
SEMANA DO CALOURO
SEMANA DO CALOURO
2/2016
2/2016
BEM-VINDOS A
BEM-VINDOS A
QUE O ESPETÁCULO COMECE...
APOSTILA TEORIA E EXERCÍCIOS
SEMANA DO CALOURO 2/2016

Agradecemos à Professora Elaine pelo imenso apoio prestado
durante a elaboração do nosso projeto. Poucos são aqueles que
com toda sua excelência conseguem despertar em nós a vontade
de crescer e de continuar seguindo nossos sonhos. Nossa jornada
no PET-Química tem sido algo maravilhoso, pois temos alguém
em quem nos espelhar. É uma honra tê-la como tutora e amiga.
Obrigado por suas broncas, por seu reconhecimento, por sua
amizade e dedicação, mas, principalmente, por sempre acreditar
em nós.

Sumário
1. Distribuição Eletrônica .................................................................................................... 1
1.1. Orbitais e Números Quânticos ..................................................................................... 1
1.2. Orbitais e Suas Energias .............................................................................................. 2
1.3. Spin Eletrônico e o Princípio de Exclusão de Pauli .................................................... 2
1.4. Configurações Eletrônicas ........................................................................................... 3
1.5. Regra de Hund ............................................................................................................. 4
1.6. Configurações Eletrônicas Condensadas ..................................................................... 4
1.7. Exercícios .................................................................................................................... 4
2. Conceitos Básicos de Ligação Química .......................................................................... 9
2.1. Ligações Químicas, símbolos de Lewis e regra do octeto. .......................................... 9
2.2. Polaridade da Ligação e Eletronegatividade ............................................................... 9
2.3. Desenhando Estruturas de Lewis ............................................................................... 10
2.4. Carga Formal ............................................................................................................. 11
2.5. Exceções à Regra do Octeto ...................................................................................... 12
2.6. Exercícios .................................................................................................................. 12
3. Geometria Molecular ..................................................................................................... 15
3.1. Modelo VSEPR Básico ............................................................................................. 16
3.2. Teoria da Ligação de Valência .................................................................................. 17
3.3. Exercícios .................................................................................................................. 23
4. Funções Inorgânicas ...................................................................................................... 24
4.1. Ácidos ........................................................................................................................ 24
4.2. Bases .......................................................................................................................... 29
4.3. Sais............................................................................................................................. 31
4.4. Óxidos ........................................................................................................................ 33
4.5. Exercícios .................................................................................................................. 36
5. Teorias Ácido-Base ....................................................................................................... 42

5.1. Teoria de Arrhenius ................................................................................................... 42
5.2. Teoria de Brᴓnsted-Lowry ........................................................................................ 42
5.3. Teoria de Lewis ......................................................................................................... 43
5.4. Teoria de Pearson: Ácidos e Bases Duros e Macios ................................................. 46
5.5. Exercícios .................................................................................................................. 47
6. Equilíbrio Químico ........................................................................................................ 49
6.1. Princípio de Le Chatelier ........................................................................................... 50
6.2. Exercícios .................................................................................................................. 53
7. Limite ............................................................................................................................ 55
7.1. Conceito ..................................................................................................................... 55
7.2. Propriedades .............................................................................................................. 57
7.2.1. Regra da soma ........................................................................................................ 57
7.2.2. Regra da diferença: ................................................................................................ 57
7.2.3. Regra do produto: .................................................................................................. 58
7.2.4. Regra da multiplicação por constante: ................................................................... 58
7.2.5. Regra do quociente: ............................................................................................... 58
7.2.6. Regra da potenciação: ............................................................................................ 58
7.3. Limites Laterais ......................................................................................................... 58
7.4. Definição de limite .................................................................................................... 61
7.5. Limites que envolvem infinito ................................................................................... 64
7.6. Exercícios .................................................................................................................. 70
8. Derivada ........................................................................................................................ 72
8.1. Definição ................................................................................................................... 72
8.2. Propriedades .............................................................................................................. 74
8.2.1. Regra da constante ................................................................................................. 74
8.2.2. Regra da soma ........................................................................................................ 74
8.2.3. Regra da subtração ................................................................................................. 75

8.2.4. Regra da multiplicação por um escalar .................................................................. 75
8.2.5. Regra do produto .................................................................................................... 75
8.2.6. Regra do quociente ................................................................................................ 75
8.2.7. Regra da potência ................................................................................................... 77
8.3. Derivadas Importantes ............................................................................................... 77
8.4. Aplicações ................................................................................................................. 78
8.4.1. Extremos de Funções ............................................................................................. 78
8.4.1.1. Teorema do Valor Extremo ................................................................................ 79
8.4.2. Teorema do Valor Médio ....................................................................................... 82
8.4.3. Teste da Primeira Derivada .................................................................................... 83
8.4.4. Teste da Segunda Derivada .................................................................................... 84
8.4.5. Problemas de Otimização ...................................................................................... 85
8.4.6. Outras Aplicações .................................................................................................. 88
8.5. Regra de L’Hospital ................................................................................................... 91
8.6. Exercícios .................................................................................................................. 92
9. Integral .......................................................................................................................... 95
9.1. Integral indefinida e definida ..................................................................................... 95
9.1.1. Integral Indefinida .................................................................................................. 95
9.1.1.1. Técnicas de Integração ....................................................................................... 97
9.1.2. Integral Definida .................................................................................................... 99
9.2. Integrais Importantes ............................................................................................... 102
9.3. Teorema Fundamental do Cálculo ........................................................................... 104
9.4. Aplicações ............................................................................................................... 105
9.4.1. Área ...................................................................................................................... 105
9.4.2. Comprimento de arco ........................................................................................... 107
9.5. Exercícios ................................................................................................................ 108
10. Regressão ................................................................................................................. 111

11. Referências Bibliográficas ....................................................................................... 113

1
1. Distribuição Eletrônica
Em 1926, o físico austríaco Erwin Schrödinger (1887-1961) propôs uma equação,
conhecida atualmente como a equação de Schrödinger, que incorpora tanto o comportamento
ondulatório como o de partícula do elétron. Seu trabalho abriu uma nova maneira de lidar com
partículas subatômicas conhecidas como mecânica quântica ou mecânica ondulatória.
A resolução da equação de Schrödinger leva a uma série de funções matemáticas
chamadas funções de onda que descrevem a questão ondulatória do elétron. Para o átomo de
hidrogênio, as energias permitidas são as mesmas previstas pelo modelo de Bohr. Contudo, o
modelo de Bohr supõe que o elétron está em órbitas específicas enquanto que o modelo da
mecânica quântica considera o princípio da incerteza para o comportamento do elétron e
introduz o conceito de probabilidade do elétron ser encontrado em certa região do espaço em
determinado momento. A densidade eletrônica é outra maneira de expressar probabilidade: as
regiões onde existe alta probabilidade de encontrar o elétron são regiões de alta densidade
eletrônica.
1.1. Orbitais e Números Quânticos
A solução da equação de Schrödinger para o átomo de hidrogênio produz um conjunto
de funções de onda e energias correspondentes. Essas funções são chamadas orbitais. Cada
orbital descreve uma distribuição específica de densidade eletrônica no espaço, como
determinado pela probabilidade de densidade. Cada orbital, consequentemente, tem energia e
forma características. O modelo da mecânica quântica não pode ser descrito por órbitas
porque o movimento do elétron em um átomo não pode ser medido ou localizado com
precisão (princípio da incerteza de Heisenberg).
O modelo de Bohr utilizada apenas um número quântico, n, para descrever certa
órbita. O modelo da mecânica quântica usa três números quânticos, n, l e ml, para descrever
um orbital.
I. O número quântico principal, n, pode ter valores positivos inteiros (1, 2, 3,...). À
medida que n aumenta, o orbital torna-se maior, e o elétron passa mais tempo distante

2
do núcleo, o que significa também que esse elétron tem energia alta e, por isso, está
menos fortemente ligado ao núcleo.
II. O segundo número quântico - número quântico azimutal, l - pode ter valores inteiros
de 0 a n-1 para cada valor de n. Esse número quântico define o formato do orbital. O
valor de l para determinado orbital é normalmente assinalado pelas letras s, p, d e f,
correspondendo aos valores de l de 0, 1, 2 e 3, respectivamente.
III. O número quântico magnético pode ter valores inteiros entre l e -l, inclusive zero.
Esse número quântico descreve a orientação do orbital no espaço.
O conjunto de orbitais com o mesmo valor de n é chamado nível eletrônico e o
conjunto de que tem os mesmo valores de n e l é chamado de subnível.
1.2. Orbitais e Suas Energias
O modelo da mecânica quântica não seria muito útil se não pudéssemos estender aos
outros átomos o que aprendemos sobre o hidrogênio. Apesar das formas dos orbitais dos
átomos serem as mesmas daquelas para o hidrogênio, a presença de mais de um elétron muda
bastante a energia dos orbitais. Em um átomo polieletrônico, a repulsão elétron-elétron faz
com que os diferentes subníveis estejam em diferentes níveis de energia. Por exemplo, o
subnível 2s é mais baixo em energia que o 2p. A ideia importante é esta: em um átomo
polieletrônico, para certo valor de n, a energia de um orbital aumenta com o aumento do
valor de l. Vale ressaltar que os orbitais com a mesma energia são chamados de orbitais
degenerados.
1.3. Spin Eletrônico e o Princípio de Exclusão de Pauli
Em 1925, os físicos holandeses George Uhlenbeck e Samuel Goudsmit postularam que
os elétrons tinham uma propriedade intrínseca, chamada spin eletrônico. O elétron
comportava-se como se fosse uma esfera minúscula rodando em torno de seu próprio eixo.
O spin eletrônico é quantizado e essa observação levou a atribuição de um novo
número quântico para o elétron. Esse novo número quântico, o número quântico magnético

3
de spin, é simbolizado por ms, e tem apenas dois valores possíveis, +1/2 e -1/2, que foi
inicialmente interpretado como indicador dos dois sentidos opostos nos quais o elétron pode
girar.
O spin eletrônico é crucial para o entedimento das estruturas eletrônicas dos átomos.
Em 1925, o físico austríaco Wolfgang Pauli descobriu o princípio que governa a distribuição
de elétrons em átomos polieletrônicos. O Princípio da Exclusão de Pauli afirma que dois
elétrons em um átomo não podem ter o conjunto de quatro números quânticos n, l, ml e ms,
iguais. Se quisermos colocar mais de um elétron em um orbital e satisfazer o princípio da
exclusão de Pauli, a única escolha é assinalar valores distintos de ms para os elétrons. Como
existem apenas dois destes valores, concluímos que um orbital pode receber o máximo de
dois elétrons, e eles devem ter spins opostos.
1.4. Configurações Eletrônicas
A maneira na qual os elétrons são distribuídos entre os vários orbitais de um átomo é
chamada configuração eletrônica. A mais estável configuração eletrônica, ou estado
fundamental, de um átomo é aquela na qual os elétrons estão nos estados mais baixos
possíveis de energia. Se não existissem restrições nos possíveis valores para os números
quânticos dos elétrons, todos os elétrons se aglomerariam no orbital 1s por ser o mais baixo
em energia. Entretanto, o Princípio da Exclusão de Pauli limita o máximo de dois elétrons por
orbital, assim os orbitais são preenchidos por ordem crescente de energia, com não mais que
dois elétrons por orbital.

4
Tabela 01 – Configurações Eletrônicas
Nível Camada Nº máximo de elétrons Subníveis conhecidos
1º K 2 1s
2º L 8 2s e 2p
3º M 18 3s, 3p e 3d
4º N 32 4s, 4p, 4d e 4f
5º O 32 5s, 5p, 5d e 5f
6º P 18 6s, 6p e 6d
7º Q 2 (alguns autores admitem até 8) 7s 7p
1.5. Regra de Hund
A regra de Hund afirma que para orbitais degenerados, a menor energia será obtida
quando o número de elétrons com o mesmo spin for maximizado. Isso significa que os
elétrons ocuparão individualmente os orbitais até a máxima extensão possível, com o mesmo
número quântico magnético de spin. Diz-se que os elétrons distribuídos dessa forma possuem
spin paralelos.
1.6. Configurações Eletrônicas Condensadas
Ao escrever a configuração eletrônica condensada de um elemento, a configuração eletrônica
do gás nobre de menor número atômico mais próximo é representada por seu símbolo
químico entre colchetes. Por exemplo, podemos escrever a configuração eletrônica do lítio
como: [He] 2s1.
1.7. Exercícios
01) (ITA-2002) Considere as seguintes configurações eletrônicas de espécies no estado
gasoso:
I. 1s2 2s2 2p1.

5
II. 1s2 2s2 2p3 .
III. 1s2 2s2 2p4 .
IV. 1s2 2s2 2p5 .
V. 1s2 2s2 2p5 3s1 .
Assinale a alternativa ERRADA.
a) As configurações I e IV podem representar estados fundamentais de cátions do segundo
período da Tabela Periódica.
b) As configurações II e III podem representar tanto um estado fundamental como um estado
excitado de átomos neutros do segundo período da Tabela Periódica.
c) A configuração V pode representar um estado excitado de um átomo neutro do segundo
período da Tabela Periódica.
d) As configurações II e IV podem representar estados excitados de átomos neutros do
segundo período da Tabela Periódica.
e) As configurações II, III e V podem representar estados excitados de átomos neutros do
segundo período da Tabela Periódica.
02) (Uniube-2001) Dos íons, abaixo, aquele(s) que possui(em) o seu último elétron
representado em 2 p6 , de acordo com o diagrama de Pauling, é (são):
I) 11 Na+
II) 19 K+
III) 20 Ca2+
IV) 9 F–
Assinale a afirmativa correta:
a) II, III e IV
b) I e IV
c) I e III
d) II e III
3) (FGV - SP-2007) O titânio e seus compostos são amplamente empregados tanto na área
metalúrgica como na produção de cosméticos e fármacos. No Brasil, os minérios são

6
extraídos na forma de óxidos, rutilo (TiO2) e ilmenita (FeTiO3). O titânio apresenta o mesmo
estado de oxidação nesses dois minérios. O número de oxidação do titânio e a configuração
eletrônica da camada de valência do ferro no estado de oxidação em que se encontra na
ilmenita são, respectivamente,
a) +2 e 3d6 4s2 .
b) +2 e 3d4 4s2 .
c) +3 e 3d5 .
d) +4 e 3d6 .
e) +4 e 3d4 .
4) (Mack-2003) Uma distribuição eletrônica possível para um elemento X, que pertence à
mesma família do elemento bromo, cujo número atômico é igual a 35, é:
a) 1s2 , 2s2 , 2p5
b) 1s2 , 2s2 , 2p6 , 3s2 , 3p1
c) 1s2 , 2s2 , 2p2
d) 1s2 , 2s2 , 2p6 , 3s1
e) 1s2 , 2s2 , 2p6 , 3s2 , 3p6 , 4s2 , 3d5
5) (ITA-2003) Considere as seguintes espécies químicas no estado gasoso, bem como os
respectivos átomos assinalados pelos algarismos romanos:
I. ONNO2 II. FClO2 III. ICl3 IV. F4ClO-
Os orbitais híbridos dos átomos assinalados por I, II, III e IV são respectivamente:
a) sp2 , sp3 , dsp3 e d2 sp3 .
b) sp2 , sp2 , sp3 e dsp3 .
c) sp3 , dsp3, d2 sp3 e sp3 .
d) sp3 , sp2 , dsp3 e d2 sp3 .
6) (Faculdades Positivo-1998) A análise da distribuição eletrônica dos elementos, ao longo da
Classificação Periódica, fornece-nos uma série de características quanto ao comportamento

7
químico destes elementos. Sendo dadas as distribuições eletrônicas para os átomos dos
elementos genéricos A, B, C e D, no estado fundamental, é correto afirmar:
A – 1s2 2s2 2p6 3s1
B – 1s2 2s2 2p6 3s2 3p5
C – 1s2 2s2 2p6 3s2 3p6 4s2 3d10 4p5
D – 1s2 2s2 2p6 3s2 3p1
a) O átomo do elemento A possui o maior valor para o primeiro potencial de ionização.
b) A distribuição eletrônica do átomo do elemento B corresponde a um metal do grupo dos
metais alcalinoterrosos.
c) O íon estável correspondente ao átomo do elemento A possui distribuição eletrônica 1s2
2s2 2p6 .
d) O átomo do elemento C possui 5 elétrons em sua camada de valência.
e) O átomo do elemento D apresenta o maior valor relativo à eletronegatividade.
7) (UFF/1-2000) Conhece-se, atualmente, mais de cem elementos químicos que são, em sua
maioria, elementos naturais e, alguns poucos, sintetizados pelo homem. Esses elementos estão
reunidos na Tabela Periódica segundo suas características e propriedades químicas. Em
particular, os Halogênios apresentam:
a) o elétron diferenciador no antepenúltimo nível
b) subnível f incompleto
c) o elétron diferenciador no penúltimo nível
d) subnível p incompleto
e) subnível d incompleto
8) (UPE-2001) Entre as alternativas abaixo, relacionadas com as ligações químicas, escolha a
verdadeira.
a) Os orbitais híbridos sp3d2 direcionam-se para os vértices de uma pirâmide pentagonal.
b) O ângulo entre os átomos de hidrogênio na molécula do gás sulfídrico é maior que entre os
átomos de hidrogênio na molécula da água.

8
c) O CF62- tem uma geometria octaédrica com os átomos de flúor direcionados para os
vértices do octaedro.
d) O paramagnetismo da molécula do oxigênio experimentalmente comprovado sinaliza para
uma estrutura de Lewis com elétrons desemparelhados.
e) A molécula do oxigênio é diamagnética e não paramagnética, de tal forma que a estrutura
de Lewis, na qual todos os elétrons aparecem em pares, está correta.

9
2. Conceitos Básicos de Ligação Química
2.1. Ligações Químicas, símbolos de Lewis e regra do octeto.
Sempre que átomos ou íons estão muito ligados uns aos outros, dizemos que existe
uma ligação química entre eles. Existem três tipos gerais de ligações químicas: iônica,
covalente e metálica. O termo ligação iônica refere-se às forças eletrostáticas que existem
entre íons de cargas de sinais contrários. Os íons devem ser formados a partir átomos pela
transferência de elétrons de um átomo para o outro. Uma ligação covalente resulta do
compartilhamento de elétrons entre os átomos. As ligações metálicas são encontradas em
metais como o cobre, ferro e alumínio. Os elétrons ligantes estão relativamente livres para
mover-se pela estrutura tridimensional do metal.
Os elétrons envolvidos em ligações químicas são os elétrons de valência, os
localizados no nível incompleto mais externo de um átomo. O químico americano G. N.
Lewis (1875-1946) sugeriu uma maneira simples de mostrar os elétrons de valência dos
átomos e seguir o rastro deles durante a formação da ligação, usando o que conhecemos hoje
como símbolos de Lewis. O símbolo de Lewis para um elemento consiste do símbolo
químico do elemento mais um ponto para cada elétron de valência.
Os átomos frequentemente ganham, perdem ou compartilham seus elétrons para
atingir o número de elétrons do gás nobre mais próximo na tabela periódica. Os gases nobres
tem distribuições eletrônicas muito estáveis, como evidenciado por suas altas energias de
ionização, baixas afinidades por elétrons adicionais e deficiência geral de reatividade química.
Como todos os gases nobres (exceto o He), possuem oito elétrons de valência, e muitos
átomos sofrendo reações também terminam com oito elétrons de valência. Essa observação
levou a uma norma conhecida como a regra do octeto: os átomos tendem a ganhar, perder
ou compartilhar elétrons até que eles estejam circundados por oito elétrons de valência.
Existem muitas exceções a regra do octeto, mas ela fornece uma estrutura útil para introduzir
muitos conceitos importantes de ligação.
2.2. Polaridade da Ligação e Eletronegatividade

10
O conceito de polaridade da ligação ajuda a descrever o compartilhamento de
elétrons entre os átomos. Uma ligação covalente apolar é aquela na qual os elétrons estão
igualmente compartilhados entre dois átomos. Em uma ligação covalente polar um dos
átomos exerce maior atração pelos elétrons ligantes que o outro. Se a diferença na habilidade
relativa em atrair elétrons é grande o suficiente, uma ligação iônica é formada.
A eletronegatividade é definida como a habilidade de um átomo em atrair elétrons
para si em certa molécula. A eletronegativade de um átomo em um molécula está relacionada
a sua energia de ionização e a sua afinidade eletrônica, que são propriedades de átomos
isolados. A energia de ionização mede quão fortemente um átomo segura seus elétrons. De
forma similara, a afinidade eletrônica é uma medida de quão facilmente um átomo atrai
elétrons adicionais. Um átomo com afinidade eletrônica muito negativa e alta energia de
ionização tanto atrairá elétrons de outros átomos quanto resistirá em ter seus elétrons atraídos
por outros, além do que será altamente eletronegativo. Vale ressaltar que quanto maior a
diferença na eletronegatividade entre os átomos, mais polares serão suas ligações.
2.3. Desenhando Estruturas de Lewis
As estruturas de Lewis podem nos ajudar a entender as ligações em muitos compostos
e são bastante usadas quando discutimos as propriedades das moléculas. Para desenhá-las, se
deve seguir um procedimento:
I. Some os elétrons de valência de todos os átomos. Para um ânion, adicione um elétron
para cada carga negativa. Para um cátion, subtraia um elétron para cada carga positiva.
Apenas o número total de elétrons é importante.
II. Escreva os símbolos para os átomos a fim de mostrar quais átomos estão ligados
entre si e una-os com uma ligação simples.
III. Complete os octetos dos átomos ligados ao átomo central.
IV. Coloque qualquer sobra de elétrons no átomo central, mesmo que ao fazer isso você
provoque mais de um octeto.
V. Se não existem elétrons suficientes para dar ao átomo central um octeto, tente
ligações múltiplas. Use um ou mais dos pares de elétrons não compartilhados nos
átomos ligados ao átomo central para formar ligações duplas ou triplas.

11
Como exemplo, tente desenhar a molécula de PCl3.
2.4. Carga Formal
Quando desenhamos a estrutura de Lewis, estamos descrevendo como os elétrons
estão distribuídos em uma molécula ou íon. Em alguns casos podemos desenhar várias
estruturas de Lewis diferentes que obedecem à regra do octeto. Para decidir qual estrutura é a
mais razoável, pode ser feito um tipo de 'contabilidade' dos elétrons de valência para
determinar a carga formal de cada átomo em cada estrutura. A carga formal de um átomo é a
carga que um átomo teria em uma molécula se todos os outros átomos tivessem a mesma
eletronegatividade. Para calcular a carga formal em qualquer átomo em uma estrutura de
Lewis, atribuímos os elétrons aos átomos como a seguir:
I. Todos os elétrons não compartilhados (não-ligantes) são atribuídos ao átomo no qual
estão localizados.
II. Metade dos elétrons ligantes é atribuída a cada átomo na ligação.
A carga formal de um átomo é igual ao número de elétrons de valência no átomo
isolado menos o número de elétrons atribuídos ao átomo na estrutura de Lewis. Vamos
ilustrar as regras calculando as cargas formais nos átomos de C e de N no íon cianeto, CN-,
que tem a seguinte estrutura de Lewis:
Para o átomo de C, existem dois elétrons não-ligantes e três elétrons dos seis na
ligação tripla, dando um total de 5. O número de elétrons de valência em um átomo neutro de
C é 4. Portanto, a carga formal no carbono é 4 - 5 = -1. Para N, existem 2 elétrons não-
ligantes e 3 elétrons da ligação tripla. Como o número de elétrons de valência em um átomo

12
neutro de N é 5, sua carga formal é igual a 0. A soma das cargas formais deve ser igual a
carga do íon, quando for íon, e igual a zero, quando tivermos uma molécula.
Algumas vezes uma única estrutura de Lewis é inadequada para representar uma
molécula (ou íon) em particular. Em tais situações descrevemos a molécula como usando duas
ou mais estruturas de ressonância para a molécula. A molécula é vista como uma mistura
dessas estruturas múltiplas de ressonância. As estruturas de ressonância são importantes na
descrição das ligações na molécula orgânica do benzeno, C6H6.
2.5. Exceções à Regra do Octeto
A regra do octeto é tão simples e útil em introduzir conceitos básicos de ligação que
poderíamos afirmar que ela é sempre obedecida. A regra do octeto é falha para compostos
iônicos, covalentes e metálicos. As exceções covalentes nas quais a regra do octeto atua de
forma precária, são:
I. moléculas com número ímpar de elétrons.
II. moléculas nas quais um átomo tem menos de um octeto, ou seja, moléculas deficientes
em elétrons.
III. moléculas nas quais um átomo tem mais de um octeto, ou seja, moléculas com
expansão do octeto.
Nesse último caso, visualizamos os orbitais vazios d do átomo grande sendo usados
para 'expandir' o nível de valência do átomo. Octetos expandidos são observados para átomos
do terceiro período e dos períodos subsquentes da tabela periódica, para os quais os orbitais d
de baixa energia estão disponíveis.
2.6. Exercícios
01)
a) O que são elétrons de valência?
b) Quantos elétrons de valência um átomo de nitrogênio possui?

13
c) Um átomo tem a configuração eletrônica 1s2 2s2 2p6 3p2. Quantos elétrons de valência o
átomo tem?
02) Usando os símbolos de Lewis, faça um diagrama da reação entre os átomos de magnésio e
oxigênio para formar a substância iônica MgO.
03)
a) Qual o significado de ligação covalente? Dê três exemplos de ligação covalente.
b) Usando os símbolos e as estruturas de Lewis, faça um diagrama da formação do SiCl4 a
partir dos átomos Si e Cl.
04)
a) Como uma molécula polar difere de uma apolar? Os átomos X e Y tem diferentes
eletronegatividades. A molécula diatômica X-Y será necessariamente polar? Explique.
05) Quais das seguintes ligações são polares? Qual é o átomo mais eletronegativo em cada
ligação polar?
a) P-O;
b) S-F;
c) Br-Br;
d) O-Cl?
06) Desenhe as estruturas de Lewis para os seguintes compostos:
a) SiH4;
b) CO;
c) SF2;
d) H2SO4;
e) ClO2-2;
f) NH2OH.
07)

14
a) Use o conceito de ressonância para explicar por que as seis ligações C-C no benzeno são
iguais em comprimento.
b) Os comprimentos das ligações no C-C no benzeno são mais curtos que os de ligações
simples, mas mais longos que os de ligações duplas. Use o modelo de ressonância para
explicar essa observação,
08) Desenhe as estruturas de Lewis para cada um dos seguintes íons ou moléculas. Identifique
os que não obedecem a regra do octeto e explique por que isso ocorre.
a) NO;
b) ICl2;
c) SO2;
d) BCl3;
e) XeF4.

15
3. Geometria Molecular
As estruturas de Lewis mostram apenas as ligações entre os átomos e a presença de
pares isolados. Elas não mostram o arranjo tridimensional dos átomos no espaço.
Figura 1 – Nomes das formas de moléculas e a representação da estrutura com os pares isolados (arranjo
eletrônico) e sem os pares isolados (geometria molecular).

16
3.1. Modelo VSEPR Básico
Começa-se por moléculas simples formadas por um átomo central ao qual se unem os
demais átomos. O modelo da repulsão dos pares de elétrons da camada de valência (modelo
VSEPR) amplia a teoria da ligação química de Lewis para explicar as formas das moléculas
(figura 1), adicionando regras que explicam os ângulos das ligações. O modelo baseia-se na
ideia de que, como os elétrons se repelem, os pares de elétrons de ligação tendem a se afastar
o máximo possível.
Tendo identificado o arranjo que localiza os pares de elétrons na posição “mais
distante” (ligações e pares isolados do átomo central), que é chamado de arranjo eletrônico na
molécula, há a determinação da posição dos átomos e identificação da forma da molécula.
O BeCl2 é molécula com apenas dois átomos ligados ao átomo central. A estrutura de
Lewis é:
Não há pares isolados no átomo central. A posição na qual os pares ligantes estão os
mais afastados possível é quando eles se encontram em lados opostos do átomo de Be e o
arranjo dos elétrons é linear. O modelo VSEPR prediz a forma linear para a molécula de
BeCl2, essa forma é confirmada experimentalmente.
No modelo de VSEPR, não existe distinção entre ligações simples ou múltiplas: uma
ligação múltipla é tratada como uma região de alta concentração de elétrons. Em outras
palavras, os dois pares de elétrons de uma ligação dupla permanecem juntos e repelem outras
ligações ou pares isolados como se fossem uma unidade. Da mesma forma funciona para uma
ligação tripla. Assim, a molécula de dióxido de carbono (CO2) :
Tem estrutura semelhante à da molécula de BeCl2, mesmo com ligações duplas.
Quando existe mais de um átomo central, as ligações de cada átomo são tratadas
independentemente. Como por exemplo, para predizer a forma de uma molécula de eteno

17
(etileno), CH2=CH2, consideramos cada átomo de carbono separadamente. O arranjo ao redor
de cada átomo de carbono é trigonal planar.
De acordo com o modelo VSEPR, as regiões de alta concentração de elétrons se
posicionam de forma a ficar mais afastadas possível. Os pares de elétrons de uma ligação
múltipla são tratados com uma unidade. A forma da molécula é identificada pela posição
relativa de seus átomos.
Todas as regiões de densidade eletrônica elevada, pares de elétrons isolados e ligantes,
são incluídas na descrição do arranjo eletrônico. Todavia, somente a posição dos átomos são
considerados quando descrevemos a forma de uma molécula.
Para explicar o fato de que os ângulos de ligação em moléculas com pares isolados são
geralmente menores do que o esperado, o modelo VSEPR trata os pares isolados como
exercendo maior repulsão do que os pares de ligação. Em outras palavras, o par isolado
empurra os átomos ligados ao átomo central uns contra os outros. Uma possível explicação
desse fato é que a nuvem eletrônica de um par isolado ocupa um volume maior do que a de
um par ligado que está preso a dois átomos, não a um. A repulsão é exercida na ordem: par
isolado – par isolado > par isolado – átomo > átomo – átomo.
Exceções das regras de VSEPR também são encontradas para átomos tão grandes que
os pares de elétrons isolados não afetam a forma da molécula. Por exemplo, o íon SeCl62- é
octaédrico, mesmo com átomo de Se contendo um par isolado além de ligações para seis
átomos.
3.2. Teoria da Ligação de Valência
O modelo de Lewis trata-se de uma modelo com elétrons localizados. Entretanto, a
partir da dualidade onda – partícula do elétron, que a posição de um elétron em um átomo não
pode ser descrita de forma precisa, mas somente em termos de probabilidade de encontra-lo
em algum lugar do espaço definido por orbitais. O mesmo princípio se aplica aos elétrons das
moléculas, excerto que eles estão distribuídos por mais de um átomo.
A primeira descrição da ligação covalente em termos de orbitais atômicos foi chamada
de Teoria da Ligação de Valência, modelo quanto-mecânico da distribuição dos elétrons pelas
ligações que ultrapassa a teoria de Lewis e o modelo VSEPR.

18
Ligações Sigma e Pi
Na teoria da ligação de valência supomos que, quando dois átomos de hidrogênio (H)
se aproximam, o par de elétrons 1s (descrito como ↿⇂) e os orbitais atômicos se fundem. A
distribuição de elétrons resultante apresenta forma de uma salsicha, tem densidade eletrônica
acumulada entre os dois núcleos e é chamada de “ligação σ (sigma) ”. Formalmente, uma
ligação σ é simetricamente cilíndrica (é igual em todas as direções ao longo do eixo) e não
tem um plano nodal contendo o eixo internuclear. A fusão dos orbitais atômicos é chamada
superposição de orbitais, e quando maior a superposição mais forte é a ligação. Os dois
elétrosn
Figura 2 – Superposição dos orbitais na molécula de H2, F2e HF.
Todas as ligações covalentes simples são ligações σ. Os elétrons que se ligam se
espalham por toda a região do espaço ao redor da superfície-limite.
Quando analisamos a molécula de N2 dois dos orbitais 2p de cada átomo de nitrogênio
(2px e 2py) são perpendiculares ao eixo internuclear e cada um deles contém um elétron
desemparelhado. Quando esses elétrons, um de cada orbital p, se emparelharem, seus orbitais
só podem se sobrepor lado a lado (paralelamente). Esse tipo de ligação leva o nome de

19
“ligação π (pi), uma ligação em que dois elétrons estão em dois lobos, um de cada eixo
internuclear. Mas formalmente, uma ligação π tem um único plano nodal sobre os eixos
internucleares.
Na teoria da ligação de valência, supomos que as ligações se formam quando elétrons
desemparelhados de orbitais atômicos da camada de valência formam pares. Os orbitais
atômicos que eles ocupam se sobrepõem cabeça-cabeça para formar ligações σ ou
lateralmente para formar ligações π.
Figura 3 – Comportamento dos orbitais da molécula de N2.
Promoção de elétrons e hibridização dos orbitais.
O átomo de carbono tem configuração [He]2s22px12py
1 com quatro elétrons de
valência. No entanto, dois elétrons já estão emparelhados e somente os dois orbitais 2p
incompletos estão disponíveis para ligação. Porém, sabemos experimentalmente que o
carbono realiza quatro ligações. Para entender e solucionar a questão, os orbitais s e p são
ondas de densidade eletrônica centradas no núcleo do átomo. Pode-se imaginar que os quatros
orbitais interferem uns nos outros e produzem novos arranjos quando se cruzam. Esses novos
arranjos são chamados orbitais hibridizados. Cada um dos orbitais híbridos formam-se pela

20
combinação linear de quatro orbitais atômicos. Os quatro orbitais híbridos só diferem na
orientação, cada um apontando para o vértice de um tetraedro (no caso do carbono).Esses
quatro orbitais hibridizados são chamados de sp3 porque são formados a partir de um orbital s
e três orbitais p. Esses orbitais sp3 são degenerados, isto é, possuem a mesma quantidade
energia.
Figura 4 – Diagrama de energia dos orbitais hibridizados.
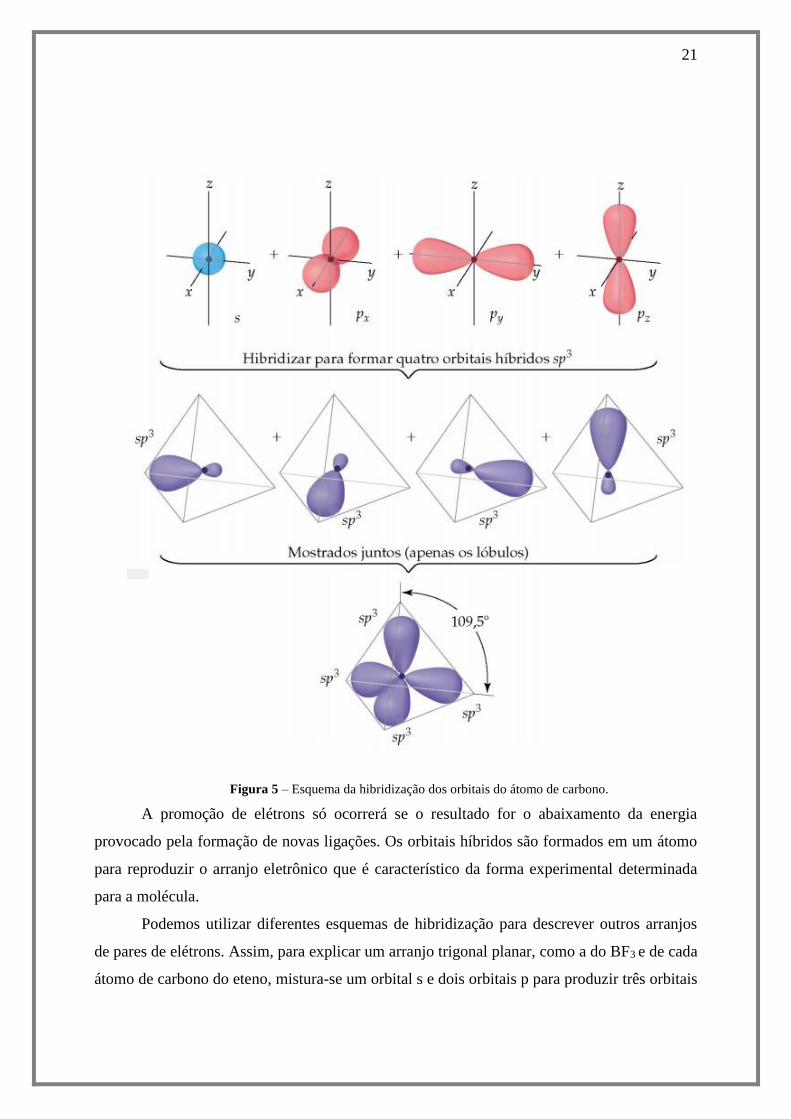
21
Figura 5 – Esquema da hibridização dos orbitais do átomo de carbono.
A promoção de elétrons só ocorrerá se o resultado for o abaixamento da energia
provocado pela formação de novas ligações. Os orbitais híbridos são formados em um átomo
para reproduzir o arranjo eletrônico que é característico da forma experimental determinada
para a molécula.
Podemos utilizar diferentes esquemas de hibridização para descrever outros arranjos
de pares de elétrons. Assim, para explicar um arranjo trigonal planar, como a do BF3 e de cada
átomo de carbono do eteno, mistura-se um orbital s e dois orbitais p para produzir três orbitais

22
híbridos sp2. Um arranjo linear de pares de elétrons requer dois orbitais híbridos, então há a
mistura de um orbital s com um orbital p para produzir dois orbitais sp.
Não importa quantos orbitais atômicos são misturados, o número de orbitais híbridos é
sempre igual ao de orbitais atômicos utilizados. O esquema de hibridização é adaptado para
descrever o arranjo de elétrons de uma molécula. A expansão do octeto implica o
envolvimento de orbitais d.
Tabela 2 – Tabela de Hibridizações
A presença de uma ligação dupla ou tripla indica que os elétrons não hibridizados que
formam a ligação π por superposição lateral dos orbitais. As ligações duplas e sua influência
sobre a forma das moléculas são extremamente importantes para os organismo vivos.
Nas ligações múltiplas, um átomo forma uma ligação σ, usando um orbital híbrido sp
ou sp2, e uma ou mais ligações π, usando os orbitais p não hibridizados. A superposição
lateral que produz uma ligação π restringe a rotação das moléculas, resulta em ligações mais
fracas do que as ligações σ e impede que átomos com raios maiores formem ligações
múltiplas.

23
3.3. Exercícios
1. Qual a forma do íon SbF52-? Quantos ângulos FSbF diferentes existem na molécula?
Quais são os valores esperados para o ângulo FSbF?
2. Utilize as estruturas de Lewis e a teoria VSEPR para dizer a forma de cada uma das
seguintes espécies: a) PF4-; b) ICl4
+; c) pentafluoreto de fósforo; d) tetrafluoreto de
xenônio.
3. O xenônio forma XeO3, XeO4 e XeO64-, que são poderosos oxidantes. Escreva suas
estruturas de Lewis, ângulos de ligação e dê a hibridização dos átomos de Xe. Qual
deles teria a maior distância Xe–O?
4. Para cada uma das seguintes moléculas ou íons, escreva a forma, liste o número de
pares isolados do átomo central e estime os ângulos de ligação: a) PBr5; b) XeOF2; c)
SF5; d) IF3; e) BrO3-.
5. Prediga a forma de: a) TeF4; b) NH2-; c) NO2
+; d) NH4+; e) OCS; f) SnH4.
6. Dê a hibridização do átomo em negrito das seguintes moléculas: a) SF6; b) O3Cl-O-
ClO3; c) H2N-CH2-COOH; d) OC(NH2)2 (uréia).
7. O fósforo branco (P4) é tão reativo que inflama ao ar. Os quatro átomos de P4 formam
um tetraedro no qual cada átomo de P está ligado a três outros átomos de P. Sugira um
esquema de hibridização para a molécula de P4. A molécula de P4 é polar ou apolar?
8. NH2- e NH2
+ são espécies angulares, mas o ângulo de ligação em NH2- é menor do que
em NH2+. Qual a razão da diferença? Faça o eixo x perpendicular ao plano da
molécula. Será que o orbital N2px participa da hibridização nessas espécies?
9. Descreva a estrutura da molécula de formaldeído (CH2O) em termos de orbitais
híbridos, ângulos de ligação e ligações σ e. O átomo de C é o átomo central ao quais
os outros três átomos estão ligados.
10. A composição dos híbridos pode ser discutida quantitativamente. O resultado é que, se
dois híbridos equivalentes compostos de um orbital s e dois orbitais p formam um
ângulo θ, então os híbridos podem ser considerados spλ com λ=-cosθ/cos2(1
2𝜃). Qual é
a hibridização dos orbitais de O que formam as duas ligações O-H em H2O?

24
4. Funções Inorgânicas
4.1. Ácidos
Os ácidos são um dos tipos de substâncias mais presentes em nosso cotidiano. Eles
estão nos alimentos, nas indústrias, em laboratórios, na vida doméstica e até mesmo nos
fluidos do corpo humano (suco gástrico, por exemplo).
Os ácidos já foram definidos, de forma bem simples, como substâncias que
apresentam sabor azedo. No século XIX, alguns químicos observaram que, em algumas
substâncias, o aumento da quantidade de oxigênio aumentava sua acidez. Lavoisier foi um
desses químicos e chegou a propor uma primeira definição para os ácidos, associando a acidez
à presença de oxigênio na substância, considerando este elemento como o gerador da acidez.
Contudo, essa definição foi insuficiente para descrever os ácidos, pois outras substâncias que
não contém oxigênio, como o HCl, são mais ácidas do que outras que contém esse elemento.
i. Definição dos ácidos
Definição de Arrhenius
Para Arrhenius, os ácidos são substâncias que na presença de água se ionizam, e
liberam íons H+.
Ex.: HCl(g)+ H2O(l)→H+ (aq)+ Cl-(aq)
O íon H+ tende a se ligar a uma molécula de água, formando um íon mais estável que
ele que é H3O+:
H2O(l) + H2O(l)⇋ H3O+
(aq) + OH-(aq)

25
Como característica comum, essas substâncias apresentam o átomo de hidrogênio
ligado a um átomo eletronegativo, como o cloro ou o oxigênio.
ii. Classificação dos ácidos
Quanto à presença de oxigênio:
- Hidrácidos: são aqueles que não apresentam oxigênio na molécula.
Ex.: HCl, HBr, H4Fe(CN)6 .
- Oxiácidos: são aqueles que apresentam oxigênio na molécula.
Ex.: HClO , H2SO4 .
Quanto ao número de hidrogênios ionizáveis:
Nos hidrácidos, todos os hidrogênios são ionizáveis. Nos oxiácidos, só são ionizáveis
os átomos de hidrogênio ligados a oxigênio.
Ex.: H3PO4 tem apenas dois H ligados a O, portanto tem somente dois H ionizáveis.
- Monoácidos: têm apenas um átomo de hidrogênio ionizável. Ex.: HCl, HNO3
- Diácidos: têm dois átomos de hidrogênio ionizáveis. Ex.: H2S ,H3PO3
- Triácidos: têm três átomos de hidrogênio ionizáveis. Ex.: H3PO4
- Tetrácidos: têm quatro átomos de hidrogênio ionizáveis. Ex.: H4P2O7
Quanto ao número de elementos químicos:
- Binários: têm dois elementos. Ex.: HF, HCl, HBr.
- Ternários: têm três elementos. Ex.: H2SO4, H3PO4, HCN
- Quaternários: têm quatro elementos. Ex.: (H4[Fe(CN)6])
Quanto ao grau de ionização:
Grau de ionização (α) é a relação entre o número de mols ionizados e o número de
mols inicial.

26
Pode-se dizer que um ácido é forte quando ele está em grande partedesprotonado em
solução. Já um ácido fraco está incompletamente desprotonado em solução.
- Ácidos fortes: são aqueles que ionizam 50% ou mais das moléculas dissolvidas.
- Ácidos moderados: são aqueles que ionizam entre 5% e 50% das moléculas dissolvidas.
- Ácidos fracos: são aqueles que ionizam menos de 5% das moléculas dissolvidas.
Dos hidrácidos, são fortes o HCl, HBr e HI, em ordem crescente; e HF é moderado.
Dos óxiácidos pode-se classificá-los pelo grau de ionização através da regra empírica: HaBbOc
sendo “a” o número de hidrogênios ionizáveis, “b” o número do outro elemento e “c” o
número de oxigênios.
x ≥2 – oxiácido forte;
𝑥 = 𝑐−𝑎
𝑏1 ≤x ≤ 2 – oxiácido moderado;
x < 1 – oxiácido fraco;
Quanto à volatilidade:
- Ácidos voláteis: são aqueles que têm ponto de ebulição por volta da temperatura ambiente
(na faixa de 25° a 35°C).
- Ácidos fixos: são aqueles que têm ponto de ebulição muito acima da temperatura ambiente.
Se o número de átomos da molécula for ≥ a 7, o ácido é fixo.
Nomenclatura
o Hidrácidos
Forma geral:
Ácido+ nome do ânion com a terminação ídrico.
Ex.: HCl – ácido clorídrico
HBr – ácido bromídrico

27
o Oxiácidos
- Nomenclatura segundo os ânions que são liberados na ionização:
Forma geral:
Ácido + nome do elemento + oso ou ico
oso – menor número de átomos de oxigênio
ico – maior número de átomos de oxigênio
Para relacionar o nome do ácido com o nome do seu ânion, usamos a seguinte regra:
Tabela 03 - Relação do nome do ácido com o nome do ânion.
Ex.:
Tabela 04 – Exemplo de relação entre o nome do ácido com o nome do ânion.
- Nomenclatura segundo o NOx do elemento central:
Forma geral:
Ácido + prefixo (se necessário) + elemento central + sufixo

28
De acordo com o elemento central (o primeiro é o hidrogênio e o terceiro é o
oxigênio), temos o sufixo OSO para o menor NOx e ICO para o maior NOx (ver tabela 1).
Tabela 05 – Prefixo e sufixo de acordo com o NOx
Ex:
- Regra quando varia o grau de hidratação:
O prefixo orto é usado para o ácido fundamental; o prefixo meta é usado quando do
ácido orto retira-se 1H2O; o prefixo piro é usado para indicar a retirada de 1 H2O das duas
moléculas de orto.
Ex.:
NOx Prefixo Sufixo
+1, +2 hipo oso
+3, +4 - oso
+5, +6 - ico
+7 (hi)per ico

29
Figura 06 – Tabela de Ânions
4.2. Bases
Definição de Arrhenius
De acordo com Arrhenius, base é toda substância que libera OH- (hidroxila), em
solução aquosa, como único ânion.
Exemplos:
𝑁𝑎𝑂𝐻(𝑠)𝐻2𝑂→ 𝑁𝑎+(𝑎𝑞) + 𝑂𝐻−(𝑎𝑞)
𝑆𝑟(𝑂𝐻)2(𝑠)𝐻2𝑂→ 𝑆𝑟2+(𝑎𝑞) + 2𝑂𝐻−(𝑎𝑞)
𝐹𝑒(𝑂𝐻)3(𝑠)𝐻2𝑂→ 𝐹𝑒3+(𝑎𝑞) + 3𝑂𝐻−(𝑎𝑞)
Nomenclatura
Para a nomenclatura de bases, segue-se o seguinte modelo:

30
Hidróxido de nome do cátion
Exemplos:
Hidróxido de potássio:
𝑐á𝑡𝑖𝑜𝑛: 𝐾+
â𝑛𝑖𝑜𝑛: 𝑂𝐻−}𝐾+𝑂𝐻− → 𝐾𝑂𝐻
Hidróxido de magnésio:
𝑐á𝑡𝑖𝑜𝑛: 𝑀𝑔2+
â𝑛𝑖𝑜𝑛: 𝑂𝐻−}𝑀𝑔2+𝑂𝐻− → 𝑀𝑔(𝑂𝐻)2
Hidróxido de alumínio:
𝑐á𝑡𝑖𝑜𝑛: 𝐴𝑙3+
â𝑛𝑖𝑜𝑛: 𝑂𝐻−}𝐴𝑙3+𝑂𝐻− → 𝐴𝑙(𝑂𝐻)3
Entretanto, alguns elementos podem existir como diferentes espécies catiônicas. Para
estes, é necessário indicar o estado de oxidação em algarismos romanos após o nome do
cátion. Outra maneira para nomear é o uso do sufixo-oso para o íon de menor carga e –ico
para o de maior carga.
Cobre:
{
𝐶𝑢+: 𝐶𝑢𝑂𝐻
ℎ𝑖𝑑𝑟ó𝑥𝑖𝑑𝑜𝑑𝑒𝑐𝑜𝑏𝑟𝑒(𝐼)𝑜𝑢ℎ𝑖𝑑𝑟ó𝑥𝑖𝑑𝑜𝑐𝑢𝑝𝑟𝑜𝑠𝑜.
𝐶𝑢2+: 𝐶𝑢(𝑂𝐻)2ℎ𝑖𝑑𝑟ó𝑥𝑖𝑑𝑜𝑑𝑒𝑐𝑜𝑏𝑟𝑒(𝐼𝐼)𝑜𝑢ℎ𝑖𝑑𝑟ó𝑥𝑖𝑑𝑜𝑐ú𝑝𝑟𝑖𝑐𝑜.
Ouro:
{
𝐴𝑢+: 𝐴𝑢𝑂𝐻
ℎ𝑖𝑑𝑟ó𝑥𝑖𝑑𝑜𝑑𝑒𝑜𝑢𝑟𝑜(𝐼)𝑜𝑢ℎ𝑖𝑑𝑟ó𝑥𝑖𝑑𝑜𝑎𝑢𝑟𝑜𝑠𝑜.
𝐴𝑢3+: 𝐴𝑢(𝑂𝐻)3ℎ𝑖𝑑𝑟ó𝑥𝑖𝑑𝑜𝑑𝑒𝑜𝑢𝑟𝑜(𝐼𝐼𝐼)𝑜𝑢ℎ𝑖𝑑𝑟ó𝑥𝑖𝑑𝑜á𝑢𝑟𝑖𝑐𝑜.
Força e solubilidade de bases
A solubilidade das bases inorgânicas está relacionada diretamente ao metal que a
compõe. Hidróxidos de metais alcalinos e metais alcalinos terrosos, com exceção dos
hidróxidos de berílio e magnésio, são solúveis em água. As demais bases são insolúveis ou
praticamente insolúveis.
A força de uma base está relacionada com a sua solubilidade. Ou seja, as bases fortes são:
LiOH, NaOH, KOH, RbOH, CsOH, Ca(OH)2, Sr(OH)2 e Ba(OH)2. As demais bases são

31
fracas. Dentro de uma mesma família, a força da base cresce com o aumento do raio atômico
do metal.
Dentre as bases inorgânicas, há ainda uma muito importante que não apresenta um
metal em sua composição: a amônia (NH3). A amônia é uma base muito solúvel em água que
se ioniza, em meio aquoso, gerando íons hidroxila. Entretanto, apesar da sua solubilidade, a
amônia é uma base fraca.
𝑁𝐻3(𝑔) + 𝐻2𝑂(𝑙) ⇌ 𝑁𝐻4𝑂𝐻(𝑎𝑞) ⇌ 𝑁𝐻4+(𝑎𝑞) + 𝑂𝐻−(𝑎𝑞)
Principais bases e aplicações
i. Hidróxido de sódio (NaOH): conhecido como soda cáustica, é utilizado na produção de
sabões a partir de óleos e gorduras (saponificação);
ii.Hidróxido de magnésio (Mg(OH)2): utilizado na forma de uma suspensão como antiácido
estomacal;
iii.Hidróxido de amônio (NH4OH): a partir da amônia são produzidos fertilizantes para produção
de alimentos e também produtos de limpeza.
4.3. Sais
De acordo com SvanteArrhenius, pai dos conceitos clássicos de funções inorgânicas,
um sal é uma substância que, ao se dissociar em solução aquosa, libera pelo menos um cátion
que não seja o cátion hidrônio e pelo menos um ânion que não seja o ânion hidroxônio nem o
ânion superóxido (O2-).
A maioria dos sais é resultante de uma reação ácido-base, reações denominadas
reações de neutralização. Além de um sal, esta também libera um óxido (geralmente, água).
O caráter iônico do sal justifica o fato de estes conduzirem corrente elétrica se em solução
aquosa ou em estado líquido ou vapor. Os pontos de fusão e ebulição de sais são bem
elevados devido às fortes interações entre os íons, de modo que os íons se organizam de
maneira simétrica e estável, formando um cristal bem compacto (cada íon com vários íons de

32
carga oposta ao redor e baixo grau de liberdade dos mesmos). Veja alguns exemplos de
reações de neutralização abaixo:
HCl(aq) + NaOH(aq) NaCl(aq) + H2O(l)
H2SO4(aq) + 2 KOH(aq) K2SO4(aq) + 2 H2O(l)
A nomenclatura de sais é dada da seguinte maneira: inicialmente, é indicado o nome
do ânion pertencente ao sal, seguido pela preposição “de” e, por fim, o nome do cátion
pertencente ao sal. Geralmente, o nome do cátion é o nome de um elemento, e o cátion,
proveniente da base (pela teoria de Arrhenius); já o nome do ânion depende do nome do ácido
do qual ele foi originado, e o ânion, proveniente da base. Alguns exemplos:
CaCO3: carbonato de cálcio
KMnO4: permanganato de potássio
Enquanto o nome do cátion se encontra explícito na base de origem, o nome do ânion
deriva do nome do ácido da seguinte maneira: somente o sufixo muda, de acordo com a tabela
a seguir.
Tabela 06 – Comparação entre o sufixo do ânion com o sufixo do cátion correspondente.
Sufixo do ácido -ídrico -oso -ico
Sufixo do ânion -eto -ito -ato
Os sais podem ser classificados de acordo com a natureza dos íons de origem:
Sal neutro: não possui hidrogênio ionizável nem o ânion OH-;
Sal ácido ou hidrogenossal: possui dois cátions, sendo um deles o íon hidrônio
ionizável, e somente um ânion;
Sal básico ou hidroxissal: possui apenas um cátion e dois ânions, sendo um deles o íon
hidroxila;
Sal duplo ou misto: possui dois cátions diferentes (que não sejam o íon hidrônio) e
dois ânions diferentes (que não sejam o íon hidroxila);

33
Sal hidratado: possui em seu retículo cristalino moléculas de água (com estequiometria
definida).
4.4.Óxidos
Óxidos são compostos formados por dois elementos, sendo o oxigênio o mais
eletronegativo deles.
Exemplos:
- Na2O, BaO, H2O, ZnO.
Obs.: como o flúor é mais eletronegativo que o oxigênio, os compostos OF2 e O2F2
não são considerados óxidos, conforme dito acima.
Nomenclatura
A nomenclatura dos óxidos de acordo com as normas estabelecidas pela IUPAC leva
em consideração se estes são iônicos ou moleculares.
i. Óxidos moleculares
São aqueles formados por ametais ligados a oxigênio, e são nomeados de acordo com a
seguinte regra:
[
𝑀𝑜𝑛𝑜 −𝐷𝑖 −𝑇𝑟𝑖 −…
] + Óxido de [
𝐷𝑖 −𝑇𝑟𝑖 −𝑇𝑒𝑡𝑟𝑎 −…
]elemento
Exemplos:
CO = Monóxido de carbono SO3 = Trióxido de enxofre
CO2 = Dióxido de carbono Cl2O7 = Heptóxido de dicloro
ii. Óxidos iônicos

34
São óxidos formados por metais ligados a oxigênio. São nomeados da seguinte forma:
Óxido de elemento. + carga do cátion (em algarismos romanos)
Exemplos
Na2O = Óxido de sódio
Cu2O = Óxido de cobre I
CaO = Óxido de cálcio
Fe2O3 = Óxido de ferro III
Classificação dos óxidos
Os óxidos podem ser classificados de acordo com o seu comportamento na presença
de água, bases e ácidos. Dessa forma, eles podem ser classificados em óxidos: ácidos, básicos,
neutros, anfóteros ou mistos.
i. Óxidos ácidos
Apresentam caráter covalente e geralmente são formados por ametais, esses óxidos ao
reagirem com água produzem ácidos e com base produzem sal e água.
Exemplo:
SO2 + H2O → H2SO3
SO2 + NaOH → Na2SO3
ii. Óxidos básicos
Apresentam caráter iônico e geralmente são formados por metais, esses óxidos ao
reagirem com água produzem bases e com ácidos produzem sal e água.
Exemplo:
Na2O + H2O → 2NaOH

35
Na2O + H2SO4 → Na2SO4 + H2O
iii. Óxidos neutros
São óxidos covalentes e não reagem com água, nem com ácidos ou bases. Alguns
exemplos são CO, NO e N2O.
iv. Óxidos anfóteros
São óxidos que se comportam como óxidos básicos na presença de um ácido, e como
óxidos ácidos na presença de uma base.
Exemplo:
ZnO + 2HCl → ZnCl2 + H2O
ZnO + 2NaOH → Na2ZnO2 + H2O
v. Óxidos duplos ou mistos
São óxidos que resultam da combinação de dois óxidos de um mesmo elemento.
Exemplo:
FeO + Fe2O3 → Fe3O4
vi. Peróxidos
Os peróxidos são compostos que apresentam fórmula estrutural R-O-O-R ou (O2)2-.
São geralmente formados por hidrogênio, metais alcalinos e metais alcalinos terrosos. Em
solução aquosa, os peróxidos reagem produzindo uma base e água oxigenada, e com ácidos
produzem sal e água oxigenada.
Exemplo:
K2O2 + H2O → 2KOH + H2O2

36
4.5. Exercícios
1. (ITA-SP) Qual dos ácidos a seguir é o menos volátil?
a) HCl
b) HI
c) H2SO3
d) H2SO4
e) CH3CH2COOH
2. (Mackenzie-SP) Um ácido, quanto à força, classifica-se como forte, moderado e fraco,
conforme a escala de grau de ionização dada abaixo.
Assim, comparando-se o ácido A, cujo grau de ionização é de 40%, com outro B, no
qual, na ionização de 1 mol de moléculas, somente 2,4 ·1023 moléculas não ionizam,
podemos dizer que:
a) A é mais forte que B.
b) A e B são igualmente moderados.
c) A é tão fraco quanto B.
d) B é mais forte que A.
e) B é tão forte quanto A.
3. (Mackenzie-SP) Associe corretamente as duas colunas e assinale a alternativa correta.
i. H2SO4
ii. HI
iii. HNO2
iv. HClO4

37
v. H2S
(A) Hidrácido, monoácido, forte, volátil.
(B) Hidrácido, diácido, fraco, volátil.
(C) Oxiácido, monoácido, forte, volátil.
(D) Oxiácido, diácido, forte, fixo.
(E) Oxiácido, monoácido, semiforte, volátil.
a) I - A; II - B; III - C; IV - D; V - E
b) I - D; II - B; III - E; IV - C; V - A
c) I - D; II - A; III - E; IV - C; V - B
d) I - E; II - D; III - C; IV - B; V – A
e) I - C; II - A; III - D; IV - E; V – B
4. (EEM-SP) Têm-se os três ácidos e os valores da tabela, que foram obtidos dissolvendo-se
em água à temperatura constante:
a) Calcule o grau de ionização para cada ácido e coloque-os em ordem crescente de sua
força de ionização.
b) Equacione a ionização do HNO3 em água.

38
5. (UFSC-SC) Soluções ácidas e soluções alcalinas exibem propriedades importantes,
algumas delas ligadas à força do ácido ou da base. Uma solução aquosa de um ácido
genérico HA poderá ser classificada como "solução de um ácido fraco" quando:
i. (01) não se alterar na presença de uma base.
ii. (02) apresentar coloração avermelhada na presença do indicador fenolftaleína.
iii. (04) apresentar uma concentração de íons H+ maior que a concentração de íons
A-.
iv. (08) mantiver uma concentração de HA muito maior que a concentração dos íons
H+.
v. (16) a solução for altamente condutora de corrente elétrica.
Soma: ___
6. Coloque as seguintes bases em ordem crescente de força: KOH, Mg(OH)2, Cr(OH)3 e
Ba(OH)2.
7. Nomeie as seguintes bases e escreva suas equações de dissociação em meio aquoso:
Zn(OH)2, Fe(OH)2, Sr(OH)2, Co(OH)2. Qual é, dentre as bases listadas, a mais forte?
8. Um aluno dissolveu cloreto de amônio (NH4Cl) em água e posteriormente adicionou à
solução gotas de hidróxido de sódio. Após a adição da base, ele sentiu um cheiro muito
forte e irritante. A que se deve esse cheiro? Explique com reações químicas.
9. Ao aquecer hidróxidos metálicos a elevadas temperaturas, os mesmos se decompõem em
seus óxidos metálicos e água. Sabendo disso, escreva as equações de decomposição
térmica para os hidróxidos niqueloso, plúmbico e férrico.
10. Coloque as seguintes bases em ordem crescente de força: KOH, Mg(OH)2, Cr(OH)3 e
Ba(OH)2.
11. Nomeie as seguintes bases e escreva suas equações de dissociação em meio aquoso:
Zn(OH)2, Fe(OH)2, Sr(OH)2, Co(OH)2. Qual é, dentre as bases listadas, a mais forte?

39
12. Um aluno dissolveu cloreto de amônio (NH4Cl) em água e posteriormente adicionou à
solução gotas de hidróxido de sódio. Após a adição da base, ele sentiu um cheiro muito
forte e irritante. A que se deve esse cheiro? Explique com reações químicas.
13. Ao aquecer hidróxidos metálicos a elevadas temperaturas, os mesmos se decompõem em
seus óxidos metálicos e água. Sabendo disso, escreva as equações de decomposição
térmica para os hidróxidos niqueloso, plúmbico e férrico.
14. (PUC Rio-2008)Cloreto de sódio é um composto iônico que se encontra no estado sólido.
Dissolvido em água, se dissocia completamente. Acerca desse sal, é INCORRETO
afirmar que:
a) Tem Fórmula NaCl;
b) No estado sólido, a atração entre seus íons é muito forte e por essa razão possui
elevado ponto de fusão;
c) Em solução aquosa, conduz corrente elétrica muito bem;
d) A ligação entre os seus íons é por covalência;
e) HCl e NaOH são o ácido e a base que dão origem a esse sal.
15. (Cesgranrio-RJ) Os fertilizantes com potássio são muito utilizados na agricultura. As
formas mais comuns de fertilização são o cloreto, o sulfato, o nitrato e o fosfato de
potássio. Suas fórmulas moleculares são representadas, respectivamente, por:
a) KCl, K2SO3, KNO3, K3PO4
b) KCl, K2SO3, KNO2, K2PO3;
c) KCl, K2SO4, KNO3, K3PO4;KClO, K2SO3, KNO2, K2PO3;
d) KClO, K2SO4, KNO3, K3PO4.
16. (G1 - IFSC 2011) A azia é uma sensação de “queimação” no estômago, relacionada à
acidez do suco gástrico, e pode ser provocada por alimentação em excesso, alimentação
deficiente, estresse, entre outros motivos. Alguns medicamentos indicados para o alívio

40
dos sintomas contêm, normalmente, substâncias como Al(OH)3 e Mg(OH)2. Nesse
contexto e com relação a ácidos, bases e reações de neutralização, é correto afirmar que:
a) as substâncias: H2SO4, NaHSO4, H2CO3 e NaHCO3 podem ser classificadas como
ácidos, conforme a definição de Arrhenius;
b) Al(OH)3 e Mg(OH)2 podem ser classificados como sais básicos;
c) como produto da neutralização do ácido clorídrico, presente no suco gástrico, por
hidróxido de alumínio ter-se-á uma solução aquosa de AlCl3;
d) as bases como o hidróxido de alumínio e o hidróxido de magnésio são substâncias
moleculares e, portanto, não se dissolvem bem na água;
e) os ácidos formam soluções aquosas não condutoras de eletricidade.
17. (UDESC SC/2009)Os calcários são rochas sedimentares que contêm minerais de
carbonato de cálcio (aragonita ou calcita). Quando esses minerais são aquecidos a
altastemperaturas (calcinação), ocorre a decomposição térmica do carbonato, com
liberação de gás carbônico eformação de uma outra substância sólida. As fórmulas eas
funções químicas dessas substâncias envolvidas são, respectivamente:
a) CaCO3 (óxido), CO2 (óxido) e CaO2 (base).
b) CaCO3 (sal), CO2 (óxido) e CaO (óxido).
c) CaC2O4 (sal), CO2 (óxido) e CaC2 (sal).
d) CaCO4 (sal), CO (óxido) e CaO (óxido).
e) CaCO2 (sal), CO2 (óxido) e CaO (sal).
18. (UECE/2009)Quando o monóxido de carbono é inalado ele pode substituir o oxigênio e
combinar com as moléculas de hemoglobina, impedindo a respiração dos tecidos.
Sobreo monóxido de carbono, um estudante registrou as seguintes informações:
i. É um gás incolor e inodoro.
ii. Pode ser obtido pela reação do carvão com o vapord’água.
iii. É usado na indústria química, porque a partir delesão obtidas moléculas orgânicas
mais complexas.

41
iv. É um óxido ácido.
v. É um dos produtos da combustão completa dealcanos.
São verdadeiras apenas as informações:
a) I, III e IV.
b) II, IV e V.
c) I, II e III.
d) II, III e V.
19. (UNESP SP/2009)Considere as seguintes afirmações a respeitos dos óxidos:
i. Óxidos de metais alcalinos são tipicamente iônicos.
ii. Óxidos de ametais são tipicamente covalentes.
iii. Óxidos básicos são capazes de neutralizar um ácidoformando sal mais água.
iv. Óxidos anfóteros não reagem com ácidos ou com base.
Estão corretas as afirmativas:
a) I, II e III, apenas.
b) II e III, apenas.
c) I, II e IV, apenas.
d) II, III e IV, apenas.
e) I e III, apenas.
20. (UFCG PB/2009)Dois metais X e Y se combinam com o nitrogênio formando os
seguintes compostos: X3N e Y3N2. Assinaledentre as alternativas abaixo aquela que
contém as 2 fórmulas corretas dos óxidos e superóxidos destes dois elementos:
a) X2O XO2 YO YO4;
b) X2O XO2 Y2O3 YO4;
c) XO XO2 Y2O3 YO4;
d) X2O XO YO YO2;
e) XO2 XO YO YO2.

42
5. Teorias Ácido-Base
5.1. Teoria de Arrhenius
A definição de Arrhenius é umas teoria útil e simples para classificar essas
substâncias, todavia se restringe apenas para o meio aquoso e não pode ser empregado em
outros solventes. Segundo esse químico sueco, ácidos são compostos que em meio aquoso
liberam o cátion exclusivo H3O+ e bases liberam o ânion exclusivo OH-, em meio aquoso.
Exemplos:
HCl (g) + H2O (l) H3O+ (aq)+ Cl- (aq)
NaOH (aq) + H2O (l) OH- (aq)+ Na+ (aq)
5.2. Teoria de Brᴓnsted-Lowry
Para Johannes Brᴓnsted e Thomas Lowry o caráter ácido e básico está ligado a
transferência protônica. Ou seja, ácidos são espécies químicas capazes de doar prótons e bases
são capazes de receber prótons. Como as reações não se referem ao meio em que há essa troca
de prótons essa teoria pode ser empregada em qualquer solvente ou mesmo na ausência dele.
B: + H-A ↔ B+- H + :A-
Exemplo:
HCl + NH3 NH4+ + Cl-
Outro conceito importante nessa teoria é o conceito de par ácido-base conjugado. Essa
definição esclarece justamente a relação que há entre os compostos dos reagentes e dos
produtos. Considere um ácido genérico HX que irá doar um próton para a água e formar a
espécie X-. Esse ânion pode em seguida desprotonar o H3O+ e voltar a ser HX. Ou seja, HX e

43
X- formam um par ácido-base, respectivamente. Do mesmo modo ocorre com a água, cujo par
ácido-base é H3O+ e H2O.
Exemplo 01: Identifique os pares ácido-base conjugado da seguinte reação:
HClO4 + H2O ↔ H3O+ + ClO4
-
SOLUÇÃO – Os pares ácido-base conjugados são dados a seguir
HClO4 – ácido /// ClO4 - - base conjugada
H3O+ - ácido conjugado /// H2O – base
Uma base e seu ácido conjugado diferem em um único próton. A mesma coisa ocorre
para o ácido e sua base conjugada.
5.3. Teoria de Lewis
Segundo Lewis a definição de ácido e base está ligada diretamente a densidade
eletrônica, ou seja, da capacidade em doar pares de elétrons. Lewis definiu o ácido, ou
eletrófilo, como sendo uma substância aceptora de par de elétrons e base, ou nucleófilo, como
doador de par de elétrons. Assim, diferentemente de Arrhenius e Brᴓnsted-Lowry a definição
de Lewis não limitava o solvente e não falava apenas de transferência protônica, mas sim do
arranjo eletrônico entre duas espécies. Ou seja, é uma teoria mais ampla no que concerne a
explicação de outras reações que também são entre um ácido e uma base.
A+ + B- ↔ A-B
A interpretação da definição de Lewis com base nos orbitais de fronteira é justamente
a combinação do LUMO do ácido (A) com o HOMO da base (B) formando um complexo (A-
B).

44
Figura 07 – HOMO e LUMO
HOMO: orbital ocupado de mais baixa energia.
LUMO: orbital desocupado de mais baixa energia.
Exemplos:
I. HClO4 + NH3 NH4+ + ClO4
- : ataque do par de elétrons, isolado, do nitrogênio
no hidrogênio do ácido perclórico, formando como produtos os íons perclorato e
amônio.
II. Propeno + brometo de hidrogênio 2-iodopropano : regra de Markovnikov é
satisfeita na adição de haleto de hidrogênio em alcenos. O mecanismo da reação
ocorre da seguinte maneira: cisão heterolítica do haleto de hidrogênio; protonação
da ligação dupla carbono-carbono pelo haleto de hidrogênio; combinação
carbocátion-ânion (ácido = carbocátion; base = íon haleto).
Os fatores que influenciam na força de um ácido de Lewis são:
1) Força da ligação com o átomo ligado diretamente ao próton que é perdido
2) Eletronegatividade
3) Deslocalização eletrônica da base conjugada

45
1) A força de ligação, geralmente, diminui ao descermos em um grupo na tabela periódica.
Como é o caso dos halogênios. A ligação com o hidrogênio se torna mais comprida e menos
fortalecida, logo a força do ácido aumenta. No caso do HF mesmo com a alta
eletronegatividade do flúor um fator contribui para que esse ácido se torne fraco comparado
com os outros haletos de hidrogênio. Esse fator é a alta razão entre a carga e o tamanho do F-.
Tabela 07 – pKa de ácidos de halogênios
Ácidos HF HCl HBr HI
pKa 3,1 -3,9 -5,8 -10,4
2) A medida que o átomo ligado diretamente ao hidrogênio, que será desprotonado, é mais
eletronegativo a polarização da ligação se torna bem mais pronunciada e consequentemente a
formação do próton será favorecida.
Tabela 08 – pKa de alguns ácidos
Substância CH4 NH3 H2O HF
pKa 60 36 15,7 3,1
A eletronegatividade de átomos presentes em uma estrutura e que não estão ligados
diretamente ao H que será desprotonado podem também contribuir para a acidez dessa
substância. Como? Pelo efeito indutivo. Este efeito estrutural é transmitido pelas ligações da
estrutura e induz uma polarização nas ligações. E trata-se de um efeito que decresce com a
distância.
Exemplo: Comparação entre o etanol (pKa= 16) e o 2,2,2-trifluoroetanol (pKa= 11,3).
3) A deslocalização eletrônica da base conjugada é outro fator fundamental que contribui para
o estudo da acidez de uma substância. O ácido nítrico é um bom exemplo para explicarmos
esse efeito. Sabemos que o HNO3 é um ácido forte e possui como sua base conjugado o íon
nitrato (NO3-). Este íon pode ser representado com três estruturas de ressonância, ou seja,
evidenciando a deslocalização eletrônica em sua estrutura. Assim, a base conjugada é

46
estabilizada por este fenômeno e consequentemente aumenta a constante de equilíbrio da
ionização deste ácido.
5.4. Teoria de Pearson: Ácidos e Bases Duros e Macios
Observou-se que existem alguns ligantes (bases) que preferem se complexar com
certos metais (ácidos) e essa ligação refletia na estabilidade do produto. S. Ahrland, J. Chatt e
N. R. Davies registraram e denominaram dois tipos de categorias para os íons metálicos com
base nas suas ligações preferenciais com bases (íons haletos). Essas categorias eram chamadas
de forma genérica de classes a e b. A mesma classificação foi feita para as basess. Após esse
levantamento foi possível prever a estabilidade de complexos com base nessa classificação
dos ácidos e bases. R. G. Pearson nomeou essas classes como sendo espécies duras e macias.
Espécies duras: tendem a ser pequenos; pouco polarizáveis e ligações eletrostáticas
predominam.
Espécies macias: tendem a ser maiores; mais polarizáveis e as ligações covalentes
predominam.
Abaixo se encontram duas tabelas que mostram ácidos e bases com suas respectivas
classificações: duro, macio ou fronteira.
Figura 08 – Bases duras e macias

47
Figura 09 – Ácidos Duros e Macios
Exemplos:
I. bases como a fosfina (R3P) e tioéteres (R2S) possuem uma tendência voltada em se
coordenar com íons metálicos como Hg2+, Pd2+ e Pt2+.
II. amônia, aminas, água e o fluoreto (F-) já preferem se coordenadar com Be2+, Ti4+ e
Co3+.
Princípio de Pearson: ácidos duros tendem a se ligar a bases duras e ácidos macios
tendem a se ligar a bases macias.
5.5.Exercícios
01) Qual é o ácido mais forte? (CH3)3N+H ou (CH3)3O
+H ? Explique.
02) Calcule o Ka dos ácidos abaixo. Em seguida coloque as substâncias em ordem de acidez
crescente.
Substância pKa
Aspirina 3,48
Vitamina C (ácido ascórbico) 4,17

48
Ácido fórmico 3,75
Ácido oxálico 1,19
03) Preveja a estabilidade da formação de complexos a partir desses reagentes de partida:
a) NH3 com Co3+
b) CO com Na+
c) CN- com Pt2+
04) Em relação a formamida, responda:
a) desenhe a estrutura de Lewis que corresponde a sua base conjugada.
b) desenhe a(s) estrutura(s) de Lewis de ressonância da formamida.

49
6. Equilíbrio Químico
A lei da Ação das Massas de Guldberg-Waage aborda que a velocidade de uma reação
química depende do produto da concentração do reagente elevado a um expoente determinado
empiricamente e de uma constante k que depende exclusivamente da temperatura. Para uma
reação genérica do tipo:
aA bB (I)
Lei de Velocidade: v= k[A]α
Caso a reação acima seja elementar o valor de α é o próprio coeficiente
estequiométrico, ou seja, “a”. Mas, o que viria ser uma reação elementar? Reação elementar é
aquela reação que se desenvolve em uma única etapa e os expoentes da lei de velocidade
serão os seus respectivos coeficientes estequiométricos. Todavia, uma reação não elementar é
aquela que se desenvolve em mais de uma etapa. Logo, a lei de velocidade não será
comandada pela reação global, mas sim pela etapa mais lenta desse processo. Ou seja, a etapa
mais lenta será determinante na velocidade dessa reação e com base será analisado a
velocidade que irá levar a cabo a reação química.
Considere a reação química genérica (elementar) abaixo:
Aa +bB ↔ cC + Dd (II)
As principais características do equilíbrio químico é a reversibilidade da reação e
dinamicidade desse equilíbrio. Ou seja, comparando (I) com a (II) verificamos que a primeira
é unidirecional e a segunda bidirecional. O que isso quer dizer? A reação (I) se trata da
formação apenas dos produtos. Já a segunda trata-se de uma reversibilidade (indicada pela
dupla seta) no sistema, isto é, reagentes formam produtos como também os produtos podem
formar novamente os reagentes. É claro satisfazendo as condições de energia, colisão
favorável e afinidade química. O equilíbrio químico é dinâmico, pois mesmo quando o
sistema atinge o equilíbrio as concentrações dos produtos e reagentes permanecem constantes,
ou seja, a formação de reagentes e de produtos ocorrem de forma simultânea mesmo no
equilíbrio.

50
E quando é atingido o equilíbrio químico? Quando as velocidades inversa e direta são
iguais. As expressões dessas velocidades são descritas abaixo:
v DIRETA = k [A]a [B]b
v INVERSA= k’ [C]c [D]d
Igualando as expressões encontraremos a razão das constantes de velocidade que irá
gerar uma nova constante que é chamada de constante de equilíbrio (K). Qualquer operação
matemática básica realizada com constantes irá gerar uma nova constante. Logo a expressão
da constante de equilíbrio irá ficar:
K = [C]c [D]d / [A]a [B]b
K>1: produto favorecido
0<K<1: reagente favorecido
A expressão acima é baseado nas concentrações (KC), todavia podemos expressar a
constante de equilíbrio em termos da pressão utilizando a equação de Clayperon (PV=nRT).
A equação que relaciona o Kp com o KC ficará:
Kp= Kc (RT)∆n
R= constante universal dos gases
T= temperatura na escala absoluta
∆n= variação na quantidade de mols (produtos-reagentes)
6.1. Princípio de Le Chatelier
Uma vez que o sistema atinge o equilíbrio químico este pode ser perturbado,
posteriormente, por meio de uma ação externa. Logo, o sistema, imediatamente, irá tentar

51
minimizar esta perturbação para que um novo equilíbrio químico seja alcançado novamente.
Três principais fatores que deslocam o equilíbrio são: efeito da concentração; temperatura e
pressão.
Considere a seguinte equação genérica:
2A(g) + C(g) ↔ 3W(s) + 5G(g) ∆H>0
- Efeito da concentração
Uma forma de perturbarmos esse sistema é adicionando a ele mais reagentes/produtos
ou mesmo os retirar do sistema. Assim, a reação irá deslocar para uma das direções de tal
forma a restabelecer o equilíbrio. Por exemplo: caso seja adicionado A o equilíbrio seria
deslocado pros produtos (W e G) de tal forma a consumir o excesso de A e alcançar outro
equilíbrio.
- Efeito da temperatura
Reações químicas são caracterizadas também por meio da variação de entalpia (∆H).
Reações que absorvem calor (endotérmicas) possuem ∆H>0 e reações que liberam calor
(exotérmicas) ∆H<0. Ao aumentarmos a temperatura a reação endotérmica é favorecida e ao
diminuirmos exotérmica. Logo, caso se aumentarmos a temperatura na reação acima os
produtos serão favorecidos.
- Efeito da pressão
Sabemos que volume e pressão são inversamente proporcionais e que quantidade de
matéria e volume são diretamente. Assim, o efeito da pressão em reações químicas consiste
justamente no deslocamento do equilíbrio para o sentido que há mais ou menos quantidade de
matéria. Lembrando que espécies sólidas e líquidas são incompressíveis, logo a concentração
dessas espécies não são alteradas pelo efeito da pressão. Diferentemente dos gases. Logo, se
aumentarmos a pressão na reação acima o equilíbrio será deslocado para os reagentes, ou seja,
para o sentido que há menor quantidade de matéria.

52
Cálculo das concentrações no equilíbrio a partir da constante de equilíbrio
Ionização do ácido carbônico: H2CO3 + H2O ↔ H3O+ + HCO3
-
Tabela 09 – Tabela para a determinação das concentrações no equilíbrio da reação especificada
H2CO3 H3O+ HCO3
-
Início 2mol.L-1 0 0
Variação -x +x +x
Equilíbrio 2-x x x
32
33 ]][[
COH
HCOOHKa
Ka= 4,45 x 10-7
Colocar os valores na expressão de Ka e encontrar o valor de x em mol.L-1.
Exemplo do cotidiano de reação em equilíbrio: lentes fotossensíveis.
2 Ag+ + 2 Cl- ↔ 2 Ag0 + Cl2 presença de luz
2 Cu+ + Cl2 ↔ 2 Cu2+ + 2 Cl- e
2 Cu2+ + 2 Ag0 ↔ 2 Ag+ + 2Cu+ ausência de luz

53
Figura 10 – Lentes Fotossensíveis.
A constante de equilíbrio é definida em inúmeros sistemas que se estende desde a
ionização de um ácido inorgânico até a formação do complexo enzima-substrato no
mecanismo de catálise enzimática que ocorre em nosso organismo.
6.2. Exercícios
01)Escreva a expressão da constante de equilíbrio (Kc) para a seguinte reação:
CO(g) + ½ O2(g) ↔ CO2 (g)
02) Calcule o KPS do sulfato de estrôncio, cuja solubilidade, a uma determinada temperatura,
é 91,8mg.L-1.
03) Considere a seguinte equação química:
2 BrF5 (g) ↔ Br2 (g) + 5 F2 (g)
Responda os seguintes itens abaixo sobre a decomposição do pentafluoreto de bromo:
a) se ao decorrer da reação fosse retirado o gás Br2 do sistema. O que iria ocorrer com o
equilíbrio químico?
b) e se a pressão fosse reduzida, o que iria ocorrer no equilíbrio?

54
c) 0,200 mol de BrF5 é colocado em um recipiente de 20L a 1500K. Ao final do equilíbrio foi
encontrado 0,180 mol de F2. Considere que no equilíbrio a pressão total é 2,12 atm. Calcule
Kp.
04) Calcule as concentrações de equilíbrio da ionização do ácido genérico, fraco e
monoprótico, HG. Sabendo-se que a concentração inicial é 4x10-3 mol.L-1 e que a constante
Ka= 2,20 x 10-5.

55
7. Limite
7.1. Conceito
O limite é a base para definirmos derivada e integral, além de ser essencial para todo o
cálculo diferencial. Suponhamos que queremos determinar para qual ponto a função f(x) = x²-
4 vai quando x se aproxima de 3, veja a tabela a seguir:
Tabela 10 – Valores de x e f(x) para a função especificada anteriormente
x 2,9 2,99 2,999 2,9999 2,99999
f(x) 4,41 4,9401 4,994001 4,99940001 4,99994
Percebe-se que, quando a x se aproxima de 3, a função se aproxima cada vez mais do
número 5. Portanto, assim, concluímos que:
54lim 2
3
x
x
Lemos: “Limite de x² - 4 quando x tende a 3 é igual a 5”
É levantada uma pergunta a partir do cálculo desse limite: por que precisamos calcular
o limite se, ao substituirmos o valor de x na função, conseguimos o valor de f(x)?
Bom, a definição de limite se mostra importante para funções que não são definidas
em algum ponto. Por exemplo, no caso da função
67²5
252)(
2
xx
xxxf
Quando tentamos calcular o valor da função quando x = 2, temos que
0
0
6272.5
22522)(
2
2
xf

56
Como sabemos, não se pode dividir uma quantia por zero, ainda mais zero. O
quociente 0/0 constitui uma indeterminação e torna uma função indefinida nesse ponto.
Assim, como sabemos para qual ponto a função está tendendo quando ela se aproxima
do ponto x = 2?
Quando uma função possui uma indeterminação, significa que há um termo que zera
ela em cima e embaixo. Sabendo que a função acima é composta por dois polinômios sendo
divididos, pela definição de polinômio, sabemos que eles podem ser fatorados. Como há a
indeterminação 0/0, significa que dois é uma das raízes dos dois polinômios (do polinômio do
numerador e do polinômio do denominador). Portanto, para resolvermos o problema, temos
que, inicialmente, dividir ambos os polinômios por (x – 2), que é o termo que zera os
polinômios em x = 2.
Dividindo os polinômios como uma forma de fatoração, temos que:
)35)(2(
)12)(2(lim
67²5
252lim
2
2
2
xx
xx
xx
xx
xx
Agora, basta cancelarmos o termo que zera o numerador e o denominador para
resolvermos o problema.
13
3
325
122
)35(
)12(lim
67²5
252lim
2
2
2
x
x
xx
xx
xx
Assim, somente por meio do limite podemos indicar a tendência de funções que
possuem indeterminações aos pontos nos quais elas são indeterminadas.
Existem vários tipos de indeterminações, e várias maneiras de fugir de cada uma delas
na resolução do limite. Serão discutidas algumas delas nessa apostila, e outras serão
discutidas no decorrer do semestre da disciplina “Cálculo 1”.
Exemplo 01: Determine o valor de 3
9lim
9
x
x
x

57
SOLUÇÃO – Percebemos, calculando o valor de f(x) quando x tende a 9, que o ponto não
pertence ao domínio da função, pois percebe-se a indeterminação 0/0. Para resolver o
problema, neste caso em que temos um número não-racional no denominador, racionalizá-lo
usando o produto da soma pela diferença de dois termos. Temos então:
3
9lim)(lim
99 x
xxf
xx
3
3
3
9lim
9 x
x
x
x
x
9
)3)(9(lim
9 x
xx
x
Agora, devemos cancelar o termo que zera o numerador e o denominador da função e
resolver o limite:
63lim)(lim99
xxfxx
7.2. Propriedades
Se L, M e k são números reais e lim𝑥→𝑐
𝑓(𝑥) = 𝐿 e lim𝑥→𝑐
𝑔(𝑥) = 𝑀, então:
7.2.1. Regra da soma: o limite da soma de duas funções é a soma de seus limites.
MLxgxfcx
))()((lim
7.2.2. Regra da diferença: o limite da diferença de duas funções é a diferença de seus
limites.
MLxgxfcx
))()((lim

58
7.2.3. Regra do produto: o limite do produto de duas funções é o produto de seus
limites.
MLxgxfcx
))()((lim
7.2.4. Regra da multiplicação por constante: o limite de uma constante multiplicado
pela função é a constante multiplicada pelo limite da função.
Lkxfkcx
))((lim
7.2.5. Regra do quociente: o limite do quociente de duas funções é o quociente de seus
limites, desde que o limite do denominador não seja zero.
0,)(
)(lim
M
M
L
xg
xf
cx
7.2.6. Regra da potenciação: se r e s são inteiros e não têm um fator comum, e s ≠ 0,
então s
r
s
r
cxLxf
))((lim , desde que s
r
L seja um número real. (Se s é par,
pressupomos que L > 0). O limite de uma potência racional de uma função é a
potência do limite da função, desde que esse último seja um número real.
7.3. Limites Laterais
Quando analisamos lim𝑥→𝑎
𝑓(𝑥) estamos pensando que 𝑥 → 𝑎, isto é, x se aproxima de a,
por valores maiores ou menores que a.
Escrevemos lim𝑥→𝑎−
𝑓(𝑥) = 𝐿 e dizemos que o limite à esquerda de f(x) quando x tende a
a é igual a L se pudermos tornar os valores de f(x) arbitrariamente próximos de L, para x
suficientemente próximo de a e x menor que a.

59
Entretanto, podemos fazer x se aproximar de a apenas por valores maiores do que a.
Nesse caso, dizemos que x tende a a pela direita e indicamos lim𝑥→𝑎+
𝑓(𝑥).
Figura 11 – Limite à esquerda Figura 12 – Limite à direita.
Existe lim𝑥→𝑎
𝑓(𝑥) se e somente se os limites laterais são iguais. Em outras palavras,
lim𝑥→𝑎
𝑓(𝑥) = 𝐿 se e somente se lim𝑥→𝑎−
𝑓(𝑥) = L e lim𝑥→𝑎+
𝑓(𝑥) = L.
Exemplo 1 : Considere a seguinte função:
1,2
1,2
1,4
)(
xx
x
xx
xf
Calcule os limites laterais com x tendendo a 1.
SOLUÇÃO – Se atribuirmos a x valores próximos de 1, porém menores que 1 (à esquerda de
1), temos que os valores da função ficam próximos de 3; e atribuindo a x valores próximos de
1, porém maiores que 1 (à direita de 1), temos que os valores da função ficam próximos de –
1.
Exemplo 2: Seja a função f definida por

60
1,4
1,4
1,1
)(
2
xx
x
xx
xf
Determinar:
a) lim𝑥→1−
𝑓(𝑥)
b) lim𝑥→1+
𝑓(𝑥)
c) Esboce o gráfico de f(x)
SOLUÇÃO – Pela definição de limite à esquerda, você responde a letra a). Observe que a
função f(x) está definida por f(x) = x² + 1 se x < 1. Logo,
211)1(lim)(lim 22
11
xxf
xx
Agora, pela definição de limite à direita você responde a letra b). Observe que a função f(x)
está definida por f(x) = 4-x se x>1. Logo,
314)4(lim)(lim11
xxfxx
Como solução da letra c), note que f(1) = 4. Com estas informações, de que f (1) = 4,
2)(lim1
xfx
e 3)(lim1
xfx
, você consegue perceber como f(x) se comporta quando x está
próximo de 1. Para esboçar o gráfico de f(x), dê valores para x, x < 1 e calcule os valores de
f(x) correspondentes através da expressão x² + 1; dê valores para x > 1 e calcule os valores de
f(x) correspondentes através da expressão 4 – x e veja o gráfico de f(x), abaixo.
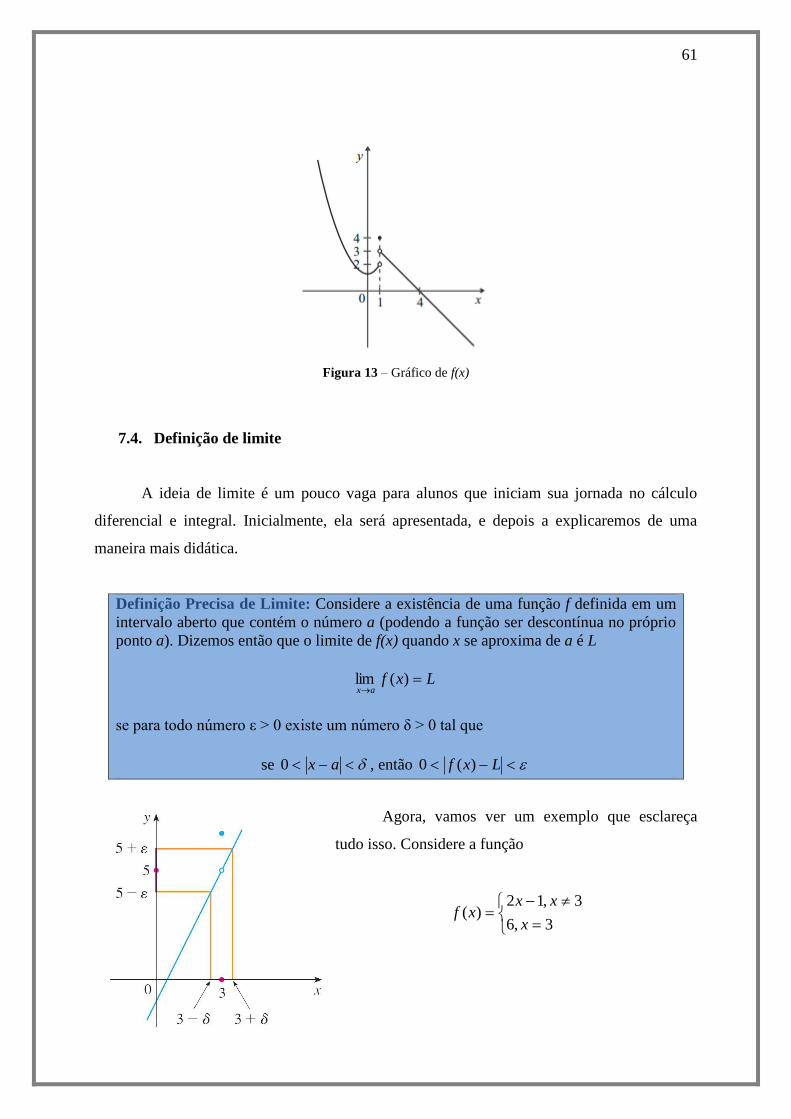
61
Figura 13 – Gráfico de f(x)
7.4. Definição de limite
A ideia de limite é um pouco vaga para alunos que iniciam sua jornada no cálculo
diferencial e integral. Inicialmente, ela será apresentada, e depois a explicaremos de uma
maneira mais didática.
Definição Precisa de Limite: Considere a existência de uma função f definida em um
intervalo aberto que contém o número a (podendo a função ser descontínua no próprio
ponto a). Dizemos então que o limite de f(x) quando x se aproxima de a é L
Lxfax
)(lim
se para todo número ε > 0 existe um número δ > 0 tal que
se ax0 , então Lxf )(0
Agora, vamos ver um exemplo que esclareça
tudo isso. Considere a função
3,6
3,12)(
x
xxxf

62
Figura 14 – Gráfico de f(x). Retirado de [2]
Como percebemos, ela é uma função com uma descontinuidade em x = 3, porém é
uma função definida em todo o domínio dos reais. Como vimos anteriormente, o limite dessa
função existe e é igual a 5. Porém, como, por meio da definição, provar que esse limite
existe?
No caso, o que deve ser feito é estabelecer um desvio para a variável no eixo x, assim,
poderemos observar o desvio no eixo y. No caso, definiremos o desvio δ = 0,01. Devemos
descobrir qual é o desvio em y ε gerado por esse desvio em x. Para x = 3,01 e para x = 2,99,
02,5101,32)01,3( f
98,4199,22)99,2( f
Percebe-se, por aí, que o desvio ε, para δ = 0,01, é
02,0))99,2(),01,3(max()01,0(
02,0)(lim)99,2()99,2(
02,0)(lim)01,3()01,3(
3
3
xx
xffx
xffx
x
x
É possível definirmos uma função que descreva o desvio para o ponto x = 3. Para um
ponto x = 3 ± δ,
251261)3(2)3(f
Assim, temos que
2)(

63
Um fator interessante que deve ser considerado para que seja comprovada a existência do
limite no ponto x = 3 é que, quando δ tende zero, ε também tende a zero.
Exemplo 1: Prove, por meio da definição, que 065²lim2
xxx
SOLUÇÃO – Inicialmente, é importante vermos que a função f(x)=x²-5x+6 é contínua, o que
nos livra de algumas preocupações por meio de notações. Temos que, inicialmente, calcular o
valor da função para um valor de x deslocado do valor onde o limite é definido. Assim, para x
= 2 ± δ, temos que
6510²446)2(5)²2()2( f
)²(0)2( f
Assim, concluímos que o erro ε associado ao desvio em y é dado por
²)(
Quando δ tende a zero, ε tende a zero, o que comprova que o limite do enunciado está
correto.
Exemplo 2: Prove, por meio da definição, que o limite da função Heaviside H(t) não existe no
ponto x = 0.
0,1
0,0)(
t
ttH
Figura 15 – Função Heaviside. Retirado de [2]

64
SOLUÇÃO – Percebe-se, primeiramente, que a função Heaviside não é contínua, pois há um
salto na função no ponto x = 0. Assim, teremos que calcular os desvios para cada um dos
trechos (antes e depois do zero) separadamente. Para x = 0 + δ, temos que
1)0( f
Já para x = 0 – δ, temos que
0)0( f
Assim, temos que
0,1
0,0)0(
f
Assim, temos que a função ε(δ) é dada por
1)(
Como, ao tendermos δ a zero, ε não tende a
zero, o limite não existe.
7.5. Limites que envolvem infinito
Quando estamos lidando com limites laterais em algumas funções tendendo a um valor
definido, pode ocorrer que essas funções cresçam ou decresçam indefinidamente. Por
exemplo, tomemos a função
3
2)(
x
xxf

65
Figura 16 – Gráfico de f(x). Retirado de [2]
Inicialmente, percebemos que
3
2lim
3 x
x
x não existe, já que os limites laterais são distintos.
Ao calcularmos o limite lateral pela esquerda da função tendendo a x = 3, percebe-
se que a função decresce rapidamente.
Tabela 11 – Valores de f(x) para diferentes valores de x
x 2 2,9 2,99 2,999 2,9999 2,99999
f(x) -4 -58 -598 -5998 -59998 -599998
Assim, podemos escrever que
3
2lim
3 x
x
x
É importante ressaltar que o símbolo infinito não é um número, é uma notação para
representar um número demasiadamente grande. Ou seja, se o limite de uma função tende a
infinito, na verdade, ele não existe, já que a função tende a valores absurdamente extremos
(extremo positivo – infinito – ou extremo negativo – menos infinito) mas não a um valor
definido.
Já quando calculamos o limite lateral da função pela direita, temos que
Tabela 12 – Valores de f(x) para diferentes valores de x
x 4 3,1 3,01 3,001 3,0001 3,00001
f(x) 8 62 602 6002 60002 600002
Dessa forma, temos que

66
3
2lim
3 x
x
x
ou seja, o limite lateral pela esquerda também não existe.
A linha x = 3 é definida como a assíntota vertical do gráfico da função f(x), pois a
função tende a beirar a linha, mesmo que nunca a toque.
Exemplo 1: Circuitos elétricos são geralmente tratados como um circuito no qual se acopla
uma resistência elétrica (geralmente, um equipamento elétrico) para que energia seja
dissipada nesta. Porém, sabe-se que existe uma resistência interna do próprio filamento que
conecta os componentes do circuito. Considere um circuito formado apenas por uma fonte de
tensão e um fio conectando as duas entradas da fonte. Pela lei de Ohm, temos que a corrente
que passa por esse filamento é dada por
R
VI
em que R é a resistência interna do filamento e V é a tensão exercida pela fonte. Determine, se
existir, o limite da corrente elétrica que percorre o circuito quando a resistência tende a zero.
SOLUÇÃO – Percebe-se, inicialmente, que o limite tende pelo lado direito, já que a
resistência de um circuito elétrico é maior do que zero. Percebe-se, também, que quanto
menor o valor da resistência, como ela está no denominador da função, maior o valor da
corrente, e valores ínfimos de resistência levam a valores gigantescos de corrente. Assim,
conclui-se que
R
VI
RR 00limlim
ou seja, o limite não existe. A reta x = 0 é a assíntota vertical do gráfico.

67
Exemplo 2: Calcule o limite a seguir: 2²
²2lim
1 xx
x
x
SOLUÇÃO – Para calcularmos o limite, inicialmente, os limites laterais da função:
2²
²2lim
1 xx
x
x
2²
²2lim
1 xx
x
x
Percebe-se, já por aí, que os limites laterais não coincidem. Assim, o limite
2²
²2lim
1 xx
x
x não existe.
Obs.: Caso ambos os limites laterais de uma função arbitrária f(x) tendessem a infinito ou a
menos infinito quando x tendesse a a, poderíamos escrever
)(lim xfax
mesmo que, teoricamente, o limite não exista.
Assim como são objetos de estudo do cálculo diferencial e integral os limites
infinitos, também são os limites que tendem a infinito. Consideremos a seguinte função:
xxf
15)(
Vejamos o comportamento da função à medida que x tende a infinito.
Tabela 13 – Valores de f(x) para os valores de x especificados
x 1 10 100 1000 10000 100000
f(x) 6 5,1 5,01 5,001 5,0001 5,00001

68
Percebe-se, por meio dos valores, que à medida que a variável aumenta, a função
tende a 5. Assim, concluímos que
51
5lim
xx
Para a resolução de limites tendendo a infinito, é muito importante o seguinte
teorema:
Teorema: Se k é um número racional positivo e c é um número real arbitrário, então
0lim kx x
c e 0lim
kx x
c
desde que xk seja sempre definido.
A importância desse teorema é dedicada à resolução de limite de funções racionais.
Para resolvermos um limite desse tipo, devemos primeiro dividir numerador e denominador
por xn, em que n é a mais alta potência de x que aparece no denominador e, em seguida,
aplicamos o teorema acima.
Exemplo 3: Determine 2²3
5²2lim
xx
x
x
SOLUÇÃO: A maior potência de x no denominador é x². Assim, dividimos numerador e
denominador pelo termo e aplicamos o teorema acima.
²
2²3²
52
lim2²3
52lim
2
2
x
xxx
x
xx
x
xx
²
213
²
52
lim
xx
xx

69
²
213lim
²
52lim
xx
x
x
x
3
2
003
02
Exemplo 4: Determine 34
2²9lim
x
x
x
SOLUÇÃO – Para iniciar a solução do problema, temos que dar um jeito de retirar a potência
de dentro da raiz. Assim, fatoraremos o polinômio da raiz em função do x²
34
²
29²
lim34
²
29²
lim34
2²9lim
x
xx
x
xx
x
x
xxx
34
²
29
lim
x
xx
x
Agora, podemos continuar com o método tradicionalmente.
x
xx
xx
x
x
xx 34
²
29
lim34
2²9lim
x
xx 3
4
²
29
lim
²
2lim
1lim3lim
²
5lim2lim
xx
x
xxx
xx

70
4
3
04
09
7.6. Exercícios
01) Calcule os limites abaixo
a)
b)
c)
d)
e)
f)
g)
h)
i)
j)
k)
l)
m)

71
n)
o)
p)
q)
r)
s)
t)
02) Dada a função f definida por
1,5
1,3
1,23
)(
xax
x
xx
xf . Determinar a є R para que exista
)(lim1
xfx
.
03) Calcule os limites a seguir:
a) 742
135lim
2
2
xx
xx
x
b) 726
13lim
23
3
xx
xx
x
c) xx
x
x 54
32lim
3
2

72
8. Derivada
8.1. Definição
Para entender o que é a derivada, é preciso que se conheça o conceito de limite.
Relembrando: O limite é uma aproximação infinitesimal de x a um valor de x, mas sem que x
seja exatamente aquele valor.
DEFINIÇÃO: É o coeficiente de inclinação da reta tangente ao gráfico de uma função y =
f(x) em um determinado ponto P=(x0, f(x0)). Também pode ser interpretada como o quanto y
varia em função de x. E é calculada como:
0
0
0
)()(lim)(
xx
xfxfxf
dx
d
x
Se considerarmos 0xxh , pode-se utilizar:
h
xfhxfxf
dx
d
h
)()(lim)( 00
0

73
Agora, você deve estar se perguntando: Qual maneira é usada para calcular? Use
aquela que for mais conveniente na resolução do problema.
Exemplo 1: Use a definição de derivada via limite para calcular a derivada f'(x), para
735)( 2 xxxf
SOLUÇÃO –
h
xfhxfxf
dx
d
h
)()(lim)( 00
0
h
xxhxhxxf
dx
d
h
]7)(3)(5[]7)(3)(5[lim)( 0
2
00
2
0
0
h
xxhxhhxxxf
dx
d
h
7357335)(105lim)( 0
2
00
2
0
2
0
0
Simplificando,
h
hhhxxf
dx
d
h
35)(10lim)(
2
0
0
Colocando h em evidência,
h
hxhxf
dx
d
h
)3510(lim)( 0
0
Simplificando novamente,
3510lim)( 00
hxxfdx
d
h
Finalmente, resolvendo o limite acima,

74
310)( 0 xxfdx
d
Existe outra forma de representar )(xfdx
d, que é da forma explicitada a seguir:
')( yxfdx
d
)(')( xfxfdx
d
A do lado esquerdo da equação é a notação formal e do lado direito a notação usual.
8.2. Propriedades
Considere que f e g são funções de x deriváveis.
8.2.1. Regra da constante
A derivada de uma função constante é igual a zero.
cf
0'f
8.2.2. Regra da soma
A derivada da soma é a soma das derivadas.
'')'( gfgf

75
8.2.3. Regra da subtração
A derivada da subtração é a subtração das derivadas.
'')'( gfgf
8.2.4. Regra da multiplicação por um escalar
Considere c uma constante não nula (diferente de zero). A derivada de uma função que
multiplica uma constante é a constante vezes a derivada da função.
')'( cfcf
8.2.5. Regra do produto
A derivada do produto é derivada da primeira função vezes a segunda função mais a
primeira função vezes a derivada da segunda função. (Resumindo: derivada da primeira vez a
segunda mais primeira vezes a derivada da segunda).
'')'( fggffg
8.2.6. Regra do quociente
A derivada do quociente é a derivada da primeira função vezes a segunda função
menos a primeira função vezes a derivada da segunda função, tudo sobre o quadrado da
segunda função; Sendo que neste caso g(x) tem que ser não nulo.
2
''
g
fggf
g
f

76
Exemplo 01: Utilizando as regras acima, resolva a derivada no ponto 𝑥0 utilizando a definição
por limite.
103)( 2 xxxf
Podemos dividir a função em duas:
)()()( xhxgxf
xxxg 23)(
10)( xh
Como )(xh é uma função constante, então h’(x) = 0. Agora, vamos resolver a derivada da
função g(x):
h
xghxgg
h
)()(lim' 00
0
h
xxhxhxg
h
)3()]()(3[lim' 0
2
00
2
0
0
h
xxhxhhxxg
h
0
2
00
2
0
2
0
0
3363lim'
Simplificando,
h
hhhxg
h
2
0
0
36lim'
Colocando h em evidência,
h
hxhg
h
)136(lim' 0
0

77
Simplificando novamente
)136(lim' 00
hxgh
Resolvendo o limite,
16' 0 xg
Utilizando a regra da soma, finalmente,
'')'(' hghgf
16' 0 xf
8.2.7. Regra da potência
A derivada de uma função do tipo xr, sendo r um número racional, é dada por
1 rr rxxdx
d
8.3. Derivadas Importantes
Funções exponenciais e logarítmicas
𝑎)𝑑(𝑎𝑐𝑥)
𝑑𝑥= 𝑎𝑐𝑥 ln 𝑎 . 𝑐, 𝑎 > 0 𝑑)
𝑑(ln 𝑥)
𝑑𝑥=1
𝑥, 𝑥 > 0
𝑏)𝑑(𝑒𝑎𝑥)
𝑑𝑥= 𝑎𝑒𝑎𝑥 𝑒)
𝑑(ln|𝑥|)
𝑑𝑥=1
𝑥
𝑐)𝑑(log𝑎 𝑥)
𝑑𝑥=
1
𝑥𝑙𝑛 𝑎, 𝑎 > 0, 𝑎 ≠ 1 𝑓)
𝑑(𝑥𝑥)
𝑑𝑥= 𝑥𝑥(1 + 𝑙𝑛𝑥)

78
Funções trigonométricas e suas inversas
𝑎)𝑑(𝑠𝑒𝑛 𝑥)
𝑑𝑥= cos 𝑥 𝑔)
𝑑(𝑎𝑟𝑐 𝑠𝑒𝑛 𝑥)
𝑑𝑥=
1
√1 − 𝑥2
𝑏)𝑑(cos 𝑥)
𝑑𝑥= −𝑠𝑒𝑛 𝑥 ℎ)
𝑑(arc cos 𝑥)
𝑑𝑥= −
1
√1 − 𝑥2
𝑐)𝑑(𝑡𝑔 𝑥)
𝑑𝑥= 𝑠𝑒𝑐2𝑥 𝑖)
𝑑(𝑎𝑟𝑐 𝑡𝑔 𝑥)
𝑑𝑥=
1
1 + 𝑥2
𝑑)𝑑(sec 𝑥)
𝑑𝑥= sec 𝑥 𝑡𝑔 𝑥 𝑗)
𝑑(arc sec 𝑥)
𝑑𝑥=
1
|𝑥|√𝑥2 − 1
𝑒)𝑑(𝑐𝑜𝑠𝑠𝑒𝑐 𝑥)
𝑑𝑥= −cossec 𝑥 𝑐𝑜𝑡𝑔 𝑥 𝑘)
𝑑(𝑎𝑟𝑐 𝑐𝑜𝑠𝑠𝑒𝑐 𝑥)
𝑑𝑥= −
1
|𝑥|√𝑥2 − 1
𝑓)𝑑(𝑐𝑜𝑡𝑔 𝑥)
𝑑𝑥= −cossec2 𝑥 𝑙)
𝑑(𝑎𝑟𝑐 𝑐𝑜𝑡𝑔 𝑥)
𝑑𝑥= −
1
1 + 𝑥2
Funções hiperbólicas e suas inversas
𝑎)𝑑(𝑠𝑒𝑛ℎ 𝑥)
𝑑𝑥= cosh 𝑥 𝑔)
𝑑(𝑎𝑟𝑐 𝑠𝑒𝑛ℎ 𝑥)
𝑑𝑥=
1
√𝑥2 + 1
𝑏)𝑑(cosh 𝑥)
𝑑𝑥= 𝑠𝑒𝑛ℎ 𝑥 ℎ)
𝑑(arc cosh 𝑥)
𝑑𝑥=
1
√𝑥2 − 1
𝑐)𝑑(𝑡𝑔ℎ 𝑥)
𝑑𝑥= 𝑠𝑒𝑐ℎ2𝑥 𝑖)
𝑑(𝑎𝑟𝑐 𝑡𝑔ℎ 𝑥)
𝑑𝑥=
1
1 − 𝑥2
𝑑)𝑑(sech 𝑥)
𝑑𝑥= −sech 𝑥 𝑡𝑔ℎ 𝑥 𝑗)
𝑑(arc sech 𝑥)
𝑑𝑥= −
1
𝑥√1 − 𝑥2
𝑒)𝑑(𝑐𝑜𝑠𝑠𝑒𝑐ℎ 𝑥)
𝑑𝑥= −cossec 𝑥 𝑐𝑜𝑡𝑔ℎ 𝑥 𝑘)
𝑑(𝑎𝑟𝑐 𝑐𝑜𝑠𝑠𝑒𝑐 𝑥)
𝑑𝑥= −
1
|𝑥|√1 + 𝑥2
𝑓)𝑑(𝑐𝑜𝑡𝑔ℎ 𝑥)
𝑑𝑥= −cossech2 𝑥 𝑙)
𝑑(𝑎𝑟𝑐 𝑐𝑜𝑡𝑔ℎ 𝑥)
𝑑𝑥=
1
1 − 𝑥2
8.4. Aplicações
8.4.1. Extremos de Funções
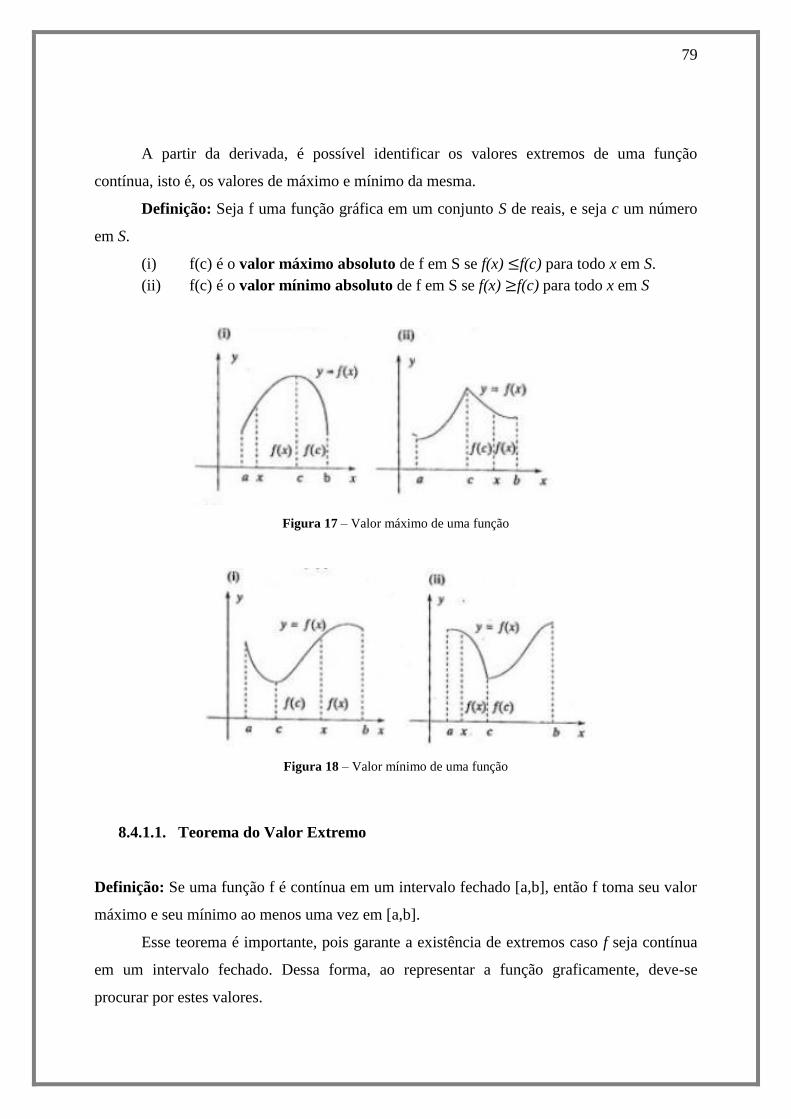
79
A partir da derivada, é possível identificar os valores extremos de uma função
contínua, isto é, os valores de máximo e mínimo da mesma.
Definição: Seja f uma função gráfica em um conjunto S de reais, e seja c um número
em S.
(i) f(c) é o valor máximo absoluto de f em S se f(x) ≤f(c) para todo x em S.
(ii) f(c) é o valor mínimo absoluto de f em S se f(x) ≥f(c) para todo x em S
Figura 17 – Valor máximo de uma função
Figura 18 – Valor mínimo de uma função
8.4.1.1. Teorema do Valor Extremo
Definição: Se uma função f é contínua em um intervalo fechado [a,b], então f toma seu valor
máximo e seu mínimo ao menos uma vez em [a,b].
Esse teorema é importante, pois garante a existência de extremos caso f seja contínua
em um intervalo fechado. Dessa forma, ao representar a função graficamente, deve-se
procurar por estes valores.
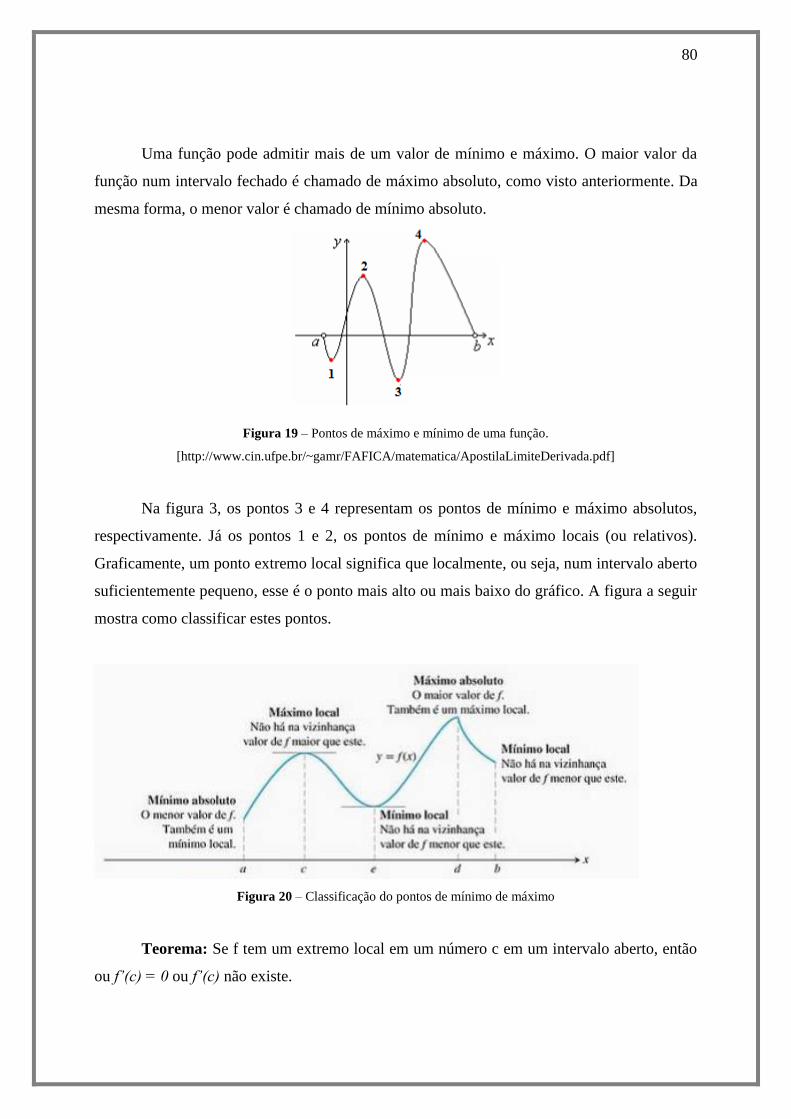
80
Uma função pode admitir mais de um valor de mínimo e máximo. O maior valor da
função num intervalo fechado é chamado de máximo absoluto, como visto anteriormente. Da
mesma forma, o menor valor é chamado de mínimo absoluto.
Figura 19 – Pontos de máximo e mínimo de uma função.
[http://www.cin.ufpe.br/~gamr/FAFICA/matematica/ApostilaLimiteDerivada.pdf]
Na figura 3, os pontos 3 e 4 representam os pontos de mínimo e máximo absolutos,
respectivamente. Já os pontos 1 e 2, os pontos de mínimo e máximo locais (ou relativos).
Graficamente, um ponto extremo local significa que localmente, ou seja, num intervalo aberto
suficientemente pequeno, esse é o ponto mais alto ou mais baixo do gráfico. A figura a seguir
mostra como classificar estes pontos.
Figura 20 – Classificação do pontos de mínimo de máximo
Teorema: Se f tem um extremo local em um número c em um intervalo aberto, então
ou f’(c) = 0 ou f’(c) não existe.

81
Como consequência, temos que se a derivada em um ponto existir ou for diferente de
zero, o valor de f(c) não é um extremo local.
Ponto crítico: Um número c no domínio de uma função é um número crítico de f se
f’(c) = 0 ou se f’(c) não existe.
Recapitulando...
Passos para encontrar os extremos de um função contínua em um intervalo fechado
[a,b]:
I. Determinar todos os números críticos da função em (a,b)
II. Calcular f(c) para cada número crítico obtido anteriormente
III. Calcular os valores extremos f(a) e f(b)
IV. Os valores máximo e o mínimo de f em [a,b] são o maior e o menor valores da
função calculados em 2 e 3.
Exemplo 1: Determine os valores de máximo e mínimo absolutos de f(x) = x² no intervalo [-
2,1].
SOLUÇÃO – A função é derivável em todo o seu domínio, portanto o único ponto crítico é
onde a derivada é igual à zero:
f’(x) = 2.x = 0
x = 0
Precisamos verificar os valores da função no ponto crítico (x=0) e nas extremidades
x= -2 e x=1
f(0) = 0² = 0
f(-2) = (-2)² = 4
f(1) = 1² = 1
Assim, a função apresenta um valor máximo absoluto de 4 em x=-2 e um mínimo
absoluto de 0 em x=0.

82
8.4.2. Teorema do Valor Médio
A determinação dos pontos críticos nem sempre é fácil. Assim, há um teorema que dá
condições suficientes para a existência de pelo menos um ponto crítico:
Teorema de Rolle: Se f é contínua em um intervalo fechado [a,b] e diferenciável no
intervalo aberto (a,b) e se f(a) = f(b), então f’(c) = 0 para ao menos um número c em (a,b).
A principal aplicação deste teorema é na demonstração do Teorema do Valor Médio,
um dos mais importantes no Cálculo.
Teorema do valor médio: Se f é contínua em um intervalo fechado [a,b] e
diferenciável no intervalo aberto (a,b), então existe um número c em (a,b) tal que:
ab
afbfcf
)()()('
Basicamente, este teorema garante que se há uma função f derivável e sendo dados
dois pontos (a, f(a)) e (b, f(b)) do gráfico de f, existe pelo menos um ponto c, tal que a<c<b,
de modo que a reta tangente ao gráfico de f traçada pelo ponto (c, f(c)) é paralela à reta
traçada entre os pontos (a, f(a)) e (b, f(b)).
Figura 21 – Representação gráfica do teorema do valor médio.
Exemplo 2: Se 14
1)( 2 xxf , mostre que f verifica as hipóteses do teorema do valor médio
no intervalo [-1,4], e determine um número c em (-1,4) que satisfaça a conclusão do teorema.

83
SOLUÇÃO – A função quadrática f é contínua em [-1,4] e diferenciável em (-1,4); logo, pelo
teorema do valor médio, existe um número c em (-1,4) tal que:
)1(4
)1()4()('
ffcf
Com f(4) = 5, f(-1) = 5/4 e f’(x) = ½.x, temos
)1(4
4
55
2
1
c
4
3
2
1 c
2
3c
8.4.3. Teste da Primeira Derivada
O estudo do sinal da primeira derivada é importante, pois com isso é possível
determinar onde uma função cresce ou decresce e, assim, classificar os extremos locais.
Seja f uma função contínua em [a,b] e diferenciável em (a,b): ela será crescente se para
todo x pertencente ao intervalo (a,b), sua derivada for maior que zero em todo o intervalo
[a,b]. Analogamente, f será decrescente se para todo x em (a,b), sua derivada for menor que
zero em [a,b].
Quando estudamos extremos de funções (vide 1.1), vimos que quando há um extremo
local a derivada neste ponto ou é zero ou não existe, e neste ponto temos um número crítico.
Porém, a recíproca não é verdadeira: Nem todo ponto crítico conduz a um extremo local, por
isso temos que testar todos para saber onde ocorre ou não um extremo local.
Há varias formas de teste, mas neste capítulo vamos focar em um em especial: o teste
do sinal da primeira derivada!

84
Definição: Seja c um número crítico de f, e suponhamos f contínua em c e diferenciável em
um intervalo aberto I contendo c, exceto possivelmente no próprio c.
(i). Se f’ passa de positiva para negativa em c, então f(c) é máximo local de f.
(ii). Se f’ passa de negativa a positiva em c, então f(c) é mínimo local de f.
(iii). Se f’(x)>0 ou se f’(x)<0 para todo x em I exceto x=c, então f(c) não é extremo
local de f.
Exemplo 3 – Determine os pontos críticos de f(x) = x³ - 12.x – 5 e identifique os intervalos
onde f é crescente e decrescente.
SOLUÇÃO – A função f é contínua e derivável em qualquer ponto. A primeira derivada:
f’(x) = 3.x² - 12 = 3.(x+2).(x-2)
possui como raízes x = -2 e x =2. Esses pontos subdividem o domínio de f nos intervalos (-
∞,-2), (-2,2) e (2,∞) nos quais f’ é ou positiva ou é negativa. Determinamos o sinal de f’
calculando f em um ponto conveniente em cada subintervalo. O comportamento de f é
determinado, então, aplicando-se o teste da primeira derivada a cada subintervalo temos que:
Intervalo -∞<x<-2 -2<x<2 2<x<∞
Valor calculado de f’ f’(-3) = 15 f’(0) = -12 f’(3) = 15
Sinal de f’ + - +
Comportamento de f Crescente Decrescente Crescente
8.4.4. Teste da Segunda Derivada
Da mesma forma que estudamos o sinal da primeira derivada para descobrir onde uma
função cresce ou decresce, o sinal da segunda derivada também nos dá uma informação
importante. Se f’’(x)>0 é crescente em um intervalo aberto I, então f’(x) é crescente em I, ou
seja, o coeficiente angular da tangente ao gráfico I aumenta com x, dizemos então que o
gráfico é côncavo para cima.
Definição: Se f for diferenciável em um intervalo aberto I. O gráfico de f é:

85
(i) Côncavo para cima em I se f’ é crescente em I.
(ii) Côncavo para baixo em I se f’ é decrescente em I.
Teste da segunda derivada: Se a derivada segunda f’’ de f existe em um intervalo aberto I,
então o gráfico de f é
(i) Côncavo para cima em I se f’’(x)>0 em I.
(ii) Côncavo para baixo em I se f’’(x)<0 em I.
Figura 22 – Concavidade do gráfico
Exemplo 4: Se f = x³ + x² - 5.x – 5, determine intervalos em que o gráfico de f é côncavo para
cima ou côncavo para baixo.
SOLUÇÃO – Como a primeira derivada da função f é: f’(x) = 3.x² + 2.x – 5, a segunda
derivada será: f’’(x) = 6.x + 2 = 2(3.x+1). Logo, f’’(x) < 0 se 3.x+1 < 0, isto é, se x < -1/3.
Analogamente, f’’(x) > 0 se x> -1/3. Aplicando o teste da segunda derivada:
Intervalo (-∞,-1/3) (-1/3, -∞)*
Sinal de f’ + -
Concavidade Para baixo Para cima
8.4.5. Problemas de Otimização
“Otimização é o processo de otimizar, de tornar ótimo. É a busca da excelência. É
o emprego de técnicas para seleção das melhores alternativas, com o propósito de alcançar os
objetivos determinados.” Aqui, buscaremos os melhores ou mais favoráveis valores para
diversas situações.

86
Exemplo 5: Uma caixa sem tampa será feita recortando-se pequenos quadrados congruentes
dos cantos de uma folha de estanho 12x12 pol e dobrando-se os lados para cima. Que
tamanho os quadrados das bordas devem ter para que a caixa chegue à sua capacidade
máxima?
SOLUÇÃO –
Atenção: Neste tipo de problema de otimização, é importante ler várias vezes o
enunciado e depois fazer um esquema a fim de retirar todas as informações para
determinar as quantidades desconhecidas.
Veja o esquema abaixo:
Figura 23 – Esboço do Problema
Nele, os quadrados nos cantos tem x polegadas de lado. O volume da caixa é uma
função dessa variável:
32 448144)212()212()( xxxxxxxV
Como os lados da folha de estanho medem só 12 pol, 6x , o domínio de V é o
intervalo 60 x .
Para descobrir mais, examina-se a primeira derivada de V em relação à x:
)6()2(12)812(121296144 22 xxxxxxdx
dV

87
Das duas raízes, x=2 e x=6, apenas x =2 está contida no domínio da função, fazendo
parte da lista de pontos críticos. Os valores de V neste único ponto crítico e nas extremidades
são
V(2) = 128
V(0) = 0
V(6) = 0
O volume máximo é 128 pol³. Os quadrados a serem recortados devem ter 2 pol de
lado.
Exemplo 6: Um recipiente cilíndrico, aberto em cima, deve ter a capacidade de 375.π cm³. O
custo do material usado para a base do recipiente é de 15 centavos por cm² e o custo do
material usado para a parte curva é de 5 centavos por cm². Se não há perda de material,
determine as dimensões que minimizem o custo do material.
SOLUÇÃO – Primeiro faz-se um esboço do recipiente, onde h é a altura do cilindro e r o raio
da base, ambos em centímetro.
Figura 24 – Esboço do Problema
A quantidade a minimizar é o custo C do material. Como os custos, por centímetro
quadrado, da base e da parte curva são 15 e 5 centavos, respectivamente, temos, em termos de
reais:
Custo do recipiente = 15.área da base + 5.área lateral
Assim,

88
)23(5)2(5)(15 22 rhrhrrC
Podemos expressar C como função de uma variável, r, escrevendo h em termos de r.
Como o volume do recipiente é 375.π cm³, vemos que:
3752 hr , ou 2
375
rh
Substituindo h por 2
375
r em C, temos:
rr
rrrC
75035
375235 2
2
2
Para achar os pontos críticos basta derivar C em relação à r:
2
3
22
12530
12530
75065
r
r
rr
rr
dr
dC
Como 𝑑𝐶
𝑑𝑟= 0, se r=5, então 5 é um número crítico. E como
𝑑𝐶
𝑑𝑟 < 0, se r<5 e
𝑑𝐶
𝑑𝑟 >0, se
r>5, segue-se do teste da primeira derivada que C tem seu mínimo quando o raio do cilindro é
igual a 5 cm. O valor correspondente da altura é h = 375
𝑟² = 15 cm.
8.4.6. Outras Aplicações
A utilização da derivada vai além do cálculo, esta também tem aplicações na física e
na economia, como veremos a seguir.
Seja S(t) uma função de deslocamento de uma partícula P. Esta função fornece o local
que a partícula está num determinado instante t. A velocidade de P no instante t é a derivada
S’(t), ou seja, a taxa de variação da partícula em função do tempo. De modo análogo, a

89
aceleração é a taxa de variação da velocidade em relação ao tempo, isto é, a derivada da
velocidade v’(t), ou ainda, a derivada segunda do deslocamento S’’(t).
dt
dSv e
dt
dS
dt
d
dt
dva
Exemplo 7: Uma bola de basquete é lançada verticalmente para cima, por um menino, e tem
posições S no decorrer do tempo t, dadas pela função horária S(t) = 60.t – 5.t² (s em metros e t
em segundos).
a) Calcule o tempo gasto para atingir a altura máxima.
b) Determine a altura máxima em relação ao solo. Ver Figura 6.
Figura 25 – Lançamento Vertical
SOLUÇÃO – Como a função horária é a função que determina o espaço em função do tempo,
ao derivar a expressão S(t) = 60.t – 5.t² , tem-se a velocidade instantânea em função do tempo,
assim, S’(t) = 60 – 10.t, igualando a zero, já que ao atingir a altura máxima a velocidade é
nula, temos que:
v(t) = 0
60 – 10.t = 0
t = 6 s
Para determinar a altura é preciso substituir o tempo na expressão do espaço, tem-se:
S(6) = 60.6 – 5.6²
S(6) = 180 m

90
Assim, conclui-se que o tempo gasto para atingir a altura máxima é 6 segundos e a
altura máxima em relação ao solo é de 180 metros.
Na economia, Segundo MUNEM & FOULIS (1982), o termo “marginal” é
freqüentemente usado como um sinônimo virtual para “derivada de”. Assim, se x é o número
de unidades de um determinado produto, C(x) é o custo da produção de x unidades e, C’(x) o
custo marginal, que pode ser interpretado como a taxa que o custo varia em relação ao número
de x unidades produzidas.
Da mesma forma, c(x) é o custo médio da produção de uma unidade e c’(x) é custo
médio marginal. R(x), a receita proveniente da venda de x unidades e R’(x), a receita
marginal, e por fim, P(x) o lucro na venda de x unidades e P’(x), o lucro marginal.
Exemplo 8 – No cinema, o preço de um pacote de pipoca é de R$ 4,50. O pipoqueiro pode
vender 500 pacotes de pipocas com o custo de R$1,40 por pacote. Para cada centavo que o
pipoqueiro baixar no preço do pacote, a quantidade vendida pode aumentar de 50 unidades
(pacotes). Que preço de venda maximizará o lucro?
SOLUÇÃO – Inicialmente, observe que o lucro é de R$ 3,10 por pacote. Se x é o número de
centavos que o pipoqueiro baixa no preço de cada pacote; o lucro na venda de cada pacote de
pipoca será então de (310 – x) centavos, e a quantidade vendida será (500 + 50x). O lucro
total é, portanto, o lucro por unidade (pacote) vezes a quantidade vendida, ou seja,
P = P(x) = (310 – x) (500 + 50x) = 155000 + 15500x – 500x – 50x²
P(x) = 155000 + 15000x – 50x²
Agora, deve-se maximizar a função P(x). Como P é uma função polinomial, basta
igualar sua derivada à zero (uma vez que a derivada sempre existe) e resolver a equação
resultante. Sendo:
P’(x) = 15000 – 100x
15000 – 100x = 0
x = 150

91
Como a derivada segunda de P é igual a P’’(x) = − 100, portanto negativa para
qualquer valor de x, segue que x = 150 é um ponto de máximo. Assim, o preço de venda que
dará o maior lucro é de R$ 3,00.
8.5. Regra de L’Hospital
Muitas vezes nos deparamos com limites indeterminados, da forma 0/0 ou ∞/∞. Para
resolvê-los podemos utilizar a derivada de acordo com uma regra especial, chamada de Regra
de L’Hospital.
Teorema: Regra de L’Hospital 1ª forma
Suponha que f(a) = g(a) = 0, que f’(a) e g’(a) existam e que g’(a) ≠ 0. Então
)('
)('
)(
)(lim
ag
af
xg
xf
ax
Exemplo 1: Calcule o 4
16lim
2
4
x
x
x
SOLUÇÃO – Nesse caso temos uma indeterminação da forma 0/0, então aplica-se a regra de
L’Hospital, dividindo a derivada da função de cima pela derivada da função de baixo. Assim:
81
2lim
4
16lim
4
2
4
x
x
x
xx
Às vezes, mesmo após a primeira derivação, os novos numerador e denominador
continuam iguais a zero em x=a. Para isso, usamos uma forma mais forte da regra de
L’Hospital.
Teorema: Forma mais forte da Regra de L’Hospital:

92
Suponha que f(a) = g(a) = 0, que f e g sejam deriváveis em um intervalo aberto I
contendo a e que g’(x) ≠ 0 em I se x ≠ a. Então
)('
)('lim
)(
)(lim
xg
xf
xg
xf
axax
desde que o limite exista do lado direito da igualdade.
Exemplo 2: Calcule o 30
limx
senxx
x
limx→0
x − senx
x³
0
0
limx→0
1 − cosx
3. x² Ainda
0
0
limx→0
senx
6. x Ainda
0
0
limx→0
cosx
6= 1
6
8.6. Exercícios
01) Ache os números críticos das funções:
a) f(x) = 4.x² - 3.x + 2
b) g(x) = 2.t³ + t² - 20.t + 4
02) Determine se f satisfaz as hipóteses do teorema do valor médio em [a,b] e, em caso
afirmativo, ache todos os números c em (a,b) tais que:
f(b) – f(a) = f’(c).(b – a)
a) f(x) = 5.x² - 3.x +1; [1,3]
b) f(x) = 𝑥+3
𝑥−2; [-2;3]
c) f(x) = 4+√𝑥 − 1; [1,5]

93
03) Determine os extremos locais de f e os intervalos em que f é crescente ou decrescente.
a) f(x) = 5 – 7.x – 4.x²
b) f(x) = 6.x² - 9.x + 5
c) f(x) = (x² - 10.x)4
04) Use o teste da derivada segunda para achar os extremos locais de f no intervalo [0,2π]
a) f(x) = cos x + sen x
b) ½ x – sen x
05) Uma caixa de base quadrada, sem tampa, deve ter 1m³ de volume. Determine as
dimensões que exigem o mínimo de material. (Desprezar a espessura do material e as perdas
na construção da caixa).
06) Uma página de livro deve ter uma área de 580 cm², com margens de 2,5 cm em baixo e
dos lados e 1,25 em cima. Determine as dimensões da página com maior área imprensa.
07) Um automóvel desce uma ladeira percorrendo s(t) metros em t segundos.
a) Ache a velocidade quando t=3.
b) Após quantos segundo a velocidade será de k m/s.
Resolva os exercícios acima para as seguintes equações de deslocamento:
s(t) = 5.t²+2 k = 28
s(t) = 3.t² + 7 k = 88
08) Use a regra de L’Hospital para encontrar os limites abaixo:
a) limt→0
sen t²
t
b) limx→∞
5x³−2x
7x³+3
c) limx→0
8𝑥²
cos𝑥−1
09) Qual está correto qual está errado? Justifique sua resposta.

94
a) limx→3
x−3
x²−3= lim
x→3
1
2𝑥=
1
6
b) limx→3
x−3
x2−3=0
6= 0

95
9. Integral
9.1. Integral indefinida e definida
9.1.1. Integral Indefinida
Considere uma função f. Chamamos uma função F de antiderivada de f, em dado
intervalo, se vale a igualdade F’(x)=f(x) para todo x no intervalo. Assim, F(x)=x4+5 é
antiderivada de f(x)=4x3, pois F’(x)=Dx(x4)+Dx(5)=4x3. Observe que o mesmo resultado seria
obtido se F(x)= x4 + 6. Assim, podemos generalizar essa antiderivada por F(x)=x4+C onde C
é uma constante arbitrária que sempre será anulada no processo de derivação.
A partir do processo de integração indefinida, podemos obter as antiderivadas da
função f(x). A integral é dita indefinida porque não especifica apenas uma antiderivada, mas
sim uma família delas, já que C pode assumir qualquer valor real.
CxFdxxf )()(
Na maioria dos casos, elas podem ser resolvidas através da regra da potência:
1,1
1
rCr
xdxx
rr
Algumas propriedades são utilizadas no cálculo de integrais indefinidas, entre elas:
Rxdxxfcdxxcf ,)()(
dxxgdxxfdxxgxf )()()]()([
Exemplo 1: Determine a dxx 22

96
SOLUÇÃO – Utilizando a regra da potência,
Cx
Cx
dxx
2
)12(22
122
Exemplo 2: Determine a dxx )2(3 5
SOLUÇÃO – Utilizando as regras da subtração e da potência,
dxdxxdxx 2)2( 3/53 5
2
10
1
13/5
102
13
5C
xC
x
Cxx 28
3 3/8
Observe que as constantes de integração C1 e C2 foram somadas, resultando em apenas
uma constante C. É comum acrescentá-la apenas ao final e não a cada soma da integral, pois
ela conterá todas as outras constantes.
Exemplo 3: Você foi encarregado de determinar a função custo total da produção de drogas
farmacêuticas. Sabe-se que o custo marginal (derivada do custo total CT em relação à
quantidade produzida Q) é dado pela função CM= 150Q+35(R$). Também determine CT para
a produção de 20 unidades dessa droga, se cada uma pode ser produzida à R$210.
SOLUÇÃO – Utilizando a definição de integral indefinida vista anteriormente e o conceito de
antiderivada (F’(x)=f(x)), podemos rearranjar os termos de forma que CxFdxxF )()(' .
Aplicando ao problema:
dQQdQCT )35150('

97
dQQdQ 35150
CQQ 3575 2
Portanto a função procurada é CQQCT 3575' 2
Para Q=1, CT=75+25+C=210 → C=110 e reescrevemos CT=75Q2+35Q+110.
CT(20)=75(20)2+35(20)+110=R$ 30810.
9.1.1.1. Técnicas de Integração
Muitas integrais não podem ser rearranjadas inicialmente de forma a se aplicar a regra
da potência. Por isso, podemos utilizar artifícios que facilitem as operações, como
apresentado a seguir.
MUDANÇA DE VARIÁVEIS OU MÉTODO DA SUBSTITUIÇÃO
A integração e diferenciação indefinida são operações inversas, ou seja, integrar a
derivada de uma função resulta na própria função. Esse enunciado foi utilizado no exemplo c
da seção anterior e ficará mais claro quando estudarmos o teorema fundamental do cálculo.
Seja h(x) uma função composta de F e g, ou seja: h(x)=F(g(x)). Sua integral indefinida
é dada abaixo:
CxgFdxxgFDx ))(())](([
Aplicando a regra da cadeia ao lado esquerdo da equação e considerando F’=f:
dxxgxgFdxxgFDx )('))(('))](([
dxxgxgf )('))((
CxgF ))((

98
Ou, de maneira mais simplificada:
CuFduuf )()(
onde g(x) = u e g’(x)dx = du
Exemplo 4: Determine a dxx 56
SOLUÇÃO – A substituição será u = 6x+5; portanto, du = 6dx. Multiplicamos e dividimos a
integral inicial por um fator de 6, a fim de explicitar o 6dx e em seguida utilizamos a regra da
potência para calcular a integral. Ao final, desfazemos a substituição:
dxxdxx 5666
156
duu 2/1
6
1
CxCu
32/3
)56(9
1
2
36
1
Exemplo 5: Determine a
dx
xx
x23
2
)16(
2
SOLUÇÃO – A substituição será u=x3-6x+1; portanto du=3x2-6 dx=3(x2-2) dx. Nesse caso, é
necessário apenas multiplicar e dividir por um fator de 3, pois (x2-2) já está explícito na
equação original. Utilizamos novamente a regra da potência e desfazemos a substituição:
)2(
)2(3
3
1
)16(
222
2
23
2
x
dx
u
xdx
xx
x
Cxx
Cu
duu )16(3
1
3
11
3
132

99
9.1.2. Integral Definida
Antes de conceituarmos a integral definida, devemos ter em mente o conceito de soma
de Riemann. Inicialmente, consideremos uma região R em um plano coordenado, delimitada
por x=a e x=b e pelo gráfico de uma função f contínua e não negativa no intervalo fechado
[a,b]. Como determinamos a área A da região abaixo do gráfico de f? Podemos dividir o
intervalo [a,b] em n subintervalos iguais tais que ∆𝑥 =𝑏−𝑎
𝑛= 𝑥𝑘 − 𝑥𝑘−1. Graficamente,
teríamos vários retângulos de mesma largura inscritos no gráfico (também poderíamos
realizar a dedução com retângulos circunscritos), interceptando-o em pelo menos um ponto.
Consideremos um retângulo de largura ∆x e altura f(uk). A área de cada retângulo é f(uk)∆x e,
portanto, a área total do polígono é representada por:
𝐴𝑃 =∑𝑓(𝑢𝑘)∆𝑥𝑘
𝑛
𝑘=1
Se fizermos a largura dos retângulos tender a zero, a área total do polígono formado é
aproximadamente a área A procurada. Ou seja:
𝐴 = lim∆𝑥→0
𝐴𝑃 = lim∆𝑥→0
∑𝑓(𝑢𝑘)∆𝑥𝑘
𝑛
𝑘=1
Agora, consideremos que a função f possa ser descontínua e negativa em alguns
pontos do intervalo [a,b]. Consideremos também que os comprimentos dos subintervalos
possam ser diferentes e o valor uk possa ser qualquer número em [xk-1,xk]. Sendo f definida em
um intervalo fechado [a,b] e P uma partição desse intervalo, a soma de Riemann de f para P é
dada por:
𝑅𝑝 =∑𝑓(𝑢𝑘)∆𝑥𝑘
𝑛
𝑘=1
,

100
onde uk está em [xk-1, xk] e k=1,2,3...,n.
Como f(x) pode ser negativo, devemos estudar as implicações desse fato na soma de
Riemann. Se f(uk) é positiva, o retângulo estará acima do eixo x; se f(uk) é negativa, ele estará
abaixo. Sendo assim, Rp é a soma das áreas dos retângulos acima do eixo e dos negativos das
áreas dos retângulos abaixo do eixo.
Retirando a especificação de um número n de subintervalos de uma partição P, a
integral definida de f , de a a b, pode ser entendida como o limite de uma soma:
∫ 𝑓(𝑥)𝑑𝑥𝑏
𝑎
= lim‖𝑃‖→0
∑𝑓(𝑢𝑘)∆𝑥𝑘𝑘
= 𝐿 ,
desde que o limite exista. A expressão f(x) é o integrando, a e b são os limites de integração,
sendo a o limite inferior e b o superior, dx refere-se ao incremento ∆xk e L é número real.
PROPRIEDADES DA INTEGRAL DEFINIDA
Apresentamos algumas das propriedades mais utilizadas no cálculo integral,
considerando funções contínuas no intervalo [a,b], sendo algumas delas já apresentadas
anteriormente para a integral indefinida:
𝑎)∫ 𝑐 𝑑𝑥 = 𝑐(𝑏 − 𝑎), 𝑐 𝜖 𝑅 𝑏
𝑎
𝑏)∫ 𝑐𝑓(𝑥)𝑏
𝑎
𝑑𝑥 = 𝑐∫ 𝑓(𝑥)𝑏
𝑎
𝑑𝑥, 𝑐 𝜖 𝑅
𝑐)∫ 𝑓(𝑥)𝑑𝑥 = −∫ 𝑓(𝑥)𝑑𝑥𝑐
𝑑
𝑑
𝑐
𝑑)∫ [𝑓(𝑥) + 𝑔(𝑥)]𝑑𝑥 = ∫ 𝑓(𝑥)𝑑𝑥 + ∫ 𝑔(𝑥)𝑑𝑥𝑏
𝑎
𝑏
𝑎
𝑏
𝑎
𝑒)∫ 𝑓(𝑥)𝑑𝑥 = ∫ 𝑓(𝑥)𝑑𝑥 + ∫ 𝑓(𝑥), 𝑐𝑏
𝑐
𝑐
𝑎
𝑏
𝑎
𝜖[𝑎, 𝑏]
Exemplo 1: Calcule a dxx )52(
4
2

101
SOLUÇÃO – Ao lado, há o esboço do
gráfico f(x)=2x+5. Vemos que a área sob o
gráfico entre o intervalo [2,4] delimita um
trapézio. Portanto, sua área é
222
2)913(
A
Logo,
22)52(
4
2
dxx
Figura 26 – Gráfico de f(x)
Exemplo 2: Se
3
2
4 2,42dxx e
3
2
5,2xdx , determine dxxx
3
2
4 )7123( .
SOLUÇÃO – Aplicamos inicialmente a propriedade d, seguida da a para as duas primeiras
integrais e da b para a última:
3
2
3
2
3
2
4
3
2
4 753753 dxxdxdxxdxxx
1,132)23(75,252,423
Exemplo 3: Se 996
7
4
xdx e 726
7
5
xdx , determine 5
4
6xdx .
SOLUÇÃO – Aplicamos a propriedade e:
7
5
5
4
7
4
666 xdxxdxxdx
5
4
2772996xdx
0
4
8
12
1 2 3 4 5
(2,9)
(4,13)
A

102
9.2. Integrais Importantes
Funções racionais Funções exponenciais e
logarítmicas
𝑎)∫𝑎 𝑑𝑥 = 𝑎𝑥 + 𝐶 𝑎)∫𝑎𝑥𝑑𝑥 =𝑎𝑥
ln 𝑎+ 𝐶
𝑏)∫𝑥𝑎𝑑𝑥 =𝑥𝑎+1
𝑎 + 1, 𝑎 ≠ −1 𝑏)∫𝑓′(𝑥)𝑒𝑓(𝑥)𝑑𝑥 = 𝑒𝑓(𝑥) + 𝐶
𝑐)∫(𝑎𝑥 + 𝑏)𝑛 𝑑𝑥 =(𝑎𝑥 + 𝑏)𝑛+1
𝑎(𝑛 + 1)+ 𝐶,
𝑛 ≠ 0
𝑐)∫ 𝑒𝑎𝑥𝑑𝑥 =1
𝑎𝑒𝑎𝑥 + 𝐶
𝑑)∫1
𝑥 𝑑𝑥 = 𝑙𝑛|𝑥| + 𝐶 𝑑)∫ log𝑎 𝑥 𝑑𝑥 = 𝑥 log𝑎 𝑥 −
𝑥
ln 𝑎+ 𝐶
𝑒)∫𝑐
𝑎𝑥 + 𝑏 𝑑𝑥 =
𝑐
𝑎𝑙𝑛|𝑎𝑥 + 𝑏| + 𝐶 𝑒)∫ ln 𝑥 𝑑𝑥 = 𝑥 ln 𝑥 − 𝑥 + 𝐶
Funções trigonométricas
𝑎)∫ 𝑠𝑒𝑛 𝑥𝑑𝑥 = − cos 𝑥 + 𝐶 ℎ)∫ 𝑐𝑜𝑠𝑠𝑒𝑐2 𝑥𝑑𝑥 = −𝑐𝑜𝑡𝑔 𝑥 + 𝐶
𝑏)∫ 𝑐𝑜𝑠 𝑥𝑑𝑥 = sen 𝑥 + 𝐶 𝑖)∫ sec 𝑥 𝑡𝑔 𝑥𝑑𝑥 = sec 𝑥 + 𝐶
𝑐)∫ 𝑡𝑔 𝑥 𝑑𝑥 = − ln|cos 𝑥| + 𝐶 𝑗)∫ 𝑐𝑜𝑠𝑠𝑒𝑐𝑥 𝑐𝑜𝑡𝑔𝑥 𝑑𝑥 = −𝑐𝑜𝑠𝑠𝑒𝑐 𝑥 + 𝐶
𝑑)∫ 𝑠𝑒𝑐 𝑥𝑑𝑥 = ln|sec 𝑥 + 𝑡𝑔 𝑥| + 𝐶 𝑘)∫ 𝑠𝑒𝑛2 𝑥𝑑𝑥 =1
2(𝑥 −
𝑠𝑒𝑛2𝑥
2) + 𝐶
𝑒)∫𝑐𝑜𝑠𝑠𝑒𝑐𝑥 𝑑𝑥
= ln|cossec 𝑥 − 𝑐𝑜𝑡𝑔𝑥|+ 𝐶
𝑙)∫ 𝑐𝑜𝑠2 𝑥𝑑𝑥 =1
2(𝑥 +
𝑠𝑒𝑛2𝑥
2) + 𝐶
𝑓)∫𝑐𝑜𝑡𝑔 𝑥𝑑𝑥 = ln|sen 𝑥| + 𝐶 𝑚)∫𝑡𝑔2 𝑥𝑑𝑥 = 𝑡𝑔 𝑥 − 𝑥 + 𝐶
𝑔)∫𝑠𝑒𝑐2 𝑥𝑑𝑥 = 𝑡𝑔 𝑥 + 𝐶
Funções trigonométricas inversas
𝑎)∫𝑎𝑟𝑐𝑠𝑒𝑛 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑠𝑒𝑛 𝑥 + √1 − 𝑥2 + 𝐶, |𝑥| ≤ 1

103
𝑏)∫𝑎𝑟𝑐𝑐𝑜𝑠 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑐𝑜𝑠 𝑥 − √1 − 𝑥2 + 𝐶, |𝑥| ≤ 1
𝑐)∫𝑎𝑟𝑐𝑡𝑔 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑡𝑔 𝑥 −1
2ln|1 + 𝑥2| + 𝐶, 𝑥 𝜖 𝑅
𝑑)∫𝑎𝑟𝑐𝑠𝑒𝑐 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑠𝑒𝑐 𝑥 − ln |𝑥 (1 + √1 − 𝑥−2)| + 𝐶, |𝑥| ≥ 1
𝑒)∫𝑎𝑟𝑐𝑐𝑜𝑠𝑠𝑒𝑐 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑐𝑜𝑠𝑠𝑒𝑐 𝑥 + ln |𝑥 (1 + √1 − 𝑥−2)| + 𝐶, |𝑥| ≥ 1
𝑓)∫𝑎𝑟𝑐𝑐𝑜𝑡𝑔 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑐𝑜𝑡𝑔 𝑥 +1
2ln|1 + 𝑥2| + 𝐶, 𝑥 𝜖 𝑅
Funções hiperbólicas
𝑎)∫ 𝑠𝑒𝑛ℎ 𝑥𝑑𝑥 = cosh 𝑥 + 𝐶 𝑑)∫ 𝑠𝑒𝑐ℎ 𝑥𝑑𝑥 = 𝑎𝑟𝑐𝑡𝑔(𝑠𝑒𝑛ℎ 𝑥) + 𝐶
𝑏)∫ 𝑐𝑜𝑠ℎ 𝑥𝑑𝑥 = senh𝑥 + 𝐶 𝑒)∫𝑐𝑜𝑠𝑠𝑒𝑐ℎ 𝑥𝑑𝑥 = ln |𝑡𝑎𝑛ℎ𝑥
2| + 𝐶 ,
𝑥 ≠ 0
𝑐)∫ 𝑡𝑔ℎ 𝑥𝑑𝑥 = ln cosh 𝑥 + 𝐶 𝑓)∫𝑐𝑜𝑡𝑔𝑥𝑑𝑥 = ln|senh𝑥| + 𝐶,
𝑥 ≠ 0
Funções hiperbólicas inversas
𝑎)∫𝑎𝑟𝑐𝑠𝑒𝑛ℎ 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑠𝑒𝑛ℎ 𝑥 − √𝑥2 + 1 + 𝐶, 𝑥 𝜖 𝑅
𝑏)∫𝑎𝑟𝑐𝑐𝑜𝑠ℎ 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑐𝑜𝑠ℎ 𝑥 − √𝑥2 − 1 + 𝐶, 𝑥 ≥ 1
𝑐)∫𝑎𝑟𝑐𝑡𝑔ℎ 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑡𝑔ℎ 𝑥 +1
2ln(1 − 𝑥2) + 𝐶, |𝑥| < 1
𝑑)∫𝑎𝑟𝑐𝑠𝑒𝑐ℎ 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑠𝑒𝑐ℎ 𝑥 + 𝑎𝑟𝑐𝑠𝑒𝑛 𝑥 + 𝐶, 0 < 𝑥 ≤ 1
𝑒)∫𝑎𝑟𝑐𝑐𝑜𝑠𝑠𝑒𝑐ℎ 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑐𝑜𝑠𝑠𝑒𝑐ℎ 𝑥 + |𝑎𝑟𝑐𝑠𝑒𝑛ℎ 𝑥| + 𝐶, 𝑥 ≠ 0
𝑓)∫𝑎𝑟𝑐𝑐𝑜𝑡𝑔ℎ 𝑥 𝑑𝑥 = 𝑥𝑎𝑟𝑐𝑐𝑜𝑡𝑔ℎ 𝑥 +1
2ln(𝑥2 − 1) + 𝐶, |𝑥| > 1

104
9.3. Teorema Fundamental do Cálculo
O Teorema Fundamental do Cálculo é o teorema que define tanto a relação entre
derivada e integral quanto a maneira de se solucionar uma integral definida.
Teorema Fundamental do Cálculo: Suponhamos f contínua em um intervalo fechado [a,
b].
Se a função G é definida por
tdtfxG
x
a
)()(
Para todo x em [a,b], então G é uma antiderivada de f em [a, b].
Se F é qualquer antiderivada de f em [a, b], então
b
a
aFbFdxxf )()()(
Exemplo 1: Calcular
2
1
23 )1( dxx
SOLUÇÃO – Desenvolveremos primeiro o produto notável do integrando e, depois,
aplicaremos a regra da potência.
2
1
2
1
3623 )12()1( dxxxdxx
2
1
47
42
7x
xx
)1(
4
)1(2
7
)1(2
4
22
7
2 4747
=14
405

105
9.4. Aplicações
9.4.1. Área
Para calcularmos a área entre duas curvas f(x) e g(x) no intervalo entre a e b, sendo,
nesse intervalo, f(x) sempre maior que g(x) , segundo o conceito de integral, basta apenas
calcularmos a integral a seguir:
b
a
dxxgxfA )]()([
É sempre importante termos um esboço das funções para que possamos calcular
esse tipo de integral, já que o cálculo de área sempre envolverá a função superior menos a
função inferior.
Assim, no caso, se quisermos calcular a área entre as curvas f(x) e g(x) no intervalo
entre a e c, sendo f(x) maior que g(x) no intervalo [a,b) e menor do que g(x) no intervalo (b,c],
calculamos a área como:
b
a
c
b
dxxfxgdxxgxfA )]()([)]()([
Outra coisa importante que percebemos é que, quando estamos calculando a b
a
dxxf )( de uma
função qualquer, estamos calculando a integral da função entre a função f(x) e a função y = 0.
Assim, caso a função seja negativa no intervalo (a,b), para se calcular a área entre ela e o eixo
x, temos que calcular a área como:
b
a
b
a
dxxfdxxfA )()](0[

106
Exemplo 1: Qual é a área da região delimitada pelos gráficos 62 xy e 032 xy .
SOLUÇÃO – Para que calculemos a área corretamente, devemos primeiro fazer um esboço
do gráfico
Figura 27 – Gráficos do problema
Percebemos, pelo esboço, que a parábola se situa acima da reta. Consequentemente, na
resolução da integral, 62 xy é f(x) e 032 xy é g(x). Assim, temos que
3
1
2 )]23()6[( dxxxA
3
1
2 )23( dxxx
3
1
23
33 x
xx
3
321
3
139
3
279

107
Um exemplo de aplicação do cálculo de área entre curvas é a teoria da elasticidade.
Para testar a resistência de um material, são registrados os valores de tensão registrados para
diferentes cargas (esforços). A figura a seguir é um diagrama típico tensão-carga para uma
amostra de um material elástico como borracha vulcanizada.
Percebemos pela figura que, à medida que a
carga aplicada aumenta, a tensão aumenta. Isso ocorre
até que a distensão do material atinja seis vezes seu
tamanho original. À medida que a carga decresce, o
material volta ao seu estado original, porém as tensões
pelas quais ele passa não são as mesmas, e sim
menores do que anteriormente. O nome do fenômeno
que ocorre é histerese elástica, e a área entre as curvas
é numericamente igual à energia dissipada durante o
teste pelo material elástico. [1]
Figura 28 – Diagrama tensão esforço para
material elástico. Retirado de [1]
9.4.2. Comprimento de arco
O comprimento do arco de um gráfico de f de um ponto A (a, f(a)) até B (b, f(b)) é dado por:
b
a
b
a dxxfL2
)('1
Exemplo 02: Determine o comprimento do arco da função 103)( 32
xxf do ponto A(8,2)
ao ponto B(27,17).
SOLUÇÃO – A primeira coisa que deve ser feita é se encontrar a derivada da função f(x):

108
31
31 2
2)('x
xxf
Agora, podemos aplicar na fórmula:
dx
xdx
xL
27
83
2
27
8
2
31
27
8
41
21
dxx
x27
83
2
32
4
dxx
x
27
83
13
2 14
Resolvendo a integral pelo método de substituição, 2,2427
8 L .
9.5. Exercícios
01) Resolva as seguintes integrais indefinidas:
𝑎)∫(𝑥 + 1)2𝑑𝑥
𝑏)∫√𝑥 +3
√𝑥𝑑𝑥
𝑐)∫2𝑥1/3 + 𝑥−3 +1
𝑥2𝑑𝑥
𝑑)∫𝑥2 − 2𝑥 + 2
𝑥 − 1𝑑𝑥
𝑒)∫𝑥2 − 4
𝑥 − 2𝑑𝑥
𝑓)∫𝑥3(−2𝑥 + 𝑥−5)𝑑𝑥
𝑔)∫𝑥 + 1
𝑥5

109
02) Resolvas as seguintes integrais indefinidas pelo método de substituição:
𝑎)∫√1 + 𝑥2. 2𝑥𝑑𝑥
𝑏)∫1
(7𝑥 − 2)5𝑑𝑥
𝑐)∫𝑥2
√1 − 𝑥3𝑑𝑥
𝑑)∫(2𝑥2 − 3)5𝑥𝑑𝑥
𝑒)∫(1 + √𝑥)
3
√𝑥𝑑𝑥
𝑓)∫𝑥2𝑒−4𝑥3𝑑𝑥
𝑔)∫𝑠𝑒𝑛 3𝑥
2 𝑑𝑥
ℎ)∫𝑠𝑒𝑐2√𝑥
√𝑥𝑑𝑥
𝑖)∫ 𝑐𝑜𝑠3𝑥 𝑠𝑒𝑛 𝑥 𝑑𝑥
03) Expresse a área sob o gráfico como uma integral definida e dê seu valor:
-3
-2
-1
0
1
2
3
-4 -2 0 2 4
0
1
2
3
4
-4 -2 0 2 4
a) b)

110
04) Resolva com base nas propriedades de integrais definidas:
𝑎)∫ −7
3
6
4
𝑑𝑥
𝑆𝑒 ∫ 𝑥3𝑑𝑥4
1
= 63,75 𝑒 ∫ 𝑥−2𝑑𝑥 = 0,75 ∶4
1
𝑏)∫ 2𝑥3 −2
3𝑥2+
4
1
12𝑑𝑥
𝑐)∫ 𝑥3 −5
𝑥2𝑑𝑥
1
4
05) Calcule a área das funções do exercício 01 com x variando de zero a dez.

111
10. Regressão
A análise de regressão consiste em um processo estatístico que objetiva verificar a
relação funcional entre uma variável dependente com uma ou mais variáveis independentes.
Por meio desse processo podem ser escolhidos os melhores parâmetros de uma curva que
passa por um determinado conjunto de pontos.
Frequentemente a análise de regressão envolve um conjunto de dados medidos
experimentalmente. Dessa forma, baseado no estudo teórico do fenômeno estudado, sabe-se
que tipo de comportamento aquele conjunto de dados terá.
A fim de verificar o comportamento dos valores da variável dependente (Y) em
função da variável independente (X), pode-se fazer um fazer um gráfico denominado
diagrama de dispersão.
Os dados podem apresentar comportamento similar a diversas funções: linear,
quadrática, cúbica, logarítmica, exponencial... Portanto é necessário verificar qual tipo de
curva e modelo matemático que mais se aproxima dos pontos representados no diagrama de
dispersão.
A escolha do modelo deve ser coerente com o comportamento dos dados em análise.
Dessa forma, o modelo escolhido deve ser coerente no aspecto da curva e o grau para
representar corretamente o fenômeno em estudo e ele deve ainda conter apenas variáveis
relevantes para explicar o fenômeno em questão.
Entretanto, são raras as situações em que o ajuste dos dados à equação será perfeito.
Esta situação ideal está representada na figura 1. Usualmente, haverá uma distância entre os
pontos e a curva ajustada. Isso ocorre, pois o fenômeno em estudo é, substancialmente, um
fenômeno físico sujeito a influências externas e não uma situação puramente matemática.
Assim, por meio da regressão dos dados espera-se obter o melhor ajuste possível que
correlacione as variáveis X e Y e o fato de que um determinado ajuste não é perfeito não o
torna descartável. A figura 2 apresenta situações em que o ajuste é imperfeito.
A fim de verificar o quão bem feito é um determinado ajuste existe um parâmetro
estatístico denominado “coeficiente de correlação (R2)”. Os valores de R2 variam de 0 a 1,
sendo que 1 indica uma correlação perfeita.

112
Figura 29 – Situações ideais de correlação perfeita.
Figura 30 – Situações onde o ajuste é imperfeito.
y = 3x - 5
R² = 1
-10
-5
0
5
10
15
0 2 4 6 8
y = x2
R² = 1
0
10
20
30
40
0 2 4 6 8
y = LOG(X)
R² = 1
-1,5
-1
-0,5
0
0,5
1
0 2 4 6 8
y = 3,6321x - 6,9821
R² = 0,9088
-10
0
10
20
0 2 4 6 8
y = 1,0476x2 - 0,8929x + 0,4167
R² = 0,9237
0
10
20
30
40
0 2 4 6 8
y = 0,4267ln(x) + 0,0217
R² = 0,9373
0
0,2
0,4
0,6
0,8
1
0 2 4 6 8

113
11. Referências Bibliográficas
[1] – SWOKOWSKI, Earl W. Cálculo com Geometria Analítica. Tradução Alfredo Alves
de Faria. São Paulo: Makron Books, 1994.
[2] – STEWART, James. Calculus. Tradução: Cyro de Carvalho Patarra et al. São Paulo:
Pioneira Thomson Learning, 2001.
[3] – THOMAS, George B. Cálculo - Volume 1. Tradução: Paulo Boschcov. São Paulo:
Pearson Education do Brasil, 2002.
[4] – USBERCO, João; Salvador, Edgard. Química Geral. 12ª.ed. São Paulo: Saraiva, 2006.
480 p.
[5] – ATKINS, P.W.; JONES, Loretta. Princípios de química: questionando a vida
moderna e o meio ambiente. 3.ed. Porto Alegre: Bookman, 2006. 965 p.
Todo o conteúdo da seção de Química da apostila foi baseado na leitura das
referências [4] e [5]. Todo o conteúdo da seção de cálculo da apostila foi baseado na leitura
das referências [1], [2] e [3].







