resolution of bronchial hyperresponsiveness and pulmonary inflammation is associated with il3 and...
TRANSCRIPT

of December 11, 2013.This information is current as
with IL-3 and Tissue Leukocyte Apoptosisand Pulmonary Inflammation Is Associated Resolution of Bronchial Hyperresponsiveness
Jose-Carlos Gutierrez-RamosDelaney, Jane Tian, Hans Oettgen, Anthony J. Coyle and Clare M. Lloyd, Jose-Angel Gonzalo, Trang Nguyen, Tracy
http://www.jimmunol.org/content/166/3/20332001; 166:2033-2040; ;J Immunol
Referenceshttp://www.jimmunol.org/content/166/3/2033.full#ref-list-1
, 16 of which you can access for free at: cites 35 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2001 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on Decem
ber 11, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on D
ecember 11, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from

Resolution of Bronchial Hyperresponsiveness and PulmonaryInflammation Is Associated with IL-3 and Tissue LeukocyteApoptosis
Clare M. Lloyd, 1* Jose-Angel Gonzalo,* Trang Nguyen,* Tracy Delaney,* Jane Tian,*Hans Oettgen,† Anthony J. Coyle,* and Jose-Carlos Gutierrez-Ramos2*
We have used two models of murine pulmonary inflammation to investigate the signals responsible for the resolution of bronchialhyperresponsiveness (BHR). Both protocols involved two sensitizations with OVA followed by serial aerosolized challenge withOVA. We determined that administration of the second sensitization by aerosol (model A) was associated with a transientresponse, whereas administration by the i.p. route (model B) induced a sustained response, in the form of BHR and eosinophilia.This difference in kinetics was due solely to the route of the second Ag administration and was not associated with Ag dose oradjuvant. Differences in kinetics of lung eosinophilia/BHR were shown to be independent of IgE levels and IL-4 or IL-5. However,IL-3 levels in model A closely correlated with the rate of leukocyte clearance by apoptosis and were observed concomitant witha decline in BHR. Blockage of IL-3 in model B increased leukocyte apoptosis but reduced tissue eosinophilia and BHR. The useof mouse models in which a single different administration of allergen is associated with a failure/success to resolve inflammationand BHR by 72 h postchallenge indicates a link between IL-3 production, leukocyte apoptosis, and BHR responses.The Journalof Immunology,2001, 166: 2033–2040.
A sthma is a disease characterized by reversible airflowand is associated with episodic increases in bronchialhyperreactivity (BHR)3 to nonspecific stimuli. Various
studies have shown that this BHR occurs as a consequence ofleukocytic infiltration of the airways (1–3). The cellular composi-tion of the inflammatory infiltrate in the lung is characterized byincreased numbers of eosinophils, T lymphocytes, neutrophils,monocytes, and mast cells (4, 5). The recruitment of these cells tothe site of inflammation and their subsequent activation is thoughtto be strictly controlled by a complex series of cellular interactionsmediated by cytokines and chemokines, secreted by resident lungcells as well as by the infiltrating inflammatory cells (6–9). Fol-lowing migration to the site of inflammation, leukocytes releaseproinflammatory mediators (histamines, PGs, leukotrienes, plate-let-activating factor, reactive oxygen intermediates, cytokines, andchemokines) that initiate tissue injury, augment the immune re-sponse, and ultimately result in BHR (10–12).
Increased BHR in asthmatic individuals has been reported asearly as 2 h after provocation, persisting for up to 7 days (13–15).Clinical and experimental studies have outlined mediators that are
thought to be responsible for the initiation of BHR, but there islittle information regarding the factors responsible for the resolu-tion of this lung dysfunction. Animal models of the allergic re-sponse to inhaled Ags show many similarities to those in human,including the induction of BHR and inflammation (7, 16, 17). Ingeneral there is correlation between the degree of BHR and eo-sinophil infiltration in the majority of models (18, 19). The specificimportance of selected mediators, such as particular IgE, IL-4 andIL-5, in the development of the allergic response has been high-lighted by various investigators (17, 19–21). However, despitethese findings the signals responsible for determining whetherBHR resolves or is maintained remain unclear. In the presentstudy, we have addressed some of the issues surrounding the de-velopment of airway hyperresponsiveness by investigating thechronological response to inhaled Ag. We have identified twomodels of murine allergic airway disease (AAD) that, althoughusing similar immunization protocols for disease induction, resultin profound differences in BHR, as well as inflammation. Thisstudy highlights the importance of the route of Ag exposure indetermining the kinetics of BHR and assesses the relative impor-tance of mediators produced by leukocytes in the resolution of thispathophysiology.
Materials and MethodsInduction of AAD
Eight- to 10-wk-old BALB/c mice were purchased from The Jackson Lab-oratory (Bar Harbor, ME) and kept in Millennium Pharmaceuticals spe-cific-pathogen free mouse facility (Cambridge, MA). Mice treated withmodel A were sensitized by OVA, 0.1 mg/mouse i.p. on day 0 (Sigma, St.Louis, MO) and challenged on day 8 (2% OVA aerosolized for 5 min) anddaily between day 15 and 21 (1% OVA, aerosolized for 20 min). PBS (i.p.and/or aerosolized) was administered to mice as a negative control. Micetreated with model B were sensitized by OVA, 0.01 mg/mouse in 0.2 mlalum (Au-Gel-S; Boehringer Ingelheim, Ridgefield, CT) i.p. on day 0.Mice were then challenged on day 10 with 0.01 mg/mouse in 0.2 ml alumand daily between day 19 and 24 (5% OVA aerosolized for 20 min). Thescheme for both treatment protocols is depicted in Fig. 1. Variations of this
*Millennium Pharmaceuticals Inc., Cambridge MA 02139; and†Division of Immu-nology, Children’s Hospital, Boston, MA 02115
Received for publication October 13, 1999. Accepted for publication November1, 2000.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 Address correspondence and reprint requests to Dr. Clare M. Lloyd at her currentaddress: Leukocyte Biology Section, Biomedical Sciences Division, Sir AlexanderFleming Building, Imperial College School of Medicine, London SW7 2AZ, U.K.E-mail address: [email protected] Address correspondence and reprint request to Dr. Jose-Carlos Gutierrez-Ramos,Millennium Pharmaceuticals Inc., 75 Sidney Street, Cambridge, MA 02139. E-mailaddress: [email protected] Abbreviations used in this paper: BHR, bronchial hyperreactivity; AAD, allergicairway disease; BAL, bronchoalveolar lavage; MCh, methacholine; Penh, enhancedpause.
Copyright © 2001 by The American Association of Immunologists 0022-1767/01/$02.00
by guest on Decem
ber 11, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

protocol were used, which included injection of 0.01 mg OVA/mouse or0.1 mg OVA/mouse in the absence of alum on days 0 and 10. Mice weresacrificed by CO2 asphyxiation at 6, 24, 48, or 72 h after OVA adminis-tration on day 21 (model A) or day 24 (model B) and analyzed using thefollowing parameters: bronchoalveolar lavage (BAL), BHR, and histology.
Mice deficient in IgE were obtained from Dr. H. Oettgen (Children’sHospital, Boston, MA) and were subjected to protocol B as describedabove (22). Control age- and sex-matched mice of the same strain (129/SVEV) were obtained from Taconic (Germantown, NY).
For IL-3 blockage experiments, mice were treated with protocol B asdescribed above, until 24 h after the final OVA challenge on day 24 whenone group was given 50mg/mouse of goat anti-mouse IL-3 Ab (R&DSystems, Minneapolis, MN) intranasally. Another OVA-treated group wasgiven 50mg of goat Ig (Jackson ImmunoResearch, West Grove, PA) as acontrol. BHR was measured at 6, 24, 48, and 72 h postchallenge when micewere sacrificed and their lungs analyzed.
Bronchoalveolar lavage
BAL was performed as described (6). Briefly, the airways of the mice werelavaged via a trachea cannula with 1 ml PBS. The resulting BAL fluid wasimmediately centrifuged (7003 g, 5 min at 4°C) and cells were thenwashed and resuspended in 1 ml PBS. Total BAL cell counts were per-formed, and aliquots (53 105 cells/slide) were pelleted onto glass slides bycytocentrifugation. Differential counts were performed on Geimsa-stainedcytospins, and percentages of eosinophils, lymphocytes, neutrophils, andmacrophages was determined by counting their number in eight high-power fields (340 magnification; total area 0.5 mm2) per area randomlyselected and dividing this number by the total number of cells per high-power field (7). To obtain the absolute number of each leukocyte subtypein the lavage, these percentages were multiplied by the total number ofcells recovered from the BAL fluid.
Broncial hyperreactivity
The degree of bronchoconstriction (BHR) was measured at 6, 24, 48, and72 h in the same mice after the last aerosolized Ag challenge by recordingrespiratory pressure curves by whole-body plethysmography (Buxco Tech-nologies, Sharon, CT) in response to inhaled methacholine (MCh; AldrichChemical, Milwaukee, WI) at a concentration of 33 1022 M for 1 min, asdescribed previously (18). BHR was expressed as enhanced pause (Penh),a calculated value, which correlates with measurement of airway resis-tance, impedance, and intrapleural pressure in the same mouse: Penh5(Te/Tr 2 1) 3 (Pef/Pif ), whereTe is the expiration time,Tr is the relaxationtime,Pef is the peak expiratory flow, andPif is the peak inspiratory flow30.67 coefficient) (23). The relaxation time is the time it takes for the boxpressure to change from a maximum to a user-defined percentage of themaximum. Here,Tr measurement begins at the maximum box pressure andends at 40%.Values were expressed as the percentage shift from baseline,which was measured by comparing the Penh of mice before (i.e., baseline)and after stimulation with MCh.
Histology
Lung sections from the different experimental groups of mice were pre-pared as described (7). Briefly, lungs were fixed in 10% neutral-bufferedformalin (J.T. Baker, Phillipsburg, NJ) and paraffin embedded, and sections(4 mm) were stained with hematoxylin/eosin according to standard proto-cols. A semiquantitative scoring system was used to grade the size of lunginfiltrates, where15 signified a large (.3 cells deep) widespread infiltratearound the majority of vessels and bronchioles and11 signifies a smallnumber of inflammatory foci.
Cytokines
Levels of IL-4, IL-5, GM-CSF, IL-3, and IFN-g were assessed in bron-chiolar lavage samples by ELISA according to the manufacturers instruc-tions (Endogen, Woburn, MA).
IgE
Serum levels of total IgE were measured by ELISA using paired Abs ac-cording to the manufacturer’s instructions (BD PharMingen, San Jose,CA). Serum levels of anti-OVA IgE were measured by ELISA, and Abtiters were related to pooled standards generated in the laboratory, thenwere assigned the arbitrary values U/ml.
Apoptosis staining
An estimation of the degree of apoptosis was made in paraffin-embeddedlung sections from mice after protocol A or B at 6, 24, 48, and 72 h
following the final challenge, using an Apotag in situ detection kit (Oncor,Gaithersburg, MD) according to the manufacturer’s instructions. Briefly,sections were rehydrated, digested with proteinase K (20mg/ml, 15 min),and treated to block endogenous peroxidase (3% hydrogen peroxidase inPBS, 5 min). Sections were then incubated with TdT enzyme (37°C, 1 h),washed and incubated with antidigoxigenin conjugate (30 min), and colorwas developed with diaminobenzidine substrate kit (6 min) and counter-stained with hematoxylin. Apoptotic cells were visualized by the brownreaction product on a blue background. Apoptosis was quantified by count-ing the number of positive and negatively stained leukocytes within peri-bronchiolar infiltrates in five high-power fields per section (each field5 0.5mm2 at 3400). Apoptosis was then expressed as a percentage in 2.5 mm2
for each mouse.
Statistics
All results are expressed as mean1 SEM. Student’st test or ANOVA wereused to determine statistical significance between groups of mice, usingbetween 12 and 20 mice in a group. Values ofp , 0.05 were consideredsignificant.
ResultsModel A induces transient AAD whereas model B results in asustained AAD
We examined different models of AAD from the literature withrespect to the pathophysiological parameters BHR and eosinophilinflammation and identified two models that differed profoundly inthe kinetics of BHR. Subsequently, these models were termedmodel A (6) and model B (24). Both models were induced by twosensitizations with OVA followed by multiple aerosolized chal-lenges and resulted in BHR with associated pulmonary inflamma-tion (Fig. 1 and data not shown). However, when BHR was mea-sured at timed intervals after the final aerosolized challenge, wedetermined that model A resulted in an increase in BHR thatpeaked at 24 h but that had returned to baseline by 72 h (Fig. 1).In contrast, model B resulted in a sustained in BHR that was main-tained for at least 72 h (Fig. 1). The magnitude of the responses atpeak BHR was similar in both models with a 700% shift frombaseline achieved in model A (baseline Penh5 0.43 6 0.01, in-creasing to 2.76 0.2 at 24 h) and a 900% shift in baseline seen inmodel B (baseline Penh5 0.396 0.01, moving to 2.966 0.5 at24 h).Although the difference in magnitude was not statisticallydifferent at 24 h (p . 0.1), it was highly significant at 72 h (p ,0.001).
The presence of alum did not account for the difference in ki-netics of BHR development between model A and B. This wasdetermined in mice sensitized with either high (100mg) dose OVA(termed model B/100) or low (10mg) dose OVA (termed modelB/10) in the absence of alum. Both groups developed sustainedhyperreactivity to MCh over 72 h (data not shown), similar tothose mice given protocol B proper. The degree of hyperreactivitywas most pronounced in those mice receiving the higher doseOVA, with the percentage change from baseline reaching compa-rable levels to those achieved in model B proper (baseline Penh50.416 0.01, moving to 2.816 0.41 at 72 h). Thus, although alumseemed to compensate for a lower dose of OVA (model B/100compared with model B), it did not account for differences inkinetics.
Kinetics of eosinophilic inflammation reflects kinetics of BHR
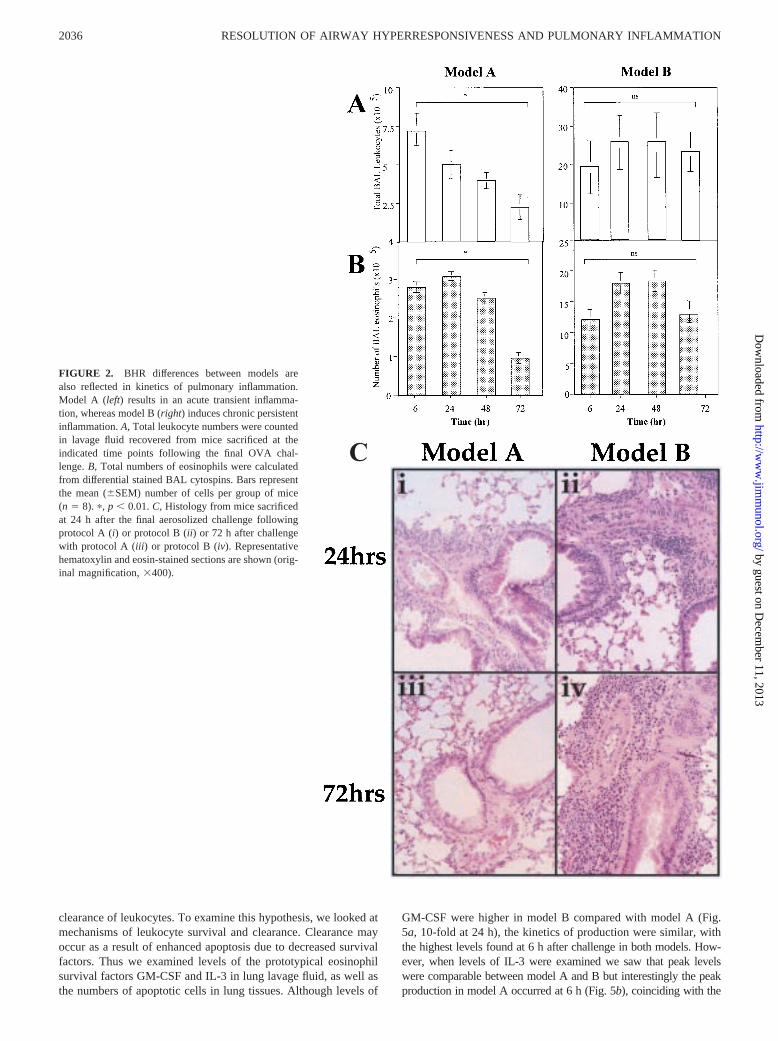
To determine whether the difference in kinetics of BHR observedbetween model A and B correlated with the pattern of inflamma-tion, we determined the extent of leukocytic inflammation in bothlavage and tissue in mice at the same time intervals used to ex-amine BHR. Model A resulted in an increase in total leukocytesfrom as early as 1 h post challenge as previously described in B6mice (7). These numbers peaked at 6 h but declined to prechal-lenge levels by 72 h (Fig. 2A). This increase in cells was primarily
2034 RESOLUTION OF AIRWAY HYPERRESPONSIVENESS AND PULMONARY INFLAMMATION
by guest on Decem
ber 11, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

due to a pronounced eosinophilia and neutrophilia at 6 h, witheosinophils predominating at 24 h (Fig. 2B and data not shown). Incontrast, model B resulted in a prolonged accumulation of cells inthe lavage, where there was no significant decrease in cells overthe 72-h period (Fig. 2A). Again the primary cell type responsiblefor this increase in cellularity in response to aerosolized Ag waseosinophils (Fig. 2B). The difference in cell numbers betweenmodel A and B at 72 h was highly significant with p, 0.0001.
The profile of BAL cell infiltration was found to be independentof either the dose of OVA used for immunization or the use ofalum. Low-dose OVA in the absence of alum still resulted in aneosinophilia that was maintained at 72 h, with 6.96 1.2 3 105
eosinophils remaining at this time point.The extent and anatomical location of eosinophilia in the airway
interstitium was examined in sections from mice sacrificed at timepoints following the last aerosolized Ag challenge. Leukocyteswere observed around vessels and airways from as early as 6 hpostchallenge after protocol A as well as after protocol B (Fig.2C). However, the peak eosinophilia was reached at 24 h for pro-tocol A and 72 h for model B. The primary cell type making up theinfiltrates were eosinophils, although some neutrophils were ob-served with both protocols at 6 h. Although the anatomical locationof infiltrates did not differ discernibly between protocols, the peakinfiltrate was considerably larger in model B than in A (averagescore of15 vs 13, respectively). Most importantly, the infiltrateremained at 72 h in model B, but was cleared by 72 h in model A.
In considering the extent of cellular inflammation between mod-els, model B resulted in a greater degree of eosinophilia comparedwith model A, but interestingly this was not reflected in a signif-icantly greater peak in BHR. However, persistence of inflamma-tory infiltrate did correlate with the persistence of hyperreactivity.
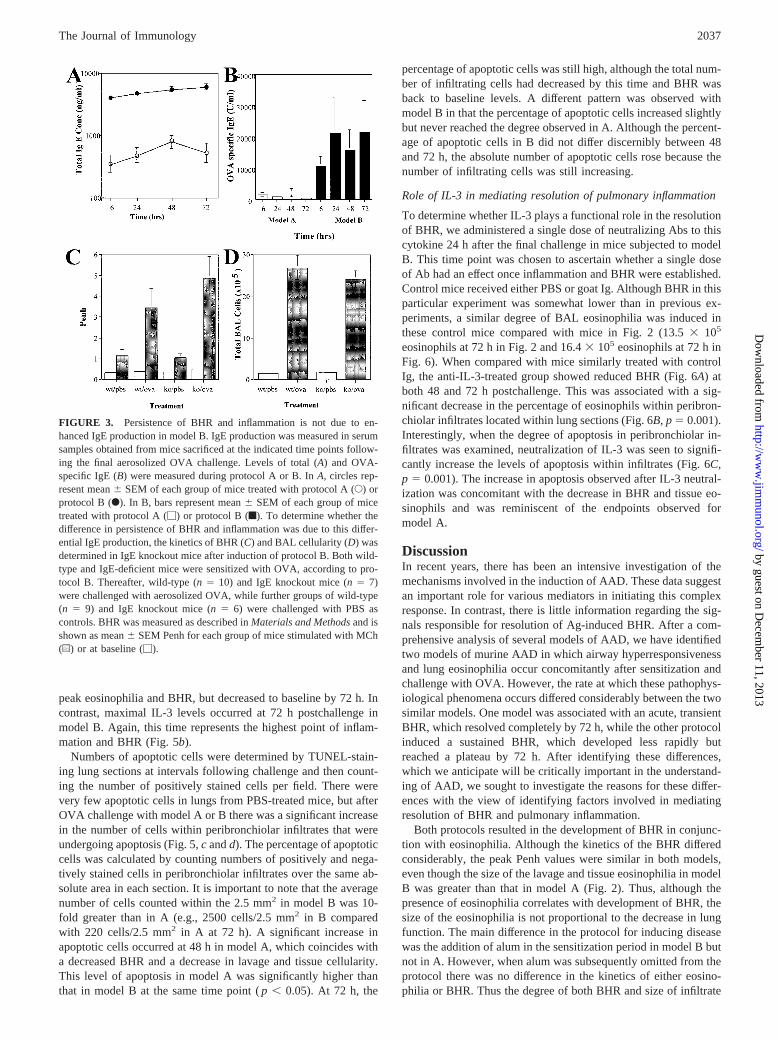
Persistence of BHR and inflammation is not due to enhancedIgE production in model B
Elevated levels of serum IgE have been reported to be important inthe development of an allergic pulmonary response. Therefore, weevaluated whether the difference in kinetics between models couldbe explained by IgE production. Both model A and B resulted inthe secretion of serum IgE, which did not change over the 72-hperiod following the final aerosolized Ag challenge (Fig. 3A).However, the administration of the second OVA challenge via theperitoneal route (model B) induced 10-fold higher levels of IgEthan when administered via aerosol (model A). This difference wasnot due to the presence of alum because comparable levels of IgE
were induced even in the absence of alum (data not shown). OVA-specific IgE varied significantly between the models, with model Beliciting several thousandfold more U/ml of OVA-IgE than modelA (Fig. 3B). We hypothesized that the difference in specific IgElevels might be responsible for the different kinetics of the models.Thus, we took advantage of gene-targeted mice that lack the fa-cility to produce IgE. We induced model B in these knockout miceto determine whether their response to OVA followed the kineticsof model B or A due to their lack of IgE. In fact, absence of IgEdid not change the kinetics of the response, with maximal BHRand inflammation still occurring at 72 h postchallenge (Fig. 3,CandD, respectively). Thus it seems that the delayed resolution inmodel B is not due to the heightened IgE response observed withthis protocol.
The kinetics of inflammation is not dependent on T cell cytokineresponses
For the generation of an efficient immune response, the inductionof a strong T cell response is critical. We compared T cell re-sponses during both protocols by measuring levels of T cell cyto-kines during the challenge phase of both models. Levels of IL-4,IL-5, and IFN-g were measured in BAL fluid collected at differenttime points following the final aerosolized challenge during modelA or B. Both models induced the secretion of IL-4 and IL-5 intothe BAL, with the peak of cytokine production occurring in bothcases at 6 h and decreasing to control levels by 72 h (Fig. 4).However, model A induced 10-fold less of both IL-4 and IL-5compared with model B, and both models showed 10-fold moreIL-5 than IL-4. Levels of IFN-g were comparable between models.Importantly, the kinetics of production followed a similar patternin both models throughout the 72-h period following challenge.Similarly, in vitro-stimulated splenocytes from mice treated withprotocol B showed a 300-fold increase in IL-4, a 5-fold increase inIL-5, and a 1.5-fold increase in IFN-g compared with those pre-pared from model A (data not shown). Our data suggest that sen-sitization solely via the peritoneal route in model B leads to aresponse that is more Th2-like compared with that elicited aftersensitization via the peritoneum plus aerosol as in model A.
Role of leukocyte survival and clearance in determining kineticsof inflammation
The early peak in eosinophils observed at 24 h in model A com-pared with a sustained eosinophilia in model B at 72 h may rep-resent a more efficient resolution of the inflammatory response by
FIGURE 1. BHR and protocols for induction ofAAD. Female BALB/c mice were treated with eitherprotocol A or B, and hyperreactivity to MCh wasmeasured at time points following the final allergenchallenge (6, 24, 48, or 72 h). Results shown arefrom four different experiments with either protocolwith four mice in each group during each experi-ment. Model A (left) results in an acute transientBHR, whereas model B (right) induces chronicmaintained BHR. The percentage change in Penhwas calculated from values taken before and afterprovocation with 20 mg/ml MCh for mice immu-nized with OVA and challenged with PBS (M) orimmunized and challenged with OVA (E). Bars rep-resent SEM.
2035The Journal of Immunology
by guest on Decem
ber 11, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
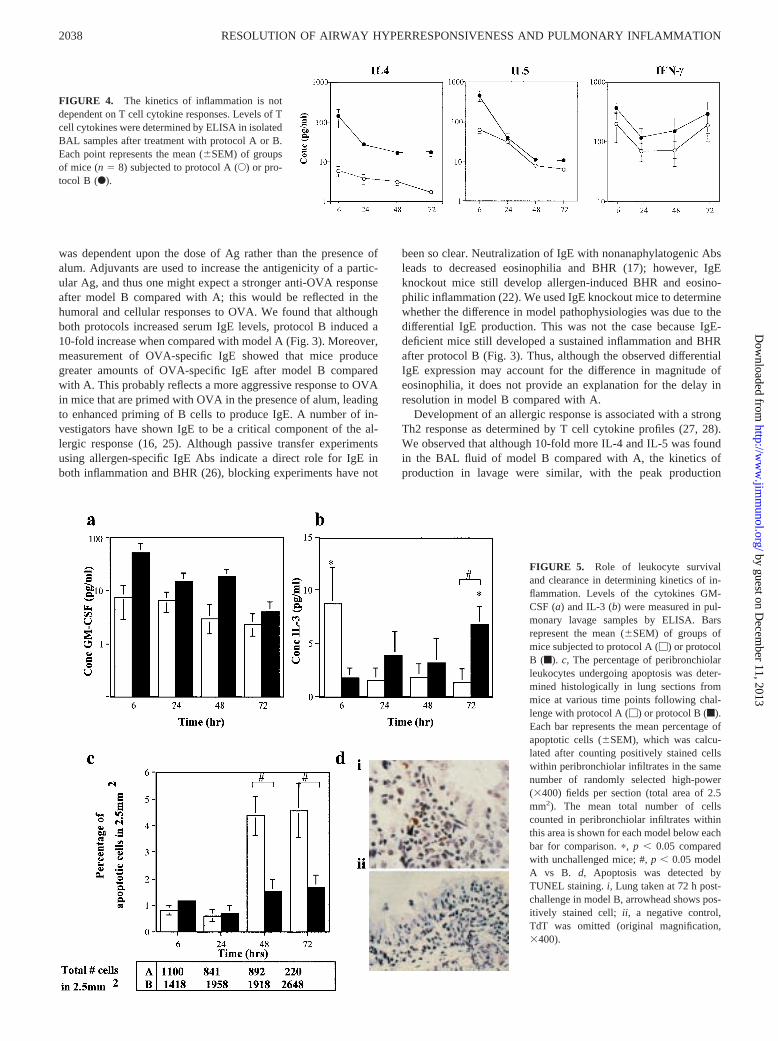
clearance of leukocytes. To examine this hypothesis, we looked atmechanisms of leukocyte survival and clearance. Clearance mayoccur as a result of enhanced apoptosis due to decreased survivalfactors. Thus we examined levels of the prototypical eosinophilsurvival factors GM-CSF and IL-3 in lung lavage fluid, as well asthe numbers of apoptotic cells in lung tissues. Although levels of
GM-CSF were higher in model B compared with model A (Fig.5a, 10-fold at 24 h), the kinetics of production were similar, withthe highest levels found at 6 h after challenge in both models. How-ever, when levels of IL-3 were examined we saw that peak levelswere comparable between model A and B but interestingly the peakproduction in model A occurred at 6 h (Fig. 5b), coinciding with the
C
FIGURE 2. BHR differences between models arealso reflected in kinetics of pulmonary inflammation.Model A (left) results in an acute transient inflamma-tion, whereas model B (right) induces chronic persistentinflammation.A, Total leukocyte numbers were countedin lavage fluid recovered from mice sacrificed at theindicated time points following the final OVA chal-lenge.B, Total numbers of eosinophils were calculatedfrom differential stained BAL cytospins. Bars representthe mean (6SEM) number of cells per group of mice(n 5 8). p, p , 0.01.C, Histology from mice sacrificedat 24 h after the final aerosolized challenge followingprotocol A (i) or protocol B (ii) or 72 h after challengewith protocol A (iii) or protocol B (iv). Representativehematoxylin and eosin-stained sections are shown (orig-inal magnification,3400).
2036 RESOLUTION OF AIRWAY HYPERRESPONSIVENESS AND PULMONARY INFLAMMATION
by guest on Decem
ber 11, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
peak eosinophilia and BHR, but decreased to baseline by 72 h. Incontrast, maximal IL-3 levels occurred at 72 h postchallenge inmodel B. Again, this time represents the highest point of inflam-mation and BHR (Fig. 5b).
Numbers of apoptotic cells were determined by TUNEL-stain-ing lung sections at intervals following challenge and then count-ing the number of positively stained cells per field. There werevery few apoptotic cells in lungs from PBS-treated mice, but afterOVA challenge with model A or B there was a significant increasein the number of cells within peribronchiolar infiltrates that wereundergoing apoptosis (Fig. 5,c andd). The percentage of apoptoticcells was calculated by counting numbers of positively and nega-tively stained cells in peribronchiolar infiltrates over the same ab-solute area in each section. It is important to note that the averagenumber of cells counted within the 2.5 mm2 in model B was 10-fold greater than in A (e.g., 2500 cells/2.5 mm2 in B comparedwith 220 cells/2.5 mm2 in A at 72 h). A significant increase inapoptotic cells occurred at 48 h in model A, which coincides witha decreased BHR and a decrease in lavage and tissue cellularity.This level of apoptosis in model A was significantly higher thanthat in model B at the same time point (p , 0.05). At 72 h, the
percentage of apoptotic cells was still high, although the total num-ber of infiltrating cells had decreased by this time and BHR wasback to baseline levels. A different pattern was observed withmodel B in that the percentage of apoptotic cells increased slightlybut never reached the degree observed in A. Although the percent-age of apoptotic cells in B did not differ discernibly between 48and 72 h, the absolute number of apoptotic cells rose because thenumber of infiltrating cells was still increasing.
Role of IL-3 in mediating resolution of pulmonary inflammation
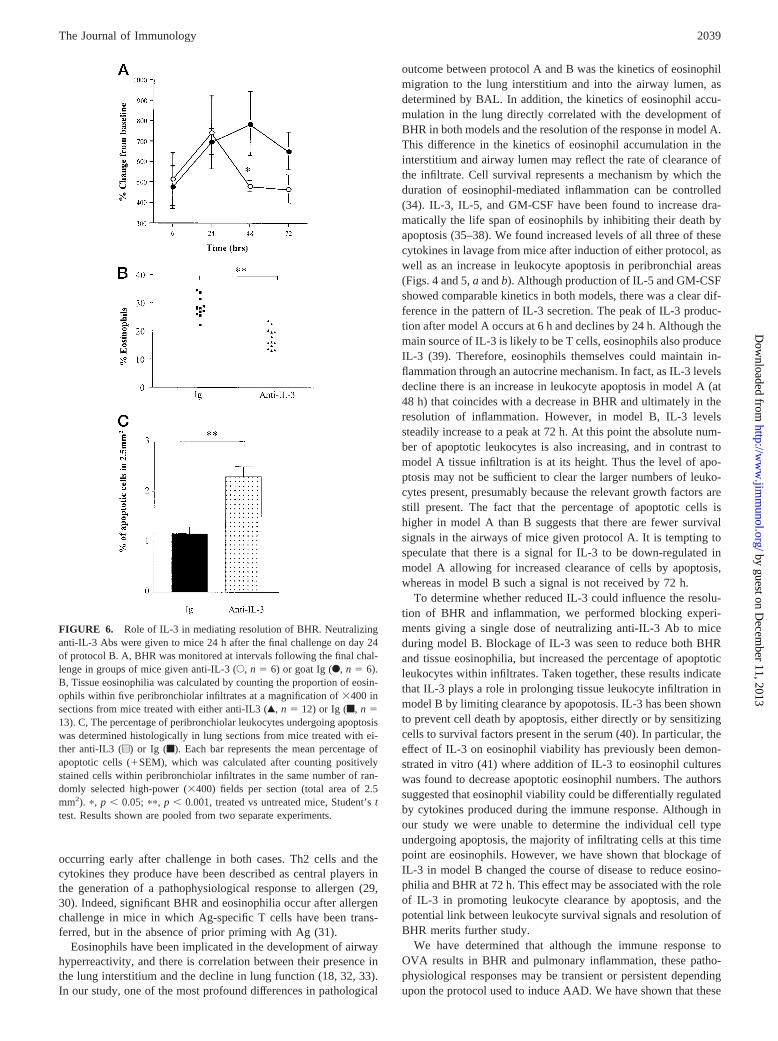
To determine whether IL-3 plays a functional role in the resolutionof BHR, we administered a single dose of neutralizing Abs to thiscytokine 24 h after the final challenge in mice subjected to modelB. This time point was chosen to ascertain whether a single doseof Ab had an effect once inflammation and BHR were established.Control mice received either PBS or goat Ig. Although BHR in thisparticular experiment was somewhat lower than in previous ex-periments, a similar degree of BAL eosinophilia was induced inthese control mice compared with mice in Fig. 2 (13.53 105
eosinophils at 72 h in Fig. 2 and 16.43 105 eosinophils at 72 h inFig. 6). When compared with mice similarly treated with controlIg, the anti-IL-3-treated group showed reduced BHR (Fig. 6A) atboth 48 and 72 h postchallenge. This was associated with a sig-nificant decrease in the percentage of eosinophils within peribron-chiolar infiltrates located within lung sections (Fig. 6B,p 5 0.001).Interestingly, when the degree of apoptosis in peribronchiolar in-filtrates was examined, neutralization of IL-3 was seen to signifi-cantly increase the levels of apoptosis within infiltrates (Fig. 6C,p 5 0.001). The increase in apoptosis observed after IL-3 neutral-ization was concomitant with the decrease in BHR and tissue eo-sinophils and was reminiscent of the endpoints observed formodel A.
DiscussionIn recent years, there has been an intensive investigation of themechanisms involved in the induction of AAD. These data suggestan important role for various mediators in initiating this complexresponse. In contrast, there is little information regarding the sig-nals responsible for resolution of Ag-induced BHR. After a com-prehensive analysis of several models of AAD, we have identifiedtwo models of murine AAD in which airway hyperresponsivenessand lung eosinophilia occur concomitantly after sensitization andchallenge with OVA. However, the rate at which these pathophys-iological phenomena occurs differed considerably between the twosimilar models. One model was associated with an acute, transientBHR, which resolved completely by 72 h, while the other protocolinduced a sustained BHR, which developed less rapidly butreached a plateau by 72 h. After identifying these differences,which we anticipate will be critically important in the understand-ing of AAD, we sought to investigate the reasons for these differ-ences with the view of identifying factors involved in mediatingresolution of BHR and pulmonary inflammation.
Both protocols resulted in the development of BHR in conjunc-tion with eosinophilia. Although the kinetics of the BHR differedconsiderably, the peak Penh values were similar in both models,even though the size of the lavage and tissue eosinophilia in modelB was greater than that in model A (Fig. 2). Thus, although thepresence of eosinophilia correlates with development of BHR, thesize of the eosinophilia is not proportional to the decrease in lungfunction. The main difference in the protocol for inducing diseasewas the addition of alum in the sensitization period in model B butnot in A. However, when alum was subsequently omitted from theprotocol there was no difference in the kinetics of either eosino-philia or BHR. Thus the degree of both BHR and size of infiltrate
FIGURE 3. Persistence of BHR and inflammation is not due to en-hanced IgE production in model B. IgE production was measured in serumsamples obtained from mice sacrificed at the indicated time points follow-ing the final aerosolized OVA challenge. Levels of total (A) and OVA-specific IgE (B) were measured during protocol A or B. InA, circles rep-resent mean6 SEM of each group of mice treated with protocol A (E) orprotocol B (F). In B,bars represent mean6 SEM of each group of micetreated with protocol A (M) or protocol B (f). To determine whether thedifference in persistence of BHR and inflammation was due to this differ-ential IgE production, the kinetics of BHR (C) and BAL cellularity (D) wasdetermined in IgE knockout mice after induction of protocol B. Both wild-type and IgE-deficient mice were sensitized with OVA, according to pro-tocol B. Thereafter, wild-type (n5 10) and IgE knockout mice (n5 7)were challenged with aerosolized OVA, while further groups of wild-type(n 5 9) and IgE knockout mice (n5 6) were challenged with PBS ascontrols. BHR was measured as described inMaterials and Methodsand isshown as mean6 SEM Penh for each group of mice stimulated with MCh(u) or at baseline (M).
2037The Journal of Immunology
by guest on Decem
ber 11, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
was dependent upon the dose of Ag rather than the presence ofalum. Adjuvants are used to increase the antigenicity of a partic-ular Ag, and thus one might expect a stronger anti-OVA responseafter model B compared with A; this would be reflected in thehumoral and cellular responses to OVA. We found that althoughboth protocols increased serum IgE levels, protocol B induced a10-fold increase when compared with model A (Fig. 3). Moreover,measurement of OVA-specific IgE showed that mice producegreater amounts of OVA-specific IgE after model B comparedwith A. This probably reflects a more aggressive response to OVAin mice that are primed with OVA in the presence of alum, leadingto enhanced priming of B cells to produce IgE. A number of in-vestigators have shown IgE to be a critical component of the al-lergic response (16, 25). Although passive transfer experimentsusing allergen-specific IgE Abs indicate a direct role for IgE inboth inflammation and BHR (26), blocking experiments have not
been so clear. Neutralization of IgE with nonanaphylatogenic Absleads to decreased eosinophilia and BHR (17); however, IgEknockout mice still develop allergen-induced BHR and eosino-philic inflammation (22). We used IgE knockout mice to determinewhether the difference in model pathophysiologies was due to thedifferential IgE production. This was not the case because IgE-deficient mice still developed a sustained inflammation and BHRafter protocol B (Fig. 3). Thus, although the observed differentialIgE expression may account for the difference in magnitude ofeosinophilia, it does not provide an explanation for the delay inresolution in model B compared with A.
Development of an allergic response is associated with a strongTh2 response as determined by T cell cytokine profiles (27, 28).We observed that although 10-fold more IL-4 and IL-5 was foundin the BAL fluid of model B compared with A, the kinetics ofproduction in lavage were similar, with the peak production
FIGURE 5. Role of leukocyte survivaland clearance in determining kinetics of in-flammation. Levels of the cytokines GM-CSF (a) and IL-3 (b) were measured in pul-monary lavage samples by ELISA. Barsrepresent the mean (6SEM) of groups ofmice subjected to protocol A (M) or protocolB (f). c, The percentage of peribronchiolarleukocytes undergoing apoptosis was deter-mined histologically in lung sections frommice at various time points following chal-lenge with protocol A (M) or protocol B (f).Each bar represents the mean percentage ofapoptotic cells (6SEM), which was calcu-lated after counting positively stained cellswithin peribronchiolar infiltrates in the samenumber of randomly selected high-power(3400) fields per section (total area of 2.5mm2). The mean total number of cellscounted in peribronchiolar infiltrates withinthis area is shown for each model below eachbar for comparison.p, p , 0.05 comparedwith unchallenged mice; #,p , 0.05 modelA vs B. d, Apoptosis was detected byTUNEL staining.i, Lung taken at 72 h post-challenge in model B, arrowhead shows pos-itively stained cell; ii, a negative control,TdT was omitted (original magnification,3400).
FIGURE 4. The kinetics of inflammation is notdependent on T cell cytokine responses. Levels of Tcell cytokines were determined by ELISA in isolatedBAL samples after treatment with protocol A or B.Each point represents the mean (6SEM) of groupsof mice (n5 8) subjected to protocol A (E) or pro-tocol B (F).
2038 RESOLUTION OF AIRWAY HYPERRESPONSIVENESS AND PULMONARY INFLAMMATION
by guest on Decem
ber 11, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
occurring early after challenge in both cases. Th2 cells and thecytokines they produce have been described as central players inthe generation of a pathophysiological response to allergen (29,30). Indeed, significant BHR and eosinophilia occur after allergenchallenge in mice in which Ag-specific T cells have been trans-ferred, but in the absence of prior priming with Ag (31).
Eosinophils have been implicated in the development of airwayhyperreactivity, and there is correlation between their presence inthe lung interstitium and the decline in lung function (18, 32, 33).In our study, one of the most profound differences in pathological
outcome between protocol A and B was the kinetics of eosinophilmigration to the lung interstitium and into the airway lumen, asdetermined by BAL. In addition, the kinetics of eosinophil accu-mulation in the lung directly correlated with the development ofBHR in both models and the resolution of the response in model A.This difference in the kinetics of eosinophil accumulation in theinterstitium and airway lumen may reflect the rate of clearance ofthe infiltrate. Cell survival represents a mechanism by which theduration of eosinophil-mediated inflammation can be controlled(34). IL-3, IL-5, and GM-CSF have been found to increase dra-matically the life span of eosinophils by inhibiting their death byapoptosis (35–38). We found increased levels of all three of thesecytokines in lavage from mice after induction of either protocol, aswell as an increase in leukocyte apoptosis in peribronchial areas(Figs. 4 and 5,a andb). Although production of IL-5 and GM-CSFshowed comparable kinetics in both models, there was a clear dif-ference in the pattern of IL-3 secretion. The peak of IL-3 produc-tion after model A occurs at 6 h and declines by 24 h. Although themain source of IL-3 is likely to be T cells, eosinophils also produceIL-3 (39). Therefore, eosinophils themselves could maintain in-flammation through an autocrine mechanism. In fact, as IL-3 levelsdecline there is an increase in leukocyte apoptosis in model A (at48 h) that coincides with a decrease in BHR and ultimately in theresolution of inflammation. However, in model B, IL-3 levelssteadily increase to a peak at 72 h. At this point the absolute num-ber of apoptotic leukocytes is also increasing, and in contrast tomodel A tissue infiltration is at its height. Thus the level of apo-ptosis may not be sufficient to clear the larger numbers of leuko-cytes present, presumably because the relevant growth factors arestill present. The fact that the percentage of apoptotic cells ishigher in model A than B suggests that there are fewer survivalsignals in the airways of mice given protocol A. It is tempting tospeculate that there is a signal for IL-3 to be down-regulated inmodel A allowing for increased clearance of cells by apoptosis,whereas in model B such a signal is not received by 72 h.
To determine whether reduced IL-3 could influence the resolu-tion of BHR and inflammation, we performed blocking experi-ments giving a single dose of neutralizing anti-IL-3 Ab to miceduring model B. Blockage of IL-3 was seen to reduce both BHRand tissue eosinophilia, but increased the percentage of apoptoticleukocytes within infiltrates. Taken together, these results indicatethat IL-3 plays a role in prolonging tissue leukocyte infiltration inmodel B by limiting clearance by apopotosis. IL-3 has been shownto prevent cell death by apoptosis, either directly or by sensitizingcells to survival factors present in the serum (40). In particular, theeffect of IL-3 on eosinophil viability has previously been demon-strated in vitro (41) where addition of IL-3 to eosinophil cultureswas found to decrease apoptotic eosinophil numbers. The authorssuggested that eosinophil viability could be differentially regulatedby cytokines produced during the immune response. Although inour study we were unable to determine the individual cell typeundergoing apoptosis, the majority of infiltrating cells at this timepoint are eosinophils. However, we have shown that blockage ofIL-3 in model B changed the course of disease to reduce eosino-philia and BHR at 72 h. This effect may be associated with the roleof IL-3 in promoting leukocyte clearance by apoptosis, and thepotential link between leukocyte survival signals and resolution ofBHR merits further study.
We have determined that although the immune response toOVA results in BHR and pulmonary inflammation, these patho-physiological responses may be transient or persistent dependingupon the protocol used to induce AAD. We have shown that these
FIGURE 6. Role of IL-3 in mediating resolution of BHR. Neutralizinganti-IL-3 Abs were given to mice 24 h after the final challenge on day 24of protocol B. A, BHR was monitored at intervals following the final chal-lenge in groups of mice given anti-IL-3 (E, n 5 6) or goat Ig (F,n 5 6).B, Tissue eosinophilia was calculated by counting the proportion of eosin-ophils within five peribronchiolar infiltrates at a magnification of3400 insections from mice treated with either anti-IL3 (Œ,n 5 12) or Ig (f,n 513). C, The percentage of peribronchiolar leukocytes undergoing apoptosiswas determined histologically in lung sections from mice treated with ei-ther anti-IL3 (u) or Ig (f). Each bar represents the mean percentage ofapoptotic cells (1SEM), which was calculated after counting positivelystained cells within peribronchiolar infiltrates in the same number of ran-domly selected high-power (3400) fields per section (total area of 2.5mm2). p, p , 0.05; pp, p , 0.001, treated vs untreated mice, Student’sttest. Results shown are pooled from two separate experiments.
2039The Journal of Immunology
by guest on Decem
ber 11, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

differences in Ag administration lead to a differential T cell cyto-kine response. In conclusion, i.p. administration of allergen is as-sociated with a failure to resolve inflammation and BHR by 72 hpostchallenge, which is, at least in part, mediated by a low IL-3production and apoptosis. Investigation of these differences maylead investigators in the field to a greater understanding of how theimmune response to inhaled Ags develops and, perhaps more im-portantly, the mechanisms by which hyperreactivity and inflam-mation resolves.
References1. Beasley, R., W. R. Roche, J. A. Roberts, and S. T. Holgate. 1989. Cellular events
in the bronchi in mild asthma and after bronchial provocation.Am. Rev. Respir.Dis. 139:806.
2. Gleich, G. J. 1990. The eosinophil and bronchial asthma: current understanding.J. Allergy Clin. Immunol. 85:422.
3. Sedgwick, J. B., W. J. Calhoun, G. J. Gleich, H. Kita, J. S. Abrams,L. B. Schwartz, B. Volovitz, M. Ben-Yaakov, and W. W. Busse. 1991. Immediateand late airway response of allergic rhinitis patients to segmental antigen chal-lenge: characterization of eosinophil and mast cell mediators.Am. Rev. Respir.Dis. 144:1274.
4. Djukanovic, R., W. R. Roche, J. W. Wilson, C. R. Beasley, O. P. Twentyman,R. H. Howarth, and S. T. Holgate. 1990. Mucosal inflammation in asthma.Am.Rev. Respir. Dis. 142:434.
5. Bradley, B. L., M. Azzawi, M. Jacobson, B. Assoufi, J. V. Collins,A.-M. A. Irani, L. B. Schwartz, S. R. Durham, P. K. Jeffrey, and A. B. Kay. 1991.Eosinophils, T-lymphocytes, mast cells, neutrophils and macrophages in bron-chial biopsy specimens from atopic subjects with asthma: comparison with bi-opsy specimens from atopic subjects without asthma and normal control subjectsand relationship to bronchial hyperresponsiveness.J. Allergy Clin. Immunol. 88:661.
6. Gonzalo, J.-A., G.-Q. Jia, V. Aguirre, D. Friend, A. J. Coyle, N. A. Jenkins,G. S. Lin, H. Katz, A. Litchman, N. Copeland, M. Kopf, andJ. C. Gutierrez-Ramos. 1996. Mouse eotaxin expression parallels eosinophil ac-cumulation during lung allergic inflammation but it is not restricted to a Th2-typeresponse.Immunity 4:1.
7. Gonzalo, J. A., C. M. Lloyd, L. Kremer, E. Finger, C. Martinez-A,M. H. Siegelman, M. Cybulski, and J. C. Gutierrez-Ramos. 1996. Eosinophilrecruitment to the lung in a murine model of allergic inflammation: the role of Tcells, chemokines and endothelial adhesion receptors.J. Clin. Invest. 98:2332.
8. Lukacs, N. W., R. M. Strieter, S. W. Chensue, and S. L. Kunkel. 1996. Activationand regulation of chemokines in allergic airway inflammation.J. Leukocyte Biol.59:13.
9. Ohkawara, Y., X. F. Lei, M. R. Stampfli, J. S. Marshall, Z. Xing, and M. Jordana.1997. Cytokine and eosinophil responses in the lung, peripheral blood, and bonemarrow compartments in a murine model of allergen-induced airways inflam-mation.Am. J. Respir. Cell Mol. Biol. 16:510.
10. Drazen, J. M., K. F. Austen, R. A. Lewis, D. A. Clark, G. Goto, A. Marfat, andE. J. Corey. 1980. Comparative airway and vascular activities of leukotrienes C-1and D in vivo and in vitro.Proc. Natl. Acad. Sci. USA 77:4354.
11. Casale, T. B., D. Wood, H. B. Richerson, S. Trapp, W. J. Metzger, Z. D, andG. W. Hunninghake. 1987. Elevated bronchiolar lavage fluid histamine levels inallergic asthmatics are associated with methacholine bronchial hyperresponsive-ness.J. Clin. Invest. 79:1197.
12. Gleich, G. J., N. A. Flavahan, T. Fujisawa, and P. M. Vanhoutte. 1988. Theeosinophil as a mediator of damage to respiratory epithelium: a model for bron-chial hyperreactivity.J. Allergy Clin. Immunol. 81:776.
13. Cockroft, D. W., and K. Y. Murdock. 1987. Changes in bronchial responsivenessto histamine at intervals after allergen challenge.Thorax 42:302.
14. Smith, H. R., G. L. Larsen, R. M. Cherniak, S. E. Wenzel, N. F. Voelkel,J. Y. Westcott, and R. A. Bethel. 1992. Inflammatory cells and eicosanoid me-diators in subjects with late asthmatic responses and increases in airway respon-siveness.J Allergy Clin. Immunol. 86:1076.
15. Bernstein, D. I., Y. Ploysongsang, R. J. Mittman, A. Piyamahunt, andI. L. Bernstein. 1992. The relationship between airway responsiveness measuredbefore and after the allergen-induced late asthmatic response.Chest 101:437.
16. Renz, H., H. R. Smith, J. E. Henson, B. S. Ray, C. G. Irvin, and E. W. Gelfand.1992. Aerosolized antigen exposure without adjuvant causes increased IgE pro-duction and increased airway responsiveness in the mouse.J Allergy Clin. Im-munol. 89:1127.
17. Coyle, A. J., K. Wagner, C. Bertrand, S. Tsuyuki, J. Bews, and C. Heusser. 1996.Central role of immunoglobulin IgE in the induction of lung eosinophil infiltra-tion and T helper 2 cell cytokine production: Inhibition by a non-anaphylacto-genic anti-IgE antibody.J. Exp. Med. 183:1303.
18. Eum, S.-Y., S. Haile, J. Lefort, M. Huerre, and B. B. Vargaftig. 1995. Eosinophilrecruitment into the respiratory epithelium following antigenic challenge in hy-
per-IgE mice is accompanied by interleukin 5-dependent bronchial hyperrespon-siveness.Proc. Natl. Acad. Sci. USA 92:12290.
19. Corry, D. B., H. G. Folkesson, M. L. Warnock, D. J. Erle, M. A. Matthay,J. P. Wiener-Kronish, and R. M. Locksley. 1996. Interleukin 4, but not interleukin5 or eosinophils, is required in a murine model of acute airway hyperreactivity.J. Exp. Med. 183:109.
20. Oshiba, A., E. Hamelmann, K. Takeda, K. L. Bradley, J. E. Loader, G. L. Larsen,and E. W. Gelfand. 1996. Passive transfer of immediate hypersensitivity andairway hyperresponsiveness by allergen-specific immunoglobulin IgE and IgG1in mice.J. Clin. Invest. 97:1398.
21. Foster, P. S., S. P. Hogan, A. J. Ramsay, K. I. Matthaei, and I. G. Young. 1996.Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lungdamage in a mouse asthma model.J. Exp. Med. 183:195.
22. Mehlop, P. D., M. van de Rijn, A. B. Goldberg, J. P. Brewer, V. P. Kurup,T. R. Martin, and H. C. Oettgen. 1997. Allergen-induced bronchial hyperreac-tivity and eosinophilic inflammation occur in the absence of IgE in a mousemodel of asthma.Proc. Natl. Acad. Sci. USA 94:1344.
23. Gelfand, E. W., and C. G. Irvin. 1997. T lymphocytes: setting the tone of theairways.Nat. Med. 3:382.
24. Tsuyuki, S., J. Tsuyuki, K. Einsle, M. Kopf, and A. J. Coyle. 1997. Costimulationthrough B7-2 (CD86) is required for the induction of a lung mucosal T helper cell2 (TH2) immune response and altered airway responsiveness.J. Exp. Med. 185:9:1671.
25. Oshiba, A., E. Hamelmann, A. Haczku, K. Takeda, D. H. Conrad, H. Kikutani,and E. W. Gelfand. 1997. Modulation of antigen-induced B and T cell responsesby antigen-specific IgE antibodies.J. Immunol. 159:4056.
26. Oshiba, A., E. Hamelmann, K. Takeda, K. L. Bradley, J. E. Loader, G. L. Larsen,and E. W. Gelfand. 1996. Passive transfer of immediate hypersensitivity andairway hyperresponsiveness by allergen-specific immunoglobulin IgE and IgG1in mice.J. Clin. Invest. 97:1398.
27. Robinson, D. S., Q. Hamid, S. Ying, A. Tsicopoulus, J. Barkans, A. M. Bentley,C. Corrigan, S. R. Durham, and A. B. Kay. 1992. Predominant Th2-like bron-choalveolar T-lymphocyte population in atopic asthma.N. Engl. J. Med. 326:298.
28. Nakajima, H., A. Nakao, Y. Watanabe, S. Yoshida, and I. Iwamoto. 1994. IFN-ainhibits antigen-induced eosinophil and CD41 T cell recruitment into tissue.J. Immunol. 1264.
29. Anderson, G. P., and A. J. Coyle. 1994. Th2 and “Th2-like” cells in allergy andasthma: pharmacological perspectives.Trends Pharmacol. Sci. 15:324.
30. Kay, A. B. 1996. Th2-type cytokines in asthma.Ann. NY Acad. Sci. 796:1.31. Cohn, L., R. J. Homer, A. Marinov, J. Rankin, and K. Bottomly. 1997. Induction
of airway mucus production By T helper 2 (Th2) cells: a critical role for inter-leukin 4 in cell recruitment but not mucus production.J. Exp. Med. 186:1737.
32. Gonzalo, J. A., C. M. Lloyd, D. Wen, J. P. Albar, T. N. Wells, A. Proudfoot,C. Martinez-A, M. Dorf, T. Bjerke, A. J. Coyle, and J. C. Gutierrez-Ramos. 1998.The coordinated action of CC chemokines in the lung orchestrates allergic in-flammation and airway hyperresponsiveness.J. Exp. Med. 188:no.1:157.
33. Gerwin, N., J. A. Gonzalo, C. Lloyd, A. J. Coyle, Y. Reiss, N. Banu, B. Wand,H. Xu, H. Avraham, B. Engelhardt, T. A. Springer, and J. C. Gutierrez-Ramos.1998. Severely impaired eosinophil accumulation in the airways of intercellularadhesion molecule-2 (ICAM-2) deficient mice during the development of allergiclung inflammation, does not prevent bronchial hyperresponsiveness.Immunity10:9.
34. Stern, M., L. Meagher, J. Savill, and C. Haslett. 1992. Apoptosis in human eo-sinophils: programmed cell death in the eosinophil leads to phagocytosis by mac-rophages and is modulated by IL-5.J. Immunol. 148:3543.
35. Rothenberg, M. E., W. F. Owen, D. S. Silberstein, J. Woods, R. J. Soberman,K. F. Austen, and R. L. Stevens. 1988. Human eosinophils have prolonged sur-vival, enhanced functional properties, and become hypodense when exposed tohuman interleukin 3.J. Clin. Invest. 816:1986.
36. Yamaguchi, Y., T. Suda, S. Ohta, K. Tominaga, Y. Miura, and T. Kasahara. 1991.Analysis of the survival of mature human eosinophils: interleukin-5 preventsapoptosis in mature human eosinophils.Blood 78:2542.
37. Walker, C., J. C. Virchow, P. L. B. Bruijnzeel, and K. Blaser. 1991. T cell subsetsand their soluble products regulate eosinophilia in allergic and non-allergicasthma.J. Immunol. 146:1829.
38. Simon, H. U., S. Yousefi, C. Schranz, A. Schapowal, C. Bachert, and K. Blaser.1997. Direct demonstration of delayed eosinophil apoptosis as a mechanism caus-ing tissue eosinophilia.J. Immunol. 158:3902.
39. Kita, H., T. Ohnishi, Y. Okubo, D. Weiler, J. S. Abrams, and G. J. Gleich. 1991.Granulocyte/macrophage colony-stimulating factor and interleukin 3 release fromhuman peripheral blood eosinophils and neutrophils.J. Exp. Med. 174:745.
40. Shi, Y., R. Wang, A. Sharma, C. Gao, M. Collins, L. Penn, and G. Mills. 1997.Dissociation of cytokine signals for proliferation and apoptosis.J. Immunol. 159:5318.
41. Morita, M., B. Lamkhioued, A. Soussi Gounni, D. Aldebert, E. Delaporte,A. Capron, and M. Capron. 1996. Induction by interferons of human eosinophilapoptosis and regulation by interleukin-3, granulocyte/macrophage-colony stim-ulating factor and interleukin-5.Eur. Cytokine Network 7:725.
2040 RESOLUTION OF AIRWAY HYPERRESPONSIVENESS AND PULMONARY INFLAMMATION
by guest on Decem
ber 11, 2013http://w
ww
.jimm
unol.org/D
ownloaded from