
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
Nº
1 IAinA.1. Cambio del nombre o dirección del titular de la autorización de
comercialización
2.4.1 /
2.4.6.SI SI
IAin a)a) para productos autorizados con arreglo al procedimiento centralizado
2.1.1 SI SI SI SI
IB b) para productos autorizados con arreglo al procedimiento nacional 2.1.1 SI SI
3 IAin A.3. Cambio de denominación de la sustancia activa o de un excipiente2.6.1.(P
y E)SI SI SI SI
4 IA
A.4 Cambio del nombre o dirección de: un fabricante (incluyendo, en su caso, los centros de pruebas de control de la calidad); o un titular del archivo principal de sustancia activa; o un
proveedor de la sustancia activa, el material de partida, el reactivo
o agente intermedio utilizados en la fabricación de la sustancia
activa (cuando se indique en el expediente técnico), cuando en el
expediente aprobado no figure un certificado de conformidad con la
Farmacopea Europea; o un fabricante de un excipiente nuevo (cuando se indique en el expediente técnico)
2.5.3.
IAin a) Fabricante responsable de la liberación de lotes
IA b) Todos los demás
6 IA A.6. Cambio del Código ATC/Código veterinario ATC 2.1.3. SI SI
A.7. Deletion of manufacturing sites for an active substance, intermediate or
finished product, packaging site, manufacturer responsible for batch release,
site where batch control takes place, or supplier of a starting material, reagent
or excipient (when mentioned in the dossier) *
A.8 Cambios hasta la fecha de la auditoría para comprobar el cumplimiento de las buenas prácticas de fabricación del fabricante de la sustancia activa*
*Nota: Esta modificación no se aplica si la información ha sido transmitida de
otro modo a las autoridades (por ejemplo, mediante la denominada
«declaración de la persona cualificada»).
IA
SI
A. C
AM
BIO
S A
DM
INIS
TR
AT
IVO
S
SI
7
IA
A.5. Cambio del nombre o dirección del fabricante / importador del
producto terminado (incluyendo los centros de pruebas de control de la calidad
o liberación de los lotes)5
2.5.1.a
/
2.5.1.1.
/
2.5.1.2/
* Nota: Cuando las autoridades hayan comunicado la intención de realizar una
inspección, se notificará inmediatamente la supresión del sitio pertinente.
SI SI
8
SI
2.5.1.2
/2.5.1.a
/2.5.1.b
/2.5.2
/2.5.3
SE
INC
LU
YE
N
CA
MB
IOS
D
OC
UM
EN
TA
CIO
N A
A
PO
RT
AR
DEFINICIÓNDEFINICIÓN
SE
INC
LU
YE
N
CA
MB
IOS
EN
C
ON
DIC
ION
ES
A
CU
MP
LIR
Se
envi
a F
T a
l TA
C
con
la r
esu
luci
ón
Mo
dif
ica
FT
, P. y
/o
MA
TIP
O
Pto
s d
e R
aefa
r
X
A.2. Cambio de la denominación (arbitraria) del medicamento2
Se
no
tifi
ca a
D.G
.de
Car
tera
Bás
ica
(SN
S
y F
arm
acia
)
OBJETIVO DEL CAMBIO
N
SU
B
T
IPO
x n
SI SI
División de Gestión y Procedimientos de Registro (DMUH) 1 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IAin a)El fabricante propuesto es parte del mismo grupo farmacéutico que el
fabricante aprobado actual2.5.3 SI
II b)Introducción de un nuevo fabricante de la sustancia activa soportado con un
ASMF2.5.3
II c)
El fabricante propuesto deberá utilizar una ruta de síntesis o condiciones de
fabricación sustancialmente distintas, que puedan cambiar características
cualitativas importantes de la sustancia activa, como el perfil cualitativo o
cuantitativo de impurezas que requieran cualificación, o propiedades
fisicoquímicas que influyan sobre la biodisponibilidad
2.5.3
II d)Nuevo fabricante de material para el que se exija una evaluación de la
seguridad viral o del riesgo de EET2.5.3
II e)
El cambio se refiere a una sustancia activa biológica o un material de partida,
reactivo o producto intermedio utilizado en la fabricación de un producto
biológico o inmunológico
IA f)
Cambios de las pruebas de control de la calidad para la sustitución de la
sustancia activa o adición de un sitio en el que se realicen pruebas o controles
de lotes
2.5.3 SI
II g)
g) Introducción de un nuevo fabricante de la sustancia activa que no cuenta con un archivo principal de sustancia activa y requiera una actualización considerable de la sección del expediente correspondiente a la sustancia activa pertinente
2.5.3
IB h)Adición de un centro de esterilización alternativo para la sustancia activa siguiendo un método conforme a la Farmacopea Europea
2.5.3 SI
IA i) Introducción de un nuevo sitio de micronización 2.5.3 SI SI
II j)
Cambios de las pruebas de control de la calidad para una sustancia activa biológica: sustitución o adición de un sitio donde se realizan pruebas o controles de los lotes, incluyendo un método biológico, inmunológico o inmunoquímico
2.5.3
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
B.I.a.1. Cambio del fabricante de un material de partida, reactivo o producto
intermedio utilizado en el proceso de fabricación de la sustancia activa o
cambio del fabricante (incluyendo, en su caso, los centros de pruebas de control de calidad) de la sustancia activa, cuando en el expediente
aprobado no figure un certificado de conformidad con la Farmacopea
Europeapruebas
I. S
ust
anci
a ac
tiva
a) F
abri
caci
on
IB k)Nuevo lugar de almacenamiento de bancos de células primarios o bancos de células de trabajo
1
SI2.5.3
División de Gestión y Procedimientos de Registro (DMUH) 2 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a) Cambio de importancia menor del proceso de fabricación de la sustancia activa
II b)
Cambio sustancial del proceso de fabricación de la sustancia activa, que pueda
tener efectos significativos para la calidad, seguridad o eficacia del
medicamento
II c)
El cambio se refiere a una sustancia biológica o inmunológica o al uso de una
sustancia obtenida por medios químicos diferentes para la fabricación de una
sustancia biológica / inmunológica que puede tener repercusiones significativas en la calidad, seguridad y eficacia del medicamento y
que no esté relacionada con un protocolo
II d)El cambio se refiere a un medicamento a base de plantas y existe un cambio en
el origen geográfico, la ruta de fabricación o la producción
IA a) a) Hasta un aumento de 10 veces del tamaño de lote aprobado actualmente
IA b) Reducción del tamaño del lote
II c)El cambio requiere una evaluación de la comparabilidad de una sustancia
activa biológica o inmunológica
IB d) Un aumento de más de 10 veces del tamaño de lote aprobado actualmente
IB e)
La escala de una sustancia activa biológica o inmunológica se aumenta o
disminuye sin modificar el proceso (por ejemplo, duplicación de la línea de
producción)
a) F
abri
caci
on
2
3
I. S
ust
anci
a ac
tiva
IB e)Cambio de importancia menor a la parte restringida de un archivo principal de
sustancia activa
B.I.a.2. Cambios del proceso de fabricación de la sustancia activa
B.I.a.3. Cambio del tamaño del lote (incluyendo los intervalos de tamaños de
lotes) de sustancia activa o el producto intermedio utilizado en el proceso de fabricaciónde la sustancia activa
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
División de Gestión y Procedimientos de Registro (DMUH) 3 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a) Reducción de los límites en proceso
IA b) Adición de una nueva prueba y límites en proceso
IA c) Supresión de una prueba en proceso no significativa SI
II d)Ampliación de las pruebas y límites en proceso que puedan tener un efecto
importante sobre la calidad de la sustancia activa en general
II e)Supresión de una prueba en proceso que pueda tener un efecto importante
sobre la calidad de la sustancia activa en general
5 IIB.I.a.5. Cambios de la sustancia activa de una vacuna estacional,
prepandémica o pandémica contra la gripe humanaa)
Sustitución de la cepa o cepas de una vacuna estacional, prepandémica o
pandémica contra la gripe humana2.6.1.P SI
IB
a) F
abri
caci
on
4
Adición o sustitución de una prueba en proceso como resultado de un problema
de seguridad o calidad
B.I.a.4. Cambio de las pruebas o límites en proceso aplicados durante la
fabricación de la sustancia activa
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
I. S
ust
anci
a ac
tiva
f)
División de Gestión y Procedimientos de Registro (DMUH) 4 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IAin a)
Reducción de los límites de especificación de medicamentos con arreglo a la aprobación oficial del lote por parte de la autoridad de control
IA c)Adición de un nuevo parámetro a la especificación con su correspondiente
método de pruebaSI
IA d)Supresión de un parámetro de especificación no significativo (por ejemplo,
supresión de un parámetro obsoleto)SI SI
II e)
Supresión de un parámetro de especificación que pueda tener un efecto
importante sobre la calidad de la sustancia activa o el producto terminado en
general
II f)
Cambio fuera de los límites de las especificaciones
aprobados para la sustancia activa
II g)
Ampliación de los límites de las especificaciones aprobados para los materiales
de partida o productos intermedios, que puedan tener un efecto importante
sobre la calidad de la sustancia activa o el producto terminado en general
IB h)
Adición o sustitución (excepto sustancias biológicas o inmunológicas) de un
parámetro de especificación con su correspondiente método de prueba como resultado de un problema de seguridad o calidad
IB i)
Cuando no exista monografía en la Farmacopea Europea o la farmacopea nacional de un Estado miembro para la sustancia activa, un cambio en la especificación de la farmacopea interna a una no oficial o una farmacopea de un tercer país
SI
1B.I.b.1. Cambio de los parámetros o límites de especificaciónde una sustancia
activa, material de partida, producto intermedio o reactivo utilizado en el
proceso de fabricación de la sustancia activa
IAin
I. S
ust
anci
a ac
tiva
b)
Co
ntr
ol d
e la
su
stan
cia
acti
va
Reducción de los límites de especificación
B. C
AM
BIO
S D
E C
AL
IDA
D
b)
División de Gestión y Procedimientos de Registro (DMUH) 5 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a) Cambios de importancia menor de un procedimiento de prueba aprobado
IA c)
Otros cambios de un procedimiento de prueba (incluyendo sustitución o
adición) para un reactivo, que no tenga un efecto significativo sobre la calidad
de la sustancia activa en general
II d)Cambio sustancial o sustitución de un método de prueba biológico,
inmunológico o inmunoquímico o un método que utilice un reactivo biológico
para una sustancia activa biológica
IB e)Otros cambios de un procedimiento de prueba (incluyendo sustitución o
adición) para la sustancia activa o un material de partida o producto intermedio
IA a) Composición cualitativa o cuantitativa SI SI
II b)Composición cualitativa o cuantitativa para sustancia activas biológicas o
inmunológicas estériles no conge ladas
IB c) Sustancias activas líquidas (no estériles) SI
IA a) Reducción de los límites de especificación
IA b)Adición de un nuevo parámetro a la especificación con su correspondiente
método de pruebaSI
IA c)Supresión de un parámetro de especificación no sig-ificativo (por ejemplo,
supresión de un parámetro obsoleto)SI
IB d)Adición o sustitución de un parámetro de especificación como resultado de un
problema de seguridad o calidad
IA a) Cambios de importancia menor de un procedimiento de prueba aprobado
IA b)Otros cambios de un procedimiento de prueba (incluyendo sustitución o
adición)
I. S
ust
anci
a ac
tiva
B.I.c.1. Cambio del acondicionamiento primario de la sustancia activa
b)
B.I.c.3. Cambio del procedimiento de prueba para el acondicionamiento
primario de la sustancia activa
IA
b)
Co
ntr
ol d
e la
su
stan
cia
acti
va
B.I.b.2. Cambio del procedimiento de prueba de la sustancia activa o material
de partida, reactivo o producto intermedioutilizado en el proceso de fabricación
de la sustancia activa
Supresión de un procedimiento de prueba para la sustancia activa o un material
de partida, reactivo o producto intermedio, si ya se ha autorizado
procedimiento de prueba alternativo
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
I. S
ust
anci
a ac
tiva
c) S
iste
ma
de
cier
re d
el e
nva
se
Supresión de un procedimiento de prueba si ya se ha autorizado un
procedimiento de prueba alternativo
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
3
IA
1
2
B.I.c.2. Cambio de los parámetros o límites de las especificaciones del
acondicionamiento primario de la sustancia activa2
c)
División de Gestión y Procedimientos de Registro (DMUH) 6 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a.1) Reducción
Extensión del período de reanálisis basada en la extrapolación de datos de
estabilidad no conformes con las directrices ICH/VICH*
* Nota: período de reanálisis no aplicable a las sustancias activas biológicas o
inmunológicas
II a.3) Extensión del período de almacenamiento de una sustancia activa biológica o
inmunológica no conforme con un protocolo de estabilidad aprobado
IB a.4) Extensión o introducción de un período de reanálisis o almacenamiento
basado en datos en tiempo real
IA b.1) Adopción de condiciones de almacenamiento más estrictas para la sustancia
activa
II b.2)
Cambio de las condiciones de almacenamiento de sustancias activas
biológicas o inmunológicas, cuando los estudios de estabilidad no se han
realizado de conformidad con el protocolo de estabilidad aprobado vigente
IB b.3) Cambio de las condiciones de almacenamiento de la sustancia activa
IA
B.I.d.1. Cambio del período de reanálisis o de almacenamiento o las
condiciones de almacenamiento de la sustancia activa, cuando en el
expediente aprobado no figure un certificado de conformidad con la
Farmacopea Europea que abarque el período de reanálisis c) Cambio a un protocolo de estabilidad aprobado
c) Cambio a un protocolo de estabilidad aprobado SI SI
II a.2)
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
d)
Est
abili
dad
B.I.d.1. Cambio del período de reanálisis o de almacenamiento o las
condiciones de almacenamiento de la sustancia activa, cuando en el
expediente aprobado no figure un certificado de conformidad con la
Farmacopea Europea que abarque el período de reanálisis
b) Condiciones de almacenamiento
I. S
ust
anci
a ac
tiva
1
B.I.d.1. Cambio del período de reanálisis o de almacenamiento o las
condiciones de almacenamiento de la sustancia activa, cuando en el
expediente aprobado no figure un certificado de conformidad con la
Farmacopea Europea que abarque el período de reanálisis
a) Período de reanálisis o almacenamiento
División de Gestión y Procedimientos de Registro (DMUH) 7 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
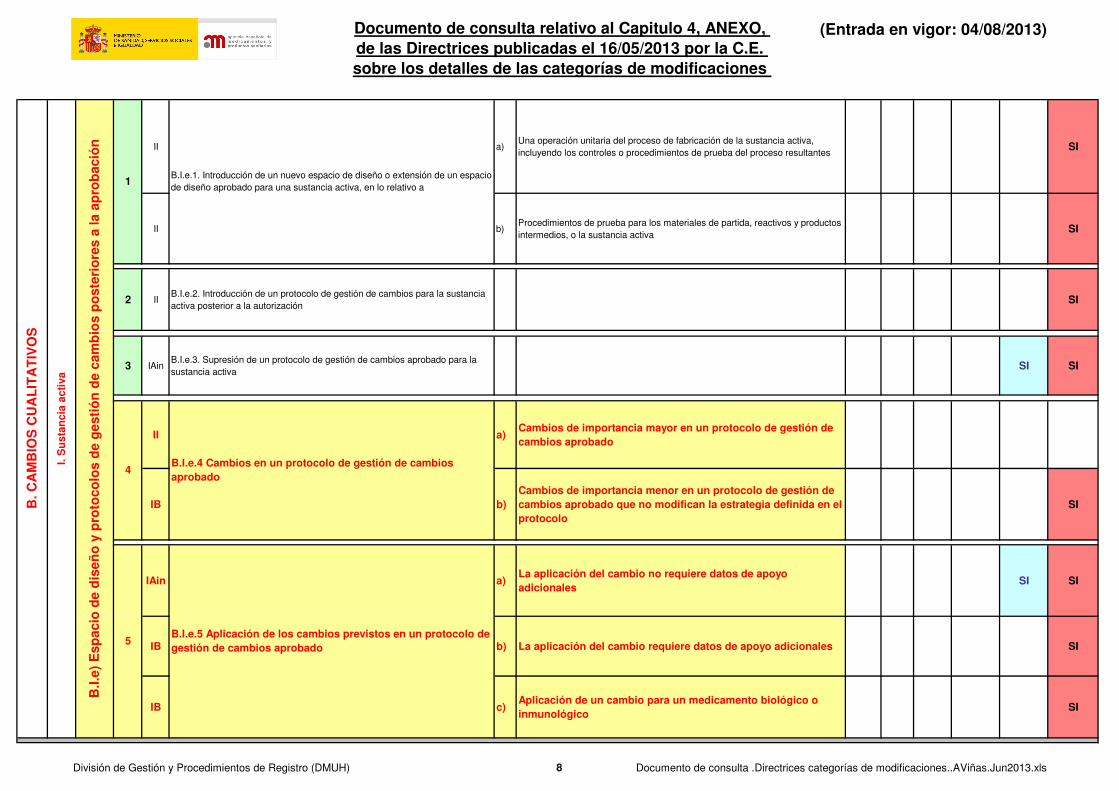
II a)Una operación unitaria del proceso de fabricación de la sustancia activa,
incluyendo los controles o procedimientos de prueba del proceso resultantesSI
II b)Procedimientos de prueba para los materiales de partida, reactivos y productos
intermedios, o la sustancia activaSI
2 IIB.I.e.2. Introducción de un protocolo de gestión de cambios para la sustancia
activa posterior a la autorizaciónSI
3 IAinB.I.e.3. Supresión de un protocolo de gestión de cambios aprobado para la
sustancia activaSI SI
II a)Cambios de importancia mayor en un protocolo de gestión de cambios aprobado
IB b)Cambios de importancia menor en un protocolo de gestión de cambios aprobado que no modifican la estrategia definida en el protocolo
SI
IAin a)La aplicación del cambio no requiere datos de apoyo adicionales
SI SI
IB b) La aplicación del cambio requiere datos de apoyo adicionales SI
IB c)Aplicación de un cambio para un medicamento biológico o inmunológico
SI
4B.I.e.4 Cambios en un protocolo de gestión de cambios aprobado
1B.I.e.1. Introducción de un nuevo espacio de diseño o extensión de un espacio
de diseño aprobado para una sustancia activa, en lo relativo a
B.I.e.5 Aplicación de los cambios previstos en un protocolo de gestión de cambios aprobado
5
I. S
ust
anci
a ac
tiva
B.I.
e) E
spac
io d
e d
iseñ
o y
pro
toco
los
de
ges
tió
n d
e ca
mb
ios
po
ster
iore
s a
la a
pro
bac
ión
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
División de Gestión y Procedimientos de Registro (DMUH) 8 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IAin a) Cambios de las marcas de impresión, relieve y otras marcas SI SI
IB b)Cambios de las incisiones o líneas de puntos destinadas a dividir el producto en
dosis igualesSI
IAin a) Comprimidos, cápsulas, supositorios y óvulos vaginales de liberación inmediata SI
IB b)
Formas farmacéuticas gastrorresistentes modificadas o de liberación
prolongada y comprimidos con incisiones destinadas a dividirlos en dosis
iguales
SI
II c)Adición de un nuevo equipo reactivo para un preparado radiofarmacéutico con otro volumen de llenado
IAin a.1) Adición, supresión o sustitución 2.6.1.E SI SI
IA a.2) Aumento o reducción 2.6.1.E SI SI
IA b.1)Cualquier ajuste de importancia menor de la composición cuantitativa de los
excipientes del producto terminado2.6.1.E SI
II b.2)Cambios cualitativos o cuantitativos de uno o más excipientes que puedan
tener efectos importantes para la seguridad, calidad o eficacia del medicamento2.6.1.E SI
II b.3) Cambio relacionado con un producto biológico o inmunológico 2.6.1.E SI
II b.4)
Cualquier nuevo excipiente que incluya el uso de materiales de origen humano
o animal para loscuales deba realizarse una evaluación de los datos relativos a
seguridad viral o riesgo de EET
2.6.1.E SI
II b.5) Cambios que se basen en un estudio de bioequivalencia 2.6.1.E SI
B.II.a.1. Cambio o adición de las marcas de impresión, relieve u otras marcas,
incluyendo sustitución o adición de las tintas utilizadas para marcar el producto
IB
3
2.6.1.E
B.II.a.2. Cambio de la conformación o dimensiones de la forma farmacéutica
Medicamentos veterinarios biológicos para administración oral en los cuales el
agente aromatizante o colorante es importante para la ingestión por parte de
las especies animales
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
2
2.6.1.E
SISustitución de un único excipiente por un excipiente comparable con las
mismas características funcionales y a un nivel similar
a.3)II
B.II.a.3. Cambios de la composición (excipientes) del producto terminado
b) Otros excipientes
1
a) D
escr
ipci
ón
y c
om
po
sici
ón
II. P
rod
uct
o t
erm
inad
o
B.II.a.3. Cambios de la composición (excipientes) del producto terminado
a) Cambios de los componentes del sistema de aromatizantes y colorantes
b.6)
SI
División de Gestión y Procedimientos de Registro (DMUH) 9 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
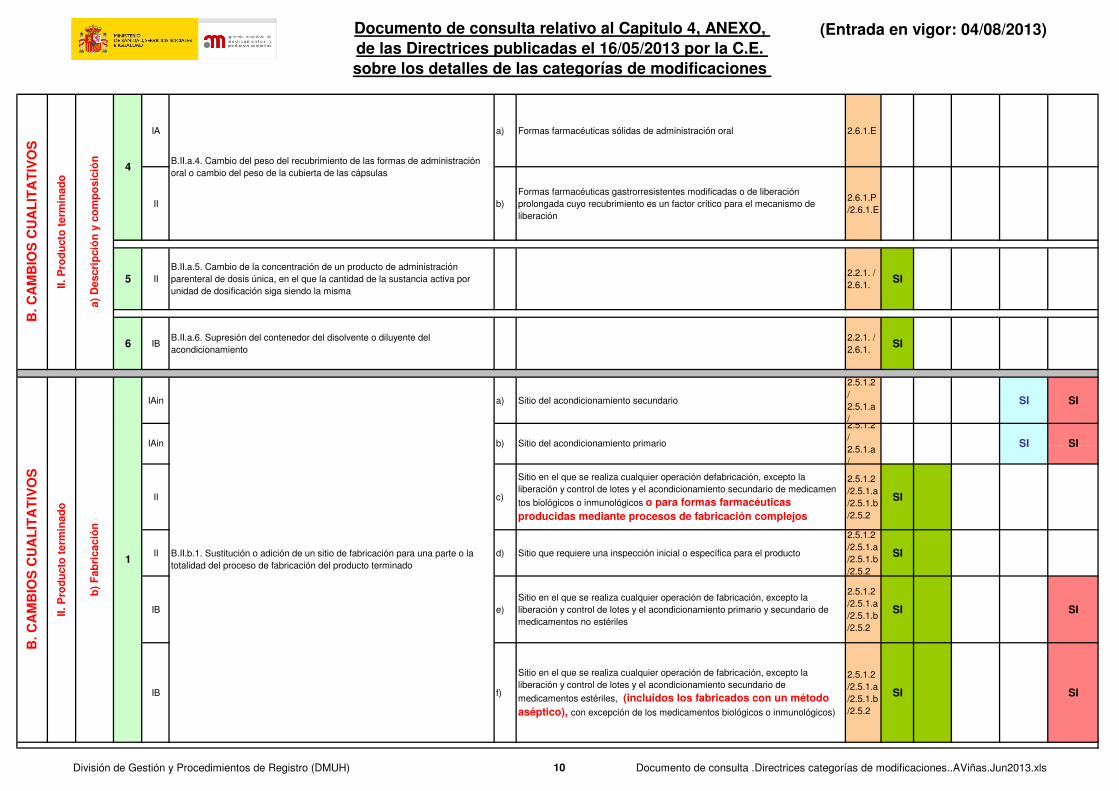
IA a) Formas farmacéuticas sólidas de administración oral 2.6.1.E
II b)
Formas farmacéuticas gastrorresistentes modificadas o de liberación
prolongada cuyo recubrimiento es un factor crítico para el mecanismo de
liberación
2.6.1.P
/2.6.1.E
5 II
B.II.a.5. Cambio de la concentración de un producto de administración
parenteral de dosis única, en el que la cantidad de la sustancia activa por
unidad de dosificación siga siendo la misma
2.2.1. /
2.6.1.SI
6 IBB.II.a.6. Supresión del contenedor del disolvente o diluyente del
acondicionamiento
2.2.1. /
2.6.1.SI
IAin a) Sitio del acondicionamiento secundario
2.5.1.2
/
2.5.1.a
/
SI SI
II c)
Sitio en el que se realiza cualquier operación defabricación, excepto la
liberación y control de lotes y el acondicionamiento secundario de medicamen
tos biológicos o inmunológicos o para formas farmacéuticas producidas mediante procesos de fabricación complejos
2.5.1.2
/2.5.1.a
/2.5.1.b
/2.5.2
SI
II d) Sitio que requiere una inspección inicial o específica para el producto
2.5.1.2
/2.5.1.a
/2.5.1.b
/2.5.2
SI
IB e)
Sitio en el que se realiza cualquier operación de fabricación, excepto la
liberación y control de lotes y el acondicionamiento primario y secundario de
medicamentos no estériles
2.5.1.2
/2.5.1.a
/2.5.1.b
/2.5.2
SI SI
IB f)
Sitio en el que se realiza cualquier operación de fabricación, excepto la
liberación y control de lotes y el acondicionamiento secundario de
medicamentos estériles, (incluidos los fabricados con un método aséptico), con excepción de los medicamentos biológicos o inmunológicos)
2.5.1.2
/2.5.1.a
/2.5.1.b
/2.5.2
SI SI
SI SI
B.II.a.4. Cambio del peso del recubrimiento de las formas de administración
oral o cambio del peso de la cubierta de las cápsulas
b)
B.II.b.1. Sustitución o adición de un sitio de fabricación para una parte o la
totalidad del proceso de fabricación del producto terminado
4
2.5.1.2
/
2.5.1.a
/
1
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
a) D
escr
ipci
ón
y c
om
po
sici
ón
b)
Fab
rica
ció
n
II. P
rod
uct
o t
erm
inad
o
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
II. P
rod
uct
o t
erm
inad
o
Sitio del acondicionamiento primarioIAin
División de Gestión y Procedimientos de Registro (DMUH) 10 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a)Sustitución o adición de un sitio en el que se realizan el control o las pruebas
de los lotes2.5.1.2 SI SI
II b)
Sustitución o adición de un sitio en el que se realizan el control o las pruebas de los lotes de productos biológicos o inmunológicos y cualquiera de los métodos de prueba realizados en el sitio es un método biológico o inmunológico
2.5.1.2 SI
IAin c.1)
Sustitución o adición de un fabricante responsable de la importación o
liberación de los lotes
1. Sin incluir control o prueba de los lotes
2.5.1.2
/2.5.1.a
/2.5.1.b
SI SI
IAin c.2)
Sustitución o adición de un fabricante responsable de la importación o
liberación de los lotes
2. Incluyendo control o prueba de los lotes
2.5.1.2
/2.5.1.a
/2.5.1.b
SI SI SI
II c.3)
Sustitución o adición de un fabricante responsable de la importación o
liberación de los lotes 3.
Incluyendo control o prueba de los lotes de un producto biológico o
inmunológico y uno de los métodos de prueba realizados en el sitio es un
método biológico, inmunológico o inmunoquímico
2.5.1.2
/2.5.1.a
/2.5.1.b
SI
IA a)Cambio de importancia menor del proceso de fabricación de una forma sólida o
soluciones de administración oral de liberación inmediata
II b)Cambios sustanciales del proceso de fabricación que puedan tener efectos
significativos para la calidad, seguridad o eficacia del medicamento
II c)El producto es un medicamento biológico o inmunológico y el cambio requiere
una evaluación de comparabilidad
II d) Introducción de un método de esterilización terminal no estándar
II e)Introducción o aumento de la sobredosificación utilizada para la sustancia
activa
B.II.b.3. Cambios del proceso de fabricación del producto terminado,
incluyendo un producto intermedio utilizado en la fabricación del producto terminado
2
II. P
rod
uct
o t
erm
inad
o
IB f) SI
b)
Fab
rica
ció
n
B.II.b.2. Cambio en el importador, las medidas para la liberación de
lotes y las pruebas de control de calidad del producto terminado
3
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
Cambio de importancia menor del proceso de fabricación de una suspensión
acuosa de administración oralSI
División de Gestión y Procedimientos de Registro (DMUH) 11 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
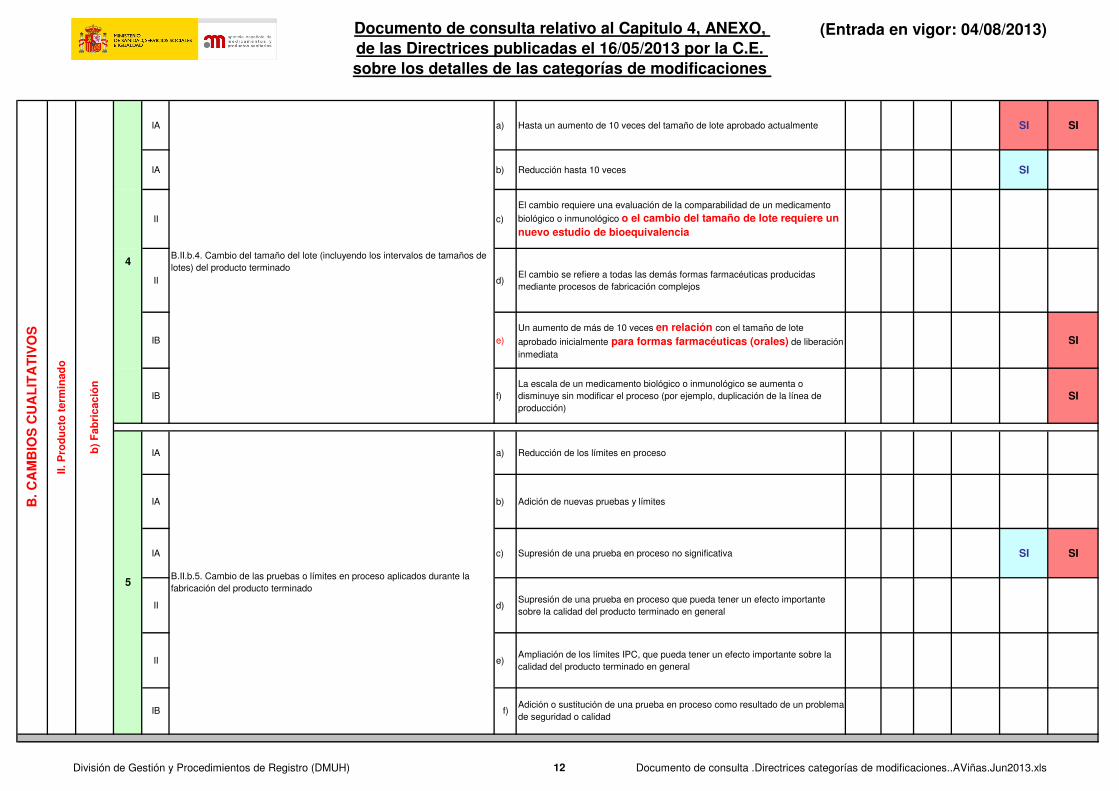
IA a) Hasta un aumento de 10 veces del tamaño de lote aprobado actualmente SI SI
IA b) Reducción hasta 10 veces SI
II c)
El cambio requiere una evaluación de la comparabilidad de un medicamento
biológico o inmunológico o el cambio del tamaño de lote requiere un nuevo estudio de bioequivalencia
II d)El cambio se refiere a todas las demás formas farmacéuticas producidas
mediante procesos de fabricación complejos
IB e)
Un aumento de más de 10 veces en relación con el tamaño de lote
aprobado inicialmente para formas farmacéuticas (orales) de liberación
inmediata
SI
IB f)
La escala de un medicamento biológico o inmunológico se aumenta o
disminuye sin modificar el proceso (por ejemplo, duplicación de la línea de
producción)
SI
IA a) Reducción de los límites en proceso
IA b) Adición de nuevas pruebas y límites
IA c) Supresión de una prueba en proceso no significativa SI SI
II d)Supresión de una prueba en proceso que pueda tener un efecto importante
sobre la calidad del producto terminado en general
II e)Ampliación de los límites IPC, que pueda tener un efecto importante sobre la
calidad del producto terminado en general
B.II.b.4. Cambio del tamaño del lote (incluyendo los intervalos de tamaños de
lotes) del producto terminado4
B.II.b.5. Cambio de las pruebas o límites en proceso aplicados durante la
fabricación del producto terminado
f)
5
IBAdición o sustitución de una prueba en proceso como resultado de un problema
de seguridad o calidad
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
II. P
rod
uct
o t
erm
inad
o
b)
Fab
rica
ció
n
División de Gestión y Procedimientos de Registro (DMUH) 12 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
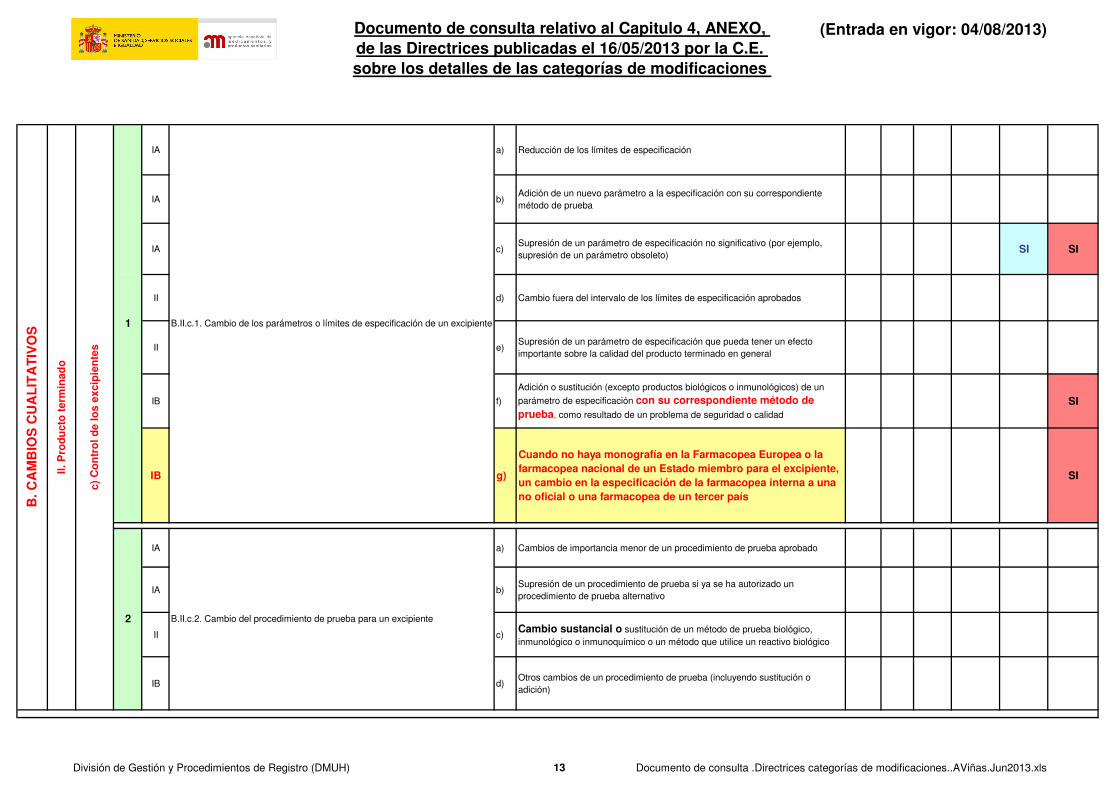
IA a) Reducción de los límites de especificación
IA b)Adición de un nuevo parámetro a la especificación con su correspondiente
método de prueba
IA c)Supresión de un parámetro de especificación no significativo (por ejemplo,
supresión de un parámetro obsoleto)SI SI
II d) Cambio fuera del intervalo de los límites de especificación aprobados
II e)Supresión de un parámetro de especificación que pueda tener un efecto
importante sobre la calidad del producto terminado en general
IB f)
Adición o sustitución (excepto productos biológicos o inmunológicos) de un
parámetro de especificación con su correspondiente método de prueba, como resultado de un problema de seguridad o calidad
SI
IB g)
Cuando no haya monografía en la Farmacopea Europea o la farmacopea nacional de un Estado miembro para el excipiente, un cambio en la especificación de la farmacopea interna a una no oficial o una farmacopea de un tercer país
SI
IA a) Cambios de importancia menor de un procedimiento de prueba aprobado
IA b)Supresión de un procedimiento de prueba si ya se ha autorizado un
procedimiento de prueba alternativo
II c)Cambio sustancial o sustitución de un método de prueba biológico,
inmunológico o inmunoquímico o un método que utilice un reactivo biológico
IB d)Otros cambios de un procedimiento de prueba (incluyendo sustitución o
adición)
II. P
rod
uct
o t
erm
inad
o
2
1
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
c) C
on
tro
l de
los
exci
pie
nte
s
B.II.c.2. Cambio del procedimiento de prueba para un excipiente
B.II.c.1. Cambio de los parámetros o límites de especificación de un excipiente
División de Gestión y Procedimientos de Registro (DMUH) 13 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a.1)
De material de riesgo de EET de origen vegetal o sintético Para excipientes o
reactivos no utilizados en la fabricación de una sustancia activa biológica o
inmunológica o en un medicamento biológico o inmunológico
2.6.2.
IB a.2)
De material de riesgo de EET de origen vegetal o sintético Para excipientes o
reactivos utilizados en la fabricación de una sustancia activa biológica o
inmunológica o en un medicamento biológico o inmunológico
2.6.2.
II b)
Cambio o introducción de un material de riesgo de EET o sustitución de un
material de riesgo de EET por otro material de riesgo de EET, no incluido en un
certificado de conformidad EET
2.6.2.
IA a)Cambio de importancia menor de la síntesis o recuperación de un excipiente no
incluido en la farmacopea o un excipiente nuevo SI SI
II b)
Las especificaciones se ven afectadas o se produce un cambio de las
propiedades fisicoquímicas del excipiente que podría influir sobre la calidad del
producto terminado
II c) El excipiente es una sustancia biológica o inmunológica
B.II.c.3. Cambio del origen de un excipiente o reactivo con riesgo de EET
c) C
on
tro
l de
los
exci
pie
nte
s
4B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
II. P
rod
uct
o t
erm
inad
o
3
5
B.II.c.4 Cambio de la síntesis o recuperación de un excipiente no incluido en la
farmacopea (si se describe en el expediente) o un excipiente nuevo
Esta varIacion aparecia en el borrador (Nov 2012) y en el indice pero ahora aparece como eliminada
División de Gestión y Procedimientos de Registro (DMUH) 14 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a) Reducción de los límites de especificación
IAIN b)
Reducción de los límites de especificación de medicamentos con arreglo a la aprobación oficial del lote por parte de la autoridad de control
IA c)Adición de un nuevo parámetro a la especificación con su correspondiente
método de prueba
IA d)
Supresión de un parámetro de especificación no significativo (por ejemplo, supresión de un parámetro obsoleto como olor y sabor o prueba de identificación de un agente colorante o aromatizante)
SI SI
II e) Cambio fuera del intervalo de los límites de especificación aprobados
II f)Supresión de un parámetro de especificación que pueda tener un efecto
importante sobre la calidad del producto terminado en general
IB g)
Adición o sustitución (excepto productos biológicos o inmunológicos) de un
parámetro de especificación con su correspondiente método de prueba como como resultado de un problema de seguridad o calidad
SI
h)Actualización del expediente para cumplir con las disposiciones de una monografía general actualizada de la Farmacopea Europea para el producto terminado*
SI SI
* Nota: No es necesario notificar a las autoridades competentes la
actualización de la monografía de la Farmacopea Europea o de la farmacopea
nacional de un Estado miembro si se hace referencia a la «edición vigente» en
el expediente de un medicamento autorizado. Así pues, esta modificación se
aplica a los casos en que el expediente técnico no contenga referencia alguna
a la monografía actualizada de la farmacopea y por lo tanto, se realiza para
hacer referencia a la versión actualizada.
IB i)
Farmacopea Europea 2.9.40 Se introduce la Uniformidad de dosificación para sustituir el método actualmente registrado, tanto Farmacopea Europea 2.9.5 (Uniformidad de masa) como Farmacopea Europea 2.9.6 (Uniformidad de contenido)
SI SI
1
IAin
B.II.d.1. Cambio de los parámetros de especificación o límites de
especificación del producto terminado
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
II. P
rod
uct
o t
erm
inad
o
d)
Co
ntr
ol d
el p
rod
uct
o t
erm
inad
o
División de Gestión y Procedimientos de Registro (DMUH) 15 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA b)Supresión de un procedimiento de prueba si ya se ha autorizado un método
alternativo
II c)
Cambio sustancial o sustitución de un método de prueba biológico,
inmunológico o inmunoquímico o un método que utilice un reactivo biológico o sustitución de un preparado de referencia biológico que no esté cubierto por un protocolo aprobado
IB d)Otros cambios de un procedimiento de prueba (incluyendo sustitución o
adición)
IA e)Actualización del procedimiento de prueba para ajustarse a la monografía general actualizada en la Farmacopea Europea
SI SI
Para reflejar el cumplimiento de la Farmacopea Europea y eliminar la referencia al método de prueba interno y al número del método de prueba obsoletos*
SI SI
* Nota: No es necesario notificar a las autoridades competentes la
actualización de la monografía de la Farmacopea Europea si se
hace referencia a la «edición vigente» en el expediente de un
medicamento autorizado.
3 IIB.II.d.3. Modificaciones relativas a la introducción de la liberación en tiempo
real o la liberación paramétrica en la fabricación del producto terminado
f)
B.II.d.2. Cambio del procedimiento de prueba para el producto terminado
IA
2
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
II. P
rod
uct
o t
erm
inad
o
d)
Co
ntr
ol d
el p
. ter
min
ado
IA a) Cambios de importancia menor de un procedimiento de prueba aprobado
División de Gestión y Procedimientos de Registro (DMUH) 16 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a.1) Composición cualitativa y cuantitativa: Formas farmacéuticas sólidas 2.2.3.T
IB a.2)Composición cualitativa y cuantitativa: Formas farmacéuticas semisólidas y no
estériles2.2.3.T
II a.3) Composición cualitativa y cuantitativa 2.2.3.T
II a.4)Composición cualitativa y cuantitativa: Medicamentos estériles y biológicos o
inmunologicos2.2.3.T
IB b.1)Cambio en el tipo de envase o adición de un nuevo envase: Formas
farmacéuticas sólidas, semisólidas y líquidas no estériles2.2.3.M
II b.2)Cambio en el tipo de envase o adición de un nuevo envase: Medicamentos
estériles y biológicos o inmunológicos2.2.3.M
Supresión de un envase de acondicionamiento primario que no implique la supresión total de una dosificación o forma farmacéutica
Nota: En el caso del procedimiento B.II.e.1.b) se recuerda a los solicitantes que
cualquier cambio que tenga por resultado una «nueva forma farmacéutica»
requiere la presentación de una solicitud de extensión.
IA b)Adición de un nuevo parámetro a la especificación con su correspondiente
método de prueba
IA c)Supresión de un parámetro de especificación no significativo (por ejemplo,
supresión de un parámetro obsoleto)SI
IB d)Adición o sustitución de un parámetro de especificación como resultado de un
problema de seguridad o calidad
IA a) Cambios de importancia menor de un procedimiento de prueba aprobado
IA b)Otros cambios de un procedimiento de prueba (incluyendo sustitución o
adición)
IA c)Supresión de un procedimiento de prueba si ya se ha autorizado un
procedimiento de prueba alternativo
IA
2
B.II.e.3. Cambio del procedimiento de prueba para el acondicionamiento
primario del producto terminado
1
SI
B.II.e.1. Cambio del acondicionamiento primario del producto terminado
IA SIb.3
)
a) Reducción de los límites de especificación
B.II.e.2. Cambio de los parámetros o límites de las especificaciones del
acondicionamiento primario del producto terminado
3
II. P
rod
uct
o t
erm
inad
o
2.2.3
e) S
iste
ma
de
cier
re d
el e
nva
se
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
División de Gestión y Procedimientos de Registro (DMUH) 17 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a) Medicamentos no estériles 2.2.3.T
II b)
El cambio de forma o dimensiones se refiere a una parte esencial del material
de acondicionamiento que pueda tener efectos importantes para la
administración, uso, seguridad o estabilidad del producto terminado
2.2.3.M
IB c) Medicamentos estériles 2.2.3.M
IAin a.1)
Cambio del número de unidades (por ejemplo, com¬primidos, ampollas, etc.)
en un envase Cambio dentro de los límites de los tamaños deenvase
aprobados actualmente
2.2.3.M SI SI
IB a.2)Cambio del número de unidades (por ejemplo, com¬primidos, ampollas, etc.)
Cambio fuera de los límites de los tamaños deenvase aprobados actualmente2.2.3.M SI SI SI
IA b) Supresión de tamaños de envases 2.2.3.B SI SI SI
II c)
Cambio del peso o volumen de llenado de medicamentos estériles estériles de
administración parenteral para múltiples dosis (o dosis única para uso parcial)
incluidos los medicamentos biológicos o inmunológicos
2.2.3.M SI SI
Cambio del peso o volumen de llenado de medicamentos de administración no
parenteral para múlti-ples dosis (o de dosis única para uso parcial)2.2.3.M SI SI SI
Nota: En el caso del procedimiento B.II.e.5.c) y d), se recuerda a los
solicitantes que cualquier cambio de la «dosificación» del medicamento
requiere la presentación de una solicitud de extensión.
IAin a) Cambio que afecte a la información sobre el producto 2.2.3.T SI
IA b) Cambio que no afecte a la información sobre el producto 2.2.3. SI
IB
B.II.e.5. Cambio del tamaño del envase del producto terminado
4
B.II.e.6. Cambio de cualquier parte del material del acondicionamiento
(primario) que no entre en contacto con la fórmula del producto terminado
[como color de los tapones a presión, códigos de color de las anillas de las
ampo llas, cambio de la protección de las agujas (uso de un plástico
diferente)]
d)
5
B.II.e.4. Cambio de la forma o dimensiones del envase o cierre
(acondicionamiento primario)
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
6
II. P
rod
uct
o t
erm
inad
o
e) S
iste
ma
de
cier
re d
el e
nva
se
División de Gestión y Procedimientos de Registro (DMUH) 18 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a) Supresión de un proveedor
IA b) Sustitución o adición de un proveedor
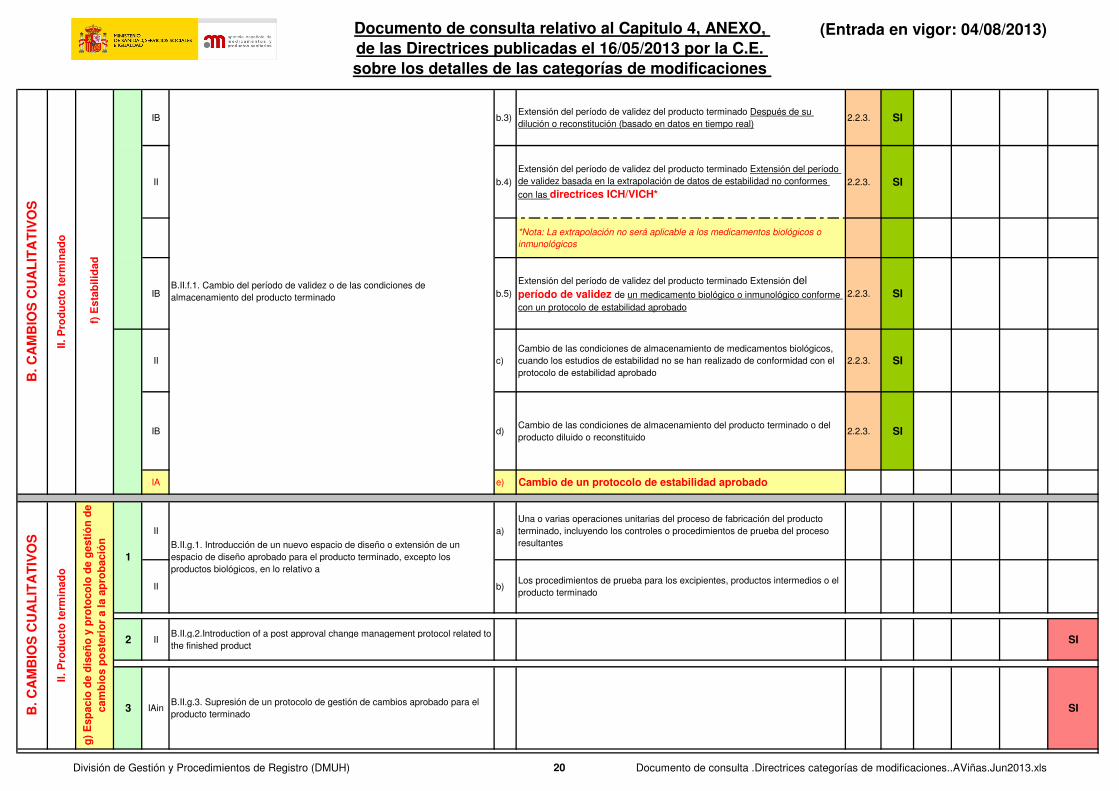
IAin a.1)Reducción del período de validez del producto terminado 1. Envasado para su
venta2.2.3. SI
IAin a.2) Reducción del período de validez del producto terminado Tras ser abierto 2.2.3. SI
IAin a.3)Reducción del período de validez del producto terminado Después de su
dilución o reconstitución2.2.3. SI
IB b.1)
Extensión del período de validez del producto terminado. Envasado para su
venta (basado en datos en tiempo real) 2.2.3. SI
IB b.2)Extensión del período de validez del producto terminado Tras ser abierto
(basado en datos en tiempo real2.2.3. SI
II
B.II.e.7. Cambio del proveedor de los componentes o dispositivos del
acondicionamiento (si se menciona en el expediente)
Cualquier cambio de los proveedores de separadores para inhaladores
dosificadores
B.II.f.1. Cambio del período de validez o de las condiciones de
almacenamiento del producto terminado
e) S
iste
ma
de
cier
re d
el e
nva
se
c)
f)
Est
abili
dad
1
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
II. P
rod
uct
o t
erm
inad
o
7
División de Gestión y Procedimientos de Registro (DMUH) 19 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IB b.3)Extensión del período de validez del producto terminado Después de su
dilución o reconstitución (basado en datos en tiempo real)2.2.3. SI
II b.4)
Extensión del período de validez del producto terminado Extensión del período
de validez basada en la extrapolación de datos de estabilidad no conformes
con las directrices ICH/VICH*2.2.3. SI
*Nota: La extrapolación no será aplicable a los medicamentos biológicos o
inmunológicos
IB b.5)
Extensión del período de validez del producto terminado Extensión del
período de validez de un medicamento biológico o inmunológico conforme
con un protocolo de estabilidad aprobado
2.2.3. SI
II c)
Cambio de las condiciones de almacenamiento de medicamentos biológicos,
cuando los estudios de estabilidad no se han realizado de conformidad con el
protocolo de estabilidad aprobado
2.2.3. SI
IB d)Cambio de las condiciones de almacenamiento del producto terminado o del
producto diluido o reconstituido2.2.3. SI
IA e) Cambio de un protocolo de estabilidad aprobado
II a)
Una o varias operaciones unitarias del proceso de fabricación del producto
terminado, incluyendo los controles o procedimientos de prueba del proceso
resultantes
II b)Los procedimientos de prueba para los excipientes, productos intermedios o el
producto terminado
2 IIB.II.g.2.Introduction of a post approval change management protocol related to
the finished product SI
3 IAinB.II.g.3. Supresión de un protocolo de gestión de cambios aprobado para el
producto terminadoSI
B.II.g.1. Introducción de un nuevo espacio de diseño o extensión de un
espacio de diseño aprobado para el producto terminado, excepto los
productos biológicos, en lo relativo a
B.II.f.1. Cambio del período de validez o de las condiciones de
almacenamiento del producto terminado
1
g)
Esp
acio
de
dis
eño
y p
roto
colo
de
ges
tió
n d
e ca
mb
ios
po
ster
ior
a la
ap
rob
ació
n
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
II. P
rod
uct
o t
erm
inad
o
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
II. P
rod
uct
o t
erm
inad
o
f)
Est
abili
dad
División de Gestión y Procedimientos de Registro (DMUH) 20 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
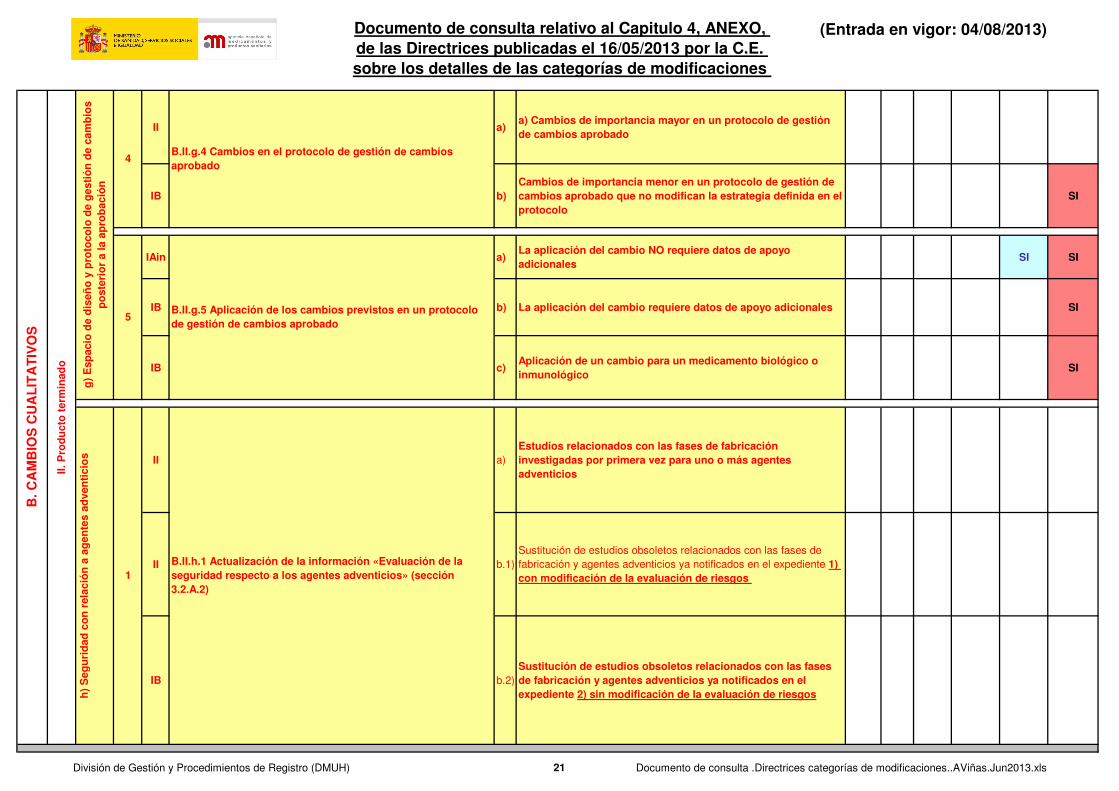
II a)a) Cambios de importancia mayor en un protocolo de gestión de cambios aprobado
IB b)Cambios de importancia menor en un protocolo de gestión de cambios aprobado que no modifican la estrategia definida en el protocolo
SI
IAin a)La aplicación del cambio NO requiere datos de apoyo adicionales
SI SI
IB b) La aplicación del cambio requiere datos de apoyo adicionales SI
IB c)Aplicación de un cambio para un medicamento biológico o inmunológico
SI
II a)
Estudios relacionados con las fases de fabricación investigadas por primera vez para uno o más agentes adventicios
II b.1)
Sustitución de estudios obsoletos relacionados con las fases de
fabricación y agentes adventicios ya notificados en el expediente 1) con modificación de la evaluación de riesgos
IB b.2)
Sustitución de estudios obsoletos relacionados con las fases de fabricación y agentes adventicios ya notificados en el expediente 2) sin modificación de la evaluación de riesgos
B.II.h.1 Actualización de la información «Evaluación de la seguridad respecto a los agentes adventicios» (sección 3.2.A.2)
h)
Seg
uri
dad
co
n r
elac
ión
a a
gen
tes
adve
nti
cio
s
B.II.g.4 Cambios en el protocolo de gestión de cambios aprobado
1
4
5B.II.g.5 Aplicación de los cambios previstos en un protocolo de gestión de cambios aprobado
II. P
rod
uct
o t
erm
inad
o
g)
Esp
acio
de
dis
eño
y p
roto
colo
de
ges
tió
n d
e ca
mb
ios
po
ster
ior
a la
ap
rob
ació
n
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
División de Gestión y Procedimientos de Registro (DMUH) 21 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
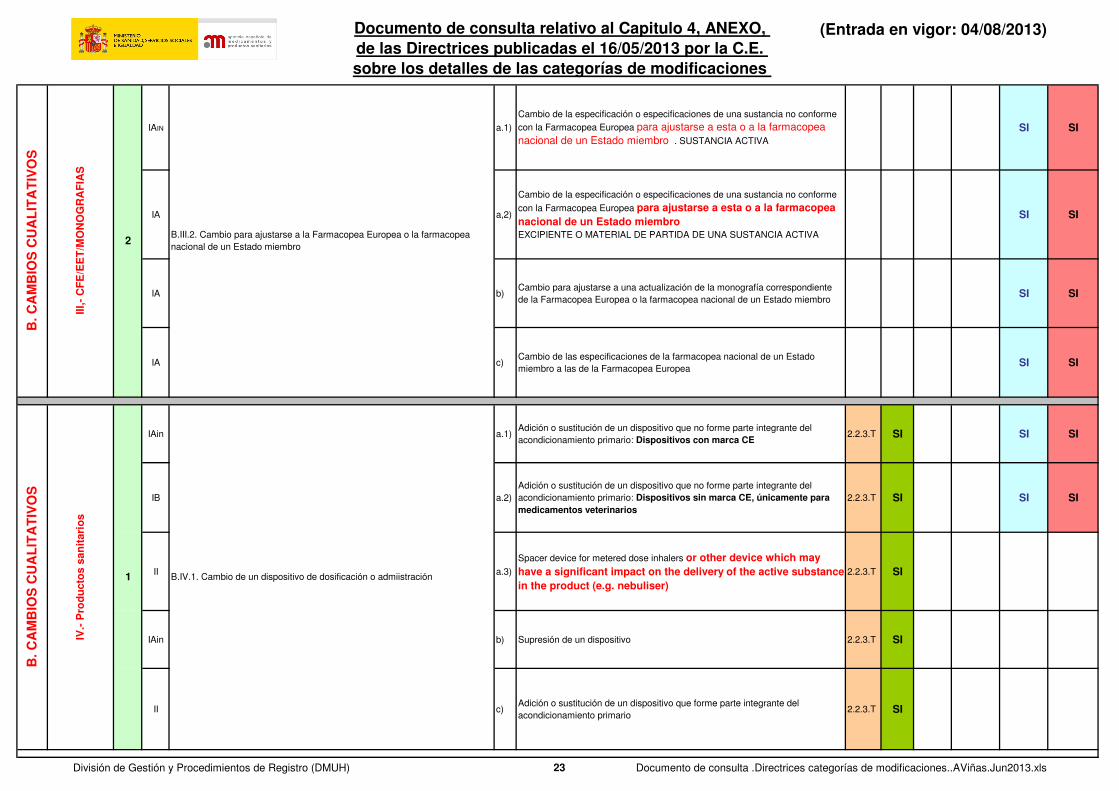
IAIN a.1)
Certificado de conformidad con la Farmacopea Europea para la monografía de
Farmacopea Europea correspondiente: Nuevo certificado de un fabricante ya
aprobado
2.5.3 SI
IA a.2)
Certificado de conformidad con la Farmacopea Europea para la monografía de
Farmacopea Europea correspondientec Certificado actualizado de un fabricante
ya aprobado
2.5.3
IA a.4)Supresión de certificados (en caso de que existan múltiples certificados por material)
2.5.3 SI SI
IB a.5)
Nuevo certificado para una sustancia activa no estéril que se pretenda utilizar en un medicamento estéril cuando se utilice agua en las últimas fases de la síntesis y no se indique que el material esté libre de endotoxinas
2.5.3 SI
IAin b.1)
Certificado de conformidad EET de la Farmacopea Europea para una sustancia
activa, material de partida, reactivo, producto intermedio o excipiente Nuevo
certificado para una sustancia activa de un nuevo fabricante o uno ya aprobado
2.6.2 SI SI
IA b.2)
Certificado de conformidad EET de la Farmacopea Europea para una sustancia
activa, material de partida,reactivo, producto intermedio o excipiente Nuevo
certificado para un material de partida, reactivo, producto intermedio o
excipiente de un nuevo fabricante o uno ya aprobado
2.6.2 SI SI
IA b.3)
Certificado de conformidad EET de la Farmacopea Europea para una sustancia
activa, material de partida, reactivo, producto intermedio o excipiente
Certificado actualizado de un fabricante ya aprobado
2.6.3 SI SI
IAb.4)
Supresión de certificados (en caso de que existan múltiples certificados por material)
2.6.4 SI SI
IIb.5)
Certificado nuevo o actualizado de un fabricante nuevo o ya aprobado que utilice material de origen humano o animal que exija la realización de una evaluación de riesgos con respecto a la contaminación potencial con agentes adventicios
2.6.5
a.3) 2.5.3 SIIAin
1
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
B.III.1. Presentación de un certificado de conformidad con la Farmacopea
Europea nuevo o actualizado o supresión del certificado de conformidad con la
Farmacopea Europea:
Para una sustancia activaPara un material de partida, reactivo o producto intermedio utilizado en el proceso de fabricación de la sustancia activaPara un excipiente
III,-
CF
E/E
ET
/MO
NO
GR
AF
IAS
Certificado de conformidad con la Farmacopea Europea para la monografía de
Farmacopea Europea correspondiente Nuevo certificado de un nuevo
fabricante (sustitución o adición)
División de Gestión y Procedimientos de Registro (DMUH) 22 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
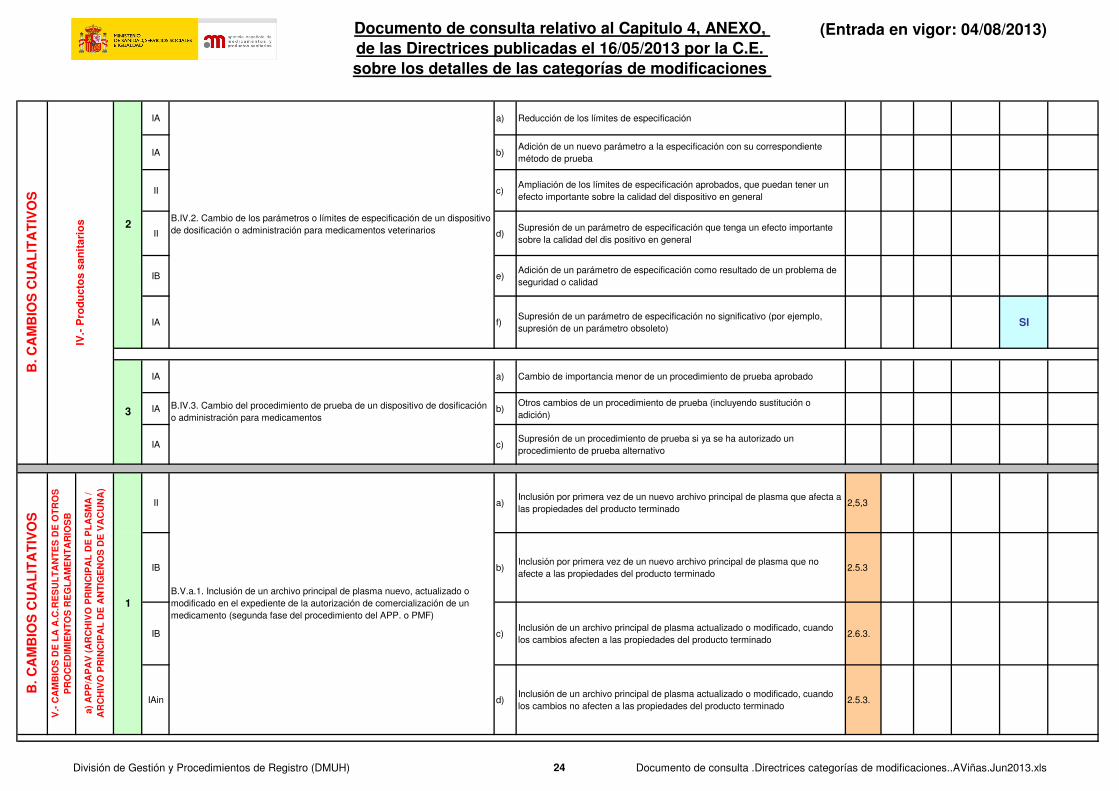
IAIN a.1)
Cambio de la especificación o especificaciones de una sustancia no conforme
con la Farmacopea Europea para ajustarse a esta o a la farmacopea
nacional de un Estado miembro . SUSTANCIA ACTIVA
SI SI
IA a,2)
Cambio de la especificación o especificaciones de una sustancia no conforme
con la Farmacopea Europea para ajustarse a esta o a la farmacopea nacional de un Estado miembroEXCIPIENTE O MATERIAL DE PARTIDA DE UNA SUSTANCIA ACTIVA
SI SI
IA b)Cambio para ajustarse a una actualización de la monografía correspondiente
de la Farmacopea Europea o la farmacopea nacional de un Estado miembroSI SI
IA c)Cambio de las especificaciones de la farmacopea nacional de un Estado
miembro a las de la Farmacopea EuropeaSI SI
IAin a.1)Adición o sustitución de un dispositivo que no forme parte integrante del
acondicionamiento primario: Dispositivos con marca CE2.2.3.T SI SI SI
IB a.2)
Adición o sustitución de un dispositivo que no forme parte integrante del
acondicionamiento primario: Dispositivos sin marca CE, únicamente para medicamentos veterinarios
2.2.3.T SI SI SI
II a.3)
Spacer device for metered dose inhalers or other device which may have a significant impact on the delivery of the active substance in the product (e.g. nebuliser)
2.2.3.T SI
IAin b) Supresión de un dispositivo 2.2.3.T SI
II c)Adición o sustitución de un dispositivo que forme parte integrante del
acondicionamiento primario 2.2.3.T SI
1 B.IV.1. Cambio de un dispositivo de dosificación o admiistración
2B.III.2. Cambio para ajustarse a la Farmacopea Europea o la farmacopea
nacional de un Estado miembro
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
IV.-
Pro
du
cto
s sa
nit
ario
s
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
III,-
CF
E/E
ET
/MO
NO
GR
AF
IAS
División de Gestión y Procedimientos de Registro (DMUH) 23 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IA a) Reducción de los límites de especificación
IA b)Adición de un nuevo parámetro a la especificación con su correspondiente
método de prueba
II c)Ampliación de los límites de especificación aprobados, que puedan tener un
efecto importante sobre la calidad del dispositivo en general
II d)Supresión de un parámetro de especificación que tenga un efecto importante
sobre la calidad del dis positivo en general
IB e)Adición de un parámetro de especificación como resultado de un problema de
seguridad o calidad
IA f)Supresión de un parámetro de especificación no significativo (por ejemplo,
supresión de un parámetro obsoleto)SI
IA a) Cambio de importancia menor de un procedimiento de prueba aprobado
IA b)Otros cambios de un procedimiento de prueba (incluyendo sustitución o
adición)
IA c)Supresión de un procedimiento de prueba si ya se ha autorizado un
procedimiento de prueba alternativo
II a)Inclusión por primera vez de un nuevo archivo principal de plasma que afecta a
las propiedades del producto terminado2,5,3
IB b)Inclusión por primera vez de un nuevo archivo principal de plasma que no
afecte a las propiedades del producto terminado2.5.3
IB c)Inclusión de un archivo principal de plasma actualizado o modificado, cuando
los cambios afecten a las propiedades del producto terminado2.6.3.
IAin d)Inclusión de un archivo principal de plasma actualizado o modificado, cuando
los cambios no afecten a las propiedades del producto terminado2.5.3.
1
IV.-
Pro
du
cto
s sa
nit
ario
s
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
B.IV.2. Cambio de los parámetros o límites de especificación de un dispositivo
de dosificación o administración para medicamentos veterinarios2
B.V.a.1. Inclusión de un archivo principal de plasma nuevo, actualizado o
modificado en el expediente de la autorización de comercialización de un
medicamento (segunda fase del procedimiento del APP. o PMF)
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
B.IV.3. Cambio del procedimiento de prueba de un dispositivo de dosificación
o administración para medicamentos3
V.-
CA
MB
IOS
DE
LA
A.C
.RE
SU
LT
AN
TE
S D
E O
TR
OS
P
RO
CE
DIM
IEN
TO
S R
EG
LA
ME
NT
AR
IOS
B
a) A
PP
/AP
AV
(A
RC
HIV
O P
RIN
CIP
AL
DE
PL
AS
MA
/ A
RC
HIV
O P
RIN
CIP
AL
DE
AN
TIG
EN
OS
DE
VA
CU
NA
)
División de Gestión y Procedimientos de Registro (DMUH) 24 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
II a) Inclusión por primera vez de un nuevo archivo principal de antígenos de vacuna 2.5.3. SI
IB b)
Inclusión de un archivo principal de antígenos de vacuna actualizado o
modificado, cuando los cambios afecten a las propiedades del producto
terminado
2.5.3. SI
IAin c)
Inclusión de un archivo principal de antígenos de vacuna actualizado o
modificado, cuando los cambios no afecten a las propiedades del producto
terminado
2.5.3. SI
IAin a) El cambio aplica los resultados deL arbitraje SI SI SI
II b)La armonización del expediente de calidad no era parte del referral y la
actualización tiene por finalidad armonizarloSI
IAIN a)Implementation of agreed change(s) for which no new additional data are submission by the MAH
II b)Implementation of change(s) which require to be further substantiated by new additional data to be submitted by the MAH
IB c)Aplicación e un cambio para un medicamento biológico o inmunológico
Co
mp
rob
ar:
Est
e su
bg
rup
o a
par
ecía
en
el u
ltim
o b
orr
ado
r y
aho
ra h
a d
esap
arec
ido
. Sin
em
bar
go
p
erm
anec
e aú
n e
n e
l ín
dic
e P
EN
DIE
NT
E D
E C
OM
FIR
MA
CIO
N
1B.V.b.1 Actualización del expediente de calidad destinado a aplicar el resultado de un procedimiento de consulta de la Unión
B. C
AM
BIO
S C
UA
LIT
AT
IVO
S
B.V.c.1 Update of the quality dossier requested by the Competent Authority outside a Union referral procedure
a) A
PP
/AP
AV
(A
RC
HIV
O P
RIN
CIP
AL
DE
PL
AS
MA
/ A
RC
HIV
O P
RIN
CIP
AL
DE
AN
TIG
EN
OS
DE
VA
CU
NA
)
2
B.V.a.2. Inclusión de un archivo principal de antígenos de vacuna nuevo,
actualizado o modificado en el expediente de la autorización de
comercialización de un medicamento (segunda fase del procedimiento del
APAV. O VAMF)
1V.-
CA
MB
IOS
DE
LA
A.C
.RE
SU
LT
AN
TE
S D
E O
TR
OS
PR
OC
ED
IMIE
NT
OS
RE
GL
AM
EN
TA
RIO
SB
c) O
ther
ch
ang
es t
o t
he
qu
alit
y d
oss
ier
req
ues
ted
by
the
Co
mp
eten
t A
uth
ori
tyb
) C
on
sult
a
División de Gestión y Procedimientos de Registro (DMUH) 25 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
IAin a) El medicamento está incluido en el ámbito definido en el procedimiento SI SI SI SI
IB b)
El medicamento no está incluido en el ámbito definido en el procedimiento pero el cambio o cambios aplican al resultado del procedimiento y el
titular de la autorización de comercialización no ha presentado nuevos datos
complementarios
SI SI SI
II c)
El medicamento no está incluido en el ámbito definido en el procedimiento, pero el cambio o cambios aplican el resultado de este
con nuevos datos complementarios presentados por el titular de la autorización
de comercialización
SI SI SI
IB a)
Aplicación del cambio o cambios para los cuales no es necesario que
el titular de la autorización de comercialización presente nuevos datos
complementarios
SI SI SI
II b)
Aplicación del cambio o cambios que deben sustanciarse mediante nuevos
datos complementarios presentados por el titular de la autorización de
comercialización (por ejemplo, comparabilidad)
SI SI
IAIN a)Aplicación de la redacción acordada por la autoridadcompetente
SI SI SI SI
II b)Aplicación del cambio o cambios que deben sustanciarsemediante nuevos datos complementarios presentados por eltitular de la autorización de comercialización
SI SI SI
1
SI SI SISISI
C.I.4 Cambio o cambios en el resumen de las características del producto, del etiquetado o del prospecto debido a la existencia de nuevas informaciones clínicas o preclínicas o referidas a la calidad o la farmacovigilancia.
2.5.4
Nota: Esta modificación no se aplica cuando la información nueva se presenta
con arreglo a la modificación C.I.13. En dichos casos, el cambio o los cambios
del resumen de características del producto, del etiquetado o del prospecto del
envase están cubiertos por la modificación C.I.13.
II
C.I.1 Cambio o cambios en el resumen de las características del producto, del etiquetado o del prospecto a fin de aplicar el resultado de un procedimiento de consulta a la Unión
4
3
I.- M
ED
ICA
ME
NT
OS
PA
RA
US
O H
UM
AN
O Y
ME
DIC
AM
EN
TO
S V
ET
ER
INA
RIO
S
C.I.2. Cambio del resumen de las características del producto, etiquetado o
prospecto de medicamentos genéricos,híbridos o biosimilares a raíz de la
evaluación del mismo cambio en el producto de referencia
2
C.I.3 Cambio o cambios en el resumen de las características del producto, del etiquetado o del prospecto de medicamentos de uso humano con fin de aplicar el resultado de un procedimiento relativo a los informes periódicos actualizados en materia de seguridad (PSUR) o los estudios de seguridad posteriores a la autorización (PASS), o el resultado de la evaluación realizada por la autoridad competente en virtud de los artículos 45 o 46 del Reglamento (CE) nº 1901/2006.
C. C
AM
BIO
S E
N S
EG
UR
IDA
D, E
FIC
AC
IA Y
FA
RM
AC
OV
IGIL
AN
CIA
División de Gestión y Procedimientos de Registro (DMUH) 26 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
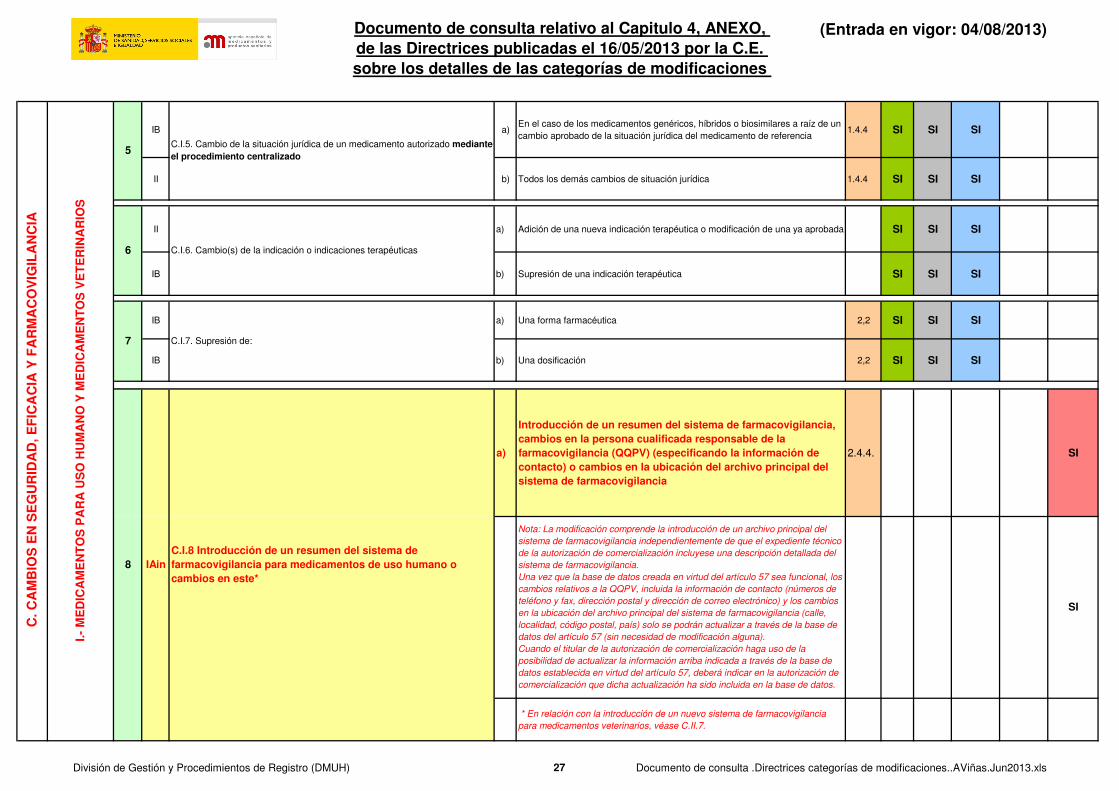
IB a)En el caso de los medicamentos genéricos, híbridos o biosimilares a raíz de un
cambio aprobado de la situación jurídica del medicamento de referencia1.4.4 SI SI SI
II b) Todos los demás cambios de situación jurídica 1.4.4 SI SI SI
II a) Adición de una nueva indicación terapéutica o modificación de una ya aprobada SI SI SI
IB b) Supresión de una indicación terapéutica SI SI SI
IB a) Una forma farmacéutica 2,2 SI SI SI
IB b) Una dosificación 2,2 SI SI SI
a)
Introducción de un resumen del sistema de farmacovigilancia, cambios en la persona cualificada responsable de la farmacovigilancia (QQPV) (especificando la información de contacto) o cambios en la ubicación del archivo principal del sistema de farmacovigilancia
2.4.4. SI
* En relación con la introducción de un nuevo sistema de farmacovigilancia
para medicamentos veterinarios, véase C.II.7.
7 C.I.7. Supresión de:
IAin8
SI
C. C
AM
BIO
S E
N S
EG
UR
IDA
D, E
FIC
AC
IA Y
FA
RM
AC
OV
IGIL
AN
CIA
5
I.- M
ED
ICA
ME
NT
OS
PA
RA
US
O H
UM
AN
O Y
ME
DIC
AM
EN
TO
S V
ET
ER
INA
RIO
S
C.I.5. Cambio de la situación jurídica de un medicamento autorizado mediante el procedimiento centralizado
C.I.6. Cambio(s) de la indicación o indicaciones terapéuticas6
C.I.8 Introducción de un resumen del sistema de farmacovigilancia para medicamentos de uso humano o cambios en este*
Nota: La modificación comprende la introducción de un archivo principal del
sistema de farmacovigilancia independientemente de que el expediente técnico
de la autorización de comercialización incluyese una descripción detallada del
sistema de farmacovigilancia.
Una vez que la base de datos creada en virtud del artículo 57 sea funcional, los
cambios relativos a la QQPV, incluida la información de contacto (números de
teléfono y fax, dirección postal y dirección de correo electrónico) y los cambios
en la ubicación del archivo principal del sistema de farmacovigilancia (calle,
localidad, código postal, país) solo se podrán actualizar a través de la base de
datos del artículo 57 (sin necesidad de modificación alguna).
Cuando el titular de la autorización de comercialización haga uso de la
posibilidad de actualizar la información arriba indicada a través de la base de
datos establecida en virtud del artículo 57, deberá indicar en la autorización de
comercialización que dicha actualización ha sido incluida en la base de datos.
División de Gestión y Procedimientos de Registro (DMUH) 27 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
9 IAin a)Cambio de la persona cualificada encargada de la farmacovigilancia (QPPV) o de su información de contacto o del procedimiento de suplencia
2.4.4. SI SI
IAin b)
Cambio o cambios de la base de datos de seguridad o de los principales acuerdos contractuales relacionados con el cumplimiento de las obligaciones en materia de farmacovigilancia o cambio del sitio que realiza actividades de farmacovigilancia
2.4.4. SI SI
IA c)
Otros cambios de la DDSF que no influyan en el funcionamiento del sistema de farmacovigilancia (por ejemplo, cambio del principal lugar de almacenamiento o archivo, cambios administrativos)
SI SI
IAin d)Cambios de una DDSF a raíz de la evaluación de la misma DDSF en relación con otro medicamento del mismo titular de la autorización de comercialización
SI SI
Nota: C.I.9 comprende los cambios de un sistema de farmacovigilancia
existente 1) para medicamentos veterinarios y 2) para medicamentos de uso
humano que todavía no hayan introducido un archivo principal del sistema de
farmacovigilancia.
Nota para a): Una vez que la base de datos creada en virtud del artículo 57
sea funcional, los cambios relativos a la QPPV, incluida la información de
contacto (números de teléfono y fax, dirección postal y dirección de correo
electrónico) solo se podrán actualizar a través de la base de datos del artículo
57 (sin necesidad de modificación alguna). Cuando el titular de la autorización
de comercialización haga uso de la posibilidad de actualizar esta información
través de la base de datos establecida en virtud del artículo 57, deberá indicar
en la autorización de comercialización que dicha actualización ha sido incluida
en la base de datos.
Nota para d): La evaluación de una DDSF presentada como parte de una nueva
10 IAinC.I.10 Cambio en la frecuencia o la fecha de presentación de los informes periódicos actualizados en materia de seguridad (PSUR) para medicamentos de uso humano
Nota: Esta modificación se aplicará solo cuando el ciclo del PSUR se
especifique en la autorización de comercialización a través de un medio que no
sea una referencia a la lista de las fechas de referencia de la Unión y cuando
sea preciso presentar el PSUR.
SI SI
I.- M
ED
ICA
ME
NT
OS
PA
RA
US
O H
UM
AN
O Y
ME
DIC
AM
EN
TO
S V
ET
ER
INA
RIO
S
C.I.9 Cambio o cambios de un sistema de farmacovigilancia existente
descrito en la descripción detallada del sistema de farmacovigilancia
(DDSF)
C. C
AM
BIO
S E
N S
EG
UR
IDA
D, E
FIC
AC
IA Y
FA
RM
AC
OV
IGIL
AN
CIA
División de Gestión y Procedimientos de Registro (DMUH) 28 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
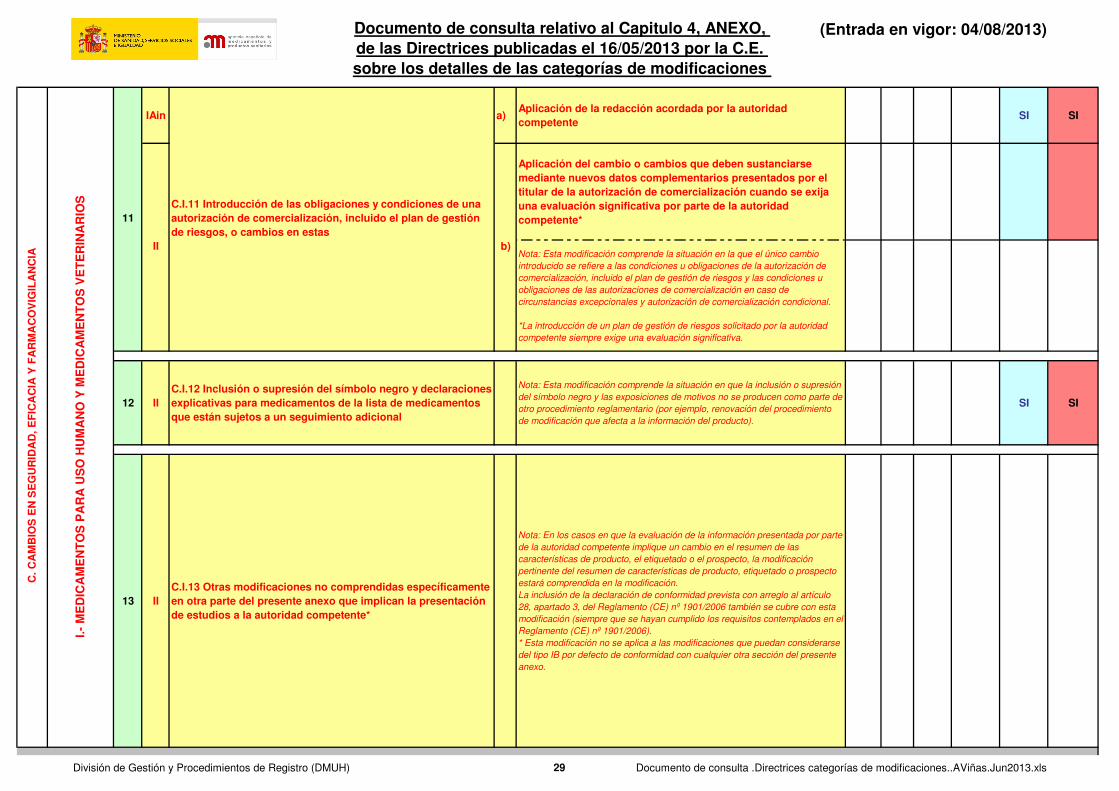
IAin a)Aplicación de la redacción acordada por la autoridad competente
SI SI
Aplicación del cambio o cambios que deben sustanciarse mediante nuevos datos complementarios presentados por el titular de la autorización de comercialización cuando se exija una evaluación significativa por parte de la autoridad competente*
Nota: Esta modificación comprende la situación en la que el único cambio
introducido se refiere a las condiciones u obligaciones de la autorización de
comercialización, incluido el plan de gestión de riesgos y las condiciones u
obligaciones de las autorizaciones de comercialización en caso de
circunstancias excepcionales y autorización de comercialización condicional.
*La introducción de un plan de gestión de riesgos solicitado por la autoridad
competente siempre exige una evaluación significativa.
12 IIC.I.12 Inclusión o supresión del símbolo negro y declaraciones explicativas para medicamentos de la lista de medicamentos que están sujetos a un seguimiento adicional
Nota: Esta modificación comprende la situación en que la inclusión o supresión
del símbolo negro y las exposiciones de motivos no se producen como parte de
otro procedimiento reglamentario (por ejemplo, renovación del procedimiento
de modificación que afecta a la información del producto).
SI SI
13 IIC.I.13 Otras modificaciones no comprendidas específicamente en otra parte del presente anexo que implican la presentación de estudios a la autoridad competente*
Nota: En los casos en que la evaluación de la información presentada por parte
de la autoridad competente implique un cambio en el resumen de las
características de producto, el etiquetado o el prospecto, la modificación
pertinente del resumen de características de producto, etiquetado o prospecto
estará comprendida en la modificación.
La inclusión de la declaración de conformidad prevista con arreglo al artículo
28, apartado 3, del Reglamento (CE) nº 1901/2006 también se cubre con esta
modificación (siempre que se hayan cumplido los requisitos contemplados en el
Reglamento (CE) nº 1901/2006).
* Esta modificación no se aplica a las modificaciones que puedan considerarse
del tipo IB por defecto de conformidad con cualquier otra sección del presente
anexo.
C.I.11 Introducción de las obligaciones y condiciones de una autorización de comercialización, incluido el plan de gestión de riesgos, o cambios en estas
II b)
11
I.- M
ED
ICA
ME
NT
OS
PA
RA
US
O H
UM
AN
O Y
ME
DIC
AM
EN
TO
S V
ET
ER
INA
RIO
S
C. C
AM
BIO
S E
N S
EG
UR
IDA
D, E
FIC
AC
IA Y
FA
RM
AC
OV
IGIL
AN
CIA
División de Gestión y Procedimientos de Registro (DMUH) 29 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
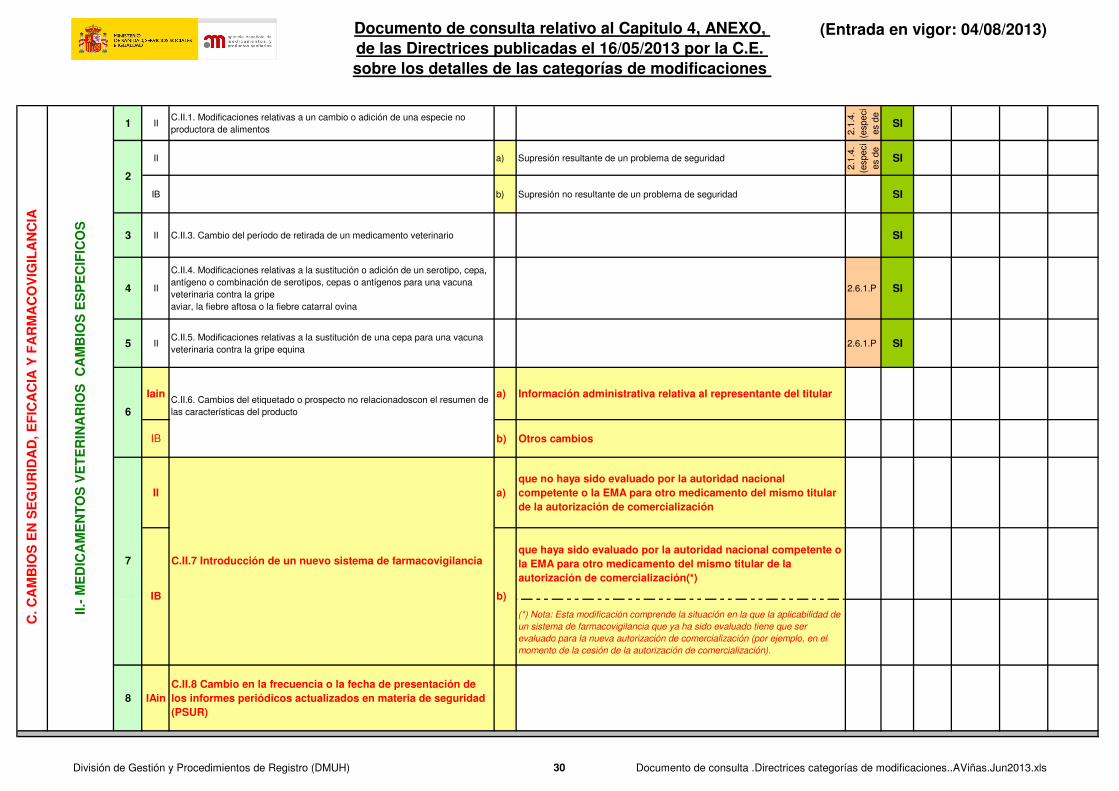
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
1 IIC.II.1. Modificaciones relativas a un cambio o adición de una especie no
productora de alimentos 2.1
.4.
(especi
es d
e
SI
II a) Supresión resultante de un problema de seguridad
2.1
.4.
(especi
es d
e
SI
IB b) Supresión no resultante de un problema de seguridad SI
3 II C.II.3. Cambio del período de retirada de un medicamento veterinario SI
4 II
C.II.4. Modificaciones relativas a la sustitución o adición de un serotipo, cepa,
antígeno o combinación de serotipos, cepas o antígenos para una vacuna
veterinaria contra la gripe
aviar, la fiebre aftosa o la fiebre catarral ovina
2.6.1.P SI
5 IIC.II.5. Modificaciones relativas a la sustitución de una cepa para una vacuna
veterinaria contra la gripe equina2.6.1.P SI
Iain a) Información administrativa relativa al representante del titular
IB b) Otros cambios
II a)que no haya sido evaluado por la autoridad nacional competente o la EMA para otro medicamento del mismo titular de la autorización de comercialización
que haya sido evaluado por la autoridad nacional competente o la EMA para otro medicamento del mismo titular de la autorización de comercialización(*)
(*) Nota: Esta modificación comprende la situación en la que la aplicabilidad de
un sistema de farmacovigilancia que ya ha sido evaluado tiene que ser
evaluado para la nueva autorización de comercialización (por ejemplo, en el
momento de la cesión de la autorización de comercialización).
8 IAinC.II.8 Cambio en la frecuencia o la fecha de presentación de los informes periódicos actualizados en materia de seguridad (PSUR)
II.-
ME
DIC
AM
EN
TO
S V
ET
ER
INA
RIO
S C
AM
BIO
S E
SP
EC
IFIC
OS
2
6C.II.6. Cambios del etiquetado o prospecto no relacionadoscon el resumen de
las características del producto
b)
C.II.7 Introducción de un nuevo sistema de farmacovigilancia
IB
C. C
AM
BIO
S E
N S
EG
UR
IDA
D, E
FIC
AC
IA Y
FA
RM
AC
OV
IGIL
AN
CIA
7
División de Gestión y Procedimientos de Registro (DMUH) 30 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls
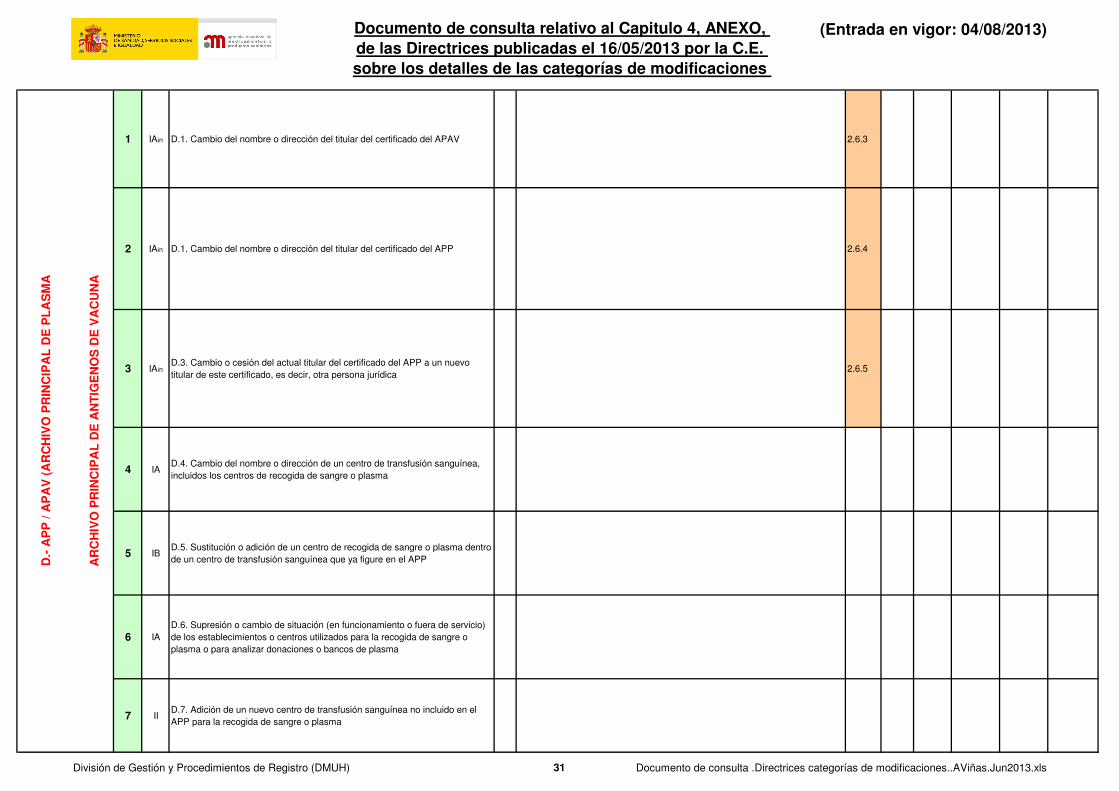
Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
1 IAin D.1. Cambio del nombre o dirección del titular del certificado del APAV 2.6.3
2 IAin D.1. Cambio del nombre o dirección del titular del certificado del APP 2.6.4
3 IAinD.3. Cambio o cesión del actual titular del certificado del APP a un nuevo
titular de este certificado, es decir, otra persona jurídica2.6.5
4 IAD.4. Cambio del nombre o dirección de un centro de transfusión sanguínea,
incluidos los centros de recogida de sangre o plasma
5 IBD.5. Sustitución o adición de un centro de recogida de sangre o plasma dentro
de un centro de transfusión sanguínea que ya figure en el APP
6 IA
D.6. Supresión o cambio de situación (en funcionamiento o fuera de servicio)
de los establecimientos o centros utilizados para la recogida de sangre o
plasma o para analizar donaciones o bancos de plasma
7 IID.7. Adición de un nuevo centro de transfusión sanguínea no incluido en el
APP para la recogida de sangre o plasma
AR
CH
IVO
PR
INC
IPA
L D
E A
NT
IGE
NO
S D
E V
AC
UN
A
D.-
AP
P /
AP
AV
(A
RC
HIV
O P
RIN
CIP
AL
DE
PL
AS
MA
División de Gestión y Procedimientos de Registro (DMUH) 31 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
8 IBD.8. Sustitución o adición de un centro de recogida de sangre para analizar
donaciones o bancos de plasma dentro de un centro que ya figure en el APP
9 IID.9. Adición de un nuevo centro de transfusión sanguínea no incluido en el
APP para analizar las donaciones o bancos
10 IBD.10. Sustitución o adición de un nuevo centro de transfusión sanguínea en el
que se almacene plasma
11 IAD.11. Supresión de un centro de transfusión sanguínea en el que se almacene
plasma
12 IBD.12. Sustitución o adición de una organización que interviene en el transporte
de plasma
13 IAD.13. Supresión de una organización que interviene en el transporte de
plasma
14 IAD.14. Adición de un kit de prueba provisto de la marca CE para analizar
donaciones, cuando sea nuevo o una sustitución de uno ya existente
D.-
AP
P /
AP
AV
(A
RC
HIV
O P
RIN
CIP
AL
DE
PL
AS
MA
AR
CH
IVO
PR
INC
IPA
L D
E A
NT
IGE
NO
S D
E V
AC
UN
A
División de Gestión y Procedimientos de Registro (DMUH) 32 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
II a)El nuevo kit no ha sido aprobado previamente en el APP para analizar
donaciones en un centro de transfusión sanguínea
IA b)El nuevo kit ha sido aprobado previamente en el APP para analizar donaciones
en un centro de transfusión sanguínea
16 IID.16. Cambio del kit o método utilizado para analizar bancos (análisis de
anticuerpos o antígenos, o pruebas de NAT)
17 IAD.17. Introducción o extensión de un procedimiento para el período de
retención
18 IB D.18. Retirada de un período de retención o reducción de su duración
IA a) Los nuevos contenedores de sangre llevan la marca CE
II b) Los nuevos contenedores de sangre no llevan la marca CE
IA a) Condiciones de almacenamiento o transporte
IA b) Período máximo de almacenamiento del plasma
D.-
AP
P /
AP
AV
(A
RC
HIV
O P
RIN
CIP
AL
DE
PL
AS
MA
D.15. Adición de un kit de prueba no provisto de la marca CE para analizar
donaciones, cuando sea nuevo o una sustitución de uno ya existente
D.20. Cambio de almacenamiento o transporte20
15
19D.19. Sustitución o adición de contenedores de sangre (por ejemplo, bolsas,
botellas)
AR
CH
IVO
PR
INC
IPA
L D
E A
NT
IGE
NO
S D
E V
AC
UN
A
División de Gestión y Procedimientos de Registro (DMUH) 33 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
21 IID.21. Introducción de una prueba para los marcadores virales, cuando esta
introducción tenga efectos importantes para la evaluación del riesgo viral
22 IB
D.22. Cambio de la preparación del banco de plasma (por ejemplo, método de
fabricación, tamaño del banco, almacenamiento de las muestras del banco de
plasma)
23 II
D.23. Cambio de las medidas que se adoptarían si se descubriera
posteriormente que algunas donaciones deberían haberse excluido del
procesamiento (procedimiento retrospectivo)
D.-
AP
P /
AP
AV
(A
RC
HIV
O P
RIN
CIP
AL
DE
PL
AS
MA
AR
CH
IVO
PR
INC
IPA
L D
E A
NT
IGE
NO
S D
E V
AC
UN
A
División de Gestión y Procedimientos de Registro (DMUH) 34 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls

Documento de consulta relativo al Capitulo 4, ANEXO, de las Directrices publicadas el 16/05/2013 por la C.E. sobre los detalles de las categorías de modificaciones
(Entrada en vigor: 04/08/2013)
Se incluyen cambios en condiciones a cumplir
Se incluyen cambios documentación a aportar
Capítulos del anexo (A, B, C o D)
Tipo IA, IAin, IB o II
Celdas en fondo amarillo y texto en rojo:
Celdas con alguna parte del texto en rojo:
Se notifica a D.G. de Cartera Básica (SNS y Farmacia)
SUBTIPO
El solicitante deberá incorporar los cambios en los puntos de Raefar afectados
Ptos de Raefar
Modifica FT, P. y/o MA
Se envía FT al Titular de la Autorización de comercialización junto a la resolución
INTERPRETACIÓN DEL CONTENIDO
OBJETIVO DEL CAMBIO
TIPO
DEFINICIÓN Lista de los distintos tipos de modificaciones
En las nuevas Directrices se modifica parte o totalidad del texto relativo a "Condiciones que deben cumplirse"
En las nuevas Directrices se modifica parte o totalidad del texto relativo a "Documentación que debe presentarse"
NUEVA Variación
a), b), c),….
Cambia alguna parte de la redacción del texto
Se modifican materiales en el momento de la resolución, pero no se envían a los titulares. Estos pueden consultar la información en la ultima versión en CIMA (ej. Condiciones de conservación)
Se modifican materiales y se envía la resolución junto a FT, P y MA modificados (ej. nueva indicación)
Se notifica a la DG de Cartera básica (SNS y Farmacia) / (precios)
Si no se indica ningún punto , el solicitante deberá incorporar los cambios en las cajas de texto definidas como SITUACIÓN ACTUAL Y SITUACIÓN PROPUESTA
División de Gestión y Procedimientos de Registro (DMUH) 35 Documento de consulta .Directrices categorías de modificaciones..AViñas.Jun2013.xls







![es.unesco.org · Web viewLos futuros de la educación: Aprender a transformarse [NUEVAS] DIRECTRICES DE CONSULTA PARA GRUPOS DE DISCUSIÓN DE PARTES INTERESADAS (Julio 2020](https://cdn.vdocuments.mx/doc/165x107/5fdef7ffa39bda3c9e2dbb4b/es-web-view-los-futuros-de-la-educacin-aprender-a-transformarse-nuevas-directrices.jpg)