pharmacophore and quantitative structure activity relationship modelling of...
TRANSCRIPT

Pharmacophore and quantitative structure activityrelationship modelling of UDP-glucuronosyltransferase 1A1(UGT1A1) substratesMichael J. Soricha, Paul A. Smithb, Ross A. McKinnona and John O. Minersb
UDP-glucuronosyltransferase 1A1 (UGT1A1) is a
polymorphic enzyme responsible for the glucuronidation
of structurally diverse drugs, non-drug xenobiotics and
endogenous compounds (e.g. bilirubin). Thus, definition of
UGT1A1 substrate and inhibitor selectivities and binding
affinities assumes importance for the identification of
compounds whose elimination may be impaired in
subjects with variant genotypes, and for the prediction of
potentially inhibitory interactions involving xenobiotics and
endogenous compounds metabolized by UGT1A1. We
report the generation of two- and three-dimensional (2D
and 3D) quantitative structure activity relationships (QSAR)
and pharmacophore models for 23 known UGT1A1
substrates with diverse structure and binding affinity.
Initially, a simple procedure was developed to determine
apparent inhibition constants (K i,app) for these compounds.
Eighteen substrates were subsequently used to construct
models and the remaining five to validate the predictive
ability of the models. Three different models were
constructed: (i) three feature pharmacophore model able
to predict the K i,app on the basis of the degree to which a
substrate can fit to the arrangement of 3D features
(r2 0.87, K i,app for all five test substrates predicted within
log unit); (ii) 3D-QSAR using a ‘common features’
pharmacophore to align the substrates (r2 0.71, K i,app for
four out of five test substrates predicted within one log
unit); (iii) 2D-QSAR constructed with six chemical
descriptors (r2 0.92, K i,app of all five test substrates
predicted within one log unit). The common features
pharmacophore demonstrated the importance of two
hydrophobic domains separated from the glucuronidation
site by 4 A and 7 A, respectively. These models, which
represent the first generalized predictive models for a UGT
isoform, complement each other and are an important first
step towards computer based (in silico) models of
UGT1A1 for high throughput prediction of metabolism.
Pharmacogenetics 12:635–645 & 2002 Lippincott Williams
& Wilkins
Pharmacogenetics 2002, 12:635–645
Keywords: UGT1A1, QSAR, pharmacophore, UDP-glucuronosyltransferase
aSchool of Pharmaceutical, Molecular and Biomedical Sciences, University ofSouth Australia, South Australia and bDepartment of Clinical Pharmacology,Flinders University of South Australia, South Australia, Australia
Correspondence to Professor John Miners, Department of ClinicalPharmacology, Flinders Medical Centre, Bedford Park, SA 5042, AustraliaTel: +61 8 8204 4131; fax: +61 8 8204 5114;e-mail: [email protected]
Received 6 March 2002Accepted 31 July 2002
IntroductionConjugation with glucuronic acid, a reaction catalysed
by the microsomal enzyme UDP-glucuronosyltransfer-
ase (UGT), is an essential clearance mechanism for
drugs from almost all therapeutic classes [1]. Moreover,
glucuronidation serves as an elimination pathway for a
myriad of endogenous compounds, dietary chemicals
and environmental pollutants (including some chemical
carcinogens), and facilitates excretion of the products of
phase I metabolism. Endogenous compounds metabo-
lized by glucuronidation include bilirubin, bile acids,
fatty acids, hydroxysteroids and thyroid hormones. Con-
sistent with this substrate diversity, UGT exists as a
superfamily of enzymes. UGT gene products character-
ized to date have been classified into two gene families,
UGT1 and UGT2, based on sequence identity and
evolutionary divergence [2]. Fifteen functional UGT
proteins (isoforms) have been identified in humans.
These isoforms tend to exhibit distinct, but broadly
overlapping, substrate selectivities [3,4].
Extensive polymorphism has been described for UGTgenes [5]. Notable in this regard is UGT1A1. Because
UGT1A1 is the sole isoform involved in bilirubin
glucuronidation, and additionally has the capacity to
metabolize hydroxyoestrogens, thyroid hormones and
numerous drug and non-drug xenobiotics [6–8], genetic
polymorphism of UGT1A1 may be of physiological,
pharmacological and toxicological significance. More
than 50 lesions of UGT1A1 are associated with inherited
disorders of bilirubin conjugation, namely the Crigler–
Najjar syndromes (types 1 and 2) and Gilbert syndrome
[9]. Additionally, there is increasing evidence to suggest
that Gilbert syndrome, which arises from insertional
mutations in the TATAA element upstream of UGT1A1or from a limited number of structural mutations, may
be a risk factor in drug related toxicity. For example,
patients with variant UGT1A1 genotypes are over-
represented amongst those experiencing severe toxicity
to the anticancer drug irinotecan due to impaired
glucuronidation of the active metabolite SN-38 [10].
Original paper 635
0960–314X & 2002 Lippincott Williams & Wilkins

Indinavir, which appears to be a UGT1A1 substrate,
may precipitate jaundice in patients with Gilbert syn-
drome variant TATAA element alleles as a result of
competitive inhibition [11]. In general, reduced clear-
ance of drug substrates of UGT1A1 might be expected
in subjects with Gilbert syndrome [12].
It is apparent from such observations that the ability to
predict the interaction between UGT1A1 and any
newly developed drug or non-drug chemical may be of
therapeutic or toxicological importance, and could influ-
ence the further development of a new chemical entity.
Analogous arguments have been applied to poly-
morphic xenobiotic metabolizing cytochromes P450
(CYP), such as CYP2D6 [13]. More generally, elucida-
tion of CYP or UGT isoform substrate and inhibitor
specificity further assists the prediction of drug–drug
interactions and definition of those structural and
physicochemical features of compounds that confer
substrate and inhibitor selectivity.
Computational methods have found widespread use in
recent years for the investigation of CYP active sites
[14]. In particular, two-, three- and four-dimensional
(2D, 3D and 4D) quantitative structure activity rela-
tionships (QSAR), pharmacophores and homology mod-
els have been developed to infer CYP isoform active
site binding requirements [14,15]. QSAR and pharma-
cophore models are generated from the structures of
known substrates/inhibitors, whereas homology models
are constructed from experimentally determined 3D
coordinates of a crystallized protein. The 2D, 3D and
4D QSAR methodologies differ in respect to the
properties used as predictors in the model. In general,
two dimensional properties are independent of the 3D
conformation of the chemical, 3D properties are depen-
dent on conformation and are derived from a single
conformation, and 4D properties involve sampling 3D
properties of multiple conformations [16,17]. A pharma-
cophore is a defined 3D arrangement of chemical
features, whereas a homology model is the predicted
3D structure of the enzyme based on the structure of a
similar enzyme [18,19]. Models may be developed for
multiple purposes, such as predicting kinetic constants,
understanding the nature of the substrate–enzyme
interaction, searching a chemical database for new leads
and lead structure optimization [17,18,20].
Although the lack of an UGT crystal structure pre-
cludes homology modelling, it is apparent that compu-
tational methodologies are available which permit the
development of QSAR and pharmacophore models to
predict UGT isoform substrate selectivity and provide
a quantitative measure of substrate binding. We de-
scribe the validation of a simple experimental proce-
dure to determine the apparent inhibition constant
(Ki,app) of structurally diverse UGT1A1 substrates and
the use of these data to develop a number of models
capable of predicting the Ki,app of UGT1A1 substrates
based on their chemical and physicochemical proper-
ties. This represents the first report of the development
of generalized predictive models for substrate binding
to UGT1A1 and, together with models for UGT1A4
generated in this laboratory (Smith et al., unpublished
observations), the first generalized 2D and 3D QSAR
and pharmacophore models for UGT.
Materials and methodsChemicals and UGT1A1 expression
For inclusion in the model generation dataset, it was
necessary that chemicals were known substrates of
UGT1A1 and were significantly different to each other
in either structure or UGT1A1 activity. Thus, the 23
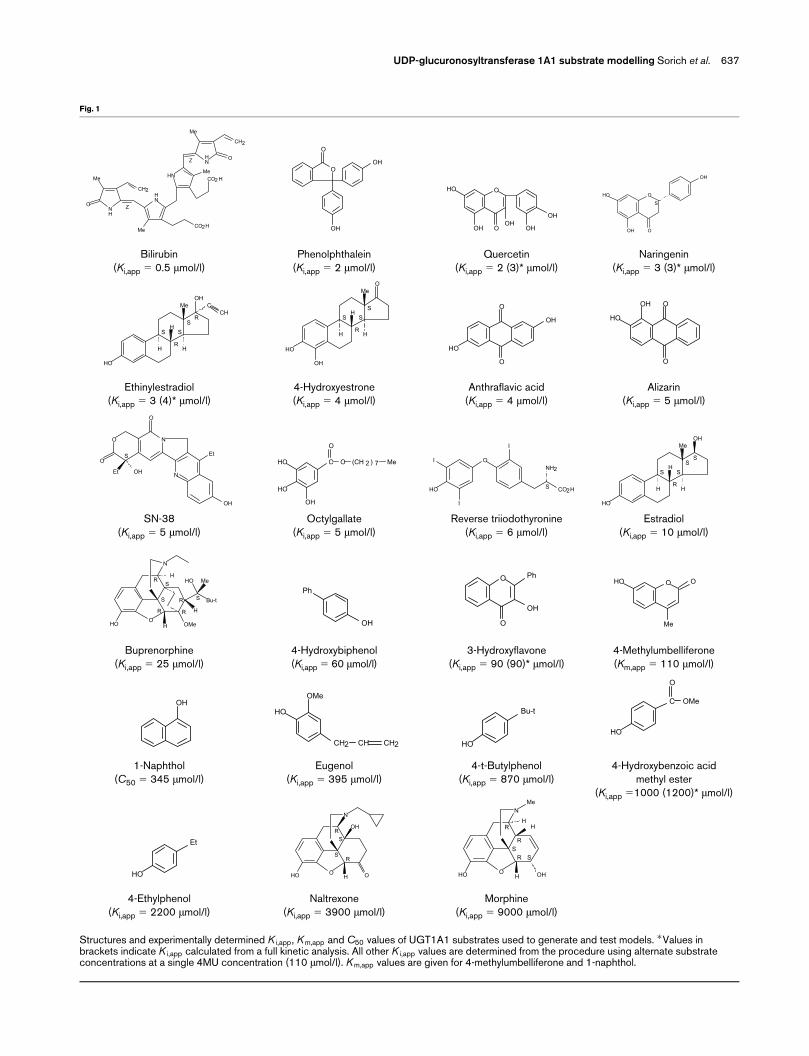
UGT1A1 substrates included in the dataset (Fig. 1)
exhibited a wide range of activities [8,21–24]. Except
for those chemicals referred to below, all of these
compounds and UDP-glucuronic acid (UDPGA; so-
dium salt), 4-methylumbelliferone �-D-glucuronide
(4MUG) and 1-naphthol �-D-glucuronide (1NPG) were
purchased from Sigma-Aldrich (St Louis, Missouri,
USA). The following compounds were provided as
gifts: SN-38 from Dr M. Kuboki (Yakult Honsha Co.,
Ltd, Tokyo, Japan), naltrexone from Dr G. Gourlay
(Flinders Medical Centre, Bedford Park, Australia), and
buprenorphine from Dr A. Somogyi (University of
Adelaide, Adelaide, Australia). All other chemicals and
reagents were of analytical reagent grade.
HK293 cells stably expressing UGT1A1 were grown in
Dulbecco’s modified Eagle’s medium (GibcoBRL) with
10% fetal calf serum and gentamicin (80 mg/l) in a
humidified incubator, with an atmosphere of 5% CO2,
at 37 8C. Microsomes were prepared by differential
centrifugation. Cells were probe sonicated and the
cellular homogenate was centrifuged at 9000 g for 10
min. The supernatant fraction was subsequently recen-
trifuged at 120 000 g for 2 h to obtain the microsomal
pellet, which was suspended in distilled water and
stored at –80 8C until use.
Assays for 4-methylumbelliferone (4MU) and 1-naphthol
(1NP) glucuronidation
4MU and 1NP glucuronidation by HK293 cell micro-
somes was measured according to a previously pub-
lished procedure [25]. Briefly, incubation mixtures
contained 4MU (20–750 �mol/l) or 1NP (50–1000
�mol/l), UDPGA (5 mmol/l), MgCl2 (5 mmol/l) and
HK293 microsomes (0.2 mg protein; 0.33 mg/ml) in
phosphate buffer (0.1 mol/l, pH 7.4) in a total volume
of 0.6 ml. Reactions were initiated by the addition of
UDPGA and performed in air at 37 8C for 2 h (shaking
water bath). Incubations were terminated by the addi-
tion of 0.6 mol/l glycine/0.4 mol/l trichloroacetic acid
(0.14 ml) and cooling on ice. Following the addition of
636 Pharmacogenetics 2002, Vol 12 No 8
Me
Me
Me
Me
CO2H
CO2 H
NH
NH
NH
NH
CH2
O
O
CH2
Z
Z
O
OOH
OH OOH
O
OH
HO
OH
OH
O
O
OH
HO
OH
S
Me COH
CH
HO
H
H H
S
S
R
R
S
MeO
OH
HO
H
H H
S
S
R
S
O
O
OH
HO
OOH
O
HO
Et
Et N
N
OH
O
OH
O
OS
(CH 2 ) 7 MeC O
O
OH
HO
HO CO2H
I
I
I
O
HO
NH2
S
MeOH
HO
H
H H
S
S
S
R
S
Bu-t
OMe
Me
O
HO
N
HO H
H
HRR
RS S
SR
Ph
OH
Ph
OH
O
O Me
O OHO
OHOMe
HO
CH2 CH CH2
Bu-t
HO
OMeC
O
HO
Et
HO
OH
N
O OHO H
S
S
R
R
Me
OHO
N
HO
H
H
H
SRS
R
R
Bilirubin(Ki,app � 0.5 µmol/l)
Phenolphthalein(Ki,app � 2 µmol/l)
Quercetin(Ki,app � 2 (3)* µmol/l)
Naringenin(Ki,app � 3 (3)* µmol/l)
Ethinylestradiol(Ki,app � 3 (4)* µmol/l)
4-Hydroxyestrone(Ki,app � 4 µmol/l)
Anthraflavic acid(Ki,app � 4 µmol/l)
Alizarin(Ki,app � 5 µmol/l)
SN-38(Ki,app � 5 µmol/l)
Octylgallate(Ki,app � 5 µmol/l)
Reverse triiodothyronine(Ki,app � 6 µmol/l)
Estradiol(Ki,app � 10 µmol/l)
Buprenorphine(Ki,app � 25 µmol/l)
4-Hydroxybiphenol(Ki,app � 60 µmol/l)
3-Hydroxyflavone(Ki,app � 90 (90)* µmol/l)
4-Methylumbelliferone(Km,app � 110 µmol/l)
1-Naphthol(C50 � 345 µmol/l)
Eugenol(Ki,app � 395 µmol/l)
4-t-Butylphenol(Ki,app � 870 µmol/l)
4-Hydroxybenzoic acidmethyl ester
(Ki,app �1000 (1200)* µmol/l)
4-Ethylphenol(Ki,app � 2200 µmol/l)
Naltrexone(Ki,app � 3900 µmol/l)
Morphine(Ki,app � 9000 µmol/l)
Fig. 1
Structures and experimentally determined K i,app, K m,app and C50 values of UGT1A1 substrates used to generate and test models. �Values inbrackets indicate K i,app calculated from a full kinetic analysis. All other K i,app values are determined from the procedure using alternate substrateconcentrations at a single 4MU concentration (110 �mol/l). K m,app values are given for 4-methylumbelliferone and 1-naphthol.
UDP-glucuronosyltransferase 1A1 substrate modelling Sorich et al. 637

0.1 ml of phosphate buffer (1 mol/l, pH 7.4), the mix-
ture was extracted with chloroform (7 ml) using a rotary
mixer and then centrifuged (1500 g for 10 min). A
0.6 ml aliquot of the aqueous phase was separated for
measurement of fluorescence (Perkin-Elmer model
3000 fluorescence spectrometer; Perkin-Elmer, Foster
City, California, USA). Excitation/emission wavelengths
were 315/365 nm and 290/330 nm for 4MUG and
1NPG, respectively. Aqueous 4MUG and 1NPG stan-
dards in the range 0.1–20 and 1–60 �mol/l were treated
in the same manner as incubation samples, and un-
known concentrations were determined by comparison
of fluorescence measurements with those of the appro-
priate standard curve.
Reaction rates for both 4MU and 1NP were linear for
incubation times to 2 h and for microsomal protein
concentrations to at least 0.5 mg/ml. The limit of
sensitivity of both assays was 0.1 �mol/l. Within-day
overall 4MUG assay imprecision, assessed by measuring
4MUG formation in five separate incubations of the
same batch of microsomes, was 2.9% and 1.6% for
substrate concentrations of 20 and 200 �mol/l, respec-
tively. Similar imprecision data have been reported
previously for the 1NP glucuronidation assay [25].
Inhibition experiments with alternate UGT1A1 substrates
The apparent inhibitor constant (Ki,app) of each of the
known UGT1A1 substrates (excluding 1NP) shown in
Fig. 1 was determined using 4MU as the probe
substrate. Assays performed for the calculation of Ki,app
contained 4MU at a concentration approximately equal
to its Km,app (110 �mol/l). Inhibition of 4MU glucuroni-
dation was measured for three alternate substrate con-
centrations. This approach was validated by calculation
of the Ki,app for five selected alternate substrates, using
three 4MU concentrations and four concentrations of
the alternate substrate at each 4MU concentration.
Analysis of kinetic data
All data points represent the mean of duplicate estima-
tions. Kinetic data were model-fitted using a nonlinear
regression method implemented by EnzFitter (Biosoft,
Cambridge, UK). The choice of model (Michaelis–
Menten or Hill function) for the calculation of Km,app
and Vmax values for 4MU and 1NP glucuronidation was
confirmed by F-test and coefficients of determination.
Similarly, Ki,app values for alternate UGT1A1 substrates
were calculated using EnzFitter, assuming a competi-
tive inhibition model.
Model generation
The molecular modelling studies were performed using
Silicon Graphics Octane2 (Silicon Graphics, Mountain
View, California, USA) and x86 Intel workstations.
Models were constructed using three different method-
ologies, as described below.
In order to test the predictive ability of the models, the
23 substrates were split into two groups: one to gener-
ate the models (18 chemicals) and one to test the
models (five chemicals). The test set of five chemicals
was selected to span a wide range of Ki,app values and
for diversity of chemical structure, so as to stringently
test the models generated. In general, prediction within
one log unit of the experimental Ki,app value is consid-
ered satisfactory [26]. Randomization tests were em-
ployed to assess statistical significance of the models.
Alternate training sets were generated by randomiza-
tion of the association between Ki,app to the substrate,
such that the Ki,app values were not associated with the
correct substrate. The model generation procedure was
repeated with these randomized datasets to calculate
the probability that the model was a result of a chance
correlation. To achieve a 95% confidence interval (95%
CI), 19 randomization tests were required, while 99
randomization tests were necessary to achieve a 99%
confidence level.
Discriminative features pharmacophore
The discriminative features pharmacophore was gener-
ated using modules of the Catalyst software suite
(Accelrys Inc., San Diego, California, USA). Chemical
structures of the UGT1A1 substrates were built using
the Visualizer module. The ConFirm module was used
to sample a maximum of 255 representative conforma-
tions for each substrate, all within a 20 kcal/mol energy
limit of the minimum energy conformation. Each sub-
strate and its associated conformers were subsequently
input to the Hypogen module to search for a 3D
arrangement of abstract chemical features (hydrogen
bond donor, hydrogen bond acceptor, hydrophobic
region, aromatic ring, glucuronidation feature) that
could be used to predict the Ki,app. The fit of the
substrates to this putative pharmacophore (i.e. how
closely the features of each chemical match the features
of the pharmacophore) was correlated with the ex-
perimentally determined –logKi,app (pKi,app) of the
substrates [27]. The glucuronidation feature was con-
structed specifically for use with UGT substrates to
recognize functional groups which could be glucuroni-
dated (i.e. –OH, –NH, –COOH) (Smith et al., unpub-
lished observations). Features in the pharmacophore
were assigned variable weighting. Finally, the Compare
module was used to align the test set substrates onto
the pharmacophore and to predict their Ki,app values.
Self organizing molecular field analysis (SOMFA) using a
common features pharmacophore to align substrates
The substrates (and associated set of sampled confor-
mations) were input to the HipHop module of Catalyst.
This module was used to search for 3D arrangements
of chemical features (hydrophobic region, aromatic ring,
nucleophile) common to the 23 substrates [28]. The
minimum spacing allowed between features in the
638 Pharmacogenetics 2002, Vol 12 No 8
pharmacophore was set to 1.5 A to allow for the small
molecules in the training set. This pharmacophore was
then used with the Compare module to align the
substrates for the 3D QSAR.
SOMFA [29] is a 3D QSAR algorithm, related to both
CoMFA (Comparative Molecular Field Analysis) [30]
and similarity analysis [31]. SOMFA is able to deter-
mine the influence of shape and electrostatic field on
the activity of a dataset. By sampling the steric and
electrostatic fields of the aligned substrates at points on
a 3D grid surrounding the substrates, it was possible to
find the grid points where the steric and electrostatic
fields influenced the Ki,app of the substrates.
The aligned substrates were input to the program along
with their –logKi,app values. The shape and electrostatic
field for each chemical were sampled at 1 A intervals
on a 3D grid, and a set of points in 3D where the shape
and/or electrostatic field of the chemical influenced the
Ki,app was reported. Models generated were used subse-
quently to predict the Ki,app of the test set of substrates.
2D QSAR
A wide range of 2D descriptors were calculated from
the structure of each chemical using the Dragon
(Milano Chemometrics and QSAR Research Group,
Milan, Italy) and Cerius2 (Accelrys Inc.) programs.
These included topological, 2D autocorrelation, consti-
tutional, BCUT, thermodynamic and electronic de-
scriptors [16]. Prior to generating the model, the most
relevant and least collinear subsets of descriptors were
selected. Initially, all descriptors without a statistically
significant correlation to –logKi,app were removed. The
significantly correlated descriptors were input to a
program implementing the Unsupervised Forward Se-
lection (UFS) algorithm [32]. This method initially
selects the two descriptors which are least well corre-
lated, and then selects additional variables on the basis
of their multiple correlation with those already chosen,
thus selecting a subset of variables that are as close to
orthogonal as possible. Subsets of descriptors were
selected using this procedure with varying degrees of
collinearity allowed (r2max ¼ 0.1, 0.2, . . . , 0.9, 0.99).
A regression model was built for each descriptor subset
using partial least squares (PLS) regression [33]. The
variable subset giving the best leave-one-out (LOO)
cross-validated r2, a measure of model predictive ability
of the model, was chosen for further optimization. Any
descriptor found to have a significant correlation with
the residuals of the model was included if it improved
the LOO cross-validated r2. Descriptors were omitted
from the model if removal resulted in an increase in
the LOO cross-validated r2.
ResultsValidation of the experimental approach used to calculate
K i,app
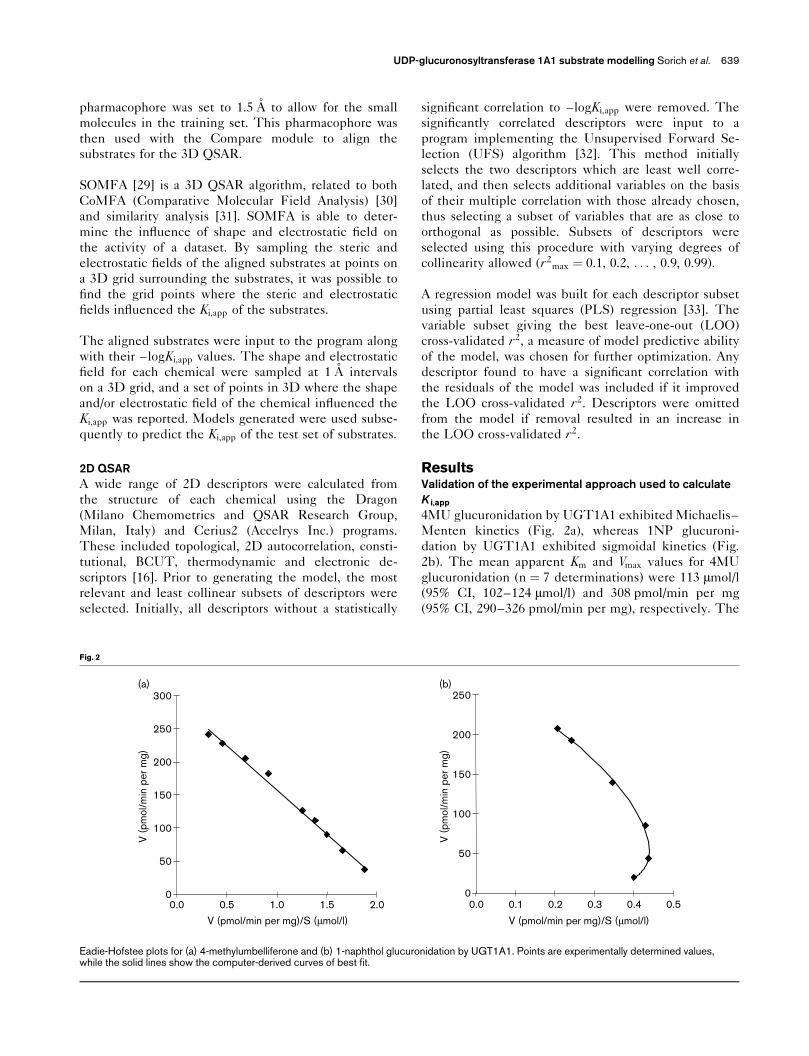
4MU glucuronidation by UGT1A1 exhibited Michaelis–
Menten kinetics (Fig. 2a), whereas 1NP glucuroni-
dation by UGT1A1 exhibited sigmoidal kinetics (Fig.
2b). The mean apparent Km and Vmax values for 4MU
glucuronidation (n ¼ 7 determinations) were 113 �mol/l
(95% CI, 102–124 �mol/l) and 308 pmol/min per mg
(95% CI, 290–326 pmol/min per mg), respectively. The
300
250
200
150
100
50
0
V (p
mol
/min
per
mg)
(a)
0.0 0.5 1.0 1.5 2.0
V (pmol/min per mg)/S (µmol/l)
250
200
150
100
50
00.0 0.1 0.2 0.3 0.4 0.5
V (p
mol
/min
per
mg)
(b)
V (pmol/min per mg)/S (µmol/l)
Fig. 2
Eadie-Hofstee plots for (a) 4-methylumbelliferone and (b) 1-naphthol glucuronidation by UGT1A1. Points are experimentally determined values,while the solid lines show the computer-derived curves of best fit.
UDP-glucuronosyltransferase 1A1 substrate modelling Sorich et al. 639

derived C50 and Vmax values for 1NP glucuronidation
were 345 �mol/l and 260 pmol/min per mg, respec-
tively, with a sigmoidocity factor (n) of 1.25.
Since 4MU glucuronidation exhibited Michaelis–
Menten kinetics, 4MU was used as the substrate for
the determination of Ki,app values of alternate UGT1A1
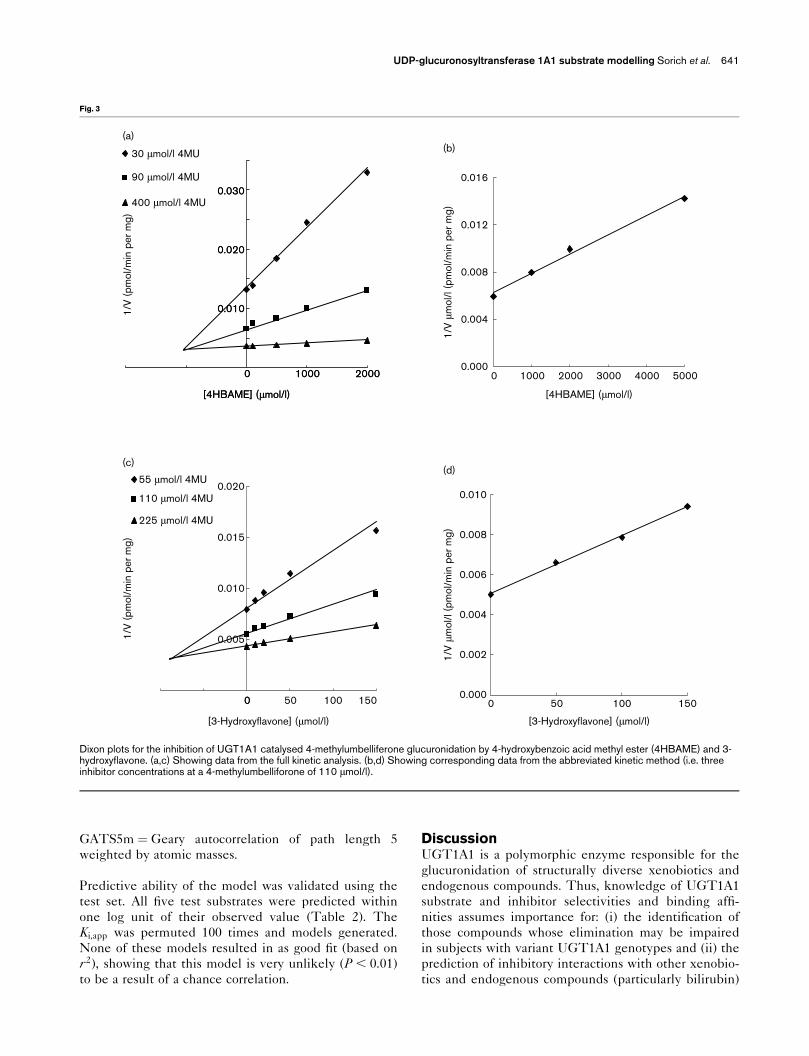
substrates. To validate the use of only three inhibitor
concentrations and a single concentration of 4MU to
determine the Ki,app, data generated using this approach
were compared to Ki,app determined using full kinetic
analysis for five compounds. Ki,app values determined
using the two methods are shown in Table 1, and
representative kinetic plots are shown in Fig. 3. Ki,app
values determined by the two approaches were close in
value, and hence the abbreviated kinetic method (i.e.
three inhibitor concentrations at a 4MU concentration
of 110 �mol/l) was subsequently used for the remaining
16 chemicals comprising the dataset. All experimentally
determined Ki,app values are shown in Fig. 1. These
data, along with the apparent Km values for 4MU and
1NP, were used for model generation. Nine other
alternate substrates were investigated, but not used for
model generation, due either to activation of 4MUG
formation (2-phenylphenol, paracetamol, carvacrol), in-
solubility in the incubation mixture (fisetin, all-trans
retinoic acid, retigabine) or interference with 4MUG
fluorescence (4-nitrophenol, 7-hydroxyflavone, 4-amino-
biphenyl).
Discriminative features pharmacophore
The best pharmacophore generated contained three
features; an aromatic ring, a hydrophobic region and a
hydrogen bond donor (Fig. 4a), and gave a fit of
r ¼ 0.93 (r2 ¼ 0.87). By way of example, Figure 4(b–d)
shows bilirubin (high affinity substrate), 3-hydroxy-
flavone (intermediate affinity substrate) and morphine
(low affinity substrate), respectively, aligned to the
pharmacophore. As shown in Table 2, all five of the
test set (i.e. those substrates not used for model
generation) were predicted to have a Ki,app within one
log unit of the experimentally determined value. In
order to demonstrate that the model was not likely to
be the result of a chance correlation, pharmacophore
generation was repeated with 19 randomized datasets.
All of the models generated by these training sets
exhibited an r2 , 0.87, indicating that it is highly
unlikely (P , 0.05) that the original model was the
result of a chance correlation.
SOMFA using a common features pharmacophore
alignment
The common features pharmacophore used to align the
substrates was based on a nucleophilic feature and two
hydrophobic regions (Fig. 5a). When aligned on this
pharmacophore, the sites of glucuronidation on each
substrate were overlaid. The best SOMFA model was
generated using the shape field alone (r ¼ 0.84,
r2 ¼ 0.71) and was capable of predicting the Ki,app for
four of the five test set chemicals within one log unit of
their experimental value (Table 2). Figure 5(b,c) shows
morphine (a low affinity substrate) and bilirubin (a high
affinity substrate), respectively, aligned on the SOMFA
model. Bilirubin predominantly occupies the cyan area,
where steric bulk enhances binding affinity. However,
a large section of morphine occupies the red area,
where steric bulk is associated with reduced binding
affinity.
2D QSAR
Of the 319 descriptors calculated for each chemical
using the Dragon and Cerius2 software, 161 were found
to be significantly correlated to the –logKi,app at the
95% CI. The UFS with an r2max of 0.9 gave a 12
descriptor model with the best LOO cross-validated r2
of 0.64. One other descriptor (GATS5m) was found to
be highly correlated with the residual of this model,
and to significantly increase the cross-validated r2.
Seven descriptors in the model were found to increase
the LOO cross-validated r2 when removed from the
model generation process. Thus, the optimal model
was a six descriptor, two-component model with an r2
of 0.92 and a LOO cross-validated r2 of 0.77. The
model is shown below in terms of the standardized
descriptors (� ¼ 0, � ¼ 1). The relative importance of
each descriptor is represented by the magnitude of its
coefficient:
–logKi,app ¼
4:5 þ 0:49�AlogP98–0:55 3 nRO8 þ 0:50 3 JGI7 þ 0:44
3 MATS1p–0:26 3 MATS7p þ 0:29 3 GATS5m
where: AlogP98 ¼ log of the octanol/water partition
coefficient calculated by an atom based method;
nR08 ¼ number of eight-membered rings; JGI7 ¼ mean
topological charge index of order 7; MATS1p ¼ Moran
autocorrelation of path length 1 weighted by atomic
polarizabilities; MATS7p ¼ Moran autocorrelation of
path length 7 weighted by atomic polarizabilities;
Table 1. Comparison of experimentally determined K i,app values forfive substrates using a single 4MU concentration and full kineticanalysis
Substrate
K i,app (�mol/l)Single 4MU
concentration(110 �mol/l)
K i,app (�mol/l)Full kinetic
analysis
Quercetin 2 3Naringenin 3 3Ethinylestradiol 3 43-Hydroxyflavone 90 904-Hydroxybenzoic acid methyl ester 1000 1200
640 Pharmacogenetics 2002, Vol 12 No 8
GATS5m ¼ Geary autocorrelation of path length 5
weighted by atomic masses.
Predictive ability of the model was validated using the
test set. All five test substrates were predicted within
one log unit of their observed value (Table 2). The
Ki,app was permuted 100 times and models generated.
None of these models resulted in as good fit (based on
r2), showing that this model is very unlikely (P , 0.01)
to be a result of a chance correlation.
DiscussionUGT1A1 is a polymorphic enzyme responsible for the
glucuronidation of structurally diverse xenobiotics and
endogenous compounds. Thus, knowledge of UGT1A1
substrate and inhibitor selectivities and binding affi-
nities assumes importance for: (i) the identification of
those compounds whose elimination may be impaired
in subjects with variant UGT1A1 genotypes and (ii) the
prediction of inhibitory interactions with other xenobio-
tics and endogenous compounds (particularly bilirubin)
1/V
µm
ol/l
(pm
ol/m
in p
er m
g)
(d)
0.010
0.008
0.006
0.002
0.0000 50 100 150
0.004
[3-Hydroxyflavone] (µmol/l)
1/V
µm
ol/l
(pm
ol/m
in p
er m
g)
(b)
0.016
0.012
0.008
0.004
0.0000 1000 2000 3000 4000 5000
[4HBAME] (µmol/l)
55 µmol/l 4MU
110 µmol/l 4MU
225 µmol/l 4MU
0.015
0.010
0.005
0 100 150
[3-Hydroxyflavone] (µmol/l)
(c)
0 50
1/V
(pm
ol/m
in p
er m
g)
0.020
30 µmol/l 4MU
90 µmol/l 4MU
400 µmol/l 4MU0.030
0.020
0.010
0 1000 2000
[4HBAME] (µmol/l)
(a)
0.030
0.020
0.010
0 1000 2000
[4HBAME] (µmol/l)
1/V
(pm
ol/m
in p
er m
g)
Fig. 3
Dixon plots for the inhibition of UGT1A1 catalysed 4-methylumbelliferone glucuronidation by 4-hydroxybenzoic acid methyl ester (4HBAME) and 3-hydroxyflavone. (a,c) Showing data from the full kinetic analysis. (b,d) Showing corresponding data from the abbreviated kinetic method (i.e. threeinhibitor concentrations at a 4-methylumbelliforone of 110 �mol/l).
UDP-glucuronosyltransferase 1A1 substrate modelling Sorich et al. 641
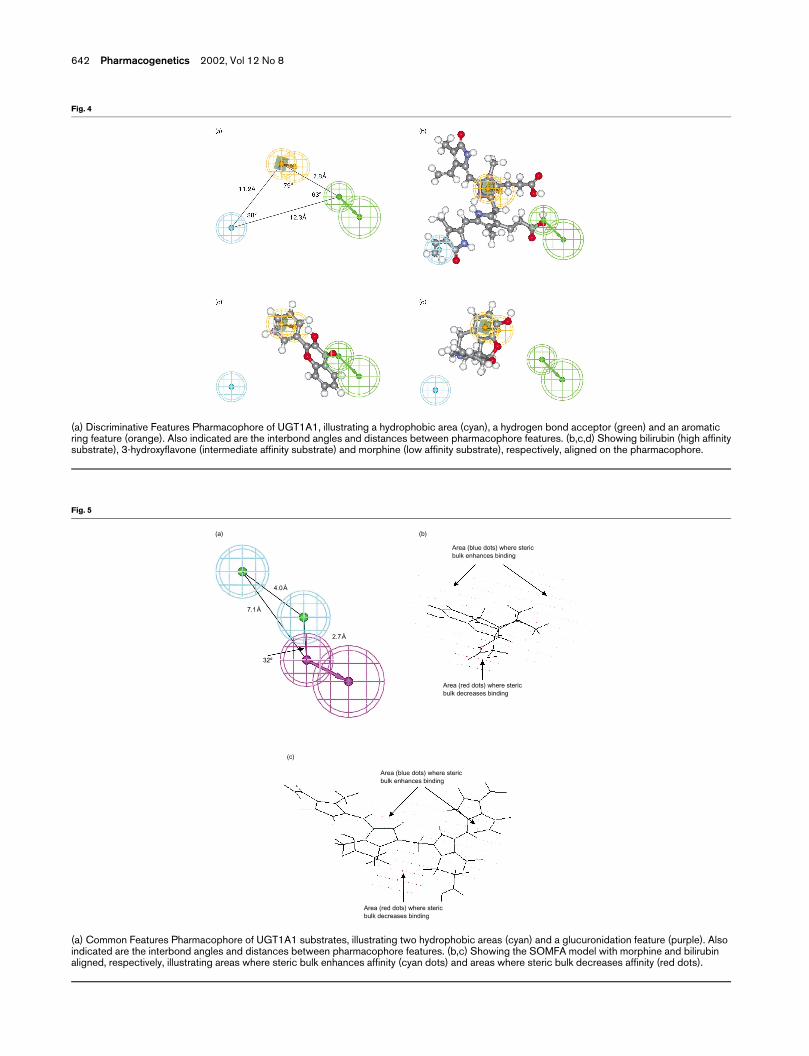
Fig. 4
(a) Discriminative Features Pharmacophore of UGT1A1, illustrating a hydrophobic area (cyan), a hydrogen bond acceptor (green) and an aromaticring feature (orange). Also indicated are the interbond angles and distances between pharmacophore features. (b,c,d) Showing bilirubin (high affinitysubstrate), 3-hydroxyflavone (intermediate affinity substrate) and morphine (low affinity substrate), respectively, aligned on the pharmacophore.
(a)
4.0Å
7.1Å
32º
2.7Å
Area (blue dots) where stericbulk enhances binding
Area (red dots) where stericbulk decreases binding
Area (blue dots) where stericbulk enhances binding
Area (red dots) where stericbulk decreases binding
(c)
(b)
Fig. 5
(a) Common Features Pharmacophore of UGT1A1 substrates, illustrating two hydrophobic areas (cyan) and a glucuronidation feature (purple). Alsoindicated are the interbond angles and distances between pharmacophore features. (b,c) Showing the SOMFA model with morphine and bilirubinaligned, respectively, illustrating areas where steric bulk enhances affinity (cyan dots) and areas where steric bulk decreases affinity (red dots).
642 Pharmacogenetics 2002, Vol 12 No 8
metabolized by this enzyme. In the present study,
three different methodologies were used to develop
models which predict UGT1A1 substrate selectivity
and binding affinity. The predictive ability of models
was tested with compounds withheld from the training
set. Along with models generated concurrently for
UGT1A4 (Smith et al., unpublished observations),
these represent the first generalized 2D and 3D QSAR
and pharmacophore models developed for UGT.
A rapid and simple procedure was initially developed
here to calculate the Ki,app values of UGT1A1 sub-
strates. In the context of the range of values used to
construct the models (Ki,app 0.5–9000 �mol/l), the dif-
ferences in values between the abbreviated method
and full kinetic analysis are insignificant. The proce-
dure increases the viability of characterizing sufficient
substrates to construct a reliable predictive model
(i.e. . 20 compounds). Previous pharmacophores and
3D QSAR developed for CYP isoforms have generally
relied on kinetic constants generated in multiple
laboratories using different assay procedures, which
decreases data reliability and hence model predictive-
ness [34,35].
The common features pharmacophore (Fig. 5a) indi-
cates that hydrophobic regions on the substrate are
commonly found close to the glucuronidation site of
the chemical. This is very similar to the common
features pharmacophore generated in this laboratory for
substrates of UGT1A4 (Smith et al., unpublished ob-
servations). The discriminative features pharmacophore
is more difficult to analyse due to the absence of the
glucuronidation feature in the model, which is useful as
a common reference point. It indicates a geometrical
arrangement of an aromatic ring, a hydrogen bond
donor and a hydrophobic region, which may be impor-
tant in binding to the active site of the enzyme.
Bilirubin is a high affinity substrate for UGT1A1. Thus,
not unexpectedly, bilirubin fits the discriminative fea-
tures pharmacophore well (Fig. 4b). The 2D QSAR
model suggests that the logP of a substrate has an
important influence on the ability of that compound to
bind to the active site of UGT1A1. Previous analyses
have also indicated that this is an important property of
substrates [36–38]. This may be due to properties of
the active site of the enzyme and/or the membrane
environment of the enzyme. In this regard, it should be
noted that the UGT active site is believed to be
located on the luminal face of the microsomal mem-
brane, and hence substrates must traverse the endoplas-
mic reticulum to gain access to the active site. The
other descriptors from the 2D QSAR model are calcu-
lated from the atomic properties and connectivity of
the molecules. These descriptors are commonly found
to be useful in predicting ligand–protein interactions,
however, their physical interpretation is very difficult
[16].
As shown by statistical evaluation and test set predic-
tions, all three models have useful predictive ability,
with the 2D QSAR performing best. The ability of a
model to predict the Ki,app of a novel chemical is
dependent on the substrates used in the training set. If
the training set substrates are similar in chemical
structure, the models are easier to construct, but the
generalizability of the model is greatly reduced. Such a
training set is unlikely to be able to reliably predict the
Ki,app value of diverse chemical structures. Having been
trained on a diverse set of known UGT1A1 substrates,
both in terms of structure and Ki,app, the models
constructed here should be well placed to predict the
Ki,app of new substrates of UGT1A1, at least within one
order of magnitude (the ‘acceptable’ standard for QSAR
developed for CYP isoforms [26]). The accuracy of
predicted Ki,app values is likely to improve as additional
(novel) substrates are incorporated into the model.
Furthermore, the three models were constructed using
very different properties of the substrates and very
different methods. The discriminative features pharma-
cophore predicts the Ki,app on the basis of abstract
chemical features (e.g. hydrogen bond donors, hydro-
phobic regions) in 3D. The algorithm includes a search
of conformational space (i.e. set of possible conforma-
tions of the chemical in 3D) for each substrate. This
procedure can be classed as a pharmacophore (3D
arrangement of chemical features) based 4D QSAR.
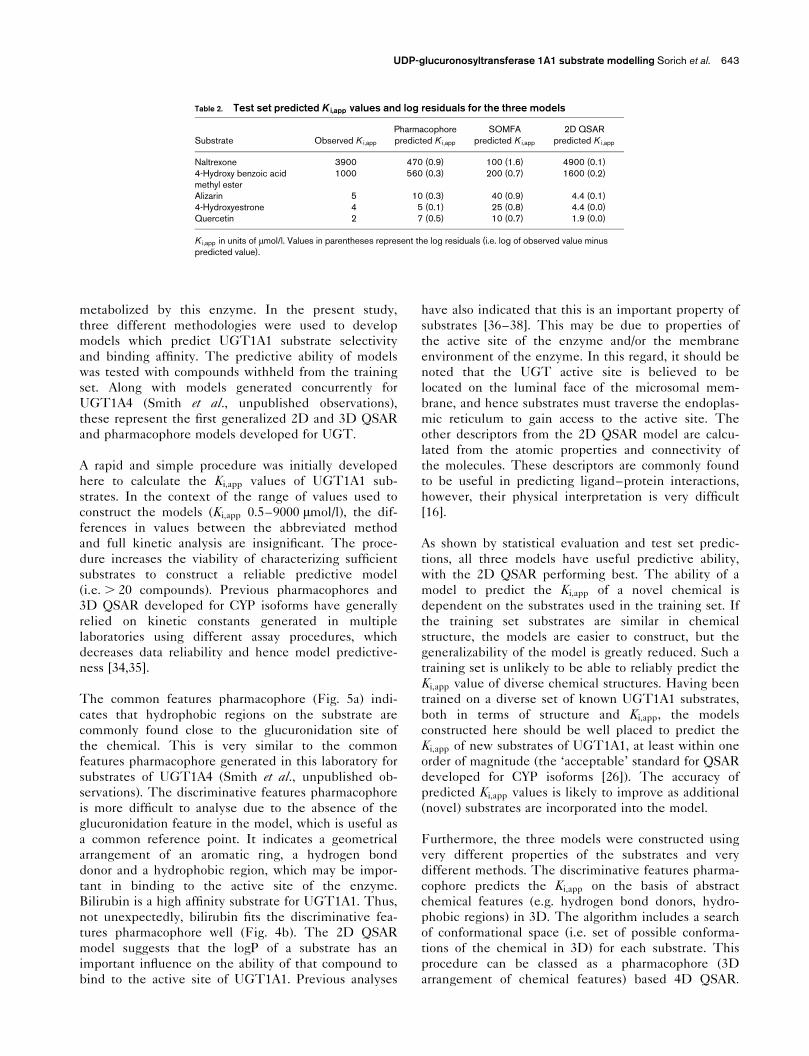
Table 2. Test set predicted K i,app values and log residuals for the three models
Substrate Observed K i,app
Pharmacophorepredicted K i,app
SOMFApredicted K i,app
2D QSARpredicted K i,app
Naltrexone 3900 470 (0.9) 100 (1.6) 4900 (0.1)4-Hydroxy benzoic acidmethyl ester
1000 560 (0.3) 200 (0.7) 1600 (0.2)
Alizarin 5 10 (0.3) 40 (0.9) 4.4 (0.1)4-Hydroxyestrone 4 5 (0.1) 25 (0.8) 4.4 (0.0)Quercetin 2 7 (0.5) 10 (0.7) 1.9 (0.0)
K i,app in units of �mol/l. Values in parentheses represent the log residuals (i.e. log of observed value minuspredicted value).
UDP-glucuronosyltransferase 1A1 substrate modelling Sorich et al. 643

The SOMFA methodology uses 3D field (steric and
electrostatic) properties to predict the Ki,app. The
SOMFA algorithm requires the input of the aligned
substrates and does not use the conformational space of
the substrates. This can be classed as a field-based 3D
QSAR. The 2D QSAR is generated from 2D properties
of the substrates, which require no conformational or
alignment searching. Each of these models will contain
unique information and they can be considered to be
complementary.
The SOMFA model reported here had useful predic-
tive ability, but was inferior to the two other models
(discriminative features pharmacophore and 2D QSAR).
It has been recognized for some time that the major
obstacle to generating a 3D QSAR for a diverse set of
compounds is the selection of the bioactive conforma-
tion and superposition method [20]. The great struc-
tural diversity of the substrates, which makes the
predictions of the models so useful, also makes the
search for the correct pharmacophore and alignment
very difficult. The alignment may be aided by incor-
poration of the reaction site on the substrate. When
bound to the active site of the enzyme, the substrates
should align the functional groups to be glucuronidated,
since this part of the molecule would need to be
located in the region adjacent to the bound UDPGA
(cosubstrate). An attempt to use this information in the
Catalyst software met with mixed success. A feature
that recognized potential glucuronidation sites (e.g.
–OH, –NH) was constructed, but software limitations
meant it was not possible to ensure that all chemicals
fitted this feature. Similarly, there was no way of
selecting the preferred site of glucuronidation when
more than one nucleophilic site was present on a
substrate. It may be possible to improve the predictiv-
ity by using Comparative Molecule Similarity Analysis
(CoMSIA) molecular fields. CoMSIA is a variation of
CoMFA, in which the molecular fields are based on
‘soft’ Gaussian functions that are less likely to be
influenced by small errors in the alignment. They also
explicitly take into account the hydrophobicity of the
chemicals, in addition to the electrostatics and shape
[39].
Few previous studies have applied molecular modelling
approaches to UGT. A CoMFA model has been
reported for triphenylalkyl carboxylic acid analogue
inhibitors of UGT1A1 [40]. These structures are all
very similar and this model is therefore only useful for
predicting the binding of closely related compounds. A
QSAR using 2D descriptors has been derived to predict
the glucuronidation of benzoic acid analogues in the
rat, but does not consider the involvement of individual
isoforms [41]. As noted in the Introduction, molecular
modelling has been used widely in recent years to infer
active site binding requirements of CYP. Many of the
models generated for CYP isoforms have incorporated
enzyme structure, but this is currently not an option for
UGT. However, in general, the UGT1A1 models gen-
erated here are of at least comparable statistical power
and predictive ability to those QSAR generated for
CYP isoforms, including the polymorphic CYP2C9 and
CYP2D6 which do not incorporate direct information
from the protein structure [13,34,35].
In summary, three different methodologies have been
used to construct predictive models from the Ki,app
values of 18 known structurally diverse substrates of
UGT1A1. The predictive capability of the models was
validated primarily using a test set of five substrates not
used in the model construction process. When used
together, the models are capable of predicting whether
a chemical is a likely substrate for UGT1A1 through
inspection of the 3D positioning of important func-
tional groups, and estimating an in-vitro Ki,app for
putative substrates of UGT1A1. The predictive ability
of these models should improve with increasing num-
bers of newly discovered UGT1A1 substrates, greater
understanding of the 3D structure of UGT and new
model generation algorithms.
AcknowledgementsThe authors gratefully acknowledge Daniel Robinson
and the Computational Chemistry Research Group
(Oxford University, UK) and Milano Chemometrics
and QSAR Research Group (University of Milano-
Bicocca, Italy) for making available the SOMFA and
Dragon software, respectively. This work was funded
by a grant from the National Health and Medical
Research Council of Australia. M.J.S. is the recipient of
an Australian Postgraduate Award.
References1 Miners JO, Mackenzie PI. Drug glucuronidation in humans. Pharmacol
Ther 1991; 51:347–369.2 Mackenzie PI, Owens IS, Burchell B, Bock KW, Bairoch A, Belanger A,
et al. The UDP glycosyltransferase gene superfamily: recommendednomenclature update based on evolutionary divergence. Pharmaco-genetics 1997; 7:255–269.
3 Radominska-Pandya A, Czernik PJ, Little JM, Battaglia E, Mackenzie PI.Structural and functional studies of UDP-glucuronosyltransferases. DrugMetab Rev 1999; 31:817–899.
4 Tukey RH, Strassburg CP. Human UDP-glucuronosyltransferases: meta-bolism, expression, and disease. Ann Rev Pharmacol Toxicol 2000;40:581–616.
5 Miners JO, McKinnon RA, Mackenzie PI. Genetic polymorphism of UDP-glucuronosyltransferases and their functional significance. Toxicology2002; in press.
6 Ritter JK, Crawford JM, Owens IS. Cloning of two human liver bilirubinUDP-glucuronosyl-transferase cDNAs with expression in COS-1 cells.J Biol Chem 1991; 266:1043–1047.
7 Ebner T, Remmel RP, Burchell B. Human bilirubin UDP-glucuronosyltrans-ferase catalyzes the glucuronidation of ethinylestradiol. Mol Pharmacol1993; 43:649–654.
8 Senafi SB, Clarke DJ, Burchell B. Investigation of the substrate specificityof a cloned expressed human bilirubin UDP-glucuronosyltransferase:UDP-sugar specificity and involvement in steroid and xenobiotic glucur-onidation. Biochem J 1994; 303:233–240.
9 Kadakol A, Ghosh SS, Sappal BS, Sharma G, Chowdhury JR,Chowdhury NR. Genetic lesions of bilirubin uridine-diphospho-glucuro-
644 Pharmacogenetics 2002, Vol 12 No 8

nate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gil-bert syndromes: correlation of genotype to phenotype. Hum Mut 2000;16:297–306.
10 Ando Y, Saka H, Ando M, Sawa T, Muro K, Ueoka H, et al. Polymorphismof UDP-glucuronosyltransferase gene and irinotecan toxicity: A pharmaco-genetic analysis. Cancer Res 2000; 60:6921–6926.
11 Zucker SD, Qin X, Rouster, Yu F, Green RM, Keshaven P, et al.Mechanism of indinivir-induced hyperbilirubinemia. Proc Natl Acad SciUSA 2001; 98:12671–12676.
12 Posner J, Cohen HF, Land G, Winton C, Peck AW. The pharmacokineticsof lamotrigine (BW430C) in healthy subjects with unconjugated hyper-bilirubinaemia (Gilbert’s Syndrome). Br J Clin Pharmacol 1989; 28:117–120.
13 Ekins S, Bravi G, Binkley S, Gillespie JS, Ring BJ, Wilkel JH, WrightonSA. Three and four dimensional-quantitative structure activity relationship(3D/4D-QSAR) analyses of CYP2D6 inhibitors. Pharmacogenetics 1999;9:477–489.
14 Ekins S, De Groot MJ, Jones JP. Pharmacophore and three-dimensionalquantitative structure activity relationship methods for modeling cyto-chrome P450 active sites. Drug Metab Dispos 2001; 29:936–944.
15 Lewis DFV, Ioannides C, Parke DV. An improved and updated version ofthe compact procedure for the evaluation of P450-mediated chemicalactivation. Drug Metab Rev 1998; 30:709–737.
16 Todeschini R, Consonni V. Handbook of molecular descriptors.Weinheim: Wiley-VCH; 2000.
17 Livingstone DJ. The characterization of chemical structures using molecu-lar properties. A survey. J Chem Inf Comput Sci 2000; 40:195–209.
18 Clement OO, Mehl AT. HipHop: pharmacophores based on multiplecommon-feature alignments. In: Guner O, ed. Pharmacophore perception,development, and use in drug design. La Jolla: International UniversityLine; 2000. pp. 69–84.
19 Schafferhans A, Klebe G. Docking ligands onto binding site representa-tions derived from proteins built by homology modeling. J Mol Biol 2001;307:407–427.
20 Martin YC, Kim KH, Lin CT. Comparative molecular field analysis:CoMFA. In: Charton M, ed. Advances in quantitative structure-propertyrelationships, vol. 1. Greenwich: Jai Press Inc; 1996. pp. 1–52.
21 King CD, Green MD, Rios GR, Coffman BL, Owens IS, Bishop WP,Tephly TR. The glucuronidation of exogenous and endogenous com-pounds by stably expressed rat and human UDP-glucuronosyltransferase1.1. Arch Biochem Biophys 1996; 332:92–100.
22 Visser TJ, Kaptein E, Gijzel AL, de Herder WW, Ebner T, Burchell B.Glucuronidation of thyroid hormone by human bilirubin and phenol UDP-glucuronosyltransferase isoenzymes. FEBS Lett 1993; 324:358–360.
23 Iyer L, King CD, Whitington PF, Green MD, Roy SK, Tephly TR, et al.Genetic predisposition to the metabolism of irinotecan (CPT-11). Role ofuridine diphosphate glucuronosyltransferase isoform 1A1 in the glucur-onidation of its active metabolite (SN-38) in human liver microsomes.J Clin Invest 1998; 101:847–854.
24 King CD, Rios GR, Green MD, Mackenzie PI, Tephly TR. Comparison ofstably expressed rat UGT1.1 and UGT2B1 in the glucuronidation ofopioid compounds. Drug Metab Dispos 1997; 25:251–255.
25 Miners JO, Lillywhite KJ, Matthews AP, Jones ME, Birkett DJ. Kinetic andinhibitor studies of 4-methylumbelliferone and 1-naphthol glucuronidationin human liver microsomes. Biochem Pharmacol 1988; 37:665–671.
26 Ekins S, Ring BJ, Bravi G, Winkel JH, Wrighton SA. Predicting drug-druginteractions in silico using pharmacophores: a paradigm for the nextmillennium. In: Guner, ed. Pharmacophore perception, development, anduse in drug design. La Jolla: International University Line; 2000. pp.270–299.
27 Sprague PW. Automated chemical hypothesis generation and databasesearching with catalyst. Perspect Drug Discov Des 1995; 3:1–20.
28 Barnum D, Greene J, Smellie A, Sprague P. Identification of commonfunctional configurations among molecules. J Chem Inf Comput Sci1996; 36:563–571.
29 Robinson DD, Winn PJ, Lyne PD, Richards G. Self-organizing molecularfield analysis: A tool for structure-activity studies. J Med Chem 1999;42:573–583.
30 Cramer RD III, Patterson DE, Bunce JD. Comparative molecular fieldanalysis (CoMFA). 1. Effect of shape on binding of steroids to carrierproteins. J Am Chem Soc 1988; 110:5959–5967.
31 Good AC, So S, Richards G. Structure-activity relationships frommolecular similarity matrices. J Med Chem 1993; 36:433–438.
32 Whitley DC, Ford MG, Livingstone DJ. Unsupervised forward selection:a method for eliminating redundant variables. J Chem Inf Comput Sci2000; 40:1160–1168.
33 Wold S, Ruhe A, Wold H, Dunn WJ III. The collinear problem in linear
regression. The partial least squares (PLS) approach to generalizedinverses. Siam J Sci Stat Comput 1984; 5:735–743.
34 Ekins S, Bravi G, Wikel JH, Wrighton SA. Three-dimensional-quantitativestructure activity relationship analysis of cytochrome P-450 3A4 sub-strates. J Pharmacol Exp Ther 1999; 291:424–433.
35 Ekins S, Bravi G, Ring BJ, Gillespie TA, Gillespie JS, Vandenbranden M,et al. Three-dimensional quantitative structure activity relationshipanalyses of substrates for CYP2B6. J Pharmacol Exp Ther 1999;288:21–29.
36 Resetar A, Minick D, Spector T. Glucuronidation of 39-azido-39-deoxythy-midine catalyzed by human liver UDP-glucuronosyltransferase. Signifi-cance of nucleoside hydrophobicity and inhibition by xenobiotics.Biochem Pharmacol 1991; 42:559–568.
37 Yin H, Bennett G, Jones JP. Mechanistic studies of uridine diphosphateglucuronosyltransferase. Chem Biol Interact 1994; 90:47–58.
38 Kim KH. Quantitative structure-activity relationships of the metabolism ofdrugs by uridine diphosphate glucuronosyltransferase. J Pharm Sci1991; 80:966–970.
39 Klebe G, Abraham U, Mietzner T. Molecular similarity indices in acomparative analysis (CoMSIA) of drug molecules to correlate andpredict their biological activity. J Med Chem 1994; 37:4130–4146.
40 Said M, Ziegler J, Magdalou J. Inhibition of bilirubin UDP-glucuronosyl-transferase: a comparative molecular field analysis (CoMFA). QuantStruct-Act Relat 1996; 15:382–388.
41 Cupid BC, Holmes E, Wilson ID, Lindon JC, Nicholson JK. Quantitativestructure-metabolism relationships (QSMR) using computational chemis-try: pattern recognition analysis and statistical prediction of phase IIconjugation reactions of substituted benzoic acids in the rat. Xenobiotica1999; 29:27–42.
UDP-glucuronosyltransferase 1A1 substrate modelling Sorich et al. 645