redundancy in b cell developmental pathways: c-cbl inactivation rescues early b cell development...
TRANSCRIPT

of June 18, 2013.This information is current as
Protein-Independent PathwayCell Development through a B Cell Linker
BPathways: c-Cbl Inactivation Rescues Early Redundancy in B Cell Developmental
Siraganian and Richard J. HodesHaifeng Song, Juan Zhang, Y. Jeffrey Chiang, Reuben P.
http://www.jimmunol.org/content/178/2/9262007; 178:926-935; ;J Immunol
Referenceshttp://www.jimmunol.org/content/178/2/926.full#ref-list-1
, 22 of which you can access for free at: cites 51 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2007 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

Redundancy in B Cell Developmental Pathways: c-CblInactivation Rescues Early B Cell Development througha B Cell Linker Protein-Independent Pathway1
Haifeng Song,* Juan Zhang,† Y. Jeffrey Chiang,* Reuben P. Siraganian,†
and Richard J. Hodes2*‡
Deficiency in the adaptor protein B cell linker protein (BLNK) results in a substantial but incomplete block in B cell development,suggesting that alternative pathways exist for B lineage differentiation. Another adaptor protein, c-Cbl, plays a negative regulatoryrole in several BCR-signaling pathways. We therefore investigated the role of c-Cbl during B cell development and addressed thepossibility that redundancies in pathways for B cell differentiation could be further revealed by eliminating negative effectsmediated by c-Cbl. Strikingly, c-Cbl inactivation reversed a number of the critical defects in early B cell differentiation that areseen in BLNK-deficient mice. c-Cbl�/�BLNK�/� mice exhibited normalized down-regulation of pre-BCR and CD43, up-regula-tion of MHC class II, and augmented L chain rearrangement, resulting in a successful transition from pre-B cells to immature Bcells. c-Cbl inactivation also reversed the potentially tumor-predisposing hyperproliferative response of BLNK�/� pre-B cells toIL-7. Pre-BCR cross-linking induced enhanced and prolonged tyrosine phosphorylation in c-Cbl�/�BLNK�/� pre-BCR� pre-Bcells compared with c-Cbl�/�BLNK�/� cells, including elevated phosphorylation of Lyn, Syk, Btk, and phospholipase C-�2. Ourstudies suggest that some, but not all, pre-BCR-triggered developmental events can be mediated by BLNK-independent pathwaysthat are negatively regulated by c-Cbl, and further suggest that different events during early B cell development require differentstrength or duration of pre-BCR signaling. The Journal of Immunology, 2007, 178: 926–935.
S ignaling from the BCR/precursor BCR (BCR/pre-BCR)plays a pivotal role in B cell development and function.Both BCR and pre-BCR signaling are initiated by activa-
tion of Src kinases, such as Lyn and Blk, upon receptor engage-ment. Activated Lyn phosphorylates tyrosines in the ITAM regionof Ig� and Ig�, and phosphorylated Ig� and Ig� in turn recruit Sykkinase, which is then activated by Lyn (1, 2). Syk further activatesseveral down-stream protein tyrosine kinases (PTK)3 and adaptorproteins. One of the substrates of Syk is B cell linker protein(BLNK; also termed SLP-65, or BASH). BLNK is an adaptor inbridging Syk to downstream signaling pathways by recruiting sig-naling molecules, such as Btk, phospholipase C (PLC)-�2, Vav,and Grb2 to the cell membrane to form a signalosome complex,where downstream events, including the PLC-�2 and Ca2� mobi-lization pathway are activated (3–5). Pre-BCR signaling mediatesimportant developmental events during pre-B cell transition, in-cluding early pre-B cell expansion, allelic exclusion, and modula-
tion of cell surface molecules, as well as induction of cell cyclearrest and L chain rearrangement (6–9). BCR signaling mediatespositive and negative selection at the immature B cell stage and thedevelopment from transitional stage B cells (T1 and T2) to matureB cells in the spleen (10). In addition, BCR signaling is essentialfor the survival, maintenance, and functions of mature B cells (11).Although it has not been clearly demonstrated how the individualevents that occur during B development are induced by specificsignaling pathways, increasing evidence suggests that B cell-sig-naling pathways are redundant. One example of this apparent re-dundancy is the alteration in B cell development that occurs inBLNK-deficient mice. BLNK-deficient mice display a major blockduring the transition from early pre-B cells to small resting pre-Bcell stage, manifested by the accumulation of large pre-B cellsexpressing surface pre-BCR and reduction in the number of smallpre-B cells and immature B cells in the bone marrow (BM). Nev-ertheless, a small proportion of pre-B cells bypass the BLNK-dependent pathway and differentiate into immature B cells thatmigrate into the spleen, where most cells maintain an immaturephenotype (12–15). The incomplete block of B cell development inBLNK�/� mice suggests that there exists a BLNK-independentpathway(s) capable of supporting B cell development. The findingthat B cell development is more severely defective in doubleknockout mice in which BLNK deficiency is combined with inac-tivation of linker for activation of T cells (LAT), Btk, or CD19indicates that these molecules are involved in one or more BLNK-independent pathways of B cell development (16–19).
In addition to triggering positive pathways for differentiationand activation, BCR/pre-BCR engagement also activates negativeregulatory molecules capable of inhibiting these responses. Onesuch negative signaling molecule is c-Cbl (Casitas B-lineage lym-phoma proto-oncogene c). c-Cbl is an adaptor protein containingmultiple functional domains. A phosphotyrosine-binding (PTB)
*Experimental Immunology Branch, National Cancer Institute, †Oral Infection andImmunity Branch, National Institute of Dental and Craniofacial Research, and ‡Na-tional Institute on Aging, National Institutes of Health, Bethesda, MD 20892
Received for publication May 22, 2006. Accepted for publication November 8, 2006.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the Intramural Research Program of the National Can-cer Institute, Center for Cancer Research and the National Institute of Dental Re-search, National Institutes of Health.2 Address correspondence and reprint requests to Dr. Richard J. Hodes, NationalInstitute on Aging, National Institutes of Health, Building 10, Room 4B10, 9000Rockville Pike, Bethesda, MD 20892. E-mail address: [email protected] Abbreviations used in this paper: PTK, protein tyrosine kinase; BLNK, B cell linkerprotein; PLC, phospholipase C; LAT, linker for activation of T cells; SH, Src ho-mology; BM, bone marrow; MFI, mean fluorescence intensity; MZ, marginal zone;FO, follicular; pre-BCR, precursor BCR; c-Cbl, Casitas B-lineage lymphoma proto-oncogene c; SLC, surrogate L chain.
The Journal of Immunology
www.jimmunol.org
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

region on its N terminus binds to the Src homology (SH) 2 regionof PTKs; a conserved ring finger region has been demonstrated toplay a negative regulatory role as a ubiquitin ligase E3 (20); aproline-rich region interacts with SH3 domain-containing proteins;and multiple tyrosine phosphorylation sites at the C terminus me-diate interactions with SH2-domain-containing proteins (21, 22).Several molecules functioning in BCR-signaling pathways, includ-ing Syk, Lyn, and Vav, have been shown to be targets of c-Cbl forubiquitination and consequent inactivation or degradation (23–25).B cell development and function in c-Cbl�/� mice are grosslynormal, although a slightly altered distribution of B cell subsetswas observed in peripheral B cells (26, 27). However, the functionof c-Cbl during discrete stages of B cell development has not beeninvestigated in detail.
In the current study, it was hypothesized that the deficiency ofB cell development in BLNK�/� mice may be circumvented byenhancing BLNK-independent pathways. Our results demon-strated that c-Cbl inactivation substantially corrected the defect inB cell development in BLNK-deficient mice, and that rescue wasmost apparent in the pre-B cell transition of BM B cell develop-ment, indicating a previously unappreciated role for c-Cbl in in-hibiting BLNK-independent B cell developmental pathways.
Materials and MethodsMice
Mice aged 8–12 wk were used in all experiments. BLNK-deficient miceand c-Cbl-deficient mice have been described previously (14, 28). BothBLNK�/� mice and c-Cbl�/� mice are of a mixed S129/C57BL/6 back-ground. c-Cbl�/�BLNK�/� and control mice heterozygous (�/�) for c-Cbl or BLNK inactivation were generated by breeding c-Cbl�/� andBLNK�/� mice. All mice were maintained in the Bioqual facility.
Reagents and Abs
The following Abs and reagents were purchased from BD Pharmingen:biotinylated Abs specific for B220 (RA3-6B2), I-Ab, CD2, �5 (LM34),pre-BCR (SL156), CD19, IgM (II/41); FITC-conjugated Abs againstB220 (RA3-6B2), IgM (II/41), BP-1, CD43 (S7), Ig� (187.7), CD5;PE-conjugated Abs against B220 (RA3-6B2), CD19, CD43, CD25, IL-7R; and allophycocyanin-labeled Abs against B220 (RA3-6B2), IgM(II/41), streptavidin-PerCP. Anti-IgD-biotin and anti-�-FITC were pur-chased from Southern Biotechnology Associates. Streptavidin-AlexaFluor 594 was purchased from Molecular Probes. Anti-Syk, anti-Lyn,and anti-PLC-�2 Abs were from Santa Cruz Biotechnology, anti-phosphotyrosine Ab (4G10) from Upstate Biotechnology anti-Btk fromGenWay and the anti-Btk (pY551) phosphospecific Abs were from BDBiosciences.
Flow cytometry
A total of 1 � 106 cells were incubated with 2.4G2 Ab for 5 min on ice toblock Fc�R, followed by addition of 50 �l of diluted biotinylated Abs and20 min of incubation on ice. After washing with FACS buffer, the cellswere stained for another 20 min in a combination of FITC-, PE-, allophy-cocyanin-conjugated Abs and streptavidin-PerCP. In staining for FACSsorting, streptavidin-Alexa Fluor 594 was used to replace streptavidin-PerCP. For intracellular �5 and �-chain staining, the cells were first stainedfor cell surface markers, then fixed with Cyto/perm (BD Pharmingen),followed by staining with anti-�5 and anti-� Abs. To stain intracellularIg�, fresh BM cells were incubated with excess purified anti-Ig� mAb toblock surface Ig�, followed by surface staining for other markers. Thencells were fixed and stained for intracellular � chain. Stained samples wereanalyzed using FACSCalibur with CellQuest software (BD Biosciences).
In vitro BM cell culture
BM B cell culture was performed with either IgM-depleted CD19� BM Bcells or with FACS-sorted CD19�CD43�IgM� pro-B cells. Briefly, BMsuspensions were depleted of sIgM� cells by negative selection using bi-otinylated anti-IgM and streptavidin microbeads (Miltenyi Biotec). Then,CD19� cells were positively selected with CD19 microbeads (MiltenyiBiotec). CD19�CD43�IgM� pro-B cells were sorted with the FACS Van-tage SE system (BD Biosciences). To measure the BM B cell response toIL-7 stimulation, 1–2.5 � 104 cells/well were cultured in 96-well flat-
bottom plates with different concentration of IL-7 (R&D Systems) in Opti-Mem I medium (Invitrogen Life Technologies) supplemented with 10%FCS, 5 � 10�5M 2-ME, and penicillin/streptomycin for 4 and 6 days. Thecultures were pulsed with 1 �Ci/well of [3H]thymidine 8 h before harvest.[3H]Incorporation was measured using a beta counter system (1205 betaplates; PerkinElmer).
Establishment of pre-B cell lines
BM and fetal liver cells were cultured on S17 stromal cell layers in 96-wellplate with 10 cells/well in IL-7 containing (3T3-IL-7 culture supernatant)Opti-Mem I medium. Ten days later, large cell colonies were observed andtransferred to 6-well plates to expand the culture. The cultures were main-tained for 5–8 mo with periodic analysis of surface markers. Colonies withsimilar phenotypes from each group were used for biochemical studies.
Biochemical studies
Pre-BCR� pre-B cells were stimulated with anti-IgM F(ab�)2 (�-chain spe-cific; The Jackson Laboratory) at 37°C and reaction was stopped at dif-ferent time points by adding ice-cold PBS containing 5 mM EDTA, 2 mMNa3VO4, and protease inhibitors (2 mM PMSF, 90 mU/ml aprotinin, 50�g/ml leupeptin, 50 �g/ml pepstatin). Cells were solubilized in lysis buffer(1% Triton X-100, 0.1% SDS, 50 mM Tris, pH 7.4, 50 mM NaCl, 50 mMNaF, plus protease inhibitors and Na3VO4), and postnuclear supernatantswere immunoprecipitated with Abs bound to protein A-, or protein G-agarose beads. After rotation at 4°C for 1 h, the beads were washed fourtimes with ice-cold lysis buffer and the proteins eluted by boiling for 10min with SDS-PAGE sample buffer. In some experiments, total cell lysateswere immunoprecipitated with anti-phosphotyrosine Ab 4G10 coupled toagarose beads, and after washing the bound proteins were eluted with 100mM phenyl phosphate and analyzed by immunoblotting with the indicatedAbs. Whole cell lysates or immunoprecipitated proteins were separatedby SDS-PAGE and electrotransferred to polyvinylidene difluoridemembranes (Millipore). The blots were probed with anti-phosphotyrosine or other specific Abs as indicated. Signals were revealedwith ECL system (PerkinElmer). The signal intensity was analyzed withImagequant program (Molecular Dynamics) and the phosphorylation sig-nal was normalized based on total protein levels of the indicated molecules.Level of activation was expressed as ratio of normalized phosphorylationof stimulated samples vs unstimulated samples.
Immunofluorescence microscopy
Pre-BCR� pre-B cells were settled on cover slides on ice for 40 min,warmed up at 37oC and stimulated with goat F(ab�)2 Ab specific to mouse�-chain. At different time points of the stimulation, cells were fixed with4% paraformaldehyde for 15 min, quenched with 50 �M NH4Cl for 10min, followed by 30 min of permeabilization with 0.05% saponin. The cellswere then blocked with 5% goat serum in PBS for 30 min and incubatedwith rabbit anti-Syk or phospho-Syk (Y519/520-specific) Abs at 4°C forovernight. Staining was resolved with goat anti-rabbit IgG-Alexa 488 atroom temperature for 40 min. The cells were imaged with a Zeiss LSM 510META confocal microscope using a 1.4 oil planapochromat �63 objective.The mean fluorescence intensity (MFI) of phospho-Syk (Alexa 488 stain-ing) from different time points was analyzed with Meta Morph softwareand the MFI was calculated based on the intensity of over 200 cells fromfive different view fields. The statistical confidence limit value was calcu-lated by Excel software.
Resultsc-Cbl inactivation corrects the impaired early B celldevelopment in BLNK-deficient mice
B cell development is deficient in BLNK�/� mice, manifested asa major block at the transition from late pro-B cells to pre-B cells,and a block in maturation from transitional B cells to mature Bcells, with a resultant overall reduction in the number of peripheralB cells. This defect in B cell development, while profound, is notcomplete, suggesting that alternative pathways exist for B lineagedifferentiation. It has been demonstrated that c-Cbl plays a nega-tive regulatory role in several BCR signaling pathways and thatthese signaling pathways are thus up-regulated upon c-Cbl-inacti-vation (21, 29–31). We therefore addressed the possibility thatc-Cbl inhibits BLNK-independent B cell differentiation by assessingthe effect of c-Cbl inactivation on the defect in B cell development in
927The Journal of Immunology
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
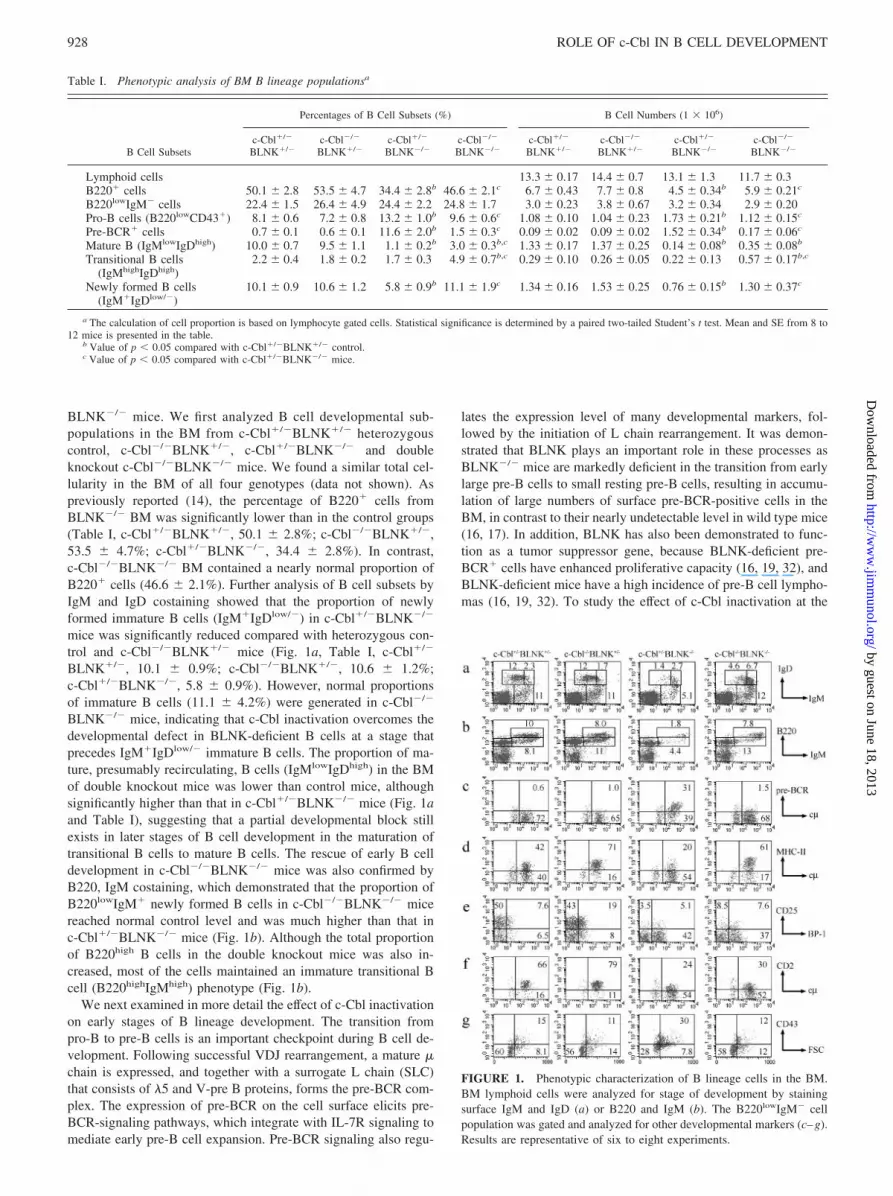
BLNK�/� mice. We first analyzed B cell developmental sub-populations in the BM from c-Cbl�/�BLNK�/� heterozygouscontrol, c-Cbl�/�BLNK�/�, c-Cbl�/�BLNK�/� and doubleknockout c-Cbl�/�BLNK�/� mice. We found a similar total cel-lularity in the BM of all four genotypes (data not shown). Aspreviously reported (14), the percentage of B220� cells fromBLNK�/� BM was significantly lower than in the control groups(Table I, c-Cbl�/�BLNK�/�, 50.1 � 2.8%; c-Cbl�/�BLNK�/�,53.5 � 4.7%; c-Cbl�/�BLNK�/�, 34.4 � 2.8%). In contrast,c-Cbl�/�BLNK�/� BM contained a nearly normal proportion ofB220� cells (46.6 � 2.1%). Further analysis of B cell subsets byIgM and IgD costaining showed that the proportion of newlyformed immature B cells (IgM�IgDlow/�) in c-Cbl�/�BLNK�/�
mice was significantly reduced compared with heterozygous con-trol and c-Cbl�/�BLNK�/� mice (Fig. 1a, Table I, c-Cbl�/�
BLNK�/�, 10.1 � 0.9%; c-Cbl�/�BLNK�/�, 10.6 � 1.2%;c-Cbl�/�BLNK�/�, 5.8 � 0.9%). However, normal proportionsof immature B cells (11.1 � 4.2%) were generated in c-Cbl�/�
BLNK�/� mice, indicating that c-Cbl inactivation overcomes thedevelopmental defect in BLNK-deficient B cells at a stage thatprecedes IgM�IgDlow/� immature B cells. The proportion of ma-ture, presumably recirculating, B cells (IgMlowIgDhigh) in the BMof double knockout mice was lower than control mice, althoughsignificantly higher than that in c-Cbl�/�BLNK�/� mice (Fig. 1aand Table I), suggesting that a partial developmental block stillexists in later stages of B cell development in the maturation oftransitional B cells to mature B cells. The rescue of early B celldevelopment in c-Cbl�/�BLNK�/� mice was also confirmed byB220, IgM costaining, which demonstrated that the proportion ofB220lowIgM� newly formed B cells in c-Cbl�/�BLNK�/� micereached normal control level and was much higher than that inc-Cbl�/�BLNK�/� mice (Fig. 1b). Although the total proportionof B220high B cells in the double knockout mice was also in-creased, most of the cells maintained an immature transitional Bcell (B220highIgMhigh) phenotype (Fig. 1b).
We next examined in more detail the effect of c-Cbl inactivationon early stages of B lineage development. The transition frompro-B to pre-B cells is an important checkpoint during B cell de-velopment. Following successful VDJ rearrangement, a mature �chain is expressed, and together with a surrogate L chain (SLC)that consists of �5 and V-pre B proteins, forms the pre-BCR com-plex. The expression of pre-BCR on the cell surface elicits pre-BCR-signaling pathways, which integrate with IL-7R signaling tomediate early pre-B cell expansion. Pre-BCR signaling also regu-
lates the expression level of many developmental markers, fol-lowed by the initiation of L chain rearrangement. It was demon-strated that BLNK plays an important role in these processes asBLNK�/� mice are markedly deficient in the transition from earlylarge pre-B cells to small resting pre-B cells, resulting in accumu-lation of large numbers of surface pre-BCR-positive cells in theBM, in contrast to their nearly undetectable level in wild type mice(16, 17). In addition, BLNK has also been demonstrated to func-tion as a tumor suppressor gene, because BLNK-deficient pre-BCR� cells have enhanced proliferative capacity (16, 19, 32), andBLNK-deficient mice have a high incidence of pre-B cell lympho-mas (16, 19, 32). To study the effect of c-Cbl inactivation at the
FIGURE 1. Phenotypic characterization of B lineage cells in the BM.BM lymphoid cells were analyzed for stage of development by stainingsurface IgM and IgD (a) or B220 and IgM (b). The B220lowIgM� cellpopulation was gated and analyzed for other developmental markers (c–g).Results are representative of six to eight experiments.
Table I. Phenotypic analysis of BM B lineage populationsa
B Cell Subsets
Percentages of B Cell Subsets (%) B Cell Numbers (1 � 106)
c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�
Lymphoid cells 13.3 � 0.17 14.4 � 0.7 13.1 � 1.3 11.7 � 0.3B220� cells 50.1 � 2.8 53.5 � 4.7 34.4 � 2.8b 46.6 � 2.1c 6.7 � 0.43 7.7 � 0.8 4.5 � 0.34b 5.9 � 0.21c
B220lowIgM� cells 22.4 � 1.5 26.4 � 4.9 24.4 � 2.2 24.8 � 1.7 3.0 � 0.23 3.8 � 0.67 3.2 � 0.34 2.9 � 0.20Pro-B cells (B220lowCD43�) 8.1 � 0.6 7.2 � 0.8 13.2 � 1.0b 9.6 � 0.6c 1.08 � 0.10 1.04 � 0.23 1.73 � 0.21b 1.12 � 0.15c
Pre-BCR� cells 0.7 � 0.1 0.6 � 0.1 11.6 � 2.0b 1.5 � 0.3c 0.09 � 0.02 0.09 � 0.02 1.52 � 0.34b 0.17 � 0.06c
Mature B (IgMlowIgDhigh) 10.0 � 0.7 9.5 � 1.1 1.1 � 0.2b 3.0 � 0.3b,c 1.33 � 0.17 1.37 � 0.25 0.14 � 0.08b 0.35 � 0.08b
Transitional B cells(IgMhighIgDhigh)
2.2 � 0.4 1.8 � 0.2 1.7 � 0.3 4.9 � 0.7b,c 0.29 � 0.10 0.26 � 0.05 0.22 � 0.13 0.57 � 0.17b,c
Newly formed B cells(IgM�IgDlow/�)
10.1 � 0.9 10.6 � 1.2 5.8 � 0.9b 11.1 � 1.9c 1.34 � 0.16 1.53 � 0.25 0.76 � 0.15b 1.30 � 0.37c
a The calculation of cell proportion is based on lymphocyte gated cells. Statistical significance is determined by a paired two-tailed Student’s t test. Mean and SE from 8 to12 mice is presented in the table.
b Value of p � 0.05 compared with c-Cbl�/�BLNK�/� control.c Value of p � 0.05 compared with c-Cbl�/�BLNK�/� mice.
928 ROLE OF c-Cbl IN B CELL DEVELOPMENT
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

pre-B cell stage, BM B220lowIgM� populations were gated andanalyzed for B cell developmental markers. As previously reported(16), BLNK-deficient early pre-B cells failed to down-regulate sur-face pre-BCR and CD43 or to up-regulate MHC-II. However,pre-B cells from c-Cbl�/�BLNK�/� mice successfully did so(Fig. 1, c, d, and g), and the accumulation of pre-BCR� cells inBLNK�/� BM was reversed to normal levels (Fig. 1c, Table I). Inaddition, while the proportion of pro-B cells (B220lowCD43�) wasincreased in c-Cbl�/�BLNK�/� BM, this population was reducedto normal level in c-Cbl�/�BLNK�/� mice (Table I). Further-more, compared with the enhanced level of large cycling earlypre-B cells in c-Cbl�/�BLNK�/� BM, the number of cells in cellcycle as assessed by flow cytometric light scatter is also reduced tonormal level in c-Cbl�/�BLNK�/� BM (Fig. 1g). In contrast tothe normalization of these parameters in c-Cbl�/�BLNK�/�mice,early pre-B cells in these double knockouts failed to down-regulateBP-1 or up-regulate CD2 and CD25 expression (Fig. 1, e and f).Intracellular � staining showed that c-Cbl-inactivation also par-tially corrected the impaired L chain rearrangement in BLNK�/�
mice. Although �4.5% of surface IgM�B220low B cells were pos-itive for intracellular � staining in BLNK�/� mice, a higher pro-portion of intracellular �-positive cells (9.6%) were observed inc-Cbl�/�BLNK�/� mice, although this number was still lowerthan that from control mice (Fig. 2, Cbl�/�BLNK�/�, 17%;c-Cbl�/�BLNK�/�, 17%). In contrast to � staining, intracellular�5 protein, which is markedly elevated in BLNK�/� mice, was sig-nificantly down-regulated in c-Cbl�/�BLNK�/� mice (c-Cbl�/�
BLNK�/�, 12%; c-Cbl�/�BLNK�/�, 17%; c-Cbl�/�BLNK�/�,63%; c-Cbl�/�BLNK�/�, 34%). Overall, these data confirmed theaccumulation of early pre-BCR� pre-B cells in BLNK�/� BM anddemonstrated that c-Cbl inactivation reduced the number of latepro-B/early pre-B cells, and enhanced the number of more maturesmall pre-B cells, consistent with an effect in selectively correctingsome of the critical developmental events in BLNK�/� mice in thetransition from large cycling to small resting pre-B cells.
c-Cbl inactivation corrects the impaired in vitro development ofBLNK-deficient B cells
The observed effects of c-Cbl inactivation on B cell developmentin vivo could reflect cell autonomous effects on B lineage devel-opment and/or the effects of c-Cbl inactivation on stromal or othercells that influence B cell maturation. To determine whether c-Cblinactivation directly affects the development of BLNK�/� B cells,we used an in vitro culture system for the IL-7-dependent differ-
entiation of sorted CD19�IgM� BM cells. At day 4 of culture,15.8 and 18.5% of total live cells became double positive for sur-face IgM and Ig� expression in control and c-Cbl�/�BLNK�/�
groups, respectively; whereas only 2.6% of cells were IgM and Ig�positive in c-Cbl�/�BLNK�/� cultures, confirming their intrinsicdeficiency in B cell development. In contrast, in c-Cbl�/�
BLNK�/� cultures, 12.7% IgM, Ig� double-positive cells weredetected (Fig. 3a). Enumeration of total cell number from eachculture showed that the cells from c-Cbl�/�BLNK�/� culture in-creased �12-fold from the cell number of initial culture, whichwas much higher than the increases in cell number of the c-Cbl�/�
BLNK�/� and c-Cbl�/�BLNK�/� groups (Fig. 3b, 5.6-fold inc-Cbl�/�BLNK�/� group and 5.1-fold in c-Cbl�/�BLNK�/�
group). The total cell number in day 4 culture from double knock-out mice was similar to that from control and c-Cbl�/�BLNK�/�
FIGURE 2. Inactivation of c-Cbl in BLNK-deficient mice partially res-cued � chain rearrangement. BM cells were incubated with excess of anti-Ig� mAb to block surface Ig�, followed by surface staining of IgM andB220, then fixed and permeabilized, followed by intracellular staining for�5 and �-chain. Data were analyzed and presented by gating on surfaceB220lowIgM� populations. The result shown is representative of three in-dependent experiments.
FIGURE 3. c-Cbl inactivation rescued B cell development in in vitroculture. FACS-sorted CD19�IgM� BM cells were cultured in IL-7-sup-plemented medium in duplicate wells (3.8 � 105 cells/well) in 24-wellplates. At day 4 of culture, B cell differentiation was examined by stainingfor surface expression of IgM and Ig� (a). Total cell number of day 4cultures was enumerated using optical microscopy and the increase in cellnumber was expressed as fold increase of total cell number of day 4 cultureover the initial culture cell number (b). Differentiated IgM�Ig�� B cellnumber was calculated by multiplying the percentages of IgM�Ig�� Bcells and total cell number (c). Results shown are representative of fourindependent experiments.
929The Journal of Immunology
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
mice (Fig. 3b). When the total differentiated cell (IgM�Ig��)number was calculated, c-Cbl�/�BLNK�/� culture gave �3-foldfewer IgM�Ig�� cells than the yield from control group andc-Cbl�/�BLNK�/� group. In contrast, the number of IgM�Ig��
cells in the c-Cbl�/�BLNK�/� group was restored to control level(Fig. 3c). These data confirmed that BLNK�/� pro-B and pre-Bcells are defective in in vitro differentiation and demonstrated thatc-Cbl-inactivation substantially corrected this defect. Thus, in vitrocultures recapitulated the effect of c-Cbl inactivation observed invivo in rescuing early B cell development.
c-Cbl�/�BLNK�/� pre-B cells display normalized proliferationto IL-7 stimulation
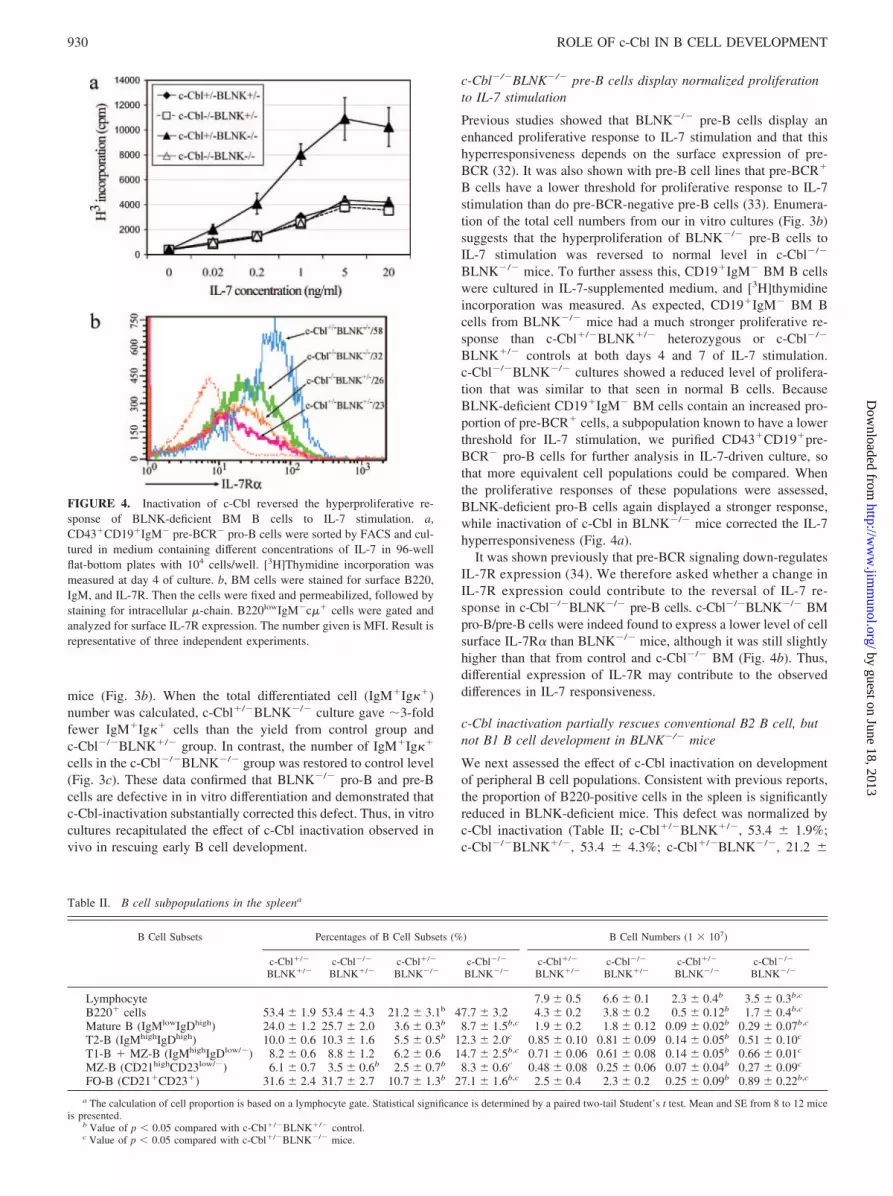
Previous studies showed that BLNK�/� pre-B cells display anenhanced proliferative response to IL-7 stimulation and that thishyperresponsiveness depends on the surface expression of pre-BCR (32). It was also shown with pre-B cell lines that pre-BCR�
B cells have a lower threshold for proliferative response to IL-7stimulation than do pre-BCR-negative pre-B cells (33). Enumera-tion of the total cell numbers from our in vitro cultures (Fig. 3b)suggests that the hyperproliferation of BLNK�/� pre-B cells toIL-7 stimulation was reversed to normal level in c-Cbl�/�
BLNK�/� mice. To further assess this, CD19�IgM� BM B cellswere cultured in IL-7-supplemented medium, and [3H]thymidineincorporation was measured. As expected, CD19�IgM� BM Bcells from BLNK�/� mice had a much stronger proliferative re-sponse than c-Cbl�/�BLNK�/� heterozygous or c-Cbl�/�
BLNK�/� controls at both days 4 and 7 of IL-7 stimulation.c-Cbl�/�BLNK�/� cultures showed a reduced level of prolifera-tion that was similar to that seen in normal B cells. BecauseBLNK-deficient CD19�IgM� BM cells contain an increased pro-portion of pre-BCR� cells, a subpopulation known to have a lowerthreshold for IL-7 stimulation, we purified CD43�CD19�pre-BCR� pro-B cells for further analysis in IL-7-driven culture, sothat more equivalent cell populations could be compared. Whenthe proliferative responses of these populations were assessed,BLNK-deficient pro-B cells again displayed a stronger response,while inactivation of c-Cbl in BLNK�/� mice corrected the IL-7hyperresponsiveness (Fig. 4a).
It was shown previously that pre-BCR signaling down-regulatesIL-7R expression (34). We therefore asked whether a change inIL-7R expression could contribute to the reversal of IL-7 re-sponse in c-Cbl�/�BLNK�/� pre-B cells. c-Cbl�/�BLNK�/� BMpro-B/pre-B cells were indeed found to express a lower level of cellsurface IL-7R� than BLNK�/� mice, although it was still slightlyhigher than that from control and c-Cbl�/� BM (Fig. 4b). Thus,differential expression of IL-7R may contribute to the observeddifferences in IL-7 responsiveness.
c-Cbl inactivation partially rescues conventional B2 B cell, butnot B1 B cell development in BLNK�/� mice
We next assessed the effect of c-Cbl inactivation on developmentof peripheral B cell populations. Consistent with previous reports,the proportion of B220-positive cells in the spleen is significantlyreduced in BLNK-deficient mice. This defect was normalized byc-Cbl inactivation (Table II; c-Cbl�/�BLNK�/�, 53.4 � 1.9%;c-Cbl�/�BLNK�/�, 53.4 � 4.3%; c-Cbl�/�BLNK�/�, 21.2 �
FIGURE 4. Inactivation of c-Cbl reversed the hyperproliferative re-sponse of BLNK-deficient BM B cells to IL-7 stimulation. a,CD43�CD19�IgM� pre-BCR� pro-B cells were sorted by FACS and cul-tured in medium containing different concentrations of IL-7 in 96-wellflat-bottom plates with 104 cells/well. [3H]Thymidine incorporation wasmeasured at day 4 of culture. b, BM cells were stained for surface B220,IgM, and IL-7R. Then the cells were fixed and permeabilized, followed bystaining for intracellular �-chain. B220lowIgM�c�� cells were gated andanalyzed for surface IL-7R expression. The number given is MFI. Result isrepresentative of three independent experiments.
Table II. B cell subpopulations in the spleena
B Cell Subsets Percentages of B Cell Subsets (%) B Cell Numbers (1 � 107)
c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�c-Cbl�/�
BLNK�/�
Lymphocyte 7.9 � 0.5 6.6 � 0.1 2.3 � 0.4b 3.5 � 0.3b,c
B220� cells 53.4 � 1.9 53.4 � 4.3 21.2 � 3.1b 47.7 � 3.2 4.3 � 0.2 3.8 � 0.2 0.5 � 0.12b 1.7 � 0.4b,c
Mature B (IgMlowIgDhigh) 24.0 � 1.2 25.7 � 2.0 3.6 � 0.3b 8.7 � 1.5b,c 1.9 � 0.2 1.8 � 0.12 0.09 � 0.02b 0.29 � 0.07b,c
T2-B (IgMhighIgDhigh) 10.0 � 0.6 10.3 � 1.6 5.5 � 0.5b 12.3 � 2.0c 0.85 � 0.10 0.81 � 0.09 0.14 � 0.05b 0.51 � 0.10c
T1-B � MZ-B (IgMhighIgDlow/�) 8.2 � 0.6 8.8 � 1.2 6.2 � 0.6 14.7 � 2.5b,c 0.71 � 0.06 0.61 � 0.08 0.14 � 0.05b 0.66 � 0.01c
MZ-B (CD21highCD23low/�) 6.1 � 0.7 3.5 � 0.6b 2.5 � 0.7b 8.3 � 0.6c 0.48 � 0.08 0.25 � 0.06 0.07 � 0.04b 0.27 � 0.09c
FO-B (CD21�CD23�) 31.6 � 2.4 31.7 � 2.7 10.7 � 1.3b 27.1 � 1.6b,c 2.5 � 0.4 2.3 � 0.2 0.25 � 0.09b 0.89 � 0.22b,c
a The calculation of cell proportion is based on a lymphocyte gate. Statistical significance is determined by a paired two-tail Student’s t test. Mean and SE from 8 to 12 miceis presented.
b Value of p � 0.05 compared with c-Cbl�/�BLNK�/� control.c Value of p � 0.05 compared with c-Cbl�/�BLNK�/� mice.
930 ROLE OF c-Cbl IN B CELL DEVELOPMENT
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

3.1%; c-Cbl�/�BLNK�/�, 47.7 � 3.2%). However, because theabsolute lymphocyte numbers in the spleens of both c-Cbl�/�
BLNK�/� and c-Cbl�/�BLNK�/� mice were lower than controlmice, the total B cell number in the spleen of c-Cbl�/�BLNK�/�
mice was still �3-fold lower than the heterozygous control, al-though 3- to 4-fold higher than c-Cbl�/�BLNK�/� mice (Table II;c-Cbl�/�BLNK�/�, 4.3 � 0.2 � 107; c-Cbl�/�BLNK�/�, 3.8 �0.2 � 107; c-Cbl�/�BLNK�/�, 0.5 � 0.12 � 107; c-Cbl�/�
BLNK�/�, 1.7 � 0.4 � 107). Examination of B cell subsets byIgM and IgD costaining for T1, T2, and mature follicular (FO) Bcells (Fig. 5a), and CD21 combined with CD23 staining for mar-ginal zone (MZ) and FO B cells (Fig. 5b) showed that all subsetswere, to different extents, increased in c-Cbl�/�BLNK�/� micecompared with c-Cbl�/�BLNK�/� mice (Table II). It is notewor-thy that the proportion of T2 B cells (IgMhighIgDhigh) in c-Cbl�/�
BLNK�/� mice was lower than that in control mice, while thispopulation in c-Cbl�/�BLNK�/� mice reached normal levels(Fig. 5a, Table II: c-Cbl�/�BLNK�/�, 10.0 � 0.6%; c-Cbl�/�
BLNK�/�, 10.3 � 1.6%; c-Cbl�/�BLNK�/�, 5.5 � 0.5%;c-Cbl�/�BLNK�/�, 12.3 � 2.0%). The splenic IgMhighIgDlow/�
population contains T1 immature B cells and mature MZ B cells.When the IgMhighIgDlow/� population was analyzed with AA4.1staining to distinguish immature (AA4.1�) and mature (AA4.1�)B cells, it was found that the percentages of both T1 and MZ Bcells in the double knockout mice were normalized to control lev-els (data not shown). Although the proportions of T1, T2, and MZB cells from c-Cbl�/�BLNK�/� mouse spleen were normalized,the percentage of mature FO B cells (IgMlowIgDhigh) was stillsignificantly lower than control group (Fig. 5a and Table II), in-dicating that a developmental block still exists at the transitionfrom T2 to mature FO B cells, but not to MZ B cells
As previously reported (14), peritoneal B1 B cells (CD5�IgM�)are completely absent in BLNK�/� mice. We found that there wasno rescue of B1 B cell development by c-Cbl-inactivation (Fig.5c). Taken together, these findings indicate that c-Cbl-deficiencypartially rescued conventional B2 B cell, but not B1 B celldevelopment.
c-Cbl inactivation induces elevated and prolonged pre-BCRsignaling
In B cell lines, c-Cbl has been shown to negatively regulate severalsignaling molecules downstream of BCR, including Syk and Lyn(31, 35, 36). However, studies of c-Cbl-deficient splenic B cellshave given different results: c-Cbl positively regulates Syk, Btk,
and ERK activation and Ca2� mobilization, but negatively regu-lates Lyn, PI3K, and Akt (27). To determine how c-Cbl modulatespre-BCR-signaling in BLNK-deficient pre-B cells, pre-BCR�
pre-B cells derived from c-Cbl�/�BLNK�/� and c-Cbl�/�
BLNK�/� fetal liver were stimulated by pre-BCR cross-linkingwith anti-� F(ab�)2 Ab, and total tyrosine phosphorylation wasanalyzed by immunoblotting with a phosphotyrosine-specific Ab(4G10). Pre-B cells from Cbl�/�BLNK�/� and Cbl�/�BLNK�/�
mice could not be included in this analysis due to difficulty ingenerating pre-BCR� cell lines from these genotypes. Pre-B cellsfrom Cbl�/�BLNK�/� and Cbl�/�BLNK�/� mice expressed
FIGURE 5. Phenotypic analysis of peripheral B cells. a, IgM and IgDcostaining was performed to distinguish different developmental stages ofsplenic B cells. b, MZ B cells were distinguished from FO B cells by CD21and CD23 costaining. c, Peritoneal B1 B cells were revealed by CD5 andIgM staining. All analysis was based on lymphocyte-gated cells. The resultfrom one of five independent experiments is shown.
FIGURE 6. Pre-BCR cross-linking in c-Cbl�/�BLNK�/� comparedwith BLNK�/� pre-B cells induced increased and prolonged tyrosine-phosphorylation signaling. A total of 2 � 107 pre-BCR� pre-B cells of 5–8mo of culture from c-Cbl�/�BLNK�/� and c-Cbl�/�BLNK�/� mice wereincubated at 37°C for the indicated time with F(ab�)2 Abs specific formouse � chain. The cells were then washed and lysed, and a portion of thetotal cell lysates analyzed for tyrosine phosphorylation by immunoblottingwith mAb 4G10 (a). For specific measurement of Btk, total cell lysateswere blotted with phosphospecific anti-Btk (pY551) and anti-Btk Abs (d).For measurements of Syk and PLC-�2, lysates were precipitated with Absspecific for Syk or PLC-�2, and the precipitates were analyzed by immu-noblotting with anti-phosphotyrosine mAb 4G10, anti-Syk or anti-PLC-�2as indicated (b and e). For measurement of tyrosine phosphorylated Lyn,lysates were precipitated with anti-phosphotyrosine mAb 4G10 and immu-noblotted with anti-Lyn Ab (c). The signal intensity was analyzed withImageQuant program (Molecular Dynamics) and the phosphorylation sig-nals of Syk, Btk, and PLC-�2 were normalized based on total protein levelof the molecules. Level of activation was expressed as ratio of stimulatedsamples vs unstimulated samples. In the experiment shown in c, the in-tensity of blotting of the total cell lysates with anti-Lyn was 1.3 times in thec-Cbl�/�BLNK�/� compared with the c-Cbl�/�BLNK�/� cells (data notshown). Results shown are representative of two independent experiments.
931The Journal of Immunology
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

equivalent cell surface levels of pre-BCR/�5 and CD19 (data notshown). In both Cbl�/�BLNK�/� and Cbl�/�BLNK�/� pre-Bcells, pre-BCR engagement consistently induced tyrosine phos-phorylation of multiple bands (Fig. 6a). For both groups, the peakintensity of overall phosphorylation signaling was 2 min afterstimulation. However, pre-BCR signaling in c-Cbl�/�BLNK�/�
pre-B cells was more sustained (at 5, 10, and 20 min) than that inBLNK�/� pre-B cells. One exception was a band at �110 kDathat appears prominently in BLNK�/� and is absent in c-Cbl�/�
BLNK�/� pre-B cells. Immunoprecipitation with anti-c-Cbl Abdemonstrated that this band was c-Cbl (data not shown).
Immunoprecipitation was then used to assess tyrosine phosphor-ylation of specific molecules in response to pre-BCR engagement.Phosphorylation of Syk, a critical mediator of pre-BCR functionwas first assessed by precipitation of lysates with anti-Syk Abfollowed by immunoblotting with anti-phosphotyrosine mAb4G10. Pre-BCR cross-linking triggered stronger and more sus-tained phosphorylation of Syk in c-Cbl�/�BLNK�/� comparedwith the response of BLNK�/� pre-B cells (Fig. 6b). The en-hanced and prolonged phosphorylation of Syk in c-Cbl�/�
BLNK�/� pre-B cells was confirmed by confocal microscopy
(Fig. 7) and by flow cytometry (data not shown). These data in-dicate that Cbl negatively regulates tyrosine phosphorylated Syk inresponse to pre-BCR signaling. Because Syk phosphorylation canbe regulated by Lyn kinase, Lyn activation was analyzed by im-munoprecipitation with anti-phosphotyrosine mAb 4G10, fol-lowed by immunoblotting for Lyn. Both basal and the receptorstimulated phosphorylation of Lyn were enhanced in c-Cbl�/�
BLNK�/� pre-B cells, indicating an increase in this very proximalcomponent of pre-BCR signaling (Fig. 6c).
It has been proposed that, in addition to a BLNK-dependentmechanism for pre-BCR signaling, there exists a distinct BLNK-independent but Btk-mediated pathway (37). We therefore exam-ined the possibility that the ability of c-Cbl inactivation to restorepre-BCR signaling in BLNK-deficient cells might be mediatedthrough Btk activation. Baseline phosphorylation of Btk, before invitro activation, was elevated in c-Cbl�/�BLNK�/� pre-B cellsrelative to BLNK�/� pre-B cells. Cross-linking of the pre-BCRresulted in increased tyrosine phosphorylation of Btk, and thestrength of this response was increased in c-Cbl�/�BLNK�/�
pre-B cells over the response in BLNK�/� pre-B cells, consistentwith a role for Btk in mediating the effect of c-Cbl inactivation(Fig. 6d). Phosphorylation of PLC-�2, proposed to be involved inboth BLNK-dependent and BLNK-independent pathways of pre-BCR signaling (37), was also increased in c-Cbl�/�BLNK�/�
pre-B cells (Fig. 6e).
DiscussionPrevious studies of c-Cbl-deficient mice suggested that c-Cbl mayaffect B cell development (27). However, it has not previouslybeen determined at which stage and how c-Cbl carries out its rolein regulating this process. In the current study, we analyzed B celldevelopment in c-Cbl�/�BLNK�/� mice, and found that c-Cblmost strongly affects early B cell development in the BM. Specif-ically, c-Cbl inactivation corrected several components of the im-paired B cell developmental process in BLNK�/� mice, including:1) down-regulation of the expression of pre-BCR and CD43 mol-ecules on early pre-B cell surface; 2) up-regulation of cell surfaceMHC class II expression on pre-B cells; 3) initiation of �-chainrearrangement in small pre-B cells; and 4) transition of pre-B cellsto surface IgM� immature B cells. However, c-Cbl appears to haveless effect on other developmental events, such as down-regulationof surface BP-1 and up-regulation of CD25 and CD2 expression.In addition, the transition from T2 B cells to mature B cells was notrescued in c-Cbl�/�BLNK�/� mice. c-Cbl thus differentially reg-ulates distinct B cell development events.
The expression of pre-BCR on the surface of early pre-B cellselicits signals that are essential for B cell development. A criticalproximal event in pre-BCR signaling is mediated by the tyrosinekinase Syk, as evidenced by the essentially complete abrogation ofB cell development that results from inactivation of Syk. However,it appears that at least two independent pathways exist downstreamof Syk, each of which is able to support a significant though re-duced level of B cell development (37, 38). One of these pathwaysis BLNK dependent, and its function is reflected in the substantialthough incomplete block in B cell development observed inBLNK-deficient mice. The existence of a second, BLNK-indepen-dent, pathway(s) is suggested by the incomplete nature of this de-velopmental block, and is further supported by analysis of com-pound genetic ablations. It has been demonstrated that Btkdeficiency results in a partial developmental defect, but that inac-tivation of both BLNK and Btk results in a profound and essen-tially complete arrest, resembling that which results from Syk de-ficiency. The effect of c-Cbl inactivation presented here can beassessed in the context of these multiple pathways of pre-BCR
FIGURE 7. Pre-BCR cross-linking in c-Cbl�/�BLNK�/� pre-B cellsinduced a stronger and more prolonged Syk-phosphorylation thanBLNK�/� pre-B cells by confocal microscopic study. Pre-BCR� pre-Bcells were allowed to settle on cover slides on ice for 40 min, warmed to37°C, and stimulated as described in Fig. 6. At the indicated time pointsafter stimulation, cells were fixed with 4% paraformaldehyde for 15 min,quenched with 50 �M NH4Cl for 10 min, followed by 30 min of perme-abilization with 0.05% saponin. The cells were then blocked with 5% goatserum in PBS for 30 min and incubated overnight at 4°C with rabbit anti-Syk or anti-phospho-Syk (Y519/520-specific) Abs. Staining was resolvedwith goat anti-rabbit IgG-Alexa 488 at RT for 40 min. The cells wereimaged with a Zeiss LSM 510 META confocal microscope. a-d, Repre-sentative images at 5 min of stimulation. e, The MFI of phosphor-Syk(Alexa 488 staining) from different time points was analyzed with Meta-Morph software and the MFI was calculated based on the intensity of over200 cells from five different view fields. Results are expressed as mean �SE as calculated with Excel software.
932 ROLE OF c-Cbl IN B CELL DEVELOPMENT
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

signaling. The observation that c-Cbl inactivation enhances ty-rosine phosphorylation of Syk and Lyn in BLNK-deficient pre-Bcells suggests a proximal effect on strength of signaling that couldenhance early B cell development through multiple downstreampathways and signaling events. The inactivation of Cbl could re-move a negative feedback loop thereby increasing the activation ofLyn and the molecules that are to its downstream such as Syk, Btkand PLC-�2. In addition, however, the finding of increased Btkphosphorylation suggests that enhancement of a Btk-dependentpathway might be a specific mechanism by which c-Cbl inactiva-tion affects signaling and B cell development. The enhancement ofBtk response could reflect a direct interaction of c-Cbl and Btkand/or an indirect consequence of enhanced proximal events. Ad-ditional evidence supporting a critical role of Btk in the restorationof B cell development by c-Cbl inactivation has been generated inour recent finding that the ability of c-Cbl inactivation to restore Bcell development in BLNK-deficient mice is completely abrogatedby inactivation of Btk (data not shown).
Two independent pre-BCR signaling pathways downstream ofSyk have been proposed to mediate specific events important in Bcell development (37, 38). A BLNK-independent pathway exists,which converges with IL-7R signaling to activate ERK and thusconfers on pre-BCR� B cells a lower threshold for proliferativeresponse to IL-7 stimulation than that observed in pre-BCR-neg-ative pre-B cells (33). A Syk-dependent, BLNK-independent path-way also mediates allelic exclusion (19, 39). In contrast, it hasbeen proposed that a second signaling pathway downstream of Sykis BLNK-dependent and that this pathway is essential for down-regulation of pre-BCR expression, and thus for suppression ofpre-B cell expansion (16, 32, 38, 40). BLNK-dependent pathwayshave also been shown to induce the modulation of other cell sur-face molecules and to stimulate L chain rearrangement (16, 34, 38,41, 42). In BLNK�/� mice, pre-BCR down-regulation is blocked,leading to a prominent accumulation of pre-BCR� pre-B cells inthe BM (32). Our studies revealed that c-Cbl inactivation correctsthe deficiency in down-regulation of pre-BCR in BLNK�/� mice,indicating that surface pre-BCR expression can be regulated notonly by a BLNK-dependent pathway, but also by a BLNK-inde-pendent, c-Cbl-modulated pathway. Similarly, the expression ofCD43 and MHC-II, as well as the induction of L chain rearrange-ment, another critical step in B cell differentiation, are also mod-ulated by BLNK-independent, c-Cbl-modulated pathways,whereas the expression of CD25, CD2, and BP-1 is relatively un-affected by c-Cbl expression. The results of our studies reinforcethe differential effects of specific signaling pathways on distinctaspects of differentiation and indicate the complex plasticity ofsignaling regulation during B cell development. When BLNK ispresent, the role of c-Cbl in early B cell development is not ap-parent. However, in BLNK-deficient mice, c-Cbl-modulated sig-naling pathways can regulate critical B cell development events.
It has been suggested that BLNK functions as a tumor suppres-sor, as BLNK-deficient pre-B cells display a hyperproliferativeresponse to IL-7 signaling, and pre-B cell lymphomas appear atincreased frequency in BLNK-deficient mice (32). Furthermore,BLNK mutations have also been observed in �50% of the humanpre-B cell lymphomas, further supporting the conclusion thatBLNK acts as a tumor suppressor gene (43). Because inactivationof c-Cbl reversed the hyperproliferative responses of BLNK-defi-cient pre-B cells to IL-7 stimulation in our studies, it can be pos-tulated that the pre-B cell tumors induced by BLNK-deficiencywould be prevented by c-Cbl-inactivation. In addition, pre-B celltumors have also been observed in BLNK�/�Btk�/� mice at afrequency higher than that in BLNK�/� mice, suggesting Btk maycooperate with BLNK in mediating tumor suppressor function
(16). Studies on possible protective effects of c-Cbl inactivation onpre-B cell tumors induced by impaired BCR signaling in thosemice are in progress. A number of mechanisms may explain thereversal of IL-7 hyperresponsiveness of BLNK�/� pro-B/pre-Bcells by c-Cbl inactivation. The decreased proportion of pre-BCR�
cells in c-Cbl�/�BLNK�/� BM could reduce the frequency of cellsthat are hyperresponsive to IL-7. The reduced density of cell surfaceIL-7R� that we have observed on c-Cbl�/�BLNK�/� BM B cellsrelative to that expressed in BLNK�/� mice may also contribute tothe decreased response.
c-Cbl is an adaptor protein with multiple functions. One majorrole of c-Cbl is to mediate the ubiquitination and subsequent deg-radation of its target proteins. In B cells, several signaling proteinsincluding Syk and Lyn have been described as targets of c-Cbl.Syk-c-Cbl association is induced by BCR ligation and c-Cbl-phos-phorylation (31). c-Cbl is itself a substrate for phosphorylation bySyk and Lyn (2, 25, 44), and it is conceivable that activated Sykand Lyn induce a feedback down-regulation of their own proteinlevels by mediating c-Cbl phosphorylation and in turn enhancingc-Cbl-dependent degradation of target molecules. Our studiesshowed that tyrosine phosphorylation of the proximal signalingmolecules Lyn and Syk, and their downstream substrates, Btk andPLC-�2, together with additional unidentified proteins, was ele-vated in c-Cbl�/�BLNK�/� pre-B cells compared with c-Cbl�/�
BLNK�/� pre-B cells. As noted above, it is possible that a Btk-PLC-�2 pathway mediates the corrected phenotypes that weobserved in c-Cbl�/�BLNK�/� mice, a possibility supported byour recent finding that inactivation of Btk abrogates the ability ofc-Cbl inactivation to restore B cell development in BLNK-defi-cient mice (data not shown). These results suggest that differentevents downstream of pre-BCR signaling have different thresholdsin the strength and duration of signaling and that elevated and/orprolonged Lyn and Syk activation resulting from c-Cbl inactiva-tion can bypass the BLNK-dependent signaling pathway to inducecritical differentiation events including the down-regulation of pre-BCR and up-regulation of MHC-II, as well as initiation of L chainrearrangement. In contrast, activation of BLNK-dependent path-ways may be essential for the up-regulation of CD25 and CD2, anddown-regulation of BP-1. It has been widely proposed that BCRsignaling strength determines peripheral B cell fate, specificallythat B1 B cell development requires the strongest BCR signaling;that an intermediate-high level of BCR signaling favors follicularB cell development; and that weak BCR signaling directs B celldevelopment to marginal zone B cells (10, 45, 46). We proposethat pre-BCR signaling strength and duration similarly determinedistinct downstream events during pre-B cell transition.
Studies using genetic approaches suggest that pre-BCR signal-ing pathways may be particularly redundant. Many molecules,such as Zap70, LAT, and SLP-76, that are expressed in T cells, butnot in mature B cells, are expressed in pre-B cells; and it appearsthat these molecules can partially replace their B lineage counter-parts in mediating signaling transduction during pre-B cell transi-tion (18, 47–50). Therefore, a finely tuned and redundant signalingnetwork may be particularly important in pre-BCR signaling andearly B cell development, and it is possible that c-Cbl plays a moreimportant role in this early signaling network than in mature Bcells.
Although c-Cbl inactivation selectively corrected some criticalevents in the early stages of BM B cell development in BLNK�/�
mice, it did not correct the block from T2 B cells to mature B cellsin the spleen of those mice. Splenic B cell development takes placein discrete steps from transitional T1 stage to T2 stage, then to themature B cell stage (10). The sequential progression from T1 to T2to mature B cells may require an increasingly higher threshold
933The Journal of Immunology
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

level of BCR signaling. T1 cells require a low-threshold “tonicBCR signal” to generate T2. In contrast, a relatively higher level ofBCR signaling is required for the transition from T2 B cells tomature B cells (10). Thus, the failure of c-Cbl inactivation to res-cue development of mature B cells in BLNK�/� mice may reflecta failure to achieve the strength of BCR-dependent signaling re-quired for this transition. c-Cbl deficiency enhances the positiveselection of TCR-transgenic CD4� T cells (28), probably due toelevated Lck and Zap70 signaling. The role of c-Cbl in CD4� Tcell development and selection is also strongly supported by stud-ies of c-Cbl�/�SLP-76�/� mice, in which the lethality as well asdefective development of CD4� T cells observed in SLP-76�/�
mice is rescued. In contrast, inactivation of another close memberof the c-Cbl family, Cbl-b, did not rescue CD4� T cell develop-ment (51). In parallel to what has been observed in early T celldifferentiation, we show here that c-Cbl regulates important eventsin early B cell development, whereas we have observed that Cbl-binactivation did not correct the impaired B cell development inBLNK�/� mice (our unpublished data). Thus, we conclude thatc-Cbl, but not Cbl-b, regulates B cell development, prominently ina BLNK-independent manner during early stages of B celldevelopment.
AcknowledgmentsWe thank Genevieve Sanchez and staff at Bioqual for excellent animalcare; Susan Sharrow, Larry Granger, and Tony Adams for expert assistancewith FACS sorting; and Drs. Rudi W. Hendriks, Susan Pierce, and JanCerny for critical reading and comments on the manuscript. We also thankMichael Kruhlak for assistance with the Zeiss confocal laser scanning mi-croscope manipulation and data analysis; Steve Bauer and Craig Milne foradvice on culture of pre-B cell lines; Hae Won Sohn for advice on immu-nofluorescence staining; and Mary Robinson for technical assistance withbiochemistry studies.
DisclosuresThe authors have no financial conflict of interest.
References1. Kurosaki, T., M. Takata, Y. Yamanashi, T. Inazu, T. Taniguchi, T. Yamamoto,
and H. Yamamura. 1994. Syk activation by the Src-family tyrosine kinase in theB cell receptor signaling. J. Exp. Med. 179: 1725–1729.
2. Kurosaki, T. 1998. Molecular dissection of B cell antigen receptor signaling(review). Int. J. Mol. Med. 1: 515–527.
3. Fu, C., C. W. Turck, T. Kurosaki, and A. C. Chan. 1998. BLNK: a central linkerprotein in B cell activation. Immunity 9: 93–103.
4. Ishiai, M., M. Kurosaki, R. Pappu, K. Okawa, I. Ronko, C. Fu, M. Shibata,A. Iwamatsu, A. C. Chan, and T. Kurosaki. 1999. BLNK required for couplingSyk to PLC�2 and Rac1-JNK in B cells. Immunity 10: 117–125.
5. Zhang, Y., J. Wienands, C. Zurn, and M. Reth. 1998. Induction of the antigenreceptor expression on B lymphocytes results in rapid competence for signalingof SLP-65 and Syk. EMBO J. 17: 7304–7310.
6. Kitamura, D., J. Roes, R. Kuhn, and K. Rajewsky. 1991. A B cell-deficient mouseby targeted disruption of the membrane exon of the immunoglobulin � chaingene. Nature 350: 423–426.
7. Kraus, M., L. I. Pao, A. Reichlin, Y. Hu, B. Canono, J. C. Cambier, M. C.Nussenzweig, and K. Rajewsky. 2001. Interference with immunoglobulin (Ig)� im-munoreceptor tyrosine-based activation motif (ITAM) phosphorylation modulates orblocks B cell development, depending on the availability of an Ig� cytoplasmic tail.J. Exp. Med. 194: 455–469.
8. Reichlin, A., Y. Hu, E. Meffre, H. Nagaoka, S. Gong, M. Kraus, K. Rajewsky,and M. C. Nussenzweig. 2001. B cell development is arrested at the immature Bcell stage in mice carrying a mutation in the cytoplasmic domain of immuno-globulin �. J. Exp. Med. 193: 13–23.
9. Reth, M., and J. Wienands. 1997. Initiation and processing of signals from the Bcell antigen receptor. Annu. Rev. Immunol. 15: 453–479.
10. Loder, F., B. Mutschler, R. J. Ray, C. J. Paige, P. Sideras, R. Torres, M. C. Lamers,and R. Carsetti. 1999. B cell development in the spleen takes place in discrete stepsand is determined by the quality of B cell receptor-derived signals. J. Exp. Med. 190:75–89.
11. Lam, K. P., R. Kuhn, and K. Rajewsky. 1997. In vivo ablation of surface im-munoglobulin on mature B cells by inducible gene targeting results in rapid celldeath. Cell 90: 1073–1083.
12. Xu, S., J. E. Tan, E. P. Wong, A. Manickam, S. Ponniah, and K. P. Lam. 2000.B cell development and activation defects resulting in xid-like immunodeficiencyin BLNK/SLP-65-deficient mice. Int. Immunol. 12: 397–404.
13. Jumaa, H., B. Wollscheid, M. Mitterer, J. Wienands, M. Reth, and P. J. Nielsen.1999. Abnormal development and function of B lymphocytes in mice deficientfor the signaling adaptor protein SLP-65. Immunity 11: 547–554.
14. Pappu, R., A. M. Cheng, B. Li, Q. Gong, C. Chiu, N. Griffin, M. White, B. P.Sleckman, and A. C. Chan. 1999. Requirement for B cell linker protein (BLNK)in B cell development. Science 286: 1949–1954.
15. Hayashi, K., R. Nittono, N. Okamoto, S. Tsuji, Y. Hara, R. Goitsuka, andD. Kitamura. 2000. The B cell-restricted adaptor BASH is required for normaldevelopment and antigen receptor-mediated activation of B cells. Proc. Natl.Acad. Sci. USA 97: 2755–2760.
16. Kersseboom, R., S. Middendorp, G. M. Dingjan, K. Dahlenborg, M. Reth,H. Jumaa, and R. W. Hendriks. 2003. Bruton’s tyrosine kinase cooperates withthe B cell linker protein SLP-65 as a tumor suppressor in Pre-B cells. J. Exp. Med.198: 91–98.
17. Jumaa, H., M. Mitterer, M. Reth, and P. J. Nielsen. 2001. The absence of SLP65and Btk blocks B cell development at the preB cell receptor-positive stage. Eur.J. Immunol. 31: 2164–2169.
18. Su, Y. W., and H. Jumaa. 2003. LAT links the pre-BCR to calcium signaling.Immunity 19: 295–305.
19. Hayashi, K., M. Yamamoto, T. Nojima, R. Goitsuka, and D. Kitamura. 2003.Distinct signaling requirements for Dmu selection, IgH allelic exclusion, pre-Bcell transition, and tumor suppression in B cell progenitors. Immunity 18:825–836.
20. Zheng, N., P. Wang, P. D. Jeffrey, and N. P. Pavletich. 2000. Structure of ac-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell102: 533–539.
21. Lupher, M. L., Jr., N. Rao, M. J. Eck, and H. Band. 1999. The Cbl protoonco-protein: a negative regulator of immune receptor signal transduction. Immunol.Today 20: 375–382.
22. Rao, N., I. Dodge, and H. Band. 2002. The Cbl family of ubiquitin ligases: criticalnegative regulators of tyrosine kinase signaling in the immune system. J. Leu-kocyte Biol. 71: 753–763.
23. Lupher, M. L., Jr., N. Rao, N. L. Lill, C. E. Andoniou, S. Miyake, E. A. Clark,B. Druker, and H. Band. 1998. Cbl-mediated negative regulation of the Syktyrosine kinase: a critical role for Cbl phosphotyrosine-binding domain binding toSyk phosphotyrosine 323. J. Biol. Chem. 273: 35273–35281.
24. Ota, S., K. Hazeki, N. Rao, M. L. Lupher, Jr., C. E. Andoniou, B. Druker, andH. Band. 2000. The RING finger domain of Cbl is essential for negative regu-lation of the Syk tyrosine kinase. J. Biol. Chem. 275: 414–422.
25. Tezuka, T., H. Umemori, N. Fusaki, T. Yagi, M. Takata, T. Kurosaki, andT. Yamamoto. 1996. Physical and functional association of the cbl protooncogenproduct with an src-family protein tyrosine kinase, p53/56lyn, in the B cell antigenreceptor-mediated signaling. J. Exp. Med. 183: 675–680.
26. Murphy, M. A., R. G. Schnall, D. J. Venter, L. Barnett, I. Bertoncello, C. B.Thien, W. Y. Langdon, and D. D. Bowtell. 1998. Tissue hyperplasia and en-hanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol. Cell Biol. 18:4872–4882.
27. Shao, Y., C. Yang, C. Elly, and Y. C. Liu. 2004. Differential regulation of the Bcell receptor-mediated signaling by the E3 ubiquitin ligase Cbl. J. Biol. Chem.279: 43646–43653.
28. Naramura, M., H. K. Kole, R. J. Hu, and H. Gu. 1998. Altered thymic positiveselection and intracellular signals in Cbl-deficient mice. Proc. Natl. Acad. Sci.USA 95: 15547–15552.
29. Keshvara, L. M., C. C. Isaacson, T. M. Yankee, R. Sarac, M. L. Harrison, andR. L. Geahlen. 1998. Syk- and Lyn-dependent phosphorylation of Syk on mul-tiple tyrosines following B cell activation includes a site that negatively regulatessignaling. J. Immunol. 161: 5276–5283.
30. Yankee, T. M., L. M. Keshvara, S. Sawasdikosol, M. L. Harrison, andR. L. Geahlen. 1999. Inhibition of signaling through the B cell antigen receptorby the protooncogene product, c-Cbl, requires Syk tyrosine 317 and the c-Cblphosphotyrosine-binding domain. J. Immunol. 163: 5827–5835.
31. Panchamoorthy, G., T. Fukazawa, S. Miyake, S. Soltoff, K. Reedquist, B. Druker,S. Shoelson, L. Cantley, and H. Band. 1996. p120cbl is a major substrate oftyrosine phosphorylation upon B cell antigen receptor stimulation and interacts invivo with Fyn and Syk tyrosine kinases, Grb2 and Shc adaptors, and the p85subunit of phosphatidylinositol 3-kinase. J. Biol. Chem. 271: 3187–3194.
32. Flemming, A., T. Brummer, M. Reth, and H. Jumaa. 2003. The adaptor proteinSLP-65 acts as a tumor suppressor that limits pre-B cell expansion. Nat. Immunol.4: 38–43.
33. Fleming, H. E., and C. J. Paige. 2001. Pre-B cell receptor signaling mediatesselective response to IL-7 at the pro-B to pre-B cell transition via an ERK/MAPkinase-dependent pathway. Immunity 15: 521–531.
34. Schebesta, M., P. L. Pfeffer, and M. Busslinger. 2002. Control of pre-BCR sig-naling by Pax5-dependent activation of the BLNK gene. Immunity 17: 473–485.
35. Liu, Y. C., and A. Altman. 1998. Cbl: complex formation and functional impli-cations. Cell Signal. 10: 377–385.
36. Miyake, S., M. L. Lupher, Jr., C. E. Andoniou, N. L. Lill, S. Ota, P. Douillard,N. Rao, and H. Band. 1997. The Cbl protooncogene product: from an enigmaticoncogene to center stage of signal transduction. Crit. Rev. Oncog. 8: 189–218.
37. Jumaa, H., R. W. Hendriks, and M. Reth. 2005. B cell signaling and tumorigen-esis. Annu. Rev. Immunol. 23: 415–445.
38. Hendriks, R. W., and S. Middendorp. 2004. The pre-BCR checkpoint as a cell-autonomous proliferation switch. Trends Immunol. 25: 249–256.
39. Xu, S., and K. P. Lam. 2002. Delayed cellular maturation and decreased immu-noglobulin � light chain production in immature B lymphocytes lacking B celllinker protein. J. Exp. Med. 196: 197–206.
934 ROLE OF c-Cbl IN B CELL DEVELOPMENT
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

40. Grawunder, U., T. M. Leu, D. G. Schatz, A. Werner, A. G. Rolink, F. Melchers,and T. H. Winkler. 1995. Down-regulation of RAG1 and RAG2 gene expressionin preB cells after functional immunoglobulin heavy chain rearrangement. Im-munity 3: 601–608.
41. Karasuyama, H., A. Rolink, Y. Shinkai, F. Young, F. W. Alt, and F. Melchers.1994. The expression of Vpre-B/�5 surrogate light chain in early bone marrowprecursor B cells of normal and B cell-deficient mutant mice. Cell 77: 133–143.
42. Torres, R. M., H. Flaswinkel, M. Reth, and K. Rajewsky. 1996. Aberrant B celldevelopment and immune response in mice with a compromised BCR complex.Science 272: 1804–1808.
43. Jumaa, H., L. Bossaller, K. Portugal, B. Storch, M. Lotz, A. Flemming,M. Schrappe, V. Postila, P. Riikonen, J. Pelkonen, C. M. Niemeyer, and M. Reth.2003. Deficiency of the adaptor SLP-65 in pre-B-cell acute lymphoblastic leu-kaemia. Nature 423: 452–456.
44. Feshchenko, E. A., W. Y. Langdon, and A. Y. Tsygankov. 1998. Fyn, Yes, andSyk phosphorylation sites in c-Cbl map to the same tyrosine residues that becomephosphorylated in activated T cells. J. Biol. Chem. 273: 8323–8331.
45. Cariappa, A., M. Tang, C. Parng, E. Nebelitskiy, M. Carroll, K. Georgopoulos,and S. Pillai. 2001. The follicular versus marginal zone B lymphocyte cell fatedecision is regulated by Aiolos, Btk, and CD21. Immunity 14: 603–615.
46. Martin, F., and J. F. Kearney. 2000. Positive selection from newly formed tomarginal zone B cells depends on the rate of clonal production, CD19, and btk.Immunity 12: 39–49.
47. Schweighoffer, E., L. Vanes, A. Mathiot, T. Nakamura, and V. L. Tybulewicz.2003. Unexpected requirement for ZAP-70 in pre-B cell development and allelicexclusion. Immunity 18: 523–533.
48. Su, Y. W., S. Herzog, M. Lotz, N. Feldhahn, M. Muschen, and H. Jumaa. 2004.The molecular requirements for LAT-mediated differentiation and the role ofLAT in limiting pre-B cell expansion. Eur. J. Immunol. 34: 3614–3622.
49. Oya, K., J. Wang, Y. Watanabe, R. Koga, and T. Watanabe. 2003. Appearanceof the LAT protein at an early stage of B-cell development and its possible role.Immunology 109: 351–359.
50. Wong, J., M. Ishiai, T. Kurosaki, and A. C. Chan. 2000. Functional complemen-tation of BLNK by SLP-76 and LAT linker proteins. J. Biol. Chem. 275:33116–33122.
51. Chiang, Y. J., C. L. Sommers, M. S. Jordan, H. Gu, L. E. Samelson, G. A.Koretzky, and R. J. Hodes. 2004. Inactivation of c-Cbl reverses neonatal lethalityand T cell developmental arrest of SLP-76-deficient mice. J. Exp. Med. 200:25–34.
935The Journal of Immunology
by guest on June 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from