
Antiviral Agents

Introduction Because viruses are obligate intracellular parasites,
identification of safe and effective antiviral therapies is difficult.
The best antiviral drugs inhibit a specific step in viral replication or pathogenesis.
Drug discovery can be accomplished by screening or rational design.
The emergence of virus mutants resistant to antiviral drugs is a serious problem.
Combination of targeted delivery strategies to control toxicities and resistance.

Drug Discovery/Development Pipeline
• Multifaceted, complicated, lengthy process
O
OH
OO
HO
O
O
O
N
N
OOH
HOOH
NHCH3
Cl
Cl
IdeaIdea DrugDrug12 -15 Years12 -15 Years
DiscoveryDiscovery Exploratory DevelopmentExploratory Development Full DevelopmentFull Development
Phase IPhase I Phase IIPhase II Phase IIIPhase III
0 155 10
Pre-c
linic
al
Pharm
acolo
gy
Pre-c
linic
al
Pharm
acolo
gy
Pre-c
linic
al S
afet
y
Pre-c
linic
al S
afet
y
Clinic
al P
harm
acolo
gy
& S
afet
y
Clinic
al P
harm
acolo
gy
& S
afet
y
ProductsProductsNH2
CO2HNN
OHN
N N
N
F
F
NH
OCH3O
O
O
ClO
NH2
NNH
OO-
OH OH O
F
O2SN
N
HNO
N
NN
O
N NCF3
SO
OH2N
Today's Focus

The pathway for drug discovery

Drug Development Viruses are now becoming better
understood and several viral genomes have been properly mapped.
Scientists are now looking for the best drug targets
The main point of interest is any viral protein that the host organism does not normally produce
Once these viral proteins are identified they are tested using a large scale screening process to test for effectiveness

Drug Development
Antiviral candidates are tested in mass quantities
Antiviral drugs generally have strange side effects and a high toxicity
As with any pathogenic agent, Viruses evolve and develop resistance.
Thus the need for new drugs always exists

Drug Development
There are several known methods that the makers of Antiviral drugs are looking at, including:
Prevention of Viral Entry Targeting the RNA/DNA replication in
the cell Targeting the transcriptase factors for
Viral DNA Destroying Viral proteases so that viral
proteins are not cut and rearranged in optimal order
Stopping the release of the mature viruses from the host cell

The development of antiviral agents lagged significantly behind the development of antibacterial drugs.
Early drugs were highly toxic.
Analysis of the steps of viral replication has identified potential targets for antiviral drugs (e.g. structures, enzymes or processes).
Inhibitors of Attachment include anti-receptor antibody, natural ligands and synthetic ligands.

Inhibitors of Penetration and uncoating
Amantadine and Rimantadine:-
They are hydrophobic amines (weak organic bases) with clinical efficacy against influenza A only.
They concentrate in and buffer the
contents of the endosomal vesicles preventing uncoating.
Their specificity stems from their ability
to bind to and block the proton channel formed by the M2 matrix protein.

Influenza Treatment with Ion Channel Blockers Amantadine & Rimantadine
Prevent seasonal influenza A in 70-80% of cases
Can reduce severity & duration of illness if started within 48 hrs of onset of symptoms.
Treated persons may shed resistant virus after 5-7 days of treatment (sometimes as early as 2-3 days). All pandemic H1N1 strains are resistant.
Treatment should be discontinued after 3-5 days of treatment or within 24-48 hrs after disappearance of signs/symptoms).

Pleconaril It is a broad spectrum antipicorna virus
agent.
It is a small cyclic drug which binds to a canyon pore of the virus.
In doing so it blocks attachment and
uncoating of the viral particle. It is orally bioavailable and can reduce
peak viral titers by more than 99%.

Inhibitors of Genome Replication
Many viruses have evolved their own specific enzymatic mechanisms to preferentially replicate virus nucleic acids at the expense of cellular molecules.
There is often sufficient specificity in virus polymerases to provide a target for a specific antiviral agent and this method has produced the majority of the specific antiviral agents currently in use.
The majority of these drugs function as polymerase substrate, i.e. nucleoside analogues.
Toxicity varies considerably.
There is a serious problem with the pharmacokinetics of these nucleoside analogues (typically short serum half lives of 1-4 hours).
Nucleoside analogues are in fact pro-
drugs, since they need to be phosphorylated before becoming effective. This is the key to their selectivity.

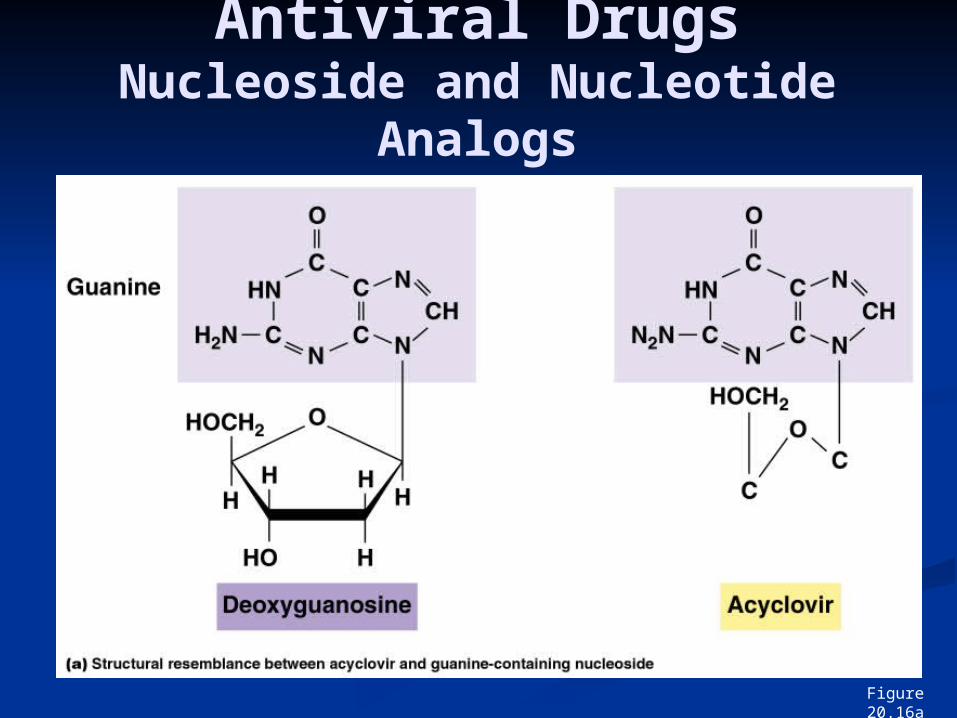
Nucleoside AnaloguesAcyclovir (acycloguanosine )- ACV.
Close to a perfect antiviral drug (specific, nontoxic).
Highly effective against herpes simplex virus (HSV), less so against varicella -zoster virus (VZV).
Highly selective and extremely safe.
Acyclic guanine derivative (differs from guanosine by having an acyclic side chain) that inhibits viral DNA synthesis.
Antiviral DrugsNucleoside and Nucleotide
Analogs
Figure 20.16a

It is a prodrug, a precursor of the antiviral compound.
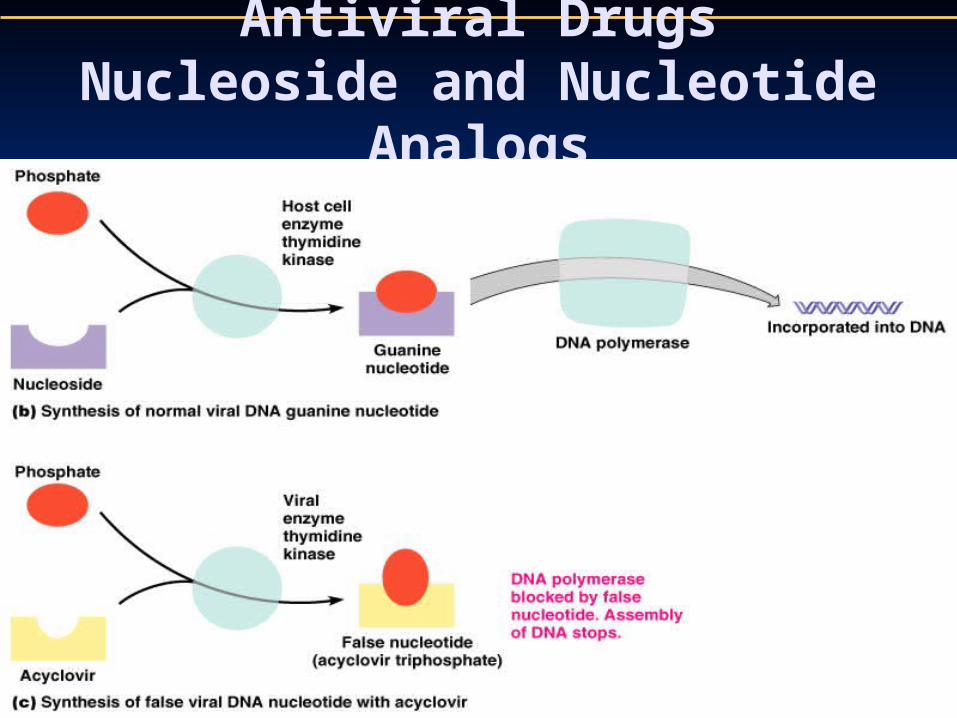
Activation of the drug requires three kinase activities to be present in the cell to convert acyclovir to a triphosphate derivative, the actual antiviral drug.
It is phosphorylated by a virus thymidine kinase (200 times more efficiently than by cellular enzymes) producing a monophosphate form.
Cellular enzymes complete phosphorylation to the di - and triphosphate forms.

The triphosphorylated form competes with GTP inhibiting the enzyme (DNA polymerase) and causing termination of the growing viral DNA chain because of lack of 3' OH group.
ACV affinity to viral polymerase is more than 100 folds that to cellular polymerase.
Acyclovir has no effect on host DNA replication because the first kinase activity is not found in an noninfected cell.
Antiviral DrugsNucleoside and Nucleotide
Analogs

Valacyclovir
othe valyl ester derivative of ACV is more efficiently absorbed and rapidly converted to ACV increasing its bioavailability.
oPenciclovir and famciclovir are related drugs.
Ganciclovir
oIt differs from ACV by the addition of a single hydroxymethyl group in the acyclic side chain; the result is a remarkable activity against CMV.
oIt is phosphorylated by a virus-encoded kinase (not thymidine kinase).

Adenine arabinoside (vidarabine)- Ara -A
oA purine analogue (identical to adenosine but arabinose is substituted for ribose).
oPhosphorylated by cellular enzymes ( toxicity?) to inhibit both viral and cellular polymerases but viral is 6-12 times more sensitive.
oIt was used for HSV and VZV before ACV.

Azidothymidine (Zidovudine) AZT
Dideoxy analog of thymidine
(a synthetic thymidine analogue) that Inhibits viral DNA synthesis by inhibiting the reverse transcriptase enzyme.
It has higher affinity to RT (100 times) than to cellular DNA polymerase.

Efficiently phosphorylated (several steps of phosphorylation) to triphosphate by cellular kinases
AZT monophosphate competes with thymidine monophosphate
Much less selective than acylovir and has side effects
Does not eliminate previously incorporated provirus

Dideoxyinosine (Didanosine, ddI) Dideoxycytidine (Zalcitabine, ddc) Stavudine (d4t) Lamivudine (3Tc).
oAll are inhibitors of reverse transcriptase used for the treatment of HIV infection.
Ribavarin
oAn analogue of guanosine but the base ring is incomplete and open.
oIt is active against DNA and RNA viruses by inhibiting inosine monophosphate dehydrogenase and the synthesis of the mRNA 5- cap and RNA polymerase.

Iododeoxyuridine (Idoxyuridine) Trifluorothymidine (Trifluridine) Fluorouracil
oAll are analogues of thymidine and they inhibit the biosynthesis of thymidine or replace it and become incorporated in DNA.
Nucleotide Analogue (cidofovir)
oIt has the phosphate group attached and it inhibits DNA polymerase .

Nonnucleoside polymerase Inhibitors
Foscarnet: anti-herpes viruses. Nevirapine and delaviridine : anti HIV
Protease Inhibitors Anti HIV: Saquinavir, Indinavir, and
Ritonavir
oAnti HCV: Boceprevir and Telaprevir
oThe Unique structure of HIV protease and its essential role in the production of a functional virion has made this enzyme a good target for antiviral drugs.

Uncleavable mimics of gag- pol polyprotein
Inhibits HIV protease
Does not eliminate previously incorporated provirus but does prevent further spread
Resistance due to protease alterations

Inhibitors of Assembly, Maturation and Release
Zanamivir/ Relenza (aerosol) Oseltamivir / Tamiflu (tables) Peramivir/ IV for emergency use in hospitalized adults or children
oActive against influenza as they are inhibitors of neuraminidase preventing the release of budded viruses from the cell.
oBecause they act late in the life cycle of the
virus it is hoped that problems with resistance emergence will be minimized.

Neuraminidase Inhibitors Zanamivir & Oseltamivir
Mechanism: blocking of the active site of neuraminidase; prevents removal of sialic acid residues and results in clumping of viral progeny
Effective against influenza A & B.
Effective when flu symptoms are < 2 days old.
Inhibitors reduce disease syndrome by 1 day.
May decrease influenza secondary complications
Antiviral resistance can occur, but much less frequently than with the ion channel blockers amantadine or rimantadine

Neuraminidase inhibitors appear to have similar efficacy to the amantidine & rimantidine ion channel blockers for prevention & treatment of influenza
Neuraminidase inhibitors have Less Central Nerveous System side effects, but more Gastro-Intestinal effects
Neuraminidase Inhibitors are more expensive, but there is less risk of inducing virus resistance.

Methisazone
oIt is of historical importance as an inhibitor of poxviruses.
oIt was highly virus specific and did not
affect cellular metabolism. oIt blocked a late stage in viral replication
resulting in the formation of immature, noninfectious virus particles.







