the fact that 97% of tumor-associated mutations cluster in this
domain (Sigal and Rotter, 2000). The mutations situated in the DBD can disrupt specific
DNA binding by several possible ways. Mutant at some positions (e.g. Arg273His) lose
direct contacts with DNA and are therefore denominated “contact mutants” (Bullock et al.,
1997; Cho et al., 1994). Some other mutants, defined “conformational mutants” (e.g.
Arg175His, Gly245Ser, Arg249Ser, Arg282Trp) show reduced binding due to
destabilization of the tertiary structure of p53 DBD (Bullock et al., 2000; Wong et al.,
1999). Mutation at position 248 (Arg248Trp), in addition to breaking DNA-protein
contacts, also introduces extensive structural changes into the DBD ( Wong et al., 1999).
Notably the p53 DBD requires a significant level of flexibility within the tetramer in order
to grant recognition and contact with the different bases of a consensus RE. The same
flexibility, though, seems to be responsible for the ability of some p53 mutants to bind and
inactivate the other two p53 relatives, p63 and p73, as well as to force wild-type p53
molecules (translated from the remaining wild-type p53 allele) into a mutant conformation
(Bensaad et al., 2003; Di Como et al., 1999; Gaiddon et al., 2001).
It has been recently found that at least three DNA-binding defective molecules of p53 are
needed in order to effectively inactivate the tetramer (Chan et al., 2004; Shieh et al.,
1999).The existence of partially active tetramers in the cell may lead to a new phenotype
via appearance of an unusual binding surface on a p53 molecule, which may change the
pattern of coactivators/corepressors interacting with p53. Alternatively, mutations in the
DBD may change the relative affinity for target genes either resulting in loss of binding or
in recognition of novel DNA sequences with consequent alterations in the normal global
transcriptional response to p53.
4.4. Oligomerization Domain
The tetramerization required for high-affinity DNA binding and transcriptional activation
is mediated through the oligomerization domain (OD, aa 326-356). Tetramerization
appears to be essential for p53 tumor suppressor activities in fact it has been reported that
DNA damage-induced signalling to p53 mediated by phosphorylation of Ser15, Ser20, and
Ser33 requires the OD but not other domains (Shieh et al., 1999). Moreover p53 is unable
16

INTRODUCTION
to bind DNA in vitro if the hydrophobic core that stabilizes the dimer-dimer interaction is
disrupted. The OD’s are highly conserved among of the p53 family proteins, however they
show distinct properties that prevent the heterotetramerization of the p53 family proteins.
This is significant for the function of these proteins since p53, p63, and p73 are likely to be
expressed simultaneously, at least in some tissues (Chene, 2001). Interestingly, mutant
forms of p53 are able to heterotetramerize with p73 thus inhibiting its apoptotic functions
(Strano et al., 2000) . Moreover, the heterotetramerization of wild-type and mutant p53 is
likely causative of the dominant-negative activities of mutant p53. It will be interesting to
learn how heterotetramerization of full-length p53 and the other recently identified
isoforms could affect p53 activity and if the transcriptional activity of the heterotetramer is
different from that of the homotetramer.
4.5. C-terminal Domain
The last 30 amino acids of p53 constitute a basic C-terminal domain (CT, aa 364-393) that
has been regarded as a regulatory domain due to its ability to influence p53 activity and to
the great number of post-translational modifications it undergoes upon stress-signalling.
Nearly every residue within this domain is in fact subjected to at least one post-
translational modification. This domain does not form a regular secondary structure and is
able to interact directly with DNA and RNA (Ayed et al., 2001; Lee et al., 1995). In
particular CT is able to bind ssDNA ends, insertion/deletion mismatches, recombination
intermediates and γ-irradiated DNA in vitro thus implying an ability to recognize damaged
DNA and DNA repair intermediates in vivo (Bakalkin et al., 1995; Lee et al., 1995;
Zotchev et al., 2000). Moreover some recent works demonstrated that the CT is important
for binding to non-linear DNAs (Fojta et al., 2004; McKinney and Prives, 2002; Palecek et
al., 2004) and is involved in the ability of p53 to diffuse linearly on DNA (McKinney et al.,
2004). These studies also revealed that the CT is required for efficient promoter activation
by p53. The role of the CT and its post-translational modifications in directing p53
activities will be treated more in detail below.
4.6. Nuclear localization and nuclear export signals
p53 is known to shuttle between the nucleus and the cytoplasm. Since tetrameric p53 is too
large to passively diffuse across the nuclear pore, its nucleo-cytoplasmic shuttling is
facilitated by nuclear import and export signals (Middeler et al., 1997). The C terminus of
17

INTRODUCTION
the p53 molecule contains a cluster of three nuclear localization signals (NLS) that mediate
the migration of the protein into the cell nucleus. NLSI (aa 316–322), the most active
domain, is highly conserved in genetically diverged species and shares perfect homology
with consensus NLS sequences found in other nuclear proteins (Shaulsky et al., 1990). This
NLS has a bipartite structure (Liang and Clarke, 1999a) consisting in the canonical NLS
sequence (aa 315-322) and a basic sequence (aa 305-306) separated by a spacer (Liang and
Clarke, 1999b). The basic residues at the N- and C-termini of the NLS (Lys305-
Arg306/Lys319-Lys320-Lys321) are necessary and sufficient for the complete nuclear
localization of a cytoplasmic reporter protein (Liang and Clarke, 1999a) The other two
NLSs, II and III ( aa 370-384) appear to be less effective and less conserved (Shaulsky et
al., 1990). Moreover, p53 has two putative nuclear export signals NES, one in the N-
terminus and another in the oligomerization domain (aa 11–27 and 340–351, respectively).
As the NLS is adjacent to the OD and the NES is contained within the OD, it has been
proposed that the oligomerization of p53 may regulate its nucleo-cytoplasmic transport by
affecting the accessibility of the NLS and/or NES to their respective receptors (Liang and
Clarke, 2001; Stommel et al., 1999).
5. Upstream events engaging the p53 pathway
p53 is the central node of a complex web of incoming stress signals and outgoing effector
pathways. The great variety of stress signals that lead to p53 activation illustrate the
multiplicity of p53 functions in responding to potentially oncogenic insults to prevent
tumor development (Fig. 4)
Fig 4. Schematic representation of the upstream events that induce p53 activation
18

INTRODUCTION
5.1. DNA damage
DNA damage was the first type of stress found to activate p53 and, based on this, p53 has
been widely regarded as “the guardian of the genome” (Lane, 1992). DNA damage
signalling is triggered by a great variety of exogenous and endogenous events that might
compromise genome integrity both by altering the primary structure of DNA thus
generating mutations, and by causing double strand breaks (DSB) with consequent
genomic rearrangements or loss of genetic information. Exogenous damage might be
caused by external agents such as UV radiation, ionizing radiations or chemical mutagenic
compounds. Endogenous DNA damage derives instead from normal cellular processes
linked to metabolism and replication. For instance, reactive oxygen species (ROS)
produced from normal metabolic by-products can lead to nucleotide oxidation. Replication
stress due to premature termination of replication fork progression can result in fork
collapse and DNA breakage (Branzei and Foiani, 2005). Moreover, telomere erosion,
consequent to continuous replication, is perceived by mammalian cells as a form of DSB
(d'Adda di Fagagna et al., 2003; Takai et al., 2003).
Remarkably, in human cancers the DNA damage signalling cascade is permanently
activated, suggesting that the cancerous state is intrinsically associated to the generation of
DNA damage (Bartkova et al., 2005; Gorgoulis et al., 2005). The constitutive DNA
damage present in cancer cells is shown to emanate primarily from the DNA replication
stress due to aberrant firing of DNA replication origins that occurs upon activation of
oncogenes or loss of tumor suppressors (Bartkova et al., 2006; Di Micco et al., 2006;
DiTullio et al., 2002; Mallette and Ferbeyre, 2007). The strong generation of reactive
oxygen species detected in cells transformed by various oncogene (Mallette and Ferbeyre,
2007) may also contribute to activation and maintenance of the DNA damage response.
DNA damage signals of different nature are transduced to p53 through a cascade of
Ser/Thr kinases that play key roles in p53 activation by mediating the phosphorylations
necessary to promote its stabilization and its transcriptional activity (Lambert et al., 1998).
ATR and ATM, the two DNA damage sensor kinases and their respective downstream
kinases Chk1 and Chk2, phosphorylate p53 at different sites. Specifically, ATM and Chk2
act in response to ionizing radiation and DSBs leading to phosphorylation of p53 at Ser15
Thr 18, and Ser20. ATR and Chk1 appear to be required in UV damage response (Banin et
al., 1998; Chehab et al., 1999; Hirao et al., 2000) but are also involved in response to
hypoxia (Hammond et al., 2002), to fork stalling and single strand DNA formation (Zou
19

INTRODUCTION
and Elledge, 2003). Upon activation ATR phosphorylates p53 at Ser15 and Ser37 while
Chk1 at Ser6, Ser9 and Ser20.
Ionizing radiations induce also the activation of DNA-dependent protein kinase (DNA-
PK), another member of a protein kinase family that includes ATR and ATM. DNA-PK
has been shown phosphorylate p53 at Ser15 and Ser37 and to interact with it at sites of
DNA-damage (Okorokov et al., 2002). Interestingly, when the severity of DNA-damage is
elevated, DNA-PK activation is required in combination with Chk2 to induce an apoptotic
response (Woo et al., 2002)
p38, is activated in response to stress stimuli and cytokines (Pearson et al., 2001) and has
been shown to phosphorylate p53 at ser 15, ser33, ser37, ser46 and ser 389 upon different
stimuli and with different outcomes. Activation of p38 upon UV irradiation or nitric oxide
treatment leads to apoptosis that is abrogated upon treatment with p38 inhibitors (She et
al., 2002). Upon osmotic shock p38 phosphorylates p53 at Ser33 causing a G1 arrest (Kishi
et al., 2001), while its inhibition during UV irradiation leads to a decrease in
phosphorylation at Ser15, Ser33, Ser37 and Ser46 and to a reduced p53-dependent
apoptotic response (Bulavin et al., 1999). Moreover, during UV radiation p38 has been also
reported to phosphorylate p53 at Ser389 (Huang et al., 1999). Notably, the phosphatase
Wip1, a p53 transcriptional target, is known to inhibit p38 activity upon UV radiation
(Fiscella et al., 1997) by participating in a negative feedback loop that controls the MAPK
signalling to p53 (Takekawa et al., 2000).
The stress-activated protein kinase JNK phosphorylates p53 at Ser15 upon UV radiation
(Buschmann et al., 2000a) and at Ser20 under oxidative stress (Buschmann et al., 2000c)
leading to an increase in p53 transcriptional activity. In addition, JNK-mediated
phosphorylation at Thr 81 (Buschmann et al., 2001) is important for p53 stabilization
through a mechanism involving the prolyl-isomerase Pin1 (See also below).
The Ser/Thr kinase HIPK2 is an important inducer of p53 apoptotic response (D'Orazi et
al., 2002). Indeed, high doses of DNA‑damaging agents lead to HIPK2‑mediated
phosphorylation of human p53 at Ser46, a modification that is required for p53 to engage
an effective apoptotic response (Di Stefano et al., 2004b; Mayo et al., 2005; Oda et al.,
2000). HIPK2 stimulates the apoptotic response by stabilizing p53 (D'Orazi et al., 2002).
Moreover, it leads to p53 accumulation by antagonizing Mdm2-mediated nuclear export
and ubiquitination of p53 (Di Stefano et al., 2004a) thus granting the presence of high
levels of p53 necessary to activate apoptotic promoters (Chen et al., 1996b). It has recently
20

INTRODUCTION
been reported that upon mild DNA-damage, p53 may act in negative feedback loop
inducing HIPK2 degradation by Mdm2 (Rinaldo et al., 2007). Thus, under conditions that
do not require the triggering of the apoptotic response, p53 may indirectly repress its
phosphorylation at Ser46 and consequently its pro-apoptotic activation.
5.2. Oncogenic signalling
Oncogenic signalling activates p53 not only through the DDR but also through the
transcriptional activation of p14ARF (ARF) (de Stanchina et al., 1998; Palmero et al.,
1998). The expression of ARF is up-regulated by E2F-1 (Zhu et al., 1999) and beta-
catenin (Damalas et al., 2001). Moreover, the levels of ARF protein are found to be
increased upon Ras (Lin and Lowe, 2001) and Myc (Zindy et al., 1998) activation. One of
the key roles of the ARF protein is to bind to Mdm2 and inhibit its ubiquitin ligase activity
thus favouring p53 stabilization (Stott et al., 1998). In human tumors ARF is inactivated
with an extraordinarily high frequency. However loss of ARF occurs almost invariably in
combination with loss of p16INK4a thus generating ambiguity on which is the key targeted
tumor suppressor. It was demonstrated that p16INK4A is the major tumor suppressor of the
human INK4A locus. In detail, p16INK4A synergizes with p53 to protect primary cells
from unrestricted growth and from oncogenic transformation. Instead, ARF regulates
growth of primary human cells in normal culture conditions through p53 but loss of ARF
has little tumorigenic effect in human cells transformed with oncogenes (Voorhoeve and
Agami, 2003). This finding is consistent with the observation that mutations that inactivate
p16INK4a only, sparing ARF, outnumbers of a factor of 20 those which inactivate ARF
alone (Kim and Sharpless, 2006). In mice, instead , lack of ARF leads to a remarkable
tumor-prone phenotype, although not to the extent of p53-deficient mice (Jacks et al.,
1994; Kamijo et al., 1999).
The great relevance of oncogenic signalling in inducing the tumor suppressor functions of
p53 was recently highlighted by the observation that in mice, the cancer-protective activity
of p53 was abolished in the absence of ARF even in the presence of an additional p53
allele (“super”p53 mice) leading to an increased response to DNA damage (Efeyan et al.,
2006). Moreover, in a study exploiting p53-null mice ingegnerized so that p53 expression
could be conditionally restored, it was observed that reinstatement of p53 during the phase
of acute genotoxic damage did not lead to increased tumor protection respect to its
complete absence. Conversely, p53 restoration at later times, after the acute DNA damage
21

INTRODUCTION
response had subseded, lead to decreased tumorigenesis that was abolished in the absence
of ARF (Christophorou et al., 2006). Although these evidences suggest that oncogenic
signalling play a fundamental role in inducing p53 tumor suppressive functions, yet this
does not call into question the relevance of DDR. Indeed, other studies demonstrated that
mice deficient in DDR are tumor-prone that DNA-damage signalling in premalgnacies
activates p53 preventing the formation of tumors (Bartkova et al., 2005; Gorgoulis et al.,
2005). Taken together this data indicate that acute DNA damage does not lead to a
persistent signalling and might result predominantly in tossicity that eliminate most of the
damaged cells. The surviving cells may acquire oncogenic mutations and generate an
incipient tumor. On the one hand, sustained oncogenic signalling leads to ARF activation
while on the other hand results in replication stress and DNA damage that activate the
DDR through the ATR–Chk1 and ATM–Chk2 kinase signalling pathways respectively.
These two responses converge in p53 activation and are both necessary for sustained p53
tumor suppressive activity in order to prevent tumor progression to malignancy.
5.3. Other kinds of stimuli
Other kinds of stimuli have been shown to participate in p53 activation. Interestingly,
nucleolar disruption has shown to be a key element in the signalling to p53. A model has
been proposed according to which the nucleolus is a central stress sensor for a variety of
agents and nucleolar disruption is required besides DNA-damage for p53 stabilization
(Rubbi and Milner, 2003a). A growing body of literature has demonstrated that nucleolar
morphology is diagnostic for the general metabolism of the cell and that nucleolar structure
depends on rDNA transcription (Schwarzacher and Wachtler, 1991). After inhibition of
rDNA transcription by drugs blocking PolI, such as actinomycin D, or by physiological
stimuli that include serum starvation and cell-cell contact growth inhibition, the nucleolar
components are more or less rapidly rearranged and the nucleolar structure disintegrates.
This causes the release of ribosomal proteins, such as L5, L11, or L23, all of which bind to
Mdm2 and stabilize p53 through inhibiting the E3 ligase activity of Mdm2 (Bhat et al.,
2004; Dai et al., 2004; Gilkes et al., 2006; Lohrum et al., 2003; Zhang et al., 2003).
To further support the role of the nucleolus in p53 activation there is the observation that
the nucleolar protein nucleophosmin binds to p53 after leaving the nucleolus upon DNA-
damage and mediates p53 stabilization (Colombo et al., 2002).
22

INTRODUCTION
Hypoxia, a condition faced by the cells at the centre of expanding tumors, has shown to
activate p53 to its apoptotic response through a yet not clear mechanism that involves
ATR, HIF1α (Hypoxia inducible factor) and VHL (Von Hippel-Lindau) (Liu et al., 2007;
Roe et al., 2006; Roe and Youn, 2006). Interestingly, after hypoxia induction p53 shows a
different pattern of target gene activation than seen following DNA-damage suggesting that
under hypoxic signalling p53 might mediate a quite different response compared to other
stress signals (Hammond and Giaccia, 2005; Krieg et al., 2006)
In addition to acute insults, p53 responds to a variety of milder, constitutive stresses such
as the generation of ROS by normal metabolism. p53 was known to be a potent pro-oxidant
protein, inducing a set of ROS-generating genes that contribute to p53-mediated apoptosis
(Achanta and Huang, 2004; Macip et al., 2003).Yet an unexpected anti-oxidant function
mediated by p53 has recently emerged Through the activation of antioxidant target genes
p53 functions to allow survival and repair of the damage rather than to eliminate damaged
cells. In this way, p53 exerts its tumor suppression function by decreasing the incidence of
genetic alterations even contributing to the longevity of the organism. (Sablina et al.,
2005).
In addition, many other stresses that will result in a loss of fidelity in the cellular
duplication process have recently been shown to communicate with the p53 pathway. Heat
and cold shock (Ohnishi et al., 1998a; Ohnishi et al., 1998b), presence of denaturated
proteins, depletion of ribonucleotidephosphate pool in the cell (Khan et al., 2000), spindle
damage leading to faulty chromosomal segregation(Lanni and Jacks, 1998; Peng et al.,
2007), nitric oxide production associated with infections and inflammation (Hofseth et al.,
2003) all activate p53 protein and its response.
6. Outcomes of p53 activation
Once the p53 protein is activated, the ultimate outcome can be quite different, ranging from
the induction of reversible cell cycle arrest, apoptosis and senescence to protective
antioxidant activities and DNA repair. How the choice can be driven toward a specific
response depends on different factors. First, the induction of p53 activation has different
outcomes depending on cell type. In primary fibroblasts it is in fact usually associated with
cell cycle arrest (Di Leonardo et al., 1994; Kuerbitz et al., 1992) whereas the activation of
23

INTRODUCTION
p53 in hematopoietic cells (e.g., thymocytes) generally results in apoptosis (Lowe et al.,
1993). Moreover, even within a particular cell type, the p53 response can be influenced by
many factors such as the nature of the stimuli that activate p53 and a plethora of protein
partners that affect its stability and activity.
Probably the best understood way by which p53 mediates its response is to act as a
transcription factor with sequence-specific DNA binding ability and the potential to induce
the expression of a large number of genes (Table I). Jointed bio-informatic and ChIP based
studies have suggested that the number of genes containing p53 binding sites may vary
between 500 and 1600 (Cawley et al., 2004; Wei et al., 2006). Certainly, genes involved in
the well-established responses of cell-cycle arrest and apoptosis are largely represented, yet
the identification of genes involved in other processes such as metabolism or cell adhesion,
indicate that p53 might play further roles in governing cellular homeostasis (Wei et al.,
2006). Moreover p53 has shown to play also roles that are not related to transcriptional
activation, such as the induction of apoptosis through direct interaction with mitochondrial
proteins
6.1. Cell cycle arrest
The ability of p53 to induce cell-cycle arrest mostly depends on three critical target genes :
p21, 14-3-3σ and GADD45 (el-Deiry et al., 1993; Hermeking et al., 1997; Kastan et al.,
1992). The cyclin-dependent kinase inhibitor p21Waf1/Cip1 was the first transcriptional target
identified and its transactivation results in cell cycle arrest in G1 phase due to inhibition of
cyclinE/CDK2, CyclinA/CDK2 and cyclinD/CDK4 (Harper et al., 1993; Prives and Hall,
1999). The role of p21 in G1-arrest is further underlined by the observation that cells
lacking p21 fail to arrest in response to DNA-damage, yet they can still undergo cell death
(Brugarolas et al., 1995). Although the most prominent function of p21 is the mediation of
G1 arrest, evidence has been presented that it also participates in the G2/M arrest after
DNA damage, presumably by blocking PCNA function at replication forks (Ando et al.,
2001; Bunz et al., 1998). However, the p53-induced G2 arrest is mostly mediated by the
activation of genes such as GADD45 and 14-3-3σ. 14-3-3σ has been shown to prevent
nuclear import of cyclin B1 and CDC2, through their sequestration in the cytoplasm (Chan
et al., 1999), whereas GADD45 destabilizes CDC2/cyclinB complexes and these two
processes cooperate to prevent initiation of mitosis (Jin et al., 2002a; Zhan et al., 1999).
24

INTRODUCTION
Table I. Classes of genes induced by p53. Adapted from The Biology of Cancer (© Garland Science 2007)
6.2. DNA-damage repair
Part of tumor suppressor functions of p53 is exerted by preventing propagation of
deleterious mutations arising from DNA damage. Indeed, p53 plays an indirect role also in
DNA repair through the induction of ribonucleotide reductase subunits (Hwang et al.,
1999; Xue et al., 2003). Another p53-regulated gene, GADD45, that was originally
proposed to participate in global genomic repair GGR downstream of p53 (Smith et al.,
1994) has been later reported to have more likely functions in remodelling chromatin to
give access to the sites of DNA (Smith et al., 2000). Furthermore, two mismatch repair
genes MLH1 and PMS2 have recently been shown to contain p53-response elements (p53
25

INTRODUCTION
RE) within their first intron and to be responsive to p53 activation after DNA-damage.
These two genes may provide a sensor in DNA repair mechanisms and constitute a critical
determinant for the decision between cell-cycle arrest and apoptosis (Chen and Sadowski,
2005).
A direct participation of p53 in DNA repair was suggested by a number of biochemical
observations. For instance, the C-terminal 30 amino acids of p53 were shown to recognise
several DNA damage-related structures, such as DNA ends, gaps, and insertion/deletion
mismatches (Bakalkin et al., 1995; Jayaraman and Prives, 1995; Lee et al., 1995). p53 was
also demonstrated to catalyze reannealing of short stretches of single- and double-stranded
DNA and to promote strand exchange between them (Oberosler et al., 1993; Brain and
Jenkins, 1994). In addition to p53’s biochemical activities, numerous reports on physical
and functional protein interactions further strengthened the proposal of a direct role of p53
in nucleotide excision repair (NER), base excision repair (BER), and double-strand break
(DSB) repair (Albrechtsen et al., 1999; Bertrand et al., 2004).
Although it is well documented that efficient nucleotide excision repair (NER) requires p53
its exact role has been difficult to define (Hanawalt, 2001). It has been suggested that p53
may function in NER by facilitating access to the chromatin to the repair machinery thus
favoring DNA repair. It was further demonstrated that p53 is required for global chromatin
relaxation induced by UV-irradiation (Rubbi and Milner, 2003b). In concordance with this,
the histone deacetylase inhibitor trichostatin A overcomes the requirement for p53
suggesting that p53 may induce global chromatin relaxation through changes in histone
acetylation (Rubbi and Milner, 2003b). Moreover the histone acetyltransferase p300 co
localizes with p53 to sites of NER and inhibition of p300 by antibody microinjection
inhibits NER suggesting that p53-dependent recruitment of p300 histone acetyl transferase
(HAT) activity may be mechanistically involved in the ability of p53 to induce global
chromatin relaxation to foster DNA repair (Rapic-Otrin et al., 2002).
p53 is also directly involved in inhibiting homologous recombination (HR). Two studies
demonstrated that p53 inhibits HR in response to replication fork stalling (Janz and
Wiesmuller, 2002; Saintigny and Lopez, 2002). Consistently, it was further noticed that
p53 prevents the accumulation of DSBs at stalled-replication forks induced by UV or
hydroxyurea treatment Kumari et al.,2004; Squires et al., 2004).When DNA replication is
blocked, p53 becomes phosphorylated on serine 15 and associates with key enzymes of HR
(Linke et al., 2003; Sengupta et al., 2003; Zink et al., 2002). Notably, during replication
26

INTRODUCTION
arrest p53 remains inactive in transcriptional transactivation (Gottifredi et al., 2001; Restle
et al., 2005) supporting the idea that p53 is involved in HR regulatory functions unrelated
to transcriptional transactivation activities.
6.3. Senescence
Several lines of evidence support the idea that p53 tumor suppressor activity is partly
mediated through the induction of senescence, a program leading to irreversible arrest of
cell growth accompanied by a characteristic set of phenotypic changes in the cell.
Senescence can be triggered by the shortening of telomeres due to proliferation (replicative
senescence) or by other exogenous or endogenous acute and chronic stress signals
(telomere-independent or premature senescence) such as cytokine signalling (TGFβ)
oxidative damage (Chen et al., 2000; Di Leonardo et al., 1994), mitogenic oncogene over-
expression (Ras, Raf) (Ferbeyre et al., 2002; Michaloglou et al., 2005; Serrano et al., 1997;
Zhu et al., 1998), loss of anti-oncogenic tumor suppressors (PTEN) (Chen et al., 2005b) or
supra-physiological mitogenic signals (over expressed MAPK or E2F1) (Dimri et al.,
2000; Lin et al., 1998). In both replicative and premature senescence a key role is mediated
by tumor suppressor pathways involving p53 and p16-pRB as demonstrated by a general
refractoriness of human cells to multiple senescence-inducing stimuli upon loss of p53 and
pRB function (Serrano et al., 1997; Dimri et al., 2000). These two pathways interact but
can also independently halt cell-cycle progression and to some extent they respond to
different stimuli in a cell-type or species-specific fashion. Recent work has revealed the
importance of DNA-damage response (DDR) in initiating both replicative and premature
senescence. A common signal is the occurrence of double strand breaks caused by telomere
erosion or by oncogene activation through DNA hyper replication (Bartkova et al., 2006;
d'Adda di Fagagna et al., 2003; Di Micco et al., 2006; Hemann and Narita, 2007; Herbig et
al., 2004; Mallette and Ferbeyre, 2007; Takahashi et al., 2006) Consistently, in models of
cellular senescence induced by DNA damaging agents causing double strand breaks,
ATR/ATM mediates the activation of cell-cycle checkpoints via CHK1/CHK2 and p53,
with the participation of p21, p16 and Rb (Itahana et al., 2004).
While p53 involvement in senescence and the signals that trigger it are well established
much less is known on the actual mechanisms that contribute to this outcome. p21 is
certainly a crucial transcription target in mediating p53-induced senescence (Brown et al.,
1997) Interestingly, at early phases p21 levels are transiently elevated and gradually fall
27

INTRODUCTION
while p16 levels rise (Alcorta et al., 1996; Stein et al., 1999). This suggests that p53 is
crucial for the onset of senescence at least by inducing p21, then p16 may maintain the
growth arrest. What needs to be defined in much detail is how p53 can mediate both
reversible cell-cycle arrest and senescence through the same effector, p21. One possibility
is that rapid DNA-repair quickly terminates p53-mediated p21 induction, while slow,
incomplete or faulty repair results in sustained signalling and senescence. This might be
sustained by the fact that proteins involved in DNA-repair mechanisms, such as PARP,
play a role in post-translational activation of p53 during senescence (Vaziri et al., 1997).
An indication that signalling to p53 may be different depending on the response is given by
the observation that p53 phosphorylation pattern in senescence is distinct from that of DNA
damage. In fact, in both cases there is an increase in Ser-15 phosphorylation but senescent
cells have additional phosphorylation in Thr18 and Ser-376 and decrease in Ser-392
phosphorylation (Webley et al., 2000). This signature might be fundamental in recruiting
co-factors that specifically direct p53 response.
Some key p53 regulators involved in senescence have been identified. Association among
p53 senescence and PML has been reported by several groups. PML is up regulated upon
oncogenic Ras expression and induces senescence in a p53-dependent manner by
promoting p53 acetylation at Lys-382 by p300 in the nuclear bodies (NBs) (Pearson et al.,
2000; Webley et al., 2000). In addition, the deacetylase SIRT1 also co-localizes in PML
NBs and inhibits PML- and p53- induced senescence by deacetylating p53 (Langley et al.,
2002).
Finally, the recent identification of two chromatin remodelling proteins, namely p400 and
BS69, whose knock-down causes premature senescence starts to shed light on the
regulation of p53 transcriptional activity within the senescence programme (Chan et al.,
2005; Zhang et al., 2007).
6.4. Apoptosis
Clearly the main role of p53 is to prevent the outgrowth of damaged or stressed cells that
may develop into malignancies if left unchallenged. This can be achieved by eliminating
any aberrant cells through apoptosis (Fig.5), indeed the more ancient and evolutionarily
conserved function also found in p53 orthologues from simpler organisms. Indeed, the best
known transcriptional targets of p53 include a large number of pro-apoptotic genes that can
be divided into several categories depending on their specific functions. These target genes
28

INTRODUCTION
are generally classified on the basis of their involvement either in the extrinsic or in the
intrinsic apoptotic pathway. The extrinsic apoptotic pathway is triggered upon the
engagement of particular death-receptors belonging to TNF-receptor family by their
specific ligands and leads to the induction of a cascade of caspase activation which in turn
induces apoptosis (Attardi et al., 2000; Nagata and Golstein, 1995; Wu et al., 1997). The
intrinsic pathway instead is activated in response to different signals such as DNA damage,
oncogenic signalling, hypoxia or endoplasmic reticulum stress, and is associated with
mitochondrial depolarization and release of cytochrome C (CytC) from the mitochondrial
intermembrane space into the cytoplasm. This event leads to the formation of the
apoptosome, a complex of CytC, APAF-1 and pro-caspase-9, that activates the caspase
cascade, thus converging in the effector phase with the extrinsic pathway (Cory and
Adams, 2002)
p53 can promote apoptosis via the extrinsic pathway by activating the transcription of the
death receptors located at the plasma membrane, including Fas, DR4 and KILLER/DR5,
PERP and PIDD. Both DR5 and DR4 can trigger apoptosis or enhance apoptosis induced
by their ligand TRAIL and by chemotherapeutic agents (Liu et al., 2004b). Fas can be
activated by p53, yet its induction upon DNA damage is tissue specific and often does not
require p53 (Bouvard et al., 2000). Moreover, Fas is dispensable for p53-dependent
apoptosis in most tissues (O'Connor et al., 2000). PERP, a PMP-22/gas family protein, is
activated in transformed MEFs following DNA damage (Attardi et al., 2000) and
contributes to the p53-dependent apoptosis induced by γ-irradiation in thymocytes and
neurons, but not to that induced by oncogene activation (Ihrie et al., 2003; Reczek et al.,
2003). PIDD was identified as a p53-regulated gene in mouse erythroleukemia cells and
shown to promote apoptosis. Its induction by ionizing radiation is p53-dependent also in
MEFs (Lin et al., 2000). It has been described that PIDD can form an activating complex
with caspase 2 (Tinel and Tschopp, 2004). Yet, it remains unclear whether PIDD is
required for p53-dependent apoptosis as caspase 2 deficiency does not abrogate p53
responses in vivo.
Several other p53-regulated genes such as Bax, Noxa and PUMA enhance the release of
cytochrome c into cytoplasm from mitochondria to initiate the intrinsic apoptotic pathway.
Bax was the first identified p53-regulated pro-apoptotic Bcl-2 family member (Miyashita
and Reed, 1995). Loss of Bax accounts for nearly half of the accelerated tumor growth
which resulted from the loss of p53 in a brain tumor model (Schmitt et al., 2002).Bax is
29

INTRODUCTION
also responsible for nearly half of p53-dependent apoptosis induced by 5-FU in colorectal
cancer cells (Zhang et al., 2000). Nevertheless, Bax is dispensable for the apoptosis
induced by γ-irradiation in thymocytes and intestinal epithelial cells and its induction is not
strictly dependent on p53 in many tissues (Bouvard et al., 2000).
p53 regulates the expression of several BH3 domain-only proteins that function upstream
of Bax to induce apoptosis. Some of these proteins are shown to be critical mediators of
p53-dependent apoptosis. PUMA and Noxa are activated in a p53-dependent manner
following DNA damage (Han et al., 2001; Nakano and Vousden, 2001; Yu et al., 2001).
PUMA mediates apoptosis induced by p53 in response to hypoxia, DNA damaging agents,
and endoplasmic reticulum (ER) stress in human colorectal cancer cells (Reimertz et al.,
2003; Yu et al., 2003). Remarkably, PUMA-knockout mice recapitulated several key
apoptotic deficiencies observed in the p53-knockout mice, including deficiencies in the
apoptosis induced by γ-irradiation in thymocytes, by oncogenes in MEFs, and by DNA
damage in developing neurons (Jeffers et al., 2003, Villunger et al., 2003). This suggests
that the ability of PUMA to mediate apoptosis and tumor suppression is context-dependent.
Indeed, other studies demonstrated that the ability of PUMA to act as a tumor suppressor
can be dependent on other oncogenic events such as myc or E1A activation. Notably,
PUMA depletion cooperated with myc and E1A induced tumorigenesis but not with Ras as
this last oncogene induces a senescence response rather than apoptosis. Similarly, Noxa-
deficient mice develop normally but their MEFs are strongly resistant to apoptosis induced
by oncogenes and UV radiation while only slightly resistant to etoposide induced cell death
(Shibue et al., 2003; Villunger et al., 2003). In vivo, the absence of Noxa resulted in
resistance to X-ray-induced apoptosis in the small intestinal crypts (Shibue et al., 2003;
Villunger et al., 2003). Moreover, p53 contributes to the formation of the apoptosome also
through the transcriptional activation of APAF-1 (Kannan et al., 2001; Moroni et al.,
2001)and is involved in the more downstream phases of apoptosis by activating the
transcription of caspase-6 (MacLachlan and El-Deiry, 2002).
p53 can also induce the expression of a mitochondrial protein encoded by p53AIP1 gene
following severe DNA damage (Oda et al., 2000). Interestingly, this induction is regulated
by the phosphorylation of p53 at Serine 46 which might be mediated by several kinases
such as p38, HIPK2, PKC and another p53 target p53DINP1 (Taira et al., 2007; Yoshida et
al., 2006). p53 may sense the alterations in ROS levels and can activate numerous
REDOX genes like PIG3, POX2/PIG6 and ferredoxin reductase (Donald et al., 2001;
30

INTRODUCTION
Hwang et al., 2001). This results in an increased generation of ROS and originates a
positive loop feeding p53 activation that further contributes to apoptosis (Johnson et al.,
1996; Li et al., 1999; Martindale and Holbrook, 2002; Polyak et al., 1997). Moreover, p53
can contribute to the apoptotic pathway also through repressing the transactivation of anti-
apoptotic genes such as Bcl-2 (Haldar et al., 1994; Miyashita et al., 1994a; Miyashita et al.,
1994b), Bcl-XL (Cherbonnel-Lasserre and Dosanjh, 1997) and survivin (Hoffman et al.,
2002).
Interestingly p53 appears also to provide a connection between the extrinsic death receptor
pathway and the triggering of mitochondrial disruption processes through the activity of its
transcriptional target Bid. In fact Bid is activated by caspase-8 upon triggering of the
extrinsic pathway and translocates to the mitochondria where it activates Bax and
consequently the initiation of the intrinsic pathway (Sax et al., 2002).Among the pro-
apoptotic p53 target proteins Scotin (Bourdon et al., 2002), that is located in the ER and the
nuclear membrane, has been shown to be required for ER-stress mediated apoptosis. As a
protein-folding compartment, the ER is extremely sensitive to alterations in homeostasis
that disrupt its functions (Ferri and Kroemer, 2001; Kaufman, 1999). Prolonged ER stress
can result in the activation of caspase-12 that in turn activates caspase-9 thus executing
apoptosis. It has been proposed that, upon ER-stress, the release of calcium from the ER
triggers mitochondrial depolarization thus allowing the release of CytC to the cytosol and
the activation of the caspase cascade (Ichas et al., 1997; Ichas and Mazat, 1998; Jouaville
et al., 1998). Notably, upon ER-stress p53 is up regulated and induces transcription of two
other pro-apoptotic genes, Noxa and Puma (Li et al., 2006). These evidences clearly
suggest that p53 mediates the apoptotic response at multiple levels and that the apoptotic
pathways are not independent but rather involved in a complex interplay.
p53 participates in apoptosis also in a transcription-independent fashion by acting directly
at mitochondria where it can perturb or modulate the functions of proteins implicated in the
apoptotic machinery. This was thought to account for the reported ability of a
transactivation incompetent p53 mutant to trigger apoptosis via the mitochondrial death
pathway (Regula and Kirshenbaum, 2001). A fraction of p53 protein rapidly translocates to
mitochondria in response to genotoxic, hypoxic, and oxidative stresses (Marchenko et al.,
2000) and it has recently been demonstrated that this translocation is dependent on its
monoubiquitination by Mdm2 (Marchenko et al., 2007). At the mitochondria, p53 has been
found to interact with the Bcl-XL and Bcl-2 protective proteins and to prevent them from
31

INTRODUCTION
inhibiting on the oligomerization among Bax and Bak (Mihara et al., 2003).There is a
crosstalk among p53 functions at the mitochondria and its transcriptional activity, in fact
upon stress-induced-stabilization and activation within the nucleus, p53 induces the
transcription of Puma and this one is able to release cytoplasmic p53 from the inhibitory
interaction with Bcl-XL, thus allowing it to directly activate Bax (Chipuk et al., 2005). p53
interacts also with Bad and the mitochondrial p53/Bad complex promotes apoptosis via
activation and oligomerization of Bak (Jiang et al., 2006). Moreover, p53 acts directly on
the pro-apoptotic Bak promoting its dissociation from the anti-apoptotic protein MCL1
(Leu et al., 2004). Once the inhibitory interactions upon Bax and Bak are relieved, they
oligomerize to form a transmembrane pore for the release of cytochrome C from
mitochondria.
Figure 5 Schematic representation of the different apoptotic pathways controlled by p53. In response to cellular stress, p53 transactivates proapoptotic genes activating (1) the death receptor pathway (pidd, DR4, DR5, Bid), (2) the mitochondrial pathway (Bax, Noxa, PUMA, p53AIP1, Apaf-1) and (3) the ER pathway (Scotin). All the different pathways converge to a common downstream pathway (4), where caspase-6 is directly transactivated by p53, thus modulating the sensitivity of the cell to die. p53 can also repress the transcription of relevant prosurvival genes such as Bcl2 (5). Moreover, p53 can mediate apoptosis by acting directly at mitochondria. (6). Finally, it has been shown that p53 participates in maintaining mtDNA stability by interacting with Polγ and mtDNA
32

INTRODUCTION
Notably, upon translocation at mitochondria, p53 has been shown to play a role in
maintaining mitochondrial genetic stability. Indeed, in response to mitochondrial damage
p53 interacts with Polγ and enhances its DNA replication functions (Achanta et al., 2005) .
Moreover it has been proposed that p53 might interact with mtDNA (Heyne et al., 2004)
and that it might participate in mitochondrial base excision repair (mtBER) (de Souza-
Pinto et al., 2004).
The precise contribution of the transcriptional-independent apoptotic activities of p53 to
the overall apoptotic response needs to be figured out much more in detail. However they
seem to be important, as demonstrated by the fact that the stronger capacity of the isoform
p53Arg72 to activate apoptosis as compared to the Pro72 form, is associated at least in part
with the greater ability of Arg72 isoform to localize to mitochondria (Dumont et al., 2003).
Recent reports showed that p53 can also modulate autophagy, a cellular process triggered
by nutrient starvation and genotoxic agents that may function in different contexts to either
promote or inhibit cell survival (Crighton et al., 2006). DRAM (damage-regulated
autophagy modulator), a p53-induced protein, has been identified to be a likely linker
between p53-dependent apoptosis and autophagy. In fact p53 induces autophagy in a
DRAM-dependent manner and DRAM, which has been found to be down regulated in
human cancer, has proven to be also critical for p53-induced cell death.
6.5. Pro-survival and antioxidant functions
Growing evidence of pro-survival roles for p53 suggests that it has key functions not only
under condition of exceptionally severe stress, but also in response to the milder but more
constitutive stress encountered during everyday life of the organism. Indeed, pro-survival
genes induced by p53 have been identified. Among these, SLUG acts as inhibitor of
PUMA expression (Wu et al., 2005) while Myosin VI, an unconventional motor protein, is
induced by p53 to aid survival by maintaining Golgi-complex integrity (Jung et al., 2006).
Many recent studies have also proposed a role for p53 in determining cell-survival through
the regulation of intracellular reactive oxygen species (ROS) (Macip et al., 2003). It has
been suggested that p53 may exert this function by inducing the expression of proteins,
such as sestrins or ALDH4, that function to lower ROS levels. This antioxidant function of
p53 is important in preventing DNA damage and tumor development under low-stress
conditions (Budanov et al., 2004; Sablina et al., 2005). Furthermore, the recently
discovered p53-inducible protein TIGAR (TP53-induced glycolysis and apoptosis
33

INTRODUCTION
regulator), has shown to regulate glycolysis and protect against oxidative stress (Bensaad et
al., 2006). TIGAR can lower ROS levels and decrease sensitivity to p53 and other ROS-
associated apoptotic signals and is likely to be an important component in mediating the
pro-survival and tumor-suppressive effects of p53.
An attractive model would be that the low levels of p53 that are induced under conditions
of normal proliferation and daily exposure to stress promote the expression of genes that
induce cell cycle arrest, lower ROS levels and promote DNA damage repair. This will
temporarily halt the proliferation of the stressed cell and allow damage repair to occur thus
granting cell survival and organismal longevity. More severe and irreparable damage, or
stress that is associated with oncogene activation or loss of survival signals, would
conversely result either in activation of the apoptotic target genes, and elimination of the
errant cell or in activation of the senescence program thus preventing its proliferation
(Vousden, 2006).
7. Regulation of p53
When cells do respond adequately to p53-activating signals, the consequent biological
outcome may vary considerably. As it appears from a large number of studies, much of the
choice does not depend on p53 itself. Rather it is the cellular context, as defined by the
balance of intracellular and extra cellular signalling events which dictates the direction
undertaken by p53 response. Multiple proteins contribute to the activation of p53 in
response to diverse stress stimuli. These include proteins that modify p53 for both
stabilization and transcriptional activation, that reverse these modifications, that enhance
the translation of p53 mRNA or that alter its sub cellular localization.
7.1. Regulation of translation
Regulation of p53 abundance has long been thought to rely exclusively on its stabilization
as a consequence of reduced ubiquitination and proteasome degradation. However,
regulation of p53 mRNA translation has also been shown to play a role in determining p53
expression levels. A negative auto-regulation of p53 mRNA translation is mediated by both
a secondary structure of the 5’UTR in p53 mRNA and an element within its 3’UTR (Fu
and Benchimol, 1997; Fu et al., 1996). Besides, other proteins such as tymidilate-syntase
34

INTRODUCTION
inhibit translation of p53 mRNA through direct binding while Hu antigen R stimulates it
after UV radiation (Mazan-Mamczarz et al., 2003). A recent study showed that the
translational regulation of p53 is modified in response to DNA damage by the ribosomal
protein L26 (RPL26) and nucleolin. These two proteins are part of the nucleolin-containing
ribonucleocomplex and compete for binding to the 5’UTR of p53 mRNA co-regulating its
abundance with opposing effects (Takagi et al., 2005). RPL26 preferentially binds to the
5'UTR after DNA damage, and its over-expression enhances association of p53 mRNA
with heavier polysomes, increases the rate of p53 translation, induces G1 cell-cycle arrest,
and augments irradiation-induced apoptosis. In contrast, nucleolin over-expression
suppresses p53 translation and induction after DNA damage.
7.2. Regulation by the Mdm2/MdmX proteins.
In the absence of stress, p53 abundance is maintained at very low levels. Multiple lines of
evidence indicate that Mdm2 and MdmX play a major role in regulating p53 stability.
Mdm2 was first isolated from a mouse double minute chromosome (hence the name) that
was present at a high copy number in a spontaneously transformed derivative of mouse
3T3 cells (Cahilly-Snyder et al., 1987). Its role as an oncogene was assessed after the
observation that its over-expression leads to immortalization of rodent primary fibroblasts
and induced a fully transformed phenotype in cultured cells. Early studies ascribed the
transforming capabilities of Mdm2 to its ability to form a complex with p53 (Momand et
al., 1992) and to the observation that its over-expression lead to abrogation of both p53-
mediated G1 phase cell cycle arrest and induction of apoptosis in cultured cells (Chen et al.,
1996a; Haupt et al., 1996). The MdmX oncogene was identified because of its ability to
interact with p53 (Shvarts et al., 1996) and later as a Mdm2 partner (Tanimura et al., 1999).
Like Mdm2, MdmX acts as an oncogene and both are found amplified in many cancers
including soft tissue, breast, lung and small intestine cancer (Danovi et al., 2004; Momand
et al., 1998). The two protein share a great structural similarity and are able to omo- and
heterodimerize through their RING-finger domain (Tanimura et al., 1999). Importantly, the
highest similarity among the two proteins resides in the region encompassing the p53
binding domain where the amino acid residues necessary for the interaction p53 are strictly
conserved (Freedman et al., 1997). Conversely, the same amino acids on p53 are required
for both the interaction with Mdm2 and with MdmX (Bottger et al., 1997). Yet, an
important difference between Mdm2 and MdmX is that the RING domain of Mdm2 is
35

INTRODUCTION
essential for its action as an E3-ubiquitin ligase, whereas MdmX apparently has no intrinsic
ubiquitin-ligase activity.
Genetic studies revealed that germ line inactivation of either Mdm2 or MdmX leads to
early embryonic lethal phenotypes that are completely overcome by concomitant
inactivation of the p53 gene (Finch et al., 2002; Jones et al., 1995; Migliorini et al., 2002;
Montes de Oca Luna et al., 1995; Parant et al., 2001) These studies also indicated that
Mdm2 and MdmX are non-redundant p53 inhibitors, as normal levels of either regulator
cannot compensate for the loss of the other. These evidences clearly indicate that p53 is a
key downstream target of Mdm2/MdmX and that Mdm2/MdmX are essential negative
regulators of p53.
Mdm2 and MdmX mediate the inactivation of p53 through different molecular
mechanisms. Both Mdm2 and MdmX interfere with the transcriptional activity of p53. This
was initially thought to occur by virtue of their binding to the N-terminal domain of p53
that masks the transactivation domain of p53 (Oliner et al., 1993), disabling its interactions
with the basal transcriptional machinery and essential co-factors (Chen et al., 1995).
Indeed, Mdm2/MdmX bind to the N-terminus of p53 within a domain that serves to recruit
the histone acetyltransferases p300 and CBP (Dumaz and Meek, 1999; Lambert et al.,
1998) thus interfering with p53 acetylation by p300 (Kobet et al., 2000; Sabbatini and
McCormick, 2002). Mdm2 also associates with the histone deacetylase HDAC1, targeting
it toward p53 deacetylation (Ito et al., 2002); Mdm2 has also been shown to localize with
p53 at its responsive elements on the p21 and Mdm2 genes in normal growth conditions
and to dissociate upon DNA damage (White et al., 2006) thus suggesting that the
recruitment of a Mdm2-HDAC1 complex to chromatin might potentially promote histone
deacetylation and transcriptional silencing. In addition, it has been reported that Mdm2 is
capable of binding directly to histones and promoting their monoubiquitylation leading to a
transcriptional repression (Minsky and Oren, 2004). Moreover, Mdm2 further inhibits
p53’s transcriptional functions by catalyzing the conjugation of NEDD8, a small ubiquitin-
like protein, to p53 Lys-370, 372 and 373 (Xirodimas et al., 2004).
The most important role played by Mdm2 is however the regulation of p53 turn-over.
Mdm2 is in fact an E3-ubiquitin ligase that upon binding to p53 can catalyze either mono-
and poly-ubiquitination of p53, leading to nuclear export or proteasomal degradation,
respectively (Li et al., 2003). In contrast to Mdm2, MdmX does not act as an E3 ubiquitin
ligase and by itself cannot stimulate p53 ubiquitination and proteasome-dependent
36

INTRODUCTION
degradation (Jackson and Berberich, 2000; Stad et al., 2001). However MdmX can
stabilize Mdm2 preventing its auto-ubiquitination (Sharp et al., 1999; Stad et al., 2001),
while Mdm2 mediates MdmX poly-ubiquitination (Linares et al., 2003; Pan and Chen,
2003).
It has been shown that Mdm2 mediates both monoubiquitination or polyubiquitination of
p53 depending on its relative amounts respect to p53 (Li et al., 2003): when Mdm2 is
abundant, it promotes effective p53 polyubiquitination, leading to subsequent p53
degradation by the 26S proteasome. Several proteins have been demonstrated to assist the
ubiquitination of p53 by Mdm2. Gankyrin increases the ubiquitin-ligase activity of Mdm2
and p53 degradation by favouring the association of the Mdm2-p53 complex to the 26S
proteasome (Higashitsuji et al., 2005). Notably, gankyrin is frequently over expressed in
hepatocellular carcinomas (HCC), therefore it may constitute an important mechanism for
inactivating the p53 pathway in these tumors usually retaining wild-type p53 (Fu et al.,
2002). Moreover, transcription factor Yin Yang 1 (YY1), a protein that plays a key role in
development, increases p53 degradation by stabilizing the complex p53-Mdm2 (Sui et al.,
2004). Intriguingly, it has been shown that p300 cooperates with Mdm2 to promote p53
poly-ubiquitination (Grossman, 2001) by acting as a E4 ubiquitin ligase (Grossman et al.,
2003). This reveals a complex interplay between the two proteins that needs further
elucidation.
Conversely, when Mdm2 is scarce, p53 becomes monoubiquitinated and rather than being
degraded, p53 is exported into the cytoplasm. Notably, Mdm2 and p53 do not leave the
nucleus together, it seems rather that mono-ubiquitination of p53 by Mdm2 leads to an
unmasking of the NES within the C-terminus of p53 (Gu et al., 2001; Li et al., 2003).
Movement of p53 into the cytoplasm has been thought for long to have an inhibitory effect
on p53 activity. Clearly this translocation prevents p53 from transcriptional activation of its
target genes, however it might be important for transcription-independent functions of p53
such as interactions with mitochondrial proteins in the apoptosis response (Chipuk and
Green, 2004; Mihara et al., 2003). Indeed it has recently been reported that Mdm2-
mediated monoubiquitylation of p53 greatly promotes its mitochondrial translocation and
thereby its direct mitochondrial apoptosis (Marchenko et al., 2007). Upon arrival at the
mitochondria, p53 undergoes rapid deubiquitination by mitochondrial HAUSP via a stress-
induced mitochondrial p53–HAUSP complex, thus generating the apoptotically active non-
ubiquitinated p53. A novel model for mitochondrial p53 targeting has been proposed,
37

INTRODUCTION
whereby a distinct cytoplasmic pool of stabilized monoubiquitinated p53, generated in
resting cells by basal levels of Mdm2, is subject to a binary switch from a fate of
inactivation via subsequent polyubiquitination and degradation in unstressed cells, to a fate
of activation via mitochondrial trafficking upon stress stimuli (Marchenko et al., 2007).
Mdm2-mediated monoubiquitination and subsequent cytoplasmic translocation of p53 may
represent an important aspect in p53 regulation in unstressed cells, where Mdm2 is
maintained at low levels (Boyd et al., 2000; Geyer et al., 2000; Jimenez et al., 1999;
Stommel et al., 1999). On the other hand, Mdm2-mediated polyubiquitination and nuclear
degradation may play a critical role in suppressing p53 function during the later stages of
DNA damage response or when Mdm2 is over expressed in tumors (Shirangi et al., 2002;
Xirodimas et al., 2001). Indeed, in many tumors bearing wild type p53, the p53 pathway is
attenuated due to Mdm2 over-expression that may derive either from gene amplification or
from the presence of a single nucleotide polymorphism in the Mdm2 promoter (SNP309
T/G). The G-allele of this polymorphism originates a stronger binding site for the
transcriptional activator SP1 leading to a 2-3 fold increase in Mdm2 expression and
correlates with accelerated tumor formation. Notably, this occurs in a gender-specific and
hormonal dependent manner (Bond et al., 2006a; Bond et al., 2006b) probably due to the
cooperation of ER with SP1 on Mdm2 promoter.
Under stress condition p53 activity increases and an early step in this process is the
abrogation of Mdm2-mediated inhibition. Stress signalling directly communicates with the
p53/Mdm2/MdmX axis and the activation of ATM-Chk2/ATR-Chk1 kinases leads to
phosphorylation of p53, Mdm2 and MdmX (Kawai et al., 2003; Meek, 2002; Pereg et al.,
2005). On the one hand, ATM-mediated phosphorylation of p53 at Ser15 and indirectly
(through Chk2) at Ser20 inhibits the interaction between p53 and its ubiquitin-ligase
allowing the stabilization of p53 (Sakaguchi et al., 1998; Shieh et al., 1997; Unger et al.,
1999). Moreover, in response to DNA-damage the Jun NH2-term Kinase (JNK)
phosphorylates p53 within the PRD at Thr81 (Buschmann et al., 2001). Upon
phosphorylation of this residue (Phospho-Thr81/Pro82) p53 can be recognized and
isomerized by the prolyl isomerase Pin1 to promote Chk2-dependent phosphorylation of
p53 on Ser20, thereby stimulating its dissociation from Mdm2 (Berger et al., 2005).
On the other hand phosphorylation of Mdm2 and MdmX enhances their degradation by
auto-ubiquitination and by reduction of their association with the de-ubiquitinating enzyme
HAUSP, further releasing p53 from its negative regulation (Brooks et al., 2007; Li et al.,
38

INTRODUCTION
2004; Li et al., 2002). Notably HAUSP can de-ubiquitinate p53 directly, yet its main
function in the p53 pathway seems to be in the de-ubiquitination and stabilization of Mdm2
and MdmX (Cummins and Vogelstein, 2004; Li et al., 2004; Li et al., 2002) resulting in
enhanced degradation of p53.
The p53-Mdm2 interplay is tightly regulated in a negative feedback loop wherein p53
stimulates Mdm2 synthesis through transactivation of its promoter (which contains two
adjacent p53 binding sites) while Mdm2 inhibits p53 activity. Interestingly MdmX is not a
p53 transcriptional target and its abundance is mainly regulated through the interaction
with HAUSP.
The interplay between p53 and Mdm2/MdmX determining p53 stability and activation has
been elegantly summarized by G. Wahl into a model (Fig. 6) that divides p53 regulation in
four phases (Wahl, 2006). In unstressed cells p53 is maintained at low levels and in an
inactive state by interaction with Mdm2 and MdmX (homeostasis). In this phase Mdm2
levels are sustained through the interaction with HAUSP, through mitogen-dependent post-
translational modifications (Ashcroft et al., 2002; Gottlieb et al., 2002) and, through
transcriptional activation (Lahav et al., 2004); (Ries et al., 2000). Mdm2 can ubiquitinate
MdmX that is however de-ubiquitinated and stabilized via interaction with HAUSP.
Moreover, Mdm2-mediated ubiquitination of histones may provide an additional means of
controlling p53 transcription functions. DNA damage leads to the activation of ATM and
other kinases within minutes (early activation), as well as phosphorylation of p53’s N-
terminal serines that may modulate association with Mdm2/X and co activators. Of equal
importance, these kinases also rapidly phosphorylate Mdm2 and MdmX thus accelerating
their degradation by preventing their interaction with HAUSP. In this phase MdmX levels
do not decline immediately thus p53 transcriptional activity is still inhibited to some extent
and p53 target genes are activated weakly.
Full p53 activation occurs only after a lag of about 2h and requires Mdm2 and MdmX
phosphorylation to decrease HAUSP interaction with both proteins, the net effect of which
is to increase Mdm2-mediated self-ubiquitylation and ubiquitylation of MdmX. This
decreases the levels of Mdm2 at early time points and as a consequence activates p53. As a
consequence of p53 transcriptional activation, the increased levels of Mdm2 can then
degrade MdmX, resulting in full activation of p53. Notably p53 activation requires a
positive feedback loop in which increasing Mdm2 abundance titrates the amount of MdmX
degradation to assure the fine-tuning of the timing and magnitude of the p53 transcriptional
39

INTRODUCTION
40

INTRODUCTION
response (see Figure 6). The components of this system also provide the mechanism for
attenuating p53 signalling after stress dissipation or damage repair. ATM and p53
phosphorylation at Ser15 return to background levels within 4–6 h of induction of DNA
damage (Stommel and Wahl, 2004). In normal human fibroblasts, Mdm2 levels are high at
4 h, and start to decline thereafter, along with a parallel decrease in p53 transactivation.
Reasonably, the damage-phosphorylated Mdm2 pool is replaced with a non-
phosphorylated pool as a consequence of new synthesis and diminished abundance of
activated ATM. The elevated levels of Mdm2 should be able to interact with and inactivate
p53. Furthermore, in the absence of DNA damage-activated kinases, MdmX will be
allowed to interact with HAUSP, leading to deubiquitination and stabilization thus
providing a second barrier to continued p53 activation.
p53 stabilization and activation is furthermore regulated by other pathways that crosstalk
with the p53-Mdm2 axis. A feedback loop governing p53 activity involves the AKT kinase
and the PIP-3 phosphatase PTEN. In detail, AKT phosphorylates Mdm2 and induces its
translocation into the nucleus, where it down-regulates p53 (Mayo et al., 2002; Zhou et al.,
2003a). By inhibiting AKT and by blocking the nuclear entry of Mdm2, PTEN sustains
p53 activity (Mayo and Donner, 2002). On the other hand, in damaged cells, p53 activates
the transcription of PTEN (Stambolic et al., 2001) thus facilitating formation of a cycle in
which p53 induces PTEN and PTEN stabilizes p53.
Upon increased oncogenic signalling, p53 activation and stability is regulated also by the
tumor suppressor ARF. ARF increases p53 stability as it down-modulates Mdm2 ubiquitin
ligase activity both by sequestering it to the nucleolus (Damalas et al., 2001; Honda and
Yasuda, 1999; Lowe, 1999) and by interfering with its interaction with YY1 (Chen et al.,
2005). Notably p53 itself has been shown to down-regulate ARF expression (Robertson
and Jones, 1998). In this way increased levels of p53 act in a negative feedback, resulting
in a down-regulation of ARF transcription and concomitant decreases in ARF levels. This
will then allow MDM2 to bind to p53 and reduce its stability by targeting it for degradation
Fig. 6. Model of the attenuation and destabilization of p53 before and after stress stimuli, and for its activation and stabilization in response to stress. p53 regulation is presented in four phases as described in detail in the text. (From Wahl, 2006)
41

INTRODUCTION
7.3. Regulation by other ubiquitin ligases
The ubiquitination of p53 was first discovered in papilloma-virus-infected cells, where the
HPV protein E6 was found to associate to an E3 ubiquitin ligase, E6-AP thus mediating
p53 degradation. E6-AP-mediated disruption of p53 is important in the aetiology of HPV
associated cervical cancer and anogenital malignancies as it prevents p53 from activating
the apoptotic program, that would be induced upon oncogenic signalling by E7 and that
constitute a major obstacle to viral replication. Other oncogenic viruses exploit this
mechanism to prevent p53 response, for instance the adenoviral proteins E1B55K and
E4orf6 have shown to cooperate in promoting p53 ubiquitination and degradation through
a SCF E3 ubiquitin-ligase (Querido et al., 2001).
c-JUN NH2-terminal kinase (JNK) mediates another Mdm2-independent p53 degradation
pathway that seems to have a dual role. Under unstressed conditions, JNK binds to p53
preferentially in the G0/G1 cell-cycle phase and the interaction results in the ubiquitylation
and degradation of p53 Conversely, under stress conditions, JNK phosphorylates p53 at
Thr81, resulting in its stabilization and activation (Buschmann et al., 2001; Fuchs et al.,
1998a; Fuchs et al., 1998b). Other proteins that act as E3 ubiquitin ligase have been shown
to target p53 and promote its proteasome-mediated degradation. Pirh2, a RING-H2
domain-containing protein, interacts with p53 and promotes Mdm2-independent p53
ubiquitination and degradation (Leng et al., 2003). Similar to Mdm2, Pirh2 is a p53
responsive gene and participates in a comparable auto regulatory negative feedback loop.
Another E3 ligase, COP1, has also been described recently as a direct ubiquitin ligase for
p53 (Dornan et al., 2004). COP1 is also a p53-inducible gene and can ubiquitinate and
degrade p53. Further, COP1 depletion by siRNA enhances p53-mediated G1 arrest and can
sensitize cells to ionizing radiation. COP1 is phosphorylated by ATM upon DNA damage
thus leading to its auto-degradation (Dornan et al., 2006).
ARF-BP1/Mule/HectH9 was recently identified as a HECT domain E3 ligase that can
ubiquitinate and degrade p53 (Chen et al., 2005a). ARF interacts with ARF-
BP1/Mule/HectH9 and inhibits its activity thus leading to p53 stabilization.
Another route to degradation for p53 is mediated by the ubiquitin ligase CHIP (chaperone-
associated ubiquitin ligase), that has been shown to poly-ubiquitinate p53 upon transient
binding to HSP90 and HSP70. This mechanism might contribute to maintaining low levels
of p53 under physiological conditions as CHIP depletion significatively augment p53 basal
42

INTRODUCTION
levels (Esser et al., 2005). Notably CHIP has been recently shown to play a role in mutant
p53 ubiquitin-mediated degradation (Muller et al., 2008).
Recently, p53 was shown to be substrate of another E3 ubiquitin ligase, TOPORS
(Rajendra et al., 2004; Zhong et al., 2005). Interestingly, TOPORS has shown to localize
into PML nuclear bodies and to enhance the conjugation of the small ubiquitin-like
modifier 1 (SUMO-1) to p53 fashion (Rasheed et al., 2002). The role of TOPORS in
regulating p53 activity is controversial. As there are reports indicating that TOPORS has an
inhibitory effect on p53 transcriptional activity (Shinbo et al., 2005) while others
suggesting that it is a p53 co-activator (Lin et al., 2005).
Together, Mdm2, COP1, Pirh2, ARF-BP1 and Topors represent an array of E3 ligases that
act to regulate and maintain p53 levels (Fig.7). This redundancy suggests that multiple
mechanisms are used cooperatively by the cell for tight p53 regulation. It might be
suggested that the Mdm2/MdmX-independent pathways of degradation might serve for
Fig. 7. Schematic representation of the regulation of p53 degradation.
43

INTRODUCTION
fine tuning of p53 response. For example, they might be activated in different cellular
contexts or upon specific stimuli. Alternatively, they might backup Mdm-2/MdmX during
the phases of p53 full activation, when Mdm-2/MdmX levels are low, in order to avoid
unbalancing of the system and to favour the attenuation phase when the stress stimuli are
dissolved. However, further investigation on the specific regulation of these proteins is
needed in order to fully understand their actual role.
Two other ubiquitin- ligases, namely Cullin 7 (CUL7) and WWP1 can promote the
cytoplasmic localization of p53 yet without targeting p53 for proteasomal degradation
(Andrews et al., 2006; Laine and Ronai, 2007). Similarly, regulation of p53 ubiquitination
by the E2 ubiquitin-conjugating enzyme Ubc13 has been shown to drive nuclear export of
p53 (Laine et al., 2006). Curiously, ubiquitination of p53 by E4F1 results in neither
degradation nor nuclear export, but enhances transcriptional activation resulting selectively
in cell cycle arrest (Le Cam et al., 2006).
In addition to ubiquitin-conjugation it has been shown that p53 can be degraded as well
through an ubiquitin-independent mechanism. This pathway involves the 20S proteasome
and is regulated by the NAD(P)H quinone oxido-reductase 1 (NQO1) (Asher et al., 2001).
NQO1 can bind directly to p53 thus preventing it from being degraded by 20S proteasome
Binding of NQO1 to p53 is augmented in the presence of NADH and disrupted by
dicoumarol, an inhibitor of NQO1 (Asher et al., 2005). Genotoxic insults of various nature
lead to increased association between P53 and NQO1 and to consequent p53 stabilization
(Asher and Shaul, 2005). Although NQO1 regulates p53 stability via a distinct pathway
that is both Mdm2 and ubiquitin independent, this pathway is still responsive to some of
the regulators of Mdm2-ubiquitin dependent degradation such as ARF, which in addition to
inhibiting the ability of Mdm2 to target p53 for degradation also inhibits dicoumarol-
induced p53 degradation (Asher et al., 2002).
7.4. Localization at the Nuclear Bodies (NBs)
The promyelocytic leukaemia (PML) tumour-suppressor protein is localized either in the
nucleoplasm or in subnuclear structures referred to as nuclear bodies (NBs). Although their
function is not totally clear, NBs are dynamic multiprotein-complexes comprising
numerous transient or permanently localized proteins among which figure p53 and many of
its effectors (Borden, 2002; Guo et al., 2000; Zhong et al., 2000a; Zhong et al., 2000b) The
co localization of p53, CBP, and HIPK2 in PML NBs contributes to the regulated
44

INTRODUCTION
phosphorylation (by HIPK2) and acetylation (by CBP) of p53 in response to DNA damage
(D'Orazi et al., 2002; Guo et al., 2000; Hofmann et al., 2002). Protein inhibitor of activated
STAT (PIAS) and HAUSP also accumulate in NBs, further indicating that NBs could
provide a scaffold for the regulation of p53 post-translational modification
It has been reported that NBs are required for Ras-induced senescence through a
mechanism involving p53 acetylation at Lys382 by CBP (Pearson et al., 2000). In addition,
SIRT1, a p53 deacetylase, also co-localises with acetylated p53 in NBs and seems to be
involved in mediating cellular senescence (Langley et al., 2002). PML protects p53 from
Mdm2-mediated ubiquitylation and degradation by inducing the auto-phosphorylation and
activation of CHK2 that consequently phosphorylates p53 on Ser20 (Yang et al., 2006a).
7.5. Post-translational modifications
p53 undergoes a great variety of post-translational modifications that influence its stability
and its transcriptional activity. These include phosphorylation of serines and threonines,
acetylation, mono- and poly-ubiquitination, sumoylation, neddylation and methylation of
lysines. The actual pattern of post-translational modifications is complicated since the same
residue might be modified in different ways by different enzymes. Notably nearly all of
these modifications occur in the N-terminal or in the C-terminal domains. In general N-
terminal phosphorylations seem to be important for stabilizing p53 and crucial for
acetylation to occur in the C-terminal sites which in combination lead to full p53
activation. An intriguing model for the function of the C-terminal region of the p53 protein,
corroborated by the observation that nearly every residue of it is subject to a post-
translational modification, is that it acts like a histone tail, progressively accumulating
modifications that permit the sequential recruitment of chromatin modifying enzymes, for
example through binding of acetylated p53 residues to bromodomain proteins (Agalioti et
al., 2002).
.
7.5.1. Phosphorylation
Many kinases, including ATM, ATR, Chk1, Chk2, casein kinase1 and 2 (CK1, CK2), c-
JUN NH2-terminal kinase (JNK), Erk, p38, Aurora Kinase A (AurKA), glycogen synthase
kinase-3β (GSK3β), HIPK2 and DYRK2, have been shown to phosphorylate p53 after
DNA damage. So far, 17 phosphorylation/dephosphorylation sites have been detected in
45

INTRODUCTION
human cells following DNA damage induced by ionizing radiation or ultraviolet (UV)-light
irradiation (Fig 8). In humans, these include serines 6, 9, 15, 20, 33, 37 and 46 and
threonines 18 and 81 in the amino-terminal region; Ser315 and Ser392 in the C-terminal
domain; and Thr150, Thr155 and Ser215 in the central core. In addition, Thr55, Ser376 and
Ser378 seem to be constitutively phosphorylated in unstressed cells (Gatti et al., 2000;
Waterman et al., 1998). At this time, only a few p53 sites are reported to be phosphorylated
by one specific protein kinase. Ser6, Ser9 and Thr18 are phosphorylated only by casein
kinase 1 (CK1). Furthermore, the phosphorylation of Thr18 requires previous
phosphorylation of Ser15 (Dumaz et al., 1999; Sakaguchi et al., 2000). Moreover, Thr81 is
reported to be phosphorylated only by JNK (Buschmann et al., 2001). Clearly, significant
redundancies are observed, yet this multiplicity may provide a failsafe mechanism and a
distinctive combination of phosphorylated residues could be required for further
modifications, leading to maximal activation.
Fig. 8. Schematic rapresentation of p53 phosphorylation sites and of the enzyme involved in p53 phosphorylation upon different stimuli.
The phosphorylation of the amino-acid residues Ser15, Thr18 and Ser20 in the N-terminal
domain of human p53 is the most extensively studied. These residues are located in, or
close to, the region of p53 that also binds to Mdm2/MdmX (Appella and Anderson, 2001;
Bode and Dong, 2004; Ito et al., 2001; Sabbatini and McCormick, 2002) Ser15 is
phosphorylated in an ATM-dependent manner early in response to γ-irradiation but is not
phosphorylated upon exposure to UV-light (Banin et al., 1998; Waterman et al., 1998).
46

INTRODUCTION
Conversely p38 phosphorylates p53 at this residue in response to UV radiation (Bulavin et
al., 1999). Data from in vitro or over-expression studies indicate that phosphorylation at
Ser15 stimulates p53-dependent transactivation, growth arrest and apoptosis in response to
DNA damage (Fiscella et al., 1993; Shieh et al., 1997). However, there exist conflicting
data on whether Ser15 phosphorylation affects Mdm2 binding or not (Dumaz and Meek,
1999; Shieh et al., 1997) but it is known that it increases the interaction with the
acetyltransferase CBP (Lambert et al., 1998). Phosphorylation of other residues, including
Thr18 and Ser20, which occurs later in the response to DNA damage, was shown to depend
on initial phosphorylation at Ser15 and seems to interfere with the interaction of p53 with
Mdm2, thus promoting p53 stabilization (Chehab et al., 1999; Dumaz et al., 1999; Unger et
al., 1999). Notably, mice with a serine-to-alanine substitution at serine 23 (corresponding
to Ser20 in humans) develop a wide spectrum of B-cell tumours thus indicating that B-cells
are most sensitive to activating p53 through phosphorylation of serine 23 (MacPherson et
al., 2004).
Phosphorylation of Ser46 by the kinases HIPK2, p38MAPK and DYRK2 was reported to
mediate selectivity in promoter binding by p53 and to specifically promote the induction of
apoptosis inducing genes, such as p53-regulated apoptosis-inducing protein1 (p53AIP1)
(D'Orazi et al., 2002; Oda et al., 2000; Taira et al., 2007). Moreover, HIPK2-mediated
phosphorylation of p53 at Ser 46 is required for the acetylation of p53 at Lys 382 by CBP
(Hofmann et al., 2002). Notably, the stronger apoptotic potential of the polymorphic
variant p53-Arg72 as compared to p53-Pro72 has been associated with phosphorylation of
Ser46 as mutation of this residue abolishes the differences between these two isoforms
(Sullivan et al., 2004). Another coding region polymorphism exists in p53 just at codon 47
encoding either a proline or, less frequently, a serine residue (Felley-Bosco et al., 1993).
The Ser47 polymorphism shows impaired phosphorylation on Ser46 as proline-directed
kinases like p38-MAPK and HIPK2 require an adjacent proline to direct phosphorylation.
Consistently, the p53-Ser47 variant has impaired ability to transactivate the p53 target
genes p53AIP1 and PUMA, further confirming that reduced phosphorylation on Ser46
leads to impaired ability to induce apoptosis (Li et al., 2005).
Phosphorylation at Ser46 has been recently shown to be important in p53 promoter
selection. It is in fact sufficient for p53 to induce the PTEN tumor suppressor protein in
preference to Mdm2 (Mayo et al., 2005). The shift of p53 gene targeting from Mdm2 to
47

INTRODUCTION
PTEN results in sustained p53 activation and diminished survival as p53 and PTEN
coordinate cell death by facilitating one another’s expression and functions (see above).
Conversely, in normal conditions p53 represses its own phosphorylation at Ser46 due to
Mdm2-mediated HIPK2 degradation (Rinaldo et al., 2007). This creates another positive
feedback loop in which Mdm2 degradates HIPK2 thus decreasing p53 phosphorylation at
Ser46. Under this condition p53 will activate Mdm2 transcription rather than that of PTEN
These findings support the notion that the cell-cycle-arresting functions of p53 include
active inhibition of the apoptotic ones, and furthermore indicates that phosphorylation at
Ser46 is an important determinant of p53 choice as it acts like a switch between two
opposite feedback loops.
Phosphorylation at Thr81 by JNK generates a binding site for the prolyl isomerase Pin1
(Thr81/Pro82) that influences p53 stabilization and activation (Buschmann et al., 2001).
The relevance of this modification will be treated in detail below.
Within the DNA binding domain Ser 215 phosphorylation by means of Aurora Kinase A,
abolishes the DNA binding ability of p53 (Liu et al., 2004a). Besides, Thr 150 and Thr 155
are phosphorylated by two COP9 signalosome-associated kinases, CK2 and protein kinase
D, leading to ubiquitin-dependent degradation of p53 (Uhle et al., 2003). Interestingly, a
recent report demonstrated that Ser 149 of p53 is O-GlcNAcylated and that this
modification is associated with decreased phosphorylation of p53 at Thr 155, resulting in
decreased p53 ubiquitination (Yang et al., 2006b). This offers another demonstration of the
dynamic interplay between different post-translational modifications in p53.
Phosphorylation of p53 at serine 315 and serine 376, mediated by GSK-3β upon ER stress
has been shown to increase its cytoplasmic localization and reduce its stability (Pluquet et
al., 2005; Qu et al., 2004; Qu and Koromilas, 2004). In this way the cell may adapt to ER-
stress and engage mechanism that limit accumulation of unfolded proteins in the ER.
However, when ER stress is prolonged the apoptotic pathway is triggered, both by direct
activation of caspase-12 and by mechanisms that involve the p53 target gene Scotin (see
above).
Two serine residues are phosphorylated in the CTD of p53. Phosphorylation of Ser315 by
the cyclinB-dependent kinase, p34(Cdc2), was reported to increase the transactivation
potential of human p53 in response to irradiation damage, possibly by promoting nuclear
retention, mediated via interaction of p53 with E2F1 (Blaydes et al., 2001; Fogal et al.,
2005a). Phosphorylation at the same site by aurora kinase A, however, was suggested to
48

INTRODUCTION
promote Mdm2-dependent ubiquitination and proteolysis of p53, arguing for an inhibitory
role of this modification (Katayama et al., 2004). Ser392 (Ser389 in mouse p53) is
phosphorylated by p38 MAPK or casein kinase 2 in response to UV-irradiation, but only
very inefficiently after γ-irradiation. An UV-activated protein kinase complex that
phosphorylates Ser-392 of p53 in vitro has been identified. This kinase complex contains
casein kinase 2 (CK2) and the chromatin transcriptional elongation factor FACT (a
heterodimer of hSpt16 and SSRP1). FACT seems to alter the specificity of CK2 in the
complex such that it selectively phosphorylates p53 over other substrates including casein.
Phosphorylation by the kinase complex enhances p53 activity (Keller et al., 2001). It has
been demonstrated that damage-stimulated phosphorylation of p53 at Ser392 by CK2 has
an important impact on p53 structure as it can increase intrinsic thermostability of the core
DNA-binding domain thus enhancing p53 affinity for its consensus DNA binding site.
Mice with a serine-to-alanine mutation of p53amino acid 389 reveal a slight decrease in
DNA binding, reduced and delayed transcriptional activation of several p53-responsive
genes, and decreased apoptosis in response to UV (Bruins et al., 2004).
In addition to stress-induced phosphorylation there are also a few sites in p53 which are de-
phosphorylated in response to stress signals, such as Ser-376 and Thr-55. The loss of
phosphorylation from Ser-376 creates a consensus binding site for 14-3-3σ and in turn
increases the affinity of p53 for sequence-specific binding sites on DNA (Waterman et al.,
1998). Dephosphorylation by PP2A at Thr55 instead has been shown to stabilize p53 (Li et
al., 2007b). It is likely that there exists an ordered pattern and interdependence of stress-
induced modifications to p53. However, the relevance and the precise role of
phosphorylations on p53 in its stabilization and activation remain controversial. While it
seems likely that the complex network of stress-induced modifications to p53 is important
in generating a functional molecule, there is also evidence that p53 can be activated
without these changes. It was demonstrated that p53 stabilization occurs as well upon
mutation of a series of known stress-induced phosphorylation sites. (Blattner et al., 1999;
Fuchs et al., 1995). Further evidence pointing to a lack of requirement for post-translational
modification in p53 stabilization comes from the use of small molecule inhibitors of the
p53–Mdm2 interaction. In fact, p53 accumulates in cells treated with Nutlin-1 (a cis-
imidazoline analogue that displaces p53 from its complex with Mdm2) and this is followed
by an increase in the levels of both p21 and Mdm2 consistent with activation of the p53
pathway. Yet, unlike DNA damage, nutlins did not induce phosphorylation of p53 but this
49

INTRODUCTION
stabilized, unphosphorylated form of p53 was equally efficient at sequence-specific DNA
binding and the induction of apoptosis (Thompson et al., 2004).
These controversial results does not mean however that phosphorylations on p53 are
irrelevant or do not occur in vivo but nature appears to be able to compensate quite easily
for the loss of one or even two or more of these modifications in p53.
7.5.2. Prolyl-isomerization
Tightly connected to phosphorylation is the prolyl-isomerization mediated by Pin1. Pin1 is
a phosphorylation-dependent prolyl isomerase (PPIase) that specifically recognizes and
catalyzes cis/trans isomerization at Ser-Pro or Thr-Pro motifs in which the first amino acid
is phosphorylated (pSer/pThr-Pro). These motifs are present in many proteins involved in
controlling cell proliferation and transformation, such as Cyclin D1, β-catenin, NF-κB,
p53, p73 and p66Shc (Mantovani et al., 2004a; Ryo et al., 2002; Zacchi et al., 2002; Zheng
et al., 2002). It has been demonstrated that Pin1 has a central role in transducing
phosphorylation of p53 into conformational changes that affect its stability and function.
Indeed, p53 stabilization and activation upon genotoxic stresses are impaired in the absence
of Pin1 (Mantovani et al., 2004b; Zacchi et al., 2002; Zheng et al., 2002). Within the p53
sequence six Ser/Thr-Pro sites are present (namely Ser33-Pro34, Ser46-Pro47, Thr81-
Pro82, Ser127-Pro128, Thr150-Pro151 and Ser315-Pro316). Ser-33, Thr-81 and Ser-315
residues are phosphorylated upon UV radiation while Ser-46 upon ionizing radiation (See
above) thus generating consensus sites for Pin1 The structural change mediated by Pin1
can trigger various functional outcomes. Data from Berger et al. suggest that the residues
Thr81-Pro82 within the PRD are crucial for Pin1 to promote Chk2-dependent
phosphorylation on Ser-20 (Berger et al., 2005) and this might explain the ability of Pin1 to
induce dissociation of the p53-Mdm2 complex and favor p53 stabilization. Moreover,
mutations of Thr81-Pro82 have been reported in cancer patients with Li-Fraumeni
syndrome and sporadic breast tumors (Berger et al., 2005; Sun et al., 1996). In addition to a
role in the dissociation of Mdm2 from p53, in a recent report from our group it has been
demonstrated that Pin1 is involved also in regulation of p53 acetylation by p300
(Mantovani et al., 2007) It has been shown that mutants of p53 unable to bind to Pin1
(upon substitution of Ser/Thr residues at Pin1 consensus binding sites with alanines)
interact with p300 markedly less than wild type p53 and are consequently less acetylated.
Notably, in an in vitro assay it was demonstrated that a Pin1 catalytic mutant (C113A,
50

INTRODUCTION
described in Zhou et al., 2000) that binds p53 but cannot isomerize it, is not able to
increase p53 acetylation by p300 as its wild type counterpart does, thus indicating that the
conformational change induced by Pin1 on p53 upon prolyl-isomerization is required for
effective acetylation by p300. It is likely that Pin1 might regulate p53 acetylation by p300
through phosphorylation induced isomerization of Thr81-Pro82 within p53 PRD as this
domain has been proven to be essential for p300-mediated acetylation of p53 upon DNA
binding (Dornan et al., 2003a). This is also supported by the evidence that tumor-
associated mutations at Thr81 or Pro82 result in impaired acetylation at Lys 373 and
Lys382.
Notably, it has been also demonstrated that the prolyl-isomerase Pin1 is required for
efficient binding of p53 to its REs on p21 and Bax promoters upon stress (Mantovani et al.,
2007). Moreover, it was shown that Pin1 itself is recruited by p53 on its cognate promoters
on chromatin where it acts enhancing p53 acetylation by p300. Indeed, it was observed that
acetylation of chromatin-bound p53 on Lys373 and Lys382 increased upon stress only in
the presence of Pin1. This effect of Pin1 may be mediated through isomerization of the
Thr81-Ser82 site within p53 PRD as, as described above, the integrity of these residues
proved to be fundamental to allow p53 acetylation (Bushmann et al., 2001; Berger et al.,
2005).
Another site which may be important for Pin1 effect on p53-mediated apoptosis is Ser46-
Pro47, as also confirmed by the reduced apoptotic potential displayed by the Ser47 allele of
the Pro/Ser47 polymorphism (Li et al., 2005).
Pin1 is also an essential co-factor of the p53-family member p73 pro-apoptotic function.
Indeed, it has been demonstrated by our group that the binding between Pin1 and p73 is
stimulated upon stress conditions by c-Abl and p38 kinases and that this association
increases p73 acetylation by p300, thereby stimulating its transcriptional activity towards
apoptotic target genes, such as p53AIP1 (Mantovani et al., 2004b).
7.5.3. Acetylation/Deacetylation
p53 can be acetylated at several lysines by different HAT. Acetylation of p53 by
p300/CBP and PCAF occurs in response to DNA damaging agents, such as UV- and γ-
irradiation. CBP and p300 acetylate p53 at lysines within the C-terminal domain (Lys370,
372, 373, 381, 382) (Avantaggiati et al., 1997; Gu et al., 1997; Lill et al., 1997). Moreover,
Lys320 and Lys305 in the nuclear localization domain are acetylated by PCAF (p300/CBP-
51

INTRODUCTION
associated factor) and p300, respectively (Liu et al., 1999; Sakaguchi et al., 1998). Finally,
the MYST family acetyl transferases, hMOF and TIP60, were recently shown to acetylate
p53 at Lys120 in the DBD (Berns et al., 2004; Doyon et al., 2004; Legube et al., 2002)
(Fig. 9).
The role of acetylation in mediating p53 binding to DNA has been largely debated and is
still controversial (Gu and Roeder, 1997) yet, what is commonly assessed is that it
potentiates the recruitment of co-factors thus favoring p53 transcriptional activation.
Consistently, reports showing that acetylated p53 is enriched at target promoters in a stress
dependent manner (Liu et al., 2003; Mantovani et al., 2007) suggest a role for p53
acetylation in affecting its transcriptional activation. While some studies could show that
acetylation favours the recruitment of co-factors and histone acetyltransferases (TTRAP
and CBP) thus increasing p53 transcriptional activity (Barlev et al., 2001) other suggested
that the enhancing effect of p300/CBP on p53 transactivation function was independent of
p53 acetylation (Espinosa and Emerson, 2001)
Nevertheless, a number of recent studies have indicated that acetylation of specific lysine
residues on p53 has differential effects on the choice of the target genes activated by p53.
Using acetylation-mimicking lysine to glutamine mutations, functional differences between
acetylation of Lys320 versus Lys373 were reported. Acetylation of Lys320 in p53 was
shown to favour interaction with high-affinity p53-binding sites in target genes, promoting
cell survival and cell cycle arrest. In contrast, acetylation of Lys373 led to a stronger
interaction of p53 with low-affinity binding sites, which are found in pro-apoptotic target
genes and therefore promoted cell death (Knights et al., 2006).
Acetylation on Lys120 by the MYST family acetyl transferase Tip60 has also been shown
to modulate p53 transcriptional activity and to be necessary for apoptotic response.
Notably, Lys120 is specifically acetylated by Tip60 (Legube et al., 2004). Lys120-
acetylated p53 was reported to accumulate upon DNA damage preferentially on the
promoters of pro-apoptotic target genes, including Bax and Puma (Tang et al., 2006).
Moreover, mutants of p53 that can no longer be modified at this residue showed impaired
pro-apoptotic activity, whereas cell-cycle arrest and induction of Mdm2 were not affected.
Interestingly, Lys120 is conserved in all species that contain functional p53 genes and,
more importantly, it is mutated in human tumors (IARC TP53 Mutation Database).
Acetylation might also enhance p53 transcriptional activity through promoting the
assembly of an active transcriptional apparatus. It is known that TFIID binding to the core
52

INTRODUCTION
promoter element is required for assembly of a functional transcriptional initiation complex
(Thomas and Chiang, 2006). TAF1, the larger subunit of TFIID has been shown to interact
with the C-terminal domain of p53 on p21 promoter (Espinosa et al., 2003; Li and Wu,
2004). In a recent study it has been demonstrated that the acetylation of two lysine
residues in p53, Lys373 and Lys382, leads to a direct interaction with the Double
Bromodomain (DBrD, a structural protein module that recognizes acetylated lysines) of
TAF1. Transcription of the human p21 gene is regulated through two p53 binding elements
at -2.3 kb (5’ site) and -1.4 kb (3’ site) relative to the transcription start site (+1). Upon
DNA damage, TAF1 is initially recruited to the 3’ site and then through DNA looping is
brought to the TATA-box-containing core promoter. Site-directed mutants in p53 or in the
TAF1 bromodomains disrupt acetyl- p53/bromodomain interaction and fail to recruit TAF1
to both the distal p53-binding site and the core promoter. These results suggest that the
diacetyllysine/DBrD interaction is required for the recruitment of TAF1 to the p21 core
promoter and consequent assembly of a complete TFIID complex. Notably, also the
recruitment of CBP is mediated through the binding of its single bromodomain to p53
acetylated on Lys382 (Mujtaba et al., 2004). Recruitment of HATs has been shown both to
enhance p53 acetylation and to favour the acetylation of histones on p53 responsive
promoters thus favouring transcriptional activation.
Interestingly, it has been proposed that DNA-binding is required to allow p53 acetylation
by p300. In the absence of DNA the C-terminal domain of p53 is not accessible for
acetylation by p300 and this provides an intrinsic negative regulatory mechanism to
prevent acetylation until the tetramer is promoter bound. After DNA binding, allosteric
effects mediate an exposure of the acetylation motif to allow DNA-dependent acetylation
of the tetramer. Notably, the conformational change allows p300 to contact p53 on its PRD
and this interaction is essential for DNA-dependent acetylation of p53 (Dornan et al.,
2003a). Indeed, upon PRD deletion, p300 can still bind to p53 on its N-terminal LXXLL
motif encompassing Thr18-Ser20, but cannot acetylate DNA-bound p53. It has been
suggested that the prolyl-isomerase Pin1 may assist structural rearrangements that occur
upon DNA binding thus promoting the DNA-dependent acetylation of p53 by p300. This
may occur at least in part through phosphorylation induced isomerization of Thr81-Pro82
within the PRD. Indeed, it has been demonstrated that mutations affecting this site, result in
impaired acetylation of p53 at Lys 373 and Lys382 (Mantovani et al., 2007). Moreover,
Pin1-mediated Pro82 isomerization within the PRD has been shown to be required for
53
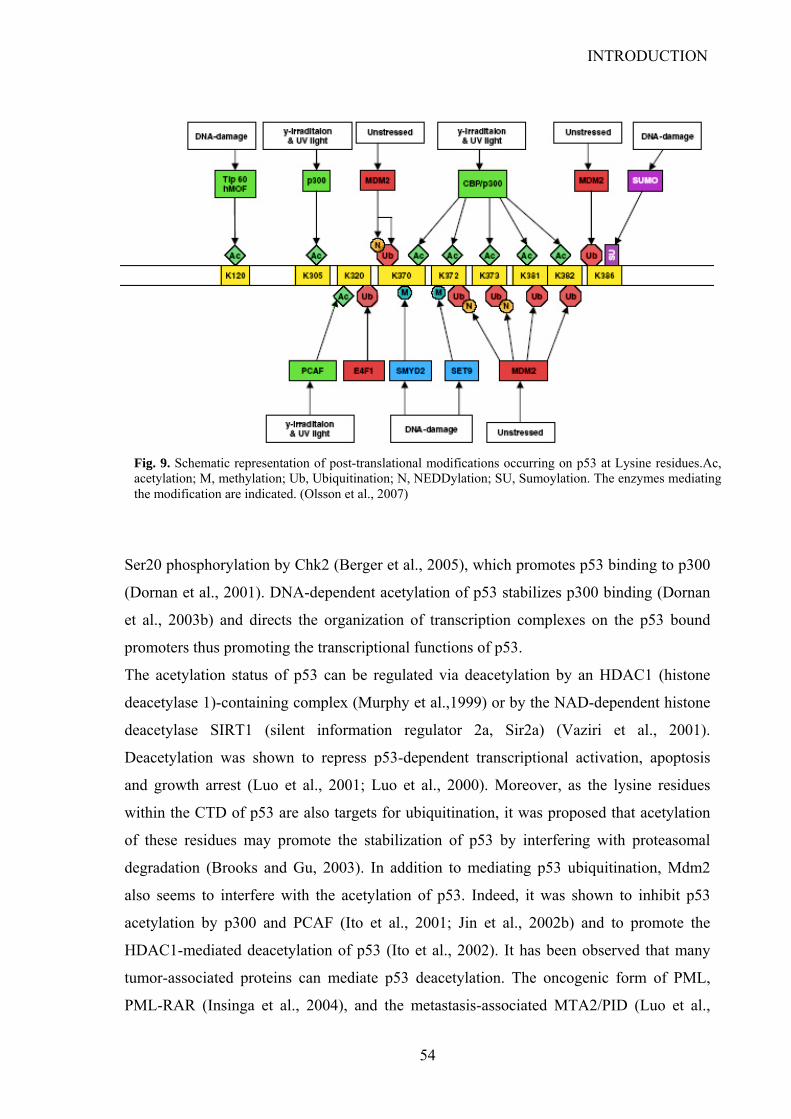
INTRODUCTION
Fig. 9. Schematic representation of post-translational modifications occurring on p53 at Lysine residues.Ac, acetylation; M, methylation; Ub, Ubiquitination; N, NEDDylation; SU, Sumoylation. The enzymes mediating the modification are indicated. (Olsson et al., 2007)
Ser20 phosphorylation by Chk2 (Berger et al., 2005), which promotes p53 binding to p300
(Dornan et al., 2001). DNA-dependent acetylation of p53 stabilizes p300 binding (Dornan
et al., 2003b) and directs the organization of transcription complexes on the p53 bound
promoters thus promoting the transcriptional functions of p53.
The acetylation status of p53 can be regulated via deacetylation by an HDAC1 (histone
deacetylase 1)-containing complex (Murphy et al.,1999) or by the NAD-dependent histone
deacetylase SIRT1 (silent information regulator 2a, Sir2a) (Vaziri et al., 2001).
Deacetylation was shown to repress p53-dependent transcriptional activation, apoptosis
and growth arrest (Luo et al., 2001; Luo et al., 2000). Moreover, as the lysine residues
within the CTD of p53 are also targets for ubiquitination, it was proposed that acetylation
of these residues may promote the stabilization of p53 by interfering with proteasomal
degradation (Brooks and Gu, 2003). In addition to mediating p53 ubiquitination, Mdm2
also seems to interfere with the acetylation of p53. Indeed, it was shown to inhibit p53
acetylation by p300 and PCAF (Ito et al., 2001; Jin et al., 2002b) and to promote the
HDAC1-mediated deacetylation of p53 (Ito et al., 2002). It has been observed that many
tumor-associated proteins can mediate p53 deacetylation. The oncogenic form of PML,
PML-RAR (Insinga et al., 2004), and the metastasis-associated MTA2/PID (Luo et al.,
54

INTRODUCTION
2000), have been shown to recruit HDACs to exert their negative control on p53 function.
Moreover, the tumor antigen MAGEA2 interacts with p53 and represses its transactivation
functions by recruiting HDAC3 to the MAGEA2-p53 complex (Monte et al., 2006).
Notably, mouse mutants that express p53 in which the C-terminal 6 or 7 lysines (Feng et
al., 2005; Krummel et al., 2005) are mutated to arginine residues, result almost similar to
mice with wild-type p53, thus suggesting that lysines in the CTD are not essential for p53
function, but instead fine-tune stress responses (Krummel et al., 2005).
.
7.5.4. Other modifications affecting Lysine residues (Fig. 9) Lysine residues are also
targeted by methyl transferases. Methylation of p53 can occur at least at two different sites,
reported to lead to opposing effects on p53 function. Methylation at Lys372 by the methyl-
transferase Set9, increases the stability of p53, restricts it to the nucleus and enhances p53
dependent transcription, whereas methylation of Lys370, mediated by another methyl
transferase, Smyd2, leads to repression of transcriptional activity (Chuikov et al., 2004;
Huang et al., 2006). In human cells, the histone lysine-specific demethylase LSD1 interacts
with p53 to repress p53-mediated transcriptional activation and to inhibit the role of p53 in
promoting apoptosis (Huang et al., 2007). While in vitro LSD1 removes both
monomethylation (K370me1) and dimethylation (K370me2) at K370, in vivo, LSD1 shows
a strong preference to reverse K370me2, which is performed by an unknown
methyltransferase, distinct from Smyd2. Interestingly, methylation at K370me2 has a
different role in regulating p53 from that of K370me1: K370me1 represses p53 function,
whereas K370me2 promotes association with the co activator 53BP1 (p53-binding protein
1) through tandem Tudor domains in 53BP1. Thus, LSD1 represses p53 function through
the inhibition of interaction of p53 with 53BP1. Taken together, these observations show
that p53 is dynamically regulated by lysine methylation and demethylation and that the
methylation status at a single lysine residue confers distinct regulatory output.
Lysine residues are also subjected to other post-translational modifications such as
ubiquitination, sumoylation and NEDDylation. p53 is ubiquitinated by several E3
ubiquitin-ligase among which Mdm2 is the best characterized (see above). Mdm2 can
induce both monoubiquitination, leading to nuclear export (Gu et al., 2001) and
mitochondrial localization (Marchenko et al., 2007) of p53 and polyubiquitination that
leads to degradation by the 26S proteasome (Li et al., 2003).
55

INTRODUCTION
Three lysines targeted for ubiquitination (Lys 370, Lys372 and Lys373) are also subjected
to NEDDylation. The C-terminal glycine residue of the ubiquitin-like protein NEDD8 is
covalently linked to p53 by Mdm2 thus inducing transcriptional repression (Xirodimas et
al., 2004). Sumoylation is also similar to ubiquitylation in that an isopeptide bond is
formed between the C-terminal carboxy group of the small ubiquitin-like protein SUMO1
and the ε-amino group of Lys386 (Gostissa et al., 1999; Rodriguez et al., 1991).
Sumoylation was reported to positively modulate p53 transcriptional activity (Gostissa et
al., 1999; Rodriguez et al., 1991). Other data indicate that sumoylation of p53 has a
repressive effect (Buschmann et al., 2000b; Chen and Chen, 2003), leaving the issue of
whether sumoylation of p53 results in activation or repression of p53 activities
controversial. Although the effect on p53 is not completely elucidated several other key
regulators of p53 activation are subjected to sumoylation, including PML, Mdm2, HIPK2
and p300 (Girdwood et al., 2003; Kim et al., 1999; Xirodimas et al., 2002). In particular
sumoylation of PML is important in the formation of the nuclear bodies (Ishov et al.,
1999).
7.6. Regulation by co-factors
An increasing array of co-factors is known to influence p53 activity in different ways. Co-
activator and co-repressors may affect p53 transcriptional activity by inducing
modifications in chromatin surrounding the p53RE, by mediating or preventing the
assembly of the transcriptional machinery or by directing p53 activation towards a
particular subset of target genes thus leading to a specific response. It has been suggested
that co-factors may also influence p53 loading on its target promoters.
The need for additional partners that assist p53 loading on its target promoters might be of
particular importance in the case of genes harbouring low-affinity p53BS, which may fail
to be engaged effectively by the limited concentrations of p53 attained under physiological
conditions (Szak et al., 2001). It is noteworthy that many, albeit not all, pro-apoptotic p53
target genes harbour p53RE of rather low binding affinity that need elevated and sustained
p53 activation. It might be hypothesized that the requirement of specific rate-limiting co-
factors may serve well the need to call on the apoptotic option sparingly, only under
conditions where death is the best solution, rather than any time that a cell is exposed to a
p53-activating stress.
56

INTRODUCTION
An important role in regulating the apoptotic functions of p53 at the transcriptional level is
played by the highly conserved ASPP family of proteins, composed of three members
ASPP1, ASPP2 and the inhibitory iASPP that all bind to p53 both on its DNA-binding and
its Proline Rich domain (Bergamaschi et al., 2006). Notably ASPP1/2 bind preferentially to
the DBD while iASPP to the PRD of p53. The importance of the ASPP family in human
malignancies is underscored by the observation that down-regulation of ASPP1 and ASPP2
is a frequent event in human tumors expressing wild-type p53, and to a lesser extent, in
tumor cells expressing mutant p53 (Samuels-Lev et al., 2001). Notably, studies conducted
on ASPP2-/- and ASPP2+/- demonstrated that ASPP2 is a haploinsufficient tumor suppressor
gene that cooperates with p53 in tumor suppression (Vives et al., 2006). ASPP2 is activated
upon DNA damage while in the absence of damage, p53 suppresses transcription of the
ASPP2 gene. Significantly, after UV treatment the ability of ASPP2 to interact with p53
increases (Samuels-Lev et al., 2001). Notably, ASPP1/2 transcription is up regulated upon
oncogenic signalling by the E2F family of transcription factors (Fogal et al., 2005b;
Hershko et al., 2005). When ASPP1 or ASPP2 bind to p53, they can specifically enhance
the ability of p53 to induce apoptosis but not cell cycle arrest (Samuels-Lev et al., 2001).
Chromatin immunoprecipitation assays have shown that the binding of ASPP1 and ASPP2
to p53 can selectively stimulate the loading of p53 on promoters of the pro-apoptotic genes,
such as Bax and PIG3, but not to the promoters of the cell cycle arrest gene p21 or the
Mdm2 gene (Samuels-Lev et al., 2001). Similarly, the binding of ASPP1 and ASPP2 to
the p53 family members, p63 and p73, also enhance their apoptotic function by selectively
stimulating their transcriptional activities on pro-apoptotic promoters (Bergamaschi et al.,
2004). Moreover, ectopic ASPP2 is predominantly cytoplasmic where it partially co-
localises and interacts with Bcl-2. Some evidences suggest that Bcl-2 and p53 are not
capable of simultaneously binding to ASPP2, rather Bcl-2 may compete with ASPP2 for
p53 binding (Naumovski et al., 1996).
Another member of the ASPP protein family, iASPP (inhibitory member of the ASPP
family), an oncoprotein that cooperates with Ras, E1A and E7 to transform cells in vitro
(Bergamaschi et al., 2003). iASPP shows opposite effects to ASPP1 and ASPP2, in fact
depletion of iASPP expression increases p53-dependent apoptosis while its over-expression
inhibits it. Moreover, iASPP is a nuclear protein that competes with ASPP1 and ASPP2
for binding to p53 (Bergamaschi et al., 2003b). Interestingly, iASPP binds preferentially
and inhibits the isoform wt p53 Pro72, that indeed shows lower apoptotic potential
57

INTRODUCTION
compared with wt p53 Arg72 in cells with elevated iASPP levels (Bergamaschi et al.,
2003a; Bergamaschi et al., 2006). It is thus likely that the greater apoptosis-inducing
activity of p53Arg72 results from its ability to escape iASPP inhibition in addition to a
greater ability to locate to the mitochondria (Dumont et al., 2003). Notably, iASPP is
frequently over expressed in tumors retaining wt p53 Pro72 but only rarely in those
expressing wt p53 Arg 72. This indicates that there would be no advantage for tumor cells
to select for iASPP over-expression in presence of wt p53 Arg72. Conversely, in tumors
over expressing iASPP the isoform p53 Pro 72 is rarely mutated, while the p53 Arg72
allele is preferentially targeted for mutations that inactivate its functions (Bergamaschi et
al., 2003a; Bergamaschi et al., 2006).
In a report by Das et al. hematopoietic zinc finger (Hzf), a p53 target gene, was identified
as a modulator of p53 activity (Das et al., 2007). Hzf is induced by p53 and interacts with
its DNA-binding domain, resulting in preferential transactivation of pro-arrest p53 target
genes over its pro-apoptotic target genes. It has been demonstrated that Hzf facilitates p53
binding to the p53REs in p21waf1 and 14-3-3σ promoters, while it inhibits p53 loading on
Bax, Perp and Noxa genes. Consistently, Hzf knockdown has shown to compromises p53’s
ability to promote expression of cell-cycle arrest target genes whilst enhancing the
transactivation of pro-apoptotic genes. Hzf expression levels may play a critical role in this
decision, in fact following prolonged DNA damage both the total protein levels and the
amount of Hzf bound to p53 rapidly declined and this correlated with the up-regulation of
pro-apoptotic genes and induction of apoptosis. Notably, Hzf does not influence its own
expression levels by acting in a loop with p53 on its promoter. It would be therefore
interesting to determine how Hzf protein abundance is regulated and which are the
mechanisms through which it determines p53’s choice. As Hzf effects are dependent on its
abundance it might be supposed that it competes with other factors for the interaction with
p53 and the binding of either leads to different outcomes. Putative competitors of Hzf are
ASPP1/2 protein that also interact with p53 DBD and that lead to selective activation of
pro-apoptotic genes.
In apparent contrast with the above observations many studies highlighted that there is no
shift in p53 loading on promoters that control growth arrest versus those implied in
apoptosis upon specific stimuli. Instead, high affinity sites (p21, Mdm2, Puma) are
efficiently bound by p53 in conditions leading to either cell cycle arrest or apoptosis,
whereas low affinity sites (Bax, Pig3, p53Aip1) are poorly bound regardless of the ultimate
58

INTRODUCTION
cellular outcome adopted (Kaeser and Iggo, 2002; Szak et al., 2001). This suggested that
selective binding of p53 to a promoter is not a determinant for choice of cell fate. It is
therefore likely that recruitment of a specific array of transcriptional co activators dictates
selective activation of p53 target genes in response to specific stimuli (Fig. 10)
A number of transcriptional co-activator and co-repressor complexes that bind p53 in cells
possess histone modifying activities suggesting that targeted chromatin remodelling may
be important for p53 to function as a transcription factor. In fact, although euchromatin (in
which most promoters are located) is transcriptionally competent, access of the basal
transcriptional machinery to the DNA is severely restricted and transcription of genes
located within euchromatin still requires local chromatin remodelling in order to proceed.
p53-dependent changes in histone acetylation have been observed at the promoters of a
number of p53 target genes (Kaeser and Iggo, 2004; Shan et al., 2003).
The role of CBP/p300 in regulating p53-dependent transcription is well established.
p300/CBP belong to a group of ubiquitous transcriptional co-activators with HAT activity
that interact with a plethora of sequence-specific transcription factors (Avantaggiati et al.,
1997; Gu and Roeder, 1997; Lill et al., 1997; Scolnick et al., 1997). As described above,
the C-terminal region of p53 contains two sites for acetylation by CBP/p300 and one for
P/CAF (Liu et al., 1999). Chromatin immunoprecipitation (ChIP) experiments suggest that
p53 recruits p300 to the p21 promoter thereby enabling targeted acetylation of chromatin-
assembled core histones H2A, H2B, H3 and H4 (Espinosa and Emerson, 2001).
Consistently, recruitment of CBP and TRRAP (a component of multiple co-activator
complexes with HAT activity) by acetylated p53 correlated with elevated levels of
acetylated H3 and H4 at the endogenous p21 promoter (Barlev et al., 2001). In another
study, ChIP analysis revealed that variations in p53 ability to activate transcription at the
p21 promoter correlate directly with the extent of histone acetylation and levels of
p300/CBP (Liu et al., 1999).
Many reports indicate that p300 takes part into multi-component co activator complexes.
The p300 cofactor JMY regulates the physiological response to p53 activation by
augmenting p53-dependent transcription and apoptosis (Shikama et al., 1999). JMY and
p300 associate in physiological conditions and the p300/JMY complex is recruited to
59

INTRODUCTION
60
Fig 10 Model for p53 transcriptional activation in different chromatin contexts accounting for the differential induction kinetics among subsets of target genes. p53 binding to most response elements does not result in altered gene expression, as only a minor fraction of p53-bound loci display changes in their transcriptional status. Before p53 activation, p53 target loci display many differences, as manifested by different concurring cis-regulatory elements, distinct combinations of histone modifications and DNA methylation (blue, red and turquoise lollipops), even different amounts of preloaded paused RNAP II. These differences impose gene-specific requirements for transcriptional coregulators, such as chromatin modifying and remodeling activities, variants of the Mediator and SAGA complexes, specific subunits of general transcription factors (GTFs), even defined sets of elongation and RNA processing factors. For example, the presence of a repressive histone methylation mark at a given locus would impose the requirement for a specific histone demethylase, whereas the presence of fixed nucleosomes at the promoter would impose a requirement for a chromatin remodeling complex. Alternatively, promoters not carrying preloaded RNAP II will require the recruitment of GTFs and assembly of a pre-initiation complex (PIC) before they could be readily activated. In turn, the requirement for specific subunits of the PIC may be dictated by the core promoter elements (CPEs) present at a given locus (for example, TATAbox, Initiator, downstream promoter element) (blue, red and turquoise barrels). Finally, effective gene activation will require conversion of paused RNAP II into an elongating form, as well as correct processing of the nascent mRNA, all of which may be also regulated in a gene-specific manner. Importantly, the quality of these combinatorial ‘filters’ could vary with the signaling scenario (that is, cell type or type of p53 activating stimuli), as the levels and activity of coregulators in each category could be modulated, thus allowing for great flexibility within the p53 transcriptional program. (Espinosa, 2008)

INTRODUCTION
activated p53 during the stress response. Furthermore, protein isoforms of JMY that arise
through alternative splicing display selective activation of distinct p53 target genes
(Shikama et al., 1999). In fact an isoform lacking an N-terminal stretch rich in Prolines
(ΔP) showed impaired ability to induce p53-dependent apoptosis but increased G1-arrest.
Besides, the tandem tetra-tricopeptide (TPR) co-factor Strap is involved in regulation of the
p300/JMY complex formation (Demonacos et al., 2001). As Strap undergoes stress-
responsive protein stabilization upon exposure to diverse genotoxic insults it has been
suggested that it might be a mediator of the DNA-damage signalling pathway directly
communicating with p53 to promote its transcriptional activation by p300 (Demonacos et
al., 2004). In further support of the importance of chromatin remodelling-proteins in
mediating regulation of p21 transcription, is the observation that p53 and the histone
deacetylase HDAC1 are antagonistic regulators of p21 gene expression and that p53 can
displace HDAC1 from the p21 promoter (Lagger et al., 2003). Besides p21, also
transcriptional activation of Mdm2 promoter is dependent on recruitment of TRRAP (Ard
et al., 2002), and correlates with increased histone acetylation suggesting that chromatin
remodelling is quite a general feature in regulation of p53-dependent transcription.
p53 also interacts with histone deacetylase complexes in vivo and this markedly influences
its transcriptional repression activity. p53 recruits the transcriptional co-repressor mSin3a,
bound to HDAC1, to Map4 promoter. This correlates with histone deacetylation and
reduced transcription (Murphy et al., 1999). Moreover, the histone deacetylase inhibitor
trichostatin A (TSA) can abrogate the ability of p53 to repress transcription of the p53
target genes Map4 and stathmin.
The arginine methyltransferases CARM1 and PRMT1 have also been implicated in p53
transcriptional activity, in that they co-operate with p300 in activating the GADD45 gene
and assemble with it on the GADD45 promoter during the p53 response (An et al., 2004).
Finally, several subunits of the ATP-dependent remodelling complex SWI/SNF have been
shown to bind p53. p53 is able to recruit hSNF and hBRG1 to p21 promoter. While over-
expression of hSNF5 and BRG1 stimulated p53-dependent transcription of a reporter
construct, dominant-negative forms of hSNF5 and BRG1 repressed transcription,
inhibiting p53-induced growth arrest and apoptosis (Lee et al., 2002).
It is apparent from these and other studies that regulation of local chromatin structure,
mediated through the varying actions of histone acetyltransferases and deacetylases,
histone methyltransferases and chromatin remodelling complexes, is important for p53
61

INTRODUCTION
function as a transcription factor. This might be a general mechanism occurring at p53-
responsive promoters, yet the factors determining the selection between different cellular
outcomes, are far less understood.
A recent study (Wei et al., 2005) revealed a mechanism by which growth arrest and
survival responses may be selected in respect of cell death. It was shown that upon DNA
damage the oncoprotein MUC1 is loaded with p53 on the p53REs in the p21 promoter.
Thus, MUC1 co activates p21 transcription at least in part through recruitment of CBP (and
displacing of HDAC1) to the p21 promoter with consequent increase in acetylation of
histone H4. Interestingly, MUC1 also binds to Bax proximal promoter in response to stress
and attenuates Bax transcription in a p53-independent way. In tumors MUC1 may provide
a survival advantage against genotoxic stresses in that activation of MUC1 might
contribute to a transient p53-mediated growth arrest, p53-independent suppression of Bax
activation and attenuation of the intrinsic apoptotic pathway as it is confirmed by the high
incidence of MUC1 over-expression in human tumors.
The molecular mechanism for selective pro-apoptotic promoter activation was
concomitantly addressed by another group (Tanaka et al., 2007). A complex experimental
approach combining ChIP and sucrose gradient ultracentrifugation followed by mass
spectrometry allowed isolating functionally distinct promoter-bound p53 assemblies in
vivo. This assessed that different promoter-bound p53 multiprotein complexes are present
in the cell and that each fraction, identified by differential sedimentation, may contain
unique proteins. This study also led to the identification of hCAS/CSE1L as a component
of some chromatin-bound p53 transcriptional complexes associated to PIG3, p53AIP1 and
p53R2 but not p21, PUMA and 14-3-3σ promoters. hCAS/CSE1L is required in these sites
to reduce repressive Lys27 methylation on histone H3 possibly by inhibiting an unknown
histone methyltransferase (HMT). A putative model suggests that hCAS/CSE1L co
regulates a subset of genes with p53 and the two proteins function together to facilitate the
formation of active chromatin at selected target promoter in human cells.
In another study aimed at investigating the mechanisms of stimulus-specific p53
transcriptional responses leading to cell-cycle arrest or apoptosis it was observed that
sustained transcriptional activation of p21 occurs in Nutlin-treated cells that undergo cell-
cycle arrest, but not in cells that undergo apoptosis in a stress-specific fashion (Donner et
al., 2007). This turned out to depend on differential assembly of the Mediator complex, a
multiprotein complex that functions as a coactivator and binds to the C-terminal domain of
62

INTRODUCTION
RNA polymerase II holoenzyme, acting as a bridge between this enzyme and transcription
factors (Bjorklund and Gustafsson, 2005). In particular, CDK8, a subunit of the CDK
module of Mediator, proved to function as a stimulus-specific transcriptional co-activator
within the p53 transcriptional program. Histone acetylation and association with core
Mediator subunits MED1 and MED17 is observed with in the p21 locus under both
conditions. In contrast, CDK8, cyclin C and MED12 are recruited to the p21 locus
exclusively after Nutlin 3 treatment, when p53-dependent p21 transcription is strongly
activated. CDK8 recruitment is also observed during p53-dependent activation of the
Mdm2 locus, and the levels of CDK8 occupancy at p53 target genes correlate positively
with gene activity. These data indicate that p53 binding to chromatin and p53-mediated
histone acetylation is not sufficient for transcriptional activation of p21, suggesting that
downstream events define the ultimate transcriptional status of this gene. A recent report
(Mattia et al., 2007) reached similar conclusions. Upon hydroxyurea (HU) treatment, p53
showed to be stabilized, to bind to the p21 locus, and to mediate histone acetylation.
However, p21 transcriptional activation was drastically impaired in HU-treated cells as
compared to cells in which p53 was activated by the DNA-damaging agent daunorubicin.
RNA Pol II was effectively recruited to the p21 promoter in response to HU treatment but
it failed to elongate into the intragenic region, revealing the importance of post-initiation
mechanisms in regulation of p21 transcription.
An elegant study by Espinosa et al. demonstrated that p53 is present before activation on
p21 promoter together with different amounts of poised PolII (Espinosa et al., 2003).
Although PolII is already loaded on promoters before activation, it is transcriptionally
paused and recruitment of elongation factors is needed in order to allow efficient
transcription. It has been proposed that in a stepwise process, p53 recognizes high-affinity
chromatin binding sites and mediates pre-initiation complex (PIC) assembly at some
promoters, but this action is not sufficient to achieve maximum transcriptional activation
due to RNA Pol II arrest. After DNA damage, increased p53 activity leads to completion of
the transactivation process by stimulating elongation of arrested RNA Pol II. Notably,
among p53 target promoters there are differences in RNA Pol II occupancy which might
establish different thresholds for activation of each particular gene. Indeed, growth arrest
genes like p21 and GADD45 show rapid induction kinetics and require little increase in
p53 levels to achieve activation, whereas most pro-apoptotic are expressed with
intermediate or late kinetics (Zhao et al., 2000).
63

INTRODUCTION
Taken together all these evidences suggest that many regulatory forces are involved in p53
transcriptional activation. The signal generated by p53 binding is filtered by the availability
of chromatin modifying/remodelling complexes and other coregulators of RNA Pol II
activity at a given promoter. Consequently, only a small subset of p53 target genes is
expressed in a given context. It might be hypothesized that p53 post-translational
modifications provide the means for selective coregulator recruitment. This is also
supported by the evidence of a correlation between particular post-translational
modifications and specific cellular outcomes. Indeed, besides the well established
association of Ser46 phosphorylation with apoptotic response, two recent reports indicated
that upon p53 binding to DNA, acetylation of p53 at Lys120 by MOF and TIP60 favors
selective activation of apoptotic genes (Sykes et al., 2006; Tang et al., 2006)
7.7. Cooperation with other transcription factors
The cooperation between p53 and other transcription factors that interact with discrete
DNA-binding sites within different promoters has shown to hugely influence the pattern of
gene expression in response to p53. An example of such cooperation is seen between p53
and NF-κB, another transcriptional activator that plays an important role in the regulation
of apoptosis. The cooperation between p53 and NF-κB reflects their ability to function
together to induce expression of apoptotic target genes regulated by promoters containing
both p53- and NF-κB-binding sites, such as that of the death receptor DR5 (Shetty et al.,
2005). Similarly, both p53 and Miz are required for the activation of expression of
p21WAF1/CIP1 (Seoane et al., 2002; Vousden, 2002).
An important contribution to the regulation of p53 activity is made by E2F1, a transcription
factor often implicated in apoptosis. It was previously known that E2F1 associates directly
to p53 thus modifying its biochemical properties and that the two cooperate in the
induction of apoptosis (Hsieh et al., 2002; Wu and Levine, 1994). In addition, E2F1 can
synergize with p53 to activate particular pro-apoptotic genes containing adjacent binding
sites for each of these two transcription factors, as best illustrated for the Apaf1 gene
(Moroni et al., 2001). This is probably an example of a broader generic mechanism, where
the availability of a particular transcription factor is expected to affect selectively the
ability of p53 to trigger transcription from genes containing responsive elements for both
p53 and that factor. This is further confirmed by the observation that, on the other way
64

INTRODUCTION
round, p53 is required for TGF-β response. p53 and SMADs in fact interact on separate cis
binding elements on a target promoter thus synergistically activating TGF-beta induced
transcription (Cordenonsi et al., 2003). Moreover p53 and Smad proteins have been found
to simultaneously occupy overlapping p53- and Smad-regulatory elements (SRE/p53RE) to
repress transcription of the onco-developmental tumor marker alpha-fetoprotein (AFP)
(Wilkinson et al., 2005; Wilkinson et al., 2008).
65

INTRODUCTION
8. Bromodomain proteins in transcriptional regulation
Bromodomains comprise an extensive family of evolutionarily conserved protein modules
of 110aa that have the ability to bind to acetylated lysines with high specificity (Haynes et
al., 1992; Zeng and Zhou, 2002). Bromodomain are found in many chromatin-associated
proteins and derive their name from the Drosophila protein Brahma, where it was
identified first (Elfring et al., 1998; Haynes et al., 1992). The finding that this domain have
specific acetyl-lysine binding properties (Dhalluin et al., 1999; Hudson et al., 2000;
Jacobson et al., 2000; Owen et al., 2000) highlighted how some protein-protein interaction
can be modulated by lysine-acetylation, with broad implications in a wide variety of
cellular processes including chromatin remodelling and transcriptional activation (Dyson et
al., 2001; Winston and Allis, 1999). Histone acetylation emerges as a central switch that
allows interconversion between repressive and permissive chromatin domains in terms of
transcriptional competence. Acetylation can loosen chromatin structure by neutralizing the
positive charges of histones and attenuating their interaction with (negatively charged)
DNA. This possibly increases the exposure of DNA to transcription factors and thus
correlates with gene activation (Horn and Peterson, 2002). Consistent with this notion,
transcriptional co-activators and co-repressors have been found to posses intrinsic HAT
and HDAC activities, respectively (Carrozza et al., 2003; Grozinger and Schreiber, 2002;
Sterner and Berger, 2000). Moreover, HATs and HDACs have shown to exert fundamental
roles in developmental processes and their deregulation has been linked to the progression
of diverse human disorders, including cancer.
Bromodomain containing proteins are important for sequential chromatin modifications by
HATs, HDACs and other chromatin remodelling proteins such as Swi2/Snf2, the ATPase
subunit of the SWI/SNF complex (Carrozza et al., 2003; Fry and Peterson, 2001; Pazin and
Kadonaga, 1997). Some bromodomain proteins in fact present additional domains with
specific enzymatic activity, through which they may directly mediate chromatin
modifications. Among these p300/CBP (Kraus et al., 1999; Manning et al., 2001), PCAF
(Dhalluin et al., 1999), TAFII250 (Jacobson et al., 2000) have HAT activity, the
transcriptional silencer Ash1L (Gregory et al., 2007) and the tumor suppressor RIZ1
(Steele-Perkins et al., 2001) have histone methyl transferase (HMT) activity and the SNF2α
and β subunits of the SWI/SNF complex have ATP-dependent remodelling functions
(Smith and Peterson, 2005). Conversely, other bromodomain proteins such as MTA1
(Nicolson et al., 2003) and ACF1 (Eberharter et al., 2001), that have no catalytic activity,
66

INTRODUCTION
may mediate communication between chromatin and multiprotein remodelling complexes.
These scaffolding functions contribute to epigenetic regulation of transcription through a
mechanism called the histone code (Rea et al., 2000; Strahl and Allis, 2000; Turner, 2000),
wherein combination of specific modifications of histones, such as acetylation, mediate the
recruitment of different chromatin-remodelling machinery.
By recognizing acetylated lysines at the flexible N-terminal tails of histones bromodomains
can also contribute at a larger scale, to the physical organization of chromosomes and
chromatin domains. An example of this last function is given by Bdf1, a yeast member of
the Brd/BET family of double bromodomain proteins, that binds to acetylated histone H4
and imposes a physical barrier between euchromatin and heterochromatin
(Matangkasombut and Buratowski, 2003). Brd2 and Brd4, two other members of the
Brd/BET family, are instead involved in cell cycle regulation. Brd2 activates cyclinA
transcription synergistically with oncogenic Ras leading to increased proliferation (Sinha et
al., 2005) while Brd4 arrests cell-cycle progression to S-phase through inhibitory
interaction with Replication factor C (Maruyama et al., 2002).
Lysine acetylation is also known to occur on many sequence-specific transcription factors
such as p53, c-Myb, and MyoD affecting their DNA-binding activity, association with co-
regulator, nuclear localization and stability (Avantaggiati et al., 1997; Barlev et al., 2001;
Polesskaya and Harel-Bellan, 2001; Sano and Ishii, 2001). Bromodomain-containing
proteins might mediate these processes by interacting with acetylated lysines on
transcription complexes. Interestingly it has been recently demonstrated that the acetylation
of Lys373 and Lys382 on p53 promotes the recruitment of the TFIID subunit TAF1 to the
p21 promoter through its double bromodomain. In this way TAF1 may function as a
specific transcriptional co-activator to bridge enhancer-binding proteins to the core
transcription machinery (Li et al., 2007a).
An important characteristic of bromodomain-acetyl lysine interaction is its specificity in
fact the presence of acetylated lysines is necessary but not sufficient for efficient binding
by bromodomains. Indeed, the bromodomain of CBP, but not that of p300 or PCAF,
specifically binds to Lys382-acetylated p53 being the flanking residues His380, Lys381
and Leu383 crucial for efficient and selective binding (Mujtaba et al., 2004).
Similar to other protein-interaction modules, such as SH2 domains for phospho-tyrosines,
the specific association of bromodomains with acetylated lysines might be a further mean
to grant dynamicity and regulability to the interaction networks existing within the cell.
67

INTRODUCTION
9. The Bromodomain-containing protein Brd7
Brd7 (Fig. 11) is a 75kDa protein, highly conserved from C.elegans to humans, that
displays a characteristic bromodomain moiety. A Brd7 homolog in C.elegans is called
C01H6.7 and two stretches of high homology with C01H6.7 are present in the C-terminal
portion of Brd7.
Brd7 bromodomain
shares the highest
similarity to the
bromodomains of a
human zinc finger
protein, Peregrin or
BR140, abundant in testes and spermatogonia (Cuppen et al., 1999; Thompson et al., 1994)
Fig. Schematic representation of the Brd7 protein
Fig. 11. Schematic representation of Brd7 domains.
The gene is located on chromosome 16q12, a region frequently deleted in Nasal-
Pharyngeal Carcinoma, an epithelial neoplasm whose pathogenesis is related to Epstein-
Barr virus infection and genetic abnormalities. Interestingly these tumors bear wild-type
p53 and gain metastatic properties upon p53 mutation (Chan et al., 2002).
Brd7 mRNA is ubiquitously expressed (Staal et al., 2000) and its expression has been
found reduced in NPC in respect to normal tissue (Yu et al., 2000). Brd7 localization is
predominantly nuclear due to the two bipartite NLS present at the N-terminal portion of the
protein within a basic domain rich in lysine residues (Zhou et al., 2006a). Brd7 contains a
single Bromodomain located between aa 129 and 239 and that suggests a putative function
in chromatin remodelling and transcriptional regulation.
It has been demonstrated that Brd7 co-localizes with hyper acetylated histones H3 and H4
(Peng et al., 2006; Staal et al., 2000) and that binds strongly to histone H3 acetylated on
Lys14 while only weakly to non-acetylated or to Lys9 acetylated form. The bromodomain
is fundamental for the selective interaction as a mutant depleted of the bromodomain is
impaired in binding to acetylated histones (Peng et al., 2006). Brd7 has also been shown to
co-localize with active PolII but not with BRG1, a component of a SWI/SNF chromatin
remodelling complex (Staal et al., 2000). Brd7 has no HAT activity and its over-expression
does not lead to an increase in histone acetylation thus its involvement in chromatin
remodelling at large scale is not likely (Peng et al., 2007). Moreover, Brd7 interacts with
68

INTRODUCTION
proteins involved in transcriptional regulation such as IRF-2, a transcription factor that
binds to ISRE and modulates cell-cycle dependent histone H4 gene expression (Staal et
al.,2000) and E1B-AP5, a member of the hnRNP family that negatively regulates
transcription (Kzhyshkowska et al., 2003). Thus the interaction with “open” chromatin
rather than chromatin modification might be the means by which Brd7 exerts its biological
function possibly by mediating the assembly of a functional transcriptional apparatus. Yet,
there are no strong evidences confirming this hypothesis.
An involvement of Brd7 in cell cycle regulation is suggested by a report where inducible
expression of Brd7 in a Nasal-Pharyngeal carcinoma cell line (HNE1) resulted in inhibition
of cell growth and cell cycle progression from G1 to S phase, therefore suggesting that
down-regulation of Brd7 found in this tumor type may provide a proliferative advantage.
Brd7 induction affected the expression of 13 genes belonging to ras/MEK/ERK and
Rb/E2F pathways. Among them P19-INK4D was up regulated while MEK, ERK1/2,
cyclinD1 and E2F3 were down regulated (Peng et al., 2007; Zhou et al., 2004). The
presence of the bromodomain was demonstrated to be essential for transcriptional
inhibition of E2F3 promoter in reporter assays, yet Brd7 could not be detected on the
endogenous E2F3 promoter by ChIP assay, possibly suggesting an indirect role for Brd7 in
regulation of E2F3 (Peng et al., 2006). Interestingly, in a study aimed at investigating the
mechanisms regulating Brd7 gene expression it emerged that binding sites for E2F and the
repressive E2F6 are present (together with SP1 binding site) within Brd7 promoter. This
suggests that Brd7 might be transcriptionally regulated during cell-cycle and that an auto-
regulatory loop may occur among Brd7 and E2F.
Brd7 interacts with two other bromodomain containing proteins, Brd2 and Brd3 (Zhou et
al., 2003b; Zhou et al., 2006b). These two proteins belong to the BET family and contain a
double bromodomain. While little is known about Brd3 several reports have characterized
Brd2 as a serine/threonine kinase that has elevated activity in human leukaemias. Brd2
transforms NIH/3T3 cells and is activated by mitogenic signals. Moreover its over-
expression leads to transactivation of the promoters of cyclin D1, cyclin A, cyclin E, and
dihydrofolate reductase (dhfr) genes in a manner dependent on Ras signalling, probably
through the formation of a complex with E2F1 and E2F2. All these evidence suggest that,
together with Brd7, it may play an opposite role in cell cycle-responsive transcription.
In agreement with the anti-proliferative role indicated for Brd7, a recent report showed that
Brd7 could block accumulation of β-catenin in the nucleus through up-regulation of α-
69

INTRODUCTION
70
catenin (Peng et al., 2007) Indeed, abnormal nuclear β-catenin accumulation is a common
event in NPC and this might be a consequence of Brd7 down-expression (Morrison et al.,
2004; Zheng et al., 1999)
In contrast, in a different cell context, it has been shown that Brd7 can enhance Wnt
signalling. Upon binding to the DIX domain of Dvl-1, Brd7 induces dephosphorylation of
GSK-3β at Tyr216. This negatively regulates kinase activity thus favouring β-catenin
activation (Kim et al., 2003). The actual mechanism is not known but it has been
hypothesized that Brd7 could target a Tyr-Phosphatase to the Dvl-1/Axin/APC/GSK-3β/β-
catenin complex to mediate dephosphorylation of GSK-3β. A candidate phosphatase is
PTP-BL that has been demonstrated to interact both with Brd7 and with APC (Cuppen et
al., 1999; Erdmann et al., 2000). Notably, these data suggest that Brd7 might exert
functions also in the cytoplasm, independent of its activities as transcriptional regulator. .
Therefore, it appears that Brd7 can exert different roles in control of proliferation
depending on cell context and further analysis is needed to better define Brd7 biological
functions.

AIM OF THE THESIS
The p53 tumor suppressor plays key roles in preventing cancer development, as
underscored by the fact that its pathway is compromised to some extent in almost all
human tumors. Significant advances have been made in understanding p53 functions, yet a
greater insight is needed on the specificity of p53 response.
Aim of this thesis is to understand the molecular mechanisms by which p53 response is
finely orchestrated and regulated. The first part of this study will focus on the role of the
prolyl isomerase Pin1 in regulating p53 apoptotic activity. In the second part, the functional
interaction between p53 and the novel interactor Brd7 will be characterized, as the
presence of the bromodomain and evidences from literature make it a promising candidate
for modulating the p53 pathway at the transcriptional level.
These studies are conducted in the light to achieving greater knowledge on the regulation
of the p53 pathway. This might be of relevance for understanding the tumorigenesis
process and could be exploited to design new targeted therapies for cancer treatment.
71

RESULTS - Part1
RESULTS - Part1 The prolyl-isomerase Pin1 has been shown to interact with p53 upon phosphorylation
induced by DNA damage (Zacchi et al., 2002; Zheng et al., 2002). Six Ser/Thr-Pro are
present on p53 sequence, namely Ser33/Pro34, Ser46/Pro47, Thr81/Pro82, Ser127/Pro128,
Thr150/Pro151 and Ser315/Pro316. Among these, Ser33, Ser46, Thr 81 and Ser315 are
phosphorylated by DNA damage-activated kinases (Bulavin et al., 1999; Bushmann et al.,
2001; Hofmann et al., 2001; D’Orazi et al., 2001, Radhakrishnan and Gartel, 2006)
therefore generating putative binding sites for Pin1. Pin1 has been shown to generate
conformational changes in p53 and to enhance its stability and transcriptional activity (Wulf
et al., 2002; Zacchi et al., 2002; Zheng et al., 2002). Both mouse and human cells lacking
Pin1 show impaired accumulation of p53 upon DNA damage (Zacchi et al., 2002; Zheng et
al., 2002; Mantovani et al., 2004) owing to its inability to efficiently dissociate from Mdm2.
Pin1 isomerase activity is in fact required for p53 stabilization (Zacchi et al, 2002). Notably,
the residues Thr81-Pro82 within the PRD have been shown to be crucial for Pin1 to promote
Chk2-dependent phosphorylation on Ser-20 (Berger et al., 2005), that favors dissociation of
the p53-Mdm2 complex (Sakaguchi et al., 1998; Shieh et al., 1997; Unger et al., 1999). In a
recent report from our group it has been demonstrated that the conformational change
induced by Pin1 on p53 upon prolyl-isomerization promotes p53 interaction with p300.
Indeed, p53 mutants unable to bind Pin1 were impaired in binding and acetylation by p300
as compared to wild type p53. Notably, the Thr81-Pro82 site within p53 PRD appeared to
be important for p53 mediated acetylation. Although these events are important for general
p53 activation, Pin1 appears to be particularly required for p53-dependent apoptosis.
Indeed, MEF Pin1-/- are strikingly impaired in apoptotic response and in the induction of the
pro-apoptotic p53 target genes Bax and Killer/DR5 upon genotoxic-stress and oncogene
expression (Zacchi et al., 2002). Thus it was of interest to investigate which critical steps in
the activation of p53-dependent apoptotic response are controlled by Pin1.
72
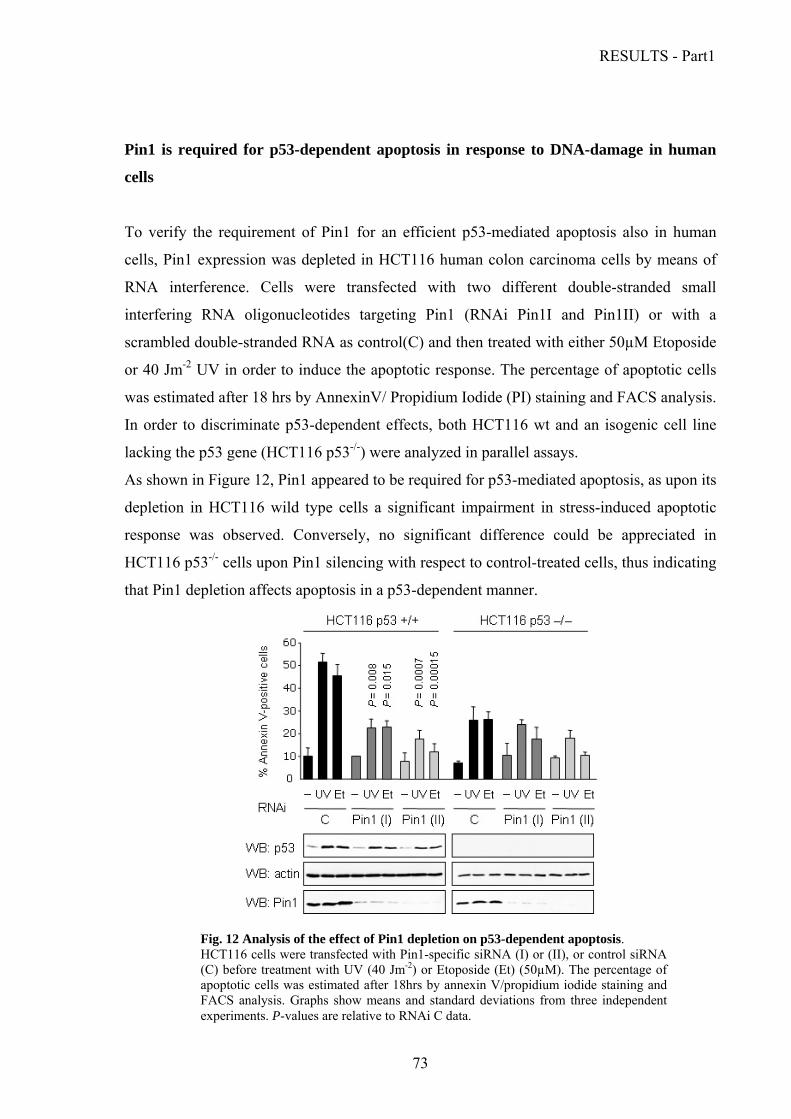
RESULTS - Part1
Pin1 is required for p53-dependent apoptosis in response to DNA-damage in human
cells
To verify the requirement of Pin1 for an efficient p53-mediated apoptosis also in human
cells, Pin1 expression was depleted in HCT116 human colon carcinoma cells by means of
RNA interference. Cells were transfected with two different double-stranded small
interfering RNA oligonucleotides targeting Pin1 (RNAi Pin1I and Pin1II) or with a
scrambled double-stranded RNA as control(C) and then treated with either 50µM Etoposide
or 40 Jm-2 UV in order to induce the apoptotic response. The percentage of apoptotic cells
was estimated after 18 hrs by AnnexinV/ Propidium Iodide (PI) staining and FACS analysis.
In order to discriminate p53-dependent effects, both HCT116 wt and an isogenic cell line
lacking the p53 gene (HCT116 p53-/-) were analyzed in parallel assays.
As shown in Figure 12, Pin1 appeared to be required for p53-mediated apoptosis, as upon its
depletion in HCT116 wild type cells a significant impairment in stress-induced apoptotic
response was observed. Conversely, no significant difference could be appreciated in
HCT116 p53-/- cells upon Pin1 silencing with respect to control-treated cells, thus indicating
that Pin1 depletion affects apoptosis in a p53-dependent manner.
Fig. 12 Analysis of the effect of Pin1 depletion on p53-dependent apoptosis. HCT116 cells were transfected with Pin1-specific siRNA (I) or (II), or control siRNA
(C) before treatment with UV (40 Jm-2) or Etoposide (Et) (50µM). The percentage of apoptotic cells was estimated after 18hrs by annexin V/propidium iodide staining and FACS analysis. Graphs show means and standard deviations from three independent experiments. P-values are relative to RNAi C data.
73

RESULTS - Part1
Pin1 is required for dissociation of the iASPP-p53 complex upon DNA damage It was of interest to investigate the mechanisms by which Pin1 could specifically assist p53
in mediating apoptosis. It is known from literature that the inhibitory factor iASPP leads to
downregulation of p53-mediated apoptosis by interacting with p53 both on its DBD and,
with higher affinity, on its PRD (Bergamaschi et al., 2006). Yet, the mechanism by which
this inhibition is relieved upon stress stimuli that induce p53 apoptotic response is not clear.
Interestingly, it has been demonstrated that iASPP interacts preferentially with the
polymorphic variant p53-Pro72 as compared with p53-Arg72. Besides the effect of the
aminoacid substitution on direct contact between the iASPP SH3 domain and p53 PRD
predicted by docking analysis (Bergamaschi et al., 2006), this might also depend on
different conformation of the two isoforms, that has been suggested by their different
electrophoretic mobility (Matlashewski et al., 1987). Since a Pin1 binding site within the
PRD has been shown to be important for p53 activation (see above), it was hypothesized
that a conformational change induced by Pin1 on p53 upon cytotoxic stress, might affect the
p53-iASPP interaction. To test this hypothesis a co-immunoprecipitation assay of
endogenous p53 and iASPP proteins was performed by F. Mantovani in the lab upon
blocking Pin1 expression by RNA interference in U2OS cells either treated with UV
radiation or not. In order to compare similar input levels of p53 in different conditions, p53
was stabilized in unstressed cells with the CBZ proteasome inhibitor. Immunoprecipitation
was performed with a polyclonal anti-iASPP antibody and the immunoprecipitated
complexes were analyzed for p53 presence by western blot with anti-p53 monoclonal
antibody (DO-I). Interestingly, in cells transfected with control siRNA (C) the interaction
between p53 and iASPP was decreased upon DNA damage (Figure 13). This suggests that
pro-apoptotic stress stimuli induce dissociation of p53 from iASPP in order to achieve an
effective apoptotic response. Most notably, in the absence of Pin1 stress-induced
dissociation of p53 from iASPP was impaired, indicating that Pin1 is required for this
process. Similar results were also observed upon Etoposide treatment (data not shown).
74

RESULTS - Part1
Fig 13. Analysis of the effect of Pin1 depletion on the dissociation of p53 from iASPP upon cytotoxic stress The interaction of endogenous p53 and iASPP proteins was analyzed by co-immunoprecipitation (IP). U2OS cells were transfected with Pin1-specific siRNA (Pin1) or control (C) siRNA and treated with either cytotoxic UV dosage (40 Jm-2) or with the proteasome inhibitor CBZ as a control. Immunoprecipitation of endogenous iASPP was performed with a polyclonal anti-iASPP antibody. Co-immunoprecipitated p53 was analyzed by western blot with monoclonal anti p53 antibody (DO-1). An aliquot of each protein lysate used for IP (5% of input) was also loaded as a control.
Pin1 directly mediates dissociation of p53 from iASPP
To understand whether the effect of Pin1 on p53 dissociation from iASPP requires direct
interaction with p53, p53 mutants in Pin1 binding sites were employed.
To verify that individual consensus binding sites for Pin1 are effectively recognized by Pin1
when phosphorylated, mutant p53 proteins with only one intact Pin1-consensus site (p53-
5M: Ser33-wt, Ser46-wt, Thr81-wt, or Ser315-wt) were generated. This was obtained by
disrupting the consensus sites for Pin1 binding (Ser/Thr-Pro) outside the DBD, that are
known to be phosphorylated by stress-activated kinases, through substitution of the Ser/Thr
residue to Ala by means of site-directed mutagenesis. A schematic representation of the
constructs is given in Figure 14A. The p53-5M proteins were then expressed in H1299 cells
75

RESULTS - Part1
and tested for binding to Pin1 in a GST pull-down assay. The mutant p53-6M, where all
Pin1 consensus sites are mutated, was used as negative control (the construct for p53 6M
was generated by A.Bisso in the lab). Cell lysates were incubated with either GST-Pin1 (or
GST alone as negative control) bound to Glutatione-sepharose beads. Parallel experiments
were also performed by treating protein lysates with λ-phosphatase prior to GST-pulldown.
Proteins associated with the beads were loaded on a SDS-PAGE gel and analyzed for the
presence of p53 by western blot with a monoclonal antibody (DO-1). The result indicated
that all the sites examined are bound by Pin1, however this binding is lost when these sites
are dephosphorylated (Figure 14B).
Therefore, a mutant at all four Pin1 binding sites outside the DBD (p53 4M) was
constructed by site-directed mutagenesis as previously described (performed by F.Agostini
in the lab). And is shown in Figure 14A. p53 4M and p53 6M were then compared for
binding to p53. As expected, it was observed that substitution of Ser33, Ser46, Thr81 and
Ser315 to alanine (p53-4M) greatly reduced the binding of p53 to glutathione S-transferase
(GST)-tagged Pin1 upon stress. The interaction was completely abolished when Ser127 and
Thr150 were additionally mutated to alanine (p53-6M) (Figure 14C) Notably, the residual
binding of p53 4M to GST-Pin1 indicates that at least one of the two Pin1 consensus sites
located within the DBD (Ser127 and/or Thr150) is also bound by Pin1.
Then, the ability of p53-4M and p53-6M to bind to iASPP was compared to that of p53 wt
upon stress treatment. To do this, H1299 cells were transiently transfected with either
p53wt, p53-4M or p53-6M expression constructs and treated with etoposide in order to
stimulate the dissociation of p53 from iASPP. Upon immunoprecipitation of endogenous
iASPP from cell lysates, immunoprecipitated complexes were analyzed by western blot with
anti-p53 antibody. As shown in Figure 15A it was observed that p53 mutants impaired in
binding to Pin1 displayed a stronger interaction with iASPP as compared to wt p53 upon
stress stimuli. This suggests that Pin1 might be directly involved in dissociation of p53 from
iASPP.
76

RESULTS - Part1
Subsequently, the ability of p53-4M and p53-6M mutants to induce apoptosis was compared
with that of wild-type p53. SaOS-2 p53-null cells were transiently transfected with either
wild type p53 (p53-wt), p53-4M or p53-6M expression constructs. pCDNA3 empty vector
was also transfected as control. The percentage of apoptotic cells was evaluated by
AnnexinV/PI staining and FACS analysis. The result presented in Figure 15 B showed that
the ability of p53-4M and p53-6M to promote apoptosis was greatly impaired with respect
to that of p53-wt, consistent with the different capacity to dissociate from iASPP.
Interestingly, the ability to promote apoptosis of p53 mutants was decreased proportionally
with their ability to bind to Pin1.
Fig. 14 Analysis of the requirement of Pin1 binding sites for p53 mediated apoptosis A. schematic representation of p53 indicating the positions of Pin1 consensus sites (phospho-Ser/Thr-Pro), TA, transactivation domain; DBD, DNA binding domain; NLS, nuclear localization signal. The p53 5M mutants with only one intact Pin1-consensus site (p53-5M: Ser33-wt, Ser46-wt, Thr81-wt, or Ser315-wt), the p53-4M with the four consensus sites outside the DBD mutated to Ala and the p53 6M mutant where all Pin1 consensus binding sites are mutated to Ala are also shown. B. The p53-5M constructs and p53-6M (described in A) were expressed in H1299 cells and were tested for binding to GST-Pin1. Control experiments were also performed by treating protein lysates with λ-phosphatase prior to GST pulldown; Binding reactions were compared with 10% of protein inputs by western blotting (WB). C. H1299 cells were transiently transfected with p53-wt, p53-4M or p53 6M and treated with 50µM etoposide for 12 hrs. GST pulldown was then performed using either GST-Pin1(P) or GST alone (G) as control; Binding reactions were compared with 10% of protein inputs by western blotting (WB).
77

RESULTS - Part1
Ser46 is required for Pin1-mediated regulation of p53-iASPP interaction
Fig. 15 Analysis of the requirement of Pin1 binding sites for p53 dissociation from iASPP and p53-mediated apoptosis A. The binding of iASPP with p53-wt, p53-4M or p53-6M was compared by co-IP upon transfection in H1299 cells and UV treatment (40 Jm-2). After 12 hrs the lysates were immunoprecipitated with anti-iASPP polyclonal antibody and the binding of p53 was evaluated by western blot with monoclonal anti p53 antibody (DO-1). The expression levels of iASPP and p53 in the input lysates were also determined by western blot. B. SaOS-2 cells were transiently transfected with p53-wt, p53-4M and p53-6M or with empty vector, and then treated with 40Jm-2 UV The percentage of apoptotic cells was estimated after 18 hrs. by AnnexinV/propidium-iodide staining and FACS analysis. The result of a representative experiment is shown, with western blots of total cell lysates confirming equal expression levels of the different p53 proteins.
To identify which Pin1 binding site(s) is directly involved in mediating p53 dissociation
from iASPP, we decided to focus on Ser46 and Thr 81. Ser46 has been largely demonstrated
to play a key role in p53-mediated apoptotic response as phosphorylation at this site is
specifically associated with the induction of pro-apoptotic genes. Thr81 is located within the
PRD where the main iASPP binding site lies and is important for Pin1 induced activation of
78

RESULTS - Part1
p53. We speculated that a conformational change at this site could be the means by which
Pin1 mediated p53 dissociation from iASPP. To indagate the involvement of Ser46 and
Thr81 in the dissociation of p53 from iASPP, H1299 cells were transfected with either wild-
type p53 or with two point mutants at Ser46 or Thr81 where Ser or Thr were substituted
with Ala (p53-S46A and p53-T81A). Cells were then treated with etoposide to stimulate
iASPP dissociation and co-immunoprecipitation with iASPP was performed as described
above. As can be observed in Figure 16 A, p53-S46A appeared to bind to iASPP more
strongly as compared to wt-p53. In contrast p53-T81A bound iASPP to the same extent as
wt-p53. This suggests that phosphorylation at Ser46 might reduce the interaction with
iASPP.
In order to confirm these results and to prove that Pin1 is able to induce p53 dissociation
from iASPP upon stress-dependent phosphorylation of Ser46, co-immunoprecipitation of
wt-p53, p53-S46A or p53-T81A with iASPP upon overexpressing Pin1 was performed.
(This experiment was performed by F.Mantovani). As reported in Figure 16B the
overexpression of Pin1 resulted in an increased dissociation of wt p53 and p53-T81A from
iASPP while it had only a slight effect on p53-S46A. These results together indicate that
Ser46 is required for Pin1-mediated regulation of p53-iASPP interaction.
Fig 16. Analysis of the requirement of specific Pin1 binding sites to mediate p53 dissociation from iASPP A. H1299 cells were transfected with p53wt, p53 S46A or p53 T81A and treated with Etoposide 10µM for 12 hrs. The lysates were immunoprecipitated with anti-iASPP polyclonal antibody. Bound p53 was detected by western blot with DO-1 monoclonal antibody B. H1299 cells were transfected with either wt-p53 or mutant S46A or T81A p53, and with vector encoding HA-Pin1 (+) or empty vector (–) and treated with UV (40 Jm-2). After 12 hrs, the interaction of p53 with endogenous iASPP was analyzed by co-immunoprecipitation with anti iASPP polyclonal antibody and western blot with anti-p53 (DO-1) and anti-iASPP monoclonal antibodies.
79

RESULTS - Part1
80
Results previously obtained in the lab (Mantovani et al., 2007) have demonstrated that Pin1
directly enhances the acetylation of p53 at Lys382 and Lys373 by p300. This evidence,
together with the data from literature indicating that phosphorylation at Ser 46 is required
for the acetylation of p53 at Lys 382 by CBP (Hofmann et al., 2002), lead to hypothesize
that Pin1-mediated dissociation of p53 from iASPP might depend on the increased
acetylation of p53. To test this possibility, a co-immunoprecipitation was performed by
comparing the binding of iASPP to p53-wt and a p53 mutant where the C-terminal lysines
had been mutated to arginines (p53-9KR, described in Ard et al. 2002). p53-wt and p53-
9KR expression constructs were transiently transfected in H1299 cells and p300 was also
transfected in order to induce p53 acetylation. The acetylation status of p53 was evaluated
by western blot with a polyclonal anti-AcK382 antibody. Co-immunoprecipitation was
performed with anti iASPP monoclonal antibody and with an unrelated antibody as a
control. As shown in Figure 17 it was observed that iASPP could bind to the same extent to
both wild type p53 and non-acetylable p53-9KR (compare lanes 2 and 4) and that
acetylation of p53 by p300 did not alter its binding to iASPP (compare lanes 2 and 3). These
results indicate that the interaction between p53 and iASPP occurs irrespectively of p53
acetylation on C-terminal lysines. Therefore it can be concluded that Pin1-dependent
acetylation of p53 and its dissociation from iASPP are independent events.
Fig. 17 Analysis of the effect of p53 C-terminus acetylation on iASPP binding. H1299 cells were transfected with either wild-type (WT) or 9KR p53 and with GFP-p300 as indicated. The ability of wt and p53-9KR to interact with endogenous iASPP was then compared by co-IP. C: control IP with non-immune serum. Expression of GFP-p300 and acetylation of p53 on Lys382 were analyzed by western blot.

RESULTS - Part2
RESULTS - Part 2
The overall picture of p53 regulation is highly complex yet the discovery of novel co-factors
that modulate its functions allows to gain insights on the multiple regulatory levels within
the p53 pathway. In order to increase the knowledge on p53 family members’ regulation by
analyzing their interaction profiles, a yeast two-hybrid screening of a human foetal brain
cDNA library has been previously performed in our laboratory. Wild-type p53, mutant
p53R175H, p63 and p73, deleted of their transactivation domains were used as baits.
Among the protein partners identified, the Bromodomain containing protein Brd7
demonstrated the capability to interact with all the three members of the p53 family and
with mutant p53 R175H. The presence of the bromodomain and the evidences from
literature that support a role in transcriptional activation rendered Brd7 an appealing
candidate for modulating the p53 pathway.
Analysis of Brd7 expression levels and half-life
Brd7 has been reported to be ubiquitously expressed in mouse tissues (Cuppen et al., 1999).
Brd7 expression levels were thus analyzed by western blot in human cell lines of different
origin, either transformed or not, that were commonly used in our lab. As shown in Figure
18 it was observed that Brd7 protein could be detected in all cell lines analyzed, with lower
expression levels in all primary (WI38) and immortalized (IMR90, HaCat) fibroblasts tested
as compared to tumor-derived cell lines (MCF-7, SkBr3, MDAMB468, HeLa, SHSY5Y,
293T, RKO, U2OS, HCT116, H1299, SaOS2 and HL60). Then, HCT1116 human colon
carcinoma cell line, expressing high levels of Brd7 protein, was chosen to analyze Brd7
protein stability. Brd7 half life was estimated by means of blocking protein synthesis in
HCT116 cells by treatment with ciclohexymide 50µM (CHX) and analyzing the amount of
Brd7 protein remaining at different time points by western blotting. p53 half life, known to
be of about 3-4 hours in the tumor cell lines examined was also evaluated as a control. As
shown in Figure 19, Brd7 protein is very stable, as its protein levels declined only upon 18
hours of CHX treatment and were reduced to a half after 24 hours. Similar results have been
obtained also in U2OS osteosarcoma cells (data not shown).
81

RESULTS - Part2
82
Fig. 19 Analysis Brd7 protein expression and stability A. Analysis of Brd7 protein levels in different cell lines. Total lysates from various primary cells and immortalized or tumor-derived cell lines were analyzed for Brd7 expression by western blotting. B. Analysis of Brd7 stability. A time course upon CHX treatment was performed in HCT116 cell lines. Cells were treated with CHX 50 µM and harvested at the time points indicated. Brd7 and p53 protein levels were analyzed by western blotting. Actin was used as a control.
Since the Brd7 promoter contains several E2F binding sites it was sought to test whether
Brd7 expression may be influenced by E2F transcription factors. To this aim, analysis of
Brd7 mRNA levels upon E2F1 induction was performed. An U2OS/ER-E2F1 inducible cell
line, expressing inducible E2F1 in fusion with estrogen receptor ligand binding domain
(ER) (kindly provided by K.Helin, Muller et al. 2001) was employed. Cells were treated
with 4-hydroxy-tamoxyfen (4OHT) for 24 hours to induce E2F1 activity or left uninduced
as control. Upon extraction of total mRNA and retro-transcription, RealTime PCR
amplification was performed with primers specific for Brd7 coding sequence and GAPDH
as a control. The result reported in Figure 20 indicated that Brd7 mRNA levels increase of
about two-fold 24 hrs after E2F1 induction.

RESULTS - Part2
Figure 20. Analysis of the induction of Brd7 expression by E2F1. U2OS/ER-E2F1 cells were treated with 4-hydroxy-tamoxyfen (4OHT) for 24 hours to induce E2F1 activity or left uninduced as control.Brd7 mRNA levels relative to GAPDH were analyzed by Real Time RT PCR.
Brd7 acetylation and oligomerization
Analysis of Brd7 protein sequence by means of the PROSITE protein motif database
predicted the presence of a Lysine-rich domain at its N-terminus (aa 3-91) which might be
subjected to acetylation. With the aim of determining whether Brd7 is acetylated pCDNA3-
Flag Brd7 expression vector was overexpressed in H1299 cells and purified by
immunoprecipitation. Subsequently, western blot analysis was performed with a monoclonal
antibody that specifically recognizes acetylated lysines (AcK). To discriminate background
signal the p53 non-acetylable mutant p53 9KR (see above) was used as a negative control.
The result presented in Figure 20 shows that Brd7 is indeed subject to acetylation.
Since lysine acetylation can provide a means for recognition and binding by bromodomain
motifs, it was hypothesized that upon acetylation Brd7 might oligomerize through its
bromodomain. To verify this possibility, in vitro pull-down assays were performed by
incubating full-length Brd7 transcribed and translated in vitro in the presence of 35S-
methionine (IVTT) with bacterially expressed MBP-fused N-terminal portion of Brd7
containing amino acids 1-236 and comprising the bromodomain (MBP Brd7 1-BD),
schematically represented in Figure 21B (upper panel). The result presented in figure 21B
demonstrates that Brd7 is able to oligomerize through its N-terminal domain.
83

RESULTS - Part2
84
Fig 20. Analysis of the acetylation and oligomerization of Brd7 A. H1299 cells were transiently transfected with either pCDNA3 Flag Brd7 or p53 9KR expression constructs. Upon immunoprecipitation with specific polyclonal antibodies, the acetylation status of Brd7 and p53-9KR was evaluated by western blot with a monoclonal antibody recognizing acetylated lysines (AcK). (The non-acetylable p53-9KR was employed as negative control) B. (Upper panel) Schematic representation of the MBP-fusion protein used in the MBP pull down assay. (Lower panel) Full length Brd7 expression construct was transcribed-translated in vitro (IVTT) in the presence of 35S-Met, and its interaction with MBPBrd7 1-BD was estimated by MBP pull down. 10 μl of the IVTT reaction were assayed for interaction with a recombinant protein encoding the N-term portion of Brd7 up to the bromodomain in fusion with MBP (MB-Brd7 1-BD) or MBP as control. Bound Brd7 was detected by autoradiography. The amounts of MBP proteins employed in the experiment were evaluated by Coomassie staining (right panel)
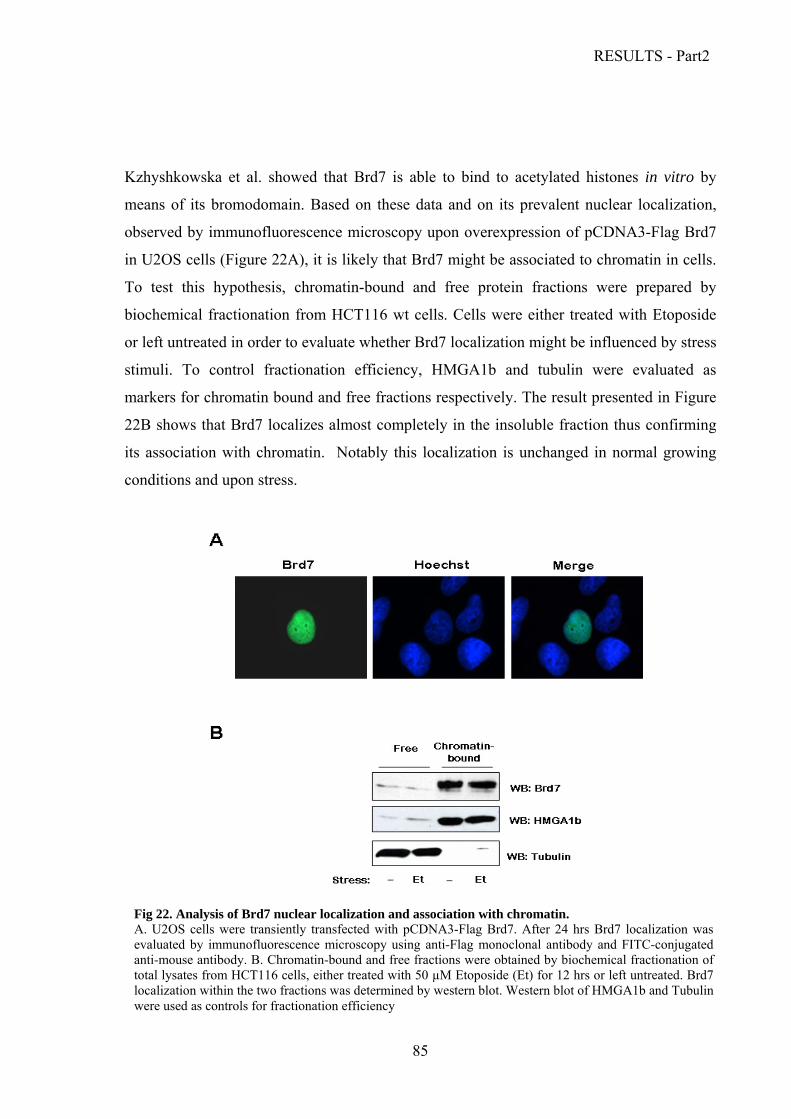
RESULTS - Part2
Kzhyshkowska et al. showed that Brd7 is able to bind to acetylated histones in vitro by
means of its bromodomain. Based on these data and on its prevalent nuclear localization,
observed by immunofluorescence microscopy upon overexpression of pCDNA3-Flag Brd7
in U2OS cells (Figure 22A), it is likely that Brd7 might be associated to chromatin in cells.
To test this hypothesis, chromatin-bound and free protein fractions were prepared by
biochemical fractionation from HCT116 wt cells. Cells were either treated with Etoposide
or left untreated in order to evaluate whether Brd7 localization might be influenced by stress
stimuli. To control fractionation efficiency, HMGA1b and tubulin were evaluated as
markers for chromatin bound and free fractions respectively. The result presented in Figure
22B shows that Brd7 localizes almost completely in the insoluble fraction thus confirming
its association with chromatin. Notably this localization is unchanged in normal growing
conditions and upon stress.
Fig 22. Analysis of Brd7 nuclear localization and association with chromatin. A. U2OS cells were transiently transfected with pCDNA3-Flag Brd7. After 24 hrs Brd7 localization was
evaluated by immunofluorescence microscopy using anti-Flag monoclonal antibody and FITC-conjugated anti-mouse antibody. B. Chromatin-bound and free fractions were obtained by biochemical fractionation of total lysates from HCT116 cells, either treated with 50 µM Etoposide (Et) for 12 hrs or left untreated. Brd7 localization within the two fractions was determined by western blot. Western blot of HMGA1b and Tubulin were used as controls for fractionation efficiency
85

RESULTS - Part2
Characterization of the interaction between Brd7 and the p53 family proteins.
Validation of the binding between Brd7 and the p53 family proteins in vitro
Brd7 was identified as a common interactor of all the three members of the p53 family and
of mutant p53 (R175H) in a yeast two-hybrid screening conducted in our laboratory. The
interaction was observed in three independent screenings using as a bait respectively p53,
mutp53 (R175H) or p73α lacking the N-terminal domain and was subsequently confirmed
also for p63 in secondary screening.
To confirm the interaction of Brd7 with full-length p53 family proteins an MBP-pull down
was performed between IVTT human Brd7 and bacterially produced MBP-fused full length
p53, p63 and p73. The result shown in Fig. 23 demonstrated that Brd7 is able to interact in
vitro with all the p53 family members.
Fig.23 Analysis of the interaction between Brd7 and the p53 family members
A. Full length Brd7 was in vitro transcribed-translated (IVTT) in presence of 35S-Met, and their interaction with the p53 family members was estimated by MBP pull down. 10 μl of each IVTT reaction were pulled down with MBP-fused p53, p63 or p73 recombinant proteins. Bound Brd7 was detected by autoradiography (upper panel). The amounts of MBP proteins used in the experiment were evaluated by Coomassie staining (lower panel)
86
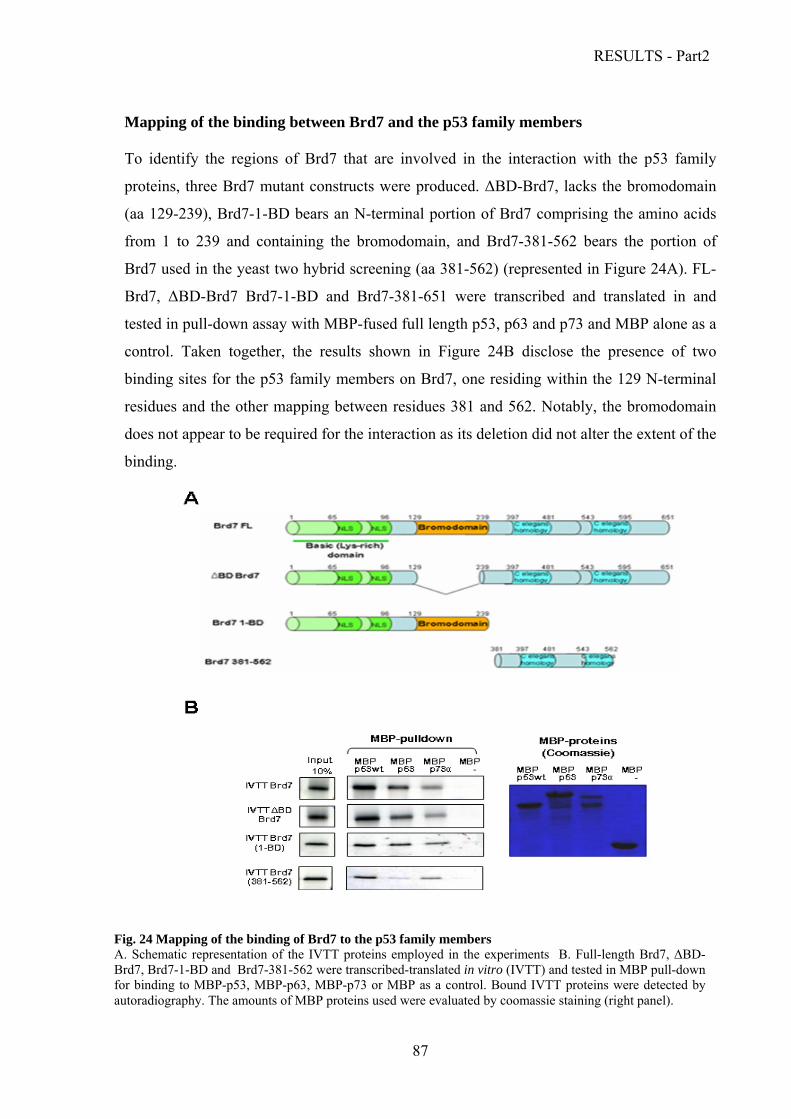
RESULTS - Part2
Mapping of the binding between Brd7 and the p53 family members
To identify the regions of Brd7 that are involved in the interaction with the p53 family
proteins, three Brd7 mutant constructs were produced. ΔBD-Brd7, lacks the bromodomain
(aa 129-239), Brd7-1-BD bears an N-terminal portion of Brd7 comprising the amino acids
from 1 to 239 and containing the bromodomain, and Brd7-381-562 bears the portion of
Brd7 used in the yeast two hybrid screening (aa 381-562) (represented in Figure 24A). FL-
Brd7, ΔBD-Brd7 Brd7-1-BD and Brd7-381-651 were transcribed and translated in and
tested in pull-down assay with MBP-fused full length p53, p63 and p73 and MBP alone as a
control. Taken together, the results shown in Figure 24B disclose the presence of two
binding sites for the p53 family members on Brd7, one residing within the 129 N-terminal
residues and the other mapping between residues 381 and 562. Notably, the bromodomain
does not appear to be required for the interaction as its deletion did not alter the extent of the
binding.
Fig. 24 Mapping of the binding of Brd7 to the p53 family members A. Schematic representation of the IVTT proteins employed in the experiments B. Full-length Brd7, ΔBD-Brd7, Brd7-1-BD and Brd7-381-562 were transcribed-translated in vitro (IVTT) and tested in MBP pull-down for binding to MBP-p53, MBP-p63, MBP-p73 or MBP as a control. Bound IVTT proteins were detected by autoradiography. The amounts of MBP proteins used were evaluated by coomassie staining (right panel).
87
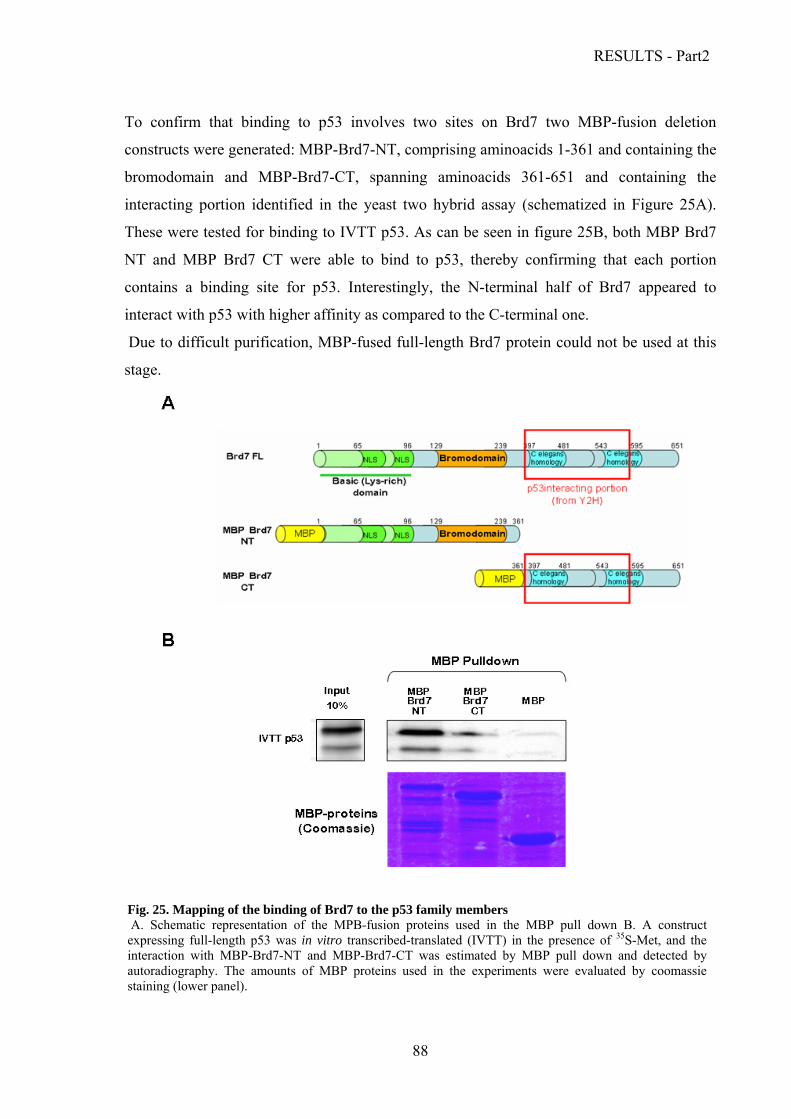
RESULTS - Part2
To confirm that binding to p53 involves two sites on Brd7 two MBP-fusion deletion
constructs were generated: MBP-Brd7-NT, comprising aminoacids 1-361 and containing the
bromodomain and MBP-Brd7-CT, spanning aminoacids 361-651 and containing the
interacting portion identified in the yeast two hybrid assay (schematized in Figure 25A).
These were tested for binding to IVTT p53. As can be seen in figure 25B, both MBP Brd7
NT and MBP Brd7 CT were able to bind to p53, thereby confirming that each portion
contains a binding site for p53. Interestingly, the N-terminal half of Brd7 appeared to
interact with p53 with higher affinity as compared to the C-terminal one.
Due to difficult purification, MBP-fused full-length Brd7 protein could not be used at this
stage.
88
Fig. 25. Mapping of the binding of Brd7 to the p53 family members A. Schematic representation of the MPB-fusion proteins used in the MBP pull down B. A construct expressing full-length p53 was in vitro transcribed-translated (IVTT) in the presence of 35S-Met, and the interaction with MBP-Brd7-NT and MBP-Brd7-CT was estimated by MBP pull down and detected by autoradiography. The amounts of MBP proteins used in the experiments were evaluated by coomassie staining (lower panel).

RESULTS - Part2
To start defining more in detail the regions of p53 involved in the binding to Brd7, MBP
pull-down assays were performed with different N-terminal and C-terminal deletions of p53
(schematically represented in Figure 26C).
MBP Brd7 NT and MBP Brd7 CT were tested for binding to 35S-labeled IVTT p53 full-
length (p53 FL), p53 1-298 (deleted of the oligomerization domain and the C-terminal
domain), p53 1-175 (encoding the N-terminal 175 residues) and p53 175-393 (deleted of the
first 175 residues). MBP Brd7 NT was also tested for binding to p53 251-393 (deleted of the
first 251 residues).
The results are shown in Figure 26. From these data, it was possible to conclude that p53 C-
terminal aa 298-393 are involved in the interaction with Brd7 N-terminal part, containing
the bromodomain, while p53 N-terminal aa 1-175 are involved in the interaction with Brd7
C-terminal portion.
As the region of Brd7 containing the bromodomain interacts with p53 C-terminus, where
most of the lysine residues known to be acetylated reside, it was hypothesized that this
interaction might be mediated by the bromodomain through binding to the acetylated lysines
of p53. To assess the role of p53 acetylation in this interaction, Brd7 binding to p53-wt and
the p53-9KR non acetylable was compared by pull-down. Quite surprisingly, p53-9KR
appeared to be still able to bind both Brd7-NT and Brd7-CT, thus suggesting that
acetylation of p53 is not required for binding to Brd7 (data not shown).
89

RESULTS - Part2
90
Figu
re
26:
Map
ping
of
th
e do
mai
ns
invo
lved
in
th
e in
tera
ctio
n be
twee
n B
rd7
and
p53
Con
stru
cts
expr
essi
ng
full-
leng
th
p53
or
p53
dele
ted
of
incr
easi
ng p
ortio
ns a
t its
C-te
rm o
r N
-term
(de
pict
ed i
n C
) w
ere
in v
itro
trans
crib
ed-tr
ansl
ated
(IV
TT)
in th
e pr
esen
ce o
f 35
S-M
et, a
nd th
eir i
nter
actio
n w
ith (A
) the
reco
mbi
nant
pro
tein
en
codi
ng e
ither
the
N-te
rm p
ortio
n (M
BP
Brd
7 N
T) o
r (B
) the
C
-term
inal
por
tion
(MB
P B
rd7
CT)
of
Brd
7 in
fus
ion
with
M
BP
was
est
imat
ed b
y M
BP
pull-
dow
n an
d de
tect
ed b
y au
tora
diog
raph
y. T
he a
mou
nts
of M
BP
prot
eins
use
d in
the
ex
perim
ents
wer
e ev
alua
ted
by C
oom
assi
e st
aini
ng (
low
er
pane
ls)
C S
chem
atic
rep
rese
ntat
ion
of t
he p
53 d
elet
ion
mut
ants
and
su
mm
ary
of t
heir
inte
ract
ion
with
MB
P B
rd7
NT
and
MB
P B
rd7
CT

RESULTS - Part2
Analysis of the interaction between Brd7 and the p53 family members in human cells.
Co-immunoprecipitation assays were performed on cell lysates from p53-null H1299 cells
upon over expression of wt p53, mutant p53R175H, p63α or p73α together with Flag-tagged
Brd7 or pCDNA3 as negative control. The lysates were immunoprecipitated with an anti-
Flag antibody hence analyzed by western blot with antibodies specific for p53, p63 and p73.
Results shown in Figure 27 confirmed that Brd7 interacts with p53 (A), mut p53 R175H
(B), p63 (C) and p73 (D) also in cells.
Fig. 27 Analysis of the interaction between Brd7 and the p53 family members in human cells H1299 cells were transiently transfected with either p53wt (A), mutant p53 R175H (B), p63 (C) or p73 (D) together with Flag-tagged Brd7. Brd7 was immunoprecipitated by means of the monoclonal antibody α Flag. Lysates of immunoprecipitated proteins were analyzed by SDS-PAGE followed by western blotting. Immunoprecipitated Brd7 was detected with anti-Flag antibody. Co-precipitated p53 (wt and mutant R175H), p63 and p73 were analyzed with the corresponding polyclonal antibodies. An aliquot of total lysate (5% input) was also loaded and analyzed by western blot.
91

RESULTS - Part2
The ability of Brd7 to interact with wild-type p53 and mutant p53 R175H was directly
compared by co-immunoprecipitation upon transient over expression in H1299 cells of
either p53 wt or mutant p53R175H together with Flag-tagged Brd7. As shown in Figure 28,
Brd7 appeared to bind more strongly to mutant p53 R175H with respect to wild type p53.
Thus it might be hypothesized that the altered conformation of p53R175H leads to the
disclosure of a stronger binding site for Brd7.
Fig.28 Comparison of the ability of Brd7 to bind to wt and mutant p53 (R175H) H1299 cells were transiently transfected with either p53 wt or mutant p53 R175H expression vectors, together with Flag-tagged Brd7 or pCDNA3 expression constructs. The interaction was then evaluated by immunoprecipitating Brd7 with the monoclonal antibody α-Flag and detecting bound p53 by western blot with the polyclonal antibody FL-393.
Finally, to assess that the binding of Brd7 to p53 occurs in cells among endogenous proteins
a co-immunoprecipitation of Brd7 and p53 from HCT116 cell lysates was performed. To
this purpose an anti-Brd7 polyclonal antibody was produced (described in Materials and
Methods section) and used for co-immunoprecipitation. The result presented in Figure 29
confirms that endogenous Brd7 and p53 interact in cells.
Fig. 29 Analysis of the interaction among endogenous p53 and Brd7 proteins in HCT116 cells. Total lysates from HCT116 cells were immunoprecipitated with anti Brd7 polyclonal antibody. Control immunoprecipitation was performed with non-immune serum (NRS). The immunoprecipitated proteins were analyzed by western blot with anti p53 and anti Brd7 monoclonal antibodies
92
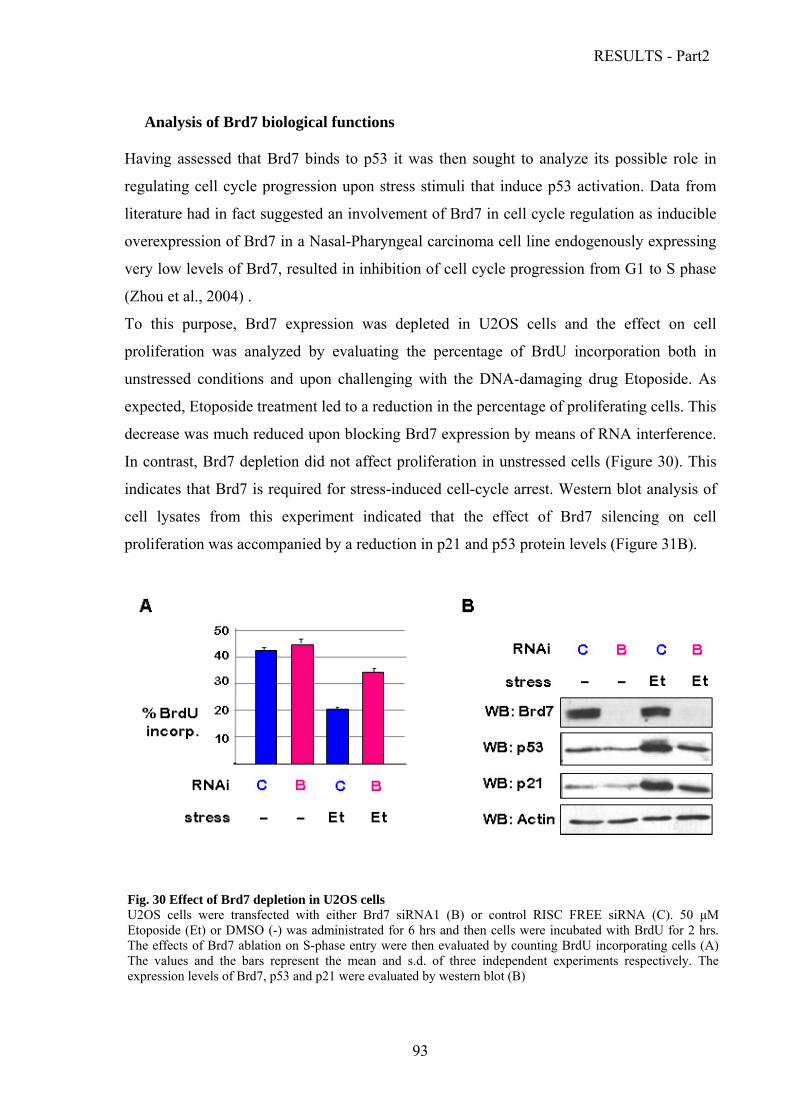
RESULTS - Part2
Analysis of Brd7 biological functions
Having assessed that Brd7 binds to p53 it was then sought to analyze its possible role in
regulating cell cycle progression upon stress stimuli that induce p53 activation. Data from
literature had in fact suggested an involvement of Brd7 in cell cycle regulation as inducible
overexpression of Brd7 in a Nasal-Pharyngeal carcinoma cell line endogenously expressing
very low levels of Brd7, resulted in inhibition of cell cycle progression from G1 to S phase
(Zhou et al., 2004) .
To this purpose, Brd7 expression was depleted in U2OS cells and the effect on cell
proliferation was analyzed by evaluating the percentage of BrdU incorporation both in
unstressed conditions and upon challenging with the DNA-damaging drug Etoposide. As
expected, Etoposide treatment led to a reduction in the percentage of proliferating cells. This
decrease was much reduced upon blocking Brd7 expression by means of RNA interference.
In contrast, Brd7 depletion did not affect proliferation in unstressed cells (Figure 30). This
indicates that Brd7 is required for stress-induced cell-cycle arrest. Western blot analysis of
cell lysates from this experiment indicated that the effect of Brd7 silencing on cell
proliferation was accompanied by a reduction in p21 and p53 protein levels (Figure 31B).
93
Fig. 30 Effect of Brd7 depletion in U2OS cells U2OS cells were transfected with either Brd7 siRNA1 (B) or control RISC FREE siRNA (C). 50 μM Etoposide (Et) or DMSO (-) was administrated for 6 hrs and then cells were incubated with BrdU for 2 hrs. The effects of Brd7 ablation on S-phase entry were then evaluated by counting BrdU incorporating cells (A) The values and the bars represent the mean and s.d. of three independent experiments respectively. The expression levels of Brd7, p53 and p21 were evaluated by western blot (B)

RESULTS - Part2
To verify that the reduction observed on p21 protein levels upon using Brd7 small
interfering RNA (siRNA B1) was a specific effect of Brd7 depletion, a different si RNA
was tested (siRNA B2). As can be seen in Figure 31, a reduction in p21 levels could be
observed with both siRNAs thus indicating that there was no off-target effect. siRNA B1
was then used to perform the experiment described below.
Figure 31. Analysis of the specificity of the effect of Brd7 depletion. U2OS cells were transfected with either Brd7 siRNA1 (B1), Brd7 siRNA 2 (B2) or control RISC FREE siRNA (C). 50 μM Etoposide (Et) or DMSO (-) was administrated for 12 hrs. The levels of p21, p53 and Brd7 were evaluated by western blot
To investigate whether Brd7 affects the induction of p21 at the transcriptional level, Brd7
expression was blocked by RNA interference in U2OS cells either treated with etoposide or
left untreated. Real Time RT-PCR of different p53 target genes was then performed, using
GAPDH housekeeping gene mRNA levels as control. The result shown in Figure 32
highlighted that induction of p21 transcription upon DNA damage was reduced when Brd7
expression was blocked by means of RNA interference. In contrast, there was only little
variation in Mdm2 and Puma mRNA levels. Under normal conditions, slight inhibition of
p21, Mdm2 and Puma transcription was observed.
94
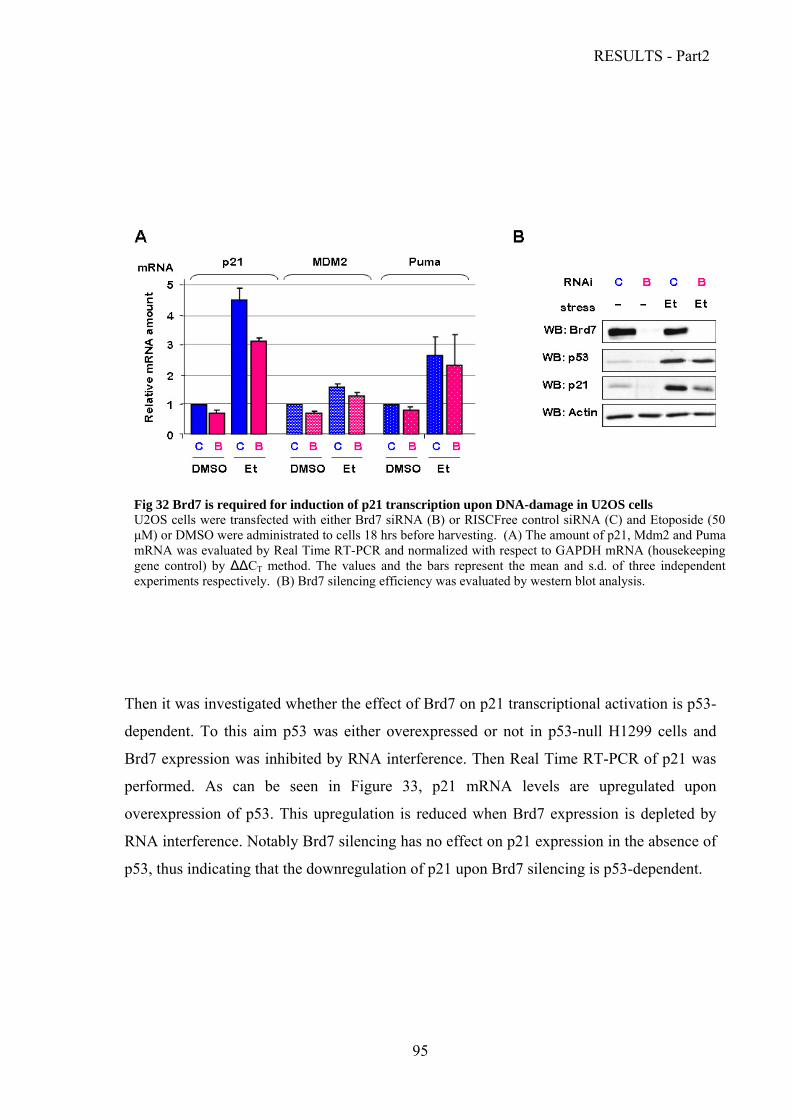
RESULTS - Part2
95
Fig 32 Brd7 is required for induction of p21 transcription upon DNA-damage in U2OS cells U2OS cells were transfected with either Brd7 siRNA (B) or RISCFree control siRNA (C) and Etoposide (50 μM) or DMSO were administrated to cells 18 hrs before harvesting. (A) The amount of p21, Mdm2 and Puma mRNA was evaluated by Real Time RT-PCR and normalized with respect to GAPDH mRNA (housekeeping gene control) by ΔΔCT method. The values and the bars represent the mean and s.d. of three independent experiments respectively. (B) Brd7 silencing efficiency was evaluated by western blot analysis.
Then it was investigated whether the effect of Brd7 on p21 transcriptional activation is p53-
dependent. To this aim p53 was either overexpressed or not in p53-null H1299 cells and
Brd7 expression was inhibited by RNA interference. Then Real Time RT-PCR of p21 was
performed. As can be seen in Figure 33, p21 mRNA levels are upregulated upon
overexpression of p53. This upregulation is reduced when Brd7 expression is depleted by
RNA interference. Notably Brd7 silencing has no effect on p21 expression in the absence of
p53, thus indicating that the downregulation of p21 upon Brd7 silencing is p53-dependent.

RESULTS - Part2
96
It is noteworthy that the above experiments (see western blots in Figures 30B and 32B)
highlighted a reduction in p53 levels upon Brd7 depletion. Preliminary data indicated that
this reduction does not occur at the transcriptional level, as p53 mRNA levels do not vary
upon block of Brd7 expression by means of RNA interference as compared to mock treated
control (not shown).This suggests that Brd7 depletion might influence p53 stability and that
the effect on p21 expression might, at least in part, be exerted through reduction of p53
levels.
e observed upon Brd7 depletion (not shown).
luciferase reporter assay was also performed upon overexpression of increasing amounts
Fig 33. The effect of Brd7 depletion on p21 transcription is p53-dependent H1299 cells were transfected with either Brd7 siRNA (B) or RISCFree control siRNA (C) together with pCDNA3-p53 or pCDNA3. The amount of p21 mRNA was evaluated by Real Time RT-PCR and normalized with respect to GAPDH mRNA (housekeeping gene control) by ΔΔCT method. Transfection efficiency was evaluated by western blot analysis. The values represent the mean of two independent experiments.
In order to confirm the effect of Brd7 on p21 transcription and to determine whether this
effect involves regulation of induction of the p21 promoter by p53 a luciferase reporter
assay was conducted. p53-null H1299 cells were co-transfected with constructs containing
p21 or Bax promoter upstream of the luciferase gene (p21-prom-luc and Bax-prom-luc)
along with p53 expression vectors and with either Brd7 siRNA or control siRNA. However
in these conditions no significant variation neither in p21 nor in Bax promoter induction
could b
A
of Brd7 together with p53 or pCDNA3 (Figure 34). From the result showed in Figure 34 it
appeared that Brd7 over-expression does not significantly alter p53 transcriptional activity
towards p21 promoter. Interestingly, it instead caused a reduction of Bax promoter trans-
activation by p53. This suggests that Brd7, when present at high levels, might participate in
transcriptional repression of Bax promoter.
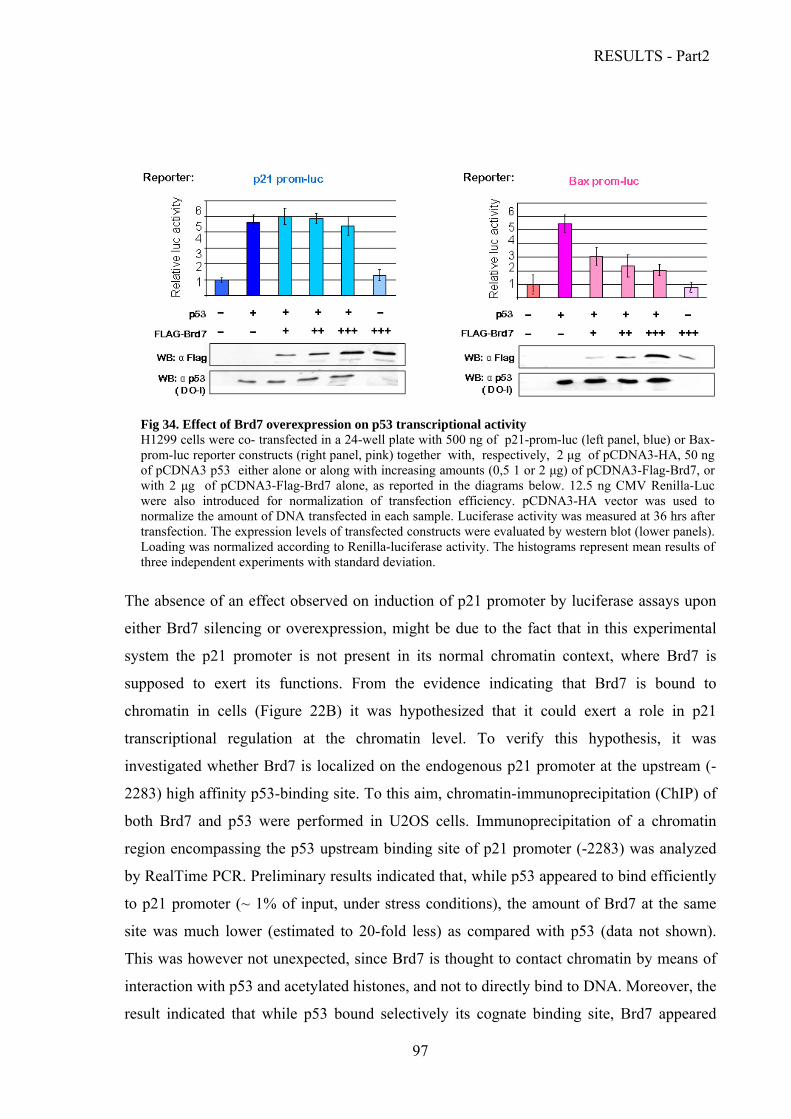
RESULTS - Part2
97
The absence of an effect observed on induction of p21 promoter by luciferase assays upon
either Brd7 silencing or overexpression, might be due to the fact that in this experimental
sy
su
ch
tra
in
2283) high affinity p53-binding site. To this atin-immunoprecipitation (ChIP) of
Fig 34. Effect of Brd7 overexpression on p53 transcriptional activity H1299 cells were co- transfected in a 24-well plate with 500 ng of p21-prom-luc (left panel, blue) or Bax-prom-luc reporter constructs (right panel, pink) together with, respectively, 2 μg of pCDNA3-HA, 50 ng of pCDNA3 p53 either alone or along with increasing amounts (0,5 1 or 2 μg) of pCDNA3-Flag-Brd7, or with 2 μg of pCDNA3-Flag-Brd7 alone, as reported in the diagrams below. 12.5 ng CMV Renilla-Luc were also introduced for normalization of transfection efficiency. pCDNA3-HA vector was used to normalize the amount of DNA transfected in each sample. Luciferase activity was measured at 36 hrs after transfection. The expression levels of transfected constructs were evaluated by western blot (lower panels). Loading was normalized according to Renilla-luciferase activity. The histograms represent mean results of three independent experiments with standard deviation.
stem the p21 promoter is not present in its normal chromatin context, where Brd7 is
pposed to exert its functions. From the evidence indicating that Brd7 is bound to
romatin in cells (Figure 22B) it was hypothesized that it could exert a role in p21
nscriptional regulation at the chromatin level. To verify this hypothesis, it was
vestigated whether Brd7 is localized on the endogenous p21 promoter at the upstream (-
aim, chrom
both Brd7 and p53 were performed in U2OS cells. Immunoprecipitation of a chromatin
region encompassing the p53 upstream binding site of p21 promoter (-2283) was analyzed
by RealTime PCR. Preliminary results indicated that, while p53 appeared to bind efficiently
to p21 promoter (~ 1% of input, under stress conditions), the amount of Brd7 at the same
site was much lower (estimated to 20-fold less) as compared with p53 (data not shown).
This was however not unexpected, since Brd7 is thought to contact chromatin by means of
interaction with p53 and acetylated histones, and not to directly bind to DNA. Moreover, the
result indicated that while p53 bound selectively its cognate binding site, Brd7 appeared

RESULTS - Part2
98
tion was performed on U2OS cells
matin-bound p53 at this residue.
instead to be loaded both on p21 promoter and on GAPDH genomic sequence, thus
indicating that it might be widely distributed on chromatin. Nonetheless, a preference for
p21 over GAPDH was apparent. Further investigation is needed in order to asses if Brd7
recruitment on p21 promoter is p53 or stress-dependent
.Moreover to investigate whether Brd7 depletion could influence p53 loading on p21
promoter at its upstream (-2283) binding site, ChIP was performed with anti-p53 antibody
from U2OS cells transfected with Brd7 specific siRNA or control siRNA and either treated
with etoposide or left untreated. Preliminary results indicated that Brd7 is not required for
recruitment of p53 to its upstream (-2383) high affinity binding site on p21 promoter upon
stress conditions (not shown).
To investigate whether Brd7 could affect p53 transcriptional activity by altering its
acetylation status on chromatin, a biochemical fractiona
transfected with either Brd7 specific siRNA or with control siRNA. Cells were either treated
with etoposide to induce p53 activation or left untreated. Chromatin-bound fraction was
analyzed by SDS-PAGE. Western blot was then performed with a polyclonal antibody that
specifically recognizes p53 acetylated at Lys382 as this modification is associated with p53
transcriptional activation. The result shown in Figure 35 indicates that Brd7 is not required
for effective acetylation of chro
Fig. 35 Brd7 does not influence p53 acetylation at Lys-382 on chromatin.
Biochemical fractionation was performed on lysates from U2OS cells either transfected with Brd7 siRNA (B) or control siRNA (C) and treated with etoposide 50 µM for 12 hrs. Chromatin bound fraction was analyzed by western blot with anti p53acetyl-lys382 .Protein loading was normalized in order to obtain equal levels of total p53 in the samples transfected with Brd7siRNA or control siRNA that were subjected to the same treatment. Western blot of HMGA1b was used as control for fractionation efficiency

DISCUSSION
DISCUSSION The p53 signal transduction pathway is a complex network finely coordinated to sense a
plethora of intrinsic and extrinsic stress signals and to mediate a response in order to
preserve genomic stability and cellular homeostasis. When activated in response to
different kind of stimuli, p53 may induce the expression of a large number of genes that
may be divided into categories depending on their functions. Notably, p53 activities are not
only circumscribed within the nucleus where it acts as a transcription factor. Indeed, it has
been demonstrated that p53 translocates to mitochondria where it directly mediates the
activation of the apoptotic program (Marchenko et al., 2000). Depending on the cellular
context and on the nature of the stimuli that activate p53, the ultimate outcome can be quite
different, ranging from the induction of reversible cell cycle arrest, apoptosis and
senescence to protective antioxidant activities and DNA repair. p53 activities are finely
regulated through a large number of post-translational modifications and interactions of
other proteins that direct the execution of the appropriate cellular response. Throughout
many years of research, considerable knowledge on p53 activities has been achieved.
However, less is understood on the mechanisms that dictates the specificity of its response.
New insights on the regulation of p53 apoptotic response have been gained with the
discovery of new interactors such as the ASPP protein family, composed by ASPP1,
ASPP2 and iASPP. ASPP1/2 interact with p53 and specifically enhance p53-induced
apoptosis, while iASPP competes with ASPP1/2 for binding to p53 and has an inhibitory
effect on p53-mediated apoptotic response, yet the mechanisms at the basis of these effects
are still unclear.
The identification of different co-factors and the discovery of novel mechanisms that
regulate p53 activity at different levels by modulating its functions will thus be of great
help to understand how p53 activation can lead to different cellular outcomes in response
to various stimuli.
Among the various p53 modulators, the prolyl isomerase Pin1 plays key roles in
transducing stress signalling-induced phosphorylations into conformational changes that
99

DISCUSSION
determine p53 activation (Zacchi et al., 2002; Zheng et al., 2002; Wulf et al., 2002). Upon
DNA damage and phosphorylation at Ser/Thr-Pro sites, Pin1 has been shown to interact
with p53 and to influence its stability and activity at different levels (Zacchi et al., 2002;
Zheng et al., 2002; Wulf et al., 2002). Pin1 isomerase activity is in fact required for p53
accumulation upon stress stimuli, by favoring dissociation of the p53-Mdm2 complex
(Zacchi et al., 2002). Recently, data from our group highlighted also a role for Pin1 in
assisting p53 efficient loading on its cognate promoters (Mantovani et al., 2007).
Moreover, it was demonstrated that Pin1 promotes binding of p300 and subsequent
acetylation of p53, and in this way it might enhance p53 transcriptional activity (Mantovani
et al., 2007). Notably, data from literature highlighted that the presence of Pin1 is
particularly required for p53-dependent apoptosis (Zacchi et al., 2002; Zheng et al., 2002).
In this work a new mechanism was identified by which Pin1 specifically influences p53
apoptotic activity. It has been demonstrated that Pin1 directly binds to its consensus sites at
Ser33/Pro34, Ser46/Pro47, Thr81/Pro82, and Ser315/Pro316, and likely also to
Ser127/Pro128 and/or Thr150/Pro151 when these sites are phosphorylated. Most
importantly it has been shown that upon stress conditions Pin1 directly modulates the
dissociation of p53 from the inhibitory factor iASPP. This sheds light on the mechanism
through which inhibition of p53-mediated apoptotic response by iASPP is relieved in
response to stress stimuli and indicates that Pin1 plays a direct role in this process. We
speculated that a conformational change mediated by Pin1 at its Thr81-Pro82 within the
PRD, where the main binding site for iASPP lies, could be the means by which Pin1
mediated p53 dissociation from iASPP. Yet this proved not to be the case as mutating
Thr81 to Ala did not alter the interaction between p53 and iASPP as compared with wild
type p53. Interestingly, it emerged instead that Ser46 is required for Pin1-mediated
dissociation of the p53-iASPP complex and this might, at least in part, account for the
relevance of this site for p53 mediated apoptosis (Feng et al., 2006). Indeed,
phosphorylation of p53 at Ser46 is specifically induced by severe or persistent stress
conditions and represents a major event in the specific commitment of the p53-mediated
response to apoptotic cell death (D'Orazi et al., 2002; Oda et al., 2000; Taira et al., 2007).
The data presented in this thesis together disclose a mechanism by which, upon genotoxic
stress-induced phosphorylation of Ser46, Pin1 is recruited at p53 Ser46-Thr47 site and it
mediates dissociation of the p53-iASPP complex thus relieving the inhibition to p53-
mediated apoptosis exerted by iASPP. Interestingly, iASPP does not contact p53 in the
100
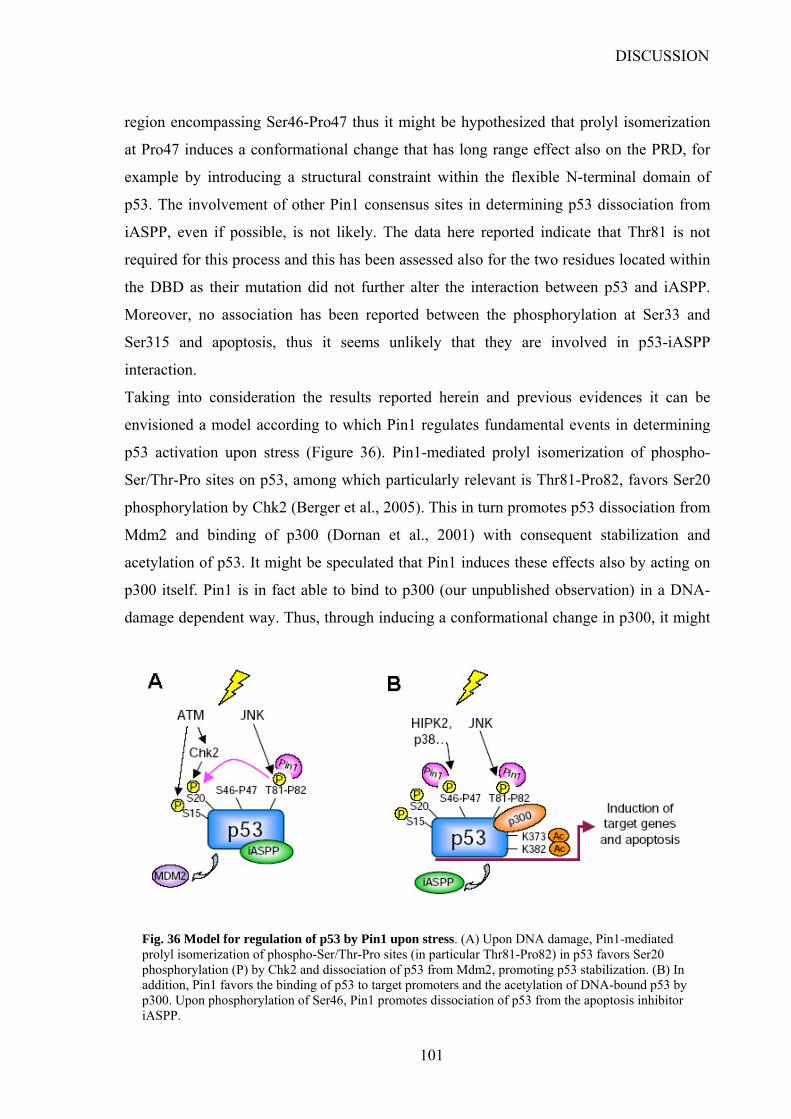
DISCUSSION
region encompassing Ser46-Pro47 thus it might be hypothesized that prolyl isomerization
at Pro47 induces a conformational change that has long range effect also on the PRD, for
example by introducing a structural constraint within the flexible N-terminal domain of
p53. The involvement of other Pin1 consensus sites in determining p53 dissociation from
iASPP, even if possible, is not likely. The data here reported indicate that Thr81 is not
required for this process and this has been assessed also for the two residues located within
the DBD as their mutation did not further alter the interaction between p53 and iASPP.
Moreover, no association has been reported between the phosphorylation at Ser33 and
Ser315 and apoptosis, thus it seems unlikely that they are involved in p53-iASPP
interaction.
Taking into consideration the results reported herein and previous evidences it can be
envisioned a model according to which Pin1 regulates fundamental events in determining
p53 activation upon stress (Figure 36). Pin1-mediated prolyl isomerization of phospho-
Ser/Thr-Pro sites on p53, among which particularly relevant is Thr81-Pro82, favors Ser20
phosphorylation by Chk2 (Berger et al., 2005). This in turn promotes p53 dissociation from
Mdm2 and binding of p300 (Dornan et al., 2001) with consequent stabilization and
acetylation of p53. It might be speculated that Pin1 induces these effects also by acting on
p300 itself. Pin1 is in fact able to bind to p300 (our unpublished observation) in a DNA-
damage dependent way. Thus, through inducing a conformational change in p300, it might
Fig. 36 Model for regulation of p53 by Pin1 upon stress. (A) Upon DNA damage, Pin1-mediated prolyl isomerization of phospho-Ser/Thr-Pro sites (in particular Thr81-Pro82) in p53 favors Ser20 phosphorylation (P) by Chk2 and dissociation of p53 from Mdm2, promoting p53 stabilization. (B) In addition, Pin1 favors the binding of p53 to target promoters and the acetylation of DNA-bound p53 by p300. Upon phosphorylation of Ser46, Pin1 promotes dissociation of p53 from the apoptosis inhibitor iASPP.
101

DISCUSSION
favour its association with p53 or even determine a switch from E4-ubiquitin-ligase to
acetyl-transferase activity. In this way, upon DNA damage Pin1 would favour p53 stability
both by enhancing its dissociation from Mdm2 and by preventing p53 poly-ubiquitination
by p300.
Moreover, Pin1 favors the binding of p53 to its target promoters and, being recruited on
chromatin with p53 it might assist structural rearrangements of p53 that occur upon DNA
binding, exposing its C terminus to p300 (Ceskova et al., 2006) and thus promoting the
DNA-dependent acetylation and transcriptional function of p53 as well as the organization
of transcription complexes on the promoter (Barlev et al., 2001). Thr81-Pro82 site within
p53 PRD seems to play an important role also in these steps as its mutation leads to a
decrease in acetylation of p53 bound to its target promoters (Mantovani et al., 2007). In
addition, Ser33 and Ser315 have been recently reported to be phosphorylated by CDK9
(Radhakrishnan and Gartel, 2006), a component of the transcription elongation factor
pTEFb, thus it might e suggested that if Pin1 directly regulates p53 transcriptional activity
upon recruitment on chromatin (Mantovani et al., 2007) also through coordinating the
recruitment of specific elongation factors on p53 responsive promoters
In addition to these effects that mediate general activation of p53 response, Pin1 plays a
role in specifically regulating p53 dependent apoptosis. Upon Ser46 phosphorylation in
response to severe stress stimuli, Pin1 binds to its consensus site at Ser46-Pro47 and may
provide the switch that allows p53 dissociation from iASPP thus activating p53 apoptotic
response. Interestingly, this effect appears to be independent from Pin1-induced acetylation
of p53, further confirming that Pin1 may modulate p53 activity at different levels.
Moreover it might not be excluded that Pin1 exerts its effect by acting directly also on
iASPP. It might be speculated that Pin1 favors p53 apoptotic activity also by assisting p53
functions at mitochondria were both can localize (Marchenko et al, 2000; our unpublished
observations). Interestingly, the pro-apoptotic member of the ASPP family ASPP2 has
been found to localize at mitochondria were it interacts with Bax stimulating Cytochrome
C release into cytoplasm. It is thus likely that Pin1 might favor the interaction between p53
and ASPP2 to promote apoptosis. Otherwise it is possible that also iASPP localizes at
mitochondria thereby preventing p53 from triggering apoptosis. Thus, through the
mechanism described in this work, Pin1 would relieve iASPP-mediated inhibition and
favor p53 apoptotic activity.
102

DISCUSSION
Ser46 phosphorylation (Sullivan et al., 2004) together with iASPP interaction
(Bergamaschi et al., 2006) have been indicated as important determinants of the differential
apoptotic response of p53 Pro72 and p53 Arg72 isoforms at the transcriptional level. Work
from our group demonstrated that more efficient Ser46 phosphorylation occurs on p53
Arg72 than on p53 Pro72 upon stress and indeed Pin1 binds more to p53 Arg72
(Mantovani et al., 2007). This indicates that Pin1 plays a role also in determining the
differential apoptotic potential of the two isoforms. In fact, higher efficiency in
phosphorylation at Ser 46 would determine an increased Pin1-mediated dissociation of the
p53-iASPP complex in the case of p53Arg72 with respect to p53 Pro72. This would add to
the already weaker interaction with iASPP of the p53 Arg72 isoform due to the effect of
the amino acid substitution on direct contact between p53 PRD and iASPP SH3 domain.
Interestingly, in breast cancers Pin1 over-expression correlates inversely with iASPP over-
expression. These evidences favours a model where overexpression of Pin1 would be
selected against in tumours bearing wt p53 Pro72, as it would counteract iASPP-dependent
inhibition of p53-mediated apoptosis. Conversely, in cancers with wt-p53 72 Arg/Arg,
there would be no requirement to select against Pin1 over-expression because iASPP does
not efficiently target this p53 isoform, thus cancer cells would take advantage of the pro-
tumorigenic effects of Pin1 (as the amplification of pro-oncogenic pathways through cyclin
D or β-catenin) without being affected by its pro-apoptotic function.
As Pin1 governs the functions of several substrates, and it is itself under control of
signalling pathways that could become aberrantly activated in cancers (Wulf et al., 2005) is
not surprising that its abundance has been associated both positively and negatively with
cancer, highlighting the relevance of the cellular context in determining whether Pin1
inhibits or amplifies cell-growth pathways. Due to its pleiotropic effects, Pin1 might play
contrasting roles in different phases of tumorigenesis. In normal cells or at early stages it
might be involved in the regulation of cell-cycle check-points (Atchison et al., 2003; You
et al., 2002) and its overexpression would be favourable to contrast tumor initiation by
enhancing p53-dependent apoptotic response. Yet, at more advanced tumor stages,
concomitant with deregulation of signalling pathways, it may instead contribute to the
amplification of proliferative signals (Wulf et al., 2005) thus promoting tumor progression
and maintenance, in particular in p53wt Arg/Arg contexts. In this case, development of
specific Pin1 inhibitors would be potentially effective in cancer treatment to counteract the
amplification of proliferative signals. Yet, based on our observations, at early stages,
103

DISCUSSION
inhibition of Pin1 would be predicted to inhibit cytotoxicity and hence the clinical activity
of agents inducing p53-dependent apoptosis. Thus the disclosure of novel mechanisms in
the interplay between p53 and its co-regulators, such as the one reported in this thesis,
would be of relevance in designing targeted therapies for cancer treatment.
Analyzing the protein interaction profiles is a powerful system to gain insight on the
mechanisms governing p53 regulation. From a yeast two-hybrid screening previously
performed in our laboratory, the bromodomain containing protein Brd7 emerged as a
common interactor of the p53 family members (as well as of mutant p53 R175H). The
presence of the Bromodomain and the evidences from literature supporting a role in
transcriptional activation made Brd7 a promising candidate for modulating the p53
pathway at the transcriptional level. This protein and its functional interaction with p53
have been therefore characterized in the second part of this thesis.
The interaction emerged among Brd7 and the p53 family members has been validated both
in vitro and in cells, disclosing the presence of two binding sites for p53 on Brd7. One,
located in the C-terminal region of Brd7, interacts with p53 N-terminal portion spanning
from aa 74 to aa 175. Interestingly, in these assays the p53 deletion mutant 1-175 appear to
bind even strongly than p53 full length, it might be hypothesized that a region present in
p53 C-terminal portion could exert an inhibitory effect on this interaction, or, most likely,
that this deletion, occurring within the p53DBD might cause an improper folding leading to
the disclosure of a fictitiously stronger binding site.
A second binding site for p53 maps at the N-terminus of Brd7, in a region that contains the
bromodomain. Interestingly, this region has shown to interact with p53 CTD where most of
the acetylable lysine residues are located. It was thus supposed that Brd7 bromodomain
might bind to acetylated lysines at p53 CTD. Yet, preliminary investigations suggested that
the interaction between Brd7 N-terminus and p53 occurs irrespective of p53 acetylation
status. Conversely, in vitro assays indicated that the bromodomain might mediate Brd7
oligomerization. This might involve acetylated lysines within Brd7 lysine-rich domain.
However, as this interaction has been tested only in vitro, further investigation is needed to
asses whether it actually occurs in cells. The functional significance of Brd7
oligomerization has not been investigated, yet an auto-inhibitory effect might be speculated
in preventing the interaction with acetylated histones by keeping the bromodomain
104

DISCUSSION
inaccessible. Conversely it might stabilize multi-protein complexes on chromatin.
Moreover it would be interesting to determine whether Brd7 is constitutively acetylated or
this modification is subjected to regulation.
Data from literature suggested an involvement of Brd7 in cell cycle regulation (Peng et al.,
2007; Zhou et al., 2004). Overexpression of BRD7 could inhibit NPC cell growth and
block cell cycle progression from G1 to S phase. This was ascribed to modulation of the
expression of factors involved in Ras/MEK/ERK and Rb/E2F pathways, such as E2F3,
cyclin D1 and MEK1 (Zhou et al., 2004). Notably, Brd7 could significantly inhibit E2F3
promoter activity in luciferase assays, yet ChIP analysis did not show the presence of Brd7
on the endogenous promoter. The role for Brd7 as a transcriptional regulator had also been
suggested by other reports indicating that Brd7 could interact with other transcription
factors such as E1B-AP5 (early adenovirus E1B-55kD association protein) (Kzhyshkowska
et al., 2003) and IRF2 (Interferon Regulatory Factor-2) (Staal et al., 2000).
In this thesis it was demonstrated that Brd7 is required for efficient cell-cycle arrest in
U2OS cells upon challenging with genotoxic stimuli. This effect was, at least in part, due
to a reduction in p21 expression that occurred upon Brd7 depletion and under stress
condition. The down-regulation of p21 as a consequence of Brd7 silencing occurred at the
transcriptional level and proved to be p53-depedent. Notably, this effect seemed to be
specific for p21 as the stress-dependent induction of two other p53 targets, Mdm2 and
Puma, appeared to be almost unaffected. Interestingly, Brd7 depletion lead to a decrease in
p53 protein levels, thus it might be hypothesized that it exerts its effect also regulating p53
abundance.
On the basis of these data and of the evidence demonstrating that Brd7 is localized within
the nucleus in the chromatin bound fraction, it was hypothesized that Brd7 might be
involved in regulation of p21 promoter induction by p53. Reporter assays showed no
significant variation in p21 promoter induction by p53 upon Brd7 silencing. However the
role of Brd7 might require an endogenous chromatin context, and this might explain the
lack of effect on a p21 promoter fragment cloned into an expression vector.
We therefore sought to verify the ability of Brd7 to bind endogenous p21 promoter in
conditions where its expression is induced by p53 by performing chromatin
immunoprecipitation assays. The results highlighted lower levels of Brd7 binding to p21
promoter with respect to p53. This however was not surprising as Brd7 is supposed to
contact DNA indirectly, by means of interaction with acetylated histones or with p53. The
105

DISCUSSION
precipitation of lower amounts of chromatin might thus be due to a limitation of this
technique. Indeed, ChIP experiments conducted on p53 transcriptional co-factors such as
p300 lead to similar results (our unpublished observations).
Notably, it also appeared that Brd7 is not required for p53 recruitment on its high affinity
binding site on p21 promoter neither it influenced the acetylation status of chromatin-
associated p53. However, Brd7 might exert its effect on p53 transcriptional activity by
assisting the assembly of a functional transcriptional apparatus or by recruiting other
chromatin remodelling factors on chromatin surrounding p53 responsive element thus it
would be worth analyzing the chromatin modifications occurring at this site upon Brd7
depletion.
In contrast to p53, Brd7 might be widely distributed on chromatin, consistent with binding
to acetylated histones. However, binding to p21 promoter appeared to be preferential with
respect to other sites. Thus, it would be interesting to investigate whether Brd7 is recruited
on p21 promoter by p53 and if this occurs in response to specific stimuli. Brd7 might be
also bound at other sites within p21 promoter or at enhancers governing p21 expression,
and this would also explain the lack of effect observed in the reporter assays. It has been
demonstrated (Cordenonsi et al., 2003) that p53 and Smad2 interact on separate cis binding
elements present on target promoters and synergistically activate various genes, among
which p21. Thus it might be speculated that Brd7 may play a role in the cooperation
between p53 and the TGFβ signalling pathway in the induction of p21 transcription.
Therefore, it would be interesting to investigate whether Brd7 influences also TGFβ-
induced transcription of p21 and if it localizes with p53 and Smad2 on p21 promoter.
Consistently, data from literature indicating a role for Brd7 in downregulation of the Wnt
pathway (Peng et al., 2007; Kim et al., 2003) suggest that Brd7 may have pleiotropic
effects in regulating cell growth and it might be highly interconnected with different
signalling pathways.
However, data regarding Brd7 involvement in the Wnt pathways are contrasting. It seems
in fact that Brd7 might up-regulate α-catenin expression thus favoring β-catenin
sequestering while, it was reported to bind to dishevelled-1 (Dvl-1) and positively regulate
Wnt signalling (Kim et al, 2003). As this last function would be exerted within the
cytoplasm it might be supposed that Brd7 plays different roles depending on its localization
within the cells and that there might be mechanisms that induce its cytoplasmic localization
106

DISCUSSION
and that prevent it from mediating its growth suppressive functions. It would thus be of
interest to determine if its localization might be altered in some tumor context.
Taken together the data reported in this thesis suggest a role for Brd7 as a positive
regulator of p53 transcriptional activity during cell-cycle arrest response. Interestingly it
was observed that Brd7 expression is higher in tumor-derived cell lines with respect to
normal or immortalized cells. In the light of its role in mediating cell-cycle arrest this
seems surprising, as it would be expected that it is generally downregulated in tumors (as
in the case of NPC). Yet, it might be supposed that its upregulation is an upstream event in
response to altered growth signals and that the disruption of downstream pathways
bypasses the induction of cell-cycle arrest mediated by Brd7. Notably, Brd7 seems to be
regulated by E2F1, thus it might be speculated that it plays also a role in response to
oncogenic signalling (Berkovich and Ginsberg, 2001) leading to the onset of the
senescence program. Notably, the p53-p21 axis plays key roles in mediating senescence.
Thus, upon activation of oncogenes such as Ras, Brd7 might be upregulated through E2F1
and assist p53 in the induction of p21 to favor the onset of senescence thus offering a
barrier to unscheduled cell proliferation. Mutations of this pathway are frequently present
in cancer and this might explain why Brd7 overexpression is not selected against. It would
thus be of interest to investigate whether Brd7 is required for Ras-induced senescence.
It has also been observed that, when present at high levels (overexpression) Brd7 might
cause a reduction in p53-mediated transactivation of Bax promoters. Thus it might be
supposed that Brd7 plays a role in p53 choice towards different subset of promoters. Thus
its overexpression, when cell-cycle arrest response is impaired, might favor tumor
progression by mediating a by-pass of apoptosis.
Interestingly, Brd7 has proven to interact also with mutant p53 R175H and the binding
appeared to be stronger as compared to that of wild type p53. This suggests that the altered
conformation of p53R175H might disclose a stronger binding site as hypothesized for what
observed in the in vitro binding of p53 1-175 deletion mutant with Brd7 C-terminal
portion. It might thus be speculated that, as mutant p53R175H binds much strongly to Brd7
with respect to wt p53, it might compete for interaction with Brd7 and lead to a phenotype
similar to Brd7 depletion in cells expressing mutant p53 R175H thus Brd7 overexpression
would be selected in tumors bearing mutant p53. However, it would be of interest to
investigate whether Brd7 might directly influence also mutant p53 functions.
107

DISCUSSION
108
In conclusion, the role of Brd7 in regulating p53 activity needs further investigation in
order to be elucidated, yet from the data here reported it seems a promising candidate to
better understand how p53 response might be directed towards specific outcomes as a
result of the complex cross-talk among different signalling pathways.
The work described in Results - Part1 is contained in the following article (see
APPENDIX):
Mantovani F, Tocco F, Girardini J, Smith P, Gasco M, Lu X, Crook T and Del Sal G.
(2007)
The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis
inhibitor iASPP. Nat Struct Mol Biol. 14(10); 912-20.

APPENDIX
APPENDIX
The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation
from the apoptosis inhibitor iASPP. Fiamma Mantovani, UUFrancesca ToccoUU, Javier Girardini, Paul Smith, Milena Gasco, Xin Lu, Tim Crook & Giannino Del Sal. The tumor-suppressor function of p53 relies on its transcriptional activity, which is modulated by post-translational modifications and interactions with regulatory proteins. The prolyl isomerase Pin1 has a central role in transducing phosphorylation of p53 into conformational changes that affect p53 stability and function. We found that Pin1 is required for efficient loading of p53 on target promoters upon stress. In addition, Pin1 is recruited to chromatin by p53 and stimulates binding of the p300 acetyltransferase and consequent p53 acetylation. Accordingly, tumor-associated mutations at Pin1-binding residues within the p53 proline-rich domain hamper acetylation of p53 by p300. After phosphorylation of p53 at Ser46 triggered by cytotoxic stimuli, Pin1 also mediates p53’s dissociation from the apoptosis inhibitor iASPP, promoting cell death. In tumors bearing wild-type p53, expression of Pin1 and iASPP are inversely correlated, supporting the clinical relevance of these interactions.
109

The prolyl isomerase Pin1 orchestrates p53 acetylationand dissociation from the apoptosis inhibitor iASPPFiamma Mantovani1, Francesca Tocco1, Javier Girardini1, Paul Smith2, Milena Gasco3, Xin Lu4, Tim Crook2
& Giannino Del Sal1
The tumor-suppressor function of p53 relies on its transcriptional activity, which is modulated by post-translational modificationsand interactions with regulatory proteins. The prolyl isomerase Pin1 has a central role in transducing phosphorylation of p53into conformational changes that affect p53 stability and function. We found that Pin1 is required for efficient loading ofp53 on target promoters upon stress. In addition, Pin1 is recruited to chromatin by p53 and stimulates binding of the p300acetyltransferase and consequent p53 acetylation. Accordingly, tumor-associated mutations at Pin1-binding residues within thep53 proline-rich domain hamper acetylation of p53 by p300. After phosphorylation of p53 at Ser46 triggered by cytotoxic stimuli,Pin1 also mediates p53’s dissociation from the apoptosis inhibitor iASPP, promoting cell death. In tumors bearing wild-type p53,expression of Pin1 and iASPP are inversely correlated, supporting the clinical relevance of these interactions.
Prevention of cancer development depends on the ability of p53 toeither arrest proliferation or induce apoptosis of damaged cells, andrelies substantially on its transcriptional function. The high frequencyof mutations in the p53 DNA-binding domain in tumors highlightsthe importance of loss of p53’s transcriptional activity for the ability ofcancer cells to evade stress-induced growth suppression. However, themechanisms by which p53 function is subverted in cancers lackingthese mutations remain poorly defined.
The orchestration of appropriate cellular responses to genotoxic andcytotoxic stimuli depends on the ability of stress pathways to finelymodulate p53’s functions through regulation of its post-translationalmodifications, conformation and interactions with other proteins1,2. Inparticular, whereas stress-induced phosphorylation of N-terminalresidues promotes p53 stabilization, full activation of its transcriptionalfunction also involves acetylation of the C-terminal3–5 and core6,7
domains as well as conformational changes5,8. Phosphorylation-dependent prolyl isomerization induced by Pin1 is a well-conservedand extremely efficient mechanism for transducing post-translationalmodifications into conformational changes in key cellular proteins.Upon recognizing phosphorylated serine or threonine preceding pro-line in its substrates, Pin1 catalyzes the switch of the interveningpeptide from the cis to the trans conformation or vice versa9. Theresulting structural rearrangements control the stabilities and functionsof several proteins governing cell proliferation and transformation10,among which is p53. Upon stress, different kinases phosphorylate p53at Ser33, Ser46, Thr81 and Ser315 (refs. 11–16), generating sites forrecognition by Pin1, which greatly affects p53 stability and activity17–19.
In particular, Thr81-Pro82 is a crucial site through which Pin1promotes Chk2-dependent phosphorylation of p53 on Ser20 (ref.20), thereby stimulating dissociation of p53 from Mdm2 (ref. 17). Thissite lies within p53’s proline-rich domain (PRD)21, which modulatesthe stability22, acetylation23, transcriptional activity24–26 and directapoptotic function of p53 at mitochondria27.
The tumor-suppressor activity of p53 is also controlled by theapoptosis inhibitor iASPP28, which interacts with the PRD and blocksp53 binding to cell death–related promoters29. We recently reportedthat a common single-nucleotide polymorphism within the PRD,encoding either a proline or arginine at residue 72 (refs. 30–32),determines differential binding of iASPP29 and that this correlateswith the different apoptotic potentials of the p53-Pro72 andp53-Arg72 isoforms32.
In this report, we dissect the multiple levels at which Pin1 mightfine-tune the transcriptional activity of p53 in human cells subjectedto genotoxic stress. We investigate the requirement of Pin1 for efficientrecruitment of p53 to its target promoters and for acetylationmediated by p300. In addition, we analyze how Pin1 might affectiASPP-dependent regulation of p53 apoptotic activity.
RESULTSPin1 directly favors p53 binding on target promotersTo assess the impact of Pin1-induced prolyl isomerization on thetranscriptional activity of p53, we performed chromatin immunoprecipitation (ChIP) assays of endogenous p53 from HCT116 cells.Under stress conditions promoting the interaction of p53 with Pin1,
©20
07 N
atu
re P
ub
lish
ing
Gro
up
htt
p:/
/ww
w.n
atu
re.c
om
/nsm
b
Received 26 February; accepted 29 August; published online 30 September 2007; doi:10.1038/nsmb1306
1Laboratorio Nazionale Consorzio Interuniversitario Biotecnologie (LNCIB), Area Science Park, and Dipartimento di Biochimica Biofisica e Chimica delleMacromolecole, Universita di Trieste, Trieste, 34100, Italy. 2The Breakthrough Breast Cancer Research Centre, The Institute of Cancer Research, 237 Fulham Road,London, SW3 6JB, UK. 3Department of Medical Oncology, S. Croce e Carle Hospital, 12100 Cuneo, Italy. 4Ludwig Institute for Cancer Research, University CollegeLondon, 91 Riding House Street, London W1W 7BS, UK. Correspondence should be addressed to G.D.S. ([email protected]).
NATURE STRUCTURAL & MOLECULAR BIOLOGY ADVANCE ONLINE PUBLICATION 1
ART IC L E S
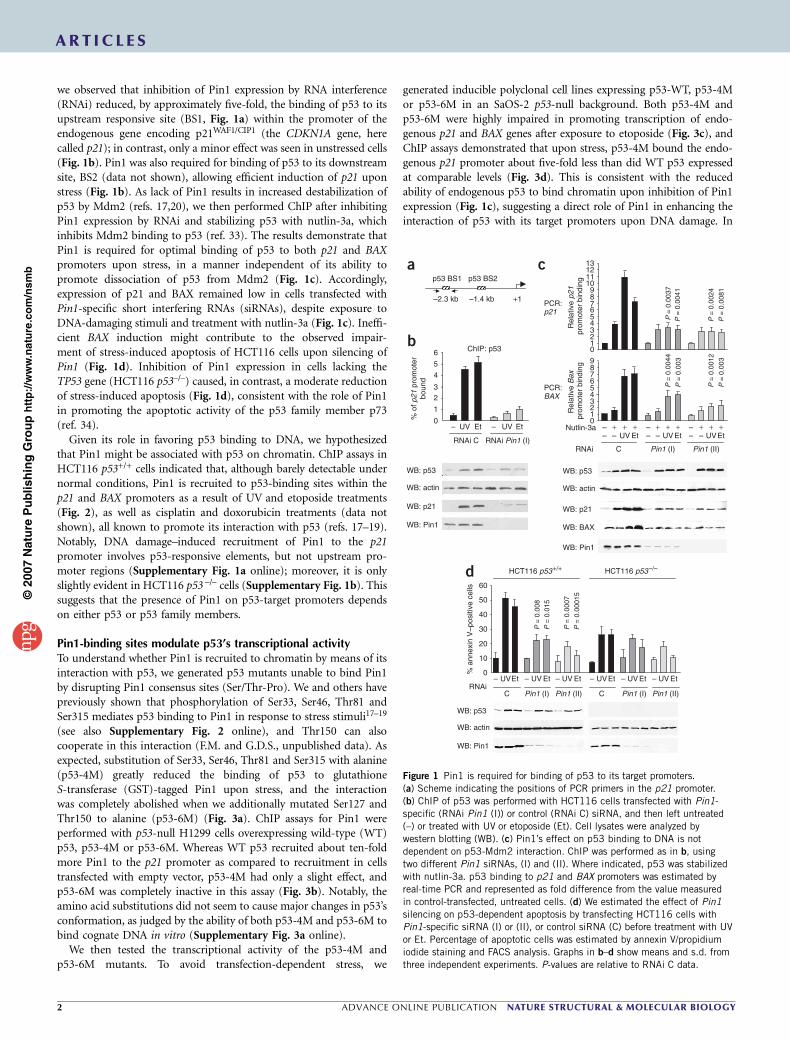
we observed that inhibition of Pin1 expression by RNA interference(RNAi) reduced, by approximately five-fold, the binding of p53 to itsupstream responsive site (BS1, Fig. 1a) within the promoter of theendogenous gene encoding p21WAF1/CIP1 (the CDKN1A gene, herecalled p21); in contrast, only a minor effect was seen in unstressed cells(Fig. 1b). Pin1 was also required for binding of p53 to its downstreamsite, BS2 (data not shown), allowing efficient induction of p21 uponstress (Fig. 1b). As lack of Pin1 results in increased destabilization ofp53 by Mdm2 (refs. 17,20), we then performed ChIP after inhibitingPin1 expression by RNAi and stabilizing p53 with nutlin-3a, whichinhibits Mdm2 binding to p53 (ref. 33). The results demonstrate thatPin1 is required for optimal binding of p53 to both p21 and BAXpromoters upon stress, in a manner independent of its ability topromote dissociation of p53 from Mdm2 (Fig. 1c). Accordingly,expression of p21 and BAX remained low in cells transfected withPin1-specific short interfering RNAs (siRNAs), despite exposure toDNA-damaging stimuli and treatment with nutlin-3a (Fig. 1c). Ineffi-cient BAX induction might contribute to the observed impair-ment of stress-induced apoptosis of HCT116 cells upon silencing ofPin1 (Fig. 1d). Inhibition of Pin1 expression in cells lacking theTP53 gene (HCT116 p53–/–) caused, in contrast, a moderate reductionof stress-induced apoptosis (Fig. 1d), consistent with the role of Pin1in promoting the apoptotic activity of the p53 family member p73(ref. 34).
Given its role in favoring p53 binding to DNA, we hypothesizedthat Pin1 might be associated with p53 on chromatin. ChIP assays inHCT116 p53+/+ cells indicated that, although barely detectable undernormal conditions, Pin1 is recruited to p53-binding sites within thep21 and BAX promoters as a result of UV and etoposide treatments(Fig. 2), as well as cisplatin and doxorubicin treatments (data notshown), all known to promote its interaction with p53 (refs. 17–19).Notably, DNA damage–induced recruitment of Pin1 to the p21promoter involves p53-responsive elements, but not upstream pro-moter regions (Supplementary Fig. 1a online); moreover, it is onlyslightly evident in HCT116 p53 –/– cells (Supplementary Fig. 1b). Thissuggests that the presence of Pin1 on p53-target promoters dependson either p53 or p53 family members.
Pin1-binding sites modulate p53’s transcriptional activityTo understand whether Pin1 is recruited to chromatin by means of itsinteraction with p53, we generated p53 mutants unable to bind Pin1by disrupting Pin1 consensus sites (Ser/Thr-Pro). We and others havepreviously shown that phosphorylation of Ser33, Ser46, Thr81 andSer315 mediates p53 binding to Pin1 in response to stress stimuli17–19
(see also Supplementary Fig. 2 online), and Thr150 can alsocooperate in this interaction (F.M. and G.D.S., unpublished data). Asexpected, substitution of Ser33, Ser46, Thr81 and Ser315 with alanine(p53-4M) greatly reduced the binding of p53 to glutathioneS-transferase (GST)-tagged Pin1 upon stress, and the interactionwas completely abolished when we additionally mutated Ser127 andThr150 to alanine (p53-6M) (Fig. 3a). ChIP assays for Pin1 wereperformed with p53-null H1299 cells overexpressing wild-type (WT)p53, p53-4M or p53-6M. Whereas WT p53 recruited about ten-foldmore Pin1 to the p21 promoter as compared to recruitment in cellstransfected with empty vector, p53-4M had only a slight effect, andp53-6M was completely inactive in this assay (Fig. 3b). Notably, theamino acid substitutions did not seem to cause major changes in p53’sconformation, as judged by the ability of both p53-4M and p53-6M tobind cognate DNA in vitro (Supplementary Fig. 3a online).
We then tested the transcriptional activity of the p53-4M andp53-6M mutants. To avoid transfection-dependent stress, we
generated inducible polyclonal cell lines expressing p53-WT, p53-4Mor p53-6M in an SaOS-2 p53-null background. Both p53-4M andp53-6M were highly impaired in promoting transcription of endo-genous p21 and BAX genes after exposure to etoposide (Fig. 3c), andChIP assays demonstrated that upon stress, p53-4M bound the endo-genous p21 promoter about five-fold less than did WT p53 expressedat comparable levels (Fig. 3d). This is consistent with the reducedability of endogenous p53 to bind chromatin upon inhibition of Pin1expression (Fig. 1c), suggesting a direct role of Pin1 in enhancing theinteraction of p53 with its target promoters upon DNA damage. In
©20
07 N
atu
re P
ub
lish
ing
Gro
up
htt
p:/
/ww
w.n
atu
re.c
om
/nsm
b
p53 BS1
–2.3 kb –1.4 kb +1
p53 BS2
a c
d
RNAiC
– UV – UV – UV
C
– UV – UV – UV0
10
20
30
40
50
60
% a
nnex
in V
–pos
itive
cel
ls
WB: p53
WB: actin
WB: Pin1
HCT116 p53+/+ HCT116 p53–/–
b
% o
f p21
pro
mot
erbo
und
0
1
23
4
5
6
WB: p53
WB: actin
WB: Pin1
WB: p21
– UV
RNAi C
UV
RNAi Pin1 (I)
ChIP: p53
–
Rel
ativ
e p2
1pr
omot
er b
indi
ngR
elat
ive
Bax
prom
oter
bin
ding
PCR:p21
0123456789
10111213
RNAi C Pin1 (I) Pin1 (II)
Pin1 (I) Pin1 (II) Pin1 (I) Pin1 (II)
Nutlin-3a0123456789
PCR:BAX
WB: p53
WB: actin
WB: Pin1
WB: p21
WB: BAX
P =
0.0
03
P =
0.0
0015
P =
0.0
15P
= 0
.008
P =
0.0
007
P =
0.0
012
P =
0.0
081
P =
0.0
024
P =
0.0
041
P =
0.0
037
P =
0.0
03
P =
0.0
044
UV–– – – – – –
+ + +UV
– + + +UV
– + + +EtEtEt Et Et
EtEtEtEtEtEt
Figure 1 Pin1 is required for binding of p53 to its target promoters.
(a) Scheme indicating the positions of PCR primers in the p21 promoter.
(b) ChIP of p53 was performed with HCT116 cells transfected with Pin1-
specific (RNAi Pin1 (I)) or control (RNAi C) siRNA, and then left untreated
(–) or treated with UV or etoposide (Et). Cell lysates were analyzed by
western blotting (WB). (c) Pin1’s effect on p53 binding to DNA is not
dependent on p53-Mdm2 interaction. ChIP was performed as in b, using
two different Pin1 siRNAs, (I) and (II). Where indicated, p53 was stabilized
with nutlin-3a. p53 binding to p21 and BAX promoters was estimated by
real-time PCR and represented as fold difference from the value measured
in control-transfected, untreated cells. (d) We estimated the effect of Pin1
silencing on p53-dependent apoptosis by transfecting HCT116 cells withPin1-specific siRNA (I) or (II), or control siRNA (C) before treatment with UV
or Et. Percentage of apoptotic cells was estimated by annexin V/propidium
iodide staining and FACS analysis. Graphs in b–d show means and s.d. from
three independent experiments. P-values are relative to RNAi C data.
ART IC L E S
2 ADVANCE ONLINE PUBLICATION NATURE STRUCTURAL & MOLECULAR BIOLOGY

agreement with the requirement of Pin1 for p53-dependent apoptosis(Fig. 1c), the ability of p53-4M and p53-6M to promote apoptosis inresponse to stress was greatly impaired as compared to WT p53(Supplementary Fig. 3b,c).
Pin1 enhances p300-mediated acetylation of p53We hypothesized that prolyl isomerization might affect subsequentpost-translational modifications of p53, thereby increasing its bindingto chromatin and its transcriptional activity. In particular, as acetyla-tion of p53 C-terminal lysines at positions 373 and 382 by p300 (or itshomolog CBP) stimulates these activities4,35,36, we tested whether p53mutants unable to bind Pin1 were modified by p300 differently fromWT p53. Indeed, p53-4M and p53-6M bound p300 markedly less thanWT p53 (Fig. 4a) and were consequently acetylated much lessefficiently by p300 (Fig. 4b), particularly on Lys373 and Lys382(Supplementary Fig. 4a online). Notably, overexpression of p300did not increase the transcriptional activity of p53-6M (Supplemen-tary Fig. 4b). Moreover, RNAi-mediated inhibition of Pin1 expressionin HCT116 cells reduced the interaction of endogenous p53 and p300proteins (Fig. 4c) as well as the stress-induced acetylation of p53Lys373 and Lys382 (Fig. 4d), suggesting a direct role of Pin1 inthese events.
Finally, we sought to prove that the contribution of Pin1 to p300-mediated acetylation of p53 depends on its direct activity toward p53,rather than on indirect effects on p300 or other components of stresspathways. Accordingly, we immunoprecipitated in vitro–translatedp53 and incubated it with purified catalytically competent Pin1
protein37. A control reaction was set up using a catalytically inactivePin1 mutant (C113A)37 that binds p53 but cannot isomerize it17. Afterremoval of bound Pin1, p53 was subjected to acetylation by purifiedp300 protein. Treatment of p53 with WT Pin1, but not mutant Pin1,increased the efficiency of subsequent acetylation by p300 (Fig. 4e),suggesting that this depends on isomerization of p53.
Pin1 favors the acetylation of p53 by p300 on chromatinAcetylation of p53 C-terminal lysines by p300 or CBP has beenreported to increase binding of p53 to its target genes4,35, but otherstudies have indicated that the prevalent role of this acetylation is inpromoting the recruitment of transcription cofactors on p53-targetpromoters36. We sought to understand the contribution of Pin1-dependent acetylation of p53 to each of these processes.
To assess whether Pin1 promotes binding of p53 to target chromatinsites by favoring its acetylation, we assayed the effect of silencing Pin1on promoter binding by WT p53 and p53-9KR, a mutant where nineC-terminal lysines, including those acetylated by p300, are substitutedby arginines38. This mutant protein bound Pin1 to a similar extent asdid WT p53 (Supplementary Fig. 5a online). Notably, in ChIP assays,p53-9KR showed reduced binding to p21 promoter as compared toWT p53 expressed at similar levels (Fig. 5a), suggesting that acetyla-tion of C-terminal lysines modulates DNA binding in vivo. RNAi-mediated inhibition of Pin1 expression reduced the binding ofp53-9KR to the p21 promoter, although to a lesser extent than seenwith WT p53 (Fig. 5b). This suggests that Pin1 affects binding of p53to chromatin independently of its impact on p53 acetylation.
To estimate the extent to which Pin1’s ability to promote p53acetylation affects p53 transcriptional activity, we inhibited either Pin1expression or isomerase activity and used ChIP assays to analyze theacetylation of p53 bound to p21 promoter under these conditions.Similar amounts of DNA-bound p53 were compared in these experi-ments, as detailed in Supplementary Figure 5b. The results showedthat acetylation of promoter-bound p53 on Lys373 and Lys382increased upon stress only in the presence of normal levels of Pin1,and could be blocked by inhibition of Pin1’s catalytic activity with thesmall molecule PiB39 (Fig. 5c). Acetylation of Lys373 and Lys382 isknown to induce the recruitment of p300 to p53-target promoters35,and indeed, the binding of p300 to the upstream p53-responsiveelement of the p21 promoter was reduced upon silencing of Pin1(Fig. 5d).
Finally, we performed biochemical fractionation of HCT116 celllysates after inhibiting Pin1 expression by RNAi. This showed thatacetylation of mostly the chromatin-bound pool of p53 was reduced;there was only a smaller effect on free p53 protein (Fig. 5e).
The above results suggest that Pin1 favors the recruitment of p300and the acetylation of p53 on chromatin, which activate transcription.In addition, Pin1 also affects the acetylation of soluble p53, and thismight further stimulate its binding to chromatin.
Interplay of Pin1 and PRD in p53 acetylationIt has been shown that the p300-dependent acetylation of p53 thatoccurs upon binding to cognate DNA involves p53’s PRD domain23,which contains a Pin1 target site at residues Thr81-Pro82. We soughtto verify the requirement of these residues for p53 acetylation bytransfecting cells with plasmids encoding WT or mutant p53 andp300 together with the pG13 plasmid, which contains 13 tandemp53-binding sites40. In agreement with previous reports8,41, thisconsensus DNA sequence stimulated WT p53 acetylation, whereas aplasmid containing mutated p53-binding sites (pM13) did not(Fig. 6a, left blots). Notably, p53 acetylation was reduced by the
©20
07 N
atu
re P
ub
lish
ing
Gro
up
htt
p:/
/ww
w.n
atu
re.c
om
/nsm
b
ChIP: control Ab
ChIP: Pin1
Time (h)
% o
f p21
DN
A b
ound
% o
f p21
DN
A b
ound
% o
f p21
DN
A b
ound
% o
f BA
X D
NA
bou
nd%
of B
AX
DN
A b
ound
% o
f BA
X D
NA
bou
nd
ChIP: Pin1
ChIP: control Ab
Time (h)
UV Et
ChIP: p53ChIP: p53
0 5 8 12 5 8 12
0 5 8 12 5 8 12
Time (h) 0 5 8 12 5 8 12 Time (h) 0 5 8 12 5 8 12
Time (h) 0 5 8 12 5 8 12
Time (h) 0 5 8 12 5 8 12
UV Et
00.20.40.60.8
11.21.41.6
00.1
0.20.3
0.40.5
0.6
0.7
UV Et
0
0.1
0.2
0.3
0.4
0.5
0.6
UV Et
0
1
2
3
4
5
UV Et
00.20.40.60.8
11.21.41.6
UV Et
0
0.10.20.3
0.40.5
0.60.7
Figure 2 Pin1 is recruited to p53-responsive promoters upon DNA damage.HCT116 cells were either left untreated or treated with UV or etoposide (Et)
before ChIP with Pin1, p53 or control antibodies (HA; control Ab). The
percentage of input p21 and BAX promoters bound by p53 and Pin1 at
indicated times after DNA damage was estimated by real-time PCR. Graphs
represent means and s.d. from four independent experiments.
ART IC L E S
NATURE STRUCTURAL & MOLECULAR BIOLOGY ADVANCE ONLINE PUBLICATION 3

mutations T81A and P82I (Fig. 6a, right blots), which also weakenp53 binding to Pin1 (refs. 17,20). We then performed ChIP assays withSaOS-2 cell lines expressing either WT p53 or p53-T81A. As expected,lower amounts of acetylated p53-T81A than of WT p53 were found onthe p21 promoter (Fig. 6b). The above results reinforce the previousobservation that the Thr81-Pro82 site is important for p53 transcrip-tional and apoptotic activity12,20, and in particular for its activation byPin1 (ref. 20).
To confirm the role of the PRD in p53 acetylation, we then assayedthe relevance of the codon 72 proline/arginine polymorphism, whichalso affects the functions of this domain31,32. Upon stress, p53-Arg72was more rapidly and efficiently acetylated on Lys373 and Lys382 thanwas p53-Pro72, and this was observed when comparing cell linesendogenously expressing either p53-Pro72 or p53-Arg72 (Fig. 6c), aswell as otherwise isogenic p53-inducible H1299 cell lines (Supple-mentary Fig. 6a online). The stronger apoptotic potential ofp53-Arg72 has been associated with phosphorylation of Ser46(ref. 32), which enhances p53 acetylation at Lys382 by CBP (homologof p300)13. Notably, we observed more Ser46 phosphorylation onp53-Arg72 than on p53-Pro72 (Fig. 6d and Supplementary Fig. 6b).Consistent with this, p53-Arg72 bound GST-Pin1 more strongly thandid p53-Pro72 upon stress (Supplementary Fig. 6c).
Pin1 stimulates p53 dissociation from iASPPOur results indicate that Pin1 controls p53’s transcriptional andapoptotic activities. However, although acetylation of Lys373 orLys382 can promote binding of p53 to cell death–related promoters5,it is not sufficient to activate its apoptotic function, as dissociationfrom inhibitory factors is also required. Among these inhibitoryfactors, iASPP28 binds the PRD, interacting preferentially with the
p53-Pro72 variant29. We therefore asked whether, upon cytotoxicstress, Pin1 might regulate the p53-iASPP interaction.
Using co-immunoprecipitation experiments, we found that theinteraction of iASPP with ectopically expressed p53 was reducedupon irradiation of H1299 cells with cytotoxic UV dosage (Fig. 7a),even though p53 levels were equalized by treatment of unstressed cellswith a proteasome inhibitor (CBZ). iASPP, ASPP1 and ASPP2 proteinlevels remained unchanged (Fig. 7a and data not shown). This resultsuggests that stress-induced post-translational modifications mightregulate the interaction of p53 with iASPP. Indeed, the phosphoryla-tion mutants p53-4M and p53-6M bound iASPP more strongly thandid WT p53 upon DNA damage (Supplementary Fig. 7a online).Notably, disruption of single Pin1 target sites on p53 demonstratedthat stress-induced dissociation of p53 from iASPP required Ser46and, to a lesser extent, Thr81 (Fig. 7a).
We therefore reasoned that, by modifying the conformation of p53in response to cytotoxic stress, Pin1 could specifically modulate p53’sdissociation from iASPP in addition to affecting its acetylation. Insupport of this hypothesis, we found that in the absence of Pin1, thedissociation of p53 from iASPP upon cytotoxic stress was impaired,despite the fact that Ser46 was phosphorylated (Fig. 7b). Conversely,Pin1 overexpression enhanced p53 dissociation from iASPP in aSer46-dependent fashion (Fig. 7c). This effect required Pin1 isomeraseactivity, as it was not observed either upon expression of catalyticallyinactive Pin1 mutants (Fig. 7d) or upon inhibition of prolyl isomeraseactivity (Supplementary Fig. 7b). Together, these observations suggestthat, upon stress-induced phosphorylation of p53 on Ser46, Pin1favors its dissociation from iASPP. However, this does not depend onPin1-induced acetylation of p53, as iASPP bound p53 irrespective ofits acetylation status (Supplementary Fig. 8a online). It also does not
©20
07 N
atu
re P
ub
lish
ing
Gro
up
htt
p:/
/ww
w.n
atu
re.c
om
/nsm
b
Figure 3 Pin1-binding sites are essential for p53
functions. (a) Top, p53 scheme indicating the
positions of Pin1 consensus sites (phospho-Ser/
Thr-Pro). TA, transactivation domain; DBD, DNA
binding domain; NLS, nuclear localization signal.
p53 mutants have Ser/Thr-to-Ala substitutions in
Pin1 consensus sites, either at residues 33, 46,
81 and 315 (in bold, p53-4M) or at all six
positions (p53-6M). Bottom, binding of p53
variants to Pin1 upon overexpression in SaOS-2
cells, etoposide treatment and GST-Pin1 pull-
down. G, GST alone; P, GST-Pin1. Binding
reactions were compared with 10% of protein
inputs by western blotting (WB). (b) Pin1
recruitment to chromatin requires binding top53. p53-null H1299 cells transfected with
plasmids encoding WT p53, p53-4M or p53-6M,
or vector alone, were used for ChIP with Pin1-
specific or control (HA) antibodies, and the
percentage of input p21 promoter bound by Pin1
was estimated by real-time PCR. Graph shows
means and s.d. from three independent
experiments. p53 levels were determined by
western blotting. (c) To assess the role of Pin1-
binding sites in p53’s transcriptional activity,
polyclonal SaOS-2 cell lines were generated for
PonA-inducible expression of WT p53, p53-4M
and p53-6M. Upon induction with PonA, cells
were treated with etoposide (Et) or left untreated.
Induction of p53 target genes p21 and BAX was
evaluated by semiquantitative RT-PCR, with GAPDH serving as a control for efficiency of RNA extraction and transcription. (d) Binding of WT p53 and
p53-4M to p21 promoter in inducible SaOS-2 cell lines treated as in c was analyzed by ChIP with antibodies (Ab) to p53 or HA (control, C). Bound p21
promoter was estimated by semiquantitative PCR, with relative band intensities reported below.
a 33 46 31581 127 150
TA PRD DBD NLSp53
GST pull-down
WT 4M 6M
WB:p53
Inputs
WT 4M 6Mp53
G P G PPG
EtPonA
WT p53 p53-4M p53-6M
RT-PCR: p21
RT-PCR: BAX
RT-PCR: GAPDH
WB: p53
WB: P-S15 p53
b
c
d
ChIP
WT p53 p53-4M
EtPonA
Cp53 p53CAb
Chromatininputs
–
–
– – – –+ ++
+ + +–– –
–+
+ +––
–+
+ +––
–+
+ +––
+ –– – +–+ + +
+ – + +Et
PonA
WT p53 p53-4M
1 4.6 0.5 0.8
WB: p53
00.10.20.30.40.50.60.70.8
% o
f p21
DN
A b
ound
p53
ChIP: Pin1
– WT 4M 6M – WT 4M 6M
ChIP: control Ab
WB: p53
WB: actin
WB: Pin1
ART IC L E S
4 ADVANCE ONLINE PUBLICATION NATURE STRUCTURAL & MOLECULAR BIOLOGY

depend on DNA binding (data not shown). Conversely, modu-lation of iASPP abundance by either overexpression or RNAi didnot influence p53 acetylation (Supplementary Fig. 8b,c). Theseobservations suggest that Pin1 regulates p53 acetylation and bindingto iASPP independently.
Our data also imply that, under certain circumstances, overexpres-sion of Pin1, reported in several tumors42, might prevent iASPP frombinding p53. Overexpression of iASPP, which preferentially bindsp53-Pro72 (ref. 29), is significantly (P ¼ 0.037) more common inhuman breast tumors arising in individuals with germline 72 Pro/Pro
(that is, proline at codon 72 on each chromosome) than 72 Arg/Arg(ref. 29, Fig. 7e and Supplementary Table 1 online). In contrast, Pin1overexpression was significantly less frequent in 72 Pro/Pro than 72Arg/Arg cancers (4 of 15 tumors, or 26.7%, compared with 26 of 40tumors, or 65%; P ¼ 0.04; Fig. 7e and Supplementary Table 1). Thissuggests that overexpression of Pin1 might be selected against incancers with WT p53 72 Pro/Pro, as it would counteract iASPP-dependent inhibition of p53-mediated apoptosis. In cancers with WTp53 72 Arg/Arg, there may be no selection against Pin1 overexpressionbecause iASPP does not efficiently target this p53 isoform.
©20
07 N
atu
re P
ub
lish
ing
Gro
up
htt
p:/
/ww
w.n
atu
re.c
om
/nsm
b
WT C113A
WB: p53 acLys382
WB: p53
++–
– –
+p300
GST-Pin1
GST-Pin1
WT C113A
p53p300
WT 4M+ +– –
6M+–
IP:p53
WB: acLys
WB: p53
WB: p300Inputs
IP:a b c d e
WB: p53
WB: p300
WB: p53
WB: GFP(p300)
Inputs
p53
GFP-p300
WT 4M
+ +
6M
+
WT
+
GFP NRS
WB: Pin1
RNAi Pin1RNAi C
UV UV
WB: p53
WB: p53(DO-1)
WB: Pin1
WB: p53acLys373
acLys382
WB: p300
IP:
WB: p53
WB: p300
p300 NRS
WB: Pin1
RNAi Pin1 CC
WB: p53
WB: p300 EtEt– –
Figure 4 Pin1 directly stimulates p53 acetylation by p300. (a) The interaction of WT p53, p53-4M and p53-6M with GFP-p300 was determined by
co-immunoprecipitation after overexpression in SaOS-2 cells. NRS, control immunoprecipitation with nonimmune serum. (b) SaOS-2 cells were transfected
as in a and acetylation of immunoprecipitated (IP) p53 was analyzed by western blotting (WB) with antibody to acetyllysine. (c) Interaction of endogenous
p53 and p300 proteins in HCT116 cells transfected with Pin1-specific or control (C) siRNAs was evaluated by co-immunoprecipitation. (d) Acetylation (ac) of
p53 on Lys373 and Lys382 was evaluated by western blotting of HCT116 cells, transfected as in c and then either left untreated or treated with UV or
etoposide (Et). To compare similar amounts of p53 protein, we loaded excess cell lysate from Pin1-silenced cells relative to control-transfected cells.
(e) Modification by Pin1 increases p53 acetylation in vitro. In vitro-translated, purified HA-p53 was incubated with either catalytically competent
recombinant GST-Pin1 (WT) or catalytically inactive Pin1 mutant (C113A) and then acetylated in vitro using purified p300. p53 acetylation on Lys382
was detected by western blotting. The amounts of Pin1 proteins were compared by Coomassie staining.
a b c d
e
WB: p53
WB: Pin1
acLys373
WB: HMGA1b
WB: α-tubulin
Pin1 (I)C Pin1 (II)RNAi
Pin1 (I)C Pin1 (II)
Et Et Et Et Et Et
Chromatin-bound FreeFraction:
WTp53
0
0.20.40.60.8
11.21.4
0
0.2
0.4
0.6
0.8
1
1.2
0
0.2
0.4
0.6
0.8
1
1.2
prom
oter
bou
nd%
of p
21
prom
oter
bin
ding
Rel
ativ
e p2
1
prom
oter
bin
ding
Rel
ativ
e p2
1
P =
0.0
2
P =
0.0
002
P =
0.0
002
P =
0.0
014
P =
0.0
08
P =
0.0
1
0
0.2
0.4
0.6
0.8
1
1.2
RNAi PCPiBDMSO
UV Et UV Et
Dam
aged
/und
amag
edC
hlP
dat
a
123456789
RNAiPin1
RNAiC
UV Et UV Et
ChIP: p53 acLys373/382 ChlP: p3009KR WT
WT 9KR
acLys382
WB: acLys373
WB: p53
WB: Pin1
C P C PRNAi CP PC
P =
0.0
006
P =
0.0
036
WB: p53
WB: p53
9KR
– – – – – –
Figure 5 Pin1 promotes p300 recruitment and p53 acetylation on chromatin. (a) Results
of ChIP of WT p53 or p53-9KR overexpressed in H1299 cells. Percentage of input p21
promoter bound was estimated by real-time PCR. Graphs represent means and s.d. from
three independent experiments. (b) Pin1 affects p53 binding to chromatin independently of
acetylation. ChIP was done as in a, after expression of WT p53 or p53-9KR together with
either Pin1-specific (P) or control (C) siRNAs. p53 expression and acetylation (ac) on Lys373
and Lys382 was analyzed by western blotting (WB). (c) Pin1 affects p53 acetylation on target
DNA. ChIP was performed with HCT116 cells treated with UV or etoposide (Et) and with
antibodies specific for p53 acetylated on Lys373 and Lys382. Using real-time PCR, weanalyzed the increase in acetylation of p53 bound to p21 promoter (upstream binding site)
upon DNA damage, after either blocking Pin1’s expression by RNAi (RNAi Pin1 compared
with control RNAi C; black bars) or inhibiting its prolyl isomerase activity (PiB treatment
compared with DMSO treatment; gray bars). Similar amounts of DNA-bound p53 were compared in all experiments (see text). Histograms show means and
s.d. from three independent experiments. P-values are for results from RNAi Pin1 compared with RNAi C and from PiB compared with DMSO. (d) Pin1
affects p300 recruitment to p21 promoter. ChIP of p300 was performed with HCT116 cells transfected with either Pin1-specific (P) or control (C) siRNAs
and treated with Et. Binding of p300 to p21 promoter (upstream p53-binding site) was evaluated by real-time PCR as in c. (e) Pin1 controls primarily the
acetylation of the chromatin-bound p53 pool. Chromatin-bound and free protein fractions were prepared from HCT116 cells transfected with Pin1-specific
siRNA (I) or (II) or with control siRNA, and then treated with Et or left untreated. Acetylation (ac) of p53 on Lys373 was evaluated by western blotting. We
loaded excess protein from Pin1-silenced cells relative to control-transfected cells to allow comparison of similar amounts of p53. Western blots of HMGA1b
and tubulin were used as controls for fractionation efficiency. Graphs in a–d represent means and s.d. from three independent experiments.
ART IC L E S
NATURE STRUCTURAL & MOLECULAR BIOLOGY ADVANCE ONLINE PUBLICATION 5

DISCUSSIONAs knowledge of the mechanisms of p53 activation increases, differentlevels of regulation are revealed, which imply ordered sequences ofpost-translational modifications and protein interactions, ultimatelyleading to adaptation and differential susceptibility to stress. Previousdata have indicated that, upon DNA damage and phosphorylation onSer/Thr-Pro sites (in particular Thr81), prolyl isomerization of p53 byPin1 precedes its stabilization and full activation17–20. In this report,we have further highlighted different levels at which Pin1 stimulatesp53 functional activation (summarized in Fig. 8). Pin1 is required forp53 loading on target promoters, and is itself recruited to chromatinby p53 to promote binding of p300 and acetylation of p53 on targetDNA. Moreover, upon cytotoxic stress, Pin1 induces dissociation ofp53 from the apoptosis inhibitor iASPP.
On the basis of our results and previous evidences8,23, we hypothe-size that Pin1 might assist structural rearrangements of p53 that occurupon DNA binding, exposing its C terminus to p300 (ref. 8) and thus
promoting the DNA-dependent acetylation and transcriptional func-tion of p53. In fact, our data suggest that Pin1 regulates these events atleast in part through phosphorylation-induced isomerization ofThr81-Pro82 within p53 PRD, supporting the notion that this domainis required for DNA-dependent acetylation of p53 by p300 (ref. 23).This, in turn, stabilizes p300 binding41 and directs the organization oftranscription complexes on the promoter36. Indeed, Pin1-mediatedPro82 isomerization is required for Ser20 phosphorylation by Chk2(ref. 20), which promotes p53 binding to p300 (ref. 43). Thr81regulates the transcriptional and apoptotic activity of p53, and,notably, mutations of Thr81 and Pro82 have been described in severalcancers12,44. Mutations at Pin1 target sites are less frequent than thoseat residues involved in DNA binding. This is to be expected, however,as we show here that alteration of multiple Pin1 consensus sites isrequired for complete abrogation of p53 acetylation and transcrip-tional activity, suggesting cooperative actions of Pin1 on differentdomains of p53.
©20
07 N
atu
re P
ub
lish
ing
Gro
up
htt
p:/
/ww
w.n
atu
re.c
om
/nsm
b
Figure 6 Pin1-target site Thr81-Pro82 and the
codon 72 polymorphism affect p53 acetylation.
(a) DNA-dependent acetylation of WT p53 and
p53 mutants T81A and P82I overexpressed in
SaOS-2 cells was induced by cotransfection of a
plasmid encoding p300 and either the pG13 or
pM13 plasmid, which contain 13 WT or mutated
p53-binding sites, respectively. Acetylation (ac)
of p53 on Lys382 was estimated by western
blotting (WB). (b) ChIP was performed, using an
antibody specific for p53 acetylated on Lys382,
with PonA-inducible polyclonal SaOS-2 cell lines
expressing WT p53 or p53-T81A and either
treated with UV or etoposide (Et) or left
untreated. The increase in acetylation of p53bound to p21 promoter (upstream binding
site) after DNA damage was estimated by
semiquantitative PCR (right gels). Relative band intensities are reported below gel. (c) Acetylation of p53 on Lys382 was estimated by western blotting of
WM278 and WM115 melanoma cells, homozygous for alleles encoding p53-Pro72 and p53-Arg72, respectively, upon treatment with either UV, Et,
doxorubicin (Dox) or deacetylase inhibitors (DI). (d) Phosphorylation of p53 on Ser46 (pSer46) was estimated by western blotting of doxycycline-inducible
H1299 cell lines expressing p53-Pro72 and p53-Arg72, upon treatment with doxorubicin for different times.
ChIP:p53AcK382
Chromatininputs
T81AWT
EtUV–EtUV–
1 3.2 4.1 0.71.4 2.2
WB: p53
T81A
+–
WT
+–PonA
WB: actin
p53
WB: p53acLys382
WB: p53
WM278 (Pro72) WM115 (Arg72)
DIDoxEtUV– – DIDoxEtUV 2418126024181260
H1299 p53-Pro72 H1299 p53-Arg72
WB: p53pSer46
WB: p53
Time (h) after dox
WB: p53
p53
GFP-p300
WT
++
P82IT81A
+++++++ pG13 DNA
WB: GFP(p300)
a b
c d
pM13 DNA
WT
+++
+
+–––––
–– –– ––––
WB: p53acLys382
a b c d
e
T81AS46AWTp53
UVUVUVCBZ CBZ CBZ CBZ UVUVUVCBZ CBZ CBZ
IP
Inputs InputsInputs
Inputs
(mAb)
WB: p53
WB: iASPP
WB: iASPP
WB: p53
iASPP (pAb)C
T81AS46AWTp53
+HA-Pin1 + +
WB: iASPPWB: iASPP
(mAb)
WB: p53
WB: HA (Pin1)
WB: HA (Pin1)
0
20
40
60
80
001
% o
f tum
ors
p53P/P
n = 15R/R
n = 40
Pin1 (I)CRNAi Pin1 (II)
IP:iASPP
IP:iASPP
WB: p53
WB: iASPP
WB: iASPP WB: iASPP
WB: iASPP
iASPP (pAb)
WB: Pin1
WB: p53
WB: p53WB: p53
WB: p53
WB: p53
PSer46
–––
IP: C
HA-Pin1
p53 +++ ++
– WT
S67
E
C11
3A–
(pAb)
72 p53 72
Tumors with iASPP overexpression
Tumors with Pin1 overexpression
Figure 7 Pin1 is required for p53 dissociation from iASPP upon cytotoxic stress. (a) The interaction of WT p53 or mutant
S46A or T81A p53 expressed in H1299 cells with endogenous iASPP was analyzed by co-immunoprecipitation (IP)
after treatment with either cytotoxic UV dosage or the proteasome inhibitor CBZ (as a control). WB, western blot; mAb,
monoclonal antibody. (b) The interaction of endogenous p53 and iASPP proteins was analyzed, as in a, in HCT116 cells
transfected with Pin1-specific siRNA (I) or (II) or control (C) siRNA and treated with either UV or CBZ. (c) H1299 cells
were transfected with either WT p53 or mutant S46A or T81A p53, and with vector encoding HA-Pin1 (+) or empty vector
(–). After UV treatment, the interaction of p53 with endogenous iASPP was analyzed as in a. (d) Vector encoding WT p53
was transfected into H1299 cells with plasmids encoding HA-tagged WT Pin1 or HA-tagged catalytically inactive Pin1
mutants S67E or C113A, or empty vector (–). After UV treatment, the interaction of p53 with endogenous iASPP was
analyzed as in a. (e) Histograms show the percentage of tumors overexpressing iASPP (gray bars) and Pin1 (black bars),
assessed by comparing mRNA abundance in breast tumor samples homozygous for the allele encoding WT p53-Pro72
(P/P) or WT p53-Arg72 (R/R) to mRNA abundance in their matched normal samples.
ART IC L E S
6 ADVANCE ONLINE PUBLICATION NATURE STRUCTURAL & MOLECULAR BIOLOGY

Given its pleiotropic effects on numerous substrates45,46, oncerecruited to chromatin, Pin1 might enhance p53-dependent transcrip-tion at several levels—for example, by modifying transcription coac-tivators (including p300), chromatin-remodeling factors or structuralcomponents. In addition, acetylation of Lys120 has recently beenreported to direct p53 to low-affinity apoptotic promoters6,7, and itwould be interesting to determine whether Pin1-mediated isomeriza-tion influences this event as well.
However, acetylation is not sufficient to activate the apoptoticfunction of p53 unless it dissociates from the iASPP inhibitor,whose main binding site also lies within the PRD29. Our dataimplicate Pin1 prolyl isomerase activity in stimulating the release ofp53 from iASPP upon cytotoxic stress. Although phosphorylation ofThr81 also seems to be involved in this process, phosphorylation ofSer46 is essential in allowing Pin1 to trigger p53 dissociationfrom iASPP. This finding is biologically relevant, as Ser46 phos-phorylation is specifically induced by severe or persistent stressconditions and represents a major event in shifting the p53 responsefrom cell-cycle arrest to apoptosis47,48. Notably, Ser46 phosphoryla-tion32 and iASPP interaction29 have been shown to determine thedifferential apoptotic potentials of p53 Pro72 and Arg72 isoforms atthe transcriptional level, and our data indicate that these events areindeed correlated, as we observed more efficient Lys373 and Lys382acetylation and Ser46 phosphorylation of p53-Arg72 than of p53-Pro72 upon stress. This is in agreement with the reported induction ofCBP binding to p53 upon HIPK2-mediated Ser46 phosphorylation13.It is tempting to speculate that Pin1 might also have a role inthis process.
As overexpression of iASPP has been found in several tumortypes28,49, the molecular basis of the interplay between p53, Pin1and iASPP is of potential clinical interest. We have previously reportedthat iASPP overexpression is common in breast carcinomas bearingWT p53 that arise in a 72 Pro/Pro genetic background29. We havefurther shown that Pin1 is overexpressed in a substantial proportion ofprimary breast cancers bearing WT p53; however, this correlatesinversely with iASPP overexpression and is more common in cancersarising in 72 Arg/Arg individuals. It is conceivable that, in tumors withp53-Pro72, overexpression of Pin1 might be selected against, as itwould relieve p53 inhibition by iASPP. In contrast, the moderateinteraction of iASPP with p53-Arg72 might not lead to strongselection pressure against Pin1 in cancers bearing this p53 isoform.Notably, homozygosity for the allele encoding WT p53-Arg72 isassociated with greater sensitivity of tumor cells to anticancer drugsand is predictive of a more favorable clinical response to chemo-therapy in individuals with head and neck cancer32.
Pin1, together with Pro-directed kinases, governs the functions ofseveral substrates, and it is itself under control of signaling pathwaysthat could become aberrantly activated in cancers45. Indeed, Pin1abundance has been associated both positively and negatively withcancer, underscoring the relevance of the cellular context in dictatingwhether Pin1 inhibits or amplifies cell-growth pathways10. In normalcells or at early stages of cancer, Pin1 could be part of a checkpointsystem that surveys cell proliferation. At more advanced tumorstages, concomitant with deregulation of signaling pathways, it mayinstead contribute to the amplification of proliferative signals45.In the first case, high levels of Pin1 could improve the apoptoticperformance of WT p53 (and of p73; see ref. 34) upon drug treatment,particularly when levels of iASPP are low. Our data support thishypothesis. However, at later stages of tumor progression, whencancer cells have acquired additional changes (among them p53mutations), high levels of Pin1 could amplify proliferative signals.Our results predict that inhibition of Pin1 may counteract thecytotoxicity and hence the clinical activity of drugs that induce p53-dependent apoptosis (for example, the effect of etoposide on p53acetylation and dissociation from iASPP) if administered at earlystages. Conversely, Pin1 inhibition could be beneficial at later stages intumors that may have also acquired p53 mutations. Therefore, inaddition to providing a clearer view into the dynamics of themodifications that allow p53 to mount an efficient response to stress,our observations may also be relevant in designing new strategies forcancer treatment.
METHODSCell lines and treatments. Human HCT116 p53+/+ and p53 –/– colon carcino-
ma, U2OS osteosarcoma, WM115 and WM278 melanoma, p53-null SaOS-2
osteosarcoma and H1299 non–small cell lung carcinoma cell lines were cultured
as described34. Doxycycline-inducible H1299 cell lines expressing p53-Pro72 or
p53-Arg72 have been described32. For generation of ponasterone A (PonA)-
inducible SaOS-2 cell lines expressing p53 mutants, see Supplementary Meth-
ods online. UV dosage was 20 J m–2 for ChIP assays and 40 J m–2 for apoptosis
and iASPP-binding assays; etoposide was added to cells at 50 mM and
doxorubicin at 1 mM. Where not otherwise specified, DNA damage was
administered to cells for 12 h for ChIP and binding assays and for 18 h for
apoptosis assays. Other treatments were as follows: N-CBZ-Leu-Leu-Leu-al
proteasome inhibitor (Sigma), 50 mM for 4 h; peptidylprolyl isomerase–parvulin
inhibitor PiB (diethyl-1,3,6,8-tetrahydro-1,3,6,8-tetraoxobenzo[lmn][3,8]phe-
nanthroline-2,7-diacetate; Calbiochem), 1.5 mM for 48 h; PonA (Invitrogen),
2.5 mM for 12 h. Deacetylase inhibitors (1 mM trichostatin A and 5 mM
nicotinamide) were added to cells for 4 h before analysis of p53 acetylation.
Cells were transfected with Lipofectamine 2000 (Invitrogen). For RNAi,
cells were transfected with 50 nM of the following double-stranded siRNA
©20
07 N
atu
re P
ub
lish
ing
Gro
up
htt
p:/
/ww
w.n
atu
re.c
om
/nsm
b
a b
HIPK2,
p38...
Induction of
target genes
and apoptosis
Ser15
Ser20P
P
Pin1
PSer46-Pro47 Thr81-Pro82
p53p300
AcLys373
Lys382 Ac
P
Pin1
ATM
Chk2
JNK
Ser15
Ser20P
P
Pin 1
PSer46-Pro47 Thr81-Pro82
p53
Mdm2
iASPP
iASPP
JNK
Figure 8 Model for regulation of p53 by Pin1
upon stress. (a) Upon DNA damage, Pin1-
mediated prolyl isomerization of phospho-Ser/
Thr-Pro sites (in particular Thr81-Pro82) in p53
favors Ser20 phosphorylation (P) by Chk2 and
dissociation of p53 from Mdm2, promoting
p53 stabilization. (b) In addition, Pin1 favors
the binding of p53 to target promoters and the
acetylation of DNA-bound p53 by p300. Upon
phosphorylation of Ser46, Pin1 promotes
dissociation of p53 from the apoptosis inhibitor
iASPP. The codon 72 polymorphism influences
these events, the p53-Arg72 isoform being more
efficiently acetylated (Ac) on Lys373 and Lys382
and phosphorylated on Ser46, and less inhibitedby iASPP.
ART IC L E S
NATURE STRUCTURAL & MOLECULAR BIOLOGY ADVANCE ONLINE PUBLICATION 7

oligonucleotides: siRNA Pin1 (I), sense 5¢-GCCAUUUGAAGACGCCUC
GdTdT-3¢ siRNA Pin1 (II), sense 5¢-CGGGAGAGGAGGACUUUGAdTdT-3¢;or scrambled control double-stranded RNA. Where not otherwise specified,
siRNA Pin1 (I) was used. For iASPP RNAi, Dharmacon SMARTpool siRNA
was used.
Plasmids. pcDNA3-Pin1 and pcDNA3-Pin1-C113A, pGEX-Pin1 (ref. 17),
pGEX-Pin1-C113A37 (also described as C109A)17, pcDNA3-Pin1-S67E37,
pEGFP-Pin1 (ref. 34), pcDNA3-p53-T81A and pcDNA3-p53-S46A17,
pcDNA3-p53-P82I20 and pcDNA3-iASPP-V5 (ref. 28) have been described
previously. pGFP-p300 was a gift, as were pcDNA3-p53-Pro72, pcDNA3-p53-
Arg72 and pcDNA3-p53-R175H (see Acknowledgments). Ser/Thr-to-Ala p53
mutants were generated by PCR-based site-directed mutagenesis (Supplemen-
tary Methods).
RNA extraction and reverse-transcription PCR. Total RNA was extracted with
TRIzol (Invitrogen), and 5 mg was reverse-transcribed using SuperscriptIII
reverse transcriptase and random primers (Invitrogen). Reverse-transcription
(RT)-PCR primer sequences are listed in Supplementary Methods.
In vitro binding, co-immunoprecipitation assays and western blotting. GST
pull-down assays were done as described17. For p53-p300 co-immunoprecipi-
tation, cells were lysed in 25 mM HEPES-KOH (pH 8), 150 mM KCl, 2 mM
EDTA, 1 mM DTT and 0.1% (v/v) Nonidet P-40, and cleared lysates were
incubated with antibodies specific to p300 or GFP. For p53-iASPP co-
immunoprecipitation, cells were lysed in 50 mM Tris-HCl (pH 8), 150 mM
NaCl, 1 mM EDTA, 1% (v/v) Nonidet P-40 and 0.5% (v/v) sodium deox-
ycholate with protease inhibitor cocktail (Sigma), 1 mM PMSF, 5 mM NaF and
1 mM Na3VO4, and incubated with anti-p53 (FL-393, Santa Cruz Biotech),
anti-iASPP (pAbiASPPN1) or anti-V5 (Invitrogen). Where required, antibodies
were covalently bound to protein A– or protein G–Sepharose (Amersham)
using 5 mg ml–1 dimethylpimelimidate (Pierce). Pin1-specific rabbit polyclonal
antiserum17, pAbiASPPN1 rabbit polyclonal antibody and the mAbiASPP49.3
monoclonal antibody to iASPP29 have been described previously. GFP-specific
polyclonal antiserum (see Acknowledgments) was affinity-purified. Poly-
clonal antibody to HMGA1b was a gift (see Acknowledgments). Other
antibodies were anti-p53 DO-1 and FL-393 (Santa Cruz Biotech); anti–
phospho-Ser15-p53 and anti–acetyl-Lys382-p53 (Cell Signaling Technology);
anti–acetyl-Lys373-p53 (Upstate); anti-p300 NM11 (BD-Pharmingen); anti-
acetyllysine (Ab409/11A1 Abcam); rabbit polyclonal anti-actin (Sigma); and
anti-p21 C-19 (Santa Cruz Biotechnology). Band intensities on autoradiograms
were quantified using ImageJ software (http://rsb.info.nih.gov/ij/).
Apoptosis assays. Apoptosis was determined by cytofluorimetry (Bio-Rad
Bryte HS) using the Annexin V-FITC Apoptosis detection kit (Sigma).
Cell fractionation. Proteins were separated into chromatin-bound and free
fractions by standard methods50. Briefly, 106 cells were incubated for 20 min on
ice in 10 mM PIPES (pH 7.0), 100 mM NaCl, 3 mM MgCl2, 300 mM sucrose
and 0.1% (v/v) Nonidet P-40. Soluble and insoluble fractions were collected by
centrifugation at 2,500g for 5 min at 4 1C and added to Laemmli sample buffer.
Chromatin immunoprecipitation. ChIP was performed as described51 (Sup-
plementary Methods). Antibodies were as follows: Pin1-specific antiserum (see
above); anti-p53 FL-393 (Santa Cruz Biotechnology) and polyclonal antibody
421 (Ab-1 Calbiochem); anti–acetyl-Lys373-p53 (Upstate) and anti–acetyl-
Lys382-p53 (Cell Signaling Technology); and anti-p300 NM11 (BD-Pharmin-
gen). Control antibody was anti-HA Y11 (Santa Cruz Biotechnology).
DNA–protein complexes were recovered with protein A/G PLUS-Agarose
(Santa Cruz Biotechnology). Semiquantitative PCR products were quantified
with Kodak Digital Science 1d 2.0.2 software. Real-time PCR was performed on
an ABI PRISM 7000 cycler, using TaqMan Universal PCR Master Mix (Applied
Biosystems) as described52. Promoter occupancy was calculated as the percen-
tage of input chromatin immunoprecipitated using the 2–DCT method53. Primer
sequences and PCR settings are available upon request.
In vitro acetylation. HA-p53 was produced in vitro using the TNT T7 Coupled
Transcription/Translation System (Promega) and immunoprecipitated using
anti-HA linked to protein G–Sepharose. Beads were washed in 50 mM Tris-HCl
(pH 7.5), 10 mM MgCl2, 5 mM NaF and 1mM Na3VO4 and incubated with
500 ng of recombinant GST-Pin1 or GST–Pin1-C113A for 20 min at room
temperature. Beads were washed extensively with 50 mM Tris-HCl (pH 7.5),
1 mM DTT and 10% (v/v) glycerol and then resuspended in 40 ml of the same
buffer. The acetylation reaction contained 100 mM acetyl-CoA, and 200 ng
purified recombinant p300 (see Acknowledgments) was added. After 1 h
incubation at 30 1C, the beads were washed and resuspended in Laemmli
sample buffer. p53 acetylation and total abundance were detected using the
antibodies anti–acetyl-Lys382-p53 (Cell Signaling) and FL-393 (Santa Cruz).
Recombinant WT GST-Pin1 and GST-Pin1-C113A were produced in bacteria
and purified using glutathione-Sepharose as described17.
Nucleic acid isolation, gene analysis and immunohistochemistry. Primary
breast cancers were all ductal carcinomas of no special type. Tumor tissue
was obtained and used with fully informed patient consent and approval
of the S. Croce e Carle hospital ethical committee. For each cancer, the
diagnosis of invasive ductal carcinoma, not otherwise specified (NOS), was
confirmed by independent histopathology review. Normal and tumor
tissue were obtained from tissue sections by microdissection. The presence
of an adequate proportion of tumor tissue was confirmed histologically
before analysis.
Genomic DNA and messenger RNA were isolated using Qiagen kits.
Germline p53 codon 72 single-nucleotide polymorphism genotypes were
determined by amplification of individual exons and sequencing of multiple
clones. Expression of iASPP and Pin1 were analyzed using quantitative PCR.
Primer sequences and reaction conditions are described in Supplementary
Methods. For both Pin1 and iASPP, overexpression was defined as at least a
three-fold greater mRNA abundance in the cancer relative to matched normal
tissue. In additional experiments in cell lines and primary cancers, we verified
that Pin1 mRNA levels measured in the quantitative PCR assay correlated with
protein expression determined by western blotting and immunocytochemistry,
concluding that a cutoff of three-fold overexpression of mRNA accurately
predicts an increase in Pin1 protein.
Statistical analysis. For ChIP and apoptosis assays, P-values were obtained
from two-tailed unpaired t-tests. For tissue analysis, P values were obtained
from z-tests with continuity corrections.
Note: Supplementary information is available on the Nature Structural & MolecularBiology website.
ACKNOWLEDGMENTSWe thank C. Schneider, M. Monte and our colleagues at the LaboratorioNazionale Consorzio Interuniversitario Biotecnologie for advice and discussion;A. Grison, A. Bisso, M. Mioni and A. Soldano for technical assistance; B. Amati(European Institute of Oncology) for critical reading of the manuscript;M. Murphy (Fox Chase Cancer Center), Y. Haupt (Hadassah University Hospital),M. Oren (Weizmann Institute) and G. Blandino (Regina Elena Cancer Institute)for providing reagents; and M. Giacca, M. Lusic, M. Bestagno, N. Gammohand L. Banks for access to International Centre for Genetic Engineering andBiotechnology facilities. pGFP-p300 was provided by B. Amati; pcDNA3-p53-Pro72, pcDNA3-p53-Arg72 and pcDNA3-p53-R175H were provided by L. Banks;GFP-specific polyclonal antiserum was provided by C. Brancolini (UdineUniversity); polyclonal antibody to HMGA1b was provided by G. Manfioletti(Trieste University); purified recombinant p300 was provided by R. Mantovani(Milan University). J.G. is an International Centre for Genetic Engineering andBiotechnology Fellow. T.C. is a clinical research fellow of Cancer Research UK.This work was supported by grants from the Associazione Italiana per la Ricercasul Cancro, Association for International Cancer Research UK and Ministerodell’Universita e della Ricerca to G.D.S. and by European Community SixthFramework Programme funding (contract 503576). This publication reflects theauthors’ views and not necessarily those of the European Community. TheEuropean Community is not liable for any use that may be made of theinformation contained herein.
AUTHOR CONTRIBUTIONSF.M., F.T. and J.G. performed biochemical and cell biology experiments, P.S. andM.G. analyzed tissue samples, X.L. produced constructs and reagents, and T.C.supervised P.S. and M.G. and produced reagents. G.D.S. was responsible for theoverall project.
©20
07 N
atu
re P
ub
lish
ing
Gro
up
htt
p:/
/ww
w.n
atu
re.c
om
/nsm
bART IC L E S
8 ADVANCE ONLINE PUBLICATION NATURE STRUCTURAL & MOLECULAR BIOLOGY

Published online at http://www.nature.com/nsmb
Reprints and permissions information is available online at http://npg.nature.com/
reprintsandpermissions
1. Appella, E. & Anderson, C.W. Post-translational modifications and activation of p53 bygenotoxic stresses. Eur. J. Biochem. 268, 2764–2772 (2001).
2. Lavin, M.F. & Gueven, N. The complexity of p53 stabilization and activation. Cell DeathDiffer. 13, 941–950 (2006).
3. Gu, W. & Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylationof the p53 C-terminal domain. Cell 90, 595–606 (1997).
4. Luo, J. et al. Acetylation of p53 augments its site-specific DNA binding both in vitroand in vivo. Proc. Natl. Acad. Sci. USA 101, 2259–2264 (2004).
5. Knights, C.D. et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 173, 533–544 (2006).
6. Tang, Y., Luo, J., Zhang, W. & Gu, W. Tip60-dependent acetylation of p53 modulatesthe decision between cell-cycle arrest and apoptosis. Mol. Cell 24, 827–839 (2006).
7. Sykes, S.M. et al. Acetylation of the p53 DNA-binding domain regulates apoptosisinduction. Mol. Cell 24, 841–851 (2006).
8. Ceskova, P., Chichger, H., Wallace, M., Vojtesek, B. & Hupp, T.R. On the mechanism ofsequence-specific DNA-dependent acetylation of p53: the acetylation motif is exposedupon DNA binding. J. Mol. Biol. 357, 442–456 (2006).
9. Yaffe, M.B. et al. Sequence-specific and phosphorylation-dependent proline isomeriza-tion: a potential mitotic regulatory mechanism. Science 278, 1957–1960 (1997).
10. Yeh, E.S. & Means, A.R. PIN1, the cell cycle and cancer. Nat. Rev. Cancer 7, 381–388(2007).
11. Bulavin, D.V. et al. Phosphorylation of human p53 by p38 kinase coordinatesN-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 18,6845–6854 (1999).
12. Buschmann, T. et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81is important for p53 stabilization and transcriptional activities in response to stress.Mol. Cell. Biol. 21, 2743–2754 (2001).
13. Hofmann, T.G. et al. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 4, 1–10 (2002).
14. D’Orazi, G. et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser46 and mediates apoptosis. Nat. Cell Biol. 4, 11–19 (2002).
15. Radhakrishnan, S.K. & Gartel, A.L. CDK9 phosphorylates p53 on serine residues 33,315 and 392. Cell Cycle 5, 519–521 (2006).
16. Wang, Y. & Prives, C. Increased and altered DNA binding of human p53 by S and G2/Mbut not G1 cyclin-dependent kinases. Nature 376, 88–91 (1995).
17. Zacchi, P. et al. The prolyl isomerase Pin1 reveals a mechanism to control p53functions after genotoxic insults. Nature 419, 853–857 (2002).
18. Zheng, H. et al. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response.Nature 419, 849–853 (2002).
19. Wulf, G.M., Liou, Y.C., Ryo, A., Lee, S.W. & Lu, K.P. Role of Pin1 in the regulation ofp53 stability and p21 transactivation, and cell cycle checkpoints in response to DNAdamage. J. Biol. Chem. 277, 47976–47979 (2002).
20. Berger, M., Stahl, N., Del Sal, G. & Haupt, Y. Mutations in proline 82 of p53 impair itsactivation by Pin1 and Chk2 in response to DNA damage. Mol. Cell. Biol. 25,5380–5388 (2005).
21. Walker, K.K. & Levine, A.J. Identification of a novel p53 functional domain that isnecessary for efficient growth suppression. Proc. Natl. Acad. Sci. USA 93,15335–15340 (1996).
22. Berger, M., Vogt Sionov, R., Levine, A.J. & Haupt, Y. A role for the polyproline domain ofp53 in its regulation by Mdm2. J. Biol. Chem. 276, 3785–3790 (2001).
23. Dornan, D., Shimizu, H., Burch, L., Smith, A.J. & Hupp, T.R. The proline repeatdomain of p53 binds directly to the transcriptional coactivator p300 and allostericallycontrols DNA-dependent acetylation of p53. Mol. Cell. Biol. 23, 8846–8861(2003).
24. Venot, C. et al. The requirement for the p53 proline-rich functional domain formediation of apoptosis is correlated with specific PIG3 gene transactivation and withtranscriptional repression. EMBO J. 17, 4668–4679 (1998).
25. Zhu, J., Jiang, J., Zhou, W., Zhu, K. & Chen, X. Differential regulation of cellular targetgenes by p53 devoid of the PXXP motifs with impaired apoptotic activity. Oncogene18, 2149–2155 (1999).
26. Toledo, F. et al. A mouse p53 mutant lacking the proline-rich domain rescues Mdm4deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory network. CancerCell 9, 273–285 (2006).
27. Chipuk, J.E. et al. Direct activation of Bax by p53 mediates mitochondrial membranepermeabilization and apoptosis. Science 303, 1010–1014 (2004).
28. Bergamaschi, D. et al. iASPP oncoprotein is a key inhibitor of p53 conserved fromworm to human. Nat. Genet. 33, 162–167 (2003).
29. Bergamaschi, D. et al. iASPP preferentially binds p53 proline-rich region andmodulates apoptotic function of codon 72-polymorphic p53. Nat. Genet. 38,1133–1141 (2006).
30. Matlashewski, G.J. et al. Primary structure polymorphism at amino acid residue 72 ofhuman p53. Mol. Cell. Biol. 7, 961–963 (1987).
31. Thomas, M. et al. Two polymorphic variants of wild-type p53 differ biochemically andbiologically. Mol. Cell. Biol. 19, 1092–1100 (1999).
32. Sullivan, A. et al. Polymorphism in wild-type p53 modulates response to chemotherapyin vitro and in vivo. Oncogene 23, 3328–3337 (2004).
33. Vassilev, L.T. et al. In vivo activation of the p53 pathway by small-molecule antagonistsof MDM2. Science 303, 844–848 (2004).
34. Mantovani, F. et al. Pin1 links the activities of c-Abl and p300 in regulating p73function. Mol. Cell 14, 625–636 (2004).
35. Zhao, Y. et al. Acetylation of p53 at lysine 373/382 by the histone deacetylaseinhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol. Cell. Biol. 26,2782–2790 (2006).
36. Barlev, N.A. et al. Acetylation of p53 activates transcription through recruitment ofcoactivators/histone acetyltransferases. Mol. Cell 8, 1243–1254 (2001).
37. Zhou, X.Z. et al. Pin1-dependent prolyl isomerization regulates dephosphorylation ofcdc25 and tau proteins. Mol. Cell 6, 873–883 (2000).
38. Ard, P.G. et al. Transcriptional regulation of the mdm2 oncogene by p53requires TRRAP acetyltransferase complexes. Mol. Cell. Biol. 22, 5650–5661(2002).
39. Uchida, T. et al. Pin1 and Par14 peptidyl prolyl isomerase inhibitors block cellproliferation. Chem. Biol. 10, 15–24 (2003).
40. Kern, S.E. et al. Oncogenic forms of p53 inhibit p53-regulated gene expression.Science 256, 827–830 (1992).
41. Dornan, D., Shimizu, H., Perkins, N.D. & Hupp, T.R. DNA-dependent acetylation ofp53 by the transcription coactivator p300. J. Biol. Chem. 278, 13431–13441(2003).
42. Bao, L. et al. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am.J. Pathol. 164, 1727–1737 (2004).
43. Dornan, D. & Hupp, T.R. Inhibition of p53-dependent transcription by BOX-I phospho-peptide mimetics that bind to p300. EMBO Rep. 2, 139–144 (2001).
44. Sun, X.F. et al. A novel p53 germline alteration identified in a late onset breast cancerkindred. Oncogene 13, 407–411 (1996).
45. Wulf, G., Finn, G., Suizu, F. & Lu, K.P. Phosphorylation-specific prolyl isomerization: isthere an underlying theme? Nat. Cell Biol. 7, 435–441 (2005).
46. Xu, Y.X., Hirose, Y., Zhou, X.Z., Lu, K.P. & Manley, J.L. Pin1 modulates thestructure and function of human RNA polymerase II. Genes Dev. 17, 2765–2776(2003).
47. Oda, K. et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and itsregulation by Ser-46-phosphorylated p53. Cell 102, 849–862 (2000).
48. Mayo, L.D. et al. Phosphorylation of human p53 at serine 46 determines promoterselection and whether apoptosis is attenuated or amplified. J. Biol. Chem. 280,25953–25959 (2005).
49. Samuels-Lev, Y. et al. ASPP proteins specifically stimulate the apoptotic function ofp53. Mol. Cell 8, 781–794 (2001).
50. Geng, Y. et al. Cyclin E ablation in the mouse. Cell 114, 431–443 (2003).51. Monte, M. et al. MAGE-A tumor antigens target p53 transactivation function through
histone deacetylase recruitment and confer resistance to chemotherapeutic agents.Proc. Natl. Acad. Sci. USA 103, 11160–11165 (2006).
52. Kaeser, M.D. & Iggo, R.D. Chromatin immunoprecipitation analysis fails to support thelatency model for regulation of p53 DNA binding activity in vivo. Proc. Natl. Acad. Sci.USA 99, 95–100 (2002).
53. Livak, K.J. & Schmittgen, T.D. Analysis of relative gene expression data using real-timequantitative PCR and the 2-DDC(T) method. Methods 25, 402–408 (2001).
©20
07 N
atu
re P
ub
lish
ing
Gro
up
htt
p:/
/ww
w.n
atu
re.c
om
/nsm
bART IC L E S
NATURE STRUCTURAL & MOLECULAR BIOLOGY ADVANCE ONLINE PUBLICATION 9

MATHERIALS and METHODS
MATHERIALS and METHODS Cell lines and treatments. WI38 are human lung fibroblasts; IMR90 are immortalized
human lung fibroblast; HaCat are immortalized human keratinocytes; HCT116 p53+/+ and
RKO are human colon-carcinoma cells, HCT116 p53–/– ,derived from wt p53-containing
HCT116 by gene disruption (Bunz et al., 1998); U2OS are human osteosarcoma cells;
U2OS/ER-E2F1 inducible cell line was kindly provided by K. Helin and is described in
(Muller et al., 2001); SaOS-2 are human p53-null osteosarcoma cells; H1299 are human
p53-null non–small cell lung carcinoma cells; MCF-7, SkBR-3 and MDAMB468 are
human breast carcinoma cells; HeLa are human cervical carcinoma cells; 293T are a
derivative of the 293 kidney carcinoma cells and constitutively express the simian virus 40
(SV40) large T antigen; SHSY5Y are human neuroblastoma cells; HL 60 are human
promyelocytic leukemia cells.
UV dosage was 40 J m–2 for 18 h in apoptosis assays and for 12 h in iASPP-binding assays;
Etoposide was added to cells at the concentration and time indicated. N-CBZ-Leu-Leu-
Leu-al proteasome inhibitor (Sigma), 50 µM for 4 h; Ciclohexymide (CHX), 50 µM for the
time indicated.
Cells were transfected with Lipofectamine 2000 (Invitrogen). For RNAi, cells were
transfected with Lipofectamine RNAiMAX (Invitrogen) with 50 nM of the following
double-stranded siRNA oligonucleotides:
siRNA Pin1 (I), sense 5’-GCCAUUUGAAGACGCCUCGdTdT-3’
siRNA Pin1 (II), sense 5’-CGGGAGAGGAGGACUUUGAdTdT-3’;
siRNA Brd7-1, sense 5’-GCACGTATGGAGTTCGAAAdTdT-3’
siRNA Brd7-2, sense 5’-CCAGAGACCATTTATTATAdTdT-3’
as control a siCONT RISC Free (Dharmacon) double stranded RNA oligonucleotide was
used
Plasmids. pcDNA3-p53wt and pcDNA3-p53-R175H were provided by L. Banks;
pCDNA3-p53-4M and pCDNA3-p53 6M, pCDNA3-p53T81A, pCDNA3-p53S46A and all
the pCDNA3 p535M were produced by site directed mutagenesis from pCDNA3 p53wt;
pCDNA3-HA-Pin1 was cloned in the lab by P.Zacchi; pcDNA3-HAp73α and pcDNA3-
110

MATHERIALS and METHODS
mycp63α have been kindly provided by G. Melino and F. McKeon respectively; pCDNA3
Flag Brd7 and pCDNA3-Flag ΔBD Brd7 were kindly provided by T.Dobner;
Brd7 expression plasmid for the expression of MBP-Brd7 fusion proteins in bacteria were
all obtained by cloning the entire Brd7 CDS, or its portions, in frame with Maltose binding
protein (MBP) gene of pMAL-2X expression vector. To obtain Brd7 deletion derivatives,
defined portions of the Brd7 CDS were PCR-amplified with specific primers using as a
template pCDNA3-Flag Brd7.
Antibodies. Endogenous and overexpressed p53 were detected using DO-1 monoclonal
antibody or FL-393 polyclonal antibody (Santa Cruz). Other commercial antibodies were
rabbit polyclonal anti–acetyl-Lys382-p53 (Cell Signaling Technology); rabbit polyclonal
anti- p63α H-129 (Santa Cruz); mouse monoclonal anti-p300 NM11 (BD-Pharmingen);
mouse monoclonal anti-acetyllysine (Ab409/11A1 Abcam); mouse monoclonal anti-Flag
(M2 Sigma); rabbit polyclonal anti-actin (Sigma); mouse monoclonal anti-BrdU (GE
Healthcare)and anti-p21 C-19 (Santa Cruz Biotechnology). Purification and production of
p73 rabbit polyclonal serum and Pin1 polyclonal serum is described in Mantovani et al.,
2004 and Zacchi et al., 2002, respectively. Anti iASPP pAbiASPPN1 rabbit polyclonal
antibody and the mAbiASPP49.3 monoclonal antibody to were kindly provided by X. Lu
and are described in Bergamaschi et al., 2006; polyclonal antibody against HMGA1b was
kindly provided by G. Manfioletti.
Anti Brd7 rabbit polyclonal serum was raised against the C-terminal portion (aa361-651)
of human Brd7. A recombinant protein bearing Brd7 C-terminal portion (aa 361-651) in
fusion with MBP was expressed in BL-21 E.coli bacterial strain and affinity-purified with
amiloyd resin (NEB E8021S). The purified protein has been injected in a rabbit to induce
immunization. The sera deriving from the bleedings have been tested for the presence of
Brd7 antibody by western blot (Figure 36 A). The antiserum derived from the 5th bleed has
been affinity purified. Briefly, bacterially expressed Brd7 C-terminal portion (aa 361-651)
in fusion with GST was cross-linked to glutathione Sepharose4B beads (GST was used
instead of MBP to avoid purification of anti-MBP antibodies) and incubated over-night
with the serum. Upon washing with 10 ml PBS, 10 ml Tris pH8, 10 ml SDS 0,1% in PBS,
20 ml PBS, 10 ml NaCl 400mM in PBS and 20 ml PBS, the antibody was eluted from the
resin with glycine 0,1M pH 2.8 and pH was adjusted to 7.5-8 with Tris pH8. The antibody
purified was then tested for selectivity and affinity in western blot (Figure 36B).
111

MATHERIALS and METHODS
Figure.36 . Production and purification of anti-Brd7 antibody. A.The bleeds were tested for the presence of anti-Brd7 antibody by western blot on lysates from H1299 transfected with pCDNA3-Flag-Brd7. (PIS, Pre immune serum; FB, final bleed) B. The purified antibody fracrions were tested for specificity and affinity by western blot on lysates from H1299 either transfected with pCDNA3-Flag-Brd7or not.
In vitro pull-down assays. Cells were lysed in lysis buffer (LY) (300 mM NaCl , 50 mM
Tris pH 7.5, 0,5% NP40, 10% glycerol, 1 mM PMSF, 5mM NaF and 1mM Na3VO4) for 20
min at 4°C rocking and lysates were cleared by centrifuging for 10 min at 13000 rpm at
4°C. The supernatant was diluted 1:2 with pull down buffer (PD) (50 mM Tris pH=7.5,
0,5% NP40, 10% glycerol, 1 mM PMSF, 5mM NaF and 1mM Na3VO4). GST-Pin1 fusion
protein and GST were extracted from bacteria in GST purification buffer (250mM Tris pH
7.5, 1% Triton X-100, 0,5% NP40, 0,1% Tween-20, 0,2%SDS) and purified by incubation
with glutathione- Sepharose 4B beads 4hrs at 4°C and washed three times at 4°C with PBS.
The beads were equilibrated with binding buffer (150mM Tris pH 7.5, 0,5% NP40, 10%
glycerol). Cell lysates were precleared with glutathione Sepharose 4B beads for 1 h at 4°C
and then incubated with GST or GST-Pin1 proteins bound to glutathione Sepharose 4B
beads. After incubation at 4°C for 3 hrs, the beads were washed five times in binding
buffer, and proteins bound to the beads were eluted into sample buffer, followed by
SDS/PAGE, and Western blots. λ-phosphatase treatment was performed where indicated
by incubating half of the cell lysate (w/o phosphatase inhibitors) for 1h with 100 units of
λ-phosphatase at 30°C.
IVTT proteins were obtained by incubating 1µg of the indicated expression vectors with
12,5 µl rabbit reticulocyte lysate, 5 µl IVTT buffer (2mM MgCl2, 50mM KOAc, 1mM
NTP, 1mM creatine phosphate), 20 units of T7 RNA Pol, , aminoacid mix (minus Met) and 35S-Met to a final volume of 25 µl, for 1h at 30°C (Promega) following the manifacturer’s
instructions. IVTT proteins were diluted in MBP-PD buffer (150mM Tris pH 0, 50 mM
Tris pH=7.5, 1% NP40, 10% glycerol) and pull down was performed as described above.
MBP-fusion proteins and MBP were purified as described above with amiloyd resin (NEB
112

MATHERIALS and METHODS
E8021S) in Column buffer (20mM Tris-HCl pH7.5, 200mM NaCl, 1mM EDTA pH8, and
10mM Beta-mercaptoethanol)
Co-immunoprecipitation assays. For p53-iASPP coimmunoprecipitation, cells were
lysed in 50 mM Tris-HCl (pH 8), 150 mM NaCl, 1 mM EDTA, 1% NP-40 and 0.5%
sodium deoxycholate with protease inhibitor cocktail (Sigma), 1 mM PMSF, 5 mM NaF
and 1 mM Na3VO4, and incubated with anti-iASPP (pAbiASPPN1) or anti-V5 (Invitrogen)
covalently bound to protein A/G–Sepharose (Amersham) using 5
mg/mldimethylpimelimidate (Pierce).
For p53-Brd7 coimmunoprecipitation, cells were lysed in 50 mM Tris-HCl (pH 8), 150
mM NaCl, 1% NP-40, 10% glycerol, with protease inhibitor cocktail (Sigma), 1 mM
PMSF, 5 mM NaF, 1 mM Na3 VO4, 1µM TSA and 5 µM nicotinamide and incubated with
anti-Flag monoclonal antibody (Sigma) or anti-Brd7 polyclonal antibody covalently bound
to protein A- or protein G–Sepharose (Amersham) using 5 mg/ml dimethylpimelimidate
(Pierce). For the co-immunprecipitation of the endogenous p53 and Brd7 pre-immune
rabbit serum was used as a control.
Chromatin immunoprecipitation (ChIP). Cells were crosslinked with 1% formaldehyde
in DMEM for 10 min, neutralized with 125 mM glycine pH 2.5 in PBS and washed in
PBS. Nuclei were prepared by hypotonic lysis (5mM Pipes pH 6.8, 85 mM KCl, 0,5%
NP40) and centrifugation, and resuspended in RIPA-100 buffer (20 mM Tris HCl, pH 7.5,
100 mM NaCl 1 mM EDTA 0,5% NP-40 0,5% deoxycholate 0,1% SDS) with protease
inhibitor cocktail (Sigma), 1mM PMSF and phosphatase inhibitors (NaF 5mM and Na3VO4
1mM). Chromatin was sonicated with Bioruptor (Diagenode) to 500-1000 bp average
fragment size and cleared by centrifugation. IP was performed overnight at 4°C with either:
anti-Brd7 antiserum (see above) or anti-p53 FL-393 (Santa Cruz Biotechnology) a negative
control was performed in the presence of Y-11 anti-HA polyclonal antibody (Santa Cruz).
DNA–protein complexes were recovered by protein A/G PLUS-Agarose (Santa Cruz
Biotech.) and washed sequentially with RIPA-100 buffer, RIPA-250 buffer (20 mM Tris
HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 0,5% NP-40, 0,5% deoxycholate, 0,1% SDS)
and LiCl solution (10 mM Tris-HCl pH 8, 1mM EDTA, 250 mM LiCl, 0.5% Na-
Deoxycholate, 0.5% NP40), then resuspended in TE, digested with 2U DNase-free RNase
(Calbiochem) for 30 min at 37°C, and incubated o/n at 68°C with 300 mg/ml Proteinase K
113

MATHERIALS and METHODS
(Invitrogen) in 0.5% SDS, 100 mM NaCl to digest proteins and reverse crosslinks. After
purification by phenol-chloroform extraction and ethanol precipitation, DNA was
resuspended in H2O and 1/10 volume was used for quantification. Real Time PCR was
performed on ABI PRISM 7000, using TaqMan Universal PCR Master Mix (Applied
Biosystems), with primers and probes (Applied Biosystems) and amplification settings as
described in Kaeser and Iggo, 2002.
p21 fw: 5’- GTGGCTCTGATTGGCTTTCTG -3’,
p21 rv:5’-CTGAAAACAGGCAGCCCAAG-3’,
p21 probe: 5’-TGGCATAGAAGAGGCTGGTGGCTATTTTGT-3’
GAPDH fw: 5’- GTATTCCCCCAGGTTTACAT-3’,
GAPDH rv: 5’-TTCTGTCTTCCACTCACTCC-3’
GAPDH Probe: 5’-TGGCATAGAAGAGGCTGGTGGCTATTTTG-3’
Diluted inputs (1/10 and 1/100) were used as standards for each primer set. Promoter
occupancy was calculated as a percent of input chromatin immunoprecipitated using the 2-
ΔCT method.
RNA extraction and RT-PCR. Total RNA was extracted with TRI Reagent (Sigma)
according to manufacturer's instructions. RNA treated with RQ1 RNase-free DNAse
(Promega) for 30 min at 37°C and reaction was stopped by incubating with 1mM EGTA
pH8 for 10 min at 65°C. 500 ng of total RNA were reverse-transcribed using ImProm-II
Reverse Transcriptase(Promega) according to the manufacturer’s instructions. Real Time
PCR was performed on cDNAs on ABI PRISM 7000, using TaqMan Universal PCR
Master Mix (Applied Biosystems) or with Fast SYBR Green Master Mix (Applied
Biosystems). Primers and probes for p21 mRNA are
p21 fw 5’- CTGGAGACTCTCAGGGTCGAAA-3’,
p21 rv: 5’GATTAGGGCTTCCTCTTGGAGAA-3’,
p21 probe: 5’-GGCAGACCAGCATGACAGATTTCTACC-3’
GAPDH primers and probe sequences are:
FW: 5'-TTTGGAGGGATCTCGCTCCT-3',
RV: 5'-CAACTACATGGTTTACATGTTC-3';
Probe:5’-TGGCATAGAAGAGGCTGGTGGCTATTTTG-3’;
For Mdm2 PRIMER&PROBE 20X mix (Hs00242813_m1) from Applied Biosystems; for
PUMA(BBC3): PRIMER&PROBE 20X mix (Hs0o248075_m1) from Applied Biosystems;
114

MATHERIALS and METHODS
Brd7 amplification was performed with Fast SYBR Green Master Mix (Applied
Biosystems) with primers:
FW: 5'-CTGGAGATGCCGAAGCACAC-3'
RV: 5'-TGGGATCCACAGGATGGAGA-3'.
mRNA abundance was calculated relative to GAPDH housekeeping gene using the 2-ΔΔCT
method (Livak and Schmittgen, 2001).
Transient Transactivation Assays. H1299 cells were co- transfected in a 24-well plate
with 500 ng of p21-prom-luc or Bax-prom-luc reporter constructs together with,
respectively, 2 μg of pCDNA3-HA, 50 ng of pCDNA3 p53 either alone or along with
increasing amounts (0,5 1 or 2 μg) of pCDNA3-Flag-Brd7, or with 2 μg of pCDNA3-Flag-
Brd7. For normalization of transfection efficiency, 12.5 ng of pRL-CMV reporter
(Promega), constitutively expressing the Renilla reniformis luciferase, was included. After
36 hrs, cells were lysed and assayed for luciferase activity using the Dual Luciferase kit
(Promega). H1299 cells in a 60 mm plate were transfected by RNAiMAX (Promega) with
2 µl of 20 µM double-stranded siRNA oligonucleotides specific for Brd7 (siBrd7) or with
the same amount of RISC Free siRNA control (siC) (Dharmacon). After 12hrs the cells
were splitted to a 24 well plate and transfected by Lipofectamine 2000 (Invitrogen) with
500 ng of Luc reporters, 12.5 ng of pRL-CMV (Promega) together with either siBrd7 or
siC. After 36 hrs, cells were lysed and assayed for luciferase activity using the Dual
Luciferase kit (Promega).
Apoptosis assays. Apoptosis was determined by cytofluorimetry (Bio-Rad Bryte HS)
using the Annexin V-FITC Apoptosis detection kit (Sigma).
BrdU Incorporation Assays. Cells were plated on a slide in 35 mm dish and transfected
with either 6 µl of 20 µM double-stranded siRNA oligonucleotides specific for Brd7 or
with the same amount of RISC Free siRNA control. After 60 hrs cells were treated with
etoposide 50 µM or left untreated for 12 hrs. Cells were pulsed with 3 µl of 20 mM
bromodeoxyuridine (BrdU, Sigma) for 3 hrs. After pulsing, cells were fixed, permeabilized
and denatured. BrdU incorporation was measured by immunofluorescence using an anti-
BrdU antibody (GE Healthcare) and the nuclei were stained with Hoechst. The stained
115

MATHERIALS and METHODS
116
cells were visualized under a fluorescence microscope and at least 300 cells were scored
for BrdU incorporation.
Cell fractionation. Proteins were separated into chromatin-bound and free fractions by
standard methods (Geng et al., 2003). Briefly, 106 cells were incubated for 20 min on ice in
10 mM PIPES (pH 7.0), 100 mM NaCl, 3 mM MgCl2, 300 mM sucrose and 0.1% (v/v)
Nonidet P-40. Soluble and insoluble fractions were collected by centrifugation at 2,500g
for 5 min at 4°C and added to Laemmli sample buffer.

REFERENCES
REFERENCES
Achanta, G., and Huang, P. (2004). Role of p53 in sensing oxidative DNA damage in response to reactive oxygen
species-generating agents. Cancer research 64, 6233-6239.
Achanta, G., Sasaki, R., Feng, L., Carew, J.S., Lu, W., Pelicano, H., Keating, M.J., and Huang, P. (2005). Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. The EMBO journal 24, 3482-3492.
Agalioti, T., Chen, G., and Thanos, D. (2002). Deciphering the transcriptional histone acetylation code for a human gene. Cell 111, 381-392.
Ahomadegbe, J.C., Tourpin, S., Kaghad, M., Zelek, L., Vayssade, M., Mathieu, M.C., Rochard, F., Spielmann, M., Tursz, T., Caput, D., et al. (2000). Loss of heterozygosity, allele silencing and decreased expression of p73 gene in breast cancers: prevalence of alterations in inflammatory breast cancers. Oncogene 19, 5413-5418.
Albrechtsen, N., Dornreiter, I., Grosse, F., Kim, E., Wiesmuller, L., and Deppert, W. (1999). Maintenance of genomic integrity by p53: complementary roles for activated and non-activated p53. Oncogene 18, 7706-7717.
Alcorta, D.A., Xiong, Y., Phelps, D., Hannon, G., Beach, D., and Barrett, J.C. (1996). Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America 93, 13742-13747.
An, W., Kim, J., and Roeder, R.G. (2004). Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 117, 735-748.
Anderson, L., Henderson, C., and Adachi, Y. (2001). Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Molecular and cellular biology 21, 1719-1729.
Ando, T., Kawabe, T., Ohara, H., Ducommun, B., Itoh, M., and Okamoto, T. (2001). Involvement of the interaction between p21 and proliferating cell nuclear antigen for the maintenance of G2/M arrest after DNA damage. The Journal of biological chemistry 276, 42971-42977.
Andrews, P., He, Y.J., and Xiong, Y. (2006). Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizing p53 function. Oncogene 25, 4534-4548.
Appella, E., and Anderson, C.W. (2001). Post-translational modifications and activation of p53 by genotoxic stresses. European journal of biochemistry / FEBS 268, 2764-2772.
Ard, P.G., Chatterjee, C., Kunjibettu, S., Adside, L.R., Gralinski, L.E., and McMahon, S.B. (2002). Transcriptional regulation of the mdm2 oncogene by p53 requires TRRAP acetyltransferase complexes. Molecular and cellular biology 22, 5650-5661.
Ashcroft, M., Ludwig, R.L., Woods, D.B., Copeland, T.D., Weber, H.O., MacRae, E.J., and Vousden, K.H. (2002). Phosphorylation of HDM2 by Akt. Oncogene 21, 1955-1962.
Asher, G., Lotem, J., Cohen, B., Sachs, L., and Shaul, Y. (2001). Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proceedings of the National Academy of Sciences of the United States of America 98, 1188-1193.
Asher, G., Lotem, J., Sachs, L., Kahana, C., and Shaul, Y. (2002). Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proceedings of the National Academy of Sciences of the United States of America 99, 13125-13130.
Asher, G., and Shaul, Y. (2005). p53 proteasomal degradation: poly-ubiquitination is not the whole story. Cell cycle (Georgetown, Tex 4, 1015-1018.
Asher, G., Tsvetkov, P., Kahana, C., and Shaul, Y. (2005). A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes & development 19, 316-321.
117

REFERENCES
Atchison, F.W., Capel, B., and Means, A.R. (2003). Pin1 regulates the timing of mammalian primordial germ cell proliferation. Development (Cambridge, England) 130, 3579-3586.
Attardi, L.D., Reczek, E.E., Cosmas, C., Demicco, E.G., McCurrach, M.E., Lowe, S.W., and Jacks, T. (2000). PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes & development 14, 704-718.
Avantaggiati, M.L., Ogryzko, V., Gardner, K., Giordano, A., Levine, A.S., and Kelly, K. (1997). Recruitment of p300/CBP in p53-dependent signal pathways. Cell 89, 1175-1184.
Ayed, A., Mulder, F.A., Yi, G.S., Lu, Y., Kay, L.E., and Arrowsmith, C.H. (2001). Latent and active p53 are identical in conformation. Nature structural biology 8, 756-760.
Bakalkin, G., Selivanova, G., Yakovleva, T., Kiseleva, E., Kashuba, E., Magnusson, K.P., Szekely, L., Klein, G., Terenius, L., and Wiman, K.G. (1995). p53 binds single-stranded DNA ends through the C-terminal domain and internal DNA segments via the middle domain. Nucleic acids research 23, 362-369.
Banin, S., Moyal, L., Shieh, S., Taya, Y., Anderson, C.W., Chessa, L., Smorodinsky, N.I., Prives, C., Reiss, Y., Shiloh, Y., et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science (New York, NY 281, 1674-1677.
Barlev, N.A., Liu, L., Chehab, N.H., Mansfield, K., Harris, K.G., Halazonetis, T.D., and Berger, S.L. (2001). Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Molecular cell 8, 1243-1254.
Bartkova, J., Horejsi, Z., Koed, K., Kramer, A., Tort, F., Zieger, K., Guldberg, P., Sehested, M., Nesland, J.M., Lukas, C., et al. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864-870.
Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N., Vassiliou, L.V., Kolettas, E., Niforou, K., Zoumpourlis, V.C., et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633-637.
Bensaad, K., Le Bras, M., Unsal, K., Strano, S., Blandino, G., Tominaga, O., Rouillard, D., and Soussi, T. (2003). Change of conformation of the DNA-binding domain of p53 is the only key element for binding of and interference with p73. The Journal of biological chemistry 278, 10546-10555.
Bensaad, K., Tsuruta, A., Selak, M.A., Vidal, M.N., Nakano, K., Bartrons, R., Gottlieb, E., and Vousden, K.H. (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107-120.
Bergamaschi, D., Gasco, M., Hiller, L., Sullivan, A., Syed, N., Trigiante, G., Yulug, I., Merlano, M., Numico, G., Comino, A., et al. (2003a). p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer cell 3, 387-402.
Bergamaschi, D., Samuels, Y., Jin, B., Duraisingham, S., Crook, T., and Lu, X. (2004). ASPP1 and ASPP2: common activators of p53 family members. Molecular and cellular biology 24, 1341-1350.
Bergamaschi, D., Samuels, Y., O'Neil, N.J., Trigiante, G., Crook, T., Hsieh, J.K., O'Connor, D.J., Zhong, S., Campargue, I., Tomlinson, M.L., et al. (2003b). iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nature genetics 33, 162-167.
Bergamaschi, D., Samuels, Y., Sullivan, A., Zvelebil, M., Breyssens, H., Bisso, A., Del Sal, G., Syed, N., Smith, P., Gasco, M., et al. (2006). iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nature genetics 38, 1133-1141.
Berger, M., Stahl, N., Del Sal, G., and Haupt, Y. (2005). Mutations in proline 82 of p53 impair its activation by Pin1 and Chk2 in response to DNA damage. Molecular and cellular biology 25, 5380-5388.
Berger, M., Vogt Sionov, R., Levine, A.J., and Haupt, Y. (2001). A role for the polyproline domain of p53 in its regulation by Mdm2. The Journal of biological chemistry 276, 3785-3790.
Berkovich, E., and Ginsberg, D. (2001). Ras induces elevation of E2F-1 mRNA levels. The Journal of biological chemistry 276, 42851-42856.
118

REFERENCES
Berns, K., Hijmans, E.M., Mullenders, J., Brummelkamp, T.R., Velds, A., Heimerikx, M., Kerkhoven, R.M., Madiredjo, M., Nijkamp, W., Weigelt, B., et al. (2004). A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428, 431-437.
Bertrand, P., Saintigny, Y., and Lopez, B.S. (2004). p53's double life: transactivation-independent repression of homologous recombination. Trends Genet 20, 235-243.
Bhat, K.P., Itahana, K., Jin, A., and Zhang, Y. (2004). Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. The EMBO journal 23, 2402-2412.
Bjorklund, S., and Gustafsson, C.M. (2005). The yeast Mediator complex and its regulation. Trends in biochemical sciences 30, 240-244.
Blattner, C., Tobiasch, E., Litfen, M., Rahmsdorf, H.J., and Herrlich, P. (1999). DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene 18, 1723-1732.
Blaydes, J.P., Luciani, M.G., Pospisilova, S., Ball, H.M., Vojtesek, B., and Hupp, T.R. (2001). Stoichiometric phosphorylation of human p53 at Ser315 stimulates p53-dependent transcription. The Journal of biological chemistry 276, 4699-4708.
Bode, A.M., and Dong, Z. (2004). Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 4, 793-805.
Bonafe, M., Salvioli, S., Barbi, C., Trapassi, C., Tocco, F., Storci, G., Invidia, L., Vannini, I., Rossi, M., Marzi, E., et al. (2004). The different apoptotic potential of the p53 codon 72 alleles increases with age and modulates in vivo ischaemia-induced cell death. Cell death and differentiation 11, 962-973.
Bond, G.L., Hirshfield, K.M., Kirchhoff, T., Alexe, G., Bond, E.E., Robins, H., Bartel, F., Taubert, H., Wuerl, P., Hait, W., et al. (2006a). MDM2 SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner. Cancer research 66, 5104-5110.
Bond, G.L., Menin, C., Bertorelle, R., Alhopuro, P., Aaltonen, L.A., and Levine, A.J. (2006b). MDM2 SNP309 accelerates colorectal tumour formation in women. Journal of medical genetics 43, 950-952.
Borden, K.L. (2002). Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies. Molecular and cellular biology 22, 5259-5269.
Bottger, A., Bottger, V., Garcia-Echeverria, C., Chene, P., Hochkeppel, H.K., Sampson, W., Ang, K., Howard, S.F., Picksley, S.M., and Lane, D.P. (1997). Molecular characterization of the hdm2-p53 interaction. Journal of molecular biology 269, 744-756.
Bourdon, J.C. (2007). p53 and its isoforms in cancer. British journal of cancer 97, 277-282.
Bourdon, J.C., Fernandes, K., Murray-Zmijewski, F., Liu, G., Diot, A., Xirodimas, D.P., Saville, M.K., and Lane, D.P. (2005). p53 isoforms can regulate p53 transcriptional activity. Genes & development 19, 2122-2137.
Bourdon, J.C., Renzing, J., Robertson, P.L., Fernandes, K.N., and Lane, D.P. (2002). Scotin, a novel p53-inducible proapoptotic protein located in the ER and the nuclear membrane. The Journal of cell biology 158, 235-246.
Bouvard, V., Zaitchouk, T., Vacher, M., Duthu, A., Canivet, M., Choisy-Rossi, C., Nieruchalski, M., and May, E. (2000). Tissue and cell-specific expression of the p53-target genes: bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene 19, 649-660.
Boyd, S.D., Tsai, K.Y., and Jacks, T. (2000). An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nature cell biology 2, 563-568.
Brooks, C.L., and Gu, W. (2003). Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Current opinion in cell biology 15, 164-171.
Brooks, C.L., Li, M., Hu, M., Shi, Y., and Gu, W. (2007). The p53--Mdm2--HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene 26, 7262-7266.
119

REFERENCES
Brown, J.P., Wei, W., and Sedivy, J.M. (1997). Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science (New York, NY 277, 831-834.
Brugarolas, J., Chandrasekaran, C., Gordon, J.I., Beach, D., Jacks, T., and Hannon, G.J. (1995). Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377, 552-557.
Bruins, W., Zwart, E., Attardi, L.D., Iwakuma, T., Hoogervorst, E.M., Beems, R.B., Miranda, B., van Oostrom, C.T., van den Berg, J., van den Aardweg, G.J., et al. (2004). Increased sensitivity to UV radiation in mice with a p53 point mutation at Ser389. Molecular and cellular biology 24, 8884-8894.
Brunner, H.G., Hamel, B.C., and Van Bokhoven, H. (2002). The p63 gene in EEC and other syndromes. Journal of medical genetics 39, 377-381.
Budanov, A.V., Sablina, A.A., Feinstein, E., Koonin, E.V., and Chumakov, P.M. (2004). Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science (New York, NY 304, 596-600.
Bulavin, D.V., Saito, S., Hollander, M.C., Sakaguchi, K., Anderson, C.W., Appella, E., and Fornace, A.J., Jr. (1999). Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. The EMBO journal 18, 6845-6854.
Bullock, A.N., Henckel, J., DeDecker, B.S., Johnson, C.M., Nikolova, P.V., Proctor, M.R., Lane, D.P., and Fersht, A.R. (1997). Thermodynamic stability of wild-type and mutant p53 core domain. Proceedings of the National Academy of Sciences of the United States of America 94, 14338-14342.
Bullock, A.N., Henckel, J., and Fersht, A.R. (2000). Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19, 1245-1256.
Bunz, F., Dutriaux, A., Lengauer, C., Waldman, T., Zhou, S., Brown, J.P., Sedivy, J.M., Kinzler, K.W., and Vogelstein, B. (1998). Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science (New York, NY 282, 1497-1501.
Buschmann, T., Adler, V., Matusevich, E., Fuchs, S.Y., and Ronai, Z. (2000a). p53 phosphorylation and association with murine double minute 2, c-Jun NH2-terminal kinase, p14ARF, and p300/CBP during the cell cycle and after exposure to ultraviolet irradiation. Cancer research 60, 896-900.
Buschmann, T., Fuchs, S.Y., Lee, C.G., Pan, Z.Q., and Ronai, Z. (2000b). SUMO-1 modification of Mdm2 prevents its self-ubiquitination and increases Mdm2 ability to ubiquitinate p53. Cell 101, 753-762.
Buschmann, T., Potapova, O., Bar-Shira, A., Ivanov, V.N., Fuchs, S.Y., Henderson, S., Fried, V.A., Minamoto, T., Alarcon-Vargas, D., Pincus, M.R., et al. (2001). Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Molecular and cellular biology 21, 2743-2754.
Buschmann, T., Yin, Z., Bhoumik, A., and Ronai, Z. (2000c). Amino-terminal-derived JNK fragment alters expression and activity of c-Jun, ATF2, and p53 and increases H2O2-induced cell death. The Journal of biological chemistry 275, 16590-16596.
Bykov, V.J., Issaeva, N., Shilov, A., Hultcrantz, M., Pugacheva, E., Chumakov, P., Bergman, J., Wiman, K.G., and Selivanova, G. (2002). Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nature medicine 8, 282-288.
Cahilly-Snyder, L., Yang-Feng, T., Francke, U., and George, D.L. (1987). Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somatic cell and molecular genetics 13, 235-244.
Carrozza, M.J., Utley, R.T., Workman, J.L., and Cote, J. (2003). The diverse functions of histone acetyltransferase complexes. Trends Genet 19, 321-329.
Cawley, S., Bekiranov, S., Ng, H.H., Kapranov, P., Sekinger, E.A., Kampa, D., Piccolboni, A., Sementchenko, V., Cheng, J., Williams, A.J., et al. (2004). Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116, 499-509.
120

REFERENCES
Ceskova, P., Chichger, H., Wallace, M., Vojtesek, B., and Hupp, T.R. (2006). On the mechanism of sequence-specific DNA-dependent acetylation of p53: the acetylation motif is exposed upon DNA binding. Journal of molecular biology 357, 442-456.
Chan, A.T., Teo, P.M., and Johnson, P.J. (2002). Nasopharyngeal carcinoma. Ann Oncol 13, 1007-1015.
Chan, H.M., Narita, M., Lowe, S.W., and Livingston, D.M. (2005). The p400 E1A-associated protein is a novel component of the p53 --> p21 senescence pathway. Genes & development 19, 196-201.
Chan, T.A., Hermeking, H., Lengauer, C., Kinzler, K.W., and Vogelstein, B. (1999). 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 401, 616-620.
Chan, W.M., Siu, W.Y., Lau, A., and Poon, R.Y. (2004). How many mutant p53 molecules are needed to inactivate a tetramer? Molecular and cellular biology 24, 3536-3551.
Chehab, N.H., Malikzay, A., Stavridi, E.S., and Halazonetis, T.D. (1999). Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America 96, 13777-13782.
Chen, D., Kon, N., Li, M., Zhang, W., Qin, J., and Gu, W. (2005a). ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 121, 1071-1083.
Chen, J., Lin, J., and Levine, A.J. (1995). Regulation of transcription functions of the p53 tumor suppressor by the mdm-2 oncogene. Molecular medicine (Cambridge, Mass 1, 142-152.
Chen, J., and Sadowski, I. (2005). Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements. Proceedings of the National Academy of Sciences of the United States of America 102, 4813-4818.
Chen, J., Wu, X., Lin, J., and Levine, A.J. (1996a). mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Molecular and cellular biology 16, 2445-2452.
Chen, L., and Chen, J. (2003). MDM2-ARF complex regulates p53 sumoylation. Oncogene 22, 5348-5357.
Chen, Q.M., Liu, J., and Merrett, J.B. (2000). Apoptosis or senescence-like growth arrest: influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. The Biochemical journal 347, 543-551.
Chen, X., Ko, L.J., Jayaraman, L., and Prives, C. (1996b). p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes & development 10, 2438-2451.
Chen, Z., Trotman, L.C., Shaffer, D., Lin, H.K., Dotan, Z.A., Niki, M., Koutcher, J.A., Scher, H.I., Ludwig, T., Gerald, W., et al. (2005b). Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725-730.
Chene, P. (2001). The role of tetramerization in p53 function. Oncogene 20, 2611-2617.
Cherbonnel-Lasserre, C., and Dosanjh, M.K. (1997). Suppression of apoptosis by overexpression of Bcl-2 or Bcl-xL promotes survival and mutagenesis after oxidative damage. Biochimie 79, 613-617.
Chipuk, J.E., and Green, D.R. (2004). Cytoplasmic p53: bax and forward. Cell cycle (Georgetown, Tex 3, 429-431.
Chipuk, J.E., Kuwana, T., Bouchier-Hayes, L., Droin, N.M., Newmeyer, D.D., Schuler, M., and Green, D.R. (2004). Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science (New York, NY 303, 1010-1014.
Cho, Y., Gorina, S., Jeffrey, P.D., and Pavletich, N.P. (1994). Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science (New York, NY 265, 346-355.
Christophorou, M.A., Ringshausen, I., Finch, A.J., Swigart, L.B., and Evan, G.I. (2006). The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 443, 214-217.
121

REFERENCES
Chuikov, S., Kurash, J.K., Wilson, J.R., Xiao, B., Justin, N., Ivanov, G.S., McKinney, K., Tempst, P., Prives, C., Gamblin, S.J., et al. (2004). Regulation of p53 activity through lysine methylation. Nature 432, 353-360.
Colombo, E., Marine, J.C., Danovi, D., Falini, B., and Pelicci, P.G. (2002). Nucleophosmin regulates the stability and transcriptional activity of p53. Nature cell biology 4, 529-533.
Contente, A., Dittmer, A., Koch, M.C., Roth, J., and Dobbelstein, M. (2002). A polymorphic microsatellite that mediates induction of PIG3 by p53. Nature genetics 30, 315-320.
Cordenonsi, M., Dupont, S., Maretto, S., Insinga, A., Imbriano, C., and Piccolo, S. (2003). Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell 113, 301-314.
Cory, S., and Adams, J.M. (2002). The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2, 647-656.
Crighton, D., Wilkinson, S., O'Prey, J., Syed, N., Smith, P., Harrison, P.R., Gasco, M., Garrone, O., Crook, T., and Ryan, K.M. (2006). DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121-134.
Cummins, J.M., and Vogelstein, B. (2004). HAUSP is required for p53 destabilization. Cell cycle (Georgetown, Tex 3, 689-692.
Cuppen, E., van Ham, M., Pepers, B., Wieringa, B., and Hendriks, W. (1999). Identification and molecular characterization of BP75, a novel bromodomain-containing protein. FEBS letters 459, 291-298.
d'Adda di Fagagna, F., Reaper, P.M., Clay-Farrace, L., Fiegler, H., Carr, P., Von Zglinicki, T., Saretzki, G., Carter, N.P., and Jackson, S.P. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194-198.
D'Orazi, G., Cecchinelli, B., Bruno, T., Manni, I., Higashimoto, Y., Saito, S., Gostissa, M., Coen, S., Marchetti, A., Del Sal, G., et al. (2002). Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nature cell biology 4, 11-19.
Dai, M.S., Zeng, S.X., Jin, Y., Sun, X.X., David, L., and Lu, H. (2004). Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Molecular and cellular biology 24, 7654-7668.
Damalas, A., Kahan, S., Shtutman, M., Ben-Ze'ev, A., and Oren, M. (2001). Deregulated beta-catenin induces a p53- and ARF-dependent growth arrest and cooperates with Ras in transformation. The EMBO journal 20, 4912-4922.
Danovi, D., Meulmeester, E., Pasini, D., Migliorini, D., Capra, M., Frenk, R., de Graaf, P., Francoz, S., Gasparini, P., Gobbi, A., et al. (2004). Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Molecular and cellular biology 24, 5835-5843.
Das, S., Raj, L., Zhao, B., Kimura, Y., Bernstein, A., Aaronson, S.A., and Lee, S.W. (2007). Hzf Determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell 130, 624-637.
Dawson, R., Muller, L., Dehner, A., Klein, C., Kessler, H., and Buchner, J. (2003). The N-terminal domain of p53 is natively unfolded. Journal of molecular biology 332, 1131-1141.
de Souza-Pinto, N.C., Harris, C.C., and Bohr, V.A. (2004). p53 functions in the incorporation step in DNA base excision repair in mouse liver mitochondria. Oncogene 23, 6559-6568.
de Stanchina, E., McCurrach, M.E., Zindy, F., Shieh, S.Y., Ferbeyre, G., Samuelson, A.V., Prives, C., Roussel, M.F., Sherr, C.J., and Lowe, S.W. (1998). E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes & development 12, 2434-2442.
Demma, M.J., Wong, S., Maxwell, E., and Dasmahapatra, B. (2004). CP-31398 restores DNA-binding activity to mutant p53 in vitro but does not affect p53 homologs p63 and p73. The Journal of biological chemistry 279, 45887-45896.
Demonacos, C., Krstic-Demonacos, M., and La Thangue, N.B. (2001). A TPR motif cofactor contributes to p300 activity in the p53 response. Molecular cell 8, 71-84.
122

REFERENCES
Demonacos, C., Krstic-Demonacos, M., Smith, L., Xu, D., O'Connor, D.P., Jansson, M., and La Thangue, N.B. (2004). A new effector pathway links ATM kinase with the DNA damage response. Nature cell biology 6, 968-976.
Dhalluin, C., Carlson, J.E., Zeng, L., He, C., Aggarwal, A.K., and Zhou, M.M. (1999). Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491-496.
Di Como, C.J., Gaiddon, C., and Prives, C. (1999). p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Molecular and cellular biology 19, 1438-1449.
Di Leonardo, A., Linke, S.P., Clarkin, K., and Wahl, G.M. (1994). DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes & development 8, 2540-2551.
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C., Schurra, C., Garre, M., Nuciforo, P.G., Bensimon, A., et al. (2006). Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638-642.
Di Stefano, V., Blandino, G., Sacchi, A., Soddu, S., and D'Orazi, G. (2004a). HIPK2 neutralizes MDM2 inhibition rescuing p53 transcriptional activity and apoptotic function. Oncogene 23, 5185-5192.
Di Stefano, V., Rinaldo, C., Sacchi, A., Soddu, S., and D'Orazi, G. (2004b). Homeodomain-interacting protein kinase-2 activity and p53 phosphorylation are critical events for cisplatin-mediated apoptosis. Experimental cell research 293, 311-320.
Dimri, G.P., Itahana, K., Acosta, M., and Campisi, J. (2000). Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Molecular and cellular biology 20, 273-285.
DiTullio, R.A., Jr., Mochan, T.A., Venere, M., Bartkova, J., Sehested, M., Bartek, J., and Halazonetis, T.D. (2002). 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nature cell biology 4, 998-1002.
Donald, S.P., Sun, X.Y., Hu, C.A., Yu, J., Mei, J.M., Valle, D., and Phang, J.M. (2001). Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer research 61, 1810-1815.
Donner, A.J., Szostek, S., Hoover, J.M., and Espinosa, J.M. (2007). CDK8 is a stimulus-specific positive coregulator of p53 target genes. Molecular cell 27, 121-133.
Dornan, D., Shimizu, H., Burch, L., Smith, A.J., and Hupp, T.R. (2003a). The proline repeat domain of p53 binds directly to the transcriptional coactivator p300 and allosterically controls DNA-dependent acetylation of p53. Molecular and cellular biology 23, 8846-8861.
Dornan, D., Shimizu, H., Mah, A., Dudhela, T., Eby, M., O'Rourke, K., Seshagiri, S., and Dixit, V.M. (2006). ATM engages autodegradation of the E3 ubiquitin ligase COP1 after DNA damage. Science (New York, NY 313, 1122-1126.
Dornan, D., Shimizu, H., Perkins, N.D., and Hupp, T.R. (2003b). DNA-dependent acetylation of p53 by the transcription coactivator p300. The Journal of biological chemistry 278, 13431-13441.
Dornan, D., Wertz, I., Shimizu, H., Arnott, D., Frantz, G.D., Dowd, P., O'Rourke, K., Koeppen, H., and Dixit, V.M. (2004). The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429, 86-92.
Doyon, Y., Selleck, W., Lane, W.S., Tan, S., and Cote, J. (2004). Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Molecular and cellular biology 24, 1884-1896.
Druker, B.J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G.M., Fanning, S., Zimmermann, J., and Lydon, N.B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine 2, 561-566.
Dumaz, N., and Meek, D.W. (1999). Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. The EMBO journal 18, 7002-7010.
123

REFERENCES
Dumaz, N., Milne, D.M., Jardine, L.J., and Meek, D.W. (2001). Critical roles for the serine 20, but not the serine 15, phosphorylation site and for the polyproline domain in regulating p53 turnover. The Biochemical journal 359, 459-464.
Dumaz, N., Milne, D.M., and Meek, D.W. (1999). Protein kinase CK1 is a p53-threonine 18 kinase which requires prior phosphorylation of serine 15. FEBS letters 463, 312-316.
Dumont, P., Leu, J.I., Della Pietra, A.C., 3rd, George, D.L., and Murphy, M. (2003). The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nature genetics 33, 357-365.
Dyson, M.H., Rose, S., and Mahadevan, L.C. (2001). Acetyllysine-binding and function of bromodomain-containing proteins in chromatin. Front Biosci 6, D853-865.
Eberharter, A., Ferrari, S., Langst, G., Straub, T., Imhof, A., Varga-Weisz, P., Wilm, M., and Becker, P.B. (2001). Acf1, the largest subunit of CHRAC, regulates ISWI-induced nucleosome remodelling. The EMBO journal 20, 3781-3788.
Efeyan, A., Garcia-Cao, I., Herranz, D., Velasco-Miguel, S., and Serrano, M. (2006). Tumour biology: Policing of oncogene activity by p53. Nature 443, 159.
el-Deiry, W.S., Kern, S.E., Pietenpol, J.A., Kinzler, K.W., and Vogelstein, B. (1992). Definition of a consensus binding site for p53. Nature genetics 1, 45-49.
el-Deiry, W.S., Tokino, T., Velculescu, V.E., Levy, D.B., Parsons, R., Trent, J.M., Lin, D., Mercer, W.E., Kinzler, K.W., and Vogelstein, B. (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817-825.
Elfring, L.K., Daniel, C., Papoulas, O., Deuring, R., Sarte, M., Moseley, S., Beek, S.J., Waldrip, W.R., Daubresse, G., DePace, A., et al. (1998). Genetic analysis of brahma: the Drosophila homolog of the yeast chromatin remodeling factor SWI2/SNF2. Genetics 148, 251-265.
Erdmann, K.S., Kuhlmann, J., Lessmann, V., Herrmann, L., Eulenburg, V., Muller, O., and Heumann, R. (2000). The Adenomatous Polyposis Coli-protein (APC) interacts with the protein tyrosine phosphatase PTP-BL via an alternatively spliced PDZ domain. Oncogene 19, 3894-3901.
Espinosa, J.M. (2008). Mechanisms of regulatory diversity within the p53 transcriptional network. Oncogene.
Espinosa, J.M., Verdun, R.E., and Emerson, B.M. (2003). p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Molecular cell 12, 1015-1027.
Esser, C., Scheffner, M., and Hohfeld, J. (2005). The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. The Journal of biological chemistry 280, 27443-27448.
Evans, S.C., and Lozano, G. (1997). The Li-Fraumeni syndrome: an inherited susceptibility to cancer. Molecular medicine today 3, 390-395.
Fang, J.Y., Yang, L., Zhu, H.Y., Chen, Y.X., Lu, J., Lu, R., Cheng, Z.H., and Xiao, S.D. (2004). 5-Aza-2'-deoxycitydine induces demethylation and up-regulates transcription of p16INK4A gene in human gastric cancer cell lines. Chinese medical journal 117, 99-103.
Felley-Bosco, E., Weston, A., Cawley, H.M., Bennett, W.P., and Harris, C.C. (1993). Functional studies of a germ-line polymorphism at codon 47 within the p53 gene. American journal of human genetics 53, 752-759.
Felsher, D.W., and Bishop, J.M. (1999). Reversible tumorigenesis by MYC in hematopoietic lineages. Molecular cell 4, 199-207.
Feng, L., Hollstein, M., and Xu, Y. (2006). Ser46 phosphorylation regulates p53-dependent apoptosis and replicative senescence. Cell cycle (Georgetown, Tex 5, 2812-2819.
Feng, L., Lin, T., Uranishi, H., Gu, W., and Xu, Y. (2005). Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Molecular and cellular biology 25, 5389-5395.
124

REFERENCES
Ferbeyre, G., de Stanchina, E., Lin, A.W., Querido, E., McCurrach, M.E., Hannon, G.J., and Lowe, S.W. (2002). Oncogenic ras and p53 cooperate to induce cellular senescence. Molecular and cellular biology 22, 3497-3508.
Ferri, K.F., and Kroemer, G. (2001). Organelle-specific initiation of cell death pathways. Nature cell biology 3, E255-263.
Fields, S., and Jang, S.K. (1990). Presence of a potent transcription activating sequence in the p53 protein. Science (New York, NY 249, 1046-1049.
Finch, R.A., Donoviel, D.B., Potter, D., Shi, M., Fan, A., Freed, D.D., Wang, C.Y., Zambrowicz, B.P., Ramirez-Solis, R., Sands, A.T., et al. (2002). mdmx is a negative regulator of p53 activity in vivo. Cancer research 62, 3221-3225.
Fiscella, M., Ullrich, S.J., Zambrano, N., Shields, M.T., Lin, D., Lees-Miller, S.P., Anderson, C.W., Mercer, W.E., and Appella, E. (1993). Mutation of the serine 15 phosphorylation site of human p53 reduces the ability of p53 to inhibit cell cycle progression. Oncogene 8, 1519-1528.
Fiscella, M., Zhang, H., Fan, S., Sakaguchi, K., Shen, S., Mercer, W.E., Vande Woude, G.F., O'Connor, P.M., and Appella, E. (1997). Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proceedings of the National Academy of Sciences of the United States of America 94, 6048-6053.
Flores, E.R., Sengupta, S., Miller, J.B., Newman, J.J., Bronson, R., Crowley, D., Yang, A., McKeon, F., and Jacks, T. (2005). Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer cell 7, 363-373.
Flores, E.R., Tsai, K.Y., Crowley, D., Sengupta, S., Yang, A., McKeon, F., and Jacks, T. (2002). p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416, 560-564.
Fogal, V., Hsieh, J.K., Royer, C., Zhong, S., and Lu, X. (2005a). Cell cycle-dependent nuclear retention of p53 by E2F1 requires phosphorylation of p53 at Ser315. The EMBO journal 24, 2768-2782.
Fogal, V., Kartasheva, N.N., Trigiante, G., Llanos, S., Yap, D., Vousden, K.H., and Lu, X. (2005b). ASPP1 and ASPP2 are new transcriptional targets of E2F. Cell death and differentiation 12, 369-376.
Fojta, M., Pivonkova, H., Brazdova, M., Nemcova, K., Palecek, J., and Vojtesek, B. (2004). Investigations of the supercoil-selective DNA binding of wild type p53 suggest a novel mechanism for controlling p53 function. European journal of biochemistry / FEBS 271, 3865-3876.
Freedman, D.A., Epstein, C.B., Roth, J.C., and Levine, A.J. (1997). A genetic approach to mapping the p53 binding site in the MDM2 protein. Molecular medicine (Cambridge, Mass 3, 248-259.
Fry, C.J., and Peterson, C.L. (2001). Chromatin remodeling enzymes: who's on first? Curr Biol 11, R185-197.
Fu, L., and Benchimol, S. (1997). Participation of the human p53 3'UTR in translational repression and activation following gamma-irradiation. The EMBO journal 16, 4117-4125.
Fu, L., Minden, M.D., and Benchimol, S. (1996). Translational regulation of human p53 gene expression. The EMBO journal 15, 4392-4401.
Fu, X.Y., Wang, H.Y., Tan, L., Liu, S.Q., Cao, H.F., and Wu, M.C. (2002). Overexpression of p28/gankyrin in human hepatocellular carcinoma and its clinical significance. World J Gastroenterol 8, 638-643.
Fuchs, B., O'Connor, D., Fallis, L., Scheidtmann, K.H., and Lu, X. (1995). p53 phosphorylation mutants retain transcription activity. Oncogene 10, 789-793.
Fuchs, S.Y., Adler, V., Buschmann, T., Yin, Z., Wu, X., Jones, S.N., and Ronai, Z. (1998a). JNK targets p53 ubiquitination and degradation in nonstressed cells. Genes & development 12, 2658-2663.
Fuchs, S.Y., Adler, V., Pincus, M.R., and Ronai, Z. (1998b). MEKK1/JNK signaling stabilizes and activates p53. Proceedings of the National Academy of Sciences of the United States of America 95, 10541-10546.
Fujita, M., Norris, D.A., Yagi, H., Walsh, P., Morelli, J.G., Weston, W.L., Terada, N., Bennion, S.D., Robinson, W., Lemon, M., et al. (1999). Overexpression of mutant ras in human melanoma increases invasiveness, proliferation and
125

REFERENCES
anchorage-independent growth in vitro and induces tumour formation and cachexia in vivo. Melanoma research 9, 279-291.
Gaiddon, C., Lokshin, M., Ahn, J., Zhang, T., and Prives, C. (2001). A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Molecular and cellular biology 21, 1874-1887.
Gatti, A., Li, H.H., Traugh, J.A., and Liu, X. (2000). Phosphorylation of human p53 on Thr-55. Biochemistry 39, 9837-9842.
Geng, Y., Yu, Q., Sicinska, E., Das, M., Schneider, J.E., Bhattacharya, S., Rideout, W.M., Bronson, R.T., Gardner, H., and Sicinski, P. (2003). Cyclin E ablation in the mouse. Cell 114, 431-443.
Geyer, R.K., Yu, Z.K., and Maki, C.G. (2000). The MDM2 RING-finger domain is required to promote p53 nuclear export. Nature cell biology 2, 569-573.
Ghosh, A., Stewart, D., and Matlashewski, G. (2004). Regulation of human p53 activity and cell localization by alternative splicing. Molecular and cellular biology 24, 7987-7997.
Gilkes, D.M., Chen, L., and Chen, J. (2006). MDMX regulation of p53 response to ribosomal stress. The EMBO journal 25, 5614-5625.
Girdwood, D., Bumpass, D., Vaughan, O.A., Thain, A., Anderson, L.A., Snowden, A.W., Garcia-Wilson, E., Perkins, N.D., and Hay, R.T. (2003). P300 transcriptional repression is mediated by SUMO modification. Molecular cell 11, 1043-1054.
Gorgoulis, V.G., Vassiliou, L.V., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou, T., Venere, M., Ditullio, R.A., Jr., Kastrinakis, N.G., Levy, B., et al. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907-913.
Gostissa, M., Hengstermann, A., Fogal, V., Sandy, P., Schwarz, S.E., Scheffner, M., and Del Sal, G. (1999). Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. The EMBO journal 18, 6462-6471.
Gostissa, M., Hofmann, T.G., Will, H., and Del Sal, G. (2003). Regulation of p53 functions: let's meet at the nuclear bodies. Current opinion in cell biology 15, 351-357.
Gottifredi, V., Shieh, S., Taya, Y., and Prives, C. (2001). p53 accumulates but is functionally impaired when DNA synthesis is blocked. Proceedings of the National Academy of Sciences of the United States of America 98, 1036-1041.
Gottlieb, T.M., Leal, J.F., Seger, R., Taya, Y., and Oren, M. (2002). Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene 21, 1299-1303.
Gregory, G.D., Vakoc, C.R., Rozovskaia, T., Zheng, X., Patel, S., Nakamura, T., Canaani, E., and Blobel, G.A. (2007). Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Molecular and cellular biology 27, 8466-8479.
Grossman, H.B. (2001). Biomarkers for transitional cell carcinoma-pro. Urology 57, 847-848.
Grossman, S.R., Deato, M.E., Brignone, C., Chan, H.M., Kung, A.L., Tagami, H., Nakatani, Y., and Livingston, D.M. (2003). Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science (New York, NY 300, 342-344.
Grozinger, C.M., and Schreiber, S.L. (2002). Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chemistry & biology 9, 3-16.
Grunwald, V., and Hidalgo, M. (2002). The epidermal growth factor receptor: a new target for anticancer therapy. Current problems in cancer 26, 109-164.
Gu, J., Nie, L., Wiederschain, D., and Yuan, Z.M. (2001). Identification of p53 sequence elements that are required for MDM2-mediated nuclear export. Molecular and cellular biology 21, 8533-8546.
Gu, W., and Roeder, R.G. (1997). Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595-606.
126

REFERENCES
Gu, W., Shi, X.L., and Roeder, R.G. (1997). Synergistic activation of transcription by CBP and p53. Nature 387, 819-823.
Guo, A., Salomoni, P., Luo, J., Shih, A., Zhong, S., Gu, W., and Pandolfi, P.P. (2000). The function of PML in p53-dependent apoptosis. Nature cell biology 2, 730-736.
Haldar, S., Negrini, M., Monne, M., Sabbioni, S., and Croce, C.M. (1994). Down-regulation of bcl-2 by p53 in breast cancer cells. Cancer research 54, 2095-2097.
Hammond, E.M., Denko, N.C., Dorie, M.J., Abraham, R.T., and Giaccia, A.J. (2002). Hypoxia links ATR and p53 through replication arrest. Molecular and cellular biology 22, 1834-1843.
Hammond, E.M., and Giaccia, A.J. (2005). The role of p53 in hypoxia-induced apoptosis. Biochemical and biophysical research communications 331, 718-725.
Han, J., Flemington, C., Houghton, A.B., Gu, Z., Zambetti, G.P., Lutz, R.J., Zhu, L., and Chittenden, T. (2001). Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proceedings of the National Academy of Sciences of the United States of America 98, 11318-11323.
Hanawalt, P.C. (2001). Controlling the efficiency of excision repair. Mutation research 485, 3-13.
Harms, K.L., and Chen, X. (2006). The functional domains in p53 family proteins exhibit both common and distinct properties. Cell death and differentiation 13, 890-897.
Harper, J.W., Adami, G.R., Wei, N., Keyomarsi, K., and Elledge, S.J. (1993). The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805-816.
Haupt, Y., Barak, Y., and Oren, M. (1996). Cell type-specific inhibition of p53-mediated apoptosis by mdm2. The EMBO journal 15, 1596-1606.
Haupt, Y., Maya, R., Kazaz, A., and Oren, M. (1997). Mdm2 promotes the rapid degradation of p53. Nature 387, 296-299.
Haynes, S.R., Dollard, C., Winston, F., Beck, S., Trowsdale, J., and Dawid, I.B. (1992). The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic acids research 20, 2603.
Hemann, M.T., and Narita, M. (2007). Oncogenes and senescence: breaking down in the fast lane. Genes & development 21, 1-5.
Herbig, U., Jobling, W.A., Chen, B.P., Chen, D.J., and Sedivy, J.M. (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Molecular cell 14, 501-513.
Hermeking, H., Lengauer, C., Polyak, K., He, T.C., Zhang, L., Thiagalingam, S., Kinzler, K.W., and Vogelstein, B. (1997). 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Molecular cell 1, 3-11.
Hershko, T., Chaussepied, M., Oren, M., and Ginsberg, D. (2005). Novel link between E2F and p53: proapoptotic cofactors of p53 are transcriptionally upregulated by E2F. Cell death and differentiation 12, 377-383.
Heyne, K., Mannebach, S., Wuertz, E., Knaup, K.X., Mahyar-Roemer, M., and Roemer, K. (2004). Identification of a putative p53 binding sequence within the human mitochondrial genome. FEBS letters 578, 198-202.
Higashitsuji, H., Higashitsuji, H., Itoh, K., Sakurai, T., Nagao, T., Sumitomo, Y., Masuda, T., Dawson, S., Shimada, Y., Mayer, R.J., et al. (2005). The oncoprotein gankyrin binds to MDM2/HDM2, enhancing ubiquitylation and degradation of p53. Cancer cell 8, 75-87.
Hirao, A., Kong, Y.Y., Matsuoka, S., Wakeham, A., Ruland, J., Yoshida, H., Liu, D., Elledge, S.J., and Mak, T.W. (2000). DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science (New York, NY 287, 1824-1827.
Hoffman, W.H., Biade, S., Zilfou, J.T., Chen, J., and Murphy, M. (2002). Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. The Journal of biological chemistry 277, 3247-3257.
127

REFERENCES
Hofmann, T.G., Moller, A., Sirma, H., Zentgraf, H., Taya, Y., Droge, W., Will, H., and Schmitz, M.L. (2002). Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nature cell biology 4, 1-10.
Hofseth, L.J., Saito, S., Hussain, S.P., Espey, M.G., Miranda, K.M., Araki, Y., Jhappan, C., Higashimoto, Y., He, P., Linke, S.P., et al. (2003). Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proceedings of the National Academy of Sciences of the United States of America 100, 143-148.
Honda, R., and Yasuda, H. (1999). Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. The EMBO journal 18, 22-27.
Horn, P.J., and Peterson, C.L. (2002). Molecular biology. Chromatin higher order folding--wrapping up transcription. Science (New York, NY 297, 1824-1827.
Hsieh, J.K., Yap, D., O'Connor, D.J., Fogal, V., Fallis, L., Chan, F., Zhong, S., and Lu, X. (2002). Novel function of the cyclin A binding site of E2F in regulating p53-induced apoptosis in response to DNA damage. Molecular and cellular biology 22, 78-93.
Huang, C., Ma, W.Y., Maxiner, A., Sun, Y., and Dong, Z. (1999). p38 kinase mediates UV-induced phosphorylation of p53 protein at serine 389. The Journal of biological chemistry 274, 12229-12235.
Huang, J., Perez-Burgos, L., Placek, B.J., Sengupta, R., Richter, M., Dorsey, J.A., Kubicek, S., Opravil, S., Jenuwein, T., and Berger, S.L. (2006). Repression of p53 activity by Smyd2-mediated methylation. Nature 444, 629-632.
Huang, J., Sengupta, R., Espejo, A.B., Lee, M.G., Dorsey, J.A., Richter, M., Opravil, S., Shiekhattar, R., Bedford, M.T., Jenuwein, T., et al. (2007). p53 is regulated by the lysine demethylase LSD1. Nature 449, 105-108.
Hudson, B.P., Martinez-Yamout, M.A., Dyson, H.J., and Wright, P.E. (2000). Solution structure and acetyl-lysine binding activity of the GCN5 bromodomain. Journal of molecular biology 304, 355-370.
Huettner, C.S., Zhang, P., Van Etten, R.A., and Tenen, D.G. (2000). Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nature genetics 24, 57-60.
Hwang, B.J., Ford, J.M., Hanawalt, P.C., and Chu, G. (1999). Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proceedings of the National Academy of Sciences of the United States of America 96, 424-428.
Hwang, P.M., Bunz, F., Yu, J., Rago, C., Chan, T.A., Murphy, M.P., Kelso, G.F., Smith, R.A., Kinzler, K.W., and Vogelstein, B. (2001). Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nature medicine 7, 1111-1117.
Ichas, F., Jouaville, L.S., and Mazat, J.P. (1997). Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89, 1145-1153.
Ichas, F., and Mazat, J.P. (1998). From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochimica et biophysica acta 1366, 33-50.
Ihrie, R.A., Reczek, E., Horner, J.S., Khachatrian, L., Sage, J., Jacks, T., and Attardi, L.D. (2003). Perp is a mediator of p53-dependent apoptosis in diverse cell types. Curr Biol 13, 1985-1990.
Inga, A., Storici, F., Darden, T.A., and Resnick, M.A. (2002). Differential transactivation by the p53 transcription factor is highly dependent on p53 level and promoter target sequence. Molecular and cellular biology 22, 8612-8625.
Insinga, A., Monestiroli, S., Ronzoni, S., Carbone, R., Pearson, M., Pruneri, G., Viale, G., Appella, E., Pelicci, P., and Minucci, S. (2004). Impairment of p53 acetylation, stability and function by an oncogenic transcription factor. The EMBO journal 23, 1144-1154.
Irwin, M.S., and Kaelin, W.G., Jr. (2001). Role of the newer p53 family proteins in malignancy. Apoptosis 6, 17-29.
Ishov, A.M., Sotnikov, A.G., Negorev, D., Vladimirova, O.V., Neff, N., Kamitani, T., Yeh, E.T., Strauss, J.F., 3rd, and Maul, G.G. (1999). PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. The Journal of cell biology 147, 221-234.
128

REFERENCES
Itahana, K., Campisi, J., and Dimri, G.P. (2004). Mechanisms of cellular senescence in human and mouse cells. Biogerontology 5, 1-10.
Ito, A., Kawaguchi, Y., Lai, C.H., Kovacs, J.J., Higashimoto, Y., Appella, E., and Yao, T.P. (2002). MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. The EMBO journal 21, 6236-6245.
Ito, A., Lai, C.H., Zhao, X., Saito, S., Hamilton, M.H., Appella, E., and Yao, T.P. (2001). p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. The EMBO journal 20, 1331-1340.
Iwabuchi, K., Basu, B.P., Kysela, B., Kurihara, T., Shibata, M., Guan, D., Cao, Y., Hamada, T., Imamura, K., Jeggo, P.A., et al. (2003). Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. The Journal of biological chemistry 278, 36487-36495.
Iwabuchi, K., Li, B., Massa, H.F., Trask, B.J., Date, T., and Fields, S. (1998). Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. The Journal of biological chemistry 273, 26061-26068.
Jacks, T., Remington, L., Williams, B.O., Schmitt, E.M., Halachmi, S., Bronson, R.T., and Weinberg, R.A. (1994). Tumor spectrum analysis in p53-mutant mice. Curr Biol 4, 1-7.
Jackson, E.L., Willis, N., Mercer, K., Bronson, R.T., Crowley, D., Montoya, R., Jacks, T., and Tuveson, D.A. (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes & development 15, 3243-3248.
Jackson, M.W., and Berberich, S.J. (2000). MdmX protects p53 from Mdm2-mediated degradation. Molecular and cellular biology 20, 1001-1007.
Jacobson, R.H., Ladurner, A.G., King, D.S., and Tjian, R. (2000). Structure and function of a human TAFII250 double bromodomain module. Science (New York, NY 288, 1422-1425.
Janz, C., and Wiesmuller, L. (2002). Wild-type p53 inhibits replication-associated homologous recombination. Oncogene 21, 5929-5933.
Jayaraman, J., and Prives, C. (1995). Activation of p53 sequence-specific DNA binding by short single strands of DNA requires the p53 C-terminus. Cell 81, 1021-1029.
Jett, S.D., Cherny, D.I., Subramaniam, V., and Jovin, T.M. (2000). Scanning force microscopy of the complexes of p53 core domain with supercoiled DNA. Journal of molecular biology 299, 585-592.
Jiang, D., Srinivasan, A., Lozano, G., and Robbins, P.D. (1993). SV40 T antigen abrogates p53-mediated transcriptional activity. Oncogene 8, 2805-2812.
Jiang, P., Du, W., Heese, K., and Wu, M. (2006). The Bad guy cooperates with good cop p53: Bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Molecular and cellular biology 26, 9071-9082.
Jimenez, G.S., Khan, S.H., Stommel, J.M., and Wahl, G.M. (1999). p53 regulation by post-translational modification and nuclear retention in response to diverse stresses. Oncogene 18, 7656-7665.
Jin, S., Tong, T., Fan, W., Fan, F., Antinore, M.J., Zhu, X., Mazzacurati, L., Li, X., Petrik, K.L., Rajasekaran, B., et al. (2002a). GADD45-induced cell cycle G2-M arrest associates with altered subcellular distribution of cyclin B1 and is independent of p38 kinase activity. Oncogene 21, 8696-8704.
Jin, Y., Zeng, S.X., Dai, M.S., Yang, X.J., and Lu, H. (2002b). MDM2 inhibits PCAF (p300/CREB-binding protein-associated factor)-mediated p53 acetylation. The Journal of biological chemistry 277, 30838-30843.
Johnson, R.A., Ince, T.A., and Scotto, K.W. (2001). Transcriptional repression by p53 through direct binding to a novel DNA element. The Journal of biological chemistry 276, 27716-27720.
Johnson, T.M., Yu, Z.X., Ferrans, V.J., Lowenstein, R.A., and Finkel, T. (1996). Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proceedings of the National Academy of Sciences of the United States of America 93, 11848-11852.
129

REFERENCES
Jones, S.N., Roe, A.E., Donehower, L.A., and Bradley, A. (1995). Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206-208.
Jouaville, L.S., Ichas, F., and Mazat, J.P. (1998). Modulation of cell calcium signals by mitochondria. Molecular and cellular biochemistry 184, 371-376.
Jung, E.J., Liu, G., Zhou, W., and Chen, X. (2006). Myosin VI is a mediator of the p53-dependent cell survival pathway. Molecular and cellular biology 26, 2175-2186.
Kaeser, M.D., and Iggo, R.D. (2002). Chromatin immunoprecipitation analysis fails to support the latency model for regulation of p53 DNA binding activity in vivo. Proceedings of the National Academy of Sciences of the United States of America 99, 95-100.
Kaeser, M.D., and Iggo, R.D. (2004). Promoter-specific p53-dependent histone acetylation following DNA damage. Oncogene 23, 4007-4013.
Kaghad, M., Bonnet, H., Yang, A., Creancier, L., Biscan, J.C., Valent, A., Minty, A., Chalon, P., Lelias, J.M., Dumont, X., et al. (1997). Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90, 809-819.
Kamijo, T., Bodner, S., van de Kamp, E., Randle, D.H., and Sherr, C.J. (1999). Tumor spectrum in ARF-deficient mice. Cancer research 59, 2217-2222.
Kannan, K., Kaminski, N., Rechavi, G., Jakob-Hirsch, J., Amariglio, N., and Givol, D. (2001). DNA microarray analysis of genes involved in p53 mediated apoptosis: activation of Apaf-1. Oncogene 20, 3449-3455.
Kastan, M.B., Zhan, Q., el-Deiry, W.S., Carrier, F., Jacks, T., Walsh, W.V., Plunkett, B.S., Vogelstein, B., and Fornace, A.J., Jr. (1992). A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71, 587-597.
Katayama, H., Sasai, K., Kawai, H., Yuan, Z.M., Bondaruk, J., Suzuki, F., Fujii, S., Arlinghaus, R.B., Czerniak, B.A., and Sen, S. (2004). Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nature genetics 36, 55-62.
Kaufman, R.J. (1999). Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes & development 13, 1211-1233.
Kawai, H., Wiederschain, D., Kitao, H., Stuart, J., Tsai, K.K., and Yuan, Z.M. (2003). DNA damage-induced MDMX degradation is mediated by MDM2. The Journal of biological chemistry 278, 45946-45953.
Kay, B.K., Williamson, M.P., and Sudol, M. (2000). The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. Faseb J 14, 231-241.
Keller, D.M., Zeng, X., Wang, Y., Zhang, Q.H., Kapoor, M., Shu, H., Goodman, R., Lozano, G., Zhao, Y., and Lu, H. (2001). A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Molecular cell 7, 283-292.
Khan, S.H., Moritsugu, J., and Wahl, G.M. (2000). Differential requirement for p19ARF in the p53-dependent arrest induced by DNA damage, microtubule disruption, and ribonucleotide depletion. Proceedings of the National Academy of Sciences of the United States of America 97, 3266-3271.
Kim, C.A., and Bowie, J.U. (2003). SAM domains: uniform structure, diversity of function. Trends in biochemical sciences 28, 625-628.
Kim, S., Lee, J., Park, J., and Chung, J. (2003). BP75, bromodomain-containing M(r) 75,000 protein, binds dishevelled-1 and enhances Wnt signaling by inactivating glycogen synthase kinase-3 beta. Cancer research 63, 4792-4795.
Kim, W.Y., and Sharpless, N.E. (2006). The regulation of INK4/ARF in cancer and aging. Cell 127, 265-275.
Kim, Y.H., Choi, C.Y., and Kim, Y. (1999). Covalent modification of the homeodomain-interacting protein kinase 2 (HIPK2) by the ubiquitin-like protein SUMO-1. Proceedings of the National Academy of Sciences of the United States of America 96, 12350-12355.
130

REFERENCES
Kishi, H., Nakagawa, K., Matsumoto, M., Suga, M., Ando, M., Taya, Y., and Yamaizumi, M. (2001). Osmotic shock induces G1 arrest through p53 phosphorylation at Ser33 by activated p38MAPK without phosphorylation at Ser15 and Ser20. The Journal of biological chemistry 276, 39115-39122.
Knights, C.D., Catania, J., Di Giovanni, S., Muratoglu, S., Perez, R., Swartzbeck, A., Quong, A.A., Zhang, X., Beerman, T., Pestell, R.G., et al. (2006). Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. The Journal of cell biology 173, 533-544.
Kobet, E., Zeng, X., Zhu, Y., Keller, D., and Lu, H. (2000). MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proceedings of the National Academy of Sciences of the United States of America 97, 12547-12552.
Kraus, W.L., Manning, E.T., and Kadonaga, J.T. (1999). Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Molecular and cellular biology 19, 8123-8135.
Krieg, A.J., Hammond, E.M., and Giaccia, A.J. (2006). Functional analysis of p53 binding under differential stresses. Molecular and cellular biology 26, 7030-7045.
Krummel, K.A., Lee, C.J., Toledo, F., and Wahl, G.M. (2005). The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proceedings of the National Academy of Sciences of the United States of America 102, 10188-10193.
Kubbutat, M.H., Jones, S.N., and Vousden, K.H. (1997). Regulation of p53 stability by Mdm2. Nature 387, 299-303.
Kuerbitz, S.J., Plunkett, B.S., Walsh, W.V., and Kastan, M.B. (1992). Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proceedings of the National Academy of Sciences of the United States of America 89, 7491-7495.
Lagger, G., Doetzlhofer, A., Schuettengruber, B., Haidweger, E., Simboeck, E., Tischler, J., Chiocca, S., Suske, G., Rotheneder, H., Wintersberger, E., et al. (2003). The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Molecular and cellular biology 23, 2669-2679.
Lahav, G., Rosenfeld, N., Sigal, A., Geva-Zatorsky, N., Levine, A.J., Elowitz, M.B., and Alon, U. (2004). Dynamics of the p53-Mdm2 feedback loop in individual cells. Nature genetics 36, 147-150.
Laine, A., and Ronai, Z. (2007). Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 26, 1477-1483.
Laine, A., Topisirovic, I., Zhai, D., Reed, J.C., Borden, K.L., and Ronai, Z. (2006). Regulation of p53 localization and activity by Ubc13. Molecular and cellular biology 26, 8901-8913.
Lambert, P.F., Kashanchi, F., Radonovich, M.F., Shiekhattar, R., and Brady, J.N. (1998). Phosphorylation of p53 serine 15 increases interaction with CBP. The Journal of biological chemistry 273, 33048-33053.
Lane, D.P. (1992). Cancer. p53, guardian of the genome. Nature 358, 15-16.
Langley, E., Pearson, M., Faretta, M., Bauer, U.M., Frye, R.A., Minucci, S., Pelicci, P.G., and Kouzarides, T. (2002). Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. The EMBO journal 21, 2383-2396.
Lanni, J.S., and Jacks, T. (1998). Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Molecular and cellular biology 18, 1055-1064.
Le Cam, L., Linares, L.K., Paul, C., Julien, E., Lacroix, M., Hatchi, E., Triboulet, R., Bossis, G., Shmueli, A., Rodriguez, M.S., et al. (2006). E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell 127, 775-788.
Lee, D., Kim, J.W., Seo, T., Hwang, S.G., Choi, E.J., and Choe, J. (2002). SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. The Journal of biological chemistry 277, 22330-22337.
131

REFERENCES
Lee, S., Elenbaas, B., Levine, A., and Griffith, J. (1995). p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell 81, 1013-1020.
Legube, G., Linares, L.K., Lemercier, C., Scheffner, M., Khochbin, S., and Trouche, D. (2002). Tip60 is targeted to proteasome-mediated degradation by Mdm2 and accumulates after UV irradiation. The EMBO journal 21, 1704-1712.
Legube, G., Linares, L.K., Tyteca, S., Caron, C., Scheffner, M., Chevillard-Briet, M., and Trouche, D. (2004). Role of the histone acetyl transferase Tip60 in the p53 pathway. The Journal of biological chemistry 279, 44825-44833.
Leng, R.P., Lin, Y., Ma, W., Wu, H., Lemmers, B., Chung, S., Parant, J.M., Lozano, G., Hakem, R., and Benchimol, S. (2003). Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112, 779-791.
Li, A.G., Piluso, L.G., Cai, X., Gadd, B.J., Ladurner, A.G., and Liu, X. (2007a). An acetylation switch in p53 mediates holo-TFIID recruitment. Molecular cell 28, 408-421.
Li, H., and Wu, X. (2004). Histone deacetylase inhibitor, Trichostatin A, activates p21WAF1/CIP1 expression through downregulation of c-myc and release of the repression of c-myc from the promoter in human cervical cancer cells. Biochemical and biophysical research communications 324, 860-867.
Li, H.H., Cai, X., Shouse, G.P., Piluso, L.G., and Liu, X. (2007b). A specific PP2A regulatory subunit, B56gamma, mediates DNA damage-induced dephosphorylation of p53 at Thr55. The EMBO journal 26, 402-411.
Li, J., Lee, B., and Lee, A.S. (2006). Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. The Journal of biological chemistry 281, 7260-7270.
Li, M., Brooks, C.L., Kon, N., and Gu, W. (2004). A dynamic role of HAUSP in the p53-Mdm2 pathway. Molecular cell 13, 879-886.
Li, M., Brooks, C.L., Wu-Baer, F., Chen, D., Baer, R., and Gu, W. (2003). Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science (New York, NY 302, 1972-1975.
Li, M., Chen, D., Shiloh, A., Luo, J., Nikolaev, A.Y., Qin, J., and Gu, W. (2002). Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416, 648-653.
Li, P.F., Dietz, R., and von Harsdorf, R. (1999). p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. The EMBO journal 18, 6027-6036.
Li, X., Dumont, P., Della Pietra, A., Shetler, C., and Murphy, M.E. (2005). The codon 47 polymorphism in p53 is functionally significant. The Journal of biological chemistry 280, 24245-24251.
Liang, S.H., and Clarke, M.F. (1999a). A bipartite nuclear localization signal is required for p53 nuclear import regulated by a carboxyl-terminal domain. The Journal of biological chemistry 274, 32699-32703.
Liang, S.H., and Clarke, M.F. (1999b). The nuclear import of p53 is determined by the presence of a basic domain and its relative position to the nuclear localization signal. Oncogene 18, 2163-2166.
Liang, S.H., and Clarke, M.F. (2001). Regulation of p53 localization. European journal of biochemistry / FEBS 268, 2779-2783.
Lill, N.L., Grossman, S.R., Ginsberg, D., DeCaprio, J., and Livingston, D.M. (1997). Binding and modulation of p53 by p300/CBP coactivators. Nature 387, 823-827.
Lin, A.W., Barradas, M., Stone, J.C., van Aelst, L., Serrano, M., and Lowe, S.W. (1998). Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes & development 12, 3008-3019.
Lin, A.W., and Lowe, S.W. (2001). Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proceedings of the National Academy of Sciences of the United States of America 98, 5025-5030.
Lin, L., Ozaki, T., Takada, Y., Kageyama, H., Nakamura, Y., Hata, A., Zhang, J.H., Simonds, W.F., Nakagawara, A., and Koseki, H. (2005). topors, a p53 and topoisomerase I-binding RING finger protein, is a coactivator of p53 in growth suppression induced by DNA damage. Oncogene 24, 3385-3396.
132

REFERENCES
Lin, Y., Ma, W., and Benchimol, S. (2000). Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nature genetics 26, 122-127.
Linares, L.K., Hengstermann, A., Ciechanover, A., Muller, S., and Scheffner, M. (2003). HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proceedings of the National Academy of Sciences of the United States of America 100, 12009-12014.
Linke, S.P., Sengupta, S., Khabie, N., Jeffries, B.A., Buchhop, S., Miska, S., Henning, W., Pedeux, R., Wang, X.W., Hofseth, L.J., et al. (2003). p53 interacts with hRAD51 and hRAD54, and directly modulates homologous recombination. Cancer research 63, 2596-2605.
Liu, G., Xia, T., and Chen, X. (2003). The activation domains, the proline-rich domain, and the C-terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB-binding protein. The Journal of biological chemistry 278, 17557-17565.
Liu, L., Scolnick, D.M., Trievel, R.C., Zhang, H.B., Marmorstein, R., Halazonetis, T.D., and Berger, S.L. (1999). p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Molecular and cellular biology 19, 1202-1209.
Liu, Q., Kaneko, S., Yang, L., Feldman, R.I., Nicosia, S.V., Chen, J., and Cheng, J.Q. (2004a). Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. The Journal of biological chemistry 279, 52175-52182.
Liu, T., Laurell, C., Selivanova, G., Lundeberg, J., Nilsson, P., and Wiman, K.G. (2007). Hypoxia induces p53-dependent transactivation and Fas/CD95-dependent apoptosis. Cell death and differentiation 14, 411-421.
Liu, X., Miller, C.W., Koeffler, P.H., and Berk, A.J. (1993). The p53 activation domain binds the TATA box-binding polypeptide in Holo-TFIID, and a neighboring p53 domain inhibits transcription. Molecular and cellular biology 13, 3291-3300.
Liu, X., Yue, P., Khuri, F.R., and Sun, S.Y. (2004b). p53 upregulates death receptor 4 expression through an intronic p53 binding site. Cancer research 64, 5078-5083.
Livak, K.J., and Schmittgen, T.D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif 25, 402-408.
Lohrum, M.A., Ludwig, R.L., Kubbutat, M.H., Hanlon, M., and Vousden, K.H. (2003). Regulation of HDM2 activity by the ribosomal protein L11. Cancer cell 3, 577-587.
Lowe, S.W. (1999). Activation of p53 by oncogenes. Endocrine-related cancer 6, 45-48.
Lowe, S.W., Schmitt, E.M., Smith, S.W., Osborne, B.A., and Jacks, T. (1993). p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362, 847-849.
Luo, J., Nikolaev, A.Y., Imai, S., Chen, D., Su, F., Shiloh, A., Guarente, L., and Gu, W. (2001). Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107, 137-148.
Luo, J., Su, F., Chen, D., Shiloh, A., and Gu, W. (2000). Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408, 377-381.
Ma, B., Pan, Y., Gunasekaran, K., Venkataraghavan, R.B., Levine, A.J., and Nussinov, R. (2005). Comparison of the protein-protein interfaces in the p53-DNA crystal structures: towards elucidation of the biological interface. Proceedings of the National Academy of Sciences of the United States of America 102, 3988-3993.
Macip, S., Igarashi, M., Berggren, P., Yu, J., Lee, S.W., and Aaronson, S.A. (2003). Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Molecular and cellular biology 23, 8576-8585.
MacLachlan, T.K., and El-Deiry, W.S. (2002). Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proceedings of the National Academy of Sciences of the United States of America 99, 9492-9497.
MacPherson, D., Kim, J., Kim, T., Rhee, B.K., Van Oostrom, C.T., DiTullio, R.A., Venere, M., Halazonetis, T.D., Bronson, R., De Vries, A., et al. (2004). Defective apoptosis and B-cell lymphomas in mice with p53 point mutation at Ser 23. The EMBO journal 23, 3689-3699.
133

REFERENCES
Mallette, F.A., and Ferbeyre, G. (2007). The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell cycle (Georgetown, Tex 6, 1831-1836.
Manning, E.T., Ikehara, T., Ito, T., Kadonaga, J.T., and Kraus, W.L. (2001). p300 forms a stable, template-committed complex with chromatin: role for the bromodomain. Molecular and cellular biology 21, 3876-3887.
Mantovani, F., Gostissa, M., Collavin, L., and Del Sal, G. (2004a). KeePin' the p53 family in good shape. Cell cycle (Georgetown, Tex 3, 905-911.
Mantovani, F., Piazza, S., Gostissa, M., Strano, S., Zacchi, P., Mantovani, R., Blandino, G., and Del Sal, G. (2004b). Pin1 links the activities of c-Abl and p300 in regulating p73 function. Molecular cell 14, 625-636.
Mantovani, F., Tocco, F., Girardini, J., Smith, P., Gasco, M., Lu, X., Crook, T., and Del Sal, G. (2007). The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nature structural & molecular biology 14, 912-920.
Marchenko, N.D., Wolff, S., Erster, S., Becker, K., and Moll, U.M. (2007). Monoubiquitylation promotes mitochondrial p53 translocation. The EMBO journal 26, 923-934.
Marchenko, N.D., Zaika, A., and Moll, U.M. (2000). Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. The Journal of biological chemistry 275, 16202-16212.
Martindale, J.L., and Holbrook, N.J. (2002). Cellular response to oxidative stress: signaling for suicide and survival. Journal of cellular physiology 192, 1-15.
Martins, C.P., Brown-Swigart, L., and Evan, G.I. (2006). Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 127, 1323-1334.
Maruyama, T., Farina, A., Dey, A., Cheong, J., Bermudez, V.P., Tamura, T., Sciortino, S., Shuman, J., Hurwitz, J., and Ozato, K. (2002). A Mammalian bromodomain protein, brd4, interacts with replication factor C and inhibits progression to S phase. Molecular and cellular biology 22, 6509-6520.
Matangkasombut, O., and Buratowski, S. (2003). Different sensitivities of bromodomain factors 1 and 2 to histone H4 acetylation. Molecular cell 11, 353-363.
Matlashewski, G.J., Tuck, S., Pim, D., Lamb, P., Schneider, J., and Crawford, L.V. (1987). Primary structure polymorphism at amino acid residue 72 of human p53. Molecular and cellular biology 7, 961-963.
Mattia, M., Gottifredi, V., McKinney, K., and Prives, C. (2007). p53-Dependent p21 mRNA elongation is impaired when DNA replication is stalled. Molecular and cellular biology 27, 1309-1320.
Mayo, L.D., Dixon, J.E., Durden, D.L., Tonks, N.K., and Donner, D.B. (2002). PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. The Journal of biological chemistry 277, 5484-5489.
Mayo, L.D., and Donner, D.B. (2002). The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends in biochemical sciences 27, 462-467.
Mayo, L.D., Seo, Y.R., Jackson, M.W., Smith, M.L., Rivera Guzman, J., Korgaonkar, C.K., and Donner, D.B. (2005). Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. The Journal of biological chemistry 280, 25953-25959.
Mazan-Mamczarz, K., Galban, S., Lopez de Silanes, I., Martindale, J.L., Atasoy, U., Keene, J.D., and Gorospe, M. (2003). RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proceedings of the National Academy of Sciences of the United States of America 100, 8354-8359.
McKinney, K., Mattia, M., Gottifredi, V., and Prives, C. (2004). p53 linear diffusion along DNA requires its C terminus. Molecular cell 16, 413-424.
McKinney, K., and Prives, C. (2002). Efficient specific DNA binding by p53 requires both its central and C-terminal domains as revealed by studies with high-mobility group 1 protein. Molecular and cellular biology 22, 6797-6808.
134

REFERENCES
Meek, D.W. (2002). p53 Induction: phosphorylation sites cooperate in regulating. Cancer biology & therapy 1, 284-286.
Michaloglou, C., Vredeveld, L.C., Soengas, M.S., Denoyelle, C., Kuilman, T., van der Horst, C.M., Majoor, D.M., Shay, J.W., Mooi, W.J., and Peeper, D.S. (2005). BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720-724.
Middeler, G., Zerf, K., Jenovai, S., Thulig, A., Tschodrich-Rotter, M., Kubitscheck, U., and Peters, R. (1997). The tumor suppressor p53 is subject to both nuclear import and export, and both are fast, energy-dependent and lectin-inhibited. Oncogene 14, 1407-1417.
Migliorini, D., Lazzerini Denchi, E., Danovi, D., Jochemsen, A., Capillo, M., Gobbi, A., Helin, K., Pelicci, P.G., and Marine, J.C. (2002). Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Molecular and cellular biology 22, 5527-5538.
Mihara, M., Erster, S., Zaika, A., Petrenko, O., Chittenden, T., Pancoska, P., and Moll, U.M. (2003). p53 has a direct apoptogenic role at the mitochondria. Molecular cell 11, 577-590.
Minsky, N., and Oren, M. (2004). The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Molecular cell 16, 631-639.
Miyashita, T., Harigai, M., Hanada, M., and Reed, J.C. (1994a). Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer research 54, 3131-3135.
Miyashita, T., Krajewski, S., Krajewska, M., Wang, H.G., Lin, H.K., Liebermann, D.A., Hoffman, B., and Reed, J.C. (1994b). Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 9, 1799-1805.
Miyashita, T., and Reed, J.C. (1995). Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80, 293-299.
Momand, J., Jung, D., Wilczynski, S., and Niland, J. (1998). The MDM2 gene amplification database. Nucleic acids research 26, 3453-3459.
Momand, J., Zambetti, G.P., Olson, D.C., George, D., and Levine, A.J. (1992). The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237-1245.
Monte, M., Simonatto, M., Peche, L.Y., Bublik, D.R., Gobessi, S., Pierotti, M.A., Rodolfo, M., and Schneider, C. (2006). MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proceedings of the National Academy of Sciences of the United States of America 103, 11160-11165.
Montes de Oca Luna, R., Wagner, D.S., and Lozano, G. (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203-206.
Moroni, M.C., Hickman, E.S., Lazzerini Denchi, E., Caprara, G., Colli, E., Cecconi, F., Muller, H., and Helin, K. (2001). Apaf-1 is a transcriptional target for E2F and p53. Nature cell biology 3, 552-558.
Morrison, J.A., Gulley, M.L., Pathmanathan, R., and Raab-Traub, N. (2004). Differential signaling pathways are activated in the Epstein-Barr virus-associated malignancies nasopharyngeal carcinoma and Hodgkin lymphoma. Cancer research 64, 5251-5260.
Mujtaba, S., He, Y., Zeng, L., Yan, S., Plotnikova, O., Sachchidanand, Sanchez, R., Zeleznik-Le, N.J., Ronai, Z., and Zhou, M.M. (2004). Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Molecular cell 13, 251-263.
Muller, H., Bracken, A.P., Vernell, R., Moroni, M.C., Christians, F., Grassilli, E., Prosperini, E., Vigo, E., Oliner, J.D., and Helin, K. (2001). E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes & development 15, 267-285.
Muller, P., Hrstka, R., Coomber, D., Lane, D.P., and Vojtesek, B. (2008). Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene.
135

REFERENCES
Murphy, M., Ahn, J., Walker, K.K., Hoffman, W.H., Evans, R.M., Levine, A.J., and George, D.L. (1999). Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes & development 13, 2490-2501.
Nagata, S., and Golstein, P. (1995). The Fas death factor. Science (New York, NY 267, 1449-1456.
Nakano, K., and Vousden, K.H. (2001). PUMA, a novel proapoptotic gene, is induced by p53. Molecular cell 7, 683-694.
Nicolson, G.L., Nawa, A., Toh, Y., Taniguchi, S., Nishimori, K., and Moustafa, A. (2003). Tumor metastasis-associated human MTA1 gene and its MTA1 protein product: role in epithelial cancer cell invasion, proliferation and nuclear regulation. Clinical & experimental metastasis 20, 19-24.
Nomoto, S., Haruki, N., Kondo, M., Konishi, H., Takahashi, T., Takahashi, T., and Takahashi, T. (1998). Search for mutations and examination of allelic expression imbalance of the p73 gene at 1p36.33 in human lung cancers. Cancer research 58, 1380-1383.
O'Connor, L., Harris, A.W., and Strasser, A. (2000). CD95 (Fas/APO-1) and p53 signal apoptosis independently in diverse cell types. Cancer research 60, 1217-1220.
Oda, K., Arakawa, H., Tanaka, T., Matsuda, K., Tanikawa, C., Mori, T., Nishimori, H., Tamai, K., Tokino, T., Nakamura, Y., et al. (2000). p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102, 849-862.
Ohnishi, K., Wang, X., Takahashi, A., and Ohnishi, T. (1998a). Contribution of protein kinase C to p53-dependent WAF1 induction pathway after heat treatment in human glioblastoma cell lines. Experimental cell research 238, 399-406.
Ohnishi, T., Wang, X., Ohnishi, K., and Takahashi, A. (1998b). p53-dependent induction of WAF1 by cold shock in human glioblastoma cells. Oncogene 16, 1507-1511.
Okorokov, A.L., Warnock, L., and Milner, J. (2002). Effect of wild-type, S15D and R175H p53 proteins on DNA end joining in vitro: potential mechanism of DNA double-strand break repair modulation. Carcinogenesis 23, 549-557.
Oliner, J.D., Pietenpol, J.A., Thiagalingam, S., Gyuris, J., Kinzler, K.W., and Vogelstein, B. (1993). Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362, 857-860.
Olsson, A., Manzl, C., Strasser, A., and Villunger, A. (2007). How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell death and differentiation 14, 1561-1575.
Owen, D.J., Ornaghi, P., Yang, J.C., Lowe, N., Evans, P.R., Ballario, P., Neuhaus, D., Filetici, P., and Travers, A.A. (2000). The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. The EMBO journal 19, 6141-6149.
Palecek, E., Brazda, V., Jagelska, E., Pecinka, P., Karlovska, L., and Brazdova, M. (2004). Enhancement of p53 sequence-specific binding by DNA supercoiling. Oncogene 23, 2119-2127.
Palmero, I., Pantoja, C., and Serrano, M. (1998). p19ARF links the tumour suppressor p53 to Ras. Nature 395, 125-126.
Pan, Y., and Chen, J. (2003). MDM2 promotes ubiquitination and degradation of MDMX. Molecular and cellular biology 23, 5113-5121.
Parant, J., Chavez-Reyes, A., Little, N.A., Yan, W., Reinke, V., Jochemsen, A.G., and Lozano, G. (2001). Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nature genetics 29, 92-95.
Park, B.J., Lee, S.J., Kim, J.I., Lee, S.J., Lee, C.H., Chang, S.G., Park, J.H., and Chi, S.G. (2000). Frequent alteration of p63 expression in human primary bladder carcinomas. Cancer research 60, 3370-3374.
Park, H.R., Kim, Y.W., Park, J.H., Maeng, Y.H., Nojima, T., Hashimoto, H., and Park, Y.K. (2004). Low expression of p63 and p73 in osteosarcoma. Tumori 90, 239-243.
136

REFERENCES
Pazin, M.J., and Kadonaga, J.T. (1997). SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein-DNA interactions? Cell 88, 737-740.
Pearson, L.L., Castle, B.E., and Kehry, M.R. (2001). CD40-mediated signaling in monocytic cells: up-regulation of tumor necrosis factor receptor-associated factor mRNAs and activation of mitogen-activated protein kinase signaling pathways. International immunology 13, 273-283.
Pearson, M., Carbone, R., Sebastiani, C., Cioce, M., Fagioli, M., Saito, S., Higashimoto, Y., Appella, E., Minucci, S., Pandolfi, P.P., et al. (2000). PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406, 207-210.
Pellegrini, G., Dellambra, E., Golisano, O., Martinelli, E., Fantozzi, I., Bondanza, S., Ponzin, D., McKeon, F., and De Luca, M. (2001). p63 identifies keratinocyte stem cells. Proceedings of the National Academy of Sciences of the United States of America 98, 3156-3161.
Peng, C., Liu, H.Y., Zhou, M., Zhang, L.M., Li, X.L., Shen, S.R., and Li, G.Y. (2007). BRD7 suppresses the growth of Nasopharyngeal Carcinoma cells (HNE1) through negatively regulating beta-catenin and ERK pathways. Molecular and cellular biochemistry 303, 141-149.
Peng, C., Zhou, J., Liu, H.Y., Zhou, M., Wang, L.L., Zhang, Q.H., Yang, Y.X., Xiong, W., Shen, S.R., Li, X.L., et al. (2006). The transcriptional regulation role of BRD7 by binding to acetylated histone through bromodomain. Journal of cellular biochemistry 97, 882-892.
Pereg, Y., Shkedy, D., de Graaf, P., Meulmeester, E., Edelson-Averbukh, M., Salek, M., Biton, S., Teunisse, A.F., Lehmann, W.D., Jochemsen, A.G., et al. (2005). Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America 102, 5056-5061.
Pluquet, O., Qu, L.K., Baltzis, D., and Koromilas, A.E. (2005). Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3beta. Molecular and cellular biology 25, 9392-9405.
Polesskaya, A., and Harel-Bellan, A. (2001). Acetylation of MyoD by p300 requires more than its histone acetyltransferase domain. The Journal of biological chemistry 276, 44502-44503.
Polyak, K., Xia, Y., Zweier, J.L., Kinzler, K.W., and Vogelstein, B. (1997). A model for p53-induced apoptosis. Nature 389, 300-305.
Prives, C., and Hall, P.A. (1999). The p53 pathway. The Journal of pathology 187, 112-126.
Puig, P., Capodieci, P., Drobnjak, M., Verbel, D., Prives, C., Cordon-Cardo, C., and Di Como, C.J. (2003). p73 Expression in human normal and tumor tissues: loss of p73alpha expression is associated with tumor progression in bladder cancer. Clin Cancer Res 9, 5642-5651.
Qu, L., Huang, S., Baltzis, D., Rivas-Estilla, A.M., Pluquet, O., Hatzoglou, M., Koumenis, C., Taya, Y., Yoshimura, A., and Koromilas, A.E. (2004). Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes & development 18, 261-277.
Qu, L., and Koromilas, A.E. (2004). Control of tumor suppressor p53 function by endoplasmic reticulum stress. Cell cycle (Georgetown, Tex 3, 567-570.
Querido, E., Blanchette, P., Yan, Q., Kamura, T., Morrison, M., Boivin, D., Kaelin, W.G., Conaway, R.C., Conaway, J.W., and Branton, P.E. (2001). Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes & development 15, 3104-3117.
Radhakrishnan, S.K., and Gartel, A.L. (2006). CDK9 phosphorylates p53 on serine residues 33, 315 and 392. Cell cycle (Georgetown, Tex 5, 519-521.
Rajendra, R., Malegaonkar, D., Pungaliya, P., Marshall, H., Rasheed, Z., Brownell, J., Liu, L.F., Lutzker, S., Saleem, A., and Rubin, E.H. (2004). Topors functions as an E3 ubiquitin ligase with specific E2 enzymes and ubiquitinates p53. The Journal of biological chemistry 279, 36440-36444.
137

REFERENCES
Rapic-Otrin, V., McLenigan, M.P., Bisi, D.C., Gonzalez, M., and Levine, A.S. (2002). Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic acids research 30, 2588-2598.
Rappold, I., Iwabuchi, K., Date, T., and Chen, J. (2001). Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. The Journal of cell biology 153, 613-620.
Rasheed, Z.A., Saleem, A., Ravee, Y., Pandolfi, P.P., and Rubin, E.H. (2002). The topoisomerase I-binding RING protein, topors, is associated with promyelocytic leukemia nuclear bodies. Experimental cell research 277, 152-160.
Rea, S., Eisenhaber, F., O'Carroll, D., Strahl, B.D., Sun, Z.W., Schmid, M., Opravil, S., Mechtler, K., Ponting, C.P., Allis, C.D., et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593-599.
Reczek, E.E., Flores, E.R., Tsay, A.S., Attardi, L.D., and Jacks, T. (2003). Multiple response elements and differential p53 binding control Perp expression during apoptosis. Mol Cancer Res 1, 1048-1057.
Regula, K.M., and Kirshenbaum, L.A. (2001). p53 activates the mitochondrial death pathway and apoptosis of ventricular myocytes independent of de novo gene transcription. Journal of molecular and cellular cardiology 33, 1435-1445.
Reimertz, C., Kogel, D., Rami, A., Chittenden, T., and Prehn, J.H. (2003). Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. The Journal of cell biology 162, 587-597.
Resnick, M.A., and Inga, A. (2003). Functional mutants of the sequence-specific transcription factor p53 and implications for master genes of diversity. Proceedings of the National Academy of Sciences of the United States of America 100, 9934-9939.
Restle, A., Janz, C., and Wiesmuller, L. (2005). Differences in the association of p53 phosphorylated on serine 15 and key enzymes of homologous recombination. Oncogene 24, 4380-4387.
Ries, S., Biederer, C., Woods, D., Shifman, O., Shirasawa, S., Sasazuki, T., McMahon, M., Oren, M., and McCormick, F. (2000). Opposing effects of Ras on p53: transcriptional activation of mdm2 and induction of p19ARF. Cell 103, 321-330.
Rinaldo, C., Prodosmo, A., Mancini, F., Iacovelli, S., Sacchi, A., Moretti, F., and Soddu, S. (2007). MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis. Molecular cell 25, 739-750.
Rippin, T.M., Bykov, V.J., Freund, S.M., Selivanova, G., Wiman, K.G., and Fersht, A.R. (2002). Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 21, 2119-2129.
Robertson, K.D., and Jones, P.A. (1998). The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Molecular and cellular biology 18, 6457-6473.
Rodriguez, M.A., Ford, R.J., Goodacre, A., Selvanayagam, P., Cabanillas, F., and Deisseroth, A.B. (1991). Chromosome 17- and p53 changes in lymphoma. British journal of haematology 79, 575-582.
Roe, J.S., Kim, H., Lee, S.M., Kim, S.T., Cho, E.J., and Youn, H.D. (2006). p53 stabilization and transactivation by a von Hippel-Lindau protein. Molecular cell 22, 395-405.
Roe, J.S., and Youn, H.D. (2006). The positive regulation of p53 by the tumor suppressor VHL. Cell cycle (Georgetown, Tex 5, 2054-2056.
Rohaly, G., Chemnitz, J., Dehde, S., Nunez, A.M., Heukeshoven, J., Deppert, W., and Dornreiter, I. (2005). A novel human p53 isoform is an essential element of the ATR-intra-S phase checkpoint. Cell 122, 21-32.
Rubbi, C.P., and Milner, J. (2003a). Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. The EMBO journal 22, 6068-6077.
138

REFERENCES
Rubbi, C.P., and Milner, J. (2003b). p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. The EMBO journal 22, 975-986.
Ryo, A., Liou, Y.C., Wulf, G., Nakamura, M., Lee, S.W., and Lu, K.P. (2002). PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Molecular and cellular biology 22, 5281-5295.
Sabbatini, P., Lin, J., Levine, A.J., and White, E. (1995). Essential role for p53-mediated transcription in E1A-induced apoptosis. Genes & development 9, 2184-2192.
Sabbatini, P., and McCormick, F. (2002). MDMX inhibits the p300/CBP-mediated acetylation of p53. DNA and cell biology 21, 519-525.
Sablina, A.A., Budanov, A.V., Ilyinskaya, G.V., Agapova, L.S., Kravchenko, J.E., and Chumakov, P.M. (2005). The antioxidant function of the p53 tumor suppressor. Nature medicine 11, 1306-1313.
Saintigny, Y., and Lopez, B.S. (2002). Homologous recombination induced by replication inhibition, is stimulated by expression of mutant p53. Oncogene 21, 488-492.
Sakaguchi, K., Herrera, J.E., Saito, S., Miki, T., Bustin, M., Vassilev, A., Anderson, C.W., and Appella, E. (1998). DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes & development 12, 2831-2841.
Sakaguchi, K., Saito, S., Higashimoto, Y., Roy, S., Anderson, C.W., and Appella, E. (2000). Damage-mediated phosphorylation of human p53 threonine 18 through a cascade mediated by a casein 1-like kinase. Effect on Mdm2 binding. The Journal of biological chemistry 275, 9278-9283.
Sakamuro, D., Sabbatini, P., White, E., and Prendergast, G.C. (1997). The polyproline region of p53 is required to activate apoptosis but not growth arrest. Oncogene 15, 887-898.
Samuels-Lev, Y., O'Connor, D.J., Bergamaschi, D., Trigiante, G., Hsieh, J.K., Zhong, S., Campargue, I., Naumovski, L., Crook, T., and Lu, X. (2001). ASPP proteins specifically stimulate the apoptotic function of p53. Molecular cell 8, 781-794.
Sano, Y., and Ishii, S. (2001). Increased affinity of c-Myb for CREB-binding protein (CBP) after CBP-induced acetylation. The Journal of biological chemistry 276, 3674-3682.
Sax, J.K., Fei, P., Murphy, M.E., Bernhard, E., Korsmeyer, S.J., and El-Deiry, W.S. (2002). BID regulation by p53 contributes to chemosensitivity. Nature cell biology 4, 842-849.
Schmitt, C.A., Fridman, J.S., Yang, M., Baranov, E., Hoffman, R.M., and Lowe, S.W. (2002). Dissecting p53 tumor suppressor functions in vivo. Cancer cell 1, 289-298.
Schultz, L.B., Chehab, N.H., Malikzay, A., and Halazonetis, T.D. (2000). p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. The Journal of cell biology 151, 1381-1390.
Schwarzacher, H.G., and Wachtler, F. (1991). The functional significance of nucleolar structures. Annales de genetique 34, 151-160.
Scolnick, D.M., Chehab, N.H., Stavridi, E.S., Lien, M.C., Caruso, L., Moran, E., Berger, S.L., and Halazonetis, T.D. (1997). CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer research 57, 3693-3696.
Sengupta, S., Linke, S.P., Pedeux, R., Yang, Q., Farnsworth, J., Garfield, S.H., Valerie, K., Shay, J.W., Ellis, N.A., Wasylyk, B., et al. (2003). BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. The EMBO journal 22, 1210-1222.
Seoane, J., Le, H.V., and Massague, J. (2002). Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 419, 729-734.
Serrano, M., Lin, A.W., McCurrach, M.E., Beach, D., and Lowe, S.W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593-602.
139

REFERENCES
Shan, B., Xu, J., Zhuo, Y., Morris, C.A., and Morris, G.F. (2003). Induction of p53-dependent activation of the human proliferating cell nuclear antigen gene in chromatin by ionizing radiation. The Journal of biological chemistry 278, 44009-44017.
Sharp, D.A., Kratowicz, S.A., Sank, M.J., and George, D.L. (1999). Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. The Journal of biological chemistry 274, 38189-38196.
Shaulsky, G., Goldfinger, N., Ben-Ze'ev, A., and Rotter, V. (1990). Nuclear accumulation of p53 protein is mediated by several nuclear localization signals and plays a role in tumorigenesis. Molecular and cellular biology 10, 6565-6577.
She, Q.B., Huang, C., Zhang, Y., and Dong, Z. (2002). Involvement of c-jun NH(2)-terminal kinases in resveratrol-induced activation of p53 and apoptosis. Molecular carcinogenesis 33, 244-250.
Shetty, S., Graham, B.A., Brown, J.G., Hu, X., Vegh-Yarema, N., Harding, G., Paul, J.T., and Gibson, S.B. (2005). Transcription factor NF-kappaB differentially regulates death receptor 5 expression involving histone deacetylase 1. Molecular and cellular biology 25, 5404-5416.
Shibue, T., Takeda, K., Oda, E., Tanaka, H., Murasawa, H., Takaoka, A., Morishita, Y., Akira, S., Taniguchi, T., and Tanaka, N. (2003). Integral role of Noxa in p53-mediated apoptotic response. Genes & development 17, 2233-2238.
Shieh, S.Y., Ikeda, M., Taya, Y., and Prives, C. (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325-334.
Shieh, S.Y., Taya, Y., and Prives, C. (1999). DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. The EMBO journal 18, 1815-1823.
Shikama, N., Lee, C.W., France, S., Delavaine, L., Lyon, J., Krstic-Demonacos, M., and La Thangue, N.B. (1999). A novel cofactor for p300 that regulates the p53 response. Molecular cell 4, 365-376.
Shimizu, H., Burch, L.R., Smith, A.J., Dornan, D., Wallace, M., Ball, K.L., and Hupp, T.R. (2002). The conformationally flexible S9-S10 linker region in the core domain of p53 contains a novel MDM2 binding site whose mutation increases ubiquitination of p53 in vivo. The Journal of biological chemistry 277, 28446-28458.
Shinbo, Y., Taira, T., Niki, T., Iguchi-Ariga, S.M., and Ariga, H. (2005). DJ-1 restores p53 transcription activity inhibited by Topors/p53BP3. International journal of oncology 26, 641-648.
Shirangi, T.R., Zaika, A., and Moll, U.M. (2002). Nuclear degradation of p53 occurs during down-regulation of the p53 response after DNA damage. Faseb J 16, 420-422.
Shvarts, A., Steegenga, W.T., Riteco, N., van Laar, T., Dekker, P., Bazuine, M., van Ham, R.C., van der Houven van Oordt, W., Hateboer, G., van der Eb, A.J., et al. (1996). MDMX: a novel p53-binding protein with some functional properties of MDM2. The EMBO journal 15, 5349-5357.
Sigal, A., and Rotter, V. (2000). Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer research 60, 6788-6793.
Sinha, A., Faller, D.V., and Denis, G.V. (2005). Bromodomain analysis of Brd2-dependent transcriptional activation of cyclin A. The Biochemical journal 387, 257-269.
Smith, C.L., and Peterson, C.L. (2005). A conserved Swi2/Snf2 ATPase motif couples ATP hydrolysis to chromatin remodeling. Molecular and cellular biology 25, 5880-5892.
Smith, M.L., Chen, I.T., Zhan, Q., Bae, I., Chen, C.Y., Gilmer, T.M., Kastan, M.B., O'Connor, P.M., and Fornace, A.J., Jr. (1994). Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science (New York, NY 266, 1376-1380.
Smith, M.L., Ford, J.M., Hollander, M.C., Bortnick, R.A., Amundson, S.A., Seo, Y.R., Deng, C.X., Hanawalt, P.C., and Fornace, A.J., Jr. (2000). p53-mediated DNA repair responses to UV radiation: studies of mouse cells lacking p53, p21, and/or gadd45 genes. Molecular and cellular biology 20, 3705-3714.
140

REFERENCES
Staal, A., Enserink, J.M., Stein, J.L., Stein, G.S., and van Wijnen, A.J. (2000). Molecular characterization of celtix-1, a bromodomain protein interacting with the transcription factor interferon regulatory factor 2. Journal of cellular physiology 185, 269-279.
Stad, R., Little, N.A., Xirodimas, D.P., Frenk, R., van der Eb, A.J., Lane, D.P., Saville, M.K., and Jochemsen, A.G. (2001). Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO reports 2, 1029-1034.
Stambolic, V., MacPherson, D., Sas, D., Lin, Y., Snow, B., Jang, Y., Benchimol, S., and Mak, T.W. (2001). Regulation of PTEN transcription by p53. Molecular cell 8, 317-325.
Steele-Perkins, G., Fang, W., Yang, X.H., Van Gele, M., Carling, T., Gu, J., Buyse, I.M., Fletcher, J.A., Liu, J., Bronson, R., et al. (2001). Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes & development 15, 2250-2262.
Stein, G.H., Drullinger, L.F., Soulard, A., and Dulic, V. (1999). Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Molecular and cellular biology 19, 2109-2117.
Sterner, D.E., and Berger, S.L. (2000). Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev 64, 435-459.
Stiewe, T., Zimmermann, S., Frilling, A., Esche, H., and Putzer, B.M. (2002). Transactivation-deficient DeltaTA-p73 acts as an oncogene. Cancer research 62, 3598-3602.
Stommel, J.M., Marchenko, N.D., Jimenez, G.S., Moll, U.M., Hope, T.J., and Wahl, G.M. (1999). A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. The EMBO journal 18, 1660-1672.
Stommel, J.M., and Wahl, G.M. (2004). Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. The EMBO journal 23, 1547-1556.
Stott, F.J., Bates, S., James, M.C., McConnell, B.B., Starborg, M., Brookes, S., Palmero, I., Ryan, K., Hara, E., Vousden, K.H., et al. (1998). The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. The EMBO journal 17, 5001-5014.
Strahl, B.D., and Allis, C.D. (2000). The language of covalent histone modifications. Nature 403, 41-45.
Strano, S., Munarriz, E., Rossi, M., Cristofanelli, B., Shaul, Y., Castagnoli, L., Levine, A.J., Sacchi, A., Cesareni, G., Oren, M., et al. (2000). Physical and functional interaction between p53 mutants and different isoforms of p73. The Journal of biological chemistry 275, 29503-29512.
Sui, G., Affar el, B., Shi, Y., Brignone, C., Wall, N.R., Yin, P., Donohoe, M., Luke, M.P., Calvo, D., Grossman, S.R., et al. (2004). Yin Yang 1 is a negative regulator of p53. Cell 117, 859-872.
Sullivan, A., Syed, N., Gasco, M., Bergamaschi, D., Trigiante, G., Attard, M., Hiller, L., Farrell, P.J., Smith, P., Lu, X., et al. (2004). Polymorphism in wild-type p53 modulates response to chemotherapy in vitro and in vivo. Oncogene 23, 3328-3337.
Sun, X.F., Johannsson, O., Hakansson, S., Sellberg, G., Nordenskjold, B., Olsson, H., and Borg, A. (1996). A novel p53 germline alteration identified in a late onset breast cancer kindred. Oncogene 13, 407-411.
Sykes, S.M., Mellert, H.S., Holbert, M.A., Li, K., Marmorstein, R., Lane, W.S., and McMahon, S.B. (2006). Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Molecular cell 24, 841-851.
Szak, S.T., Mays, D., and Pietenpol, J.A. (2001). Kinetics of p53 binding to promoter sites in vivo. Molecular and cellular biology 21, 3375-3386.
Taira, N., Nihira, K., Yamaguchi, T., Miki, Y., and Yoshida, K. (2007). DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Molecular cell 25, 725-738.
Takagi, M., Absalon, M.J., McLure, K.G., and Kastan, M.B. (2005). Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 123, 49-63.
141

REFERENCES
Takahashi, A., Ohtani, N., Yamakoshi, K., Iida, S., Tahara, H., Nakayama, K., Nakayama, K.I., Ide, T., Saya, H., and Hara, E. (2006). Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nature cell biology 8, 1291-1297.
Takai, H., Smogorzewska, A., and de Lange, T. (2003). DNA damage foci at dysfunctional telomeres. Curr Biol 13, 1549-1556.
Takekawa, M., Adachi, M., Nakahata, A., Nakayama, I., Itoh, F., Tsukuda, H., Taya, Y., and Imai, K. (2000). p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. The EMBO journal 19, 6517-6526.
Tan, T.H., Wallis, J., and Levine, A.J. (1986). Identification of the p53 protein domain involved in formation of the simian virus 40 large T-antigen-p53 protein complex. Journal of virology 59, 574-583.
Tanaka, T., Ohkubo, S., Tatsuno, I., and Prives, C. (2007). hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell 130, 638-650.
Tang, Y., Luo, J., Zhang, W., and Gu, W. (2006). Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Molecular cell 24, 827-839.
Tanimura, S., Ohtsuka, S., Mitsui, K., Shirouzu, K., Yoshimura, A., and Ohtsubo, M. (1999). MDM2 interacts with MDMX through their RING finger domains. FEBS letters 447, 5-9.
Thomas, M., Kalita, A., Labrecque, S., Pim, D., Banks, L., and Matlashewski, G. (1999). Two polymorphic variants of wild-type p53 differ biochemically and biologically. Molecular and cellular biology 19, 1092-1100.
Thomas, M.C., and Chiang, C.M. (2006). The general transcription machinery and general cofactors. Critical reviews in biochemistry and molecular biology 41, 105-178.
Thompson, K.A., Wang, B., Argraves, W.S., Giancotti, F.G., Schranck, D.P., and Ruoslahti, E. (1994). BR140, a novel zinc-finger protein with homology to the TAF250 subunit of TFIID. Biochemical and biophysical research communications 198, 1143-1152.
Thompson, T., Tovar, C., Yang, H., Carvajal, D., Vu, B.T., Xu, Q., Wahl, G.M., Heimbrook, D.C., and Vassilev, L.T. (2004). Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. The Journal of biological chemistry 279, 53015-53022.
Tinel, A., and Tschopp, J. (2004). The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science (New York, NY 304, 843-846.
Toledo, F., Lee, C.J., Krummel, K.A., Rodewald, L.W., Liu, C.W., and Wahl, G.M. (2007). Mouse mutants reveal that putative protein interaction sites in the p53 proline-rich domain are dispensable for tumor suppression. Molecular and cellular biology 27, 1425-1432.
Turner, B.M. (2000). Histone acetylation and an epigenetic code. Bioessays 22, 836-845.
Uhle, S., Medalia, O., Waldron, R., Dumdey, R., Henklein, P., Bech-Otschir, D., Huang, X., Berse, M., Sperling, J., Schade, R., et al. (2003). Protein kinase CK2 and protein kinase D are associated with the COP9 signalosome. The EMBO journal 22, 1302-1312.
Unger, T., Juven-Gershon, T., Moallem, E., Berger, M., Vogt Sionov, R., Lozano, G., Oren, M., and Haupt, Y. (1999). Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. The EMBO journal 18, 1805-1814.
Unger, T., Nau, M.M., Segal, S., and Minna, J.D. (1992). p53: a transdominant regulator of transcription whose function is ablated by mutations occurring in human cancer. The EMBO journal 11, 1383-1390.
Urist, M.J., Di Como, C.J., Lu, M.L., Charytonowicz, E., Verbel, D., Crum, C.P., Ince, T.A., McKeon, F.D., and Cordon-Cardo, C. (2002). Loss of p63 expression is associated with tumor progression in bladder cancer. The American journal of pathology 161, 1199-1206.
Vaziri, H., Dessain, S.K., Ng Eaton, E., Imai, S.I., Frye, R.A., Pandita, T.K., Guarente, L., and Weinberg, R.A. (2001). hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149-159.
142

REFERENCES
Vaziri, H., West, M.D., Allsopp, R.C., Davison, T.S., Wu, Y.S., Arrowsmith, C.H., Poirier, G.G., and Benchimol, S. (1997). ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. The EMBO journal 16, 6018-6033.
Venot, C., Maratrat, M., Dureuil, C., Conseiller, E., Bracco, L., and Debussche, L. (1998). The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. The EMBO journal 17, 4668-4679.
Venot, C., Maratrat, M., Sierra, V., Conseiller, E., and Debussche, L. (1999). Definition of a p53 transactivation function-deficient mutant and characterization of two independent p53 transactivation subdomains. Oncogene 18, 2405-2410.
Ventura, A., Kirsch, D.G., McLaughlin, M.E., Tuveson, D.A., Grimm, J., Lintault, L., Newman, J., Reczek, E.E., Weissleder, R., and Jacks, T. (2007). Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661-665.
Villunger, A., Michalak, E.M., Coultas, L., Mullauer, F., Bock, G., Ausserlechner, M.J., Adams, J.M., and Strasser, A. (2003). p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science (New York, NY 302, 1036-1038.
Vives, V., Slee, E.A., and Lu, X. (2006). ASPP2: a gene that controls life and death in vivo. Cell cycle (Georgetown, Tex 5, 2187-2190.
Vousden, K.H. (2002). Switching from life to death: the Miz-ing link between Myc and p53. Cancer cell 2, 351-352.
Vousden, K.H. (2006). Outcomes of p53 activation--spoilt for choice. Journal of cell science 119, 5015-5020.
Vousden, K.H., and Lu, X. (2002). Live or let die: the cell's response to p53. Nat Rev Cancer 2, 594-604.
Wahl, G.M. (2006). Mouse bites dogma: how mouse models are changing our views of how P53 is regulated in vivo. Cell death and differentiation 13, 973-983.
Walker, K.K., and Levine, A.J. (1996). Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proceedings of the National Academy of Sciences of the United States of America 93, 15335-15340.
Walter, K., Warnecke, G., Bowater, R., Deppert, W., and Kim, E. (2005). tumor suppressor p53 binds with high affinity to CTG.CAG trinucleotide repeats and induces topological alterations in mismatched duplexes. The Journal of biological chemistry 280, 42497-42507.
Wang, W., Kim, S.H., and El-Deiry, W.S. (2006). Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proceedings of the National Academy of Sciences of the United States of America 103, 11003-11008.
Waterman, M.J., Stavridi, E.S., Waterman, J.L., and Halazonetis, T.D. (1998). ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nature genetics 19, 175-178.
Webley, K., Bond, J.A., Jones, C.J., Blaydes, J.P., Craig, A., Hupp, T., and Wynford-Thomas, D. (2000). Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Molecular and cellular biology 20, 2803-2808.
Wei, C.L., Wu, Q., Vega, V.B., Chiu, K.P., Ng, P., Zhang, T., Shahab, A., Yong, H.C., Fu, Y., Weng, Z., et al. (2006). A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207-219.
Wei, X., Xu, H., and Kufe, D. (2005). Human MUC1 oncoprotein regulates p53-responsive gene transcription in the genotoxic stress response. Cancer cell 7, 167-178.
Weinstein, I.B., and Joe, A.K. (2006). Mechanisms of disease: Oncogene addiction--a rationale for molecular targeting in cancer therapy. Nature clinical practice 3, 448-457.
White, D.E., Talbott, K.E., Arva, N.C., and Bargonetti, J. (2006). Mouse double minute 2 associates with chromatin in the presence of p53 and is released to facilitate activation of transcription. Cancer research 66, 3463-3470.
143

REFERENCES
Wilkinson, D.S., Ogden, S.K., Stratton, S.A., Piechan, J.L., Nguyen, T.T., Smulian, G.A., and Barton, M.C. (2005). A direct intersection between p53 and transforming growth factor beta pathways targets chromatin modification and transcription repression of the alpha-fetoprotein gene. Molecular and cellular biology 25, 1200-1212.
Wilkinson, D.S., Tsai, W.W., Schumacher, M.A., and Barton, M.C. (2008). Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer TGF{beta}-mediated transcription repression. Molecular and cellular biology.
Winston, F., and Allis, C.D. (1999). The bromodomain: a chromatin-targeting module? Nature structural biology 6, 601-604.
Wong, K.B., DeDecker, B.S., Freund, S.M., Proctor, M.R., Bycroft, M., and Fersht, A.R. (1999). Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proceedings of the National Academy of Sciences of the United States of America 96, 8438-8442.
Woo, R.A., Jack, M.T., Xu, Y., Burma, S., Chen, D.J., and Lee, P.W. (2002). DNA damage-induced apoptosis requires the DNA-dependent protein kinase, and is mediated by the latent population of p53. The EMBO journal 21, 3000-3008.
Wu, G.S., Burns, T.F., McDonald, E.R., 3rd, Jiang, W., Meng, R., Krantz, I.D., Kao, G., Gan, D.D., Zhou, J.Y., Muschel, R., et al. (1997). KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nature genetics 17, 141-143.
Wu, W.S., Heinrichs, S., Xu, D., Garrison, S.P., Zambetti, G.P., Adams, J.M., and Look, A.T. (2005). Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell 123, 641-653.
Wu, X., and Levine, A.J. (1994). p53 and E2F-1 cooperate to mediate apoptosis. Proceedings of the National Academy of Sciences of the United States of America 91, 3602-3606.
Wulf, G., Finn, G., Suizu, F., and Lu, K.P. (2005). Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nature cell biology 7, 435-441.
Wulf, G.M., Liou, Y.C., Ryo, A., Lee, S.W., and Lu, K.P. (2002). Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. The Journal of biological chemistry 277, 47976-47979.
Xirodimas, D.P., Chisholm, J., Desterro, J.M., Lane, D.P., and Hay, R.T. (2002). P14ARF promotes accumulation of SUMO-1 conjugated (H)Mdm2. FEBS letters 528, 207-211.
Xirodimas, D.P., Saville, M.K., Bourdon, J.C., Hay, R.T., and Lane, D.P. (2004). Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 118, 83-97.
Xirodimas, D.P., Stephen, C.W., and Lane, D.P. (2001). Cocompartmentalization of p53 and Mdm2 is a major determinant for Mdm2-mediated degradation of p53. Experimental cell research 270, 66-77.
Xue, L., Zhou, B., Liu, X., Qiu, W., Jin, Z., and Yen, Y. (2003). Wild-type p53 regulates human ribonucleotide reductase by protein-protein interaction with p53R2 as well as hRRM2 subunits. Cancer research 63, 980-986.
Xue, W., Zender, L., Miething, C., Dickins, R.A., Hernando, E., Krizhanovsky, V., Cordon-Cardo, C., and Lowe, S.W. (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656-660.
Yang, A., Kaghad, M., Caput, D., and McKeon, F. (2002). On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet 18, 90-95.
Yang, A., Kaghad, M., Wang, Y., Gillett, E., Fleming, M.D., Dotsch, V., Andrews, N.C., Caput, D., and McKeon, F. (1998). p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Molecular cell 2, 305-316.
Yang, A., Walker, N., Bronson, R., Kaghad, M., Oosterwegel, M., Bonnin, J., Vagner, C., Bonnet, H., Dikkes, P., Sharpe, A., et al. (2000). p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404, 99-103.
144

REFERENCES
Yang, S., Jeong, J.H., Brown, A.L., Lee, C.H., Pandolfi, P.P., Chung, J.H., and Kim, M.K. (2006a). Promyelocytic leukemia activates Chk2 by mediating Chk2 autophosphorylation. The Journal of biological chemistry 281, 26645-26654.
Yang, W.H., Kim, J.E., Nam, H.W., Ju, J.W., Kim, H.S., Kim, Y.S., and Cho, J.W. (2006b). Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nature cell biology 8, 1074-1083.
Yoshida, K., Liu, H., and Miki, Y. (2006). Protein kinase C delta regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. The Journal of biological chemistry 281, 5734-5740.
You, H., Zheng, H., Murray, S.A., Yu, Q., Uchida, T., Fan, D., and Xiao, Z.X. (2002). IGF-1 induces Pin1 expression in promoting cell cycle S-phase entry. Journal of cellular biochemistry 84, 211-216.
Yu, J., Wang, Z., Kinzler, K.W., Vogelstein, B., and Zhang, L. (2003). PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proceedings of the National Academy of Sciences of the United States of America 100, 1931-1936.
Yu, J., Zhang, L., Hwang, P.M., Kinzler, K.W., and Vogelstein, B. (2001). PUMA induces the rapid apoptosis of colorectal cancer cells. Molecular cell 7, 673-682.
Yu, Y., Zhang, B.C., Xie, Y., Cao, L., Zhou, M., Zhan, F.H., and Li, G.Y. (2000). Analysis and Molecular Cloning of Differentially Expressing Genes in Nasopharyngeal Carcinoma. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 32, 327-332.
Zacchi, P., Gostissa, M., Uchida, T., Salvagno, C., Avolio, F., Volinia, S., Ronai, Z., Blandino, G., Schneider, C., and Del Sal, G. (2002). The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 419, 853-857.
Zeng, L., and Zhou, M.M. (2002). Bromodomain: an acetyl-lysine binding domain. FEBS letters 513, 124-128.
Zhan, Q., Antinore, M.J., Wang, X.W., Carrier, F., Smith, M.L., Harris, C.C., and Fornace, A.J., Jr. (1999). Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene 18, 2892-2900.
Zhang, L., Yu, J., Park, B.H., Kinzler, K.W., and Vogelstein, B. (2000). Role of BAX in the apoptotic response to anticancer agents. Science (New York, NY 290, 989-992.
Zhang, W., Chan, H.M., Gao, Y., Poon, R., and Wu, Z. (2007). BS69 is involved in cellular senescence through the p53-p21Cip1 pathway. EMBO reports 8, 952-958.
Zhang, Y., Wolf, G.W., Bhat, K., Jin, A., Allio, T., Burkhart, W.A., and Xiong, Y. (2003). Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Molecular and cellular biology 23, 8902-8912.
Zhao, R., Gish, K., Murphy, M., Yin, Y., Notterman, D., Hoffman, W.H., Tom, E., Mack, D.H., and Levine, A.J. (2000). Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes & development 14, 981-993.
Zheng, H., You, H., Zhou, X.Z., Murray, S.A., Uchida, T., Wulf, G., Gu, L., Tang, X., Lu, K.P., and Xiao, Z.X. (2002). The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419, 849-853.
Zheng, X., and Chen, X. (2001). Aquaporin 3, a glycerol and water transporter, is regulated by p73 of the p53 family. FEBS letters 489, 4-7.
Zheng, Z., Pan, J., Chu, B., Wong, Y.C., Cheung, A.L., and Tsao, S.W. (1999). Downregulation and abnormal expression of E-cadherin and beta-catenin in nasopharyngeal carcinoma: close association with advanced disease stage and lymph node metastasis. Human pathology 30, 458-466.
Zhong, Q., Gao, W., Du, F., and Wang, X. (2005). Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121, 1085-1095.
Zhong, S., Muller, S., Ronchetti, S., Freemont, P.S., Dejean, A., and Pandolfi, P.P. (2000a). Role of SUMO-1-modified PML in nuclear body formation. Blood 95, 2748-2752.
145

REFERENCES
Zhong, S., Salomoni, P., and Pandolfi, P.P. (2000b). The transcriptional role of PML and the nuclear body. Nature cell biology 2, E85-90.
Zhou, J., Ahn, J., Wilson, S.H., and Prives, C. (2001). A role for p53 in base excision repair. The EMBO journal 20, 914-923.
Zhou, J., Ma, J., Zhang, B.C., Li, X.L., Shen, S.R., Zhu, S.G., Xiong, W., Liu, H.Y., Huang, H., Zhou, M., et al. (2004). BRD7, a novel bromodomain gene, inhibits G1-S progression by transcriptionally regulating some important molecules involved in ras/MEK/ERK and Rb/E2F pathways. Journal of cellular physiology 200, 89-98.
Zhou, M., Gu, L., Findley, H.W., Jiang, R., and Woods, W.G. (2003a). PTEN reverses MDM2-mediated chemotherapy resistance by interacting with p53 in acute lymphoblastic leukemia cells. Cancer research 63, 6357-6362.
Zhou, M., Liu, H., Xu, X., Zhou, H., Li, X., Peng, C., Shen, S., Xiong, W., Ma, J., Zeng, Z., et al. (2006a). Identification of nuclear localization signal that governs nuclear import of BRD7 and its essential roles in inhibiting cell cycle progression. Journal of cellular biochemistry 98, 920-930.
Zhou, M., Peng, C., Nie, X.M., Zhang, B.C., Zhu, S.G., Yu, Y., Li, X.L., and Li, G.Y. (2003b). [Expression of BRD7-interacting proteins,BRD2 and BRD3, in nasopharyngeal carcinoma tissues]. Ai zheng = Aizheng = Chinese journal of cancer 22, 123-127.
Zhou, M., Xu, X.J., Zhou, H.D., Liu, H.Y., He, J.J., Li, X.L., Peng, C., Xiong, W., Fan, S.Q., Lu, J.H., et al. (2006b). BRD2 is one of BRD7-interacting proteins and its over-expression could initiate apoptosis. Molecular and cellular biochemistry 292, 205-212.
Zhu, H., Wu, L., and Maki, C.G. (2003). MDM2 and promyelocytic leukemia antagonize each other through their direct interaction with p53. The Journal of biological chemistry 278, 49286-49292.
Zhu, J., Woods, D., McMahon, M., and Bishop, J.M. (1998). Senescence of human fibroblasts induced by oncogenic Raf. Genes & development 12, 2997-3007.
Zhu, J., Zhang, S., Jiang, J., and Chen, X. (2000). Definition of the p53 functional domains necessary for inducing apoptosis. The Journal of biological chemistry 275, 39927-39934.
Zhu, J.W., DeRyckere, D., Li, F.X., Wan, Y.Y., and DeGregori, J. (1999). A role for E2F1 in the induction of ARF, p53, and apoptosis during thymic negative selection. Cell Growth Differ 10, 829-838.
Zindy, F., Eischen, C.M., Randle, D.H., Kamijo, T., Cleveland, J.L., Sherr, C.J., and Roussel, M.F. (1998). Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & development 12, 2424-2433.
Zink, D., Mayr, C., Janz, C., and Wiesmuller, L. (2002). Association of p53 and MSH2 with recombinative repair complexes during S phase. Oncogene 21, 4788-4800.
Zotchev, S.B., Protopopova, M., and Selivanova, G. (2000). p53 C-terminal interaction with DNA ends and gaps has opposing effect on specific DNA binding by the core. Nucleic acids research 28, 4005-4012.
Zou, L., and Elledge, S.J. (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science (New York, NY 300, 1542-1548.
146

REFERENCES
147

ACKNOWLEDGEMENTS
ACKNOWLEDGEMENTS I would like to acknowledge Professor Giannino Del Sal who gave me the opportunity to work on this project and sent me its great enthusiasm and passion for science. I want to acknowledge Fiamma Mantovani for her indispensable contribution to this work. I thank all the people of the “GDS group” with whom I shared beautiful moments in the lab. My gratitude goes also to all the colleagues at L.N.C.I.B. and in particular to Andrea Lunardi for the helpful discussion on science and life. Last but not least, I really want to thank Enio Klaric for its valuable aid and for patiently sustaining me in good and bad times.
148







