retinoic acid fails to induce cell cycle arrest with myogenic differentiation in rhabdomyosarcoma
TRANSCRIPT

Pediatr Blood Cancer 2012;58:877–884
Retinoic Acid Fails to Induce Cell Cycle Arrest With MyogenicDifferentiation in Rhabdomyosarcoma
Alaa Al-Tahan, MD,1 Omar Sarkis, BS,1 Mohamad Harajly, MS,1 Omar Kebbe Baghdadi, BS,1 Kazem Zibara, PhD,2
Fouad Boulos, MD,3 Dipti Dighe, MD,4 Steven Kregel, BS,4 Ali Bazarbachi, MD, PhD,5 Marwan El-Sabban, PhD,6
Stephen X. Skapek, MD,4 and Raya Saab, MD1*
INTRODUCTION
Rhabdomyosarcoma (RMS) is the most common soft tissue
sarcoma in children [1]. Despite multimodality therapy, approxi-
mately 30% of treated children will experience relapse, with
overall survival being particularly poor in those originally diag-
nosed with high risk disease (reviewed in Ref. 2). By definition,
rhabdomyosarcoma cells express some skeletal myogenic pro-
teins, such as MyoD and Myogenin, and certain structural pro-
teins like desmin and vimentin [3–5]. Despite this lineage
commitment, the normal myogenic differentiation program is
stalled in RMS cells because many of the later aspects of differ-
entiation (e.g., expression of functional proteins, morphological
changes, cell fusion) are absent and, unlike terminally differenti-
ated myocytes, rhabdomyosarcoma cells proliferate [6]. There has
been a long-standing interest in promoting differentiation in can-
cers such as RMS because such differentiation could, in principle,
irreversibly arrest cell proliferation to control the disease with less
side effects than conventional therapies [7,8].
Retinoic acid is a morphogen that has been shown to induce
differentiation of normal myoblasts, neuroblasts, and epithelial
cells [9–11]. Pre-clinical studies show that retinoic acid fosters
differentiation in neuroblastoma, another primitive and aggressive
childhood tumor [12]. When used in the setting of minimal resid-
ual disease, retinoic acid improves disease-free survival for chil-
dren with metastatic neuroblastoma [13]. Whether such an
approach is useful for children with other poorly differentiated
tumors is not known. Several in vitro studies have demonstrated
that retinoic acid influences cell proliferation and muscle gene
expression in human RMS cell lines [14,15]. However, in vivo
studies of retinoic acid therapy in rhabdomyosarcoma are lacking.
We assessed the effect of retinoic acid on RMS in vitro and
using an in vivo minimal residual disease model, using two cell
lines representing the major subtypes of RMS: embryonal and
alveolar histology. We report that All-Tans Retinoic Acid
(ATRA) slowed RMS cell proliferation and cell accumulation in
culture. In addition, prolonged in vitro exposure to ATRA altered
cell morphology and augmented the expression of Myosin Heavy
Chain (MyHC), an indicator of terminal differentiation. However,
maintenance therapy following chemotherapy-induced remission
of RMS xenografts augmented the expression of a more differen-
tiated myogenic phenotype in vivo, but this did not preclude
continued cell proliferation and eventual relapse. When ATRA
and Cis-Retinoic Acid (CRA) were applied to normal myoblasts
in vitro, only an early marker of muscle differentiation was
enhanced, and RB protein activation and cell cycle arrest did
not occur. These findings suggest that retinoic acid alone is
unlikely to be beneficial as a single agent in inducing RMS
differentiation. Further, they provide new molecular evidence as
to why retinoids fail to fully promote myogenic differentiation
and cell cycle arrest, thereby forming a foundation from which
Background. Rhabdomyosarcoma (RMS) is the most commonsoft tissue sarcoma in children. Current treatment strategies do notcure most children with recurrent or high-risk disease, underlyingthe need for novel therapeutic approaches. Retinoic acid has beenshown to induce differentiation in a variety of cells including skele-tal myoblasts and neuroblasts. In the setting of minimal residualdisease, retinoic acid improves survival in neuroblastoma, anotherpoorly differentiated childhood tumor. Whether such an approachis useful for rhabdomyosarcoma has not yet been investigated.Several in vitro studies have demonstrated an appreciable effectof retinoic acid on human RMS cellular proliferation and differenti-ation. Procedure. We assessed the efficacy of ATRA on rhabdomyo-sarcoma, in vitro and in vivo, using cell lines and xenografts.Results. ATRA slowed RMS cell proliferation, and promoted a
more differentiated myogenic phenotype in both alveolar and em-bryonal RMS cell lines. Treatment of cultured murine myoblastswith retinoids increased Myogenin expression, but did not inducecell cycle arrest. Despite the favorable in vitro effects, ATRA failedto delay relapse of minimal residual disease using human RMSxenografts in immuno-suppressed NOD-SCID (NSG) mice. Interest-ingly, tumors that recurred after ATRA treatment showed evidenceof enhanced muscle differentiation. Conclusion. Our results indi-cate that ATRA could increase the expression of some genesassociated with muscle differentiation in rhabdomyosarcoma cells,but there was no benefit of single-agent therapy in an MRD model,likely because cell cycle arrest was uncoupled from the pro-differentiation effects of retinoids. Pediatr Blood Cancer 2012;58:877–884. � 2011 Wiley Periodicals, Inc.
Key words: differentiation; minimal residual disease; retinoic acid; rhabdomyosarcoma; therapy; xenograft
1Department of Pediatrics, American University of Beirut, Beirut,
Lebanon; 2Lebanese University, Beirut, Lebanon; 3Department of
Pathology and Laboratory Medicine, American University of Beirut,
Beirut, Lebanon; 4Division of Pediatric Hematology-Oncology,
University of Chicago, Chicago, Illinois; 5Department of Internal
Medicine, American University of Beirut, Beirut, Lebanon;6Department of Anatomy, Cell Biology and Physiological Sciences,
American University of Beirut, Beirut, Lebanon
Grant sponsor: The Lebanese National Council for Scientific
Research (CNRS). Alpha-Omega-Alpha (AOA) Carolyn L. Kuckein
Student Research Fellowship award (Alaa Al-Tahan). The American
Lebanese Syrian Associated Charities (ALSAC) and International
Outreach Program at St Jude Children’s Research Hospital, Memphis,
TN (Dr Saab’s laboratory).
Conflict of interest: Nothing to declare.
*Correspondence to: Raya Saab, MD, Department of Pediatrics,
American University of Beirut, Riad El Solh Street, Beirut 1107
2020, Lebanon. E-mail: [email protected]
Received 27 April 2011; Accepted 26 May 2011
� 2011 Wiley Periodicals, Inc.DOI 10.1002/pbc.23246Published online 13 July 2011 in Wiley Online Library(wileyonlinelibrary.com).

additional pharmacological strategies can be applied to attempt to
fully activate skeletal muscle differentiation as a therapy for
RMS.
MATERIALS AND METHODS
Cell Lines, Growth, and Treatment Conditions
Human rhabdomyosarcoma cell lines of alveolar (Rh30) and
embryonal (JR-1) histology were generously provided by Peter
Houghton; both have been previously characterized [16,17].
Mouse C2C12 myoblasts were obtained from the ATCC. Cells
were cultivated at 378C and 5% CO2 in 10% fetal bovine serum
(FBS) in DMEM (Invitrogen, Carlsbad, CA). Cells were treated
with 5 mM ATRA (Sigma–Aldrich, St. Louis, MO), or an equal
volume of DMSO vehicle. Culture medium (with fresh ATRA or
DMSO) was changed every 3 days. Because ATRA is light-
sensitive, all handling of ATRA stocks, working solution and
ATRA treatment were done in subdued light.
Cell Accumulation Assay
JR1 and Rh30 cells were seeded onto six-well plates at
1 � 103 cells per well and cultured at 378C and 5% CO2 in
10% FBS in DMEM. Six sets of triplicate wells were plated for
each cell line. The following day (day 0), half of the wells of each
cell line were refed with culture medium containing ATRA in
DMSO or an equivalent volume of vehicle. On days 3, 6, and 9,
triplicate wells were harvested, and the total number of cells was
counted via hemocytometer; remaining wells were refed with
medium containing either ATRA or DMSO as above. Phase-
contrast photomicrographs to assess cellular morphology were
taken using AxioCam HRC (Zeiss, Thornwood, NY) camera.
Data shown are representative of three or more independent
experiments.
Senescence-Associated Beta Galactosidase (SABG)Assay
SABG staining was done as described [18]. In summary, cells
were fixed using 2% paraformaldehyde, then washed and stained
in SABG staining solution (1 mg/ml X-gal, 40 mM Citric acid/
NaPhos buffer pH 6.0, 5 mM K3CN, 5 mM K4CN, 150 mM
NaCl, 2 mM MgCl2) at 378C overnight, washed in PBS, and
counterstained with eosin.
TUNEL Assay
Terminal deoxynucleotidyl transferase-mediated dUTP nick
end labeling (TUNEL) was done using the DeadEnd Cell Death
Labeling kit (Roche, Indianapolis, IN). Nuclei were labeled with
DAPI and then cover-slipped. The percentage of TUNEL-positive
cells was determined by counting representative 200� fields using
a fluorescence microscope.
BrdU Treatment and Immunostaining of CulturedRMS Cells
Rhabdomyosarcoma cells were cultured as detailed above in 8-
wells chamber slides, with ATRA or an equal volume of DMSO
vehicle, for 9 days. Cells were then treated with bromodeoxyur-
idine (BrdU) (Sigma–Aldrich) at 50 mmol/L, for 15 minutes or
4 hours. Cells were fixed by methanol/acetone (1:1), and
immunostained for either BrdU alone, or BrdU and Myosin
Heavy Chain (MyHC). For BrdU staining, fixed cells were treated
with 2N HCL for 10 minutes, then neutralized by Borate buffer
for 12 minutes. After blocking, slides were probed with anti-BrdU
antibody (Santa Cruz Biotechnology, Santa Cruz, CA), anti-
MyHC antibody (Millipore, Billerica, MA), or both. Cy3-
conjugated secondary antibody (Jackson Immunoresearch, West
Grove, PA) and/or Alexa 488-conjugated antibody (Invitrogen)
were used for detection. Stained cells were covered with aqueous
mounting medium containing DAPI (Vector Laboratories, Burlin-
game, CA), and visualized by immunofluorescence microscopy
using an Axiovert 135M microscope equipped with an Axiocam
digital camera (Zeiss). BrdU-positive cells were quantified by
counting positive cells in five high-power fields in each condition
and normalizing to total number of cells (DAPI-stained) in each
field. Statistical analysis was done using Student’s t-test. Data
shown are representative of three or more independent experi-
ments with duplicate or triplicate samples.
Proliferation and Differentiation AssaysUsing Myoblasts
C2C12 cells were cultivated as previously described [19] in
10% FBS in DMEM (Growth Medium or GM), in 2% FBS
in DMEM (differentiation medium or DM) or differentiation
medium with insulin (10 mg/ml) prior to harvest. In some experi-
ments, all-trans retinoic acid (ATRA) (5 mM) or cis-retinoic acid
(CRA) (5 mM) (or equal volume of vehicle) was added with the
GM. At harvest, cells were fixed in 4% paraformaldehyde in PBS
and processed for BrdU incorporation as described above,
or lysates of non-fixed cells were used for Western blotting as
previously described [19].
Xenograft Studies
All animal experiments were approved by the American
University of Beirut’s Institutional Animal Care and Use
Committee. NSG mice (Reference 005557, NOD.Cg-
Prkdc<scid>Il2rg<tm1Wjl>/SzJ) were obtained from the Jack-
son Laboratory (Bar Harbor, Maine). All animals were handled
under pathogen-free sterile conditions, maintained under micro-
isolators, and fed sterile food. To form xenografts, Rh30 cells
were injected subcutaneously into the right flank, at 1 � 107 cells
per injection. Mice were then monitored until tumors grew to a
volume of at least 300 mm3, assessed using the following formu-
la: [length � (width)2]/2. Mice were then treated with vincristine
(Gedeon Richter Ltd, Budapest, Hungary), at a dose of 1 mg/kg
once weekly by intra-peritoneal injection. Mice were examined
twice weekly until complete tumor regression, then randomized to
receive ATRA (3.5 mg/kg by intra-peritoneal injection once daily,
5 days/week for 4 weeks), or an equal volume of vehicle alone
(DMSO). A total of 37 mice were treated: 19 with ATRA; 18 with
vehicle. Mice were examined twice weekly to detect tumor recur-
rence. Kaplan–Meier survival estimates and box-plots were used
in statistical analyses of tumor relapse rate.
Xenograft Harvesting and ImmunohistochemicalStaining
Mice with relapsed xenografts were monitored until a tumor
volume of at least 300 mm3, at which time they were euthanized.
878 Al-Tahan et al.
Pediatr Blood Cancer DOI 10.1002/pbc

Tumors were dissected, fixed in 4% paraformaldehyde, and
embedded in paraffin. Standard histology procedures were
followed to prepare 3 mm sections for immunohistochemical
staining with the following antibodies: anti-Myogenin (Lab
Vision, Fremont, CA), anti-MyoD (Dako, Glostrup, Denmark),
anti-MyHC (BioGenex, Fremont, CA). Digital photomicrographs
were obtained using an Olympus DP12 camera and software.
Western Blot Analysis
Cells or tissue samples were lysed on ice for 20 minutes in
Universal Lysis Buffer 100 mM Tris pH 7.5, 300 mM NaCl,
4 mM EDTA, 4 mM EGTA, 0.4% Triton X-100, 1% NP40,
50 mM NaF, 20 mM b-glycerophosphate, 1 mM phenylmethyl-
sulfonyl fluoride, 10 mg/ml leupeptin, 10 mg/ml aprotinin, 1 mM
DTT. Lysates were clarified by centrifugation and protein was
quantified by Bradford assay (Bio-Rad Laboratories, Hercules,
CA). Equivalent amounts of protein (50–80 mg) were fractionatedby 10% SDS–PAGE and transferred to polyvinylidene difluoride
membranes (Bio-Rad Laboratories, Hercules, CA). Blotted
proteins were detected using the following primary antibodies:
anti-Myogenin (BD Biosciences Pharmingen, San Diego, CA);
anti-MyHC (Millipore, Billercia, MA); anti-phosphorylated RB
at Ser780 (Cell Signaling Technology, Danvers, MA); anti-
p18Ink4c, anti-p21Cip1, anti-p27Kip1, anti-HSC 70, and anti-
Cyclin A (all from Santa Cruz Biotechnology, Santa Cruz, CA).
Primary antibodies were detected with species-specific horserad-
ish peroxidase-coupled secondary antibodies (Santa Cruz
Biotechnology, Santa Cruz, CA) and visualized by enhanced
chemiluminescence (Roche, Indianapolis, IN). The level of
expression of different proteins was analyzed by using the public
Image J software. Exposures in the linear range of the X-ray film
were selected. The relative band intensity was assessed by densi-
tometric analysis of digitalized autoradiographic images, and the
ratio of the band intensity of the protein of interest (MyHC) to the
loading control protein (HSC70) was calculated.
RESULTS
ATRA decreases RMS cell proliferation in vitro: We treated
alveolar (Rh30) and embryonal (JR1) RMS cell lines with ATRA
at a final concentration of 5 mM. ATRA treatment decreased RMS
cell accumulation by day 6, and the effect was maintained at
day 9 (Fig. 1A). To investigate the mechanism by which ATRA
resulted in less cell accumulation, we evaluated treated cells for
evidence of senescence, apoptosis, or cell cycle arrest. There was
no evidence of senescence after 9 days of treatment with ATRA,
detected by staining for acidic senescence-associated beta-
galactosidase (Fig. 1B). Increased cell death was not observed
in ATRA-treated culture plates, and TUNEL staining showed less
than 1.5% cell death by day 3 of treatment, and less than 0.5%
Fig. 1. ATRA inhibits RMS cell accumulation in culture. A: Rep-resentative charts showing number of the indicated RMS cells after
cultivation for 3, 6, and 9 days in the presence of ATRA (A) or the
drug vehicle DMSO (D). B: Staining for senescence-associated beta
galactosidase (SABG) activity in RH30 and JR1 cells treated for
9 days with ATRA or vehicle (DMSO), as indicated. Central inset
demonstrates positive SABG staining in oncogene-expressing murine
primary cells. C: Percentage of Rh30 and JR1 cells that are BrdU-
positive, after 9 days of treatment with either ATRA or drug vehicle
DMSO, in high-serum (10% FBS) or low serum (1%FBS) conditions,
as indicated. Cells were treated with short pulses (15 minutes) of
BrdU. Asterisk indicates statistical significance with P-value <0.05
(student t-test).
RA Fails to Induce Cell Cycle Exit in RMS 879
Pediatr Blood Cancer DOI 10.1002/pbc
cell death by day 6 (standard deviation 0.22% and 0.003%,
respectively). However, ATRA treatment significantly decreased
the proportion of cells progressing through the S-phase of the cell
cycle, in both Rh30 and JR1 cells, assessed by pulse-labeling of
cells with BrdU. This effect was apparent regardless of whether
cells were cultivated in medium with high (10% FBS) or low
serum (1% FBS) (Fig. 1C). We conclude that ATRA treatment
in vitro slows cell proliferation in both RMS cell lines.
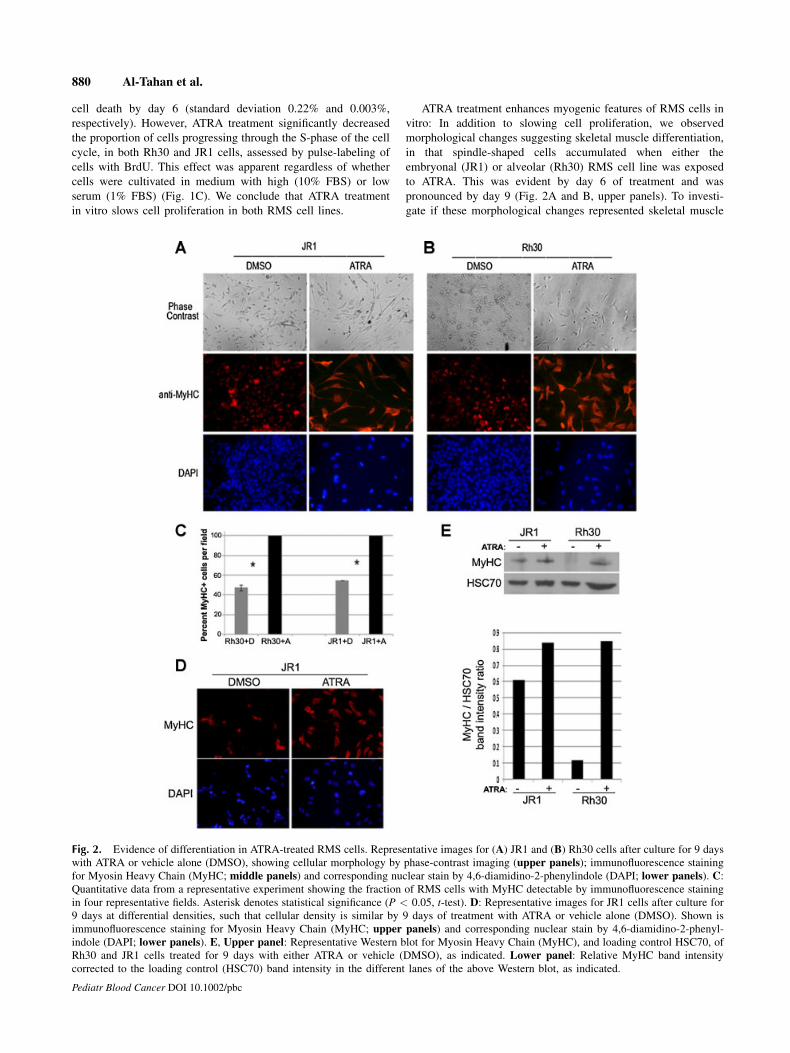
ATRA treatment enhances myogenic features of RMS cells in
vitro: In addition to slowing cell proliferation, we observed
morphological changes suggesting skeletal muscle differentiation,
in that spindle-shaped cells accumulated when either the
embryonal (JR1) or alveolar (Rh30) RMS cell line was exposed
to ATRA. This was evident by day 6 of treatment and was
pronounced by day 9 (Fig. 2A and B, upper panels). To investi-
gate if these morphological changes represented skeletal muscle
Fig. 2. Evidence of differentiation in ATRA-treated RMS cells. Representative images for (A) JR1 and (B) Rh30 cells after culture for 9 days
with ATRA or vehicle alone (DMSO), showing cellular morphology by phase-contrast imaging (upper panels); immunofluorescence staining
for Myosin Heavy Chain (MyHC; middle panels) and corresponding nuclear stain by 4,6-diamidino-2-phenylindole (DAPI; lower panels). C:Quantitative data from a representative experiment showing the fraction of RMS cells with MyHC detectable by immunofluorescence staining
in four representative fields. Asterisk denotes statistical significance (P < 0.05, t-test). D: Representative images for JR1 cells after culture for
9 days at differential densities, such that cellular density is similar by 9 days of treatment with ATRA or vehicle alone (DMSO). Shown is
immunofluorescence staining for Myosin Heavy Chain (MyHC; upper panels) and corresponding nuclear stain by 4,6-diamidino-2-phenyl-
indole (DAPI; lower panels). E, Upper panel: Representative Western blot for Myosin Heavy Chain (MyHC), and loading control HSC70, of
Rh30 and JR1 cells treated for 9 days with either ATRA or vehicle (DMSO), as indicated. Lower panel: Relative MyHC band intensity
corrected to the loading control (HSC70) band intensity in the different lanes of the above Western blot, as indicated.
880 Al-Tahan et al.
Pediatr Blood Cancer DOI 10.1002/pbc
differentiation, we assessed the expression of Myosin Heavy
Chain (MyHC), a protein expressed late in the differentiation
program. We found that individual RMS cells expressing MyHC
were much more apparent following exposure to ATRA, in both
Rh30 and JR1 lines (Fig. 2A and B, middle and lower panels; and
Fig. 2C). To account for the different cell density in ATRA-treated
versus vehicle-treated cells, these experiments were also done
with differential plating such that cell density was similar at
day 9 of treatment, and the results were reproducible (representa-
tive Fig. 2D). Western blotting confirmed that MyHC expression
was augmented in ATRA-treated cells (Fig. 2E). Thus, prolonged
exposure to ATRA induced morphological and molecular
evidence of greater skeletal muscle differentiation in both embry-
onal and alveolar RMS cell lines, and this correlated with the
decreased proliferation.
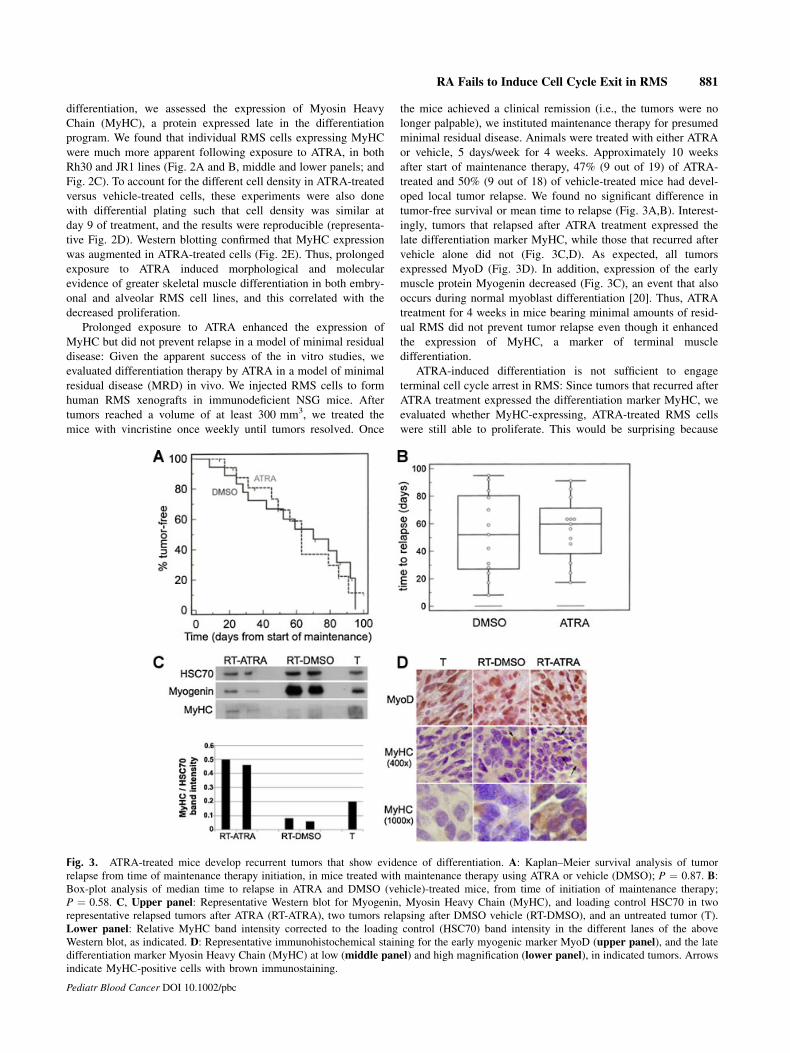
Prolonged exposure to ATRA enhanced the expression of
MyHC but did not prevent relapse in a model of minimal residual
disease: Given the apparent success of the in vitro studies, we
evaluated differentiation therapy by ATRA in a model of minimal
residual disease (MRD) in vivo. We injected RMS cells to form
human RMS xenografts in immunodeficient NSG mice. After
tumors reached a volume of at least 300 mm3, we treated the
mice with vincristine once weekly until tumors resolved. Once
the mice achieved a clinical remission (i.e., the tumors were no
longer palpable), we instituted maintenance therapy for presumed
minimal residual disease. Animals were treated with either ATRA
or vehicle, 5 days/week for 4 weeks. Approximately 10 weeks
after start of maintenance therapy, 47% (9 out of 19) of ATRA-
treated and 50% (9 out of 18) of vehicle-treated mice had devel-
oped local tumor relapse. We found no significant difference in
tumor-free survival or mean time to relapse (Fig. 3A,B). Interest-
ingly, tumors that relapsed after ATRA treatment expressed the
late differentiation marker MyHC, while those that recurred after
vehicle alone did not (Fig. 3C,D). As expected, all tumors
expressed MyoD (Fig. 3D). In addition, expression of the early
muscle protein Myogenin decreased (Fig. 3C), an event that also
occurs during normal myoblast differentiation [20]. Thus, ATRA
treatment for 4 weeks in mice bearing minimal amounts of resid-
ual RMS did not prevent tumor relapse even though it enhanced
the expression of MyHC, a marker of terminal muscle
differentiation.
ATRA-induced differentiation is not sufficient to engage
terminal cell cycle arrest in RMS: Since tumors that recurred after
ATRA treatment expressed the differentiation marker MyHC, we
evaluated whether MyHC-expressing, ATRA-treated RMS cells
were still able to proliferate. This would be surprising because
Fig. 3. ATRA-treated mice develop recurrent tumors that show evidence of differentiation. A: Kaplan–Meier survival analysis of tumor
relapse from time of maintenance therapy initiation, in mice treated with maintenance therapy using ATRA or vehicle (DMSO); P ¼ 0.87. B:Box-plot analysis of median time to relapse in ATRA and DMSO (vehicle)-treated mice, from time of initiation of maintenance therapy;
P ¼ 0.58. C, Upper panel: Representative Western blot for Myogenin, Myosin Heavy Chain (MyHC), and loading control HSC70 in two
representative relapsed tumors after ATRA (RT-ATRA), two tumors relapsing after DMSO vehicle (RT-DMSO), and an untreated tumor (T).
Lower panel: Relative MyHC band intensity corrected to the loading control (HSC70) band intensity in the different lanes of the above
Western blot, as indicated. D: Representative immunohistochemical staining for the early myogenic marker MyoD (upper panel), and the late
differentiation marker Myosin Heavy Chain (MyHC) at low (middle panel) and high magnification (lower panel), in indicated tumors. Arrows
indicate MyHC-positive cells with brown immunostaining.
RA Fails to Induce Cell Cycle Exit in RMS 881
Pediatr Blood Cancer DOI 10.1002/pbc

terminal muscle differentiation is normally coupled to cell cycle
arrest in G0/G1 phase [21]. To evaluate whether ATRA simply
delayed progression through S-phase, we exposed the cells to
longer pulses of BrdU (4 hours), then evaluated the percentage
of cells in S-phase. We found that the difference between ATRA-
treated and vehicle-treated RMS cells persisted (Fig. 4A).
We then evaluated whether ATRA-treated cells that continued
to proliferate showed evidence of differentiation. Dual staining for
BrdU as a cell proliferation marker and MyHC revealed that,
although most ATRA-treated RMS cells were either BrdU-
positive alone or MyHC-positive alone, some cells showed
positivity for both markers (Fig. 4B). This indicated that
ATRA-induced differentiation did not preclude continued cell
cycle progression and cellular proliferation in cultured RMS cells.
Previous work using genetically engineered mice has shown
that myoblasts that lack the retinoblastoma protein (RB�/� cells)
can express some terminal skeletal muscle differentiation
proteins, but can re-enter the cell cycle upon growth factor stim-
ulation [22–24]. This is due to the failure of RB to repress genes
activated by members of the E2F family of transcription factors
[25]. Thus lack of RB prevents the engagement of the terminal
differentiation program and concomitant cell cycle exit.
We considered that the RB protein may remain functionally
inactive in ATRA-treated RMS cells to foster continued prolifer-
ation. Previous studies have shown that ATRA can induce expres-
sion of CDK-inhibitors, leading to decreased phosphorylation of
RB in several types of cells [26–28]. By Western blotting, we
found that ATRA treatment increased the expression of the
CDK4-inhibitor p18Ink4c in both Rh30 and JR1 cells, and
increased the expression of the CDK2-inhibitors p21Cip1 and
p27Kip1 in JR1 cells (but not Rh30 cells; Fig. 4C). However,
this did not translate into CDK4 inhibition because the RB protein
was still phosphorylated at CDK4-dependent sites in RMS cells
regardless of ATRA treatment (Fig. 4D). In addition, Cyclin A, a
known E2F target, was still expressed in ATRA-treated RMS cells
(Fig. 4C) further demonstrating that the RB pathway was func-
tionally inactive. Thus, we conclude that, even though ATRA
treatment induced several CDK-inhibitors, this was neither suffi-
cient to repress Cyclin A nor block RB protein phosphorylation.
This provides a molecular explanation for continued cellular
proliferation despite expression of late muscle differentiation
markers.
Retinoids fail to fully engage the terminal differentiation
program in normal myoblasts: We next sought to examine the
pro-differentiation effects of retinoic acid compounds in the well-
established C2C12 murine myoblast model. The withdrawal of
serum-derived mitogens (differentiation medium, DM) and the
addition of insulin (DM þ I) is well-established to activate myo-
genic differentiation, resulting in cell elongation and myoblast
fusion, irreversible cell cycle arrest, and increased expression of
Myogenin and MyHC within 24–72 hours (Fig. 5A,C). When
either ATRA or CRA were added to mitogen-rich cell culture
medium (GM), the expression of Myogenin increased to nearly
the same level as that achieved by DM þ I (Fig. 5A). However,
we observed no induction of MyHC in this timeframe. Both DM
and DM þ I also increased the fraction of Myogenin expressing
cells by more than 10- and 25-fold over GM (Fig. 5B, left panel).
However, even though Myogenin induction by CRA
Fig. 4. ATRA-induced differentiation does not prevent RMS cellular proliferation. A: Percentage of BrdU-positive Rh30 and JR1 cells, after
9 days of treatment with either ATRA or drug vehicle DMSO; cells were treated with long pulses of BrdU (4 hours). B: Representativephotomicrograph showing dual immunofluorescence staining for Myosin Heavy Chain (MyHC) and BrdU, and corresponding DAPI staining, in
JR1 cells treated with ATRA for 9 days in culture. Small arrowhead indicates a double-positive cell; large arrowhead shows an MyHC-only
positive cell, and long arrows indicate BrdU-only positive cells. C: Representative Western blot for the indicated cell cycle proteins in Rh30 and
JR1 cells treated for 9 days with either ATRA (A) or vehicle (DMSO; D). HSC70 is a loading control. D: Representative Western blot for the
retinoblastoma (RB) protein phosphorylated at Ser780 (CDK4-dependent site) in Rh30 and JR1 cells treated for 9 days with either ATRA (A) or
vehicle (DMSO; D). HSC70 is a loading control; mature skeletal muscle (SkM) was used as a negative control; proliferating cells were used as
positive control.
882 Al-Tahan et al.
Pediatr Blood Cancer DOI 10.1002/pbc

approximated that in DM þ I when measured by Western blotting
(Fig. 5A), CRA had a very small effect on the faction of Myo-
genin-expressing cells (Fig. 5B).
We considered whether the discrepancy between the two
results was due to a failure of retinoic acid to arrest cell prolifer-
ation. Indeed, the total number of cells and the fraction of cells
incorporating BrdU were the same in cells treated with either
CRA, ATRA, or vehicle (Fig. 5B, right panel and Fig. 5C) where-
as cell number and BrdU incorporation decreased in DM or
DM þ I. These data demonstrate that retinoic acid treatment
enhanced Myogenin expression in normal myoblasts, but this
was not accompanied by the robust cell proliferation arrest that
normally accompanies myoblast differentiation.
DISCUSSION
Previous studies have shown that retinoids can slow RMS cell
proliferation and/or promote differentiation in vitro, and in certain
skeletal myoblast cell lines [9,14,15,29,30]. This prompted us to
further explore its therapeutic potential in vivo. We used a model
that closely reflects the clinical scenario in which retinoids are
likely to be applied: maintenance therapy following chemothera-
py-induced minimal residual disease. We chose the JR1 and Rh30
cell lines because previous studies with these lines had yielded
promising results [14,15] and they represented the two major
subtypes of RMS. Our results demonstrate that, despite enhancing
muscle gene expression and slowing progression of RMS cells
through the cell cycle in vitro, retinoic acid does not completely
inhibit RMS cell proliferation in vivo nor does it delay disease
recurrence when applied to minimal residual disease.
Of course, caution must be applied when drawing conclusions
from pre-clinical models, including ours. For example, it is
formally possible that longer duration of ATRA therapy could
have delayed tumor recurrence. We think this is unlikely because
some tumors had already recurred by the end of the four weeks of
treatment. Also, although the cell lines we examined represented
both major RMS subtypes, the conclusions are based on results
from only two cell lines. We chose not to further pursue this line
of investigation using a larger panel of cell lines because our
analysis of normal myoblasts revealed a disconnection between
retinoid-induced induction of myogenic proteins and cell cycle
exit. Since retinoic acid did not foster the coordinated induction of
muscle genes and cell cycle arrest in normal myoblasts, it seemed
unlikely to induce permanent cell cycle arrest in other RMS cell
lines as a single agent.
Our analyses provide some insight into why retinoic acid did
not have a more robust effect on tumor recurrence. It is well-
established that cell cycle exit in G0 phase is essential for normal
skeletal muscle differentiation, and the RB protein plays a partic-
ularly important role [21]. In cultured myoblasts, the G0 arrest
occurs approximately 24 hours after muscle differentiation begins
and is coupled to induction of Myogenin [31]. The arrest
correlates with repression of G1-cyclins such as Cyclin D1, and
induction of Cdk inhibitors like p21Cip1 and p18Ink4c [32,33].
The net effect is to activate the RB protein by preventing its
phosphorylation. Hypophosphorylated, active RB augments the
ability of muscle-specific transcription factors MyoD, Myogenin
and MEF2 to increase muscle gene expression [23,34,35] and
fosters the accompanying cycle arrest by repressing E2F-
dependent gene expression [23,24]. With these numerous layers
of control, DNA synthesis is never observed in a normal, maturing
myocyte unless RB function is compromised by phosphorylation
or mutation [23,24,36].
Our studies reveal that retinoic acid fails to orchestrate cell
cycle arrest, RB activation, and muscle gene induction in RMS
cell lines. One possible explanation is that the RMS cell lines
used may carry a mutated RB allele. This seems unlikely because
pharmacological inhibition of CDK4/6 using PD 0332991 arrests
proliferation in both JR1 and Rh30 cells [19], and cell cycle arrest
by PD 0332991 depends on wild-type RB [37]. Retinoic acid was
also unable to couple cell cycle arrest with differentiation in
normal myoblasts, supporting the conclusion that failure to link
cell cycle arrest to muscle gene induction has to do with the drug
effect rather than a defect intrinsic to the RMS cell lines.
Fig. 5. Retinoids induce Myogenin but fail to arrest cell prolifera-
tion in cultured myoblasts. A: Representative Western blot showing
Myosin Heavy Chain (MyHC), Myogenin, and Heat Shock Complex
70 (HSC70) (as a loading control) in C2C12 myoblasts cultivated for
48 hours in the indicated medium with or without 9-cis-retinoic acid
(CRA) or ATRA. B: Quantitative data showing the fraction of C2C12
cells with Myogenin detectable by immunofluorescence staining (leftpanel) and the total cells visible by DAPI staining (right panel) innine representative field. Cells harvested by fixation following
48 hours with or without CRA treatment. Data represent mean and
standard deviation from four separate wells. All values are statisti-
cally significant when compared to control (GM) (P < 0.05, t-test).
C: Quantitative data showing the fraction of C2C12 cells incorporat-
ing BrdU, measured by direct counting of 10 representative fields.
Values represent mean and standard deviation. Decreased BrdU in-
corporation in DM þ I was significantly different from GM baseline
(�P < 0.05) whereas the percent of BrdU positive cells in
GM þ ATRA and GM þ CRA was not (#P > 0.05; t-test).
RA Fails to Induce Cell Cycle Exit in RMS 883
Pediatr Blood Cancer DOI 10.1002/pbc

Treatment with retinoids enhanced Myogenin but not MyHC
in normal myoblasts, whereas it enhanced MyHC in RMS. If
retinoids were triggering a critical pro-differentiation pathway,
we would expect them to have similar effects in both models.
Instead, we suspect that a pharmacological dose of retinoic acid
likely acts as a transcriptional regulator to enhance the expression
of certain myogenic genes without activating a central regulatory
process. For example, in the C2C12 cells which are committed to
the myogenic lineage by virtue of MyoD expression, retinoic acid
may push them slightly more toward a differentiated myocyte by
enhancing Myogenin expression, but MyHC is not induced
because the RB pathway is not engaged and the cells do not
arrest. In contrast, the RMS cells already display a baseline phe-
notype that is more differentiated (i.e., expression of Myogenin)
and one in which the normal coordination of the cell cycle and
differentiation is already deranged because proliferating tumor
cells express Myogenin. In this setting, retinoic acid fosters
some maturation with increased MyHC but stops short of terminal
differentiation. Further studies identifying genes bound by reti-
noic acid receptors in different cell types seem likely to provide
insight into the differential activity of retinoids in these two cell
types.
Even though there was no obvious delay in tumor recurrence,
the RMS xenografts arising following ATRA treatment displayed
enhanced MyHC and decreased Myogenin, both of which occur in
terminal muscle cell differentiation [20]. Recent data indicate that
human rhabdomyosarcoma tumors with a differentiated pheno-
type, based on RNA expression profiling, are associated with a
better clinical outcome [38,39]. It is therefore possible that the
seemingly small effect that retinoids have on myogenic differen-
tiation in RMS may translate into substantially improved survival
when combined with standard, cytotoxic therapy. Such a possibil-
ity needs to be investigated, and may be studied using preclinical
models.
Finally, it is important to note that differentiation therapy in
RMS may still be a sound goal. Given the importance of RB
in the terminal differentiation process, the approach may not
work in the subset of embryonal RMS in which the RB gene is
mutated or deleted [40]. However, differentiation therapy may be
especially practical in the majority of alveolar RMS where the RB
gene is intact [40] but the protein is functionally compromised
such as by increased CDK4 activity [41]; or loss of CDK4/6-
inhibitors, such as p16INK4a, p15INK4b, and p18INK4c [42].
Treatment of RMS cell lines with CDK4/6 inhibitors has shown
some promise in decreasing cellular proliferation, and a positive
effect on cellular differentiation [19]. By achieving a greater
understanding of exactly how retinoids foster increased muscle
gene expression, it may be possible to rationally apply additional
targeted agents to fully engage the terminal differentiation
program.
ACKNOWLEDGMENT
The authors thank Dr Peter Houghton for providing the RMS
cell lines.
REFERENCES
1. Ries LAG, Smith MA, Gurney JG, et al., editors. Cancer incidence and survival among children and
adolescents: United States SEER Program 1975–1995. Bethesda, MD: National Cancer Institute;
SEER Program 1999.
2. Breitfeld PP, Meyer WH. Rhabdomyosarcoma: New windows of opportunity. Oncologist 2005;10:518–
527.
3. Dias P, Parham DM, Shapiro DN, et al. Myogenic regulatory protein (MyoD1) expression in childhood
solid tumors: Diagnostic utility in rhabdomyosarcoma. Am J Pathol 1990;137:1283–1291.
4. Wang NP, Marx J, McNutt MA, et al. Expression of myogenic regulatory proteins (myogenin and
MyoD1) in small blue round cell tumors of childhood. Am J Pathol 1995;147:1799–1810.
5. Wijnaendts LC, Van Der Linden JC, Van Unnik AJ, et al. The expression pattern of contractile and
intermediate filament proteins in developing skeletal muscle and rhabdomyosarcoma of childhood:
Diagnostic and prognostic utility. J Pathol 1994;174:283–292.
6. Saab R, Spunt S, Skapek SX. Myogenesis and rhabdomyosarcoma: The jekyll and hyde of skeletal
muscle. Curr Top Dev Biol 2011;94:197–234.
7. Lassar AB, Skapek SX, Novitch B. Regulatory mechanisms that coordinate skeletal muscle differenti-
ation and cell cycle withdrawal. Curr Opin Cell Biol 1994;6:788–794.
8. Merlino G, Helman LJ. Rhabdomyosarcoma-working out the pathways. Oncogene 1999;18:5340–
5348.
9. Zhu GH, Huang J, Bi Y, et al. Activation of RXR and RAR signaling promotes myogenic differentia-
tion of myoblastic C2C12 cells. Differentiation 2009;78:195–204.
10. Glaser T, Brustle O. Retinoic acid induction of ES-cell-derived neurons: The radial glia connection.
Trends Neurosci 2005;28:397–400.
11. Hansen LA, Sigman CC, Andreola F, et al. Retinoids in chemoprevention and differentiation therapy.
Carcinogenesis 2000;21:1271–1279.
12. Sidell N, Altman A, Haussler MR, et al. Effects of retinic acid (RA) on the growth and phenotypic
expression of several human neuroblastoma cell lines. Exp Cell Res 1983;148:21–30.
13. Reynolds CP, Matthay KK, Villablanca JG, et al. Retinoid therapy of high-risk neuroblastoma. Cancer
Lett 2003;197:185–192.
14. Barlow JW, Wiley JC, Mous M, et al. Differentiation of rhabdomyosarcoma cell lines using retinoic
acid. Pediatr Blood Cancer 2006;47:773–784.
15. Crouch GD, Helman LJ. All-trans-retinoic acid inhibits the growth of human rhabdomyosarcoma cell
lines. Cancer Res 1991;51:4882–4887.
16. Douglass EC, Valentine E, Parhem D, et al. A specific chromosomal abnormality in rhabdomyosarco-
ma. Cytogenet Cell Genet 1987;45:148–155.
17. Clayton J, Pincott JR, van den Berghe JA, et al. Comparative studies between a new human rhabdo-
myosarcoma cell line, JR-1 and its tumour of origin. Br J Cancer 1986;54:83–90.
18. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in
aging skin in vivo. Proc Natl Acad Sci USA 1995;92:9363–9367.
19. Saab R, Bills JL, Miceli AP, et al. Pharmacologic inhibition of cyclin-dependent kinase 4/6 activity
arrests proliferation in myoblasts and rhabdomyosarcoma-derived cells. Mol Cancer Ther 2006;5:
1299–1308.
20. Grounds MD, Garrett KL, Lai MC, et al. Identification of skeletal muscle precursor cells in vivo by use
of MyoD1 and myogenin probes. Cell Tissue Res 1992;267:99–104.
21. Lassar A, Skapek SX, Novitch B. Regulatory mechanisms that coordinate skeletal muscle differentia-
tion and cell cycle withdrawal. Curr Opin Cell Biol 1994;6:788–794.
22. Gu W, Schneider JW, Condorelli G, et al. Interaction of myogenic factors and the retinoblastoma
protein mediates muscle cell commitment and differentiation. Cell 1993;72:309–324.
23. Novitch BG, Mulligan GJ, Jacks T, et al. Skeletal muscle cells lacking the retinoblastoma protein
display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J Cell
Biol 1996;135: 441–456.
24. Schneider JW, Gu W, Zhu L, et al. Reversal of terminal differentiation mediated by p107 in RB�/�muscle cells. Science 1994;264:1467– 1471.
25. Skapek SX, Pan YR, Lee EY. Regulation of cell lineage specification by the retinoblastoma tumor
suppressor. Oncogene 2006;38:5268–5276.
26. Yen A, Soong S. Retinoic acid-induced RB hypophosphorylation enhanced by CGP 52411 (4,5-
dianilinophthalimide), an EGF family tyrosine kinase receptor inhibition. Eur J Cell Biol 1996;69:
327–334.
27. Zhang D, Vuocolo S, Masciullo V, et al. Cell cycle genes as targets of retinoid induced ovarian tumor
cell growth suppression. Onogene 2001; 20:7935–7944.
28. Lavelle D, Chen YH, Hankewych M, et al. Inhibition of myeloma cell growth by all-trans retinoic acid
is associated with upregulation of p21WAF1 and dephosphorylation of the retinoblastoma protein.
Leuk Lymphoma 1999;35:261–268.
29. Gee MF, Tsuchida R, Eichler-Jonsson C, et al. Vascular endothelial growth factor acts in an autocrine
manner in rhabdomyosarcoma cell lines and can be inhibited with all-trans-retinoic acid. Oncogene
2005; 24:8025–8037.
30. Ricaud S, Vernus B, Bonnieu A. Response of human rhabdomyosarcoma cell lines to retinoic acid:
Relationship with induction of differentiation and retinoic acid sensitivity. Exp Cell Res 2005;311:192–
204.
31. Andres V, Walsh K. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are
temporally separable events that precede cell fusion upon myogenesis. J Cell Biol 1996;132:657–
666.
32. Franklin DS, Xiong Y. Induction of the CDK inhibitor p18INK4C and its predominant association with
CDK4 and CDK6 during myogenic differentiation. Mol Biol Cell 1996;7:1587–1599.
33. Halevy O, Novitch BG, Spicer DB, et al. Correlation of terminal cell cycle arrest of skeletal muscle
with induction of p21 by MyoD. Science 1995;267:1018–1021.
34. Novitch BG, Spicer DB, Kim PS, et al. pRb is required for MEF2-dependent gene expression as well
as cell-cycle arrest during skeletal muscle differentiation. Curr Biol 1999;9:449–459.
35. Skapek SX, Rhee J, Kim PS, et al. Cyclin-mediated inhibition of muscle gene expression via a
mechanism that is independent of pRB hyperphosphorylation. Mol Cell Biol 1996;16:7043–7053.
36. Zacksenhaus E, Jiang Z, Chung D, et al. pRb controls proliferation, differentiation, and death
of skeletal muscle cells and other lineages during embryogenesis. Genes Dev 1996;10:3051–3064.
37. Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991
and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004;3:1427–
1438.
38. Davicioni E, Anderson MJ, Finckenstein FG, et al. Molecular classification of rhabdomyosarcoma—
Genotypic and phenotypic determinants of diagnosis. Am J Pathol 2009;174:550–564.
39. Davicioni E, Finckenstein FG, Shahbazian V, et al. Identification of a PAX-FKHR gene expression
signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas.
Cancer Res 2006;66:6936–6946.
40. Kohashi K, Oda Y, Yamamoto H, et al. Alterations of RB1 gene in embryonal and alveolar rhabdo-
myosarcoma: Special reference to utility of pRB immunoreactivity in differential diagnosis of rhab-
domyosarcoma subtype. J Cancer Res Clin Oncol 2008;134:1097–1103.
41. Berner JM, Forus A, Elkahloun A, et al. Separate amplified regions encompassing CDK4 and MDM2
in human sarcomas. Genes Chromosomes Cancer 1996;17:254–259.
42. Iolascon A, Faienza MF, Coppola B, et al. Analysis of cyclin-dependent kinase inhibitor genes
(CDKN2A, CDKN2B, and CDKN2C) in childhood rhabdomyosarcoma. Genes Chromosomes Cancer
1996;15:217–222.
884 Al-Tahan et al.
Pediatr Blood Cancer DOI 10.1002/pbc