doi doc url //eprints.lib.hokudai.ac.jp/dspace/bitstream/2115/65648/1/... · ptk protein-tyrosine...
TRANSCRIPT

Instructions for use
Title Studies on the function of ebolavirus glycoprotein-specific antibodies
Author(s) 古山, 若呼
Issue Date 2017-03-23
DOI 10.14943/doctoral.k12612
Doc URL http://hdl.handle.net/2115/65648
Type theses (doctoral)
File Information Wakako_Furuyama.pdf
Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP

Studies on the function of ebolavirus
glycoprotein-specific antibodies
(エボラウイルス表面糖蛋白質に対する
抗体の機能に関する研究)
Wakako FURUYAMA

i
Contents
Abbreviation -------------------------------------------------------------------------------------------- 1
Preface ---------------------------------------------------------------------------------------------------- 4
Chapter I:
Discovery of an antibody for pan-ebolavirus therapy
Introduction -------------------------------------------------------------------------------------------- 7
Materials and Methods ------------------------------------------------------------------------------- 8
Viruses and cells
Purification of virus-like particles (VLPs) for immunization
Generation of MAb 6D6
Neutralization tests
Enzyme-linked immunosorbent assay (ELISA)
Selection of escape mutants and identification of the putative epitope
Purification and fluorescent-labeling of VLPs
Imaging of attachment, internalization and membrane fusion of DiI-labeled VLPs
Passive immunization and protective efficacy in mice
Statistical analysis
Results ------------------------------------------------------------------------------------------------ 15
In vitro properties of MAb 6D6
Identification of the putative 6D6 epitope
Mechanism of the neutralizing activity of 6D6
Protective efficacy of MAb 6D6 in mouse models
Discussion -------------------------------------------------------------------------------------------- 26

ii
Summary ---------------------------------------------------------------------------------------------- 29
Chapter II:
Fcγ-receptor IIa-mediated Src signaling pathway is essential for the antibody-
dependent enhancement of Ebola virus infection
Introduction ------------------------------------------------------------------------------------------- 30
Materials and Methods ------------------------------------------------------------------------------ 32
Viruses and cells
Generation of FcγRIIa- or FcγRIIaΔCT-expressing Jurkat T cells
ADE assays
Purification and DiI-labeling of VLPs
Imaging of attachment and internalization of DiI-labeled VLPs
Dextran uptake assays
Inhibitor treatments
Generation of Src or Syk knockdown K562 cells
Phosphorylation assay
Statistical analysis
Results ------------------------------------------------------------------------------------------------ 40
Cytoplasmic tail of FcγRIIa is required for ADE of EBOV infection
Cytoplasmic tail of FcγRIIa is important for enhanced viral uptake
Intracellular signaling via Src family PTKs contributes to ADE of EBOV infection
VLP-MAb complexes activate phosphorylation of Src in K562 cells
The ADE of EBOV entry depends on phagocytosis and/or macropinocytosis mediated by
Src family PTKs
Discussion ---------------------------------------------------------------------------------------------- 64

iii
Summary ----------------------------------------------------------------------------------------------- 68
Conclusion ----------------------------------------------------------------------------------------------- 69
Acknowledgement ------------------------------------------------------------------------------------- 71
和文要旨-------------------------------------------------------------------------------------------------- 73
References ----------------------------------------------------------------------------------------------- 76

1
Abbreviations
ADE antibody-dependent enhancement
Alexa647-labeled Dx10 Dextran, Alexa Fluor 647, 10,000 MW
BDBV Bundibugyo virus
BSA bovine serum albumin
Btk Bruton’s tyrosine kinase
CT cytoplasmic tail
CTR IgG control IgG
DAPI 4',6-diamidino-2-phenylindole, dihydrochloride
DC-SIGN dendritic cell-specific ICAM-3-grabbing non-integrin
DiI 1,1'-dioctadecyl-3,3,3',3'-
tetramethylindocarbocyanine perchlorate
DMEM Dulbecco’s modified Eagle’s medium
DMSO Dimethyl sulfoxide
ELISA Enzyme-linked immunosorbent assay
EBOV Ebola virus
EVD Ebola virus disease
FCS fetal calf serum
FcγR Fcγ receptor
FFU focus forming units
GFP green fluorescent protein
GP glycoprotein
HEK human embryonic kidney
hMGL human macrophage galactose-type C-type lectin

2
HR1 heptad repeats 1
HR2 heptad repeats 2
IC50 50% inhibitory concentrations
IFL internal fusion loop
ITAM immunoreceptor tyrosine-based activation motif
IU Infectious unit
MARV Marburg virus
MAb monoclonal antibody
MLR mucin-like region
NP nucleoprotein
NPC1 Niemann-Pick C1
OD optical density
PBS phosphate-buffered saline
PI3K phosphatidylinositol 3-kinase
Plat-GP Platinum-GP
PTK protein-tyrosine kinase
Ras rat sarcoma
RBD receptor binding domain
RESTV Reston virus
RPMI Roswell Park Memorial Institute
SDS-PAGE sodium dodecyl sulfate-polyacrylamide gel
electrophoresis
Src sarcoma
SUDV Sudan virus

3
Syk spleen tyrosine kinase
TAFV Taï Forest virus
TBS Tris-buffered saline
TBST Tris-buffered saline containing 0.1% Tween 20
TIM-1 T-cell immunoglobulin and mucin domain 1
VLP virus-like particle
TM transmembrane
VSV vesicular stomatitis virus

4
Preface
Ebolaviruses, members of the family Filoviridae, cause severe hemorrhagic
fever in humans and nonhuman primates, with human case fatality rates of up to 90% 1,2.
The latest epidemic of Ebola virus disease (EVD) in West Africa, ebolaviruses pose a
significant public health concern. However, an effective prophylaxis or treatment for
EVD is not yet commercially available. Five distinct species are known in the genus
Ebolavirus; Zaire ebolavirus, Sudan ebolavirus, Taï Forest ebolavirus, Bundibugyo
ebolavirus, and Reston ebolavirus, represented by Ebola virus (EBOV), Sudan virus
(SUDV), Taï Forest virus (TAFV), Bundibugyo virus (BDBV), and Reston virus
(RESTV), respectively 3. In the last decade, EBOV, SUDV, and BDBV have caused EVD
outbreaks with increased frequency in Central and West Africa 1,2,4.
Ebolaviruses express a single transmembrane glycoprotein (GP) that is
responsible for both receptor binding and membrane fusion. GP undergoes proteolytic
cleavage by host proteases such as furin, resulting in two subunits, GP1 and GP2, which
are linked by a disulfide bond 5,6. The GP1 subunit (amino acids 33-501) contains the core
of the glycoprotein, a receptor binding domain (RBD), a glycan cap, and a large mucin-
like region (MLR) that extends around the RBD 7. The GP2 (amino acids 502-676)
subunit contains the internal fusion loop (IFL), heptad repeats 1 and 2 (HR1 and HR2),
the transmembrane region (TM), and the cytoplasmic tail (CT) 8. EBOV entry is initiated
by viral attachment to cell surface molecules such as T-cell immunoglobulin and mucin
domain 1 (TIM-1) and C-type lectins 9,10, followed by internalization of the virus particle
into cells via micropinocytosis 11-13. In the late endosome, EBOV GP is cleaved by host
proteases such as cathepsins L and B 14,15, and then binds to the receptor, Niemann-Pick
C1 (NPC1), followed by membrane fusion 8,16-19.

5
Since GP is the only surface protein of ebolaviruses and essential for viral entry
into host cells, it is the sole target of neutralizing antibodies. In the past, many GP-specific
monoclonal antibodies (MAbs) including neutralizing antibodies, have been generated 20-
23. Previous studies have shown that neutralizing antibodies against EBOV GP reduce
virus infectivity through blocking receptor binding (i.e., interaction between GP and
NPC1), cathepsin proteolysis, or membrane fusion 24,25. Importantly, passive
immunization with some GP-specific MAbs protects experimental animals, such as
rodents and nonhuman primates, from lethal EBOV infection 26-31. Indeed, a human MAb
cocktail against EBOV GP was used for the treatment of EVD in patients during the latest
outbreak in West Africa 32-35. However, most of the previously tested MAbs are EBOV-
specific and MAb therapy against other members of the genus Ebolavirus (i.e., SUDV
and BDBV) is not yet available.
On the other hand, some GP-specific MAbs reportedly induce antibody-
dependent enhancement (ADE), a phenomenon in which viral infectivity is increased by
virus-specific antibodies. ADE has been observed in vitro for a large number of viruses
36-38 and it has interfered with vaccinations against some viruses. In addition, some human
sera convalescent from EVD contain ADE antibodies 22. ADE of EBOV infection
depends on the cross-linking of virus-antibody complexes to the cellular Fcγ receptor IIa
(FcγRIIa) or complement component C1q and its ligand 22,23,39. These receptors are
known to activate various signaling pathways that lead to the reorganization of the actin
cytoskeleton and membrane remodeling 40,41. However, little is known about the
molecular mechanisms underlying ADE of EBOV infection.
In this study, I describe two MAbs with different functions: neutralization and
ADE. In chapter I, a broadly cross-reactive GP-specific MAb, 6D6, was generated and

6
analyzed for its neutralization mechanism and protective potential. In chapter II, the
contribution of FcγRIIa-mediated signaling to EBOV ADE was investigated, and the
function of the signaling pathway was analyzed.

7
Chapter I:
Discovery of an antibody for pan-ebolavirus therapy
Introduction
Several studies have demonstrated that administration of EBOV GP-specific
antibodies protects nonhuman primates from lethal EBOV infection and may constitute a
leading treatment option for EVD in humans 26-30. During the West African EVD outbreak,
EBOV GP-specific MAb cocktails (e.g., ZMapp) 26 were used to treat several patients 32-
35. However, most of characterized therapeutic MAbs to date are EBOV GP-specific and
cross-neutralizing activity against any other ebolavirus species (e.g., SUDV and BDBV)
has not been demonstrated due to antigenic differences among the species 42. Since SUDV
and BDBV have also shown their potential to cause public health emergencies during
several outbreaks in Central Africa, it is difficult to determine the priority for
development of countermeasure against those ebolaviruses.
In chapter I, I demonstrated generation of a broadly cross-reactive GP-specific
MAb. This MAb, 6D6, recognizes the putative epitope in the highly conserved IFL and
neutralizes infectivity of representative isolates of all known ebolavirus species by
inhibiting the membrane fusion. I further demonstrated its protective potential as a
therapeutic antibody in mouse models of EBOV and SUDV infections.

8
Materials and Methods
Viruses and cells. Variants Yambuku (EBOV1976), Makona (EBOV2014), Nzara
(SUDV), Butalya (BDBV), Pauléoula (TAFV), Philippines89 (RESTV) and Angola
(MARV), and mouse-adapted EBOV 43 were propagated in African green monkey kidney
Vero E6 cells and stored at -80 ˚C. Virus titters were determined as focus forming units
(FFU) by immunoplaque assays. All infectious work with filoviruses was performed in
the biosafety level 4 laboratories at the Integrated Research Facility of the Rocky
Mountain Laboratories, Division of Intramural Research, National Institute of Allergy
and Infectious Diseases, National Institutes of Health, Hamilton, Montana, USA.
Replication-competent recombinant vesicular stomatitis virus (rVSV/EBOV GP,
rVSV/SUDV GP, and rVSV/RESTV GP) and replication-incompetent pseudotyped
vesicular stomatitis viruses (VSVs) containing green fluorescent protein (GFP) instead of
the VSV G gene were generated as described previously 20,44. Infectious units (IUs) of
replication-incompetent pseudotyped VSVs were determined using Vero E6 cells as
described previously 44. Vero E6 cells and human embryonic kidney (HEK) 293T cells
were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma). The media were
supplemented with fetal calf serum (FCS) (Cell Culture Bioscience) and 100 U/ml
penicillin, 0.1 mg/ml streptomycin (Gibco).
Purification of virus-like particles (VLPs) for immunization. HEK293T cells were
transfected with equal amounts of the expression plasmids encoding GP, matrix protein
(VP40), and nucleoprotein (NP) of EBOV or SUDV using TransIT LT-1 reagent (Mirus)
according to the manufacturer's instructions. Forty-eight hours later, the culture
supernatant was harvested and centrifuged at 3,500 rpm for 15 min to remove cell debris.

9
VLPs were purified from culture supernatants by ultracentrifugation at 28,000 rpm with
an SW32Ti rotor (Beckman) at 4 ˚C for 2 h with a 25% sucrose cushion. The VLP pellets
were suspended in phosphate-buffered saline (PBS) and fractionated through a 20-50%
sucrose gradient in PBS at 28,000 rpm with an SW41 rotor (Beckman) at 4 ˚C for 2 h.
Then the VLP fractions were diluted with PBS and sedimented by ultracentrifugation at
28,000 rpm with an SW41 rotor at 4 ˚C for 2 h. Finally, the VLP pellets were resuspended
in PBS.
Generation of MAb 6D6. Fifteen-week-old female BALB/c mice were immunized
intraperitoneally with 100 μg of EBOV VLPs. At 2 and 5 weeks after the first
immunization, the mice were intraperitoneally immunized with 100 μg of EBOV VLPs.
At 10 weeks after the first immunization, the mice were immunized with 100 μg of SUDV
VLPs. Two weeks after the last immunization, the mice were boosted intraperitoneally
with 100 μg of EBOV VLPs. Three days later, the mice were euthanized and spleen cells
and mouse myeloma P3U1 cells were fused and maintained according to a standard
procedure45. The mice were treated daily with 75 µg/kg rapamycin intraperitoneally
starting 1 week prior to the primary immunization until euthanasia. Hybridomas were
screened for secretion of EBOV GP-specific MAbs by a neutralization test with VSV
pseudotyped with EBOV GP and hybridomas producing MAbs were cloned twice by
limiting dilution of the cells. Hybridomas producing neutralizing MAbs were further
screened for the cross-reactivity to the other filovirus GPs. MAb 6D6 (IgG1) was found
to be a broadly cross-neutralizing MAb and was purified from mouse ascites using protein
A agarose columns (Bio-Rad). Animal studies were carried out in strict accordance with
the Guidelines for Proper Conduct of Animal Experiments of the Science Council of

10
Japan. The protocol was approved (13-0136) by the Hokkaido University Animal Care
and Use Committee.
Neutralization tests. To evaluate the neutralizing activity a focus forming assay was used.
EBOV, SUDV, TAFV, BDBV, RESTV, and MARV was appropriately diluted to yield 20-
100 FFUs and mixed with purified MAb 6D6 for 1 h at 37 ˚C and inoculated into
confluent Vero E6 cells grown in 96-well tissue culture plates. After incubation for 3 days,
the cells infected with filoviruses were fixed and stained with a mixture of anti-GP (mouse
anti-EBOV GP ZGP42/3.7 or anti-MARV GP rabbit serum FS0505) and anti-NP (mouse
anti-EBOV NP ZNP74/7 or anti-MARV NP rabbit serum FS0609) primary antibodies 46
followed by goat anti-mouse IgG/Alexa Fluor 488 (Invitrogen) and goat anti-rabbit
IgG/Alexa Fluor 488 (Invitrogen) secondary antibodies. Virus infectivity was quantified
by counting the number of fluorescent foci. VSV pseudotyped with filovirus GPs were
appropriately diluted to yield 300 to 1,500 IUs and mixed with purified MAb 6D6,
ZGP133/3.16, or ZGP226/8.1 for 1 h at room temperature, and inoculated into confluent
Vero E6 cells grown in 96-well plates. At 20 h post-inoculation, GFP-positive cells were
counted using IN Cell Analyzer 2000 (GE Healthcare). To reduce the background
infectivity of the parent VSV G, pseudotyped viruses were treated with a neutralizing
MAb to VSV G protein (VSV-G[N]1-9) before use. The relative percentage of infectivity
was calculated by setting the number of cells infected in the absence of MAb 6D6 to
100%.
Enzyme-linked immunosorbent assay (ELISA). GP-based ELISA was performed as
described previously 42. Soluble forms of EBOV GP were purified and used as antigens.

11
MAbs were serially diluted with PBS containing 0.05% Tween 20 and 1% skim milk.
Bound antibodies were visualized with horseradish peroxidase-conjugated goat anti-
mouse IgG (H+L) (Jackson ImmunoResearch) and 3,3’,5,5’-tetramethylbenzidine
(Sigma). The reaction was stopped by adding 1 M phosphoric acid, and the optical density
at 450 nm (OD450) was measured using SoftMax Pro 6.2.1 software (Molecular Devices).
Selection of escape mutants and identification of the putative epitope. Tenfold serial
dilutions of rVSV/EBOV GP, rVSV/SUDV GP, and rVSV/RESTV GP were incubated
with 10 μg/ml MAb 6D6 for 1 h at room temperature and inoculated into confluent Vero
E6 cells grown in 6-well tissue culture plates. After adsorption for 1 h, the cells were
overlaid with Eagle’s minimal essential medium (Invitrogen) containing 0.8% Bacto Agar
(BD), 0.3% bovine serum albumin (BSA) (Sigma), 100 U/ml penicillin, 0.1 mg/ml
streptomycin, and 10 μg/ml 6D6, and then incubated for 2 days at 37 ˚C. Mutant viruses
growing in the presence of MAb 6D6 were purified from single isolated plaques at the
highest dilution of the virus and propagated in Vero E6 cells. Viral RNAs were extracted
from the supernatant, the nucleotide sequences of the GP genes of the parent viruses and
the escape mutants were determined. The deduced amino acid sequences were compared
among these viruses. The IFL amino acid sequences of EBOV, SUDV, BDBV, TAFV,
RESTV, and MARV were obtained from GenBank (Accession numbers, U23187.1,
U28134.1, NC_014373.1, U28006.1, AF522874.1, and KY047763, respectively). The
substituted amino acid positions were mapped on the trimeric structure of GPs
constructed using Discovery Studio 4.1 (Biovia) based on the crystal structure of EBOV
GP (PDB code: 3CSY). The SUDV and RESTV GP structures were generated by
homology modelling based on the EBOV GP structure. From 100 models of the GP trimer,

12
the model with the best score for probability density function (PDF) and total energy was
chosen. The model was evaluated using Profiles-3D 47.
Purification and fluorescent-labeling of VLPs. For purification of VLPs, equal
amounts of the expression plasmids for EBOV GP, VP40, and NP were transfected into
HEK293T cells by using TransIT LT-1 (Mirus). Forty-eight hours post-transfection, the
culture supernatant was harvested and centrifuged at 3,500 rpm for 15 min to remove cell
debris. VLPs were precipitated through a 25% sucrose cushion by centrifugation at 11,000
rpm for 1 h at 4 ˚C with an SW32Ti rotor (Beckman). Precipitated VLPs were suspended
in PBS, and fractionated through a 20-50% sucrose gradient in PBS at 27,000 rpm with
an SW41 rotor (Beckman) for 2.5 h at 4 ˚C. One ml of fractionated VLPs (1 μg/ml) was
incubated with 0.6 µl of 100 µM stock solution of 1,1'-dioctadecyl-3,3,3',3'-
tetramethylindocarbocyanine perchlorate (DiI) (Invitrogen) in the dark for 1 h at room
temperature with gentle agitation 11.
Imaging of attachment, internalization and membrane fusion of DiI-labeled VLPs.
Vero E6 cells expressing enhanced GFP fused to Rab7 (eGFP-Rab7) were cultured in 35
mm glass-bottom culture dishes (MatTek Corporation). DiI-labeled VLPs were treated
with 20 μg/ml 6D6 or control IgG (CTR IgG) (mouse IgG1,κ; BD Biosciences) for 1 h at
room temperature. The cells were then washed with 1 ml of phenol red-free DMEM
(Invitrogen) and incubated with either MAb 6D6-treated, CTR IgG-treated or untreated
VLPs in the same medium on ice for 30 min. Following this, the cells were washed with
the same medium to remove unbound VLPs and incubated with 200 µl of phenol red-free
DMEM containing 2% FCS and 4% BSA at 37 ˚C for 0, 2, and 6 h to analyze attachment,

13
internalization and membrane fusion, respectively. In this assay, the fluorescent signal is
enhanced once the DiI-labeled VLP envelope fuses with the endosomal membrane 11. To
count the number of DiI-labeled VLPs, the cells were fixed in 4% paraformaldehyde for
15 min at room temperature. Then nuclei were stained using 1 μg/ml of 4',6-diamidino-
2-phenylindole, dihydrochloride (DAPI) for 10 min at room temperature (Thermo Fisher
Scientific). Images were acquired with a 63 × oil objective lens on a Zeiss LSM700
inverted microscope and ZEN 2009 software (Carl Zeiss). For measurement of the
number of DiI-labeled VLPs, images of 4-20 optical sections were acquired in 0.5-1
micron steps. The number of DiI signals was determined in approximately 100 individual
cells (approximately 1-20 dots/cell) and the average number per cell was calculated for
each condition. For colocalization analysis, the percentage of DiI-labeled VLPs that
colocalized with eGFP-Rab7 (Both DiI- and eGFP-positive pixels/DiI-positive pixels ×
100) was measured using the Coloc module in ZEN 2010 software (Carl Zeiss). The
number, size, and fluorescence intensity of DiI signals were analyzed with MetaMorph
software (Molecular Devices). The relative sizes and intensities of DiI signals were
determined by defining the value of untreated cells as 1.
Passive immunization and protective efficacy in mice. BALB/c mice (female, 6-8
weeks old) were inoculated with mouse-adapted EBOV (1,000 FFU) by intraperitoneal
injection in a total volume of 200 μl. One day after infection, the mice were treated with
100 μg of MAb 6D6 intraperitoneal in a volume of 200 μl. To investigate the potential of
6D6 to protect mice from EBOV and SUDV infection, C57BL/6 IFNAR-/- mice were
chosen for this study as they are known to be susceptible to EBOV and SUDV infection.
BDBV did not cause clinical symptoms in IFNAR-/- mice (data not shown). IFNAR-/- mice

14
(male and female, 5-8 weeks old) were treated with 100 μg of MAb 6D6 intraperitoneal
in a volume of 200 μl one day after infection with EBOV1976 (1,000 FFU) or SUDV
(1,000 FFU). The animals were monitored for signs of illness and weighed daily.
Surviving mice were euthanized 28 days after infection, and serum was collected for
serology. Research was approved and conducted in compliance with the guidelines of the
National Institute of Allergy and Infectious Diseases/Rocky Mountain Laboratories
Institutional Animal Care and Use Committee. The facility where this research was
conducted is fully accredited by the Association for the Assessment and Accreditation of
Laboratory Animal Care International and has an approved Office of Laboratory Animal
Welfare Assurance (#A4149-01). All procedures were conducted by trained personnel
under the supervision of veterinarians, and all invasive clinical procedures were
performed while animals were anesthetized. Early endpoint criteria, as specified by the
Institutional Animal Care and Use Committee approved scoring parameters, were used to
determine when animals should be humanely euthanized.
Statistical analysis. All data were analyzed using the GraphPad Prism v6.0 software. For
the viral attachment, internalization, and membrane fusion experiments, Student’s t-test
was used to evaluate differences between 6D6 and CTR IgG. To assess the weight loss of
mice, I performed a 2-way repeated-measures analysis of variance (ANOVA), followed
by multiple t-tests comparing the average weights of 6D6-treated and untreated (control)
mice at each time point, using the Holm–Sidak method. P values less than 0.05 were
considered to be statistically significant.

15
Results
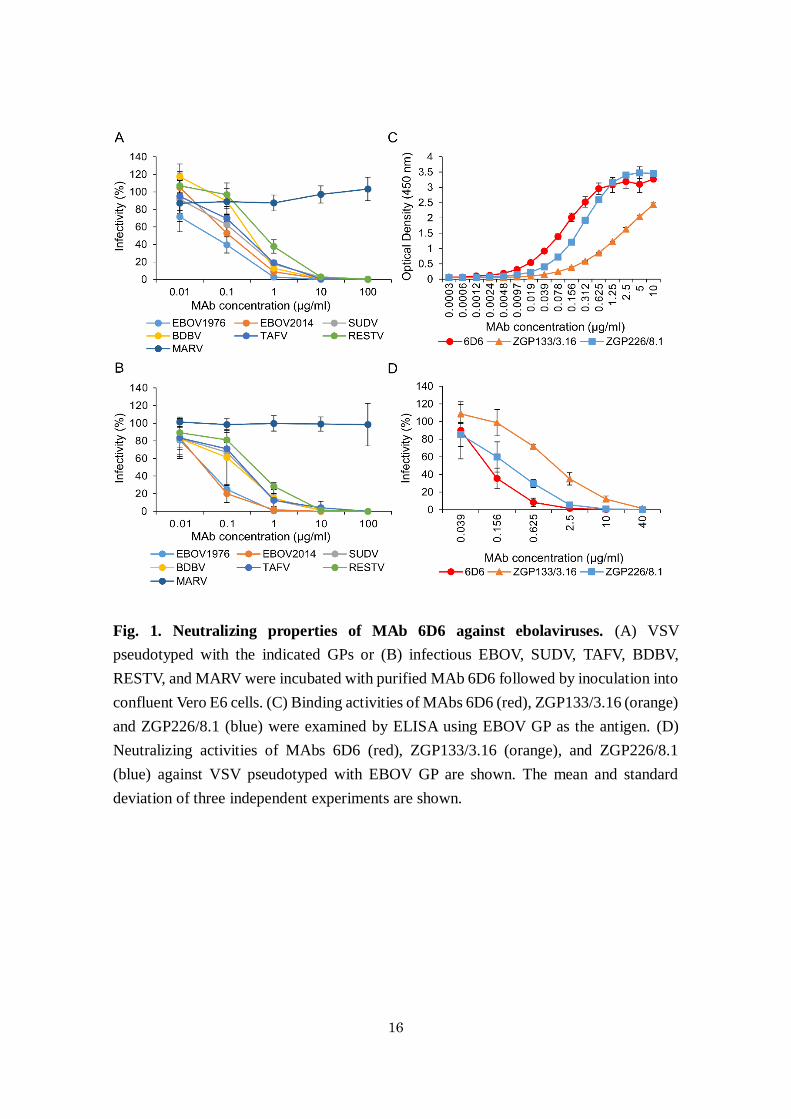
In vitro properties of MAb 6D6. The cross-reactive MAb 6D6 was selected by screening
mouse hybridoma supernatants thoroughly for the cross-neutralizing activity of GP-
specific MAbs. MAb 6D6 was found to be GP-specific and to efficiently neutralize the
infectivity of VSV pseudotyped with GPs of all known ebolavirus species (EBOV, SUDV,
TAFV, BDBV, and RESTV), including the variant that caused the latest outbreak in West
Africa (EBOV2014), but not Marburg virus (MARV) which a related filovirus and causes
human disease similar to EVD (Fig. 1A). The 50% inhibitory concentrations (IC50) of
6D6 for VSVs bearing EBOV1976, EBOV2014, SUDV, TAFV, BDBV, and RESTV GPs
were 0.05, 0.12, 0.19, 0.33, 0.24, and 0.62 μg/ml, respectively. I then confirmed that 6D6
effectively neutralized the infectivity of representative authentic isolates of all known
ebolavirus species (Fig. 1B). Furthermore, binding experiments to EBOV GP and
neutralization assays with EBOV GP-pseudotyped VSV revealed that 6D6 possessed
higher binding and neutralizing abilities than EBOV GP-specific MAbs ZGP133/3.16 and
ZGP226/8.1 (Fig. 1C and D), which have shown promising protective efficacy in animal
models of lethal EBOV infection 28,48.
Identification of the putative 6D6 epitope. To determine the putative epitope of MAb
6D6, I utilized replication-competent recombinant VSV containing the EBOV, SUDV, or
RESTV GP gene 20. The putative epitopes of ZGP133/3.16 and ZGP226/8.1 have been
successfully determined by identifying the amino acid substitutions observed in the
antigenic variants escaping from neutralization by the antibodies 20,24. I cloned 6 escape
mutants of EBOV GP and found that each mutant had a single amino acid substitution,
Gly-to-Arg (5/6) or Gly-to-Glu (1/6), at amino acid position 528 within the IFL sequence
16
Fig. 1. Neutralizing properties of MAb 6D6 against ebolaviruses. (A) VSV
pseudotyped with the indicated GPs or (B) infectious EBOV, SUDV, TAFV, BDBV,
RESTV, and MARV were incubated with purified MAb 6D6 followed by inoculation into
confluent Vero E6 cells. (C) Binding activities of MAbs 6D6 (red), ZGP133/3.16 (orange)
and ZGP226/8.1 (blue) were examined by ELISA using EBOV GP as the antigen. (D)
Neutralizing activities of MAbs 6D6 (red), ZGP133/3.16 (orange), and ZGP226/8.1
(blue) against VSV pseudotyped with EBOV GP are shown. The mean and standard
deviation of three independent experiments are shown.

17
in the GP2 subunit (Fig. 2A). One of the six SUDV GP escape mutants had a Gly-to-Arg
substitution at position 528, and other 5 SUDV GP escape mutants had an Ala-to-Thr
substitution at position 530 (Fig. 2A). Two of the six RESTV GP escape mutants had a
Gly-to-Glu substitution at position 529, which corresponded to position 528 of EBOV GP.
A total of 3 amino acid changes were found in the other 4 RESTV GP escape mutants
(Fig. 2A). Using a reverse genetics approach I verified that the Leu-to-Trp substitution at
position 530 was critical for escape from 6D6 neutralization (Fig. 3). These amino acid
positions, which are located at the tip of the IFL structures of EBOV, SUDV, and RESTV
GPs, indicate that the loop structure including these residues is important to form the
recognition site of 6D6 (Fig. 2B). I confirmed that 6D6 did not bind to the chimeric Ebola
GP whose IFL region was replaced with that of MARV; however, 6D6 showed no binding
activity to the synthetic peptide corresponding to the amino acids of the IFL of EBOV GP
(not shown), suggesting that the 6D6 epitope may partly include other conformational
structures. Importantly, the amino acid sequence of the IFL region is highly conserved
among all currently known ebolaviruses (Fig. 2A), providing a novel target for universal
antibody therapy against EVD caused by human-pathogenic ebolaviruses (EBOV, SUDV,
TAFV, and BDBV).
Mechanism of the neutralizing activity of 6D6. Since the IFL is crucial for GP-mediated
membrane fusion, I assumed that 6D6 directly inhibited the fusion step during the entry
process of ebolaviruses into cells. To confirm this, I analyzed the inhibitory effects of 6D6
on viral attachment, internalization, and membrane fusion using DiI-labeled VLPs 11. The
number of 6D6-treated VLPs attached to the surface of Vero E6 cells was not significantly
different from that of untreated or CTR IgG-treated VLPs, indicating that 6D6 did not

18
Fig. 2. Identification of the putative epitope of MAb 6D6. (A) Structure of GP and
amino acid sequences of the internal fusion loop (IFL). The GP1 subunit contains the
receptor binding domain (RBD), a glycan cap and a mucin-like domain (MLD). The GP2
subunit contains the IFL, heptad repeats 1 and 2 (HR1 and HR2), the transmembrane
region (TM), and the cytoplasmic tail (CT). Amino acid substitutions found in the EBOV,
SUDV, and RESTV GP escape mutants selected under MAb 6D6 pressure are shown in

19
red. (B) The amino acid residues (green, blue, and yellow spheres represent Gly, Ala, and
Leu, respectively) critical for escape from the MAb 6D6 neutralization are mapped on the
trimeric structure of GPs. GP1 (blue) and GP2 (red) monomers are shown as ribbon
models.

20
Fig. 3. Identification of the key amino acid residue on the escape mutant RESTV GP
selected by MAb 6D6. Two RESTV GP mutants had the same Gly-to-Glu substitution at
position 529, which is the corresponding position of the EBOV GP mutants, whereas three
amino acid changes were found in the other 4 mutants of RESTV GP: Tyr at position 517,
Val at position 522, and Leu at position 530 were replaced with His, Ala, and Trp,
respectively (Fig. 2B). To clarify which amino acid change was critical for escaping from
the 6D6 neutralization, I generated RESTV GP mutants with single amino acid
substitutions for each position (Y517H, V522A, and L530W), and investigated the
neutralizing activity of MAb 6D6 against VSV pseudotyped with these single amino acid
mutants of RESTV GP. The mean and standard deviation of three independent
experiments are shown.

21
interfere with VLP attachment (Fig. 4A and D). Likewise, the number of VLPs that
colocalized with eGFP-Rab7, a late endosome marker, was similar, suggesting that 6D6
did not affect subsequent uptake into cells (Fig. 4B and E). Finally, I analyzed membrane
fusion efficiency by detecting dequenched DiI fluorescence 11,46. I observed remarkably
enlarged and enhanced DiI signals colocalizing with Rab7 in cells incubated with
untreated and CTR IgG-treated VLPs, indicating that membrane fusion occurred
efficiently in the endosomes (Fig. 4C left and middle panels). In contrast, the size and
intensity of DiI signals from 6D6-treated VLPs were significantly reduced, indicating that
6D6 prevented GP-mediated membrane fusion (Fig. 4C right panels and F).
Protective efficacy of MAb 6D6 in mouse models. Finally, I investigated the potential
of 6D6 to protect mice from ebolavirus infections (Fig. 5). Immunocompetent BALB/c
mice were infected with a lethal dose of mouse-adapted EBOV and treated 24 h later with
100 μg of 6D6. The treated animals survived without clinical symptoms, whereas
untreated mice succumbed to infection within 9 days (Fig. 5A). I further evaluated the
cross-protective potential of 6D6 against wild-type EBOV and SUDV infections (Fig.
5B). Since immunocompetent mice do not develop disease upon infection with these
wild-type ebolavirus isolates, interferon α/β receptor knockout (IFNAR-/-) C57BL/6 mice
were used for this purpose. I found that both EBOV and SUDV caused severe weight loss
in untreated mice, whereas only EBOV uniformly caused a lethal infection. Treatment
with 6D6 24 h after infection delayed the onset of the disease caused by these ebolaviruses
and significantly reduced the weight loss in this immunocompromised mouse strain. All
6D6-treated mice survived the EBOV infection.

22
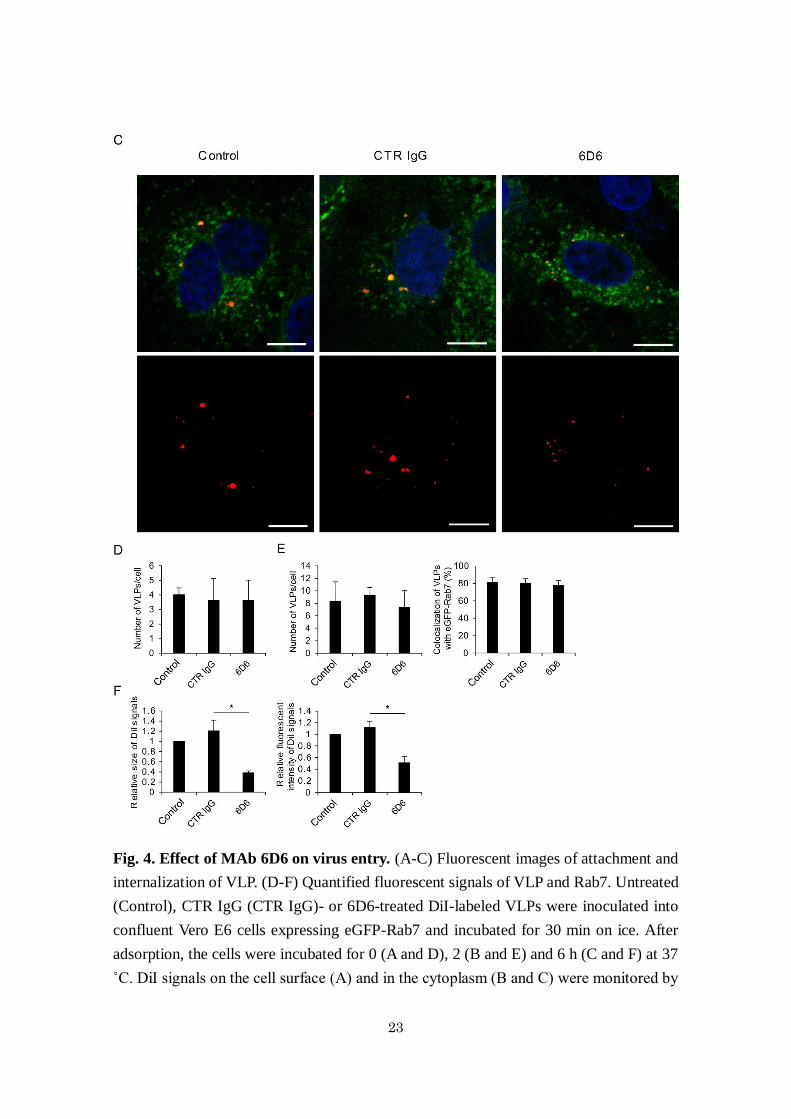
23
Fig. 4. Effect of MAb 6D6 on virus entry. (A-C) Fluorescent images of attachment and
internalization of VLP. (D-F) Quantified fluorescent signals of VLP and Rab7. Untreated
(Control), CTR IgG (CTR IgG)- or 6D6-treated DiI-labeled VLPs were inoculated into
confluent Vero E6 cells expressing eGFP-Rab7 and incubated for 30 min on ice. After
adsorption, the cells were incubated for 0 (A and D), 2 (B and E) and 6 h (C and F) at 37
˚C. DiI signals on the cell surface (A) and in the cytoplasm (B and C) were monitored by

24
confocal laser scanning microscopy. The number, size, and fluorescence intensity of DiI
signals were quantified. Scale bars represent 10 µm. The means and standard deviations
of three independent experiments are shown. Statistical analysis was performed using
Student’s t-test (*p<0.05).
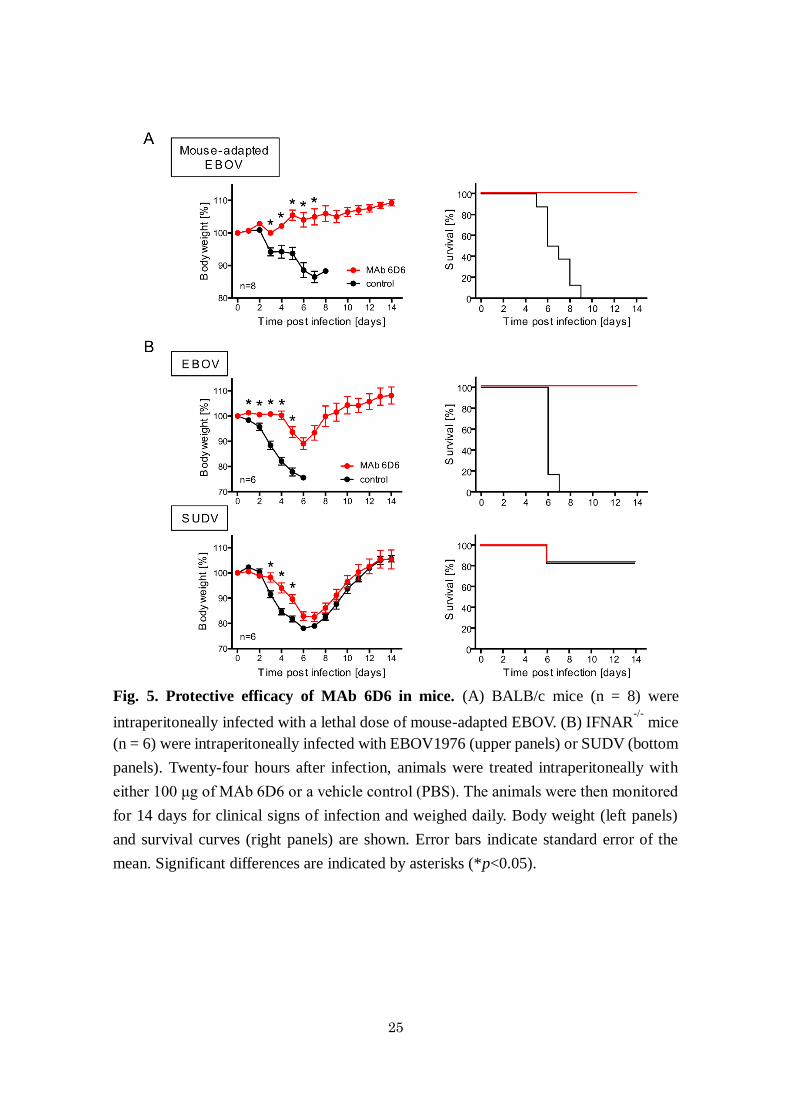
25
Fig. 5. Protective efficacy of MAb 6D6 in mice. (A) BALB/c mice (n = 8) were
intraperitoneally infected with a lethal dose of mouse-adapted EBOV. (B) IFNAR-/-
mice
(n = 6) were intraperitoneally infected with EBOV1976 (upper panels) or SUDV (bottom
panels). Twenty-four hours after infection, animals were treated intraperitoneally with
either 100 μg of MAb 6D6 or a vehicle control (PBS). The animals were then monitored
for 14 days for clinical signs of infection and weighed daily. Body weight (left panels)
and survival curves (right panels) are shown. Error bars indicate standard error of the
mean. Significant differences are indicated by asterisks (*p<0.05).

26
Discussion
Neutralizing antibody-based therapies have been tested in animal models and
clinical trials with particular attention given to highly lethal viral diseases 49. Passive
immunization with convalescent serum has also been tested for EBOV-infected patients
50,51. Recent studies have demonstrated the effectiveness of MAb treatments in nonhuman
primate models of EVD 26,28-30,52,53, and GP-specific MAb cocktails, ZMapp and ZMAb,
were used in clinical cases during the 2014 EVD outbreak caused by EBOV (belonging
to Zaire ebolavirus) 32-35. However, these MAb cocktails are not expected to be cross-
protective against the other antigenically distinct ebolaviruses. On the other hand, highly
cross-reactive MAbs against all known ebolaviruses have been generated previously, but
none of those has neutralizing activity 42,54. In this study, I generated the novel MAb 6D6,
which has cross-neutralizing activity against all known ebolaviruses.
I showed that 6D6 reduced infectivities of all known ebolaviruses in vitro with
higher neutralizing and binding activities than the EBOV GP-specific MAbs
ZGP133/3.16 and ZGP226/8.1. Indeed, the IC50 values of 6D6 were equal to or lower
than those of previously reported neutralizing MAbs 55-57, suggesting its protective
capacity as a therapuetic MAb. By analyzing the amino acid substitution observed in the
antigenic variants escaping from 6D6, I determined the putative epitope of 6D6 in the IFL
on the GP molecule, which may overlap that of a partially cross-reactive GP-specific
MAb reported recently 58. Accordingly, 6D6 directly inhibited the membrane fusion
induced by EBOV VLPs in endosomes/lysosomes. The IFL structure is highly conserved
in all species of ebolaviruses, indicating that this region can be targeted for both vaccine
and therapeutic development against ebolaviruses. Since the putative epitope of 6D6 is
different from those previously reported for other GP-specific MAbs used in passive

27
immunization studies 20,56,59-62. 6D6 may provide a promising option as a component of
antibody cocktails in combination with other previously tested MAbs.
I further demonstrated that passive immunization with 6D6 protected both
BALB/c and IFNAR-/- C57BL/6 mice from lethal infection by EBOV (i.e., mouse-
adapted and wild-type EBOV, respectively). However, the cross-protective potential of
6D6 could only be evaluated for disease severity using IFNAR-/- C57BL/6 mice since
mouse-adapted SUDV causing lethal infection in immunocompetent mice is not currently
available and SUDV did not uniformly cause lethal infection even in this
immunocompromised mouse strain. I found that passive immunization with 6D6
significantly reduced the weight loss in SUDV-infected mice, although the extent was not
as prominent as in EBOV-infected mice. The less significant protective effects seen in
SUDV-infected mice might have been due to the higher IC50 value of 6D6 against SUDV
than against EBOV. Thus, these results supported the in vitro characteristics of 6D6 and
demonstrated the effectiveness of the 6D6 treatment in vivo against multiple species of
ebolaviruses. A previous study demonstrated that immunocompromised mice treated
several times with 500 μg of MAb SUDV-specific MAbs were protected from SUDV
infection 63, whereas mice were treated once with 100 μg of 6D6 in this study. Thus, I
assume that the protective effect against SUDV could be improved by increased doses of
6D6.
The broadly cross-neutralizing antibody 6D6 recognizing the common epitope
shared among all currently known ebolaviruses, converted into a human-mouse chimeric
MAb 28, is a promising therapeutic candidate. On the other hand, the generation of 6D6
escape mutants in vitro speaks against monotherapy with this MAb and may favor the
development of antibody cocktails including 6D6 for future pan-ebolavirus therapy. For

28
other viruses, it has indeed been reported that combination of MAbs helps to avoid the
appearance of escape variants if these MAbs recognize distinct epitopes 64-66. While the
detailed mechanisms underlying antibody-mediated protection from ebolavirus infection
need to be further elucidated, the discovery of this highly cross-reactive neutralizing
antibody and its putative epitope reported here provides a promising option for the
development of a universal EVD therapy and will accelerate the design and
implementation of improved therapeutics and vaccines that can selectively elicit cross-
neutralizing antibodies against multiple species of ebolaviruses.

29
Summary
During the latest outbreak of EVD in West Africa, MAb therapy (e.g., ZMapp)
was utilized to treat patients. However, due to the antigenic differences among the five
ebolavirus species, the current therapeutic MAbs are only effective against viruses of the
species Zaire ebolavirus. Although this particular species has indeed caused the majority
of human infections in Central and, recently, West Africa, other ebolavirus species (e.g.,
Sudan ebolavirus and Bundibugyo ebolavirus) have also repeatedly caused outbreaks in
Central Africa and thus should not be neglected in the development of countermeasures
against other ebolavirus species. Here I report the generation of an ebolavirus GP-specific
MAb that effectively inhibits cellular entry of representative isolates of all known
ebolavirus species in vitro and show its protective efficacy in mouse models of ebolavirus
infections. This novel neutralizing MAb targets a highly conserved IFL in the GP
molecule and prevents membrane fusion of the viral envelope with cellular membranes.
The discovery of this highly cross-neutralizing antibody provides a promising option for
broad-acting ebolavirus antibody therapy and will accelerate the design of improved
vaccines that can selectively elicit cross-neutralizing antibodies against multiple species
of ebolaviruses.

30
Chapter II:
Fcγ-receptor IIa-mediated Src signaling pathway is essential for the
antibody-dependent enhancement of Ebola virus infection
Introduction
It has been demonstrated that EBOV exploits some GP-specific antibodies for
its entry into cells, leading to increased infectivity in vitro 22,39. This phenomenon has
been described for a number of viruses and is known as ADE 36-38. For some of these
viruses, ADE has become a great concern to disease control by vaccination. Particularly,
convalescent human sera have been shown to contain ADE antibodies 22,39, raising
concerns about potential detrimental effects of passive immunization with convalescent
human sera, which is currently under consideration for treatment of EVD. Two distinct
pathways of EBOV ADE, one mediated by Fc receptors and the other by complement
component C1q and its ligands, are known 22,23. In particular, the FcγR is commonly
involved in ADE of virus infections 67,68. However, the molecular mechanisms underlying
ADE-mediated virus entry through FcγR are not fully understood.
Three classes of FcγR, FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16), are
expressed in various human immune cells such as dendritic cells, monocytes, and B
lymphocytes 69. Among these FcγRs, FcγRII is a key molecule for EBOV ADE of
infection in human leukemia K562 cells 23. Human FcγRII exists in two isoforms, FcγRIIa
and FcγRIIb, which differ in their signal peptides and cytoplasmic tails. FcγRIIa is the
active form of FcγRII and contains an immunoreceptor tyrosine-based activation motif
(ITAM) in its cytoplasmic tail 69. The cytoplasmic tail of FcγRIIa is known to contribute
to the activitation of two structurally and functionally distinct protein-tyrosine kinase

31
(PTK) classes, the sarcoma (Src) family PTKs 70,71 and spleen tyrosine kinase (Syk) 72. In
addition, Syk is reported to participate in activation of enzymes such as rat sarcoma (Ras),
phosphatidylinositol 3-kinase (PI3K), and Bruton’s tyrosine kinase (Btk) 69,73. These
signaling pathways are known to be important for the induction of phagocytic and
endocytic processes to internalize immune complexes 69,73,74.
In the chapter II, I focused on the role of FcγRIIa and investigated the
contribution of FcγRIIa-mediated signaling to the ADE of EBOV infection. I show that
Src family PTKs are essential for EBOV ADE-mediated entry. My data indicate that
binding of antibody-virus complexes to the cell surface FcγRIIa triggers phosphorylation
of Src family PTKs and activates subsequent signaling pathways, leading to enhanced
viral uptake through phagocytosis and/or macropinocytosis.

32
Materials and Methods
Viruses and cells. EBOV expressing GFP 75 was propagated in Vero E6 cells and stored
at -80 ˚C. Replication-incompetent VSV pseudotyped with EBOV GP containing GFP
instead of the VSV G gene (VSV-EBOV GP) was generated as described previously 20,44.
Virus titers were determined as IUs by counting GFP-positive cells. All infectious work
with EBOV was performed in the biosafety level 4 laboratory at the Integrated Research
Facility of the Rocky Mountain Laboratories, Division of Intramural Research, National
Institute of Allergy and Infectious Diseases, National Institutes of Health, Hamilton,
Montana, USA. Vero E6 cells and HEK 293T cells were grown in DMEM (Sigma), and
human chronic myelogenous leukemia K562 cell line and K562 cell lines stably
expressing dendritic cell-specific ICAM-3-grabbing non-integrin (DC-SIGN) or human
macrophage galactose-type C-type lectin (hMGL) 76,77 and human leukemic Jurkat T cells
were grown in Roswell Park Memorial Institute (RPMI) 1640 medium (Sigma). These
media were supplemented with 10% FCS (Cell Culture Bioscience), 100 U/ml penicillin,
and 0.1 mg/ml streptomycin (Gibco).
Generation of FcγRIIa- or FcγRIIaΔCT-expressing Jurkat T cells. The FcγRIIa gene
was PCR-amplified from a full-length cDNA library prepared from K562 cells using the
primers, EcoRI-FcγRIIa (5’-GGGAATTCGGATGACTATGGAGACCCAA-3’) and
FcγRIIa-XhoI (5’-ATTTCTCGAGTTTGTCATCCACTCAGCAAG-3’). Mutant FcγRIIa
lacking its cytoplasmic tail (amino acid positions 241-317) (FcγRIIaΔCT) was generated
using a PrimeSTAR Mutagenesis Basal Kit (Takara). After sequence confirmation, these
PCR products were cloned into a murine leukemia virus-based retroviral vector, pMXs-
Puro Retroviral Vector (Cell Biolabs). To generate the retrovirus, 293T-derived Platinum-

33
GP (Plat-GP) cells (Cell Biolabs) were cotransfected with pMXs-puro encoding FcγRIIa
or FcγRIIaΔCT and the expression plasmid pCAGGS encoding the VSV G protein using
Lipofectamine 2000 (Invitrogen). Forty-eight h later, the culture supernatants containing
retroviruses were collected, clarified through 0.45 μm filters, and then used to infect
Jurkat T cells. Jurkat T cell lines stably expressing FcγRIIa or FcγRIIaΔCT were selected
with RPMI medium containing 10% FCS, 100 U/ml penicillin, 0.1 mg/ml streptomycin,
and 10 µg/ml puromycin (Sigma-Aldrich). For some experiments, each Jurkat T cell line
was cloned by limiting dilution to enrich the population of FcγRIIa-expressing cells. To
check the expression levels of FcγRIIa and FcγRIIaΔCT, these cells were incubated with
a mouse anti-CD32 monoclonal antibody (GeneTex) for 1 h at room temperature. After
washing 3 times with PBS, binding of the primary antibody was detected with Alexa647-
conjugated F(ab')2-goat anti-mouse IgG (H+L) (Jackson ImmunoResearch). After further
washing 3 times with PBS, the fluorescent intensity of the cells was analyzed using a
FACS Canto flow cytometer (BD Biosciences) and FlowJo software (Tree Star).
ADE assays. EBOV was appropriately diluted to yield 50-100 IUs in K562 cells and then
incubated for 30 min-1 h at 37 ˚C with or without 10 μg/ml MAbs. The anti-EBOV GP
MAb ZGP12/1.1 (IgG2a), which is known to enhance EBOV infection in vitro, was used
as the ADE MAb 39. S139/1 (IgG2a), a MAb specific to influenza A virus hemagglutinin,
was used as the control IgG (CTR IgG) 78. K562 and Jurkat T cells were inoculated with
EBOV alone or EBOV/MAb mixtures and incubated for 72 h. Virus infectivity was
measured by counting the number of GFP-positive cells in FACS and analyzed using
FlowJo software. VSV-EBOV GP was appropriately diluted to yield 50-100 IUs in K562
cells and incubated for 1 h at room temperature with or without 1 μg/ml MAbs, and then

34
inoculated into K562 and Jurkat T cells. Twenty-four h later, GFP-positive cells were
counted using an IN Cell Analyzer 2000 (GE Healthcare). To reduce the background (i.e.,
residual) infectivity of the parent VSV, VSV-EBOV GP was treated with a neutralizing
MAb to VSV G protein (VSV-G[N]1-9) before use.
Purification and DiI-labeling of VLPs. For purification of VLPs, HEK293T cells were
transfected with equal amounts of the expression plasmids encoding EBOV or SUDV GP,
VP40, and NP using TransIT LT-1 (Mirus) according to the manufacturer's instructions.
Forty-eight h after transfection, the culture supernatant was harvested and centrifuged at
3,500 rpm for 15 min at 4 ˚C to remove cell debris. VLPs were precipitated through a
25% sucrose cushion by centrifugation at 11,000 rpm for 1 h at 4 ˚C with an SW32Ti
rotor (Beckman). Pelleted VLPs were suspended in PBS, and fractionated through a 20-
50% sucrose gradient in PBS at 28,000 rpm with an SW41 rotor (Beckman) for 2 h at 4
˚C. One ml of 1 μg/ml fractionated VLPs was incubated with 0.6 µl of 100 µM DiI
(Molecular Probes) in the dark for 1 h at room temperature with gentle agitation 11.
Imaging of attachment and internalization of DiI-labeled VLPs. The eGFP-Rab7
fusion protein gene was cloned into a Moloney murine leukemia virus-based retrovirus
plasmid 11,79, and recombinant retroviruses for the expression of eGFP-Rab7 were
produced and used to infect K562 cells as described above. K562 and Jurkat T cell lines
were cultured in 35 mm glass-bottom dishes (MatTek Corporation) precoated with borate
buffer containing 0.1 mg/ml poly-L-lysine (Sigma). The cells were washed with phenol
red-free RPMI (Gibco) and inoculated with 100 μl of 1 μg/ml DiI-labeled VLPs treated
with 20 μg/ml ZGP12/1.1 or CTR IgG for 1 h at room temperature, followed by

35
incubation for 30 min on ice. They were then washed twice with the same medium to
remove unbound DiI-labeled VLPs and incubated with phenol red-free RPMI containing
2% FCS and 4% BSA for 0 and 2 h at 37 ˚C to analyze DiI-labeled VLP attachment and
internalization, respectively. To count the number of DiI-labeled VLPs, the cells were
fixed with 4% paraformaldehyde for 15 min at room temperature. Then the nuclei were
stained with 1 μg/ml DAPI (Molecular Probes) for 10 min at room temperature.
Microscopic images were acquired with a 63 × oil objective lens on a Zeiss LSM780
inverted microscope and ZEN 2010 software (Carl Zeiss). For measurement of the
number of DiI-labeled VLPs, images of 4-20 optical sections were acquired in 1 micron
steps. The number of DiI-labeled VLPs was determined in approximately 100 individual
cells using MetaMorph software (Molecular Devices) and the average number per cell
was calculated for each condition. For colocalization analysis, the percentage of DiI-
labeled VLPs that colocalized with eGFP-Rab7 (Both DiI- and eGFP-positive pixels/DiI-
positive pixels × 100) was measured in approximately 100 individual cells using the
Coloc module in ZEN 2010 software (Carl Zeiss).
Dextran uptake assays. One μg/ml DiI-labeled VLPs were treated with 20 μg/ml CTR
IgG or ZGP12/1.1 for 1 h at room temperature. K562 and Jurkat T cell lines were cultured
in poly-L-lysine-coated glass-bottom culture dishes and incubated with 100 μl untreated,
CTR IgG-, or ZGP12/1.1-treated DiI-labeled VLPs for 30 min on ice. The cells were
washed twice with phenol red-free RPMI and then incubated with phenol red-free RPMI
containing 2% FCS, 4% BSA, and 0.5 mg/ml Dextran, Alexa Fluor 647, 10,000 MW
(Alexa647-labeled Dx10) (Molecular Probes) for 1-2 h at 37 ̊ C. After washing twice with
phenol red-free RPMI to remove surface-unbound DiI-labeled VLPs and Alexa647-

36
labeled Dx10, and the cells were fixed with 4% paraformaldehyde for 15 min at room
temperature. Then, the nuclei were stained with 1 μg/ml DAPI for 10 min at room
temperature. Internalized DiI-labeled VLPs and Alexa647-labeled Dx10 were analyzed
by confocal laser scanning microscopy as described above. The percentage of DiI-labeled
VLPs that colocalized with Alexa647-labeled Dx10 (Both DiI- and Alexa647-positive
pixels/DiI-positive pixels × 100) was measured in approximately 100 individual cells
using the Coloc module in ZEN 2010 software. The number and size of Dx10-positive
vesicles were analyzed with MetaMorph software.
Inhibitor treatments. For infection assays, the Syk inhibitor R788 (Santa Cruz), Src
family PTK inhibitor PP2 (Tocris), BTK inhibitor LFM-A13 (Focus Biomolecules), PI3K
inhibitor LY294002 (Wako), and Ras inhibitor Manumycin A (Santa Cruz) were used for
treatments of K562 cells. R788, LFM-A13, and LY294002 were used at 0.15-40 μM. PP2
and Manumycin A were used at 0.15-10 μM. For imaging analysis, K562 cell lines were
cultured in 35 mm glass-bottom dishes precoated with poly-L-lysine, and then treated
with 20 μM PP2 for 1 h at 37 ˚C. PP2-treated cells were washed with phenol red-free
RPMI and inoculated with untreated, CTR IgG-, or ZGP12/1.1-treated DiI-labeled VLPs
for 30 min on ice in the presence of 20 μM PP2 in the same medium. The cells were then
washed twice with the same medium and incubated with phenol red-free RPMI containing
2% FCS, 4% BSA, and 20 μM PP2 for 0 and 2 h at 37 ˚C. Then they were fixed and
analyzed by confocal laser scanning microscopy as described above. Dimethyl sulfoxide
(DMSO, Sigma-Aldrich) or ethanol (Kanto Chemical) was used as a solvent control.
Generation of Src or Syk knockdown K562 cells. Plat-GP cells were cotransfected with

37
pRS (retroviral plasmids) encoding human Src or Syk shRNA (ORIGENE) and the
expression plasmid pCAGGS encoding the VSV G protein using Lipofectamine 2000
(Invitrogen). ShSrc target sequences were: shSrc1:5’-
GGAGGCTTCAACTCCTCGGACACCGTCAC-3’, shSrc2: 5’-
AAGAAAGGCGAGCGGCTCCAGATTGTCAA-3’, shSrc3: 5’-
GCAGTTGTATGCTGTGGTTTCAGAGGAGC-3’, shSrc4: 5’-
CTGGAGGCAATCAAGCAGACATAGAAGAG-3’. ShSyk target sequences were:
shSyk1:5’- GAATATGTGAAGCAGACATGGAACCTGCA-3’, shSyk2: 5’-
GGAGGAGGCAGAAGATTACCTGGTCCAGG-3’, shSyk3: 5’-
TGTCATTCAATCCGTATGAGCCAGAACTT-3’, shSyk4: 5’-
CTCTGGCAGCTAGTCGAGCATTATTCTTA-3’. After incubation for 48 h, culture
supernatants containing the retroviruses expressing human Src or Syk shRNAs were
collected, clarified through 0.45-μm filters, and then used to infect K562 cells.
Transduced K562 cell lines were selected with RPMI medium containing 10% FCS, 100
U/ml penicillin, 0.1 mg/ml streptomycin, and 5 µg/ml puromycin (Sigma-Aldrich). To
check the knockdown efficiency for Src and Syk, cells were collected and washed once
with PBS and treated with lysis buffer (0.1% Nonidet P-40, 150 mM NaCl, 1 mM EDTA,
10 mM Tris HCl, pH 7.8) in the presence of a protease inhibitor cocktail, Complete mini
(Roche). Then the lysates were mixed with sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) sample buffer (Bio-Rad) with 5% 2-mercaptoethanol
(Wako) and boiled for 5 min. The samples were electrophoresed by SDS-PAGE on 5 to
20% gradient polyacrylamide gel, SuperSep Ace (Wako), and separated proteins were
blotted on a polyvinylidene difluoride membrane (Millipore). The membrane was
blocked for at least 1 h at room temperature with Tris-buffered saline containing 0.1%

38
Tween 20 (TBST) and 1% BSA. Then the membrane was incubated with a rabbit anti-
Src antibody (36D10: Cell Signaling) or mouse anti-Syk antibody (4D10.1: Abcam) in
TBST containing 1% BSA, followed by incubation with horseradish peroxidase-
conjugated donkey anti-rabbit IgG (H+L) or horseradish peroxidase-conjugated goat anti-
mouse IgG (H+L) (Jackson ImmunoResearch), respectively, and visualization by
Immobilon Western (Millipore). Band intensities were analyzed with a VersaDoc Imaging
System (Bio-Rad) and quantified with Image Lab version 3.0 software (Bio-Rad).
Phosphorylation assay. One μg/ml purified VLPs were treated with 20 μg/ml ZGP12/1.1
or CTR IgG for 1 h at room temperature. K562 cells were incubated with DMSO or 20
μM PP2 for 1 h at 37 ˚C. Untreated or PP2-treated K562 cells were inoculated with
untreated, CTR IgG-, or ZGP12/1.1-treated VLPs and incubated for 0, 10, 30, or 60 min
at 37 ˚C. At each time point, cells were collected and washed once in PBS and treated
with lysis buffer (0.1% Nonidet P-40, 150 mM NaCl, 1 mM EDTA, 10 mM Tris HCl, pH
7.8) in the presence of a protease inhibitor cocktail, Complete mini (Roche), and a
phosphatase inhibitor cocktail, PhosSTOP (Roche). Then the lysates were mixed with
SDS-PAGE sample buffer (Bio-Rad) with 5% 2-mercaptoethanol (Wako) and boiled for
5 min. The samples were electrophoresed by SDS-PAGE on 5 to 20% gradient
polyacrylamide gel, SuperSep Ace (Wako), and separated proteins were blotted on a
polyvinylidene difluoride membrane (Millipore). The membrane was blocked for at least
1 h at room temperature with TBST and 1% BSA. Then the membrane was incubated
with a rabbit anti-phospho-Src family (Tyr416) antibody (Cell Signaling) in TBST
containing 1% BSA, followed by visualization using horseradish peroxidase-conjugated
donkey anti-rabbit IgG (H+L) (Jackson ImmunoResearch) and Immobilon Western

39
(Millipore). Band intensities were analyzed with a VersaDoc Imaging System (Bio-Rad)
and quantified with Image Lab version 3.0 software (Bio-Rad).
Statistical analysis. All data were analyzed using Excel software. In all experiments,
Student’s t-test was used to evaluate statistical differences. P values of less than 0.05 were
considered to be significant.

40
Results
Cytoplasmic tail of FcγRIIa is required for ADE of EBOV infection. To investigate
the role of FcγRIIa and, in particular, the importance of its cytoplasmic tail in EBOV
ADE, I compared the functions of wild-type FcγRIIa and FcγRIIaΔCT which is a deletion
mutant of FcγRIIa lacking its cytoplasmic tail. Both molecules were expressed on Jurkat
T cells, which are known to be poorly permissive for EBOV infection 80 and lack this Fc
receptor 81. Jurkat T cells were transduced with full-length FcγRIIa or FcγRIIaΔCT genes
using a retrovirus vector (Fig. 6A and B) and subsequently infected with VSV-EBOV GP
and infectious EBOV in the presence or absence of the GP-specific MAb ZGP12/1.1,
which is known to induce EBOV ADE 39 (Fig. 6C and D). I found that viral infectivity
was almost undetectable in naive and control vector-transduced Jurkat T cells but the
expression of wild-type FcγRIIa significantly enhanced the infectivity of VSV-EBOV GP
and EBOV in the presence of ZGP12/1.1, though not CTR IgG. Interestingly, the infection
rate of FcγRIIaΔCT-expressing cells was significantly lower than that of cells expressing
wild-type FcγRIIa. These results indicated that the FcγRIIa-MAb complex functioned as
a receptor-like molecule on this poorly permissive cell line and efficiently promoted
infection through the ADE of EBOV entry. More importantly, the results suggested that
signaling pathways via the FcγRIIa cytoplasmic tail were likely involved in the ADE of
EBOV entry into cells.
Cytoplasmic tail of FcγRIIa is important for enhanced viral uptake. FcγRIIa is
known to modulate phagocytosis/macropinocytosis through signaling pathways via its
cytoplasmic tail 82,83. Therefore, to analyze viral binding and intracellular uptake in more
detail, I produced DiI-labeled VLPs consisting of the major EBOV structural proteins,

41
Fig. 6. Importance of FcγRIIa and its cytoplasmic tail in EBOV ADE. (A) Schematic
representation of FcγRIIa and FcγRIIaΔCT. TM, transmembrane region; CT, cytoplasmic
tail. (B) Cell surface expression of the FcγRIIa ectodomain on Jurkat T cell lines. Jurkat
T cells stably expressing FcγRIIa and FcγRIIaΔCT were stained with an anti-CD32
antibody and analyzed by flow cytometry. Black lines represent vector-transduced,
FcγRIIa-, and FcγRIIaΔCT-expressing cells. Gray shading represents naive Jurkat T cells.
(C and D) Infectivity of VSV-EBOV GP and EBOV in Jurkat T cell lines. VSV-EBOV

42
GP (C) and EBOV (D) were incubated with medium alone (Control), CTR IgG, or
ZGP12/1.1, followed by inoculation into each Jurkat T cell line. At 24 h (C) and 72 h (D)
after inoculation, GFP-positive cells were counted. The relative percentage of infectivity
in each cell line was calculated by setting the IU value of untreated (Control), CTR IgG-,
and ZGP12/1.1-treated viruses in naive Jurkat T cells to 100%, respectively. The mean
and standard deviation of three independent experiments are shown. Statistical analysis
was performed using Student’s t-test (*p<0.05).

43
GP, VP40, and NP, and monitored the localization of VLPs in each transduced Jurkat T
cell line (Fig. 7). The number of VLPs attached to the surface of naive and empty vector-
transduced Jurkat T cells was not significantly different irrespective of the presence of
CTR IgG and ZGP12/1.1 (Fig. 7A and C). In contrast, the attachment of VLP was
significantly enhanced to similar extents in Jurkat T cells expressing FcγRIIa and
FcγRIIaΔCT in the presence of ZGP12/1.1 but not CTR IgG (Fig. 7A and C), suggesting
that the FcγRIIa ectodomain expressed on Jurkat T cells had the ability to increase the
VLP attachment mediated by ZGP12/1.1. Next, I assessed the number of VLPs
incorporated into intracellular vesicles along with Alexa Fluor 647-labeled dextran Mw
10,000 (Alexa647-labeled Dx10), a specific probe for visualizing phagocytotic and
macropinocytotic vesicles 11,84. After incubation for 2 h, ZGP12/1.1-treated VLPs
efficiently colocalized with Dx10 in Jurkat T cells expressing wild-type FcγRIIa, but not
in cells expressing FcγRIIaΔCT (Fig. 7B and D). Viral uptake was not observed
drastically in the absence of FcγRIIa and ZGP12/1.1. Furthermore, the number and size
of Dx10-positive vesicles incorporated into Jurkat T cells expressing FcγRIIaΔCT were
significantly smaller than those in cells expressing FcγRIIa, indicating the importance of
the FcγRIIa cytoplasmic tail in activating the phagocytosis/macropinocytosis (Fig. 7B
and E). These data suggested that the ADE infection of FcγRIIa-expressing Jurkat T cells
was associated with enhanced viral uptake into cellular vesicles, most likely due to the
activation of FcγRIIa-mediated signaling via its cytoplasmic tail.
Intracellular signaling via Src family PTKs contributes to ADE of EBOV infection.
To identify the intracellular signaling pathway involved in the ADE of EBOV entry, I
analyzed the effects of different inhibitors of signaling pathways in K562 cells, which

44
Fig. 7. Importance of FcγRIIa and its cytoplasmic tail in ADE-mediated VLP uptake
into cells. (A and B) Fluorescent images of attachment and internalization of VLP. (C-E)

45
Quantified fluorescent signals of VLP and Dx10. Untreated (Control), CTR IgG-, and
ZGP12/1.1-treated DiI-labeled VLPs were inoculated into each Jurkat T cell line and
incubated for 30 min on ice. After adsorption, the cells were incubated for 0 h (A and C)
or incubated with Alexa647-labeled Dx10 for 2 h at 37 ˚C (B, D, and E). VLPs (red) on
the cell surface (A and C) and VLPs (red) and Dx10 (green) in the cytoplasm (B, D, and
E) were monitored by confocal laser scanning microscopy. Scale bars represent 10 µm.
Nuclei of cells are visualized with DAPI (blue). The number of VLPs attached to the cell
surface (C), the colocalization of VLP (DiI) and Dx10 (Alexa647) signals (D), and the
number and size of Dx10 (E) were quantified. The number and size were calculated by
setting the value of FcγRIIa-expressing Jurkat T cells in the presence of ZGP12/1.1-
treated VLPs to 100%. The mean and standard deviation of three independent
experiments are shown. Statistical analysis was performed using Student’s t-test
(*p<0.05).

46
naturally express FcγRIIa. Consistent with previous studies 23,77, K562 cells were
permissive to VSV-EBOV GP and EBOV infections, and viral infection rates were
significantly enhanced in the presence of ZGP12/1.1 (Fig. 8). I then tested inhibitors of
Syk and Src family PTK (R788 and PP2, respectively) as these PTKs are known to be
principally involved in signaling pathways downstream of FcγRIIa, and in particular to
play important roles in inducing FcγR-mediated phagocytosis/micropinocytosis 74,85. I
found that the ADE of VSV-EBOV GP infection was significantly reduced in K562 cells
treated with these inhibitors in a dose-dependent manner. In contrast, only a limited
reduction was seen in non-ADE infection at the highest concentrations of the inhibitors
(Fig. 9). The ADE-specific inhibitory effect was more prominent in cells treated with PP2
than in those treated with R788.
Since Syk is reported to participate in the activation of signaling through PI3K,
Btk, and Ras, I further examined which pathways downstream of Syk contributed to the
ADE of VSV-EBOV GP infection using specific inhibitors of PI3K (LY294002), Btk
(LFM-A13), and Ras (Manumycin A) (Fig. 9). However, both ADE and non-ADE
infections by VSV-EBOV GP were dose-dependently reduced by LY294002 and
Manumycin A, respectively. LFM-A13 showed little effect on the infectivity of VSV-
EBOV GP. Subsequently, I confirmed the effects of these inhibitors using infectious
EBOV. Consistent with the data for VSV-EBOV GP, PP2 selectively reduced the ADE,
but not the non-ADE infection. Interestingly, R788 showed no effects on the ADE of
EBOV infection and rather enhanced the non-ADE infection. LFM-A13 slightly reduced
both the ADE and the non-ADE infections, whereas LY294002 and Manumycin A did
not inhibit the ADE infection, though Manumycin A slightly inhibited the non-ADE
infection.

47
Fig. 8. EBOV ADE in K562 cells. K562 cells were infected with VSV-EBOV GP or
EBOV following incubation with CTR IgG or ZGP12/1.1 for 30 min-1 h at 37 ˚C. After
incubation for 24 (VSV-EBOV GP) or 72 (EBOV) h, GFP-positive cells were counted
and IUs of viruses were determined. The relative percentage of infectivity was calculated
by setting the IU value of the viruses in CTR IgG-treated cells to 100%. The mean and
standard deviation of three independent experiments are shown.

48
Fig. 9. Identification of FcγRIIa-mediated signaling pathway for EBOV ADE. K562
cells were treated with the indicated concentrations of R788, PP2, LFM-A13, LY294002,
or Manumycin A for 1 h at 37 ˚C and inoculated with VSV-EBOV GP or EBOV
preincubated with CTR IgG, or ZGP12/1.1. The relative percentage of infectivity was
calculated by setting the IU value of the viruses in DMSO or ethanol-treated cells to 100%.
The mean and standard deviation of three independent experiments are shown. Statistical
analysis was performed using Student’s t-test (*p<0.05).

49
To further investigate the role of Src- and Syk-mediated signaling in EBOV ADE,
I generated Src and Syk knockdown K562 cells. K562 cells were transduced with
retroviral vectors expressing small hairpin RNAs (shRNAs) for silencing Src or Syk
genes (Fig. 10A and B) and infected with VSV-EBOV GP in the presence or absence of
ZGP12/1.1 (Fig. 10C). I found that transduced cells stably expressing Src shRNAs
(shSrc3 and shSrc4) showed approximately 50% reduction in protein levels (Fig. 10A)
and ZGP12/1.1-mediated ADE was significantly decreased in these cell lines (Fig. 10C).
In contrast, no significant difference was seen between ADE (ZGP12/1.1) and non-ADE
(CTR IgG) infections in Syk knockdown cells although 2 of the Syk shRNAs (shSyk3
and shSyk4) significantly reduced the expression of the Syk protein (Fig. 10B and C).
These results demonstrated that FcγRIIa-mediated signaling through the
activation of Src family PTKs contributed to the ADE of EBOV infection, but Syk-related
signaling including PI3K, Btk, and Ras did not seem to be specifically involved. I further
tested the effect of PP2 in K562/DC-SIGN or K562/hMGL, both of which have been
shown to act as attachment receptors for EBOV 77,86, and found that PP2 had limited
effects on the infectivity of VSV-EBOV GP in these cell lines (Fig. 11).
VLP-MAb complexes activate phosphorylation of Src in K562 cells. To directly detect
the activation of Src family PTKs, I quantified the phosphorylation levels of Src in K562
cells (Fig. 12). I found no significant difference in the cells inoculated with intact VLPs
alone at each time point. Likewise, inoculation of CTR IgG-treated VLPs did not enhance
Src phosphorylation levels. However, a significant increase of the Src phosphorylation
was detected at 30 and 60 min after K562 cells were exposed to ZGP12/1.1-treated VLPs
(Fig. 12 left). Furthermore, the enhanced phosphorylation was completely blocked by the

50
Fig. 10. Effects of Src and Syk knockdown on EBOV ADE. (A and B) Validation of
knockdown efficiency of Src and Syk in K562 cell lines. (C) Relative infectivity of VSV-
EBOV GP in K562 cell lines. Src (A) and Syk (B) proteins in each cell line were detected
by Western blotting. The band intensity of Src and Syk was standardized with that of the
β-actin band for each cell line. Relative intensities of the Src and Syk bands were then
calculated by setting the intensity of naive K562 cells to 100%. These cell lines were
infected with VSV-EBOV GP preincubated with CTR IgG or ZGP12/1.1 (C). The relative
percentage of infectivity was calculated by setting the IU value of the viruses in naïve
K562 cells to 100%. The mean and standard deviation of three independent experiments
are shown. Statistical analysis was performed using Student’s t-test (*p<0.05).

51
Fig. 11. Effects of PP2 on the VSV-EBOV GP infectivity in K562 cell lines stably
expressing C-type lectin. K562/DC-SIGN, K562/hMGL, and mock control K562 cells
were treated with DMSO or PP2 for 1 h at 37 ˚C and infected with VSV-EBOV GP in the
presence of the inhibitor. After incubation for 24 h, GFP-positive cells were counted. The
relative percentage of infectivity was calculated by setting the IU value of the virus in
DMSO-treated cells to 100%. The mean and standard deviation of three independent
experiments are shown.

52
Fig. 12. Increased phosphorylation of Src in K562 cells exposed to the VLP-
ZGP12/1.1 complex. Purified VLPs were preincubated with medium alone (Control),
CTR IgG, or ZGP12/1.1 and inoculated into K562 cells pretreated with DMSO or PP2
for 1 h at 37 ˚C. At 0, 10, 30, and 60 min post-inoculation, phosphorylation of Src at
Tyr416 (p-Src) was detected by Western blotting. The band intensity of p-Src was
standardized with that of the β-actin band at each time point. Relative intensities of the p-
Src bands were then calculated by setting the intensity of control cells (0 h) to 100%. The
mean and standard deviation of three independent experiments are shown. Statistical
analysis was performed using Student’s t-test (*p<0.05).

53
Src family PTK inhibitor, PP2 (Fig. 12 right). These findings suggested that Src were
activated by the interaction of the VLP-ZGP12/1.1 complex with FcγRIIa.
The ADE of EBOV entry depends on phagocytosis and/or macropinocytosis
mediated by Src family PTKs. To further characterize the role of the Src family PTK-
dependent signaling in the ADE of EBOV infection, I analyzed the effect of PP2 on the
attachment and uptake of DiI-labeled VLPs using K562 cells. I first compared the number
of VLPs attached to the cell surface among untreated, CTR IgG-, and ZGP12/1.1-treated
VLPs. Since the overexpression of FcγRIIa in Jurkat T cells increased the attachment of
VLPs to the cell surface in the presence of ZGP12/1.1 (Fig. 7), I hypothesized that
ZGP12/1.1 would enhance the VLP attachment to K562 cells. However, the number of
VLPs attached to the cell surface was not significantly different in the presence or absence
of ZGP12/1.1 (Fig. 13A left and D), and was not affected by the PP2 treatment (Fig. 13A
right and D), indicating that EBOV ADE in K562 cells did not result from increased viral
attachment to the cell surface. For the visualization of the VLP uptake into endosomes,
K562 cells expressing eGFP-Rab7, a late endosome marker, were used to analyze
colocalization of eGFP-Rab7 and internalized VLPs. I found that ZGP12/1.1-treated
VLPs were efficiently colocalized with eGFP-Rab7 in K562 cells, whereas only 10-20%
colocalization was seen in the cells inoculated with untreated or CTR IgG-treated VLPs
(Fig. 13B and E). I further found that the enhanced colocalization of eGFP-Rab7 and
ZGP12/1.1-treated VLPs was clearly blocked by the PP2 treatment (Fig. 13C and E).
These results indicated that the ADE of EBOV entry into K562 cells was dependent on
the Src family PTK activation leading to increased uptake of viral particles into cells.
Finally, I investigated whether the ADE of EBOV entry into K562 cells

54
depended on phagocytosis/macropinocytosis, which have been shown to be major
pathways of the EBOV entry through non-ADE pathways 11-13. I used Dx10 to visualize
phagocytotic and macropinocytotic vesicles in K562 cells and analyzed its colocalization
with VLPs. I found that approximately 70% of the ZGP12/1.1-treated VLP signals were
overlapped with Dx10 in intracellular vesicles (Fig. 14A and C). Furthermore, the
colocalization of Dx10 and ZGP12/1.1-treated VLPs was significantly blocked by the
PP2 treatment (Fig. 14B and C). To confirm that these observations were EBOV-specific,
I further analyzed DiI-labeled SUDV VLPs and found that ZGP12/1.1 affected neither
attachment/uptake of SUDV VLPs nor Dx10 uptake. (Fig. 15). These results suggested
that the surface-bound VLP-ZGP12/1.1 complex was incorporated into cells through Src
family PTK-dependent phagocytosis and/or macropinocytosis during the ADE of EBOV
entry.
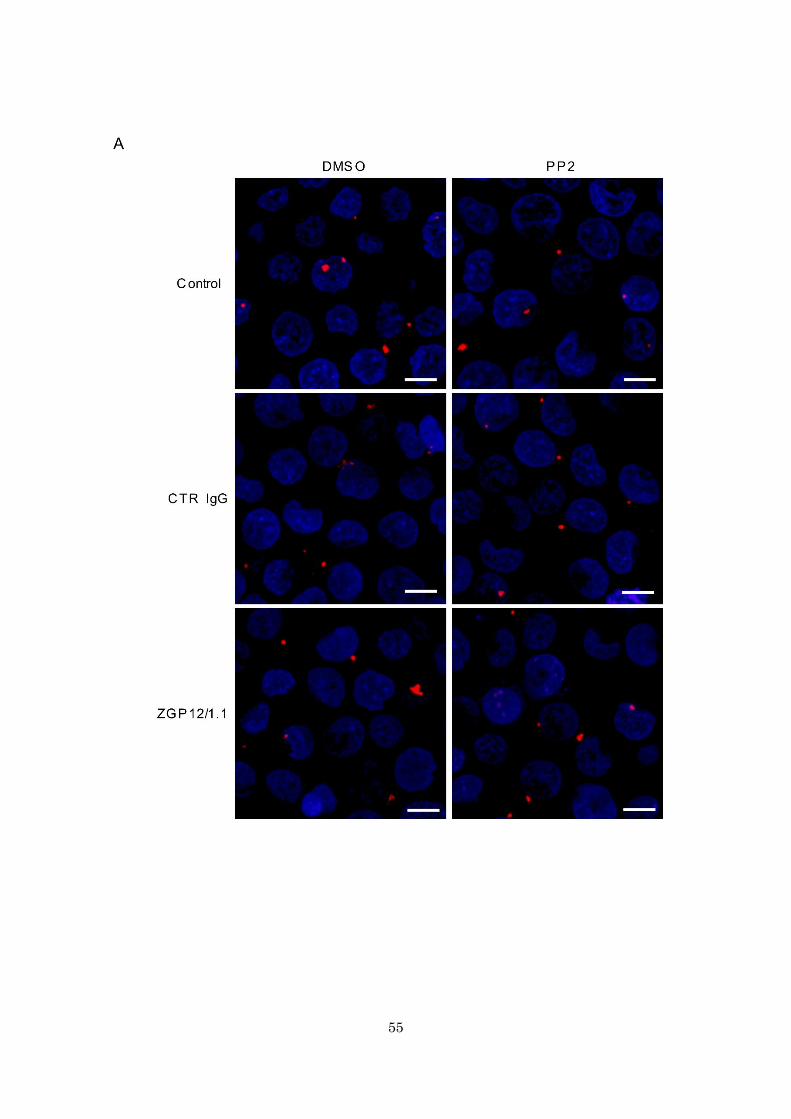
55

56

57
Fig. 13. Enhanced VLP uptake into endosomes in K562 cells during ADE. (A-C)
Fluorescent images of attachment and internalization of VLP. (D and E) Quantified
fluorescent signals of VLP and Rab7. K562 cells expressing eGFP-Rab7 were incubated
with DMSO (A and B) or PP2 (A and C) for 1 h at 37 ˚C. Untreated (Control), CTR IgG-,
and ZGP12/1.1-treated DiI-labeled VLPs were inoculated into cells and VLPs (red) on
the cell surface at 0 h (A and D) and VLPs (red) and eGFP-Rab7 (green) in the cytoplasm

58
at 2 h (B, C and E) after adsorption were monitored by confocal laser scanning microscopy.
Scale bars represent 10 µm. Nuclei of cells are visualized with DAPI (blue). The number
of VLPs on the cell surface (D) and the colocalization of VLPs (DiI) and eGFP-Rab7
signals (E) were quantified. The mean and standard deviation of three independent
experiments are shown. Statistical analysis was performed using Student’s t-test
(*p<0.05).
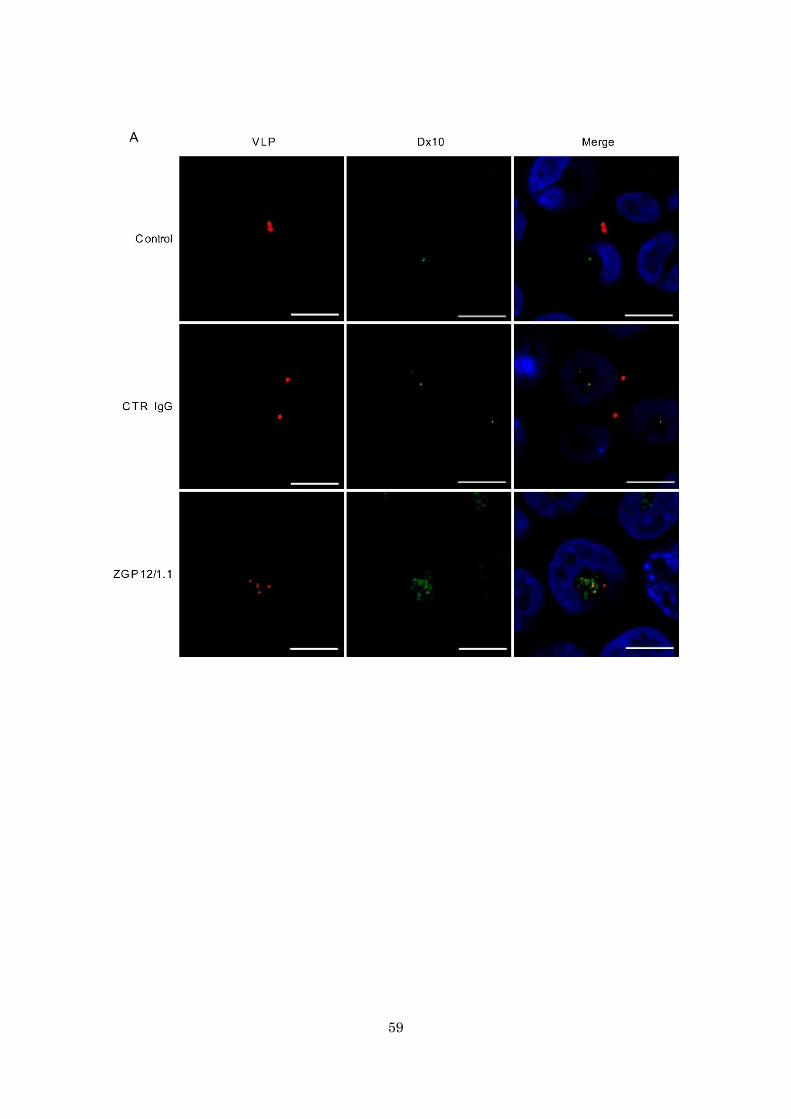
59

60
Fig. 14. Colocalization of Dx10 and ZGP12/1.1-treated VLPs. (A and B) Fluorescent
images of internalization of VLP and Dx10. (C) Quantified fluorescent signals of VLP
and Dx10. K562 cells were incubated with DMSO (A) or PP2 (B) for 1 h at 37 ˚C.
Untreated (Control), CTR IgG-, and ZGP12/1.1-treated DiI-labeled VLPs were
inoculated into cells and incubated for 30 min on ice. After adsorption, cells were
incubated with Alexa647-labeld Dx10 for 1 h at 37 ˚C in the presence of DMSO (A) or

61
PP2 (B). VLPs (red) and Dx10 (green) in the cytoplasm were monitored by confocal laser
scanning microscopy. The colocalization of VLP (DiI) and Dx10 (Alexa647) signals was
quantified (C). Scale bars represent 10 µm. Nuclei of cells are visualized with DAPI
(blue). The mean and standard deviation of three independent experiments are shown.
Statistical analysis was performed using Student’s t-test (*p<0.05).
62
Fig. 15. Attachment, uptake, and localization of DiI-labeled SUDV VLPs. Untreated
(Control), CTR IgG-, and ZGP12/1.1-treated DiI-labeled SUDV VLPs were inoculated
into K562 cell lines and SUDV VLPs (red) on the cell surface at 0 h (A and D) and VLPs
(red) and eGFP-Rab7 (B and E) (green) or Dx10 (C and F) (green) in the cytoplasm at 2
h after adsorption were monitored by confocal laser scanning microscopy. Scale bars

63
represent 10 µm. Nuclei of cells are visualized with DAPI (blue). The number of SUDV
VLPs on the cell surface (D) and the colocalization of SUDV VLPs (DiI) and eGFP-Rab7
(E) or Dx10 (F) signals were quantified. The mean and standard deviation of three
independent experiments are shown. Statistical analysis was performed using Student’s
t-test (*p<0.05).

64
Discussion
It is well established that after the attachment of virus particles to cell surface
receptors a variety of signaling pathways are activated through tyrosine and
phosphoinositol kinases, and the subsequent cellular events such as endocytosis,
including macropinocytosis and phagocytosis, are important for the entry of viruses 87,88.
Likewise, it has been suggested that signaling pathways via FcγR are involved in ADE of
virus infections 68. However, there is limited information on the detailed molecular
mechanisms of intracellular signaling pathways required for ADE of virus infection. It
has been shown that the non-ADE entry of EBOV requires host factors such as PI3K, the
Rho family, and protein kinase C 11,12,89, although the virus-specific receptor molecules
involved in these signaling pathways are not yet identified. In the present study, I focused
on the ADE of EBOV entry and found that the FcγRIIa-mediated signaling pathway was
essential for this process, which is distinct from those required for the non-ADE entry.
The FcγRIIa cytoplasmic tail has been shown to be essential for ADE of dengue
virus entry and the involvement of Syk cascade in ADE entry has been reported 90-92.
These data indicate that the cytoplasmic tail of FcγRIIa is crucial for the ADE of EBOV
infection and that Src family PTK-dependent signaling is important to enhance viral
uptake into cellular vesicles during ADE-mediated entry of EBOV. Src family PTKs are
non-receptor tyrosine kinases involved in the regulation of diverse cellular functions like
proliferation, differentiation, adhesion, and phagocytosis 93,94. Importantly, this signaling
pathway is known to regulate endocytic machinery by triggering the reorganization of the
actin cytoskeleton and membrane remodeling 69,74. Indeed, previous studies have
demonstrated that Src family PTK-dependent signaling is required for the non-ADE entry
of some viruses into host cells 95,96. These data indicate that this signaling is also used to

65
promote viral particle uptake during the FcγRIIa-mediated ADE of EBOV entry.
It was noted that there was a significant increase (approximately 400%) in
infectivity of VSV-EBOV GP when FcγRIIaΔCT is expressed on Jurkat T cells, although
it was not as high as wildtype FcγRIIa. This observation suggest that the binding provided
by the external portion of FcγRIIa may also have some importance and that the signaling
function provided by the cytoplasmic portion of FcγRIIa further enhances the ADE effect.
It might also be possible that the FcγRIIa associated with lipid rafts activates some
FcγRIIa-mediated signals through its transmembrane domain as described previously 97.
Interestingly, I found that the Syk inhibitor R788 reduced the ADE efficiency of
VSV-EBOV GP but not EBOV. While the VSV pseudotype system is widely used to study
EBOV GP functions, it has been suggested that pseudotyped VSV and authentic EBOV
can utilize different entry pathways since the particle size and structure of pseudotyped
VSV do not accurately recapitulate those of EBOV 11,98. Thus, it may be possible that the
EBOV entry primarily relies on macropinocytosis as shown previously 11-13, whereas
VSV-EBOV GP can also be incorporated into smaller vesicles. I assume that this
difference could influence the effect of the inhibitor since Syk-dependent signaling might
be associated with caveolin-mediated endocytosis 99,100. Another difference found
between VSV-EBOV GP and authentic EBOV was that while the Syk inhibitor did not
affect EBOV ADE, non-ADE infection was significantly enhanced in the presence of this
inhibitor, an effect not observed for VSV-EBOV GP. This might be due to the effect on
post-entry mechanisms such as antiviral cellular responses, as proposed by a recent study
demonstrating that dengue virus-antibody complexes decreased type-I interferon-
stimulated gene expression triggered by the FcγR-mediated signaling pathway through
Syk, leading to enhanced replication of the virus 91. Since such an antiviral response could

66
be different between VSV- and EBOV-infected cells, it is possible that the Syk inhibitor
specifically affected the replication of EBOV, but not VSV, RNA genomes.
Previous studies have demonstrated that cellular C-type lectins such as DC-
SIGN and hMGL serve as attachment receptors and promote the entry of EBOV into cells
10,77,86. These C-type lectins, as well as Fc receptors, are thought to initiate phagocytic
pathways for uptake of microorganisms, cell debris, and apoptotic cells 101,102.
Interestingly, both C-type lectins and ADE-antibodies, including ZGP12/1.1, mainly bind
to the mucin-like region of EBOV GP, which has a number of N- and O-linked
glycosylation sites in the middle portion of the protein 23,77,86, suggesting a similarity in
the mechanism of the virus entry mediated by C-type lectins and ADE. However, the Src
family PTK inhibitor PP2 showed limited effects on the infectivity of VSV-EBOV GP in
the cell lines expressing DC-SIGN or hMGL. Taken together, my data suggest that the
FcγRIIa-mediated EBOV ADE principally depends on signaling pathways distinct from
those for C-type lectin-mediated entry, while both C-type lectins and ADE-antibody-
FcγRIIa complexes are assumed to serve as attachment receptors and subsequent
processes for membrane fusion (i.e., cathepsin cleavage and NPC1 binding) appear also
to be similar.
In conclusion, these data indicate that EBOV ADE is not simply dependent on
increased viral attachment through interaction between FcγRIIa and virus-antibody
complexes, and that the induction of FcγRIIa-mediated signaling associated with the
activation of Src family PTKs is essential for EBOV ADE. This signaling pathway most
likely promotes macropinocytosis, the major entry pathway of EBOV, and leads to
enhanced viral uptake into cells. The contribution of Src family PTKs to FcγRIIa-
mediated ADE of virus entry has not been demonstrated previously. Discovery of this

67
ADE mechanism provides a novel perspective for the general understanding of ADE of
virus infection. Although the impact of ADE on disease progression remains unclear for
many viruses, my findings may offer a potential new target to develop treatments for
ADE-associated diseases such as dengue hemorrhagic fever and possibly Zika virus
infection 38,103-105, since signaling pathways are known to be essential for virus entry into
cells and some signaling inhibitors have been considered to be potential treatment options
for virus infections 106-108. However, since non-ADE entry mechanisms of these viruses
are different from EBOV, it is required to investigate whether the Src family PTK-
mediated ADE mechanism can be generally applied for other viruses known to utilize
ADE entry into cells.

68
Summary
ADE of EBOV infection has been demonstrated in vitro, raising concerns about
the detrimental potential of some anti-EBOV antibodies. ADE has been described for
many viruses and mostly depends on the cross-linking of virus-antibody complexes to
cell surface Fc receptors, leading to enhanced infection. However, little is known about
the molecular mechanisms underlying this phenomenon. Here I show that FcγRIIa-
mediated intracellular signaling through Src family PTKs is required for ADE of EBOV
infection. I found that deletion of the FcγRIIa cytoplasmic tail abolished EBOV ADE due
to decreased virus uptake into cellular endosomes. Furthermore, EBOV ADE, but not
non-ADE infection, was significantly reduced by inhibition of the Src family PTK
pathway, which was also found to be important to promote
phagocytosis/macropinocytosis for viral uptake into endosomes. I further confirmed a
significant increase of the Src phosphorylation mediated by ADE. These data suggest that
antibody-EBOV complexes bound to the cell surface FcγRIIa activate the Src signaling
pathway that leads to enhanced viral entry into cells, providing a novel perspective for
the general understanding of ADE of virus infection.

69
Conclusion
GP-specific neutralizing antibodies are known to play an important role in
protection against EBOV infection. However, the existing protective MAbs are ebolavirus
species-specific. Furthermore, some GP-specific antibodies increase EBOV infection in
vitro; indeed, ADE is considered a potential problem for the development of vaccines and
antibody therapy for EVD.
Thus, in chapter I, I developed and described MAb 6D6 which reduced
infectivities of representative isolates of all known ebolavirus species in vitro. Antigenic
variants escaping from 6D6 had amino acid substitutions within the IFL region which is
highly conserved in GPs of all ebolavirus species. MAb 6D6 inhibited the VLP-mediated
membrane fusion in endosomes, but did not affect the attachment and internalization of
VLPs. Furthermore, passive immunization with 6D6 showed protective effects against
EBOV and SUDV in mouse models. The discovery of this cross-neutralizing antibody
provides a promising option for broad-acting ebolavirus antibody therapy.
In chapter II, I demonstrated that FcγRIIa-mediated intracellular signaling is a
key factor for ADE of EBOV infection. The antibody-virus complex bound to the cell
surface FcγRIIa triggered the phosphorylation of Src family PTK that activated signaling
pathways leading to enhanced viral uptake into cells through phagocytosis and/or
macropinocytosis. These findings provide new insights into the mechanism of ADE-
mediated viral infections.
The present study provides important information useful for the design and
implementation of improved therapeutics and vaccines whose antiviral effects depend on
neutralizing antibodies. Furthermore, since for some viruses ADE has become a great
concern to disease control by vaccination and passive immunization, this study

70
demonstrates the novel perspective for the general understanding of ADE and it may also
offer a potential new cellular target to develop treatments for ADE-associated diseases.

71
Acknowledgments
I express my deepest gratitude to my supervisor, Prof. Ayato Takada (Division
of Global Epidemiology, Research Center for Zoonosis Control, Hokkaido Univ.,
Sapporo, Japan), whose comments and suggestions were innumerably valuable
throughout the course of my study. He also provided many opportunities for me to
conduct field work abroad and participate in conferences.
I sincerely appreciate the invaluable support and advice provided by Prof.
Hirofumi Sawa (Division of Molecular Pathobiology, Research Center for Zoonosis
Control, Hokkaido Univ.), Prof. Hideaki Higashi (Division of Infection and Immunity,
Research Center for Zoonosis Control, Hokkaido Univ.), and Associate Prof. Kentaro
Yoshii (Laboratory of Public Health, Graduate School of Veterinary Medicine, Hokkaido
Univ.) during my research work.
I am deeply grateful to Associate Prof. Manabu Igarashi, Specially Appointed
Assistant Prof. Reiko Yoshida, and Specially Appointed Assistant Prof. Rashid Manzoor
(Division of Global Epidemiology, Research Center for Zoonosis Control, Hokkaido
Univ.) for their technical and intellectual support.
I particularly thank Dr. Andrea Marzi, Dr. Heinz Feldmann, Ms. Key Menk, Dr.
Atsushi Okumura, Dr. Hideki Ebihara, Dr. Satoko Yamaoka (National Institute of Allergy
and Infectious Diseases, National Institutes of Health, Rocky Mountain Laboratories), Dr.
Yoshimi Tsuda (Department of Microbiology, Graduate School of Medicine, Hokkaido
Univ.), and all the employees at Rocky Mountain Laboratories for their thoughtful advice
and cooperation. Working with them proved to be a truly valuable experience.
I am grateful to the coordinator of the Program for Leading Graduate Schools,
Hokkaido Univ., Prof. Motohiro Horiuchi (Laboratory of Veterinary Hygiene, Graduate

72
School of Veterinary Medicine, Hokkaido Univ.), and the members of the Leading
Program Office for their help with my PhD coursework.
I thank Dr. Masahiro Kajihara, Dr. Mieko Muramatsu, Dr. Osamu Noyori, Dr.
Makoto Kuroda, Dr. Junki Maruyama, Dr. Naganori Nao, and Ms. Hiroko Miyamoto
(Division of Global Epidemiology, Research Center for Zoonosis Control, Hokkaido
Univ.) for their advice and suggestions. I also thank Mr. Nao Eguchi, Ms. Asako Shigeno,
Mr. Tatsunari Kondoh, Mr. Masahiro Sato, and Mr. Yoshihiro Takadate (Division of
Global Epidemiology, Research Center for Zoonosis Control, Hokkaido Univ.) for their
daily supports.
I wish to specially thank my friends, Ms. Minori Kuroda, Ms. Chiho Kaneko,
Ms. Reiko Akamatsu, Ms. Memi Muto, and Ms. Kanae Shiokawa, for their
encouragement and time. Finally, I express my gratitude to my family and my husband
for their understanding and wonderful support.
My research was funded by the Research Fellowship for Young Scientists from
the Japan Society for the Promotion of Science.

73
和文要旨
エボラウイルスは、フィロウイルス科に属し、ヒトを含む霊長動物に重
篤な出血熱を引き起こす病原体である。フィロウイルス科には、進化系統学的に
エボラウイルス属、マールブルグウイルス属、およびキュエヴァウイルス属に分
類される。マールブルグウイルス属およびキュエヴァウイルス属にはそれぞれ 1
種のみが知られているのに対し、エボラウイルス属は 5種(Zaire ebolavirus、Sudan
ebolavirus、Taï Forest ebolavirus、Bundibugyo ebolavirusおよび Reston ebolavirus)
に分けられている。2000 年以降、Zaire ebolavirus、Sudan ebolavirus および
Bundibugyo ebolavirus によるエボラウイルス病(EVD)がアフリカで頻繁に発生し
ている。これらのウイルスは抗原的に大きく異なっており、属間あるいは種間で
交差反応する免疫応答の誘導が困難なため、エボラウイルスに対する予防・治
療・診断法の開発にとって問題となっている。2014 年の西アフリカでの Zaire
ebolavirusによる EVD流行の際には、未承認薬ながら抗体療法が実施されたが、
使用されたモノクローナル抗体は Zaire ebolavirusによる EVDに対してのみ有効
である。
第 1章では、まず、Zaire ebolavirusおよび Sudan ebolavirusのウイルス様
粒子(VLP)で免疫した BALB/c マウスの脾臓を用いて、5 種のエボラウイルス全
てを中和する抗体の作出を試みた。フィロウイルスの表面糖蛋白(GP)を纏った
非増殖性シュードタイプ水胞性口炎ウイルス(VSV)に対する中和活性を指標に、
抗体産生ハイブリドーマ培養上清をスクリーニングした結果、全てのエボラウ
イルス種に対して高い中和活性を示すモノクローナル抗体(6D6)が得られた。次
に、6D6 存在下で、Zaire ebolavirus の GP 遺伝子をもつ増殖性シュードタイプ
VSVを培養することによってエスケープミュータントを選択し、GPのアミノ酸
配列を親株と比較したところ、エスケープミュータントの GPには、全てのエボ

74
ラウイルス種において高度に保存されている Internal fusion loop(IFL)のアミノ酸
配列内に変異が見られたことから、この領域がエピトープであると推察された。
共焦点顕微鏡を用いて蛍光(DiI)ラベルした VLP の細胞への吸着・取込み・膜融
合を抗体存在下および非存在下で比較した結果、6D6 はウイルスの細胞侵入過
程における膜融合を阻害することが判明した。さらに、6D6を治療的に投与した
マウスは Zaire ebolavirus の致死的感染に対し 100%が生残した。また、Sudan
ebolavirusを感染させたマウスにおいても、6D6の投与による治療効果が認めら
れた。これらの結果によって 6D6 の投与が複数のエボラウイルスに対して効果
的であることが判明した。
一方で、エボラウイルスの GPに対して誘導される抗体の一部は、in vitro
でウイルスの感染を増強することが知られている(抗体依存性感染増強現象:
ADE)。エボラウイルス感染におけるこの現象は、GPに対する抗体が Fcγ受容体
IIa(FcγRIIa)あるいは C1q と C1q 受容体を介して細胞とウイルスとを架橋するこ
とによって引き起こされると考えられているが、ADE の詳細なメカニズムは明
らかとなっていない。第 2章では、FcγRIIaを介する ADEに着目し、FcγRIIa下
流のシグナルと ADEとの関連について検討した。まず、エボラウイルス非感受
性細胞(Jurkat 細胞)を用いて、FcγRIIaまたはその細胞内領域を欠失させた変異体
(FcγRIIaΔCT)を強制発現させた細胞を作出し、ADEによる感染効率を比較した。
その結果、FcγRIIa の細胞内領域が ADE に重要であることが判明した。次に、
FcγRIIaを恒常的に発現している K562細胞を用いて、FcγRIIa下流の各種シグナ
ル阻害剤の効果を調べ ADEに必要なシグナルの特定を試みたところ、Src family
PTK特異的阻害剤である PP2 が、細胞内の Src family PTKのリン酸化を阻害す
ることによって ADEを介した感染のみを顕著に阻害することが判明した。最後
に、共焦点顕微鏡を用いて DiIラベルした Zaire ebolavirusの VLP の細胞への吸

75
着・取込みを抗体存在下および非存在下で比較した結果、ADE 抗体存在下で、
VLP の細胞への吸着数に変化は見られなかったが、細胞内への取り込み効率は
上昇していた。さらに、その上昇は PP2によって阻害された。また、細胞内に取
り込まれた VLP はマクロピノサイトーシス/ファゴサイトーシスマーカーとエ
ンドソームに共局在していた。これらの結果によって、ADE 抗体は FcγRIIa 下
流の Src family PTKsを介したシグナル伝達経路を活性化し、マクロピノサイト
ーシス/ファゴサイトーシスを誘導することによって細胞へのウイルスの取り込
み効率を上昇させ、その結果、感染増強が引き起こされていることが明らかとな
った。
本研究では、既知の全てのエボラウイルス種に対して交差中和活性をし
めすモノクローナル抗体を世界に先駆けて作出した。さらに、そのエピトープを
解析するとともに中和メカニズムを明らかにし、汎用性の高い EVD治療用抗体
医薬の開発に繋がる重要な知見を提供した。また、ワクチンや抗体医薬開発にと
って問題となる ADEのメカニズムを解析し、Src family PTKsを介したシグナル
伝達経路が重要であることを見出した。これらの知見は、今後 EVDの予防・治
療法開発に大きく貢献することが期待される。

76
References
1 Feldmann, H. & Geisbert, T. W. Ebola haemorrhagic fever. Lancet 377, 849-862 (2011).
2 Changula, K., Kajihara, M., Mweene, A. S. & Takada, A. Ebola and Marburg virus
diseases in Africa: increased risk of outbreaks in previously unaffected areas?
Microbiol. Immunol. 58, 483-491 (2014).
3 Kuhn, J. H. et al. Virus nomenclature below the species level: a standardized
nomenclature for filovirus strains and variants rescued from cDNA. Arch.Virol. 159,
1229-1237 (2014).
4 Baize, S. et al. Emergence of Zaire Ebola virus disease in Guinea. N. Engl. J. Med.
371, 1418-1425 (2014).
5 Jeffers, S. A., Sanders, D. A. & Sanchez, A. Covalent modifications of the Ebola virus
glycoprotein. J. Virol. 76, 12463-12472 (2002).
6 Volchkov, V. E., Feldmann, H., Volchkova, V. A. & Klenk, H. D. Processing of the Ebola
virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. U S A 95,
5762-5767 (1998).
7 Sanchez, A., Trappier, S. G., Mahy, B. W., Peters, C. J. & Nichol, S. T. The virion
glycoproteins of Ebola viruses are encoded in two reading frames and are expressed
through transcriptional editing. Proc. Natl. Acad. Sci. U S A 93, 3602-3607 (1996).
8 Weissenhorn, W., Carfí, A., Lee, K.-H., Skehel, J. J. & Wiley, D. C. Crystal structure
of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein
ectodomain. Mol. Cell. 2, 605-616 (1998).
9 Kondratowicz, A. S. et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a
receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci.
U S A 108, 8426-8431 (2011).
10 Alvarez, C. P. et al. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by
Ebola virus in cis and in trans. J.Virol. 76, 6841-6844 (2002).
11 Nanbo, A. et al. Ebolavirus is internalized into host cells via macropinocytosis in a
viral glycoprotein-dependent manner. PLoS Pathog. 6, e1001121 (2010).
12 Saeed, M. F., Kolokoltsov, A. A., Albrecht, T. & Davey, R. A. Cellular entry of Ebola
virus involves uptake by a macropinocytosis-like mechanism and subsequent
trafficking through early and late endosomes. PLoS Pathog. 6, e1001110 (2010).
13 Aleksandrowicz, P. et al. Ebola virus enters host cells by macropinocytosis and
clathrin-mediated endocytosis. J. Infect. Dis. 204, S957-S967 (2011).
14 Chandran, K., Sullivan, N. J., Felbor, U., Whelan, S. P. & Cunningham, J. M.
Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection.
Science 308, 1643-1645 (2005).

77
15 Schornberg, K. et al. Role of endosomal cathepsins in entry mediated by the Ebola
virus glycoprotein. J.Virol. 80, 4174-4178 (2006).
16 Carette, J. E. et al. Ebola virus entry requires the cholesterol transporter Niemann-
Pick C1. Nature 477, 340-343 (2011).
17 Cote, M. et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for
Ebola virus infection. Nature 477, 344-348 (2011).
18 White, J. M. & Schornberg, K. L. A new player in the puzzle of filovirus entry. Nat.
Rev. Microbiol. 10, 317-322 (2012).
19 Lee, J. E. & Saphire, E. O. Ebolavirus glycoprotein structure and mechanism of entry.
Future Virol. 4, 621-635 (2009).
20 Takada, A. et al. Identification of protective epitopes on Ebola virus glycoprotein at
the single amino acid level by using recombinant vesicular stomatitis viruses. J. Virol.
77, 1069-1074 (2003).
21 Saphire, E. O. & Aman, M. J. Feverish quest for Ebola immunotherapy: straight or
cocktail? Trends Microbiol. 24, 684-686 (2016).
22 Takada, A., Feldmann, H., Ksiazek, T. G. & Kawaoka, Y. Antibody-dependent
enhancement of Ebola virus infection. J. Virol. 77, 7539-7544 (2003).
23 Takada, A., Ebihara, H., Feldmann, H., Geisbert, T. W. & Kawaoka, Y. Epitopes
required for antibody-dependent enhancement of Ebola virus infection. J. Infect. Dis.
196, S347-S356 (2007).
24 Lee, J. E. & Saphire, E. O. Neutralizing ebolavirus: structural insights into the
envelope glycoprotein and antibodies targeted against it. Curr. Opini. Struct. Biol. 19,
408-417 (2009).
25 Shedlock, D. J. et al. Antibody-mediated neutralization of Ebola virus can occur by
two distinct mechanisms. Virology 401, 228-235 (2010).
26 Qiu, X. et al. Reversion of advanced Ebola virus disease in nonhuman primates with
ZMapp. Nature 514, 47-53 (2014).
27 Dye, J. M. et al. Postexposure antibody prophylaxis protects nonhuman primates
from filovirus disease. Proc. Natl. Acad. Sci. U S A 109, 5034-5039 (2012).
28 Marzi, A. et al. Protective efficacy of neutralizing monoclonal antibodies in a
nonhuman primate model of Ebola hemorrhagic fever. PLoS ONE 7, e36192 (2012).
29 Pettitt, J. et al. Therapeutic intervention of Ebola virus infection in rhesus macaques
with the MB-003 monoclonal antibody cocktail. Science Transl. Med. 5, 199ra113
(2013).
30 Olinger, G. G., Jr. et al. Delayed treatment of Ebola virus infection with plant-derived
monoclonal antibodies provides protection in rhesus macaques. Proc. Natl. Acad. Sci.

78
U S A 109, 18030-18035 (2012).
31 Matsuno, K. & Takada, A. Antibody therapy as a future treatment option for Ebola
virus infection. Future Virol. 2, 607-614 (2007).
32 McCarthy, M. US signs contract with ZMapp maker to accelerate development of the
Ebola drug. BMJ 349 (2014).
33 Lyon, G. M. et al. Clinical care of two patients with Ebola virus disease in the united
states. N. Engl. J. Med. 371, 2402-2409 (2014).
34 Bishop, B. M. Potential and emerging treatment options for Ebola virus disease. Ann.
Pharmacother. 49, 196-206 (2015).
35 Meyers, L., Frawley, T., Goss, S. & Kang, C. Ebola virus outbreak 2014: clinical
review for emergency physicians. Ann. Emerg. Med. 65, 101-108 (2015).
36 Takada, A. & Kawaoka, Y. Antibody-dependent enhancement of viral infection:
molecular mechanisms and in vivo implications. Rev. Med. Virol. 13, 387-398 (2003).
37 Halstead, S. B. Dengue antibody-dependent enhancement: knowns and unknowns.
Microbiol. Spectr. 2, 249-271 (2014).
38 Halstead, S. B. Antibody, macrophages, dengue virus infection, shock, and
hemorrhage: a pathogenetic cascade. Rev. Infect. Dis. 11, S830-S839 (1989).
39 Takada, A., Watanabe, S., Okazaki, K., Kida, H. & Kawaoka, Y. Infectivity-enhancing
antibodies to Ebola virus glycoprotein. J. Virol. 75, 2324-2330 (2001).
40 García-García, E. & Rosales, C. Signal transduction during Fc receptor-mediated
phagocytosis. J. Leukoc. Biol. 72, 1092-1108 (2002).
41 Freeman, S. A. & Grinstein, S. Phagocytosis: receptors, signal integration, and the
cytoskeleton. Immunol. Rev. 262, 193-215 (2014).
42 Nakayama, E. et al. Enzyme-linked immunosorbent assay for detection of filovirus
species-specific antibodies. Clin. Vaccine Immunol. 17, 1723-1728 (2010).
43 Bray, M., Davis, K., Geisbert, T., Schmaljohn, C. & Huggins, J. A mouse model for
evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis. 178,
651-661 (1998).
44 Takada, A. et al. A system for functional analysis of Ebola virus glycoprotein. Proc.
Natl. Acad. Sci. U S A 94, 14764-14769 (1997).
45 Shahhosseini, S. et al. Production and characterization of monoclonal antibodies
against different epitopes of Ebola virus antigens. J. Virol. Methods 143, 29-37 (2007).
46 Kuroda, M. et al. The interaction between TIM-1 and NPC1 is important for the
cellular entry of Ebola virus. J. Virol. 89, 6481-6493 (2015).
47 Luthy, R., Bowie, J. U. & Eisenberg, D. Assessment of protein models with three-
dimensional profiles. Nature 356, 83-85 (1992).

79
48 Takada, A., Ebihara, H., Jones, S., Feldmann, H. & Kawaoka, Y. Protective efficacy
of neutralizing antibodies against Ebola virus infection. Vaccine 25, 993-999 (2007).
49 Both, L. et al. Monoclonal antibodies for prophylactic and therapeutic use against
viral infections. Vaccine 31, 1553-1559 (2013).
50 Mupapa, K. et al. Treatment of Ebola hemorrhagic fever with blood transfusions from
convalescent patients. International scientific and technical committee. J. Infect. Dis.
179 Suppl 1, S18-23 (1999).
51 R&D, W. E. News on vaccines, therapies, diagnostics (2015).
52 Qiu, X. et al. Successful treatment of ebola virus-infected cynomolgus macaques with
monoclonal antibodies. Sci. Transl. Med. 4, 138ra181 (2012).
53 Qiu, X. et al. Sustained protection against Ebola virus infection following treatment
of infected nonhuman primates with ZMAb. Sci. Rep. 3, 3365 (2013).
54 Hernandez, H. et al. Development and characterization of broadly cross-reactive
monoclonal antibodies against all known ebolavirus species. J. Infec. Dis. 212 Suppl
2, S410-413 (2015).
55 Oswald, W. B. et al. Neutralizing antibody fails to impact the course of Ebola virus
infection in monkeys. PLoS Pathog. 3, e9 (2007).
56 Audet, J. et al. Molecular characterization of the monoclonal antibodies composing
ZMAb: a protective cocktail against Ebola virus. Sci. Rep. 4, 6881 (2014).
57 Davidson, E. et al. Mechanism of binding to Ebola virus glycoprotein by the ZMapp,
ZMAb, and MB-003 cocktail antibodies. J. Virol. 89, 10982-10992 (2015).
58 Reynard, O. & Volchkov, V. E. Characterization of a novel neutralizing monoclonal
antibody against Ebola virus GP. J. Infect. Dis. 212 Suppl 2, S372-378 (2015).
59 Wilson, J. A. et al. Epitopes involved in antibody-mediated protection from Ebola
virus. Science 287, 1664-1666 (2000).
60 Lee, J. E. et al. Structure of the Ebola virus glycoprotein bound to an antibody from
a human survivor. Nature 454, 177-182 (2008).
61 Dias, J. M. et al. A shared structural solution for neutralizing ebolaviruses. Nat.
Struct. Mol. Biol. 18, 1424-1427 (2011).
62 Murin, C. D. et al. Structures of protective antibodies reveal sites of vulnerability on
Ebola virus. Proc. Natl. Acad. Sci. U S A 111, 17182-17187 (2014).
63 Chen, G. et al. Synthetic antibodies with a human framework that protect mice from
lethal Sudan ebolavirus challenge. ACS Chem. Biol. 9, 2263-2273 (2014).
64 Pal, P. et al. Development of a highly protective combination monoclonal antibody
therapy against Chikungunya virus. PLoS Pathog. 9, e1003312 (2013).
65 ter Meulen, J. et al. Human monoclonal antibody combination against SARS

80
coronavirus: synergy and coverage of escape mutants. PLoS Med. 3, e237 (2006).
66 Bakker, A. B. et al. Novel human monoclonal antibody combination effectively
neutralizing natural rabies virus variants and individual in vitro escape mutants. J.
Virol. 79, 9062-9068 (2005).
67 Taylor, A. et al. Fc receptors in antibody-dependent enhancement of viral infections.
Immunol. Rev. 268, 340-364 (2015).
68 Halstead, S. B., Mahalingam, S., Marovich, M. A., Ubol, S. & Mosser, D. M. Intrinsic
antibody-dependent enhancement of microbial infection in macrophages: disease
regulation by immune complexes. Lancet Infect. Dis. 10, 712-722 (2010).
69 Nimmerjahn, F. & Ravetch, J. V. Fcγ receptors as regulators of immune responses.
Nat. Rev. Immunol. 8, 34-47 (2008).
70 Hamada, F., Aoki, M., Akiyama, T. & Toyoshima, K. Association of immunoglobulin
G Fc receptor II with Src-like protein-tyrosine kinase Fgr in neutrophils. Proc. Natl.
Acad. Sci. U S A 90, 6305-6309 (1993).
71 Ghazizadeh, S., Bolen, J. B. & Fleit, H. B. Physical and functional association of Src-
related protein tyrosine kinases with Fc gamma RII in monocytic THP-1 cells. J. Biol.
Chem. 269, 8878-8884 (1994).
72 Kiener, P. A. et al. Cross-linking of Fc gamma receptor I (Fc gamma RI) and receptor
II (Fc gamma RII) on monocytic cells activates a signal transduction pathway
common to both Fc receptors that involves the stimulation of p72 Syk protein tyrosine
kinase. J. Biol. Chem. 268, 24442-24448 (1993).
73 Joshi, T., Butchar, J. P. & Tridandapani, S. Fcγ receptor signaling in phagocytes. Int.
J. Hematol. 84, 210-216 (2006).
74 May, R. C. & Machesky, L. M. Phagocytosis and the actin cytoskeleton. J. Cell Sci.
114, 1061-1077 (2001).
75 Ebihara, H. et al. In vitro and in vivo characterization of recombinant Ebola viruses
expressing enhanced green fluorescent protein. J. Infect. Dis. 196, S313-S322 (2007).
76 Matsuno, K. et al. Different potential of C-type lectin-mediated entry between
Marburg virus strains. J. Virol. 84, 5140-5147 (2010).
77 Takada, A. et al. Human macrophage C-type lectin specific for galactose and N-
acetylgalactosamine promotes filovirus entry. J. Virol. 78, 2943-2947 (2004).
78 Yoshida, R. et al. Cross-protective potential of a novel monoclonal antibody directed
against antigenic site B of the hemagglutinin of influenza A viruses. PLoS Pathog. 5,
e1000350 (2009).
79 Kennedy, G. & Sugden, B. EBNA-1, a bifunctional transcriptional activator. Mol. Cell.
Biol. 23, 6901-6908 (2003).

81
80 Chan, S. Y., Speck, R. F., Ma, M. C. & Goldsmith, M. A. Distinct mechanisms of entry
by envelope glycoproteins of Marburg and Ebola (Zaire) viruses. J. Virol. 74, 4933-
4937 (2000).
81 Brooks, D. G., Qiu, W. Q., Luster, A. D. & Ravetch, J. V. Structure and expression of
human IgG FcRII(CD32). Functional heterogeneity is encoded by the alternatively
spliced products of multiple genes. J. Exp. Med. 170, 1369-1385 (1989).
82 Worth, R. G. et al. The cytoplasmic domain of FcγRIIA (CD32) participates in
phagolysosome formation. Blood 98, 3429-3434 (2001).
83 Araki, N., Hatae, T., Furukawa, A. & Swanson, J. A. Phosphoinositide-3-kinase-
independent contractile activities associated with Fcγ-receptor-mediated
phagocytosis and macropinocytosis in macrophages. J. Cell Sci. 116, 247-257 (2003).
84 Worth, R. G., Kim, M.-K., Kindzelskii, A. L., Petty, H. R. & Schreiber, A. D. Signal
sequence within FcγRIIA controls calcium wave propagation patterns: apparent role
in phagolysosome fusion. Proc. Natl. Acad. Sci. U S A 100, 4533-4538 (2003).
85 Kasahara, K. et al. Role of Src-family kinases in formation and trafficking of
macropinosomes. J. Cell. Physiol. 211, 220-232 (2007).
86 Simmons, G. et al. DC-SIGN and DC-SIGNR bind Ebola glycoproteins and enhance
infection of macrophages and endothelial cells. Virology 305, 115-123 (2003).
87 Greber, F. U. Signalling in viral entry. Cell. Mol. Life Sci. 59, 608-626 (2002).
88 Mercer, J. & Helenius, A. Virus entry by macropinocytosis. Nat. Cell Biol. 11, 510-
520 (2009).
89 Saeed, M. F., Kolokoltsov, A. A., Freiberg, A. N., Holbrook, M. R. & Davey, R. A.
Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS
Pathog. 4, e1000141 (2008).
90 Moi, M. L., Lim, C.-K., Takasaki, T. & Kurane, I. Involvement of the Fcγ receptor IIA
cytoplasmic domain in antibody-dependent enhancement of dengue virus infection. J.
Gen. Virol. 91, 103-111 (2010).
91 Chan, K. R. et al. Leukocyte immunoglobulin-like receptor B1 is critical for antibody-
dependent dengue. Proc. Natl. Acad. Sci. U S A 111, 2722-2727 (2014).
92 Callaway, J. B. et al. Spleen tyrosine kinase (Syk) mediates IL-1beta induction by
primary human monocytes during antibody-enhanced dengue virus infection. J. Biol.
Chem. 290, 17306-17320 (2015).
93 Berton, G., Mócsai, A. & Lowell, C. A. Src and Syk kinases: key regulators of
phagocytic cell activation. Trends Immunol. 26, 208-214 (2005).
94 Okutani, D., Lodyga, M., Han, B. & Liu, M. Src protein tyrosine kinase family and
acute inflammatory responses. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L129-

82
L141 (2006).
95 Krishnan, H. H., Sharma-Walia, N., Streblow, D. N., Naranatt, P. P. & Chandran, B.
Focal adhesion kinase is critical for entry of Kaposi's sarcoma-associated herpesvirus
into target cells. J. Virol. 80, 1167-1180 (2006).
96 Li, H. et al. Genome-wide gene expression analysis identifies the proto-oncogene
tyrosine-protein kinase Src as a crucial virulence determinant of infectious
laryngotracheitis virus in chicken cells. J. Virol. 90, 9-21 (2016).
97 Garcia-Garcia, E., Brown, E. J., Rosales, C. Transmembrane mutations to
FcgammaRIIA alter its association with lipid rafts: implications for receptor
signaling. J. Immunol. 178, 3048-3058 (2007).
98 Cureton, D. K., Massol, R. H., Whelan, S. P. J. & Kirchhausen, T. The length of
vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-
dependent endocytosis. PLoS Pathog. 6, e1001127 (2010).
99 Samaj, J. et al. Endocytosis, actin cytoskeleton, and signaling. Plant Physiol. 135,
1150-1161 (2004).
100 Utskarpen, A. et al. Shiga toxin increases formation of clathrin-coated pits through
Syk kinase. PLoS ONE 5, e10944 (2010).
101 Stuart, L. M. & Ezekowitz, R. A. B. Phagocytosis: elegant complexity. Immunity 22,
539-550 (2005).
102 Kerrigan, A. M. & Brown, G. D. C-type lectins and phagocytosis. Immunobiology 214,
562-575 (2009).
103 Dejnirattisai, W. et al. Dengue virus sero-cross-reactivity drives antibody-dependent
enhancement of infection with zika virus. Nat. Immunol. 17, 1102-1108 (2016).
104 Priyamvada, L. et al. Human antibody responses after dengue virus infection are
highly cross-reactive to Zika virus. Proc. Natl. Acad. Sci. U S A 113, 7852-7857 (2016).
105 Kawiecki, A. B. & Christofferson, R. C. Zika-induced antibody response enhances
dengue serotype 2 replication in vitro. J. Infect. Dis. 214, 1357-1360 (2016).
106 Qatsha, K. A., Rudolph, C., Marme, D., Schachtele, C., May, W. S. Go 6976, a selective
inhibitor of protein kinase C, is a potent antagonist of human immunodeficiency virus
1 induction from latent/low-level-producing reservoir cells in vitro. Proc. Natl. Acad.
Sci. U S A 90, 4674-4678 (1993).
107 Cooper, L., Longnecker, R. Inhibition of host kinase activity altered by the LMP2A
signalosome-a therapeutic target for Epstein-Barr virus latency and associated
disease. Antiviral. Res. 56, 219-231 (2002).
108 Planz, O. Development of cellular signaling pathway inhibitors as new antivirals
against influenza. Antiviral. Res. 98, 457-468 (2013).