photochemical internalization of tumor-targeted protein toxins
TRANSCRIPT

Lasers in Surgery and Medicine 43:721–733 (2011)
Photochemical Internalization of Tumor-TargetedProtein Toxins
Anette Weyergang, PhD,1�Pal K. Selbo, PhD,1,2 Maria E. B. Berstad, MPharm,1 Monica Bostad, MBiomed,1 and
Kristian Berg, PhD1
1Department of Radiation Biology, Institute for Cancer Research, Norwegian Radium Hospital, Oslo UniversityHospital, Norway
2PCI Biotech AS, 1366 Lysaker, Norway
Photochemical internalization (PCI) is a method for intra-cellular delivery of hydrophilic macromolecular drugswith intracellular targets as well as other drugs with lim-ited ability to penetrate cellular membranes. Such drugsenter cells by means of endocytosis and are to a large ex-tent degraded by hydrolytic enzymes in the lysosomes un-less they possess a mechanism for cytosolic translocation.PCI is based on photodynamic therapy (PDT) specificallytargeting the endosomes and lysosomes of the cells, sothat the drugs in these vesicles can escape into the cytosolfrom where they can reach their targets. The preferentialretention of the photosensitizer (PS) in tumor tissue incombination with controlled light delivery makes PCI rel-atively selective for cancer tissue. The tumor specificity ofPCI can be further increased by delivery of drugs thatselectively target the tumors. Indeed, this has beenshown by PCI delivery of several targeted protein toxins.Targeted protein toxins may be regarded as ideal drugsfor PCI delivery, and may represent the clinical futurefor the PCI technology. Lasers Surg. Med. 43:721–733,2011. � 2011 Wiley-Liss, Inc.
Key words: photodynamic; PDT; PCI; targeting; saporin;gelonin
BACKGROUND
A major problem with cancer therapeutics is still selec-tivity, and treatment-limiting adverse effects are often anobstacle for patient cure. Conventional cancer therapeu-tics are in general antiproliferative and are, unfortunate-ly, affecting rapidly dividing healthy cells in addition tothe cancer cells causing major adverse effects such asnausea, bone marrow suppression, impaired wound heal-ing, and in some cases secondary cancers. New advancesin cancer therapeutics include the use of drugs based onbiomolecules such as DNA, RNA, or proteins linked to acancer-targeting moiety. Such drugs can provide highercancer selectivity compared to conventional chemothera-peutics since their targets are overexpressed in malignantcells. In addition, drugs based on such biomolecules maybe less toxic than conventional chemotherapeutics caus-ing less adverse effects. Despite the high potential of bio-logic drugs in cancer treatment, very few have obtainedmarket approval, and these approved drugs mostly exert
their action on cell surface receptors or extracellulargrowth factors. Biological drugs with intracellular targetshave, however, with few exceptions, not shown sufficientefficacy to warrant clinical approval. This is to a large ex-tent due to limited cellular and intracellular drugdelivery.
The plasma membrane is semipermeable and allowsnutrients and other essential elements to enter the cell.Small and lipophilic molecules are able to diffuse freelyacross the membrane (Fig. 1A), and the passage of largerand polar molecules such as amino acids, sugars, and ionsare transported through the plasma membrane by specificchannel or carrier proteins (Fig. 1B). Large hydrophilicmolecules use a third mechanism to get into the cell; endo-cytosis, where the plasma membrane engulfs the moleculeto be taken up and allows intracellular transport withinthe endocytic vesicle compartments [1,2] (Fig. 1C). Unlessthe internalized material has a mechanism to escape theendocytic vesicles, molecules taken into the cell by meansof endocytosis is to a large extent transported to the lyso-somes where they are degraded by hydrolytic enzymes(Fig. 2A). This is a major obstacle for biomolecule-baseddrugs having intracellular targets [3].
PHOTOCHEMICAL INTERNALIZATION (PCI)
Photochemical internalization (PCI) is a method for cy-tosolic release of drugs trapped in endocytic vesicles. Themethod is based on photodynamic therapy (PDT) targetedto endosomes and lysosomes which causes the membranesof these vesicles to burst so that drugs trapped on the in-side can diffuse into the cytosol from where they can reachtheir target (Fig. 2B). PCI has in vitro been shown inmore than 80 cell lines to increase the biological activityof a large variety of clinically relevant drugs that do notreadily penetrate the plasma membrane [4]. Examplesare type I ribosome-inactivating proteins (RIPs) [5,6],
*Corresponding to: Dr. Anette Weyergang, PhD, Departmentof Radiation Biology, Institute for Cancer Research, NorwegianRadium Hospital, Oslo University Hospital, 0310 Oslo, Norway.E-mail: [email protected]
Accepted 25 May 2011Published online 15 August 2011 in Wiley Online Library(wileyonlinelibrary.com).DOI 10.1002/lsm.21084
� 2011 Wiley-Liss, Inc.

immunotoxins [7–9], gene-encoding plasmids [10,11], ade-novirus [12–14], peptides [15], PNAs [16,17], and somechemotherapeutics such as bleomycin [18], doxorubicin[19,20], and mitoxantrone [21]. The PCI principle has alsobeen demonstrated in vivo with the protein toxin gelonin[22], the p53 gene encoded on a plasmid [23], bleomycin
[18] and recently also with the targeted protein toxinscFvMEL/rGel [24]. These results show that PCI can in-duce efficient light-directed delivery of a large variety ofmolecules into the cells, indicating that PCI may be a ver-satile technology for site-specific drug delivery. The firstclinical trial with PCI is currently underway at Universi-ty College Hospital in London. This is a phase I/II trialwhere the chemotherapeutic drug bleomycin is deliveredby PCI (clinical trial ID: NCT00953512, http://clinical-trials.gov). The results of this clinical trial are very prom-ising (www.pcibiotech.no).PCI represents a method for selective drug delivery to
cancer cells. Selectivity is obtained by preferential reten-tion of the photosensitizer (PS) in tumor tissues [25] andlight application only to the desired area. Since PCIreleases drugs that would otherwise be degraded in lyso-somes, the adverse effects of the drug may be low at ther-apeutically effective doses. The selectivity of PCI towardscancer cells may be further increased by delivery of drugsthat selectively targets the tumors. Indeed, this has beenshown using targeted toxins. The ideal drug for deliveryby PCI: (i) Is taken up exclusively in tumor cells, (ii) hasan intracellular target and is not able to penetrate theplasma membrane of the cell, (iii) is taken up efficientlyby endocytosis and accumulates in endocytic vesicles, (iv)does not have an intrinsic capacity to escape from theendocytic compartment to the cytosol without PCI, whichensures no drug effects in off-target cells not treatedwith PCI. In addition, the drug must be capable of diffus-ing from the blood vessels into the tumor tissue. Tumor-targeted protein toxins can be designed to meet all thesecriteria, which is the focus of the present review.
ANTIBODIES IN CANCER TREATMENT
Unique tumor associated antigens have been difficult toidentify [26]. The last decades have, however, providedextensive knowledge on proteins over-expressed in differ-ent forms of cancers and some of these proteins, such asepidermal growth factor receptor (EGFR), human epider-mal growth factor receptor 2 (HER2), and vascular endo-thelial growth factor (VEGF) are utilized as targets formonoclonal antibodies (mAbs) in cancer therapy. Six dif-ferent antibodies for cancer treatment have obtained mar-keting authorization by FDA and EMEA, but with theexception of Rituximab, targeting CD20 in B-cell non-Hodgkin’s lymphoma, none of these are first line treat-ments. In addition, they are mostly used in combinationwith chemotherapeutic drugs or radiation. Thus, eventhough antibodies can provide high specificity for cancercells, the therapeutic efficacy of these drugs is quite lowand repeated treatments with these drugs are usuallynecessary to obtain an anticancer response. The needfor multiple injections of therapeutic antibodies overtime is associated with development of resistance [27,28].However, the capabilities of the antibodies to disrupt cel-lular functions and activate the immune system (antibodydependent cell-mediated cytotoxicity (ADCC) and comple-ment dependent cytotoxicity (CDC)) make antibodiesvery attractive as anticancer therapeutics, and several
Fig. 1. Cellular mechanisms for uptake. The mechanisms for
uptake through the plasma membrane are dependent on the
physio-chemical properties of the compound. Passive diffu-
sion (A), specific uptake channels (B), and endocytosis (C)
are the main mechanisms of cellular uptake.
Fig. 2. Schematic illustration of PCI. A: Drugs taken up
through endocytosis are transported to lysosomes where they
are degraded before they have exerted their action. B: PCI is
based on accumulation of photosensitizer (PS) in endosomes
and lysosomes. Light exposure causes rupture of the endo/ly-
sosomal membrane and releases the drug into the cytosol
where it can exert its action.
722 WEYERGANG ET AL.

strategies are therefore evaluated to make them more po-tent [29]. One of these strategies is to link mAbs to toxinsconstructing targeting protein toxins.
TARGETED PROTEIN-TOXINS
Targeted protein toxins are molecules consisting of onecell binding moiety (e.g., a targeting ligand such as amAb) and one protein toxin moiety [30–32]. The cell bind-ing part recognizes cells expressing a specific cancer asso-ciated protein, while the toxin, derived from either plantsor bacteria [31], exerts the cytotoxic effect. Targeted pro-tein toxins in cancer treatment have been studied for sev-eral decades. The first and second generation targetedprotein toxins were highly immunogenic, suffered fromlack of specificity, heterogeneous composition due to thechemical conjugation methods, and poor stability whichmade the clinical progress slow. Development of the thirdgeneration recombinant targeted protein toxins led to thefirst clinically approved (FDA 1999 and EMEA 2001) tar-geted protein toxin, denileukin diftitox for the treatmentof cutaneous T-cell lymphoma. Denileukin diftitox is a fu-sion protein between interleukin-2 (IL-2) and a truncateddiphtheria toxin [33]. Several other targeted toxins havebeen, and still are, investigated in clinical trials for bothhematologic and solid tumors [34].The cancer specificity of a targeted protein toxin is de-
pendent on the expression of the antigen to which thedrug is directed in cancer cells versus non-target/normaltissue as well as the affinity of the targeting moiety. Thetargeted protein toxins must bind to antigens expressedon the surface of the tumor cells, and the antigens need tobe endocytosed and translocated to the cytosol in order toinhibit protein synthesis. The targeting moieties mostcommonly used to construct targeting toxins are mAbs,single chain Fv fragments (scFv), and endogenous ligandssuch as growth factors and cytokines. Utilization of awhole mAb as the targeting moiety results in large sizedtargeted toxins with MW > 150 kDa, which is an obstaclefor access to the parenchyme cells of solid tissues [35,36].In addition, recombinant production of whole antibodiesis difficult to control [37]. An advantage utilizing wholemAbs in targeted toxins is the immunologic effect (ADCCand CDC) generated through the Fc part of the antibody,as these immunologic responses are claimed as importantmechanisms for the therapeutic effect of mAbs [38,39].The easier production, and smaller size of targeted toxinsbased on Fv fragments, has however, made Fv fragment-based targeted toxins the most utilized for solid as well ashematopoetic cancers. It should be pointed out that in uti-lization of growth factors and cytokines as targeting moie-ties care must be taken to avoid retention of agonisticeffects of these endogenous ligands. Such effects maystimulate to growth and proliferation of the targeted cellpopulation [40,41].
Challenges of Utilizing Targeted Protein Toxins inCancer Therapy
Targeted protein toxins are generally large sized(>>60 kDa) and are therefore slowly excreted.
Hematopoetic cancers such as leukemia and lymphomaare therefore subjected to high concentrations of targetedtoxins compared to what is achievable in solid tumors[42]. In addition, solid tumors suffer from lack of function-al lymphatic vessels resulting in reduced convective flow,which together with the leaky vasculature and the rapidtumor growth result in an increased interstitial pressure,and thereby a decrease in the tissue penetration of thetargeted toxins. The uptake of targeted protein toxins intumor cells is therefore limited by the slow diffusion ofsuch large molecules through the tumor interstitium [43].A consequence of the slow uptake of targeted toxins in sol-id tumors is that normal cells are rapidly exposed tohigher concentrations of the targeted toxins upon admin-istration. Normal cells expressing the antigen, althoughto a lesser extent than observed in the cancer cells, maytherefore be killed causing severe adverse effects of thetreatment, including vascular leak syndrome (VLS), he-molytic uremic syndrome, and liver damage. Targetedtoxins therefore generally function much better in hema-topoetic cancers compared to solid tumors [34]. Balunaet al. has shown that a structural motif of three aminoacids found both in the ricin toxin (RTA) and in IL-2 isresponsible for binding of both RTA and IL-2 to endotheli-al cells inducing VLS, and that mutation of this motifinhibits endothelial damage. Their results also indicatepresence of this structural motif in pseudomonas exotoxin(PE) and fibronectin [44]. Another problem with targetedtoxins is generation of neutralizing antibodies duringtreatment. With today’s recombinant targeted toxins thetargeting moiety of the drug is usually less immunogenic.Generation of neutralizing antibodies against the toxinpart of the drug is, however, a problem and repeatedinjections of targeted toxins are therefore often of limitedvalue. Thus, in order to optimize the therapeutic outcomeof targeted protein toxins there is a need for increasedspecificity and efficacy to reduce the side effects and thenumber of treatments.
PCI OF TARGETED PROTEIN TOXINS
Targeted protein toxins possess several characteristicsof an ideal drug for PCI delivery as pointed out above.These drugs bind to and are selectively taken up in cancercells by means of endocytosis and their site of action is inthe cytosol.
Ribosome-inactivating Protein-toxins (RIPs)From Plants
Some plants, such as Ricinus communis, Geloninummultiflorum, and Saponaria officinalis produce RIP tox-ins [45]. These plant RIPs exert N-glycosidase activity ofthe RNA unit 28S of the 60S ribosome complex, causingarrest of the protein synthesis leading to cell death[46,47]. RIPs can mainly be divided into 2 groups, type Iand type II [47,48]. Type I RIPs (e.g., gelonin, agrostin,and saporin) consist only of a cytotoxic chain with N-glycosidase activity (A-chain), while type II RIPs (e.g.,ricin, abrin, and mistelthoe lectin) have a cell binding B-chain in addition to the toxic A-chain. The toxic A-chain
PCI OF TUMOR-TARGETED PROTEIN TOXINS 723
from the different RIPs has been reported to use distinctmechanisms for cytosolic translocation [49,50]. However,once inside the cell cytosol, type I and II RIPs have similarpotency [45]. An advantage of using RIP-based targetedtoxins in cancer therapy is that they are highly toxicwhen they enter the cell cytosol making them likely to killtheir target. The lack of a cell binding B-chain in type IRIPs causes, however, poor cellular uptake. In addition,the small cellular uptake of type I RIPs are mainlythrough pinocytosis and the type I RIPs therefore end upin lysosomes where they are degraded since translocationto cytosol is exerted with very low efficacy. The cytotoxici-ty of these type I RIPs is therefore often absent or verylow [45]. Type I RIP-based targeted toxins hold, however,all the characteristics listed above for an optimal drug tobe delivered by PCI.
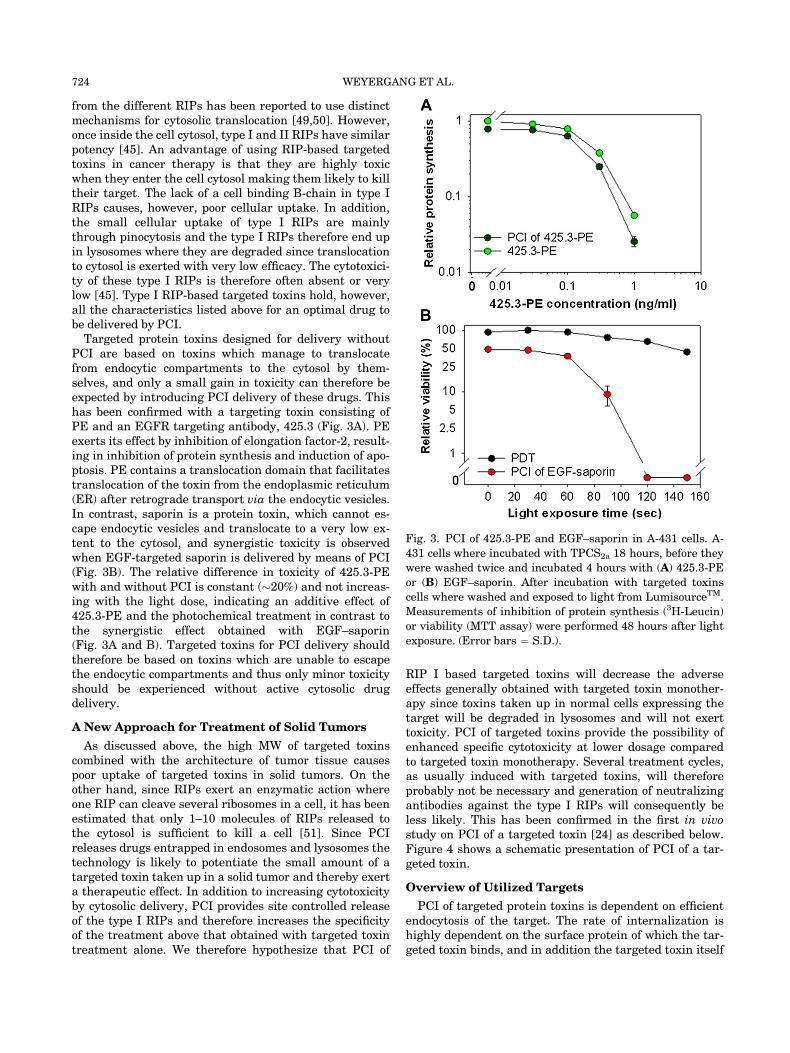
Targeted protein toxins designed for delivery withoutPCI are based on toxins which manage to translocatefrom endocytic compartments to the cytosol by them-selves, and only a small gain in toxicity can therefore beexpected by introducing PCI delivery of these drugs. Thishas been confirmed with a targeting toxin consisting ofPE and an EGFR targeting antibody, 425.3 (Fig. 3A). PEexerts its effect by inhibition of elongation factor-2, result-ing in inhibition of protein synthesis and induction of apo-ptosis. PE contains a translocation domain that facilitatestranslocation of the toxin from the endoplasmic reticulum(ER) after retrograde transport via the endocytic vesicles.In contrast, saporin is a protein toxin, which cannot es-cape endocytic vesicles and translocate to a very low ex-tent to the cytosol, and synergistic toxicity is observedwhen EGF-targeted saporin is delivered by means of PCI(Fig. 3B). The relative difference in toxicity of 425.3-PEwith and without PCI is constant (�20%) and not increas-ing with the light dose, indicating an additive effect of425.3-PE and the photochemical treatment in contrast tothe synergistic effect obtained with EGF–saporin(Fig. 3A and B). Targeted toxins for PCI delivery shouldtherefore be based on toxins which are unable to escapethe endocytic compartments and thus only minor toxicityshould be experienced without active cytosolic drugdelivery.
A New Approach for Treatment of Solid Tumors
As discussed above, the high MW of targeted toxinscombined with the architecture of tumor tissue causespoor uptake of targeted toxins in solid tumors. On theother hand, since RIPs exert an enzymatic action whereone RIP can cleave several ribosomes in a cell, it has beenestimated that only 1–10 molecules of RIPs released tothe cytosol is sufficient to kill a cell [51]. Since PCIreleases drugs entrapped in endosomes and lysosomes thetechnology is likely to potentiate the small amount of atargeted toxin taken up in a solid tumor and thereby exerta therapeutic effect. In addition to increasing cytotoxicityby cytosolic delivery, PCI provides site controlled releaseof the type I RIPs and therefore increases the specificityof the treatment above that obtained with targeted toxintreatment alone. We therefore hypothesize that PCI of
RIP I based targeted toxins will decrease the adverseeffects generally obtained with targeted toxin monother-apy since toxins taken up in normal cells expressing thetarget will be degraded in lysosomes and will not exerttoxicity. PCI of targeted toxins provide the possibility ofenhanced specific cytotoxicity at lower dosage comparedto targeted toxin monotherapy. Several treatment cycles,as usually induced with targeted toxins, will thereforeprobably not be necessary and generation of neutralizingantibodies against the type I RIPs will consequently beless likely. This has been confirmed in the first in vivostudy on PCI of a targeted toxin [24] as described below.Figure 4 shows a schematic presentation of PCI of a tar-geted toxin.
Overview of Utilized Targets
PCI of targeted protein toxins is dependent on efficientendocytosis of the target. The rate of internalization ishighly dependent on the surface protein of which the tar-geted toxin binds, and in addition the targeted toxin itself
Fig. 3. PCI of 425.3-PE and EGF–saporin in A-431 cells. A-
431 cells where incubated with TPCS2a 18 hours, before they
were washed twice and incubated 4 hours with (A) 425.3-PE
or (B) EGF–saporin. After incubation with targeted toxins
cells where washed and exposed to light from LumisourceTM.
Measurements of inhibition of protein synthesis (3H-Leucin)
or viability (MTT assay) were performed 48 hours after light
exposure. (Error bars ¼ S.D.).
724 WEYERGANG ET AL.

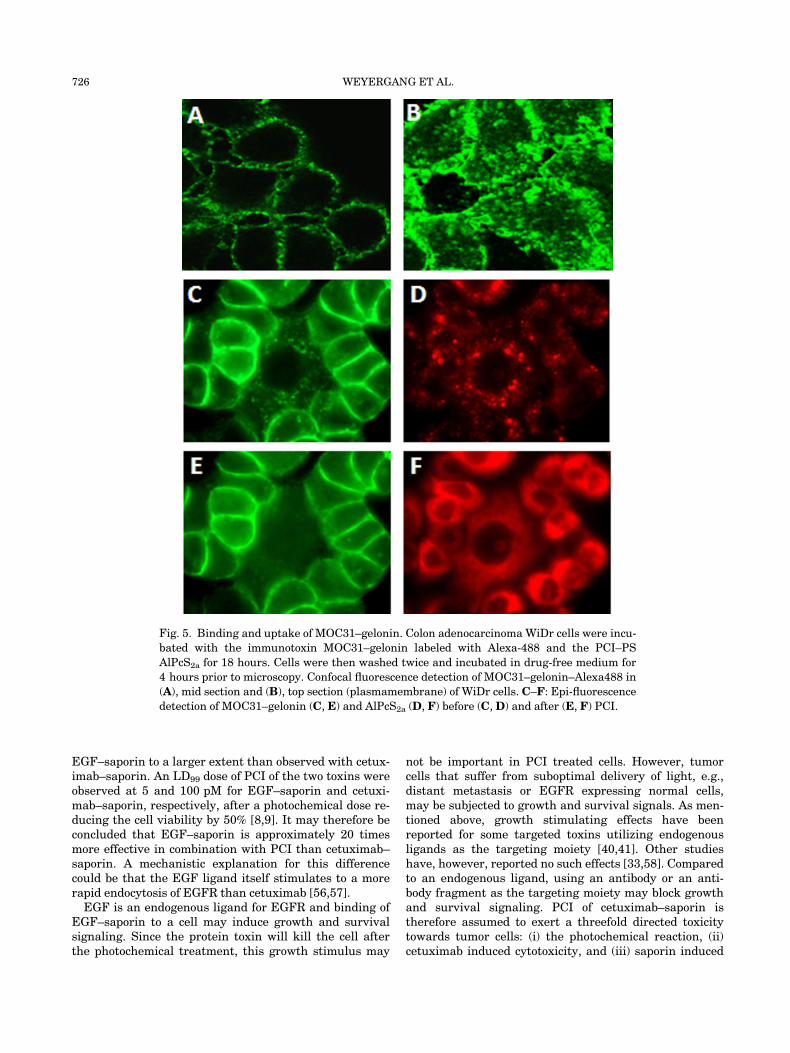
may stimulate or inhibit endocytosis of its target. In thefirst study of PCI of a targeted protein toxin a chemicalconjugation between the type I RIP gelonin and the mAb31 (MOC31), which recognizes an epithelial cell adhesionmolecule (EpCAM/EGP-2/ESA/CD326) [7,52] was used asa the targeted therapeutic. EpCAM is an attractive targetsince it is over-expressed on nearly all carcinomas. PCI ofMOC31–gelonin was shown to synergistically increasethe cytotoxic effect of the immunotoxin using bothAlPcS2a and 5-ALA-induced PPIX as PSs [52]. However,confocal fluorescence microscopy showed that even after18 hours incubation most of the MOC31–gelonin waspresent on the surface of the plasma membrane and notin endocytic vesicles (Fig. 5A and B). This was in contrastto the PS which was found to localize in endocytic vesicles(Fig. 5C). Only a minor effect on MOC31 localization wasobserved after light exposure, indicating poor relocaliza-tion of the targeted toxin to the cell cytosol (Fig. 5D) incontrast to the relocalization of the PS (Fig. 5E). Based onthis, our research group has focused on targeting-moietiesknown to be effectively endocytosed by its antigen as ex-emplified below.
PCI of EGFR-targeting Toxins
EGFR is a 170 kDa transmembrane tyrosine kinase inthe plasma membrane known to be over-expressed in sev-eral different cancers [53]. The receptor consists of an
extracellular ligand binding domain, a transmembranedomain and an intracellular domain where the kinase ac-tivity is located [53,54]. Upon activation by one of itsligands the receptor immediately undergoes endocytosiswhich makes EGFR an attractive antigen for targetedtoxins to be delivered by PCI. Two EGFR-targeted toxinsbased on the type I RIP saporin have been evaluated forPCI administration; one utilizing the endogenous ligandEGF as the targeting moiety (EGF–saporin) [8] and oneusing cetuximab, a whole EGFR-targeting antibody ap-proved for the treatment of colorectal cancer, as the tar-geting moiety (cetuximab–saporin) [9]. EGF–saporin andcetuximab–saporin were shown to be specifically taken upby EGFR indicating that utilization of EGFR-targetedtoxins can improve the specificity of PCI towards cancercells compared to delivery of untargeted therapeutics. Ac-cordingly, PCI of EGF–saporin and cetuximab–saporinwere more cytotoxic than PCI of saporin at comparabledoses of protein toxin, confirming that EGFR is a promis-ing target for PCI-delivered targeted toxins.
The affinity of EGFR for EGF is lower than for cetuxi-mab [55]. Hence, higher doses of the EGF–saporin com-plex compared to cetuximab–saporin may therefore berequired to induce comparable cytotoxicity. No direct com-parison has yet been made between EGF–saporin andcetuximab–saporin, but studies of both targeted toxins inA-431 cells indicate that PCI increases the cytotoxicity of
Fig. 4. Schematic presentation of PCI of a targeted toxin illustrated by cetuximab–saporin.
The PS has been administrated and is localizing from the plasma membrane to endocytic
vesicles following overall endocytosis. Cetuximab–saporin binds to EGFR, is internalized by
receptor-mediated endocytosis and is localized to endosomes with photosensitizer (PS) incor-
porated in the membrane. Light exposure causes membrane rupture and the toxin (saporin)
escapes into the cytosol where it binds to ribosomes and inhibits protein synthesis, which
eventually causes cell death.
PCI OF TUMOR-TARGETED PROTEIN TOXINS 725
EGF–saporin to a larger extent than observed with cetux-imab–saporin. An LD99 dose of PCI of the two toxins wereobserved at 5 and 100 pM for EGF–saporin and cetuxi-mab–saporin, respectively, after a photochemical dose re-ducing the cell viability by 50% [8,9]. It may therefore beconcluded that EGF–saporin is approximately 20 timesmore effective in combination with PCI than cetuximab–saporin. A mechanistic explanation for this differencecould be that the EGF ligand itself stimulates to a morerapid endocytosis of EGFR than cetuximab [56,57].
EGF is an endogenous ligand for EGFR and binding ofEGF–saporin to a cell may induce growth and survivalsignaling. Since the protein toxin will kill the cell afterthe photochemical treatment, this growth stimulus may
not be important in PCI treated cells. However, tumorcells that suffer from suboptimal delivery of light, e.g.,distant metastasis or EGFR expressing normal cells,may be subjected to growth and survival signals. As men-tioned above, growth stimulating effects have beenreported for some targeted toxins utilizing endogenousligands as the targeting moiety [40,41]. Other studieshave, however, reported no such effects [33,58]. Comparedto an endogenous ligand, using an antibody or an anti-body fragment as the targeting moiety may block growthand survival signaling. PCI of cetuximab–saporin istherefore assumed to exert a threefold directed toxicitytowards tumor cells: (i) the photochemical reaction, (ii)cetuximab induced cytotoxicity, and (iii) saporin induced
Fig. 5. Binding and uptake of MOC31–gelonin. Colon adenocarcinoma WiDr cells were incu-
bated with the immunotoxin MOC31–gelonin labeled with Alexa-488 and the PCI–PS
AlPcS2a for 18 hours. Cells were then washed twice and incubated in drug-free medium for
4 hours prior to microscopy. Confocal fluorescence detection of MOC31–gelonin–Alexa488 in
(A), mid section and (B), top section (plasmamembrane) of WiDr cells. C–F: Epi-fluorescence
detection of MOC31–gelonin (C, E) and AlPcS2a (D, F) before (C,D) and after (E, F) PCI.
726 WEYERGANG ET AL.
cytotoxicity. In addition, cetuximab will in vivo stimulateto ADCC which is claimed to be its main mechanism ofaction [38,39].The biotin–streptavidin linkage was used to link sap-
orin to both EGF and cetuximab in our proof of conceptstudies on PCI of EGFR targeting toxins [8,9]. This link-age rapidly forms a strong non-covalent binding(Kass: 1015 M�1), which makes it convenient for suchproof-of-concept studies. Targeted toxins based on the bio-tin–streptavidin linkage are, however, generally not suit-ed for in vivo applications. Streptavidin is a tetramer of60 kDa where each monomer has a biotin binding site.The binding reaction of biotinylated proteins to streptavi-din labeled saporin can be difficult to control due to sterichindrance, which gives the possibility of heterogeneity inthe reaction product. The ability of streptavidin to bindfour biotinylated targeted moieties also results in verylarge products. Four molecules of biotinylated cetuximabbound to streptavidin–saporin may, i.e., form a targetedtoxin of 700 kDa which is too large for efficient delivery tosolid tumors. The last decades, research on recombinanttechnologies have made it possible to synthesize targetedtoxins in transfected E. coli. Recombinant synthesis oftargeted toxins offers high control of the product, and thetechnology makes it possible to induce a peptide linker inthe product to obtain sufficient distance between the moi-eties and to achieve a desired molecular size of the fusiontoxin for optimal distribution in solid tumors.
PCI of scFvMEL/rGel; In vivo Evaluation
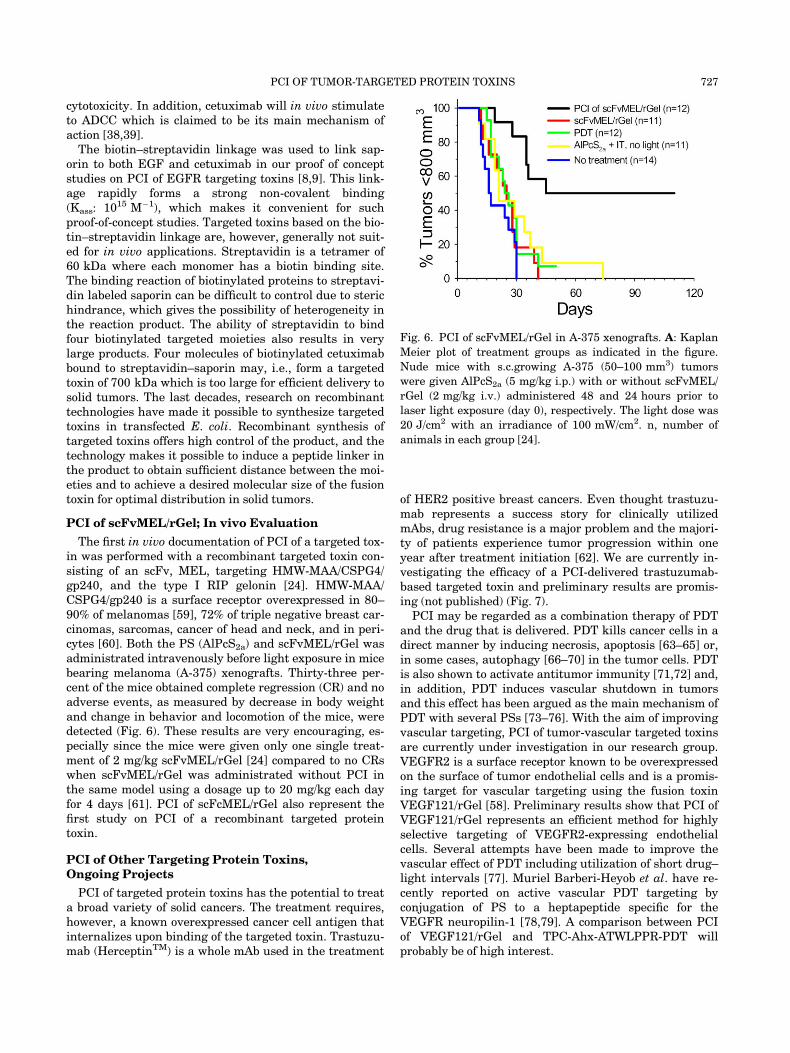
The first in vivo documentation of PCI of a targeted tox-in was performed with a recombinant targeted toxin con-sisting of an scFv, MEL, targeting HMW-MAA/CSPG4/gp240, and the type I RIP gelonin [24]. HMW-MAA/CSPG4/gp240 is a surface receptor overexpressed in 80–90% of melanomas [59], 72% of triple negative breast car-cinomas, sarcomas, cancer of head and neck, and in peri-cytes [60]. Both the PS (AlPcS2a) and scFvMEL/rGel wasadministrated intravenously before light exposure in micebearing melanoma (A-375) xenografts. Thirty-three per-cent of the mice obtained complete regression (CR) and noadverse events, as measured by decrease in body weightand change in behavior and locomotion of the mice, weredetected (Fig. 6). These results are very encouraging, es-pecially since the mice were given only one single treat-ment of 2 mg/kg scFvMEL/rGel [24] compared to no CRswhen scFvMEL/rGel was administrated without PCI inthe same model using a dosage up to 20 mg/kg each dayfor 4 days [61]. PCI of scFcMEL/rGel also represent thefirst study on PCI of a recombinant targeted proteintoxin.
PCI of Other Targeting Protein Toxins,Ongoing Projects
PCI of targeted protein toxins has the potential to treata broad variety of solid cancers. The treatment requires,however, a known overexpressed cancer cell antigen thatinternalizes upon binding of the targeted toxin. Trastuzu-mab (HerceptinTM) is a whole mAb used in the treatment
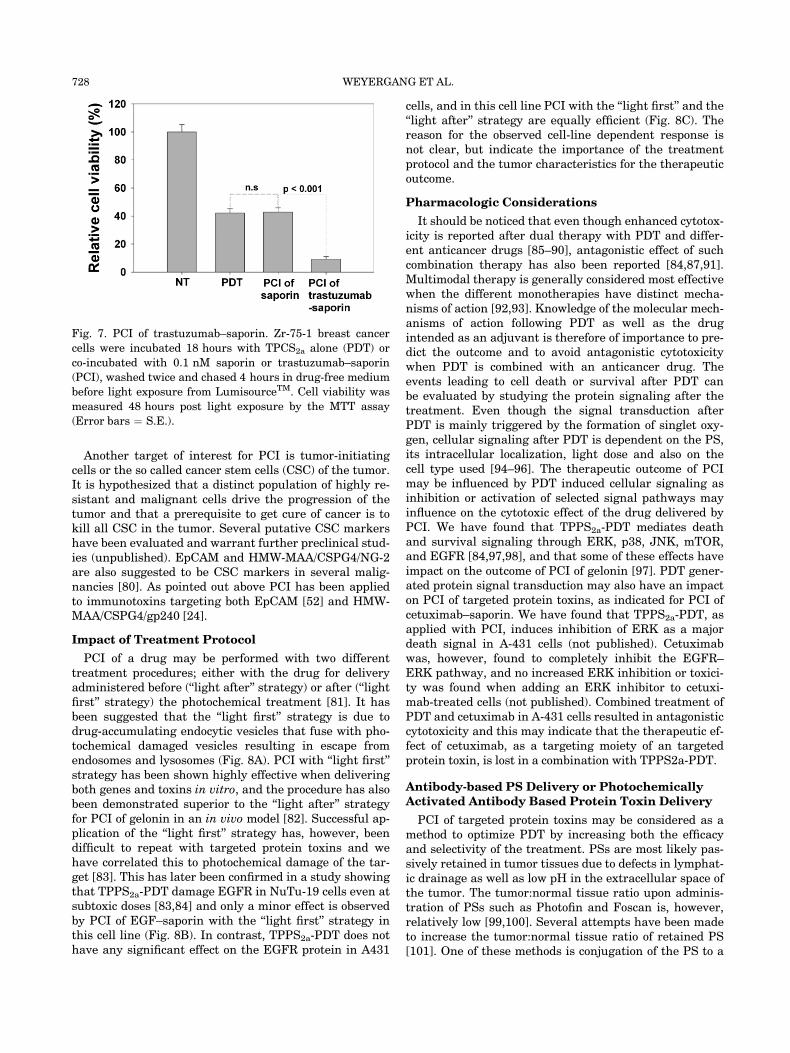
of HER2 positive breast cancers. Even thought trastuzu-mab represents a success story for clinically utilizedmAbs, drug resistance is a major problem and the majori-ty of patients experience tumor progression within oneyear after treatment initiation [62]. We are currently in-vestigating the efficacy of a PCI-delivered trastuzumab-based targeted toxin and preliminary results are promis-ing (not published) (Fig. 7).
PCI may be regarded as a combination therapy of PDTand the drug that is delivered. PDT kills cancer cells in adirect manner by inducing necrosis, apoptosis [63–65] or,in some cases, autophagy [66–70] in the tumor cells. PDTis also shown to activate antitumor immunity [71,72] and,in addition, PDT induces vascular shutdown in tumorsand this effect has been argued as the main mechanism ofPDT with several PSs [73–76]. With the aim of improvingvascular targeting, PCI of tumor-vascular targeted toxinsare currently under investigation in our research group.VEGFR2 is a surface receptor known to be overexpressedon the surface of tumor endothelial cells and is a promis-ing target for vascular targeting using the fusion toxinVEGF121/rGel [58]. Preliminary results show that PCI ofVEGF121/rGel represents an efficient method for highlyselective targeting of VEGFR2-expressing endothelialcells. Several attempts have been made to improve thevascular effect of PDT including utilization of short drug–light intervals [77]. Muriel Barberi-Heyob et al. have re-cently reported on active vascular PDT targeting byconjugation of PS to a heptapeptide specific for theVEGFR neuropilin-1 [78,79]. A comparison between PCIof VEGF121/rGel and TPC-Ahx-ATWLPPR-PDT willprobably be of high interest.
Fig. 6. PCI of scFvMEL/rGel in A-375 xenografts. A: Kaplan
Meier plot of treatment groups as indicated in the figure.
Nude mice with s.c.growing A-375 (50–100 mm3) tumors
were given AlPcS2a (5 mg/kg i.p.) with or without scFvMEL/
rGel (2 mg/kg i.v.) administered 48 and 24 hours prior to
laser light exposure (day 0), respectively. The light dose was
20 J/cm2 with an irradiance of 100 mW/cm2. n, number of
animals in each group [24].
PCI OF TUMOR-TARGETED PROTEIN TOXINS 727
Another target of interest for PCI is tumor-initiatingcells or the so called cancer stem cells (CSC) of the tumor.It is hypothesized that a distinct population of highly re-sistant and malignant cells drive the progression of thetumor and that a prerequisite to get cure of cancer is tokill all CSC in the tumor. Several putative CSC markershave been evaluated and warrant further preclinical stud-ies (unpublished). EpCAM and HMW-MAA/CSPG4/NG-2are also suggested to be CSC markers in several malig-nancies [80]. As pointed out above PCI has been appliedto immunotoxins targeting both EpCAM [52] and HMW-MAA/CSPG4/gp240 [24].
Impact of Treatment Protocol
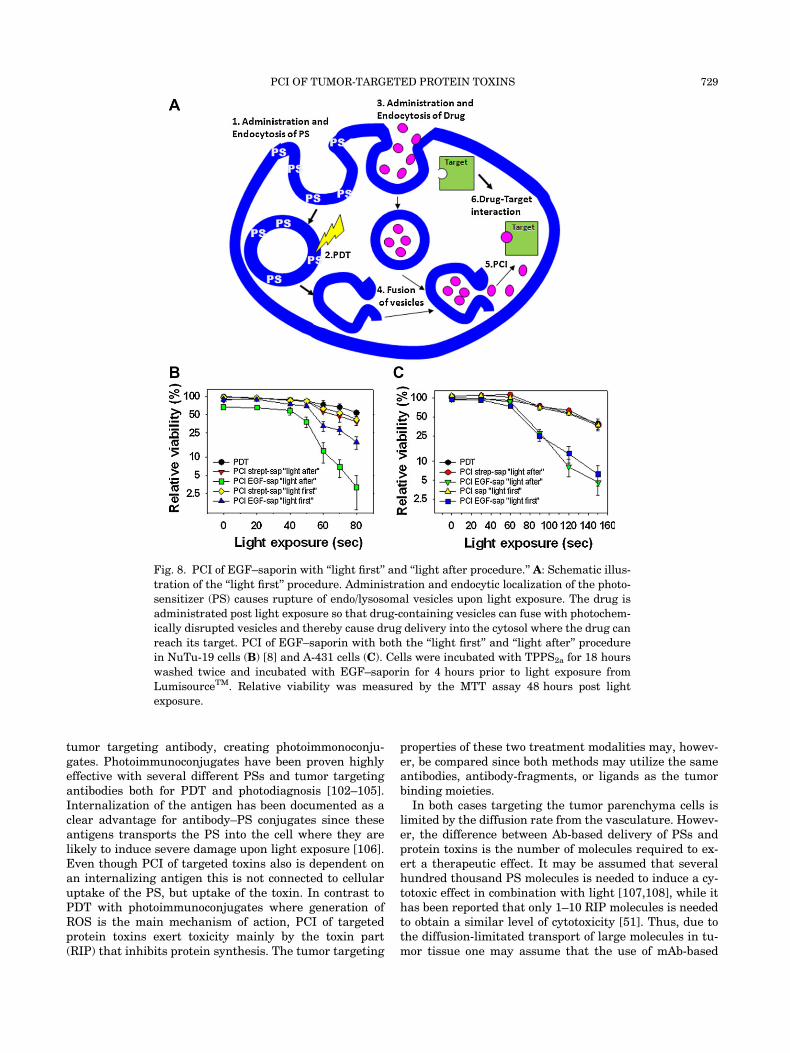
PCI of a drug may be performed with two differenttreatment procedures; either with the drug for deliveryadministered before (‘‘light after’’ strategy) or after (‘‘lightfirst’’ strategy) the photochemical treatment [81]. It hasbeen suggested that the ‘‘light first’’ strategy is due todrug-accumulating endocytic vesicles that fuse with pho-tochemical damaged vesicles resulting in escape fromendosomes and lysosomes (Fig. 8A). PCI with ‘‘light first’’strategy has been shown highly effective when deliveringboth genes and toxins in vitro, and the procedure has alsobeen demonstrated superior to the ‘‘light after’’ strategyfor PCI of gelonin in an in vivo model [82]. Successful ap-plication of the ‘‘light first’’ strategy has, however, beendifficult to repeat with targeted protein toxins and wehave correlated this to photochemical damage of the tar-get [83]. This has later been confirmed in a study showingthat TPPS2a-PDT damage EGFR in NuTu-19 cells even atsubtoxic doses [83,84] and only a minor effect is observedby PCI of EGF–saporin with the ‘‘light first’’ strategy inthis cell line (Fig. 8B). In contrast, TPPS2a-PDT does nothave any significant effect on the EGFR protein in A431
cells, and in this cell line PCI with the ‘‘light first’’ and the‘‘light after’’ strategy are equally efficient (Fig. 8C). Thereason for the observed cell-line dependent response isnot clear, but indicate the importance of the treatmentprotocol and the tumor characteristics for the therapeuticoutcome.
Pharmacologic Considerations
It should be noticed that even though enhanced cytotox-icity is reported after dual therapy with PDT and differ-ent anticancer drugs [85–90], antagonistic effect of suchcombination therapy has also been reported [84,87,91].Multimodal therapy is generally considered most effectivewhen the different monotherapies have distinct mecha-nisms of action [92,93]. Knowledge of the molecular mech-anisms of action following PDT as well as the drugintended as an adjuvant is therefore of importance to pre-dict the outcome and to avoid antagonistic cytotoxicitywhen PDT is combined with an anticancer drug. Theevents leading to cell death or survival after PDT canbe evaluated by studying the protein signaling after thetreatment. Even though the signal transduction afterPDT is mainly triggered by the formation of singlet oxy-gen, cellular signaling after PDT is dependent on the PS,its intracellular localization, light dose and also on thecell type used [94–96]. The therapeutic outcome of PCImay be influenced by PDT induced cellular signaling asinhibition or activation of selected signal pathways mayinfluence on the cytotoxic effect of the drug delivered byPCI. We have found that TPPS2a-PDT mediates deathand survival signaling through ERK, p38, JNK, mTOR,and EGFR [84,97,98], and that some of these effects haveimpact on the outcome of PCI of gelonin [97]. PDT gener-ated protein signal transduction may also have an impacton PCI of targeted protein toxins, as indicated for PCI ofcetuximab–saporin. We have found that TPPS2a-PDT, asapplied with PCI, induces inhibition of ERK as a majordeath signal in A-431 cells (not published). Cetuximabwas, however, found to completely inhibit the EGFR–ERK pathway, and no increased ERK inhibition or toxici-ty was found when adding an ERK inhibitor to cetuxi-mab-treated cells (not published). Combined treatment ofPDT and cetuximab in A-431 cells resulted in antagonisticcytotoxicity and this may indicate that the therapeutic ef-fect of cetuximab, as a targeting moiety of an targetedprotein toxin, is lost in a combination with TPPS2a-PDT.
Antibody-based PS Delivery or PhotochemicallyActivated Antibody Based Protein Toxin Delivery
PCI of targeted protein toxins may be considered as amethod to optimize PDT by increasing both the efficacyand selectivity of the treatment. PSs are most likely pas-sively retained in tumor tissues due to defects in lymphat-ic drainage as well as low pH in the extracellular space ofthe tumor. The tumor:normal tissue ratio upon adminis-tration of PSs such as Photofin and Foscan is, however,relatively low [99,100]. Several attempts have been madeto increase the tumor:normal tissue ratio of retained PS[101]. One of these methods is conjugation of the PS to a
Fig. 7. PCI of trastuzumab–saporin. Zr-75-1 breast cancer
cells were incubated 18 hours with TPCS2a alone (PDT) or
co-incubated with 0.1 nM saporin or trastuzumab–saporin
(PCI), washed twice and chased 4 hours in drug-free medium
before light exposure from LumisourceTM. Cell viability was
measured 48 hours post light exposure by the MTT assay
(Error bars ¼ S.E.).
728 WEYERGANG ET AL.
tumor targeting antibody, creating photoimmonoconju-gates. Photoimmunoconjugates have been proven highlyeffective with several different PSs and tumor targetingantibodies both for PDT and photodiagnosis [102–105].Internalization of the antigen has been documented as aclear advantage for antibody–PS conjugates since theseantigens transports the PS into the cell where they arelikely to induce severe damage upon light exposure [106].Even though PCI of targeted toxins also is dependent onan internalizing antigen this is not connected to cellularuptake of the PS, but uptake of the toxin. In contrast toPDT with photoimmunoconjugates where generation ofROS is the main mechanism of action, PCI of targetedprotein toxins exert toxicity mainly by the toxin part(RIP) that inhibits protein synthesis. The tumor targeting
properties of these two treatment modalities may, howev-er, be compared since both methods may utilize the sameantibodies, antibody-fragments, or ligands as the tumorbinding moieties.
In both cases targeting the tumor parenchyma cells islimited by the diffusion rate from the vasculature. Howev-er, the difference between Ab-based delivery of PSs andprotein toxins is the number of molecules required to ex-ert a therapeutic effect. It may be assumed that severalhundred thousand PS molecules is needed to induce a cy-totoxic effect in combination with light [107,108], while ithas been reported that only 1–10 RIP molecules is neededto obtain a similar level of cytotoxicity [51]. Thus, due tothe diffusion-limitated transport of large molecules in tu-mor tissue one may assume that the use of mAb-based
Fig. 8. PCI of EGF–saporin with ‘‘light first’’ and ‘‘light after procedure.’’ A: Schematic illus-
tration of the ‘‘light first’’ procedure. Administration and endocytic localization of the photo-
sensitizer (PS) causes rupture of endo/lysosomal vesicles upon light exposure. The drug is
administrated post light exposure so that drug-containing vesicles can fuse with photochem-
ically disrupted vesicles and thereby cause drug delivery into the cytosol where the drug can
reach its target. PCI of EGF–saporin with both the ‘‘light first’’ and ‘‘light after’’ procedure
in NuTu-19 cells (B) [8] and A-431 cells (C). Cells were incubated with TPPS2a for 18 hours
washed twice and incubated with EGF–saporin for 4 hours prior to light exposure from
LumisourceTM. Relative viability was measured by the MTT assay 48 hours post light
exposure.
PCI OF TUMOR-TARGETED PROTEIN TOXINS 729

delivery of protein toxins in a PCI regimen is preferable toAb-based delivery of PSs. However, in cases where tissuediffusion is not a limitation for therapeutic outcome suchas in endothelial cells, a combination of photoimmunocon-jugates to rupture the endocytic vesicles resulting in re-lease of Ab-linked protein toxins into the cytosol should beconsidered [109].
CONCLUSIONS
Targeted protein toxins have been a focus in cancertherapy for more than three decades. Only one targetedtoxin, denileukin diftitox, is however currently clinicallyapproved. In general, targeted toxins have been shownhighly effective in several hematopoetic cancers. Poorpenetration through malignant tissue in addition to re-duced convection are, however, obstacles for treatment ofsolid tumors [110,111]. PCI enhances the effect of tar-geted toxins that have reached the tumor cells, and there-by reduce the impact of poor tumor delivery. Anotherlimitation in the use of therapeutic targeted toxins is theformation of neutralizing antibodies due to repeated injec-tions of the drug. Since PCI may enhance the therapeuticeffect of a targeted toxin up to 1,000-fold [8], the numberof treatments is likely to be highly reduced compared totreatments with the targeted toxin alone [24]. Hence, for-mation of neutralizing antibodies will probably be lessproblematic. Another major limitation for the clinical ap-plication of targeted toxins is their uptake in normal cells.This causes severe adverse effects and damage to healthyorgans which also express the target antigen on the cellsurface [112]. These adverse effects are highly dose-de-pendent, and since PCI is expected to increase treatmentspecificity and efficiency which probably reduce the neces-sary dosage of the targeted toxin, it is to be expected thatthese adverse effects can be significantly reduced by intro-duction of the PCI technology. PCI represents a uniquehighly selective method for delivery of macromoleculardrugs to solid tumors. Overall, targeted toxins that accu-mulate in endocytic vesicles possess all the characteristicsfor optimal drugs to be delivered with PCI. Our goal istherefore to introduce this method in the clinic in nearfuture.
ACKNOWLEDGMENTS
The authors thank their former master students,Nguyen Duc Thuan and Wai Lam Yip for laboratory workwith the MOC31–gelonin and cetuximab–gelonin. Theyalso thank Dr. Olav Engebraten, Department of TumorBiology, Norwegian Radium Hospital, Norway who kindlyprovided the 425.3-PE targeted toxin. Financial supportfrom the Norwegian Cancer Society, the South-EasternNorway Regional Health Authority, and the Radium Hos-pital Research Foundation is highly appreciated.
REFERENCES
1. Mousavi SA, Malerod L, Berg T, Kjeken R. Clathrin-depen-dent endocytosis. Biochem J 2004;377(Pt 1):1–16.
2. Mayor S, Pagano RE. Pathways of clathrin-independent en-docytosis. Nat Rev Mol Cell Biol 2007;8(8):603–612.
3. Lloyd JB. Lysosome membrane permeability: Implicationsfor drug delivery. Adv Drug Deliv Rev 2000;2(41):189–200.
4. Selbo PK, Weyergang A, Hogset A, Norum OJ, Berstad MB,Vikdal M, Berg K. Photochemical internalization providestime- and space-controlled endolysosomal escape of thera-peutic molecules. J Control Release 2010;1(148):2–12.
5. Selbo PK, Sandvig K, Kirveliene V, Berg K. Release of gelo-nin from endosomes and lysosomes to cytosol by photo-chemical internalization. Biochim Biophys Acta 2000;3(1475):307–313.
6. Dietze A, Bonsted A, Hogset A, Berg K. Photochemical in-ternalization enhances the cytotoxic effect of the proteintoxin gelonin and transgene expression in sarcoma cells.Photochem Photobiol 2003;3(78):283–289.
7. Selbo PK, Sivam G, Fodstad O, Sandvig K, Berg K. Photo-chemical internalisation increases the cytotoxic effect of theimmunotoxin MOC31-gelonin. Int J Cancer 2000;6(87):853–859.
8. Weyergang A, Selbo PK, Berg K. Photochemically stimulat-ed drug delivery increases the cytotoxicity and specificity ofEGF-saporin. J Control Release 2006;1–2(111):165–173.
9. Yip WL, Weyergang A, Berg K, Tonnesen HH, Selbo PK.Targeted delivery and enhanced cytotoxicity of cetuximab-saporin by photochemical internalization in EGFR-positivecancer cells. Mol Pharm 2007;2(4):241–251.
10. Hogset A, Prasmickaite L, Tjelle TE, Berg K. Photochemi-cal transfection: A new technology for light-induced, site-di-rected gene delivery. Hum Gene Ther 2000;6(11):869–880.
11. Prasmickaite L, Hogset A, Tjelle TE, Olsen VM, Berg K.Role of endosomes in gene transfection mediated by photo-chemical internalisation (PCI). J Gene Med 2000;6(2):477–488.
12. Hogset A, Ovstebo EB, Prasmickaite L, Berg K, Fodstad O,Maelandsmo GM. Light-induced adenovirus gene transfer,an efficient and specific gene delivery technology for cancergene therapy. Cancer Gene Ther 2002;4(9):365–371.
13. Bonsted A, Engesaeter BO, Hogset A, Maelandsmo GM,Prasmickaite L, Kaalhus O, Berg K. Transgene expressionis increased by photochemically mediated transduction ofpolycation-complexed adenoviruses. Gene Ther 2004;2(11):152–160.
14. Engesaeter BO, Bonsted A, Berg K, Hogset A, EngebratenO, Fodstad O, Curiel DT, Maelandsmo GM. PCI-enhancedadenoviral transduction employs the known uptake mecha-nism of adenoviral particles. Cancer Gene Ther 2005;5(12):439–448.
15. Berg K, Sandvig K, Moan J. Transfer of molecules into thecytosol of cells. 1996; WO 96/07432.
16. Shiraishi T, Nielsen PE. Enhanced delivery of cell-pene-trating peptide-peptide nucleic acid conjugates by endoso-mal disruption. Nat Protoc 2006;2(1):633–636.
17. Oliveira S, Fretz MM, Hogset A, Storm G, Schiffelers RM.Photochemical internalization enhances silencing of epider-mal growth factor receptor through improved endosomal es-cape of siRNA. Biochim Biophys Acta 2007;5(1768):1211–1217.
18. Berg K, Dietze A, Kaalhus O, Hogset A. Site-specific drugdelivery by photochemical internalization enhances the an-titumor effect of bleomycin. Clin Cancer Res 2005;23(11):8476–8485.
19. Lou PJ, Lai PS, Shieh MJ, Macrobert AJ, Berg K, BownSG. Reversal of doxorubicin resistance in breast cancercells by photochemical internalization. Int J Cancer 2006;11(119):2692–2698.
20. Lai PS, Lou PJ, Peng CL, Pai CL, Yen WN, Huang MY,Young TH, Shieh MJ. Doxorubicin delivery by polyamido-amine dendrimer conjugation and photochemical internali-zation for cancer therapy. J Control Release2007;1(122):39–46.
21. Adigbli DK, Wilson DG, Farooqui N, Sousi E, Risley P, Tay-lor I, Macrobert AJ, Loizidou M. Photochemical internalisa-tion of chemotherapy potentiates killing of multidrug-resistant breast and bladder cancer cells. Br J Cancer2007;4(97):502–512.
22. Selbo PK, Sivam G, Fodstad O, Sandvig K, Berg K. In vivodocumentation of photochemical internalization, a novel
730 WEYERGANG ET AL.

approach to site specific cancer therapy. Int J Cancer2001;5(92):761–766.
23. Ndoye A, Dolivet G, Hogset A, Leroux A, Fifre A, ErbacherP, Berg K, Behr JP, Guillemin F, Merlin JL. Eradication ofp53-mutated head and neck squamous cell carcinoma xeno-grafts using nonviral p53 gene therapy and photochemicalinternalization. Mol Ther 2006;6(13):1156–1162.
24. Selbo PK, Rosenblum MG, Cheung LH, Zhang W, Berg K.Multi-modality therapeutics with potent anti-tumor effects:Photochemical internalization enhances delivery of the fu-sion toxin scFvMEL/rGel. PLoS One 2009;8(4):e6691.
25. Bossu E, ’Amar O, Parache RM, Notter D, Labrude P,Vigneron C, Guillemin F. Determination of the maximaltumor/normal skin ratio after HpD or m-THPC administra-tion in hairless mouse (SKh-1) by fluorescence spectrosco-py—a non-invasive method. Anticancer Drugs 1997;1(8):67–72.
26. Liu XY, Pop LM, Vitetta ES. Engineering therapeuticmonoclonal antibodies. Immunol Rev 2008;222:9–27.
27. Kruser TJ, Wheeler DL. Mechanisms of resistance to HERfamily targeting antibodies. Exp Cell Res 2010;7(316):1083–1100.
28. Mulcahy MF. Bevacizumab in the therapy for refractorymetastatic colorectal cancer. Biologics 2008;1(2):53–59.
29. Carter PJ, Senter PD. Antibody-drug conjugates for cancertherapy. Cancer J 2008;3(14):154–169.
30. Vitetta ES, Thorpe PE, Uhr JW. Immunotoxins: Magic bul-lets or misguided missiles? Trends Pharmacol Sci 1993;5(14):148–154.
31. Pastan I, Kreitman RJ. Immunotoxins for targeted cancertherapy. Adv Drug Deliv Rev 1998;1-2(31):53–88.
32. Kreitman RJ. Immunotoxins in cancer therapy. Curr OpinImmunol 1999;5(11):570–578.
33. Foss F. Clinical experience with denileukin diftitox(ONTAK). Semin Oncol 2006;33(1 Suppl 3):S11–S16.
34. Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immuno-toxin therapy of cancer. Nat Rev Cancer 2006;7(6):559–565.
35. Jain RK, Baxter LT. Mechanisms of heterogeneous distri-bution of monoclonal antibodies and other macromoleculesin tumors: Significance of elevated interstitial pressure.Cancer Res 1988;48(24 Pt 1):7022–7032.
36. Jain RK. Physiological barriers to delivery of monoclonalantibodies and other macromolecules in tumors. CancerRes 1990;3 (Suppl 50): 814s–819s.
37. Desogus A, Burioni R, Ingianni A, Bugli F, Pompei R,Fadda G. Production and characterization of a human re-combinant monoclonal Fab fragment specific for influenzaA viruses. Clin Diagn Lab Immunol 2003;4(10):680–685.
38. Kurai J, Chikumi H, Hashimoto K, Yamaguchi K, Yama-saki A, Sako T, Touge H, Makino H, Takata M, Miyata M,Nakamoto M, Burioka N, Shimizu E. Antibody-dependentcellular cytotoxicity mediated by cetuximab against lungcancer cell lines. Clin Cancer Res 2007;5(13):1552–1561.
39. Naramura M, Gillies SD, Mendelsohn J, Reisfeld RA, Muel-ler BM. Therapeutic potential of chimeric and murine anti-(epidermal growth factor receptor) antibodies in a metasta-sis model for human melanoma. Cancer Immunol Immun-other 1993;5(37):343–349.
40. Walz G, Zanker B, Brand K, Waters C, Genbauffe F, ZeldisJB, Murphy JR, Strom TB. Sequential effects of interleukin2-diphtheria toxin fusion protein on T-cell activation. ProcNatl Acad Sci USA 1989;23(86):9485–9488.
41. Ogata M, Chaudhary VK, FitzGerald DJ, Pastan I. Cytotox-ic activity of a recombinant fusion protein between interleu-kin 4 and Pseudomonas exotoxin. Proc Natl Acad Sci USA1989;11(86):4215–4219.
42. Sharkey RM, Goldenberg DM. Use of antibodies and immu-noconjugates for the therapy of more accessible cancers.Adv Drug Deliv Rev 2008;12(60):1407–1420.
43. Jain RK. Delivery of novel therapeutic agents in tumors:Physiological barriers and strategies. J Natl Cancer Inst1989;8(81):570–576.
44. Baluna R, Rizo J, Gordon BE, Ghetie V, Vitetta ES. Evi-dence for a structural motif in toxins and interleukin-2 thatmay be responsible for binding to endothelial cells and
initiating vascular leak syndrome. Proc Natl Acad Sci USA1999;7(96):3957–3962.
45. Barbieri L, Battelli MG, Stirpe F. Ribosome-inactivatingproteins from plants. Biochim Biophys Acta 1993;3–4(1154):237–282.
46. Endo Y, Mitsui K, Motizuki M, Tsurugi K. The mechanismof action of ricin and related toxic lectins on eukaryotic ribo-somes. The site and the characteristics of the modificationin 28 S ribosomal RNA caused by the toxins. J Biol Chem1987;12(262):5908–5912.
47. Barbieri L, Ferreras JM, Barraco A, Ricci P, Stirpe F. Someribosome-inactivating proteins depurinate ribosomal RNAat multiple sites. Biochem J 1992;286(Pt 1):1–4.
48. Nielsen K, Boston RS. Ribosome-inactivating proteins: Aplant perspective. Annu Rev Plant Physiol Plant Mol Biol2001;52:785–816.
49. Vago R, Marsden CJ, Lord JM, Ippoliti R, Flavell DJ, Fla-vell SU, Ceriotti A. Fabbrini MS. Saporin and ricin A chainfollow different intracellular routes to enter the cytosol ofintoxicated cells. FEBS J 2005;19(272):4983–4995.
50. Sandvig K, van DB. Delivery into cells: Lessons learnedfrom plant and bacterial toxins. Gene Ther 2005;11(12):865–872.
51. Eiklid K, Olsnes S, Pihl A. Entry of lethal doses of abrin,ricin and modeccin into the cytosol of HeLa cells. Exp CellRes 1980;2(126):321–326.
52. Selbo PK, Kaalhus O, Sivam G, Berg K. 5-Aminolevulinicacid-based photochemical internalization of the immuno-toxin MOC31-gelonin generates synergistic cytotoxic effectsin vitro. Photochem Photobiol 2001;2(74):303–310.
53. Rowinsky EK. The erbB family: Targets for therapeutic de-velopment against cancer and therapeutic strategies usingmonoclonal antibodies and tyrosine kinase inhibitors. AnnuRev Med 2004;55:433–457.
54. Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M,Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salo-mon DS. Epidermal growth factor receptor (EGFR) signal-ing in cancer. Gene 2006;1(366):2–16.
55. Huether A, Hopfner M, Baradari V, Schuppan D, ScherublH. EGFR blockade by cetuximab alone or as combinationtherapy for growth control of hepatocellular cancer. Bio-chem Pharmacol 2005;11(70):1568–1578.
56. Friedman LM, Rinon A, Schechter B, Lyass L, Lavi S,Bacus SS, Sela M, Yarden Y. Synergistic down-regulationof receptor tyrosine kinases by combinations of mAbs:Implications for cancer immunotherapy. Proc Natl Acad SciUSA 2005;6(102):1915–1920.
57. Jaramillo ML, Leon Z, Grothe S, Paul-Roc B, Abulrob A,O’Connor MM. Effect of the anti-receptor ligand-blocking225 monoclonal antibody on EGF receptor endocytosis andsorting. Exp Cell Res 2006;15(312):2778–2790.
58. Veenendaal LM, Jin H, Ran S, Cheung L, Navone N, MarksJW, Waltenberger J, Thorpe P, Rosenblum MG. In vitroand in vivo studies of a VEGF121/rGelonin chimeric fusiontoxin targeting the neovasculature of solid tumors. ProcNatl Acad Sci USA 2002;12(99):7866–7871.
59. Rentsch M, Cerny T, Geiger L, Gerber H, Brunner KW,Rosler H, Nachbur B. Radioimmunoscintigraphy with a99mTc-labeled F(ab’)2 fragment of a monoclonal antibody(HMW-MAA 225.28S) in 71 patients with malignant mela-noma. Schweiz Med Wochenschr 1989;40(119):1382–1385.
60. Wang X, Wang Y, Yu L, Sakakura K, Visus C, Schwab JH,Ferrone CR, Favoino E, Koya Y, Campoli MR, McCarthyJB, DeLeo AB, Ferrone S. CSPG4 in cancer: Multiple roles.Curr Mol Med 2010;4(10):419–429.
61. Rosenblum MG, Cheung LH, Liu Y, Marks JW III. Designexpression, purification, and characterization, in vitro andin vivo, of an antimelanoma single-chain Fv antibody fusedto the toxin gelonin. Cancer Res 2003;14(63):3995–4002.
62. Nahta R, Esteva FJ. HER2 therapy: Molecular mechanismsof trastuzumab resistance. Breast Cancer Res 2006;6(8):215.
63. Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D,Korbelik M, Moan J, Peng Q. Photodynamic therapy. J NatlCancer Inst 1998;12(90):889–905.
PCI OF TUMOR-TARGETED PROTEIN TOXINS 731

64. Kessel D, Luo Y. Mitochondrial photodamage and PDT-in-duced apoptosis. J Photochem Photobiol B 1998;2(42):89–95.
65. Plaetzer K, Kiesslich T, Oberdanner CB, Krammer B. Apo-ptosis following photodynamic tumor therapy: Induction,mechanisms and detection. Curr Pharm Des 2005;9(11):1151–1165.
66. Kessel D, Vicente MG, Reiners JJ Jr. Initiation of apoptosisand autophagy by photodynamic therapy. Autophagy2006;4(2):289–290.
67. Buytaert E, Callewaert G, Vandenheede JR, Agostinis P.Deficiency in apoptotic effectors Bax and Bak revealsan autophagic cell death pathway initiated by photodamageto the endoplasmic reticulum. Autophagy 2006;3(2):238–240.
68. Reiners JJ Jr, Agostinis P, Berg K, Oleinick NL, Kessel D.Assessing autophagy in the context of photodynamic thera-py. Autophagy 2010;1(6):7–18.
69. Ji HT, Chien LT, Lin YH, Chien HF, Chen CT. 5-ALA medi-ated photodynamic therapy induces autophagic cell deathvia AMP-activated protein kinase. Mol Cancer 2010;9:91.
70. Xue LY, Chiu SM, Oleinick NL. Atg7 deficiency increasesresistance of MCF-7 human breast cancer cells to photody-namic therapy. Autophagy 2010;2(6):248–255.
71. Castano AP, Mroz P, Hamblin MR. Photodynamic therapyand anti-tumour immunity. Nat Rev Cancer 2006;7(6):535–545.
72. Thong PS, Ong KW, Goh NS, Kho KW, Manivasager V,Bhuvaneswari R, Olivo M, Soo KC. Photodynamic-therapy-activated immune response against distant untreatedtumours in recurrent angiosarcoma. Lancet Oncol 2007;10(8):950–952.
73. Fingar VH, Kik PK, Haydon PS, Cerrito PB, Tseng M,Abang E, Wieman TJ. Analysis of acute vascular damageafter photodynamic therapy using benzoporphyrin deriva-tive (BPD). Br J Cancer 1999;11–12(79):1702–1708.
74. Engbrecht BW, Menon C, Kachur AV, Hahn SM, FrakerDL. Photofrin-mediated photodynamic therapy inducesvascular occlusion and apoptosis in a human sarcoma xeno-graft model. Cancer Res 1999;17(59):4334–4342.
75. Chen B, Roskams T, De Witte PA. Antivascular tumoreradication by hypericin-mediated photodynamic therapy.Photochem Photobiol 2002;5(76):509–513.
76. Woodhams JH, Macrobert AJ, Novelli M, Bown SG. Photo-dynamic therapy with WST09 (Tookad): Quantitative stud-ies in normal colon and transplanted tumours. Int J Cancer2006;2(118):477–482.
77. Fingar VH. Vascular effects of photodynamic therapy. JClin Laser Med Surg 1996;5(14):323–328.
78. Thomas N, Bechet D, Becuwe P, Tirand L, Vanderesse R,Frochot C, Guillemin F, Barberi-Heyob M. Peptide-conju-gated chlorin-type photosensitizer binds neuropilin-1 invitro and in vivo. J Photochem Photobiol B 2009;2(96):101–108.
79. Bechet D, Tirand L, Faivre B, Plenat F, Bonnet C, BastogneT, Frochot C, Guillemin F, Barberi-Heyob M. Neuropilin-1targeting photosensitization-induced early stages of throm-bosis via tissue factor release. Pharm Res 2010;3(27):468–479.
80. Zhu X, Bidlingmaier S, Hashizume R, James CD, BergerMS, Liu B. Identification of internalizing human single-chain antibodies targeting brain tumor sphere cells. MolCancer Ther 2010;7(9):2131–2141.
81. Prasmickaite L, Hogset A, Selbo PK, Engesaeter BO, Hel-lum M, Berg K. Photochemical disruption of endocyticvesicles before delivery of drugs: A new strategy for cancertherapy. Br J Cancer 2002;4(86):652–657.
82. Berg K, Hogset A, Prasmickaite L, Weyergang A, BonstedA, Dietze A, Lou P, Bown S, Norum O, Mollergard H, SelboPK. Photochemical internalization (PCI): A novel technolo-gy for activation of endocytosed therapeutic agents. MedLaser Appl 2006;21:239–250.
83. Weyergang A, Selbo PK, Berg K. Y1068 phosphorylation isthe most sensitive target of disulfonated tetraphenylpor-phyrin-based photodynamic therapy on epidermal growthfactor receptor. Biochem Pharmacol 2007;2(74):226–235.
84. Weyergang A, Kaalhus O, Berg K. Photodynamic targetingof EGFR does not predict the treatment outcome in combi-nation with the EGFR tyrosine kinase inhibitor TyrphostinAG1478. Photochem Photobiol Sci 2008;9(7):1032–1040.
85. Nonaka M, Ikeda H, Inokuchi T. Effect of combined photo-dynamic and chemotherapeutic treatment on lymphomacells in vitro. Cancer Lett 2002;2(184):171–178.
86. Casas A, Fukuda H, Batlle AM. Potentiation of the 5-ami-nolevulinic acid-based photodynamic therapy with cyclo-phosphamide. Cancer Biochem Biophys 1998;1–2(16):183–196.
87. Kirveliene V, Grazeliene G, Dabkeviciene D, Micke I, Kir-velis D, Juodka B, Didziapetriene J. Schedule-dependentinteraction between Doxorubicin and mTHPC-mediatedphotodynamic therapy in murine hepatoma in vitro and invivo. Cancer Chemother Pharmacol 2006;1(57):65–72.
88. Liu W, Baer MR, Bowman MJ, Pera P, Zheng X, Morgan J,Pandey RA, Oseroff AR. The tyrosine kinase inhibitor ima-tinib mesylate enhances the efficacy of photodynamic thera-py by inhibiting ABCG2. Clin Cancer Res 2007;8(13):2463–2470.
89. del Carmen MG, Rizvi I, Chang Y, Moor AC, Oliva E, Sher-wood M, Pogue B, Hasan T. Synergism of epidermal growthfactor receptor-targeted immunotherapy with photodynam-ic treatment of ovarian cancer in vivo. J Natl Cancer Inst2005;20(97):1516–1524.
90. Ferrario A, Fisher AM, Rucker N, Gomer CJ. Celecoxib andNS-398 enhance photodynamic therapy by increasing invitro apoptosis and decreasing in vivo inflammatory andangiogenic factors. Cancer Res 2005;20(65):9473–9478.
91. Zimmermann A, Walt H, Haller U, Baas P, Klein SD.Effects of chlorin-mediated photodynamic therapy com-bined with fluoropyrimidines in vitro and in a patient. Can-cer Chemother Pharmacol 2003;2(51):147–154.
92. Zhang M, Zhang Z, Goldman CK, Janik J, Waldmann TA.Combination therapy for adult T-cell leukemia-xenograftedmice: Flavopiridol and anti-CD25 monoclonal antibody.Blood 2005;3(105):1231–1236.
93. Soffietti R, Ruda R, Trevisan E. New chemotherapy optionsfor the treatment of malignant gliomas. Anticancer Drugs2007;6(18):621–632.
94. Moor AC. Signaling pathways in cell death and survival af-ter photodynamic therapy. J Photochem Photobiol B2000;1(57):1–13.
95. Piette J, Volanti C, Vantieghem A, Matroule JY, HabrakenY, Agostinis P. Cell death and growth arrest in response tophotodynamic therapy with membrane-bound photosensi-tizers. Biochem Pharmacol 2003;8(66):1651–1659.
96. Almeida RD, Manadas BJ, Carvalho AP, Duarte CB. Intra-cellular signaling mechanisms in photodynamic therapy.Biochim Biophys Acta 2004;2(1704):59–86.
97. Weyergang A, Kaalhus O, Berg K. Photodynamic therapywith an endocytically located photosensitizer cause a rapidactivation of the mitogen-activated protein kinases extra-cellular signal-regulated kinase, p38 and c-Jun NH2 termi-nal kinase with opposing effects on cell survival. MolCancer Ther 2008;6(7):1740–1750.
98. Weyergang A, Berg K, Kaalhus O, Peng Q, Selbo PK. Pho-todynamic therapy targets the mTOR signaling network invitro and in vivo. Mol Pharm 2009;1(6):255–264.
99. Cramers P, Ruevekamp M, Oppelaar H, Dalesio O, Baas P,Stewart FA. Foscan uptake and tissue distribution in rela-tion to photodynamic efficacy. Br J Cancer 2003;2(88):283–290.
100. Hahn SM, Putt ME, Metz J, Shin DB, Rickter E, Menon C,Smith D, Glatstein E, Fraker DL, Busch TM. Photofrin up-take in the tumor and normal tissues of patients receivingintraperitoneal photodynamic therapy. Clin Cancer Res2006;18(12):5464–5470.
101. Konan YN, Gurny R, Allemann E. State of the art in thedelivery of photosensitizers for photodynamic therapy. JPhotochem Photobiol B 2002;2(66):89–106.
102. Mew D, Wat CK, Towers GH, Levy JG. Photoimmunother-apy: Treatment of animal tumors with tumor-specific mono-clonal antibody-hematoporphyrin conjugates. J Immunol1983;3(130):1473–1477.
732 WEYERGANG ET AL.

103. Soukos NS, Hamblin MR, Keel S, Fabian RL, Deutsch TF,Hasan T. Epidermal growth factor receptor-targeted immu-nophotodiagnosis and photoimmunotherapy of oral pre-cancer in vivo. Cancer Res 2001;11(61):4490–4496.
104. Savellano MD, Hasan T. Targeting cells that overexpressthe epidermal growth factor receptor with polyethylene gly-colated BPD verteporfin photosensitizer immunoconju-gates. Photochem Photobiol 2003;4(77):431–439.
105. Carcenac M, Larroque C, Langlois R, van Lier JE, ArtusJC, Pelegrin A. Preparation, phototoxicity and biodistribu-tion studies of anti-carcinoembryonic antigen monoclonalantibody-phthalocyanine conjugates. Photochem Photobiol1999;6(70):930–936.
106. Carcenac M, Dorvillius M, Garambois V, Glaussel F,Larroque C, Langlois R, Hynes NE, van Lier JE, PelegrinA. Internalisation enhances photo-induced cytotoxicityof monoclonal antibody-phthalocyanine conjugates. Br JCancer 2001;11(85):1787–1793.
107. Reichenbach A, Dettmer D, Bruckner G, Neumann M, Bir-kenmeyer G. Morphological variability, lectin binding andNa,K-activated adenosine triphosphatase activity of
isolated Muller (glial) cells from the rabbit retina. NeurosciLett 1985;1(55):29–34.
108. Cauchon N, Ali H, Hassessian HM, van Lier JE.Structure-activity relationships of mono-substitutedtrisulfonated porphyrazines for the photodynamic therapy(PDT) of cancer. Photochem Photobiol Sci 2010;3(9):331–341.
109. Nishiyama N, Iriyama A, Jang WD, Miyata K, Itaka K,Inoue Y, Takahashi H, Yanagi Y, Tamaki Y, Koyama H,Kataoka K. Light-induced gene transfer from packagedDNA enveloped in a dendrimeric photosensitizer. NatMater 2005;12(4):934–941.
110. Fukumura D, Jain RK. Tumor microvasculature and micro-environment: Targets for anti-angiogenesis and normaliza-tion. Microvasc Res 2007;2–3(74):72–84.
111. Fukumura D, Jain RK. Tumor microenvironment abnor-malities: Causes, consequences, and strategies to normal-ize. J Cell Biochem 2007;4(101):937–949.
112. Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immuno-toxin treatment of cancer. Annu Rev Med 2007;(58):221–237.
PCI OF TUMOR-TARGETED PROTEIN TOXINS 733