prognostic significance of inos in hepatocellular carcinoma
TRANSCRIPT

Chapter 17Prognostic Significance of iNOSin Hepatocellular Carcinoma
Rosa M. Pascale, M. Frau, and Francesco Feo
Abstract Epidemiological research indicates a contribution of chronic inflamma-tory liver diseases to development of hepatocellular carcinoma (HCC). Mountingevidence shows a carcinogenic role of NO• produced by inflammatory or cancercells. The calcium-independent inducible isoform, iNOS, produces under inflam-matory stimulation large amounts of NO• through the conversion of l-arginine tol-citrulline. NO• and NO•-derived oxidants can oxidize biomolecules, causing DNAdamage. NO• interferes with the oxidative metabolism at different levels. It upreg-ulates the AMP-activated protein kinase, thus switching off the ATP-consumingpathways such as lipogenesis or gluconeogenesis, while switching on the ATP-producing pathways such as fatty acid and glucose oxidation. Moreover, increasein mitochondrial NO• above physiological levels affects the oxygen-binding site ofcytochrome c oxidase and induces pyruvate dehydrogenase kinase-1, an inhibitorof pyruvate dehydrogenase complex, thus inhibiting electron transport and oxygenconsumption. NO• may also interact with different signaling pathways in hepa-tocarcinogenesis, including COX2, inhibitor of κB kinase (IKK)/nuclear factor B(NF-κB), and RAS/extracellular signal-regulated kinases 1 and 2 (ERK1/2) signal-ing. NO• stimulates COX-2 activity, and COX-2 inhibitors block NO• productionin HCC cells. Furthermore, NO• and its derivatives may influence prostaglandinproduction by inducing lipid peroxidation and arachidonic acid release from cellmembranes. Stimulation of EP2 receptor by PGE2 induces the association of the α
subunit of the regulator G protein signaling and AXIN. This leads to the inactivationof glycogen synthase kinase-3β (GSK-3β) with consequent nuclear accumulationof β-catenin and increase in its transcriptional targets, c-Myc, c-Jun, and cyclinD1. PGE2 can also stimulate cell growth through the activation of several tyro-sine kinase receptors, including EGFR and the PI3K/Akt pathways. Recent researchon the interplay between iNOS and IKK/NF-κB and RAS/ERK pathways in HCC
F. Feo (B)Division of Experimental Pathology and Oncology, Department of Biomedical Sciences,University of Sassari, Sassari 07100, Italye-mail: [email protected]
309B. Bonavida (ed.), Nitric Oxide (NO) and Cancer, Cancer Drug Discoveryand Development, DOI 10.1007/978-1-4419-1432-3_17,C© Springer Science+Business Media, LLC 2010

310 R.M. Pascale et al.
showed that these interactions are highest in the highly aggressive preneoplas-tic and neoplastic liver lesions of genetically susceptible rats and c-Myc/Tgf-αtransgenic mice. The determination of iNOS expression in human HCC showedhighest values in a subtype with poorer prognosis. Interestingly, iNOS levels aredirectly correlated with genomic instability, proliferation rate, and microvessel den-sity of human HCC and inversely correlated with apoptosis and patients’ survival.These observations suggest that iNOS upregulation and changes in iNOS/NF-κBand iNOS/H-RAS/ERK cross talks are prognostic markers for HCC. Moreover, theblock of iNOS signaling by a specific inhibitor such as aminoguanidine leads to aconsistent decrease in HCC growth in c-Myc/TGF-α transgenic mice, decrease ingrowth and increase in apoptosis in human HCC cell lines, suggesting that the keycomponents of iNOS signaling could represent therapeutic targets.
Keywords Hepatocarcinogenesis · iNOS · Glycolysis · Mitochondrial activity ·Prostaglandins · Signal transduction · S-adenosylmethionine · DNA methylation
Hepatocellular carcinoma (HCC) is one of the most frequent human cancers,with ∼1 million of newly diagnosed cases each year. The highest frequencies arefound in sub-Saharan Africa and far eastern Asia, where hepatitis B virus (HBV)and hepatitis C virus (HCV) infections are endemic, and in regions where food con-taminated with aflatoxin B1 is consumed (Thorgeirsson and Grisham 2002; Bruixet al. 2004; Farazi and DePinho 2006). Other risk factors associated with the devel-opment of HCC include alcoholic steatohepatitis, high dose of androgen steroids,type 2 diabetes, and various genetic disorders such as hemochromatosis, glyco-gen storage disease (types 1 and 2), a1-antitrypsin deficiency, Wilson’s disease, andenvironmental agents (cycasin, pyrrolizidine alkaloids, etc.). HCC incidence is ris-ing, even in countries with relatively low incidence (Tanaka et al. 2002). HCC isa rapidly fatal disease, with a life expectancy of about 6 months from the time ofdiagnosis. Partial liver resection or liver transplantation is potentially curative, butonly a minority of cases are amenable to these treatments.
Hepatocarcinogenesis is a complex multistep process involving the accumulationof genetic and epigenetic events such as point mutations, chromosomal rearrange-ments, oncogene activation, and oncosuppressor gene inactivation (Feitelson et al.2002). Epidemiological evidence has shown a striking contribution of chronicinflammatory liver diseases, including hepatitis B virus or hepatitis C virus (HCV)infection, and alcoholic steatohepatitis, to HCC development through inflammation-related mechanisms (Feitelson et al. 2002). It has been shown that persistent liverinjury due to increase in nitrative and oxidative DNA damage enhances carcinogen-esis (Biegon et al. 2002; Iwai et al. 2002; Jüngst et al. 2004; Kawanishi et al. 2006).Liver infiltration by phagocytes, during liver injury, provides an important sourceof reactive oxygen species (ROS) which cause damage to DNA, proteins, and lipidswhen their generation exceeds the ability of the antioxidant systems to remove them.8-Hydroxy-2′-deoxyguanosine (8-OH-dG) was first reported as a major form ofoxidative DNA damage product which preferentially mispairs with adenine duringDNA replication, resulting in GC → TA transversion (Shibutani et al. 1991).
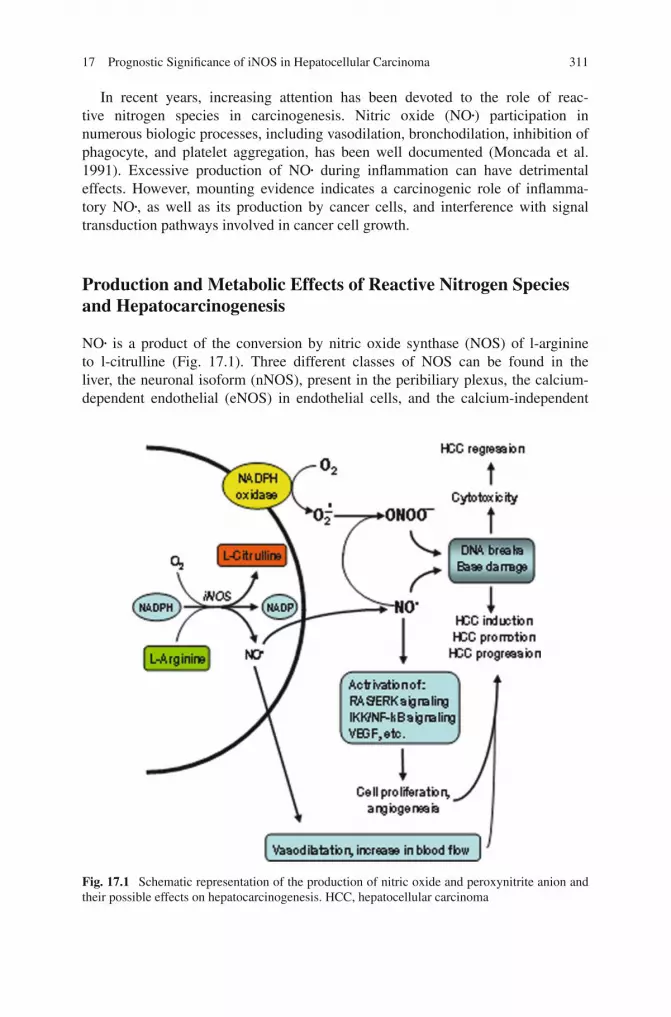
17 Prognostic Significance of iNOS in Hepatocellular Carcinoma 311
In recent years, increasing attention has been devoted to the role of reac-tive nitrogen species in carcinogenesis. Nitric oxide (NO•) participation innumerous biologic processes, including vasodilation, bronchodilation, inhibition ofphagocyte, and platelet aggregation, has been well documented (Moncada et al.1991). Excessive production of NO• during inflammation can have detrimentaleffects. However, mounting evidence indicates a carcinogenic role of inflamma-tory NO•, as well as its production by cancer cells, and interference with signaltransduction pathways involved in cancer cell growth.
Production and Metabolic Effects of Reactive Nitrogen Speciesand Hepatocarcinogenesis
NO• is a product of the conversion by nitric oxide synthase (NOS) of l-arginineto l-citrulline (Fig. 17.1). Three different classes of NOS can be found in theliver, the neuronal isoform (nNOS), present in the peribiliary plexus, the calcium-dependent endothelial (eNOS) in endothelial cells, and the calcium-independent
Fig. 17.1 Schematic representation of the production of nitric oxide and peroxynitrite anion andtheir possible effects on hepatocarcinogenesis. HCC, hepatocellular carcinoma

312 R.M. Pascale et al.
inducible isoform (iNOS) in hepatocytes, Kupffer and stellate cells, and cholan-giocytes. In general, iNOS is not expressed at a significant level in normalcells (Beckman and Koppenol 1996; Wu and Morris 1998). When induced byinflammatory/immunological stimuli (including inflammatory cytokines or bacte-rial endotoxin), iNOS is highly expressed in many cell types and produces a largeamount of NO• (Wu and Morris 1998). During inflammation, NO•-derived oxidantsare formed by inflammatory cells. Stimulation of these cells results in activation ofreduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase to formO2
– and secretion of myeloperoxidase into the phagosomes or the extracellularspace. Simultaneous production of NO• either by inflammatory cells or by adja-cent parenchymal cells causes formation of peroxynitrite anion (ONOO–) whichcan act as a substrate for myeloperoxidase with formation of nitrogen dioxide (NO2;Beckman and Koppenol 1996).
NO• and NO•-derived oxidants can oxidize biomolecules (including proteins,fatty acids, and DNA), thereby damaging cell membranes, inhibiting vital biochem-ical reactions (e.g., the mitochondrial Krebs cycle and respiratory chain), and evencausing cell death (Demple and Harrison 1994; Fang et al. 2002). NO• causes a vari-ety of DNA damages, including DNA strand breaks and oxidation, and inhibition ofDNA repair (Fig. 17.1). All of these alterations may be responsible for liver necro-sis. However, NO• at lower concentrations may favor HCC development. Notably,DNA mutations in hepatocytes surviving to oxidative stress may induce cell trans-formation, in the absence of sufficient DNA repair, and liver necrosis, when notextended to the majority of liver mass, could enhance liver regeneration and tumorpromotion (Fig. 17.1). Furthermore, NO• causes vasodilatation depending on itsreaction with the ferrous iron, in the heme prosthetic group of the soluble guanylatecyclase, that increases the concentration of guanosine-3′,5′-cyclic monophosphate(cGMP) within the respective target cell, thus mediating its relaxation (Pannen2002). Vasodilatation can favor HCC development by providing tumor cells withsufficient metabolites and oxygen.
Free radicals at physiological levels are signaling molecules involved inmetabolic regulation (Wu et al. 2004). Their overproduction leads to metabolic alter-ations which could modulate cancer development. AMP-activated protein kinase(AMPK) is an evolutionarily conserved sensor of cellular energy status, activatedby a variety of cellular stresses that deplete ATP. AMPK activation by increased[AMP]:[ATP] ratio occurs via the phosphorylation by the serine–threonine proteinkinase LKB1. The overall effect of AMPK activation is to switch off the ATP-consuming pathways such as lipogenesis or gluconeogenesis while switching onthe ATP-producing pathways such as fatty acid and glucose oxidation. Recently,NO• has been found to be an endogenous AMPK activator (Zhang et al. 2008),and low concentrations of peroxynitrite anion activate AMPK through a c-Src-mediated and phosphatidylinositol 3-kinase (PI3K)-dependent pathway in culturedbovine aortic endothelial cells and in mouse aorta and heart (Zou et al. 2003).Accordingly, NO• donors were found to inhibit the conversion of lactate and pyru-vate into glucose in rat hepatocytes (Horton et al. 1994). It has been also reportedthat in hepatocytes NO• inhibits glycogen synthase and glycogen synthesis from glu-cose (Sprangers et al. 1998). On the other hand, NO• decreases the activities of key

17 Prognostic Significance of iNOS in Hepatocellular Carcinoma 313
glycolytic enzymes such as glucokinase (Monti et al. 2000) and glyceraldehyde-3-phosphate dehydrogenase (Stadler et al. 1995). Thus, the net effect of NO•
on hepatic glucose metabolism likely could depend on the balance between twoopposing effects: AMPK-mediated inhibition of gluconeogenesis and reducedglucose utilization.
In 1926, Warburg (1956) observed that cancer cells produce most of their ATPthrough glycolysis, even under aerobic conditions. This phenomenon, subsequentlydenominated “Warburg effect,” has been the object of several studies showing thateven if mitochondria isolated from HCC may efficiently respire and produce ATP(Feo et al. 1973), intact HCC cells use prevalently glycolytic ATP for protein syn-thesis (Terranova et al. 1964), and there is a correlation between glycolytic ATPproduction and aggressiveness of the tumor cells (Kim and Dang 2006). Thus, theWarburg effect could be considered as a positive modifier of cancer, such that it maynot be causative but rather facilitates tumor progression (Kim and Dang 2006).
NO• may modulate mitochondrial activity by different mechanisms.Physiological NO• levels may increase the supply of metabolic substrates andoxygen to mitochondria by regulating blood flow. Moreover, high NO• levelsmay directly modulate the activity of the mitochondrial electron transport system(Brown 2001). Exogenous NO• concentrations, that are likely to increase NO• inmitochondria above physiological levels, reversibly bind the oxygen-binding site ofcytochrome c oxidase, thus inhibiting electron transport and oxygen consumption(Fig. 17.2). This results in a reduced supply of ATP from substrate oxidation andenhanced glycolysis as a significant alternative ATP-producing pathway (Brown2001; Nisoli et al. 2004). This situation, which should favor ADP consumingreactions of glycolysis and glucose consumption for ATP synthesis (Fig. 17.2),apparently contrasts with the above-reported inhibitory effect of NO• on somekey glycolytic enzymes. It should be noted, in this respect, that upregulation ofthe genes encoding glycolytic enzymes (see below) could overcome the enzymeactivity restriction by NO•, and thus NO• effects on glucose metabolism could playan important role in adaptation to low-oxygen conditions which may characterizeHCC (Wu et al. 2007).
The hypoxia-inducible factor 1 (HIF-1) consists of an oxygen-sensitive HIF-1α
subunit that heterodimerizes with HIF-1β to bind DNA. In high oxygen tension,HIF-1α is hydroxylated by prolyl hydroxylases (PHD; Kim and Dang 2006). Thehydroxylated HIF-1α subunit is recognized by the von Hippel–Lindau (VHL)protein and designated for degradation by the proteasome. Hypoxia is a pathophysi-ologic stimulus of anaerobic glycolysis through stabilization of HIF-1 and its directtransactivation of glycolytic enzyme genes (Kim and Dang 2006). iNOS is a targetof HIF-1 (Harris 2002). Conversely, NO• induces H-RAS activation in HCC (Calvisiet al. 2008a; Fig. 17.2), and activated H-RAS (H-RAS-GTP) has been found toincrease the level of HIF-1 (Chen et al. 2001) and PI3K signaling may stabilize HIF-1 (Semenza 2003). HIF-1 stabilization may induce angiogenesis via activation ofthe VEGF-A gene. HIF-1 also induces pyruvate dehydrogenase kinase-1 (PDK-1),an inhibitor of pyruvate dehydrogenase complex (PDH; Fig. 17.2). The consequentblock of pyruvate decarboxylation inhibits the Krebs cycle and the mitochondrialrespiratory chain.
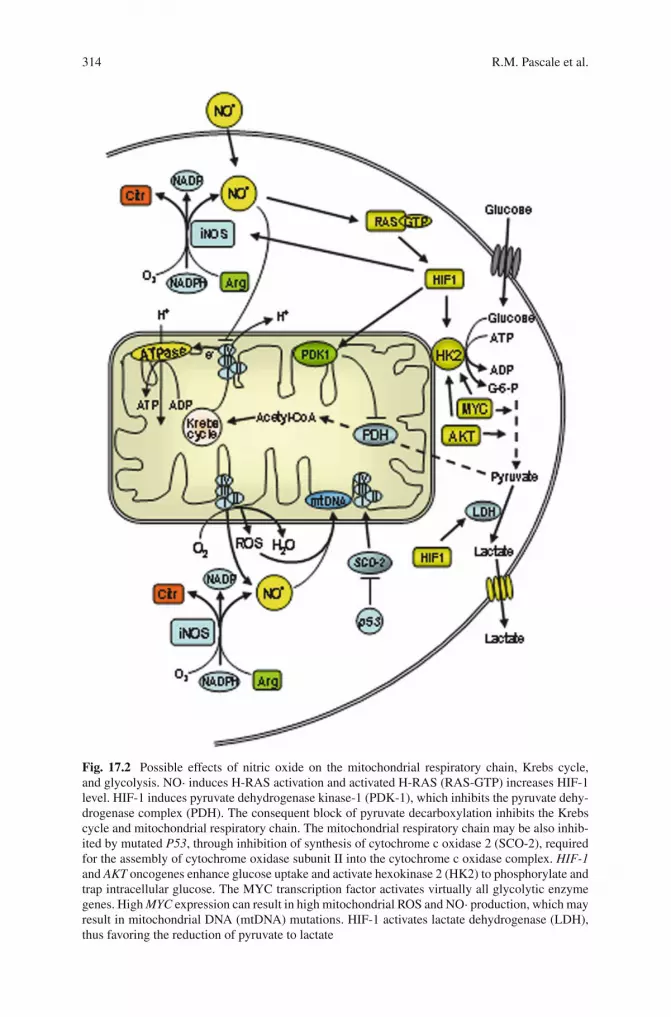
314 R.M. Pascale et al.
Fig. 17.2 Possible effects of nitric oxide on the mitochondrial respiratory chain, Krebs cycle,and glycolysis. NO· induces H-RAS activation and activated H-RAS (RAS-GTP) increases HIF-1level. HIF-1 induces pyruvate dehydrogenase kinase-1 (PDK-1), which inhibits the pyruvate dehy-drogenase complex (PDH). The consequent block of pyruvate decarboxylation inhibits the Krebscycle and mitochondrial respiratory chain. The mitochondrial respiratory chain may be also inhib-ited by mutated P53, through inhibition of synthesis of cytochrome c oxidase 2 (SCO-2), requiredfor the assembly of cytochrome oxidase subunit II into the cytochrome c oxidase complex. HIF-1and AKT oncogenes enhance glucose uptake and activate hexokinase 2 (HK2) to phosphorylate andtrap intracellular glucose. The MYC transcription factor activates virtually all glycolytic enzymegenes. High MYC expression can result in high mitochondrial ROS and NO· production, which mayresult in mitochondrial DNA (mtDNA) mutations. HIF-1 activates lactate dehydrogenase (LDH),thus favoring the reduction of pyruvate to lactate

17 Prognostic Significance of iNOS in Hepatocellular Carcinoma 315
The mitochondrial respiratory chain may be also inhibited by mutated P53,through inhibition of synthesis of cytochrome c oxidase 2 (SCO-2; Fig. 17.2).SCO-2 is required for the assembly of the COXII subunit (cytochrome oxidase sub-unit II) into the cytochrome c oxidase complex, which is integral to the respiratorychain (Leary et al. 2007). Notably, NO• was shown to induce accumulation of wild-type p53 protein in colon cancer (Hussain and Harris 2006). This could inhibit cellproliferation, thus favoring the selection of tumor cells with mutated P53. In P53mutants iNOS increases VEGF expression and promotes tumor growth, suggestingthat the activity of NO• on tumor cells may be influenced by the P53 status of thetumor (Wink et al. 2008). These observations are of particular interest for HCC inwhich frequent P53 mutations occur as an early event in areas with AFB1 exposureand as a late event in HCC associated with viral infections of other etiologies (Teufelet al. 2007).
Several oncogenes have been implicated in the Warburg effect (Fig. 17.2).The AKT oncogene, encoding a protein serine–threonine kinase, enhances glucoseuptake and activates hexokinase 2 (HK2) to phosphorylate and trap intracellu-lar glucose (Elstrom et al. 2004; Fig. 17.2). Overexpression of AKT is associatedwith glycolytic flux without affecting mitochondrial oxidative phosphorylation,thereby presumably contributing to the Warburg effect. The MYC oncogene, whichis activated in several cancer types, including HCC (Calvisi et al. 2007a), encodesa transcription factor which activates virtually all glycolytic enzyme genes anddirectly binds numerous glycolytic genes, including those encoding HK2, enolase,and lactate dehydrogenate-A (Kim and Dang 2005). MYC is involved in mitochon-drial biogenesis which, when sustained by high MYC levels, can result in highmitochondrial ROS and NO• production, which may induce mtDNA mutations thatin turn contribute to dysfunctional mitochondria (Kim and Dang 2006; Fig. 17.2).
So far, the relationships between NO• availability, cell metabolism, and cancerprogression have been prevalently evaluated in extrahepatic tumors. On the basisof these studies and of the observation that HCCs overexpress iNOS, MYC, AKT,and HIF-1 (see below), it may be reasonably suggested that NO• overproduction,in different stages of hepatocarcinogenesis, is likely to contribute to the progres-sive utilization of glycolysis as a major energy source, which may facilitate theprogression of HCC cells (Weber et al. 1977).
iNOS and Signal Transduction Pathways
Although the role of NO• has been studied in depth in inflammatory cells, chronicinflammation, oxidative damage, usually associated with viral hepatitis and HCC,and elevated NO• plasma levels are present in patients with cirrhosis and HCC(Moriyama et al. 2000). The regulation of NO• production and interactions of iNOSwith signaling pathways in hepatocarcinogenesis remain poorly investigated.
Cyclooxygenase type 2 (COX2) is an important mediator of inflammationinvolved in prostaglandin synthesis (Fig. 17.3). Arachidonic acid, released from

316 R.M. Pascale et al.
Fig. 17.3 Interplay betweenCOX2 and iNOS. Varioussignals can activate COX2and iNOS expression.Prostaglandins (PGs) and NOcontribute to theinflammatory processes.dPGJ2 (through PPAR-γ) andPGE2 decrease iNOSexpression. NO may increaseCOX2 activity and favors PGproduction by inducing therelease of arachidonic acid asa consequence of membranelipid peroxidation.Nevertheless, possibledecrease in COX2 by NO hasalso been reported. PGs favorcell growth by activatinggrowth factor receptors, suchas EGFR and MAPKsignaling
membrane phospholipids by phospholipase A2, is metabolized by cyclooxygenasesto prostaglandin H2 (PGH2) that is subsequently converted to various prostanoidsby specific synthases (Weinberg 2000; Wu 2006). PGE2, the major prostaglandin inhepatocytes, after release in the extracellular space binds the membrane receptorsEP1, EP2, EP3, and EP4, coupled with G proteins, on the same and neighboringcells. dPGJ2 (through PPAR-γ) and PGE2 induce a decrease in iNOS expression,and NO• may either increase or decrease COX2 activity and prostaglandin produc-tion (Weinberg 2000). These findings indicate the existence of a cross talk betweenCOX-2 and iNOS pathways in hepatocarcinogenesis, as also supported by the obser-vation that COX-2 inhibitors block NO• production in HCC cells (Fantappiè et al.2002). Furthermore, NO• and its derivatives may influence prostaglandin productionby inducing lipid peroxidation and arachidonic acid release from cell membranes.Stimulation of the EP2 receptor by PGE2 induces the association of the α subunitof the regulator G protein signaling and AXIN (Castellone et al. 2005). This leadsto the inactivation of glycogen synthase kinase-3β (GSK-3β), a downstream effec-tor of the WNT pathway, with consequent nuclear accumulation of β-catenin andincrease in its transcriptional targets c-MYC, C-JUN, and CYCLIN D1. PGE2 canalso stimulate cell growth through the activation of several tyrosine kinase receptors,including EGFR. This results in GRB2 and SOS phosphorylation and RAS activa-tion, which drives the mitogen-activated protein kinase (MAPK) pathway leadingto extracellular signal-regulated kinases 1 and 2 (ERK1/2; Pai et al. 2002). Finally,

17 Prognostic Significance of iNOS in Hepatocellular Carcinoma 317
PGE2 can induce actin polymerization and epithelial mesenchymal transition, lead-ing to increase in cancer metastasis through the activation of the PI3K/Akt pathway(Sheng et al. 2001).
The mechanisms described above, involved in tumor progression and forma-tion of metastases, have been mainly explored in gastrointestinal cancer. However,Rahman et al. (2001) showed, by immunohistochemical analysis, that the expressionof both COX-2 and iNOS are significantly higher in hepatitis C virus (HCV)-positive HCCs and the COX-2 expression level was significantly correlated withiNOS expression and microvascular density (MVD). The combined negative expres-sion of iNOS and COX-2 had a significant impact on patient survival, suggesting animportant role in the prognosis of HCV-positive HCC patients. In addition, EGFR isexpressed in human HCC and is a target of anticancer compounds (Hung et al. 1993;Furuse 2008). EGFR activation upregulates COX-2 expression, thereby enhancingPGE2 production (Han et al. 2006). On the other hand, PGE2 transactivates EGFRin human HCC cells, and this effect is mediated by the EP1 receptor and involvesthe c-Src protein. These findings and the observation of iNOS upregulation in HCC(Simile et al. 2005; Calvisi et al. 2008a) envisage the existence of a scenario in HCCimplying a contribution of NO• overproduction to the growth and progression of thistumor.
Other signaling pathways are involved in cross talks with iNOS, during hepato-carcinogenesis. iNOS, nuclear factor-κB (NF-κB), RAS, and ERK are upregulatedin preneoplastic rat liver lesions (Simile et al. 2005; Calvisi et al. 2008a), dysplas-tic and neoplastic liver from c-Myc-TGF-α transgenic mice (Calvisi et al. 2004b),and human HCCs (Ikeguchi et al. 2002; Sun et al. 2005). Recent research in ourlaboratory (Calvisi et al. 2008a) showed the existence of an interplay betweenthe iNOS and the inhibitor of κB kinase (IKK)/NF-κB and RAS/ERK pathwaysin HCC (Fig. 17.4). The interactions are always highest in the most aggressivepreneoplastic and neoplastic liver lesions of the genetically susceptible F344 rats,compared to the resistant BN rats, and of c-Myc-TGF-α transgenic mice much proneto hepatocarcinogenesis than TGF-α transgenic mice. Furthermore, the determina-tion of iNOS expression in human HCC showed highest values in a subtype withpoorer prognosis (based on the length of survival after partial liver resection) com-pared to a subtype with better prognosis. The suppression of iNOS signaling byaminoguanidine (Misko et al. 1993) in c-Myc/TGF-α mice and human HCC celllines resulted in significant reduction in HCC growth and NF-κB and RAS/ERKexpression and increase in apoptosis (Calvisi et al. 2008a). In contrast, NO• produc-tion by glyco-S-nitroso-N-acetylpenicillamine 2 (Glyco-Snap-2) inhibited apoptosisof in vitro growing human HCC cells. Conversely, the block of NF-κB signalingby sulfasalazine (Favata et al. 1998) or siRNA, or ERK signaling by the MAPKkinase (MEK) inhibitor UO126 (Weber et al. 2000), caused iNOS downregulationin HCC cell lines. In transgenic mice and human HCC cell lines, iNOS antiapop-totic effect seems to be mediated by the NF-κB cascade. The latter induces variousantiapoptotic proteins, such as Bcl-2-related protein, the long isoform (BCL-xL),the inhibitor of apoptosis, X-linked (XIAP), and the inhibitor of apoptosis protein 1(cIAP-1), and inhibits the proapoptotic Jun NH2 terminal kinase (JNK; Nakano et al.

318 R.M. Pascale et al.
Fig. 17.4 Schematic representation of the iNOS interplay with IKK/NF-κB and Ha-RAS/ERKsignaling cascades. iNOS activates IKK which allows proteasomal degradation of the NF-κBinhibitor, IkB-α, resulting in NF-κB activation. iNOS also activates H-RAS, thus triggering theMAPK cascade which leads to ERK activation. The activation of NF-κB by active ERK mayoccur through AURKA, which inhibits IκB-α. Thus, iNOS interplay with IKK/NF-κB and Ha-RAS/ERK cascades results in activation of genes controlling cell growth and angiogenesis andinhibiting apoptosis. Pointed and blunt arrows indicate activation and inhibition, respectively. Thedotted arrow indicate hypothetical activation
2006). Accordingly, iNOS suppression by aminoguanidine caused downregulationof NF-κB and antiapoptotic proteins, and upregulation of pJNK, in c-Myc/TGF-αand HCC cell lines (Calvisi et al. 2008a). However, these findings cannot excludethe contribution of other mechanisms to the antiapoptotic action of iNOS.
These observations assign a role to iNOS upregulation in the control of the prolif-erative phenotype of preneoplastic and neoplastic liver cells through the activationof the IKK/NF-κB axis (Fig. 17.4). They also imply a cross talk between iNOSand H-RAS/ERK. The mechanism of NF-κB regulation by pERK1/2 is still unclear.The pERK1/2 contribution to NF-κB upregulation through direct activation of iNOSis still unproved. According to recent observations, pERK1/2 activates AURORA-A(AURKA), which in turn may activate NF-κB through inhibition of the inhibitor ofκB (IKB-α) (Briassouli et al. 2007; Fig. 17.4).
Interestingly, correlative experiments showed that iNOS levels are directly cor-related with genomic instability, assessed by random amplified polymorphic DNAanalysis methodology, proliferation rate, and MVD of HCC, and inversely corre-lated with apoptosis and patients’ survival (Calvisi et al. 2008a). These observationsand the presence of highest expression of iNOS and its downstream targets in moreaggressive rodents and human HCC suggest that iNOS upregulation and changes iniNOS/NF-κB and iNOS/H-RAS/ERK cross talks are prognostic markers for HCC.These results agree with the observation of a significant association of iNOS and

17 Prognostic Significance of iNOS in Hepatocellular Carcinoma 319
metalloproteinase-9 expression with HCC recurrence (Sun et al. 2005), and iNOSoverexpression with poor prognosis for gastric cancer (Li and Xu 2005), adenoidcystic carcinoma of salivary glands (Zhang et al. 2005), fibrous histiocytoma (Hokiet al. 2007), and colorectal cancer (Cianchi et al. 2004). These iNOS effects couldat least, in part, depend on its angiogenic properties and are intensified by COX2upregulation (Rahman et al. 2001; Cianchi et al. 2004). However, no correlationbetween iNOS overexpression and prognosis has been reported for pancreatic andovarian tumors (Kong et al. 2002; Ozel et al. 2006). Furthermore, iNOS ablationdid not prevent hepatocarcinogenesis induced by a choline-deficient, l-amino acid-deficient diet in mice (Denda et al. 2007), suggesting a relatively minor role of iNOSsignaling. In this model of hepatocarcinogenesis, high production of lipid perox-ides in hepatocyte nuclei (Ghoshal 1995) may cause DNA damage and contributeto HCC development via generation of genomic instability in a iNOS-independentmanner. The contribution of iNOS overexpression to growth deregulation in pre-neoplastic and neoplastic liver cells through a cross talk with Ha-RAS/ERK andIKK-NF-κB axis does not exclude per se the activation of iNOS signaling by othermechanisms, such as inflammatory cytokines or the Wnt/β-catenin signaling (Duet al. 2006). Indeed, IL-1b, IL-6, TNF-α, and IFN-γ expressions are elevated in thedysplastic and neoplastic livers of transgenic mice, with highest values for TNF-α and IFN-γ, in double-transgenic HCC. Rise in cytokine expression also occursin human HCC and surrounding non-tumorous liver, with highest values in HCCwith poorer prognosis, for IL-1b, IL-6, and TNF-α (Calvisi et al. 2008a). However,the role of Wnt/β-catenin signaling in iNOS upregulation seems to be unlikelydue to the observation of equal β-catenin activation (nuclear localization) in HCCsfrom both F344 and BN rats (Frau, unpublished data) expressing sharply differentiNOS mRNA levels. β-catenin activation also occurs in a lower percentage of HCCcells from c-Myc/TGF-α than TGF-α transgenics (12 vs 30%; Calvisi et al. 2004a),although highest iNOS expression occurs in HCC from double transgenic mice.
According to recent research (Ying et al. 2007), NO• induces pRb hyperphospho-rylation apparently through the soluble guanylyl cyclase/cGMP/cGMP-dependentprotein kinase (sGC/cGMP/PKG) signaling pathway, during chronic inflamma-tion in the mouse colitis model. Some evidence was presented indicating that theeffect of sGC/cGMP on pRb phosphorylation is dependent on the MEK/ERK andPI3K/AKT pathways. These results reveal a role of sGC/cGMP/PKG signalingin cell cycle control, through downstream MEK/ERK and PI3K/AKT pathways.The links between sGC/cGMP/PKG signaling and MEK/ERK and PI3K/AKT sig-naling are not completely clear and should be the object of future work. pRbhyperphosphorylation occurs in preneoplastic and neoplastic rat and human liverlesions and is correlated to HCC aggressiveness (Pascale et al. 2002; Pascaleet al. 2005). Moreover, the MEK/ERK and PI3K/AKT pathways are upregulatedin HCCs (Tanaka et al. 2006). However, the interplay between NO• production andthe sGC/cGMP/PKG pathway has not been explored in hepatocarcinogenesis andthe possibility that other mechanisms are responsible for the changes in the pRbphosphorylation and the MEK/ERK and PI3K/AKT pathways in HCC cannot beexcluded.

320 R.M. Pascale et al.
NO Interference with SAM Synthesis and DNA Methylation
Early appearance of global DNA hypomethylation, associated with promoter hyper-methylation and inactivation of oncosuppressor genes, is a common feature ofhepatic carcinogenesis in rodents (Pascale et al. 1991; Teufel et al. 2007). Thisabnormality could induce genomic instability in cells throughout the different stepsof hepatic carcinogenesis and facilitate the occurrence of irreversible gene changes(Shen et al. 1994; Kim et al. 1997; Kanai et al. 1999). A body of evidence indicatesthe existence in preneoplastic and neoplastic rat liver lesions and human cirrho-sis and HCC of marked decrease in S-adenosyl-l-methionine (SAM) and SAM:S-adenosylhomocysteine (SAH) ratio, associated with global DNA hypomethyla-tion (Garcea et al. 1989; Pascale et al. 1992; Wainfan and Poirier 1992). Decreasein liver SAM content has been attributed to changes in the methionine adenosyl-transferase (MAT) isozyme pattern. In mammals, the MAT1A gene, expressed onlyin liver, encodes MATI/III isozymes, whereas widely expressed MAT2A encodesMATII isozyme (Avila et al. 2000). In response to liver injury or during repara-tive growth, MAT1A is down-regulated whereas the Mat2A, induced by NF-κB, isswitched on (Huang et al. 1998; Yang et al. 2003). Fall in MAT1A expression withconcomitant upregulation of MAT2A also occurs in hepatoma cell lines and rodentHCC as well as in human liver cirrhosis and HCC (Cai et al. 1998; Mato et al.2002). Mice lacking Mat1A show reduced SAM liver content and spontaneouslydevelop HCC (Lu et al. 2002). Differential expression of MAT1A and MAT2A genesinfluences DNA methylation and growth of human HCC (Cai et al. 1998; Avilaet al. 2000; Calvisi et al. 2007b). There is evidence that SAM interferes with theactivity of various genes and proteins. SAM treatment inhibits the expression ofc-myc and c-H-Ras of neoplastic liver nodules in rats (Garcea et al. 1989) andlipopolysaccharide-induced TNF-α and iNOS expression in rat liver and RAW 264.7cell line (Majano et al. 2001; Veal et al. 2004), and SAM binding to cystathionineβ-synthase stabilizes this protein against degradation (Prudova et al. 2006). MAT1Adownregulation in precancerous cells may contribute to maintain active NF-κB(Fig. 17.5). Since SAM enhances the synthesis of the NF-κB inhibitor, IκB-α, prob-ably by targeting IkB-α gene (Majano et al. 2001; Simile et al. 2005), low MatI/IIIactivity and SAM content in precancerous liver should contribute to the observedrelatively low levels of NF-κB/IkB-α complex as well as to NF-κB activation andoverexpression of genes targeted by the nuclear factor, such as c-myc, cyclin D1,iNos, and Vegf-A (Simile et al. 2005; Calvisi et al. 2008a). On the other hand, trans-activation of iNos by NF-κB (Majano et al. 2001) and iNos over-activity in preneo-plastic lesions (Calvisi et al. 2008a) should result in NO• overproduction that mayinhibit hepatocyte MatI/III and SAM production (Martinez-Chantar et al. 2002).Furthermore, NO• activates IκB kinase with consequent IκB-α phosphorylation andubiquitination (Zingarelli et al. 2002). Thus, NO• overproduction can contribute, bymodulating SAM level and IκB kinase activity, to decrease in MatI/III activity andincrease in NF-κB level (Fig. 17.5). NO• may also interfere with SAM metabolismby inhibiting 5,10-methyltetrahydrofolate reductase (Danishpajooh et al. 2001),the enzyme that catalyzes the synthesis of methionine from homocysteine and

17 Prognostic Significance of iNOS in Hepatocellular Carcinoma 321
Fig. 17.5 Effect of nitric oxide on the synthesis of methionine and S-adenosylmethionine andmethylation reactions. NO inhibits methyltetrahydrofolate reductase (MTR). This results in adecrease in tetrahydrofolate (FH4) and methionine. Additional reduction in the FH4 level mayoccur by the NO-induced oxidation of ferritin, a compound that inhibits the proteasomal degrada-tion of FH4. NO affects SAM synthesis not only by inducing a decrease in methionine synthesisbut also by directly inhibiting the liver-specific methyl-thioadenosyltransferase I/III (MATI/III)isozymes. The fall in SAM level cannot be fully compensated by an increase in the extrahepaticisozyme MATII, since this enzyme is inhibited by its reaction product. The reduction in homocys-teine utilization for methionine synthesis may result in homocysteine accumulation. This probablydoes not lead to a consistent rise in cystathionine and reduced glutathione synthesis, due to areduced stabilization of cystathionine β-synthase (CBS) by SAM. Consequently, an increase inSAH, associated with a decrease in the SAM/SAH ratio, inhibits methyltransferases (MT) andDNA methylation. The reduction in SAM level may decrease IκBα activation, thus favoring NF-κBactivity
5-methyltetrahydrofolate (Fig. 17.5). This may result in decrease in methionine andSAM levels, homocysteine accumulation, and reduction in the SAM/SAH ratio.Altogether, the above observations are of particular interest in light of the recentfindings indicating that the extent of DNA hypomethylation and genomic instabilityare prognostic markers for human HCC (Calvisi et al. 2007b).
Concluding Remarks
The specific roles of NO• in carcinogenesis have not yet been completely under-stood. This, at least, in part, depends on the fact that NO• effects are largelyinfluenced by its concentration, interaction with other free radicals, metal ions andproteins, and cell type and genetic background (Hussain and Harris 2006). Althoughthe results of studies on iNOS regulation and implication in hepatocarcinogenesisare still fragmentary, they depict a complex, although incomplete, scenario in which

322 R.M. Pascale et al.
the production of NO• and its derivatives insufficient to kill initiated hepatocytesis likely to affect HCC development and progression. A body of evidence indicatesthat NO• and its by-products may influence the oxidative metabolism of cancer cells,by inhibiting some key reactions of the respiratory chain, and contributing to theprogressive adaptation of cancer cells to prevalent production of glycolytic ATPand survival in hypoxic conditions. This situation could favor the progression oftransformed hepatocytes (Kim and Dang 2006).
The interplay between iNOS and signal transduction pathways is an importantaspect of the role of NO• in carcinogenesis. In vivo studies in both rodents andhumans have discovered situations characterized by NF-κB and ERK activation,prostaglandin overproduction, and interference with SAM metabolism, which mayfavor tumor cell growth and progression. Notably, highest deregulation of the inter-play of iNOS with signaling pathways involved in cell growth control occurs in moreaggressive human and experimental HCCs. Accordingly, a strong, direct correlationof iNOS expression with proliferation rate, MVD, and genomic instability has beenfound in human HCCs (Calvisi et al. 2008a). An inverse correlation occurs betweeniNOS expression and apoptosis and patients’ survival after partial liver resection.These clearly indicate that iNOS expression is potentially a prognostic marker forhuman HCC.
The association of the block of iNOS signaling by a specific inhibitor such asaminoguanidine with a consistent decrease in HCC growth in c-Myc/TGF-α trans-genic mice, and decrease in growth and increase in apoptosis in human HCC celllines (Calvisi et al. 2008a), suggests that the key components of this pathway couldrepresent therapeutic targets that may contribute to create networked biological ther-apies. Selective iNOS inhibitors have chemopreventive effects in rodent models ofcolorectal and esophageal carcinogenesis (Rao et al. 2002; Hagos et al. 2007; Stoneret al. 2007). Combined utilization of the iNOS inhibitor SC-51 and the COX-2inhibitor celecoxib appears more effective than either agent alone in suppressingcolonic aberrant crypt foci formation in the azoxymethane rat model of colon car-cinogenesis (Rao et al. 2002). The effect of these inhibitors, alone or in combination,has not been evaluated in hepatic carcinogenesis.
Another important aspect of the deregulation of iNOS signaling in hepatocar-cinogenesis is the demonstration that the susceptibility to hepatocarcinogenesis,either dependent on susceptibility genes or genomic engineering, influences theiNOS-linked signaling in liver preneoplastic and neoplastic lesions (Calvisi et al.2008a). The genetically resistant phenotype is characterized by the incapacity ofearly preneoplastic lesions to acquire autonomous growth and progress to HCC(Pascale et al. 2005). Autonomous growth of the lesions is supported, in suscep-tible F344 rats, by the deregulation of the cell cycle (Pascale et al. 2002; Pascaleet al. 2005) and the Ras/Erk pathway (Calvisi et al. 2008b; Calvisi et al. 2008c).These alterations are limited or absent in the lesions of BN-resistant rats. iNOSupregulation occurs in early stages of hepatocarcinogenesis in both F344 and BNrats. However, while iNOS protein expression sharply and progressively increasesin nodules and HCC, it undergoes lower changes in slow-growing nodules and HCCof BN rats. A link between fast growth and signaling deregulation characterizes

17 Prognostic Significance of iNOS in Hepatocellular Carcinoma 323
human HCC with poor prognosis, whereas HCC with better prognosis behaves asthe lesions of resistant rats (Pascale et al. 2005; Calvisi et al. 2008a; Calvisi et al.2008c). This does not necessarily imply a genetic regulation of signaling path-ways in humans like that found in rodents, in which polygenic inheritance withseveral low-penetrance genes and a main gene regulates the genetic predisposi-tion to HCC (Feo et al. 2006). Even if a genetic model, similar to that of rodents,can influence human hepatocarcinogenesis, further studies are needed to clarify theinfluence of susceptibility genes on signaling pathways supporting tumor growthand progression in humans.
References
Avila, M.A., Berasain, C., Torres, L., Martin-Duce, A., Corrales, F.J., Yang, H, Prieto, J., Lu,S.C., Cavalleria, J., Rodes, J., and Mato, J.M. (2000). Reduced mRNA abundance of themain enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellularcarcinoma. J. Hepatol. 33, 907–914.
Beckman, J.S. and Koppenol, W.H. (1996). Nitric oxide, superoxide, and peroxynitrite: the good,the bad, and the ugly. Am. J. Physiol. 271, C1424–C1437.
Biegon, A., Alvarado, M., Budinger, T.F., Grossman, R., Hensley, K., West, M.S., Kotake, Y.,Ono, M., and Floyd, R.A. (2002). Region-selective effects of neuroinflammation and antioxi-dant treatment on peripheral benzodiazepine receptors and NMDA receptors in the rat brain. J.Neurochem. 82, 924–934.
Briassouli, P., Chan, F., Savage, K., Reis-Filho, J.S., and Linardopoulos, S. (2007). Aurora-A reg-ulation of nuclear factor-kappaB signaling by phosphorylation of IkappaBalpha. Cancer Res.67, 1689–1695.
Brown, G.C. (2001). Regulation of mitochondrial respiration by nitric oxide inhibition ofcytochrome c oxidase. Biochim. Biophys. Acta. 1504, 46–57.
Bruix, J., Boix, L., Sala, M., and Llovet, J.M. (2004). Focus on hepatocellular carcinoma. CancerCell 5, 215–219. [PMID: 15050913]
Cai, J., Mao, Z., Hwang, J.J., and Lu, S.C. (1998). Differential expression of methionine adenosyl-transferase genes influences the rate of growth of human hepatocellular carcinoma cells. CancerRes. 58, 1444–1450.
Calvisi, D.F., Factor, V.M., Ladu, S., Conner, E.A., and Thorgeirsson, S.S. (2004a). Disruption ofbeta-catenin pathway or genomic instability define two distinct categories of liver cancer intransgenic mice. Gastroenterology 126, 1374–1386.
Calvisi, D.F., Ladu, S., Hironaka, K., Factor, V.M., and Thorgeirsson, S.S. 2004b. Vitamin E down-modulates iNOS and NADPH oxidase in c-Myc/TGF-alpha transgenic mouse model of livercancer. J. Hepatol. 41, 815–822.
Calvisi, D.F., Pascale, R.M., and Feo, F. (2007a). Dissection of signal transduction pathways as atool for the development of targeted therapies of hepatocellular carcinoma. Rev. Recent Clin.Trials. 2, 217–236.
Calvisi, D.F., Pinna, F., Ladu, S., Pellegrino, R., Muroni, M.R., Simile, M.M., Frau, M., Tomasi,M.L., De Miglio, M.R., Seddaiu, M.A., Daino, L., Sanna, V., Feo, F., and Pascale, R.M.(2008a). Aberrant iNOS signaling is under genetic control in rodent liver cancer and potentiallyprognostic for the human disease. Carcinogenesis 29, 1639–1647.
Calvisi, D.F., Pinna, F., Ladu, S., Pellegrino, R., Sanna, V., Sini, M., Daino, L., Simile, M.M.,De Miglio, M.R., Frau, M., Tomasi, M.L., Seddaiu, M.A., Muroni, M.R., Feo, F., and Pascale,R.M. (2008b). Ras-driven proliferation and apoptosis signaling during rat liver carcinogenesisis under genetic control. Int. J. Cancer 123, 2057–2064.

324 R.M. Pascale et al.
Calvisi, D.F., Pinna, F., Meloni, F., Ladu, S., Pellegrino, R., Sini, M., Daino, L., Simile, M.M., DeMiglio, M.R., Virdis, P., Frau, M., Tomasi, M.L., Seddaiu, M.A., Muroni, M.R., Feo, F., andPascale, R.M. (2008c). Dual-specificity phosphatase 1 ubiquitination in extracellular signal-regulated-kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res.68, 4192–4200.
Calvisi, D.F., Simile, M.M., Ladu, S., Pellegrino, R., De Murtas, V., Pinna, F., Tomasi, M.L., Frau,M., Virdis, P., De Miglio, M.R., Muroni, M.R., Pascale, R.M., and Feo, F. (2007b). Alteredmethionine metabolism and global DNA methylation in liver cancer: relationship with genomicinstability and prognosis. Int. J. Cancer. 121, 2410–2420.
Castellone, M.D., Teramoto, H., Williams, B.O., Druey, K.M., and Gutkind, J.S. (2005).Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signalingaxis. Science 310, 1504–1510.
Chen, C., Pore, N., Behrooz, A., Ismail-Beigi, F., and Maity, A. (2001). Regulation of glut1 mRNAby hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J. Biol. Chem. 276,9519–9525.
Cianchi, F., Cortesini, C., Fantappiè, O., Tesserini, L., Sardi, I., Lasagna, N., Perna, F., Fabbroni,V., Di Felice, A., Perigli, G., Mozzanti, R., and Masini, E. (2004). Cyclooxygenase-2 activationmediates the proangiogenic effect of nitric oxide in colorectal cancer. Clin. Cancer Res. 10,2694–2704. Clin. Chim. Acta. 296, 181–191.
Danishpajooh, I.O., Gudi, T., Chen, Y., Kharitonov, V.G., Sharma, V.S., and Boss, G.R. (2001).Nitric oxide inhibits methionine synthase activity in vivo and disrupts carbon flow through thefolate pathway. J. Biol. Chem. 276, 27296–27303.
Demple, B. and Harrison, L. (1994). Repair of oxidative damage to DNA: enzymology and biology.Ann. Rev. Biochem. 63, 915–948.
Denda, A., Kitayama, W., Kishida, H., Murata, N., Tamura, K., Kusuoka, O., Tsutsumi, M.,Nishikawa, F., Kita, E., Nakae, D., Konishi, Y., and Kuniyasu, H. (2007). Expression ofinducible nitric oxide (NO) synthase but not prevention by its gene ablation of hepatocarcino-genesis with fibrosis caused by a choline-deficient, L-amino acid defined diet in rats and mice.Nitric Oxide 16, 164–176.
Du, Q., Park, K.S., Guo, Z., He, P., Nagashima, M., Shao, L., Sahai, R., Geller, D.A., and Hussain,S.P. (2006). Regulation of human nitric oxide synthase 2 expression by Wnt beta-cateninsignaling. Cancer Res. 66, 7024–7031.
Elstrom, R.L., Bauer, D.E., Buzzai, M., Karnauskas, R., Harris, M.H., Plas, D.R., Zhuang, H.,Cinalli, R.M., Alav, A., Rudin, C.M., and Thompson, C.B. (2004). Akt stimulates aerobicglycolysis in cancer cells. Cancer Res. 64, 3892–3899.
Fang, Y.Z., Yang, S., and Wu, G. (2002). Free radicals, antioxidants, and nutrition. Nutrition 18,872–879.
Fantappié, O., Masini, E., Sardi, I., Raimondi, L., Bani, D., Solazzo, M., Vannacci, A., andMazzanti, R. (2002). The MDR phenotype is associated with the expression of COX-2 andiNOS in a human hepatocellular carcinoma cell line. Hepatology 35, 843–852.
Farazi, P.A. and DePinho, R.A. (2006). Hepatocellular carcinoma pathogenesis: from genes toenvironment. Nat. Rev. Cancer 6, 674–678.
Favata, M.F., Horiuchi, K.Y., Manos, E.J., Daulerio, A.J., Stradley, D.A., Feeser, W.S., Van Dyk,D.E., Pitts, W.J., Earl, R.A., Hobbs, F., Copeland, R.A., Magolda, R.L., Scherle, P.A., andTrzaskos, J.M. (1998). Identification of a novel inhibitor of mitogen-activated protein kinasekinase. J. Biol. Chem. 273, 18623–18632.
Feitelson, M.A., Sun, B., Satiroglu Tufan, N.L., Liu, J., Pan, J., and Lian, Z. (2002). Geneticmechanisms of hepatocarcinogenesis. Oncogene 21, 2593–2604.
Feo, F., Canuto, R.A., and Garcea, R. (1973). Acceptor control ratio of mitochondria. Factorsaffecting it in Morris hepatoma 5123 and Yoshida hepatoma AH-130. Eur. J. Cancer 9,203–214.
Feo, F., De Miglio, M.R., Simile, M.M., Muroni, M.R., Calvisi, D.F., Frau, M., andPascale, R.M. (2006). Hepatocellular carcinoma as a complex polygenic disease. Interpretive

17 Prognostic Significance of iNOS in Hepatocellular Carcinoma 325
analysis of recent developments on genetic predisposition. Biochim. Biophys. Acta. 1765,126–167.
Furuse, J. (2008). Growth factors as therapeutic targets in HCC. Crit. Rev. Oncol. Hematol. 67,8–15.
Garcea, R., Daino, L., Pascale, R.M., Simile, M.M., Puddu, M., Ruggii, M.E., Seddaiu, M.A.,Satta, G., Sequenza, M.J., and Feo, F. (1989). Protooncogene methylation and expression inregenerating liver and preneoplastic liver nodules induced in the rat by diethylnitrosamine:effect of variations of S-adenosylmethionine:S-adenosylhomocysteine ratio. Carcinogenesis10, 1183–1192.
Ghoshal, A.K. (1995). New insight into the biochemical pathology of liver in choline deficiency.Crit. Rev. Biochem. Mol. Biol. 30, 263–273.
Hagos, G.K., Carroll, R.E., Kouznetsova, T., Li, Q., Toader, V., Fernandez, P. A., Swanson, S.M.,and Thatcher, G.R. (2007). Colon cancer chemoprevention by a novel NO chimera that showsanti-inflammatory and antiproliferative activity in vitro and in vivo. Mol. Cancer Ther. 6,2230–2239.
Han, C., Michalopoulos, G., and Wu, T. (2006). Prostaglandin E2 receptor EP1 transactivatesEGFR/Met receptor tyrosine kinases and enhances invasiveness in human hepatocellularcarcinoma cells. J. Cell Physiol. 207, 261–270.
Harris, A.L. (2002). Hypoxia – a key regulatory factor in tumour growth. Nat. Rev. Cancer 2,38–47.
Hoki, Y., Hiraku, Y., Ma, N., Murata, M., Matsumine, A., Nagahama, M., Shintani, K., Uchida, A.,and Kawanishi, S. (2007). iNOS-dependent DNA damage in patients with malignant fibroushistiocytoma in relation to prognosis. Cancer Sci. 98, 163–168.
Horton, R.A., Ceppi, E.D., Knowles, R.G., and Titheradge, M.A. (1994). Inhibition of hep-atic gluconeogenesis by nitric oxide: a comparison with endotoxic shock. Biochem. J. 299,735–739.
Huang, Z.-Z., Mao, Z., Cai, J., and Lu, S.C. (1998). Changes in methionine adenosyltransferaseduring liver regeneration. Am. J. Physiol. 38, G14–G21.
Hung, W.C., Chuang, L.Y., Tsai, J.H., and Chang, C.C. (1993). Effects of epidermal growth factoron growth control and signal transduction pathways in different human hepatoma cell lines.Biochem. Mol. Biol. Int. 30, 319–328.
Hussain, S.P. and Harris, C.C. (2006). p53 biological network: at the crossroads of thecellular-stress response pathway and molecular carcinogenesis. J. Nippon Med. Sch. 73,54–64.
Ikeguchi, M., Ueta, T., Yamane, Y., Hirooka, Y., and Kaibara, N. (2002). Inducible nitric oxidesynthase and survivin messenger RNA expression in hepatocellular carcinoma. Clin. CancerRes. 8, 3131–3136.
Iwai, S., Karim, R., Kitano, M., Sukata, T., Min, W., Morimura, K., Wanibuchi, H., Seki, S., andFukushima, S. (2002). Role of oxidative DNA damage caused by carbon tetrachloride-inducedliver injury- enhancement of MeIQ-induced glutathione S-transferase placental form-positivefoci in rats. Cancer Lett. 179, 15–24.
Jüngst, C., Cheng, B., Gehrke, R., Schmitz, V., Nischalke, H.D., Ramakers, J., Schramel,P., Schirmacher, P., Sauerbruch, T., and Caselmann, W.H. (2004). Oxidative damage isincreased in human liver tissue adjacent to hepatocellular carcinoma. Hepatology 39,1663–1672.
Kanai, Y., Hui, A.M., Sun, L., Ushijima, S., Sakamoto, M., Tsuda, H., and Hirohashi, S.(1999). DNA hypermethylation at the D17S5 locus and reduced HIC-1 mRNA expression areassociated with hepatocarcinogenesis, Hepatology 29, 703–709.
Kawanishi, S., Hiraku, Y., Pinlaor, S., and Ma, N. (2006). Oxidative and nitrative DNA dam-age in animals and patients with inflammatory diseases in relation to inflammation-relatedcarcinogenesis. Biol. Chem. 387, 365–372.
Kim J.W. and Dang, C.V. (2006). Cancer’s molecular sweet tooth and the Warburg effect. CancerRes. 66, 8927–8930.

326 R.M. Pascale et al.
Kim, J.W. and Dang, C.V. (2005). Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci.30, 142–150.
Kim, Y.I., Pogribny, I.P., Basnakian, A.G., Miller, J.W., Selhub, J., James, S.J., and Mason, J. B.(1997). Folate deficiency in rats induces DNA strand breaks and hypomethylation within thep53 tumor suppressor gene. Am. J. Clin. Nutr. 65, 46–52.
Kong, G., Kim, E.K., Kim, W.S., Lee, K.T., Lee, Y.W., Lee, J.K., Paik, S.W., and Rhee, J.C.(2002). Role of cyclooxygenase-2 and inducible nitric oxide synthase in pancreatic cancer.J. Gastroenterol. Hepatol. 17, 914–921.
Leary, S.C., Cobine, P.A., Kaufman, B.A., Guercin, G.H., Mattman, A., Palaty, J., Lockitch, G.,Winge, D.R., Rustin, P., Horvath, R., and Shoubridge, E.A. (2007). The human cytochrome coxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellularcopper homeostasis. Cell Metab. 5, 9–20.
Li, L.G. and Xu, H.M. (2005). Inducible nitric oxide synthase, nitrotyrosine and apoptosis in gas-tric adenocarcinomas and their correlation with a poor survival. World J. Gastroenterol. 11,2539–2544.
Lu, S.C., Alvarez, L., Huang, Z.-Z., Chen, L., An, W., Corrales, F.J., Avila, M.T., Kanel, G., andMato, J.M. (2002). Methionine adenosyltransferase 1A knockout mice are predisposed to liverinjury and exhibit increased expression of genes involved in proliferation. Proc. Natl. Acad.Sci. USA 98, 5560–5565.
Majano, P.L., García-Monzón, C., García-Trevijano, E.R., Corrales, F.J., Cámara, J., Ortiz, P.,Mato, J.M., Avila, M.A., and Moreno-Otero, R. (2001). S-adenosylmethionine modulatesinducible nitric oxide synthase gene expression in rat liver and isolated hepatocytes. J. Hepatol.35, 692–699.
Martinez-Chantar, M.M.L., Garcıa-Trevijano, E.R., Latasa, M.U., Perez-Mmato, I., del Pino,M.M.S., Corrales, F.J., Avila, M.A., and Mato, J.M. (2002). Importance of a deficiency inS-adenosyl-L-methionine synthesis on the pathogenesis of liver injury. Am. J. Clin. Nutr. 76,1177S–1182S.
Mato J.M., Corrales, F.J., Lu, S.C., and Avila, M.A. (2002). S-Adenosylmethionine: a controlswitch that regulates liver function. FASEB J. 16, 15–26.
Misko, T.P., Moore, W.M., Kasten, T.P., Nickols, G.A., Corbett, J.A., Tilton, R.G., McDaniel,M.L., Williamson, J.R., and Currie, M.G. (1993). Selective inhibition of the inducible nitricoxide synthase by aminoguanidine. Eur. J. Pharmacol. 233, 119–125.
Moncada, S., Palmer, R.M.J., and Higgs, E.A. (1991). Nitric oxide: physiology, pathophysiology,and pharmacology. Pharmacol. Rev. 43, 109–142.
Monti, L.D., Valsecchi, G., Costa, S., Sandoli, E.P., Phan, C.V., Pontiroli, A.E., Pozza, G., andPiatti, P.M. (2000). Effects of endothelin-1 and nitric oxide on glucokinase activity in isolatedrat hepatocytes. Metabolism 49, 73–80.
Moriyama, A., Tabaru, A., Unoki, H., Abe, S., Masumoto, A., and Otsuki, M. (2000). Plasmanitrite/nitrate concentrations as a tumor marker for hepatocellular carcinoma. Clin. Chim. Acta.296, 181–291.
Nakano, H., Nakajima, A., Sakon-Komazawa, S., Piao, J.H., Xue, X., and Okumura, K. (2006).Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ.13, 730–737.
Nisoli, E., Falcone, S., Tonello, C., Cozzi, V., Palomba, L., Fiorani, M., Pisconti, A., Brunelli, S.,Cardile, A., Francolini, M., Cantoni, O., Carruba, M.O., Moncada, S., and Clementi, E. (2004).Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc.Natl. Acad. Sci. USA 101, 16507–16512.
Ozel, E., Pestereli, H.E., Simsek, T., Erdogan, G., and Karaveli, F.S. (2006). Expression ofcyclooxygenase-2 and inducible nitric oxide synthase in ovarian surface epithelial carcino-mas: is there any correlation with angiogenesis or clinicopathologic parameters? Int. J. Gynecl.Cancer 16, 549–555.
Pai, R., Soreghan, B., Szabo, I.L., Pavelka, M., Baatar, D., and Tarnawski, A.S. (2002).Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancergrowth and gastrointestinal hypertrophy. Nat. Med. 8, 289–293.

17 Prognostic Significance of iNOS in Hepatocellular Carcinoma 327
Pannen, B.H. (2002). New insights into the regulation of hepatic blood flow after ischemia andreperfusion. Anesth. Analg. 94, 1448–1457.
Pascale, R.M., Marras, V., Simile, M.M., Daino, L., Pinna, G., Bennati, S., Carta, M., Seddaiu,M.A., Massarelli, G., and Feo, F. (1992). Chemoprevention of rat liver carcinogenesis byS-adenosyl-L-methionine: a long-term study. Cancer Res. 52, 4979–4986.
Pascale, R.M., Simile, M.M., Calvisi, D.F., Frau, M., Muroni, M.R., Seddaiu, M.A., Daino, L.,Muntoni, M.D., De Miglio, M.R., Thorgeirsson, S.S., and Feo, F. (2005). Role of HSP90,CDC37, and CRM1 as modulators of P16 (INK4A) activity in rat liver carcinogenesis andand human liver cancer. Hepatology 42, 1310–1319.
Pascale, R.M., Simile, M.M., De Miglio, M.R., Muroni, M.R., Calvisi, D.F., Asara, G., Casabona,D., Frau, M., Seddaiu, M.A., and Feo. F. (2002). Cell cycle deregulation in liver lesions of ratswith and without genetic predisposition to hepatocarcinogenesis. Hepatology 35, 1341–1350.
Pascale, R.M., Simile, M.M., Satta, G., Seddaiu, M.A., Daino, L., Pinna, G., Vinci, M.A., Gaspa,L., and Feo, F. (1991). Comparative effects of L-methionine, S-adenosyl-L-methionine and5′-methylthioadenosine on the growth of preneoplastic lesions and DNA methylation in ratliver during the early stages of hepatocarcinogenesis. Anticancer Res. 11, 1617–1624.
Prudova, A., Bauman, Z., Braun, A., Vitvitsky, V., Lu, S.C., and Banerjee, R. (2006).S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity.Proc. Natl. Acad. Sci. USA 103, 6489–6494.
Rahman, M.A., Dhar, D.K., Yamaguchi, E., Maruyama, S., Sato, T., Hayashi, H., Ono, T., Yamanoi,A., Kohno, H., and Nagasue, N. (2001). Coexpression of inducible nitric oxide synthase andCOX-2 in hepatocellular carcinoma and surrounding liver: possible involvement of COX-2 inthe angiogenesis of hepatitis C virus-positive cases. Clin. Cancer Res. 7, 1325–1332.
Rao, C.V., Indranie, C., Simi, B., Manning, P.T., Connor, J.R, and Reddy, B.S. (2002).Chemopreventive properties of a selective inducible nitric oxide synthase inhibitor in colon car-cinogenesis, administered alone or in combination with celecoxib, a selective cyclooxygenase-2inhibitor. Cancer Res. 62, 165–170.
Semenza, G.L. (2003). Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732.Shen, J.C. Rideout III, W.M., and Jones, P.A. (1994). The rate of hydrolytic deamination of
5-methylcytosine in double-stranded DNA. Nucl. Acids Res. 25, 972–976.Sheng, H., Shao, J., Washington, M.K., and DuBois, R.N. (2001). Prostaglandin E2 increases
growth and motility of colorectal carcinoma cells. J. Biol. Chem. 276, 18075–18081.Shibutani, S., Takeshita, M., and Grollman, A.P. (1991). Insertion of specific bases during DNA
synthesis past the oxidation-damaged base 8-oxodG. Nature 31(349), 431–434.Simile, M.M., Pagnan, G., Pastorino, F., Brignole, C., De Miglio, M.R., Muroni, M.R., Asara,
G., Frau, M., Seddaiu, M.A., Calvisi, D.F., Feo, F., Ponzoni, M., and Pascale, R.M.(2005). Chemopreventive N-(4-hydroxyphenyl)retinamide (fenretinide) targets deregulatedNF-κB and Mat1A genes in the early stages of rat liver carcinogenesis. Carcinogenesis 26,417–427.
Sprangers, F., Sauerwein, H.P., Romijn, J.A., van Woerkom, G.M., and Meijer, A.J. (1998). Nitricoxide inhibits glycogen synthesis in isolated rat hepatocytes. Biochem. J. 330, 1045–1049.
Stadler, J., Barton, D., Beil-Moeller, H., Diekmann, S., Hierholzer, C., Erhard, W., and Heidecke,C.D. (1995). Hepatocyte nitric oxide biosynthesis inhibits glucose output and competes withurea synthesis for l-arginine. Am. J. Physiol. 268, G183–G188.
Stoner, G.D., Wang, L.S., and Chen, T. (2007). Chemoprevention of esophageal squamous cellcarcinoma. Toxicol. Appl. Pharmacol. 224, 337–349.
Sun, M.H. Han, X.C., Jia, M.K., Jiang, W.D., Wang, M., Zhang, H., Han, G., and Jiang, Y. (2005).Expressions of inducible nitric oxide synthase and matrix metalloproteinase-9 and their effectson angiogenesis and progression of hepatocellular carcinoma. World. J. Gastroenterol. 11,5931–5937.
Tanaka, H., Yamamoto, M., Hashimoto, N., Miyakoshi, M., Tamakawa, S., Yoshie, M., Tokusashi,Y., Yokoyama, K., Yaginuma, Y., and Ogawa, K. (2006). Hypoxia-independent overexpressionof hypoxia-inducible factor 1 alpha as an early change in mouse hepatocarcinogenesis. CancerRes. 66, 11263–11270.

328 R.M. Pascale et al.
Tanaka, Y., Hanada, K., Mizokami, M., Yeo, A.E., Shih, J.W., Gojobori, T., and Alter, H.J. (2002).A comparison of the molecular clock of hepatitis C virus in United States and Japan pre-dicts that hepatocellular carcinoma incidence in United States will increase over the next twodecades. Proc. Natl. Acad. Sci. USA 99, 15584–15589.
Terranova, T., Feo, F., Gravela, E., and Gabiel, L. (1964). Die wirkung der 2-desoxyglucose aufden energetischen stoffwechsel und auf proteinsynthese von tunmorzellen und normalzellen.Zeit. Krebs. 66, 41–45.
Teufel, A., Staib, F., Kanzler, S., Weinmann, A., Schulze-Bergkamen, H., and Galle, P.R. (2007).Genetics of hepatocellular carcinoma. World J. Gastroenterol. 13, 2271–2282
Thorgeirsson, S.S. and Grisham, J.W. (2002). Molecular pathogenesis of human hepatocellularcarcinoma. Nat. Genet. 31, 339–346.
Veal, N., Hsieh, C.L., Xiong, S., Mato, J.M., Lu, S., and Tsukamoto, H. (2004). Inhibitionof lipopolysaccharide-stimulated TNF-alpha promoter activity by S-adenosylmethionine and5′-methylthioadenosine. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G352–G362.
Wainfan, E. and Poirier, L.A. (1992). Methyl groups in carcinogenesis: effects on DNA methylationand gene expression. Cancer Res 2, 2071s–77s.
Warburg, O. (1956). On the origin of cancer cells. Science 123, 309–314.Weber, C.K., Liptay, S., Wirth, T., Adler, G., and Schmid, R.M. (2000). Suppression of NF-kappaB
activity by sulfasalazine is mediated by direct inhibition of IkappaB kinases alpha and beta.Gastroenterology 119, 1209–1218.
Weber, G., Kizaki, H., Shiotani, T., Tzeng, D. and Williams, J.C. (1977). The molecular correlationconcept of neoplasia: recent advances and new challenges. Adv. Exp. Med. Biol. 92, 89–116.
Weinberg, J.B. (2000). Nitric oxide synthase 2 and cyclooxygenase 2 interactions in inflammation.Immunol. Res. 22, 319–341.
Wink, D.A., Ridnour, L.A., Perwez Hussain, S., and Harris, C.C. (2008). The reemergence of nitricoxide and cancer. Nitric Oxide 19, 65–67.
Wu, G., Fang, Y.Z., Yang, S., Lupton, J.R., and Turner, N.D. (2004). Glutathione metabolism andits implications for health. J. Nutr. 134, 489–492.
Wu, G. and Morris, Jr. S.M. (1998). Arginine metabolism: nitric oxide and beyond. Biochem. J.1336, 1–17.
Wu, T. (2006). Cyclooxygenase-2 in hepatocellular carcinoma. Cancer Treat. Rev. 32, 28–44.Wu, X.Z., Xie, G.R., and Chen, D. (2007). Hypoxia and hepatocellular carcinoma: The therapeutic
target for hepatocellular carcinoma. J. Gastroenterol. Hepatol. 22, 1178–1182.Yang, H. P., Sadda, M.R., Yu, V., Zeng, Y., Lee, T.D., Ou, X.P., Chen, L.X., and Lu, S.C. (2003).
Induction of human methionine adenosyltransferase 2A expression by tumor necrosis factor a.Role of NF-κB and AP-1. J. Biol. Chem. 278, 50887–50896.
Ying, L., Hofseth, A.B., Browning, D.D., Nagarkatti, M., Nagarkatti, P.S., and Hofseth, L.J. (2007).Nitric oxide inactivates the retinoblastoma pathway in chronic inflammation. Cancer Res. 67,9286–9293.
Zhang, J., Xie, Z., Dong, Y., Wang, S., Liu, C., and Zou, M.H. (2008). Identification of nitric oxideas an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J.Biol. Chem. [Epub ahead of print]
Zhang, J., Peng, B., and Chen, X. (2005). Expressions of nuclear factor kappaB, inducible nitricoxide synthase, and vascular endothelial growth factor in adenoid cystic carcinoma of sali-vary glands: correlations with the angiogenesis and clinical outcome. Clin. Cancer Res. 11,7334–7343.
Zingarelli, B., Hake, P.W., Yang, Z., O’Connor, M., Denenberg, A., and Wong, H.R. (2002).Absence of inducible nitric oxide synthase modulates early reperfusion-induced NF-kappaBand AP-1 activation and enhances myocardial damage. FASEB J. 16, 327–342
Zou, M.H., Hou, X.Y., Shi, C.M., Kirkpatick, S., Liu, F., Goldman, M.H., and Cohen, R.A.(2003). Activation of 5 V-AMP-activated kinase is mediated through c-Src and phosphoinosi-tide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells. Role ofperoxynitrite. J. Biol. Chem. 278, 34003–34010.