pharmacodynamics and pharmacokinetics of the novel par-1 antagonist vorapaxar (formerly sch 530348)...
TRANSCRIPT

PHARMACODYNAMICS
Pharmacodynamics and pharmacokinetics of the novelPAR-1 antagonist vorapaxar (formerly SCH 530348)in healthy subjects
Teddy Kosoglou & Larisa Reyderman &
Renger G. Tiessen & André A. van Vliet &Robert R. Fales & Robert Keller & Bo Yang &
David L. Cutler
Received: 28 April 2011 /Accepted: 22 August 2011 /Published online: 21 September 2011# Springer-Verlag 2011
AbstractPurpose The aim of our study was to evaluate thepharmacology of vorapaxar (SCH 530348), an oral PAR-1antagonist, in healthy volunteers.Methods and results In two randomized, placebo-controlledstudies, subjects received either single ascending doses ofvorapaxar (0.25, 1, 5, 10, 20, or 40 mg; n=50), multipleascending doses of vorapaxar (1, 3, or 5 mg/day for 28days; n=36), a loading dose (10 or 20 mg) followed bydaily maintenance doses (1 mg) for 6 days (n=12), orplacebo. Single 20- and 40-mg doses of vorapaxarcompletely inhibited thrombin receptor activating peptide(TRAP)-induced platelet aggregation (>80% inhibition) at 1h and sustained this level of inhibition for ≥72 h. Multipledoses yielded complete inhibition on Day 1 (5 mg/day) and
Day 7 (1 and 3 mg/day). Adverse events were generallymild, transient, and unrelated to dose.Conclusion Vorapaxar provided rapid and sustained dose-related inhibition of platelet aggregation without affectingbleeding or clotting times.
Keywords Vorapaxar . SCH 530348 . PAR-1 antagonist .
Pharmacokinetics . Pharmacodynamics . Plateletaggregation . Thrombin receptor antagonist
Introduction
Platelets play a significant role in primary hemostasis andvascular repair, as well as in the formation of pathogenicthrombi, which are responsible for clinical manifestationsof atherothrombotic disease [1]. Numerous pathwaysstimulated by specific agonists, such as adenosine diphos-phate (ADP), thromboxane A2, and thrombin, contribute toplatelet activation, which is crucial for hemostasis as wellas pathologic thrombosis [1]. The key role of plateletactivation in the pathophysiology of atherothromboticdisease is supported by the well-established clinical benefitsof antiplatelet therapy, which has become the standard ofcare for the management of patients with this disease.Current oral antiplatelet agents, such as aspirin and P2Y12
receptor antagonists (e.g., clopidogrel, ticlopidine, andprasugrel), inhibit the thromboxane A2 and ADP plateletactivation pathways [2]. Although dual antiplatelet therapyhas resulted in a greater clinical benefit than either therapyalone, a considerable number of patients remain atsignificant risk for recurrent ischemic events [3–5]. Thishigh residual risk despite treatment with aspirin and a
T. Kosoglou : L. Reyderman :R. R. Fales : R. Keller : B. Yang :D. L. CutlerMerck,Whitehouse Station, NJ, USA
R. G. Tiessen :A. A. van VlietPRA International,Zuidlaren, the Netherlands
T. Kosoglou (*)Clinical Pharmacology, Merck Research Laboratories,Merck,351 N. Sumneytown Pike, UG4–D48,North Wales, PA 19454, USAe-mail: [email protected]
Present Address:L. ReydermanEisai Medical Research,Ridgefield Park, NJ, USA
Eur J Clin Pharmacol (2012) 68:249–258DOI 10.1007/s00228-011-1120-6

P2Y12 receptor antagonist can be attributed to the lack ofinhibitory effect of these therapies on other plateletactivation pathways [1]. In addition, the use of aspirin andP2Y12 receptor antagonists is associated with increased riskof bleeding [5, 6], which negatively impacts outcomesand may contribute to both short-term and long-termmorbidity and mortality [7]. Increased bleeding risk withaspirin and P2Y12 receptor antagonists may be attributedto the fact that these therapies target pathways that are notonly involved in pathologic thrombosis but also inprotective hemostasis [1]. Given the limitations of currentoral antiplatelet therapy, there is a clinical need for newtherapies with novel mechanisms of action that reducethrombosis without interfering with hemostasis.
Inhibition of protease-activated receptor 1 (PAR-1), theprincipal receptor for thrombin on human platelets, repre-sents a novel strategy for the prevention of plateletactivation and thrombosis. Notably, the binding of thrombinto PAR-1, which represents a very potent platelet activationpathway [8, 9], is necessary for thrombus formation butmay not be required for hemostasis, as suggested bypreclinical studies with PAR-1 antagonists [10–12]. There-fore, inhibition of PAR-1 may prevent thrombosis withouthaving a significant inhibitory effect on hemostasis, andthereby may potentially reduce the risk of ischemic eventswithout increasing the risk of bleeding. The novel anti-platelet agent vorapaxar (SCH 530348) is a potent, orallyactive PAR-1 antagonist [13]. Vorapaxar has demonstratedrapid absorption and distribution, with peak plasma levelsreached within 60 to 90 min following the administration ofsingle or multiple doses [14]. Vorapaxar is metabolized andeliminated primarily by biliary and gastrointestinal routes[15]. The studies reported here were undertaken to evaluatethe safety, pharmacodynamics and pharmacokinetics ofvorapaxar after the administration of single and multipledoses in healthy subjects.
Methods
Study description and conduct
The two Phase I studies included in this report wererandomized, double-blind, placebo-controlled, ascendingsingle- and multiple-dose studies of orally administeredvorapaxar in healthy subjects. The studies were con-ducted by PRA International (formerly Pharma Bio-Research Group BV), Zuidlaren, the Netherlands, inaccordance with Good Clinical Practice. The protocolwas reviewed and approved by an independent ethicscommittee, and written informed consent was obtainedfrom each potential subject before any study-relatedactivities were performed.
Subjects
Healthy male (both single-dose and multiple-dose studies)and female (multiple-dose study only) subjects aged 18–45years who had a body mass index (BMI) ranging from 19 to29 kg/m2 were eligible for enrollment. Subjects had a normalor clinically acceptable physical exam, electrocardiogram(ECG), and clinical laboratory tests [complete blood count(CBC), blood chemistries, and urinalysis]. They were free ofany clinically significant disease that would interfere withstudy evaluations, were negative for drugs with a highpotential for abuse, and had baseline blood coagulation testsand bleeding times within normal limits. Subjects with ahistory of coagulation disorders, thrombocytopenia, bleed-ing, or cardiac abnormalities, those testing positive forhuman immunodeficiency virus (HIV) antibodies, hepatitisB surface antigen, or hepatitis C antibody, and those havingmajor surgery within 3 months of study entry were excluded.Subjects should not have used any prescription or nonpre-scription drugs within 14 days prior to drug administration,excluding acetaminophen.
Study design and dosing
In the single ascending dose study, nine subjects wereenrolled in each of six sequential groups and randomizedwithin each dose level in a 2:1 ratio to receive either a singledose [capsule(s)] of vorapaxar (0.25, 1, 5, 10, 20, or 40 mg;n=6/dose level) or placebo (n=3/dose level). All subjectsreceived a single dose of the study treatment (Day 1) andwere followed for 72 h. Dose escalation was based on thedemonstration of safety and tolerability of the previous dose.
The multiple ascending dose study consisted of fourgroups. Each of three sequential groups comprised 12 subjectswho were randomized within each dose level in a 2:1 ratio toreceive either vorapaxar (1, 3, or 5 mg; n=8/dose level) orplacebo (n =4/dose level) orally, as capsules, once daily inthe morning for 28 days. Dose escalation was based on thedemonstration of safety and tolerability of the previous dose.Subjects in Group 4 (n=12), who were enrolled in parallelwith Group 3, were randomized to receive either a 10-mg (6subjects) or a 20-mg (6 subjects) dose of vorapaxar on thefirst day, followed thereafter by once-daily oral doses ofvorapaxar 1 mg on Days 2 through 7.
Pharmacokinetic analyses
Blood samples for pharmacokinetic evaluation were col-lected predose (Day 1, 0 h) and at 0.5, 1, 1.5, 2, 3, 4, 5, 6,8, 10, 12, 16, 24, 36, 48, and 72 h postdose (one 5-mLsample per time point) in pre-chilled EDTA-containingtubes in the single ascending dose study. In the multipleascending dose study, samples for pharmacokinetic evaluation
250 Eur J Clin Pharmacol (2012) 68:249–258

(one 5-mL sample per time point) were taken before dosing onDays 1, 7, 14, 21, 26, 27, and 28 and at 0.5, 1, 1.5, 2, 3, 4, 5, 6,8, 10, 12, 16, and 24 h after dosing on Days 1 and 28, as wellas 36, 48, and 72 h after the last dose for Groups 1–3. Forsubjects in Group 4, samples (one 5-mL sample per timepoint) were taken before dosing on Days 1 through 7,and 24 h after dosing on Day 1, as well as 36, 48, and72 h after the last dose. After centrifugation, plasma wasremoved from all samples collected at the time pointsindicated above, and the vorapaxar concentration wascalculated using a validated, proprietary liquid chromatogra-phy with tandem mass spectrometry (LC-MS/MS) method,with a lower limit of quantitation (LLOQ) of 0.100 ng/mL.The LC system employed a 2.0 × 50-mm (particle size 5 μm)MonoChrome MS column (MetaChem Technologies, Tor-rance, CA). The internal standard (IS) used was [3C6]-labeledvorapaxar. Vorapaxar and the IS were separated from thebulk of the plasma components using a mobile phaseconsisting of methanol:0.1% acetic acid (85:15, v:v) at aflow rate of 0.250 mL/min. The retention time for bothvorapaxar and the IS was approximately 1.4 min. Vorapaxarand the IS were detected using either a Sciex API 3000 orAPI 4000 triple quadrupole LC-MS/MS System equippedwith a TurbolonSpray source (Applied Biosystems, FosterCity, CA). The range of the standard curve using a 200-μLsample of human plasma was 0.100 to 50.0 ng/mL. At theLLOQ (0.100 ng/mL) for the vorapaxar assay, the within-daymean [standard deviation (SD)] concentration for thestandard ranged from 0.096 (0.016) to 0.119 (0.017) ng/mL,and the between-day mean (SD) concentration was 0.105(0.017) ng/mL.
For the single-dose study, the measured vorapaxar concen-trations were used to estimate the maximum observed plasmaconcentration (Cmax), area under the curve versus timefrom time zero to the final quantifiable sample (AUC(tf)),area under the curve versus time from time zero to 24 h(AUC(0-24h)), and time of maximum observed plasmaconcentration (Tmax). For the multiple-dose study, measuredvorapaxar concentrations were used to estimate Cmax,AUC(0-24h), Tmax, and minimum observed plasma concentra-tion (Cmin). The elimination half-life (t½) was calculated ast½=0.693/K, where K is the terminal-phase rate constant.The effective half-life (t½eff) is based on drug accumulationand was calculated as t½eff=0.693/Kaccum, where the accu-mulation rate constant (Kaccum) was calculated as Kaccum ¼�1=tlnf1� ½AUC 0�24hð Þ=AUC Ið Þ�g, where τ is the dosinginterval (24 h). The vorapaxar accumulation index (R) wascalculated using the equation: R ¼ AUCð0�24hÞon Day 28
� �=
AUCð0�24hÞon Day 1� �
.Pharmacokinetic analyses were performed using a
commercially available computer program (Kinetica Enter-prise Secure Edition; Kinetica version 2.5.3; Innaphase,Philadelphia, PA).
Pharmacodynamic measurements
The effect of vorapaxar on platelet activation was assessedby light-transmission aggregometry [16] using a validatedcommercial 4-channel analyzer and software (PAP-4; Bio/Data Corp, Horsham, PA). Platelet aggregometry wasperformed using thrombin receptor activating peptide-6(TRAP-6; Ser-Phe-Leu-Leu-Arg-Asn; Sigma-Aldrich, St.Louis, MO) 15 μM and ADP 10 μM (single-dose study only)as agonists. The 15-μM TRAP-6 dose was selected for exvivo platelet aggregation studies on the basis of its ability toconsistently provide potent platelet activation. Blood samples(two 4.5-mL samples per time point) were collected prior todosing and at 1, 2 ,4, 6, 12, 24, and 72 h postdose in the singleascending dose study. In the multiple ascending dose study,blood samples (one 4.5-mL sample per time point) werecollected before dosing on Days 1 (baseline), 7, 14, 21, and28, and at 1, 2, 4, 6, 12, and 24 h after dosing on Days 1 and28, for Groups 1 to 3. Additional blood samples werecollected on Day 31 (72 h after the final dose) and on Days35, 42, 49, and 56. For Group 4, blood samples (one 4.5-mLsample per time point) were collected before dosing on Days 1(baseline) to 7 and at 0.5, 1, 2, 4, 6, 12, and 24 h after dosingon Day 1. The samples for TRAP- and ADP-induced plateletaggregation assays in the single ascending dose study wereinitially planned to be collected in the anticoagulant D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone(PPACK). However, because Group 1 (vorapaxar 0.25mg) had inconsistent and generally low platelet countsin whole blood and platelet-rich plasma samplescollected in PPACK, samples from all subsequentsingle-dose groups were collected using sodium citrateas the anticoagulant. The samples for TRAP-inducedplatelet aggregation assays in the multiple ascendingdose study were also collected using the sodium citrateanticoagulant.
Soluble P-selectin concentration (both single-dose andmultiple-dose studies), membrane-bound P-selectin expres-sion (single-dose study only), and soluble CD40 ligand(sCD40L) concentration (multiple-dose study only) wereevaluated using blood samples collected on Day −1 and at2, 24, and 72 h after dosing (single-dose study), and onDays 1 [prior to dose (Baseline)], 7, 14, 21, and 28(multiple-dose study). Soluble P-selectin and sCD40Lconcentrations were measured using validated commer-cial enzyme-linked immunosorbent assays (Soluble P-selectin: Biosource Catalog No. KHS2021; Biosource Int,Camarillo, CA; sCD40L: Bender MedSystems Catalog No.BMS239; Bender MedSystems GmbH, Vienna, Austria). Forthe evaluation of membrane-bound P-selectin expression, a500-μL platelet suspension was treated with 20 μL R-PE-conjugated mouse anti-human monoclonal antibody(CD62PE; Becton Dickinson Catalog No. 348107; Becton
Eur J Clin Pharmacol (2012) 68:249–258 251

Dickinson, Franklin Lakes, NJ) for 30 min at roomtemperature in the dark. The level of membrane-boundP-selectin expression in the suspension was determinedusing a FACSCalibur immunofluorescence detector (BectonDickinson).
Bleeding time evaluation
Bleeding time was determined using the modified Ivymethod [17] at baseline (predose) and at 2, 24, and72 h postdose in the single ascending dose study, atbaseline and before dosing on Days 7, 14, 21, and 28 forGroups 1–3, and on Day 7 for Group 4 in the multipleascending dose study. For the modified Ivy method usedin our studies, a commercially available, spring-activated puncture device (Glucoject Duo; A. MenariniDiagnostics, Florence, Italy) was used to produce threeround incisions of standardized width and depth (theoriginal Ivy method did not standardize the length anddepth of the incision; this modification improves thereproducibility of the bleeding time measurement). Themedian value for the time it took for bleeding to stopfrom the three punctures performed at each time pointwas recorded as the bleeding time. If the bleeding wasprolonged more than threefold for the baseline (Day −1)value or >10 min at the 2-h time point in the single-dose study or for the Day 7 (Group 4) or Day 28(Groups 1–3) assessment in the multiple-dose study, thetest had to be repeated until the value returned towardthe baseline.
Safety evaluations
In both studies, safety was evaluated based on the incidenceof adverse events (AEs), physical examinations, vital signs,routine clinical laboratory tests (CBC, chemistry panel, andurinalysis), blood coagulation tests, including thrombintime (TT), prothrombin time (PT), activated partial throm-boplastin time (aPTT), activated coagulation time (ACT),and ecarin clotting time (ECT), as well as routine safetyECGs. In addition, digital 12-lead serial ECGs wereperformed on Day −1 (baseline) and Day 1 (single-dosestudy) or Day 28 (multiple-dose study), corresponding topredose (0 h), and 1, 2, 4, 6, 12, and 24 h after dosing.These time-matched ECGs were transferred electronically to ablinded third party for assessment of the effect of vorapaxar onthe QT/QTc interval. The National Cancer Institute’s CommonToxicity Criteria (CTC) grading system was used to grade theseverity of AEs. The following definitions were used for thoseAEs not covered by the CTC:
– mild: awareness of sign, symptom, or event, but easilytolerated;
– moderate: enough discomfort to cause interference withusual activity and may warrant intervention;
– severe: incapacitating with the inability to do usualactivities or significantly affecting clinical status andwarranting intervention;
– life-threatening: immediate risk of death.
Statistical analyses
Descriptive statistics were used to summarize demograph-ics, ECGs, clinical laboratory tests, vital signs, pharmaco-dynamic parameters, and bleeding times, by treatment anddose level. Pharmacokinetic parameters were estimatedusing summary statistics, including means, standard devia-tions, and coefficients of variation at each time point, foreach dose level. Analysis of variance (ANOVA) wasperformed on log-transformed dose-adjusted AUC andCmax extracting effects due to treatment (dose). The originalscale was used for the other pharmacokinetic parameters.The ANOVA was used as a preliminary measure of doseproportionality. Statistically significant differences amongthe treatment means was considered to be a measure of lackof dose proportionality. For the single-dose study, ratioestimates and 90% confidence intervals for the dose-normalized Cmax and C72h were calculated using an ANOVAmodel. For the multiple-dose study, ratio estimates and 90%confidence intervals were derived for dose-normalized AUCand Cmax using an ANOVA to assess dose proportionality. Arepeated-measures ANOVA was used to assess Cmin data onDays 21, 26, 27, and 28 for attainment of steady state.
Results
The demographics and baseline characteristics of studysubjects receiving the study drug in the ascending single- andmultiple-dose studies were similar to those receiving placebo(Table 1). All 50 subjects enrolled in the single ascendingdose study completed the study and 47/48 subjects completedthe multiple ascending dose study. One subject in the multipleascending dose study discontinued treatment with vorapaxar1 mg on Day 24 because of an unrelated eyelid infection.
Pharmacokinetics
Single doses of vorapaxar exhibited rapid absorptionfollowing oral administration. Mean plasma vorapaxarconcentration–time profiles after administration of singledoses are shown in Fig. 1. Individual time to maximumobserved plasma concentration (Tmax) ranged between 0.5and 2 h after dosing (Table 2). The pharmacokinetic profileof vorapaxar was characterized by a fast distribution phase(data not shown) followed by a slow terminal elimination
252 Eur J Clin Pharmacol (2012) 68:249–258

phase. To estimate the elimination t½ of vorapaxar, sampleswere collected from consenting subjects in the 20-mg and40-mg dose groups for up to 64 days after dosing. Plasmaconcentrations were quantifiable for up to 53 days in subjectsadministered 20 mg and up to 62 days in subjects adminis-tered 40 mg. The elimination t½ in these subjects ranged from126 to 269 h. Exposure to vorapaxar appeared to be doserelated up to 40 mg, with low variability in exposure.
Multiple doses of vorapaxar (1, 3, and 5 mg once daily for28 days) were rapidly absorbed following oral administration(Table 3). Tmax values ranged from 1 to 1.5 h after dosing. Theelimination of vorapaxar was slow, with a mean t½ rangingfrom 173 to 269 h. Mean R values ranged from 4.72 to 6.37,
with the effective half-live (t1/2eff) ranging from 59.8 to141 h. Exposure to vorapaxar increased in a dose-relatedmanner, with low to moderate variability. There was noconsistent trend in the fluctuations of trough plasmavorapaxar concentrations at the end of the dosing interval,and steady-state plasma concentrations were attained byDay 21.
Pharmacodynamics
Single doses of vorapaxar resulted in dose-related inhibitionof 15-μM TRAP-induced platelet aggregation. Completeinhibition (>80%) of TRAP-induced platelet aggregationwith the 20-mg and 40-mg doses of vorapaxar wasobserved at 1 h (earliest sample time) and was subsequentlysustained throughout the 72-h monitoring period (Fig. 2a).The inhibitory effects of lower doses of vorapaxar onTRAP-induced platelet aggregation were less rapid and/orless potent. The recovery of platelet aggregation to >50% ofbaseline values after the single 20- and 40-mg doses ofvorapaxar was observed after approximately 4–8 weeks.
Multiple doses of vorapaxar also produced dose-relatedinhibition of ex vivo platelet aggregation in response toTRAP 15 μM. Among the three multiple-dose regimens ofvorapaxar evaluated (Groups 1–3), the 5-mg regimen wasthe only one to provide complete (>80%) inhibition ofTRAP-induced platelet aggregation on Day 1, whereas the1-mg and 3-mg regimens achieved complete inhibition atthe next measurement time point, Day 7 (Fig. 2b). Completeinhibition of platelet aggregation was subsequently main-tained throughout the 28-day treatment period with all threeregimens (Fig. 2b). The reversal of inhibition of TRAP-induced platelet aggregation following the discontinuation ofonce-daily maintenance dosing of vorapaxar was gradual anddose related, occurring after approximately 4–8 weeks (datanot shown) [18]. Among subjects in Group 4, who were
0 12 24 36 48 60 720
100
200
300
400
Hours
Vor
apax
ar (
ng/m
L)
0.25 mg1 mg5 mg10 mg20 mg40 mg
Fig. 1 Mean vorapaxar (SCH 530348) plasma concentrations followingthe administration of single doses (linear–linear scale). n=5 or 6 at eachtime point
Table 1 Subject demographics in the single- and multiple-dose studies
Subject demographics Single ascending dose study Multiple ascending dose study
Pooled placebo (n=16) Pooled vorapaxar (n=34) Pooled placebo (n=12) Pooled vorapaxar (n=24)
Male, n (%) 16 (100) 34 (100) 8 (67) 16 (67)
Race, n (%)
Caucasian 15 (94) 30 (88) 11 (92) 21 (88)
Black 0 2 (6) 0 2 (8)
Other 1 (6) 2 (6) 1 (8) 1 (4)
Mean age, year (SD) 23 (6) 25 (6) 27 (7) 28 (8)
Range 18–40 18–40 18–40 19–45
Mean BMI, kg/m2 (SD) 23.0 (2.3) 23.5 (2.4) 25.0 (2.4) 24.1 (2.2)
Range 19.4–27.3 18.6–29.2 20.3–29.0 20.4–28.5
BMI, Body mass index; SD, standard deviation
Eur J Clin Pharmacol (2012) 68:249–258 253
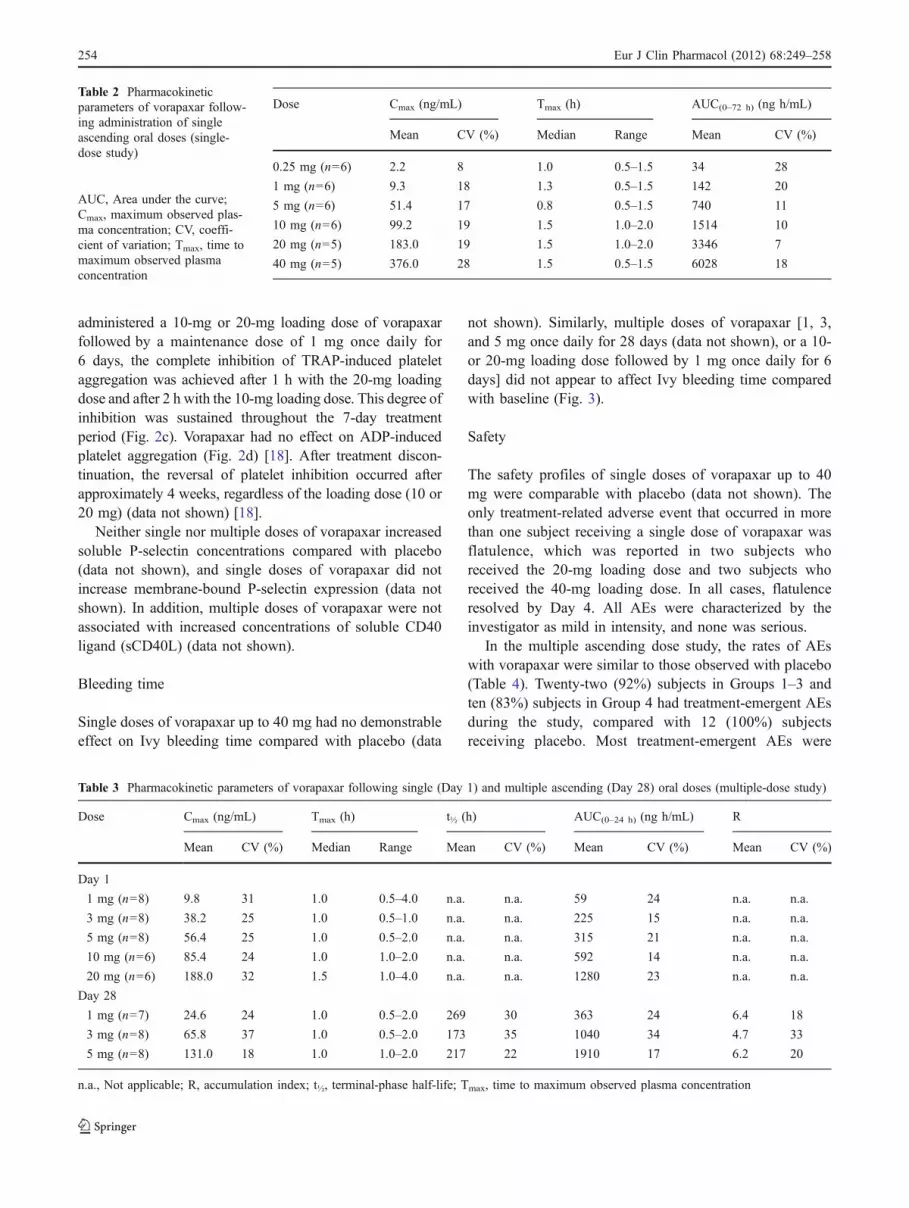
administered a 10-mg or 20-mg loading dose of vorapaxarfollowed by a maintenance dose of 1 mg once daily for6 days, the complete inhibition of TRAP-induced plateletaggregation was achieved after 1 h with the 20-mg loadingdose and after 2 h with the 10-mg loading dose. This degree ofinhibition was sustained throughout the 7-day treatmentperiod (Fig. 2c). Vorapaxar had no effect on ADP-inducedplatelet aggregation (Fig. 2d) [18]. After treatment discon-tinuation, the reversal of platelet inhibition occurred afterapproximately 4 weeks, regardless of the loading dose (10 or20 mg) (data not shown) [18].
Neither single nor multiple doses of vorapaxar increasedsoluble P-selectin concentrations compared with placebo(data not shown), and single doses of vorapaxar did notincrease membrane-bound P-selectin expression (data notshown). In addition, multiple doses of vorapaxar were notassociated with increased concentrations of soluble CD40ligand (sCD40L) (data not shown).
Bleeding time
Single doses of vorapaxar up to 40 mg had no demonstrableeffect on Ivy bleeding time compared with placebo (data
not shown). Similarly, multiple doses of vorapaxar [1, 3,and 5 mg once daily for 28 days (data not shown), or a 10-or 20-mg loading dose followed by 1 mg once daily for 6days] did not appear to affect Ivy bleeding time comparedwith baseline (Fig. 3).
Safety
The safety profiles of single doses of vorapaxar up to 40mg were comparable with placebo (data not shown). Theonly treatment-related adverse event that occurred in morethan one subject receiving a single dose of vorapaxar wasflatulence, which was reported in two subjects whoreceived the 20-mg loading dose and two subjects whoreceived the 40-mg loading dose. In all cases, flatulenceresolved by Day 4. All AEs were characterized by theinvestigator as mild in intensity, and none was serious.
In the multiple ascending dose study, the rates of AEswith vorapaxar were similar to those observed with placebo(Table 4). Twenty-two (92%) subjects in Groups 1–3 andten (83%) subjects in Group 4 had treatment-emergent AEsduring the study, compared with 12 (100%) subjectsreceiving placebo. Most treatment-emergent AEs were
Table 3 Pharmacokinetic parameters of vorapaxar following single (Day 1) and multiple ascending (Day 28) oral doses (multiple-dose study)
Dose Cmax (ng/mL) Tmax (h) t½ (h) AUC(0–24 h) (ng h/mL) R
Mean CV (%) Median Range Mean CV (%) Mean CV (%) Mean CV (%)
Day 1
1 mg (n=8) 9.8 31 1.0 0.5–4.0 n.a. n.a. 59 24 n.a. n.a.
3 mg (n=8) 38.2 25 1.0 0.5–1.0 n.a. n.a. 225 15 n.a. n.a.
5 mg (n=8) 56.4 25 1.0 0.5–2.0 n.a. n.a. 315 21 n.a. n.a.
10 mg (n=6) 85.4 24 1.0 1.0–2.0 n.a. n.a. 592 14 n.a. n.a.
20 mg (n=6) 188.0 32 1.5 1.0–4.0 n.a. n.a. 1280 23 n.a. n.a.
Day 28
1 mg (n=7) 24.6 24 1.0 0.5–2.0 269 30 363 24 6.4 18
3 mg (n=8) 65.8 37 1.0 0.5–2.0 173 35 1040 34 4.7 33
5 mg (n=8) 131.0 18 1.0 1.0–2.0 217 22 1910 17 6.2 20
n.a., Not applicable; R, accumulation index; t½, terminal-phase half-life; Tmax, time to maximum observed plasma concentration
Table 2 Pharmacokineticparameters of vorapaxar follow-ing administration of singleascending oral doses (single-dose study)
AUC, Area under the curve;Cmax, maximum observed plas-ma concentration; CV, coeffi-cient of variation; Tmax, time tomaximum observed plasmaconcentration
Dose Cmax (ng/mL) Tmax (h) AUC(0–72 h) (ng h/mL)
Mean CV (%) Median Range Mean CV (%)
0.25 mg (n=6) 2.2 8 1.0 0.5–1.5 34 28
1 mg (n=6) 9.3 18 1.3 0.5–1.5 142 20
5 mg (n=6) 51.4 17 0.8 0.5–1.5 740 11
10 mg (n=6) 99.2 19 1.5 1.0–2.0 1514 10
20 mg (n=5) 183.0 19 1.5 1.0–2.0 3346 7
40 mg (n=5) 376.0 28 1.5 0.5–1.5 6028 18
254 Eur J Clin Pharmacol (2012) 68:249–258

considered to be mild. Of the 14 moderate AEs and twosevere AEs that were reported by 12 subjects, all wereconsidered by the investigator unlikely to be treatmentrelated, with the exception of moderate transient nausea in
one subject, which was considered possibly related totreatment (vorapaxar 5 mg). Both severe AEs (braincontusion and pelvic fractures) occurred during thefollow-up period (Day 42) in one subject who previouslyreceived vorapaxar 5 mg once daily on Days 1–28. Thissubject was hospitalized for 6 days with multiple injuriesbecause of a road accident that resulted in skull, rib, andpelvic fractures and pulmonary and brain contusions. Thissubject fully recovered, without any sign of large hemato-ma, bleeding diathesis, or the need for transfusion of redblood cells, despite having >90% inhibition of TRAP-induced platelet aggregation at the time of his injuries.Hematoma at venipuncture sites and bruise/hematoma atother sites were reported at a higher rate in vorapaxar-treated subjects than in those treated with placebo,although all of these events were mild and transient,and none required intervention. Transient epistaxisconsidered possibly related to treatment was reportedin four subjects in Group 4. None of these eventsrequired an intervention.
No significant abnormality or change was observedin the laboratory tests, including liver function, vital
Vorapaxar10 mg/1 mg
Ivy
blee
ding
tim
e (s
)
0
20
40
60
80BaselineDay 7
Vorapaxar20 mg/1 mg
Fig. 3 Mean Ivy bleeding times (± SE) following the administrationof the loading/maintenance dose regimens of vorapaxar
Days0 7 14 21 28
Rel
ativ
e T
RA
P-in
duce
d (1
5 µM
) pl
atel
etag
greg
atio
n (%
) (s
odiu
m c
itrat
e an
ticoa
gula
nt)
0
10
20
30
40
50
60
70
80
90
100
110
Placebo1 mg3 mg5 mg
Hours
0 24 48 72 96 120 144
Rel
ativ
e T
RA
P-in
duce
d (1
5 µM
) pl
atel
etag
greg
atio
n (%
) (s
odiu
m c
itrat
e an
ticoa
gula
nt)
0
10
20
30
40
50
60
70
80
90
100
10 mg x 1d + 1 mg x 6d20 mg x 1d + 1 mg x 6d
Hours0 12 24 36 48 60 72
Rel
ativ
e T
RA
P-in
duce
d (1
5 µM
) pl
atel
etag
greg
atio
n (%
) (s
odiu
m c
itrat
e an
ticoa
gula
nt)
0
10
20
30
40
50
60
70
80
90
100
Placebo1 mg5 mg10 mg20 mg40 mg
Hours
b
c
a
d
0 12 24 36 48 60 72
Rel
ativ
e A
DP
-indu
ced
(10
µM)
plat
elet
aggr
egat
ion
(%)
(sod
ium
citr
ate
antic
oagu
lant
)
0102030405060708090
130
100110120
140
Placebo1 mg5 mg10 mg20 mg40 mg
Fig. 2 Mean relative percentage of ex vivo platelet aggregationinduced by thrombin receptor activating peptide (TRAP) 15 μM [±standard error (SE)] following the administration of single doses (a),multiple doses (b), and the loading/maintenance dose regimens (c) of
vorapaxar. Mean relative percentage of ex vivo platelet aggregationinduced by adenosine diphosphate (ADP) 10 μM (± SE) following theadministration of single doses of vorapaxar (d)
Eur J Clin Pharmacol (2012) 68:249–258 255

signs, ECGs (with specific evaluation for prolongationof the QT/QTc interval), and blood coagulation param-eters. Neither single nor multiple doses of vorapaxar northe combination of the loading and maintenance dosesaffected ACT (data not shown for single and multipledoses; data for combination of loading and maintenancedoses are shown in Fig. 4) or any other coagulationparameters evaluated (TT, PT, aPTT, and ECT; data notshown).
Discussion
The results of these randomized, double-blind, placebo-controlled, single- and multiple-dose Phase I studiesdemonstrated that the novel oral PAR-1 antagonist vora-
paxar provided rapid, potent, dose-dependent, and durableinhibition of TRAP-induced platelet aggregation. Single20-mg and 40-mg doses of vorapaxar completely inhibitedTRAP-induced platelet aggregation within 1 h, and thisdegree of inhibition was sustained for at least 72 h. Therecovery of platelet function following the single 20-mgand 40-mg doses was dose related and occurred graduallyover a 4- to 8-week period, an observation consistent withthe long pharmacokinetic half-life of vorapaxar. The timerequired to achieve complete platelet aggregation inhibitionand the time to recovery of platelet function with multipledoses of vorapaxar (1, 3, or 5 mg once daily for 28 days)were also dose-related. The 10- and 20-mg loading doses ofvorapaxar achieved complete inhibition of TRAP-inducedplatelet aggregation at 2 and 1 h, respectively, and thisdegree of inhibition was sustained with 1 mg once-dailydosing over a 7-day treatment period. Recovery of plateletfunction to at least 50% of baseline was observed 4 weeksafter treatment discontinuation. Notably, therapy withvorapaxar was not associated with inhibition of ADP-induced platelet aggregation, indicating that its effects onplatelets was due to selective inhibition of PAR-1. Inaddition, vorapaxar did not affect the levels of eithersoluble P-selectin or sCD40L, nor the expression ofmembrane-bound P-selectin, indicating that although itbinds to PAR-1, it is not a platelet activator and does notexhibit partial PAR-1 agonist activity.
These findings are similar to those previously reported ina Phase II trial in subjects undergoing nonurgent percuta-neous coronary intervention (the TRA-PCI study) [15].Among the loading doses of vorapaxar used in the TRA-PCI trial, the most rapid and most potent inhibitory effecton TRAP-induced platelet aggregation was most consis-tently achieved with the 40-mg dose [15]. The subsequentadministration of maintenance doses of vorapaxar 1.0 mg
Baseline Day 7 Day 10 Day 21
Vorapaxar10 mg/1 mg
Act
ivat
ed c
lotti
ng ti
me
(s)
0
40
20
80
60
120
140
100
160
Vorapaxar20 mg/1 mg
Fig. 4 Mean activated clotting time (± standard deviation) followingthe administration of the loading/maintenance dose regimens ofvorapaxar
Table 4 Pooled incidence oftreatment-emergent adverseevents in Groups 1–3 and Group4 in the multiple ascending dosestudy
Data are presented as the num-ber (n), with the percentage inparenthesis
Adverse event Number of patients (%)
Pooled placebo(n=12)
Pooled vorapaxar Groups1–3 (1, 3, 5 mg; n=24)
Pooled vorapaxarGroup 4 (10/1 mg and20/1 mg; n=12)
Any 12 (100) 22 (92) 10 (83)
Bruise 1 (8) 2 (8) 0
Dizziness 0 5 (21) 2 (17)
Epistaxis 0 0 4 (33)
Fatigue 3 (25) 3 (13) 3 (25)
Flatulence 5 (42) 5 (21) 1 (8)
Headache 6 (50) 6 (25) 3 (25)
Hematoma 1 (8) 7 (29) 2 (17)
Injection site extravasation 2 (17) 8 (33) 4 (33)
Loose stools 3 (25) 5 (21) 2 (17)
256 Eur J Clin Pharmacol (2012) 68:249–258

and 2.5 mg once daily in TRA-PCI sustained the completeinhibition of TRAP-induced platelet aggregation throughoutthe entire 60-day treatment period [15]. It is important to note,however, that a separate Phase I dose optimization study inhealthy subjects who received once-daily maintenance dosesof vorapaxar (without a loading dose) demonstrated that the2.5-mg once-daily maintenance dose consistently providedmore complete inhibition of TRAP-induced platelet aggre-gation than the 1.0-mg once-daily dose [19]. Although the2.5-mg maintenance dose was not tested in our study, thesefindings suggest that it may provide the targeted level ofinhibition of TRAP-induced platelet aggregation moreconsistently than a 1.0-mg once-daily maintenance dose,particularly in those patients who do not receive a priorloading dose. It may therefore represent an optimal mainte-nance dose for chronic outpatient treatment. Additionally,neither the loading nor the maintenance doses of vorapaxarevaluated in the TRA-PCI study interfered with plateletaggregation induced by agonists other than TRAP, includingADP, arachidonic acid, and collagen [20].
The exposure to vorapaxar was dose related in both thesingle- and multiple-dose studies, and the pharmacokineticprofile of vorapaxar showed a good concordance with thepharmacodynamic activity. Vorapaxar was rapidly absorbedand distributed, with attainment of the peak plasma levelswithin 60–90 min with all doses, whereas the terminalelimination was slow. The steady-state plasma concentra-tions in the multiple-dose study appeared to be attained byDay 21. Variability of exposure was dose related and low inboth studies. These results are similar to those reported in aseparate Phase I study, which evaluated single dosesranging from 5 to 40 mg and multiple doses ranging from0.5 to 2.5 mg [19]. These data, in addition to the findingsfrom Phase II safety studies, have been subjected to apharmacokinetic analysis, and a robust and predictive PK-PD model was developed (unpublished results).
The observation that treatment with vorapaxar does notincrease Ivy bleeding times is consistent with findings frompreviously reported preclinical [21] and clinical [15, 22]studies with vorapaxar, as well as with the results of otherpreclinical studies involving functional inactivation of theprincipal platelet receptor for thrombin [11, 12, 23]. Chintalaand colleagues have shown that vorapaxar does not increasebleeding time or surgical blood loss in cynomolgusmonkeys, either when used alone or in combination withaspirin plus clopidogrel, whereas aspirin plus clopidogrelsignificantly increased both the bleeding time and surgicalblood loss [21]. The two Phase II clinical trials that havebeen conducted with vorapaxar have demonstrated that therisk of thrombolysis in myocardial infarction (TIMI) majoror minor bleedings when this agent is added to the standardtherapy is similar to that seen with the standard therapy alone[15, 22]. The lack of increase in Ivy bleeding times with
vorapaxar is also concordant with the observation that thisagent has no significant effect on coagulation parameters,including ACT, TT, PT, aPTT, and ECT. It is also worthnoting the absence of any obvious bleeding or other safetyconcerns in a subject who had an accident approximately 2weeks after discontinuing 28-day treatment with vorapaxar 5mg once daily. This finding underscores the fact thatvorapaxar selectively inhibits the PAR-1 receptor on plate-lets, without interfering with the activity of thrombin that iscritical for normal coagulation.
The results from our studies demonstrate that the PAR-1antagonist vorapaxar, administered orally at single loadingdoses of up to 40 mg and at multiple maintenance doses ofup to 5 mg once daily, was safe and well tolerated in ourcohort of healthy subjects. There were no significantchanges in the results of laboratory tests, including liverand kidney function, vital signs, and ECGs, includingspecific changes in the QT/QTc interval. These findings aresimilar to the safety results reported in other Phase I [19]and Phase II studies of vorapaxar [15, 22].
From a clinical perspective, a single dose of vorapaxar 40mg provides complete inhibition of TRAP-induced plateletaggregation most rapidly and, therefore, is suitable foradministration in the acute setting (such as an acute coronarysyndrome), where the fast onset of action is of paramountimportance [15, 22]. On the basis of findings from our studyas well as other studies [15, 19, 22], a vorapaxar maintenancedose of 2.5 mg/day is appropriate in the chronic setting (e.g.,secondary prevention) in order to achieve potent and durableantithrombotic activity. Whereas the pharmacologic half-lifeof vorapaxar is long and the reversal of inhibition of TRAP-induced platelet aggregation occurs gradually, the rates ofmajor and minor bleeding with vorapaxar in Phase II trialshave been comparable to those of the controls [15, 22]. Thesefindings indicate that, in contrast to some of the oralantiplatelet agents currently available, vorapaxar may havea favorable safety profile despite extended antiplateletactivity, a finding that can be attributed to the observationthat the principal platelet receptor for thrombin may not becritical for vascular repair and hemostasis [23].
Acknowledgments The authors thank Joshua Barbach and SeanGregory for assisting with the electronic submission of the manuscriptand for providing editorial support. This assistance was funded bySchering-Plough Corporation (now Merck, Whitehouse Station, NJ,USA). The authors also wish to thank Alan Meehan of Merck foreditorial support.
Conflict of interest T Kosoglou, L Reyderman, RR Fales, R Keller,B Yang, and DL Cutler all declare that they are/were full-timeemployees of Schering-Plough Corporation (now Merck) at the timeof the study. RG Tiessen and AA van Vliet are full-time employees ofPRA International, and funding was provided to PRA Internationalfrom Schering-Plough Corporation (now Merck) to conduct eachstudy.
Eur J Clin Pharmacol (2012) 68:249–258 257

References
1. Davi G, Patrono C (2007) Platelet activation and atherothrombo-sis. N Engl J Med 357:2482–2494
2. Angiolillo DJ, Capodanno D, Goto S (2010) Platelet thrombinreceptor antagonism and atherothrombosis. Eur Heart J 31:17–28
3. Bhatt DL, Fox KA, Hacke W, Berger PB, Black HR, Boden WE,Cacoub P, Cohen EA, Creager MA, Easton JD, Flather MD,Haffner SM, Hamm CW, Hankey GJ, Johnston SC, Mak KH, MasJL, Montalescot G, Pearson TA, Steg PG, Steinhubl SR, WeberMA, Brennan DM, Fabry-Ribaudo L, Booth J, Topol EJ (2006)CHARISMA Investigators. Clopidogrel and aspirin versus aspirinalone for the prevention of atherothrombotic events. N Engl J Med354:1706–1717
4. Wiviott SD, Braunwald E, McCabe CH, Montalescot G, RuzylloW, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA,Riesmeyer J, Weerakkody G, Gibson CM, Antman EM (2007)TRITON-TIMI 38 investigators. Prasugrel versus clopidogrel inpatients with acute coronary syndromes. N Engl J Med 357:2001–2015
5. Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK(2001) Clopidogrel in Unstable Angina to Prevent RecurrentEvents Trial Investigators. Effects of clopidogrel in addition toaspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 345:494–502
6. García Rodríguez LA, Hernández-Díaz S, de Abajo FJ (2001)Association between aspirin and upper gastrointestinal complica-tions: systematic review of epidemiologic studies. Br J ClinPharmacol 52:563–571
7. Rao SV, O'Grady K, Pieper KS, Granger CB, Newby LK, Van deWerf F, Mahaffey KW, Califf RM, Harrington RA (2005) Impactof bleeding severity on clinical outcomes among patients withacute coronary syndromes. Am J Cardiol 96:1200–1206
8. Brummel KE, Paradis SG, Butenas S, Mann KG (2002) Thrombinfunctions during tissue factor-induced blood coagulation. Blood100:148–152
9. Mann KG (2003) Thrombin formation. Chest 124[Suppl 3]:4S–10S
10. Chintala M, Shimizu K, Ogawa M, Yamaguchi H, Doi M, JensenP (2008) Basic and translational research on proteinase-activatedreceptors: antagonism of the proteinase-activated receptor 1 forthrombin, a novel approach to antiplatelet therapy for athero-thrombotic disease. J Pharmacol Sci 108:433–438
11. Derian CK, Damiano BP, Addo MF, Darrow AL, D'Andrea MR,Nedelman M, Zhang HC, Maryanoff BE, Andrade-Gordon P(2003) Blockade of the thrombin receptor protease-activatedreceptor-1 with a small-molecule antagonist prevents thrombusformation and vascular occlusion in nonhuman primates. JPharmacol Exp Ther 304:855–861
12. Kato Y, Kita Y, Hirasawa-Taniyama Y, Nishio M, Mihara K, Ito K,Yamanaka T, Seki J, Miyata S, Mutoh S (2003) Inhibition ofarterial thrombosis by a protease-activated receptor 1 antagonist,FR171113, in the guinea pig. Eur J Pharmacol 473:163–169
13. Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS,Boykow G, Hsieh Y, Palamanda J, Agans-Fantuzzi J, Kurowski S,Graziano M, Chintala M (2008) Discovery of a novel, orallyactive himbacine-based thrombin receptor antagonist (SCH530348) with potent antiplatelet activity. J Med Chem 51:3061–3064
14. Hildemann SK, Bode C (2009) Improving antiplatelet therapy foratherothrombotic disease: preclinical results with SCH 530348,the first oral thrombin receptor antagonist selective for PAR-1.Hämostaseologie 29:349–355
15. Becker RC, Moliterno DJ, Jennings LK, Pieper KS, Pei J,Niederman A, Ziada KM, Berman G, Strony J, Joseph D,Mahaffey KW, Van de Werf F, Veltri E, Harrington RA (2009)TRA-PCI Investigators. Safety and tolerability of SCH 530348 inpatients undergoing non-urgent percutaneous coronary interven-tion: a randomised, double-blind, placebo-controlled phase IIstudy. Lancet 373:919–928
16. Ozaki Y, Satoh K, Yatomi Y, Yamamoto T, Shirasawa Y, Kume S(1994) Detection of platelet aggregates with a particle countingmethod using light scattering. Anal Biochem 218:284–294
17. Mielke CH Jr, Kaneshiro MM, Maher IA, Weiner JM, Rapaport SI(1969) The standardized normal Ivy bleeding time and itsprolongation by aspirin. Blood 34:204–215
18. Kosoglou T, Reyderman L, Tiessen R, Fales RR, Cutler DL,Keller R, Yang B, Van Lier JJ, Wadham A, Van Vliet AA (2009)TRAP-induced platelet aggregation following single and multiplerising oral doses of SCH 530348, a novel thrombin receptorantagonist, in healthy volunteers. Clin Pharmacol Ther 85(S1):S21, Abstract PI-40
19. Kosoglou T, Reyderman L, Kasserra C, Young S, Pei J, MaxwellSE, Schiller J, Cutler DL (2008) Optimizing dose of the novelthrombin receptor antagonist SCH 530348 based on pharmaco-dynamics and pharmacokinetics in healthy subjects. Clin Pharma-col Ther 83(S1):S55, Abstract PII-42
20. Jennings LK, Earhart A, Becker RC, Reyderman L, Veltri E,Strony J, Harrington RA (2007) Thrombin receptor antagonist(TRA;SCH 530348) is a selective, potent inhibitor of PAR1activity with predictable pharmacokinetics (abstract). Circulation116[Suppl]:II-674, Abstract 3010
21. Chintala M, Vemulapalli S, Kurowski S, Sabin C, Reynolds D,Prevete K, Friedrichs G (2008) SCH 530348, a novel oralantiplatelet agent, demonstrated no bleeding risk alone or incombination with aspirin and clopidogrel in cynomolgus monkeys(abstract). Arterioscl Thromb Vasc Biol 28:e138–e139, AbstractP579
22. Goto S, Yamaguchi T, Ikeda Y, Kato K, Yamaguchi H, Jensen P(2010) Safety and exploratory efficacy of the novel thrombinreceptor (PAR-1) antagonist SCH530348 for non-ST-segmentelevation acute coronary syndrome. J Atheroscler Thromb17:156–164
23. Vandendries ER, Hamilton JR, Coughlin SR, Furie B, Furie BC(2007) Par4 is required for platelet thrombus propagation but notfibrin generation in a mouse model of thrombosis. Proc Natl AcadSci USA 104:288–292
258 Eur J Clin Pharmacol (2012) 68:249–258