
UNIVERSITÀ DEGLI STUDI DI TORINO
Dipartimento di Biotecnologie Molecolari e Scienze per la Salute
SDSB – Struttura Didattica Speciale di Biotecnologie
Corso di Laurea in Biotecnologie
Tesi di Laurea di I livello
Mitochondrial Dynamism and Mitophagy in the Heart
Candidato: Relatore: Alessandra Murabito Prof.ssa Mara Brancaccio
a.a. 2014/2015

Index
Abstract....................................................................................................................3
1. Introduction..........................................................................................................4
1.1 Mitochondrial Fission....................................................................................4
1.2 Mitochondrial Fusion....................................................................................6
2. The Heart: an Unexpected Exception...................................................................7
3. How Mitochondrial Dynamism Affects Mitochondrial Quality Control.............9
4. PINK1-Parkin-Mediated Mitophagy Signaling Pathway...................................11
5. Mitochondrial Dynamism - Mitophagy Crosstalk in the Heart.........................18
6. Conclusions........................................................................................................22
Bibliography...........................................................................................................23
2

Abstract
Mitochondria are highly dynamic organelles undergoing fission, fusion and
mitophagy processes. In many cell types mitochondria form an interconnected
reticular network and dynamism is frequent, perhaps continuous, in such cells. An
exception though is present in the heart. Recent results in vivo cardiomyocyte-
specific genetic manipulation have revealed that mitochondrial dynamic factors
function in cardiomyocytes in different ways. In fact, in this cell-type there is a
partial absence of mitochondrial network and a slow mitochondrial turnover.
Nevertheless, the mitochondrial fusion proteins mitofusins (Mfn)1 and 2 and optic
atrophy (Opa)1, and the mitochondrial fission protein dynamin related protein
(Drp)1 are highly expressed in mammalian heart, wherein genetic ablation
provokes dramatic cardiac dysfunction. All these studies suggest that fission and
fusion have an important role in cardiac mitochondrial quality control. In this
work I will describe in more detail a major regulator of these processes, the
PTEN-induced putative kinase1-Parking mitophagy pathway.
3

1. Introduction
It has become axiomatic that mitochondria constantly undergo fission, fusion and
migration, collectively termed “mitochondrial dynamism”, within the cells.
Mitochondrial networks are constantly remodeling, with sections of the network
periodically breaking away and then re-estabilishing new interconnection
elsewhere. Relative frequency of fission and fusion has also a determinant role in
mitochondrial morphology. When fusion and fission are balanced, mitochondrial
aspect ratio (length/width) is stable. However, when the balance shifts in favor of
more fusion, mitochondrial elongation and network connectivity increase. On the
other hand, when an imbalance produces less fusion, this will lead to an
accumulation of smaller, shorter, fragmented organelles2. Mitochondrial
morphometry seems especially plastic during cell mitosis. In fact network
disassembly not only facilitates cytokinesis in growing and dividing cells, but also
helps to deliver equal proportions of the parental cell mitochondrial pool to each
daughter cell. In these cells, fission is an essential process that permits to populate
them with adequate numbers of mitochondria1-5.
1.1 Mitochondrial Fission
In order to replicate and expand the cellular mitochondrial pool, mitochondria
undergo fission. Through this process, from one mitochondrion, two daughters are
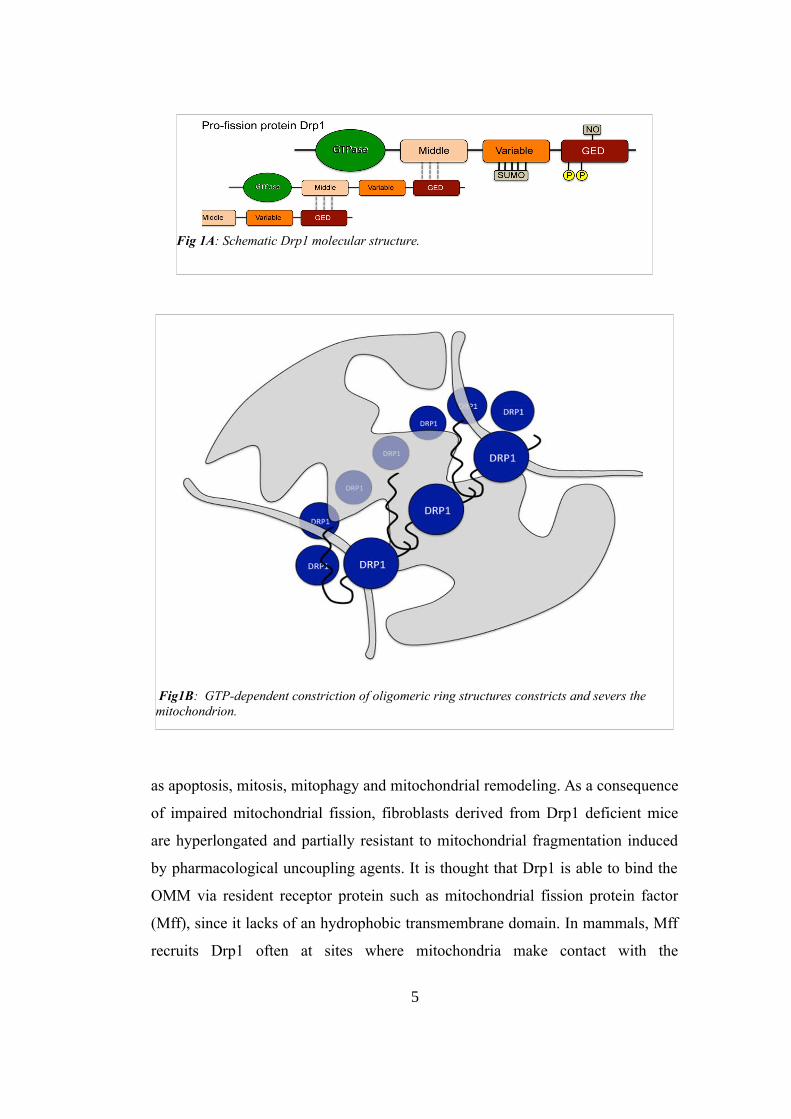
produced. Drp1 has a key role in this mitochondrial dynamic: it is a GTPase with
an amino terminal GTPase domain, a middle domain and a carboxyl terminal
GTPase effector domain (Fig 1A). This protein has to be recruited to the outer
mitochondrial membrane (OMM) to promote fission because in normal condition
is present in the cytosol. But how Drp1 is able to promote fission? After the
localization to the OMM, the GED domain (GTPase effector domain) interacts
with the middle domain of adjacent molecules to promote head-to-toe
oligomerization (Fig 1B). Following the GTPase-mediated constriction, Drp1
ligates and separates the IMM (inner mitochondrial membrane) and the OMM.
There are multiple events that stimulate Drp1 translocation form the cytosol, such
4
as apoptosis, mitosis, mitophagy and mitochondrial remodeling. As a consequence
of impaired mitochondrial fission, fibroblasts derived from Drp1 deficient mice
are hyperlongated and partially resistant to mitochondrial fragmentation induced
by pharmacological uncoupling agents. It is thought that Drp1 is able to bind the
OMM via resident receptor protein such as mitochondrial fission protein factor
(Mff), since it lacks of an hydrophobic transmembrane domain. In mammals, Mff
recruits Drp1 often at sites where mitochondria make contact with the
5
Fig 1A: Schematic Drp1 molecular structure.
Fig1B: GTP-dependent constriction of oligomeric ring structures constricts and severs the mitochondrion.

endoplasmic reticulum (ER)1. Therefore, thanks to this process cells are able to
maintain an adequate pool of mitochondria and to modify their structure before
mitosis. This is called symmetrical fission and produces from one mitochondrion
two equal daughters. Mitochondrial networks subsequently have to be re-
estabilished in each daughter cell via generalized organelle fusion.
1.2 Mitochondrial Fusion
The process of fusion is more complicated then the fission one. A complication is
compartmentalization of mitochondria that are composed by a central matrix,
enclosed by the cristae/inner mitochondrial membrane (IMM), which is separated
from the outer membrane (OMM) by intermembrane space. The integrity of the
compartments has to be maintained through mitochondrial fusion which thus
occurs layer by layer. The first step involves physical tethering of two
mitochondria via trans-interaction between the carboxyl terminal domains of
OMM mitofusins of two organelles. In fact, Mfn 1 and 2 are both GTPases like
Drp1 and both isoforms have a cytosolic N terminal GTPase domain, a
hydrophobic transmembrane domain and two cytosolic hydrophobic heptad
repeated coiled-coiled domains (HR1 and HR2). Consequently, Mitofusins insert
into the OMM with the majority of the protein being exposed to the cytosol and
available to interact with cytosolic factors. This position allows them to
participate in information exchange between the mitochondrion and its host cell.
Since Mfn1 and Mfn2 are quite similar, the interaction between mitofusins of
different organelles can occur in either a homotypic or heterotypic manner, with
the heterotypic shown to be more efficient and to yield a higher rate of successful
fusion events (Fig 2). Mitofusin-mediated physical tethering of mitochondria is
GTPase-independent and fully reversible, whereas GTP hydrolysis is essential to
irreversible OMM fusion. OMM fusion maintains the structural integrity of the
IMM and matrix, thus avoiding ROS formation. After the OMM fusion, IMM
fusion can occur. A key factor in this process is Opa1, also a GTP-ase protein.
With the loss of Opa1, mitochondrial tethering and OMM fusion still occur
(through the actions of Mfn1 and Mfn2), but absence of Opa1- mediated IMM
6
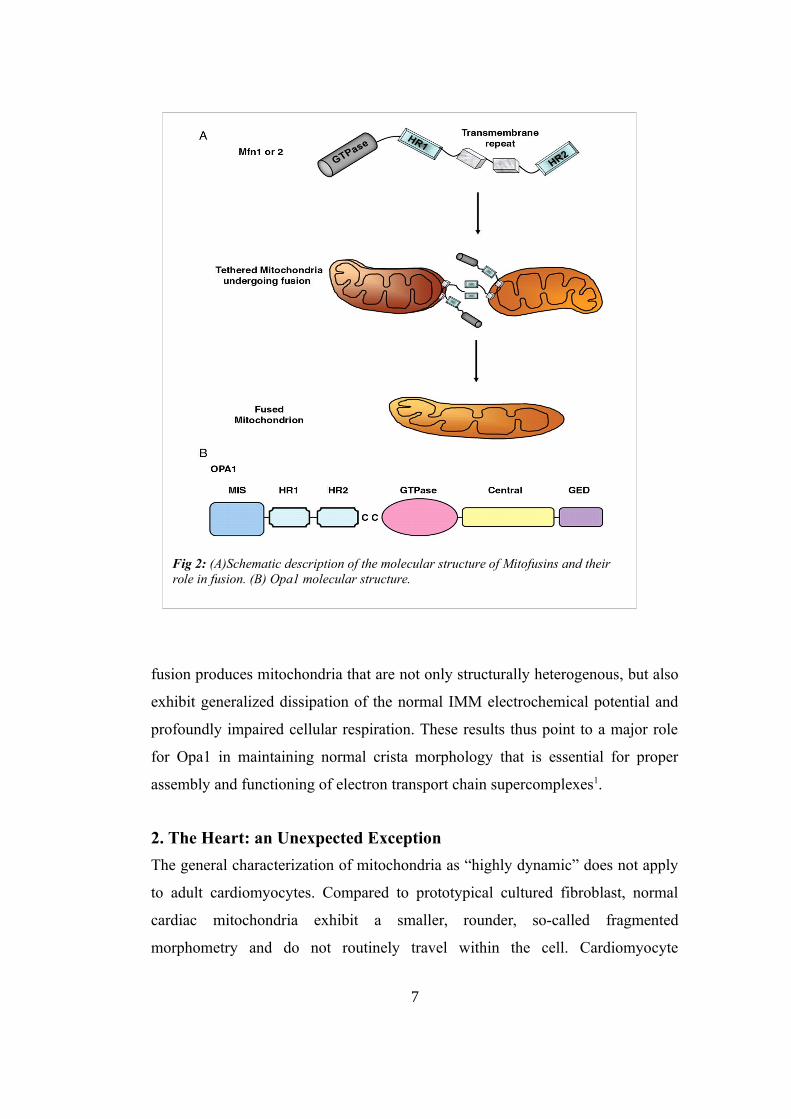
Fig 2: (A)Schematic description of the molecular structure of Mitofusins and their role in fusion. (B) Opa1 molecular structure.
fusion produces mitochondria that are not only structurally heterogenous, but also
exhibit generalized dissipation of the normal IMM electrochemical potential and
profoundly impaired cellular respiration. These results thus point to a major role
for Opa1 in maintaining normal crista morphology that is essential for proper
assembly and functioning of electron transport chain supercomplexes1.
2. The Heart: an Unexpected Exception
The general characterization of mitochondria as “highly dynamic” does not apply
to adult cardiomyocytes. Compared to prototypical cultured fibroblast, normal
cardiac mitochondria exhibit a smaller, rounder, so-called fragmented
morphometry and do not routinely travel within the cell. Cardiomyocyte
7

mitochondria appear static and no networks are observed. It appears from
ultrastructural examination of myocardium that individual cardiomyocyte
mitochondria exist as relatively short organelles, collectively grouped together
between myofibrillar elements in lanes that extend parallel to the long axis of the
cell. Even in the perinuclear region of cardiomyocytes, where mitochondria tend
to be clustered in large numbers, filamentous organelles and interconnected
networks are not observed (Fig 3).
The explanation might be that intrinsically fragmented mitochondria confer a
biomechanical advantage to striated muscle, because interconnected organelles
would constitute an internal resistance element, detrimental to myocyte
contraction. Finally, cardiomyocyte mitochondria do not undergo fusion and
fission at a rate that can be directly measured. For this reason, the widely accepted
notion that mitochondrial fragmentation is detrimental to mitochondrial and
cellular functioning likely does not apply to adult cardiomyocytes. Indeed, even
lacking interconnected mitochondrial networks, these cells have both the greatest
mitochondrial density and the highest respiratory capacity of any mammalian cell.
The maintenance of mitochondrial fitness is clearly a physiological imperative for
this organ. This thus suggests that mitochondrial dynamism plays different roles
according to the biological context2.
8
Fig 3: Murine fibroblasts and human induced pluripotent stem cells (hiPSC)-derived cells costained with anti-TOM20(green) and anti-cardiac Troponin T(cTnT,red), and mounted in DAPI. In the image of hiPSC-derived cells, the cTnT-positive cell is a cardiomyocyte with mostly fragmented mitochondria, and the cTnT-negative cell is a fibroblast with filamentous mitochondria. Mouse cardiomyocytes were isolated from an adult mouse heart expressing a mitochondrial-targeted fluorescent green marker. The picture on the left is a transmission electron microscopy image of adult mouse heart.

3. How Mitochondrial Dynamism Affects Mitochondrial QualityControl
The life cycle of mitochondria resembles the one of primordial bacteria from
which they are descendant. Mitochondria replicate, as it said before, through
symmetrical fission: a healthy parent organelle produces 2 healthy daughters that
grow by adding new components generated by mitochondrial biogenesis and by
fusing with other healthy mitochondria. But over time or as a consequence of a
stress, there are two possibilities: as long as the stress is below to a critical
threshold, the mitochondrion can go under fusion in order to mitigate the effects
of environmental damage through the exchange of proteins and lipids with other
mitochondria. If the damage is too severe for correction through biogenic or
fusion-mediated repair, the mitochondrion is removed via mitophagy. Why this
mitophagic quality control is so important? This is due to the fact that damaged
mitochondria become cytotoxic factories producing ROS. Mitochondrial ATP
synthesis depends on an electrochemical gradient generated across the inner
mitochondrial membrane through a series of redox reactions: the transfer of
electrons between complexes of the electron transport chain is coupled to
extrusion of protons (hydrogen ions) across the inner mitochondrial membrane
and into the mitochondrial intermembrane space. Reversal of this proton flow
powers the mitochondrial ATP synthase, a molecular rotor. The terminal electron
acceptor is molecular oxygen, forming superoxide anion that is normally reduced
through the sequential actions of superoxide dismutase and catalase (to form
water). When electrons prematurely leak from complexes I or III of the electron
transport chain, or when specific endogenous mitochondrial enzymes such as
Romo1 (reactive oxygen species modulator 1) are activated, damaging superoxide
radicals and hydrogen peroxide can accumulate within or escape from
mitochondria and attack critical proteins, lipids, and mitochondrial or nuclear
DNA. Because production of ROS in damaging amounts does not normally occur,
substantial ROS production is a marker of mitochondrial dysfunction. In order to
protect themselves from ROS, cells use a sophisticated surveillance and
elimination mechanism to identify and remove dysfunctional mitochondria. The
9

cellular decision to remove a dysfunctional mitochondrion is necessarily
dichotomous. Either the organelle is retained or it is targeted for autophagy. If
only severely damaged mitochondria are culled by mitophagy, the cell may have
to face toxicity deriving from damaged organelles that have not achieved high
threshold of dysfunction necessary to trigger their elimination. However if the cell
lowers its threshold for mitochondrial removal, this will lead to elimination of
still functional organelles, thus depriving the cell of ATP. Nature has addressed
this quandary by integrating mitochondrial dynamism with mitophagy in the form
of asymmetrical mitochondrial fission. It has been shown that mitochondria that
are targeted for mitophagy have a relatively depolarized membrane potential
before being targeted for mitophagy. This subpopulation also rarely is involved in
fusion and fission processes as mitofusins and Opa 1 are either cleaved or
degraded. This process is induced by either inner membrane depolarization or
reduced mitochondrial ATP production. Thus, mitochondria that are expected to
be eliminated through mitophagy are characterized by being relatively depolarized
and remaining solitary. The use of a photoactivable green fluorescent protein, that
was targeted to the matrix of mitochondria and photoactivated by a laser, showed
the existence of an asymmetrical fission event. This process produces 1 healthy
daughter, with a normal hyperpolarized potential, and 1 daughter that can have
depolarized membrane potential that may recover or persist. The latter one, who
has less chances to being involved in fusion events, will become part of the
preautophagic pool (Fig 4). Evidence supports that asymmetrical fission is the
result of uneven distribution of dysfunctional mitochondrial components,
including oxidized or older proteins. This model explains the role of fusion in
mitochondria quality control as it may allow for the redistribution of damaged
components while fission and mitophagy are responsible for the elimination. The
selectivity of the fusion process for the healthy daughter mitochondrion is
therefore key to the efficiency of the quality control. Selectivity depends on the
transition of dysfunctional daughter mitochondria from fusion-competent to
permanently solitary status. This process may be mediated by coupling the
degradation or post-translational modification of fusion proteins to the
10
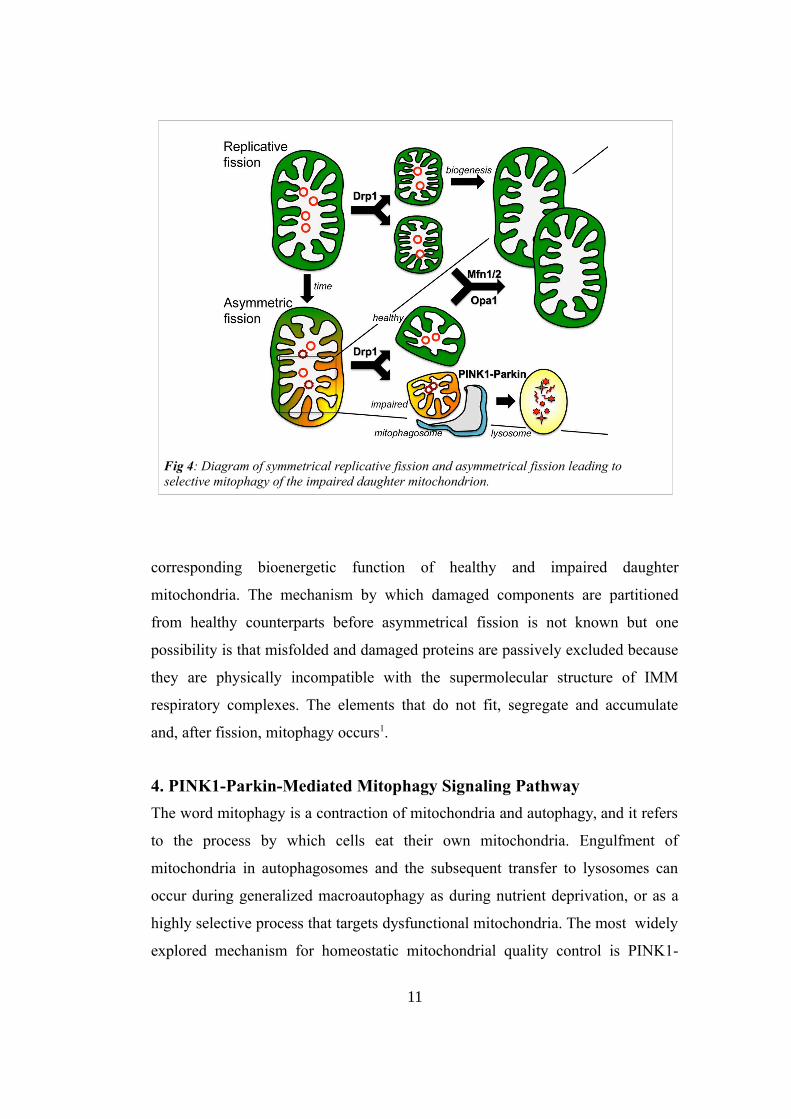
Fig 4: Diagram of symmetrical replicative fission and asymmetrical fission leading to selective mitophagy of the impaired daughter mitochondrion.
corresponding bioenergetic function of healthy and impaired daughter
mitochondria. The mechanism by which damaged components are partitioned
from healthy counterparts before asymmetrical fission is not known but one
possibility is that misfolded and damaged proteins are passively excluded because
they are physically incompatible with the supermolecular structure of IMM
respiratory complexes. The elements that do not fit, segregate and accumulate
and, after fission, mitophagy occurs1.
4. PINK1-Parkin-Mediated Mitophagy Signaling Pathway
The word mitophagy is a contraction of mitochondria and autophagy, and it refers
to the process by which cells eat their own mitochondria. Engulfment of
mitochondria in autophagosomes and the subsequent transfer to lysosomes can
occur during generalized macroautophagy as during nutrient deprivation, or as a
highly selective process that targets dysfunctional mitochondria. The most widely
explored mechanism for homeostatic mitochondrial quality control is PINK1-
11

Parkin-mediated mitophagy. One of the key proteins in this pathway is PINK1
(PTEN induced putative kinase 1). PINK1 kinase is encoded by the PINK1 gene,
which like all but 13 proteins encoded by mitochondrial DNA, is part of the
nuclear genome. For this reason, after the translocation by the ribosomal
apparatus, PINK1 has to be imported into mitochondria. In normal condition, as
fast as PINK1 is imported, it is proteolytically degraded. So healthy mitochondria
have little PINK1 protein or PINK1 kinase activity. However, if ROS,
mitochondrial depolarization, or protein misfolding occur, PINK1 senses these
inputs and initiates the appropriate response. PINK1 degradation process is
interrupted, causing its accumulation and the consequent PINK1-mediated
phosphorylation of its substrates. Conceptually, healthy mitochondria make an
effort to degrade PINK1, thus staying alive by avoiding mitophagic destruction. In
damaged mitochondria that no longer support PINK1 degradation, passive
accumulation of PINK1 triggers the mitophagic destruction of the organelles.
Unprocessed 63kDa PINK1 protein is transported across the OMM translocase
complex of the OMM, delivered to the IMM by translocase of the IMM, and
proteolytically processed in a manner typical for imported mitochondrial matrix
proteins. In healthy mitochondria, PINK1 is rapidly cleaved by presentilin-
associated rhomboid like protein in the matrix, generating a 52 kDa PINK1
fragment that escapes into the cytosol and undergoes proteosomal degradation. If
mitochondria are damaged, translocase of the IMM is not functional, protecting
the kinase from translocation. The physical association between PINK1 and the
OMM is also maintained. From this location, PINK1 phosphorylates available
substrates, such as ubiquitins, and also inhibits fusion of the damaged
mitochondrion. Among PINK1 substrates is Parkin that is another major player of
mitophagy. There are different views about the biochemical events that induce
mitochondrial Parkin localization and that interrupt mitochondrial fusion. The
importance of this mechanism in mitochondrial quality control suggests a
signaling redundancy and that multiple pathways are involved. PINK1
phosphorylates Parkin on Ser65, and Parkin polyubiquitinates mitofusins. A
former hypothesis suggested a key role of PINK1-mediated phosphorylation in
12

Parkin recruitment to damaged mitochondria. However, despite PINK1 does
phosphorylate Parkin, recent studies have revealed that PINK1-phosphorylated
ubiquitin, and not PINK1- mediated phosphorylation of Parkin itself, is essential
to enable Parkin to work as an E3 ubiquitin ligase. The observation that PINK1
can phosphorylate ubiquitin complexes on OMM proteins, and not just free
ubiquitin, has suggested that nonspecific anchoring of Parkin to phospho-
ubiquitinated OMM proteins is a mechanism for its PINK1-mediated recruitment
to mitochondria. This is consistent with the idea that Parkin binding to phospho-
ubiquitin can accelerate Parkin-mediated OMM protein ubiquitination as a
positive feedback amplification loop. However, it is unlikely that the primary
event for Parkin recruitment relies on pre-existing ubiquitinated OMM proteins1.
Two candidates that may have a crucial role in the recruitment of Parkin are
mitofusins (Mfn1 and 2). These two mitochondrial outermembrane fusion proteins
are Parkin ubiquitination substrates. Because mitophagy is stimulated by Parkin-
mediated ubiquitination of mitochondrial proteins, one possibility is that Mfn1
and/or Mfn2 are mediators of the PINK1-Parkin signaling pathway. Although
ubiquitination can explain a role of mitofusins downstream Parkin, the upstream
signaling has long remained unknown. In order to evaluate if and which
mitofusins are involved in this process, human embryonic kidneys (HEK) cells
were transfected with tagged mitofusins and Parkin. Immunoprecipitation of
Parkin showed an association with Mfn2, but not Mfn1, and this co-
immunoprecipitation was enhanced in cells also overexpressing Parkin. (Fig 5
A,B). The oxidative phosphorylation inhibitor carbonyl cyanide 4-
(trifluoromethoxy) phenylhydrazone (FCCP) depolarizes mitochondria and
stimulates PINK1-mediated translocation of Parkin to mitochondria, thus targeting
damaged organelles for mitophagy. Consistent with a role for Mfn2 in this
process, FCCP treatment stimulated Parkin binding to endogenous fibroblast
Mfn2. Overexpression of PINK1 enhanced the effect of FCCP, instead its
inhibition mediated by RNA interference lowered it. Cardiomyocytes naturally
express Mfn2 and Parkin in large amount and, because of their high endogenous
expression of these factors, no transfection is needed.
13

These cells were treated with FCCP and only in the Mfn1-null cells Parkin
translocation was stimulated. In Mfn2 null cardiomyocytes Parkin was not
distributed in punctae near mitochondrial-rich perinuclear region but remained
diffusely cytosolic (Fig 5C), demonstrating a requirement for endogenous Mfn2 to
promote Parkin localization to depolarized mitochondria. Parkin recruitment
appears to be a unique property of Mfn2. It is also known that Mfn2 is a Parkin
ubiquitination substrate. This might require PINK1-stimulated association of
Parkin and Mfn2 with mitochondria. A complete concordance of ubiquitination
14
Fig 5: Interaction of Mfn2 with Parkin in a PINK1-dependent manner and its requirement forParkin translocation to depolarized mitochondria. (A and B) Fibroblasts were transfected with Flag-Mfn1 (A) or Mfn2 (B), PINK1, and/or hemagglutinin (HA) Parkin; immunoprecipitated (IP) with anti-Flag; and immunoblotted (IB). Right-hand gels show IB ofinput homogenates. (C) Subcellular Parkin redistribution (green) induced by mitochondrial depolarization with FCCP in wild-type, Mfn1-deficient, and Mfn2-deficient mouse cardiomyocytes. KO, knockout; DAPI, 4 ,6-diamidino- 2-phenylindole.

with PINK1-stimulated Mfn2 binding to Parkin in HEK cells and with FCCP-
stimulated binding of endogenous cardiac Mfn2 to Parkin in isolated perfuse
mouse hearts has been observed, demonstrating the importance of PINK1-Mfn2-
Parkin interactions on ubiquitination. Conversely, Mfn2 (but not Mfn1) gene
ablation in cardiomyocytes decreased mitochondrial ubiquitination stimulated by
mitochondrial depolarization, following the same pattern seen for Parkin
translocation. Moreover, the mitophagic response, measured as punctal
accumulation of the mitophagy adaptor protein p62 [also called sequestosome 1
(SQSTM1)], was impaired in FCCP-treated Mfn2- deficient cardiac myocytes.
Together, these data support a model in which Mfn2 functions as a receptor to
which cytosolic Parkin binds on depolarized mitochondria, provoking
ubiquitination of mitochondrial proteins that target the organelle for autophagic
elimination. Enhancement of Mfn2- Parkin association by PINK1 also reveals
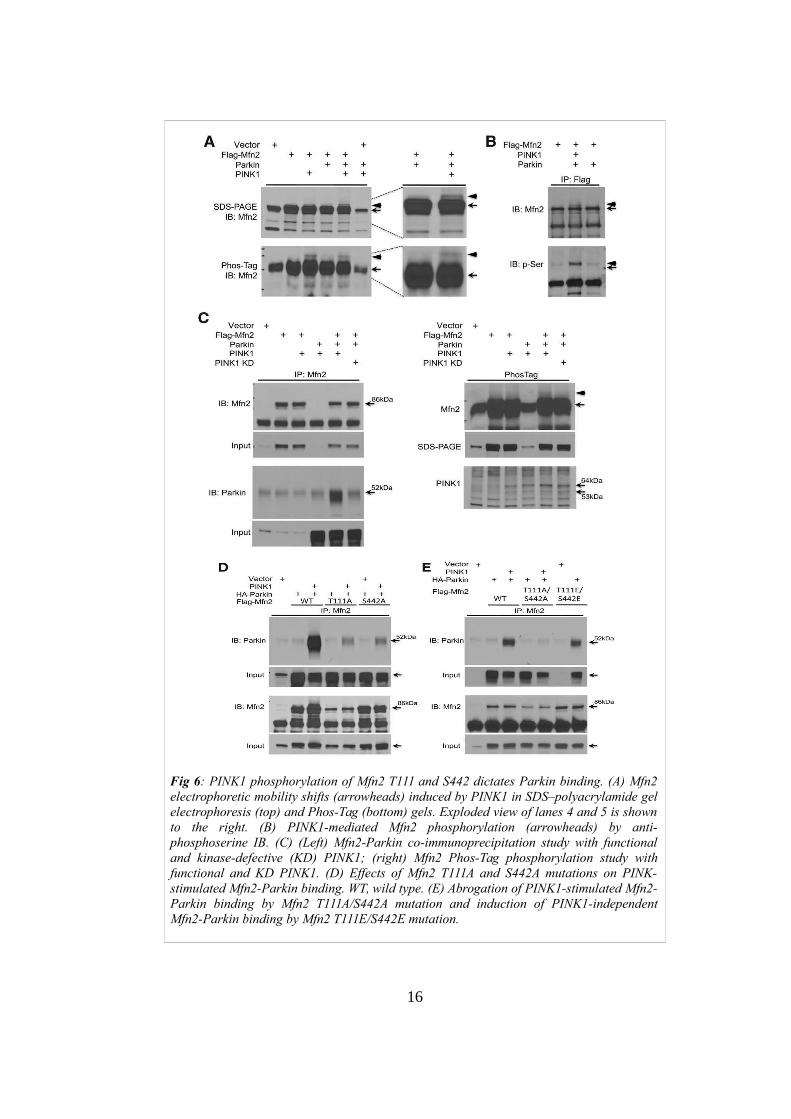
Mfn2 as a substrate for PINK1 phosphorylation. Indeed, the use of Phos-tag gels
and the mobility shifts, indicate that Mfn2 might be phosphorylated at one or
more sites (Fig 6A). Also phosphoserine immunoreactivity of immunoprecipitated
Mfn2 is increased in cells overexpressing PINK1(Fig 6B). In order to evaluate if
PINK1-mediated phosphorylation of Mfn2 is required for Mfn2-Parkin
interaction, amino acid residues Lys 219, Asp 362 and Asp 384 were mutated in
Alanine in order to inactivate PINK1 catalytic domain. These mutants failed to
promote Mfn2-Parkin binding or to induce the characteristic change in Mfn2
mobility in Phos-tag gels (Fig 6C). To establish that Mfn2 is a substrate of PINK1
and the mitochondrial binding partner for Parkin, the phosphorylation sites of
Mfn2 were mapped and then functional consequences were examined.
Bioinformatics analysis identified three highly conserved potential Mfn2
phosphorylation sites: Thr 111 (T111), Ser 442 (S442), and Tyr 448 (Y448) .
Since PINK1 is a serine threonine kinase Mfn2 T111 and S442 were mutated to
alanine, in order to prevent their phosphorylation. Both Mfn2 mutations decreased
PINK1-stimulated Mfn2-Parkin binding, whereas simultaneous mutation of both
residues (T111A/S442A) completely abrogated PINK1- stimulated interaction
between Mfn2 and Parkin (Fig 6E).
15
16
Fig 6: PINK1 phosphorylation of Mfn2 T111 and S442 dictates Parkin binding. (A) Mfn2electrophoretic mobility shifts (arrowheads) induced by PINK1 in SDS–polyacrylamide gelelectrophoresis (top) and Phos-Tag (bottom) gels. Exploded view of lanes 4 and 5 is shownto the right. (B) PINK1-mediated Mfn2 phosphorylation (arrowheads) by anti-phosphoserine IB. (C) (Left) Mfn2-Parkin co-immunoprecipitation study with functionaland kinase-defective (KD) PINK1; (right) Mfn2 Phos-Tag phosphorylation study withfunctional and KD PINK1. (D) Effects of Mfn2 T111A and S442A mutations on PINK-stimulated Mfn2-Parkin binding. WT, wild type. (E) Abrogation of PINK1-stimulated Mfn2-Parkin binding by Mfn2 T111A/S442A mutation and induction of PINK1-independentMfn2-Parkin binding by Mfn2 T111E/S442E mutation.

In agreement with these loss-of-function data, Mfn2 mutations that mimic PINK1
phosphorylation of Mfn2 (T111E/S442E) conferred PINK1-independent binding
activity to Parkin4. Collectively, these studies reveal a potential mechanism by
which Mfn2 orchestrates the PINK1-Parkin mitochondrial quality control
apparatus. In summary, stabilized PINK1 located on the OMM phosphorylates
Mfn2, transforming it into a receptor to which Parkin can bind, thereby bringing it
into physical proximity to its many mitochondrial ubiquitination substrates.
PINK1-mediated phosphorylation of the free ubiquitin activates Parkin E3
ubiquitin ligase activity, and its phosphorylation of ubiquitinated OMM proteins
amplifies mitophagy signaling (Fig 7).
A mechanistic link between PINK1, Mfn2, and Parkin can explain how the
PINK1–Parkin pathway simultaneously initiates mitophagy and shuts down
fusion, as required to preclude mitochondrial contagion. It is possible that PINK1-
mediated phosphorylation may instantaneously convert Mfn2 from a fusion
protein to a Parkin-binding protein. This mechanism of modulating mitochondrial
17
Fig 7: Molecular events leading to selective mitophagy of dysfunctionalmitochondria.

fusion in mitophagy would have the advantage of being more rapid and direct
than Parkin-mediated Mfn2 ubiquitination, extraction, and proteasomal
degradation. Indeed, the importance of Parkin-mediated ubiquitination of specific
proteins, leading to their selective elimination from soon-to-be autophagocytized
organelles, is unclear. Parkin-mediated ubiquitination of Mfn1 and Mfn2 might
deplete these fusion-promoting proteins by targeting them for proteasomal
degradation, thus interrupting mitochondrial fusion and placing the organelle in
quarantine until it can be mitophagically eliminated. Parkin does not selectively
ubiquitinate profusion proteins; it also ubiquitinates the profission protein, Drp1,
which would move the fission/fusion equilibrium in the opposite way, toward
mitochondrial fusion. Indeed, Parkin promiscuously ubiquitinates ≥100 OMM
proteins, essentially painting the organelle with a coat of ubiquitin that attracts
autophagosomes. Thus, during mitophagy at least, Parkin-mediated OMM
ubiquitination does not seem to fine tune OMM protein expression of organelles
that, in any case, are shortly headed to the graveyard. Furthermore, there is little
delay between Parkin localization to mitochondria and their engulfment by
autophagosomes. Live cell microscopy of cultured cells revealed that Parkin
localization, focal protein ubiquitination, and regional mitochondrial
fragmentation with autophagosomal engulfment all occur within minutes of
mitochondrial injury. This provides little time for selective extraction and
proteasomal degradation of mitofusins1.
5. Mitochondrial Dynamism - Mitophagy Crosstalk in the Heart
To investigate the functional impact of mitochondrial quality control mechanisms
on cardiac pathophysiology, genetic ablation of Drp1 was performed in mouse
cardiomyocyte to interrupt mitochondrial fission. A combination of Drp 1 fl/fl
alleles with myh6 promoter-driven modified estrogen receptor (MER)-Cre
transgene was used to generate mice that could be grown to adulthood prior to
cardiac-specific Drp1 deletion using tamoxifen. The same was performed for
Mfn1 and Mfn2 genes (Fig 8A). No tamoxifen effects were found on cardiac
function during the 8 weeks of the studies. Conditional deletion of both Drp1 and
18

Mfn1/Mfn2 induced progressive left ventricular enlargement and a decline in
ejection performance. Six to seven weeks after Drp1 deletion full-blown heart
failure was evident (Fig 8B), but the decline in ejection performance was less
severe in Mfn1/Mfn2 double knockout (DKO) hearts (Fig 8C).
Indeed, Drp1 deficiency induced ventricular wall thinning and chamber dilatation
and also cardiac mass (heart weight corrected for body weight) increased by only
12% 6 weeks after Drp1 deletion (Fig 8B). This is called dilated cardiomyopathy.
By comparison, combined Mfn1/Mfn2 deletion provoked eccentric ventricular
remodeling with ventricular wall thickening and an increase in cardiac mass 6
weeks after Mfn1/Mfn2 ablation (Fig 8C). This is eccentric hypertrophy. Drp 1
deletion, and the ensuring fission inhibition, should have an impact on
mitochondrial morphology, causing an enlargment of the organelles, while
Mitofusins deletion, and fusion inhibition, should lead to smaller and fragmented
19
B C
Fig 8: (A) Schematic of tamoxifen-inducible myh6-Cre-mediated Drp1 orMfn1/Mfn2 gene deletion.(B and C) Cardiac phenotypes induced by Drp1 orMfn1/Mfn2 gene deletion.(Upper left) Four-chamber view of hearts. (Upperright) Ratio of left ventricular (LV) end-diastolic radius (r) to wall thickness (h).(Lower left) Serial M-mode echocardiographic analyses. (Lower right)Quantitative group data showing % change of LV end-diastolic dimension (EDD)and fractional shortening (FS).

mitochondria. Mitochondria observed after Drp1 ablation had a relatively normal
appearance, suggesting a link between this structure and myocardial thinning.
Because the sine qua non of dilated cardiomyopathy is cardiomyocyte loss with
replacement fibrosis, examination of Drp1- and Mfn1/Mfn2-deficient hearts for
cardiomyocyte dropout was necessary. Strikingly, Drp1 deficiency provoked a
progressive increase in myocardial fibrosis that involved the 40% of the
myocardium by 6 weeks while myocardial fibrosis was not a feature of
Mfn1/Mfn2 null hearts. Replacement fibrosis is linked to cardiomyocyte death.
Accordingly, TUNELpositive cardiomyocytes were increased in Drp1 null hearts,
but this does not reliably differentiate between apoptosis and necrosis. Evans blue
staining and visualization of focal complement complex activation in Drp1-
deficient myocardium confirmed cardiomyocyte necrosis while Mfn1/Mfn2 DKO
myocardium showed no cardiomyocyte death, consistent with no myocardial
fibrosis. Mitophagic culling of damaged mitochondria is mediated in part by
mitochondrial localization of the autophagy chaperone protein p62/SQSTM1, and
binding to autophagosomal microtubule associated protein 1 light chain 3 (LC3).
Mitochondrial p62 increased up to 9-fold in Drp1 null hearts, but was unchanged
in Mfn1/Mfn2 DKO hearts. Likewise, mitochondrial LC3-II increased in Drp1
null hearts, but decreased in Mfn1/ Mfn2 DKO hearts. Levels of fibroblast growth
factor (Fgf) 21, a marker of mitochondria insufficiency, increased more rapidly
and to a greater extent after Drp1 ablation than after Mfn1/Mfn2 deletion. Thus,
myocardial autophagosome-mitochondria interactions increased after interrupting
mitochondrial fission, but not after suppressing mitochondrial fusion. By contrast,
levels of two proteases involved in the mitochondrial unfolded protein response
were normal in Drp1 null hearts, but were increased in Mfn1/Mfn2 DKO hearts.
These results raised the possibility of reciprocal defects in mitochondrial quality
control. Because of the impossibility to directly measure mitochondrial mitophagy
in vivo, an in vitro system, wherein Drp1 of Mfn1 and Mfn2 were conditionally
manipulated using adenoviral-encoded Cre and their respective floxed genes, was
developed in cultured murine embryonic fibroblasts. Confocal fluorescence
studies of Drp1- deficient MEFs stained with MitoTracker Green showed
20

mitochondrial hyperfusion with a loss of mitochondrial content within 2 days.
Even the electrochemical potential (Δψm) was not adversely impacted by
mitochondrial elongation induced by Drp1 deletion in either MEFs, or in Drp1
null hearts. Normal respiratory function and absence of increased ROS production
in isolated Drp1-deficient cardiac mitochondria further support a high fitness level
in the retained mitochondria. The same approach was used with Mfn1 and Mfn2
genes. Conditional ablation of Mfn1 and Mfn2 decreased mitochondrial aspect
ratio, reflecting the so-called fragmentation induced by unopposed fission, with no
change in mitochondrial content. Similar to Mfn1/Mfn2 DKO hearts, cell
membrane integrity was not impaired by conditional ablation of Mfn1 and Mfn2
in MEFs. Nevertheless, dissipation of mitochondrial electrochemical potential was
evident in the fragmented mitochondria of MEFs lacking both mitofusins and the
same was present in hearts with also a severe respiratory impairment and ROS
production. Taken together, the results show that conditional interruption of
mitochondrial fission or fusion has parallel effects in MEFs and mouse hearts:
mitochondrial fitness is maintained after ablating Drp1, but is severely
compromised by combined mitofusin deficiency. This is due to the fact that Drp1
ablation provokes the absence of asymmetrical fission: healty daughter
mitochondria are not generated to renew the mitochondrial pool and fully
depolarized daughter mitochondria are not generated to be culled through
mitophagy. Fusion can still occur, leading to a cell wide mitochondrial
dysfunction. It is a matter of time for the depolarized mitochondria to reach the
threshold for mitophagy activation, provoking a strong reduction in mitochondrial
content. When instead fusion is inhibited, Mfn2 absence is able to impair
mitophagy signaling independent of mitochondrial fusion, for its role in PINK1-
Parkin mediated mitophagy pathway. This prevents the normal mitochondrial
culling. Moreover, the organelle fusion renewal mechanism for reintroduction of
the healty daughter mitochondria produced by asymmetric fission cannot occur in
the absence of mitofusins (Fig 9). All these evidences show the implication of
fission and fusion proteins in quality control3.
21

6. Conclusions
Molecular crosstalk between mitochondrial dynamism and mitophagy effectors
leads to a complex and effective mechanism that exclusively eliminate damaged
mitochondria, thus ensuring fitness of the overall cellular mitochondrial
collective. The dichotomous role of Mfn2 as a mitochondrial fission factor or as a
Parkin receptor suggests that mitochondrial fusion and mitophagy are mutually
exclusive, thus protecting healty mitochondria from fusion-mediated
contamination by dysfunctional organelles.
PINK1-Parkin mediated mitophagy signaling pathway, with its function in
mitochondrial fitness, has been proven to have a crucial role in the correct
functioning of the heart, but it is involved and deregulated also in other
diseases, including degenerative pathologies such as Parkison's disease,
thus uncovering multiple opportunities for therapeutic intervention1.
22
Fig 9: Schematic representation of the role of Drp1 andMfn1/2 ablation in cardiac physiophatology.

Bibliography
1. Shirihai OS1, Song M1, Dorn GW 2nd2. How Mitochondrial Dynamism
Orchestrates Mitophagy. Circ Res. 2015 May 22;116(11):1835-1849.
2. Song M1, Dorn GW 2nd2. Mitoconfusion: noncanonical functioning of
dynamism factors in static mitochondria of the heart. Cell Metab. 2015 Feb
3;21(2):195-205. doi: 10.1016/j.cmet.2014.12.019.
3. Song M1, Mihara K2, Chen Y1, Scorrano L3, Dorn GW 2nd4. Mitochondrial
fission and fusion factors reciprocally orchestrate mitophagic culling in mouse
hearts and cultured fibroblasts. Cell Metab. 2015 Feb 3;21(2):273-85. doi:
10.1016/j.cmet.2014.12.011. Epub 2015 Jan 15.
4. Chen Y1, Dorn GW 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin
receptor for culling damaged mitochondria. Science. 2013 Apr 26;340(6131):471-
5. doi: 10.1126/science.1231031.
5. Youle RJ1, van der Bliek AM. Mitochondrial fission, fusion, and stress.
Science. 2012 Aug 31;337(6098):1062-5. doi: 10.1126/science.1219855.
23







