photoluminescence and absorption of mncl2, mnbr2 and mni2
TRANSCRIPT

Physica 144B (1987) 331-340 North-Holland, Amsterdam
P H O T O L U M I N E S C E N C E AND ABSORPTION OF MnCI2, MnBr 2 AND M n l 2
C.R. R O N D A * , H .H. SIEKMAN and C. HAAS Laboratory of Inorganic Chemistry, Materials Science Centre, University of Groningen, Nijenborgh 16, 9747 AG Groningen, The Netherlands
Received 22 December 1986
Absorption and emission spectra of MnCl2, MnBr 2 and MnI 2 in the paramagnetic temperature region are reported. The emission spectra show a broad band with a Gaussian line shape, which is assigned to the 4Tlg(G)---> 6Als(S ) crystal field transition of the Mn 2÷ ion. The corresponding absorption band 6A1~(S)---> 4Tlg(G) also has a Gaussian line shape. The observed line shape is a consequence of a strong Jahn-Teller effect due to coupling with Eg phonons, which almost completely quenches the spin-orbit splitting.
The absorption and emission bands show a strong blue shift with increasing temperature, due to the thermal expansion of the crystal. The emission intensity and the decay time of the luminescence strongly decrease with increasing temperature, because of phonon-assisted exciton hopping towards luminescence killing centres.
1. Introduction
MnC12 crystallizes in the layer-type CdC12 structure with space group R3m. The Mn 2+ ions occupy slightly trigonally distorted octahedral sites, the C1- ions form a cubic dosed packed structure. MnBr 2 and MnI 2 crystallize in the layer-type Cd(OH)2 structure with space group P3ml. In this structure the Mn 2+ ions also oc- cupy slightly trigonaUy distorted octahedral sites, but the Br- and I- ions form a hexagonal closed packed structure.
The manganese dihalides show at low tem- perature T < T N an antiferromagnetic ordering of the magnetic moments of the Mn 2+ ions; the N6el temperatures T N are 1.96 K for MnCl 2 [1, 2], 2.16K for MnBr z [3, 4] and 3.6K for MnI 2 [5].
The optical absorption spectra of the mangan- ese dihalides have been studied in considerable detail [5-7]. In spite of the fact that in octahe- dral symmetry all d -d electric dipole transitions of the high spin 3d 5 Mn 2÷ ion are both spin and parity forbidden, the d -d transitions of the crys- tal are easily observed in the absorption spectra.
* Present address: Philips Research Laboratories, P.O. Box 80.000, 5600 JA Eindhoven, The Netherlands.
This is due to the fact that the selection rules are relaxed by vibronic coupling, by exchange inter- actions between ions and by spin-orbit interac- tion [5].
The manganese dihalides MnCI 2, MnBr 2 and MnI 2 show a strong red luminescence, which is due " to the crystal field transition 4Tlg(G)---> 6Axg(S ) of the manganese ion. Only a few studies of this luminescence in the pure manganese dihalides, mixed manganese dihalides and some diluted systems have been published [8-101 .
In this paper an experimental investigation of the 6A~(S)--)4Txg(G ) absorption band and the aTlg(G)-->6Alg(S ) emission band of the man- ganese dihalides in the paramagnetic tempera- ture region is reported. The absorption and the emission bands show a large blue shift with increasing temperature. We will show that this blue shift is due to the thermal expansion of the lattice.
Both the absorption and the emission band have an almost perfectly Gaussian line shape. In particular for the absorption bands this is a rather surprising result, because one would ex- pect the 4Tlg(G ) level to be split in first order in four spin-orbit components. The emission band may have a Gaussian line shape when the emis-
0378-4363/87/$03.50 (~ Elsevier Science Publishers B.V. (North-Holland Physics Publishing Division)

sion process involves only the spin-orbit compo- nent with the lowest energy. We will show that the spin-orbit splitting is strongly reduced be- cause of the Jahn-Teller effect (JTE) [11], which results in a Gaussian line shape for absorption and emission bands.
6 4 The intensity of the Alg(S)--> Tlg(G ) ab- sorption band is temperature independent, but the intensity of the 4Tlg(G)--~ 6Alg(S ) emission band decreases strongly with increasing tempera- ture. The temperature dependence of the emis- sion intensity is caused by thermally activated hopping of the 4Tlg(G ) exciton towards luminescence killing centres. The activation for this hopping process has been determined from the temperature dependence of the emission in- tensity, and for MnI 2 also from the temperature dependence of the emission decay time.
2. Experimental methods
d d
MnC12 and MnBr 2 were prepared from Mn and HCI and HBr, respectively. The reaction products were dried using a flow of dry N 2 gas, and were sublimed in a sealed ampoule at a temperature of 525-600°C. MnI 2 was prepared from the elements in a sealed ampoule in a temperature gradient 50-600°C. The obtained crystals were small pink flakes. Large single crystals were grown using the Bridgman tech- nique. Because the manganese dihalides are very hygroscopic, all crystal handling had to be per- formed with the exclusion of moisture and air.
The absorption spectra were recorded using a Carl-Zeiss PMQII spectrophotometer equipped with a home-built flow cryostat. The direction of the incident light was parallel to the c-axis of the crystals. The thickness of the crystal was 315/zm for MnCI=, 390/xm for MnBr2, and 180/xm for MnI 2. The absorption spectra were recorded with a spectral resolution of 0.01 eV.
The emission spectra were excited using the light of a 1000 W xenon arc, dispersed by a Jobin Yvon H20 U.V. monochromator. The sample was in an Oxford Instruments MD4 cryostat. The emitted light was interrupted with a frequency of 138 Hz, dispersed with a second monochromator
(Jobin Yvon H20 VIS) and detected by an EMI 9558 QB photomultiplier. The spectral resolu- tion was about 0.01 eV. Polycrystalline MnC12 and MnBr 2 were used in the luminescence mea- surements. For MnI 2 both single crystals and polycrystalline samples were used; the emission characteristics were the same.
The decay time of the luminescence of crystal- line MnI z was measured using a Molectron DL-II dye laser, pumped by a Molectron UV-400 pulsed N 2 laser. The emitted light was detected using a Spex 1704 monochromator and an EMI 9816 photomultiplier. The decay curves were recorded using a PAR 162 boxcar averager.
3. Experimental results
The absorption spectra of the 6alg(S)---> 4Tlg(G ) transition of crystalline MnCI2, MnBr 2 and MnI 2 as a function of the temperature are given in figs. 1-3. The position of the maximum of the absorption band shows a blue shift with increasing temperature (fig. 4).
The emission spectra of the 4Tlg(G ) --~ 6Alg(S ) transition of the manganese dihalides are given
E (eV)
0.5
186 150 109
87 62 34 8.5
J J
o 2 .0
K ,
K K
K K K
/ / / / / /
J /
i !
' 2'.5
332 C.R. Ronda et al. / Photoluminescence and absorption of Mn-dihalides
Fig. 1. Absorption spectra (optical density O.D.) of MnCI 2 (d = 315 txm).
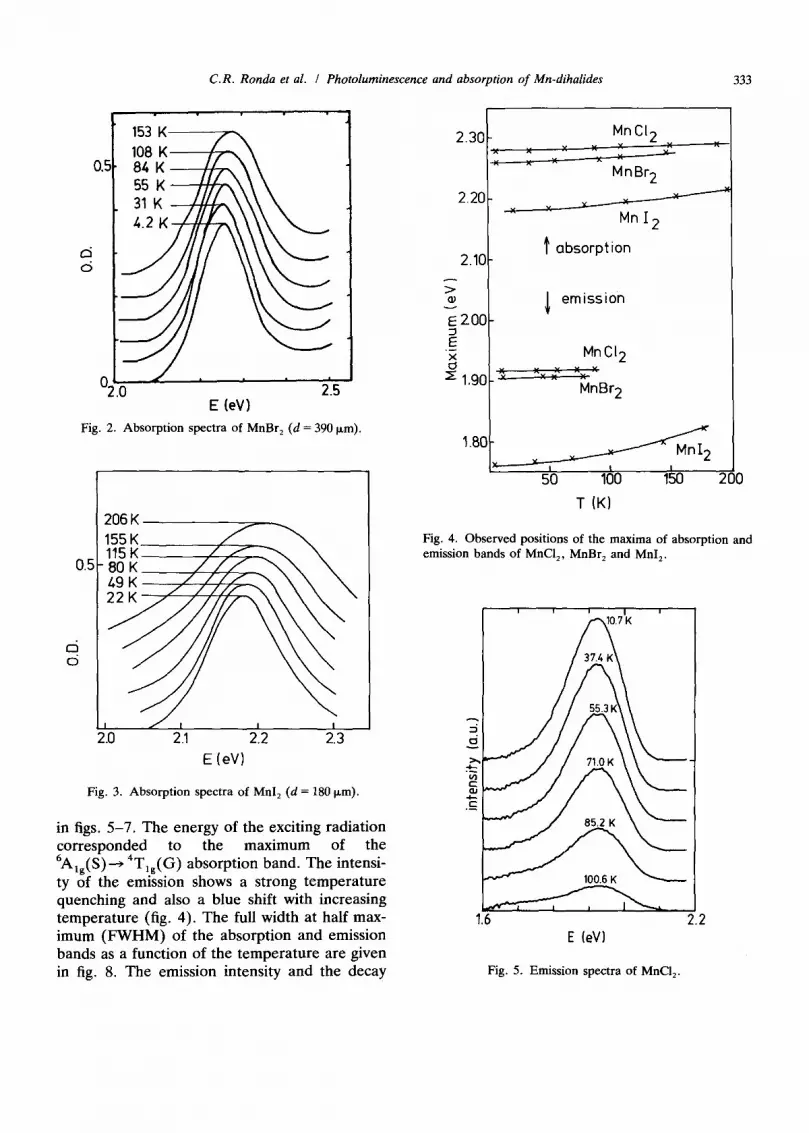
C.R. Ronda et al. / Photoluminescence and absorption of Mn-dihalides 333
153 K f \ 108 K " - o. / / . i \ \
31 K 4.2K
02 ~ 215 E (eV)
Fig. 2. Absorpt ion spectra of MnBr 2 (d = 390 Ixm).
I206 K
/Z,9K ~ \ \
'2'0 / 2Zl 2:2 • . . 2.3 E (eV)
Fig. 3. Absorpt ion spectra of Mnl 2 (d = 180 p,m).
in figs. 5-7. The energy of the exciting radiation corresponded to the maximum of the 6mlg(S)----) 4Tlg(G ) absorption band. The intensi- ty of the emission shows a strong temperature quenching and also a blue shift with increasing temperature (fig. 4). The full width at half max- imum (FWHM) of the absorption and emission bands as a function of the temperature are given in fig. 8. The emission intensity and the decay
2.3'
2.20
2.10
>
E 200 E Z
~ 1.90
1.8(
) ( 14
MnCI 2 X X x X I~
MnBr 2
v ~ INn 12
t obsorption
emission
MnCl 2
MnBr 2
I l I
50 100 150 200 T (K)
Fig. 4. Observed positions of the maxima of absorption and emission bands of MnCI 2, MnBr 2 and MnI 2.
1.6
, I i I '
2.2 E leVI
Fig. 5. Emission spectra of MnCI 2.
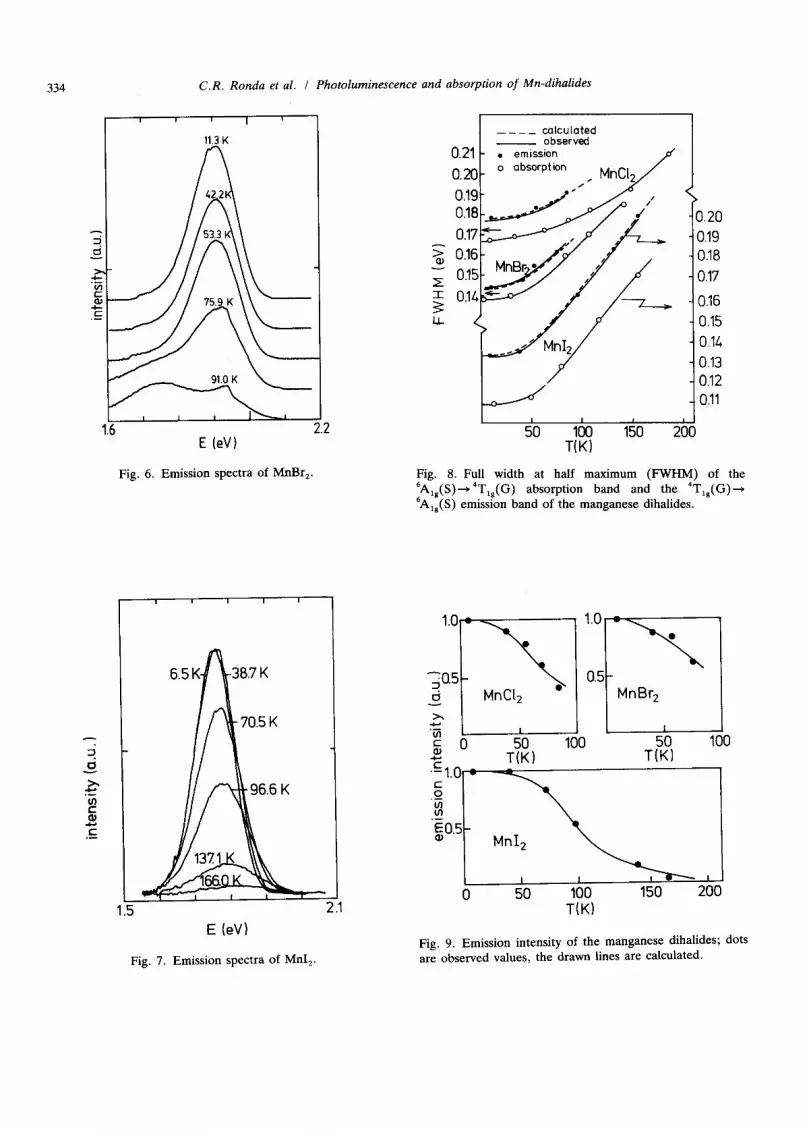
334 C.R. Ronda et al. / Photoluminescence and absorption of Mn-dihalides
' ' ' I '
11.3 K
i J i 1 ~ ' 1.6 2.2
E (eV) Fig. 6. Emission spectra of MnBr 2.
.... calculated observed
0.21 . e m i s s i o n
0 . 2 0 o absorpt ion
0.19 i , " ' " " <, 0.1e ~ _ , ~ / ° / ~ , / r e ' " 0.20 0.17 ~ , Y l-z___~ 0.19 o.16 //~/__~ o18
-
e o.14 ~ / o.16 u_ ~ ~ 0.15
0.14 0.13 0.12 0.11
I 5o lOO 1~o 260 T(K)
Fig. 8. Full width at half maximum (FWHM) of the 6Alg(S)---~4Tlg(G ) absorption band and the 4Tlg(G)---~ 6A~g(S) emission hand of the manganese dihalides.
i i i I I
6.5 K~38.7 K
_~ f / ~ 7 o s K
t-
1.5 2.1
E (eV) Fig. 7. Emission spectra of MnI 2.
001 ~'~ = MnCt2 " MnBr2
0 50 100 50 100 T(K) T(K}
" -1 . ( ; ~- "
i I I , u ~ - - - - I J
0 50 100 150 200 T(K)
Fig. 9. Emission intensity of the manganese dihalides; dots are observed values, the drawn lines are calculated.

C.R. Ronda et al. / Photoluminescence and absorption o f Mn-dihalides 335
Table I Parameters of the 6Alg(S)---+ 4Tlg(G) absorption and the 4Ttg(G)---~ 6Alg(S ) emission in the manganese dihalides. E, is the activation energy for hopping of the "T]g(G) excitons
Absorption Emission FWHM (eV) FWHM (eV) Ea (eV) maximum (eV) maximum (eV) absorption emission at 70 K at 70 K at 70 K at 70 K
MnCI2 2.282 1.918 0.174 0.186 0.015 MnBr 2 2.261 1.906 0.152 0.161 0.012 MnI 2 2.186 1.772 0.122 0.149 0.052
60C
50C
40C
"7 . 3o(
2O0
100
q
calculated
* measured
100 200 300 T(K)
Fig. 10. Calculated and observed decay time ~" of the luminescence of MnI 2.
time ~" as a function of temperature are shown in figs. 9 and 10. The decay follows an exponential law exp(-t/~-) in the measured temperature range. A survey of some of the observed data is given in table I.
4. The blue shift of the absorption and emission bands
A shift of the maxima of absorption and emis- sion bands with increasing temperature is quite common, but the shifts observed in the mangan- ese dihalides are rather large. We ascribe this to the strong dependence of the energy of the 4Tlg(G ) state of the Mn 2÷ ion on the crystal field splitting lODq.
The ground state of the Mn 2+ ion has a t~se ~ configuration, whereas the excited state has a
4 1 t2geg configuration. The blue shift can be esti- mated using the relation between Dq and the metal-ligand distance R from crystal field theory:
Dq = - Ze2r4/6R 5 , (1)
where Ze is the effective nuclear charge of the ligands, e is the unit charge, ? is the mean distance of the d-electrons from the nucleus [12]. The lattice parameters of MnI 2 were determined with neutron diffraction, those of MnC12 and MnBr 2 with X-ray diffraction (table II). The blue shift is given by
A E = ~ AR
= -9 .5 x 5Dq x ( - ~ ) . (2)
The quantity ( S E / S D q ) = - 9 . 5 was obtained from the Tanabe-Sugano diagram [12], the val- ues of Dq from absorption spectra [5-7]. For the shift A E of the absorption band we calculated
Table II Unit cell parameters a and c, metal-ligand distance R (in A) and crystal field splitting lODq (in eV) of the manganese dihalides.
MnCI 2 MnBr 2 MnI 2
a(294 K) 3.711 3.873 a(300 K) 4.148 c(294 K) 17.566 6.271 c(300 K) 6.837 R(294 K) 2.595 2.731 R(300 K) 2.942 a(123 K) 3.703 3.858 a(1.2 K) 4.128 c(123 K) 17.485 6.238 c(1.2 K) 6.784 R(123 K) 2.587 2.719 R(1.2 K) 2.925 lODq 0.955 0.842 lODq 0.750

336 C.R. Ronda et al. / Photolurninescence and absorption o f Mn-dihalides
values which are in reasonable agreement with the observed data.
The observed blue shifts of the absorption and emission bands are nearly the same in the low temperature region. This indicates that the ther- mal expansion of the interatomic distances in the ground and excited states are nearly the same. For MnI 2 the blue shift of the emission band is larger at higher temperature, indicating a larger expansion of the excited state. This effect is attributed to a larger anharmonicity in the ex- cited state.
5. The optical line shape and the dynamic Jahn- Teller effect
In spite of the fact that one expects the excited 4Tlg(G ) state to be split into four spin-orbit components, the observed line shape in both absorption and emission is almost perfectly Gaussian. The asymmetry at the high energy side at higher temperatures in the absorption spectra is caused by the 6Alg(S)----~aT2g(G ) absorption.
It has been shown theoretically that the 6Aag(S)--* 4Tag(G ) band does not split and has a Gaussian shape when the JTE quenches the orbital angular momentum as a result of coupling of the Eg vibration with the 4Tlg(G ) state; cou- pling with the T2g vibration leads to a three-fold split band [13, 14].
Even in systems where the JTE does not lead to a static distortion the orbital angular momen- tum is partly quenched [11]. The theory of this effect will be discussed briefly for the Jahn- Teller interaction of the Tag state interacting with a vibration of Eg symmetry [13, 14]. It is reason- able to assume that the Eg vibrational mode of an octahedron, which involves radial motion of the ligands and interacts with o--bonding orbitals, couples more strongly to the Tag state than the T2g vibrational mode, which involves tangential motion of the ligands and interacts only with • r-bonding orbitals [15]. The experimental obser- vation of a nearly Gaussian line shape also indi- cates that the dominant interaction is with Eg vibrational modes; strong coupling with Tzg type
vibrations would lead to a Jahn-Teller splitting of the absorption band [13, 14].
We use for the 4Tlg(G ) state a basis set of real electronic functions 0~, 0n and ~0:, transforming under rotations of the octahedral group as x, y, z, respectively (x, y, z are parallel to the fourfold axes of the octahedron).
Neglecting all effects associated with the spin, the vibronic Hamiltonian is given by:
1 2 2 2 2z.--~2 2 i H = E J + - ~ [ P o + P~ + tx to t ~d o + Q~)]
+ v [ a o e o + Q~e~], (3)
where E 0 is the energy of the degenerate state in the symmetrical configuration, Po and P, are the nuclear kinetic energy momenta conjugate to Qo and Q, , /x is the effective mass of the vibrational Eg mode and to its angular momentum, V is the Jahn-Teller coupling coefficient and i is the unit matrix. The vibrational modes Qo and Q~ trans- form as z Z - - ( 1 / 2 ) ( x Z + y 2) and (x2--y2), re- spectively, and e 0 and e, are the matrices describ- ing the coupling of the Eg vibrations (Q0, Q~) to the T~g state:
-(1/2) 0 0 ] e o = 0 (1/2) 0 ] ;
0 0 -- 1 (4)
e , = V~72 . 0 0 0
The vibronic eigenfunctions ~O~,(Q, r) of the Hamiltonian are products of one of the elec- tronic functions ~0i(r ) (i = ~, 7, if, the compo- nents of the Tag state) and harmonic oscillator functions for a displaced two dimensional oscil- lator. The equilibrium positions are:
- Veio - Vei~ Qoi = 2 , Q,i - 2 , (5)
txto p, to
where ei0 and el, are the appropriate diagonal components of the matrices e 0 and e~. The potential energy of the oscillator at these posi- tions is lowered by the Jahn-Teller energy

C.R. Ronda et al. / Photoluminescence and absorption of Mn-dihalides 337
EjT = vZ/2 tzw 2. The vibronic eignfunctions are:
( Ve i° ) qJi,,(Q, z) = ~O,('r)F,,o\Qo +
~ £ ( . . 0 2 /
x Fn~(Q~ + --Vei2) , (6) /zto
where Fn(y ) is standard harmonic oscillator function. The corresponding energies are:
V: Ei,, = Eo 2 + (no + n~ + 1) hw, (7)
2/zto
where
no, n ~ = O , 1 , 2 , 3 , . . . .
The vibronic spectrum in this case is the same as in the absence of the Jahn-Teller interaction except for the displacement V2/2/xw 2 common to all states.
However, the equilibrium position for the dis- placed oscillators is different for the three elec- tronic functions Se(~'). The separation is propor- tional to V/p,w 2 and therefore the region of overlap between corresponding oscillator states associated with different electronic functions is diminished. This means that matrix elements between these states become smaller.
Consider, for example, an electronic operator OA, independent of Qo and Q,. A vibronic matrix element of O A is
( q';...~l Ogl % , ; )
= (~,[OAl~j)(inoljn'o)o(in, l jn ' ) , , (8)
where
(inol/n;)o
f ( = deoF, ,o Q o + - ~ w z ) F , , o , I Q o - ¢,¢~
Vej )
(9)
and similarly for ( in, [ jn ' ) ~.
The evaluation of these integrals (which also appear in the theory of the optical lineshape) for the vibronic ground state leads to the following expression for the off-diagonal matrix elements of the operator OA:
( 4,1oolOAlOjoo) = ( ~,lO Al~j) exp [ --3EjT] 2hw J"
(10)
The exponential factor in (10) is the orbital reduction factor y; it is the consequence of the Jahn-Teller quenching of the operator O A. The quenching is almost complete when EjT is much larger than hw.
The considerations given above show that for the 4Tlg(G ) electronic state coupling with an Eg vibration, the vibronic state without spin-orbit splitting remains triply degenerate. The Jahn- Teller effect produces a reduction of the spin- orbit splitting.
The main line broadening mechanism is the coupling of the electron (or hole) to the lattice. The small splitting or broadening, due to the exchange interaction between the Mn 2+ can be neglected. This means that the line shape can be calculated, using the expressions of the theory of the configuration coordinates [13, 14, 16, 17]. An important contribution to the line width of the 6 4 Alg(S)--~ ~Tlg(G ) absorption band and the "Tlg(G)--) °Alg(S ) emission band is due to the very strong dependence of the energy of the 4Tlg(G ) state on Dq. If the coupling with the Eg and Aag vibrational mode is much stronger than the coupling with the Z2g vibrational mode, the intensity of the absorption or emission band as a function of the frequency is determined by the convolution of the contributions of the Alg and Eg vibrational modes [14]. In the strong elec- tron-phonon coupling limit, the FWHM of the absorption or emission band is in this case [17]
FWHM = 8 ~ [ S ~ ( h o h)2 coth(hoJ 1/2kT)
+ $2(ho~2) 2 coth(ho~2/2kT)] 0/2) , (11)

338 C.R. Ronda et al. / Photoluminescence and absorption o f Mn-dihalides
where hto~ is the frequency of t h e A l g vibrational mode and hto 2 the frequency of the Eg vibration- al mode. Sa and S 2 are average numbers of Alg and Eg phonons, involved in the optical transi- tion, respectively. We remark that eq. (11) is only valid in the harmonic approximation. The fact that the FWHM of the absorption band is smaller than the FWHM of the emission band at the same temperature indicates that the potential energy functions are not the same in the ground state and the excited state. It indicates also that the potential energy of the atoms in the excited state is less strongly dependent on the metal ligand distance than in the ground state.
We first calculate the average number of Aag and Eg phonons involved in the emission process (Sxg and S2g, respectively). The phonon frequen- cies hO)lg(Alg ) a n d htO2g(Eg ) in the ground state are known from Raman experiments [18-20] (table III). From the width of the emission band at high and at low temperature we can deduce values for Slg and SEg. The relaxation energy of the ground s t a t e SHg can be calculated from the relation
SHg = SaghtOxg + S2ghtO2g . (12)
The temperature dependence of the FWHM calculated using eq. (11), follows closely the
Table III Parameters of the 6AI,(S)--~ 4Tle(G ) and 4T1e(G ) ~ 6Alg(S ) transition in the manganese dihalides. The phonon frequen- cies are hto, the average numbers of phonons involved in the absorption and emission process are S e and S,, respectively. The relaxation energies of the 6A~e(S ) ground state and the 4Tle(G ) excited state are SH e and SH,, the Jahn-Teller stabilization energy is EjT, and the orbital reduction factor is Y
MnC12 MnBr 2 MnI 2
htOlg(A~e ) (in eV) 0.0291 0.0187 0.0139 hto2e(Ee) (in eV) 0.0179 0.0112 0.0105 Sle 2.4 6.5 11.3 S2g 11.7 12.1 9.0 SH e (in eV) 0.28 0.26 0.25 SH e (in eV) 0.09 0.10 0.15 $1o - 5 ~10 ~10 $2~ - 4 ~2 ~6 EjT (in eV) -0.06 -0.02 ~0.06 "/ ~5 × 10 _3 --5 × 10 -2 10 -4
observed curves (fig. 8). This indicates that the potential energy as a function of the metal- ligand distance in the ground state is described in good approximation by a harmonic potential.
The Stokes shift, which is the energy differ- ence between the maxima of the absorption and emission bands, is equal to the sum of the relaxa- tion energies of the ground state (SHe) and the excited state (SHe). Thus we can calculate the relaxation energy of the excited state from the Stokes shift. The results are given in table III. The smaller relaxation energy in the excited state is due to a more shallow potential energy well in the excited state.
In principle it is possible to calculate SH e also from the width of the absorption band at high temperature. In the high temperature limit (hto ~2kT) we obtain from eq. (11):
FWHM = X/-g]-n-2. V~-T[Sleh(o~e + S2ehto2e] 1/2 , (13)
where Sle and S2e a r e the average numbers of Alg a n d Eg phonons involved in the absorption process, respectively. The frequencies of Alg and Eg phonons in the excited state are htOle and h(OEe, respectively. However, the phonon fre- quencies in the excited state are not known. Moreover, one expects a larger anharmonicity for the potential energy function in the excited state. As a consequence we cannot calculate the Jahn-Teller stabilization energy nor the orbital reduction factor 3' accurately. In order to be able to estimate these quantities, we assume that the phonon frequencies in the excited state htOle(Alg ) a n d htOEe(Eg ) are 5% smaller than the corresponding phonon frequencies in the ground state. From the width of he absorption band at high and at low temperature we calculate the average number of phonons Sle and S2e involved in the absorption process. From this it is possible to calculate the Jahn-Teller stabilization energy EjT ----- S2ehtO2e of the excited state, and the orbi- tal reduction factor 3" = exp - (3EjT/2hto2e).
The results are given in table III. The values for 3" are not accurate, but the calculations clear- ly show that 3" ~ 1. The calculated spin-orbit splitting of the 4Tlg(G ) state in the absence of

C.R. Ronda et al. / Photoluminescence and absorption of Mn-dihalides 339
the Jahn-Teller effect would be about 0.1 eV. The spin-orbit splitting in the presence of JTE will be reduced to 0.1 3' eV, and will therefore be very small indeed. This is in agreement with the observed Gaussian lineshape of the absorption and emission bands for transitions involving the 4Tlg(G ) (and also the 4T2g(G ) state) of Mn 2+. For other states, such as the 4ZEg(D ) state (main-
3 2 configuration) a weak JTE is expected, ly t2geg and indeed the transitions involving these states show a pronounced spin-orbit splitting [5-7].
1 1 1 - = + - - (16) r r R rnR
The decay time r R in the absence of the thermal quenching is due to radiative processes. Because the absorption intensity of the 6Alg(s ) ~ 4Tlg(G ) transition is temperature independent, one ex- pects that also z R will be independent of the temperature. The decay due to non-radiative processes is described by r,R. These processes are thermally activated, with an activation ener- gY Ea:
6. The intensity o f the emiss ion band and the decay t ime o f the luminescence as a function of the t emperature
We first discuss the intensity of the emission band as a function of the temperature. The change of the population of the excited state with the time can be written as:
dn d'-'-[ = A I - B n - C n e x p [ - E J k T ] , (14)
where n is the population of the excited state, A is the absorption coefficient, I is the light intensi- ty, B is the emission coefficient, C is a constant of proportionality associated with the non-radia- tive transition of an electron to the ground state and E~ is the activation energy for the tempera- ture quenching. In the stationary state we obtain for the emission intensity L:
A I L = B n = 1 + ( C / B ) e x p [ - E J k T ] " (15)
The observed temperature dependence of the emission intensity follows closely the behaviour described by eq. (15) (fig. 9). From the com- parison with the observed data activation ener- gies E a of (0.015-0.002)eV for MnC12, (0.012 - 0.003) eV for MnBr 2 and (0.051 - 0.005) eV for MnI 2 are obtained.
The activation energy for the thermal quench- ing of the luminescence for MnI 2 has been ob- tained also from the decay time r of the luminescence as a function of the temperature;
r.R = T o e x p ( E J k T ) . (17)
The observed temperature dependence of the decay time follows closely the behaviour de- scribed by eqs. (16) and (17). From the data we obtain for MnI 2 the values r R =5701~s, r 0 = 7.6 ~s and E a = (0.053 - 0.005) eV. This value of Ea is in good agreement with the value deduced from the temperature dependence of the emis- sion intensity.
It is interesting to compare the temperature dependence of the luminescence with the be- haviour observed in CdI2:Mn [8, 9]. In this system the luminescence increases weakly with increasing temperature, the quantum efficiency is one, independent of the temperature. This indi- cates that non-radiative decay of the 4Tlg(G ) exciton on an isolated Mn 2÷ ion in CdI 2 has a very low probability. Therefore, the quenching of the luminescence of the pure manganese di- halides is not a single-ion effect, as this would also be present in CdI2:Mn 2+. Also thermal quenching due to dipolar energy transfer to a luminescence killer is unlikely because of the large energy difference between absorption and emission bands.
T he 4Tlg(G ) exciton states are strongly local- ized due to the strong interaction with the lat- tice. We ascribe the thermal quenching of the luminescence to phonon-assisted hopping, with an activation energy Ea, of the 4Tlg(G ) excitons towards the luminescence killers. The nature of the killers is not known, it can be an impurity ion, a native defect or a surface state. The value of r 0 is determined by the exciton diffusion rate,

340 C.R. Ronda et al. / Photoluminescence and absorption o f Mn-dihalides
but also by the concentration of the lumines- cence killers.
It is interesting to note that exciton hopping also occurs for the 4Alg(G ) and 4Eg(G) states in the manganese dihalides [5-7, 21]. In these cases the exciton transfer is not thermally activated, because the energy of the 4Alg(G ) and 4Eg(G) is almost independent of the crystal field splitting so that the exciton-lattice interaction is weak.
Acknowledgements
This investigation was supported by the Netherlands Foundation for Chemical Research (Stichting Scheikundig Onderzoek in Nederland, SON) with financial aid from the Netherlands Organization for the Advancement of Pure Re- search (Nederlandse Organisatie voor Zuiver- Wetenschappelijk Onderzoek, ZWO).
References
[1] M. Regis and Y. Farge, J. Physique 37 (1976) 627. [2] M.K. Wilkinson, J.W. Cable, E.O. Wollan and W.C.
Koehler-Ornl, Oak Ridge Nat. Lab. Rep. 2430 (1958) 65; 2501 (1958) 37.
[3] Y. Farge, M. Regis and B.S.H. Royce, J. Physique 37 (1976) 637.
[4] E. WoUan, W. Koehler and M. Wilkinson, Phys. Rev. 110 (1958) 638.
[5] H.J.W.M. Hoekstra, P.R. Boudewijn, H. Groenier and C. Haas, Physica 121B (1983) 62.
[6] H.J.W.M. Hoekstra and C. Haas, Physica 128B (1985) 327.
[7] H.J.W.M. Hoekstra, H.F. Folkersma and C. Haas, Physica 128B (1985) 133.
[8] D. Oelkrug and W. Kempny, Ber. Bunsenges. Phys. Chem. 80 (1976) 436.
[9] D. Oelkrug, A. W61pl, W. Kempny and W. Kayser, Ber. Bunsenges. Phys. Chem. 80 (1976) 441.
[10] J.C. Joshi and R. Kumar, J. Pure and Appl. Phys. 12 (1974) 315.
[11] F.S. Ham, Phys. Rev. 138 (1965) 1727. [12] S. Sugano, Y. Tanabe and H. Kamimura, Multiplets of
Transition Metal Ions in Crystals (Academic Press, New York and London, 1970).
[13] Y. Toyozawa and M. Inoue, J. Phys. Soc. Japan 20 (1965) 1289.
[141 Y. Toyozawa and M. Inoue, J. Phys. Soc. Japan 21 (1966) 1663.
[15] M.D. Sturge, Phys. Rev. B 1 (1970) 1005. [16] R. Englman, The Jahn-Teller Effect in Molecules and
Crystals (Wiley, New York, 1972). [17] J. Bourgoin and M. Lannoo, Point Defects in Semicon-
ductors II (Springer, Berlin, 1983). [18] G. Benedek and A. Frey, Phys. Rev. B 21 (1980) 2482. [19] L. Patrinieri, L. Piseri and I. Pollini, Proc. 8th Int.
Conf. Raman Spectroscopy (1982), p. 479. [20] L. Piseri and I. Pollini, J. Phys. C 17 (1984) 4519. [21] H.J.W.M. Hoekstra, H.F. Folkersma and C. Haas, Solid
State Commun. 51 (1984) 657.