dopaminergic modulation of low-mg2+-induced epileptiform activity in the intact hippocampus of the...
TRANSCRIPT

Dopaminergic Modulation of Low-Mg21-Induced Epileptiform Activity in the IntactHippocampus of the Newborn MouseIn Vitro
Salim Sharopov,1 Jochen Moser,1 Rongqing Chen,1 Sergey N. Kolbaev,1
Viviane E. Bernedo,2 Konrad J. Werhahn,2 Heiko J. Luhmann,1 and Werner Kilb1*1Institute of Physiology and Pathophysiology, University Medical Center of the Johannes-GutenbergUniversity, Mainz, Germany2Department of Neurology, University Medical Center of the Johannes-Gutenberg University,Mainz, Germany
To investigate whether epileptiform activity in the imma-ture brain is modulated by dopamine, we examined theeffects of dopaminergic agonists and antagonists in anintact in vitro preparation of the isolated corticohippo-campal formation of immature (postnatal days 3 and 4)C57/Bl6 mice using field potential recordings from CA3.Epileptiform discharges were induced by a reduction ofthe extracellular Mg21 concentration to 0.2 mM. Theseexperiments revealed that low concentrations of dopa-mine (<0.3 lM) attenuated epileptiform activity, whereas>3 lM dopamine enhanced epileptiform activity. TheD1-agonist SKF38393 (10 lM) had a strong proconvul-sive effect, and the D2-like agonist quinpirole (10 lM)mediated a weak anticonvulsive effect. The proconvul-sive effect of 10 lM dopamine was completely abol-ished by the D1-like receptor antagonist SCH39166 (2lM) or the D2-like antagonist sulpiride (10 lM), whereasthe D2 antagonist L-741626 (50 nM) and the D3 antago-nist SB-277011-A (0.1 lM) were without effect. The anti-convulsive effect of 0.1 lM dopamine could besuppressed by D1-like, D2, or D3 receptor antagonists.A proconvulsive effect of 10 lM dopamine was alsoobserved when AMPA, NMDA, or GABAA receptorswere blocked. In summary, these results suggest that 1)dopamine influences epileptiform activity already atearly developmental stages; 2) dopamine can bidirec-tionally influence the excitability; 3) D1-like receptorsmediate the proconvulsive effect of high dopamine con-centrations, although the pharmacology of the anticon-vulsive effect is less clear; and 4) dopamine-inducedalterations in GABAergic and glutamatergic systemsmay contribute to this effect. VVC 2012 Wiley Periodicals, Inc.
Key words: development; dopamine receptor; epilepsy;seizures
Epileptic seizures are a major neurological disorderwith a particular high incidence among neonates(Glauser, 1995; Cowan, 2002; Silverstein and Jensen,
2007). The higher seizure susceptibility of the immaturenervous system has been linked to a variety of immatureneuronal properties, e.g., the higher input resistance ofimmature neurons (Sanchez and Jensen, 2001), the lon-ger decay kinetics of glutamatergic inputs (Jensen, 1999;Swann and Hablitz, 2000), and immature properties ofthe GABAergic system, such as fewer GABAergic con-nections, a lower density of GABAA and GABAB recep-tors, and depolarizing GABAergic responses (Ben-Ariet al., 1989, 2007; Lang and Frotscher, 1990; Luhmannand Prince, 1991; Fukuda et al., 1993; Gaiarsa et al.,1995; Rivera et al., 1999). Because of their severity andthe observation that neonatal seizures are a risk factor forthe later development of epilepsy (Holmes, 1997; Khali-lov et al., 2003), seizures in the neonatal period requireimmediate therapeutic control. However, neonatal seiz-ures have a different etiology and therefore require analternate treatment compared with adult forms of epi-lepsy (Silverstein and Jensen, 2007). In particular, themodest pharmacological responsiveness of epileptic seiz-ures in neonates makes alternative therapeutic strategiesdesirable (Booth and Evans, 2004; Sankar and Painter,2005).
It has gradually become evident that the dopami-nergic system also plays a profound role in epileptogene-sis (Starr, 1996). Dopamine interacts with at least five
Contract grant sponsor: H.W. and J. Hector Stiftung; Contract grant
number: M46 (to K.J.W., H.J.L.); Contract grant sponsor: DFG (to
W.K., H.J.L.); Contract grant sponsor: MAIFOR program of the Uni-
versity of Medicine Mainz (to W.K.).
*Correspondence to: Werner Kilb, Institute of Physiology and Patho-
physiology, University Medical Center of the Johannes-Gutenberg Uni-
versity, Duesbergweg 6, D-55128 Mainz, Germany.
E-mail: [email protected]
Received 15 November 2011; Revised 11 April 2012; Accepted 13 April
2012
Published online in Wiley Online Library (wileyonlinelibrary.com).
DOI: 10.1002/jnr.23084
Journal of Neuroscience Research 00:000–000 (2012)
' 2012 Wiley Periodicals, Inc.

different receptors (D1–D5) that could be subdividedinto D1-like (D1 and D5) and D2-like (D2, D3, andD4) subfamilies (Missale et al., 1998). Initial clinicalobservations suggested that various dopaminergic agonistshave an anticonvulsant effect, whereas dopamine antago-nists exacerbate seizures or lower seizure thresholds(Lipka and Lathers, 1987; Clemens, 1988; Gattereauet al., 1990). More recent clinical and experimental data,however, support a complex action (Starr, 1996; Wein-shenker and Szot, 2002) in which the anticonvulsiveeffect of dopamine can be attributed to an activation ofD2-like receptors, whereas activation of D1-like recep-tors tends to promote convulsions (Loscher and Czucz-war, 1986; Altajir et al., 1990; Burke et al., 1990; Alamand Starr, 1992, 1993). In particular, in light of recentfindings of massive alterations in the density of differentdopamine receptor subtypes and transporters in variousepileptic syndromes (Werhahn et al., 2006; Ciumaset al., 2008; Fedi et al., 2008; Landvogt et al., 2010;Yakushev et al., 2010), the dopaminergic system mostprobably contributes to epileptogenesis and may thusopen a new window for therapeutic intervention. How-ever, surprisingly little information is available on theinfluence of the dopaminergic system on seizure suscep-tibility in the immature nervous system. Because monoa-minergic projections, dopamine receptors, and dopaminetransporters are already expressed in the prenatal humanbrain (Zecevic and Verney, 1995; Unis et al., 1998;Meng et al., 1999; Gurevich et al., 2000), the dopami-nergic system may substantially interfere with seizure ac-tivity and/or epileptogenesis in the immature CNS.
To elucidate the influence of dopamine on theexcitability of the immature nervous system, we exam-ined the effects of dopamine and specific dopaminergicagonists and antagonists on epileptiform discharges in anintact in vitro preparation of the corticohippocampal for-mation of newborn mice (Quilichini et al., 2002; Moseret al., 2006). Our results revealed that, already in theimmature hippocampus, dopamine mediates both anti-and proconvulsive effects, with D1-like and D2-likereceptors contributing to these opposing effects, respec-tively. These observations indicate that dopamine is ableto regulate the excitability levels of the hippocampusduring an early developmental period and that the dopa-minergic system is a potential target of anticonvulsivetherapy.
MATERIALS AND METHODS
Tissue Preparation
All experiments were conducted in accordance with EUdirective 86/609/EEC for the use of animals in research andthe NIH Guide for the care and use of laboratory animals andwere approved by the local ethical committee (Landesuntersu-chungsanstalt RLP, Koblenz, Germany). All efforts were madeto reduce the number of experimental animals and their suf-fering. The preparation procedure was performed as previ-ously described (Moser et al., 2006). Briefly, neonatal mice(C57Bl/62, P3–4) were decapitated, and the brain was trans-
ferred to oxygenated (95% O2/5% CO2), ice-cold (2–58C) ar-tificial cerebrospinal fluid (ACSF) of the following composi-tion (in mM): 124 NaCl, 5 KCl, 1.6 CaCl2, 1 MgCl2, 26NaHCO3, 1.25 NaH2PO4, and 10 glucose (pH 7.4 after CO2
equilibration). After the brains had equilibrated for 2–3 min inthe ice-cold ACSF, basal brain structures were removed usingtwo spatulas. Subsequently, the hemispheres were dissected,and the corticohippocampal formation (CHF) was isolated bya series of perpendicular cuts with a scalpel blade. The intactpreparation of the CHF includes the hippocampus and largeparts of the posterior cortex, including entorhinal and tempo-ral cortex (Quilichini et al., 2002, 2003). Each CHF wastransferred to a fully submerged chamber and superfused withACSF at 318C 6 18C at a flow rate of �5 ml/min. After anincubation period of >45 min, the CHF was superfused withlow-Mg21 solution to induce epileptiform discharges. Thelow-Mg21 solution consisted of (in mM) 124 NaCl, 5 KCl,1.6 CaCl2, 0.2 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, andglucose 10. Note that this low Mg21 concentration is mostprobably slightly underestimated because of contamination ofthe other ACSF constituents with Mg21 (Mody et al., 1987).
Data Acquisition and Analysis
Extracellular field potentials were recorded in the CA3region of the intact CHF with tungsten 4–5-MX microelectr-odes (FHC, Bowdoinham, ME). Signals were AC recordedwith extracellular amplifiers, low-pass filtered at 1 kHz, stored,and analyzed with an eight-channel PC-based software pro-gram (Tida 4.11; Heka, Lambrecht, Germany). Extracellularfield potentials recorded from up to four separate CHFs wereobtained simultaneously and were analyzed independently.Recordings were analyzed by using algorithms developed onthe basis of a Matlab environment (Matlab R2006a; Math-works, Natick, MA). This algorithm identified spikes by athreshold-crossing detector (with the threshold set manuallyabove the noise level), and subsequently the epileptiform dis-charges were analyzed according to their occurrence (i.e., thefrequency of discharges) and the average amplitude, frequency,and number of spikes per discharge. Epileptiform dischargeswere classified as seizure-like or interictal events according tothe number of spike and the duration of discharges. Powerspectrograms of epileptiform activity were calculated usingTIDA 4.11. For further analysis, the power was averaged infive frequency bands (delta 0.5–4 Hz, theta 4–8 Hz, alpha 8–14Hz, beta 14–30 Hz, and gamma 30–80 Hz). Values throughoutthis report are given as mean 6 SEM. For statistical compari-sons, the sign test and Mann-Whitney U test (Systat 11; SPSSInc., Chicago, IL) were used. Significance levels of P < 0.05,P < 0.01, and P < 0.001 were considered.
Drugs
Dopamine hydrochloride was added at 0.1, 0.3, 1, 3,10, and 30 lM in the continuous presence of 5 lM nomifen-sine (1,2,3,4-tetrahydro-2-methyl-4-phenyl-8-isoquinolinaminmaleate) and 100 lM sodium metabisulfide (Sigma, Stein-heim, Germany) to prevent endogenous dopamine reuptakeand oxidation of dopamine. Dopamine receptors were acti-vated by the subtype-specific agonists (6)-SKF-38393
2 Sharopov et al.
Journal of Neuroscience Research

(Sigma), GSK 789472 hydrochloride (Tocris, Ellisville, MO),and (2)-quinpirole hydrochloride (Sigma). The following do-pamine receptor antagonists were used: (R)-(1)-SCH-39166hydrochloride (Sigma), L-741626 (3-[[4-(4-chlorophenyl)-4-hydroxypiperidin-l-yl]methyl-1H-indole; Tocris), (2)-sulpir-ide (Sigma), and SB-277011A (Sigma). In some experiments,adrenergic receptors were blocked by the combined applica-tion of (RS)-propranolol hydrochloride (Biotrend, Zurich,Switzerland) and phentolamine mesylate (Biotrend). GABAA
and NMDA receptors were blocked by using gabazine (SR-95531) and DL-2-amino-5-phosphonopentanoic acid(6-APV), respectively. AMPA receptors were blocked by6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) or GYKI52466 (4-(8-methyl-9H-1,3-dioxolo[4,5-][2,3]benzodiazepin-5-yl)-benzenamine hydrochloride; Tocris). Dopamine wasprepared freshly in sodium metabisulfite containing ACSFevery day. (6)-APV, GSK 789472, propranolol, and phentol-amine were used from an aqueous stock solution and all otheragonists and antagonists from a stock solution in dimethylsulf-oxide (DMSO). The DMSO concentration in the bathingsolution never exceeded 0.1%.
RESULTS
Dopaminergic Modulation of Low-Mg21-InducedEpileptiform Activity
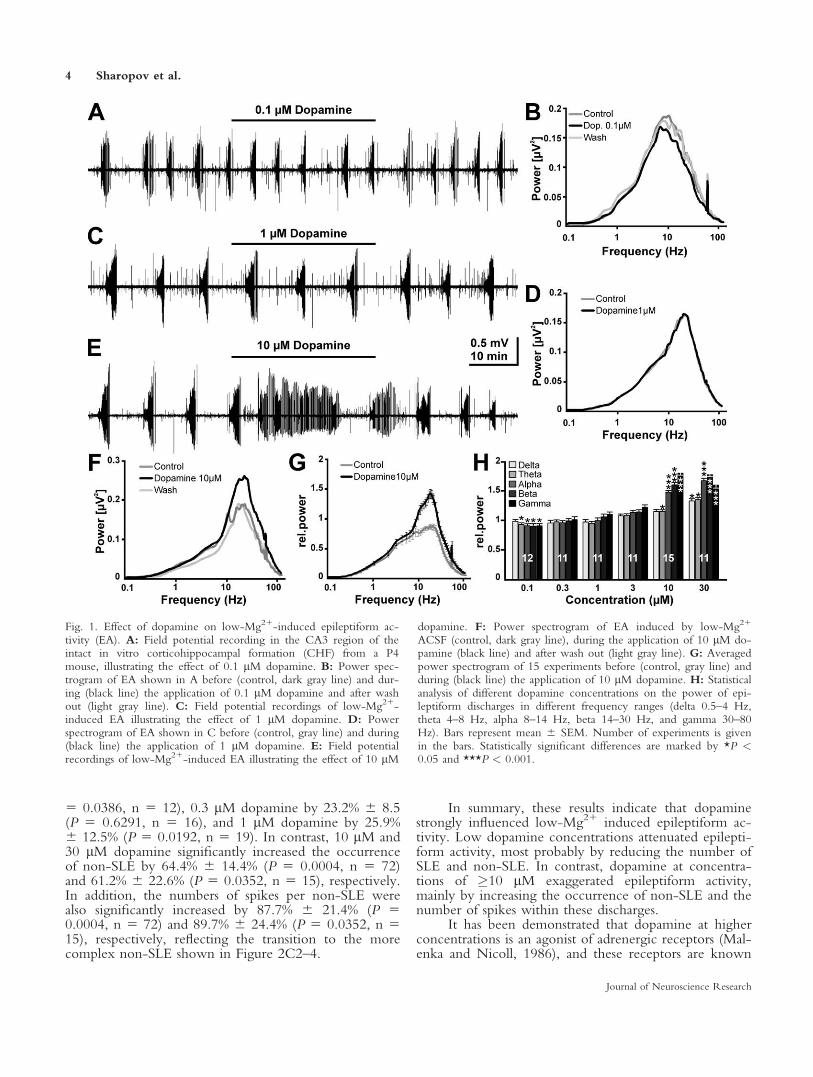
Exposure to 0.2 mM Mg21-containing ACSF (low-Mg21 solution) induced spontaneous epileptiform activityin the CHF of immature mice, as previously shown(Moser et al., 2006). In all preparations, the epileptiformactivity consisted of seizure-like and interictal-like events.To elucidate the effect of dopamine on epileptiform activ-ity, it was bath applied in concentrations ranging from 0.1to 30 lM to CHF preparations that showed stable epilepti-form discharges in low-Mg21 solution. Dopamine wasadded in the continuous presence of 5 lM nomifensine toprevent endogenous dopamine reuptake. Bath applicationof 5 lM nomifensine significantly (P < 0.01) reduced theoccurrence of seizure-like and interictal-like events by14% 6 5.9% (n 5 33) and 24% 6 3.6% (n 5 33), respec-tively (data not shown). Application of 0.1 lM dopaminein the presence of nomifensine resulted in a slight butobvious attenuation of epileptiform activity (Fig. 1A,B).At intermediate concentrations (0.3–3 lM) no changes inepileptiform activity were obvious (Fig. 1C,D), but higherdopamine concentrations (�10 lM) induced an exaggera-tion of epileptiform activity (Fig. 1E,F). The averagedpower spectrogram revealed that the dopamine effect ledto alterations in a frequency band between �3 and 50 Hz(Fig. 1G), so we analyzed dopamine’s effect on the averagepower in the classical EEG-frequency bands. These analy-ses revealed that 0.1 lM dopamine induced a significantreduction in theta (93.3% 6 0.8%, P 5 0.0386, n 5 12),alpha (90.9% 6 0.8%, P 5 0.0386), beta (90% 6 1.3%, P5 0.0386), and gamma (90.8% 6 1.5%, P 5 0.0063)bands, whereas the power in the delta frequency band wasnot altered (Fig. 1H). Bath application of 0.3 lM (n5 11),1 lM (n5 11), or 3 lM (n5 11) dopamine did not signif-icantly change the power of epileptiform activity in any
frequency band (Fig. 1H). In contrast, 10 lM dopaminesignificantly increased the power of epileptiform activity intheta (115.1% 6 1.9%, P 5 0.0352, n 5 15), alpha (147%6 1.8%, P < 0.0001), beta (159.4% 6 2.7%, P < 0.0001),and gamma (157.3% 6 2.8%, P < 0.0001) frequencybands, whereas the delta frequency range was not altered(Fig. 1H). Application of 30 lM dopamine led to a signifi-cant increase of the power in all frequency bands (delta132.5% 6 2.6%, P 5 0.0117, n 5 11; theta 134.4% 62.4%, P 5 0.0117; alpha 166.5% 6 3.6%, P 5 0.001; beta176.8% 6 3.8%, P 5 0.001; gamma 158.9% 6 4.7%, P 50.001; Fig. 1H).
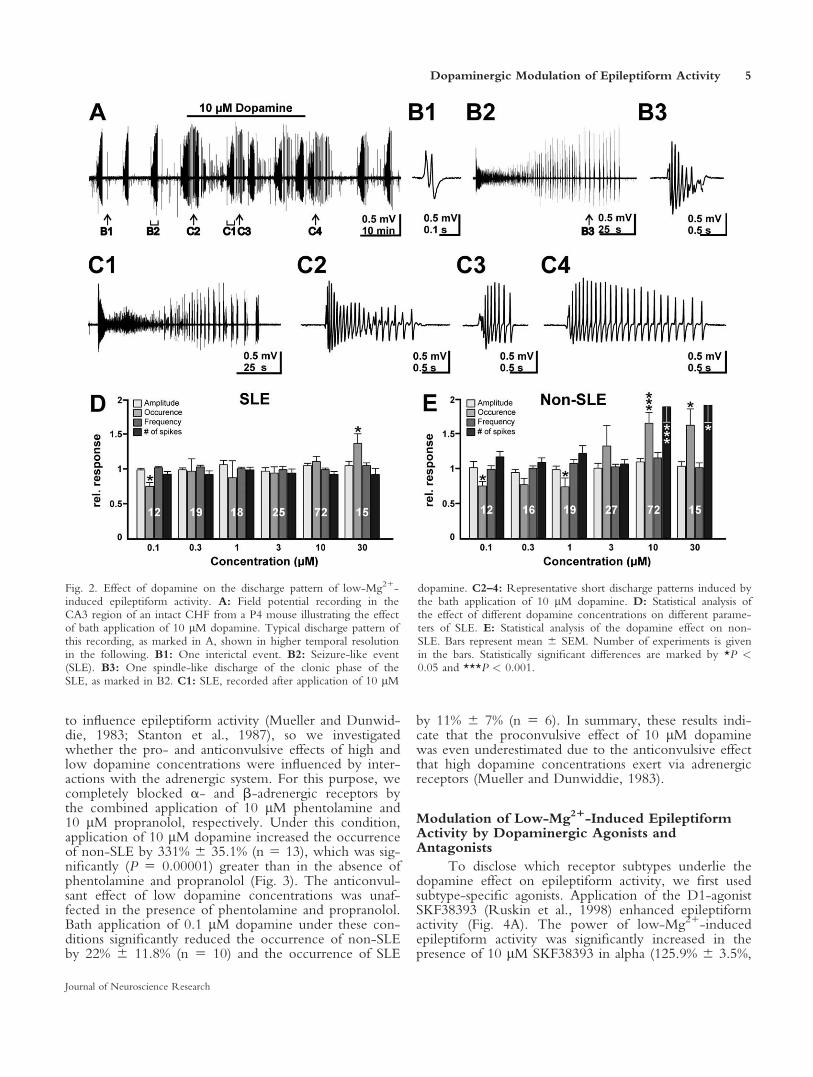
To address the question of which alterations in thedischarge pattern underlie the observed changes in thepower of epileptiform activity, we next investigated theepileptiform activity patterns in more detail. Low-Mg21-induced epileptiform activity consisted of interictalevents (IIE; Fig. 2A,B1) and seizure-like events (SLE;Fig. 2B2). IIE occurred at a frequency of 1.3 6 0.09min21 (n 5 55) and consisted on average of 4 6 0.2spikes at a frequency of 13.6 6 0.6 Hz. SLE occurred ata frequency of 0.14 6 0.006 min21 (n 5 55). Theseevents started with high-frequency spikes, followed byseparated burst discharges at the end of the SLE reminis-cent of the ‘‘clonic phase’’ in human epilepsy. These latebursts during SLE mostly had a spindle-like appearance(Fig. 2B3). In the presence of 10 lM dopamine, thestructure of epileptiform patterns changed considerably.SLE were nearly unaffected (Fig. 2C1), but virtually noIIE could be detected under this condition. On theother hand, dopamine induced short patterns (0.5–2 sec)outside of SLE that had no corresponding patterns undercontrol conditions (Fig. 2C2–4). Therefore, we classifiedall patterns outside of SLE as non-SLE. These non-SLEoccurred at a frequency of 2.2 6 0.16 min21 and con-sisted on average of 13 6 1.4 spikes at a frequency of14.8 6 0.4 Hz (n 5 72).
By using semiautomatic classification of all epilepti-form events, we analyzed the effect of different dopa-mine concentrations on SLE and non-SLE regardingtheir occurrence, amplitude, and frequency and thenumber of spikes per discharge. Application of 0.1 lMdopamine significantly (P 5 0.0386) reduced the averageoccurrence of SLE by 25.1% 6 5.1% (n 5 12; Fig. 2D),whereas amplitude, frequency, and number of spikeswere not significantly affected. At concentrationsbetween 0.3 and 10 lM, dopamine SLE were mostlyunaffected (Fig. 2D), except for a slight reduction in thefrequency of spikes by 2.2% 6 4.1% (n 5 25, P 50.0026) at 3 lM dopamine. Only in the presence of 30lM dopamine was the occurrence of SLE significantly(P 5 0.0352) increased by 36.2% 6 13.5% (n 5 15;Fig. 2D).
Dopamine exerted stronger effects on the proper-ties of non-SLE. Application of low dopamine concen-trations reduced the occurrence of non-SLE, whereasamplitude, frequency, and number of spikes per non-SLE were not affected (Fig. 2E). Specifically, 0.1 lMdopamine reduced the occurrence by 24.6% 6 6.7% (P
Dopaminergic Modulation of Epileptiform Activity 3
Journal of Neuroscience Research
5 0.0386, n 5 12), 0.3 lM dopamine by 23.2% 6 8.5(P 5 0.6291, n 5 16), and 1 lM dopamine by 25.9%6 12.5% (P 5 0.0192, n 5 19). In contrast, 10 lM and30 lM dopamine significantly increased the occurrenceof non-SLE by 64.4% 6 14.4% (P 5 0.0004, n 5 72)and 61.2% 6 22.6% (P 5 0.0352, n 5 15), respectively.In addition, the numbers of spikes per non-SLE werealso significantly increased by 87.7% 6 21.4% (P 50.0004, n 5 72) and 89.7% 6 24.4% (P 5 0.0352, n 515), respectively, reflecting the transition to the morecomplex non-SLE shown in Figure 2C2–4.
In summary, these results indicate that dopaminestrongly influenced low-Mg21 induced epileptiform ac-tivity. Low dopamine concentrations attenuated epilepti-form activity, most probably by reducing the number ofSLE and non-SLE. In contrast, dopamine at concentra-tions of �10 lM exaggerated epileptiform activity,mainly by increasing the occurrence of non-SLE and thenumber of spikes within these discharges.
It has been demonstrated that dopamine at higherconcentrations is an agonist of adrenergic receptors (Mal-enka and Nicoll, 1986), and these receptors are known
Fig. 1. Effect of dopamine on low-Mg21-induced epileptiform ac-tivity (EA). A: Field potential recording in the CA3 region of theintact in vitro corticohippocampal formation (CHF) from a P4mouse, illustrating the effect of 0.1 lM dopamine. B: Power spec-trogram of EA shown in A before (control, dark gray line) and dur-ing (black line) the application of 0.1 lM dopamine and after washout (light gray line). C: Field potential recordings of low-Mg21-induced EA illustrating the effect of 1 lM dopamine. D: Powerspectrogram of EA shown in C before (control, gray line) and during(black line) the application of 1 lM dopamine. E: Field potentialrecordings of low-Mg21-induced EA illustrating the effect of 10 lM
dopamine. F: Power spectrogram of EA induced by low-Mg21
ACSF (control, dark gray line), during the application of 10 lM do-pamine (black line) and after wash out (light gray line). G: Averagedpower spectrogram of 15 experiments before (control, gray line) andduring (black line) the application of 10 lM dopamine. H: Statisticalanalysis of different dopamine concentrations on the power of epi-leptiform discharges in different frequency ranges (delta 0.5–4 Hz,theta 4–8 Hz, alpha 8–14 Hz, beta 14–30 Hz, and gamma 30–80Hz). Bars represent mean 6 SEM. Number of experiments is givenin the bars. Statistically significant differences are marked by *P <0.05 and ***P < 0.001.
4 Sharopov et al.
Journal of Neuroscience Research
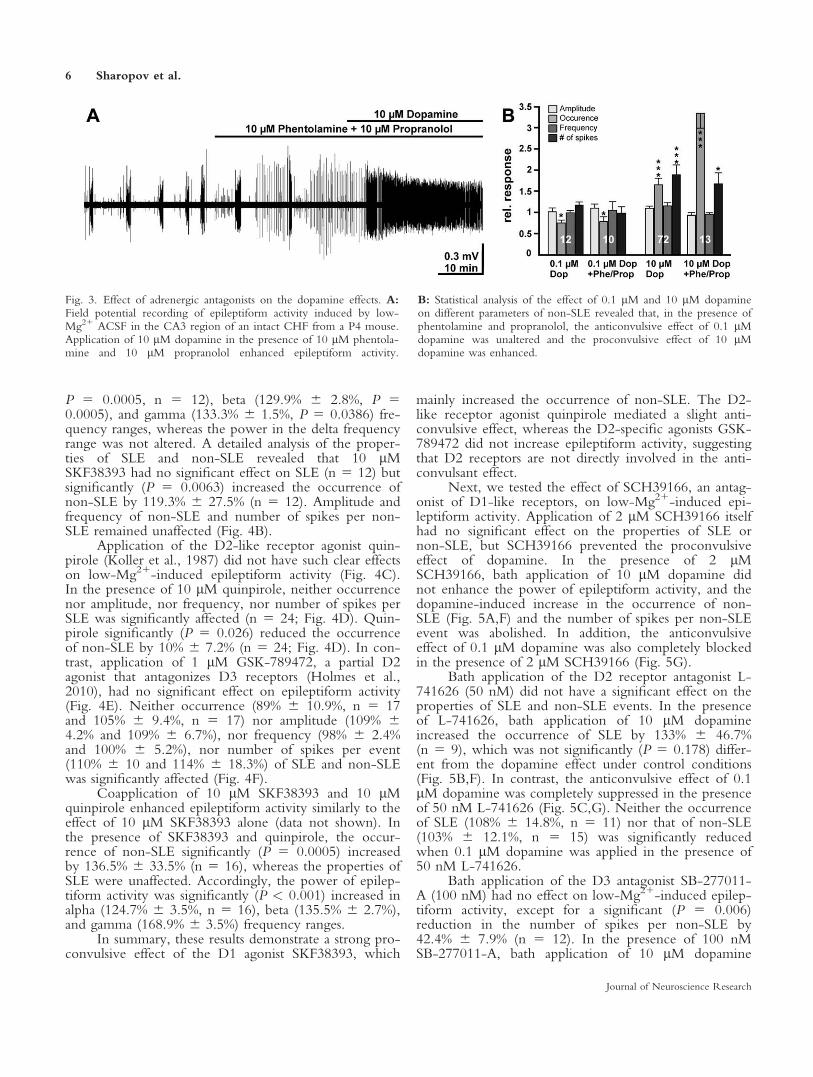
to influence epileptiform activity (Mueller and Dunwid-die, 1983; Stanton et al., 1987), so we investigatedwhether the pro- and anticonvulsive effects of high andlow dopamine concentrations were influenced by inter-actions with the adrenergic system. For this purpose, wecompletely blocked a- and b-adrenergic receptors bythe combined application of 10 lM phentolamine and10 lM propranolol, respectively. Under this condition,application of 10 lM dopamine increased the occurrenceof non-SLE by 331% 6 35.1% (n 5 13), which was sig-nificantly (P 5 0.00001) greater than in the absence ofphentolamine and propranolol (Fig. 3). The anticonvul-sant effect of low dopamine concentrations was unaf-fected in the presence of phentolamine and propranolol.Bath application of 0.1 lM dopamine under these con-ditions significantly reduced the occurrence of non-SLEby 22% 6 11.8% (n 5 10) and the occurrence of SLE
by 11% 6 7% (n 5 6). In summary, these results indi-cate that the proconvulsive effect of 10 lM dopaminewas even underestimated due to the anticonvulsive effectthat high dopamine concentrations exert via adrenergicreceptors (Mueller and Dunwiddie, 1983).
Modulation of Low-Mg21-Induced EpileptiformActivity by Dopaminergic Agonists andAntagonists
To disclose which receptor subtypes underlie thedopamine effect on epileptiform activity, we first usedsubtype-specific agonists. Application of the D1-agonistSKF38393 (Ruskin et al., 1998) enhanced epileptiformactivity (Fig. 4A). The power of low-Mg21-inducedepileptiform activity was significantly increased in thepresence of 10 lM SKF38393 in alpha (125.9% 6 3.5%,
Fig. 2. Effect of dopamine on the discharge pattern of low-Mg21-induced epileptiform activity. A: Field potential recording in theCA3 region of an intact CHF from a P4 mouse illustrating the effectof bath application of 10 lM dopamine. Typical discharge pattern ofthis recording, as marked in A, shown in higher temporal resolutionin the following. B1: One interictal event. B2: Seizure-like event(SLE). B3: One spindle-like discharge of the clonic phase of theSLE, as marked in B2. C1: SLE, recorded after application of 10 lM
dopamine. C2–4: Representative short discharge patterns induced bythe bath application of 10 lM dopamine. D: Statistical analysis ofthe effect of different dopamine concentrations on different parame-ters of SLE. E: Statistical analysis of the dopamine effect on non-SLE. Bars represent mean 6 SEM. Number of experiments is givenin the bars. Statistically significant differences are marked by *P <0.05 and ***P < 0.001.
Dopaminergic Modulation of Epileptiform Activity 5
Journal of Neuroscience Research
P 5 0.0005, n 5 12), beta (129.9% 6 2.8%, P 50.0005), and gamma (133.3% 6 1.5%, P 5 0.0386) fre-quency ranges, whereas the power in the delta frequencyrange was not altered. A detailed analysis of the proper-ties of SLE and non-SLE revealed that 10 lMSKF38393 had no significant effect on SLE (n 5 12) butsignificantly (P 5 0.0063) increased the occurrence ofnon-SLE by 119.3% 6 27.5% (n 5 12). Amplitude andfrequency of non-SLE and number of spikes per non-SLE remained unaffected (Fig. 4B).
Application of the D2-like receptor agonist quin-pirole (Koller et al., 1987) did not have such clear effectson low-Mg21-induced epileptiform activity (Fig. 4C).In the presence of 10 lM quinpirole, neither occurrencenor amplitude, nor frequency, nor number of spikes perSLE was significantly affected (n 5 24; Fig. 4D). Quin-pirole significantly (P 5 0.026) reduced the occurrenceof non-SLE by 10% 6 7.2% (n 5 24; Fig. 4D). In con-trast, application of 1 lM GSK-789472, a partial D2agonist that antagonizes D3 receptors (Holmes et al.,2010), had no significant effect on epileptiform activity(Fig. 4E). Neither occurrence (89% 6 10.9%, n 5 17and 105% 6 9.4%, n 5 17) nor amplitude (109% 64.2% and 109% 6 6.7%), nor frequency (98% 6 2.4%and 100% 6 5.2%), nor number of spikes per event(110% 6 10 and 114% 6 18.3%) of SLE and non-SLEwas significantly affected (Fig. 4F).
Coapplication of 10 lM SKF38393 and 10 lMquinpirole enhanced epileptiform activity similarly to theeffect of 10 lM SKF38393 alone (data not shown). Inthe presence of SKF38393 and quinpirole, the occur-rence of non-SLE significantly (P 5 0.0005) increasedby 136.5% 6 33.5% (n 5 16), whereas the properties ofSLE were unaffected. Accordingly, the power of epilep-tiform activity was significantly (P < 0.001) increased inalpha (124.7% 6 3.5%, n 5 16), beta (135.5% 6 2.7%),and gamma (168.9% 6 3.5%) frequency ranges.
In summary, these results demonstrate a strong pro-convulsive effect of the D1 agonist SKF38393, which
mainly increased the occurrence of non-SLE. The D2-like receptor agonist quinpirole mediated a slight anti-convulsive effect, whereas the D2-specific agonists GSK-789472 did not increase epileptiform activity, suggestingthat D2 receptors are not directly involved in the anti-convulsant effect.
Next, we tested the effect of SCH39166, an antag-onist of D1-like receptors, on low-Mg21-induced epi-leptiform activity. Application of 2 lM SCH39166 itselfhad no significant effect on the properties of SLE ornon-SLE, but SCH39166 prevented the proconvulsiveeffect of dopamine. In the presence of 2 lMSCH39166, bath application of 10 lM dopamine didnot enhance the power of epileptiform activity, and thedopamine-induced increase in the occurrence of non-SLE (Fig. 5A,F) and the number of spikes per non-SLEevent was abolished. In addition, the anticonvulsiveeffect of 0.1 lM dopamine was also completely blockedin the presence of 2 lM SCH39166 (Fig. 5G).
Bath application of the D2 receptor antagonist L-741626 (50 nM) did not have a significant effect on theproperties of SLE and non-SLE events. In the presenceof L-741626, bath application of 10 lM dopamineincreased the occurrence of SLE by 133% 6 46.7%(n 5 9), which was not significantly (P 5 0.178) differ-ent from the dopamine effect under control conditions(Fig. 5B,F). In contrast, the anticonvulsive effect of 0.1lM dopamine was completely suppressed in the presenceof 50 nM L-741626 (Fig. 5C,G). Neither the occurrenceof SLE (108% 6 14.8%, n 5 11) nor that of non-SLE(103% 6 12.1%, n 5 15) was significantly reducedwhen 0.1 lM dopamine was applied in the presence of50 nM L-741626.
Bath application of the D3 antagonist SB-277011-A (100 nM) had no effect on low-Mg21-induced epilep-tiform activity, except for a significant (P 5 0.006)reduction in the number of spikes per non-SLE by42.4% 6 7.9% (n 5 12). In the presence of 100 nMSB-277011-A, bath application of 10 lM dopamine
Fig. 3. Effect of adrenergic antagonists on the dopamine effects. A:Field potential recording of epileptiform activity induced by low-Mg21 ACSF in the CA3 region of an intact CHF from a P4 mouse.Application of 10 lM dopamine in the presence of 10 lM phentola-mine and 10 lM propranolol enhanced epileptiform activity.
B: Statistical analysis of the effect of 0.1 lM and 10 lM dopamineon different parameters of non-SLE revealed that, in the presence ofphentolamine and propranolol, the anticonvulsive effect of 0.1 lMdopamine was unaltered and the proconvulsive effect of 10 lMdopamine was enhanced.
6 Sharopov et al.
Journal of Neuroscience Research

enhanced the epileptiform activity (Fig. 5D). The proe-pileptic effect of 10 lM dopamine on the occurrenceand number of spikes per non-SLE was not significantlyaffected in the presence of 100 nM SB-277011-A (Fig.5F). In contrast, the anticonvulsive effect of 0.1 lM do-pamine was abolished in the presence of 100 nM SB-277011-A (n 5 12; Fig. 5G) These results suggest thatD3 receptors were not involved in the proconvulsiveeffect of higher dopamine concentrations but mightmediate the anticonvulsive effect of low dopamine con-centrations.
Bath application of the D2-like receptor antagonistsulpiride (10 lM) did not significantly affect the proper-ties of SLE and non-SLE, except for a slight increase inthe frequency of spikes within non-SLE by 14.7% 65.6% (n 5 22; P 5 0.004). Surprisingly, the proepilepticdopamine effect was completely abolished by 10 lM
sulpiride (Fig. 5E). In the presence of sulpiride, bathapplication of 10 lM dopamine influenced neither theaverage power of epileptiform activity nor the occur-rence and number of spikes per non-SLE (Fig. 5E,F).Sulpiride (10 lM) also suppressed the anticonvulsiveeffect of 0.1 lM dopamine (Fig. 5G).
To investigate whether the unexpected effect ofsulpiride was caused by an interaction with D1-likereceptors, we specifically activated D1-like receptorswith SKF-38393. The proconvulsive effect of SKF-38393 was virtually unaffected in the presence of 10 lMsulpiride (Fig. 6A). Under this condition, 10 lM SKF-38393 significantly (P 5 0.0078) increased the frequencyof non-SLE by 151% 6 30.4%. In contrast, 10 lMSKF-38393 had, as expected, no significant effect onepileptiform activity in the presence of the D1-likereceptor antagonist SCH39166 (2 lM, Fig. 6B). These
Fig. 4. Effect of subtype-specific dopamine receptor agonists onlow-Mg21-induced epileptiform activity. A: Field potential record-ing in the CA3 region of an intact CHF from a P3 mouse. Applica-tion of the D1 agonist SKF38393 enhanced epileptiform activity. B:Statistical analysis of SKF38393 effects on different parameters of SLEand non-SLE. Note that the properties of SLE were unaltered by 10lM SKF38393 application, whereas the occurrence of non-SLE wassignificantly increased. C: Field potential recording from a P4 mouseCHF illustrating the effect of the D2-like receptor agonist quinpir-
ole. D: Statistical analysis of the effect of 10 lM quinpirole on dif-ferent parameters of SLE and non-SLE revealed that only the occur-rence of non-SLE was significantly reduced. E: Field potentialrecording from a P3 mouse CHF illustrating the effect of the D2agonist GSK-789427. F: Statistical analysis of the effect of 1 lMGSK-789427 revealed that neither SLE nor non-SLE was affected.Bars represent mean 6 SEM. Number of experiments is given in thebars. Statistically significant differences are marked by *P < 0.05 and**P < 0.01.
Dopaminergic Modulation of Epileptiform Activity 7
Journal of Neuroscience Research

experiments illustrate that the paradoxical effect of sul-piride was most probably not mediated by an interactionwith D1-like receptors.
In summary, these results suggest that the procon-vulsive effect of high dopamine concentrations was sup-pressed by D1-like receptor antagonists, but also by sul-piride. The anticonvulsive effect of low dopamine con-centrations was suppressed by D1-like, D2, and D3receptor antagonists. These results support the hypothe-sis that D1-like receptors mediate the proconvulsiveeffect of dopamine, but the impact of the differentdopamine receptors on the anticonvulsive effect isless clear.
Identification of Synaptic Targets Mediating theDopaminergic Proconvulsive Effect
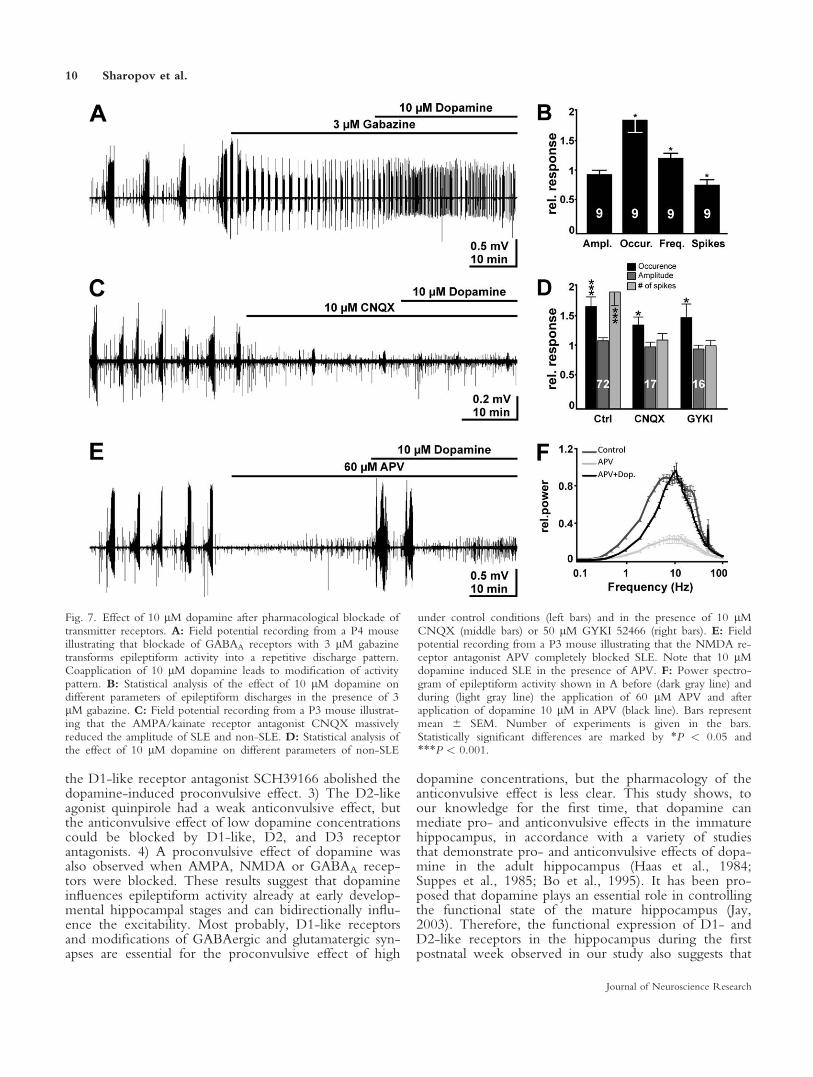
Finally, we investigated the question of whetherthe proconvulsive dopamine effect requires particularsynaptic receptors. Bath application of the GABAA an-tagonist gabazine (3 lM) to the low-Mg21 solution con-siderably changed the discharge pattern and led to an ac-tivity pattern that consisted of repetitive discharges of7.6 6 1.8 spikes (n 5 9) at a frequency of 16 6 3.5 Hz(Fig. 5A). These discharges occurred at a frequency of0.07 6 0.01 Hz (n 5 9). Under this condition, bathapplication of 10 lM dopamine changed the dischargepattern (Fig. 7A). The occurrence of epileptiform dis-
Fig. 5. Influence of dopamine receptor antagonists on the procon-vulsive and anticonvulsive effect of dopamine. A: Field potentialrecording from a P3 mouse illustrating that the D1-like receptorantagonist SCH39166 suppressed the proconvulsive effect of 10 lMdopamine. B: Field potential recording from a P4 mouse illustratingthat the D2 antagonist L-741626 had no effect on the proconvulsiveeffect of 10 lM dopamine. C: Field potential recording from a P3mouse illustrating that the D2 antagonist L-741626 suppressed theanticonvulsive effect of 0.1 lM dopamine. D: Field potential record-ing from a P4 mouse demonstrating that the D3-antagonist SB-
277011-A did not influence the proconvulsive effect of dopamine.E: Field potential recording from a P3 mouse demonstrating that theD2-like receptor antagonist sulpiride suppressed the proconvulsiveeffect of dopamine. F: Statistical analysis of the influence of dopami-nergic antagonists on the 10 lM dopamine induced proconvulsiveeffect. The key to columns is displayed in G. G: Statistical analysis ofthe influence of dopaminergic antagonists on the 0.1 lM dopamine-induced anticonvulsive effect. Bars represent mean 6 SEM. Numberof experiments is given in the bars. Statistically significant differencesare marked by *P < 0.05 and ***P < 0.001.
8 Sharopov et al.
Journal of Neuroscience Research

charges significantly (P 5 0.0391) increased from 0.076 0.01 Hz to 0.12 6 0.02 Hz (n 5 9), but the numberof spikes per discharge was significantly (P 5 0.0391)reduced from 31 6 8 to 22 6 4 (Fig. 7B). In accord-ance with these divergent effects, no significant effect of10 lM dopamine on the average power of epileptiformactivity was observed in the presence of gabazine.
Inhibition of AMPA/kainate receptors withCNQX attenuated both SLE and non-SLE. In the pres-ence of 10 lM CNQX, the occurrence and amplitudeof SLE were significantly (P < 0.001) reduced by 41%6 3.8% and 81% 6 3.1% (n 5 16), respectively (Fig.7C). CNQX reduced the occurrence and amplitude ofnon-SLE by 40% 6 6.6% and 36% 6 5.0% (n 5 17),respectively. In the continuous presence of CNQX, bathapplication of 10 lM dopamine significantly (n 50.049) increased the occurrence of non-SLE by 32% 612.5% (n 5 17). On the other hand, the other proper-ties of non-SLE such as amplitude and number of spikesper discharge were unaffected (Fig. 7D), indicating that,in the absence of functional AMPA/kainate receptors,10 lM dopamine did not mediate the typical transitionto more complex non-SLE discharges. Selective block-ade of AMPA receptors with GYKI 52466 (Paternainet al., 1995) provoked effects on epileptiform activitysimilar to those of CNQX. Bath application of 50 lMGYKI 52466 reduced the occurrence and amplitude ofSLE (by 24% 6 13% and 24% 6 6%; n 5 13) and non-SLE (by 61% 6 4.4% and 39% 6 4.4%; n 5 16). In the
continuous presence of 50 lM GYKI 52466, bath appli-cation of 10 lM dopamine induced a significant (P 50.035) increase in the occurrence of non-SLE by 44% 620.7% (n 5 16), whereas other properties of non-SLEsuch as amplitude and number of spikes per dischargewere again unaffected (Fig. 7D). Dopamine did not sig-nificantly alter the occurrence and properties of SLE inthe presence of either CNQX or GYKI 52466, exceptfor a significant (P 5 0.0034) reduction in the numberof spikes per discharge by 35% 6 6.6% (n 5 13) in thepresence of CNQX. In summary, these results demon-strate that, even in the absence of functional AMPAreceptors, dopamine can exert a proconvulsive effect.On the other hand, the typical dopamine-induced transi-tion of non-SLE to more complex discharge patternswas suppressed in the presence of both CNQX andGYKI 52466, indicating that AMPA receptor may playan important role in this proconvulsive component.
Blockade of NMDA receptors with 60 lM (6)-APV completely abolished SLE in all 10 CHF tested(Fig. 7E) and significantly (P 5 0.0215) reduced the am-plitude, frequency, and number of spikes of non-SLE,but not their occurrence. Bath application of 10 lM do-pamine in the presence of APV provoked massive epi-leptiform activity with an SLE-like appearance in all 10CHF tested (Fig. 7E). In addition, the occurrence ofnon-SLE was significantly (P 5 0.0215) increased by29% 6 7.5% (n 5 10). Accordingly, the average powerof field potential activity was significantly (all P 50.0039) increased in all frequency bands (Fig. 7F). It hasbeen shown that the prolonged application of low-Mg21
solution can lead to APV-insensitive epileptiform dis-charges (Quilichini et al., 2002), so we performed addi-tional control experiments. These experiments revealedthat, during 1 hr of APV application, a stable anticon-vulsive effect was maintained. No SLE could beobserved during this interval, and the properties of non-SLE were not significantly different between 15–30 minand 45–60 min of APV application, indicating that theproconvulsive effect of dopamine was not overestimatedby a gradual loss of APV sensitivity during this interval.In summary, these results suggest that dopamine canprovoke a proepileptic effect in the absence of functionalNMDA receptors. Overall, these experiments indicatethat dopamine mediates a reliable proconvulsive effecteven after inhibition of AMPA, NMDA, and GABAA
receptors, suggesting that dopamine enhances the excit-ability by influencing various neurotransmitter systems.
DISCUSSION
The main findings of the present study can besummarized as follows. 1) Dopamine mediated anti- andproconvulsive effects on low-Mg21-induced epileptiformactivity in the intact CHF of P3–4 mice, depending onthe applied concentration. Low concentrations of dopa-mine (<0.3 lM) attenuated and concentrations >3 lMenhanced epileptiform activity. 2) The D1-agonistSKF38393 promoted a strong proconvulsive effect, and
Fig. 6. Effect of the D2-like receptor antagonist sulpiride and theD1-like receptor antagonist SCH 39166 on the proconvulsiveSKF38393 effect. A: Field potential recording from a P3 mouse illus-trating that 10 lM sulpiride had no effect on the proconvulsive effectof 10 lM SKF38393. B: Field potential recording from a P3 mouseillustrating that 2 lM SCH39166 suppressed the proconvulsive effectof 10 lM SKF38393.
Dopaminergic Modulation of Epileptiform Activity 9
Journal of Neuroscience Research
the D1-like receptor antagonist SCH39166 abolished thedopamine-induced proconvulsive effect. 3) The D2-likeagonist quinpirole had a weak anticonvulsive effect, butthe anticonvulsive effect of low dopamine concentrationscould be blocked by D1-like, D2, and D3 receptorantagonists. 4) A proconvulsive effect of dopamine wasalso observed when AMPA, NMDA or GABAA recep-tors were blocked. These results suggest that dopamineinfluences epileptiform activity already at early develop-mental hippocampal stages and can bidirectionally influ-ence the excitability. Most probably, D1-like receptorsand modifications of GABAergic and glutamatergic syn-apses are essential for the proconvulsive effect of high
dopamine concentrations, but the pharmacology of theanticonvulsive effect is less clear. This study shows, toour knowledge for the first time, that dopamine canmediate pro- and anticonvulsive effects in the immaturehippocampus, in accordance with a variety of studiesthat demonstrate pro- and anticonvulsive effects of dopa-mine in the adult hippocampus (Haas et al., 1984;Suppes et al., 1985; Bo et al., 1995). It has been pro-posed that dopamine plays an essential role in controllingthe functional state of the mature hippocampus (Jay,2003). Therefore, the functional expression of D1- andD2-like receptors in the hippocampus during the firstpostnatal week observed in our study also suggests that
Fig. 7. Effect of 10 lM dopamine after pharmacological blockade oftransmitter receptors. A: Field potential recording from a P4 mouseillustrating that blockade of GABAA receptors with 3 lM gabazinetransforms epileptiform activity into a repetitive discharge pattern.Coapplication of 10 lM dopamine leads to modification of activitypattern. B: Statistical analysis of the effect of 10 lM dopamine ondifferent parameters of epileptiform discharges in the presence of 3lM gabazine. C: Field potential recording from a P3 mouse illustrat-ing that the AMPA/kainate receptor antagonist CNQX massivelyreduced the amplitude of SLE and non-SLE. D: Statistical analysis ofthe effect of 10 lM dopamine on different parameters of non-SLE
under control conditions (left bars) and in the presence of 10 lMCNQX (middle bars) or 50 lM GYKI 52466 (right bars). E: Fieldpotential recording from a P3 mouse illustrating that the NMDA re-ceptor antagonist APV completely blocked SLE. Note that 10 lMdopamine induced SLE in the presence of APV. F: Power spectro-gram of epileptiform activity shown in A before (dark gray line) andduring (light gray line) the application of 60 lM APV and afterapplication of dopamine 10 lM in APV (black line). Bars representmean 6 SEM. Number of experiments is given in the bars.Statistically significant differences are marked by *P < 0.05 and***P < 0.001.
10 Sharopov et al.
Journal of Neuroscience Research

dopamine most probably influences hippocampal func-tions already at early postnatal stages.
Investigations with subtype-specific dopaminergicagonists and antagonists provide compelling evidencethat activation of D1-like receptors promotes a procon-vulsive effect in many epilepsy models (Loscher andCzuczwar, 1986; Barone et al., 1990; Burke et al., 1990;Alam and Starr, 1992; O’sullivan et al., 2008; for reviewssee Starr, 1996; Weinshenker and Szot, 2002). Ourobservations that the D1 agonist SKF38393 mimickedthe proconvulsive dopamine effect and that the D1-likeantagonist SCH39166 prevented the proconvulsive effectof 10 lM dopamine strongly suggest that D1 receptorsmediate a proconvulsive effect already in the immaturehippocampus and that D1-like receptors are required forthe proconvulsive dopamine effect. The D2-like antago-nist sulpiride also attenuate the proconvulsive dopamineeffect, and D2 and D3 specific antagonists had no effect,so we could not exclude that a synergistic activation ofD4 receptors might also contribute to the proconvulsivedopamine effect. In contrast to in vivo studies (Altajiret al., 1990; Barone et al., 1990), application of the D1-like antagonist SCH39166 alone had only a negligibleeffect on the low-Mg21 induced epileptiform activity,suggesting that endogenous activation of D1-like recep-tors did not considerably affect the excitability under ourconditions. Interestingly, our study provides experimen-tal evidence that the proepileptic dopamine effect wasaugmented after blockade of adrenergic receptors. Thus,in the CA3 region, the strong proconvulsive effect ofhigher dopamine concentrations seems to be counterbal-anced by the anticonvulsive effect of dopamine mediatedvia adrenergic receptors (Mueller and Dunwiddie, 1983).In contrast, in the adult dentate gyrus, activation ofb-adrenergic receptors can even be proconvulsive(Stanton et al., 1987), which complicates an estimationof the physiological influence of adrenergic receptoractivation by dopamine.
In the adult nervous system, D2-like receptorsmediate an anticonvulsive effect (Burke et al., 1990;Alam and Starr, 1993; Bozzi et al., 2000; Clinckerset al., 2004; for reviews see Starr, 1996; Weinshenkerand Szot, 2002). In our experiments, the influence ofD2-like receptors on low-Mg21 induced epileptiformactivity is less clear. The observation that the D2-likeagonist quinpirole reduced the occurrence of non-SLEsuggests that D2-like receptors might also mediate aslight anticonvulsive effect in the immature hippocam-pus. The specific D2 agonist GSK-789472 had no anti-convulsive effect, so D3 and/or D4 receptors may bemore relevant for the anticonvulsive action. On theother hand, the D2-like antagonist sulpiride, the D2-spe-cific antagonist L-741626, and the D3-specific antagonistSB-277011-A, but also the D1-like antagonistSCH39166, completely suppressed the anticonvulsiveeffect of low dopamine concentration. From these resultswe suggest, that D3 and/or D4 receptors may mediatean anticonvulsive effect but that the anticonvulsive effectof low dopamine concentration requires the activation
of various dopamine receptors, which may reside on var-ious post- and presynaptic elements of the hippocampalcircuitry.
The application of dopamine receptor antagonistsalone had no significant effect on the epileptiform activ-ity, indicating that under our experimental conditions anintrinsic activation of dopamine receptors does not con-tribute to the control of epileptiform activity. In adultstructures with strong dopaminergic projections, such asthe striatum or the nucleus accumbens, extracellular con-centrations up to 0.5 lM had been found after stimula-tion, although the basal levels are most probably in thelow nanomolar range (Kawagoe et al., 1992; Garris andWightman, 1994; Schultz, 2007). In the hippocampus,the concentrations are probably even lower (Mokleret al., 2007) and might not be sufficient to mediate eventhe anticonvulsive effect. The observation that bathapplication of the dopamine uptake inhibitor nomifen-sine promotes a significant anticonvulsive effect suggeststhat an intrinsic release of dopamine can, however,potentially mediate an antiepileptic effect, even inreduced preparations without the ventral tegmental area,as used in our experiments. Such an anticonvulsivenomifensine effect has also been observed in the adultnervous system (Warter et al., 1988; but see Edwardsand Glenbott, 1987). During situations such as stress(Barr et al., 2009) and unpredicted reward (Schultz,2007) and during epileptic seizures (Dazzi et al., 1997),the dopamine levels increase and may even reach valueshigh enough also to stimulate D1-like receptors.
Dopamine can influence neuronal excitability bydirectly changing input resistance, action potentialthreshold, or membrane currents (see, e.g., Stanzioneet al., 1984; Gulledge and Jaffe, 1998; Chen and Yang,2007; Podda et al., 2010). D1 receptor activationincreases (Bouron, 2001; Kobayashi and Suzuki, 2007)and D2 receptor activation decreases glutamate release inthe hippocampus (Hsu, 1996). An involvement of dopa-mine receptors in transmitter release has also been dem-onstrated for GABA (Cameron and Williams, 1993;Radnikow and Misgeld, 1998). Dopamine augmentsNMDA currents via activation of D1 receptors (Flores-Hernandez et al., 2002; Gonzalez-Islas and Hablitz,2003; Tseng and O’Donnell, 2004), whereas D2 recep-tors reduce these currents (Flores-Hernandez et al.,2002; Kotecha et al., 2002). In contrast to these reportshighlighting the prominent role of NMDA receptors inmediating dopaminergic alterations of excitability, ourexperiments revealed that dopamine has a prominentproconvulsive action after inhibition of NMDA recep-tors. This result strongly suggests that other neurotrans-mitter receptors contribute to the proconvulsive effect.Dopamine can also enhance AMPA receptor functionvia D1 receptor activation (Umemiya and Raymond,1997; Gonzalez-Islas and Hablitz, 2003; O’sullivan et al.,2008). However, our experiments showed that inhibi-tion of AMPA receptors with either the AMPA/kainateantagonists CNQX or the specific AMPA receptor an-tagonist GYKI 52466 did not alter the ability of dopa-
Dopaminergic Modulation of Epileptiform Activity 11
Journal of Neuroscience Research

mine to increase the frequency of non-SLE. Instead, itprevented the dopamine-induced increase in spike num-ber per non-SLE, indicating that a dopaminergic effecton AMPA receptors may be involved in the transitionto more complex non-SLE discharges. In the presenceof the GABAA receptor antagonist gabazine, the patternof epileptiform discharges changed considerably, indicat-ing the important role of GABAergic synaptic connec-tions in controlling excitability (Farrant and Kaila, 2007).Although a direct comparison of the dopaminergic effectamong the different epileptiform patterns is not possible,our observation that dopamine enhanced epileptiformactivity in the presence of gabazine indicates that dopa-mine-induced changes in glutamate receptor functionsare sufficient to promote a proconvulsive effect. On theother hand, the proconvulsive effect of dopamine onepileptiform activity was only moderate, suggesting thata decrease in GABAergic inhibition might also substan-tially contribute to the strong proconvulsive effect of do-pamine. Overall, these experiments revealed that theeffect of dopamine on excitability is not mediated by asingle postsynaptic receptor; most probably, at least twopostsynaptic receptor systems act synergistically to medi-ate the proconvulsive effect of higher dopamine concen-trations.
In summary, our experiments demonstrate thathigher dopamine concentrations have a proconvulsanteffect in the immature hippocampus via D1-like receptoractivation, whereas lower dopamine concentrations exerta moderate anticonvulsant influence. Therefore, theoverall effect of dopamine on the excitability of theimmature hippocampus is comparable to the dopaminer-gic actions in the mature hippocampus. Although thisobservation may suggest a potential use of D1-likeantagonists in controlling seizures in children, possibleadverse developmental effects of dopaminergic agonistsand antagonists must be considered. For instance, it hasbeen shown that dopamine controls neuronal prolifera-tion (Zhang et al., 2005), migration (Crandall et al.,2007), and differentiation (Song et al., 2002) in theimmature brain. These important functions of dopaminemay complicate or exclude a pharmacological interven-tion with dopamine antagonists or agonists in the imma-ture brain.
REFERENCES
Alam AM, Starr MS. 1992. Dopaminergic modulation of pilocarpine-
induced motor seizures in the rat—the role of hippocampal dopamine-
D1 receptors. Eur J Pharmacol 222:227–232.
Alam AM, Starr MS. 1993. Dopaminergic modulation of pilocarpine-
induced motor seizures in the rat—the role of hippocampal-D2 recep-
tors. Neuroscience 53:425–431.
Altajir G, Chandler CJ, Starr BS, Starr MS. 1990. Opposite effects of
stimulation of D1 and D2 dopamine-receptors on the expression of
motor seizures in mouse and rat. Neuropharmacology 29:657–661.
Barone P, Parashos SA, Palma V, Marin C, Campanella G, Chase TN.
1990. Dopamine-D1 receptor modulation of pilocarpine-induced con-
vulsions. Neuroscience 34:209–217.
Barr GA, Moriceau S, Shionoya K, Muzny K, Gao PH, Wang SN, Sulli-
van RM. 2009. Transitions in infant learning are modulated by dopa-
mine in the amygdala. Nat Neurosci 12:1367–1369.
Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa J-L. 1989. Giant synaptic
potentials in immature rat CA3 hippocampal neurones. J Physiol
416:303–325.
Ben Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. 2007. GABA: a pioneer
transmitter that excites immature neurons and generates primitive oscil-
lations. Physiol Rev 87:1215–1284.
Bo P, Soragna D, Marchioni E, Candeloro E, Albergati A, Savoldi F.
1995. Role of dopamine D-1 and D-2 antagonists in a model of focal
epilepsy induced by electrical-stimulation of hippocampus and amygdala
in the rabbit. Prog Neuropsychopharmacol Biol Psychiatry 19:917–930.
Booth D, Evans DJ. 2004. Anticonvulsants for neonates with seizures.
Cochrane Database Syst Rev CD004218.
Bouron A. 2001. Modulation of spontaneous quantal release of neuro-
transmitters in the hippocampus. Prog Neurobiol 63:613–635.
Bozzi Y, Vallone D, Borrelli E. 2000. Neuroprotective role of dopamine
against hippocampal cell death. J Neurosci 20:8643–8649.
Burke K, Chandler CJ, Starr BS, Starr MS. 1990. Seizure promotion and
protection by D-1 and D-2 dopaminergic drugs in the mouse. Pharma-
col Biochem Behav 36:729–733.
Cameron DL, Williams JT. 1993. Dopamine D1 receptors facilitate trans-
mitter release. Nature 366:344–347.
Chen L, Yang XL. 2007. Hyperpolarization-activated cation current is
involved in modulation of the excitability of rat retinal ganglion cells
by dopamine. Neuroscience 150:299–308.
Ciumas C, Wahlin TBR, Jucaite A, Lindstrom P, Halldin C, Savic I.
2008. Reduced dopamine transporter binding in patients with juvenile
myoclonic epilepsy. Neurology 71:788–794.
Clemens B. 1988. Dopamine agonist treatment of self-induced pattern-
sensitive epilepsy—a case-report. Epilepsy Res 2:340–343.
Clinckers R, Smolders I, Meurs A, Ebinger G, Michotte Y. 2004. Anticon-
vulsant action of hippocampal dopamine and serotonin is independently
mediated by D-2 and 5-HT1A receptors. J Neurochem 89:834–843.
Cowan LD. 2002. The epidemiology of the epilepsies in children. Ment
Retard Dev Disabil Res Rev 8:171–181.
Crandall JE, McCarthy DM, Araki KY, Sims JR, Ren JQ, Bhide PG.
2007. Dopamine receptor activation modulates GABA neuron migra-
tion from the basal forebrain to the cerebral cortex. J Neurosci
27:3813–3822.
Dazzi L, Serra M, Porceddu ML, Sanna A, Chessa MF, Biggio G. 1997.
Enhancement of basal and pentylenetetrazol (PTZ)-stimulated dopa-
mine release in the brain of freely moving rats by PTZ-induced kin-
dling. Synapse 26:351–358.
Edwards JG, Glenbott M. 1987. Nomifensine and convulsive seizures.
Hum Toxicol 6:247–249.
Farrant M, Kaila K. 2007. The cellular, molecular and ionic basis of
GABAA receptor signalling. Prog Brain Res 160:59–87.
Fedi M, Berkovic SF, Scheffer IE, O’Keefe G, Marini C, Mulligan R,
Gong S, Tochon-Danguy H, Reutens DC. 2008. Reduced striatal D-1
receptor binding in autosomal dominant nocturnal frontal lobe epilepsy.
Neurology 71:795–798.
Flores-Hernandez J, Cepeda C, Hernandez-Echeagaray E, Calvert CR,
Jokel ES, Fienberg AA, Greengard P, Levine MS. 2002. Dopamine
enhancement of NMDA currents in dissociated medium-sized striatal
neurons: role of D1 receptors and DARPP-32. J Neurophysiol
88:3010–3020.
Fukuda A, Mody I, Prince DA. 1993. Differential ontogenesis of presyn-
aptic and postsynaptic GABAB inhibition in rat somatosensory cortex. J
Neurophysiol 70:448–452.
Gaiarsa J-L, Tseeb V, Ben-Ari Y. 1995. Postnatal development of pre-
and postsynaptic GABAB-mediated inhibitions in the CA3 hippocampal
region of the rat. J Neurophysiol 73:246–255.
12 Sharopov et al.
Journal of Neuroscience Research

Garris PA, Wightman RM. 1994. Different kinetics govern dopaminergic
transmission in the amygdala, prefrontal cortex, and striatum—an in-
vivo voltammetric study. J Neurosci 14:442–450.
Gattereau A, Vezina J, Rousseau S, Bielmann P. 1990. Hyperprolactine-
mia and temporal-lobe epilepsy in a woman—concomitant and persis-
tent prolactin suppression and temporal-lobe epilepsy relief. J Endocri-
nol Invest 13:247–249.
Glauser TA. 1995. Pediatric epilepsy syndromes. Curr Opin Pediatr
7:640–649.
Gonzalez-Islas C, Hablitz JJ. 2003. Dopamine enhances EPSCs in layer
II–III pyramidal neurons in rat prefrontal cortex. J Neurosci 23:
867–875.
Gulledge AT, Jaffe DB. 1998. Dopamine decreases the excitability of
layer V pyramidal cells in the rat prefrontal cortex. J Neurosci 18:
9139–9151.
Gurevich EV, Kordower JH, Joyce JN. 2000. Ontogeny of the dopamine
D2 receptor mRNA expressing cells in the human hippocampal forma-
tion and temporal neocortex. J Chem Neuroanat 20:307–325.
Haas HL, Jefferys JGR, Slater NT, Carpenter DO. 1984. Modulation of
low calcium induced field bursts in the hippocampus by monoamines
and cholinomimetics. Pflugers Arch 400:28–33.
Holmes GL. 1997. Epilepsy in the developing brain: lessons from the lab-
oratory and clinic. Epilepsia 38:12–30.
Holmes IP, Blunt RJ, Lorthioir OE, Blowers SM, Gribble A, Payne AH,
Stansfield IG, Wood M, Woollard PM, Reavill C, Howes CM,
Micheli F, Di Fabio R, Donati D, Terreni S, Hamprecht D, Arista L,
Worby A, Watson SP. 2010. The identification of a selective dopamine
D2 partial agonist, D3 antagonist displaying high levels of brain expo-
sure. Bioorg Med Chem Lett 20:2013–2016.
Hsu KS. 1996. Characterization of dopamine receptors mediating inhibi-
tion of excitatory synaptic transmission in the rat hippocampal slice. J
Neurophysiol 76:1887–1895.
Jay TM. 2003. Dopamine: a potential substrate for synaptic plasticity and
memory mechanisms. Prog Neurobiol 69:375–390.
Jensen FE. 1999. Acute and chronic effects of seizures in the developing
brain: experimental models. Epilepsia 40:S51–S58.
Kawagoe KT, Garris PA, Wiedemann DJ, Wightman RM. 1992. Regu-
lation of transient dopamine concentration gradients in the microenvir-
onment surrounding nerve-terminals in the rat striatum. Neuroscience
51:55–64.
Khalilov I, Holmes GL, Ben-Ari Y. 2003. In vitro formation of a sec-
ondary epileptogenic mirror focus by interhippocampal propagation of
seizures. Nat Neurosci 6:1079–1085.
Kobayashi K, Suzuki H. 2007. Dopamine selectively potentiates hippo-
campal mossy fiber to CA3 synaptic transmission. Neuropharmacology
52:552–561.
Koller W, Herbster G, Anderson D, Wack R, Gordon J. 1987. Quinpir-
ole hydrochloride, a potential anti-parkinsonism drug. Neuropharma-
cology 26:1031–1036.
Kotecha SA, Oak JN, Jackson MF, Perez Y, Orser BA, Van Tol HH,
MacDonald JF. 2002. A D2 class dopamine receptor transactivates a re-
ceptor tyrosine kinase to inhibit NMDA receptor transmission. Neuron
35:1111–1122.
Landvogt C, Buchholz HG, Bernedo V, Schreckenberger M, Werhahn
KJ. 2010. Alteration of dopamine D2/D3 receptor binding in patients
with juvenile myoclonic epilepsy. Epilepsia 51:1699–1706.
Lang U, Frotscher M. 1990. Postnatal development of nonpyramidal
neurons in the rat hippocampus (areas CA1 and CA3): a combined
Golgi/electron microscope study. Anat Embryol 181:533–545.
Lipka LJ, Lathers CM. 1987. Psychoactive agents, seizure production,
and sudden-death in epilepsy. J Clin Pharmacol 27:169–183.
Loscher W, Czuczwar SJ. 1986. Studies on the involvement of dopamine
D-1 and D-2 receptors in the anticonvulsant effect of dopamine ago-
nists in various rodent models of epilepsy. Eur J Pharmacol 128:55–65.
Luhmann HJ, Prince DA. 1991. Postnatal maturation of the GABAergic
system in rat neocortex. J Neurophysiol 65:247–263.
Malenka RC, Nicoll RA. 1986. Dopamine decreases the calcium-acti-
vated afterhyperpolarization in hippocampal CA1 pyramidal cells. Brain
Res 379:210–215.
Meng SZ, Ozawa Y, Itoh M, Takashima S. 1999. Developmental and
age-related changes of dopamine transporter, and dopamine D1 and D2
receptors in human basal ganglia. Brain Res 843:136–144.
Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. 1998. Dopa-
mine receptors: from structure to function. Physiol Rev 78:189–225.
Mody I, Lambert JDC, Heinemann U. 1987. Low extracellular magne-
sium induces eplileptiform activity and spreading depression in rat hip-
pocampal slices. J. Neurophysiol 57:869–888.
Mokler DJ, Torres OI, Galler JR, Morgane PJ. 2007. Stress-induced
changes in extracellular dopamine and serotonin in the medial prefron-
tal cortex and dorsal hippocampus of prenatally malnourished rats. Brain
Res 1148:226–233.
Moser J, Kilb W, Werhahn KJ, Luhmann HJ. 2006. Early developmental
alterations of low-Mg21-induced epileptiform activity in the intact cor-
ticohippocampal formation of the newborn mouse in vitro. Brain Res
1077:170–177.
Mueller AL, Dunwiddie TV. 1983. Anti-convulsant and proconvulsant
actions of alpha-noradrenergic and beta-noradrenergic agonists on epi-
leptiform activity in rat hippocampus in-vitro. Epilepsia 24:57–64.
O’sullivan GJ, Dunleavy M, Hakansson K, Clementi M, Kinsella A,
Croke DT, Drago J, Fienberg AA, Greengard P, Sibley DR, Fisone G,
Henshall DC, Waddington JL. 2008. Dopamine D1 vs D5 receptor-de-
pendent induction of seizures in relation to DARPP-32, ERK1/2 and
GluR1-AMPA signalling. Neuropharmacology 54:1051–1061.
Paternain AV, Morales M, Lerma J. 1995. Selective antagonism of
AMPA receptors unmasks kainate receptor- mediated responses in hip-
pocampal neurons. Neuron 14:185–189.
Podda MV, Riccardi E, D’Ascenzo M, Azzena GB, Grassi C. 2010. Do-
pamine D1-like receptor activation depolarizes medium spiny neurons
of the mouse nucleus accumbens by inhibiting inwardly rectifying K1
currents through a cAMP-dependent protein kinase A-independent
mechanism. Neuroscience 167:678–690.
Quilichini PP, Diabira D, Chiron C, Ben-Ari Y, Gozlan H. 2002. Persis-
tent epileptiform activity induced by low Mg21 in intact immature
brain structures. Eur J Neurosci 16:850–860.
Quilichini PP, Diabira D, Chiron C, Milh M, Ben-Ari Y, Gozlan H.
2003. Effects of antiepileptic drugs on refractory seizures in the intact
immature corticohippocampal formation in vitro. Epilepsia 44:
1365–1374.
Radnikow G, Misgeld U. 1998. Dopamine D1 receptors facilitate
GABAA synaptic currents in the rat substantia nigra pars reticulata. J
Neurosci 18:2009–2016.
Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pir-
vola U, Saarma M, Kaila K. 1999. The K1/Cl2 co-transporter KCC2
renders GABA hyperpolarizing during neuronal maturation. Nature
397:251–255.
Ruskin DN, Rawji SS, Walters JR. 1998. Effects of full D-1 dopamine
receptor agonists on firing rates in the globus pallidus and substantia
nigra pars compacta in vivo: tests for D-1 receptor selectivity and com-
parisons to the partial agonist SKF 38393. J Pharmacol Exp Ther
286:272–281.
Sanchez RM, Jensen FE. 2001. Maturational aspects of epilepsy mecha-
nisms and consequences for the immature brain. Epilepsia 42:577–585.
Sankar R, Painter MJ. 2005. Neonatal seizures: after all these years we
still love what doesn’t work. Neurology 64:776–777.
Schultz W. 2007. Multiple dopamine functions at different time courses.
Annu Rev Neurosci 30:259–288.
Silverstein FS, Jensen FE. 2007. Neonatal seizures. Ann Neurol 62:
112–120.
Dopaminergic Modulation of Epileptiform Activity 13
Journal of Neuroscience Research

Song ZM, Undie AS, Koh PO, Fang YY, Zhang L, Dracheva S, Sealfon
SC, Lidow MS. 2002. D1 dopamine receptor regulation of microtu-
bule-associated protein-2 phosphorylation in developing cerebral corti-
cal neurons. J Neurosci 22:6092–6105.
Stanton PK, Jones RSG, Mody I, Heinemann U 1987. Epileptiform ac-
tivity induced by lowering extracellular [Mg21] in combined hippo-
campal-entorhinal cortex slices: modulation by receptors for norepi-
nephrine and N-methyl-D-aspartate. Epilepsy Res 1:53–62.
Stanzione P, Calabresi P, Mercuri N, Bernardi G. 1984. Dopamine mod-
ulates Ca1 hippocampal-neurons by elevating the threshold for spike
generation—an invitro study. Neuroscience 13:1105–1116.
Starr MS. 1996. The role of dopamine in epilepsy. Synapse 22:159–194.
Suppes T, Kriegstein AR, Prince DA. 1985. The influence of dopamine
on epileptiform burst activity in hippocampal pyramidal neurons. Brain
Res 326:273–280.
Swann JW, Hablitz JJ. 2000. Cellular abnormalities and synaptic plasticity
in seizure disorders of the immature nervous system. Ment Retard Dev
Disabil Res Rev 6:258–267.
Tseng KY, O’Donnell P. 2004. Dopamine-glutamate interactions con-
trolling prefrontal cortical pyramidal cell excitability involve multiple
signaling mechanisms. J Neurosci 24:5131–5139.
Umemiya M, Raymond LA. 1997. Dopaminergic modulation of excita-
tory postsynaptic currents in rat neostriatal neurons. J Neurophysiol
78:1248–1255.
Unis AS, Roberson MD, Robinette R, Ha J, Dorsa DM. 1998. Ontog-
eny of human brain dopamine receptors—I. Differential expression of
[H-3]-SCH23390 and [H-3]-YM09151–2 specific binding. Brain Res
Dev Brain Res 106:109–117.
Warter JM, Vergnes M, Depaulis A, Tranchant C, Rumbach L, Michel-
etti G, Marescaux C. 1988. Effects of drugs affecting dopaminergic
neurotransmission in rats with spontaneous petit-mal-like seizures. Neu-
ropharmacology 27:269–274.
Weinshenker D, Szot P. 2002. The role of catecholamines in seizure sus-
ceptibility: new results using genetically engineered mice. Pharmacol
Ther 94:213–233.
Werhahn KJ, Landvogt C, Klimpe S, Buchholz HG, Yakushev I, Sies-
smeier T, Muller-Forell W, Piel M, Rosch F, Glaser M, Schrecken-
berger M, Bartenstein P. 2006. Decreased dopamine D2/D3-receptor
binding in temporal lobe epilepsy: an [18F]fallypride PET study. Epilep-
sia 47:1392–1396.
Yakushev IY, Dupont E, Buchholz HG, Tillmanns J, Debus F, Cum-
ming P, Heimann A, Fellgiebel A, Luhmann HJ, Landvogt C, Wer-
hahn KJ, Schreckenberger M, Potschka H, Bartenstein P. 2010. In vivo
imaging of dopamine receptors in a model of temporal lobe epilepsy.
Epilepsia 51:415–422.
Zecevic N, Verney C. 1995. Development of the catecholamine neurons
in human embryos and fetuses, with special emphasis on the innerva-
tion of the cerebral cortex. J Comp Neurol 351:509–535.
Zhang L, Bai J, Undie AS, Bergson C, Lidow MS. 2005. D1 dopamine
receptor regulation of the levels of the cell-cycle-controlling proteins,
cyclin D, P27 and Raf-1, in cerebral cortical precursor cells is mediated
through cAMP-independent pathways. Cereb Cortex 15:74–84.
14 Sharopov et al.
Journal of Neuroscience Research