the electronic mechanism ruling the dihydrogen bonds and halogen bonds in weakly bound systems of...
TRANSCRIPT

ORIGINAL PAPER
The electronic mechanism ruling the dihydrogen bondsand halogen bonds in weakly bound systems of H3SiH···HOXand H3SiH···XOH (X = F, Cl, and Br)
Boaz G. Oliveira & Abedin Zabardasti &Hamid Goudarziafshar & Maryam Salehnassaj
Received: 9 December 2014 /Accepted: 8 February 2015 /Published online: 10 March 2015# Springer-Verlag Berlin Heidelberg 2015
Abstract The dihydrogen bond complexes (H3SiH∙∙∙HOX)and halogen bond complexes (H3SiH∙∙∙XOH) formed betweenSiH4 and hypohalous acids HOX (X = F, Cl, and Br) havebeen studied at the MP2/6-311++G(2d,2p) computational lev-el. The analyses of structure and infrared vibration frequencieshave revealed tendencies in the red shifts and blue shifts of thestretch frequencies of the Si–H, H–O, and O–X bonds. Be-sides the computation of the interaction energies, punctualatomic charges and charge transference amounts were deter-mined at light of the Natural Bond Orbital (NBO) approach,by which the quantifications of the s- and p-characters ofhydrogen, oxygen, and silicon also were useful to unveil thefrequency shifts aforementioned. With the purpose to eluci-
date the donor/acceptor interface along the charge transfermechanism between the dihydrogen bonds and halogenbonds, the application of the hierarchical cluster analysis(HCA) and principal component analysis (PCA) chemometrictechniques were useful in this regard. Moreover, the interac-tion strengths of the H3SiH∙∙∙HOX and H3SiH∙∙∙XOH com-plexes was computed through a model that embodies the fre-quency shifts and topological parameters derived from quan-tum theory of atoms in molecules (QTAIM).
Keywords Hydrogen bond . Halogen bonds . HCA . NBO .
PCA . QTAIM
Introduction
Over the past few decades that molecular clusters with weaklybound characteristics have attracted a great interest of the con-temporary chemical science [1–10]. In line with this, a grow-ing number of experimental and theoretical evidences confirmthat hydrogen bonds and halogen bonds [11–15] play a crucialrole in research of specific areas as well as interdisciplinaryones, of which we can cite the biochemistry and, in particular,the medicinal chemistry [16–18]. On a purely theoreticalviewpoint, by means of energy decomposition scheme[19–21], scientists have an insight into the nature of hydrogenbonds and halogen bonds and think that the electrostatic inter-action, polarization, charge transfer, and dispersion are all re-sponsible for the stability of intermolecular complexes formedin light of these interactions [22]. Nevertheless, some researchgroups have appreciated a deeper recognition that halogenbonding involves the interaction of positive σ-holes (positiveregions of electrostatic potential on the extensions of the
Electronic supplementary material The online version of this article(doi:10.1007/s00894-015-2616-2) contains supplementary material,which is available to authorized users.
A. ZabardastiDepartment of Chemistry, Faculty of Science, Lorestan University,Khorramabad, Iran
H. GoudarziafsharDepartment of Chemistry, Faculty of Science, Ilam University,Ilam, Iran
H. GoudarziafsharDepartment of Chemistry, Faculty of Science SayyedJamaleddinasadabadi University, Asadabad, Iran
M. SalehnassajDepartment of Chemistry, Faculty of Science, Ilam University,Ilam, Iran
B. G. Oliveira (*)Instituto de Ciências Ambientais e Desenvolvimento Sustentável,Universidade Federal da Bahia, 47801-100 Barreiras, Brazile-mail: [email protected]
J Mol Model (2015) 21: 77DOI 10.1007/s00894-015-2616-2

covalent bond to the halogen) on the halogen with negativesites on other molecules [23, 24]. Nevertheless, some studieshave shown that halogen bonds are analogical to hydrogenbonds [25, 26], although once considered the hydrogen bondprofile [27] whose lone electron pairs of the halogen may be aLewis base to interact with proton donors [28], in this concep-tion the halogen bonds and hydrogen bonds might be oneunified interaction type. On the other hand, there is other un-usual interaction wherein two hydrogen atoms bind to eachother, known as dihydrogen bond [29, 30], which plays a keyrole in the chemistry of the solid state as well as inmany othersbranches of science. Once it has also been carried out forhydrogen bonds and halogen bonds [31, 32], the decomposi-tion energy studies reveal that stabilization of dihydrogen-bonded systems is ruled by both electrostatic as well as induc-tive effects [33], although the dispersion contributes moder-ately or even decisively in this regard [34].
About the interaction strength, it is widely known thatintermolecular systems formed by hydrogen bonds arestronger bound in comparison to those linked throughthe halogen bonds [35]. However, if the main goal is theelucidation of the interaction strengths of halogen bondsand dihydrogen bonds, a direct parallelism between themis essentially required. With this insight, this current workaims to investigate the potentiality of some molecules toform dihydrogen bonds and halogen bonds and in accordwith some molecular parameters, such as the frequenciesshifted to downward (red) or upward (blue) values in theinfrared spectrum [36–39], the determination of the inter-action strength seems reasonable to be done. In additionto that, our great goals are also focused in the investiga-tion of structural, electronic, and topological properties ofcomplexes (H3SiH∙∙∙HOX and H3SiH∙∙∙XOH) withdihydrogen bonds (H∙∙∙HO) and halogen bonds (H∙∙∙XO)formed between SiH4 and HOX, where the last one as-sumes X= F, Cl, and Br. Some time ago, Murray et al.[40] have proposed a detailed discussion about theappearing of red shifts and blue shifts in σ-hole bonds,whose effects points out that intermolecular systems areformed with higher and lower interaction energies, respec-tively. More recently, however, Clark [41] has shown amanner by which the σ-hole concept can rationalize theblue shifts on hydrogen bonds by using electrostatic per-turbations [42]. In addition, Li and collaborators [43] havepointed out that systems formed by halogen bonds withHOX are stronger rather than those with hydrogen bondinteractions. In this context of interaction strength, actu-ally this comparative work may serve us as a guide tostudy the H3SiH∙∙∙XOH and H3SiH∙∙∙HOX complexes,which differently, the interactions and related vibrationsphenomena such as frequencies shifted either to red orto blue, all of them occur via halogen bonds as well asby means of dihydrogen bonds.
Theoretical methods and chemometric techniques
In order to unveil the interaction strengths of halogen bondsand dihydrogen bonds in the H3SiH∙∙∙XOH and H3SiH∙∙∙HOXcomplexes, besides an efficient theoretical level with calcula-tions performed at the second-order perturbation theory jointlywith a complete basis set, the application of the quantum the-ory of atoms in molecules (QTAIM) [44, 45] and its topolog-ical surface descriptors can be useful. Surely, the topologicalprotocol of the QTAIM can furnish relationships with the in-teraction energies and frequency shifts, and thereby the elec-
Due to this, the protocol of the natural bond orbital (NBO)[46] will be used, by which besides the calculation of thebinding energies, the contributions of the s- and p-hybrid or-bitals of H, O, and X (X= F, Cl, and Br) may be reliable tointerpret the frequency shifts outlined after formation of thecomplexes. This is a procedure widely used in intermolecularstudies [47–49] mainly those with hydrogen bond features[50, 51]. In line with this, a comparison between the resultsof structure, electronic parameters, and infrared stretch modesobtained in this current research and those documented by thespecialized literature may be valuable as well.
As mentioned above, the NBO computations for the con-tributions of the s- and p-hybrid orbitals and binding energiessupported by the Fock operator will be performed [52]. How-ever, the NBO tools can calculate the atomic charges, which inthis current work seems useful to predict a charge transferencemechanism among the H, O and F, Cl or Br atoms. However,the occurrence of electronic cooperative effect on the covalentbonds and intermolecular interactions seems reliable. As such,the calculation of the punctual atomic charges or even thedetermination of the charge transfers [53] on the frontier or-bitals (HOMO/LUMO) likely underestimate to some extentthe real electronic behavior of the dihydrogen bonds and hal-ogen bonds in the H3SiH∙∙∙HOX and H3SiH∙∙∙XOH com-plexes. In view of this and in accord with the results reportedby the specialized literature [54, 55], a chemometric analysis[56] with pattern recognition will be also performed, specifi-cally the hierarchical cluster analysis (HCA) [57] and princi-pal component analysis (PCA) [58].
Computational methods and procedure
The GAUSSIAN 03 package of codes were used to performall calculations of this current work [59]. The geometries ofthe SiH4, HOF, HOCl, and HOBr monomers and their com-plexes were fully optimized at the MP2 theoretical method[60] with the 6-311++G(2d,2p) basis set. The calculations ofthe vibration frequencies confirm the structures as minima bywhich the zero-point energy (ZPE) [61] can be evaluated.Besides the ZPE amounts, the counterpoise procedure whose
77 Page 2 of 11 J Mol Model (2015) 21: 77
tronic dynamism of the σ-hole in HOX can be investigated.

basis is the Boys and Bernardi’s basis set superposition error(BSSE) [62] was used to correct the interaction energies. TheAIM2000 1.0 [63], AIMAll 11.12.19 [64], and AIM-UC 1.3[65, 66] suite of codes determined the topological descriptorsof the QTAIM approach. The NBO calculations served toanalyze the interactions between the occupied and unoccupiedorbitals supplied with GAUSSIAN 03. In the chemometricstudy, the HCA and PCA pattern recognitions were obtainedthrough the CHEMOFACE 1.5 program [67, 68].
Results and discussion
Structural parameters and infrared spectra
The optimized geometries of the dihydrogen-bonded com-plexes H3SiH ∙ ∙ ∙HOF (I) , H3SiH ∙ ∙ ∙HOCl (II) , andH3SiH∙∙∙HOBr (III) are illustrated in Fig. 1 whereas theha logen-bonded complexes H3SiH ∙ ∙ ∙FOH ( IV ) ,H3SiH∙∙∙ClOH (V), and H3SiH∙∙∙BrOH (VI) are depicted inFig. 2. Through the calculations performed at MP2/6-311++G(2d,2p) theoretical level, the values of the bond lengths andintermolecular distances (dihydrogen bonds and halogenbonds) of these six systems are listed in Table 1. Firstly, ana-lyzing the values of intermolecular distances of 1.9328 (I),1.9107 (II), and 1.8987 Å (III) for H∙∙∙H, it is clearly per-ceived that the dihydrogen-bonded complexes are shorterbound than those formed by the X∙∙∙H halogen bonds, whosedistances are 2.8387 (IV), 2.8041 (V), and 2.6726 Å (VI). The
dihydrogen bond distances are shorter in the range of 0.68-0.90 Å than halogen bonds. Similarly to the other systemsstudied by Li and co-authors [43], these halogen-bond dis-tances present equivalent values to those found in noncovalentinteractions. In addition to that, Riley and collaborators [1]have examined the dimeric structures of halohydrocarbons,whose halogen bond lengths reach values of up to 3.12 Å orlonger. Despite the large intermolecular difference quotedabove, it is worthy to state that the most drastic structuralchange upon the formation of the I-VI complexes seems notto be the X–O σ-hole bonds, as can be seen in the results ofrSi–H, rO–H, and rO–X gathered in Table 1. With the exception
Fig. 1 Optimized geometries of the H3SiH∙∙∙HOX dihydrogencomplexes obtained from MP2/6-311++G(2d,2p) calculations
Table 1 Values of the bond lengths and intermolecular distances of thedihydrogen bonds and halogen bonds of the H3SiH∙∙∙HOX andH3SiH∙∙∙XOH set of complexes obtained from MP2/6-311++G(2d,2p)calculations
Bonds Complexes
I II III IV V VI
RH H 1.9328 1.9107 1.8987 — — —
RX H — — — 2.8387 2.8041 2.6726
rSi–H 1.4801(6.8)
1.4803(7.0)
1.4800(6.7)
1.4727(−0.6)
1.4760(2.7)
1.4786(5.3)
rO–H 0.9690(2.5)
0.9681(2.7)
0.9679(2.1)
0.9667(0.2)
0.9655(0.4)
0.9657(−0.4)
rO–X 1.4366(1.0)
1.7167(−1.0)
1.8469(−1.4)
1.4363(0.7)
1.7204(2.7)
1.8518(3.5)
* All values are given in angstroms (Å); * Value of the Si–H bond lengthbefore complexation is 1.4733 Å; * Values of the O–H bond lengthsbefore complexation are 0.9665 (H–OF), 0.9654 (H–OCl), and0.9658 Å (H–OBr); * Values of the O–X bond lengths before complex-ation are 1.4356 (HO–F), 1.7177 (HO–Cl), and 1.8483 (HO–Br) Å; *Allvalues in parentheses must be multiplied by the factor of 10–3
J Mol Model (2015) 21: 77 Page 3 of 11 77
Fig. 2 Optimized geometries of the H3SiH∙∙∙XOH halogen complexesobtained from MP2/6-311++G(2d,2p) calculations

The results of the infrared spectrum analyses are listed inTable 2. Corroborating with the structural results, the stretchfrequencies of the dihydrogen bonds oscillate more than twicein comparison with the values of the halogen bonds, although,amazingly, an opposite behavior in the absorption intensitiesis evidenced. In an overview, on the basis of spectroscopy [69]can also be stated that the dihydrogen bond complexes arestronger bound than halogen ones. Nevertheless, the bench-marks of this insight are the frequency shifts either on HOX orSiH4 after formation of the H3SiH∙∙∙HOX and H3SiH∙∙∙XOHcomplexes. In line with this, in Fig. 3 is plotted a graph bywhich an excellent relationship supported by the correlationcoefficient (R2) of −0.99 between the variations of the bondlengths (ΔrSi–H) and frequency shifts (ΔυSi–H) of the Si–Hbonds is presented [70, 71].
ΔυSi–H ¼ −2:57ΔrSi–H−6052:9 ; R2 ¼ −0:99 ð1Þ
Not only due the more drastic structural variations followedby the largest frequency shifts, in this case those from red orblue nature, the Si–H bond behaves as the most importantrather than H–O and X–O, which are less intensively
evidenced, just like has been reported by Li et al. [43]. Also,it would be expected a total agreement between the frequencyshifts (red and blue) and variations of the H–O and X–O bondlengths (increase and decrease), although unfortunately this isnot observed. The H–O bond lengths in V and VI are slightlyincreased and decreased in 0.0004Å albeit only blue-shifts areobserved in these bonds. The σ-hole bonds have well-behavior frequency shifts, blue and red respectively todihydrogen and halogen bond complexes, although aboutthe first ones only in II and III the decrease of bond lengthare in agreement with the hypsochromic profile. It can beassumed that the poor and unsystematic relationship betweenfrequency shifts and structural variations in the H–O and X–Obonds may not be a consequence of the interaction strength.Regarding the Si–H bond, however, the useful relationshipbetween frequency shifts and bond length variations mightbe attributed to the interaction strength, e.g., the binding en-ergy [72] or intermolecular energy obtained in light of thesupermolecule approach [73].
MEP analysis, interaction energies, charge transfers,and chemometry
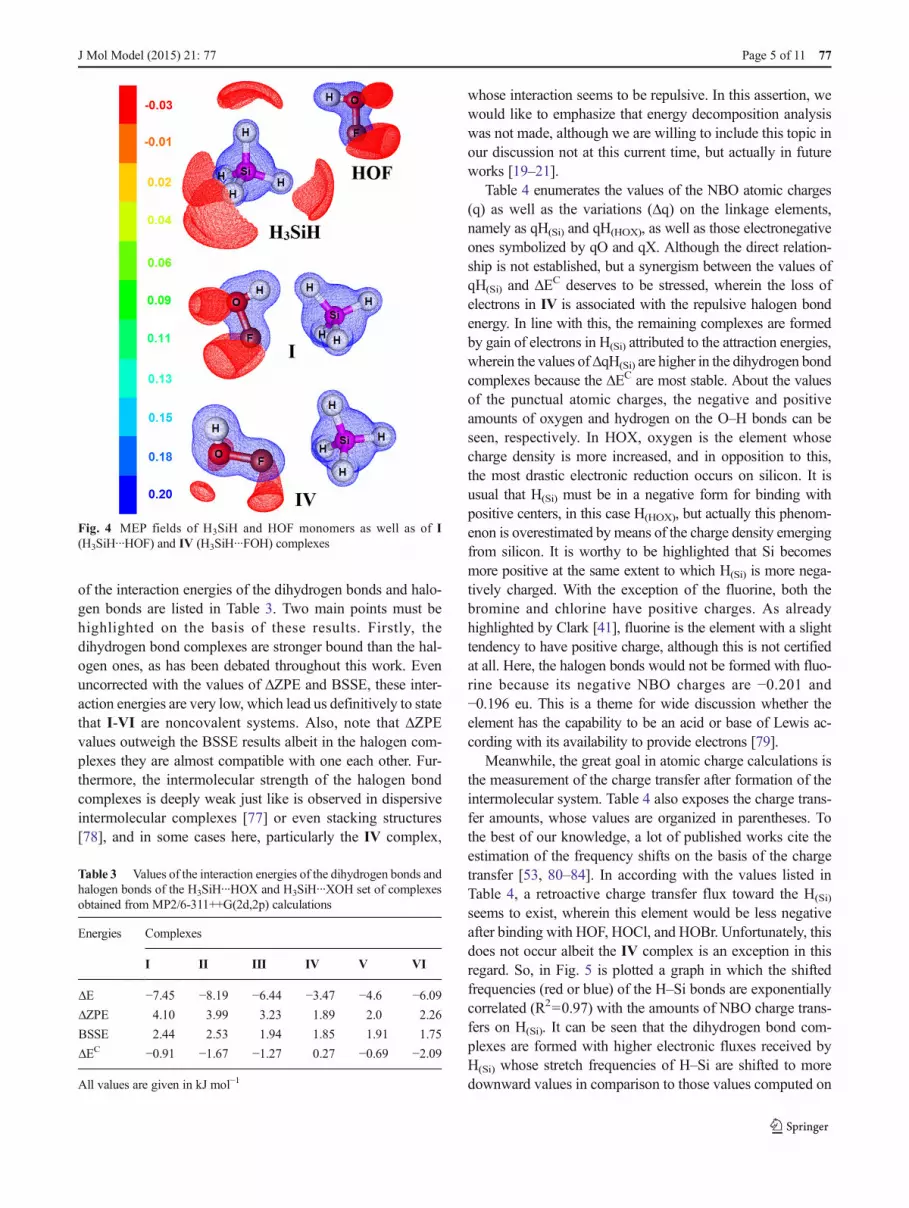
In a matter of fact, the analysis of the molecular electrostaticpotential (MEP) [70, 74–76] delineates the preferential inter-action sites upon the formation of the H3SiH∙∙∙XOH andH3SiH∙∙∙HOX complexes. Figure 4 outlines the MEP graphsof H3SiH and HOF monomers as well as of I and IV com-plexes. In H3SiH and HOF, the MEP surfaces indicate a dif-ference of −0.03 and 0.20 e.u. between the hydrogen atoms ofthe Si–H and O–H bonds. This means that preferentially thedihydrogen bonds shall be formed, although the MEP surfaceof I displays a seemingly repulsive behavior. Otherwise, thenegative MEP field of fluorine in HOF links ideally with thepositiveMEP field of hydrogen (Si–H bond) in IV. The values
Table 2 Values of the stretch frequencies and absorption intensities ofthe dihydrogen bonds and halogen bonds of the H3SiH∙∙∙HOX andH3SiH∙∙∙XOH set of complexes obtained from MP2/6-311++G(2d,2p)calculations
Modes Complexes
I II III IV V VI
υH H 102.6 114.9 164.2 — — —
IH H 13.0 3.5 2.7 — — —
υH H — — — 56.3 49.9 60.1
IX H — — — 0.26 2.0 1.3
ΔυSi–H −43.7 −44.1 −40.0 +2.4 −19.7 −39.2ISi–H,c/ISi–H,m 88.3 82.9 95.0 30.7 65.6 97.1
ΔυO–H −47.2 −47.5 −42.8 −2.1 +6.0 +3.8
IO–H,c/IO–H,m 2.8 3.9 6.6 0.9 1.8 2.1
ΔυO–X +0.5 +0.1 +2.0 −1.5 −6.9 −8.5IO–X,c/IO–X,m 0.2 6.7 1.0 1.7 1.8 2.1
* Values of Δυ and I are given in cm−1 and km mol−1 , respectively
Fig. 3 Relationship between the frequency shifts and bond lengthenhancements of the Si–H bonds
77 Page 4 of 11 J Mol Model (2015) 21: 77
of the IV complex in which a slight shortening is observed, theSi–H bond is relatively enhanced, mainly in the dihydrogen-bonded complexes. As aforementioned, the σ-hole bonds arenot affected to some extent, in particular upon the formation ofthe dihydrogen bonds. Regarding the halogen bonds, howev-er, the σ-hole bonds undergo slight and median enhancementsalbeit smaller than those computed to Si–H. Meanwhile, theH–O bond is the middle term with intermediary enhance-ments, and as such, no matter if dihydrogen bonds or halogenbonds are taken into account.
of the interaction energies of the dihydrogen bonds and halo-gen bonds are listed in Table 3. Two main points must behighlighted on the basis of these results. Firstly, thedihydrogen bond complexes are stronger bound than the hal-ogen ones, as has been debated throughout this work. Evenuncorrected with the values of ΔZPE and BSSE, these inter-action energies are very low, which lead us definitively to statethat I-VI are noncovalent systems. Also, note that ΔZPEvalues outweigh the BSSE results albeit in the halogen com-plexes they are almost compatible with one each other. Fur-thermore, the intermolecular strength of the halogen bondcomplexes is deeply weak just like is observed in dispersiveintermolecular complexes [77] or even stacking structures[78], and in some cases here, particularly the IV complex,
whose interaction seems to be repulsive. In this assertion, wewould like to emphasize that energy decomposition analysiswas not made, although we are willing to include this topic inour discussion not at this current time, but actually in futureworks [19–21].
Table 4 enumerates the values of the NBO atomic charges(q) as well as the variations (Δq) on the linkage elements,namely as qH(Si) and qH(HOX), as well as those electronegativeones symbolized by qO and qX. Although the direct relation-ship is not established, but a synergism between the values ofqH(Si) and ΔEC deserves to be stressed, wherein the loss ofelectrons in IV is associated with the repulsive halogen bondenergy. In line with this, the remaining complexes are formedby gain of electrons in H(Si) attributed to the attraction energies,wherein the values ofΔqH(Si) are higher in the dihydrogen bondcomplexes because the ΔEC are most stable. About the valuesof the punctual atomic charges, the negative and positiveamounts of oxygen and hydrogen on the O–H bonds can beseen, respectively. In HOX, oxygen is the element whosecharge density is more increased, and in opposition to this,the most drastic electronic reduction occurs on silicon. It isusual that H(Si) must be in a negative form for binding withpositive centers, in this case H(HOX), but actually this phenom-enon is overestimated by means of the charge density emergingfrom silicon. It is worthy to be highlighted that Si becomesmore positive at the same extent to which H(Si) is more nega-tively charged. With the exception of the fluorine, both thebromine and chlorine have positive charges. As alreadyhighlighted by Clark [41], fluorine is the element with a slighttendency to have positive charge, although this is not certifiedat all. Here, the halogen bonds would not be formed with fluo-rine because its negative NBO charges are −0.201 and−0.196 eu. This is a theme for wide discussion whether theelement has the capability to be an acid or base of Lewis ac-cording with its availability to provide electrons [79].
Meanwhile, the great goal in atomic charge calculations isthe measurement of the charge transfer after formation of theintermolecular system. Table 4 also exposes the charge trans-fer amounts, whose values are organized in parentheses. Tothe best of our knowledge, a lot of published works cite theestimation of the frequency shifts on the basis of the chargetransfer [53, 80–84]. In according with the values listed inTable 4, a retroactive charge transfer flux toward the H(Si)
seems to exist, wherein this element would be less negativeafter binding with HOF, HOCl, and HOBr. Unfortunately, thisdoes not occur albeit the IV complex is an exception in thisregard. So, in Fig. 5 is plotted a graph in which the shiftedfrequencies (red or blue) of the H–Si bonds are exponentiallycorrelated (R2=0.97) with the amounts of NBO charge trans-fers on H(Si). It can be seen that the dihydrogen bond com-plexes are formed with higher electronic fluxes received byH(Si) whose stretch frequencies of H–Si are shifted to moredownward values in comparison to those values computed on
Fig. 4 MEP fields of H3SiH and HOF monomers as well as of I(H3SiH∙∙∙HOF) and IV (H3SiH∙∙∙FOH) complexes
Table 3 Values of the interaction energies of the dihydrogen bonds andhalogen bonds of the H3SiH∙∙∙HOX and H3SiH∙∙∙XOH set of complexesobtained from MP2/6-311++G(2d,2p) calculations
Energies Complexes
I II III IV V VI
ΔE −7.45 −8.19 −6.44 −3.47 −4.6 −6.09ΔZPE 4.10 3.99 3.23 1.89 2.0 2.26
BSSE 2.44 2.53 1.94 1.85 1.91 1.75
ΔEC −0.91 −1.67 −1.27 0.27 −0.69 −2.09
All values are given in kJ mol−1
J Mol Model (2015) 21: 77 Page 5 of 11 77

the halogen complexes. Even though no relationship isestablished, but the largest shifted frequencies and more in-tense charge density transfers seems to be attributed to thehigher values of the dihydrogen bond energies.
ΔυSi–H ¼ −52:47þ 51:60:eΔqH Sið Þ=0:027ð Þ ; R2 ¼ −0:97 ð2Þ
In a recent work authorized by Grabowski [27], the mech-anisms of halogen bonds and hydrogen bonds were discussed.About the first one, the appearing of a positive charge onH(HOX) was demonstrated as a requirement for linking with aLewis basis. In this current research, however, H(HOX) is oftenmore positive whereas H(Si) more negative, what is in goodacceptance.
Nevertheless, it is not always easy to analyze the chargetransfer values and extract great insights from them at the sameextent. In some cases it is mandatory that the theoretical anal-ysis should be carefully made [85]. In order to rationalize
systematic tendencies, Figs. 6 and 7 illustrate the HCA dendro-gram and PCA graph [55, 86] generated bymeans of the chargetransfer values gathered in Table 4. The HCA similarity amongthe dihydrogen and halogen bond complexes was ruled by theEuclidian distance (ED). Clearly, two sub-sets of systems iden-tified as I-III and V-VI were distinctly grouped at ED valuessmaller than 0.2 and 0.4, respectively. In forward, the IV halo-gen complex cannot be embodied as similar to the remainingones because its ED reached a very long value up to 1.4. Ofcourse, this scenario should be attributed to the positive chargetransfer balance on the qH(Si). Only tomention, the dihydrogen-bonded complexes are quite similar among themselves once theED values are very short. After normalization, the PCA em-bodies both PC1 and PC2 with 75.01 and 16.84 % of the totalvariance whose sum is 91.85 %. In this overall variance, theloading contributions show that H(Si) and Si are the most im-portant variables to the functionality of the charge transfermechanism rather than X (X=F, Cl, and Br). In other words,the electronic structure of the σ-hole is preserved as well asH(HOX) is not important as expected, no matter whether
Table 4 Values of the NBO atomic charges and charge transfer amounts of the H3SiH∙∙∙HOX and H3SiH∙∙∙XOH set of complexes obtained fromMP2/6-311++G(2d,2p) calculations
Charges Complexes
I II III IV V VI
qSi 0.908 (3.0) 0.899 (2.1) 0.894 (1.6) 0.884 (0.6) 0.885 (0.7) 0.894 (1.6)
qH(Si) −0.273 (−5.4) −0.266 (−4.7) −0.259 (−4.0) −0.217 (0.2) −0.235 (−1.6) −0.247 (−2.8)qH(HOX) 0.478 (1.1) 0.490 (1.1) 0.489 (1.1) 0.468 (0.1) 0.478 (−0.1) 0.476 (−0.2)qO −0.274 (−0.3) −0.694 (−0.2) −0.783 (−0.7) −0.268 (0.3) −0.695 (−0.3) −0.788 (−1.2)qX −0.201 (−0.5) 0.209 (−0.4) 0.293 (−0.6) −0.196 (0.0) 0.219 (0.6) 0.311 (1.2)
* All values are given in electronic units (eu); * Values of the charge transfer amounts are given in parentheses: * All values in parentheses must bemultiplied by the factor of 10−2
Fig. 5 Relationship between the frequency shifts of the Si–H bonds andcharge transfer computed on the H(Si) atom
Fig. 6 Dendrogram graph of the intermolecular charge transfers onΔqH(Si) determined to the H3SiH∙∙∙HOX and H3SiH∙∙∙XOH complexes
77 Page 6 of 11 J Mol Model (2015) 21: 77

dihydrogen bonds or halogen bonds are formed. Moreover,H(Si) and Si have similar charge transfer profiles even if thesevariables are placed in opposite coordinates of the PCA graph,where the first contributes largely to PC1 whereas the secondless, but to PC2 axis.
QTAIM topography and NBO analysis
Figure 8 depicts the contour lines and internuclearconnecting paths of electronic density of the H3SiH HOFcomplex, for which the values of the QTAIM parameters(electronic density and its Laplacian, potential electronicenergy density, and kinetic electronic energy density) arelisted in Table 5. Initially, the –G/U values in the range of1.132-1.349 testify the non-covalent characters of the I-VIcomplexes [87–89]. Once the electronic density potentialoperator is outweighed by the kinetic followed by the lowelectronic density amounts and positive Laplacian values,certainly the I-VI complexes are weakly bound, just likehas been demonstrated throughout this work. According toFig. 9, the relationship between the values of ΔEC energiesand fields of ∇2ρ yields a linear tendency with coefficientR2=−0.90 [90]. Note that the non-covalent character isextremely attained in the halogen bond complexes, in ad-dition to the long intermolecular distances followed by thelow interaction energies, IV is not mentioned because itsinteraction energy is repulsive, a clear differentiation be-tween the interaction strength of the dihydrogen and halo-gen bond complexes is definitively established (Table 6).
ΔEC ¼ −111:7∇2ρþ 2:57 ; R2 ¼ −0:90 ð3Þ
The high values of electronic density of the Si–H, O–H, andO–X bonds yield covalent characters, partial or total, although
Fig. 7 PCA of the intermolecular charge transfers on ΔqH(Si) determinedto the H3SiH∙∙∙HOX and H3SiH∙∙∙XOH complexes
Table 5 Values of the QTAIM parameters of the H3SiH∙∙∙HOX andH3SiH∙∙∙XOH set of complexes
BCPs Complexes
I II III IV V VI
ρH H 0.011 0.011 0.012 — — —
∇2ρH H 0.035 0.036 0.036 — — —
-G/UH H 1.148 1.132 1.140 — — —
ρX H — — — 0.004 0.006 0.010
∇2ρX H — — — 0.021 0.027 0.037
-G/UX H — — — 1.238 1.349 1.199
ρSi–H 0.117 0.117 0.118 0.122 0.120 0.119
∇2ρSi–H 0.190 0.189 0.189 0.184 0.186 0.186
-G/USi–H 0.619 0.619 0.618 0.610 0.613 0.614
ρO–H 0.373 0.370 0.370 0.377 0.374 0.373
∇2ρO–H −3.028 −2.935 −2.906 −3.046 −2.952 −2.913-G/UO–H 0.065 0.073 0.075 0.065 0.073 0.077
ρO–X 0.272 0.196 0.158 0.373 0.194 0.155
∇2ρO–X 0.143 −0.109 0.029 0.137 −0.105 0.037
-G/UO–X 0.544 0.440 0.517 0.542 0.442 0.523
* Values of ρ and ∇2 ρ are given in e.ao−3 and e.ao
−5 , respectively
Fig. 8 Contour lines of the electronic density of the H3SiH∙∙∙HOF complex
J Mol Model (2015) 21: 77 Page 7 of 11 77

the positive Laplacian in Si–H and O–X disagree with thisstatement. Once the positive electrostatic potential is the elec-tronic requirement to form σ-holes [91], what yields an elec-tronic depletion willing to interact with centers containing highelectronic density levels, the values of ∇2ρ are positive albeit itselectronic density is relatively high. Regardless, it has beenpointed out that the Si–H bond undergoes the most incisivestructural changes and larger vibration shifts, which to someextent, can be used to evaluate the interaction strength. Sometime ago, however, some insights to predict the interaction
strength embodied by the bond length enhancements or evenfrequency shifts and topological parameters derived fromQTAIM approach were appraised as follows [92–95]:
Δmol ¼ ½ ΔυSi–H=υSi–H;m
� �2þ ΔρSi–H=ρSi–H;m� �2
þ Δ2ρSi–H=∇2ρSi–H;m
� �2�1=2 ð4Þ
It is through the relationship of Δmol versus the energies ofthe dihydrogen bonds and halogen bonds that the interactionstrength profile can be valued. It is worthy to be noticed thatthe values of ΔEC(BSSE) used in this analysis were correctedonly with the BSSE amounts because the ΔEC corrected withBSSE as well as ΔZPE yielded a very poor relationship. Ascan be seen in Fig. 10 and valued by the Eq. (5), a satisfactorylinear relationship (R2=−0.94) between ΔEC(BSSE) andΔmol was obtained. Since the Δmol embodies the frequencyshifts of the Si–H bond as well as the variations on the elec-tronic density and Laplacian, the lower halogen bond energiescause slight molecular distortions on the SiH4 structure andvice versa whether the dihydrogen bonds which are relativelystronger were taken into account.
ΔEC BSSEð Þ ¼ −80:21Δmol−1:45 ; R2 ¼ −0:94 ð5Þ
In recent papers [47, 96, 97], a suitable interpretation ofthe frequency shifts through the contributions of the hybridorbitals has been divulged. For red-shifts and blue-shifts,the contribution of the p orbital of the heavy element in-creases and reduces at the same level, which reflects in a
Table 6 Percentage values of the hybrid orbitals (s and p) of the Si, H,O, F, Cl, and Br elements obtained from NBO calculations at MP2/6-311++G(2d,2p) theoretical level
Hybridizations Complexes
I II III IV V VI
ΔsSiSi–H −1.04 −0.99 −0.97 0.27 −0.28 −0.49ΔpSiSi–H 0.98 0.93 0.90 −0.16 0.26 0.45
ΔsHSi–H 0.14 0.12 0.10 −0.02 0.02 0.05
ΔpHSi–H −0.14 −0.12 −0.10 0.02 −0.02 −0.05ΔsOO–H 1.13 1.26 1.42 −0.01 −0.04 −0.07ΔpOO–H −1.13 −1.26 −1.42 0.01 0.04 0.07
ΔsHO–H −0.01 0.00 0.00 0.00 0.00 0.00
ΔpHO–H 0.01 0.00 0.00 0.00 0.00 0.00
ΔsOO–X −0.27 −0.32 −0.26 −0.09 0.09 0.39
ΔpOO–X 0.28 0.33 0.27 0.09 −0.08 −0.38ΔsXO–X 0.03 0.01 0.00 −0.01 −0.22 −0.37ΔpXO–X −0.01 0.01 −0.01 0.02 0.25 0.42
ΔENBO 10.50 10.87 11.58 8.53 7.15 7.32
* Values of Δs and Δp are given in %; * Values of ΔENBO are given in kJmol−1
Fig. 10 Relationship between the corrected values (only with BSSE) ofthe interaction energies and molecular parameter (frequency shifts andvariations of the electronic density and Laplacians) of the H3SiH∙∙∙HOXand H3SiH∙∙∙XOH complexes
Fig. 9 Relationship between the corrected values (with BSSE and ΔZPE)of the interaction energies and Laplacians of the H3SiH∙∙∙HOX andH3SiH∙∙∙XOH complexes
77 Page 8 of 11 J Mol Model (2015) 21: 77

higher and lower polarization [98]. A relationship betweenthe variations of the p orbitals of silicon (ΔpSiSi–H) versusthe values of the red-shifts and blue-shifts of the Si–Hbonds is plotted in Fig. 11. Because an unusual relationshipbetween the frequency shifts and charge transfer isfurnished (see Fig. 5), interestingly the contributions ofthe p orbital is also exponentially enhanced in order tojustify the red-shifts of the Si–H bonds, of course withthe exception to the value of the IV complex in whichthe p orbital is suppressed caused by the blue-shift of +2.4 cm−1.
ΔυSi–H ¼ −51:3þ 41:12:e−ΔpSiSi–H=0:575ð Þ ; R2 ¼ −0:95 ð6Þ
About the O–H and O–X bonds, the variations in the s andp orbitals of oxygen are not in line with the frequency shiftspreviously discussed. For instance, the ΔsOO–H values of 1.13,1.26, and 1.42%would be used to justify the blue-shifts in theO–H bonds of the I-III dihydrogen bond complexes, althoughin opposition to this, the frequency shifts observed in thesebonds are from red nature. Regarding these results, they canbe supported by the Bent’s rule [99–101] of the chemical bondby which is affirmed that s-character of the hybrid orbitals isrelated to the contribution of the electropositive group or ele-ment. Although used successfully in recent intermolecularstudies [49, 73], we would like to consider that this conceptionof hybrid orbital enhancements should be used with somecaution because some inconsistencies were found in this cur-rent research.
Throughout this work, differently of the linear relationshipsbetween the interaction energies (ΔEC) and topological (∇2ρ),the profiles of the frequency shifts yield exponential
relationships either with charge transfer amounts or variationon hybrid orbitals. Another theoretical point of view regardingthe interaction strength is the binding energy (ENBOZ∙∙∙H) ob-tained from NBO calculations represented by the Eq. (7), inwhich Y(A) and σ*(B) represent the acceptors and donors ofcharge density, in this case, Z (H or X with X=F, Cl or Br) andH, respectively.
ENBOY Að Þ→σ* Bð Þ ¼ 2
Y Að Þ Fj jσ* Bð Þh i2εY Að Þ−εσ* Bð Þ� � ð7Þ
Conclusions
Throughout this theoretical study, some important issues re-lated to the strengths of dihydrogen bonds and halogen bondswere raised. The molecular properties of the H3SiH∙∙∙HOXand H3SiH∙∙∙XOH complexes have shown systematic tenden-cies, either those from structural or vibrational natures. Thedecrease and increase of bond lengths agree with the profilesof blue-shifts and red-shifts in the stretch frequencies, mainly
Fig. 11 Relationship between the frequency shifts (red or blue) andvariation of the silicon p hybrid orbital of the H3SiH∙∙∙HOX andH3SiH∙∙∙XOH complexes
Fig. 12 Relationship between the NBO binding energies (ENBO) andnew vibration modes of the H3SiH∙∙∙HOX and H3SiH∙∙∙XOH complexes
J Mol Model (2015) 21: 77 Page 9 of 11 77
Figure 12 illustrates the relationship between the values ofENBO
Z∙∙∙H and intermolecular stretch frequencies. As can beseen the dihydrogen complexes are stronger bound. More-over, it can be seen that the exponential behavior is once againprominent, wherein with mention to values of the bindingenergies, the dihydrogen and halogen bond complexes higherand lower than the threshold of 10 kJ mol−1, respectively.
ENBOZ⋯H ¼ 12:19−13:18 e
υZ⋯H=51:28ð Þ ; R2 ¼ 0:93 ð8Þ

the Si–H bonds. Also, the computation of the charge transferon Si was used to justify these frequency shifts, in which, anexponential relationship was modeled indicating a clear dis-tinction between the complexes of H3SiH∙∙∙HOX andH3SiH∙∙∙XOH. Interestingly, the application of the HCA andPCA chemometric techniques have served to state that siliconis the most important charge transfer center. In terms of QTAIM, the noncovalent characterization of the dihydrogen andhalogen bond complexes was definitive. In addition to that,the estimation of the interaction strength was efficiently val-ued through the compilation of frequency shifts and topolog-ical parameters, such as electronic density and its Laplacian.The binding energies of the NBO analysis also show goodconcordance with the new vibration modes of the dihydrogenbonds and halogen bonds. At last, the certifying of the fre-quency shifts regarding the contribution of the hybrid orbitalswas carried out, wherein the variations of the p orbital of Siaccord well with the frequency shifts of the Si–H bond.
References
1. Riley KE, Rezac J, Hobza P (2013) J Mol Model 19:2879–28832. Wu W, Zeng Y, Li X, Zhang X, Zheng S, Meng L (2013) J Mol
Model 19:1069–10773. Liu X, Cheng J, Li Q, Li W (2013) Spectrochim Acta A 101:172–
1774. Sanchez-Sanz G, Trujillo C, Alkorta I, Elguero J (2012) Phys Chem
Chem Phys 14:9880–98895. Zabardasti A, Kakanejadifard A, Goudarziafshar H, Salehnassaj M,
Zohrehband Z, Jaberansari F, Solimannejad M (2013) ComputTheor Chem 1014:1–7
6. Kozuch S, Martin JML (2013) J Chem Theory Comput 9:1918–1931
7. Jeffrey GA (1997) An introduction to hydrogen bonding. OxfordUniversity Press, New York
8. Scheiner S (1997) Hydrogen bonding. Oxford University Press,New York
9. Desiraju GR, Steiner T (1999) The weak hydrogen bond. OxfordUniversity Press, Oxford
10. Auffinger P, Hays FA, Westhof E, Ho PS (2014) Proc Natl Acad SciU S A 101:16789–16794
11. Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2009)Struct Chem 20:663–670
12. Oliveira BG, Araújo RCMU, Carvalho AB, RamosMN, HernandesMZ, Cavalcante KR (2007) JMol Struct (THEOCHEM) 802:91–97
13. Oliveira BG, Araújo RCMU (2007) Quim Nova 30:1167–117014. Oliveira BG, Araujo RCMU, Carvalho AB, Ramos MN (2007)
Spectrochim Acta A 68:626–63115. Oliveira BG, Araujo RCMU, Leite ES, Ramos MN (2011) Int J
Quantum Chem 111:111–11616. Voth AR, Hays FA, Ho PS (2007) Proc Natl Acad Sci U S A 104:
6188–619317. Jiang Y, Alcaraz AA, Chen JM, Kobayashi H, Lu YJ, Snyder JP
(2006) J Med Chem 49:1891–189918. Li Q, Xu X, Liu T, Jing B, Li W, Cheng J, Gong B, Sun J (2010)
Phys Chem Chem Phys 12:6837–684319. Szalewicz K (2012) WIREs Comput Mol Sci 2:254–272
20. Lao KU, Herbert JM (2014) J Chem Phys 140:0441081–044108821. Oliveira BG, Araújo RCMU (2012) Quim Nova 35:2002–201222. Lommerse JPM, Stone AJ, Taylor R, Allen FH (1996) J Am Chem
Soc 118:3108–311623. Politzer P, Lane P, Concha MC, Ma Y, Murray JS (2007) J Mol
Model 13:305–31124. Murray JS, Lane P, Politzer P (2009) J Mol Model 15:723–72925. Metrangolo P, Neukirch H, Pilati T, Resnati G (2005) Acc Chem
Res 38:386–39526. Grabowski SJ (2013) Phys Chem Chem Phys 15:7249–725927. Desiraju GR (2010) Angew Chem Int Ed 50:52–5928. Jensen WB (1980) The Lewis acid–base concepts, an overview.
Wiley-Interscience, New York29. Oliveira BG, Vasconcellos MLAA (2009) Inorg Chem Commun
12:1142–114430. Oliveira BG, VasconcellosMLAA (2009) Struct Chem 20:897–90231. Riley KE, Hobza P (2008) J Chem Theory Comput 4:232–24232. Esrafili MD, Yourdkhani S, Bahrami A (2013)Mol Phys 111:3770–
377833. Sedlák R, Fanfrlík J, Hnyk D, Hobza P, Lepsík M (2010) J Phys
Chem A 114:11304–1131134. Zierkiewicz W, Hobza P (2004) Phys Chem Chem Phys 6:5288–
529635. Zheng YZ, Wang NN, Zhou Y, Yu ZW (2014) Phys Chem Chem
Phys 16:6946–695636. Oliveira BG, Leite LFCC (2009) J Mol Struct (THEOCHEM) 915:
1–337. Oliveira BG (2014) Chem Phys 443:67–7538. Oliveira BG (2014) C R Chim 17:1041–104939. Oliveira BG (2014) Spectrochim Acta A 124:208–21540. Murray JS, Concha MC, Lane P, Hobza P, Politzer P (2008) J Mol
Model 14:699–70441. Clark T (2013) WIREs Comput Mol Sci 3:13–2042. Clark T, Hennemann M, Murray JS, Politzer P (2007) J Mol Model
13:291–29643. Li Q-Z, Jing B, Li R, Liu Z-B, LiW-Z, Luan F, Cheng J-B, Gong B-
A, Sun J-Z (2011) Phys Chem Chem Phys 13:2266–227144. Bader RFW (1990) Atoms in molecules - a quantum theory. Oxford
University Press, Oxford45. Bader RFW (1991) Chem Rev 91:893–92846. Glendening ED, Landis CR, Weinhold F (2012) WIREs Comput
Mol Sci 2:1–447. Oliveira BG (2014) Struct Chem 25:745–75348. Santos ITO, Oliveira BG (2015) J Spec Dyn 5:1–649. Santos ITO, Rego DG, Oliveira BG (2014) QuimNova 37:624–63050. Oliveira BG, deAraújo RCMU,RamosMN (2009)Orbital Electron
J Chem 1:156–16651. Oliveira BG, de Araújo RCMU, Carvalho AB, Ramos MN (2009)
Orbital Electron J Chem 1:167–18252. Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–92653. Oliveira BG, Araújo RCMU (2007) Quim Nova 30:791–79654. Arantes FFP, Barbosa LCA, Maltha CRA, Demuner AJ, Fidêncio
PH, Carneiro JWM (2011) J Chemom 25:401–40755. Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2009) J
Mol Model 15:421–43256. Branislav J, Aleksandar N, Slobodan P (2012) Hem Ind 66:1–757. Nguyen TD, Schmidt B, KwohCK (2014) Procedia Comput Sci 29:
8–1958. Abdi H, Williams LJ (2010) Wiley Interdiscip Rev Comput Stat 2:
433–45959. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA,
Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, BurantJC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B,Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, HadaM, EharaM, Toyota K, Fukuda R, Hasegawa J, IshidaM, NakajimaT, Honda Y, Kitao O, Nakai H, Klene M, Knox JE, Hratchian HP,
77 Page 10 of 11 J Mol Model (2015) 21: 77

Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE,Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, AyalaPY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ,Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O,Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV,Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G,Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T,Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, GillPMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA(2003) GAUSSIAN 03. Gaussian Inc., Pittsburgh
60. Cremer D (2011) WIREs Comput Mol Sci 1:509–53061. Rahal M, Hilali M, El Hammadi A, El Mouhtadi M, El Hajbi A
(2001) J Mol Struct (THEOCHEM) 572:73–8062. Boys SF, Bernardi F (1970) Mol Phys 19:553–56663. Biegler-König F, Bayles D, Schönbohm J (2000) AIM2000 1.0.
University of Applied Sciences, Bielefeld64. Keith TA (2011) AIMAll (Version 11.10.16). TK Gristmill
Software, Overland Park65. Vega D (2013) AIM-UC Version 1.3, Universidad de Carabobo,
Bárbula, Venezuela66. Vega D, Almeida D (2014) J ComputMethods Sci Eng 14:131–13667. http://ufla.br/chemoface/. Accessed Jun 201468. Nunes CA, Freitas MP, Pinheiro ACM, Bastos SC (2012) J Braz
Chem Soc 23:2003–201069. Biswal HS, Gloaguen E, Loquais Y, Tardivel B, Mons M (2012) J
Phys Chem Lett 3:755–75970. Zabardasti A, Goudarziafshar H, Salehnassaj M, Oliveira BG
(2014) J Mol Model 20:2403–241271. Oliveira BG, Lima MCA, Pitta IR, Galdino SL, Hernandes MZ
(2010) J Mol Model 16:119–12772. Esrafili MD, Hadipour NL (2011) Mol Phys 109:2451–246073. Nagy PI (2014) Int J Mol Sci 15:19562–1956374. Saggu M, Levinson NM, Boxer SG (2012) J Am Chem Soc 134:
18986–1899775. Azofra LM, Scheiner S (2014) Phys Chem Chem Phys 16:5142–
514976. Hennemann M, Murray JS, Politzer P, Riley KE, Clark T (2012) J
Mol Model 18:2461–246977. Nguyen TT, Nguyen PH, Tran TH, Minh TN (2011) Phys Chem
Chem Phys 13:14033–14042
78. Capim SL, Santana SR, Oliveira BG, Rocha GB, VasconcellosMLAA (2010) J Braz Chem Soc 21:1718–1726
79. Mo Y, Gao J (2001) J Phys Chem A 105:6530–653680. Wright AM, Howard AA, Howard JC, Tschumper GS, Hammer NI
(2013) J Phys Chem A 117:5435–544681. Li X, Liu L, Schlegel HB (2002) J Am Chem Soc 124:9639–964782. da Silva JBP, Silva JuniorMR, RamosMN (2005) J Braz Chem Soc
16:844–85083. Chaturvedi D, Gupta V, Tandon P, Sharma A, Baraldi C, Gamberini
MC (2012) Spectrochim Acta A 99:150–15984. Oliveira BG, Araújo RCMU, Ramos MN (2009) J Mol Struct
(THEOCHEM) 908:79–8385. Ramos-Cordoba E, Lambrecht DS, Head-Gordon M (2011)
Faraday Discuss 150:345–36286. Oliveira BG, Costa TF, Araújo RCMU (2013) J Mol Model 19:
3551–356887. Oliveira BG (2013) Phys Chem Chem Phys 15:37–7988. Grabowski SJ (2011) Chem Rev 111:2597–262589. Oliveira BG, Araújo RCMU, Pereira FS, Lima EF, Silva WLV,
Carvalho AB, Ramos MN (2008) Quim Nova 31:1673–167990. Alkorta I, Elguero J, Grabowski SJ (2008) J Phys Chem A 112:
2721–272791. McDowell SAC, Joseph JA (2014) Phys Chem Chem Phys 16:
10854–1086092. Grabowski SJ (2001) Chem Phys Lett 338:361–36693. Grabowski SJ (2004) J Phys Org Chem 17:18–3194. Oliveira BG, Pereira FS, de Araújo RCMU, Ramos MN (2006)
Chem Phys Lett 427:181–18495. Oliveira BG, Pereira FS, de Araújo RCMU, Ramos MN (2007)
Chem Phys Lett 433:390–39496. Grabowski SJ (2011) J Phys Chem A 115:12789–1279997. Grabowski SJ (2011) J Phys Chem A 115:12340–1234798. Clark T, Murray JS, Politzer P (2014) Aust J Chem 67:451–45699. Bent HA (1961) Chem Rev 61:275–311
100. Alabugin IV, Bresch S, Manoharan M (2014) J Phys Chem A 118:3663–3677
101. Alabugin IV, Bresch S, Gomes GP (2015) J Phys Org Chem 28:147–162
J Mol Model (2015) 21: 77 Page 11 of 11 77