Download - Low temperature decomposition of PCDD/PCDF, chlorobenzenes and PAHs by TiO2-based V2O5–WO3 catalysts

Low temperature decomposition of PCDD/PCDF, chlorobenzenesand PAHs by TiO2-based V2O5±WO3 catalysts
Roland Webera,*, Takeshi Sakuraia, Hanspaul Hagenmaierb
aResearch Institute, Ishikawajima-harima Heavy Industries Co., Ltd., 1, Shin-Nakahara-cho, Isogo-ku, Yokohama 235, JapanbInstitute of Organic Chemistry, University of TuÈbingen, D-72076 TuÈbingen, Germany
Received 2 October 1998; received in revised form 20 November 1998; accepted 27 November 1998
Abstract
The oxidation of representative congeners of polychlorinated dibenzo-p-dioxins (PCDDs), polychlorinated dibenzofurans
(PCDFs), polychlorinated chlorobenzenes (PCBzs), and of polyaromatic hydrocarbons (PAHs) was investigated on two
commercial V2O5±WO3/TiO2-based catalysts, optimized for the combined reduction of nitrogen oxides and decomposition of
dioxins.
The non-chlorinated polyaromatic compounds (including non-chlorinated dibenzodioxin and dibenzofuran) are destroyed at
temperatures as low as 1508C with an ef®ciency of more than 95%. PCDD and PCDF were also removed from the gas phase
with an ef®ciency of >98%. However, at 1508C they remained mainly unchanged (up to 75%) adsorbed on the catalyst.
A decrease in the oxidation rate with increasing chlorine substitution was found for the PCDD/PCDF. This could be
explained by an increasing `̀ redox potential'' with increasing chlorine substitution due to the electron withdrawing effect of
the chlorine.
For the more volatile monoaromatic PCBz, however, the effect of lowering the volatility with increasing chlorine
substitution (resulting in longer residence time on the catalyst) over-compensates the effect of the increasing `̀ redox
potential'' with higher degree of chlorination. # 1999 Elsevier Science B.V. All rights reserved.
Keywords: Vanadia±tungsta/titania; Decomposition (catalytic-); VOCs; PCDD; PCDF; Dioxins; PAHs; Chlorobenzenes;
Volatility
1. Introduction
The stringent limiting value for PCDD/PCDF emis-
sion of 0.1 ng I-TEQ/Nm3 for municipal and hazar-
dous waste incinerators has been in effect in several
European countries (e.g. Austria, Germany [1], Neth-
erlands) since the early 1990s and in Japan since
January 1997 for new municipal waste incinerators
(MWIs) [2,3].
At present primary measures such as design and
operation of the ®ring system to minimize the forma-
tion of `̀ products of incomplete combustion'' (com-
bustion technology) or boiler technology (i.e.
in¯uencing of the de novo synthesis in the cooling
of ¯ue gas) cannot guarantee compliance with this
limiting value [4]. Therefore, secondary measures are
needed to lower the emissions of PCDD/PCDF formed
Applied Catalysis B: Environmental 20 (1999) 249±256
*Corresponding author. Tel: +81-45-7592164; fax: +81-45-
7592149; e-mail: [email protected]
0926-3373/99/$ ± see front matter # 1999 Elsevier Science B.V. All rights reserved.
P I I : S 0 9 2 6 - 3 3 7 3 ( 9 8 ) 0 0 1 1 5 - 5

to an extent that the limiting value imposed is not
exceeded. Two fundamentally different technologies
are applied in waste combustion facilities:
� Adsorption on a filtration material (fixed bed or
carbon spray in the flue gas) [5].
� Catalytic oxidation with direct destruction of the
PCDD/PCDF in the flue gas [6,7].
Both systems are considered proven technologies to
enable stack gas values below 0.1 ng I-TEQ/Nm3. The
contaminated ®ltration material needs additional treat-
ment to destroy the PCDD/PCDF, e.g. low tempera-
ture treatment under oxygen de®ciency [4,8]. The
catalyst is a true sink for the PCDD/PCDF due to
destruction of the compounds resulting in CO2, H2O
and HCl [9].
It has been shown that the TiO2-based V2O5±WO3
catalysts originally designed for the removal of
nitrogen oxides (NOx) by selective catalytic re-
duction (SCR) [10±14] are very effective in the
decomposition of PCDD/PCDF at the same tempera-
tures as are used for the DeNOx-reaction. With addi-
tion of ammonia, these catalysts can therefore be used
for the combined destruction of dioxins and NOx
[6,7,15,16].
In the last few years, the commercial SCR catalysts
have been optimized for the combined dioxin/NOx
destruction. This was achieved mainly by increasing
the oxidation potential of the catalysts by a higher
vanadium content. The impact of the vanadium con-
tent on the oxidation ef®ciency was also reported
recently for the decomposition of 1,2-dichlorobenzene
in a laboratory study [17].
In previous laboratory studies of the catalytic
destruction of chlorinated VOCs on metal oxide based
catalysts, assessment of the catalytic activity of var-
ious metal oxides was made usually on the basis of
only one or two compounds. In all these experiments
with chlorinated compounds, only highly volatile
organic compounds (VOCs) such as methylene chlor-
ide [18], dichloromethane [19], ethyl chloride [20],
dichloroethane [21], tetrachloroethene [9], tetrachlor-
omethane [18,19], chlorobenzenes [9,17,19,20,22] or
chloro¯uorocarbons [23] were used. The effective
temperature for the destruction of the chlorinated
VOCs found in these studies was above 3008C. Only
the TiO2-based V2O5±WO3 catalysts show effective
destruction of tetrachloroethene at a temperature of
2308C [15]. The only laboratory study reported
for the catalytic destruction of chlorinated semi-vola-
tile organic compounds in ¯ow experiments (hexa-
chlorobenzene, 2,4,8-trichlorodibenzofuran) focused
on the temperature region between 2608C and 5008C[9].
For the effective destruction of PCDD/PCDF, how-
ever, lower temperatures might be suf®cient. In pilot
plants, temperatures of 240±2608C have already been
tested [6,7,16] and shown to be effective. For eco-
nomical reasons, especially, if the catalyst is operated
downstream of wet scrubbers, it is desirable to operate
the catalysts at the lowest temperature which guaran-
tees the required emission limits. Therefore, pilot
plant tests have been carried out with catalyst tem-
peratures around 2008C [24]. However, laboratory
studies for the interesting compounds for waste com-
bustion, e.g. PCDD, PCDF, polychlorinated biphenyls
(PCB) or PAH which unequivocally prove the ef®-
ciency of the catalysts in these temperature regions are
missing. Such tests are expensive, time consuming and
dif®cult to carry out. Another problem is the high
toxicity of some compounds of these groups. There-
fore, the destruction behavior regarding PCDD/PCDF
have been investigated mainly in ®eld tests
[6,7,16,24]. The purpose of this investigation was to
use simpli®ed testing procedures for the laboratory
study of the catalytic destruction of chlorinated poly-
aromatic compounds on commercial TiO2-based
V2O5±WO3 catalysts, developed for the combined
destruction of dioxins and NOx, in the temperature
range 150±2508C.
2. Materials and methods
2.1. Catalysts
The catalysts used in this study are commercial
catalysts (V2O5±WO3 on TiO2 basis) especially devel-
oped for the simultaneous destruction of PCDD/PCDF
and nitrogen oxides. According to the producer the
BET surface of the two catalysts was between 70 and
90 m2/g.
The two catalysts were subjected to elemental
analysis according to the Japanese industrial standard
method (JISM) with a sequential plasma spectrometer
ICPS-7500 (Shimadzu, Kyoto, Japan). The results are
shown in Table 1. According to X-ray diffraction
250 R. Weber et al. / Applied Catalysis B: Environmental 20 (1999) 249±256

analysis by a MXP-3 (Mac Science, Yokohama,
Japan), the structure of the TiO2 phase for both
catalysts is of the anatase type (Table 1).
2.2. Chemicals
All purchased from GL Science, Japan or Cam-
bridge Isotope Laboratories, USA:
Chlorobenzenes. 1,2-Dichlorobenzene (D2CBz),
1,2,3-trichlorobenzene (T3CBz), 1,2,4-T3CBz, 1,2,3,
4-tetrachlorobenzene (T4CBz), 1,2,4,5-T4CBz, and
hexachlorobenzene (H6CBz).
Polyaromatic compounds. Pyrene, biphenyl, diben-
zofuran, and dibenzodioxin.
PCDD/PCDF. 2,8-Dichlorodibenzofuran (D2CDF),
2,7-dibenzodioxin (D2CDD), 1,3,6,8/1,3,7,9-tetra-
chlorodibenzodioxin (T4CDD), 1,2,4,6,7,9-hexachlor-
odibenzodioxin (H6CDD), 1,2,4,6,8,9-hexachloro-
dibenzofuran (H6CDF), 1,2,3,4,6,7,9-heptachlorodi-
benzofuran (H7CDF), 1,2,3,4,6,7,9-heptachlorodiben-
zodioxin (H7CDD), octachlorodibenzodioxin
(O8CDD) (synthesized by condensation of the respec-
tive chlorophenols [25±27]), 2,4,8-trichlorodibenzo-
furan (T3CDF), octachlorodibenzofuran (O8CDF).
2.3. Experiments with compounds in flow-stream
The honeycomb catalysts are carefully crushed and
sieved. The particle size of the resulting ¯akes are
about 0.6 mm�1 mm�2±5 mm. Approximately 5 g
of the respective catalyst is placed in the quartz tube
(13 mm internal diameter) of the ¯ow reactor (Fig. 1).
The reactor is enclosed in a temperature controlled
furnace which provides a reactor temperature stability
of �28C. All ancillary tubing is maintained at a
temperature of 150±2008C with heating tape. The
testing substances (1,3,6,8-T4CDD, 1,3,7,9-T4CDD,
2,4,8-T3CDF, PCBz, biphenyl, dibenzodioxin, diben-
zofuran and pyrene) are applied to silica. For the
experiments, 500 mg of silica containing 1 mg of each
compound is placed in the preheating system. After
the reactor reaches stability at the respective tempera-
ture, the substances are evaporated resulting in an
average concentration of about 1 ppm in the gas phase.
Temperature program for evaporation: starts at 708C,
holds for 5 min, 108C/min to 1608C, and holds for
15 min resulting in a total time of 30 min for the
experiment. The composition of the gas mixture is
chosen analogous to MWIs (10% O2, 70% N2, and
20% H2O). The volumetric ¯ow through the catalyst
bed of 700 cm3/min corresponds to a space velocity
(SV) of 5000 hÿ1, and is comparable to that of SCR
catalyst in MWIs. The ¯ow rate is regulated using
mass ¯ow controllers (Shinagawa Seiki, Japan).
Table 1
Composition and structural data of the catalysts
Catalyst Detected
phase (XRD)
Catalyst composition
(wt%)
TiO2 V2O5 WO3
A Anatase 82.9 6.2 5.5
B Anatase 78.2 7.1 5.9
Fig. 1. Schematic of the reactor: (1) O2/N2 source, (2) flow controller, (3) water, (4) pump, (5) preheating system, (6) evaporation port for
compounds, (7) furnace, (8) pyrex reactor, (9) catalyst bed, (10) washing bottles for sampling (empty/toluene), (11) active carbon trap, and
(12) gas meter.
R. Weber et al. / Applied Catalysis B: Environmental 20 (1999) 249±256 251

2.4. Stationary experiments for rate constant
calculation
A mixture of PCDD (P
500 ng) and PCDF (P
500 ng) is applied in toluene directly on catalyst B
(500 mg). The toluene is evaporated for 10 min at
508C. The furnace is heated to the respective tem-
perature and the catalyst is ®lled into the reactor. After
5 min, the catalyst is taken out of the reactor and
rapidly cooled to room temperature.
Measurements with a thermocouple show that the
catalyst reached the desired temperature after about
60 s. The cooling process lasts about 15 s.
2.5. Analysis and quantification
After the experiment the glass tubes after the cat-
alyst were rinsed with toluene. These rinses were
combined with the toluene in the washing bottle.
The catalysts were extracted separately by 12 h Soxh-
let extraction with toluene. This procedure ensures
complete removal of any adsorbed organic com-
pounds, as was proven in one case by a second 24 h
Soxhlet extraction with toluene and toluene/acetone
(95:5). The toluene in the washing bottle and the
extracts of the catalysts were analyzed separately.
In the ¯ow experiments the destruction removal
ef®ciency (DRE) corresponds to (input less output)/
input. The destruction ef®ciency corresponds in all
experiments to (input less (output plus unchanged
material adsorbed on catalyst))/input.
The clean-up procedures are described elsewhere
[28].
Analysis was carried out on a HP 6890 gas chro-
matograph coupled to a HP 5973 mass selective
detector. The chlorobenzenes, pyrene and the biphe-
nyls were quanti®ed by external calibration. The
quanti®cation for PCDD/PCDF and the dibenzodioxin
and dibenzofuran was carried out by an isotope dilu-
tion mass spectrometry with 13C-labeled standards.
The GC columns used were a CP-SIL 88 column
(50 m, 0.25 mm i.d., 0.2 mm ®lm thickness, CHROM-
PACK, Frankfurt/FRG) and a DB-5 fused silica col-
umn (30 m, 0.32 mm i.d., 0.25 mm ®lm thickness,
J&W Scienti®c, Folsom/USA).
3. Results
The destruction ef®ciency of the catalysts for the
decomposition of PCDD/PCDF, PAHs and chloroben-
zenes (PCBzs) were compared in ¯ow experiments
over the temperature range 150±3108C.
3.1. `̀ Polyaromatic'' compounds (PCDD, PCDF,
PAH)
From the amount of polyaromatic compounds
found downstream of the catalyst, it appeared that
both catalysts decompose these molecules in the
examined temperature range 150±3108C with an ef®-
ciency of more than 95%, the PCDD/PCDF (1,3,6,8-
T4CDD, 1,3,7,9-T4CDD, 2,4,8-T3CDF) even to an
extent of more than 98% (Fig. 2). However in the
experiments carried out at 1508C, 59±75% of the
PCDD/PCDF were found unchanged on the catalysts
(Table 2), while at 1908C less than 7% of the
unchanged PCDD/PCDF were found on the catalysts
after the experiments. Therefore, at temperatures
below about 2008C, polychlorinated aromatic com-
pounds remain adsorbed on the catalyst for several
minutes without being oxidized. At temperatures
below 2008C, the oxidizing potential of V2O5±WO3
is obviously not suf®cient to decompose polychlori-
nated aromatic compounds effectively.
For the non-chlorinated polyaromatic compounds
dibenzo-p-dioxin, dibenzofuran, biphenyl and pyrene
the destruction ef®ciency is even at 1508C higher than
90% (Fig. 2 and Table 2).
Table 2
Recovery of compounds adsorbed on the catalysts after the flow stream experiments at 1508C (space velocity: 5000 hÿ1; gas phase
composition: 10% oxygen, 70% nitrogen, and 20% water)
Compounds 1,2,4,5-T4CBz H6CBz Biphenyl Pyrene DF DD 2,4,8-T3CDF 1,3,6,8-/1,3,7,9-T4CDD
Catalyst A (% recovery) 0.3 11.3 0.5 0.7 0.8 0.3 68 59
Catalyst B (% Recovery) 0.5 12.4 0.8 1.1 0.5 0.4 74 63
252 R. Weber et al. / Applied Catalysis B: Environmental 20 (1999) 249±256
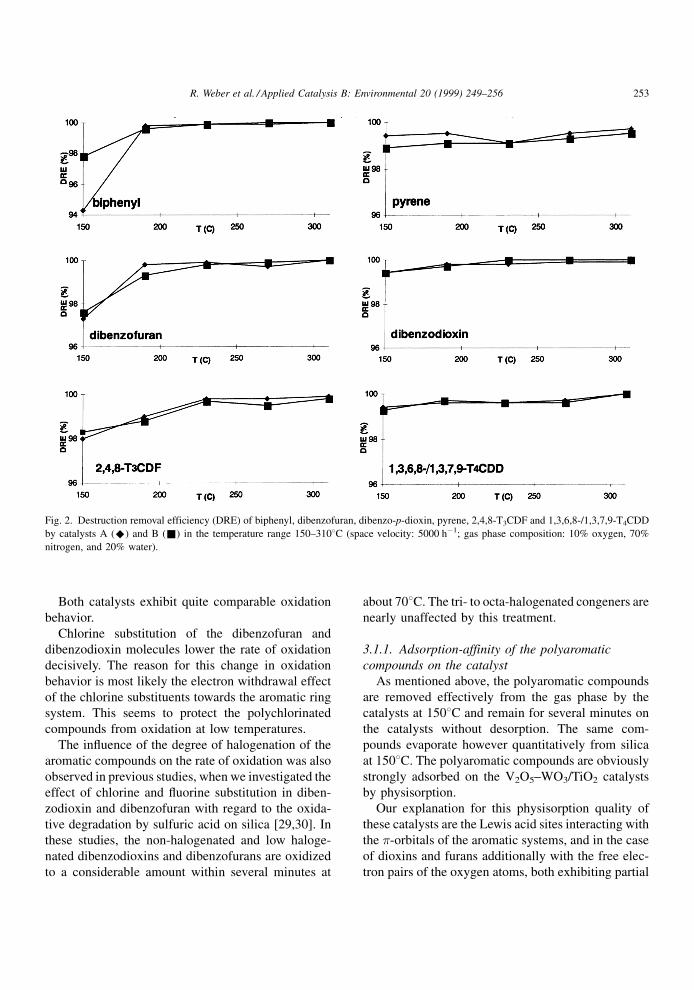
Both catalysts exhibit quite comparable oxidation
behavior.
Chlorine substitution of the dibenzofuran and
dibenzodioxin molecules lower the rate of oxidation
decisively. The reason for this change in oxidation
behavior is most likely the electron withdrawal effect
of the chlorine substituents towards the aromatic ring
system. This seems to protect the polychlorinated
compounds from oxidation at low temperatures.
The in¯uence of the degree of halogenation of the
aromatic compounds on the rate of oxidation was also
observed in previous studies, when we investigated the
effect of chlorine and ¯uorine substitution in diben-
zodioxin and dibenzofuran with regard to the oxida-
tive degradation by sulfuric acid on silica [29,30]. In
these studies, the non-halogenated and low haloge-
nated dibenzodioxins and dibenzofurans are oxidized
to a considerable amount within several minutes at
about 708C. The tri- to octa-halogenated congeners are
nearly unaffected by this treatment.
3.1.1. Adsorption-affinity of the polyaromatic
compounds on the catalyst
As mentioned above, the polyaromatic compounds
are removed effectively from the gas phase by the
catalysts at 1508C and remain for several minutes on
the catalysts without desorption. The same com-
pounds evaporate however quantitatively from silica
at 1508C. The polyaromatic compounds are obviously
strongly adsorbed on the V2O5±WO3/TiO2 catalysts
by physisorption.
Our explanation for this physisorption quality of
these catalysts are the Lewis acid sites interacting with
the �-orbitals of the aromatic systems, and in the case
of dioxins and furans additionally with the free elec-
tron pairs of the oxygen atoms, both exhibiting partial
Fig. 2. Destruction removal efficiency (DRE) of biphenyl, dibenzofuran, dibenzo-p-dioxin, pyrene, 2,4,8-T3CDF and 1,3,6,8-/1,3,7,9-T4CDD
by catalysts A (^) and B (&) in the temperature range 150±3108C (space velocity: 5000 hÿ1; gas phase composition: 10% oxygen, 70%
nitrogen, and 20% water).
R. Weber et al. / Applied Catalysis B: Environmental 20 (1999) 249±256 253
donor qualities. This is consistent with the fact that
V2O5±WO3/TiO2 catalysts show strong Lewis acid
properties [14] while the surface sites on silica are
only weakly Lewis acidic [31]. Capillary condensa-
tion phenomena seems unlikely to explain the adsorp-
tion quality hence the total amount of compounds in
one ¯ow experiment only corresponds to about 0.01±
0.1% of a monolayer on the catalyst surface. Also the
mean radius of the micropores for monolithic TiO2-
based honeycomb catalysts of 80±100 AÊ [32] seems
large compared to the size of the PCDD/PCDF mole-
cule with about 14 AÊ times 7 AÊ to explain the adsorp-
tion quality.
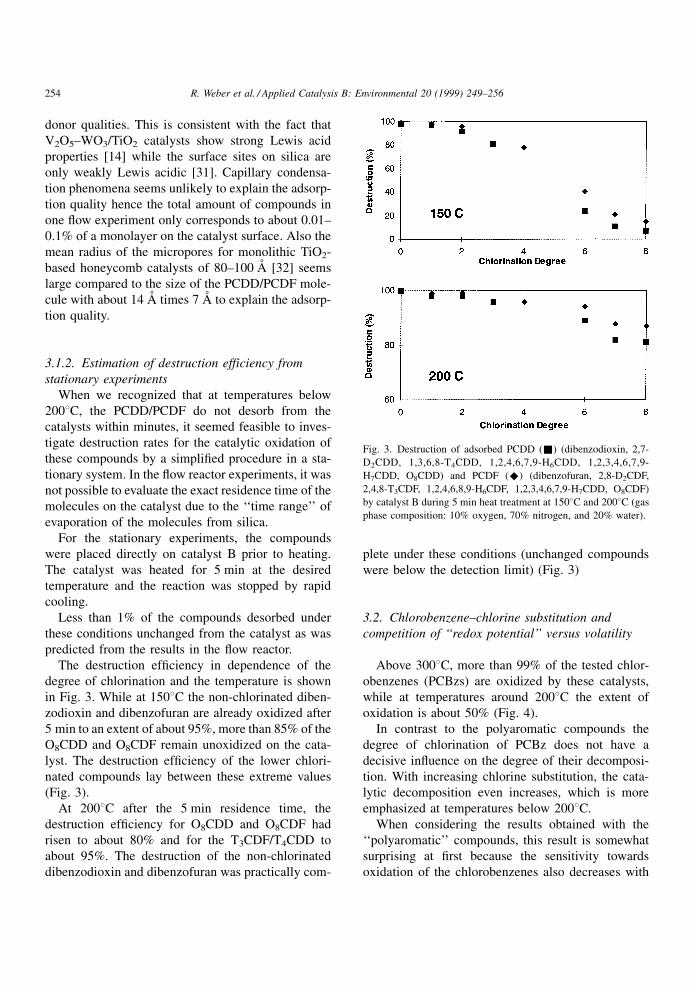
3.1.2. Estimation of destruction efficiency from
stationary experiments
When we recognized that at temperatures below
2008C, the PCDD/PCDF do not desorb from the
catalysts within minutes, it seemed feasible to inves-
tigate destruction rates for the catalytic oxidation of
these compounds by a simpli®ed procedure in a sta-
tionary system. In the ¯ow reactor experiments, it was
not possible to evaluate the exact residence time of the
molecules on the catalyst due to the `̀ time range'' of
evaporation of the molecules from silica.
For the stationary experiments, the compounds
were placed directly on catalyst B prior to heating.
The catalyst was heated for 5 min at the desired
temperature and the reaction was stopped by rapid
cooling.
Less than 1% of the compounds desorbed under
these conditions unchanged from the catalyst as was
predicted from the results in the ¯ow reactor.
The destruction ef®ciency in dependence of the
degree of chlorination and the temperature is shown
in Fig. 3. While at 1508C the non-chlorinated diben-
zodioxin and dibenzofuran are already oxidized after
5 min to an extent of about 95%, more than 85% of the
O8CDD and O8CDF remain unoxidized on the cata-
lyst. The destruction ef®ciency of the lower chlori-
nated compounds lay between these extreme values
(Fig. 3).
At 2008C after the 5 min residence time, the
destruction ef®ciency for O8CDD and O8CDF had
risen to about 80% and for the T3CDF/T4CDD to
about 95%. The destruction of the non-chlorinated
dibenzodioxin and dibenzofuran was practically com-
plete under these conditions (unchanged compounds
were below the detection limit) (Fig. 3)
3.2. Chlorobenzene±chlorine substitution and
competition of `̀ redox potential'' versus volatility
Above 3008C, more than 99% of the tested chlor-
obenzenes (PCBzs) are oxidized by these catalysts,
while at temperatures around 2008C the extent of
oxidation is about 50% (Fig. 4).
In contrast to the polyaromatic compounds the
degree of chlorination of PCBz does not have a
decisive in¯uence on the degree of their decomposi-
tion. With increasing chlorine substitution, the cata-
lytic decomposition even increases, which is more
emphasized at temperatures below 2008C.
When considering the results obtained with the
`̀ polyaromatic'' compounds, this result is somewhat
surprising at ®rst because the sensitivity towards
oxidation of the chlorobenzenes also decreases with
Fig. 3. Destruction of adsorbed PCDD (&) (dibenzodioxin, 2,7-
D2CDD, 1,3,6,8-T4CDD, 1,2,4,6,7,9-H6CDD, 1,2,3,4,6,7,9-
H7CDD, O8CDD) and PCDF (^) (dibenzofuran, 2,8-D2CDF,
2,4,8-T3CDF, 1,2,4,6,8,9-H6CDF, 1,2,3,4,6,7,9-H7CDD, O8CDF)
by catalyst B during 5 min heat treatment at 1508C and 2008C (gas
phase composition: 10% oxygen, 70% nitrogen, and 20% water).
254 R. Weber et al. / Applied Catalysis B: Environmental 20 (1999) 249±256
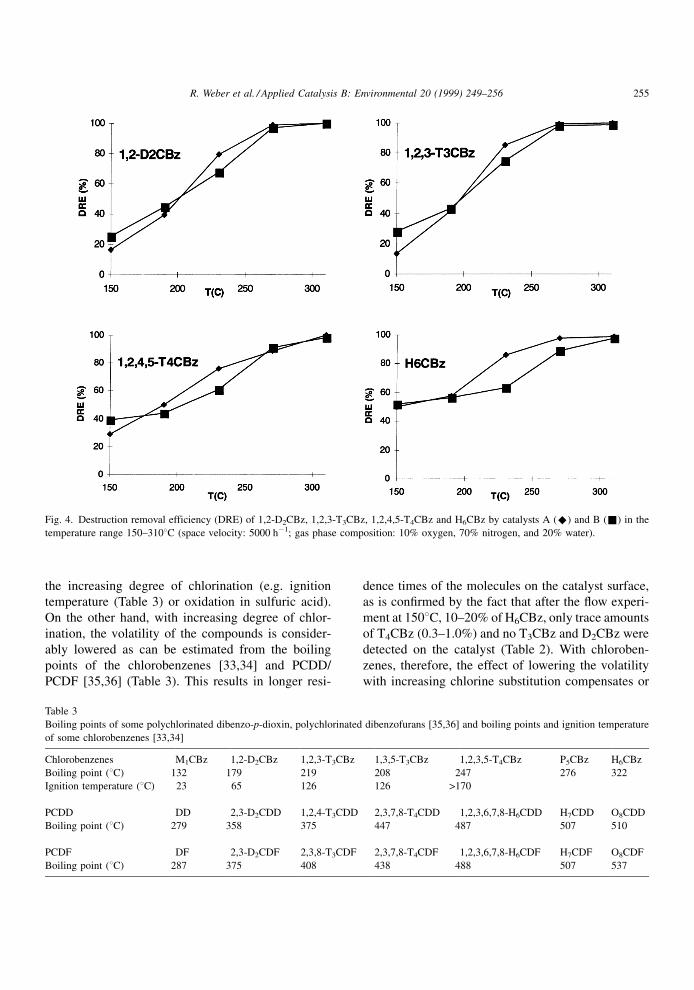
the increasing degree of chlorination (e.g. ignition
temperature (Table 3) or oxidation in sulfuric acid).
On the other hand, with increasing degree of chlor-
ination, the volatility of the compounds is consider-
ably lowered as can be estimated from the boiling
points of the chlorobenzenes [33,34] and PCDD/
PCDF [35,36] (Table 3). This results in longer resi-
dence times of the molecules on the catalyst surface,
as is con®rmed by the fact that after the ¯ow experi-
ment at 1508C, 10±20% of H6CBz, only trace amounts
of T4CBz (0.3±1.0%) and no T3CBz and D2CBz were
detected on the catalyst (Table 2). With chloroben-
zenes, therefore, the effect of lowering the volatility
with increasing chlorine substitution compensates or
Fig. 4. Destruction removal efficiency (DRE) of 1,2-D2CBz, 1,2,3-T3CBz, 1,2,4,5-T4CBz and H6CBz by catalysts A (^) and B (&) in the
temperature range 150±3108C (space velocity: 5000 hÿ1; gas phase composition: 10% oxygen, 70% nitrogen, and 20% water).
Table 3
Boiling points of some polychlorinated dibenzo-p-dioxin, polychlorinated dibenzofurans [35,36] and boiling points and ignition temperature
of some chlorobenzenes [33,34]
Chlorobenzenes M1CBz 1,2-D2CBz 1,2,3-T3CBz 1,3,5-T3CBz 1,2,3,5-T4CBz P5CBz H6CBz
Boiling point (8C) 132 179 219 208 247 276 322
Ignition temperature (8C) 23 65 126 126 >170
PCDD DD 2,3-D2CDD 1,2,4-T3CDD 2,3,7,8-T4CDD 1,2,3,6,7,8-H6CDD H7CDD O8CDD
Boiling point (8C) 279 358 375 447 487 507 510
PCDF DF 2,3-D2CDF 2,3,8-T3CDF 2,3,7,8-T4CDF 1,2,3,6,7,8-H6CDF H7CDF O8CDF
Boiling point (8C) 287 375 408 438 488 507 537
R. Weber et al. / Applied Catalysis B: Environmental 20 (1999) 249±256 255

even overcompensates the effect of an increasing
`̀ redox potential''.
4. Conclusions
The test system described for the determination of
the temperature dependence of catalytic destruction
ef®ciencies (and destruction removal ef®ciencies) is
especially useful in comparing various catalysts with
regard to their destruction ef®ciencies for compounds
of low volatility.
� The catalytic decomposition strongly depends on
the volatility of the compounds and the oxidative
behavior, both related to the degree of chlorina-
tion.
� The catalysts tested show a destruction efficiency
for PCDD/PCDF of >98% above 2008C.
� At temperatures below 2008C, part of the chlori-
nated `̀ polyaromatic'' compounds remain unox-
idized on the catalysts for several minutes. In an
incinerator with continuous gas flow, this could
result in an accumulation of these compounds on
the catalysts. Saturation could result in the dis-
placement of adsorbed compounds and a
decreased rate of adsorption, resulting in an over-
all increased emission concentration.
� Non-chlorinated polyaromatic compounds are
effectively decomposed at temperatures as low
as 1508C, while for the effective destruction of
monoaromatic compounds (chlorobenzenes and
nitrobenzenes, etc.) operating temperatures above
2508C are necessary for >95% destruction.
The results of this laboratory study obtained with
PCDD/PCDF coincide with results obtained with
catalysts installed in MWIs with operating tempera-
tures above 2008C. Therefore, the test systems used
should allow further investigations with relevance to
applications in MWIs and other thermal processes.
References
[1] 17.BImSchV vom 23.11.1990, BGBl I, 2545.
[2] The Advisory Committee for Controlling PCDDs/DFs in
MSW Management. Guidline for Controlling PCDDs/DFs in
MSW Management, 1997.
[3] M. Hiraoka, S. Sakai, T. Sakagawa, Y. Hata, Organohalogen
Compounds 31 (1997) 446.
[4] H. Hagenmaier, K. Horch, H. Fahlenkamp, G. Schetter,
Chemosphere 23 (1991) 1429.
[5] M. Eiken, J. Lambertz, G. Ritter, Umwelt ± Zeitschrift des
VDI 5 (1990) 226.
[6] H. Hagenmaier, G. Mittelbach, VGB Kraftwerkstechnik 6
(1990) 70.
[7] H. Fahlenkamp, G. Mittelbach, H. Hagenmaier, H. Brunner,
K.-H. Tichaczek, VGB Kraftwerkstechnik 7 (1991) 71.
[8] R. Trumpf, A. Christmann, H. Hagenmaier, UTA Interna-
tional 7 (1998) 82.
[9] H. Hagenmaier, VDI Berichte 730 (1989) 239.
[10] J.N. Armor, Appl. Catal. B 1 (1992) 221.
[11] N.-Y. Topsoe, J.A. Domestic, J. Catal. 151 (1995) 226.
[12] H. Schneider, S. Tschudin, M. Schneider, A. Wokaun, A.
Baiker, J. Catal. 147 (1994) 5.
[13] G.C. Bond, S.F. Tahir, Appl. Catal. 71 (1991) 1.
[14] P. Forzatti, L. Lietti, Heterogeneous Chem. Rev. 3 (1996)
33.
[15] H. Hagenmaier, K.-H. Tichaczek, H. Brunner, G. Mittelbach,
Organohalogen Compounds 3 (1990) 65.
[16] Y. Ide, K. Kashiwabara, S. Okada, T. Mori, M. Hara,
Chemosphere 32 (1996) 189.
[17] S. Krishnamoorthy, J.P. Baker, A. Amiridis, Catal. Today 40
(1998) 39.
[18] B. Ramachandran, H.L. Green, S. Chatterjee, Appl. Catal. B 8
(1996) 157.
[19] R.M. Lago, M.L.H. Green, S.C. Zang, M. Odlya, Appl. Catal.
B 8 (1996) 107.
[20] J. Jones, R.H. Ross, Catal. Today 35 (1997) 97.
[21] S. Imamura, H. Tarumoto, Ind. Eng. Chem. Res. 28 (1989)
1449.
[22] L. Jin, M.A. Abraham, Ind. Eng. Chem. Res. 30 (1991) 89.
[23] H. Nagata, T. Takakura, S. Tashiro, M. Kishida, K. Mizuno, I.
Tamori, K. Wakabayashi, Appl. Catal. B 5 (1994) 23.
[24] B. Frings, K.W. Marl, BWK/TUÈ /Umwelt-Spezial, MaÈrz
(1994) K22.
[25] O. Aniline, Adv. Chem. Ser. 120 (1973) 126.
[26] H.R. Buser, Ph.D. Dissertation, University of Umea, 1978.
[27] R. Weber, H. Hagenmaier, Chemosphere, accepted for
publication.
[28] J. HoÈckel, L. DuÈsterhoÈft, W. KoÈrner, H. Hagenmaier,
Organohalogen Compounds 23 (1995) 139.
[29] R. Weber, Ph.D. Dissertation, University of TuÈbingen, 1996.
[30] J. HoÈckel, Ph.D. Dissertation, University of TuÈbingen, 1996.
[31] V. Solinas, I. Ferino, Catal. Today 41 (1998) 179.
[32] Pio Forzatti, Daniele Ballardini, Lorenzo Sighicelli, Catal.
Today 41 (1998) 87.
[33] P.C. Weast, M.J. Astle, W.H. Beyer (Eds.), CRC Handbook of
Chemistry and Physics, CRC Press, Boca Raton, 1984.
[34] Aldrich Katalog ± Handbuch Feinchemikalien, Steinheim
(Germany), 1995.
[35] B.F. Rordorf, Thermochim. Acta 112 (1987) 117.
[36] B.F. Rordorf, Chemosphere 18 (1989) 783.
256 R. Weber et al. / Applied Catalysis B: Environmental 20 (1999) 249±256







