topiramate is likely to act outside of the trigeminocervical complex
TRANSCRIPT

Original Article
Topiramate is likely to act outsideof the trigeminocervical complex
Robin J Storer and Peter J Goadsby
Abstract
Background: To facilitate understanding the locus and mechanism of action of antimigraine preventives, we examined the
effect of topiramate on trigeminocervical activation in the cat.
Methods: Cats were anesthetized and physiologically monitored. Electrical stimulation of the superior sagittal sinus
activated nociceptive trigeminovascular afferents. Extracellular recordings were made from neurons in the trigemino-
cervical complex.
Results: Microiontophoretically delivered topiramate, applied locally at the second order synapse of the trigeminovascular
system in the trigeminocervical complex, produced significant inhibition of L-glutamate-evoked firing of neurons only at
the highest microiontophoretic currents (27� 7% at �160 nA; p< 0.05, n¼ 14 cells), but did not inhibit firing of these
neurons evoked by stimulation of the craniovascular afferents (2� 5%, p¼ 0.762, n¼ 13 cells). In contrast, systemically
administered topiramate (30 mg/kg intravenously) partly inhibited this firing (32� 10% at 15 min; F5,35¼ 3.5, p< 0.05,
n¼ 8 cats). After this systemic administration, profound inhibition (70� 10%, p< 0.001, n¼ 7) of L-glutamate-evoked
firing of cells in the trigeminocervical complex at the second order synapse of the trigeminovascular system was
observed.
Conclusions: These data suggest that topiramate acts outside of the trigeminocervical complex in the cat. Determining the
sites of action of preventive antimigraine treatments is crucial to developing laboratory models for the development of
new therapeutics, and may vary between species.
Keywords
Migraine, cluster headache, preventive, trigeminovascular
Date received: 4 March 2012; revised: 30 October 2012; 22 November 2012; accepted: 25 November 2012
Introduction
Migraine is a common (1), disabling brain disorder (2).Given the disability, many patients seek, and perhapstoo few are offered, preventive treatments (3). There is aconsiderable basis for recognizing neuronal hyperexcit-ability (4,5) in migraine and functional changes, such asthose leading to both disorders (6), which may formsome part of the basis of a comorbidity of migrainewith epilepsy. However, the change in excitation maybe through a change in synchrony (7), which mightimplicate brainstem and subcortical structures to bethe pivotal sites of migraine pathophysiology (8). Ifthat is the case, examining where and how in subcor-tical structures migraine therapeutics may act couldyield a more complete picture of their mechanismsand the pathophysiology of migraine.
Of the antiepileptics used in migraine, topiramate isclearly the best studied. It has a complex basis for its
action, interacting with channels and transmitters, eachof which may be pivotal in the antimigraine effect (9).It is effective in the prevention of episodic (10,11) andchronic (12,13) migraine. It is of comparable efficacywith propranolol (14) and is recommended by standardtreatment guidelines described by Evers et al. (15).Topiramate is effective when used acutely (16) andchronically (17,18) in animal models of cortical spread-ing depression (CSD). These models at least mimicmany aspects of migraine aura (19). In contrast,
Headache Group, Department of Neurology, University of California,
CA, USA
Corresponding author:
Peter J Goadsby, UCSF Headache Center, 1701 Divisadero St., San
Francisco, CA 94115, USA.
Email: [email protected]
Cephalalgia
33(5) 291–300
! International Headache Society 2013
Reprints and permissions:
sagepub.co.uk/journalsPermissions.nav
DOI: 10.1177/0333102412472069
cep.sagepub.com

tonabersat, which is reported to inhibit CSD (20), failedin controlled trials in migraine prevention (21), yet waseffective in blocking aura (22). The experience withtonabersat suggests models other than CSD may berequired to gain the complete picture of how preventivemedicines may work (23).
In a previous study we showed that intravenous (i.v.)administration of topiramate could inhibit nociceptivetraffic in the trigeminocervical complex in a cat modelof craniovascular nociception in which stimulation ofafferents from the superior sagittal sinus (SSS) is usedto activate neurons in the trigeminocervical complex(24). Here we used local microiontophoretic deliveryof topiramate onto second-order trigeminal neuronsto understand further how topiramate may have itseffects. This work was previously reported in prelimin-ary form at the 12th International Headache Congressin Kyoto, Japan (25).
Methods
All studies were conducted and terminated under gen-eral anesthesia in accordance with a project licenseissued by the Home Office of the United Kingdomunder the Animals (Scientific Procedures) Act, 1986,and conformed to United States National Institutes ofHealth (NIH) guidelines for animal experimentation.
Anesthesia and surgical preparation
Cats of either sex (n¼ 15) weighing 2.58� 0.17 kg(mean� SD), were anesthetized with a-chloralose(60mg/kg intraperitoneally (i.p.); Sigma, St. Louis,MO) and prepared for physiological monitoring andi.v. drug administration essentially as previouslydescribed previously (24). Isoflurane (Merial AnimalHealth, Essex, UK; 0.25%�3.0% in a 40% oxygen:aircarrier gas mixture) was administered from a calibratedvaporizer (Ohmeda-BOC Healthcare, Steeton, UK)during surgical procedures and then discontinuedbefore and during experimental protocols. Amoxicillintrihydrate (150mg; Fort Dodge Animal Health,Southampton, UK) was given by subcutaneous injectionto control prophylactically any infection during theexperiments. Saline was administered throughout theexperiment tomaintain hydration and ensure renal func-tion, initially as a 40–70ml bolus at about 1ml/min, fol-lowed by intermittent 10–20ml infusions includingduring i.v. drug administration to a total of about150–250ml per experiment. Corneal desiccation wasprevented by coating the corneas with ocular lubricantointment (Allergan Pharmaceuticals, Westport,Ireland). Core temperature was monitored and main-tained between 37�C and 39�C using a homeother-mic heater-blanket system (Harvard Apparatus,
Holliston, MA). Cats were ventilated with an oxygen-in-air mixture, and end-tidal CO2 was continuouslymonitored to maintain arterial blood pH within physio-logical limits. Depth of anesthesia was monitored peri-odically throughout the experiment by testing forsympathetic (pupillary and cardiovascular) responsesto noxious stimulation and withdrawal reflexes in theabsence of neuromuscular blockade. Supplementarydoses of i.v. a-chloralose in 2-hydroxypropyl-b-cyclo-dextrin (Sigma) were given as required at a rate of 5–10mg/kg per h (26).
Central nervous system (CNS) surgery
A midline craniotomy and C1–C2 laminectomy wereperformed in each cat, allowing access to the SSS andthe area for recording neuronal activity in the caudalextension of the trigeminal nucleus in the cervical spinalcord as previously described (24). The SSS was isolatedby dissecting the adjacent dura and falx cerebri overapproximately 15mm. The likelihood of possible arti-facts from arterial pulsation and respiratory movement,such as movement of the electrode from recording sites,was reduced by: bilateral pneumothoraces, kept patentwith polypropylene tubes; immobilization of the spineby clamping a thoracic spinous process (1780 spinalunit; Kopf); clamping the C1 transverse processes toauxiliary ear bar holders on the frame, and clampingthe remaining caudal portion of the dorsal C2 spinousprocess using a modified 1786 clamp (Kopf).
Stimulation and recording
In each cat the isolated SSS was gently lifted onto a pairof bipolar platinum hook electrodes connected to astimulus isolation unit (SIU5A; Grass Instruments,West Warwick, RI). To activate primary trigeminalafferents, the SSS was supramaximally stimulatedwith square-wave pulses from a Grass S88 stimulator(90–150V, about 50 mA; 250 ms, 0.3–0.5Hz) afterneuromuscular blockade with gallamine triethiodide(Concord Pharmaceuticals, Essex, UK; initially5–10mg/kg i.v. and maintained with 5–10mg/kgper h). The dura mater above the recording regions inthe spinal cord was reflected after a midline incisionand held to the edges of the laminectomy withN-butyl-cyanoacrylate, further stabilizing any possiblemovement of the cord in this sling-like arrangement.It is important to avoid cord movement by its physicalstabilization and the use of neuromuscular blockersduring recordings because drift or movement of elec-trodes from their recording sites may cause an apparentreduction of cell activity. Extracellular recordings weremade using carbon-fiber electrodes in a multibarreledmicroiontophoretic pipette assembly (Carbostar-7 S;
292 Cephalalgia 33(5)

Kation Scientific, Minneapolis, MN). Carbon-fiber rec-ording electrode impedances were typically 1–3M� (tipsize 17.5� 4.5; mean� SD) when measured at 1 kHz in0.9% saline (Impedance Check Module; FHC,Bowdoinham, ME). After local removal of the piamater, which was carefully dissected away from asmall area of the underlying spinal cord at the dorso-lateral sulcus above the dorsal root entry zone, the elec-trodes were lowered into the cord substance þ 1mmrostral to �3mm caudal to the C2 roots in the regionof the dorsal root entry zone (�200 mm mediolaterally).The point of contact of the electrode tip with the cordsurface was taken as the zero reference point. The elec-trodes were advanced or retracted in the cord substancein 5 mm steps using a pizoelectric motor microelectrodepositioner system (Burleigh Instruments, Harpenden,UK). Tissue culture grade agar 3% (w/v), (Sigma) inpyrogen-free saline (Baxter Healthcare) was set over theexposed cord after electrode insertion to furtherreduce cardiovascular-related movement. Signal fromthe recording electrode was amplified and filtered aspreviously described (24) and was fed to a gated amp-litude discriminator (Neurolog NL201) and analogue-to-digital converter (Power 1401; Cambridge ElectronicDesign), where it was sampled at 25 kHz to a micro-processor-based personal computer (Dell), where thesignal was processed and stored. Electrical signalsfrom action potentials were amplified audio monitoringand displayed on analogue and digital-storage oscillo-scopes to assist the isolation of single unit activity fromadjacent cell activity and noise. All electrophysiologicaldata and physiological parameters were recorded onmagnetic tape (Pulse Code Modulator; Vetter,Rebersburgh, PA) and local hard disk for documenta-tion and later review.
The position of the recording electrodes was con-trolled with reference to the mid-point of the C2 dorsalroots. Together with the depth of the recording electrodetip with respect to the surface of the spinal cord at thedorsal root entry zone, as determined by the distancetravelled display on the ULN6000 pizoelectric motorcontroller, this provided the coordinates of the recordingsites. The location of recording sites was marked bythermocoagulation using an electrolytic lesion (anodal20–50 mA, 10–30 s; D.C.LM5 constant current lesionmaker; Grass Instruments) or bymicroiontophoreticallydelivered Pontamine Sky Blue dye (�2.00 mA, 30min).Animals were euthanized with sodium pentobarbital(400mg), followed by KCl (10% w/v; 5ml). After ter-mination of experiments, sections of spinal cord contain-ing the recording sites were resected, fixed with neutralbuffered 10% formalin, stored in phosphate-buffered30% sucrose, pH 7.4 until saturated, and sectioned(40 mm; HM500 OM cryostat microtome, MicromLaborgerate, Walldorf, Germany). Electrolytic lesion
marks and Pontamine Sky Blue marks were identifiedon freshly cut sections and then counterstained withcresyl violet (Sigma) Nissl staining or nuclear fast redfor documentation and storage. The position of the rec-ording sites within the cord were determined from his-tologically identified lesions, dye marks, or by referenceto the coordinates of recording electrode positions anddye or lesion marks in the same electrode track, oftenmarking the end of the track.
Receptive fields
Cells responding to stimulation of superior sagittalsinus afferents were characterized as receiving lowthreshold mechanoreceptor input if they responded tonon-noxious input, such as brush with a cotton pledget,on cutaneous receptive fields on the face or forepaws.They were characterized as nociceptive specific (NS) ifthey responded to noxious mechanical stimuli, such aspinching with toothed forceps or pricking with a needle,or wide dynamic range (WDR), if they responded toboth (27). Receptive fields were almost exclusively ipsi-lateral to recording sites.
I.v. administration of topiramate
Topiramate (Johnson & Johnson PRD, Spring House,PA) was dissolved at 9.8mg/ml in saline for InjectionBP (Phoenix Pharma, Gloucester, UK), producing aslightly acidic (pH 6–7) solution because of topira-mate’s sulfamate moiety, and administered to cats i.v.at 30mg/kg.
Microiontophoretic administration of L-glutamateand topiramate
Multibarreled pipettes incorporating a carbon-fiber rec-ording electrode were used for microiontophoresisand the six open pipette barrels had orifices in therange 1–3 mm. The pipettes were filled with 200mMmonosodium L-glutamate (Sigma), pH 8.0 (28);30mM sodium topiramate, pH 9.0–9.5 (obtained as atrihydrate sodium salt from Johnson & Johnson.Certificate of analysis reports a sulfamate impurity<0.2mole percentage by chromatographic analysis,heavy metals <10 ppm). The sodium salt is more than70 times more soluble than the literature value of9.8mg/ml for topiramate (29). NaCl 150mM and200mM, and 2.5% (w/v) Pontamine Sky Blue (C.I.24 410; BDH Laboratory Supplies, Poole, UK) in50mM sodium acetate, pH 8.0, was used for markingrecording sites and the end of electrode tracks.Sometimes two barrels were filled with topiramate solu-tion to provide redundancy in case of barrel ‘‘blocking’’and, if not, a second barrel to allow dual delivery of this
Storer and Goadsby 293

test compound up to compliance levels. A microionto-phoresis current generator (6400A; Dagan,Minneapolis, MN) provided the current for ejectingsubstances from the micropipette barrels throughsilver wire (AGT1025; WPI, Sarasota, FL). Using thisapparatus barrel ‘‘blocking’’ can occur when the resist-ance of the channel exceeds 200M� in situ for theinstrument compliance of� 135V for a set current.If a cell was responsive to L-glutamate ejection, theejection current required to produce a stable baselineresponse across at least five epochs of L-glutamateapplication, as demanded for cell testing, was estab-lished empirically for each neuron such that the firingactivity of neurons was generally at a maximum ofaround 30–40Hz during pulses of application toavoid excitotoxicity, and was typically �10 to�60 nA. The application pulses were typically of 10 sduration alternated with a similar or longer period ofretention, so that inhibition of the cell activity, or facili-tation of firing by the test substances, could be deter-mined and distinguished from random firing and noisewithout overexciting the cells.
Responses were quantified in rate histograms using1 s bins or using 0–50ms post-stimulus histograms.L-glutamate, topiramate, and Pontamine Sky Bluewere ionized as anions and retained with small positivecurrents (approximately 3–5 nA) to restrain passive dif-fusion from barrels between ejection periods. Ejectioncurrents for topiramate and Cl� control (with a Naþ
balancing current) in directions opposite to the retain-ing currents were used. Experimental controls werebased on passing currents of the same magnitude anddirection through the barrel containing 150mM NaCl.Current balancing was always provided through abarrel containing 200mM NaCl (30,31). After filling,electrode barrels passing useful (>10 nA) iontophoreticcurrents at� 135V compliance usually had impedancesof 5–20M� tested at 10 nA peak-to-peak at 1 kHz insaline (FHC) and resistances of 20–200M� in situ.
Statistical analyses
Physiological parameters including cat weight, bloodgas parameters, and urine output are presented asmean�SD. Neuronal data are presented asmean�SEM. Inhibition by i.v. topiramate of trigem-inal neuron activation evoked by stimulation of SSSafferents was evaluated using a one-way analysis ofvariance (ANOVA) with repeated measures, followedby Bonferroni’s post hoc test. Significance was assessedat the p< 0.05 level using SPSS (SPSS, Chicago, IL) orSigmaPlot (Systat, Chicago, IL) software.
Neuronal firing evoked during each epoch ofmicroiontophoretic L-glutamate application was distin-guished from noise using the gated-amplitude
discriminator described earlier. Statistical evaluationsfor each unit were made using the mean rate of firing(in hertz). The background neuronal discharge wascalculated by averaging the period of ongoingbasal activity immediately preceding each period ofL-glutamate-evoked excitation and this value was sub-tracted from the glutamate-evoked responses. Stablebaseline response to L-glutamate application was deter-mined by examining the response to each of at least fiveepochs of L-glutamate application at the same currentto check that there was no significant difference acrossthe responses before applying test compounds. Foreach unit a minimum of five-paired baseline-responsedata sets were collected. A significant change in meanfiring rate was confirmed using a paired-samples t testor two-tailed one-sample t test vs baseline, where sig-nificance was assessed at the p< 0.05 level using SPSSor SigmaPlot software.
Results
Cats from which data are reported (n¼ 15) had cardio-respiratory parameters that were normal for ananesthetized cat. Arterial blood gas levels were mea-sured at intervals throughout experiments and werewithin normal limits for an a-chloralose anesthetizedcat: pH 7.36� 0.06; pCO2 3.16� 0.65 kPa; pO2
30.34� 4.54 kPa. Urine output was 4.3� 3.3ml/h.
Neuronal localization and characterization
Extracellular recordings were made and data collectedfrom neurons in the trigeminocervical complex of cats.Cells were locatedþ 1mm rostral to �3mm caudal tothe midpoint of the C2 rootlets,� 200 mm mediolater-ally to the dorsal root entry zone at a depth of approxi-mately �650 mm to around �3000 mm below the(dorsal) cord surface (Figure 1). Cells responded toelectrical sagittal sinus stimulation with latencies con-sistent with Ad fibers, typically 8–10ms. Cells receivedwide dynamic range or nociceptive specific mechano-receptor input from cutaneous V1 or V2 receptivefields on the face, or cutaneous receptive fields on fore-paws, or both.
Glutamate-evoked activity in cells in thetrigeminocervical complex linked to stimulationof trigeminovascular afferents
L-glutamate-evoked firing of cells in the trigeminocer-vical complex linked to electrical stimulation of affer-ents from the SSS was significantly inhibited by localmicroiontophoretic application of topiramate only atthe highest delivery current: 27� 7% inhibition of
294 Cephalalgia 33(5)

glutamate-evoked activity at �160 nA for 2.5minutescompared with current-matched controls (t¼ 2.2,p¼ 0.05, n¼ 14 cells). The maximum currents atwhich topiramate could be delivered was about�100 nA before ‘‘channel blocking’’ occurred.Therefore, two channels, each set at �80 nA, wereused to deliver the maximum topiramate current of�160 nA. We noted wide variation of the data, butthere did not appear to be any additional inhibitioneven when topiramate ejection was maintained for upto 10–15min, suggesting a steady state had beenreached by 2.5min. After i.v. administration of topir-amate (30mg/kg), profound (71� 10%) inhibition ofL-glutamate-evoked firing of the neurons was observed(p< 0.001, n¼ 7 cats; Figure 2).
Activity of cells in the trigeminocervicalcomplex evoked by stimulation oftrigeminovascular afferents
There was no significant inhibition of activity in cells inthe trigeminocervical complex evoked by stimulation of
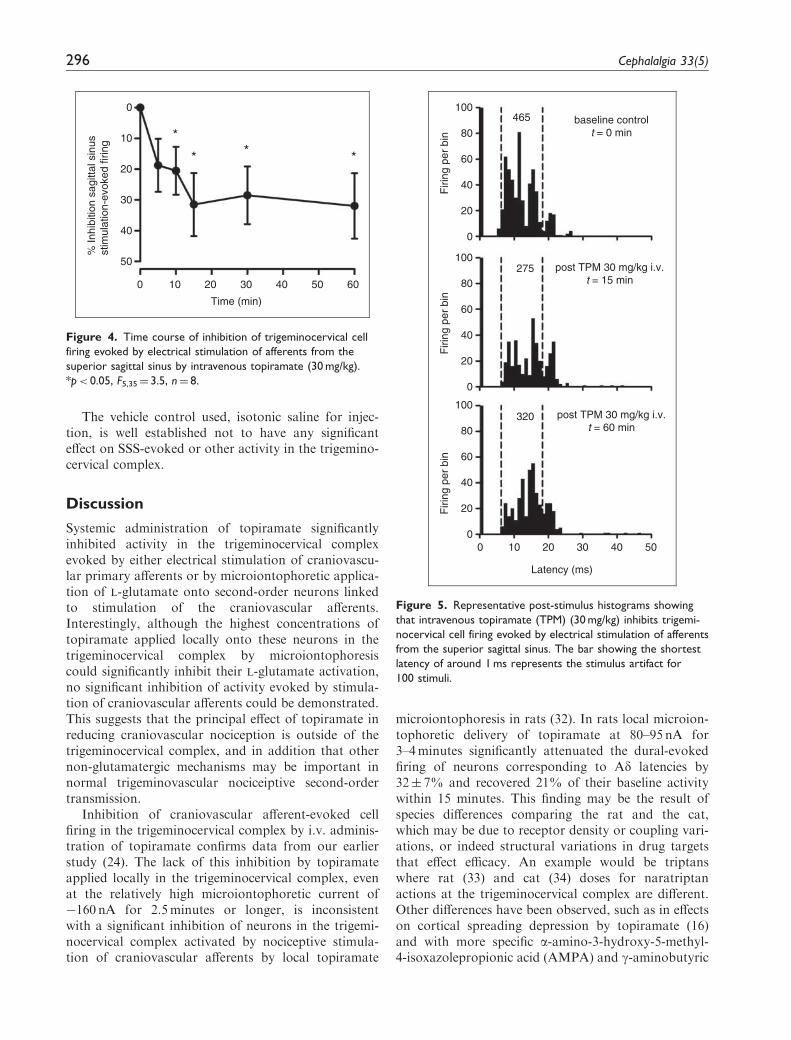
afferents from the SSS by local microiontophoreticapplication of topiramate: �2� 5% inhibition of cra-niovascular afferent evoked activity at �160 nA for upto five minutes (p¼ 0.762, n¼ 13 cells; Figure 3). Evenwhen this high microiontophoretic current was appliedfor up to 15minutes, there did not appear to beany additional affect. However, craniovascular affer-ent-evoked cell firing in the trigeminocervical complexwas significantly inhibited by i.v. topiramate at30mg/kg within 10minutes of administration(32� 10% at 15min; F5,35¼ 3.5, p< 0.05, n¼ eightcats; Figures 3–5).
recovered site
reconstructed frommicrodrive readings
1mm
Figure 1. Lesions marking recording sites were identified his-
tologically (�) or reconstructed from microdrive readings (N).
A transverse section through the spinal cord at the level of C2 is
represented. Although the positions of the recorded units are
mapped to only one side of the cord in the figure, they represent
results obtained from both the left-hand side and right-hand side
of the spinal cord. Scale bar represents a distance of 1 mm in
both directions.
100
50NaCI
–30–40 nA(n=3)
NaCI–60–80 nA
(n=8)
NaCI–160 nA
(n=4)
TPM–30–40 nA
(n=5)
TPM–60–80 nA
(n=14)
TPM–160 nA(n=14)
TPM30 mg/kg i.v.
(n=7)
0
–50
–100
% B
asel
ine
L-gl
utam
ate-
evok
ed fi
ring
Figure 2. Only maximum currents of microiontophoretically
applied topiramate (TPM) (�160 nA) significantly inhibited tri-
geminocervical cell firing evoked by L-glutamate, whereas intra-
venous (i.v.) systemic administration of topiramate (30 mg/kg)
effected profound inhibition (70� 10%) of L-glutamate-evoked
activity of second-order neurons linked to stimulation of trige-
minovascular afferents. Error bars� SEM. *p< 0.5, ***p< 0.001.
Figure 3. Trigeminocervical cell firing evoked by electrical
stimulation of afferents from the superior sagittal sinus is sig-
nificantly inhibited 15 min after 30 mg/kg intravenous topiramate
(TPM) (*p< 0.05, n¼ 8), but not when locally applied in the
trigeminocervical complex at microiontophoretic currents of up
to �160 nA for up to 5 minutes (p¼ 0.762, n¼ 13).
Storer and Goadsby 295
The vehicle control used, isotonic saline for injec-tion, is well established not to have any significanteffect on SSS-evoked or other activity in the trigemino-cervical complex.
Discussion
Systemic administration of topiramate significantlyinhibited activity in the trigeminocervical complexevoked by either electrical stimulation of craniovascu-lar primary afferents or by microiontophoretic applica-tion of L-glutamate onto second-order neurons linkedto stimulation of the craniovascular afferents.Interestingly, although the highest concentrations oftopiramate applied locally onto these neurons in thetrigeminocervical complex by microiontophoresiscould significantly inhibit their L-glutamate activation,no significant inhibition of activity evoked by stimula-tion of craniovascular afferents could be demonstrated.This suggests that the principal effect of topiramate inreducing craniovascular nociception is outside of thetrigeminocervical complex, and in addition that othernon-glutamatergic mechanisms may be important innormal trigeminovascular nociceiptive second-ordertransmission.
Inhibition of craniovascular afferent-evoked cellfiring in the trigeminocervical complex by i.v. adminis-tration of topiramate confirms data from our earlierstudy (24). The lack of this inhibition by topiramateapplied locally in the trigeminocervical complex, evenat the relatively high microiontophoretic current of�160 nA for 2.5minutes or longer, is inconsistentwith a significant inhibition of neurons in the trigemi-nocervical complex activated by nociceptive stimula-tion of craniovascular afferents by local topiramate
microiontophoresis in rats (32). In rats local microion-tophoretic delivery of topiramate at 80–95 nA for3–4minutes significantly attenuated the dural-evokedfiring of neurons corresponding to Ad latencies by32� 7% and recovered 21% of their baseline activitywithin 15 minutes. This finding may be the result ofspecies differences comparing the rat and the cat,which may be due to receptor density or coupling vari-ations, or indeed structural variations in drug targetsthat effect efficacy. An example would be triptanswhere rat (33) and cat (34) doses for naratriptanactions at the trigeminocervical complex are different.Other differences have been observed, such as in effectson cortical spreading depression by topiramate (16)and with more specific a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and g-aminobutyric
50
50 60
40
40
30
Time (min)
% In
hibi
tion
sagi
ttal s
inus
stim
ulat
ion-
evok
ed fi
ring
30
20
20
10
10
0
0
Figure 4. Time course of inhibition of trigeminocervical cell
firing evoked by electrical stimulation of afferents from the
superior sagittal sinus by intravenous topiramate (30 mg/kg).
*p< 0.05, F5,35¼ 3.5, n¼ 8.
100
80
60
40
20
465
Firi
ng p
er b
inF
iring
per
bin
Firi
ng p
er b
in
baseline controlt = 0 min
post TPM 30 mg/kg i.v.t = 15 min
post TPM 30 mg/kg i.v.t = 60 min
320
275
0
100
80
60
40
20
0
100
80
60
40
4030
Latency (ms)
10 50
20
200
0
Figure 5. Representative post-stimulus histograms showing
that intravenous topiramate (TPM) (30 mg/kg) inhibits trigemi-
nocervical cell firing evoked by electrical stimulation of afferents
from the superior sagittal sinus. The bar showing the shortest
latency of around 1 ms represents the stimulus artifact for
100 stimuli.
296 Cephalalgia 33(5)

acid (GABA) receptor modulators (35). It is clear thecat brain is more complex, for example its gyrencepha-lic cerebral cortex compared with the less-evolved lis-sencephalic rat brain, and triptan, 5-HT1B/1D
pharmacology is clearly different (36). A further differ-ence may have been anesthesia based, in rat pentobar-bital, and in cat a-chloralose; such differences canproduce complex interactions. In general terms thedata are consistent with the general caution in transla-tional research when studying therapeutic issues in non-human species.
Comparable dosing between laboratory animals andhumans is complex. The typical dose for migraine pre-vention of topiramate varies from 50 to 200mg daily,about 0.7 to 2.9mg/kg (37), compared to 30mg/kg usedhere and previously (16,24), or 60mg/kg used in oralstudies of CSD (17). In humans, free blood plasmalevels of topiramate at therapeutic oral doses of 25 to400mg/day range from 3 to 45 mM (38), although this isvariable (39). Bioavailability in humans is about 80%and not significantly affected by food intake (JanssenPharmaceuticals, 2011, Topamax (topiramate) pre-scribing information; www.topamax.com, accessed9 December 2011). At the highest therapeutic levels,a single dose of 1200mg produces a plasma concentra-tion of about 85 mM in humans (40). In male rats topir-amate administered orally is rapidly and almostcompletely absorbed with a bioavailability approaching100% (41), and plasma protein binding is about 15%; a30mg/kg oral dose will result in a plasma concentrationof �90 mM and appears to enter the CNS parenchymareadily to produce a concentration in whole brain ofabout 30 mM, despite its relatively low logP value ofabout 0.5 (9). In rats, an oral dose of 20mg/kg twicedaily is considered equivalent to a dose of about 4mg/kg in humans (9,40) and thus an oral or i.v. dose of30mg/kg in male rats is considered equivalent to a totalblood plasma (area under the curve (AUC)) exposureof about 3mg/kg in humans (Richard Shank andVirginia Smith-Swintosky, 2002, personal communica-tion to RJS). Therefore an i.v. dose of 30mg/kg in malerats is equivalent to the higher therapeutic dose of200mg orally used for migraine prevention, particularlysince all such dosing is effectively chronic. On the otherhand a single i.v. dose of 50mg/kg was reported toproduce a rat brain diasylate concentration of 10 mMand distribution studies of [14C] topiramate at 20mg/kgoral dose showed a peak whole brain concentration of11 mM (41). Renal clearance of topiramate variesbetween species and is high in rodents (42). However,to our knowledge the renal clearance in cats isunidentified.
At a molecular level, topiramate has multiple mech-anisms of action at concentrations in the human thera-peutic range of 25–1200mg per day that produce free
plasma concentrations up to around 100 mM todecrease neuronal excitation and enhance inhibition(9). These mechanisms include inhibitory effects onAMPA/kainate-type ionotropic glutamate receptors(43), blockade of voltage-activated Naþ (44) andCa2þ channels (45), positive modulatory effects onGABAA receptors (46), inhibition of carbonic anhy-drase isoenzymes (47), and inhibition of aquaporin-4water channels (APQ4) (48). Topiramate may also actto reduce abnormally high brain glutamate levels (9).The effect of topiramate may in part involvelocal inhibition of glutamatergic transmission withinthe trigeminocervical complex through its action onkainate receptors, as they are found in the trigemino-cervical complex (32), and possibly AMPA receptors,but not N-methyl-D-aspartate (NMDA) receptorsbecause topiramate does not appear to act on themeven at relatively high concentrations (100 mM) (43).Among antiepileptic drugs, the effect of topiramateon kainate-evoked currents appears to be unique to it,and consistent with a dampening of neuronalexcitability.
A number of the mechanisms of action of topira-mate involve known targets in the trigeminocervicalcomplex. These include glutamatergic (28,49),voltage-gated calcium channel (50), and GABAergicmechanisms (51). Fast Naþ channel and carbonic anhy-drase-related mechanisms have not been as clearlydescribed. Lamotrigine and oxcarbazepine, whichmay be even more effective at voltage-sensitive Naþ
channel blockade than topiramate, have been foundineffective for migraine prophylaxis (52,53), maytherefore not be important targets. The well-knowneffect of pan-carbonic anhydrase inhibition by topira-mate (47), consequently activating potassium currentsas a result of its effects on intracellular pH, extracellularpH or both (54), may be significant. Theoretically, theactivation of potassium conductance is an excellentmechanism by which to control neuronal hyperexcit-ability (55).
There are descending influences on neurons in thetrigeminocervical complex, and in higher species pre-ventives may primarily operate in supramedullaryregions such as the cortex, midbrain periaqueductalgray (PAG), and rostral ventromedial medulla(RVM) (8). An effect by topiramate in the PAG orRVM to modulate positively GABAA receptors on neu-rons with descending facilitatory influence (ON cells),damping their activity and shifting the balance of des-cending influences toward inhibitory neurons (OFFcells), would reduce activity in the trigeminocervicalcomplex (56). Ayata and colleagues (17) suggest aneffect of antiepileptic drugs at the cortex, showinginhibition of the frequency of cortical spreading depres-sion with the possibility of corticofugal influences
Storer and Goadsby 297

contributing to the modulation of migraine pain (57).In patch-clamp studies, topiramate showed a higherpotency and efficiency of inhibiting high-voltage-acti-vated Ca2þ currents in PAG neurons than in corticalneurons and inhibited P/Q-, N-, and L- channels (58).Such an action in the ventrolateral PAG would modu-late descending inhibition of the trigeminocervical com-plex (59). Braga and colleagues (60) found thattopiramate interacts with GluK1 kainate receptors onrat amygdala interneurons and that it can shape inhib-ition in the amygdala by a direct action on GABAA
receptors. The amygdala has been shown to be import-ant in trigeminal nociception (61).
If one takes the data presented here together withthat from our earlier study (24), the conclusion must bethat the principal effect of topiramate is outside thetrigeminocervical complex, at least in higher species.By providing detailed knowledge of the anatomicalpharmacology of effective antimigraine preventives,we can better target more newly recognized options.While systemic administration better mimics the clinicaluse of antimigraine compounds, further explorationwith local administration techniques, such as microion-tophoresis and microinjection, may provide clues as tothe elusive sites of action of migraine-preventivecompounds.
Clinical implications
. Topiramate inhibits activation of trigeminocervical neurons in experimental settings.
. The effect of topiramate on craniovascular afferents may be at a site other than the trigeminocervicalcomplex.
Funding
This work was supported by the Johnson & JohnsonPharmaceutical Research and Development.
Acknowledgements
The authors thank Paul Hammond and Michele Lasalandra
for technical assistance, and Simon Akerman for his criticalreading of this manuscript. We thank David Jacobs, RoyTwyman, Virginia Smith-Swintosky, Connie Bolig, and
Richard Shank for their kind gift of topiramate and itssodium salt.
Conflict of interest
RJS has nothing to disclose.PJG is on the boards of Allergan, Colucid, MAP pharma-
ceuticals, Merck, Sharpe and Dohme, eNeura, Neuroaxon,Autonomic Technologies Inc., Boston Scientific, Eli Lilly,Medtronic, Linde Gases, Arteaus, AlderBio and Bristol-
Myers Squibb. He has consulted for Gammacore, Pfizer,Nevrocorp, Lundbeck, Zogenix, Impax and Dr. Reddy, andhas been compensated for expert legal testimony. He has
received grant support from GlaxoSmithKline, MAP, MSD,eNeura, and Amgen. He has received honoraria for speakingfromMSD, Pfizer, Allergan, and Mennarini, and payment foreditorial work from Journal Watch Neurology and for
developing educational materials for the AmericanHeadache Society.
References
1. Lipton RB, Stewart WF, Diamond S, et al. Prevalence
and burden of migraine in the United States: Data fromthe American Migraine Study II. Headache 2001; 41:646–657.
2. Goadsby PJ, Lipton RB and Ferrari MD. Migraine –
current understanding and treatment. N Engl J Med2002; 346: 257–270.
3. Lipton RB, Bigal ME, Diamond M, et al. Migraine
prevalence, disease burden, and the need for preventivetherapy. Neurology 2007; 68: 343–349.
4. Welch KM. Brain hyperexcitability: The basis for anti-
epileptic drugs in migraine prevention. Headache 2005;45: S25–S32.
5. Stankewitz A and May A. The phenomenon of changesin cortical excitability in migraine is not migraine-specific
– a unifying thesis. Pain 2009; 145: 14–17.6. Dichgans M, Freilinger T, Eckstein G, et al. Mutation in
the neuronal voltage-gated sodium channel SCN1A
causes familial hemiplegic migraine. Lancet 2005; 366:371–377.
7. Niebur E, Hsiao SS and Johnson KO. Synchrony: A
neural mechanism for attentional selection? Curr OpinNeurobiol 2002; 12: 190–194.
8. Akerman S, Holland P and Goadsby PJ. Diencephalic
and brainstem mechanisms in migraine. Nat RevNeurosci 2011; 12: 570–584.
9. Shank RP, Gardocki JF, Streeter AJ, et al. An overviewof the preclinical aspects of topiramate: Pharmacology,
pharmacokinetics, and mechanism of action. Epilepsia2000; 41: S3–S9.
10. Brandes JL, Saper JR, Diamond M, et al. Topiramate for
migraine prevention: A randomized controlled trial.JAMA 2004; 291: 965–973.
11. Silberstein SD, Neto W, Schmitt J, et al. Topiramate in
migraine prevention: Results of a large controlled trial.Arch Neurol 2004; 61: 490–495.
12. Diener HC, Goadsby PJ, Bigal ME, et al. Utility of topir-amate for the treatment of patients with chronic migraine
in the presence or absence of acute medication overuse.Cephalalgia 2009; 29: 1021–1027.
298 Cephalalgia 33(5)

13. Silberstein SD, Lipton RB, Dodick DW, et al. Efficacyand safety of topiramate for the treatment of chronicmigraine: A randomized, double-blind, placebo-con-
trolled trial. Headache 2007; 47: 170–180.14. Diener HC, Tfelt-Hansen P, Dahlof C, et al. Topiramate
in migraine prophylaxis – results from a placebo-controlled trial with propranolol as an active control.
J Neurol 2004; 251: 943–950.15. Evers S, Afra J, Frese A, et al. EFNS guideline on the
drug treatment of migraine – report of an EFNS task
force. Eur J Neurol 2009; 16: 968–981.16. Akerman S and Goadsby PJ. Topiramate inhibits cortical
spreading depression in rat and cat: Impact in migraine
aura. Neuroreport 2005; 16: 1383–1387.17. Ayata C, Jin H, Kudo C, et al. Suppression of cortical
spreading depression in migraine prophylaxis. Ann
Neurol 2006; 59: 652–661.18. Bogdanov VB, Multon S, Chauvel V, et al. Migraine pre-
ventive drugs differentially affect cortical spreadingdepression in rat. Neurobiol Dis 2011; 41: 430–435.
19. Lauritzen M. Pathophysiology of the migraine aura. Thespreading depression theory. Brain 1994; 117: 199–210.
20. Smith MI, Read SJ, Chan WN, et al. Repetitive cortical
spreading depression in a gyrencephalic feline brain: Inhi-bition by the novel benzoylamino-benzopyran SB-220453. Cephalalgia 2000; 20: 546–553.
21. Goadsby PJ, Ferrari MD, Csanyi A, et al. Randomizeddouble blind, placebo-controlled proof-of-concept studyof the cortical spreading depression inhibiting agent tona-bersat in migraine prophylaxis. Cephalalgia 2009; 29:
742–750.22. Hauge AW, Asghar MS, Schytz HW, et al. Effects of
tonabersat on migraine with aura: A randomised,
double-blind, placebo-controlled crossover study.Lancet Neurol 2009; 8: 718–723.
23. Andreou AP, Summ O, Charbit AR, et al. Animal
models of headache: From bedside to bench and backto beside. Expert Rev Neurother 2010; 10: 389–411.
24. Storer RJ and Goadsby PJ. Topiramate inhibits trigemi-
novascular neurons in the cat. Cephalalgia 2004; 24:1049–1056.
25. Storer RJ and Goadsby PJ. Topiramate has a locus ofaction outside of the trigeminocervical complex.
Cephalalgia 2005; 25: 934.26. Storer RJ, Butler P, Hoskin KL, et al. A simple method,
using 2-hydroxypropyl-b-cyclodextrin, of administering
a-chloralose at room temperature. J Neurosci Methods1997; 77: 49–53.
27. Hu JW, Dostrovsky JO and Sessle BJ. Functional proper-
ties of neurons in cat trigeminal subnucleus caudalis(medullary dorsal horn). I. Responses to oral-facial nox-ious and nonnoxious stimuli and projections to thalamusand subnucleus oralis. J Neurophysiol 1981; 45: 173–192.
28. Storer RJ and Goadsby PJ. Trigeminovascular nocicep-tive transmission involves N-methyl-D-aspartate andnon-N-methyl-D-aspartate glutamate receptors.
Neuroscience 1999; 90: 1371–1376.29. Almarsson O, Remenar J, Peterson MN and inventors.
Transform Pharmaceuticals, assignee. Topiramate
sodium trihydrate. United States Patent 2003: 6559293.
30. Bloom FE. To spritz or not to spritz: The doubtful valueof aimless iontophoresis. Life Sci 1974; 14: 1819–1834.
31. Stone TW. Microiontophoresis and pressure ejection.
In: Smith AD (ed.) Methods in the neurosciences. Chich-ester: John Wiley and Sons, 1985.
32. Andreou AP and Goadsby PJ. Topiramate in the treat-ment of migraine: A kainate (glutamate) receptor antag-
onist within the trigeminothalamic pathway. Cephalalgia2011; 31: 1343–1358.
33. Akerman S, Holland PR, Summ O, et al. A translational
in vivo model of trigeminal autonomic cephalalgias –therapeutic characterization. Brain 2012. Epub ahead ofprint 11 October 2012.
34. Goadsby PJ and Knight YE. Inhibition of trigeminalneurons after intravenous administration of naratriptanthrough an action at the serotonin (5HT1B/1D) receptors.
Br J Pharmacology 1997; 122: 918–922.35. Holland PR, Akerman S and Goadsby PJ. Cortical
spreading depression associated cerebral blood flowchanges induced by mechanical stimulation are modu-
lated by AMPA and GABA receptors. Cephalalgia2010; 30: 519–527.
36. Goadsby PJ. The pharmacology of headache. Progress in
Neurobiology 2000; 62: 509–525.37. Bussone G, Diener H-C, Pfeil J, et al. Topiramate 100
mg/day in migraine prevention: A pooled analysis of
double-blinded randomised controlled trials. Int J ClinPract 2005; 59: 961–968.
38. Shank RP, Doose DR, Streeter AJ, et al. Plasma andwhole blood pharmacokinetics of topiramate: The role
of carbonic anhydrase. Epilepsy Res 2005; 63: 103–112.39. Johannessen SI, Battino D, Berry DJ, et al. Therapeutic
drug monitoring of the newer antiepileptic drugs. Ther
Drug Monit 2003; 25: 347–363.40. Doose DR, Walker SA, Gisclon LG, et al. Single-dose
pharmacokinetics and effect of food on the bioavailabil-
ity of topiramate, a novel antiepileptic drug. J ClinPharmacol 1996; 36: 884–891.
41. Masucci JA, Ortegon ME, Jones WJ, et al. In vivo micro-
dialysis and liquid chromatography/thermospray massspectrometry of the novel anticonvulsant 2,3:4,5-bis-O-(1-methylethylidene)-beta-D-fructopyranose sulfamate(topiramate) in rat brain fluid. J Mass Spectrom 1998;
33: 85–88.42. Wu WN and McKown LA. Recent advances in biotrans-
formation of CNS and cardiovascular agents. Curr Drug
Metab 2000; 1: 255–270.43. Gibbs 3rd JW, Sombati S, DeLorenzo RJ, et al. Cellular
actions of topiramate: Blockade of kainate-evoked
inward currents in cultured hippocampal neurons.Epilepsia 2000; 41: S10–S16.
44. Zona C, Ciotti MT and Avoli M. Topiramate attenuatesvoltage-gated sodium currents in rat cerebellar granule
cells. Neurosci Lett 1997; 231: 123–126.45. Zhang X, Velumian AA, Jones OT, et al. Modulation
of high-voltage-activated calcium channels in den-
tate granule cells by topiramate. Epilepsia 2000; 41:S52–S60.
46. White HS, Brown SD, Woodhead JH, et al. Topiramate
modulates GABA-evoked currents in murine cortical
Storer and Goadsby 299

neurons by a nonbenzodiazepine mechanism. Epilepsia2000; 41: S17–S20.
47. Dodgson SJ, Shank RP and Maryanoff BE. Topiramate
as an inhibitor of carbonic anhydrase isoenzymes.Epilepsia 2000; 41: S35–S39.
48. Huber VJ, Tsujita M, Yamazaki M, et al. Identificationof arylsulfonamides as aquaporin 4 inhibitors. Bioorg
Med Chem Lett 2007; 17: 1270–1273.49. Andreou AP, Holland PR and Goadsby PJ. Pre- and
post-synaptic involvement of GluR5 kainate receptors
in trigeminovascular nociceptive processing. Cephalalgia2007; 27: 605.
50. Shields KG, Storer RJ, Akerman S, et al. Calcium chan-
nels modulate nociceptive transmission in the trigeminalnucleus of the cat. Neuroscience 2005; 135: 203–212.
51. Storer RJ, Akerman S and Goadsby PJ. GABA receptors
modulate trigeminovascular nociceptive neurotransmis-sion in the trigeminocervical complex. Br J Pharmacol2001; 134: 896–904.
52. Steiner TJ, Findley LJ and Yuen AW. Lamotrigine versus
placebo in the prophylaxis of migraine with and withoutaura. Cephalalgia 1997; 17: 109–112.
53. Silberstein S, Saper J, Berenson F, et al. Oxcarbazepine in
migraine headache: A double-blind, randomized, pla-cebo-controlled study. Neurology 2008; 70: 548–555.
54. Herrero AI, Del Olmo N, Gonzalez-Escalada JR, et al.
Two new actions of topiramate: Inhibition of depolariz-ing GABA(A)-mediated responses and activation of apotassium conductance. Neuropharmacology 2002; 42:210–220.
55. Storer RJ, Yin R, Immke DC, et al. Large conductancecalcium-activated potassium channels (BKCa) modulate
trigeminovascular nociceptive transmission. Cephalalgia2009; 29: 1242–1258.
56. Fields HL, Basbaum AI and Heinricher MM. Central
nervous system mechanisms of pain modulation.In: McMahon SB, Koltzenburg M (eds) Wall and Mel-zack’s textbook of pain, 5th edn. London: ElsevierChurchill Livingston, 2006, pp.125–142.
57. Noseda R, Constandil L, Bourgeais L, et al. Changes ofmeningeal excitability mediated by corticotrigeminal net-works: A link for the endogenous modulation of migraine
pain. J Neurosci 2010; 30: 14420–14429.58. Martella G, Costa C, Pisani A, et al. Antiepileptic drugs
on calcium currents recorded from cortical and PAG
neurons: Therapeutic implications for migraine.Cephalalgia 2008; 28: 1315–1326.
59. Knight YE, Bartsch T, Kaube H, et al. P/Q-type calcium
channel blockade in the PAG facilitates trigeminal noci-ception: A functional genetic link for migraine?J Neurosci 2002; 22: 1–6.
60. Braga MF, Aroniadou-Anderjaska V, Li H, et al. Topir-
amate reduces excitability in the basolateral amygdala byselectively inhibiting GluK1 (GluR5) kainate receptorson interneurons and positively modulating GABAA
receptors on principal neurons. J Pharmacol Exp Ther2009; 330: 558–566.
61. Noseda R, Monconduit L, Constandil L, et al. Central
nervous system networks involved in the processing ofmeningeal and cutaneous inputs from the ophthalmicbranch of the trigeminal nerve in the rat. Cephalalgia2008; 28: 813–824.
300 Cephalalgia 33(5)