
COMMANDE GÉNÉTI QUE DU DÉVELOPPEMENT EMBRYONNAI RE
De la drosophile aux vertébrésSouris, homme
Tania ATTI E-BI TACHNecker - Enfants Malades

POURQUOI L ÉTUDE DES MÉCANI SMES MOLÉCULAI RESDU DÉVELOPPEMENT EMBRYONNAI RE?
I NTÉRÊT MÉDI CAL :
2-3% des grossesses pathologiquesStérilités et avortements spontanés : 20% des grossessesMalformations congénitales : 20% de la mortalité néonatale
Médecine f tale : diagnostic + + +I magerieGénétique

BI OLOGI E DU DÉVELOPPEMENTHI STORI QUE / 19°S
Le 19e siècle: aspects fondamentaux du monde du vivant:L universalité de la cellule, base du développement embryonnairedivision cellulaire, les chromosomes lieux de l hérédité, lois de Mendella théorie de la sélection, par Darwin .
La fin du 19e siècle : passage à une phase expérimentale du développement. Etablit les phénomènes fondamentaux:
» la communication cellulaire» la migration cellulaire» la différenciation contextuelle» la polarité des oeufs

Le début du 20e siècle est marqué par: la formalisation des lois mendéliennes sur l hérédité, La théorie des gènes (Morgan), la génétique /scienceLa drosophile : système expérimental cartographie génétique: analyse détaillée et quantitative des modes de transmission héréditaire, entre générations
Pendant des décennies, l embryologie et la génétiqueévoluent en parallèle sans que soit établi un rapport entre l action des gènes et les règles développementales régissant la transformation de l uf fécondé en un embryon doté d une structure particulière
BI OLOGI E DU DÉVELOPPEMENTHI STORI QUE / 20°S

Travaux d Edward Lewis (années 40) puis de Nusslein-Volhard et Wieschaus (années 70)
Analyse mutationnelle systématique de l embryogénèse de la drosophile
Le projet morphogénétique de l embryon est contrôlé, de manière hiérarchique, par l activité de gènesspécifiques.
BI OLOGI E DU DÉVELOPPEMENTHI STORI QUE / 20°S

Ed Lewis Janni Nusslein-Volhard Eric Wieschaus

ANALYSE GÉNÉTI QUE DÉVELOPPEMENTALE DE LA DROSOPHI LE
Mécanismes biologiques conservés entre espèces
Gènes et programmes conservés à différentes étapes du développement, dans différents tissus
les hiérarchies génétiques conduisant àl établissement de la polarité antéro-postérieure et à la morphogenèse chez l homme sont semblables à celles de la mouche .
Un gène de drosophile: plusieurs homologues chez les vertébrés: complexité, spécification, redondance

MOLÉCULES DU DEVELOPPEMENT EMBRYONNAI RE
Cadherin
RAS
MAPKKK
MAPK
FUSED
Wnt/ Wingless Hedgehog/ Sonic hedgehog
caténin
p120
caténin
SMADMAKK
DISHEVELD
GSK3
APC
caténin
LEF1 TCF GLISuH
Frizzeld Smoothened Patched
Cadherin
Notch
FGF-R
TGF -R
Delta FGF (9 protéines)
TGF :plus de 25 membres
NOYAU

Gènes maternels
Gènes de segmentation
Les gènes homéotiques
Les gènes maîtres
Programme & gènes du développement
Ems, otd, btd / EMX, OTX, SPGène Paired / Gènes PAXVoie hedgehog / SHH
Gènes Hox etformation du membre
Bicoid, nanos
gap pair-rulepolarité segmentaire
Gènes maternels
Gènes de segmentation
Les gènes homéotiques
Les gènes maîtres
Les gènes homéotiques
Gènes de segmentation
Les gènes maîtres
Les gènes homéotiques
Gènes maternels
Gènes de segmentation
Les gènes maîtres
Les gènes homéotiques

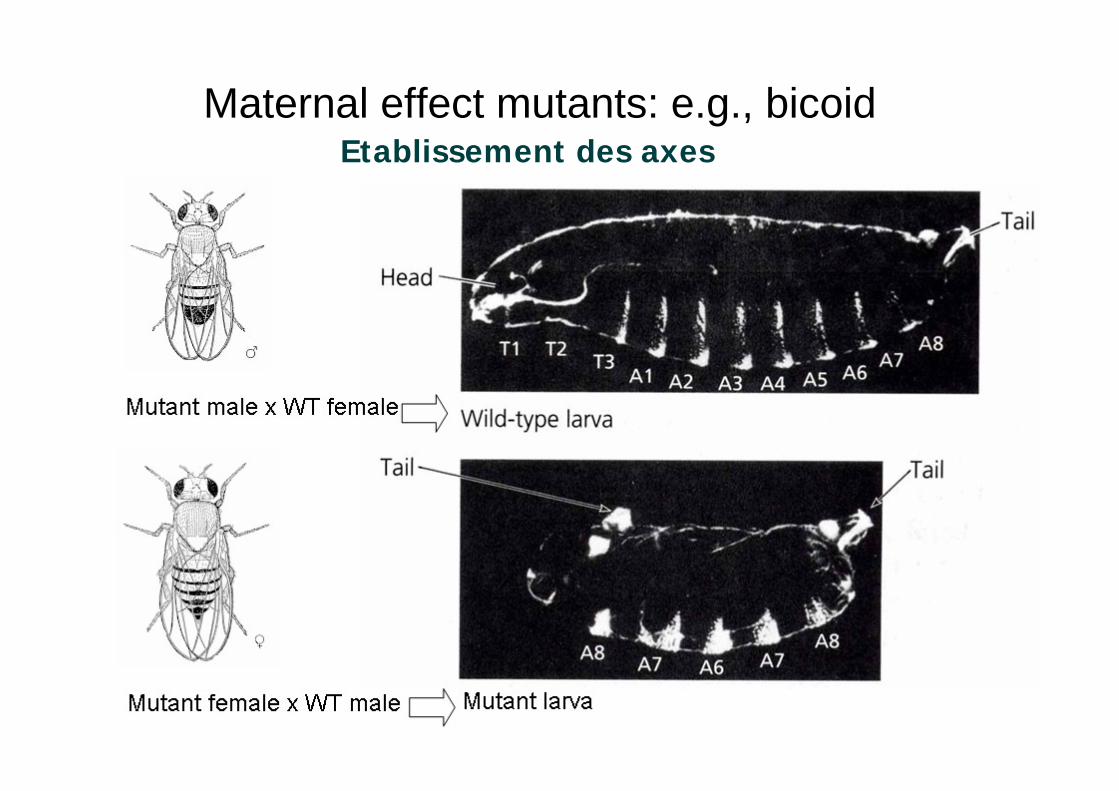
Maternal effect mutants: e.g., bicoidEtablissement des axes

Bicoid : gène de la mouche
Pas d homologues chez les autres espèces, absent chez les autres insectes.
Duplication récente d un gène homologue à Hox3 des vertébrés
a perdu sa fonction homéotiquea acquis de nouveaux rôles dans
l ovocyte et les annexes
Duplication de ce gène suivi par une divergence fonctionnelleBicoidZerknüllt (rôle dans les annexes)
PNAS January 8, 2002 vol. 99 no. 1

Traduction des ARN maternels après la fécondation
Établissement d un gradient de protéines d origine maternelle

Bicoid : gène de la mouchePas d homologues, absent chez les autres insectes.
Duplication récente d un gène homologue à Hox3: Bicoid et zerknüllt.
Les 2 gènes sont localisés dans le complexe Hox de la Drosophile , mais n ont pas de fonction homéotique
torso and hunchback seraient les organisateurs antérieurs chez les insectes primitifs
L exemple de bicoid, 1er morphogène identifié illustre comment un gène essentiel peut changer de fonction au cours de l évolution.
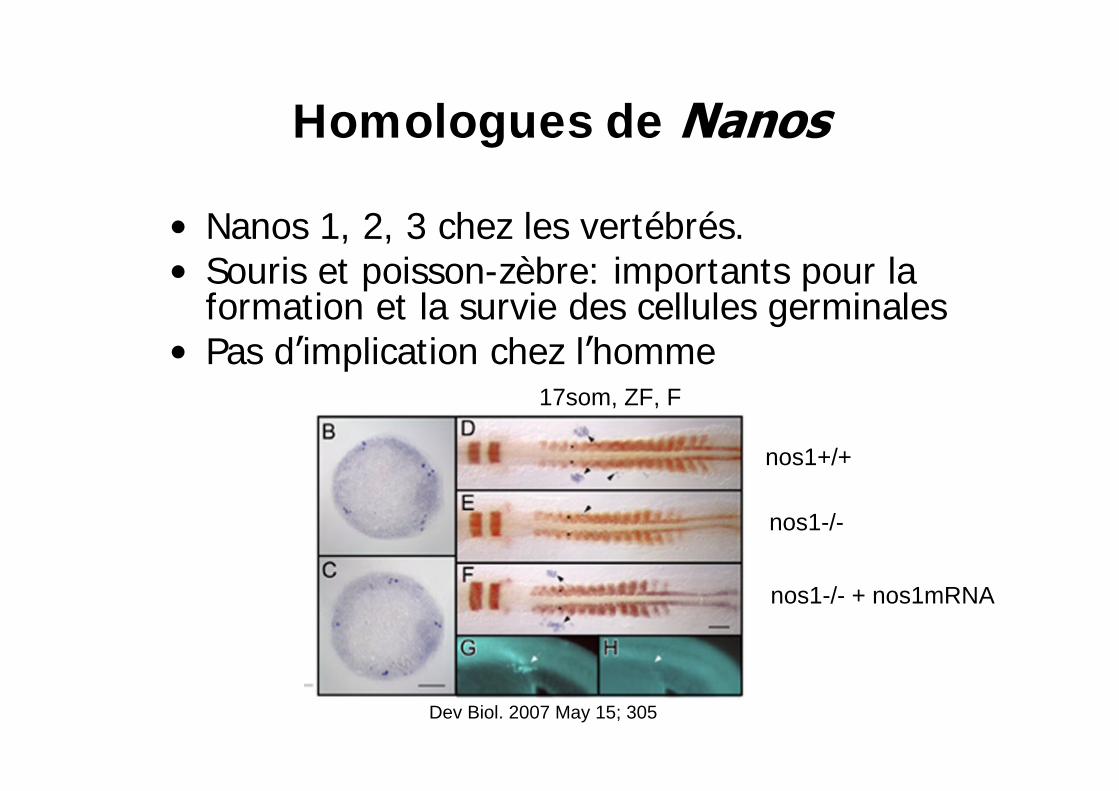
Homologues de
Nanos 1, 2, 3 chez les vertébrés.Souris et poisson-zèbre: importants pour la formation et la survie des cellules germinalesPas d implication chez l homme
nos1+/+
17som, ZF, F
nos1-/-
nos1-/- + nos1mRNA
Dev Biol. 2007 May 15; 305

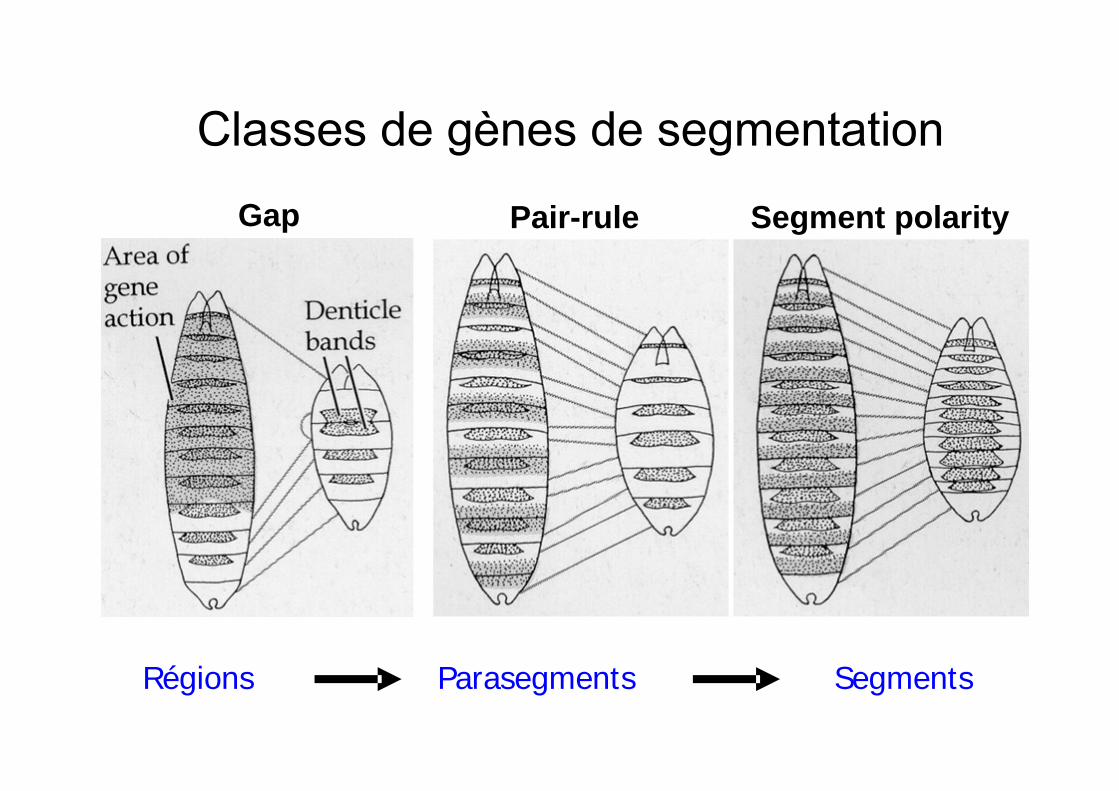
Gap Segment polarity Pair-rule
Régions Parasegments Segments

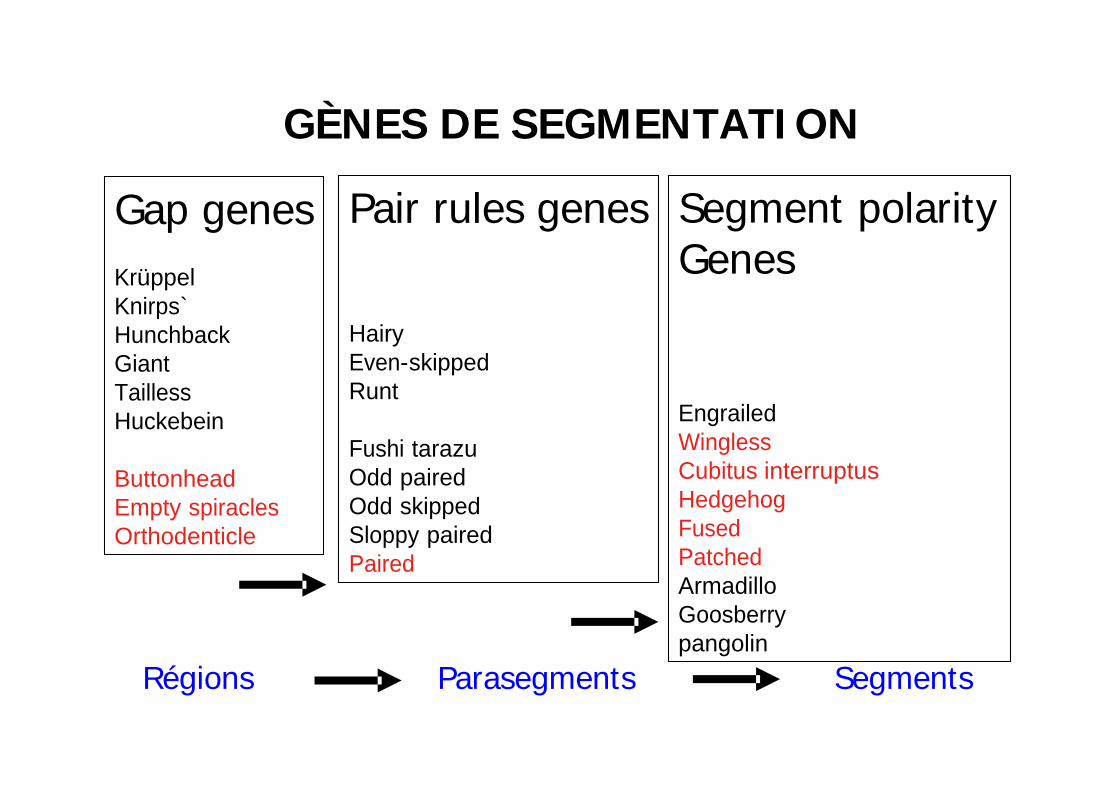
GÈNES DE SEGMENTATI ON
Gap genesKrüppelKnirps`HunchbackGiantTaillessHuckebein
ButtonheadEmpty spiraclesOrthodenticle
Pair rules genes
HairyEven-skippedRunt
Fushi tarazuOdd pairedOdd skippedSloppy pairedPaired
Segment polarityGenes
EngrailedWinglessCubitus interruptusHedgehogFusedPatchedArmadilloGoosberrypangolin
Régions Parasegments Segments

gènes gap gènes pair-rule
gènes de polarité du segment

Gènes GAP
Hunchback KrüppelKnirps`Giant
TaillessHuckebein
OrthodenticleEmpty spiraclesButtonhead

otd (orthodentide) , ems (empty spiracles)
btd (Buttonhead)
gènes "gap" qui joue un rôle dans la segmentation de la tête
Homologues vertébrés: Otx, Emx: région la plus antérieure de l'embryon (cerveau antérieur et moyen)Sp: région antérieure et jonction mesencéphale-rhombencéphale.

Des homologues des gènes Gap déterminent les grandes divisions du
cerveau des vertébrés
Boncinelli et al J. Neurobiology 24:1356-1366(1993)
Emx1Compris dans Emx2
Emx2Compris dans Otx1
Otx1Compris dans Otx2
Otx 2
Sp1

versus
Comme les gènes hox: Expression dans des régions chevauchantes du TN combinatoire de leur produits spécifie un type cellulaireExpression conservée au cours de l évolution
Contrairement aux gènes hox:expression emboitée antérieurepas d organisation en clusterséquence en défaveur d'une duplication récente d'un gène ancestral
LOCALISATION CHROMOSOMIQUE (HOMME)
2p14-p1310q26.12p1314q21-q22
EMX1EMX2OTX1OTX2

Dev Biol 1996, 178:174-178
Development 1997, 124: 1653-1664
Nature Genet 1996, 14: 218-222
Development 1995, 121: 3279-3290
Invalidation ciblée (KO)
Expression
Agénésie du corps calleux isolée
Cortex normal
(schizencéphalie)Absence de reins,
uretères, gonades, tractus génital
Epilepsie + anomalies cérébrales:
régions tel, temporale, perirhinale, hippocampe, mésencephale, cervelet, audition, vision
Absence de cerveau antérieur et moyen
(au dessus de r3)= aprosencéphalie
Te dorsalTe dorsal, petites régions Di
Te,Di,Mes dorsalDi, Mes ventral
Te,Di,Mes dorsal Di, Mes ventral
Emx1Emx2Otx1Otx2
: modèles murins
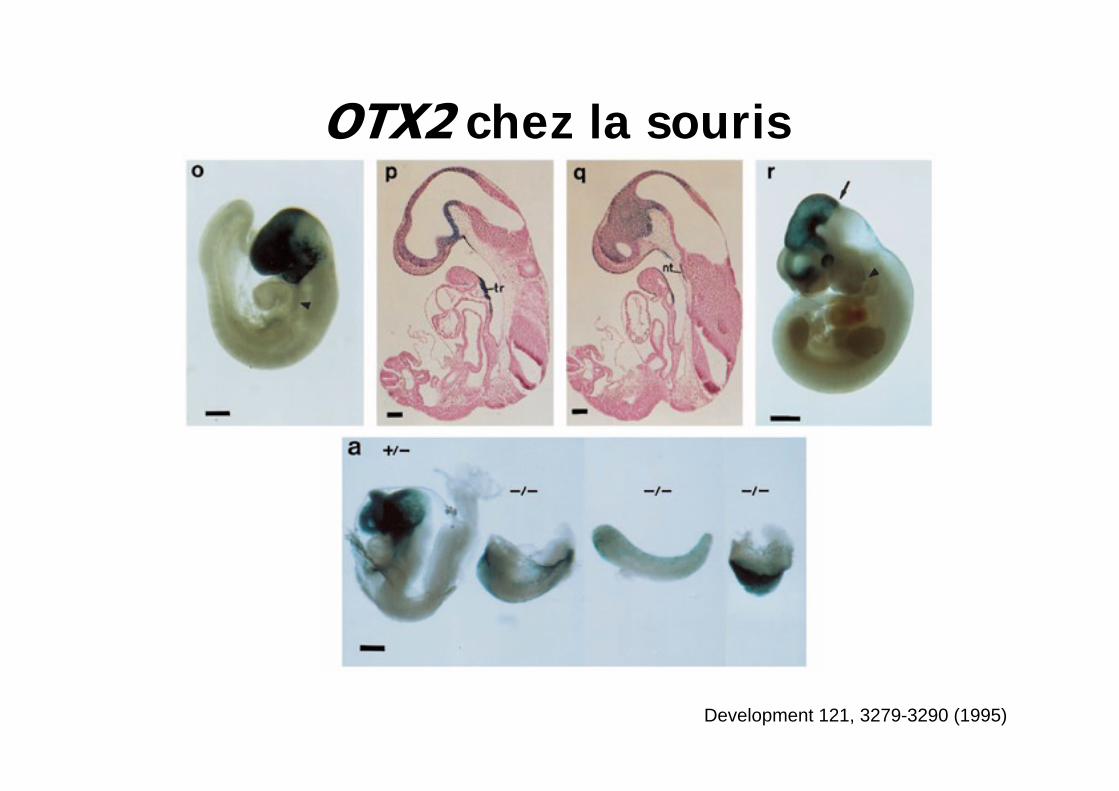
chez la souris
Development 121, 3279-3290 (1995)

en pathologie humaine
Autosomique dominantPhénotype: microphtalmie / anophtalmie+ /- anomalie hippocampe + /- retard mental, épilepsie
Autres gènes donnant une anophtalmie/ microphtalmie chez l homme:
PAX6, SOX2, RAX, CHX10
PNAS 2008 105, 5408 5413
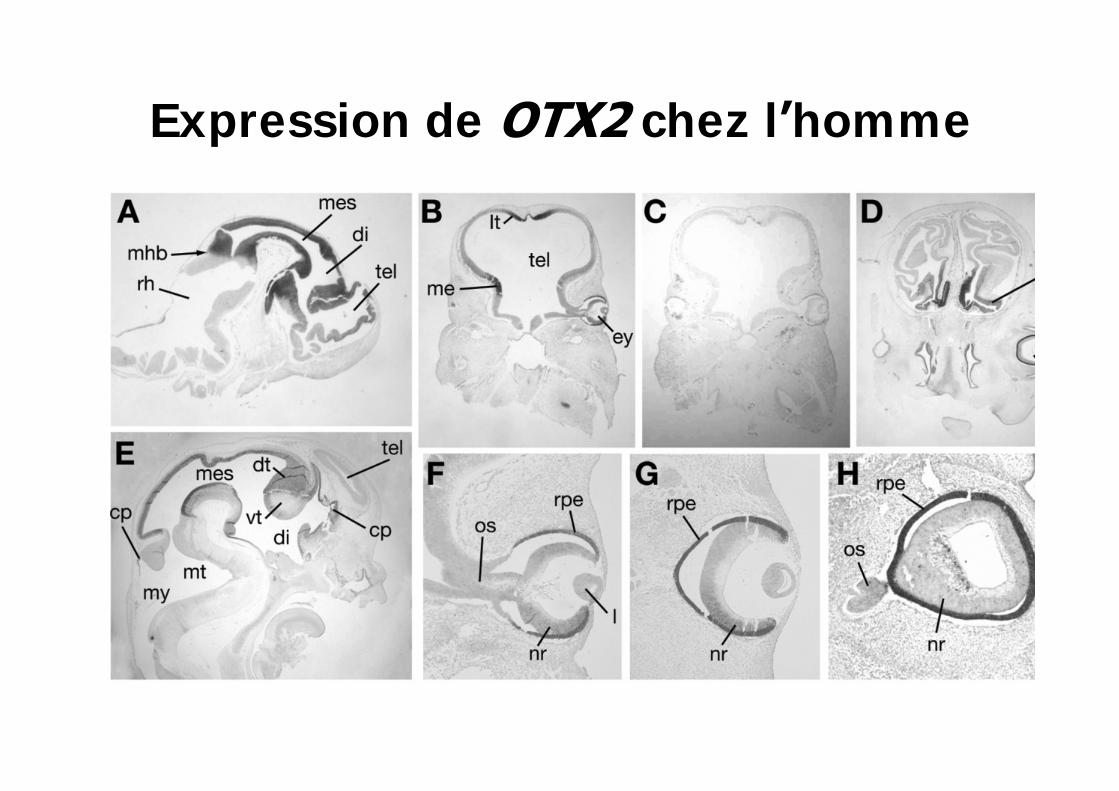
Expression de chez l homme

Mutations OTX2 chez l homme

Sp1 : expression chez la souris

Sp1 : invalidation chez la souris
Exencéphalie antérieure
Agénésie cerveletSpina bifidaAnomalie nez, palaisAnomalie réductionnelle
des membres

Développement du cerveau antérieur (prosencéphale) et moyen (mésencéphale)
N est pas sous le contrôle des gènes HOXHomologues de gènes gap ems, otd et btdimportants pour la segmentation du cerveau chez les vertébrés
OTX, EMX, SP
Combinatoire avec d'autres gènes pour la spécification des territoiresCerveau antérieur plus complexe que cerveau postérieur

gènes gapgènes pair-rule
gènes de polarité du segment
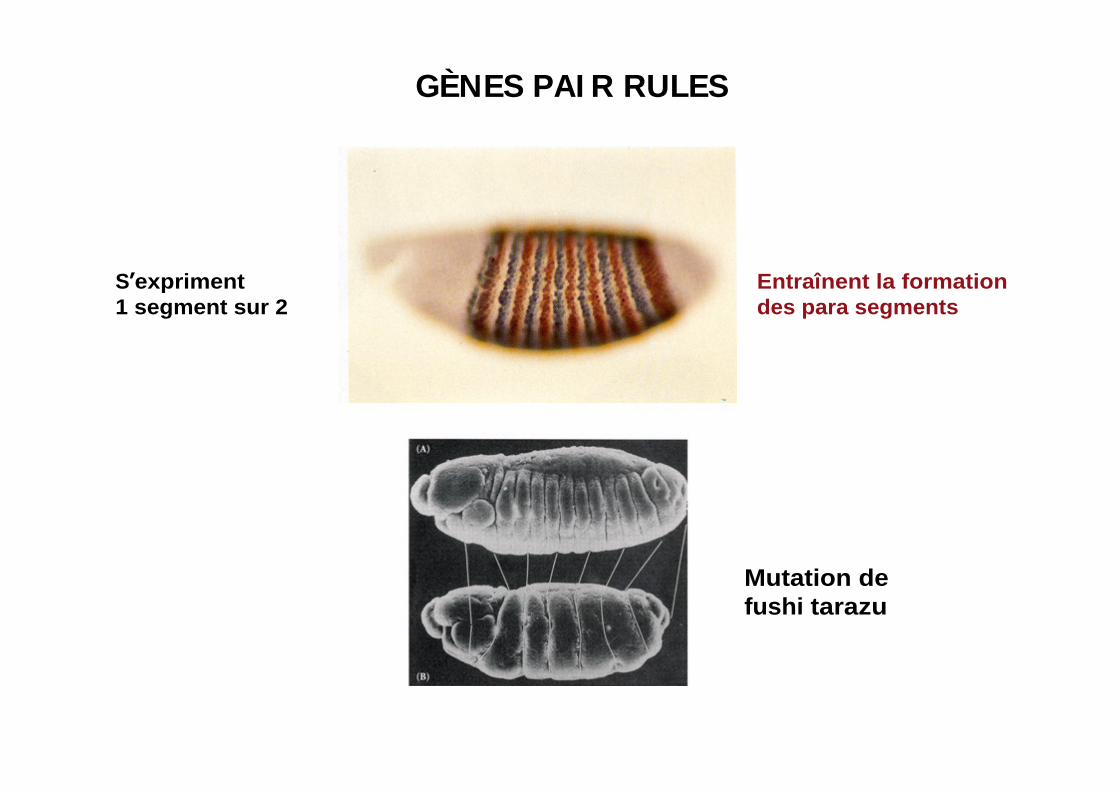
GÈNES PAI R RULES
S expriment1 segment sur 2
Entraînent la formationdes para segments
Mutation de fushi tarazu
GÈNES DE SEGMENTATI ON
Gap genesKrüppelKnirps`HunchbackGiantTaillessHuckebein
ButtonheadEmpty spiraclesOrthodenticle
Pair rules genes
HairyEven-skippedRunt
Fushi tarazuOdd pairedOdd skippedSloppy pairedPaired
Segment polarityGenes
EngrailedWinglessCubitus interruptusHedgehogFusedPatchedArmadilloGoosberrypangolin
Régions Parasegments Segments

Homologues des gènes paired: Gènes Pax
Paired box = motif protéique de 128 AA conservé entre les espècessitué à la partie NH2 terminale du gènecapable de se fixer à l ADN
DROSOPHILE (3) SOURIS (9)
HOMME (9) paired
goosberry proximal
goosberry distal Pax1-Pax9 PAX1-PAX9

Gènes : organisation structurale
C
C
C
C
Boite paired OP Homéoboite
C
C
C
C
C
C
N
C
C
C
C
N
N
Groupe 2: PAX2, PAX5, PAX8
NGroupe 1: PAX1, PAX9
Groupe 3: PAX3, PAX7
Groupe 4 : PAX4, PAX6

4 groupes de paralogues
Organisation commune de motifs protéiquesSimilarité de séquencePatron d expression similaire au cours de l embryogenèse
Gènes : organisation structurale

Gènes Localisation chromosomique
Pax/PAX
HOMME SOURIS
1 20p11 2 2 10q25 19 3 2q35 1 4 7q32 6 5 9p13 4 6 11p13 2 7 1p36.2 4 8 2q12-q14 2 9 14q12q13 12

Gènes Pax et organogénèse:interactions épithélium-mésenchyme
EPITHELIUM MESENCHYME
DENT Pax9
GLANDE SALIVAIRE
Pax9
THYMUS Pax1/Pax9
NEZ Pax6 Pax3/Pax7/Pax9
THYROIDE Pax8
REIN Pax2 Pax2/Pax8

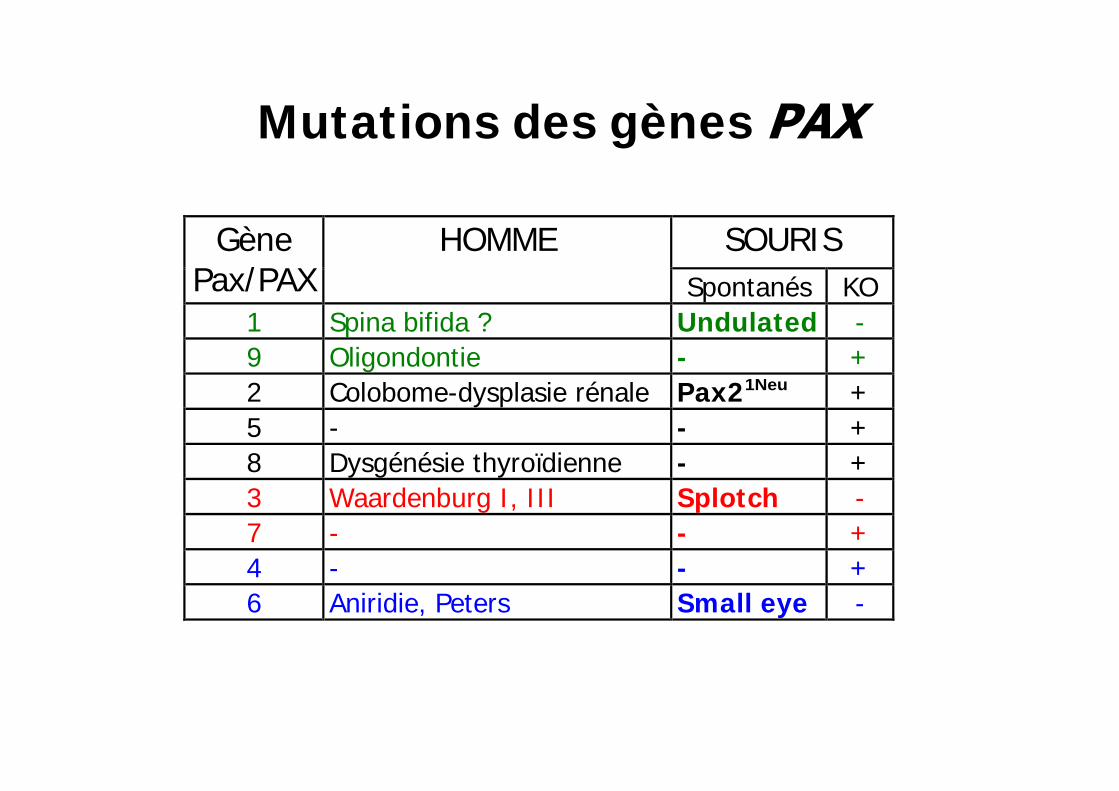
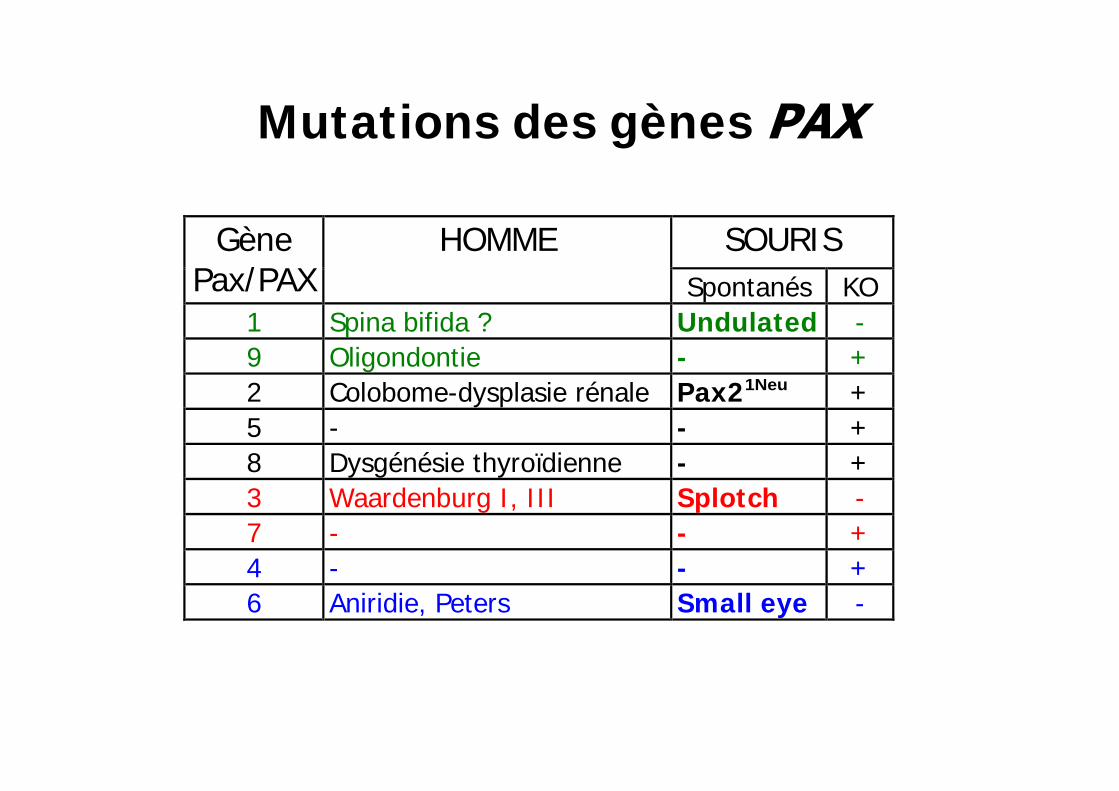
Mutations des gènes
SOURIS Gène Pax/PAX
HOMME Spontanés
KO
1 Spina bifida ? Undulated
- 9 Oligondontie - + 2 Colobome-dysplasie rénale Pax21Neu + 5 - - + 8 Dysgénésie thyroïdienne - + 3 Waardenburg I , I I I Splotch - 7 - - + 4 - - + 6 Aniridie, Peters Small eye
- 6

chez la souris
Somites
Poches pharyngées
bourgeons dentaireset palatins
Membres
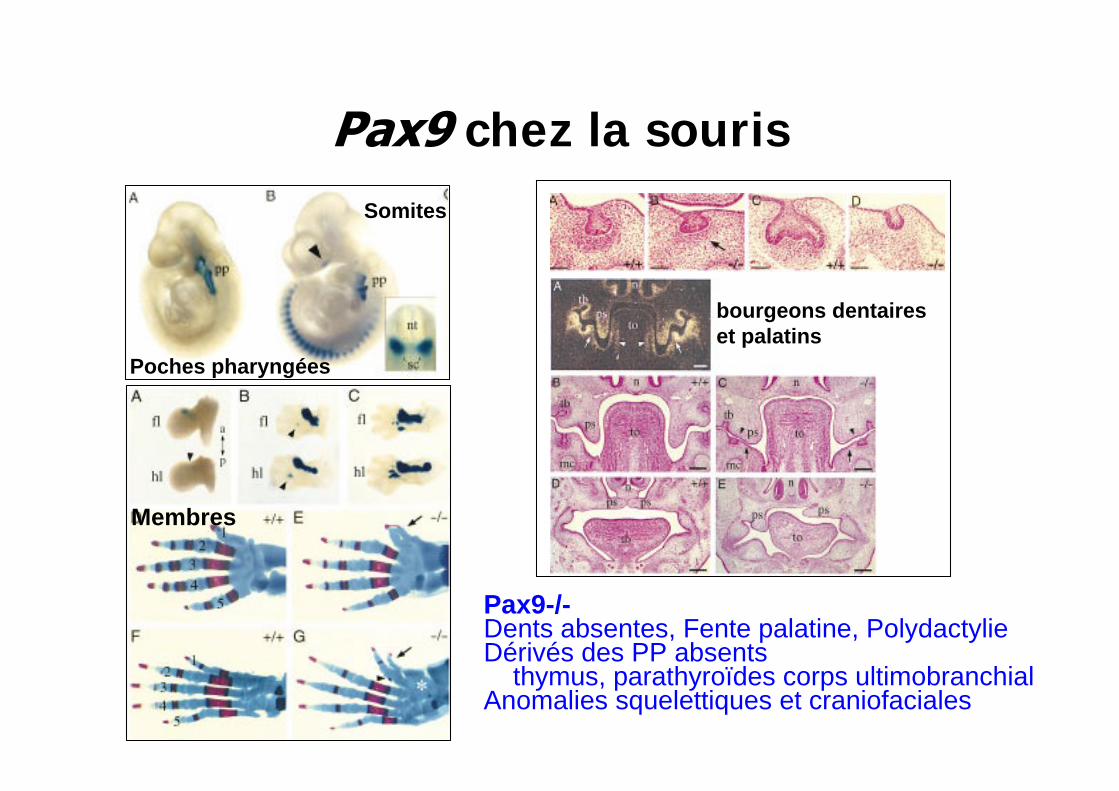
chez la souris
Pax9-/-Dents absentes, Fente palatine, Polydactylie Dérivés des PP absents
thymus, parathyroïdes corps ultimobranchialAnomalies squelettiques et craniofaciales
Somites
Poches pharyngées
bourgeons dentaireset palatins
Membres

et oligondontie
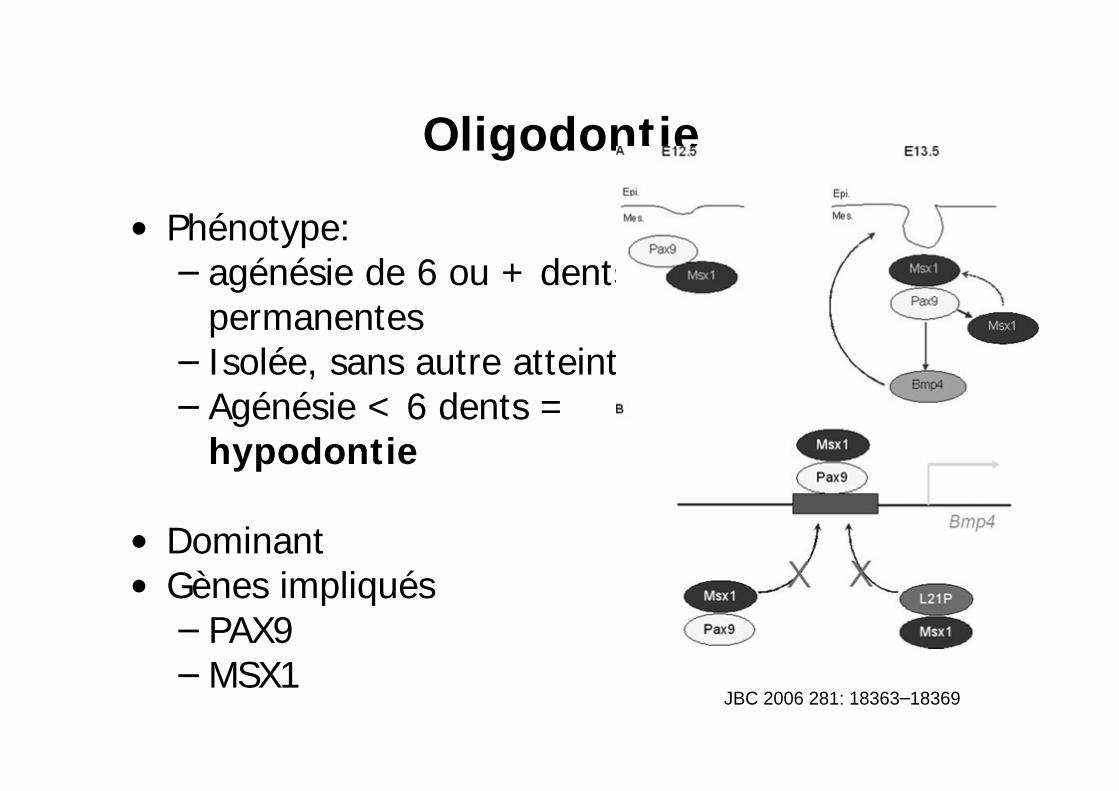
Oligodontie
Phénotype: agénésie de 6 ou + dents permanentesIsolée, sans autre atteinteAgénésie < 6 dents = hypodontie
DominantGènes impliqués
PAX9MSX1
JBC 2006 281: 18363 18369
Mutations des gènes
SOURIS Gène Pax/PAX
HOMME Spontanés
KO
1 Spina bifida ? Undulated
- 9 Oligondontie - + 2 Colobome-dysplasie rénale Pax21Neu + 5 - - + 8 Dysgénésie thyroïdienne - + 3 Waardenburg I , I I I Splotch - 7 - - + 4 - - + 6 Aniridie, Peters Small eye
- 6

modèle murin
Pax21Neu mice G insertionKrd Délétion chromosomiquePax2 Knock-out
Anomalies rénalesAnomalies de la rétine et du nerf optique

Fonctions
Rein : transition mRein : transition méésenchymesenchyme--éépitheliumpithelium
il : information positionnelle et guidance il : information positionnelle et guidance axonaleaxonale
Oreille interne : prolifOreille interne : proliféération cellulaireration cellulaire
Tube neural : compartimentation neuronaleTube neural : compartimentation neuronale
RhombencRhombencééphale : segmentationphale : segmentation

expression
Oreille interne
Nerf optique
SNC
Rein

: syndrome Rein-Colobome
Autosomique dominant
Colobome du nerf optique Hypoplasie rénaleReflux vésico-urétéralSurdité modérée
Rare: retard mental, épilepsie

Mutations des gènes
SOURIS Gène Pax/PAX
HOMME Spontanés
KO
1 Spina bifida ? Undulated
- 9 Oligondontie - + 2 Colobome-dysplasie rénale Pax21Neu + 5 - - + 8 Dysgénésie thyroïdienne - + 3 Waardenburg I , I I I Splotch - 7 - - + 4 - - + 6 Aniridie, Peters Small eye
- 6
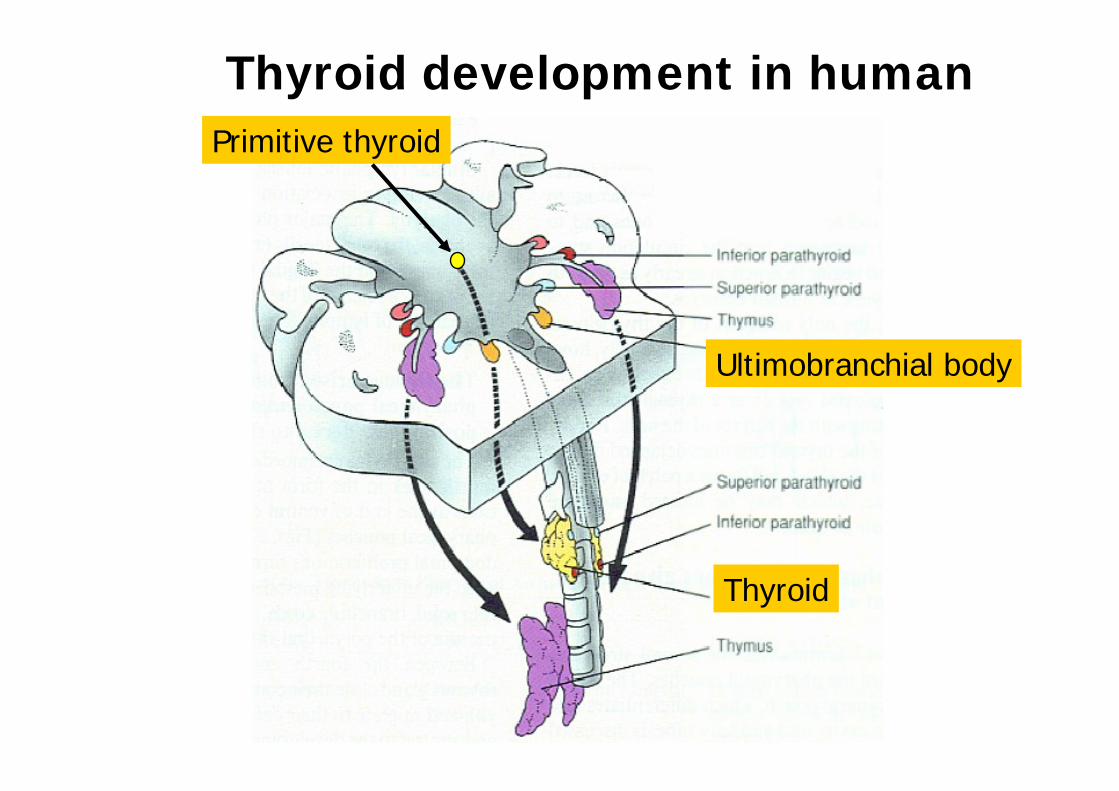
Thyroid development in humanPrimitive thyroid
Ultimobranchial body
Thyroid

chez la souris
Expression chez la souris E8,5- AdultePlancher du Pharynx primitif (ZP: Précurseurs des cellules folliculaires thyroïdiennes)ReinSystème nerveux central
Invalidation chez la sourispax8-/ -: Formation initiale de l ébauche médiane mais àE11,5 cesse de croître. Hypoplasie sévère avec absence de follicules
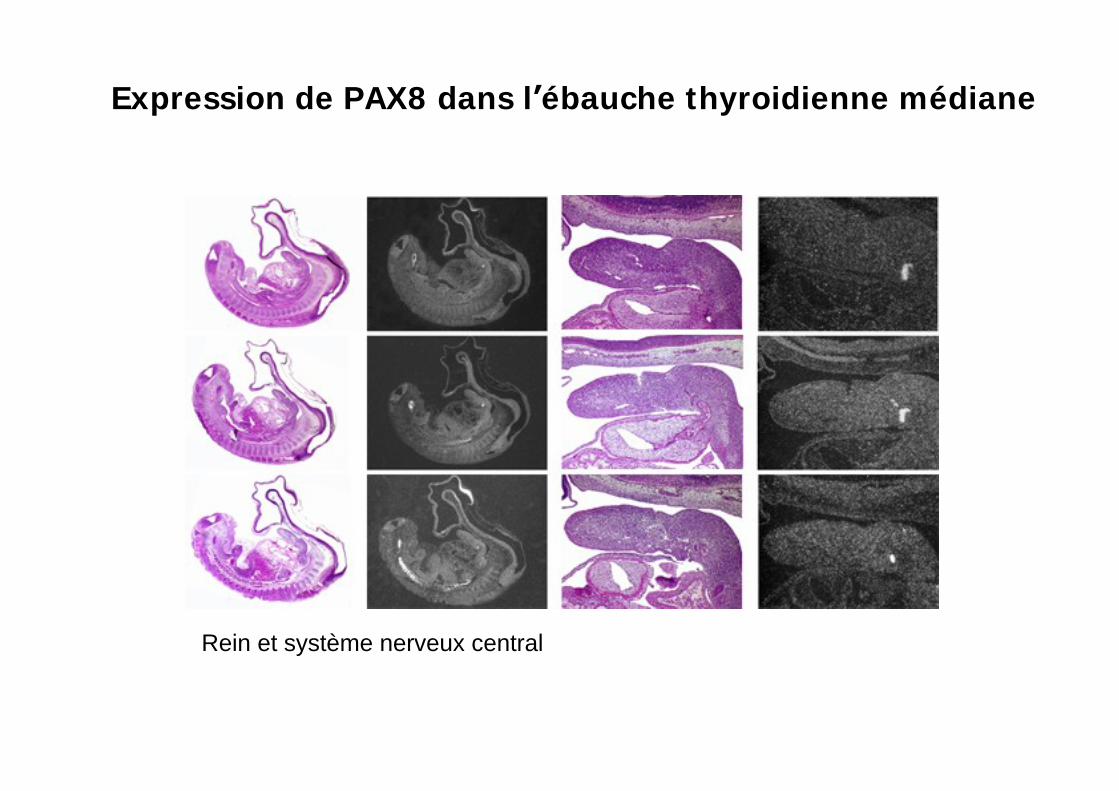
Expression de PAX8 dans l ébauche thyroidienne médiane
Rein et système nerveux central
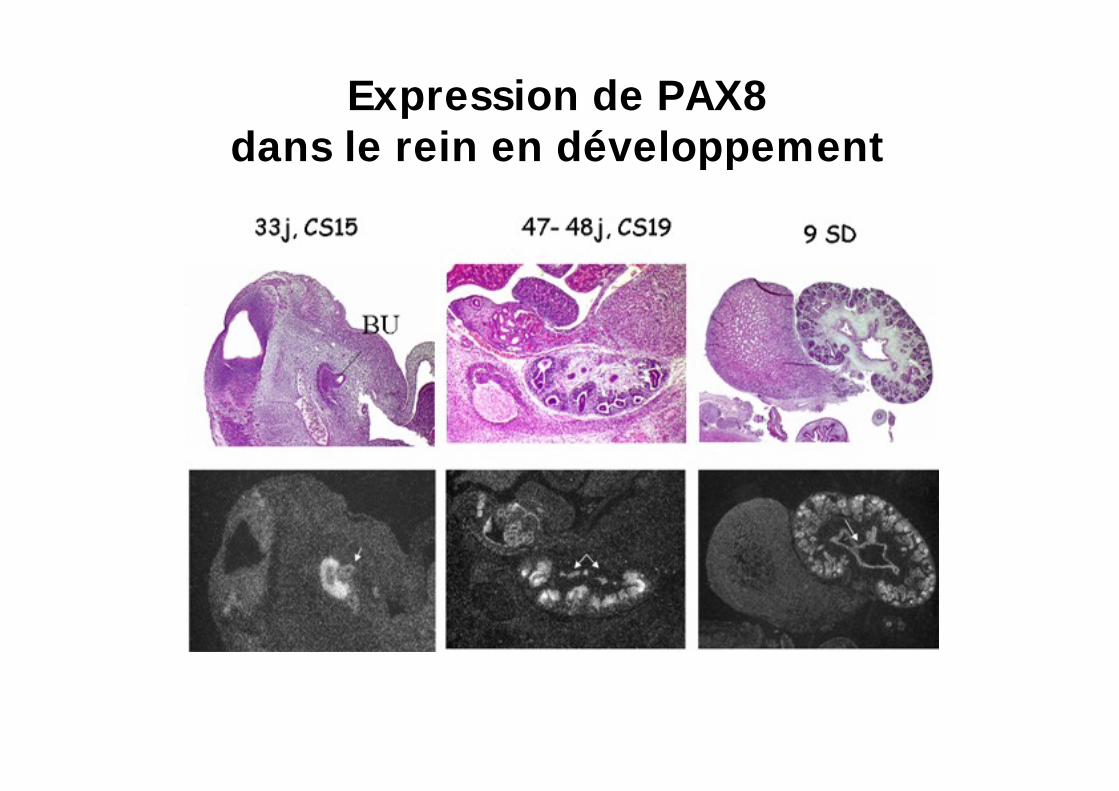
Expression de PAX8dans le rein en développement

Mutations de chez l homme
Mutations dominantes
Hypoplasies thyroidiennes ou ectopiesParfois associées à une anomalie rénale (25%)
Mutations des gènes
SOURIS Gène Pax/PAX
HOMME Spontanés
KO
1 Spina bifida ? Undulated
- 9 Oligondontie - + 2 Colobome-dysplasie rénale Pax21Neu + 5 - - + 8 Dysgénésie thyroïdienne - + 3 Waardenburg I , I I I Splotch - 7 - - + 4 - - + 6 Aniridie, Peters Small eye
- 6

Souris splotch ( )
Plusieurs lignées, létales embryonnairesSplotch retarded (spd): décès naissance
Sp-/ -Spina bifida et exencéphalieAnomalies de pigmentationCardiopathieAnomalies craniofaciales
(Sp/+ )Taches blanches



Syndrome de Waardenburg
Neurocristopathie
Surdité congénitale de perceptionAnomalies pigmentaires: peau, cheveux, iris
MITF, SLUG
EDN3, EDNRB, SOX10
PAX3
PAX3
Type 1: dystopie des canthiType 2,Type 3: anomalie des membresType 4: maladie de Hirschsprung

Relations entre les gènes WS

Mutations des gènes
SOURIS Gène Pax/PAX
HOMME Spontanés
KO
1 Spina bifida ? Undulated
- 9 Oligondontie - + 2 Colobome-dysplasie rénale Pax21Neu + 5 - - + 8 Dysgénésie thyroïdienne - + 3 Waardenburg I , I I I Splotch - 7 - - + 4 - - + 6 Aniridie, Peters Small eye
- 6
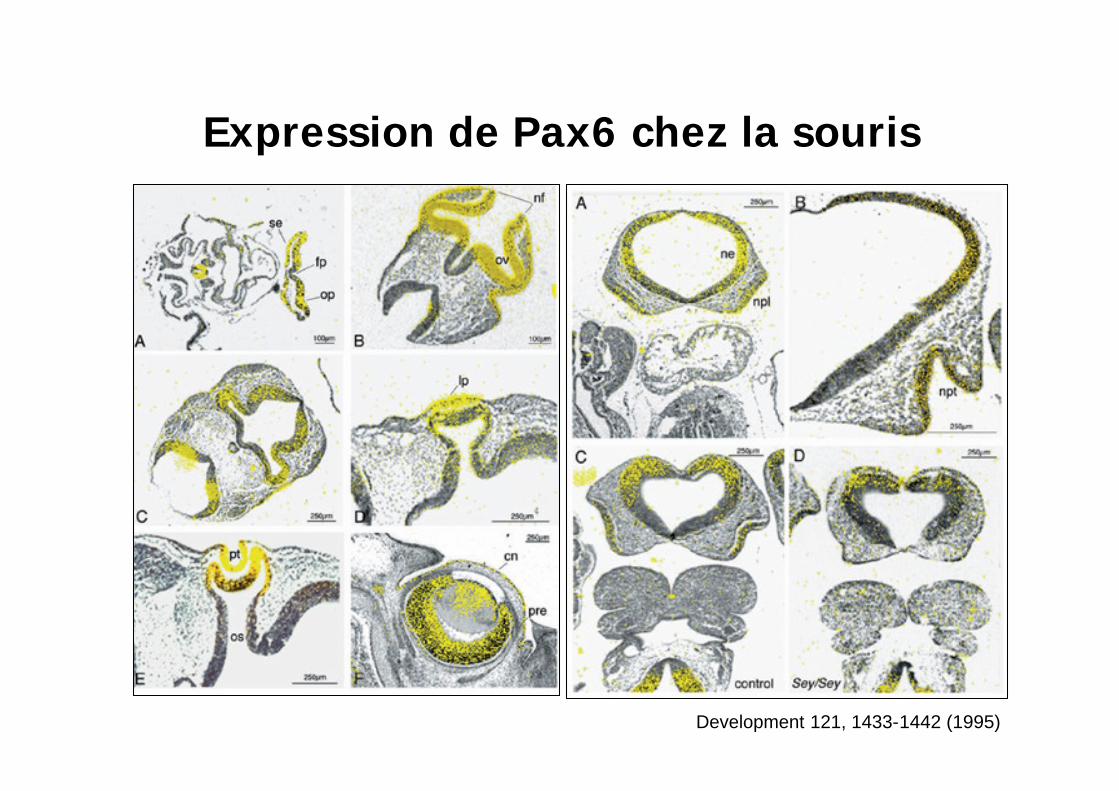
Development 121, 1433-1442 (1995)
Expression de Pax6 chez la souris
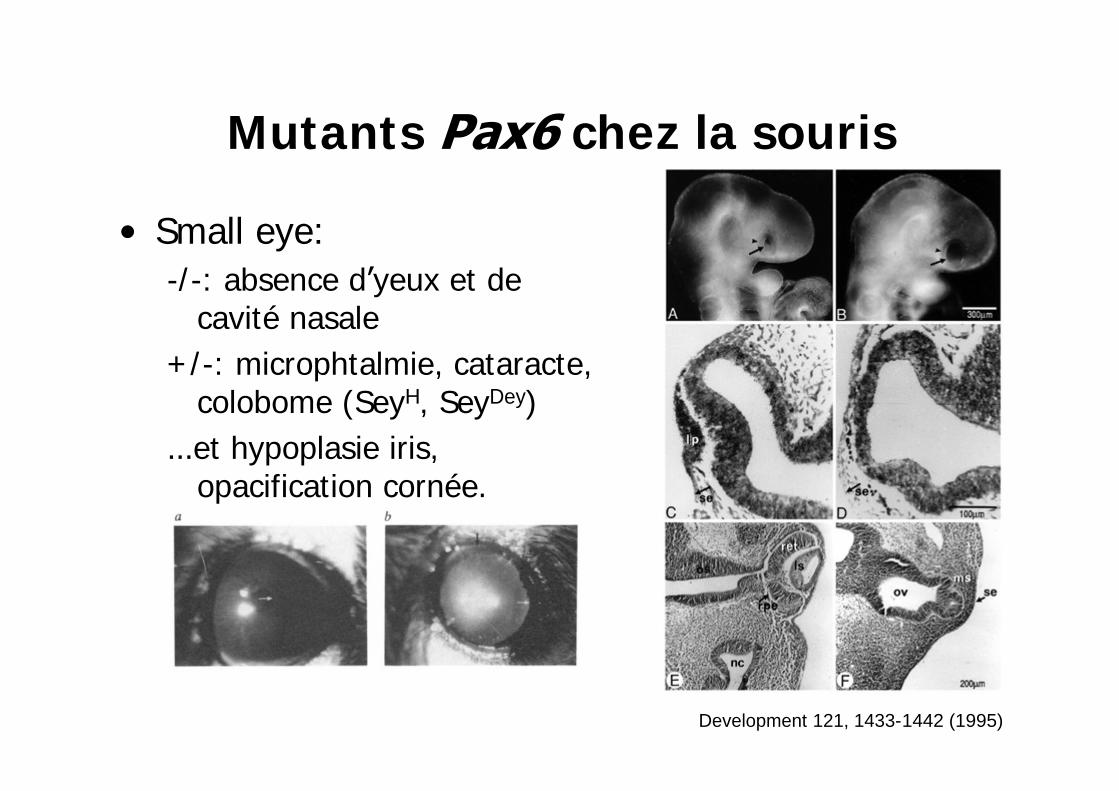
Mutants chez la souris
Small eye: -/ -: absence d yeux et de
cavité nasale+ /-: microphtalmie, cataracte,
colobome (SeyH, SeyDey)et hypoplasie iris, opacification cornée.
Development 121, 1433-1442 (1995)

Hypoplasie sévère de l'iris+ /- cataracte
opacification de la cornéehypoplasie du corps ciliaire et de la rétine
Formes familiales : autosomique dominant
11p1.13 (WAGR) Tumeur de WilmsAniridieanomalies Génito- urinairesRetard mental
spectre de l'aniridie
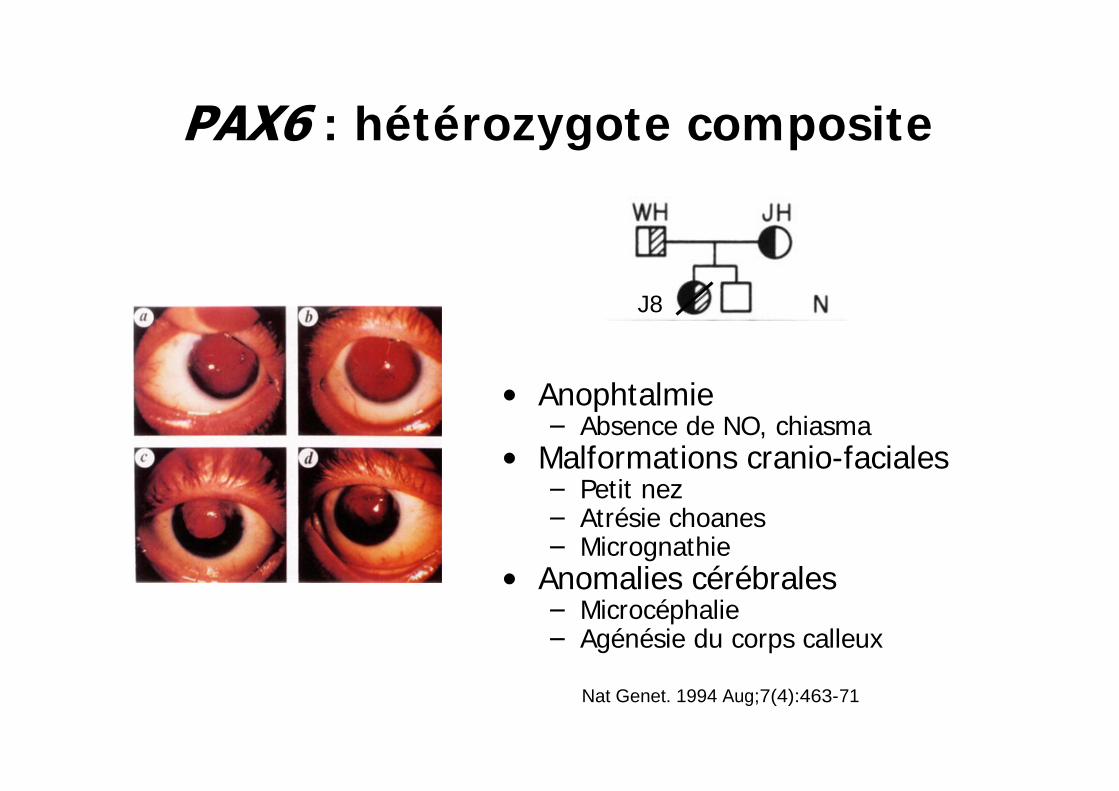
: hétérozygote composite
AnophtalmieAbsence de NO, chiasma
Malformations cranio-facialesPetit nezAtrésie choanesMicrognathie
Anomalies cérébralesMicrocéphalieAgénésie du corps calleux
J8
Nat Genet. 1994 Aug;7(4):463-71

Gènes PAX9 gènes chez l homme et la sourisFacteurs de transcription, domaine paired de liaison à l ADN Fonction essentielle dans les interactions épithélium-mésenchyme, mais dans des tissus différents4 groupes de paralogues: organisation commune de motifs protéiques, similarité de séquence et d expression embryonnairePas de clustersI mportance majeure en pathologie humaine

gènes gapgènes pair-rule
gènes de polarité du segment
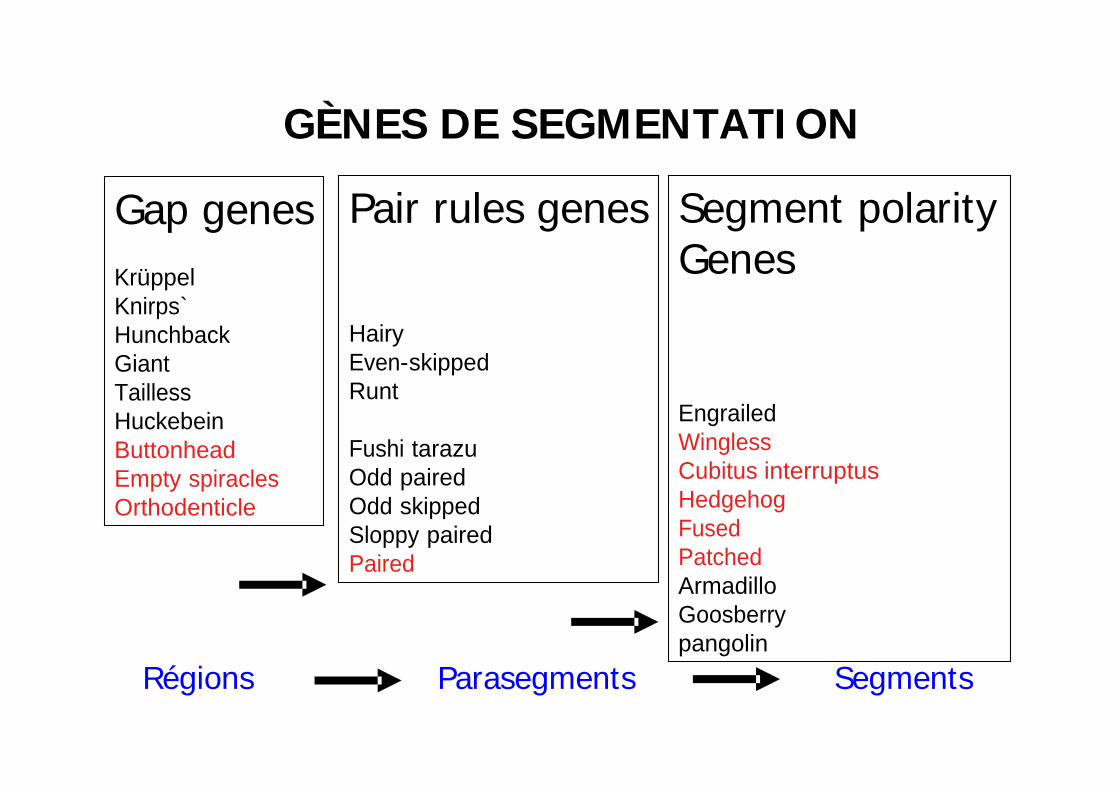
GÈNES DE SEGMENTATI ON
Gap genesKrüppelKnirps`HunchbackGiantTaillessHuckebeinButtonheadEmpty spiraclesOrthodenticle
Pair rules genes
HairyEven-skippedRunt
Fushi tarazuOdd pairedOdd skippedSloppy pairedPaired
Segment polarityGenes
EngrailedWinglessCubitus interruptusHedgehogFusedPatchedArmadilloGoosberrypangolin
Régions Parasegments Segments

GENES & DEVELOPMENT 1992 6:2635-2645 9
HH
EN
La plupart des mutants hh sont lethal embryonnaire, et les mutants ont uncuticule ventral qui a peu de polarité antéro-postérieure.
Les interactions génétiques avec d autres gènes de segmentation segmentation suggèrent un rôle dans la communication intercellulaire
Hedgehog: gène de polarité segmentaire
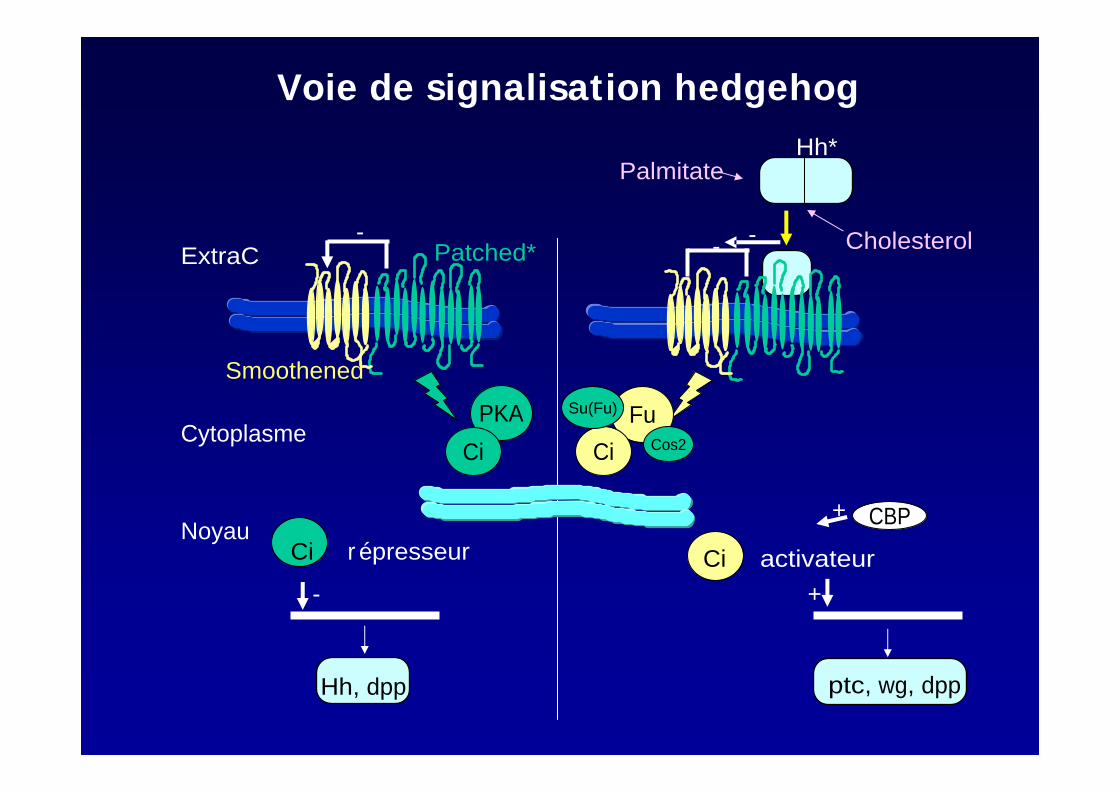
CBP+
+
ptc, wg, dpp
- -
-
Hh, dpp
Fu
Ci
Su(Fu)
Cos2
-
Smoothened
Patched*ExtraC
PKA
CiCytoplasme
Cholesterol
Hh*Palmitate
NoyauCi r épresseur Ci activateur
Voie de signalisation hedgehog

Homologues de hedgehog chez les vertébrés
3 homologues de hedgehog chez les vertébrés
Sonic Hedgehog (SHH) 7q36
Indian Hedgehog (IHH) 2q35.36 Homme
Desert Hedgehog (DHH) 12q13
Localisation chromosomique différente
Expression temporo-spatiale et fonction différentes
SHH: cerveau et membres
IHH: pathologies osseuses
DHH: dysgénésie gonadique

Conservation de Shh chez les vertébrés
Homologues de Shh hautement conservés entre les espècesPlus forte homologie dans la moitié N-terminale
Même patron d expression de Shh chez le poulet, la souris, le poisson-zèbre
-> Suggère une conservation de la fonction
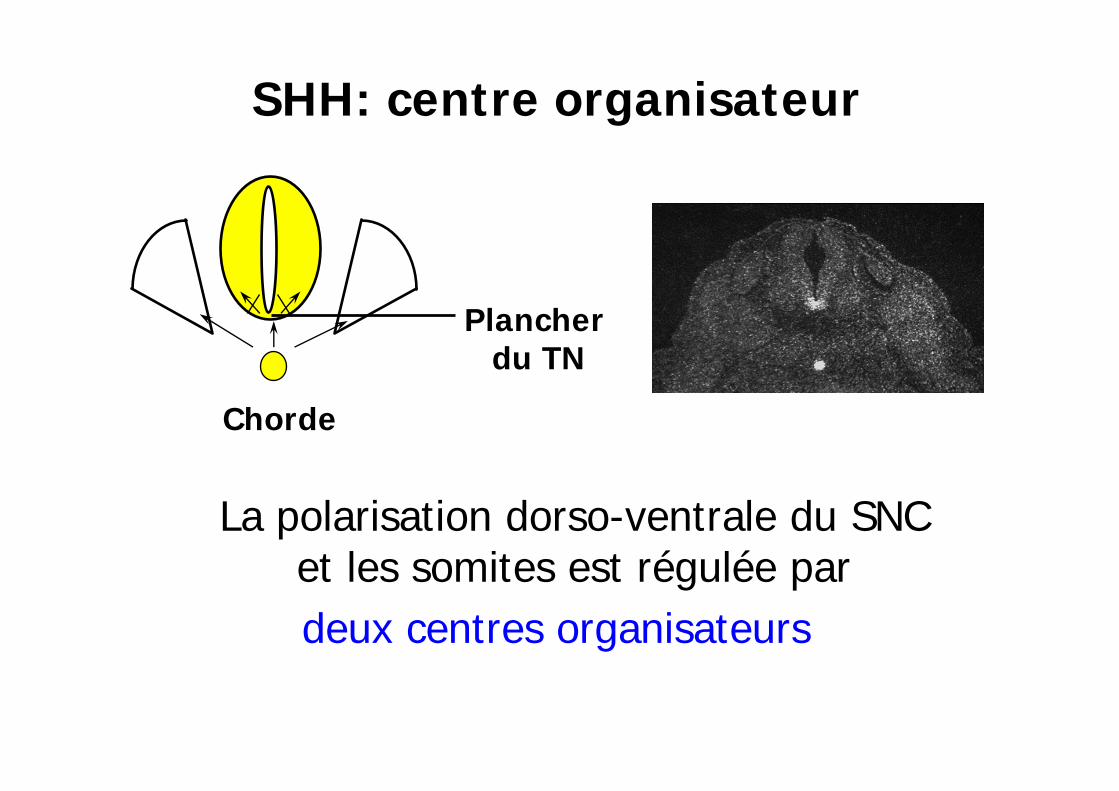
La polarisation dorso-ventrale du SNC et les somites est régulée pardeux centres organisateurs
Plancher du TN
Chorde
SHH: centre organisateur

Centre organisateur localisé dans le mésenchyme postérieur des membres
= Zone à Activité Polarisante
SHH contrôle la polarisation antéro-postérieure des membres

SHH: I dentification ventrale du SNC
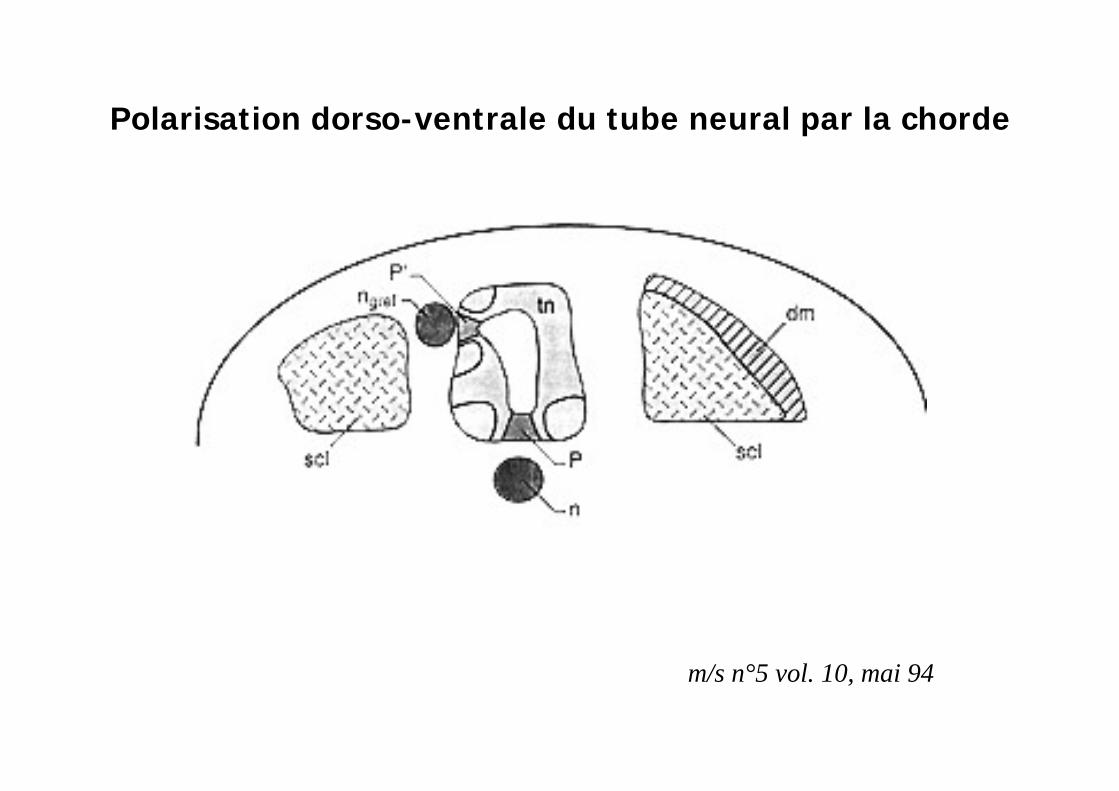
Polarisation dorso-ventrale du tube neural par la chorde
m/s n°5 vol. 10, mai 94

Polarisation antéropostérieure des membres par le
mésenchyme postérieur des bourgeons de membres
m/s n°5 vol. 10, mai 94

Polarisation antéropostérieure des membres par l acide rétinoique
Human embryology, Larsen

I nvalidation ciblée de Shh chez la souris
Hétérozygotes: normauxHomozygotes
Retard de croissance sévèreHPE, proboscis, diminution taille cerveau...Anomalies squelettiques sévères
Colonne vertébrale absente (persistance 5-6 cartilages costaux)Anomalies crâniofacialesAnomalies des membres (MI > MS): absence de doigts, absence ou fusion des os distaux
Anomalies cardiaques, pulmonaires, rénales et digestives
Chiang et al, Nature 1996; 383: 407-413

Holoprosencéphaliela face prédit le cerveau DeMyer 1964
Cyclopie Monophtalmie, Proboscis
Ethmocéphalie Hypotélorisme, Proboscis
Cébocéphalie Hypotélorisme, Narine unique
Agénésie Hypotélorisme, Fente labialemédiane
prémaxillaire
Dysmorphie atténuée Hypo- hypertélorismenez aplati, fente labialecolobome, incisive uniqueface normale
HPE alobaire
HPE semi-lobaireou lobaire
Non !

SHH, Odent et al, HMG 1999, 8: 1683
HPE3: 7q36 SHH
HPE semi-lobaireHypotélorismeFente médianeEpilepsie décès 13 mois
Q100H
HPE lobaireMicrocéphalie (-5SD)Diabète insipideRetard mental
E188Q
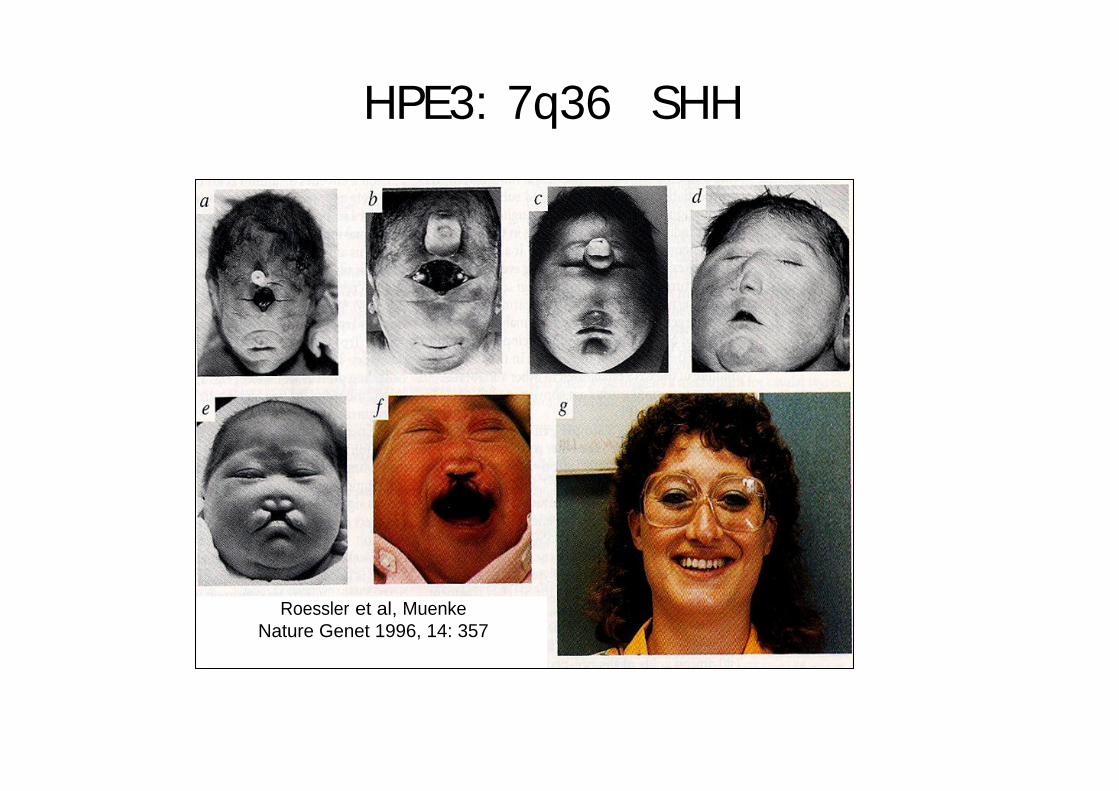
Roessler et al, MuenkeNature Genet 1996, 14: 357
HPE3: 7q36 SHH

Holoprosencéphalie: étiologies
Anomalies chromosomiques 25-45 %Trisomie 13 (50% des HPE, 70% des T13)Trisomie 18TriploidieAnomalies de structure: 12 loci
Association syndromique 18-25 %
Agents tératogènes
Isolée 30% formes familiales, 70% cas sporadiques 40%

Holoprosencéphalie: Au moins 12 loci
HPE1: 21q22.3 delHPE2: 2p21 del (dup)HPE3: 7q36 delHPE4: 18p11.3 delHPE5: 13q32 del/dupHPE6: 3p24-pter dupHPE7: 13q12-q14HPE8: 14q13 delHPE9: 20p13 HPE10: 1q42 dupHPE11: 5p HPE12: 6q26-qter dup
SIX3SHHTGIFZIC2
9q22.3 mut PATCHED1
3p21.31 mut TDGF1+
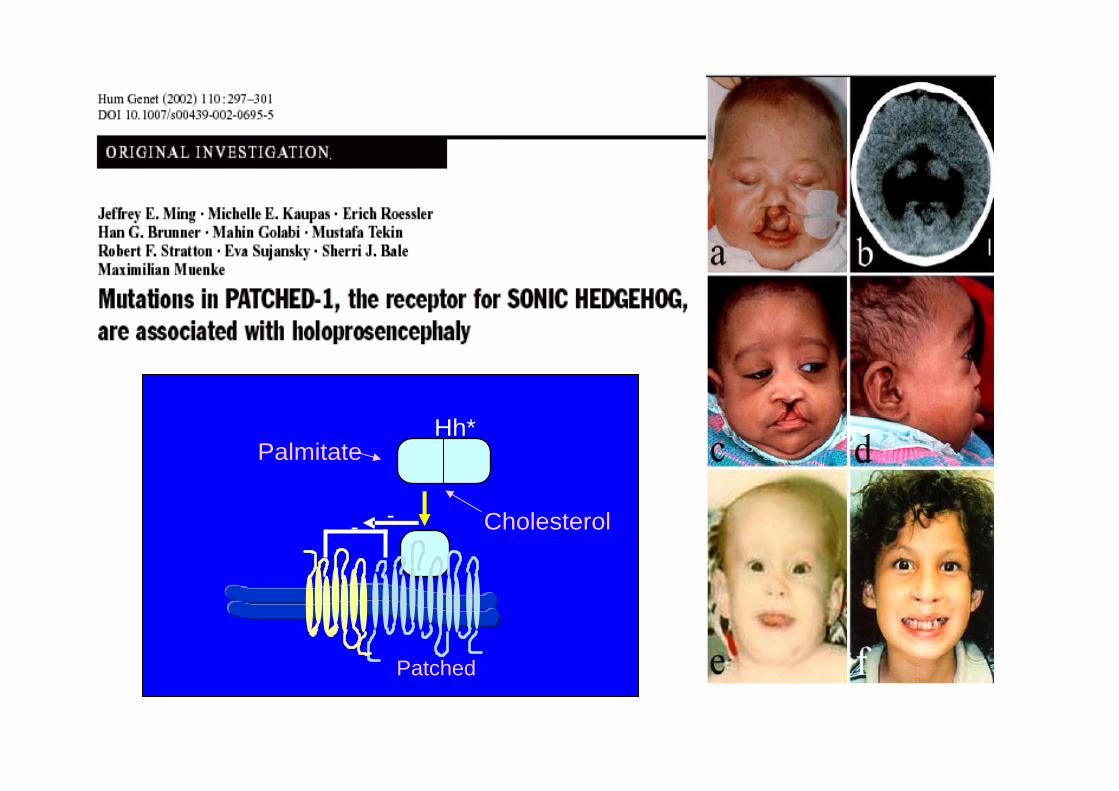
- - Cholesterol
Hh*Palmitate
Patched

DHCR7
Pallister-Hall, Greig, PPA
HoloprosencéphaliePolydactylieSmith-Lemli-OpitzHoloprosencéphalie
GorlinHoloprosencéphalie
Rubinstein-TaybiHoloprosencéphalie
GLI3
CBP
PTC1
Wnt4Wnt7A
TétramélieAnomalies membres
SHH
« hedgehogepathies »
Patched
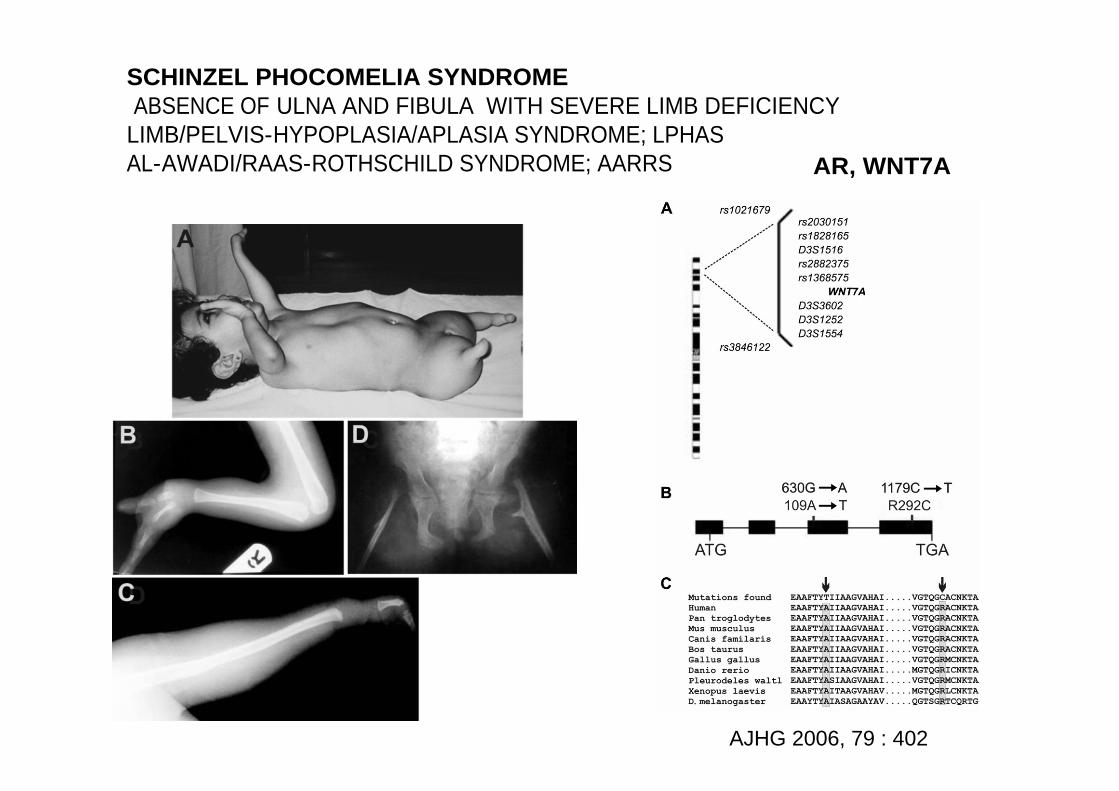
SCHINZEL PHOCOMELIA SYNDROMEABSENCE OF ULNA AND FIBULA WITH SEVERE LIMB DEFICIENCY
LIMB/PELVIS-HYPOPLASIA/APLASIA SYNDROME; LPHASAL-AWADI/RAAS-ROTHSCHILD SYNDROME; AARRS AR, WNT7A
AJHG 2006, 79 : 402



GÈNES HOMÉOTI QUES
Détermination des segments
Mutations: transformations homéotiques
Possèdent une homéoboite (180pb) dans leur partie 3 qui code pour un homéodomaine de 61 aa, qui définit la famille des gènes à boite homéo (ou homéobox)
Cet homéodomaine est formé de 3 hélices dont la 3ème est responsable de la liaison spécifique avec l ADN
Ils sont regroupés en clusters
Avec une colinéarité temporo spatialeCeux situés en 3 s expriment dans les régions antérieures,
sont les premiers à s exprimer et sont régulés par l acide rétinoïqueCeux situés en 5 s expriment dans les régions postérieures et
sont les derniers à s exprimer
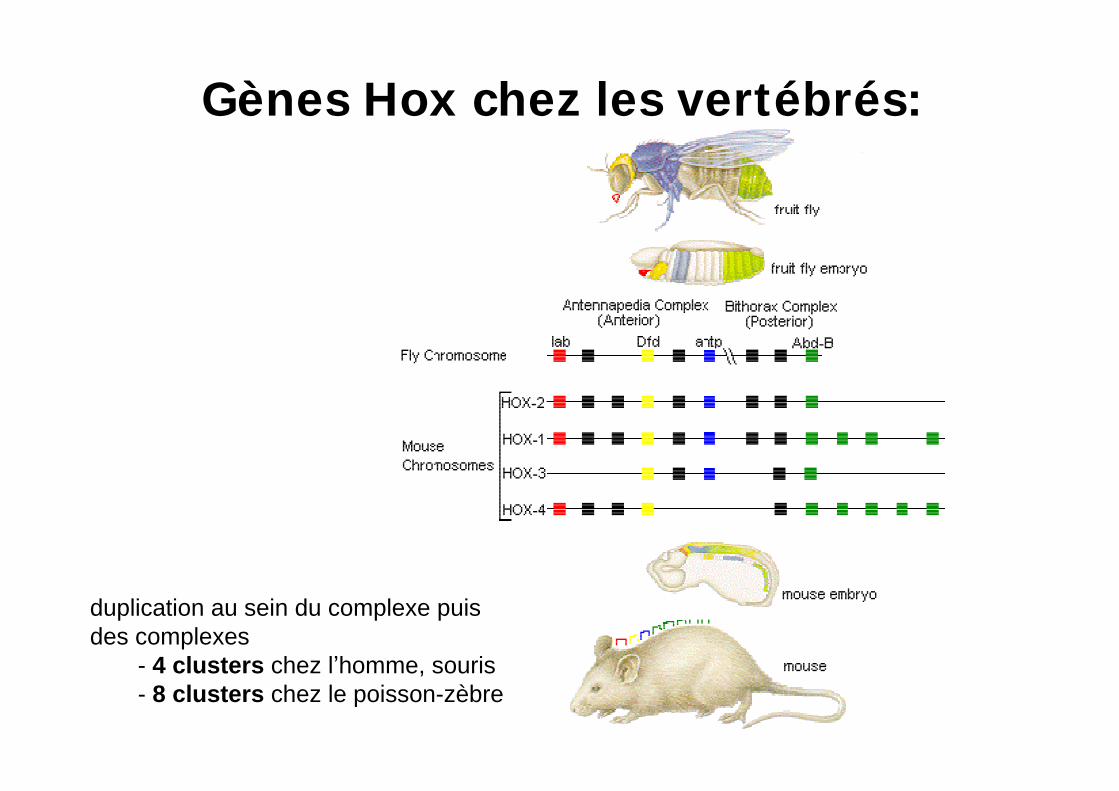
Gènes Hox chez les vertébrés:
duplication au sein du complexe puis des complexes
- 4 clusters chez l homme, souris- 8 clusters chez le poisson-zèbre

Mutations gènes HOX chez l homme et anomalies des membres
HOXA11, ADsynostose radio-cubitale avec thrombocytopénie
HOXA13 , AD HFG/HFU ou Guttmacher syndrome hypospadias , duplication génitale , pouces hypoplasiqueshypospadias polydactylie
HOXD13 , ADBrachydactylie, poysyndactylie, syndactylie

Hand-Foot-Genital (HFG)
Mortlock, Nat Genet 1997

Sympodactylie

HOXD13polysyndactylie
Goodman et al, PNAS 1997

syndactylie type V
AJHG 2007: 80:361

Développement des membresAxe proximo-distal
Fibroblast growth factors (FGFs) exprimés dans la crête apicale (AER)
P
D
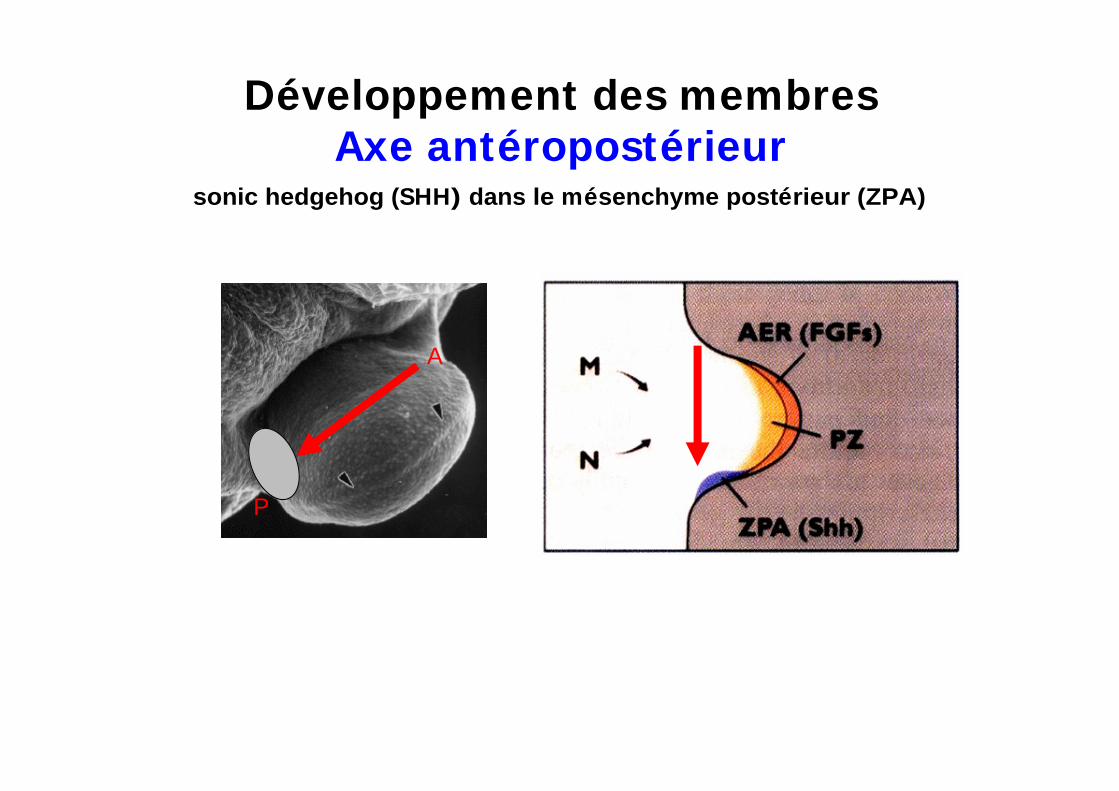
Développement des membresAxe antéropostérieur
sonic hedgehog (SHH) dans le mésenchyme postérieur (ZPA)
A
P

Développement des membresAxe dorso-ventral
BMPs, EN1 (ventral) Wnt7 (dorsal)
V
D
V
D
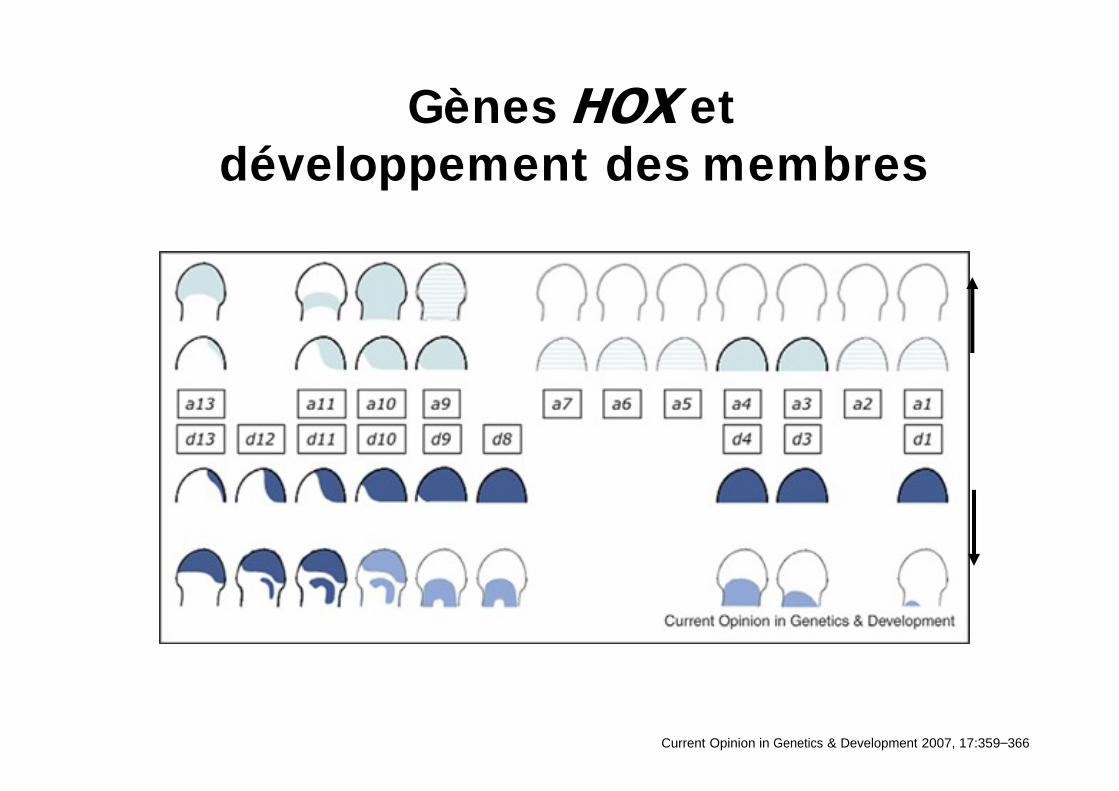
Gènes et développement des membres
Current Opinion in Genetics & Development 2007, 17:359 366
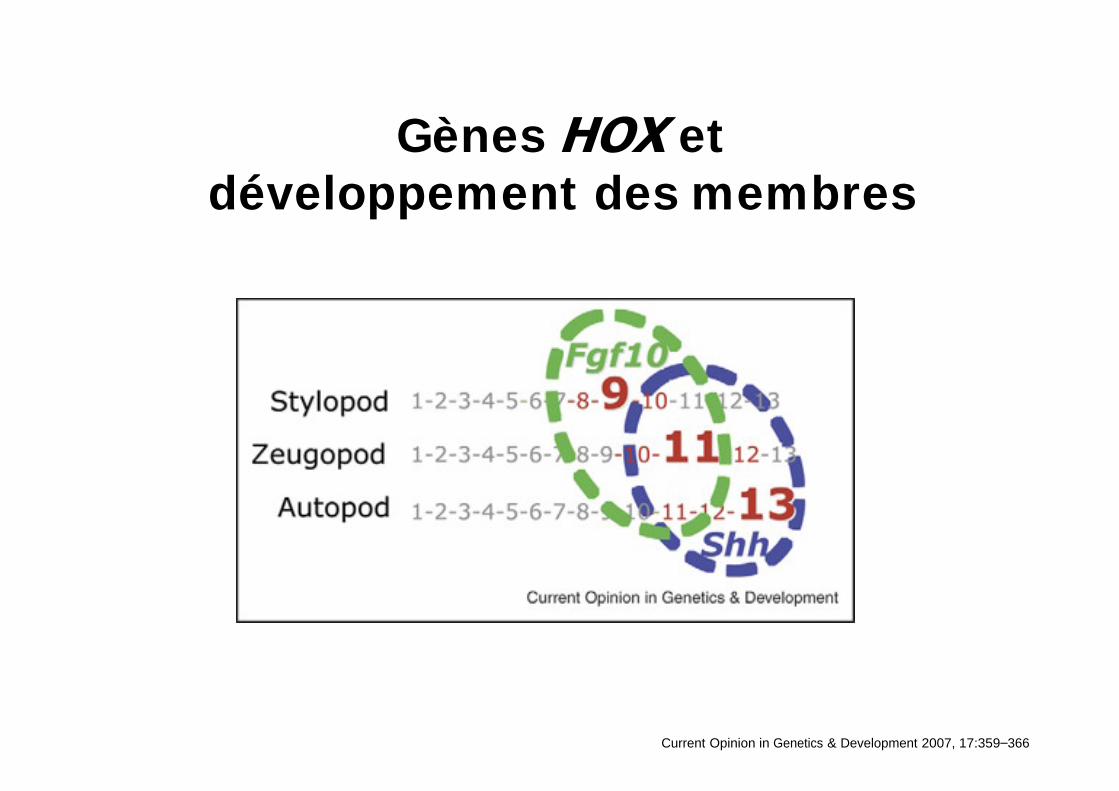
Gènes et développement des membres
Current Opinion in Genetics & Development 2007, 17:359 366

MOLÉCULES DU DEVELOPPEMENT EMBRYONNAIRE
Cadherin
RAS
MAPKKK
MAPK
FUSED
Wnt/Wingless Hedgehog/Sonic hedgehog
caténin
p120
caténin
SMADMAKK
DISHEVELD
GSK3
APC
caténin
LEF1 TCF GLISuH
Frizzeld Smoothened Patched
Cadherin
Notch
FGF-R
TGF -R
Delta FGF (9 protéines)
TGF :plus de 25 membres
NOYAU

GÈNES DU DÉVELOPPEMENT EMBRYONNAI RE
Complexité par nombre d homologues chez les vertébrés
Participent aux voies de signalisation du programme de développement embryonnaire
Voies de signalisation sélectionnées à différents moments de l embryogénèse, dans différents organes
Pathologies du développement embryonnaire : malformations
Pathologies acquises : hémopathies malignes (système embryonnaire)

Ce document à été crée avec Win2pdf disponible à http://www.win2pdf.com/frLa version non enregistrée de Win2pdf est uniquement pour évaluation ou à usage non commercial.







