pluripotent stem cells as a potential tool for disease modelling and cell therapy in diabetes
TRANSCRIPT

Pluripotent Stem Cells as a Potential Tool for Disease Modellingand Cell Therapy in Diabetes
Essam M. Abdelalim & Amélie Bonnefond &
Annelise Bennaceur-Griscelli & Philippe Froguel
Published online: 1 March 2014# Springer Science+Business Media New York 2014
Abstract Diabetes mellitus is the most prevailing diseasewith progressive incidence worldwide. To date, the pathogen-esis of diabetes is far to be understood, and there is nopermanent treatment available for diabetes. One of the prom-ising approaches to understand and cure diabetes is to usepluripotent stem cells (PSCs), including embryonic stem cells(ESCs) and induced PCSs (iPSCs). ESCs and iPSCs have agreat potential to differentiate into all cell types, and they havea high ability to differentiate into insulin-secreting β cells.Obtaining PSCs genetically identical to the patient presentingwith diabetes has been a longstanding dream for the in vitro
modeling of disease and ultimately cell therapy. For severalyears, somatic cell nuclear transfer (SCNT) was the method ofchoice to generate patient-specific ESC lines. However, thistechnology faces ethical and practical concerns. Interestingly,the recently established iPSC technology overcomes the majorproblems of other stem cell types including the lack of ethicalconcern and no risk of immune rejection. Several iPSC lineshave been recently generated from patients with differenttypes of diabetes, and most of these cell lines are able todifferentiate into insulin-secreting β cells. In this review, wesummarize recent advances in the differentiation of pancreaticβ cells from PSCs, and describe the challenges for theirclinical use in diabetes cell therapy. Furthermore, we discussthe potential use of patient-specific PSCs as an in vitro model,providing new insights into the pathophysiology of diabetes.
Keywords Diabetesmellitus . Embryonic stem cells (ESCs) .
Induced pluripotent stem cells (iPSCs) . Insulin-secretingcells . Pancreaticβ cells . Cell therapy
Introduction
Pluripotent stem cells (PSCs) including embryonic stem cells(ESCs) and induced PCSs (iPSCs) have unlimited ability forself-renewal and they can differentiate into cells of all threegerm layers [1, 2]. Human ESCs (hESCs) are derived from theinner cell mass (ICM) of blastocyst-stage embryos [1], whilehuman iPSCs (hiPSCs) are generated by transfection of so-matic cells (such as fibroblast [2], stomach cells and hepato-cytes [3], human keratinocytes [4], and blood cells [4]) withgroup of key transcription factors, including OCT4, SOX2, C-MYC, and KLF4 [2], or OCT4, SOX2, LIN28, and NANOG[5]. PSCs are characterized by rapid and symmetrical celldivision, with equal distribution of the genome [6, 7], andtheir pluripotent identity is controlled by a group of
E. M. Abdelalim (*) :A. Bonnefond : P. Froguel (*)Qatar Biomedical Research Institute, Qatar Foundation, EducationCity, 5825, Doha, Qatare-mail: [email protected]: [email protected]
E. M. AbdelalimDepartment of Cytology and Histology, Faculty of VeterinaryMedicine, Suez Canal University, Ismailia, Egypt
A. Bonnefond : P. FroguelCNRS UMR8199, Pasteur Institute of Lille, Lille, France
A. Bonnefond : P. FroguelLille 2 University, Lille, France
A. Bonnefond : P. FroguelEuropean Genomic Institute for Diabetes (EGID), Lille, France
A. Bennaceur-GriscelliINSERM UMR S935, ESTeam Paris Sud, Université Paris Sud 11,Villejuif, France
A. Bennaceur-GriscelliAP-HP Laboratory of Hematology, CHU, Bicêtre, France
P. FroguelDepartment of Genomics of Common Disease, HammersmithHospital, Imperial College London, London, UK
Stem Cell Rev and Rep (2014) 10:327–337DOI 10.1007/s12015-014-9503-6

transcription factors, such as OCT4, NANOG, and SOX2,which are the hallmark features of PSCs [2, 8, 9]. Notably,hiPSCs share similar features with hESCs including morphol-ogy, expression of pluripotency genes, cell cycle profile, self-renewal ability, differentiation potential, genomic profiles, andteratoma formation [1, 2, 5, 10].
Diabetes mellitus is the most widespread disease withprogressive incidence worldwide. The World Health Organi-zation (WHO) plans that diabetes will be the seventh leadingcause of death in 2030 (Global status report onnoncommunicable diseases 2010. Geneva, World Health Or-ganization, 2011) and it has been estimated that 347 millionpeople worldwide present with diabetes [11]. Diabetes is amajor health problem responsible for early morbidities andmortality [12]. Two different types of diabetes are well char-acterized, type 1 (T1D) and type 2 diabetes (T2D). T1D is anearly-onset autoimmune disease in which pancreatic β cellsare irreversibly destroyed by autoimmune attack, resulting in alack of insulin production; consequently, patients presentingwith T1D require lifelong insulin injections for survival [13,14]. T2D which is the most common form of diabetes(representing ~90 % of diabetic patients worldwide) occursas a result of insufficient insulin secretion from the pancreaticβ cells, and/or insulin resistance in the target tissues of theinsulin (including liver, fat, muscle), but the primary mecha-nisms involved in the development and the physiopathologyof T2D are still largely debated [15]. Some forms of diabetes,including neonatal diabetes mellitus (NDM) and maturity-onset diabetes of the young (MODY), are typically monogen-ic; they are due to modifications in a single gene or locus thatplays a major role in pancreatic β cells development and/ormature function (including reduced glucose sensing and me-tabolism of the pancreatic β cells, failure of membrane depo-larization or increased β cell apoptosis) [16]. Patients withNDM which is a very rare disorder (1:200,000 live births),present with low or even undetectable insulin levels [16].MODY which represents less than 2 % of T2D cases, usuallydevelops during childhood or young adulthood and is due toautosomal dominantly inherited mutations [17]. To date, 13MODY genes have been identified as the causes of differentMODY subtypes [18].
One of the most important strategies to treat diabetes is toproduce functional pancreatic β cells particularly for patientswith T1D, and some NDM or MODY subtypes. Furthermore,patients with T2D who need exogenous insulin may benefitfrom pancreatic β cell transplantation, considering the occur-rence of pancreatic β cell failure [19]. The recent progress inthe stem cell field showed that hPSCs are valuable sources forgenerating pancreatic β cells for rejection-free cell therapy.Moreover, it is now possible to model diabetes culturingautologous functional pancreatic β cells with a known diabe-tes related mutation and re-establishing pathogenesis in vitro.This disease modeling with such cells has the potential to give
insight into the cellular and molecular defects of diabetes andto enable new cell-based drug discovery. Recently, severalstudies have reported the ability to generate PSCs from thepatients suffering from different forms of diabetes as well astheir differentiation into insulin-secreting β cells for therapeu-tic purposes.
In this review, we discuss recent advances in the generationof insulin secreting β cells from PSCs, and discuss the poten-tial use of patient-specific PSCs as an in vitro model, provid-ing new insights into the pathophysiology of diabetes. Also,we describe challenges for their clinical use in diabetes celltherapy.
Differentiation of Pluripotent Stem Cellsinto Insulin-Secreting β Cells: 2014 State of Art
The success of β cell generation from PSCs is based onsuccessful manipulation of culture conditions and the stimu-lation of central regulatory genes involved in pancreas devel-opment (Fig. 1). The hESCs and hiPSCs share the samecharacteristics, and they can differentiate into all cell typesof the body, including pancreatic insulin-secreting β cells[20–22], using the same protocols [22–24]. Previous studiesreported that PSCs can be differentiated into insulin-secretingcells by modifying the culture conditions [23, 25–27], usingmonolayer culture [23–26, 28, 29] or an embryoid body (EB)technique [30, 31]. A previous study reported that the first stepof hESC differentiation is important for proper pancreaticdifferentiation, since the starting seeding cell density affectson the efficiency of differentiation into pancreatic endocrinecells. hESCs seeded at high density have higher ability todifferentiate into duodenal homeobox 1 gene (PDX1)- andneurogenin 3 (NGN3)-positive cells than those of low density-seeded cells [32].
Earlier studies reported that hESCs can be differentiatedinto insulin-secreting cells using a step-wise differentiationprotocol, leading to formation of up to 12 % insulin-secreting cells, but with limited glucose responsiveness [23,26, 33]. Another study has modified the differentiation proto-col of hESCs to produce insulin-secreting cells with an in vivoresponsive to glucose [25]. Recently, a higher efficiency toproduce insulin-secreting cells (up to 25 %) from hESCs hasbeen obtained by in vitro regulation of transforming growthfactor β (TGFβ) family members [30].
As a first step toward pancreatic differentiation, the PSCsshould be initially differentiated into definitive endoderm(DE) as occur in vivo (Fig. 1). The recent protocols for DEdifferentiation from hESCs have showed that at least 60–80%of differentiated cells express SOX17, FOXA2, CXCR4, andGSC, and they did not express SOX7, the visceral endodermalmarker [22, 23, 25, 34–36]. The signals of NODAL andWNThave been identified as essential signals to induce DE from
328 Stem Cell Rev and Rep (2014) 10:327–337

hESCs and hiPSCs [22, 26, 34, 37]. The activin A is used toactivate NODAL, which is endogenous endoderm inducer[38], and it has been reported that the efficient DE inductionfrom hESCs requires high concentration of activin A (50–100 ng/ml) in the absence of serum [31, 39]. Furthermore,treatment of PSCs with a combination of sodium butyrate andactivin A [23], or antagonists of PI3K pathway [22, 37],increases the efficiency of DE differentiation. Furthermore,treatment of hESCs with bone morphogeneic protein 4(BMP4) or Wnt3A enhances the efficiency of DE formation[34]. Some protocols demonstrated that during activin treat-ment, addition of WNT3A or CHIR99021, activators of Wntsignalling by suppressing glycogen synthase kinase 3 β(GSK3β) enhances endodermal differentiation [40–42]. How-ever, another study found that CHIR99021 is more potent thanWnt3A in promoting SOX17-and FOXA2-positive endoder-mal cells [29]. Also, GDF8 (myostatin), a TGFβ familymember, is effective for stimulating DE [40]. Treatment ofhESCs with small molecules such as IDE1 and IDE2 inducesapproximately 80 % of ESCs to differentiate into SOX17-expressing DE cells [43].
To further differentiate DE cells into pancreatic β cells,other growth factors should be added to the culture media soas to avoid their differentiation into hepatic lineages (Fig. 1).Treatment of DE progenitors with Noggin (BMP antagonist)and SU5402 (FGF receptor antagonist) suppresses hepaticdifferentiation [35]. It is known that inhibition of BMP inthe dorsal endoderm is essential for pancreatic differentiation[44]. However, after the pancreatic cells are formed, the BMPsignaling is required to maintain pancreatic and PDX1 expres-sion [44].
The DE cells can be further differentiated into pancreaticendocrine cells by suppressing the signaling pathways ofNOTCH and HEDGEHOG [23] (Fig. 1). Cyclopamine or
KAAD cyclopamine, which are HEDGEHOG-signalling in-hibitors, have been used in several protocols to producePDX1-expressing cells [24–26, 28, 30]. PDX1 which playsan essential role during the early development of the pancreas,is reduced during endocrine specification, and is subsequentlyupregulated inβ cells and is involved in insulin secretion [45].Fibroblast growth factor 10 (FGF10) can activate NOTCHsignaling, which is involved in the proliferation of PDX1-expressing pancreatic progenitors. However, the pancreaticexpression of NGN3 occurs due to reduction in NOTCHsignaling. Therefore, some studies use FGF10 to activateNOTCH signaling, followed by the addition of DAPT (thegamma secretase inhibitor) to block NOTCH signaling [24,26, 28]. Moreover, treatment of DE cells with retinoic acid(RA) and dorsomorphin (a BMP type 1 receptor inhibitor)enhances the differentiation into PDX1-expressing pancreaticprogenitors [29]. The epidermal growth factor (EGF) signal-ing increases the proliferation of proliferation of PDX1-positive pancreatic progenitors, derived from hESCs [22]. Asmall molecule named IndolactamV has been found to inducethe differentiation of pancreatic progenitors (PDX1-expressing cells) from hESC-derived DE [24].
Treatment of hESC-derived PDX1-positive pancreatic cellswith SB431542 (an inhibitor of the TGFβ type 1 receptor)induces their differentiation to NGN3-positive pancreatic en-docrine progenitors [29, 46]. A recent study has showed thatthe treatment of ESCs with reserpine and tetrabenazine (TBZ),the inhibitors of vesicular monoamine transporter 2(VMAT2), promotes the differentiation of PDX1-positivecells into NGN3-positive endocrine precursors, and they areable to potentiate differentiation of β cells, which are respon-sive to glucose stimulation [47].
So as to enhance pancreatic β cell maturation, pancreaticprogenitor cells are treated with forskolin (an adenylate
Fig. 1 A schematic representation of the differentiation protocol forinsulin-secreting ß cells from PSCs (hESCs and hiPSCs). PSCs can bedifferentiated into pancreatic ß cells (insulin-secreting cells) after a step-wise protocol into definitive endoderm (DE) (SOX17-positive cells),pancreatic progenitors (PDX1-positive cells), pancreatic endocrine pro-genitors (NGN3-positive cells) and ß cells. For each step of differentia-tion, the cells are treated with one or more of the reagents as indicated inthe diagram. Each step is confirmed by the expression of stage-specificmarkers. CHIR99021 (Wnt signaling activator), CYC (Cyclopamine),DAPT (the gamma secretase inhibitor), Dexamethasone (a synthetic
adrenocortical steroid), Dorsomorphin (a BMP type 1 receptor inhibitor),FGF10 (fibroblast growth factor 10), Forskolin (an adenylate cyclaseactivator), GDF8, GLP-1 (Glucagon-like peptide-1), HGF (hepatocytegrowth factor), IDEs (small molecules IDE1 and IDE2), IGF-1 (insulin-l ike growth factor 1) , Noggin (BMP antagonis t ) , PI3K(Phosphatidylinositide 3-kinases), RA (retinoic acid), Reserpine (inhibi-tor of vesicular monoamine transporter 2), SB431542 (TGFβ inhibitor),SU5402 (FGF receptor antagonist), TBZ (tetrabenazine; inhibitor ofvesicular monoamine transporter 2)
Stem Cell Rev and Rep (2014) 10:327–337 329

cyclase activator) and dexamethasone (a synthetic adrenocor-tical steroid) [29], hepatocyte growth factor (HGF), insulingrowth factor 1(IGF-1) and glucogon-like peptide 1 (GLP-1)[48]. The expression of NKX6.1 is required for in vivo mat-uration of PDX1-positive cells into functional β cells. Thetransplantation of cells expressing high levels of NKX6.1reduces hyperglycemia in diabetic mice, whereas those oflow NKX6.1 levels remain hyperglycemic [49], suggestingthe important role of NKX6.1 in β cell maturation. Theexpression of the transcription factors, which are specific formature β cells, such as C-peptide, INS (insulin), PDX1,MAFA, NKX6-1, NEUROD, ISL-1, and GLUT2, have beenreported to be expressed in insulin-secreting cells derivedfrom hESCs and hiPSCs in vitro [22].
Moreover, previous studies showed that enforced expres-sion of transcription factors, essential for pancreatic develop-ment, can induce pancreatic β cell differentiation [50–52]. Ithas been found that overexpression of PAX4 in hESCs signif-icantly enhanced their differentiation into pancreatic β cells,which expressed high mRNA levels of INS, PDX-1, GLUT2and C-peptide [53]. However, enforced expression of PDX1or FOXA2 showed low effect in enhancing pancreatic differ-entiation in hESCs.
Similar to hESCs, hiPSCs can be differentiated intoinsulin-secreting cells after a step-wise differentiation protocolinto SOX17-positive cells (DE), PDX1-positive cells (pancre-atic progenitors), and NGN3-positive cells (endocrine progen-itors) [20–22, 26, 54, 55], using the same protocols applied tohESCs [22–24]. The first report to show the differentiation ofhiPSCs into pancreatic β cells was in 2008 [21]. Using a four-stage differentiation protocol, they showed that the skinfibroblast-derived hiPSCs can differentiate into insulin-secreting cells, which are responsive to glucose stimulation[21]. Although hiPSC clones generated from T1D patientshave similar capacities to differentiate into DE cells, theyshowed intrapatient variations in their differentiation tendencyto pancreatic β cells, which are more evident in the final stageof differentiation [55]. Furthermore, the variations in pancre-atic differentiation abilities have been observed among hiPSClines [21, 28, 30, 55].
The ability of PSCs to differentiate into fully functionalpancreatic β cells, is still controversial [56]. The most impor-tant feature for defining pancreatic β cell function is glucoseresponsiveness. Also, as a sign of incomplete maturation, thehPSC-derived β cells may co-express multi-hormones, suchas INS, GCG (glucagon) and C-peptide, and absence of theexpression of specific mature pancreatic β cell markers suchas NKX6-1 and MAFA [21]. Although several studies suc-cessfully generated insulin-secreting cells in vitro from hESCsusing step-wise differentiation protocols [22, 23, 25, 26,30, 31, 33, 35, 57–59], the functionality of the pro-duced β cells is very low, since they showed littleglucose responsiveness.
An important role for the in vivo microenvironment in thematuration of pancreatic β cells has been inferred from thetransplantation studies. It has been found that the transplanta-tion of immature pancreatic β cells or pancreatic progenitorsinto experimental animals leads to the maturation of pancre-aticβ cells in vivo. Previous studies reported that when hESC-derived pancreatic progenitors are transplanted into kidneycapsule or fat pad in healthy mice [25, 31], or mice withstreptozotocin (STZ)-induced diabetes [60], they can be dif-ferentiated into functional, mature pancreatic β cells. Further-more, hESC-derived pancreatic progenitor cells are efficientlydifferentiated into mature, glucose-responsive pancreatic βcells, after their transplantation within macroencapsulationdevices into diabetic mice [61]. The transplanted iPSC-derived β cells into two mouse models of T1D and T2D, areable to efficiently secrete insulin in vivo in response to glucoseand improved hyperglycemic phenotype [62]. Furthermore,transplantation of monkey iPSC-derived β cells could rescuehyperglycemia in diabetic mouse models [46]. Moreover, theiPSC-derived insulin-secreting cells obtained from pancreaticepithelial cells in none-obese diabetic (NOD) mice, a model ofT1D, produce insulin in response to glucose stimulation, andtheir transplantation into a kidney of non-obese diabetic miceleads to a graft, with a functional response to glucose stimu-lation [63]. Taken together, these findings suggest that in vivomaturation is essential for the functionality of PSC-derivedpancreatic β cells and suggest that there are specific signals atthe transplantation sites promoting β cell differentiation andmaturation.
Generation of Pluripotent Stem Cells from DiabeticPatients and Their Interest for Molecular Characteristicsof Diabetes sub Types
The pathogenesis of diabetes subtypes is far to be understood.Therefore, obtaining cells genetically identical to diabeticpatients provide a valuable tool in basic research as anin vitro model of diabetes, which is important for understand-ing the molecular mechanisms underlying different types ofdiabetes. PSCs can be used to generate patient-specific stemcells to study diabetes in the petri dish, or engineered intopancreatic β cells to be transplanted into a donor’s own body.
Patient Specific-Embryonic Stem Cells
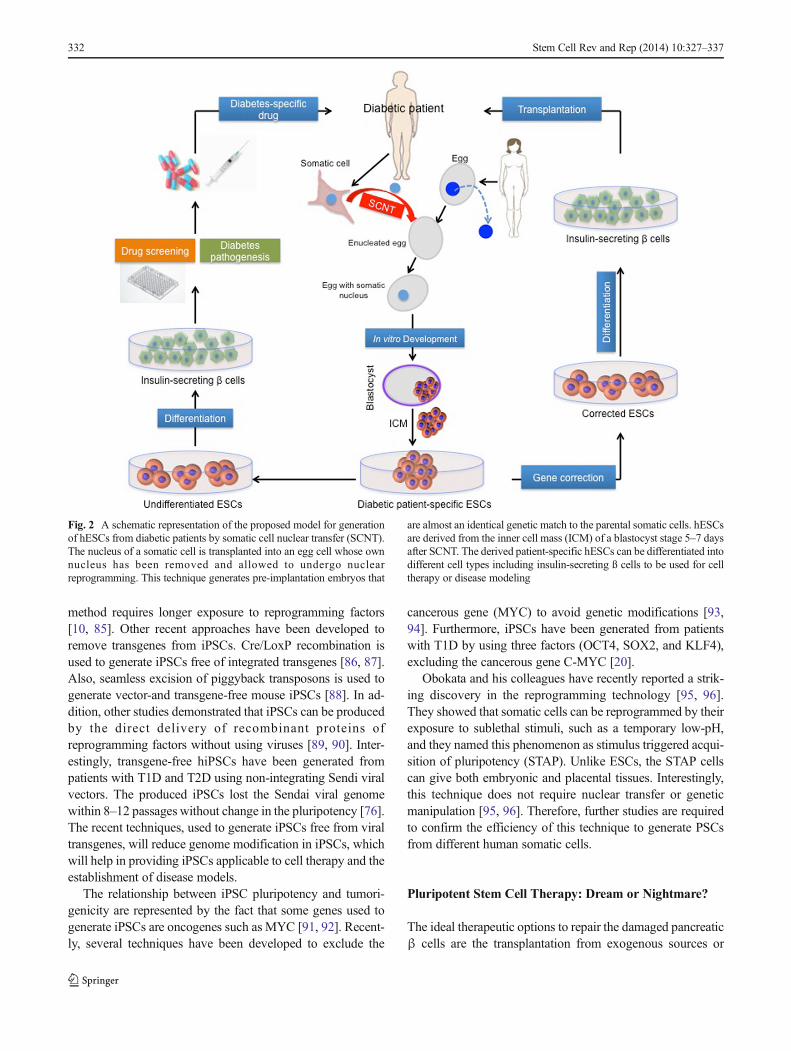
Somatic cell nuclear transfer (SCNT), which is also namedtherapeutic cloning, is considered a way of generating person-alized ESCs from somatic cells of the patients. SCNT is atechnical approach in which the nucleus of a somatic cell istransplanted into an egg cell whose nucleus has been removed,generating embryos that are almost an identical match to theparental somatic cells (Fig. 2). For several years, SCNT was
330 Stem Cell Rev and Rep (2014) 10:327–337

the only method to generate patient-specific ESC lines [64,65], which could be used to study disease mechanisms andsubsequently for autologous transplantation (Fig. 2). Al-though SCNT technology has been used to produce the firstmammals “Dolly the sheep” in the 1997 [66], it never workedin humans for several years. After past difficulties to producehESCs via SCNT, a recent study has successfullyreprogrammed human somatic cells into hESCs using SCNTapproach [67]. Interestingly, a recent study has reported thatautologous hESCs and hiPSCs are equivalent in that theydidn’t evoke the immune responses after the transplantationof the differentiated cells [68]. Although this success providesa platform to generate diabetic patient-specific ESCs, thistechnology is still facing some concerns, such as ethical andpractical troubles to obtain human oocyte.
On the other hand, there is another way to produce hESClines with genetic disorders by using embryos diagnosed asabnormal in preimplantation genetic diagnosis (PGD). PGD isa method, allowing for the discovery of a genetic defect at thelevel of an embryo fertilized in vitro [69]. Many studies havesuccessfully generated hESC lines for several genetic disor-ders from PGD embryos [70–72]. Establishing PGD-hESClines generated from embryos with mutations from monogen-ic diseases or chromosomal abnormalities offer an opportunityto produce in vitro disease models, which help in studyinghuman genetics, drug screening, and cell therapy. Althoughthis technique may be applicable to generate hESC lines frommonogenic diabetes, it cannot be used to generate patient-specific hESCs from non-inherited types of diabetes. In addi-tion to ethical concerns surround the use of hESCs, thistechnique has other limitations including, the access to pre-implanted human embryos, the restriction to study certaindiseases, which are investigated by PGD, and the technicaldifficulties [73].
Patient Specific-Induced Pluripotent Stem Cells
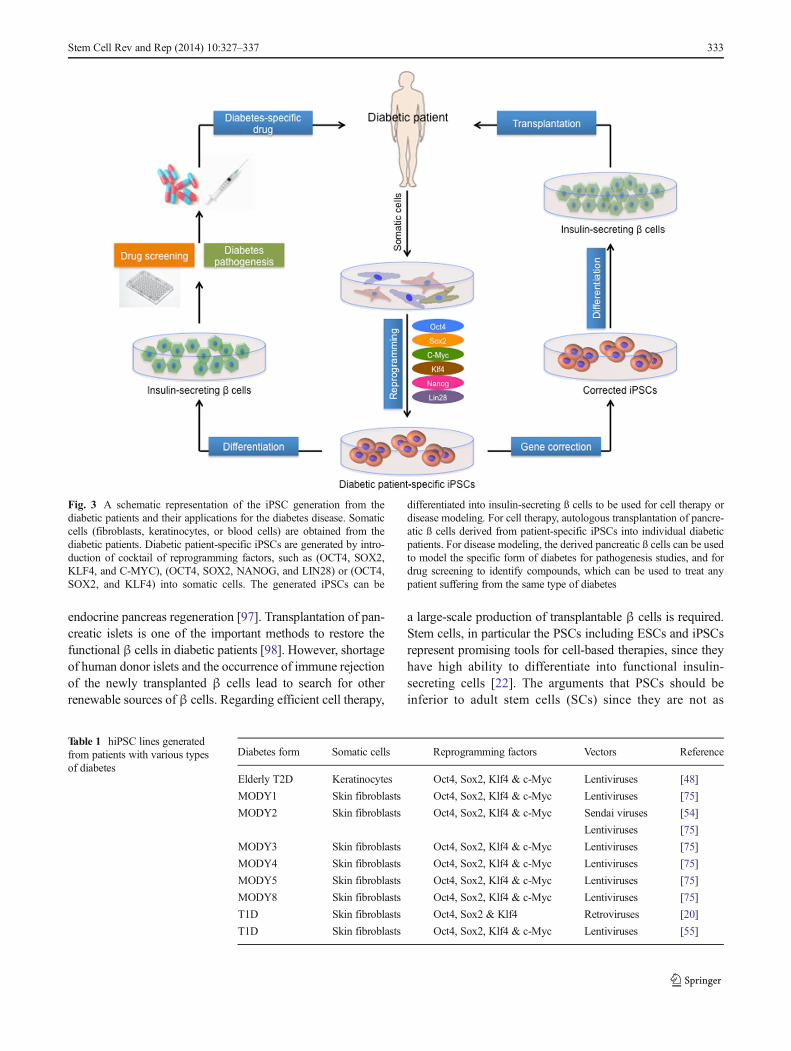
Since hESCs have a lot of restrictions to be applicable fordisease modeling, the researchers have recently focused onalternative approaches, particularly using the recently devel-oped iPSC technology (Fig. 3). The establishment of hiPSCsfrom diabetic patients and differentiation into insulin-secreting cells is important to be used as a material to studyand identify the novel pathways, contributing to the patho-genesis of diabetes disease [74]. Several studies have recentlydescribed the generation of iPSC lines from patients withdifferent types of diabetes [20, 48, 54, 55, 75, 76] (Table 1).
The first hiPSC lines have been generated from the skinfibroblasts of T1D patients using three transcription factors(OCT4, SOX2, and KLF4) [20]. Recently, hiPSC lines havebeen derived from monogenic form of diabetes, MODY tomodel the disease in vitro. Hua et al. has generated hiPSCsfrom MODY2 patients [54], which is characterized by
mutation in the gene encoding glucokinase (GCK) [77–79].The hiPSCs derived from MODY2 patients heterozygous forhypomorphic GCK mutations differentiated into insulin-secreting β cells with similar efficiency to that of controls.On the contrary, iPSCs with two inactive GCK alleles showeda reduced ability to differentiate into insulin-secreting β cells[54]. In theMODY2 patients with GCKmutations,β cells canrespond to glucose but with low sensitivity [80]. However,MODY2-specific iPSCs subjected to GCK gene correctionhave the ability to differentiate into pancreatic β cells withnormal glucose sensitivity [54]. Another study has success-fully generated hiPSC lines from patients with different typesof MODY including; MODY1 (HNF4A), MODY2 (GCK),MODY3 (HNF1A), MODY5 (HNF1B), and MODY8 (CEL)[75] (Table 1). This study used a polycistronic lentiviral vectorfor reprogramming, which gave a higher efficiency than thefrequently used retroviruses, and the derived MODY-hiPSCsshowed no karyotypic defects [75]. The generation of MODYpatient-specific iPSCs is important to investigate the role andmechanisms of MODY genes in pancreatic development andin diabetes.
Although some studies claimed that cellular senescencehinders reprogramming process [81, 82], a recent study re-ported that hiPSCs could be derived from epidermalkeratinocytes of elderly patients with T2D (from 56 to 78 yearsold) under serum free/feeder free culture conditions [48]. Thekeratinocyte-derived iPSCs from diabetic and non-diabeticpatients are highly similar to hESCs and are able to differen-tiate into all lineages, including insulin-secreting cells [48].
Previous studies reported that there are intrapatient varia-tions in the differentiation of diabetic patient-specific iPSCsinto pancreatic β cells [21, 30, 55], which may suggest adifference in the reprogramming efficiency among differentiPSC lines. Also, stem cells from different genetic back-grounds lead to distinct states of pluripotency in vitro. iPSCsfrom nonobese diabetic mice showed an unstable pluripotentstate, indicating effect of the T1D-predispositing genetic back-ground on pluripotency of iPSCs [83]. Therefore, derivationof iPSCs from different individuals suffering from the sametype of diabetes and presenting with similar clinical charac-teristics, may give essential information about certain predis-posing genes.
The reprogramming of somatic cells into iPSCs occurs byusing viral transfection of transcription factors. The majorlimitation of this technology is the use of harmful genome-integrating viruses, in which the vector backbone andtransgenes are permanently incorporated into the genome.This incorporation can cause mutations, which may hinderthe normal function of iPSCs, their differentiation ability, orcause tumorigenesis [2, 5, 84]. To overcome this problem,some studies have generated iPSCs using adenoviral-reprogramming method, in which non-integrating adenovirustransiently expressing Oct4, Sox2, Klf4 and c-Myc. This
Stem Cell Rev and Rep (2014) 10:327–337 331
method requires longer exposure to reprogramming factors[10, 85]. Other recent approaches have been developed toremove transgenes from iPSCs. Cre/LoxP recombination isused to generate iPSCs free of integrated transgenes [86, 87].Also, seamless excision of piggyback transposons is used togenerate vector-and transgene-free mouse iPSCs [88]. In ad-dition, other studies demonstrated that iPSCs can be producedby the direct delivery of recombinant proteins ofreprogramming factors without using viruses [89, 90]. Inter-estingly, transgene-free hiPSCs have been generated frompatients with T1D and T2D using non-integrating Sendi viralvectors. The produced iPSCs lost the Sendai viral genomewithin 8–12 passages without change in the pluripotency [76].The recent techniques, used to generate iPSCs free from viraltransgenes, will reduce genome modification in iPSCs, whichwill help in providing iPSCs applicable to cell therapy and theestablishment of disease models.
The relationship between iPSC pluripotency and tumori-genicity are represented by the fact that some genes used togenerate iPSCs are oncogenes such as MYC [91, 92]. Recent-ly, several techniques have been developed to exclude the
cancerous gene (MYC) to avoid genetic modifications [93,94]. Furthermore, iPSCs have been generated from patientswith T1D by using three factors (OCT4, SOX2, and KLF4),excluding the cancerous gene C-MYC [20].
Obokata and his colleagues have recently reported a strik-ing discovery in the reprogramming technology [95, 96].They showed that somatic cells can be reprogrammed by theirexposure to sublethal stimuli, such as a temporary low-pH,and they named this phenomenon as stimulus triggered acqui-sition of pluripotency (STAP). Unlike ESCs, the STAP cellscan give both embryonic and placental tissues. Interestingly,this technique does not require nuclear transfer or geneticmanipulation [95, 96]. Therefore, further studies are requiredto confirm the efficiency of this technique to generate PSCsfrom different human somatic cells.
Pluripotent Stem Cell Therapy: Dream or Nightmare?
The ideal therapeutic options to repair the damaged pancreaticβ cells are the transplantation from exogenous sources or
Fig. 2 A schematic representation of the proposed model for generationof hESCs from diabetic patients by somatic cell nuclear transfer (SCNT).The nucleus of a somatic cell is transplanted into an egg cell whose ownnucleus has been removed and allowed to undergo nuclearreprogramming. This technique generates pre-implantation embryos that
are almost an identical genetic match to the parental somatic cells. hESCsare derived from the inner cell mass (ICM) of a blastocyst stage 5–7 daysafter SCNT. The derived patient-specific hESCs can be differentiated intodifferent cell types including insulin-secreting ß cells to be used for celltherapy or disease modeling
332 Stem Cell Rev and Rep (2014) 10:327–337
endocrine pancreas regeneration [97]. Transplantation of pan-creatic islets is one of the important methods to restore thefunctional β cells in diabetic patients [98]. However, shortageof human donor islets and the occurrence of immune rejectionof the newly transplanted β cells lead to search for otherrenewable sources of β cells. Regarding efficient cell therapy,
a large-scale production of transplantable β cells is required.Stem cells, in particular the PSCs including ESCs and iPSCsrepresent promising tools for cell-based therapies, since theyhave high ability to differentiate into functional insulin-secreting cells [22]. The arguments that PSCs should beinferior to adult stem cells (SCs) since they are not as
Fig. 3 A schematic representation of the iPSC generation from thediabetic patients and their applications for the diabetes disease. Somaticcells (fibroblasts, keratinocytes, or blood cells) are obtained from thediabetic patients. Diabetic patient-specific iPSCs are generated by intro-duction of cocktail of reprogramming factors, such as (OCT4, SOX2,KLF4, and C-MYC), (OCT4, SOX2, NANOG, and LIN28) or (OCT4,SOX2, and KLF4) into somatic cells. The generated iPSCs can be
differentiated into insulin-secreting ß cells to be used for cell therapy ordisease modeling. For cell therapy, autologous transplantation of pancre-atic ß cells derived from patient-specific iPSCs into individual diabeticpatients. For disease modeling, the derived pancreatic ß cells can be usedto model the specific form of diabetes for pathogenesis studies, and fordrug screening to identify compounds, which can be used to treat anypatient suffering from the same type of diabetes
Table 1 hiPSC lines generatedfrom patients with various typesof diabetes
Diabetes form Somatic cells Reprogramming factors Vectors Reference
Elderly T2D Keratinocytes Oct4, Sox2, Klf4 & c-Myc Lentiviruses [48]
MODY1 Skin fibroblasts Oct4, Sox2, Klf4 & c-Myc Lentiviruses [75]
MODY2 Skin fibroblasts Oct4, Sox2, Klf4 & c-Myc Sendai viruses [54]
Lentiviruses [75]
MODY3 Skin fibroblasts Oct4, Sox2, Klf4 & c-Myc Lentiviruses [75]
MODY4 Skin fibroblasts Oct4, Sox2, Klf4 & c-Myc Lentiviruses [75]
MODY5 Skin fibroblasts Oct4, Sox2, Klf4 & c-Myc Lentiviruses [75]
MODY8 Skin fibroblasts Oct4, Sox2, Klf4 & c-Myc Lentiviruses [75]
T1D Skin fibroblasts Oct4, Sox2 & Klf4 Retroviruses [20]
T1D Skin fibroblasts Oct4, Sox2, Klf4 & c-Myc Lentiviruses [55]
Stem Cell Rev and Rep (2014) 10:327–337 333

frequently used in clinical trials are clearly misleading. Itshould be noted that the adult SCs have been in use for morethan 50 years, while hESCs have been established in 1998 (only16 years ago) [1] and hiPSCs have been established in 2007(only 7 years ago) [2]. It has been found that insulin-secreting βcells derived from PSCs are closer in nature to the in vivoβ cellsthan those produced from adult stem cells [26, 34].
For transplantation therapy, hESCs have to be used in anallogeneic manner, which means they are non-self transplant.Given that transplantation of hESC-derived pancreatic β cellscan evoke the immune system, it is essential to useimmunosuppressors to avoid immune rejection from the pa-tient’s body. Immunosuppressive drugs have a role in insulinresistance and suppression of insulin secretion [99]. Further-more, other problems with hESCs are the ethical concerns,and the risk of inducing cancer, occurring as a result ofcontamination of the transplanted cells with undifferentiatedcells.
Recent progress in the stem cell field suggests that onlyautologous stem cell transplantation are likely to be acceptedas a cell therapy for diabetes, while allogeneic transplantationmay be considered for certain cases, such as when associatedwith islet transplantation [100]. Therefore, iPSC-derived βcell autologous transplantation may be the solution to avoidthe risk of immune rejection and to exclude the necessity forimmunosuppression. Furthermore, iPSCs can provide treat-ment, which is personalized to the particular characteristics ofeach patient and takes into account the etiology and severity ofthe condition. However, iPSCs also faces problems for clinicalapplication such as teratoma formation. Also, some genesused to generate iPSCs (such as MYC) are oncogenes [91,92]. Therefore, before iPSCs can be considered a tool forregenerative medicine to treat diabetes, additional in vitroand in vivo studies are required, as the patient safety is themost important issue to be considered.
Challenges and Future Perspectives for Precisionand Personalized Medicine
Over recent years, our knowledge about the PSCs and theiruse for disease modeling and cell therapy has greatly expand-ed. The studies reviewed here discuss the recent progress inusing the PSCs to understand the pathogenesis of differenttypes of diabetes by generating diabetic patient-specific stemcells in vitro, their differentiation into pancreatic β cells, andhow these are applicable to cure diabetes.
Before PSCs find their way to clinical trials for treatingdiabetes, several challenges remain to be addressed. To avoidthe risk of inducing cancer, occurring as a result of contami-nation of the transplanted cells with undifferentiated cells, it isessential to develop a highly efficient protocol for differenti-ation of PSCs into pure population of pancreatic β cells. The
use of hESCs in regenerative medicine faces several problemsrelated to the ethical concerns and their immunologicallyincompatible, which restrict their clinical application. There-fore, generation of autologous stem cells including hiPSCsand hESCs from somatic cells is essential for the future ofregenerative medicine. The recent success in generatinghESCs using SCNT can be used to produce diabetic patient-specific hESCs, immunologically compatible to the patient.
Although iPSC technology represents a noteworthy dis-covery in the generation of insulin-secreting β cells as wellas patient-specific iPSCs, we believe that there are still gapsand problems in the iPSC field, which need to be clarified.Further in vivo studies are required to examine how thedifferentiated pancreatic β cells from iPSCs behave aftertransplantation. Importantly, the genomic stability of iPSCsshould be confirmed since mutations may occur, which areassociated with the reprogramming process itself as a result ofusing harmful genome-integrating viruses, and suppressing/deleting some genes to enhance the reprogramming efficiency.Furthermore, other genetic defects could be inherited from thereprogrammed somatic cells. These defects and mutationsmay negatively influence the data obtained from investigatingthe diabetic patient-iPSCs. Therefore, the effort should befocused to use safer vectors, lacking of the integration offoreign genetic materials into the host genome, to examinegenome integrity, and use more reliable methods to enhancereprogramming process.
Another important issue has been recently raised aboutwhether cells differentiated from hESCs and hiPSCs reflectmature adult phenotypes of pancreatic β cells. A previousreport showed that although most of the pluripotency genesare effectively silenced after differentiation of hESCs andhiPSCs, some genes typically associated with early embryosincluding DPPA4, LIN28A, and LIN28B remains expressedin the differentiated cells, indicating that the cells derived fromhESCs and hiPSCs are equivalent to the cells found prior to6 weeks of human development [101]. Although PSCs aredifferentiated into insulin-secreting cells, they are not fullyfunctional since they are not highly responsive to glucose,which might be as a result of incomplete maturation of pan-creatic β cells. Therefore, further work is necessary to adjustthe differentiation protocols so as to obtain fully maturepancreatic β cells equivalent to their adult counterparts,which are required for disease modeling and cell thera-py applications.
Since previous studies showed intrapatient variations in thedifferentiation of diabetic patient-specific iPSCs into pancre-atic β cells, personalized iPSC applications require the selec-tion of iPSC clones, which have the ability to differentiate intofunctional pancreatic β cells. Further studies are required toreduce the clonal variations, which would facilitate theway for new personalized medicine applications fordiabetic patients.
334 Stem Cell Rev and Rep (2014) 10:327–337

One essential question remaining to be addressed is theiPSC nature and whether hiPSCs can replace hESCs. Al-though some studies underscored the importance for generat-ing more hESCs for research purposes, the studies of hESCsconcurrently with hiPSCs may play an important role inacquiring the basic knowledge to address the problems facingthe stem cell research.
Despite above-mentioned challenges, much hope has beenderived from PSC research, since these stem cells can providecells genetically identical to the diabetic patients, which canprovide an in vitro disease model to study the pathophysiolo-gy of different forms of the disease, and consequently willfacilitate to find the way to treat diabetes. In spite of the shorthistory of PSCs, hESCs and hiPSCs are already in clinicaltrials for some other diseases. Taken together with the fact thatβ cells derived from PSCs are close in nature to true pancre-atic β cells, therefore we predict a rapid progress in the nearfuture to discover novel mechanisms underlying differentforms of diabetes and to provide a personalized medicineusing PSCs.
Conflict of Interest Statement We declare no potential conflicts ofinterest
References
1. Thomson, J. A., et al. (1998). Embryonic stem cell lines derivedfrom human blastocysts. Science, 282(5391), 1145–1147.
2. Takahashi, K., et al. (2007). Induction of pluripotent stem cells fromadult human fibroblasts by defined factors. Cell, 131(5), 861–872.
3. Aoi, T., et al. (2008). Generation of pluripotent stem cells from adultmouse liver and stomach cells. Science, 321(5889), 699–702.
4. Hanna, J., et al. (2008). Direct reprogramming of terminally differ-entiated mature B lymphocytes to pluripotency. Cell, 133(2), 250–264.
5. Yu, J., et al. (2007). Induced pluripotent stem cell lines derived fromhuman somatic cells. Science, 318(5858), 1917–1920.
6. Abdelalim, E. M. (2013). Molecular mechanisms controlling thecell cycle in embryonic stem cells. Stem Cell Reviews, 9(6), 764–773.
7. Ruiz, S., et al. (2011). A high proliferation rate is required for cellreprogramming and maintenance of human embryonic stem cellidentity. Current Biology, 21(1), 45–52.
8. Zhang, Z. N., et al. (2013). Oct4 maintains the pluripotency ofhuman embryonic stem cells by inactivating p53 through Sirt1-mediated deacetylation. Stem Cells, 32, 157–65.
9. Chin, M. H., et al. (2009). Induced pluripotent stem cells andembryonic stem cells are distinguished by gene expression signa-tures. Cell Stem Cell, 5(1), 111–123.
10. Stadtfeld, M., et al. (2008). Induced pluripotent stem cells generatedwithout viral integration. Science, 322(5903), 945–949.
11. Danaei, G., et al. (2011). National, regional, and global trends infasting plasma glucose and diabetes prevalence since 1980: system-atic analysis of health examination surveys and epidemiologicalstudies with 370 country-years and 2.7 million participants.Lancet, 378(9785), 31–40.
12. Gregg, E.W., et al. (2007).Mortality trends inmen and womenwithdiabetes, 1971 to 2000. Annals of Internal Medicine, 147(3), 149–155.
13. Tisch, R., & McDevitt, H. (1996). Insulin-dependent diabetesmellitus. Cell, 85(3), 291–297.
14. van Belle, T. L., Coppieters, K. T., & von Herrath, M. G. (2011).Type 1 diabetes: etiology, immunology, and therapeutic strategies.Physiological Reviews, 91(1), 79–118.
15. Doria, A., Patti, M. E., & Kahn, C. R. (2008). The emerging geneticarchitecture of type 2 diabetes. Cell Metabolism, 8(3), 186–200.
16. Bonnefond, A., Froguel, P., & Vaxillaire, M. (2010). The emerginggenetics of type 2 diabetes. Trends in Molecular Medicine, 16(9),407–416.
17. Vaxillaire, M., & Froguel, P. (2008). Monogenic diabetes in theyoung, pharmacogenetics and relevance to multifactorial forms oftype 2 diabetes. Endocrine Reviews, 29(3), 254–264.
18. Bonnefond, A., et al. (2012). Whole-exome sequencing and highthroughput genotyping identified KCNJ11 as the thirteenth MODYgene. PLoS One, 7(6), e37423.
19. Efrat, S. (2008). Beta-cell replacement for insulin-dependent diabe-tes mellitus. Advanced Drug Delivery Reviews, 60(2), 114–123.
20. Maehr, R., et al. (2009). Generation of pluripotent stem cellsfrom patients with type 1 diabetes. Proceedings of theNational Academy of Sciences of the United States ofAmerica, 106(37), 15768–15773.
21. Tateishi, K., et al. (2008). Generation of insulin-secreting islet-likeclusters from human skin fibroblasts. Journal of BiologicalChemistry, 283(46), 31601–31607.
22. Zhang, D., et al. (2009). Highly efficient differentiation of humanES cells and iPS cells into mature pancreatic insulin-producingcells. Cell Research, 19(4), 429–438.
23. Jiang, J., et al. (2007). Generation of insulin-producing islet-likeclusters from human embryonic stem cells. StemCells, 25(8), 1940–1953.
24. Chen, S., et al. (2009). A small molecule that directs differentiationof human ESCs into the pancreatic lineage. Nature ChemicalBiology, 5(4), 258–265.
25. Kroon, E., et al. (2008). Pancreatic endoderm derived from humanembryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nature Biotechnology, 26(4), 443–452.
26. D’Amour, K. A., et al. (2006). Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells.Nature Biotechnology, 24(11), 1392–1401.
27. Lumelsky, N., et al. (2001). Differentiation of embryonic stem cellsto insulin-secreting structures similar to pancreatic islets. Science,292(5520), 1389–1394.
28. Thatava, T., et al. (2011). Indolactam V/GLP-1-mediated differen-tiation of human iPS cells into glucose-responsive insulin-secretingprogeny. Gene Therapy, 18(3), 283–293.
29. Kunisada, Y., et al. (2012). Small molecules induce efficient differ-entiation into insulin-producing cells from human induced pluripo-tent stem cells. Stem Cell Research, 8(2), 274–284.
30. Nostro, M. C., et al. (2011). Stage-specific signaling throughTGFbeta family members and WNT regulates patterning and pan-creatic specification of human pluripotent stem cells. Development,138(5), 861–871.
31. Shim, J. H., et al. (2007). Directed differentiation of human embry-onic stem cells towards a pancreatic cell fate. Diabetologia, 50(6),1228–1238.
32. Gage, B. K., Webber, T. D., & Kieffer, T. J. (2013). Initial cellseeding density influences pancreatic endocrine development dur-ing in vitro differentiation of human embryonic stem cells. PLoSOne, 8(12), e82076.
33. Jiang, W., et al. (2007). In vitro derivation of functional insulin-producing cells from human embryonic stem cells. Cell Research,17(4), 333–344.
Stem Cell Rev and Rep (2014) 10:327–337 335

34. D’Amour, K. A., et al. (2005). Efficient differentiation of humanembryonic stem cells to definitive endoderm.Nature Biotechnology,23(12), 1534–1541.
35. Mfopou, J. K., et al. (2010). Noggin, retinoids, and fibroblastgrowth factor regulate hepatic or pancreatic fate of human embry-onic stem cells. Gastroenterology, 138(7), 2233–2245. 2245 e1–14.
36. Johannesson, M., et al. (2009). FGF4 and retinoic acid direct dif-ferentiation of hESCs into PDX1-expressing foregut endoderm in atime- and concentration-dependent manner. PLoS One, 4(3), e4794.
37. McLean, A. B., et al. (2007). Activin a efficiently specifies defini-tive endoderm from human embryonic stem cells only when phos-phatidylinositol 3-kinase signaling is suppressed. Stem Cells, 25(1),29–38.
38. Tian, T., & Meng, A. M. (2006). Nodal signals patternvertebrate embryos. Cellular and Molecular Life Sciences,63(6), 672–685.
39. Kubo, A., et al. (2004). Development of definitive endoderm fromembryonic stem cells in culture. Development, 131(7), 1651–1662.
40. Hosoya,M. (2012). Preparation of pancreatic beta-cells from humaniPS cells with small molecules. Islets, 4(3), 249–252.
41. Bruin, J. E., et al. (2013). Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells.Stem Cell Research, 12(1), 194–208.
42. Bone, H. K., et al. (2011). A novel chemically directed route for thegeneration of definitive endoderm from human embryonic stemcells based on inhibition of GSK-3. Journal of Cell Science,124(Pt 12), 1992–2000.
43. Borowiak, M., et al. (2009). Small molecules efficiently directendodermal differentiation of mouse and human embryonic stemcells. Cell Stem Cell, 4(4), 348–358.
44. Wandzioch, E., & Zaret, K. S. (2009). Dynamic signaling networkfor the specification of embryonic pancreas and liver progenitors.Science, 324(5935), 1707–1710.
45. Bernardo, A. S., et al. (2009). Biphasic induction of Pdx1 in mouseand human embryonic stem cells can mimic development of pan-creatic beta-cells. Stem Cells, 27(2), 341–351.
46. Zhu, F. F., et al. (2011). Generation of pancreatic insulin-producingcells from rhesus monkey induced pluripotent stem cells.Diabetologia, 54(9), 2325–2336.
47. Sakano, D., et al. (2014). VMAT2 identified as a regulator of late-stage beta-cell differentiation. Nat Chem Biol, 10, 141–8.
48. Ohmine, S., et al. (2012). Reprogrammed keratinocytes from elderlytype 2 diabetes patients suppress senescence genes to acquire in-duced pluripotency. Aging (Albany NY), 4(1), 60–73.
49. Rezania, A., et al. (2013). Enrichment of human embryonic stemcell-derived NKX6.1-expressing pancreatic progenitor cells accel-erates the maturation of insulin-secreting cells in vivo. Stem Cells,31, 2432–2442.
50. Blyszczuk, P., et al. (2003). Expression of Pax4 in embryonic stemcells promotes differentiation of nestin-positive progenitor andinsulin-producing cells. Proceedings of the National Academy ofSciences of the United States of America, 100(3), 998–1003.
51. Kwon, Y. D., et al. (2005). Cellular manipulation of human embry-onic stem cells by TAT-PDX1 protein transduction. MolecularTherapy, 12(1), 28–32.
52. Soria, B., et al. (2000). Insulin-secreting cells derived from embry-onic stem cells normalize glycemia in streptozotocin-induced dia-betic mice. Diabetes, 49(2), 157–162.
53. Liew, C. G., et al. (2008). PAX4 enhances beta-cell differentiation ofhuman embryonic stem cells. PLoS One, 3(3), e1783.
54. Hua, H., et al. (2013). iPSC-derived beta cells model diabetes due toglucokinase deficiency. Journal of Clinical Investigation, 123(7),3146–3153.
55. Thatava, T., et al. (2013). Intrapatient variations in type 1 diabetes-specific iPS cell differentiation into insulin-producing cells.Molecular Therapy, 21(1), 228–239.
56. Teo, A. K., Wagers, A. J., & Kulkarni, R. N. (2013). New opportu-nities: harnessing induced pluripotency for discovery in diabetesand metabolism. Cell Metabolism, 18(6), 775–791.
57. Kelly, O. G., et al. (2011). Cell-surface markers for the isolation ofpancreatic cell types derived from human embryonic stem cells.Nature Biotechnology, 29(8), 750–756.
58. Cai, J., et al. (2010). Generation of homogeneous PDX1(+) pancre-atic progenitors from human ES cell-derived endoderm cells.Journal of Molecular Cell Biology, 2(1), 50–60.
59. Xu, X., Browning, V. L., & Odorico, J. S. (2011). Activin, BMP andFGF pathways cooperate to promote endoderm and pancreaticlineage cell differentiation from human embryonic stem cells.Mechanisms of Development, 128(7–10), 412–427.
60. Rezania, A., et al. (2012). Maturation of human embryonic stemcell-derived pancreatic progenitors into functional islets capable oftreating pre-existing diabetes in mice. Diabetes, 61(8), 2016–2029.
61. Bruin, J. E., et al. (2013). Maturation and function of humanembryonic stem cell-derived pancreatic progenitors inmacroencapsulation devices following transplant into mice.Diabetologia, 56(9), 1987–1998.
62. Alipio, Z., et al. (2010). Reversal of hyperglycemia in diabeticmouse models using induced-pluripotent stem (iPS)-derived pan-creatic beta-like cells. Proceedings of the National Academy ofSciences of the United States of America, 107(30), 13426–13431.
63. Jeon, K., et al. (2012). Differentiation and transplantation of func-tional pancreatic beta cells generated from induced pluripotent stemcells derived from a type 1 diabetes mouse model. Stem Cells andDevelopment, 21(14), 2642–2655.
64. Yang, X., et al. (2007). Nuclear reprogramming of cloned embryosand its implications for therapeutic cloning. Nature Genetics, 39(3),295–302.
65. Lanza, R. P., Cibelli, J. B., & West, M. D. (1999). Prospects for theuse of nuclear transfer in human transplantation. NatureBiotechnology, 17(12), 1171–1174.
66. Wilmut, I., et al. (1997). Viable offspring derived from fetal andadult mammalian cells. Nature, 385(6619), 810–813.
67. Tachibana, M., et al. (2013). Human embryonic stem cells derivedby somatic cell nuclear transfer. Cell, 153(6), 1228–1238.
68. Araki, R., et al. (2013). Negligible immunogenicity of terminallydifferentiated cells derived from induced pluripotent or embryonicstem cells. Nature, 494(7435), 100–104.
69. Sermon, K. D., et al. (2009). Creation of a registry for humanembryonic stem cells carrying an inherited defect: joint collabora-tion between ESHRE and hESCreg. Human Reproduction, 24(7),1556–1560.
70. Verlinsky, Y., et al. (2005). Human embryonic stem cell lines withgenetic disorders. Reproductive Biomedicine Online, 10(1), 105–110.
71. Eiges, R., et al. (2007). Developmental study of fragile Xsyndrome using human embryonic stem cells derived frompreimplantation genetically diagnosed embryos. Cell StemCell, 1(5), 568–577.
72. Mateizel, I., et al. (2006). Derivation of human embryonic stem celllines from embryos obtained after IVF and after PGD for monogen-ic disorders. Human Reproduction, 21(2), 503–511.
73. Lee, G., & Studer, L. (2010). Induced pluripotent stem cell technol-ogy for the study of human disease. Nature Methods, 7(1), 25–27.
74. Maehr, R. (2011). iPS cells in type 1 diabetes research andtreatment. Clinical Pharmacology and Therapeutics, 89(5),750–753.
75. Teo, A. K., et al. (2013). Derivation of human induced pluripotentstem cells from patients with maturity onset diabetes of the young.Journal of Biological Chemistry, 288(8), 5353–5356.
76. Kudva, Y. C., et al. (2012). Transgene-free disease-specific inducedpluripotent stem cells from patients with type 1 and type 2 diabetes.Stem Cells Translational Medicine, 1(6), 451–461.
336 Stem Cell Rev and Rep (2014) 10:327–337

77. Velho, G., et al. (1992). Primary pancreatic beta-cell secretory defectcaused by mutations in glucokinase gene in kindreds of maturityonset diabetes of the young. Lancet, 340(8817), 444–448.
78. Estalella, I., et al. (2007). Mutations in GCK and HNF-1alphaexplain the majority of cases with clinical diagnosis of MODY inSpain. Clinical Endocrinology, 67(4), 538–546.
79. Froguel, P., et al. (1993). Familial hyperglycemia due to mutationsin glucokinase. Definition of a subtype of diabetes mellitus. NewEngland Journal of Medicine, 328(10), 697–702.
80. Byrne, M. M., et al. (1994). Insulin secretory abnormalities insubjects with hyperglycemia due to glucokinase mutations.Journal of Clinical Investigation, 93(3), 1120–1130.
81. Li, H., et al. (2009). The Ink4/Arf locus is a barrier for iPS cellreprogramming. Nature, 460(7259), 1136–1139.
82. Banito, A., et al. (2009). Senescence impairs successful reprogrammingto pluripotent stem cells.Genes and Development, 23(18), 2134–2139.
83. Hanna, J., et al. (2009). Metastable pluripotent states in NOD-mouse-derived ESCs. Cell Stem Cell, 4(6), 513–524.
84. Okita, K., Ichisaka, T., & Yamanaka, S. (2007). Generation ofgermline-competent induced pluripotent stem cells. Nature,448(7151), 313–317.
85. Okita, K., et al. (2008). Generation of mouse induced pluripotentstem cells without viral vectors. Science, 322(5903), 949–953.
86. Soldner, F., et al. (2009). Parkinson’s disease patient-derived in-duced pluripotent stem cells free of viral reprogramming factors.Cell, 136(5), 964–977.
87. Kaji, K., et al. (2009). Virus-free induction of pluripotency and subse-quent excision of reprogramming factors. Nature, 458(7239), 771–775.
88. Woltjen, K., et al. (2009). piggyBac transposition reprograms fibro-blasts to induced pluripotent stem cells. Nature, 458(7239), 766–770.
89. Zhou, H., et al. (2009). Generation of induced pluripotent stem cellsusing recombinant proteins. Cell Stem Cell, 4(5), 381–384.
90. Kim, D., et al. (2009). Generation of human induced pluripotentstem cells by direct delivery of reprogramming proteins. Cell StemCell, 4(6), 472–476.
91. Yamanaka, S. (2007). Strategies and new developments in thegeneration of patient-specific pluripotent stem cells. Cell StemCell, 1(1), 39–49.
92. Knoepfler, P. S. (2008). Why myc? An unexpected ingredient in thestem cell cocktail. Cell Stem Cell, 2(1), 18–21.
93. Nakagawa,M., et al. (2008). Generation of induced pluripotent stemcells without Myc from mouse and human fibroblasts. NatureBiotechnology, 26(1), 101–106.
94. Wernig, M., et al. (2008). c-Myc is dispensable for directreprogramming of mouse fibroblasts. Cell Stem Cell, 2(1), 10–12.
95. Obokata, H., et al. (2014). Stimulus-triggered fate conversion ofsomatic cells into pluripotency. Nature, 505(7485), 641–647.
96. Obokata, H., et al. (2014). Bidirectional developmental potential inreprogrammed cells with acquired pluripotency. Nature, 505(7485),676–680.
97. Santana, A., et al. (2006). Insulin-producing cells derived from stemcells: recent progress and future directions. Journal of Cellular andMolecular Medicine, 10(4), 866–883.
98. Shapiro, A. M., et al. (2000). Islet transplantation in seven patientswith type 1 diabetes mellitus using a glucocorticoid-free immuno-suppressive regimen. New England Journal of Medicine, 343(4),230–238.
99. Penfornis, A., & Kury-Paulin, S. (2006). Immunosuppressive drug-induced diabetes. Diabetes & Metabolism, 32(5 Pt 2), 539–546.
100. Fiorina, P., Voltarelli, J., & Zavazava, N. (2011). Immunologicalapplications of stem cells in type 1 diabetes. Endocrine Reviews,32(6), 725–754.
101. Patterson, M., et al. (2012). Defining the nature of human pluripo-tent stem cell progeny. Cell Research, 22(1), 178–193.
Stem Cell Rev and Rep (2014) 10:327–337 337