pichia pastoris as a host for secretion of toxic saporin chimeras
TRANSCRIPT

The FASEB Journal • Research Communication
Pichia pastoris as a host for secretion of toxic saporinchimeras
Alessio Lombardi,* Sara Bursomanno,* Teresa Lopardo,* Roberta Traini,*Marco Colombatti,† Rodolfo Ippoliti,‡ David J. Flavell,§,�,1 Sopsamorn U. Flavell,§,�
Aldo Ceriotti,*,1 and Maria Serena Fabbrini*,1,2
*Istituto di Biologia e Biotecnologia Agraria, Consiglio Nazionale delle Ricerche, Milan, Italy;†University of Verona, Policlinico G.B. Rossi, Verona, Italy; ‡University of L’Aquila, L’Aquila, Italy;§Leukaemia Busters (Clinical Trials Support Unit), Southampton, UK; and �Simon Flavell LeukaemiaResearch Laboratory, Southampton General Hospital, Southampton, UK
ABSTRACT Most of the targeting moieties, such asantibody fragments or growth factor domains, used toconstruct targeted toxins for anticancer therapy derivefrom secretory proteins. These normally fold in theoxidative environment of the endoplasmic reticulum,and hence their folding in bacterial cells can be quiteinefficient. For instance, only low amounts of properlyfolded antimetastatic chimera constituted by the amino-terminal fragment of human urokinase (ATF) fused tothe plant ribosome-inactivating protein saporin couldbe recovered. ATF-saporin was instead secreted effi-ciently when expressed in eukaryotic cells protectedfrom autointoxication with neutralizing anti-saporinantibodies. Pichia pastoris is a microbial eukaryotic hostwhere these domains can fold into a transport-compe-tent conformation and reach the extracellular medium.We show here that despite some host toxicity codon-usage optimization greatly increased the expressionlevels of active saporin but not those of an active-sitemutant SAP-KQ in GS115 (his4) strain. The lack of anytoxicity associated with expression of the latter confirmedthat toxicity is due to saporin catalytic activity. Neverthe-less, GS115 (his4) cells in flask culture secreted 3.5mg/L of a histidine-tagged ATF-saporin chimera showingan IC50 of 6 � 10�11 M against U937 cells, thus demon-strating the suitability of this expression platform forsecretion of toxic saporin-based chimeras.—Lombardi,A., Bursomanno, S., Lopardo, T., Traini, R., Colombatti,M., Ippoliti, R., Flavell, D. J., Flavell, S. U., Ceriotti, A.,Fabbrini, M. S. Pichia pastoris as a host for secretion oftoxic saporin chimeras. FASEB J. 24, 000–000 (2010).www.fasebj.org
Key Words: plant ribosome-inactivating proteins � eukaryoticexpression � codon usage � tumor-targeted therapy � humanurokinase receptor
Ribosome-inactivating proteins (RIPs) are potentinhibitors of protein synthesis that act by catalyticallydepurinating an adenine residue (A4324 in rats)present in a conserved stem-loop region in 23/26/28Slarge ribosomal RNAs, causing an irreversible arrest inprotein synthesis (1). The prototype plant N-glycosi-dase is a ricin AB dimer (2) that enters mammalian
cells by endocytosis and undergoes retrograde trans-port via the Golgi complex to the endoplasmic reticu-lum (ER) where the catalytic moiety exploits the ER-associated degradation (ERAD) pathway, normally usedfor the disposal of misfolded or unassembled polypep-tides, to reach and depurinate cytosolic ribosomes (3),finally leading to apoptotic cell death (4). This is onereason why plant RIPs, including monomeric saporin,and certain bacterial toxins, such as diphtheria toxin orPseudomonas exotoxin A, are currently used to producecytotoxic chimeras able to kill tumor cells by virtue ofthe specific targeting domain being selected to deliverthe toxic moiety (5). Most of these domains belong tosecretory proteins, such as antibody fragments andligand or growth-factor domains that normally fold inthe eukaryotic cell environment. Pichia pastoris is amicrobial host that recapitulates most of the co- andpost-translational events during protein translation andpossesses an ER quality control system allowing onlysecretion-competent polypeptides to reach the extracellu-lar medium. P. pastoris has been successfully used toproduce �700 heterologous polypeptides (6), includ-ing a type I RIP from Phytolacca americana, PAP (7). Wepreviously produced an antimetastatic chimera be-tween the amino-terminal fragment (ATF) of humanurokinase and saporin that contains 2 SS-rich domains(a growth-factor-like domain and a kringle in the ATFdomain) and demonstrated that, in contrast to whathappened during expression in bacterial host cells (8),correctly folded polypeptides were efficiently secretedwhen using as an expression system Xenopus laevisoocytes that were protected from autointoxication bycytosolic neutralizing anti-saporin antibodies (9), indi-cating that targeting to the ER was an essential step toproduce this chimera in micromolar amounts. For theclinical development of this new class of molecules, weinvestigated in the present study whether we could pro-duce active secretory saporin and saporin-based chimerasin high amounts using the methylotrophic yeast P. pastoris.
1 These authors contributed equally to this work.2 Correspondence: IBBA-CNR, via Bassini 15, 20133 Mi-
lano, Italy. E-mail: [email protected]: 10.1096/fj.08-118042
10892-6638/10/0024-0001 © FASEB
The FASEB Journal article fj.08-118042. Published online September 28, 2009.

The group of D. M. Neville, Jr. (10, 11) pioneered the useof P. pastoris host strains for expression of diphtheriatoxin-based immunotoxins. We therefore investigated dif-ferent P. pastoris strains transformed using constructssubcloned into the pPICZalpha series of vectors fromInvitrogen (Carlsbad, CA, USA), which include the tightlyregulated alcohol-oxydase-1 (AOX-1) promoter inducibleby methanol and the first 90 aa of the prepro-alpha-factordomain of S. cerevisae, to drive secretion of the inducedpolypeptides. Transformation of these strains resulted in adecrease in viable transformed cells when the activesaporin gene was used but not when a saporin KQ mutant,obtained by changing 2 key residues at the catalytic site,glutamic acid 176 and arginine 179 into lysine andglutamine, respectively, was used as a control (12). Meth-anol (MeOH) induction of wild-type transformants alsoalways resulted in growth-curve delays or arrest after 24 h,indicating that saporin expression can hardly be toleratedby the methylotrophic yeast. Nevertheless, followingcodon-usage optimization, we observed a great increase inhigh-expressor clones in the background of GS115(his4) cells expressing active saporin-optimized con-struct (SAPopt). A GS115 (his4) clone could expressup to 30 mg/L secretory saporin in flask cultures,reaching �10-fold better yields than our best bacteriaexpressors (13). Saporin was purified to homogeneityfrom the culture medium and shows identical cytotoxicactivity as the seed-extracted protein but does not seemto exert any DNA-nicking activity, a putative nucleaseactivity recently debated for RNA N-glycosidases (14,15). Finally, we also successfully expressed and purifieda secretory ATF-saporin chimera having a hexahistidinetag for single-step purification, showing the expectedcell-killing activity against human monocytic leukemiacells. Overall, our data open the way to the clinicaldevelopment of secretory targeted chimeras endowedwith ribosome-inactivating activity.
MATERIALS AND METHODS
DNA manipulations and sequencing
The codon-optimized DNA sequence encoding a seed sa-porin isoform was custom synthesized by Genscript (Piscat-away, NJ, USA): the first proposed theoretical sequence wasfurther modified based on the yeast codon usage reported bySreekrishna (16) and also compared with the available P.pastoris coding sequences (CDS) in Biomed Central (64,359codons with corresponding triplet frequencies, choosingthose most frequently represented in highly expressed P.pastoris proteins for the construction of the saporin syntheticgene that was subcloned in pUC57 and obtained as apUC57SOL1GBXbaPst construct). The pPICZalpha series ofvectors from Invitrogen was used for subcloning the amplifiedDNAs or to obtain recipient vectors. DH5-�-competent bac-teria were used for DNA transformation and large-scalepreparation, and putative positive clones were confirmed bysequencing using AOX1 forward and AOX1 reverse primersor the following SAPOPTREV2323 reverse primer: 5�-CCCAACTCCTTTCTGGAC-3� (for sequencing saporin-opti-mized constructs), synthesized by BMR Genomics (Padua,Italy), which custom performed all our sequence analyses.Sequences for synthetic primers were selected using either
Oligo 3.0 or Vector NTI software (Invitrogen), and oligonu-cleotides were synthesized by Primm (Milan, Italy). DNAamplifications were performed using Pfu Turbo-DNA poly-merase (Stratagene, La Jolla, CA, USA).
Saporin constructs
To obtain the KQopt active-site mutant, the saporin catalytic sitewas mutagenized using pUC57pSOL1GBXbaPst as a templateand the QuickChange in vitro mutagenesis system (Stratagene)with the following oligonucleotides: 5�-CTATTCAAATGAC-CGCTAAAGTTGCTCAATTCAGATACATTC-3� and 5�-GAAT-GTATCTGAATTGAGCAACTTTAGCGGTCATTTGAATAG-3�,yielding pUC57KQ mutated plasmid. Saporin constructs includ-ing an Ste13 cleavage site were obtained with primers that carryan EcoRI sequence at the 5�-end and insert a NotI sequence aftera stop codon at the 3�-end (restriction sites are underscored). Toobtain nativeSAP and nativeKQ expression constructs, the follow-ing pair of primers was used both on pDHSAP and pBSSAPKQas templates (unpublished results): 5�-CCGGAATTCGTCACAT-CAATCACATTAG-3� and 5�-CAGTAGCGGCCGCTCACTTTG-GTTTGCCCAAA-3�.
To obtain SAPopt and KQopt expression constructs, weused the following pair of primers: 5�- CCGGAATTCGT-TACCTCCATTACTTTGG-3� and 5�-CAGTAGCGGCCGCT-TACTTTGGCTTTCCCA-3� (NotIKPKrev), together withpUC57pSOL1GBXbaPst and pUC57KQ as templates. Allthe purified EcoRI-NotI fragments were subcloned intoEcoRI-NotI-cut pPICZalphaA to drive expression of maturesaporins ending with Lys253 and having, following Ste13cleavage site, an extra Glu and Phe (encoded within EcoRI)at the N terminus just before Val1.
Saporin constructs termed Kex2SAPopt and Kex2KQopt,including only the KEX-2 Lys-Arg processing site, were ob-tained using a primer that carries a XhoI site at the 5� end andrestores the KEX-2 site just ahead of the first amino acid inthe mature saporin sequence (Val1), being 5�-GCTGC-CGCTCGAGAAAAGAGTTACCTCCATTA-3� coupled to theNotIKPKrev primer. The 2 XhoI-NotI fragments were sub-cloned into XhoI-NotI-cut pPICZalphaB.
Construction of the model saporin chimeras
The SfiI-NotI fragment of a pHEN1 construct containing an�PA63 single-chain variable fragment (scFv) derived from theETH-2-Gold library (17) was purified and inserted into theSfiI-NotI-cut pPICZalphaB recipient vector, and to obtain expres-sion of the scFv�PA63-fused in-frame to the myc-histidine tag, anamber mutation was changed into a glutamine residue at the Nterminus of the VH domain with the following primers: 5�-GAGGCTTGGTACAGCCTGGGGGGTCCCTGAGACTC-3� and5�-GAGTCTCAGGGACCCCCCAGGCTGTACCAAGCCTC-3�. ANotI-NotI fragment of saporin optimized sequence was amplifiedusing the NotIVTSforPic forward primer 5�-CAGTAGCGGC-CGCCGTTACCTCCATTA-3� coupled to NotIKPKrev. The NotIpurified fragment was inserted into NotI-cut scFv�PA63 expres-sion vector to obtain �PA63-SAPopt expression construct inwhich the scFv is fused to the N terminus of saporin via analanine tripeptide linker (encoded within NotI sequence). Toobtain the recipient vector pPICSAPH, the saporin-optimizedsequence was amplified using the NotIVTSforPic primer withthe following reverse primer that includes a hexahistidine tagand a XbaI site: 5�-GCTCTAGATTAATGATGGTGGTGATGAT-GCTTTGGCTTTCCCA-3�. The NotI-XbaI fragment was sub-cloned into NotI-XbaI-cut pPICZalphaB. To obtain the ATF-SAPoptH6 construct, the human ATF fragment was amplifiedusing the plasmid pSP64TpAS (9) as a template with the5�-CGCTCGAGAAAAGAAGCAATGAACTTCAT-3� primer in-
2 Vol. 24 January 2010 LOMBARDI ET AL.The FASEB Journal � www.fasebj.org

serting XhoI and restoring the KEX-2 site just ahead of the firstamino acid in ATF sequence (Ser1) and the reverse 5�-CAG-TAGCGGCCGCTTTTCCATCTGC-3� inserting NotI. A similarconstruct, having a KEX-2 site at the N terminus of humanprourokinase, was previously successfully used for urokinasesecretion by P. pastoris KM71 cells (18). A XhoI-NotI fragment wasinserted into XhoI-NotI-cut pPICSAPH; therefore, as for our scFvfusions to saporin, the linker peptide between ATF and saporindomains is composed of 3 alanines.
Transformation, selection, and induction of P. pastorisclones
The host strains and expression constructs investigated in thisstudy are summarized in Table 1. Electrocompetent P. pastoriscells were prepared according to Invitrogen protocols foreach selected strain: GS115 (his4), KM71H (arg4; aox1:ARG4),and SMD1168 (his4; pep4). A Bio-Rad Gene pulser apparatus(Bio-Rad, Milan, Italy) was used for electroporation. TheDNA constructs were linearized for genomic integrationbefore electroporation using either HindIII, PmeI, or SacI,depending on each construct. DNAs were carefully quanti-tated in ethidium-bromide-stained agarose gels, and equiva-lent amounts of DNA (5–10 �g) resuspended in sterile waterwere used for each electroporation cuvette. Linearized emptypPICZalpha vectors were used for the mock-transformedcells. Then, 200 or 600 �l of transformed cells was plated forselection on YPD [1% (w/v) yeast extract, 2% (w/v) peptoneor tryptone, and 2% (w/v) dextrose] plates containing 18.2%sorbitol (YPDS) in the presence of 1.5% (w/v) agar and 50�g/ml Zeocin (Invitrogen). Colonies started to appear after3–4 d incubation at 30°C, and randomly selected colonieswere restreaked onto YPDS-zeocin plates. For selecting the�PA63-SAPopt best-expressor clone, 80 colonies underwent acolony-lift procedure using 0.45-�m circular Optitran BA-S 85nitrocellulose membranes and circles of prehydrated nitro-cellulose Protran (Whatmann S&S; Sigma-Aldrich, Milan,
Italy) placed onto a MMH plate [1.34% (w/v) yeast nitrogenbase, 4�10�5% (w/v) biotin; 4�10�3% (w/v) histidine; 0.5%(v/v) methanol; and 1.5% (w/v) agar] with 50 �g/m Zeocin.The Optitran BA-S 85 membrane was placed (colonies sideup) on the top of the plate and incubated at 30°C for 48 hbefore treating as for Western blotting with anti-saporin.Usually, 12–20 independent clones were grown for pilotinduction experiments in 5 ml YPD with 50 �g/ml Zeocin at30°C for 16 h. Cells were centrifuged at 1560 g for 4 min atroom temperature and resuspended in 5 ml BMMY [1%(w/v) yeast extract; 2% (w/v) peptone; 100 mM phosphatebuffer, pH 6.0; 1.34% (w/v) yeast nitrogen base, 4�10�5%(w/v) biotin; 4�10�3% (w/v) histidine; and 0.5% (v/v)methanol] to a final OD600/ml of 2.0 in 50 ml conical tubes.Noninduced cells were resuspended in 5 ml of BMDY, whichis the same as BMMY except that 2% (w/v) dextrose is presentinstead of methanol. Cultures were noninduced or inducedfor 48 h at 30°C with shaking at 250 rpm, and 0.5% (v/v)methanol was added after 24 h. At 0, 24, and 48 h, the OD600was checked by diluting cultures 1:10, and the resultinggrowth curves were compared with those of mock-inducedcontrol cultures. Equivalent amounts of induction mediumwere directly analyzed by Western or slot blots for proteinquantification, using seed-extracted saporin (SAP-S) as astandard. Best expressors were grown 16 h in liquid culture,and cells (10–15 OD600) were centrifuged at 1560 g for 5 min,resuspended in 1 ml of YPD supplemented with 15% sterileglycerol, and frozen immediately. Stocks were stored at�80°C until use. For medium-scale inductions, a freshlyplated single colony from these stocks was inoculated in 10 mlof YPD with 50 �g/ml Zeocin, and 5 ml of this overnightpreculture was inoculated in 2-L flask containing 250 ml ofthe same medium and incubated at 30°C, 250 rpm overnight.Cells were centrifuged at 1590 g for 10 min at room temper-ature, resuspended in 250 ml of BMMY (without antibiotics)at final OD600/ml of 10, and incubated at 30°C, 250 rpm for48 h. Methanol [0.5% (v/v)] was supplemented every 24 h.Samples (1 ml) were taken at different times postinductionfor protein expression analyses. At the end of the 48-hinduction period, the medium was collected after centrifuga-tion at 5000 g, 10 min at 4°C and exchanged against PBS (pH7.6; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mMKH2PO4) and concentrated 8- to 10-fold through a HydrosartVivaflow 200 membrane, 10,000 cutoff (Vivascience; SartoriusStedim Biotech, Florence, Italy). The concentrated mediumcontaining the recombinant fusion protein was supple-mented with Complete or, in the case of ATF-SAPoptH6, withEDTA-free Complete (Roche Italy, Monza, Italy), and thenmedia were kept frozen before thawing at 30°C for subse-quent protein purifications.
Protein purifications
Saporin was purified essentially as described by Fabbrini et al.(13), except that the concentrated P. pastoris medium was firstpassed through a CM-Sepharose Fast Flow column using anAKTA-prime apparatus (GE Healthcare Europe, Milan, Italy)to retain yeast contaminant proteins, dialyzing against 20 mMphosphate buffer (pH 6.5), and eluting fractions from thisfirst 2- � 20-cm column with a linear NaCl gradient from 0 to300 mM at a 5 ml/min flow rate. Fractions eluted at 150 mMNaCl were pooled, dialyzed against 20 mM phosphate buffer(pH 6.5), and then further purified onto a strong cationicexchanger Resource S column using an HPLC apparatus(LabService Analytica, Bologna, Italy) with a linear NaClgradient (0–300 mM at 1 ml/min). Saporin eluted as a singlepeak corresponding to fractions 24–27 that were subjected toboth spectrophotometric and silver-staining analyses for ac-curate protein quantifications. ATF-SAPoptH6 protein was
TABLE 1. Constructs and P. pastoris host strains
Construct
Host strain
GS115(his4)
KM71H(arg4; aox1::ARG4)
SMD1168(his4; pep4)
nativeSAP � � �nativeKQ � � �SAPopt � � �KQopt � � �Kex2SAPopt � � �Kex2KQopt � � �scFv�PA63 � � ��PA63-SAPopt � � �ATF-SAPoptH6 � � �
Saporin (SAP) was expressed using a native gene (native) or acodon-optimized construct (opt) carrying either an intact or a mutated(KQ) active site. A single-chain variable fragment (scFv�PA63) againstPA63 protective antigen was expressed alone as a fusion to a myc-histidine tag in GS115 hosts, before expressing a derived model recom-binant immunotoxin (�PA63-SAPopt) in all 3 P. pastoris host strains,GS115 (his4), KM71H (arg4; aox1:ARG4), or SMD1168 (his4; pep4), forcomparison. Constructs normally include Ste13 cleavage sites, exceptthe one for the histidine-tagged ATF-SAPoptH6 chimaera or thoseindicated as Kex2, which have the KEX2 endopeptidase-cleavage siteat the propeptide just ahead of the first amino acid in the maturepolypeptide sequences (see Materials and Methods). �, Construct/host transformations evaluated; �, construct/host transformationsnot evaluated.
3PRODUCTION OF SAPORIN-BASED CHIMERAS IN P. PASTORIS

purified from the concentrated medium with the ProteusIMAC kit (AbD Serotec, Oxford, UK), essentially followingthe manufacturer’s instructions, except that 25 mM imidazolein the binding buffer was used for sample loading and 3washes with 50 mM imidazole in the wash buffer wereperformed before elution in the presence of increasingconcentrations of imidazole (150, 300, and 500 mM); a singlepeak eluted at 150 mM imidazole. Eluates were exchangedagainst PBS (pH 7.6) and concentrated to 1 ml with Vivaspincolumn 10,000 cutoff concentrators (Vivascience; SartoriusStedim Biotech) by centrifuging at 5000 g. Samples wereanalyzed by SDS-PAGE and subjected to silver staining orWestern blotting, using SAP-S as a standard. SAP-S waspurified from Saponaria officinalis seeds as described previ-ously (19).
Soluble intracellular protein extraction
Cellular pellets from induced cultures were thawed in 100 �l(small-scale inductions) or 200 �l (medium-scale inductions)of breaking buffer [50 mM sodium phosphate, pH 7.4; 1 mMPMSF; 1 mM EDTA; 1� Complete; and 5% (w/v) glycerol].An equal volume of acid-washed glass beads (0.5 mm) wasadded, and cell lysates were obtained by vortexing 30 s withintervals of 30 s on ice for a total of 8 times. Lysates werecentrifuged at 18,000 g for 10 min at 4°C, and supernatants(S18) containing soluble proteins were analyzed (1 or 2 �l) byloading an equivalent amount of corresponding conditionedmedium (10 �l) on SDS-PAGE for Western blotting.
In vitro deglycosylation assays
Induction medium (5 or 20 �l) for Kex2KQopt andKex2SAPopt, respectively, best expressors, or rabbit IgGsdiluted in mock-induced medium, as a control (not shown)was boiled 10 min in the presence of 0.5% SDS and 1%�-mercaptoethanol, and incubated at 37°C for 1 h in thepresence of 0.05 M sodium phosphate (pH 7.5), 1% NonidetP-40, and 1000 U of peptide:N-glycosidase F (PNGase F; NewEngland BioLabs, Hitchin, UK) in a final volume of 50 �l.Finally, samples were subjected to precipitation with 10%trichloroacetic acid and resuspension in PBS (pH 7.6) beforeloading into SDS-PAGE for Western blotting.
SDS-PAGE, Western, slot blots, and silver-staining analyses
SDS-PAGE was as described by Fabbrini et al. (9). Westernblot analyses were as described by Fabbrini et al. (13),except that proteins were transferred on PVDF membranes(GE Healthcare Europe), and membranes were incubatedwith a goat anti-rabbit IgG HRP-conjugate diluted 1:15,000,or scFv�PA63 was detected by incubating with 9E10 anti-MYC antibody diluted 1:1000, followed by a goat anti-mouseIgG HRP-conjugate diluted 1:20,000 (not shown), and pro-teins were detected using the SuperSignalWest Pico Chemi-oluminescent Substrate (Pierce Biotechnology Inc., Rock-ford, IL, USA). A Bio-Dot SF Microfiltration Apparatus(Bio-Rad) with Protran nitrocellulose membranes was usedfor slot blots, essentially following manufacturer’s instruc-tions. SAP-S was loaded as a reference standard in replicates.Acrylamide gels were subjected to silver staining using Plu-sOne Silver Staining kit (GE Healthcare Europe) followingthe manufacturer’s instructions. For quantitative measure-ments, band volumes were evaluated by the TotalLab ImageAnalysis System (Phoretix, Newcastle upon Tyne, UK) onsilver-stained gels or using nonsaturated exposures of thefilms (Hyperfilm MP; GE Healthcare Europe).
Biological assays
The nuclease-like activity of secretory saporin (SAPopt) or SAP-Swas assayed using the plasmid DNAs pBR322 or pPICZalphaBexactly as reported by Ghosh and Batra (20) and usingDNaseI as a positive control. RIP activity of secretory saporinswas assayed by measuring the inhibition of BMV RNA trans-lation in nuclease-treated reticulocytes lysates, essentially asdescribed by Fabbrini et al. (13). Crude media, dialyzedagainst PBS (pH 7.6), were also assayed, indicating thatnativeKQ mock-induced media had no activity, as expected;and nativeSAP and SAPopt polypeptides showed the same RIPactivity as SAP-S, whereas boiling the medium abrogatedSAPopt-dependent RIP activity (data not shown). The dose-response curves were obtained using �3 independent exper-iments each read in duplicates, and data are reported asmean values and as a percentage incorporation of the un-treated controls. IC50 indicates the concentration that inhib-ited the incorporation of control samples by 50%. For thecytotoxicity assays, equivalent amounts of purified SAPopt orSAP-S were serially diluted in tissue culture medium, andcytotoxities were compared in HSB-2 human leukemia cellline or in Burkitt lymphoma Daudi cells, treated as in Flavellet al. (21). ATF-SAPoptH6 was assayed against human U937cells that expose both human urokinase and endocytic LRPreceptors, essentially as described by Fabbrini et al. (9) exceptthat non-acid-treated U937 cells were also exposed to seriallogarithmic dilutions ending with 10�14 M ATF-SAPoptH6that incorporated 100% as control U937 cells (data notshown). SAP-S was always assayed as a control. Each experi-ment was always performed at least in triplicates, and data arereported as means with sd. Cytotoxicity was evaluated as theconcentration inhibiting the 50% incorporation in untreatedcontrol cells and is expressed as IC50.
RESULTS
Transformation of Pichia host strains revealsimportance of codon optimization for induction ofactive saporin polypeptides and differences in hostsensitivities
Soapwort (S. officinalis) encodes several type I RIPs(22), and among them, a group of closely relatedsaporin isoforms composed of 253 residues (collectivelyknown as SO6) accumulate in the seeds. They differonly at positions 48 and 91, where either Asp or Gluand Arg or Lys, respectively, can be found, but they allshow identical RIP activity when recombinantly ex-pressed as single isoforms (13). The crystal structure ofa saporin seed isoform has been determined and foundto be highly similar to that of other type I RIPs and ofricin A chain (23). Recently, we isolated a new clone(Sol1) encoding a novel saporin precursor (unpub-lished results), which potentially codes for a matureprotein that differs from seed-extracted SO6 only byhaving Phe (instead of Ser) at position 149, a mutationhaving no effect on the catalytic activity of SO6 (20).This DNA has been first amplified using 2 oligonucle-otides inserting an EcoRI and a NotI site for subcloninginto the pPICZalphaA vector to obtain nativeSAP ex-pression vector (see Materials and Methods). Table 1summarizes the different DNA constructs that havebeen obtained and the P. pastoris host strains that have
4 Vol. 24 January 2010 LOMBARDI ET AL.The FASEB Journal � www.fasebj.org

been investigated in this study. SMD1168 strain wasrecently shown to tolerate expression of a type I RIPfrom P. americana with no apparent host toxicity, secret-ing PAP at 10 mg/L in the culture medium (7).Preliminary work done using nativeSAP, a similar con-struct for expression of the plant native DNA encodingsaporin, indicated that of several SMD1168 zeocine-resistant clones, none was able to induce saporin, withno detectable levels of protein being found in theculture media analyzed. In contrast, of several zeocine-resistant clones in GS115, almost all expressed saporinon induction, although at low levels (below 1 mg/L),with GS115 clones expressing nativeKQ, the active sitemutant SAP-KQ, at 10-fold higher levels (data notshown). These preliminary results prompted us todesign a synthetic gene that was optimized followingthe codon usage of P. pastoris (see Materials andMethods): 60% of the plant codon sequences werechanged overall, avoiding potential depletion in tRNAsfor highly frequent amino acids by choosing differentdegenerated codons in the case of lysine (which ac-counts for 10% of the amino acid sequence ofsaporin). This synthetic gene was amplified and sub-cloned using EcoRI and NotI restriction sites, as for thenativeSAP construct, to obtain the SAPopt construct. Toevaluate the potential contribution, if any, of saporintoxicity toward the host cells (due to the presence of aRIP active site), transformants expressing the active-sitemutant KQopt were also analyzed, for comparison, inGS115, SMD1168, and KM71H strains (Table 1 and Fig. 1).KM71 is a very popular strain devoid of the AOX-1endogenous gene (24) that induces expression of theless abundantly transcribed AOX-2 gene in the pres-ence of methanol, showing a methanol-utilization slow(muts) phenotype but expressing exogenous proteinsunder the AOX-1 promoter at very high levels (6). Weobserved during transformation a loss of 30% viablecolonies when SAPopt was used, as compared withmock or KQopt (Fig. 1A), as previously noticed using
nativeSAP (not shown), thus indicating some host sen-sitivity to active saporin. Next, several independentclones in GS115, SMD1168, and KM71H strains wereselected and induced for SAPopt or KQopt expression,and the minimum and maximal levels of secretedpolypeptide were evaluated in each group of clones,relative to SAP-S (see Materials and Methods). Thequantitative data are reported as shown in Fig. 1B for adirect comparison. The greatest difference in expres-sion between SAPopt (Fig. 1B, top bars) and mutantKQopt (Fig. 1B, bottom bars) is found in the back-ground of KM71H cells, which were able to express upto 120 mg/L KQopt but as low as 2 mg/L of SAPopt.SMD1168 cells, when transformed with the codon-optimized constructs, behaved in an intermediate fash-ion, showing clones expressing SAPopt between 5–10mg/L and an average 10-fold difference in expressionlevels of KQopt. Remarkably, GS115 showed essentiallyno major differences between SAPopt and KQopt ex-pression levels, thus behaving as the less sensitive strainto saporin expression. To better understand the effectsof codon optimization, GS115 cells were retransformedin parallel with the native saporin constructs (nativeSAPor nativeKQ) and the codon-optimized ones (SAPopt orKQopt), and 20 independent clones per transforma-tion were selected to directly compare yields in secretedpolypeptides after induction (Fig. 2). The comparisonin the distribution of clones expressing active saporinconstructs (Fig. 2A) clearly indicates that gene optimi-zation markedly increased the number of SAPopt highexpressors, allowing us to select a best expressor secret-ing up to 30 mg/L when induced at high cell density(see Fig. 4). Conversely, in the case of KQ mutantclones, no major differences were found among theirrelative distributions (Fig. 2B). In addition, KQ andSAP clones all grew similarly in noninduced conditions,reaching after 48 h OD600/ml between 15–20 as themock cultures (not shown), whereas the growth ofclones harboring the active constructs was clearly af-
Figure 1. Sensitivity of P. pastoris hosts to saporin expression. A) P. pastoris hoststrains (GS115, SMD1168, and KM71H) were transformed with empty vector(mock), KQopt, or SAPopt, and 600 �l (left panels) or 200 �l (right panels)of cells were plated; number of colonies obtained in each case is shown herefor KM71H transformants, as an example. B) Randomly picked 14–20 coloniesexpressing SAPopt or KQopt in the 3 host strains were induced for 48 h, andequivalent amounts of medium were quantified by Western or slot blots(Materials and Methods). Bars indicate the level of protein secreted (top bars:SAPopt; bottom bars: KQopt); intervals include levels (mg/L) found amongthe lowest- to the highest-expressing clones in the background of GS115 (blackbars), KM71H (hatched bars), and SMD1168 (white bars).
5PRODUCTION OF SAPORIN-BASED CHIMERAS IN P. PASTORIS

fected on induction of saporin expression (Fig. 2C);however, this was not observed in clones expressing KQmutant constructs (Fig. 2D).
Defective processing of saporin precursors lackingSte13 cleavage sites
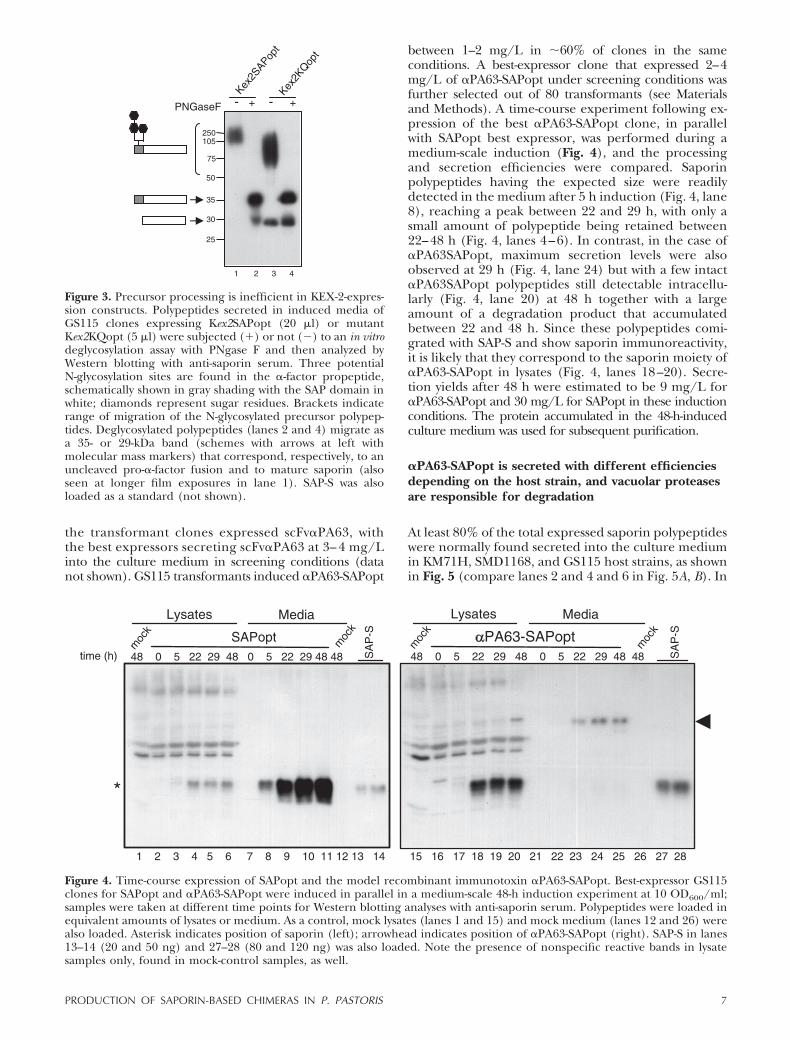
Processing of the �-mating precursor occurs in 2 stepsin the Golgi complex: KEX-2 cleaves after a Lys-Arg,and Ste13 dipeptidyl amino peptidase recognizes andcleaves after two Glu-Ala repeats. Since Ste13 wasreported to be rather inefficient in some cases (25),Kex2 constructs (Kex2SAPopt and Kex2KQopt; Table 1)were produced in which the KEX-2 processing site wasplaced just ahead of the first amino acid in saporinmature sequence (see Materials and Methods). Theprocessing of precursors either lacking (Fig. 3) orincluding Ste13 cleavage sites among GS115-trans-formed clones was compared. When induction mediaof the best expressors of Kex2SAPopt or Kex2KQoptwere analyzed, we observed secretion of high-molecu-lar-mass products, putatively corresponding to unproc-essed active (Fig. 3, lane 1) or KQ mutant (Fig. 3, lane3) precursors, most likely heavily glycosylated due tothe presence of 3 potential N-glycosylation sites in theprosequence of the �-mating factor. Indeed, deglycosy-
lation with PNGaseF allowed recovery of 35-kDa poly-peptides (Fig. 3, lanes 2 and 4), corresponding to theexpected size for unglycosylated unprocessed precur-sors. Thus, proper processing by KEX2 of saporinprecursors may occur only when Ste13 sites are present,as shown in Fig. 4.
Medium-scale expression of secretory saporin and ofa model recombinant saporin immunotoxin
Immunotoxins composed of an antibody domain con-jugated to a toxic moiety are still intensively underinvestigation as therapeutic molecules (26). Recently, apromising internalizing anti-ovarian cancer scFv se-lected from an ETH-2-Gold library (17) was ex-pressed in Escherichia coli and chemically conjugatedto saporin (27) to obtain a derived immunotoxinconstruct. Here, to demonstrate P. pastoris couldproduce a fully recombinant saporin-based immuno-toxin, we used as a model a scFv derived fromETH-2-Gold library against the protective antigen ofanthrax toxin PA63 and expressed this as a scFv aloneor as a fusion polypeptide in which the scFv domainwas placed at the N terminus of saporin via atripeptide alanine linker, generating �PA63-SAPoptchimera. Following transformation of GS115 cells, all
Figure 2. Effects of saporin codon optimization in GS115 host strain. A, B) Histograms show the distribution of 20 independentGS115 clones (y axis, number of clones) induced 48 h for expression of either native (white bars) or optimized (opt; black bars)constructs coding for active SAP (A) or KQ mutant (B). Amount of secreted polypeptide was quantified by densitometry, usingSAP-S as a reference standard. Expression levels (x axis; mg/L) are reported in different ranges from the lowest (0–4 mg/L)to the highest expressors (20–25 mg/L; C, D). Growth curves of �2 mock-induced clones (triangles) for each experiment werecompared with the averaged growth curves of the 20 clones induced for expression of either the optimized (diamonds) or nativeSaporin genes (squares) with an intact catalytic site (C) or with the KQ mutation (D). Note that independently from gene codonoptimization or the relative expression levels when GS115 cells induced active saporin polypeptides, their growth rates decreasedto same extent and arrested after 24 h induction (C).
6 Vol. 24 January 2010 LOMBARDI ET AL.The FASEB Journal � www.fasebj.org
the transformant clones expressed scFv�PA63, withthe best expressors secreting scFv�PA63 at 3– 4 mg/Linto the culture medium in screening conditions (datanot shown). GS115 transformants induced �PA63-SAPopt
between 1–2 mg/L in 60% of clones in the sameconditions. A best-expressor clone that expressed 2–4mg/L of �PA63-SAPopt under screening conditions wasfurther selected out of 80 transformants (see Materialsand Methods). A time-course experiment following ex-pression of the best �PA63-SAPopt clone, in parallelwith SAPopt best expressor, was performed during amedium-scale induction (Fig. 4), and the processingand secretion efficiencies were compared. Saporinpolypeptides having the expected size were readilydetected in the medium after 5 h induction (Fig. 4, lane8), reaching a peak between 22 and 29 h, with only asmall amount of polypeptide being retained between22–48 h (Fig. 4, lanes 4–6). In contrast, in the case of�PA63SAPopt, maximum secretion levels were alsoobserved at 29 h (Fig. 4, lane 24) but with a few intact�PA63SAPopt polypeptides still detectable intracellu-larly (Fig. 4, lane 20) at 48 h together with a largeamount of a degradation product that accumulatedbetween 22 and 48 h. Since these polypeptides comi-grated with SAP-S and show saporin immunoreactivity,it is likely that they correspond to the saporin moiety of�PA63-SAPopt in lysates (Fig. 4, lanes 18–20). Secre-tion yields after 48 h were estimated to be 9 mg/L for�PA63-SAPopt and 30 mg/L for SAPopt in these inductionconditions. The protein accumulated in the 48-h-inducedculture medium was used for subsequent purification.
�PA63-SAPopt is secreted with different efficienciesdepending on the host strain, and vacuolar proteasesare responsible for degradation
At least 80% of the total expressed saporin polypeptideswere normally found secreted into the culture mediumin KM71H, SMD1168, and GS115 host strains, as shownin Fig. 5 (compare lanes 2 and 4 and 6 in Fig. 5A, B). In
Figure 3. Precursor processing is inefficient in KEX-2-expres-sion constructs. Polypeptides secreted in induced media ofGS115 clones expressing Kex2SAPopt (20 �l) or mutantKex2KQopt (5 �l) were subjected (�) or not (�) to an in vitrodeglycosylation assay with PNgase F and then analyzed byWestern blotting with anti-saporin serum. Three potentialN-glycosylation sites are found in the �-factor propeptide,schematically shown in gray shading with the SAP domain inwhite; diamonds represent sugar residues. Brackets indicaterange of migration of the N-glycosylated precursor polypep-tides. Deglycosylated polypeptides (lanes 2 and 4) migrate asa 35- or 29-kDa band (schemes with arrows at left withmolecular mass markers) that correspond, respectively, to anuncleaved pro-�-factor fusion and to mature saporin (alsoseen at longer film exposures in lane 1). SAP-S was alsoloaded as a standard (not shown).
Figure 4. Time-course expression of SAPopt and the model recombinant immunotoxin �PA63-SAPopt. Best-expressor GS115clones for SAPopt and �PA63-SAPopt were induced in parallel in a medium-scale 48-h induction experiment at 10 OD600/ml;samples were taken at different time points for Western blotting analyses with anti-saporin serum. Polypeptides were loaded inequivalent amounts of lysates or medium. As a control, mock lysates (lanes 1 and 15) and mock medium (lanes 12 and 26) werealso loaded. Asterisk indicates position of saporin (left); arrowhead indicates position of �PA63-SAPopt (right). SAP-S in lanes13–14 (20 and 50 ng) and 27–28 (80 and 120 ng) was also loaded. Note the presence of nonspecific reactive bands in lysatesamples only, found in mock-control samples, as well.
7PRODUCTION OF SAPORIN-BASED CHIMERAS IN P. PASTORIS

contrast, the �PA63-SAPopt chimera was found mostlyretained in GS115 (Figs. 4 and 5A, lane 7) in the formof a product presumably degraded in a proteolyticcompartment, most likely corresponding to the vacu-ole. To further examine this possibility, we comparedthe behavior and secretion efficiencies of the best-expressor �PA63-SAPopt clones in the background ofKM71H and SMD1168 host strains. KM71H cells be-haved worse than GS115 by retaining and degrading
almost 90% of the total chimera produced (Fig. 5A,lane 3), with GS115 cells degrading �50% of the totalchimera and even showing some degradation productsin the culture medium in these conditions (Fig. 5A,lane 7). Interestingly, in the strain SMD1168, 25% ofchimera accumulated in the extracellular medium,with 50% accumulated as full-size intracellular�PA63-SAPopt (Fig. 5A, lane 5), with only a minorfraction being degraded. The latter is a strain defi-cient for protease A, activity of which is essential forthe activation of several other vacuolar proteases(28). Despite the intracellular accumulation of intact�PA63-SAPopt chimera, SMD1168-induced clonesbehaved similarly to GS115 or KM71H clones, de-creasing their growth rate to a similar extent asduring SAPopt induction (Fig. 5B).
SAPopt best expressor clone secretes biologicallyactive molecules devoid of nuclease-like activities
To determine whether active polypeptides are secretedby the SAPopt GS115 best clone, secretory saporin waspurified from the 48 h induction medium by ion-exchange chromatography (Fig. 4), as described inMaterials and Methods. The pure protein was analyzedand quantified by a silver-stained SDS-PAGE (Fig. 6A)before in vitro biological activities were characterized.SAPopt was found to have similar RIP activity as theseed-extracted saporin (13) in a cell-free protein inhi-bition assay and in cytotoxicity assays (Fig. 6B, C,respectively) against human leukemia HSB-2 and Daudicell lines. Among extra enzymatic activities assigned toRIPs, a putative nuclease activity was recently debated(14). Since secreted polypeptides would not mix withnuclease activities normally present in the whole-celllysates, in contrast to what may occur during proteinpreparation from total bacterial extracts (recombinant)or from seed extracts (native), we also searched for thisextra putative nuclease-like activity. To this aim,SAPopt and SAP-S were assayed exactly as reported inGhosh and Batra (20), using DNaseI as a positivecontrol and using as DNA substrates pBR322 (Fig.6D) or pPICZalphaB supercoiled/nicked plasmidDNA (not shown). As shown, no nicking or nuclease-like activity could be detected for SAPopt, and only avery faint nicking activity was detected in the case ofSAP-S by using 10 times higher concentrations thanthose originally reported (20).
Secretion and purification of active ATF-SAPoptH6chimera
�PA63-SAPopt was expressed as a first model immuno-toxin but lacks a tumor cell target. The main goal ofthis study was to produce biologically active saporin andderived cytotoxic chimeras; for this purpose, we chose awell-characterized chimera, ATF-saporin, that targetshuman urokinase receptors (CD87) involved in themetastatic spread of different tumor cells (29), being atarget receptor in acute myeloid leukemias as well (30).Proper folding of this chimeric molecule requires cor-rect SS bridge formation of the growth-factor-like and
Figure 5. Comparison between �PA63-SAPopt secretion effi-ciencies among KM71H, SMD1168, or GS115 best-expressorclones. A) Best expressors for SAPopt (lanes 2, 4, and 6) as acontrol or for �PA63-SAPopt (lanes 3, 5, and 7) in each strain,as indicated, were induced in a small-scale 48-h experiment.Lysates (top panel) or medium (bottom panel) loaded inequivalent amounts and samples were analyzed by Westernblotting with anti-saporin serum. Arrowheads indicate intact�PA63-SAPopt in SMD1168 lysate or in the induced medium.Asterisk indicates the position of saporin or saporin-deriveddegradation products. SAP-S (10 ng; top panel) or mock-induced medium (bottom panel) was loaded in lane 1. As inFig. 4, nonspecific reactive bands are present in lysates.B) Growth curves of �2 mock-induced clones (diamonds)were compared with the averaged growth curves of 12 clonesinduced for expression of either optimized saporin with anintact catalytic site (squares) or with a KQ mutation (triangle)or the PA63-SAPopt chimera (asterisk) in SMD1168, as anexample.
8 Vol. 24 January 2010 LOMBARDI ET AL.The FASEB Journal � www.fasebj.org

kringle domains. This was previously achieved by tar-geting a secretory pre-ATF-saporin polypeptide to theER in X. laevis oocytes (9). Having established here thatGS115 cells can tolerate the expression of active sa-porin polypeptides and since human ATF was effi-ciently secreted by P. pastoris (31), we prepared aconstruct to express a secretory ATF-saporin chimera inwhich a hexahistidine tag was placed after Lys253 ofSAPopt to allow affinity purification of this fusionmolecule. We isolated among 19 clones the 2 bestespressors that secreted this fusion protein at 1–2 mg/Lin screening conditions (Fig. 7A, lane 3). About 25% ofthe total expressed ATF-SAPoptH6 was found to beretained as saporin-containing degradation products(Fig. 7A, lane 2), indicating that ATF-SAPoptH6 was
more efficiently secreted by GS115 cells than the model�PA63-SAPopt chimera. In a medium-scale inductionexperiment, yields in ATF-SAPoptH6 were 3.5 mg/Lafter 48 h. The medium was concentrated, ATF-SAPoptH6 was purified by Ni-affinity chromatography(Materials and Methods), and samples were analyzedboth on silver-stained SDS-PAGE and by Western blot-ting with anti-saporin serum (Fig. 7B). The fusionprotein eluted as a single peak at 150 mM imidazole;however, in addition to an intact fusion polypeptideshowing the expected size of 44 kDa, another faintband, most likely corresponding to histidine-taggedsaporin, was copurified (Fig. 7B, lane 6). To evaluatethe cytotoxic activity of the secreted ATF-SAPoptH6,human U937 cells that expose both the human uroki-
30
20
45
66
97
100
200
300
500 M
SAP-S (ng)
SAPopt
1 2 3 4 5 6 7
10-10 10-9 10-8 10-7 10-6 10-5
Concentration [M]
0
20
40
60
80
100
110
[3 H] -
Leu
cine
Inco
rpor
atio
n (%
Con
trol
)
IC50
6 x
10-7
2 x
10-7
IC50
C
A B
D
8
Figure 6. Characterization of secretory saporin purified from the best GS115-expressor clone. A) Medium containing SAPopt wasconcentrated and purified as described in Materials and Methods, and fractions containing 2, 5, or 10 �l purified SAPopt 2(lanes 1, 2, and 3, respectively) were quantified by silver-stained SDS-PAGE, loading SAP-S in increasing amounts, as indicated(lanes 4–7). Molecular mass markers (lane 8) with positions are at right. B) In vitro inhibition translation of reporter BMV RNAwas performed assaying SAPopt and SAP-S, as a control (not shown) in equivalent serial dilutions from 1.3 nM to 0.13 pM inreplicate samples. Data are reported as mean sd percentage inhibition of protein synthesis. Dashed line indicates that SAPoptIC50 � 35 pM in these conditions. C) Cell-killing assays: SAPopt cytotoxicity (solid symbols) was compared with that of SAP-S(open symbols) in Daudi (squares) or HSB-2 cells (circles) after 48 h exposure to varying molar concentrations (x axis) of the2 toxins. Data are reported as mean sd percentage inhibition of incorporation of tritiated leucine into protein (y axis). IC50values are also shown with the dose-response curves (dashed line), indicating that these are identical for SAP-S and SAPopt(2�10�7 M in HSB-2 and 6�10�7 M in Daudi cells, respectively). D) DNase-like activity: different amounts of the purifiedSAPopt fraction or SAP-S (0–5 �g, as indicated) or 0.05 �g of DNaseI as positive control were incubated with 0.5 �g of pBR322.As a control, 0.5 �g of HindIII-linearized pBR322 was loaded in lane C. N, nicked pBR322 DNA; L, linearized pBR322 DNA; SC,supercoiled pBR322 DNA.
9PRODUCTION OF SAPORIN-BASED CHIMERAS IN P. PASTORIS

nase receptors needed for cell surface binding andendocytic LRP receptors for internalization (32) weretreated essentially as described for Fabbrini et al. (9)and exposed for 48 h to increasing concentrations ofpurified ATF-SAPoptH6 or seed-extracted saporin forcomparison. ATF-SAPoptH6 had an IC50 of 6 � 10�11
M, 4 orders of magnitude lower than that of SAP-S (Fig.7C), confirming it has the full expected biologicalactivity against promyelocytic target cells. Indeed, itshows same activity of recombinant ATF-SAP producedin bacteria and secretory ATF-saporin from the X. laevisoocyte medium (8, 9).
DISCUSSION
P. pastoris hosts are sensitive to transformation withactive saporin constructs
The main goal of this study was to produce biologicallyactive secretory saporin and derived cytotoxic chimeraswithout intoxicating the host cells. Searching for asuitable eukaryotic host, we explored different P. pasto-ris strains: SMD1168 (his4; pep4) is the protease-defi-cient strain reported to secrete a type I RIP, PAP, withno apparent host toxicity (7), and GS115 (his4) wasselected for its tolerance to diphtheria toxin (11).KM71H (arg4; aox1:ARG4) lacks transcription of theendogenous AOX1 gene, but following methanol ex-posure induces the less abundantly transcribed AOX-2
gene (24) and produces exogenous polypeptides underthe AOX1 promoter in very high yields (6). A firstunexpected observation was the lower transformationefficiency when using active saporin constructs, inde-pendently from the P. pastoris strain used. This constantloss in viable colonies suggests that integration at theAOX-1 locus could lead to leaky phenotypes, possiblydue to genomic integration events that would deregu-late the AOX-1 promoter. This observation might di-rectly correlate with the potential detrimental activity ofsaporin toward P. pastoris ribosomes. SAP-KQ mutanthelped us to establish the potential contribution of anintact RIP catalytic active site to any toxicity observedand/or detrimental effects on levels of accumulation ofsecreted polypeptides. We especially found codon op-timization was a key parameter to succeed.
Codon optimization may affect host sensitivity tosaporin
In our hands, indeed, codon optimization was found tobe necessary to obtain inducible expression of activesaporin in SMD1168 and presumably allowed recoveryof inducible SAPopt transformants in KM71H strain, aswell. Both KM71H and SMD1168 could produce mu-tant KQopt at extremely high levels (much higher thanthose obtained in GS115), but they concomitantlyshowed severalfold lower yields in active SAPopt (Figs.1 and 5), indicating the maximal expression capabilityfor the active-site mutant KQopt parallels a highersensitivity to SAPopt expression.
Figure 7. Expression and characterization of a secretory ATF-saporin chimera. A) Samples from 48-h medium-scale inductionof the best GS115-expressor clone for ATF-SAPoptH6 were analyzed by Western blotting with anti-saporin serum: mock-inducedlysate as a control (lane 1) and ATF-SAPoptH6 lysate (lane 2) together with an equivalent amount of induced medium (lane3) for comparison. B) At 48 h, induced medium containing ATF-SAPoptH6 was concentrated and ATF-SAPoptH6 Ni-purified,as described in Materials and Methods. Silver-stained gel (top panel) or Western blotting with anti-saporin serum (bottompanel) of total concentrated induced medium (Tot) in lane 4; flow-through (Flow-t.) in lane 5, and concentrated eluates (150mM or 300 mM imidazole) in lanes 6 and 7, respectively. For quantifications, SAP-S was loaded in lanes 1–3; 90, 225 and 450ng (top panel) or 30, 60 and 100 ng (bottom panel). ATF-SAPoptH6 position is shown at right (arrowhead) with the positionof a saporin-containing degradation product (asterisk). Molecular mass marker positions are at left. C) Cell-killing assay wasperformed on acid-treated U937 cells that were exposed 48 h to increasing amounts (x axis) of seed-extracted saporin (dots)as control or purified ATF-SAPoptH6 (squares), and cells were then washed and pulse-labeled with tritiated leucine. Totalincorporation into proteins is expressed as percentage control vs. non-toxin-treated samples (y axis). IC50 values (dashedlines) � 2 � 10�7 M (SAP-S); 6 � 10�11 M (ATF-SAPoptH6).
10 Vol. 24 January 2010 LOMBARDI ET AL.The FASEB Journal � www.fasebj.org

After codon optimization, SAPopt GS115 clonescould express equally well as those for KQopt, justifyingour choice of GS115 as the most tolerant strain forexpression of saporin, as also indicated by 30 mg/L ofsecretory saporin obtained with this host. Codon opti-mization should improve expression of foreign genesby increasing the rate of translation. Synthetic genescould successfully enhance protein productivity, as wellas toxin expression in P. pastoris (33–35), but no directcomparison to a noncatalytically active toxin polypep-tide was performed, as we did here. If by changing theplant codon usage we merely augmented efficiency inpolypeptide translation, we might have expected aparallel increase in protein yields both for the activeand the mutant KQopt clones (the only differenceresiding in the KQ-encoded residues, affecting RIPcatalytic activity), but this was not the case. Only SAPoptclones increased average expression levels in GS115(Fig. 2), and codon optimization was also found essen-tial to obtain inducible saporin clones in SMD1168.Although the precise mechanisms underlying theseobservations are unknown, it is well established thatwhen rare or nonoptimal codon pairs (or secondarystructures) are present within an mRNA sequence,ribosome stalling and pausing during translation elon-gation may target mRNAs to endonucleolytic cleavagein a process known as “no-go decay.” These RNAquality-control mechanisms operate in eukaryotic cellsand yeasts (36, 37), as well. In a feedback-regulatedgene of Arabidopsis thaliana, no-go decay originated5�-truncated mRNA intermediates and also gives rise toa ladder of premature polypeptides, the molecularmasses of which corresponded to products of ribo-somes in a stalled stack (38). The potential link be-tween codon optimization, host toxicity, and yields inactive saporin clearly will deserve deeper investigation.It would be tempting to speculate that when tRNAsand/or translating ribosomes become limiting, ribo-some stalling and translation-pausing events would leadto premature polypeptidyl release, potentially givingrise to cytosolic saporin-derived products endowed withresidual RIP activity.
Saporin-associated toxicity is due to intracellularevents
Although in principle a better coupling between mRNAtranscription, translation, and segregation into the ERmembranes of the SAPopt precursors would counteractany detrimental effect possibly associated with saporinexpression, SAPopt precursors should be synthesized ina translocation-competent, noncatalytically active form,since the �-factor signal peptide mediates Sec63 post-translational insertion into ER membranes (39). Whensaporin is directly added to the medium of GS115 cells,it does not affect cell growth, demonstrating thatextracellular or secreted material would not reachcytosolic ribosomes (Table 2). Host toxicity associatedwith induction of saporin expression (Fig. 2C) shouldbe due to intracellular events favoring access of this RIPmolecule to the cytosolic compartment. In certainstressed conditions, such as during methanol induc-
tion, unfolded protein response and ERAD wouldmediate cytosolic dislocation of some (toxic) polypep-tide (40). Some of these molecules may thus escapeproteasomal degradation and cause cell intoxication.Indeed, independently from the levels of secretedpolypeptide or the host strains used, induction ofexpression of active molecules (whether from nativeSAP or SAPopt constructs in GS115) equally decreasedgrowth rates of induced SAP-transformant clones, con-firming that this relies on the presence of an active site. Thiscytosolic “leakiness” should be further investigated.
Best-suited P. pastoris constructs and host strains forsecretion of saporin chimeras
When we investigated the different saporin constructsin the GS115 strain together with a first model chimera,we found that for proper processing by KEX2 ofsaporin precursors, the Ste13 Glu-Ala sites need to bepresent (see Figs. 3 and 4); therefore, these maturepolypeptides have �2 extra amino acids at the Nterminus of saporin that, however, do not seem to affectsecretory saporin biological activity (Fig. 6). While thesecretion efficiency of the mature SAPopt polypeptideswas 80% and was found to be similar in GS115 andKM71H or SMD1168 hosts, secretion efficiencies werequite different for the model immunotoxin �PA63-SAPopt (Fig. 5), with �50% of chimera retained intra-cellularly and eventually converted to major degrada-tion products in GS115, while 50% intact chimeraaccumulated intracellularly in SMD1168. Thus, vacuo-lar proteases for which SMD1168 is deficient are mostlikely involved in the intracellular degradation of thechimera (Fig. 8). We suggest that the N-terminal scFvdomain may favor targeting to the yeast vacuole of�PA63-SAPopt through the well-characterized recep-tor-mediated vacuolar pathway for protein disposal(41), and in this compartment the scFv domain mayundergo degradation (Fig. 8), giving rise to the intra-cellular accumulation of the protease-resistant saporindomain (42). SMD1168 might be a suitable host forhigh-scale fermentation processes, since it would avoidthe release of vacuolar proteases from dying cells.Moreover, the medium should be devoid of contami-nation with saporin-derived degradation products thatwere observed in GS115 cells (Fig. 5) and also duringATF-SAPoptH6 expression (Fig. 6). In Xenopus oocytes,most of the newly synthesized ATF-saporin polypeptideswere secreted, and only a small amount was found
TABLE 2. Exogenous saporin does not intoxicate P. pastorishost cells
Strain
OD600
0 h 24 h 48 h
GS115 (his4) 1.00 7.08 16.20GS115 (his4) � SAP-S 1.00 7.59 16.50
Growth rates of GS115 cells were evaluated starting with 1 OD600of cells grown up to 48 h in the absence or in the presence of 33 nM(1 mg/L) SAP-S.
11PRODUCTION OF SAPORIN-BASED CHIMERAS IN P. PASTORIS

degraded intracellularly to saporin. Besides the differ-ences among these two expression systems, the twoexpressed molecules differ by the presence of a triala-nine linker and 6 histidine residues that might havecontributed to lower secretion efficiency in yeast hostcells. Despite this, secretion efficiency was better forATF-SAPoptH6 than for �PA63-SAPopt, suggesting thata good secretory domain may be important to avoidmisfolding and/or targeting to vacuolar degradation.
New molecules will be investigated by changing thelinker residues and either expressing them without anytag or placing the hexahistidine tag at the N terminusto avoid the small contamination with the histidine-tagged saporin-derived products. While we believe thepresence of proteolytic-derived material does not affectthe results of the cytotoxicity experiment in U937 cellsbecause ATF-SAPoptH6 has a much lower IC50 ascompared with saporin, this would clearly represent athreat for clinical development. Nevertheless, histidine-tagged chimeric molecules have been produced bygood manufacturing practice (GMP) in P. pastoris,using expanded-bead adsorption technology (43) foruse in patients, while a promising bivalent anti-T-cellimmunotoxin based on diphtheria toxin was recentlybeing developed for treatment of T-cell leukemias,autoimmune diseases, and tolerance induction fortransplantation. The latter molecule has been pro-duced as a single 120-L batch of bioreactor culture,obtained using a P. pastoris mutant strain resistant todiphtheria toxin action, mutEF2JC307, for phase I/IIclinical trials (44).
CONCLUSIONS
One of the most promising biotechnological applica-tions of RIPs consists in mimicking the high cytotoxic
potential of the ricin holotoxin by constructing recom-binant fusion toxins targeted against tumor antigens ofchoice. Due to misfolding and degradation of the ATFdomain during ATF-saporin expression in bacterialhost cells, this fusion molecule could not be furtherdeveloped for therapeutic purposes. In this study, weestablished the feasibility of using P. pastoris, despiteclear evidence for the presence of some host toxicity,and demonstrated that Pichia host strains can beadapted to saporin expression. We therefore proposethis eukaryotic system as the best-suited expressionplatform, allowing cost-effective scaling up of GMPproduction in biofermentors of saporin-based thera-peutic molecules.
This work was supported by Leukaemia Busters (www.leukaemiabusters.org.uk) as part of the Recombinant Immuno-toxin Study Group coordinated by D.J.F. S.B. had a fellowshipsupported by Ingenio (Regione Lombardia). We thank AlessandroPini (University of Siena, Siena, Italy) for providing pHEN1-con-taining scFv�PA63, and Pietro Della Cristina (University of Verona,Verona, Italy) and Richard Marshall (University of Warwick, Cov-entry, UK) for fruitful discussions.
REFERENCES
1. Endo, Y., Mitsui, K., Motizuki, M., and Tsurugi, K. (1987) Themechanism of action of ricin and related toxic lectins oneukaryotic ribosomes. The site and the characteristics of themodification in 28 S ribosomal RNA caused by the toxins. J. Biol.Chem. 262, 5908–5912
2. Hartley, M., and Lord, J. (2004) Cytotoxic ribosome-inactivatinglectins from plants. Biochim. Biophys. Acta 1701, 1–14
3. Sandvig, K., and van Deurs, B. (2000) Entry of ricin and Shigatoxin into cells: molecular mechanisms and medical perspec-tives. EMBO J. 19, 5943–5950
4. Wu, Y. H., Shih, S. F., and Lin, J. Y. (2004) Ricin triggersapoptotic morphological changes through caspase-3 cleavage ofBAT3. J. Biol. Chem. 279, 19264–19275
5. Frankel, A. E., Kreitman, R., and Sausville, E. (2000) Targetedtoxins. Clin. Cancer Res. 6, 326–334
6. Cereghino, J. L., and Cregg, J. M. (2000) Heterologous proteinexpression in the methylotrophic yeast Pichia pastoris. FEMSMicrobiol. Rev. 24, 45–66
7. Rajamohan, F., Doumbia, S. O., Engstrom, C. R., Pendergras,S. L., Maher, D. L., and Uckun, F. M. (2000) Expression ofbiologically active recombinant pokeweed antiviral protein inmethylotrophic yeast Pichia pastoris. Protein Expr. Purif. 18, 193–201
8. Fabbrini, M. S., Carpani, D., Bello-Rivero, I., and Soria, M. R.(1997) The amino-terminal fragment of human urokinase di-rects a recombinant chimeric toxin to target cells: internaliza-tion is toxin mediated. FASEB J. 11, 1169–1176
9. Fabbrini, M. S., Carpani, D., Soria, M. R., and Ceriotti, A. (2000)Cytosolic immunization allows the expression of preATF-sa-porin chimeric toxin in eukaryotic cells. FASEB J. 14, 391–398
10. Liu, Y. Y., Woo, J. H., and Neville D. M., Jr. (2003) Targetedintroduction of a diphtheria toxin resistant mutation into thechromosomal EF-2 locus of Pichia pastoris and expression ofimmunotoxin in the EF-2 mutants. Protein Expr. Purif. 30,262–274
11. Woo, J. H., Liu, Y. Y., Stavrou, S., and Neville D. M., Jr. (2004)Increasing secretion of a bivalent anti-T-cell immunotoxin byPichia pastoris. Appl. Environ. Microbiol. 7, 3370–3376
12. Zarovni, N., Vago, R., Solda, T., Monaco, L., and Fabbrini, M. S.(2007) Saporin as a novel suicide gene in anticancer genetherapy. Cancer Gene Ther. 14, 165–173
13. Fabbrini, M. S., Rappocciolo, E., Carpani, D., Solinas, M.,Valsasina, B., Breme, U., Cavallaro, U., Nykjaer, A., Rovida, E.,Legname, G., and Soria, M. R. (1997) Characterization of a
Figure 8. Schematic representation of the possible fates of amodel scFv-saporin fusion. After synthesis (1), the chimera ispost-translationally translocated into the ER lumen (2), wherethe N-terminal propeptide undergoes N-glycosylation, and itis further processed in the Golgi complex (3) by KEX2/Ste13peptidases. Finally, the chimera is either secreted in theextracellular medium (4) or diverted to the vacuolar com-partment, where the scFv domain can be degraded (5).
12 Vol. 24 January 2010 LOMBARDI ET AL.The FASEB Journal � www.fasebj.org

saporin isoform with lower ribosome-inhibiting activity. Biochem.J. 322, 719–727
14. Peumans, W. J., Hao, Q., and Van Damme, E. J. (2001)Ribosome-inactivating proteins from plants: more than RNAN-glycosidases? FASEB J. 15, 1493–1506
15. Day, P. J., Lord, J. M., and Roberts, L. M. (1998) The deoxyri-bonuclease activity attributed to ribosome-inactivating proteinsis due to contamination. Eur. J. Biochem. 258, 540–545
16. Sreekrishna, K. (1993) Strategies for optimizing protein expres-sion and secretion in the methylotrophic yeast Pichia pastoris. InIndustrial Microorganism: Basic and Applied Molecular Genetics,(Baltz, R. H., Hegeman, G. D., and Skatrud, P.L., eds) pp.119–126, American Sociology of Microbiology, Washington,DC, USA
17. Silacci, M., Brack, S., Schirru, G., Mårlind, J., Ettorre, A., Merlo,A., Viti, F., and Neri, D. (2005) Design, construction, andcharacterization of a large synthetic human antibody phagedisplay library. Proteomics 5, 2340–2350
18. Wang, P., Zhang, J., Sun, Z., Chen, Y., and Liu, J. N. (2000)Glycosylation of Prourokinase produced by Pichia pastoris im-pairs enzymatic activity but not secretion. Protein. Expr. Purif. 20,179–185
19. Flavell, D. J., Boehm, D. A., Noss, A., Warnes, S. L., and Flavell,S. U. (2001) Therapy of human T-cell acute lymphoblasticleukaemia with a combination of anti-CD7 and anti-CD38-SAPORIN immunotoxins is significantly better than therapywith each individual immunotoxin. Br. J. Cancer 84, 571–578
20. Ghosh, P., and Batra, J. K. (2006) The differential catalyticactivity of ribosome-inactivating proteins saporin 5 and 6 is dueto a single substitution at position 162. Biochem. J. 400, 99–104
21. Flavell, D. J., Warnes, S., Noss, A., and Flavell, S. U. (1998)Host-mediated antibody-dependent cellular cytotoxicity contrib-utes to the in vivo therapeutic efficacy of an anti-CD7-saporinimmunotoxin in a severe combined immunodeficient mousemodel of human T-cell acute lymphoblastic leukemia. CancerRes. 58, 5787–5794
22. Benatti, L., Saccardo, M. B., Dani, M., Nitti, G., Sassano, M.,Lorenzetti, R., Lappi, D. A., and Soria, M. (1989) Nucleotidesequence of cDNA coding for saporin-6, a type-1 ribosome-inactivating protein. Eur. J. Biochem. 183, 465–470
23. Savino, C., Federici, L., Ippoliti, R., Lendaro, E., and Tserno-glou, D. (2000) The crystal structure of saporin SO6 fromSaponaria officinalis and its interaction with the ribosome. FEBSLett. 470, 239–243
24. Cregg, J. M., Madden, K. R., Barringer, K. J., Thill, G. P., andStillman, C. A. (1989) Functional characterization of the twoalcohol oxidase genes from the yeast Pichia pastoris. Mol. Cell.Biol. 9, 1316–1323
25. Emberson, L. M., Trivett, A. J., Blower, P. J., and Nicholls, P. J.(2005) Expression of an anti-CD33 single-chain antibody byPichia pastoris. J. Immunol. Methods 305, 135–151
26. Pastan, I., Hassan, R., Fitzgerald, D. J., and Kreitman, R. J.(2006) Immunotoxin therapy of cancer. Nat. Rev. Cancer 6,559–565
27. Piazza, T., Cha, E., Bongarzone, I., Canevari, S., Bolognesi, A.,Polito, L., Bargellesi, A., Sassi, F., Ferrini, S., and Fabbi, M.(2005) Internalization and recycling of ALCAM/CD166 de-tected by a fully human single-chain recombinant antibody.J. Cell Sci. 118, 1515–1525
28. Ammerer, G., Hunter, C. P., Rothman, J. H., Saari, G. C.,Valls, L. A., and Stevens, T. H. (1986) PEP4 gene of Saccha-romyces cerevisiae encodes proteinase A, a vacuolar enzyme
required for processing of vacuolar precursors. Mol. Cell. Biol.6, 2490 –2499
29. Dass, K., Ahmad, A., Azmi, A. S., Sarkar, S. H., and Sarkar, F. H.(2008) Evolving role of uPA/uPAR system in human cancers.Cancer Treat. Revs. 34, 122–136
30. Ramage, J. G., Vallera, D. A., Black, J. H., Aplan, P. D., Kees,U. R., and Frankel, A. E. (2003) The diphtheria toxin/uroki-nase fusion protein (DTAT) is selectively toxic to CD87 express-ing leukemic cells. Leuk. Res. 27, 79–84
31. Zhao, G., Yuan, C., Bian, C., Hou, X., Shi, X., Ye, X., Huang, Z.,and Huang, M. (2006) Protein expression and preliminarycrystallographic analysis of amino-terminal fragment of uroki-nase-type plasminogen activator. Protein Expr. Purif. 49, 71–77
32. Ippoliti, R., Lendaro, E., Benedetti, P. A., Torrisi, M. R.,Belleudi, F., Carpani, D., Soria, M. R., and Fabbrini, M. S.(2000) Endocytosis of a chimera between human prourokinaseand the plant toxin saporin: an unusual internalization mecha-nism. FASEB J. 14, 1335–1444
33. Yadava, A., and Ockenhouse, C. F. (2003) Effect of codonoptimization on expression levels of a functionally folded ma-laria vaccine candidate in prokaryotic and eukaryotic expressionsystems. Infect. Immun. 71, 4961–4969
34. Woo, J. H., Liu, Y., Mathias, A., Stavrou, S., Wang, Z., Thomp-son, J., and Neville, D. M., Jr. (2002) Gene optimization isnecessary to express a bivalent anti-human anti-T cell immuno-toxin in Pichia pastoris. Protein Expr. Purif. 25, 270–282
35. Gurkan, C., and Ellar, D. J. (2005) Recombinant production ofbacterial toxins and their derivatives in the methylotrophic yeastPichia pastoris. Microb. Cell Factories 4, 33
36. Doma, M. K., and Parker, R. (2006) Endonucleolytic cleavage ofeukaryotic mRNAs with stalls in translation elongation. Nature440, 561–564
37. Doma, M. K., and Parker, R. (2007) RNA quality control ineukaryotes. Cell 131, 660–668
38. Haraguchi, Y., Kadokura, Y., Nakamoto, M., Onouchi, H., andNaito, S. (2008) Ribosome stacking defines CGS1 mRNA deg-radation sites during nascent peptide-mediated translation ar-rest. Plant Cell Physiol. 49, 314–323
39. Brodsky, J. L., Goeckeler, J., and Schekman, R. (1995) BiP andSec63p are required for both co- and posttranslational proteintranslocation into the yeast endoplasmic reticulum. Proc. Natl.Acad. Sci. U. S. A. 92, 9643–9646
40. Hohenblum, H., Gasser, B., Maurer, M., Borth, N., and Mat-tanovich, D. (2004) Effects of gene dosage, promoters, andsubstances on unfolded protein stress of recombinant Pichiapastoris. Biotechnol. Bioeng. 85, 367–375
41. Hong, E., Davidson, A., and Kaiser, C. (1996) A pathway fortargeting soluble misfolded proteins to the yeast vacuole. J. CellBiol. 135, 623–633
42. Santanche, S., Bellelli, A., and Brunori, M. (1997) The unusualstability of saporin, a candidate for the synthesis of immunotox-ins. Biochem. Biophys. Res. Commun. 234, 129–132
43. Tolner, B., Smith, L., Begent, R. H., and Chester, K. A. (2006)Expanded-bead adsorption immobilized-metal affinity chroma-tography. Nat. Protoc. 1, 1213–1222
44. Woo, J. H., Liu, J. S., Kang, S. H., Singh, R., Park, S. K., Su, Y.,Ortiz, J., Neville, D. M., Jr., Willingham, M. C., and Frankel, A. E.(2008) GMP production and characterization of the bivalentanti-human T cell immunotoxin, A-dmDT390-bisFv(UCHT1)for phase I/II clinical trials. Protein Expr. Purif. 58, 1–11
Received for publication June 19, 2009.Accepted for publication August 6, 2009.
13PRODUCTION OF SAPORIN-BASED CHIMERAS IN P. PASTORIS