primary ciliary dyskinesia: kartagener syndrome with central giant cell granuloma. a case report
TRANSCRIPT

Primary ciliary dyskinesia: Kartagener syndrome with centralgiant cell granuloma. A case reportKıvanç Türkoglu, DDS,a Kaan Orhan, DDS, PhD,b Pınar Demir, DDS,c
Barıs Karabulut, DDS, PhD,d Deniz C. Can-Karabulut, DDS, PhD,e Ankara and Mersin, TurkeyUNIVERSITY OF ANKARA, GIRNE HOSPITAL, AND NEAR EAST UNIVERSITY
This paper describes a clinical case of both giant cell granuloma and Kartagener syndrome in a 15-year-oldmale patient, with emphasis on the radiographic aspects of this extremely unusual pathology. To our knowledge, thepresence of these 2 rare clinical conditions in the same patient has not been previously reported. (Oral Surg Oral Med
Oral Pathol Oral Radiol Endod 2010;110:e49-e56)Primary ciliary dyskinesia (PCD) is an autosomal re-cessive disorder that represents a significant subgroupof the multisystem disease ciliopathy1 that affects thecilia, the microtubule-based hair-like organelles thatextend from the surface of almost all cells in the humanbody.2 In �50% of cases, PCD presents with sinusitis,bronchiectasis, and mirror image arrangement (situsinversus, in which the internal organs are located on theopposite side of the body from their normal position3)in a condition known as Kartagener syndrome.
Cilia are subdivided into 2 main types: epithelial ciliaand primary cilia. Epithelial cilia are hair-like append-ages that line the human respiratory tract and contributeto the mucociliary defense mechanism.2 Primary ciliaconsist of both sensory cilia and nodal cilia. Nodal ciliahave an embryologic function in the determination oflaterality. Effective nodal flow is proposed to be a keyelement in the asymmetric expression of developmentalgenes involved in left-right determination, which ex-plains the association of situs inversus with ciliaryabnormalities.2
PCD affects the activity of proteins important to themovement of cilia, especially in the respiratory tract,3
which results in retention of mucus and bacteria.4 Sec-
aResearch Assistant, Department of Oral and Maxillofacial Surgery,Faculty of Dentistry, University of Ankara.bAssociate Professor, Department of Oral Diagnosis and Radiology,Faculty of Dentistry, University of Ankara.cResearch Assistant, Department of Orthodontics, Faculty of Den-tistry, University of Ankara.dConsultant, Department of Dentistry, Girne Hospital.eAssistant Professor, Department of Operative Dentistry, Faculty ofDentistry, Near East University.Received for publication Mar 1, 2010; returned for revision May 9,2010; accepted for publication May 20, 2010.1079-2104/$ - see front matter© 2010 Published by Mosby, Inc.
doi:10.1016/j.tripleo.2010.05.054ondary to a failure in the mucociliary defence mecha-nism, PCD patients experience recurrent respiratorytract infections that begin in early childhood and lead tochronic bronchitis and/or bronchiectasis, chronic rhino-sinusitis, and otitis media.2,5 Moreover, the congenitalreduction or absence of ciliary function responsible forsitus inversus explains the association between thiscondition and Kartagener syndrome.1,2,6,7 Patients withKartagener syndrome also have a greater incidence ofcongenital cardiovascular defects.7
Recent literature has emphasized the importance ofearly diagnosis and appropriate treatment of PCD inpreventing permanent sequelae, such as chronic rhino-sinusitis and bronchiectasis and lung damage.1,6 Thediagnosis should also be considered in older childrenwith severe otitis media, bronchiectasis, or atypicalasthma.2
Despite the substantial research on Kartagener syn-drome that exists in the medical literature,4,5,8-10 thedental literature contains few reports on the disease. Ina case report of a male patient presenting with Kart-agener syndrome, Casanova et al.3 emphasized the ra-diographic aspects and stressed the importance of earlydiagnosis by an oral radiologist. Merrett and Durning11
reported on the unusual dental morphology associatedwith the Kartagener syndrome, and Giusto and Sci-ubba12 reported on the oral findings in 2 siblings diag-nosed with the syndrome. To our knowledge, there isno previous report on the simultaneous occurrence ofKartagener syndrome and giant cell granuloma, anotherrare clinical condition, in the same patient.
Giant cell granuloma is an uncommon bony lesion inthe head and neck region that most commonly affectsthe maxilla and mandible and is usually treated withsurgery. Although it is a benign disease process, it canalso be locally destructive.13 The disease has a very lowincidence among the general population and occurs
mainly in individuals �30 years of age.14e49

OOOOEe50 Türkoglu et al. October 2010
The present paper describes a clinical case of bothgiant cell granuloma and Kartagener syndrome in a15-year-old male patient, with emphasis on the radio-graphic aspects of this extremely unusual pathology.
CASE REPORTClinical and laboratory findings
A 15-year-old male patient applied to the Oral Diagnosisand Radiology Department of the Faculty of Dentistry withcomplaints of swelling on the right side of the maxilla.Written informed consent was obtained before examinationand treatment. Extraoral examination revealed swelling on theright side of the face resulting in facial asymmetry. There wasno history of trauma, the skin was normal, and there was noelevation in local temperature at the site of the swelling. Acomplete lymph node examination of the cervical, subman-dibular, submental, supraclavicular, axillary, and epitrochlearsites revealed positive lymphadenopathy in the submandibu-lar lymph nodes, but no pain on palpation.
Intraoral examination identified a swelling in the rightmaxillary labial sulcus that was extending palatally and in-cluded the first molar, first and second premolars and canine.There was no fluctuation on palpation or paresthesia. Theright maxillary canine and first and second premolars wereslightly displaced from their sockets and highly mobile; how-ever, the teeth were insensitive to percussion, and the patientreported no pain. Electrical and thermal pulp vitality testswere performed by using an electrical pulp tester (Digitest;Parkell, Farmingdale, NY, USA) and solid carbon dioxide(CO2 ice). A negative response was obtained from all of theexamined teeth. The patient exhibited poor oral hygiene, buthad no history of smoking or systemic drug use.
Family history, history of disease, and imagingA detailed history identified consanguinity of the patient’s
parents. The patient also reported a long-standing moistcough, postnasal mucopurulent secretion, nasal congestion,and rhinitis, but no chronic secretory otitis media. An initialpanoramic radiograph showed a large radiolucent lesion inthe right maxillary region and displacement of the rightmaxillary premolar and molar teeth.
To obtain a more precise location and definition of thepathologic features of the lesion, cone-beam computerizedtomography (CBCT) images were taken using an Iluma UltraCBCT scanner (Imtec Imaging, Ardmore, OK, USA) with a24.4 � 19.5 cm amorphous silicon flat-panel image detectorand a cylindric volume of reconstruction up to 21.1 � 14.2cm. Images were obtained at 120 kVp and 3.8 mA, withultra-high voxel size of 0.09 mm and an exposure time of 40seconds. An axial CBCT scan showed a large expansivelesion in the right maxilla, and axial, coronal, and sagittalimages revealed total nasal obstruction and pansinusitis. Apanoramic reconstruction of CBCT images also showed ex-pansile lesions extending from the right side of the maxilla tothe left premolars (Fig. 1).
The lesion and surrounding soft tissue was examined usingT -, T -, and fat-saturated T -weighted (W) magnetic reso-
1 2 2nance imaging (MRI). Images were taken with a 1.5T imag-
ing unit (Magnetom Vision, Siemens, Erlangen; TR/TE500/9, 4,126/98, 512 � 256 matrix, 23° � 23° field of view,2-mm slide thickness, number of excitations (NEX) 1, 15.6kHz band width). Coronal fat-saturated T2-W images exhib-ited heterogeneous signal intensity around the maxilla andmaxillary sinus and high signal intensity in the ethmoidregion, whereas a coronal T1-W image showed hypointensesignal intensity at the maxillary and ethmoid region, with aslightly increased signal at the periphery of the lesions. Agadolinium-enhanced T1-W image revealed patchy irregularcontrast enhancement involving the maxillary sinus and in-creased signal intensity in the ethmoid region, and an axialT2-W image clearly exhibited high signal intensity that wasinterpreted as sinusitis (Fig. 2).
Based on patient history and clinical and radiographicexamination, the differential diagnosis included odontogeniccyst, odontogenic fibroma, odontogenic keratocyst, browntumor, and unilocular ameloblastoma. Central giant cell gran-uloma (CGCG) was also considered in this case, because ofthe maxillary location of the lesion. An aspiration biopsy wasperformed, and histopathologic examination confirmed aCGCG of the maxilla. Based on this finding, a treatment planwas decided on that consisted of surgical excision of thelesion and a partial maxillectomy. Figure 3 shows the pre- andpostoperative appearance of the CGCG. A definitive diagno-sis was obtained through postsurgical histopathologic analy-sis. Microscopic examination (�40 and �100) of the H&E-stained specimen revealed reactive bone and giant cells in afibroblastic stroma with extravagated red blood cells (Fig. 4).
Because of the possibility of a genetic disorder implied bythe consanguinity of the patient’s parents, Diagnostic Ser-vices was consulted, and the patient was referred to a chestconsultant and then to a genetic consultant. Blood tests re-vealed a PTH level of 37 pq/mL and no abnormalities, whichomitted a brown tumor from the differential diagnosis. Achest radiograph revealed situs inversus with dextrocardia,with the cardiac anatomy positioned in a mirror image of thenormal anatomy (Fig. 5). Nasal nitric oxide measurementsfound low levels for both nasal and exhaled nitric oxide, anda ciliary beat frequency measurement and pattern analysisfound a slow beat frequency. Radiologic images of paranasalsinuses demonstrated mucosal thickening, opacified sinuscavities, and hypoplastic frontal sinuses, and a nasal mucosabiopsy was found to be compatible with PCD. Based on theclassic triad of situs inversus, bronchiectasis, and sinusitis, thecase was defined as Kartagener syndrome. There was nofamily history of the syndrome or dental abnormalities in thiscase.
Surgical excision of the lesion and a hemi-maxillectomywere performed under general anesthesia. In view of thepatient’s bronchiectasis, pulmonary status and cardiac struc-ture and function were assessed prior to surgery in order toavoid pulmonary complications, and the patient received pro-phylactic antibiotic treatment because of the possibility ofabnormal neutrophil chemotaxis.15,16 Due to the relative con-traindication of anticholinergic and antitussive medicationsand nasal tubes, intubation anesthesia was performed usingthiopental, nitrous oxide, enflurane, and succinylcholine, as
reported by Etzel et al.16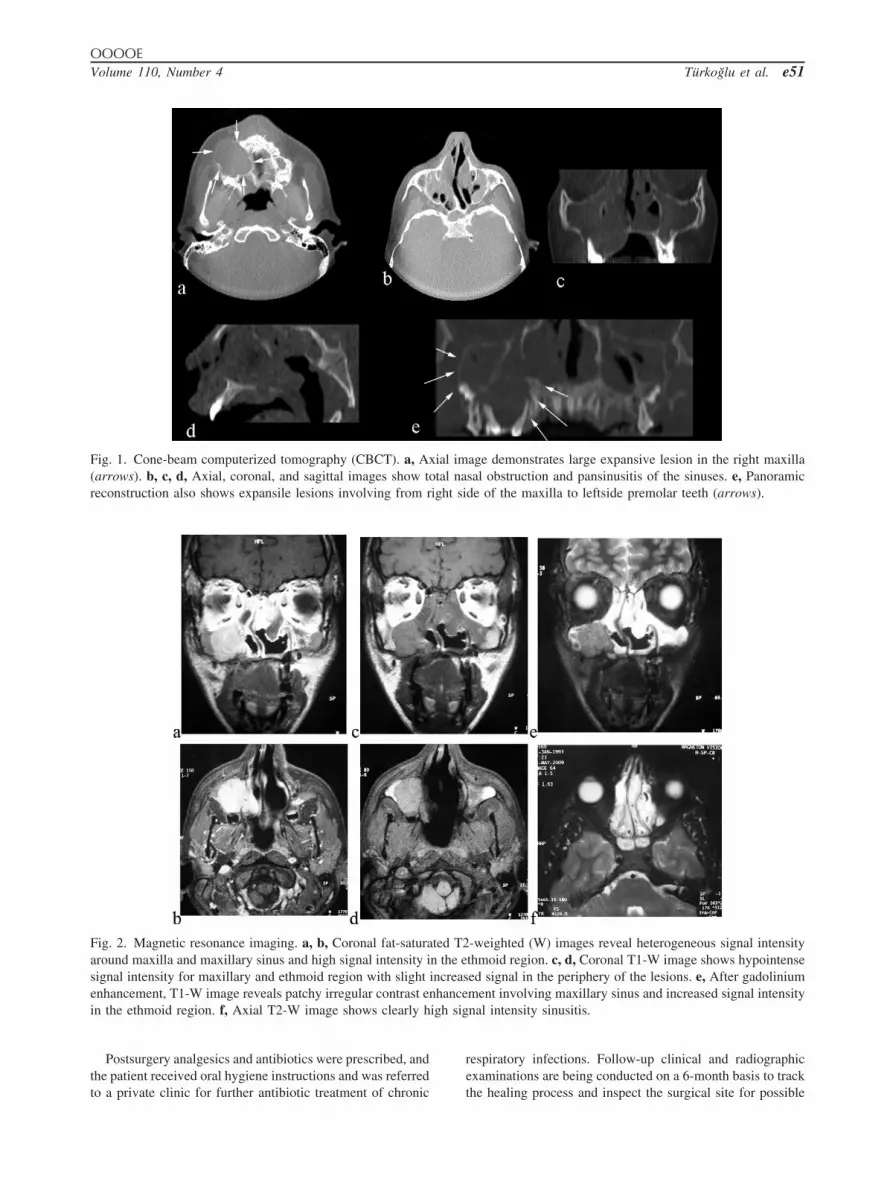
ight si
nhanceigh sig
OOOOEVolume 110, Number 4 Türkoglu et al. e51
Postsurgery analgesics and antibiotics were prescribed, andthe patient received oral hygiene instructions and was referred
Fig. 1. Cone-beam computerized tomography (CBCT). a, Ax(arrows). b, c, d, Axial, coronal, and sagittal images show toreconstruction also shows expansile lesions involving from r
Fig. 2. Magnetic resonance imaging. a, b, Coronal fat-saturaaround maxilla and maxillary sinus and high signal intensity isignal intensity for maxillary and ethmoid region with slightenhancement, T1-W image reveals patchy irregular contrast ein the ethmoid region. f, Axial T2-W image shows clearly h
to a private clinic for further antibiotic treatment of chronic
respiratory infections. Follow-up clinical and radiographicexaminations are being conducted on a 6-month basis to track
age demonstrates large expansive lesion in the right maxillasal obstruction and pansinusitis of the sinuses. e, Panoramicde of the maxilla to leftside premolar teeth (arrows).
-weighted (W) images reveal heterogeneous signal intensitythmoid region. c, d, Coronal T1-W image shows hypointenseed signal in the periphery of the lesions. e, After gadoliniumment involving maxillary sinus and increased signal intensitynal intensity sinusitis.
ial imtal na
ted T2n the eincreas
the healing process and inspect the surgical site for possible

hemim
OOOOEe52 Türkoglu et al. October 2010
recurrences. Good healing and no recurrence at the operationsite were observed at the first follow-up visit (Fig. 6). Addi-tional treatment plans include an interim obturator for max-illary resection to regain function and esthetics.
DISCUSSIONCGCG of the jaw usually presents clinically as a
painless slow-growing swelling of the jaw and radio-graphically as a radiolucent expansion.14,17 The inci-dence of the disease is rare, and it has been reported tobe highest among male individuals aged 10-14 years.
Fig. 3. a, Preoperative appearance of the giant cell granulomremoval of the lesion. d, Lesion was completely removed as
Fig. 4. Specimens with hematoxylin and eosin stain. a, �40:stroma with extravagated red blood cells.
Pain and sensory disturbances are rare.14 Differential
diagnosis may include radicular cysts, adenomatoidodontegenic tumors, brown tumor,18 fibrous dysplasia,and calcifying epithelial odontogenic cysts, as well asmore uncommon odontogenic neoplasms, such as amelo-blastomas, keratocystic odontogenic tumors, and myxo-mas. The ultimate diagnosis relies on histopathology. His-tologically, CGCGs appear as foreign body–type giantcells with irregular distribution and vacuolation. Theirhallmarks are multinucleated giant cells that are clusteredor diffusely distributed, especially in hemorrhagic areas,
e right side. b, c, Images showing the defect after completeaxillectomy including the teeth.
cells and reactive bone. b, �100: giant cells in a fibroblastic
a in th
giant
and possess collagenized or edematous stroma.17,19

OOOOEVolume 110, Number 4 Türkoglu et al. e53
CGCGs can be radiologically differentiated from otherentities containing giant cells, such as giant cell tumors(GCTs),20-22 cherubism,23-25 brown tumor of hyperpara-thyroidism,26,27 and aneurysmal bone cysts.28,29 LikeCGCGs, GCTs usually present as an aggressive, eccentric,
Fig. 5. Chest radiograph shows dextrocardia.
Fig. 6. Postoperative (a) 3-month and (b) 6-month panoramicradiographs showing no recurrence and good healing of theoperation site.
lytic lesion centered on the metaphysis and extending to
the subchondral bone with expansile remodeling; how-ever, they lack the internal mineralization seen in CGCGs.Soft tissue involvement is frequently found in GCTs.Although cherubism is microscopically indistinguishablefrom CGCG, clinicopathologic correlation (children, au-tosomal dominance, bilateral multilocular jaw lucencies)can provide helpful clues to distinguish cherubism fromCGCG. Brown tumors of hyperparathyroidism can occurin multiple areas within 1 bone or as a polyostotic process.Whereas a brown tumor of hyperparathyroidism histolog-ically resembles a CGCG,18 bone changes associated withhyperparathyroidism, such as generalized demineraliza-tion of the medullary bones of the jaws and loss oflamina dura around the roots of the teeth can helpdifferentiate a brown tumor from a CGCG, and in casesof doubt, serum calcium, phosphorus, and alkalinephosphatase levels can be measured for a differentialdiagnosis.17 Aneurysmal bone cysts appear as trabecu-lated osteolytic lesions, associated with cortical thin-ning, with a “blow out” appearance. In the present case,an aspiration biopsy was performed and the lesionhistologically identified as a CGCG of the maxilla.Definitive diagnosis of CGCG requires histopathologicanalysis.30
The literature includes several case reports describ-ing CGCG of the maxilla involving the palate.13,17,31
Roberts et al.31 reported a case of an 18-year-old maninitially treated with a partial maxillectomy thatachieved a near-total debulking of the mass and imme-diate cosmetic improvement. Steroid injections wereinitially able to stall any reappearance of the residualdisease; however, the patient experienced a recurrence8 months after surgery. As a result, a bilateral totalinferior maxillectomy was performed using a facialdegloving approach, and the patient was subsequentlyfitted with an obturator.31 The authors concluded that acombination of partial surgical resection and intrale-sional steroid injections may not be adequate for thetreatment of large CGCGs of the maxilla and suggestedthat complete surgical resection should be considered forthe initial treatment, particularly in aggressive cases.31
Ciorba et al.13 also described a case that involved themaxilla which was treated with surgical excision, fol-lowed by local injection of steroids. Dos Santos et al.32
have dealt with the usefulness of CT in the evaluationof this lesion.
A recurrence rate of up to 70% has been reported,mainly for lesions that display an aggressive biologicbehavior.19 An aggressive and extensive behavior ofthe present lesion with radiographic evidence of perfo-ration of cortex required a more radical excision, i.e.,partial maxillectomy. Follow-up sessions are very im-portant, because the recurrence rate is reported to be
high, with most occurring within the first 2 years of the
OOOOEe54 Türkoglu et al. October 2010
therapy.17 As an adjunct to surgery, the literature in-cludes reports on the use of steroids or calcitonon(which inhibits osteoclastic activity), interferon-alpha,and bisphosphonates.14,17,19,31 Combinations of �1therapeutic agent are suggested to further enhance thesynergistic effects on healing progression,19 with thepharmacologic agents adjusted to the phenotypic pro-file of the lesional cells before their administration atthe beginning and during treatment. In the present case,owing to the patient’s concurrent Kartagener syndrome,adjunct therapy was being considered in consultationwith the patient’s physician to prevent any possibleadverse affects of the prescribed antibiotics.
To the best of our knowledge, the literature containsfew case reports identifying dental anomalies withKartagener syndrome. One of them presented a girlwith Kartagener syndrome and the congenital absenceof an upper lateral incisor, enamel hypoplasia, andaberrant tooth morphology.11 The patient had beendiagnosed with the syndrome at 8 years of age, and theauthors suggested that the treatment of respiratory tractinfections with numerous courses of antibiotics mayhave played a role in the development of this unusualdental morphology and that the high incidence of cariesexperienced by the patient may have been related to theinfections themselves.11 Several studies have men-tioned disorders of the bone system associated withKartagener syndrome,11,33-35 which may have implica-tions for disorders of the dentition, because the devel-oping dentition may be affected by any alteration in thebone architecture within which the developing toothgerms are contained.11,35 Giusto and Sciubba12 docu-mented a patient with the syndrome and his sibling whoexpressed an oligosymptomatic form of the syndrome,both displaying identical dental findings: impactedmaxillary cuspids, a “gothic” or high-arched palate, anda Bolton tooth-size discrepancy. According to thoseauthors, further research is required to determinewhether the dental finding is associated with the syn-drome to a higher degree; however, the difficulty inascertaining this information relates to the syndromebeing noted so infrequently, and finding affected pa-tients who would agree to have a panoramic radiographtaken. Researchers also noted that the patients’ mothercould not recall any other family members afflictedwith the syndrome, therefore an accurate family historyand pedigree analysis to trace the lineage of the traitwas not possible. In contrast to the study mentionedabove, the present patient showed no unusual dentalmorphology, including congenitally missing teeth orother abnormalities.
Apart from intraoral findings, Casanova et al.3 men-tioned that the oral radiologist must consider the image
of the paranasal pansinusitis in association with nasalpolyposis. A high-resolution CT of the paranasal si-nuses reveals pansinusitis. The examination shows mu-cosal thickening and opacified maxillary, ethmoid andfrontal sinus cavities, besides the total nasal obstruc-tion. More recently, the presence of rhinitis, chronicotitis, nasal polyposis, and opacified sinus cavities hasbeen described. Chest radiography may be performedto confirm the syndrome components. Early detectioncan reduce or even prevent the occurrence of severerespiratory infections, thus contributing to a betterprognosis and an increase in the patient’s quality oflife.3
Gorham and Merselis36 observed the triad associatedwith Kartagener syndrome in 2 young adult siblings,both of whom had had symptoms since early childhood.The authors traced the family through 4 generations andreviewed the literature regarding familial incidence,discovering 6 cases of familial incidence, all of whichinvolved siblings only. This was in keeping with thefinding of the authors’ own cases; all 4 generationswere free of symptoms. The Kartagener triad appearedto have a high familial incidence but appeared only in1 generation, and suggesting that rather than a geneticfactor, an environmental factor present during embry-onic development may be responsible for the appear-ance of the triad.36
In contrast to this assumption, a recent study byGeremek et al.37 concluded that PCD was a mainlyautosomal-recessive genetic disorder that showed ex-tensive genetic heterogeneity and identified 4 geneswith a proven pathogenetic role in PCD. However,none of the variations alone could explain the occur-rence of the disease in these patients. The authorsconcluded that more families need to be examined toestablish the exact Kartagener syndrome region.37
Casanova et al.3 mentioned that the disorder is inheritedas an autosomal recessive trait. Male and female indi-viduals are affected equally. The complete syndromehas high familial evidence, appearing only in 1 gener-ation, and multiple siblings may have various combi-nations of its components, which do not appear in theirchildren. These features and the high incidence of con-sanguinity among the apparently normal parents ofaffected children support the contention that the geneticabnormality is carried as an autosomal recessive gene.Casanova et al.3 reported no family history of thesyndrome referred by their patient. In the present case,an extensive family history covering first- and second-degree kinship revealed no sign of Kartagener syn-drome among any other family members.
The detection of a very low nasal nitric oxide outputmay also be useful in diagnosing PCD in older chil-dren.10 The finding of immotile or dyskinetic cilia by
phase-contrast microscopy allows PCD to be diagnosed
OOOOEVolume 110, Number 4 Türkoglu et al. e55
with high sensitivity.5 Completion of the human ge-nome sequence has accelerated the identification andcharacterization of disease genes and should providenew insight into the molecular mechanisms involved inthe assembly and function of cilia, which in turn maypromote the development of new methods for the di-agnosis, prevention, and treatment of PCD, such asspecific protein or gene therapy.2 However, few pa-tients with PCD carry a well established diagnosis,which reflects the limited ability to diagnose this dis-order.7 According to Bush et al.1 and Barnes,8 althoughrespiratory disease and mirror image arrangement maysuggest the presence of PCD, diagnosis is often de-layed, in part because PCD presents with symptoms,such as rhinitis, secretory otitis media, and coughing,that are common among children. Casanova et al.3
mentioned that the oral radiologist must consider theimage of the paranasal pansinusitis in association withnasal polyposis and the patient’s clinical data. Chestradiography may be performed to confirm the syn-drome components. General pediatricians and pediatricnurses must be alert to the condition, because earlydiagnosis is essential for the prevention of permanentlung damage.2,8 The present case highlights the rolethat dentists can play in early diagnosis with appropri-ate consultations and referrals.
CONCLUSIONCGCG of the jaws is a rare benign tumor of uncertain
etiology that accounts for up to 7% of all tumors in themandible and the maxilla.38,39 The etiology of CGCG isstill not clear, except that trauma or inflammation havebeen mentioned as important factors. Once consideredto be a local reparative response of the bone, possibly tointramedullary hemorrhage or trauma,40,41 the finding ofCGCGs in patients with anomalies with a known geneticorigin, such as neurofibromatosis type 1, cherubism,Noonan syndrome, or hyperparathyroidism, suggeststhe possibility of a genetic-related etiology.14 Kartage-ner syndrome itself comprises a triad of symptomsassociated with an autosomal-recessive inherited dis-ease, and although there is insufficient proof to confirma connection between CGCG and Kartagener syn-drome, we believe a possible relationship exists be-tween them. In light of the present case report, it isrecommended that new studies be conducted to confirma possible genetic connection between CGCG and Kart-agener syndrome. Despite the rarity of the disease, oraland maxillofacial radiologists should be aware of theselesions. Moreover, the presence of CGCG in conjunc-tion with Kartagener syndrome requires additional ra-
diographic examinations.REFERENCES1. Bush A, Chodhari R, Collins N, Copeland F, Hall P, Harcourt J,
et al. Primary ciliary dyskinesia: current state of the art. Arch DisChild 2007;92:1136-40.
2. Chodhari R, Mitchison HM, Meeks M. Cilia, primary ciliarydyskinesia and molecular genetics. Paediatr Respir Rev 2004;5:69-76.
3. Casanova MS, Tuji FM, Yoo HJ, Haiter-Neto F. Kartagenersyndrome. Dentomaxillofac Radiol 2006;35:386-9.
4. Barbato A, Frischer T, Kuehni CE, Snijders D, Azevedo I, BaktaiG, et al. Primary ciliary dyskinesia: a consensus statement ondiagnostic and treatment approaches in children. Eur Respir J2009;34:1264-76.
5. Holzmann D, Ott PM, Felix H. Diagnostic approach to primaryciliary dyskinesia: a review. Eur J Pediatr 2000;159:95-8.
6. Ortega HA, Vega Nde A, Santos BQ, Maia GT. Primary ciliarydyskinesia: considerations regarding six cases of Kartagenersyndrome. J Bras Pneumol 2007;33:602-8.
7. Leigh MW, Pittman JE, Carson JL, Ferkol TW, Dell SD, DavisSD, et al. Clinical and genetic aspects of primary ciliary dyski-nesia/Kartagener syndrome. Genet Med 2009;11:473-87.
8. Barnes C. Diagnosis and management of primary ciliary dyski-nesia. Paediatr Nurs 2009;21:24-7.
9. Bouyahia O, Essadem L, Matoussi N, Gharsallah L, Fitouri Z,Mrad Mazigh S, et al. Etiology and outcome of bronchiectasis inchildren: a study of 41 patients. Tunis Med 2008;86:996-9.
10. Escudier E, Roger G, Coste A. Nasal ciliary investigations for thediagnosis of primary ciliary dyskinesia in children. Arch Pediatr2004;11:390-3.
11. Merrett SJ, Durning P. Kartagener’s syndrome: unusual dentalmorphology. Int J Paediatr Dent 2005;15:220-3.
12. Giusto TJ, Sciubba JJ. Kartagener’s syndrome: a review of theliterature and case report of oral findings in two siblings. J N JDent Assoc 2004;75:30-2.
13. Ciorba A, Altissimi G, Giansanti M. Giant cell granuloma of themaxilla: case report. Acta Otorhinolaryngol Ital 2004;24:26-9.
14. de Lange J, van den Akker HP, van den Berg H. Central giantcell granuloma of the jaw: a review of the literature with em-phasis on therapy options. Oral Surg Oral Med Oral Pathol OralRadiol Endod 2007;104:603-15.
15. Reidy J, Sischy S, Barrow V. Anaesthesia for Kartagener’ssyndrome. Br J Anaesth 2000;85:919-21.
16. Etzel S, Plötz J, Heidegger H, von Hugo R. The Kartagenersyndrome. Anaesthesist 1994;43:463-5.
17. Sholapurkar AA, Pai KM, Ahsan A. Central giant cell granulomaof the anterior maxilla. Indian J Dent Res 2008;19:78-82.
18. Slootweg PJ. Lesions of the jaws. Histopathology 2009;54:401-18.
19. Vered M, Buchner A, Dayan D. Central giant cell granuloma ofthe jawbones—new insights into molecular biology with clinicalimplications on treatment approaches. Histol Histopathol2008;23:1151-60.
20. Hoch B, Hermann G, Klein MJ, Abdelwahab IF, Springfield D.Giant cell tumor complicating Paget disease of long bone. Skel-etal Radiol 2007;36:973-8.
21. Park IH, Jeon IH. Multicentric giant cell tumor of bone: tenlesions at presentation. Skeletal Radiol 2003;32:526-9.
22. Briccoli A, Malaguti C, Iannetti C, Rocca M, Bertoni F. Giantcell tumor of the rib. Skeletal Radiol 2003;32:107-10.
23. Martinez-Tello FJ, Manjon-Luengo P, Martin-Perez M, Montes-Moreno S. Cherubism associated with neurofibromatosis type 1,and multiple osteolytic lesions of both femurs: a previouslyundescribed association of findings. Skeletal Radiol 2005;34:
793-8.
OOOOEe56 Türkoglu et al. October 2010
24. Yamaguchi T, Dorfman HD, Eisig S. Cherubism: clinicopatho-logic features. Skeletal Radiol 1999;28:350-3.
25. Bianchi SD, Boccardi A, Mela F, Romagnoli R. The computedtomographic appearances of cherubism. Skeletal Radiol 1987;16:6-10.
26. Regezi JA. Odontogenic cysts, odontogenic tumors, fibroosse-ous, and giant cell lesions of the jaws. Mod Pathol 2002;15:331-41.
27. Scholl RJ, Kellett HM, Neumann DP, Lurie AG. Cysts and cysticlesions of the mandible: clinical and radiologic-histopathologicreview. Radiographics 1999;19:1107-24.
28. Murphey MD, Nomikos GC, Flemming DJ, Gannon FH, Tem-ple HT, Kransdorf MJ. From the archives of AFIP. Imagingof giant cell tumor and giant cell reparative granuloma ofbone: radiologic-pathologic correlation. Radiographics 2001;21:1283-309.
29. Picci P, Baldini N, Sudanese A, Boriani S, Campanacci M.Giant cell reparative granuloma and other giant cell lesions ofthe bones of the hands and feet. Skeletal Radiol 1986;15:415-21.
30. Sun ZJ, Cai Y, Zwahlen RA, Zheng YF, Wang SP, Zhao YF.Central giant cell granuloma of the jaws: clinical and radiologicalevaluation of 22 cases. Skeletal Radiol 2009;38:903-9.
31. Roberts J, Shores C, Rose AS. Surgical treatment is warranted inaggressive central giant cell granuloma: a report of 2 cases. EarNose Throat J 2009;88:8-13.
32. Dos Santos LA, Campos PS, Laranjeira AL, Bonan PR, Mar-telli H Jr, Cardoso SV. Effectiveness of computed tomographyto evaluate central giant cell lesion. Dentomaxillofac Radiol2007;36:522-5.
33. Rébora ME, Cuneo JA, Marcos J, Marcos JC. Kartagener syn-
drome and rheumatoid arthritis. J Clin Rheumatol 2006;12:26-9.34. Younes M, Béjia I, Zrour-Hassen S, Touzi M, Bergaoui N.Rheumatoid arthritis and Kartagener syndrome: a rare com-bination. Joint Bone Spine 2006;73:571-2.
35. Ramotowski R, Guz W, Zieba E, Zlomaniec G. Clinical andradiological aspects of Kartagener’s syndrome. Ann Univ MariaeCurie Sklodowska Med 2001;56:151-5.
36. Gorham GW, Merselis JG Jr. Kartagener’s triad: a family study.Bull Johns Hopkins Hosp 1959;104:11-6.
37. Geremek M, Schoenmaker F, Zietkiewicz E, Pogorzelski A,Diehl S, Wijmenga C, et al. Sequence analysis of 21 geneslocated in the Kartagener syndrome linkage region on chromo-some 15q. Eur J Hum Genet 2008;16:688-95.
38. Stavropoulos F, Katz J. Central giant cell granulomas: a system-atic review of the radiographic characteristics with the additionof 20 newcases. Dentomaxillofac Radiol 2002;31:213-7.
39. Kruse-Losler B, Diallo R, Gaertner C, Mischke KL, Joos U,Kleinheinz J. Central giant cell granuloma of the jaws: a clinical,radiologic, and histopathologic study of 26 cases. Oral Surg OralMed Oral Pathol Oral Radiol Endod 2006;10:346-54.
40. Regezi JA, Sciubba JJ, editors. Oral pathology (clinical patho-logic correlations). Philadelphia: Saunders; 1993. p. 409–11.
41. Sidhu MS, Parkash H, Sidhu SS. Central giant cell granuloma ofjaws-review of 19 cases. Br J Oral Maxillofac 1995;33:43-6.
Reprint requests:
Deniz C. Can-Karabulut, PhDDepartment of Operative DentistryFaculty of DentistryNear East UniversityMersin 10Turkey
[email protected]