wip1 phosphatase modulates atm-dependent signaling pathways
TRANSCRIPT

Molecular Cell 23, 757–764, September 1, 2006 ª2006 Elsevier Inc. DOI 10.1016/j.molcel.2006.07.010
Short ArticleWip1 Phosphatase ModulatesATM-Dependent Signaling Pathways
Sathyavageeswaran Shreeram,1 Oleg N. Demidov,1
Weng Kee Hee,1 Hiroshi Yamaguchi,2
Nobuyuki Onishi,3 Calvina Kek,1 Oleg N. Timofeev,1
Crissy Dudgeon,2 Albert J. Fornace,4
Carl W. Anderson,5 Yasuhiro Minami,3
Ettore Appella,2 and Dmitry V. Bulavin1,*1 Institute of Molecular and Cell Biology61 Bioplis DriveProteos138673Singapore2Laboratory of Cell BiologyCenter for Cancer ResearchNational Cancer InstituteNational Institutes of HealthBethesda, Maryland 208923Department of Genome SciencesFaculty of Medical SciencesGraduate School of MedicineKobe University7-5-1, Kusunoki-cho, Chuo-kuKobe 650-0017Japan4Department of Genetics and Complex DiseasesHarvard School Of Public HealthBoston, Massachusetts 021155Biology DepartmentBrookhaven National LaboratoryUpton, New York 11973
Summary
Deletion of Ppm1d, the gene encoding the Wip1 phos-phatase, renders cells resistant to transformation and
mice resistant to tumor development. Here, we reportthat deficiency of Wip1 resulted in activation of the
ataxia-telangiectasia mutated (ATM) kinase. In turn,overexpression of Wip1 was sufficient to reduce acti-
vation of the ATM-dependent signaling cascade afterDNA damage. Wip1 dephosphorylated ATM Ser1981,
a site critical for ATM monomerization and activation,and was critical for resetting ATM phosphorylation as
cells repaired damaged DNA. We propose that theWip1 phosphatase is an integral component of an
ATM-dependent signaling pathway.
Introduction
Activation of the PI3-like kinases ATM and ATR initiatessignaling that inhibits cell cycle progression after DNAdamage (Kastan and Bartek, 2004). Although the ATMkinase is a master regulator of cell cycle checkpoints af-ter DNA damage, including DNA double-strand breaksthat are caused by ionizing radiation (IR), recent dataargue that ATM responds ubiquitously to several typesof stress (Kastan and Bartek, 2004; Bakkenist and
*Correspondence: [email protected]
Kastan, 2003). The ATM kinase becomes engaged tohelp cells deal with cellular stresses that affect DNAand chromatin structure through activation of numeroussignaling pathways controlling the G1/S, intra-S, andG2/M transitions, as well as apoptosis. The guardianrole of ATM in blocking cell proliferation and initiatingapoptosis, especially in the presence of oncogenes,may be critical for preventing tumorigenesis, as ATM-deficient mice are prone to an early onset of lymphomas(Barlow et al., 1996; Xu et al., 1996); in addition, germ-line mutations in the ATM gene cause ataxia-telangiec-tasia (A-T), a multisystem disorder that is associatedwith a predisposition to lymphoma and acute leukemia(Shiloh and Kastan, 2001; Gumy-Pause et al., 2004).
The protein phosphatase Wip1 was identified as theproduct of a gene whose expression is induced in re-sponse to IR and ultraviolet (UV) light in a p53-depen-dent manner (Fiscella et al., 1997). The first specific tar-get of the Wip1 phosphatase to be identified was the p38mitogen-activated protein kinase (MAPK) that is criticalfor mediating the activation of p53 following exposureof cells to UV light (Takekawa et al., 2000). This functionof Wip1 phosphatase resembles that of the MDM2 pro-tein, and, similar to MDM2’s established role as an onco-gene (Oliner et al., 1993), accumulating evidence alsohas implicated Wip1 as an oncogene (Bulavin et al.,2002; Li et al., 2002). Wip1 is encoded by the PPM1D(protein phosphatase magnesium-dependent 1 delta)gene, which was mapped to chromosome 17q22/q24,a hotspot for gene amplifications in multiple primary hu-man cancers. The regional gain on chromosome 17q22/q24 was initially observed using a comparative genomichybridization (CGH) technique that detected amplifica-tion in 18% of primary breast tumors (Kallioniemi et al.,1994). In addition to breast tumors, gain and amplifica-tion of the 17q22/q24 region also have been detectedin tumors of the lung (Ried et al., 1994), pancreas (Soli-nas-Toldo et al., 1996), bladder (Voorter et al., 1995),and liver (Wong et al., 1999). Gain and amplificationalso were detected in neuroblastomas (Plantaz et al.,1997) and meningiomas (Khan et al., 1998). A combina-tion of genetic and molecular studies established theoncogenic function of Wip1 (Bulavin et al., 2002; Liet al., 2002). Recently, we found that Wip1 null miceare resistant to tumorigenesis in several mouse models(Bulavin et al., 2004). Disruption of the murine Wip1phosphatase gene (Ppm1d) activated the p53 andInk4a/ARF pathways and suppressed transformationof mouse embryo fibroblasts (MEFs) by oncogenes, asmeasured by the ability of transformed MEFs to formtumors when explanted into nude mice. In vivo, deletionof Wip1 in mice bearing MMTV-driven c-Neu or cHa-ras oncogenes impaired mammary carcinogenesis,whereasreduced expression of p16Ink4a and p19Arf viamethylation-induced silencing or inactivation of p38MAPK correlated with tumor appearance (Bulavin et al.,2004). Thus, our recent results indicated that either in-activation or depletion of the Wip1 phosphatase couldsuppress tumorigenesis (Bulavin et al., 2004; Belovaet al., 2005).

Molecular Cell758
In this work, we identify Ser1981 of the human ataxia-telangiectasia mutated (ATM) kinase as a substrate,both in vivo and in vitro, of the Wip1 phosphatase. Wefurther showed that, while depletion of Wip1 results inactivation of ATM, overexpression attenuates the ATM-dependent signaling pathways. We propose that theWip1 phosphatase is an essential component of an ATM-dependent signaling pathway that is crititical for regulat-ing the ATM-mediated tumor surveillance network.
Results
Activation of the ATM-p53-Dependent SignalingPathway in Wip1-Deficient Cells
We previously showed that deficiency of the Wip1 phos-phatase resulted in resistance to tumor formation, asWip1 null MEFs expressing different oncogenes didnot form tumors after injection into nude mice. Further-more, Wip1-deficient mice displayed a p38 MAPK- andCdkn2a-dependent delay in the onset of mammarygland tumors driven by the cHa-ras and c-Neu onco-genes (Bulavin et al., 2004). To identify physiologicaltargets for the Wip1 phosphatase, in addition to p38MAPK, that may play a role in regulating tumor resis-tance, Wip1-deficient MEFs were challenged with eitherIR or exposure to UV light at 254 nm (UVC). Severalsignaling cascades then were examined for activationusing phosphospecific antibodies to appropriate pro-teins within specific pathways. Our rationale was that,as a stress-induced phosphatase, Wip1 could be criticalfor dephosphorylating molecules that are involved inregulating stress-responsive signaling pathways, theactivation of which ultimately may contribute to sup-pression of tumorigenesis in Wip1 null mice (Bulavinet al., 2004). We found that phosphorylation of theATM kinase at the key regulatory site, Ser1987 (humanSer1981, Bakkenist and Kastan [2003]), was increasedin E1A+Ras-expressing Wip1 null MEFs comparedwith wt MEFs under nonstress conditions, as well asafter IR (Figure 1A). Phosphorylation of the ATM sub-strate, p53, at Ser18 (human Ser15) also was increasedin Wip1-deficient cells under nonstress conditions, andexposure of cells to 5–20 Gy of IR induced robust phos-phorylation of p53 both in wt and, to a slightly higherdegree, in Wip1 null MEFs (Figure 1A). We did not ob-serve a substantial difference in the phosphorylation ofH2AX (Figure 1A) in wt versus Wip1 null MEFs, suggest-ing that the increased activation of ATM in cells was nota result of persistent DNA damage. Further, no apparentdifference was observed for other ATM substrates,including Rad17, NBS1 (Figure 1A), or Chk1 (see Fig-ure S1 in the Supplemental Data available with this arti-cle online), supporting the idea that deficiency of theWip1 phosphatase contributes primarily to ATM/p53-dependent signaling. Importantly, inclusion of the p38MAPK chemical inhibitor SB202190 had no apparenteffect on ATM phosphorylation in Wip1-deficient cells,suggesting that activation of ATM in these cells wasnot due to higher levels of p38 MAPK activity (data notshown).
We next analyzed ATM phosphorylation at Ser1987 inwt and Wip1 null MEFs at early time points (Figure 1B,left panel). If the Wip1 involvement in ATM phosphoryla-tion was indirect, then a difference between these cells
should be observed only at later time points but notimmediately after IR. The data in Figure 1B show, how-ever, that phosphorylation of ATM Ser1987 was high inWip1-deficient MEFs compared to wt MEFs as early as15 min after IR. However, ATM became substantially de-phosphorylated on Ser1987 in both wt and Wip1-defi-cient cells by 8 hr (Figure 1B, right panel). The replace-ment of phosphorylated ATM with newly synthesizedATM that does not become autophosphorylated lateafter IR exposure may explain these results (ATM hasa relatively short [w6 hr] half-life in IR-treated wt andWip1-deficient E1A+Ras MEFs; Figure S2).
To understand whether Wip1 attenuates ATM activity,we carried out in vitro kinase reactions with ATM immu-noprecipitated from either wt or Wip1-deficient cells. Asshown in Figure 1C, the level of ATM activity was in-creased in Wip1-deficient cells under both normal con-ditions and after IR (quantitative analysis is shown inthe right panel of Figure 1C). Importantly, in our ATMprecipitates, no phosphatase activity toward p53 Ser15was observed (phosphorylation of GST-p53 was accom-plished with IR-treated nuclear extracts), suggestingthat the difference in ATM activity between wt andWip1-deficient cells was not due to coprecipitation ofWip1 or any other phosphatase (data not shown). Thus,deficiency of Wip1 increased both ATM phosphoryla-tion at Ser1987 and the kinase activity of ATM (Figures1A–1C).
As the effects seen in Wip1-deficient cells could beindirect and could reflect a role of Wip1 in a feedbackcontrol of the DNA damage response through p53 (Take-kawa et al., 2000; Bulavin et al., 2004), but not reflect theprimary regulatory function toward ATM, we establishedMEFs from p532/2 and double p53/Wip12/2 mice. Early-passage MEFs then were infected with a cHa-Ras-ex-pressing retrovirus to eliminate the difference in prolifer-ation rates (Bulavin et al., 2004), treated with differentdoses of IR, and analyzed for ATM phosphorylation (Fig-ure 1D). Similar to Wip12/2 MEFs, removal of p53 did notaffect the extent of ATM phosphorylation in p53/Wip12/2
MEFs. This suggests that increased phosphorylation ofATM in Wip1-deficient cells is not regulated through afeedback mechanism involving p53.
Wip1 Phosphatase Resets ATM Phosphorylation
after DNA DamageNext we asked whether ATM is a target for the Wip1phosphatase in vivo. We infected wt E1A+Ras MEFswith either PINCO-neo or PINCO-neo-Wip1-expressingretroviruses. G418-resistant wt E1A+Ras MEFs weresubsequently irradiated with different doses of IR andthen analyzed for ATM and p53 phosphorylation. Asshown in Figure 1E, overexpression of Wip1 reducedthe levels of phosphorylation of ATM and p53 at theATM site (mouse Ser18) after IR.
A recent study suggested that Wip1 is involved innegatively regulating Chk1 activity, but not ATM activity,in human cells (Lu et al., 2005). As we observed a clearsuppression of ATM phosphorylation in mouse cellsupon overexpression of Wip1, we next extended ouranalysis to human Saos-2 cells expressing tet-inducibleWip1 phosphatase. The fact that Wip1 is commonlyoverexpressed in human cancers provides a rationalefor the use of our inducible system as physiologically
Wip1 Regulates ATM-Dependent Signaling Pathways759
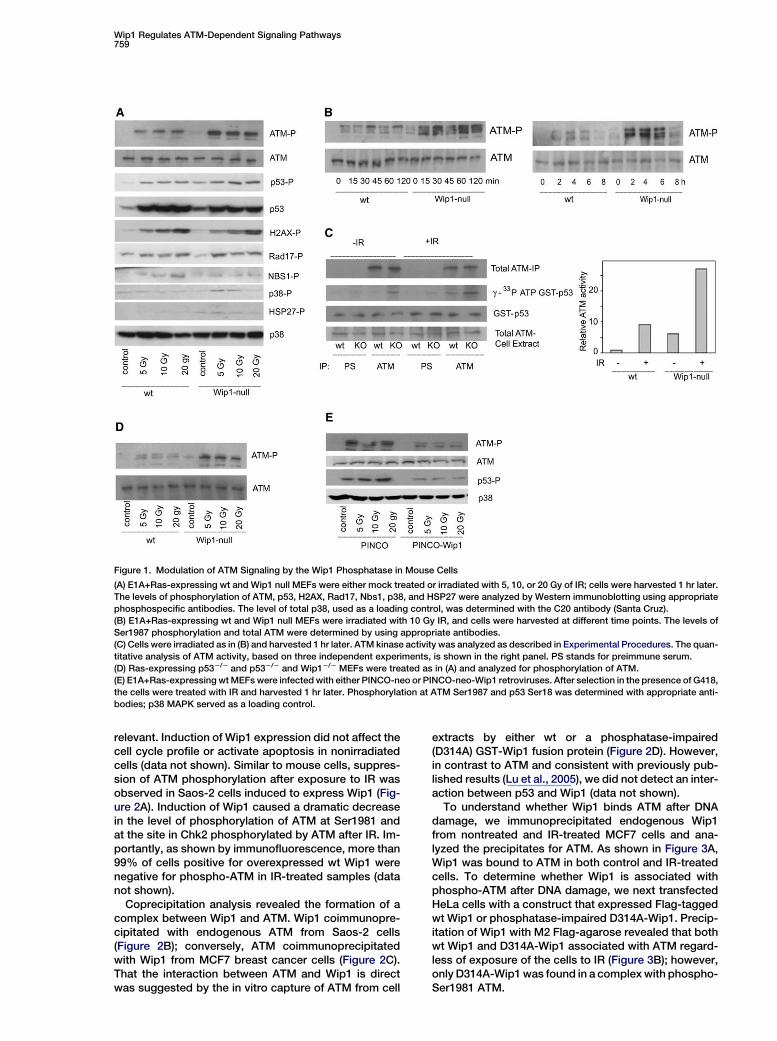
Figure 1. Modulation of ATM Signaling by the Wip1 Phosphatase in Mouse Cells
(A) E1A+Ras-expressing wt and Wip1 null MEFs were either mock treated or irradiated with 5, 10, or 20 Gy of IR; cells were harvested 1 hr later.
The levels of phosphorylation of ATM, p53, H2AX, Rad17, Nbs1, p38, and HSP27 were analyzed by Western immunoblotting using appropriate
phosphospecific antibodies. The level of total p38, used as a loading control, was determined with the C20 antibody (Santa Cruz).
(B) E1A+Ras-expressing wt and Wip1 null MEFs were irradiated with 10 Gy IR, and cells were harvested at different time points. The levels of
Ser1987 phosphorylation and total ATM were determined by using appropriate antibodies.
(C) Cells were irradiated as in (B) and harvested 1 hr later. ATM kinase activity was analyzed as described in Experimental Procedures. The quan-
titative analysis of ATM activity, based on three independent experiments, is shown in the right panel. PS stands for preimmune serum.
(D) Ras-expressing p532/2 and p532/2 and Wip12/2 MEFs were treated as in (A) and analyzed for phosphorylation of ATM.
(E) E1A+Ras-expressing wt MEFs were infected with either PINCO-neo or PINCO-neo-Wip1 retroviruses. After selection in the presence of G418,
the cells were treated with IR and harvested 1 hr later. Phosphorylation at ATM Ser1987 and p53 Ser18 was determined with appropriate anti-
bodies; p38 MAPK served as a loading control.
relevant. Induction of Wip1 expression did not affect thecell cycle profile or activate apoptosis in nonirradiatedcells (data not shown). Similar to mouse cells, suppres-sion of ATM phosphorylation after exposure to IR wasobserved in Saos-2 cells induced to express Wip1 (Fig-ure 2A). Induction of Wip1 caused a dramatic decreasein the level of phosphorylation of ATM at Ser1981 andat the site in Chk2 phosphorylated by ATM after IR. Im-portantly, as shown by immunofluorescence, more than99% of cells positive for overexpressed wt Wip1 werenegative for phospho-ATM in IR-treated samples (datanot shown).
Coprecipitation analysis revealed the formation of acomplex between Wip1 and ATM. Wip1 coimmunopre-cipitated with endogenous ATM from Saos-2 cells(Figure 2B); conversely, ATM coimmunoprecipitatedwith Wip1 from MCF7 breast cancer cells (Figure 2C).That the interaction between ATM and Wip1 is directwas suggested by the in vitro capture of ATM from cell
extracts by either wt or a phosphatase-impaired(D314A) GST-Wip1 fusion protein (Figure 2D). However,in contrast to ATM and consistent with previously pub-lished results (Lu et al., 2005), we did not detect an inter-action between p53 and Wip1 (data not shown).
To understand whether Wip1 binds ATM after DNAdamage, we immunoprecipitated endogenous Wip1from nontreated and IR-treated MCF7 cells and ana-lyzed the precipitates for ATM. As shown in Figure 3A,Wip1 was bound to ATM in both control and IR-treatedcells. To determine whether Wip1 is associated withphospho-ATM after DNA damage, we next transfectedHeLa cells with a construct that expressed Flag-taggedwt Wip1 or phosphatase-impaired D314A-Wip1. Precip-itation of Wip1 with M2 Flag-agarose revealed that bothwt Wip1 and D314A-Wip1 associated with ATM regard-less of exposure of the cells to IR (Figure 3B); however,only D314A-Wip1 was found in a complex with phospho-Ser1981 ATM.
Molecular Cell760
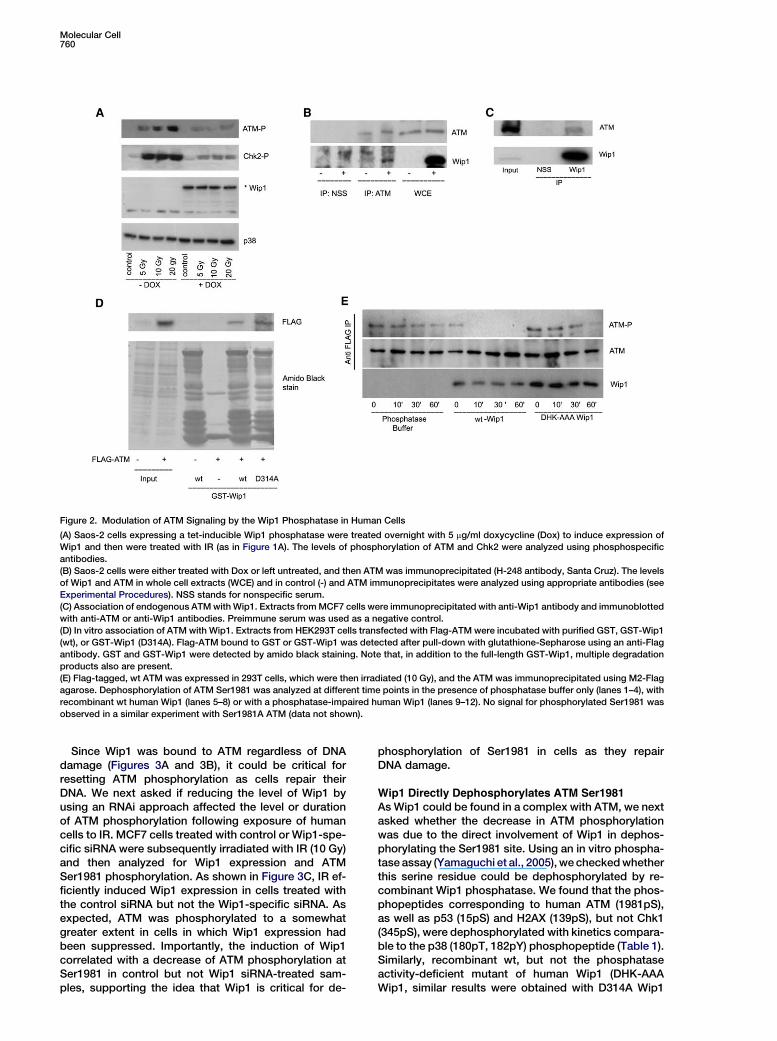
Figure 2. Modulation of ATM Signaling by the Wip1 Phosphatase in Human Cells
(A) Saos-2 cells expressing a tet-inducible Wip1 phosphatase were treated overnight with 5 mg/ml doxycycline (Dox) to induce expression of
Wip1 and then were treated with IR (as in Figure 1A). The levels of phosphorylation of ATM and Chk2 were analyzed using phosphospecific
antibodies.
(B) Saos-2 cells were either treated with Dox or left untreated, and then ATM was immunoprecipitated (H-248 antibody, Santa Cruz). The levels
of Wip1 and ATM in whole cell extracts (WCE) and in control (-) and ATM immunoprecipitates were analyzed using appropriate antibodies (see
Experimental Procedures). NSS stands for nonspecific serum.
(C) Association of endogenous ATM with Wip1. Extracts from MCF7 cells were immunoprecipitated with anti-Wip1 antibody and immunoblotted
with anti-ATM or anti-Wip1 antibodies. Preimmune serum was used as a negative control.
(D) In vitro association of ATM with Wip1. Extracts from HEK293T cells transfected with Flag-ATM were incubated with purified GST, GST-Wip1
(wt), or GST-Wip1 (D314A). Flag-ATM bound to GST or GST-Wip1 was detected after pull-down with glutathione-Sepharose using an anti-Flag
antibody. GST and GST-Wip1 were detected by amido black staining. Note that, in addition to the full-length GST-Wip1, multiple degradation
products also are present.
(E) Flag-tagged, wt ATM was expressed in 293T cells, which were then irradiated (10 Gy), and the ATM was immunoprecipitated using M2-Flag
agarose. Dephosphorylation of ATM Ser1981 was analyzed at different time points in the presence of phosphatase buffer only (lanes 1–4), with
recombinant wt human Wip1 (lanes 5–8) or with a phosphatase-impaired human Wip1 (lanes 9–12). No signal for phosphorylated Ser1981 was
observed in a similar experiment with Ser1981A ATM (data not shown).
Since Wip1 was bound to ATM regardless of DNAdamage (Figures 3A and 3B), it could be critical forresetting ATM phosphorylation as cells repair theirDNA. We next asked if reducing the level of Wip1 byusing an RNAi approach affected the level or durationof ATM phosphorylation following exposure of humancells to IR. MCF7 cells treated with control or Wip1-spe-cific siRNA were subsequently irradiated with IR (10 Gy)and then analyzed for Wip1 expression and ATMSer1981 phosphorylation. As shown in Figure 3C, IR ef-ficiently induced Wip1 expression in cells treated withthe control siRNA but not the Wip1-specific siRNA. Asexpected, ATM was phosphorylated to a somewhatgreater extent in cells in which Wip1 expression hadbeen suppressed. Importantly, the induction of Wip1correlated with a decrease of ATM phosphorylation atSer1981 in control but not Wip1 siRNA-treated sam-ples, supporting the idea that Wip1 is critical for de-
phosphorylation of Ser1981 in cells as they repairDNA damage.
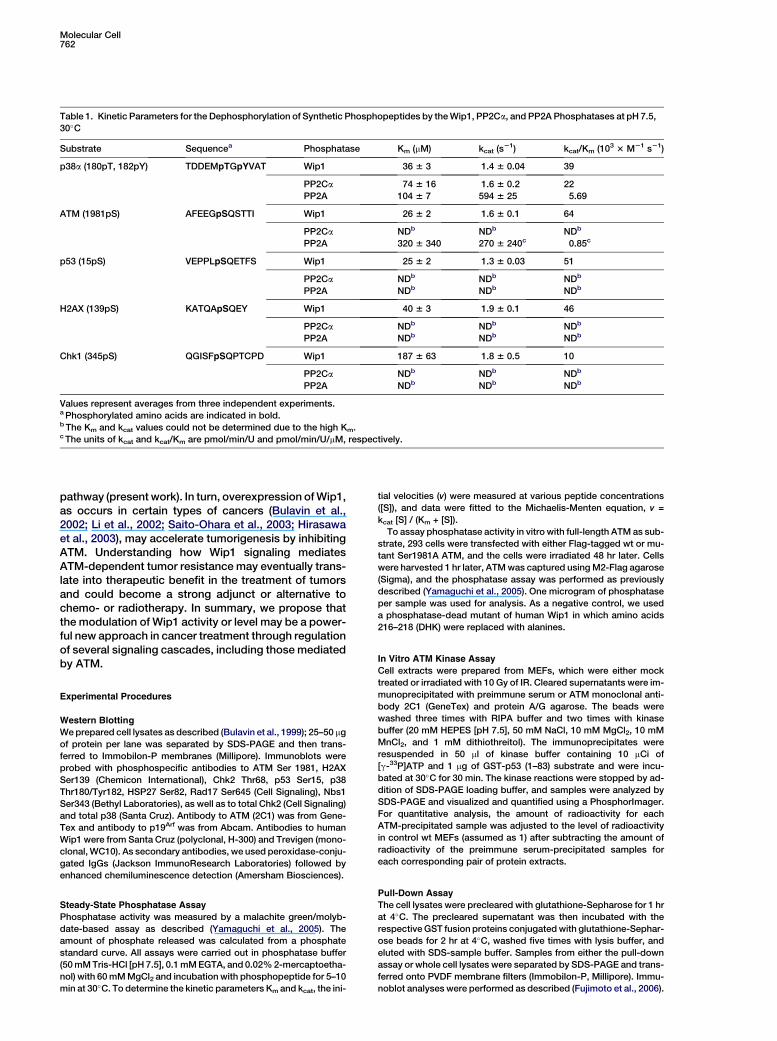
Wip1 Directly Dephosphorylates ATM Ser1981As Wip1 could be found in a complex with ATM, we nextasked whether the decrease in ATM phosphorylationwas due to the direct involvement of Wip1 in dephos-phorylating the Ser1981 site. Using an in vitro phospha-tase assay (Yamaguchi et al., 2005), we checked whetherthis serine residue could be dephosphorylated by re-combinant Wip1 phosphatase. We found that the phos-phopeptides corresponding to human ATM (1981pS),as well as p53 (15pS) and H2AX (139pS), but not Chk1(345pS), were dephosphorylated with kinetics compara-ble to the p38 (180pT, 182pY) phosphopeptide (Table 1).Similarly, recombinant wt, but not the phosphataseactivity-deficient mutant of human Wip1 (DHK-AAAWip1, similar results were obtained with D314A Wip1

Wip1 Regulates ATM-Dependent Signaling Pathways761
Figure 3. Control of ATM Phosphorylation at
Ser1981 by Wip1, and Their Binding after IR
(A) MCF7 cells were exposed to IR (10 Gy, 1
hr), and Wip1 was immunoprecipitated 1 hr
later with polyclonal antibody (Fujimoto
et al., 2006). The level of ATM in precipitates
was analyzed using a monoclonal antibody
(2C1). MCF7 cells treated with Wip1 siRNA
were used as a control (lane 4). NSS stands
for nonspecific serum.
(B) HeLa cells were transfected with either
a Flag-tagged wt Wip1 or D314A-Wip1. After
28 hr, cells were irradiated (10 Gy), and 1 hr
later Wip1 was precipitated with M2 Flag-
agarose. The level of ATM and phospho-
ATM was analyzed.
(C) Control or Wip1 siRNA-transfected MCF7
cells were exposed to IR (10 Gy) and incu-
bated for the indicated times. Extracts were
then analyzed by immunoblotting with the
indicated antibodies.
[data not shown]) was able to efficiently dephosphory-late Ser1981 on full-length ATM (Figure 2E). As Wip1belongs to the family of PP2C phosphatases, we alsocarried out a dephosphorylation analysis using PP2Ca,as well as PP2A. Both phosphatases showed somesubstrate specificity toward the p38 phosphopeptide;nevertheless, PP2A, but not PP2Ca, was able to dephos-phorylate the phospho-ATM peptide, although not effi-ciently (Table 1). No activity was detectable for othertested peptides. Thus, while the Wip1 phosphatasehad broad substrate specificity in vitro, for the sub-strates examined in vivo, phosphorylation was increasedonly for ATM and p53 in Wip1-deficient cells (Figure 1).Given that we observed an interaction between Wip1and ATM, attenuated phosphorylation of Ser1981 incells that overexpressed Wip1, increased Ser1981 phos-phorylation in Wip1-depleted cells, and dephosphoryla-tion of human ATM Ser1981 in vitro by Wip1, it seemslikely that ATM is a direct target for Wip1 phosphatasein vivo.
Discussion
Here we show that deficiency of Wip1 phosphataseresults in activation of the ATM kinase, while its overex-pression is sufficient to reduce activation of the ATM-dependent signaling cascade after DNA damage. TheWip1 phosphatase binds ATM and dephosphorylatesSer1981 in vivo and in vitro (Figures 1 and 2; Table 1),a site critical for ATM monomerization and activation(Bakkenist and Kastan, 2003). As Wip1 is associatedwith ATM in unstressed cells, it may set a threshold forthe initial phosphorylation and activation of ATM, which,in turn, would modulate p53 and Chk2 activity (Fujimotoet al., 2006). There is compelling evidence that the basallevel of p53 in both mice and humans, as set by theactivity of Mdm2, is critical for suppressing tumorigene-sis (Alt et al., 2003; Bond et al., 2006). By altering thethreshold for activation of ATM in response to intracellu-lar events resulting from expression of different onco-genes, Wip1 would affect the amount and/or activity ofp53 in a manner analogous to changes in the level ofMdm2.
We note that the absence of Wip1 alone does not re-sult in a full activation of all ATM-dependent signalingpathways, as phosphorylation of several downstreamtargets that accumulate at DNA damage foci was notsignificantly increased in Wip1 null MEFs after DNAdamage (Figure 1A). The phosphorylation of these sub-strates thus may require additional factors regulatingthe magnitude of ATM autophosphorylation and itsactivation. The PP2A phosphatase, which dissociatesfrom ATM following DNA damage (Goodarzi et al.,2004) could be one of the factors regulating the magni-tude of ATM phosphorylation at Ser1981. On the otherhand, under conditions when Wip1 is depleted or over-expressed, as in certain human cancers, the level ofATM autophosphorylation and its duration after DNAdamage is changed (Figures 1 and 3C). As Wip1 remainsassociated with ATM after DNA damage, its presencecould be especially critical for allowing the resumptionof cell growth or to prevent the initiation of apoptosisas cells attempt to complete the repair of DNA damage.
Recent studies have shown that activation of ATM ismore complex than the initial model proposed by Bak-kenist and Kastan (2003). Autophosphorylation of ATMis not required for its monomerization by MRN in vitro(Lee and Paull, 2004); however, acetylation by Tip60 isessential for activation in response to DNA damage(Sun et al., 2005). Furthermore, additional ATM auto-phosphorylation sites recently have been identified(Lavin et al., 2006). Although an increase in ATM auto-phosphorylation at Ser1987 was accompanied by anincrease in ATM’s activity in Wip1 null MEFs (Figure 1),and the original data from Kastan’s lab suggested thatSer1981 is important for regulating ATM activity througha dissociation of ATM oligomers (Bakkenist and Kastan,2003), it remains to be determined whether Wip1 mayalso affect ATM activity through the dephosphorylationof other sites.
While suppression of tumorigenesis in breast epitheliain the presence of the ras or c-neu oncogenes is medi-ated through p38 MAPK-dependent signaling, presum-ably by controlling the association of Bmi-1 with chro-matin (Voncken et al., 2005), Wip1-deficient mice alsocould be resistant to cancer through the ATM-p53
Molecular Cell762
Table 1. Kinetic Parameters for the Dephosphorylation of Synthetic Phosphopeptides by the Wip1, PP2Ca, and PP2A Phosphatases at pH 7.5,
30�C
Substrate Sequencea Phosphatase Km (mM) kcat (s21) kcat/Km (103 3 M21 s21)
p38a (180pT, 182pY) TDDEMpTGpYVAT Wip1 36 6 3 1.4 6 0.04 39
PP2Ca 74 6 16 1.6 6 0.2 22
PP2A 104 6 7 594 6 25 5.69
ATM (1981pS) AFEEGpSQSTTI Wip1 26 6 2 1.6 6 0.1 64
PP2Ca NDb NDb NDb
PP2A 320 6 340 270 6 240c 0.85c
p53 (15pS) VEPPLpSQETFS Wip1 25 6 2 1.3 6 0.03 51
PP2Ca NDb NDb NDb
PP2A NDb NDb NDb
H2AX (139pS) KATQApSQEY Wip1 40 6 3 1.9 6 0.1 46
PP2Ca NDb NDb NDb
PP2A NDb NDb NDb
Chk1 (345pS) QGISFpSQPTCPD Wip1 187 6 63 1.8 6 0.5 10
PP2Ca NDb NDb NDb
PP2A NDb NDb NDb
Values represent averages from three independent experiments.a Phosphorylated amino acids are indicated in bold.b The Km and kcat values could not be determined due to the high Km.c The units of kcat and kcat/Km are pmol/min/U and pmol/min/U/mM, respectively.
pathway (present work). In turn, overexpression of Wip1,as occurs in certain types of cancers (Bulavin et al.,2002; Li et al., 2002; Saito-Ohara et al., 2003; Hirasawaet al., 2003), may accelerate tumorigenesis by inhibitingATM. Understanding how Wip1 signaling mediatesATM-dependent tumor resistance may eventually trans-late into therapeutic benefit in the treatment of tumorsand could become a strong adjunct or alternative tochemo- or radiotherapy. In summary, we propose thatthe modulation of Wip1 activity or level may be a power-ful new approach in cancer treatment through regulationof several signaling cascades, including those mediatedby ATM.
Experimental Procedures
Western Blotting
We prepared cell lysates as described (Bulavin et al., 1999); 25–50 mg
of protein per lane was separated by SDS-PAGE and then trans-
ferred to Immobilon-P membranes (Millipore). Immunoblots were
probed with phosphospecific antibodies to ATM Ser 1981, H2AX
Ser139 (Chemicon International), Chk2 Thr68, p53 Ser15, p38
Thr180/Tyr182, HSP27 Ser82, Rad17 Ser645 (Cell Signaling), Nbs1
Ser343 (Bethyl Laboratories), as well as to total Chk2 (Cell Signaling)
and total p38 (Santa Cruz). Antibody to ATM (2C1) was from Gene-
Tex and antibody to p19Arf was from Abcam. Antibodies to human
Wip1 were from Santa Cruz (polyclonal, H-300) and Trevigen (mono-
clonal, WC10). As secondary antibodies, we used peroxidase-conju-
gated IgGs (Jackson ImmunoResearch Laboratories) followed by
enhanced chemiluminescence detection (Amersham Biosciences).
Steady-State Phosphatase Assay
Phosphatase activity was measured by a malachite green/molyb-
date-based assay as described (Yamaguchi et al., 2005). The
amount of phosphate released was calculated from a phosphate
standard curve. All assays were carried out in phosphatase buffer
(50 mM Tris-HCl [pH 7.5], 0.1 mM EGTA, and 0.02% 2-mercaptoetha-
nol) with 60 mM MgCl2 and incubation with phosphopeptide for 5–10
min at 30�C. To determine the kinetic parameters Km and kcat, the ini-
tial velocities (v) were measured at various peptide concentrations
([S]), and data were fitted to the Michaelis-Menten equation, v =
kcat [S] / (Km + [S]).
To assay phosphatase activity in vitro with full-length ATM as sub-
strate, 293 cells were transfected with either Flag-tagged wt or mu-
tant Ser1981A ATM, and the cells were irradiated 48 hr later. Cells
were harvested 1 hr later, ATM was captured using M2-Flag agarose
(Sigma), and the phosphatase assay was performed as previously
described (Yamaguchi et al., 2005). One microgram of phosphatase
per sample was used for analysis. As a negative control, we used
a phosphatase-dead mutant of human Wip1 in which amino acids
216–218 (DHK) were replaced with alanines.
In Vitro ATM Kinase Assay
Cell extracts were prepared from MEFs, which were either mock
treated or irradiated with 10 Gy of IR. Cleared supernatants were im-
munoprecipitated with preimmune serum or ATM monoclonal anti-
body 2C1 (GeneTex) and protein A/G agarose. The beads were
washed three times with RIPA buffer and two times with kinase
buffer (20 mM HEPES [pH 7.5], 50 mM NaCl, 10 mM MgCl2, 10 mM
MnCl2, and 1 mM dithiothreitol). The immunoprecipitates were
resuspended in 50 ml of kinase buffer containing 10 mCi of
[g-33P]ATP and 1 mg of GST-p53 (1–83) substrate and were incu-
bated at 30�C for 30 min. The kinase reactions were stopped by ad-
dition of SDS-PAGE loading buffer, and samples were analyzed by
SDS-PAGE and visualized and quantified using a PhosphorImager.
For quantitative analysis, the amount of radioactivity for each
ATM-precipitated sample was adjusted to the level of radioactivity
in control wt MEFs (assumed as 1) after subtracting the amount of
radioactivity of the preimmune serum-precipitated samples for
each corresponding pair of protein extracts.
Pull-Down Assay
The cell lysates were precleared with glutathione-Sepharose for 1 hr
at 4�C. The precleared supernatant was then incubated with the
respective GST fusion proteins conjugated with glutathione-Sephar-
ose beads for 2 hr at 4�C, washed five times with lysis buffer, and
eluted with SDS-sample buffer. Samples from either the pull-down
assay or whole cell lysates were separated by SDS-PAGE and trans-
ferred onto PVDF membrane filters (Immobilon-P, Millipore). Immu-
noblot analyses were performed as described (Fujimoto et al., 2006).

Wip1 Regulates ATM-Dependent Signaling Pathways763
siRNA-Mediated Suppression of Wip1
RNAi experiments were performed as described (Fujimoto et al.,
2006). The sequence of the Wip1 siRNA oligo was UUGGCCUUGU
GCCUACUAA. The control siRNA oligo used (Scramble II Duplex)
was GCGCGCUUUGUAGGAUUCG. Cells were transfected with
siRNA duplexes using GeneSilencer (Gene Therapy System), follow-
ing the manufacturer’s instructions.
Supplemental Data
Supplemental Data include two figures and can be found with this
article online at http://www.molecule.org/cgi/content/full/23/5/757/
DC1/.
Acknowledgments
We are very grateful to F. Vikhanskaya for help with irradiation of
cells. This work was supported by the Agency for Science, Technol-
ogy and Research (Singapore). C.W.A. was supported in part by
a grant from the Low Dose Program of the Office of Biological and
Environmental Research of the U.S. Department of Energy and by
a Laboratory Directed Research and Development Award at The
Brookhaven National Laboratory under contract with the U.S. De-
partment of Energy. The research for H.Y., C.D., and E.A. was sup-
ported by the Intramural Research Program of the National Institutes
of Health, National Cancer Institute.
Received: December 4, 2005
Revised: March 14, 2006
Accepted: July 8, 2006
Published: August 31, 2006
References
Alt, J.R., Greiner, T.C., Cleveland, J.L., and Eischen, C.M. (2003).
Mdm2 haplo-insufficiency profoundly inhibits Myc-induced lympho-
magenesis. EMBO J. 22, 1442–1450.
Bakkenist, C.J., and Kastan, M.B. (2003). DNA damage activates
ATM through intermolecular autophosphorylation and dimer disso-
ciation. Nature 421, 499–506.
Barlow, C., Hirotsune, S., Paylor, R., Liyanage, M., Eckhaus, M., Col-
lins, F., Shiloh, Y., Crawley, J.N., Ried, T., Tagle, D., and Wynshaw-
Boris, A. (1996). Atm-deficient mice: a paradigm of ataxia telangiec-
tasia. Cell 86, 159–171.
Belova, G.I., Demidov, O.N., Fornace, A.J., and Bulavin, D.V. (2005).
Chemical inhibition of Wip1 phosphatase contributes to suppres-
sion of tumorigenesis. Cancer Biol. Ther. 4, 1154–1158.
Bond, G.L., Hirshfield, K.M., Kirchhoff, T., Alexe, G., Bond, E.E.,
Robins, H., Bartel, F., Taubert, H., Wuerl, P., Hait, W., et al. (2006).
MDM2 SNP309 accelerates tumor formation in a gender-specific
and hormone-dependent manner. Cancer Res. 66, 5104–5110.
Bulavin, D.V., Saito, S., Hollander, M.C., Sakaguchi, K., Anderson,
C.W., Appella, E., and Fornace, A.J., Jr. (1999). Phosphorylation of
human p53 by p38 kinase coordinates N-terminal phosphorylation
and apoptosis in response to UV radiation. EMBO J. 18, 6845–6854.
Bulavin, D.V., Demidov, O.N., Saito, S., Kauraniemi, P., Phillips, C.,
Amundson, S.A., Ambrosino, C., Sauter, G., Nebreda, A.R., Ander-
son, C.W., et al. (2002). Amplification of PPM1D in human tumors
abrogates p53 tumor-suppressor activity. Nat. Genet. 31, 210–215.
Bulavin, D.V., Phillips, C., Nannenga, B., Timofeev, O., Donehower,
L.A., Anderson, C.W., Appella, E., and Fornace, A.J., Jr. (2004). Inac-
tivation of the Wip1 phosphatase inhibits mammary tumorigenesis
through p38 MAPK-mediated activation of the p16Ink4a-p19Arf path-
way. Nat. Genet. 36, 343–350.
Fiscella, M., Zhang, H., Fan, S., Sakaguchi, K., Shen, S., Mercer,
W.E., Vande Woude, G.F., O’Connor, P.M., and Appella, E. (1997).
Wip1, a novel human protein phosphatase that is induced in re-
sponse to ionizing radiation in a p53-dependent manner. Proc.
Natl. Acad. Sci. USA 94, 6048–6053.
Fujimoto, H., Onishi, N., Kato, N., Takekawa, M., Xu, X.Z., Kosugi, A.,
Kondo, T., Oishi, I., Yoda, A., Imamura, M., and Minami, Y. (2006).
Regulation of the anti-oncogenic Chk2 kinase by the oncogenic
Wip1 phosphatase. Cell Death Differ. 13, 1170–1180.
Goodarzi, A.A., Jonnalagadda, J.C., Douglas, P., Young, D., Ye, R.,
Moorhead, G.B., Lees-Miller, S.P., and Khanna, K.K. (2004). Auto-
phosphorylation of ataxia-telangiectasia mutated is regulated by
protein phosphatase 2A. EMBO J. 23, 4451–4461.
Gumy-Pause, F., Wacker, P., and Sappino, A.-P. (2004). ATM gene
and lymphoid malignancies. Leukemia 18, 238–242.
Hirasawa, A., Saito-Ohara, F., Inoue, J., Aoki, D., Susumu, N., Yo-
koyama, T., Nozawa, S., Inazawa, J., and Imoto, I. (2003). Associa-
tion of 17q21-q24 gain in ovarian clear cell adenocarcinomas with
poor prognosis and identification of PPM1D and APPBP2 as likely
amplification targets. Clin. Cancer Res. 9, 1995–2004.
Kallioniemi, A., Kallioniemi, O.-P., Piper, J., Tanner, M., Stokke, T.,
Chen, L., Smith, H.S., Pinkel, D., Gray, J.W., and Waldman, F.M.
(1994). Detection and mapping of amplified DNA sequences in
breast cancer by comparative genomic hybridization. Proc. Natl.
Acad. Sci. USA 91, 2156–2160.
Kastan, M.B., and Bartek, J. (2004). Cell-cycle checkpoints and can-
cer. Nature 432, 316–323.
Khan, J., Parsa, N.Z., Harada, T., Meltzer, P.S., and Carter, N.P.
(1998). Detection of gains and losses in 18 meningiomas by compar-
ative genomic hybridization. Cancer Genet. Cytogenet. 103, 95–100.
Lavin, M.F., Delia, D., and Chessa, L. (2006). ATM and the DNA dam-
age response. Workshop on ataxia-telangiectasia and related syn-
dromes. EMBO Rep. 7, 154–160.
Lee, J.H., and Paull, T.T. (2004). Direct activation of the ATM protein
kinase by the Mre11/Rad50/Nbs1 complex. Science 304, 93–96.
Li, J., Yang, Y., Peng, Y., Austin, R.J., van Eyndhoven, W.G., Nguyen,
K.C.Q., Gabriele, T., McCurrach, M.E., Marks, J.R., Hoey, T., et al.
(2002). Oncogenic properties of PPM1D located within a breast can-
cer amplification epicenter at 17q23. Nat. Genet. 31, 133–134.
Lu, X., Nannenga, B., and Donehower, L.A. (2005). PPM1D dephos-
phorylates Chk1 and p53 and abrogates cell cycle checkpoints.
Genes Dev. 19, 1162–1174.
Oliner, J.D., Pietenpol, J.A., Thiagalingam, S., Gyuris, J., Kinzler,
K.W., and Vogelstein, B. (1993). Oncoprotein MDM2 conceals the
activation domain of tumour suppressor p53. Nature 362, 857–860.
Plantaz, D., Mohapatra, G., Matthay, K.K., Pellarin, M., Seeger, R.,
and Feuerstein, B.G. (1997). Gain of chromosome 17 is the most
frequent abnormality detected in neuroblastoma by comparative
genomic hybridization. Am. J. Pathol. 150, 81–89.
Ried, T., Petersen, I., Holtgreve-Grez, H., Speicher, M.R., Schrock,
E., du Manoir, S., and Cremer, T. (1994). Mapping of multiple DNA
gains and losses in primary small cell lung carcinomas by compara-
tive genomic hybridization. Cancer Res. 54, 1801–1806.
Saito-Ohara, F., Imoto, I., Inoue, J., Hosoi, H., Nakagawara, A., Sugi-
moto, T., and Inazawa, J. (2003). PPM1D is a potential target for 17q
gain in neuroblastoma. Cancer Res. 63, 1876–1883.
Shiloh, Y., and Kastan, M.B. (2001). ATM: genome stability, neuronal
development, and cancer cross paths. Adv. Cancer Res. 83, 209–
254.
Solinas-Toldo, S., Wallrapp, C., Muller-Pillasch, F., Bentz, M., Gress,
T., and Lichter, P. (1996). Mapping of chromosomal imbalances in
pancreatic carcinoma by comparative genomic hybridization. Can-
cer Res. 56, 3803–3807.
Sun, Y., Jiang, X., Chen, S., Fernandes, N., and Price, B.D. (2005). A
role for the Tip60 histone acetyltransferase in the acetylation and ac-
tivation of ATM. Proc. Natl. Acad. Sci. USA 102, 13182–13187.
Takekawa, M., Adachi, M., Nakahata, A., Nakayama, I., Itoh, F., Tsu-
kuda, H., Taya, Y., and Imai, K. (2000). p53-inducible Wip1 phospha-
tase mediates a negative feedback regulation of p38 MAPK-p53
signaling in response to UV radiation. EMBO J. 19, 6517–6526.
Voncken, J.W., Niessen, H., Neufeld, B., Rennefahrt, U., Dahlmans,
V., Kubben, N., Holzer, B., Ludwig, S., and Rapp, U.R. (2005).
MAPKAP kinase 3pK phosphorylates and regulates chromatin
association of the polycomb group protein Bmi1. J. Biol. Chem.
280, 5178–5187.
Voorter, C., Joos, S., Bringuier, P.P., Vallinga, M., Poddighe, P.,
Schalken, J., du Manoir, S., Ramaekers, F., Lichter, P., and Hopman,
A. (1995). Detection of chromosomal imbalances in transitional cell

Molecular Cell764
carcinoma of the bladder by comparative genomic hybridization.
Am. J. Pathol. 146, 1341–1354.
Wong, N., Lai, P., Lee, S.-W., Fan, S., Pang, E., Liew, C.-T., Sheng, Z.,
Lau, J.W.-Y., and Johnson, P.J. (1999). Assessment of genetic
changes in hepatocellular carcinoma by comparative genomic hy-
bridization analysis: relationship to disease stage, tumor size, and
cirrhosis. Am. J. Pathol. 154, 37–43.
Xu, Y., Ashley, T., Brainerd, E.E., Bronson, R.T., Meyn, M.S., and Bal-
timore, D. (1996). Targeted disruption of ATM leads to growth retar-
dation, chromosomal fragmentation during meiosis, immune de-
fects, and thymic lymphoma. Genes Dev. 10, 2411–2422.
Yamaguchi, H., Minopoli, G., Demidov, O.N., Chatterjee, D.K., An-
derson, C.W., Durell, S.R., and Appella, E. (2005). Substrate specific-
ity of the human protein phosphatase 2Cd, Wip1. Biochemistry 44,
5285–5294.