oxidation of methanol on platinum, ruthenium and mixed pt–m metals (m=ru, sn): a theoretical study
TRANSCRIPT

Surface Science 463 (2000) 66–80www.elsevier.nl/locate/susc
Oxidation of methanol on platinum, ruthenium and mixed Pt–M metals (M=Ru, Sn): a theoretical study
Yasuyuki Ishikawa *, Meng-Sheng Liao, Carlos R. CabreraDepartment of Chemistry, University of Puerto Rico, P.O. Box 23346, San Juan, PR 00931-3346, USA
Received 31 January 2000; accepted for publication 8 May 2000
Abstract
A relativistic density-functional study of the dehydrogenation of CH3OH and H2O on pure platinum, ruthenium,and mixed Pt–M (M=Ru, Sn) metals is reported. Cluster models of Pt
nM10−n were used to simulate the metal
surfaces. Calculated adsorption energies of a series of intermediate species agree well with available experimentalvalues. Reaction energies for the elementary steps involved are determined and their activation energies estimated bythe analytic unity bond index–quadratic exponential potential (UBI-QEP) formula of Shustorovich and Sellers [Surf.Sci. Rep. 31 (1998) 1]. On pure platinum, the dehydrogenation of surface-adsorbed methanol, CH3OHs, is energeticallyfavorable, with a hydrogen atom first stripped off the carbon end. However, the dehydrogenation of H2Os to OHs,crucial in oxidative removal of poisoning CO, has the highest activation energy (E1) among all of the reaction steps.Dehydrogenation of H2Os on pure ruthenium is considerably more favorable than on pure platinum, but thecombination reaction, COs+OHs�COOHs, for the oxidative removal of COs by OHs possesses the highest E1. Onmixed Pt–M metals, the effect of M is to promote the formation of OHs and a combined effect of platinum and Matoms favors oxidative removal of COs and formation of COOHs. The relative importance of a ‘ligand effect’ versusa bifunctional mechanism for oxidative removal of COs is discussed. A ligand effect plays some role in the oxidationof CO on Pt–Ru, but it is unimportant on Pt–Sn. © 2000 Elsevier Science B.V. All rights reserved.
Keywords: Alcohols; Carbon monoxide; Chemisorption; Density functional calculations; Platinum; Ruthenium; Surface chemicalreaction; Tin; Water
1. Introduction the poisoning species formed during EOM.Methanol oxidation has been reported to involvethe adsorption of CH3OH and its successive dehy-The electro-oxidation of methanol (EOM ) has
been the subject of numerous studies because of drogenation, yielding linearly bonded CO [1,2]:its importance in fuel cell application [1–16 ]. So
CH3OH�CH3OHs (1)far, the best catalyst for adsorption and dehydroge-nation of methanol is platinum. However, the CH
xOHs�CH
x−1OHs+Hs (x=3, 2, 1) (2)adsorbed residues poison the platinum anode and
CHOs�COs+Hs (3)impede the catalytic performance for methanoloxidation. It is now generally accepted that CO is where (s) indicates adsorption on a surface. The
dissociative mechanism for EOM agrees with anumber of in situ infrared (IR) spectroscopy inves-* Corresponding author. Fax: +1 787 751 0625.
E-mail address: [email protected] (Y. Ishikawa) tigations performed on adsorbed methanol during
0039-6028/00/$ - see front matter © 2000 Elsevier Science B.V. All rights reserved.PII: S0039-6028 ( 00 ) 00600-2

67Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
electro-oxidation [2–9]. For further oxidation of superposition and electron delocalization molecu-lar orbital (ASED-MO) studies of alloys of plati-COs to CO2, a second oxygen atom is needed. This
oxygen atom can be supplied by splitting H2O on num, focusing on OHs as the oxidizing species. Ina recent study we performed an extensive density-the metal surfacefunctional study of the adsorption behavior of CO
H2Os�OHs+Hs . (4)on platinum and mixed Pt–M clusters (M=Ru,Sn, Ge) [27]. This study also examined the effectThe adsorbed COs is believed to react with an
adsorbed OH intermediate according to [10,11] of M alloying atoms in H2O dissociation. We haveshown that the presence of M atoms reduces the
COs+OHs�COOHs�CO2+Hs . (5)PtMCO bond strength by 0.3–0.5 eV and pro-motes H2O dissociation more strongly than doesThe poor performance of pure platinum for metha-
nol oxidation results from the strong adsorption pure platinum. The results [27] support the viewthat the promoting effect of alloying atoms mayshown by CO on the metal, and the formation of
OHs on platinum is claimed to be difficult (i.e., a involve both platinum modification and wateractivation [28].relatively high positive potential is needed to ‘acti-
vate’ H2Os). In order to overcome the blocking Besides the promoting effects of mixed Pt–Mclusters, a number of mechanistic details in EOMeffect of CO on platinum, platinum has been
alloyed with secondary metals, M. The alloying need to be investigated to identify cluster composi-tions that enhance dissociation of water and COmetals used most [3] have been ruthenium and tin.
Binary Pt–Ru and Pt–Sn catalysts have been oxidation. Such compositions are a prerequisite toimproving activity by the subnanostructuring ofshown to exhibit considerably higher methanol
electro-oxidation rates than pure platinum. catalysts for EOM. Three important questions areaddressed in this study. First, what is the relativeThe promoting effect of M may be explained
with a bifunctional mechanism for CO oxidation importance of a ligand effect versus a bifunctionalmechanism? Second, what are the possible reactive[12,13], in which H2O and OH are bound preferen-
tially to M surface atoms. On Pt–M, adsorbed OH intermediates and their adsorption energies inmethanol decomposition? The problem of interme-on the M sites facilitates oxidative removal of CO
residues on platinum diates in EOM has been addressed by in situ IRspectroscopy [2–9], but controversy exists as to
Pt–CO+M–OH�Pt–COOH+M. (6)the nature of the ‘reactive’ intermediates. Becauseof the similarity in stretching frequencies of manyThis mechanism successfully explains many aspects
of the catalysis for methanol and CO oxidation of the postulated intermediates, IR spectroscopymay not be able to give an unequivocal answer.and is supported by a number of experimental
electrochemical studies [11,14–16 ]. Last, which reaction step is the rate-determiningstep (RDS)? We examine theoretically severalAn alternative explanation for the enhanced
behavior is the so-called ligand or electronic effect, alternative RDSs that experimental studies havepostulated. The principal aims of the present studyin which the substituted metal M is assumed to
reduce the strength of adsorption of CO on plati- are to examine the energetics of methanol decom-position, PtMC bond length, OH formation andnum. Such an effect has been observed in the gas-
phase adsorption of CO on Pt(111)–Sn [17,18], CO oxidation on platinum, ruthenium, and mixedPt–M (M=Ru, Sn) metals, and attempt to providewhere the PtMCO bond strength was found to
weaken in the presence of tin. Some experimental a broader understanding of the catalysis of EOM.electrochemical results have also been presented insupport of this mechanism [19–22].
Quantum chemical methods have proved to be 2. Computational methoduseful tools with which to study chemisorptionand reactions on surfaces. Anderson et al. [23–26 ] The quantum chemical calculations were carried
out using the Amsterdam density functionalpioneered their application with a number of atom

68 Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
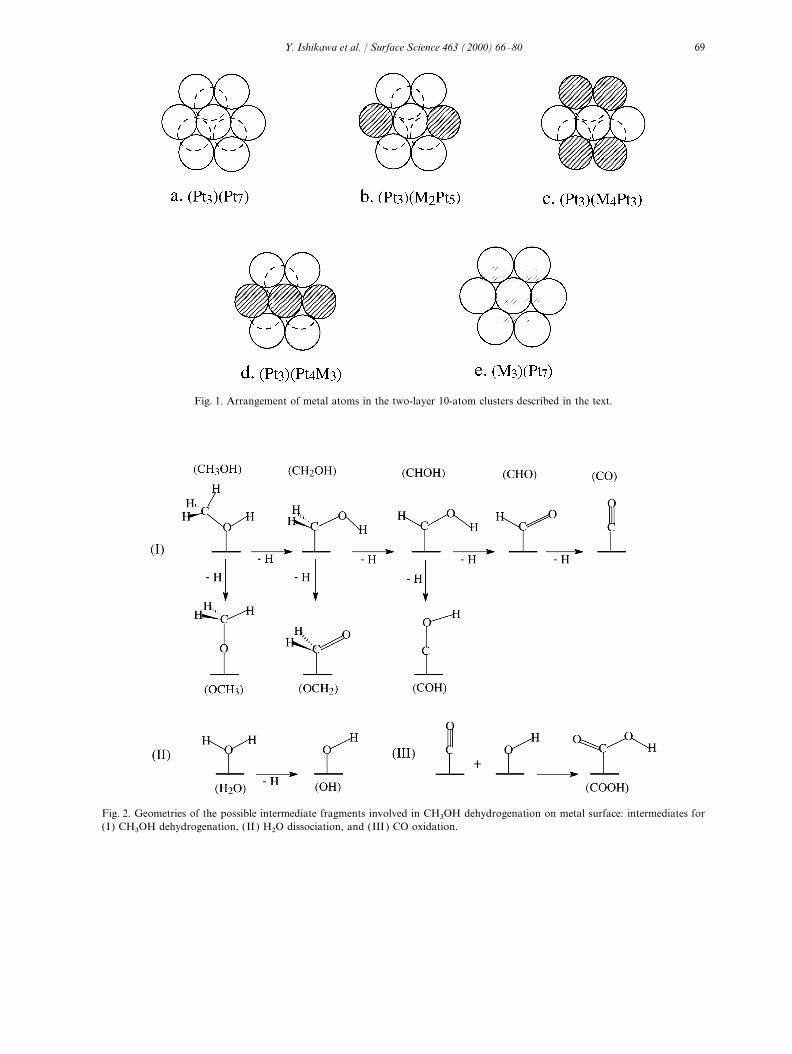
(ADF) program package (version 2.0.1) developed cluster models: (Pt3) (M2Pt5), (Pt3) (M4Pt3),(Pt3) (Pt4M3) and (M3) (Pt7). A cluster denotedby Baerends et al. [29,30] and the quasi-relativistic
extension of Ziegler et al. [31]. The frozen core (Am
) (BnA10−m−n) comprises n B atoms and
10−m−n A atoms in the first layer, m A atomsapproximation [29] was used to reduce the compu-tational cost. In order to obtain accurate results, in the second layer, and the adsorbate species
attached to the central A atom (for an on-top sitetriple-zeta STO basis sets were used for the valenceshells, single-zeta STO for the inner core. The model ). The geometrical structures of the different
clusters are shown in Fig. 1. For pure ruthenium,frozen core preserves the nodal structure of inner-shell orbitals. For platinum and ruthenium, the a (001) plane was selected as the adsorption surface
and was modeled by a (Ru3) (Ru7) cluster. Because(n−1)d and ns were considered valence shells, andone np polarization function was added. For tin, the cluster model of Ru(001) is two layers thick,
it is in fact equivalent to the cluster model of5s and 5p were the valence shell, and one 5dpolarization function was added. For carbon and Pt(111).
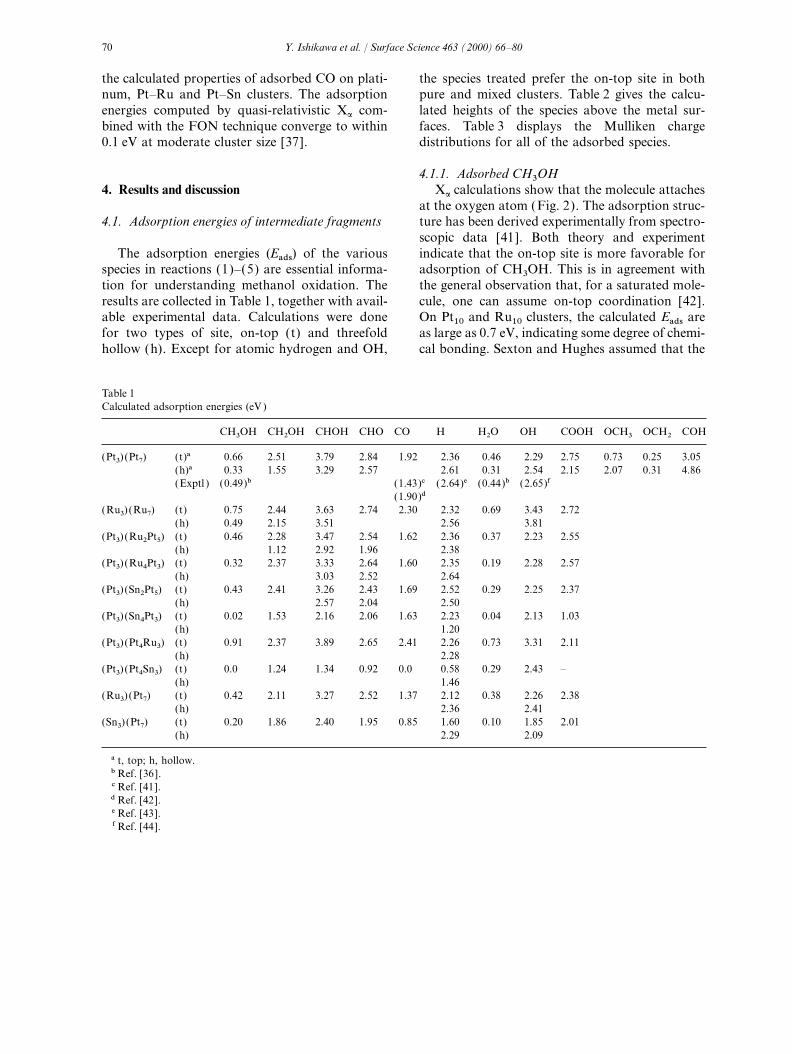
The geometries of the possible intermediateoxygen, 2s and 2p were the valence shell, and a 3dpolarization function was added. For hydrogen, fragments adsorbed on the metal surface are shown
in Fig. 2. The structures of the adsorbed speciesthe 1s shell was taken to be the valence shell, andone 2p polarization function was added. The shells on the cluster surface were fully optimized under
certain symmetries. The PtnM10−n cluster geome-of lower energy were treated as core and frozen.
The simple Xa
functional (a=0.7) was adopted tries were held fixed in the calculations and nearest-neighbor metal–metal distances were based onfor this study. The use of the X
afunctional in
quasi-relativistic density-functional calculations bulk crystal data; they are 2.77, 2.74, 2.83 and2.68 A for platinum, Pt–Ru [33], Pt–Sn [34] andhas been justified in our previous study [27] on
heavy metal–carbon monoxide complexes; the ruthenium, respectively. The alloying of platinumwith ruthenium decreases the PtMPt bond length.functional reproduces available experimental CO
adsorption energy, stretching force constants and The opposite is the case for M=Sn.In a large metal cluster, a number of low-lyingvibrational frequencies well. So far, a universally
‘best’ functional for accurate prediction of energies (unoccupied) orbitals lie energetically very close(within ~0.1 eV ). In our calculations the frac-of varieties of systems has not been found,
although some general conclusions have been tional occupation number (FON ) technique [35]was employed, whereby electrons are ‘smeared’ bydrawn based on the performance on a number of
light-atom systems [32]. an energy width of 0.15 eV over the orbitals aroundthe Fermi energy. The resulting total energy maybe viewed as an average over configurations lyingenergetically close to the ground state of the clus-3. Metal surface modelingter. The FON technique is useful in suppressingthe finite size effect of discrete level spacings inPt(111) was chosen as the surface for adsorp-
tion. The Pt(111) surface was simulated by a two- clusters, and in achieving convergence of the self-consistent field (SCF ). Although the finite sizelayer (Pt3) (Pt7) cluster model, containing seven
platinum atoms in the first layer and three in the effect in small clusters is partially compensated bythe FON technique, one must be aware of thesecond. In the case of mixed Pt–M metal (M=
Ru, Sn), the cluster model of Pt(111) was used limitations of the cluster approximation in model-ing bulk surfaces. The most notable problemwith some platinum atoms being substituted by M
atoms. Experimentally, EOM on Pt–M alloys with involves the cluster-size dependence of the calcu-lated properties of adsorbates. Local propertiesseveral M/Pt atom ratios have been examined
[3,14–16]. However, the microstructures of these such as bond lengths and vibrational frequenciesshow little size effect, while chemisorption energiesalloys are unknown. In our calculations,
PtnM10−n clusters with different n were used and are usually sensitive to cluster size [36–40]. We
have recently examined the effect of cluster size oncompared. We have considered the following
69Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
Fig. 1. Arrangement of metal atoms in the two-layer 10-atom clusters described in the text.
Fig. 2. Geometries of the possible intermediate fragments involved in CH3OH dehydrogenation on metal surface: intermediates for(I ) CH3OH dehydrogenation, (II ) H2O dissociation, and (III ) CO oxidation.
70 Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
the calculated properties of adsorbed CO on plati- the species treated prefer the on-top site in bothpure and mixed clusters. Table 2 gives the calcu-num, Pt–Ru and Pt–Sn clusters. The adsorption
energies computed by quasi-relativistic Xa
com- lated heights of the species above the metal sur-faces. Table 3 displays the Mulliken chargebined with the FON technique converge to within
0.1 eV at moderate cluster size [37]. distributions for all of the adsorbed species.
4.1.1. Adsorbed CH3OH
Xa
calculations show that the molecule attaches4. Results and discussionat the oxygen atom (Fig. 2). The adsorption struc-ture has been derived experimentally from spectro-4.1. Adsorption energies of intermediate fragmentsscopic data [41]. Both theory and experimentindicate that the on-top site is more favorable forThe adsorption energies (Eads) of the various
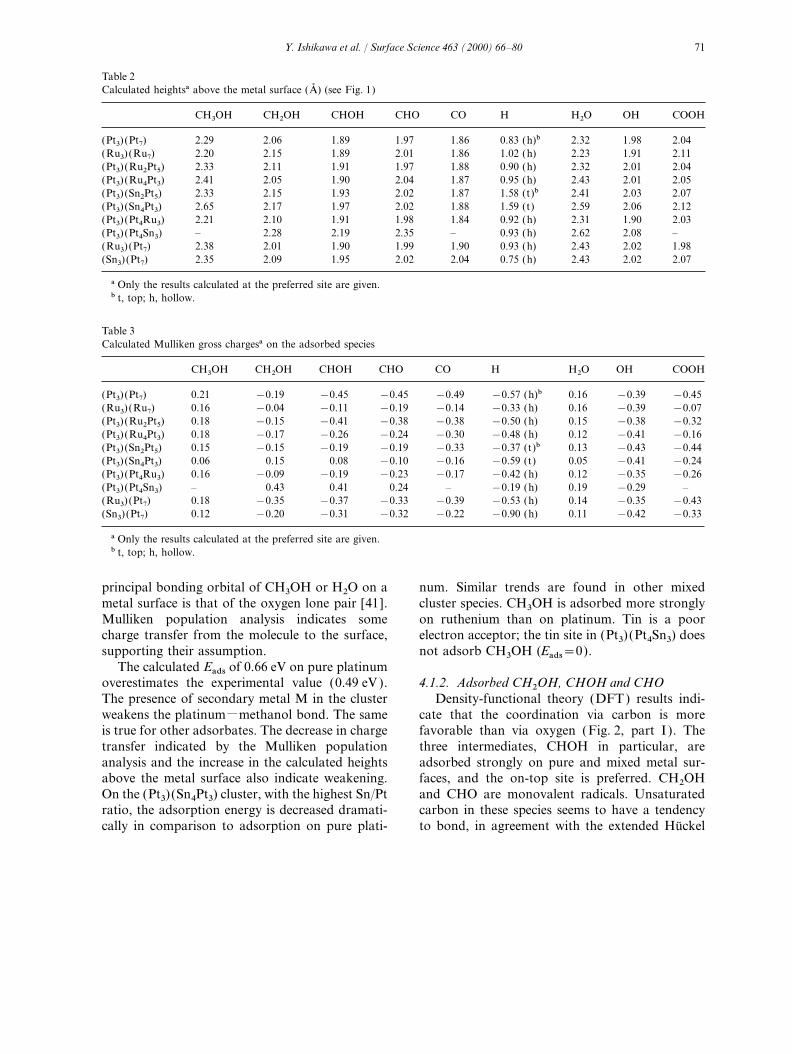
species in reactions (1)–(5) are essential informa- adsorption of CH3OH. This is in agreement withthe general observation that, for a saturated mole-tion for understanding methanol oxidation. The
results are collected in Table 1, together with avail- cule, one can assume on-top coordination [42].On Pt10 and Ru10 clusters, the calculated Eads areable experimental data. Calculations were done
for two types of site, on-top (t) and threefold as large as 0.7 eV, indicating some degree of chemi-cal bonding. Sexton and Hughes assumed that thehollow (h). Except for atomic hydrogen and OH,
Table 1Calculated adsorption energies (eV )
CH3OH CH2OH CHOH CHO CO H H2O OH COOH OCH3 OCH2 COH
(Pt3) (Pt7) (t)a 0.66 2.51 3.79 2.84 1.92 2.36 0.46 2.29 2.75 0.73 0.25 3.05(h)a 0.33 1.55 3.29 2.57 2.61 0.31 2.54 2.15 2.07 0.31 4.86(Exptl ) (0.49)b (1.43)c (2.64)e (0.44)b (2.65)f
(1.90)d(Ru3) (Ru7) (t) 0.75 2.44 3.63 2.74 2.30 2.32 0.69 3.43 2.72
(h) 0.49 2.15 3.51 2.56 3.81(Pt3) (Ru2Pt5) (t) 0.46 2.28 3.47 2.54 1.62 2.36 0.37 2.23 2.55
(h) 1.12 2.92 1.96 2.38(Pt3) (Ru4Pt3) (t) 0.32 2.37 3.33 2.64 1.60 2.35 0.19 2.28 2.57
(h) 3.03 2.52 2.64(Pt3) (Sn2Pt5) (t) 0.43 2.41 3.26 2.43 1.69 2.52 0.29 2.25 2.37
(h) 2.57 2.04 2.50(Pt3) (Sn4Pt3) (t) 0.02 1.53 2.16 2.06 1.63 2.23 0.04 2.13 1.03
(h) 1.20(Pt3) (Pt4Ru3) (t) 0.91 2.37 3.89 2.65 2.41 2.26 0.73 3.31 2.11
(h) 2.28(Pt3) (Pt4Sn3) (t) 0.0 1.24 1.34 0.92 0.0 0.58 0.29 2.43 –
(h) 1.46(Ru3) (Pt7) (t) 0.42 2.11 3.27 2.52 1.37 2.12 0.38 2.26 2.38
(h) 2.36 2.41(Sn3) (Pt7) (t) 0.20 1.86 2.40 1.95 0.85 1.60 0.10 1.85 2.01
(h) 2.29 2.09
a t, top; h, hollow.b Ref. [36 ].c Ref. [41].d Ref. [42].e Ref. [43].f Ref. [44].
71Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
Table 2Calculated heightsa above the metal surface (A) (see Fig. 1)
CH3OH CH2OH CHOH CHO CO H H2O OH COOH
(Pt3) (Pt7) 2.29 2.06 1.89 1.97 1.86 0.83 (h)b 2.32 1.98 2.04(Ru3) (Ru7) 2.20 2.15 1.89 2.01 1.86 1.02 (h) 2.23 1.91 2.11(Pt3) (Ru2Pt5) 2.33 2.11 1.91 1.97 1.88 0.90 (h) 2.32 2.01 2.04(Pt3) (Ru4Pt3) 2.41 2.05 1.90 2.04 1.87 0.95 (h) 2.43 2.01 2.05(Pt3) (Sn2Pt5) 2.33 2.15 1.93 2.02 1.87 1.58 (t)b 2.41 2.03 2.07(Pt3) (Sn4Pt3) 2.65 2.17 1.97 2.02 1.88 1.59 (t) 2.59 2.06 2.12(Pt3) (Pt4Ru3) 2.21 2.10 1.91 1.98 1.84 0.92 (h) 2.31 1.90 2.03(Pt3) (Pt4Sn3) – 2.28 2.19 2.35 – 0.93 (h) 2.62 2.08 –(Ru3) (Pt7) 2.38 2.01 1.90 1.99 1.90 0.93 (h) 2.43 2.02 1.98(Sn3) (Pt7) 2.35 2.09 1.95 2.02 2.04 0.75 (h) 2.43 2.02 2.07
a Only the results calculated at the preferred site are given.b t, top; h, hollow.
Table 3Calculated Mulliken gross chargesa on the adsorbed species
CH3OH CH2OH CHOH CHO CO H H2O OH COOH
(Pt3) (Pt7) 0.21 −0.19 −0.45 −0.45 −0.49 −0.57 (h)b 0.16 −0.39 −0.45(Ru3) (Ru7) 0.16 −0.04 −0.11 −0.19 −0.14 −0.33 (h) 0.16 −0.39 −0.07(Pt3) (Ru2Pt5) 0.18 −0.15 −0.41 −0.38 −0.38 −0.50 (h) 0.15 −0.38 −0.32(Pt3) (Ru4Pt3) 0.18 −0.17 −0.26 −0.24 −0.30 −0.48 (h) 0.12 −0.41 −0.16(Pt3) (Sn2Pt5) 0.15 −0.15 −0.19 −0.19 −0.33 −0.37 (t)b 0.13 −0.43 −0.44(Pt3) (Sn4Pt3) 0.06 0.15 0.08 −0.10 −0.16 −0.59 (t) 0.05 −0.41 −0.24(Pt3) (Pt4Ru3) 0.16 −0.09 −0.19 −0.23 −0.17 −0.42 (h) 0.12 −0.35 −0.26(Pt3) (Pt4Sn3) – 0.43 0.41 0.24 – −0.19 (h) 0.19 −0.29 –(Ru3) (Pt7) 0.18 −0.35 −0.37 −0.33 −0.39 −0.53 (h) 0.14 −0.35 −0.43(Sn3) (Pt7) 0.12 −0.20 −0.31 −0.32 −0.22 −0.90 (h) 0.11 −0.42 −0.33
a Only the results calculated at the preferred site are given.b t, top; h, hollow.
principal bonding orbital of CH3OH or H2O on a num. Similar trends are found in other mixedcluster species. CH3OH is adsorbed more stronglymetal surface is that of the oxygen lone pair [41].
Mulliken population analysis indicates some on ruthenium than on platinum. Tin is a poorelectron acceptor; the tin site in (Pt3) (Pt4Sn3) doescharge transfer from the molecule to the surface,
supporting their assumption. not adsorb CH3OH (Eads=0).The calculated Eads of 0.66 eV on pure platinum
overestimates the experimental value (0.49 eV ). 4.1.2. Adsorbed CH2OH, CHOH and CHO
Density-functional theory (DFT) results indi-The presence of secondary metal M in the clusterweakens the platinumMmethanol bond. The same cate that the coordination via carbon is more
favorable than via oxygen (Fig. 2, part I ). Theis true for other adsorbates. The decrease in chargetransfer indicated by the Mulliken population three intermediates, CHOH in particular, are
adsorbed strongly on pure and mixed metal sur-analysis and the increase in the calculated heightsabove the metal surface also indicate weakening. faces, and the on-top site is preferred. CH2OH
and CHO are monovalent radicals. UnsaturatedOn the (Pt3)(Sn4Pt3) cluster, with the highest Sn/Ptratio, the adsorption energy is decreased dramati- carbon in these species seems to have a tendency
to bond, in agreement with the extended Huckelcally in comparison to adsorption on pure plati-

72 Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
band calculations of Hoffmann et al. [43]. Because (Pt3) (M2Pt5) and (Pt3) (M4Pt3) clusters, Eads islower by ~0.3 eV than that on pure platinum.CHOH is divalent, it might be expected to prefer
a bridging site to either of those considered here. Such an effect has been observed in gas-phaseexperiments on CO adsorption on a Pt(111)–SnThe facts that Eads differs little from on-top to
hollow, and that on-top is the preferred position, alloy [17,18], where the PtMC bond strength wasfound to weaken by 5 kcal mol−1 (0.22 eV ) in thelead to the conclusion that any preference for a
bridge site is small. No experimental Eads data are presence of tin. On an (M3) (Pt7) cluster, whereseven platinum atoms are in the first layer and allavailable for these species. The calculated adsorp-
tion energies on pure platinum and ruthenium are of the M atoms are in the second, the CO adsorp-tion energy is significantly reduced and PtMCsimilar. The adsorption energies at a tin site are
comparatively small. bond length increased, especially for M=Sn. It isshown that the adsorption energy of CO on pureruthenium is higher than on pure platinum.4.1.3. Adsorbed OCH
3, OCH
2and COH
Successive dehydrogenation of CH3OH may Anderson and Grantscharova found a similartrend in ASED-MO calculations [24]. For Sn–COalso produce OCH3, OCH2 and COH (see Fig. 2,
part I ). We have evaluated adsorption energies of interaction on a (Pt3) (Pt4Sn3) cluster, the calcula-tion does not yield bound CO, in agreement withthese species only on pure platinum. An examina-
tion of the energetics in Section 4.2 will show that experimental evidence that CO does not chemisorbon tin in Pt(111)–Sn surface alloys [18].the formation of these species is less favorable
than the formation of CH2OH, CHOH and CHO.OCH2 is a saturated molecule and can bond only 4.1.5. Adsorbed hydrogen
Atomic hydrogen favors the hollow site on pureweakly to the metal surface; the calculated Eads(hollow site) is 0.31 eV. A hollow site is clearly platinum, ruthenium, and most Pt–M clusters.
The (Pt3) (Sn4Pt3) cluster is an exception, wherepreferred for adsorption of OCH3 and COH. Thedifferences in Eads between the on-top and hollow the on-top site is preferred. On (Pt3) (Ru2Pt5),
(Pt3) (Sn2Pt5) and (Pt3) (Pt5Ru3), there is but asites for the two species are large. COH is atrivalent radical possessing a large adsorption small difference (0.02 eV ) in Eads between on-top
and hollow sites. The hydrogen-atom adsorptionenergy (4.86 eV ).energy at substituted ruthenium is smaller than atplatinum. The calculated Eads of 2.61 eV on Pt104.1.4. Adsorbed CO
It is well established that, on pure platinum and is in excellent agreement with experimental valueof 2.64 eV for atomic hydrogen on Pt(111) [46 ].alloyed Pt–M electrodes, CO is adsorbed mainly
at on-top sites at low coverage [2,16 ]. In an earlylow-energy electron diffraction (LEED) study [44], 4.1.6. Adsorbed H
2O and OH
Like CH3OH, H2O is bound to the surface viathe adsorption energy of CO atop Pt(111) wasmeasured to be 1.43 eV. More recently, using a the oxygen atom and prefers the on-top site.
H2O is more strongly adsorbed on ruthenium thanmicro-calorimetric technique, King et al. [45] mea-sured a value of 1.90±0.07 eV for the low-coverage on platinum, while tin adsorbs H2O more weakly
than platinum. The OH radical is stronglyadsorption energy of CO on Pt(110). Althoughthe two values refer to different metal surfaces, the adsorbed on the metal surface, whether platinum,
ruthenium or tin. The MMOH bond lengths areadsorption energies of CO and other small mole-cules do not vary much with crystal facial struc- smaller than those of MMOH2. On pure platinum
and ruthenium clusters, OH favors the hollow site.ture. The calculated Eads of 1.92 eV for CO onPt10 overestimates the experimental values. However, there is no significant difference in Eads
between the on-top and hollow sitesIn all mixed Pt–M clusters, the secondary Matoms are shown to weaken the PtMC bond. This (Dhol−top~0.3 eV ). Because H2O is adsorbed at an
on-top site, it is reasonable to assume that whenindicates that a ‘ligand effect’ exists in electro-oxidation of CO on Pt–M alloy electrodes. On an OMH bond in H2Os breaks, OH will remain at

73Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
the original site to react with COs [reaction (5)]. to determine De,s and the energy of combination,Ce,s.Therefore we consider only the on-top site for
In addition to reaction energies, activation bar-OHs. The OH adsorption energy, Eads(OH), toriers are also important in interpreting reactionruthenium is considerably larger, by more thanprocesses. It is, of course, difficult to locate trans-1 eV, than that to platinum. The Eads(OH) to theition states for surface reactions. In order to cir-secondary tin is slightly larger than that to plati-cumvent the problem, Shustorovich [42] andnum. The calculated adsorption energies on pureShustorovich and Sellers [48] have developed theplatinum agree well with experiment [41,47] forbond order conservation–Morse potentialthese adsorbates.(BOC-MP) and unity bond index–quadratic expo-nential potential (UBI-QEP) methods, respec-4.1.7. Adsorbed COOHtively, to predict accurately activation energies forReaction of COs with OHs results in formationdissociations or combinations of adsorbates onof COOH. In situ IR spectroscopy [3] has demon-metal surfaces. For the dissociation reaction ofstrated the presence of COOH, suggesting that theCH
xOHs or H2O, the activation energy is given byreaction involves a COOH intermediate. COOH is
a monovalent species that binds more stably at anE1=
1
2 ADe,g+E2EH
E2+EH
+E1−E
2−EHBon-top site. The surface–carbon distance is longer
for COOHs than for COs in each cluster, whereas,on most clusters, the adsorption energy of COOH
=1
2 A E2EH
E2+EH
+De,sB, (9)is larger than that of CO.
where the energy terms are shown in reaction4.2. The methanol oxidation mechanism
scheme (7). The UBI-QEP method [48] employsexperimental atomic adsorption energies to evalu-
The EOM on a metal surface is known to ate the energy terms of Eq. (9). The adsorptionproceed via a three-step mechanism, namely and activation energies evaluated with the UBI-CH3OH dissociation [reactions (2) and (3)], OH QEP method are typically accurate to withinformation [reaction (4)] and CO oxidative removal 1–3 kcal mol−1 [48] if accurate experimental[reaction (5)]. The calculated adsorption energies atomic adsortion energies are used. In the presentgiven in Table 1 together with the calculated CMH study, the X
amethod is employed to calculate the
or OMH bond energy in gas-phase CHxOHg can energy terms in Eq. (9). For combination reaction
be used to determine the dissociation energy, De,s, (5), the appropriate equation isof CH
xOHs. The scheme is
E1=1
2 A ECOEOHECO+EOH
+Ce,sB. (10)CH
xOHs CADe,s CH
x−1OHs + Hs(−E
1(−E
2(−EH
CHxOHg CA
De,g
CHx−1OHg + Hg
(7) Here, ECO and EOH are, respectively, the adsorptionenergies of CO and OH computed by X
a. Ce,s is
the combination energy of COs+OHs�COOHs.According to Eqs. (9) and (10), the formulae
Thus reveal a correlation between the activation energy(E1) and dissociation energy (De,s) or combinationDe,s=De,g+E
1−E
2−EH , (8)
energy (Ce,s) for the reaction. The Xa-calculated
where E1, E2 and EH are, respectively, the adsorp- De,s or Ce,s, and E1 on the various clusters aretion energies of CH
xOH, CH
x−1OH and atomic given in Tables 4–6.hydrogen. The term De,g represents the dissociationenergy of CH
xOH in the gas phase. For H2Os 4.2.1. CH
3OH oxidation on pure platinum
dissociation [reaction (4)] and COs+OHs combi- Dehydrogenations of CHxOH to CH
x−1OH arehighly endothermic in the gas phase (Table 4). Thenation [reaction (5)], similar schemes can be used
74 Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
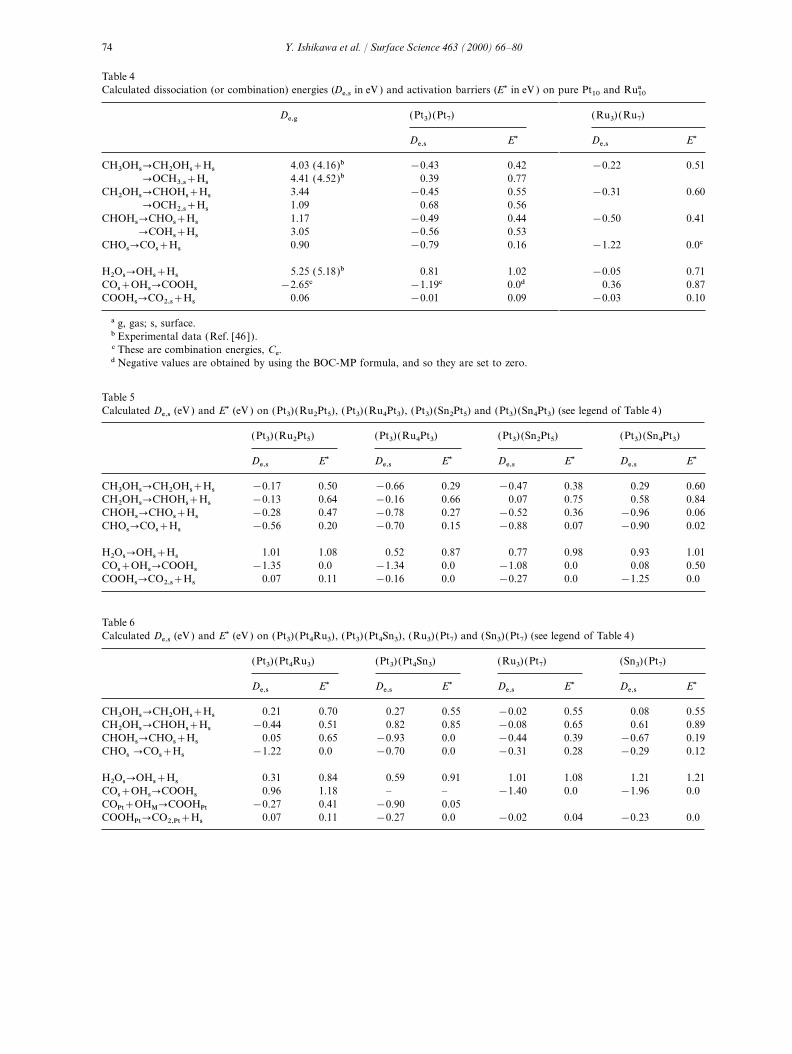
Table 4Calculated dissociation (or combination) energies (De,s in eV ) and activation barriers (E1 in eV ) on pure Pt10 and Ru10a
De,g (Pt3) (Pt7) (Ru3) (Ru7)
De,s E1 De,s E1
CH3OHs�CH2OHs+Hs 4.03 (4.16)b −0.43 0.42 −0.22 0.51�OCH3,s+Hs 4.41 (4.52)b 0.39 0.77
CH2OHs�CHOHs+Hs 3.44 −0.45 0.55 −0.31 0.60�OCH2,s+Hs 1.09 0.68 0.56
CHOHs�CHOs+Hs 1.17 −0.49 0.44 −0.50 0.41�COHs+Hs 3.05 −0.56 0.53
CHOs�COs+Hs 0.90 −0.79 0.16 −1.22 0.0c
H2Os�OHs+Hs 5.25 (5.18)b 0.81 1.02 −0.05 0.71COs+OHs�COOHs −2.65c −1.19c 0.0d 0.36 0.87COOHs�CO2,s+Hs 0.06 −0.01 0.09 −0.03 0.10
a g, gas; s, surface.b Experimental data (Ref. [46 ]).c These are combination energies, Ce.d Negative values are obtained by using the BOC-MP formula, and so they are set to zero.
Table 5Calculated De,s (eV) and E1 (eV ) on (Pt3)(Ru2Pt5), (Pt3) (Ru4Pt3), (Pt3) (Sn2Pt5) and (Pt3) (Sn4Pt3) (see legend of Table 4)
(Pt3) (Ru2Pt5) (Pt3)(Ru4Pt3) (Pt3) (Sn2Pt5) (Pt3) (Sn4Pt3)
De,s E1 De,s E1 De,s E1 De,s E1
CH3OHs�CH2OHs+Hs −0.17 0.50 −0.66 0.29 −0.47 0.38 0.29 0.60CH2OHs�CHOHs+Hs −0.13 0.64 −0.16 0.66 0.07 0.75 0.58 0.84CHOHs�CHOs+Hs −0.28 0.47 −0.78 0.27 −0.52 0.36 −0.96 0.06CHOs�COs+Hs −0.56 0.20 −0.70 0.15 −0.88 0.07 −0.90 0.02
H2Os�OHs+Hs 1.01 1.08 0.52 0.87 0.77 0.98 0.93 1.01COs+OHs�COOHs −1.35 0.0 −1.34 0.0 −1.08 0.0 0.08 0.50COOHs�CO2,s+Hs 0.07 0.11 −0.16 0.0 −0.27 0.0 −1.25 0.0
Table 6Calculated De,s (eV) and E1 (eV ) on (Pt3)(Pt4Ru3), (Pt3) (Pt4Sn3), (Ru3) (Pt7) and (Sn3)(Pt7) (see legend of Table 4)
(Pt3) (Pt4Ru3) (Pt3) (Pt4Sn3) (Ru3) (Pt7) (Sn3) (Pt7)
De,s E1 De,s E1 De,s E1 De,s E1
CH3OHs�CH2OHs+Hs 0.21 0.70 0.27 0.55 −0.02 0.55 0.08 0.55CH2OHs�CHOHs+Hs −0.44 0.51 0.82 0.85 −0.08 0.65 0.61 0.89CHOHs�CHOs+Hs 0.05 0.65 −0.93 0.0 −0.44 0.39 −0.67 0.19CHOs �COs+Hs −1.22 0.0 −0.70 0.0 −0.31 0.28 −0.29 0.12
H2Os�OHs+Hs 0.31 0.84 0.59 0.91 1.01 1.08 1.21 1.21COs+OHs�COOHs 0.96 1.18 – – −1.40 0.0 −1.96 0.0COPt+OHM�COOHPt −0.27 0.41 −0.90 0.05COOHPt�CO2,Pt+Hs 0.07 0.11 −0.27 0.0 −0.02 0.04 −0.23 0.0

75Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
calculated dehydrogenation energies of CH3OH, intermediate fragments discussed in the presentstudy. For the reaction CH3OHs�CH2OHs+Hsfor both CH3OHg�CH2OHg+Hg and CH3OHg�
OCH3,g+Hg, and of H2O in the gas phase are in on Pt(111), the activation energy computed withthe BOC-MP method is 14.4 kcal mol−1 (0.63 eV ),good agreement with the available experimental
data [49]. On a metal surface [3], there is a 0.2 eV higher than our Xa-calculated value.
Table 4 shows that the dehydrogenation stepsconsiderable reduction in the dehydrogenationenergies De,s due to the formation of strong of CH3OH on pure platinum are all exothermic
processes with low activation energies. The dehy-metalMadsorbate bonds.On pure platinum, two alternative elementary drogenation of CHOs is exothermic, and CHOs
can decompose rapdily (E1=0) to form COs andreaction steps that may be involved in methanoldissociation were examined. The dehydrogenation Hs in agreement with earlier BOC-MP calculations
[50]. Previous studies have suggested that adsorbedof CH3OHs may produce CH2OHs or OCH3,s. Thereaction to yield OCH3,s is endothermic, while that CHO [6 ] and COH [52] are poisoning species.
Others have thought it more likely that they arefor producing CH2OHs is exothermic. A lowerdissociation energy in general corresponds to a short-lived reaction intermediates [2]. Our calcula-
tions support the latter view.lower activation energy according to Eq. (9)(E1=0.42 eV and 0.77 eV, respectively, for forma- The dissociation reaction of H2Os is endother-
mic by 0.8 eV, and its activation energy is estimatedtion of CH2OHs and OCH3,s). Therefore, the for-mation of CH2OHs should be the preferred to be 1 eV. The combination reaction,
COs+OHs�COOHs, should be rapid; it is exo-process. Similarly, the dehydrogenation ofCH2OHs may produce either CHOHs or OCH2,s, thermic by −1.2 eV and there is no activation
barrier. COOHs should decompose readily tothe former being preferred. It has been shown thatin the gas phase OCH2 is more stable than CHOH. CO2,s and Hs. No energy is required to break the
COOMH bond on the metal surface; the estimatedHowever, the calculated adsorption energy ofCHOHs is lower than that of OCH2,s (Table 4). dissociation and activation energies are, respec-
tively, −0.01 eV and 0.09 eV. Among the dehydro-In situ Fourier transform infrared (FTIR) studieshave identified CHOHs as an intermediate [9]. genation processes, the X
acalculations clearly
indicate that the H2Os dissociation to generateDehydrogenation of CHOHs can produceCHOs and COHs. The formation of COHs is OHs is the rate-determining step (RDS) for metha-
nol oxidation on pure platinum. The Xa
studyslightly more exothermic (by ~0.1 eV ) than thatof CHOs, but the formation of COHs requires a supports the conjecture made earlier by Anderson
and Grantscharova [24].slightly higher activation energy (by ~0.1 eV ).Experimentally, controversy exists about thenature of the adsorbates. In situ electrochemically 4.2.2. CH
3OH oxidation on pure ruthenium
In the electrochemical environment, pure ruthe-modulated infrared (EMIR) investigations claimevidence for CHOs [2,6,7], while the adsorbate is nium is found to be inactive for methanol dissoci-
ation at low voltages [5,22]. This situation issuggested to be COHs according to in situ FTIRstudies [4,5,8]. The present study indicates that ascribed to the fact that adsorbed H2O and OH
block the ruthenium surface and inhibit thethe formations of both CHOs and COHs areenergetically feasible, and likely. The preferred adsorption of CH3OH. In Section 4.1.6 it was
shown that ruthenium adsorbs H2O and OH morespecies depends on the experimental conditions.Shustorovich and Bell [50] employed the strongly than does platinum (see Table 1). There
have been a few experimental studies on theBOC-MP method to examine the energetics of theelementary steps in methanol synthesis by hydro- electro-oxidation of CO on pure ruthenium
electrodes [5,15,22,53], where CO and COOH weregenation of CO on copper and palladium. Sellersand co-workers [51] examined the elementary steps found to form adsorbates on the ruthenium sur-
face. In ultrahigh vacuum (UHV ), methanol isin methanol synthesis from CH4 on platinum.These studies have identified some of the same found to decompose on Ru(001), CH3OHs�

76 Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
COs+4Hs, at temperatures of 220–280 K [54]. In the combination reaction is the RDS for CH3OHoxidation on pure ruthenium.this subsection we examine the energetics of
CH3OH oxidation on pure ruthenium.The dehydrogenation steps of CH
xOH are exo- 4.2.3. CH
3OH oxidation at platinum on mixed Pt–
Ru surfacesthermic on both platinum and ruthenium and haverelatively low activation energies (Table 4). Two clusters, (Pt3) (Ru2Pt5) and (Pt3)-
(Ru4Pt3), were used to simulate the platinum sitePlatinum and ruthenium under UHV behave simi-larly for CH3OH decomposition [54,55], and the in mixed Pt–Ru clusters. The dehydrogenation
energies of CHxOH on (Pt3) (Ru2Pt5) are slightlycalculated results are consistent with the experi-
mental observations. The greatest differences occur less exothermic than on pure platinum. H2Os disso-ciation on (Pt3) (Ru2Pt5) is also slightly more endo-in the first (methanol dissociation) and last
[CHOs�COs+Hs, reaction (3)] steps. thermic and has a slightly higher activation energythan on (Pt3) (Pt7). On (Pt3) (Ru4Pt3), the dehydro-The H2Os dissociation (H2Os�OHs+Hs) on
ruthenium is slightly exothermic (De,s= genation energies of CH3OH and CHOH becomesignificantly more exothermic than they are on−0.05 eV ), in contrast with dissociation on pure
platinum, which is quite endothermic (Pt3) (Ru2Pt5). De,s and E1 for H2Os dissociationon (Pt3) (Ru4Pt3) are also lower, by 0.5 eV and(De,s=0.81 eV ). E1 is also notably smaller on
ruthenium than on platinum. Thus H2O dissoci- 0.2 eV, respectively, than on (Pt3) (Ru2Pt5). Ingeneral then, the mixed Pt–Ru cluster with theation on ruthenium is more favorable than on
platinum. The COs+OHs combination reaction on higher ruthenium percentage exhibits the greateractivity in EOM. Experimental studies have indi-ruthenium is endothermic and requires a high
activation energy (0.87 eV ); again this is in con- cated that the optimum Pt/Ru composition liesbetween 10 and 50 at% [14]. Fig. 3 compares thetrast to the situation on platinum, where the
activation energy is zero. E1 for this reaction is the De,s and E1 for CHxOH dehydrogenation on pure
platinum with those on (Pt3) (Ru4Pt3). The dehy-highest among the dehydrogenation steps. Thus
Fig. 3. Potential energy diagrams for the CH3OH dehydrogenation, water dissociation and COs+OHs combination reactions on pureplatinum and mixed Pt–Ru surfaces.

77Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
drogenation of CHxOH on (Pt3)(Ru4Pt3) is more ruthenium atoms in the surface provide nucleation
sites only for OHs formation. Like pure Ru10,favorable than on pure platinum.substituted ruthenium shows smaller De,s and E1
for H2O dissociation than does platinum. The4.2.4. CH3OH oxidation at platinum on Pt–Sn
clusters calculated De,s of H2O on (Pt3) (Pt4Ru3) is 0.31 eV,0.5 eV less endothermic than on pure platinumWe have also examined (Pt3) (Sn2Pt5) and
(Pt3) (Sn4Pt3) cluster models. The dehydrogenation (Fig. 3). The activation energy for H2O dissoci-ation on (Pt3) (Pt4Ru3) is also smaller by 0.18 eVof CH2OH on (Pt3) (Sn2Pt5) has an activation
energy of 0.75 eV, 0.2 eV higher than that, but all than on pure platinum, in agreement with earlierwork of Anderson et al. [23–26 ]. Assuming thatother calculated De.s and E1 are comparable to
those, on pure platinum. On (Pt3) (Sn4Pt3), the the pre-exponential factors A in the Arrheniusequation, k=A exp(−E1/RT ), are approximatelydehydrogenation of CH2OH has even higher acti-
vation energy (0.84 eV ). The dehydrogenation the same for both pure platinum and mixed Pt–Ru clusters, a decrease of 0.18 eV in E1 wouldenergies of CH3OH and CH2OH on the cluster
also become endothermic. The results indicate that increase the rate of H2O dissociation reaction bya factor of 1000.a Pt–Sn surface with a high atomic percentage of
tin is not conducive to CH3OH dissociation. The combination reaction COs+OHs�COOHs on the secondary ruthenium is endother-Hence, in EOM, the optimal tin surface coverage
must be low. At a surface with a high Sn/Pt ratio mic with high E1, again similar to the case withpure Ru10. On a Pt–Ru electrode, however, theof 3:4, catalytic activity for methanol oxidation
was found to be lower than on pure platinum [56 ]. initial dehydrogenation of methanol (yielding CO)proceeds only through platinum sites and ruthe-The De,s and E1 values for H2Os dissociation on
(Pt3) (Sn2Pt5) and (Pt3) (Sn4Pt3) are similar to those nium provides sites solely for OHs. Reaction (6)is the process of interest, and we find thaton Pt10. The combination reaction of COs+OHs
on (Pt3)(Sn4Pt3) has an activation energy of COPt+OHRu�COOHPt is exothermic with lowE1, and should proceed readily.0.5 eV, while the corresponding E1 is zero on (P
t3) (Sn2Pt5). On substituted tin, the dehydrogenation ofCH2OH is endothermic and has a high E1, indicat-ing that the tin atoms in mixed Pt–Sn metal do4.2.5. CH
3OH oxidation at M on Pt–M (M=Ru,
Sn) not facilitate CH3OH dissociation. Because CO isnot adsorbed at the tin site (Eads=0), no COOHsA (Pt3)(Pt4M3) cluster (Fig. 1d) was used to
simulate adsorption at the M site in a Pt–M mixed will form on tin in Pt–Sn. H2Os dissociation onalloyed tin is 0.3 eV less endothermic and has ametal surface. It is shown in Table 1 that the
adsorption energy of hydrogen atoms on the smaller E1 (by 0.1 eV ) than does the process onpure Pt10. This implies that tin in Pt–Sn is morehollow site of (Pt3) (Pt4M3) is lower than on the
platinum site of (Pt3)(M2Pt5) or (Pt3) (M4Pt3). active than pure platinum, but less so than ruthe-nium in Pt–Ru. The combination reactionTherefore atomic hydrogen species should adsorb
preferentially at platinum. Dissociation of H2O on COPt+OHSn�COOHPt is strongly exothermicwith a low activation energy.M would lead to MMOH and PtMH bond forma-
tions. The adsorption energy of atomic hydrogen(EH) at the on-top site of (Pt3) (M2Pt5) was 4.2.6. CH
3OH oxidation on (M
3)(Pt
7) clusters
It has been shown [27] that when the atomsemployed in calculating De,s and E1 on (Pt3) (Pt4M3). substituted for platinum are placed in the second
cluster layer, the adsorption of CO to platinumThe calculations show that the substitutionalruthenium should be active for CH3OH dissoci- can be weakened, especially when M=Sn. We
therefore examined CH3OH and H2O dissociationsation. As mentioned earlier, experimentally metha-nol does not bind at ruthenium because the sites on (M3) (Pt7) clusters. For M=Ru the dissociation
of CH3OH on the cluster can proceed readily, butare preferentially covered by H2O and OH. The

78 Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
each step in the dehydrogenation of CHxOH is 5. Conclusions
less exothermic than on pure Pt10. On the otherThe present theoretical study provides insightshand, the dissociation of H2O is more difficult on
into the energetics of the elementary steps of(Ru3) (Pt7) than on pure Pt10. For M=Sn, bothCH3OH oxidation on pure platinum, ruthenium,CH3OH and H2O dissociations on the cluster areand mixed Pt–M (M=Ru, Sn) metals. Theenergetically unfavorable. So, although M3 in thefollowing conclusions may be drawn from thesecond layer greatly reduces the adsorption energycomputational study.of CO, it is not conducive to CH3OH and H2O 1. On pure platinum, the dissociation of CH3OHdissociations.
to form COs occurs easily, in agreement withexperiment [54]. CH2OH, CHOH and CHO(or COH) are intermediates in methanol decom-4.2.7. Ligand effect versus bifunctional mechanismposition. The formations of OCH3,s andThere has been a longstanding controversy inOCH2,s are much less favorable than the forma-EOM studies over whether the effect of alloys ontions of CH2OHs and CHOHs. The highestelectrode performance is due solely to the bifuncti-activation energy for the stepwise dehydrogena-onality provided by the substituted metal, ortion of CH3OH is that for the second step,whether there are extra electronic effects that needCH2OHs�CHOHs+Hs, while the last step,to be considered [8,11,19,20]. From the calculatedCHOs (or COHs)�COs+Hs, has the lowestdissociation and activation energies of reactionsactivation energy. The dissociation reaction of(4), (5) and (6), we are able to conjecture as toH2Os is endothermic by 0.8 eV, and its activa-the relative importance of a ligand electronic effecttion energy is estimated to be 1 eV. The dissoci-versus the bifunctional mechanism. On pure Pt10, ation of H2O to form OHs is rate-determining.the dissociation of H2O is the RDS, while the
2. Theoretically, the dissociation of CH3OH onformation of COOHs from COs+OHs is exother-pure ruthenium is as favorable as on puremic. So, once OHs is generated, the poison COs isplatinum. This is in agreement with experimen-easily removed from the surface despite the facttal results on methanol dissociation in UHV on
that CO is strongly adsorbed on platinum. Theboth platinum and ruthenium single-crystal sur-
calculated results for (Pt3) (Ru2Pt5) show that a faces [54,55]. In the electrochemical environ-reduction in Eads for CO on platinum gives a more ment, ruthenium atoms can be blocked bynegative De,s for the COs+OHs combination reac- strong H2O and OH adsorptions. This may betion, but reduction in Eads should not affect the the reason why methanol does not undergorate of reaction. For a mixed Pt–M metal, the oxidation on ruthenium electrodes at low volt-effect of M is to promote the activation of water. age. The RDS on ruthenium is the formationThere are decreases in De,s and E1 for the genera- of COOHs. H2O dissociation on ruthenium istion of OHs from H2O on M site as compared significantly more favorable than on platinum.with a platinum site. Nevertheless, H2O dissoci- 3. Energetically, there is not much differenceation at M is still the RDS for CH3OH oxidation between single ruthenium atoms in Pt–Ru andon Pt–M. For M=Ru, the combination reaction in pure ruthenium for H2O dissociation.COPt+OHRu�COOHPt is slightly exothermic (ca. Ruthenium in Pt–Ru promotes the dissociation−0.3 eV ) and has an activation energy of of CH3OH and the formation of OHs from~0.4 eV. Were not Eads for CO on platinum adsorbed H2O (Fig. 3). The dissociation ofreduced, this reaction would have an E1 of 0.62 eV. H2O at a tin site in Pt–Sn is only slightly moreThus the ligand effect does play some role in CO favorable than at a platinum site. The presenceoxidation. In the case of M=Sn the reaction of M atoms reduces the PtMCO bond strengthCOPt+OHSn�COOHPt is exothermic, and substantially, indicating the existence of aelectronic effects on the PtMCO bond should not ‘ligand effect’. Because H2O dissociation is
the RDS and the combination reactionaffect the rate of oxidation.

79Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
[12] M. Watanabe, S. Motoo, J. Electroanal. Chem. 60COPt+OHM�COOHPt can proceed readily on(1975) 267.Pt–M, a ligand effect is not to be expected to
[13] M. Watanabe, S. Motoo, J. Electroanal. Chem. 60play a significant role in the rate of CO oxida-(1975) 275.
tion. However, a ligand effect may reduce the [14] H.A. Gasteiger, N.M. Markovic, P.N. Ross Jr., J. Phys.accumulation of surface CO. Chem. 99 (1995) 16757.
[15] H.A. Gasteiger, N.M. Markovic, P.N. Ross Jr.,4. The activity of a platinum site in mixed Pt–ME.J. Cairns, J. Phys. Chem. 97 (1993) 12020.for CH3OH dissociation should vary somewhat
[16 ] R. Ianniello, V.M. Schmidt, U. Stimming, J. Stumper, A.with atomic M/Pt ratio. With more tin, theWallau, Electrochim. Acta 39 (1994) 1863.activity at platinum becomes less. Because [17] M.T. Paffett, S.C. Gebhard, R.G. Windham, B.E. Koel,
another effect of tin is to impede methanol J. Phys. Chem. 94 (1990) 6831.adsorption, the optimal tin surface composition [18] C. Xu, B.E. Koel, Surf. Sci. 304 (1994) L505.
[19] M.M.P. Janssen, J. Moolhuysen, Electrochim. Acta 21must be low. In contrast, the CH3OH dissoci-(1976) 861.ation on a ruthenium-rich surface can proceed
[20] M.M.P. Janssen, J. Moolhuysen, J. Catal. 46 (1977) 289.effectively. A metal with a relatively high Ru/Pt[21] A. Wieckowski, J. Electroanal. Chem. 78 (1977) 229.ratio should be beneficial for CH3OH oxidation. [22] M. Krausa, W. Vielstich, J. Electroanal. Chem. 379
(1994) 307.[23] A.B. Anderson, E. Grantscharova, J. Phys. Chem. 99
(1995) 9143.Acknowledgements[24] A.B. Anderson, E. Grantscharova, J. Phys. Chem. 99
(1995) 9149.This work was supported by DOD-EPSCoR [25] A.B. Anderson, E. Grantscharova, P. Shiller,
grant DAAH04-96-1-0199, DOE-HiCREST grant J. Electrochem. Soc. 142 (1995) 1880.3-49811-7840, and by the Office of Naval Research. [26 ] A.B. Anderson, E. Grantscharova, S. Seong,
J. Electrochem. Soc. 143 (1996) 2075.The authors gratefully acknowledge computational[27] M.-S. Liao, C.R. Cabrera, Y. Ishikawa, Surf. Sci. 445facilities provided by the RCMI Center for
(2000) 267.Molecular Modeling and Computational[28] T. Frelink, W. Visscher, J.A.R. van Veen, Surf. Sci. 335Chemistry. (1995) 353.[29] E.J. Baerends, D.E. Ellis, P. Ros, Chem. Phys. 2 (1973) 41.[30] G. te Velde, E.J. Baerends, J. Comp. Phys. 99 (1992) 84.[31] T. Ziegler, V. Tschinke, E.J. Baerends, J.G. Snijders, W.References
Ravenek, J. Phys. Chem. 93 (1989) 3050.[32] B.G. Johnson, P.M.W. Gill, J.A. Pople, J. Chem. Phys. 98
[1] V.S. Bagotzky, Yu.B. Vassil’en, O.A. Khazova, J. Electroa- (1993) 5612.nal. Chem. 81 (1977) 229.
[33] A.J. Aldykiewicz, Jr., D. Zurawski, S. Baxter, R. Kumar,[2] J.-M. Leger, C. Lamy, Ber. Bunsenges. Phys. Chem. 94
M. Krumpelt, G. Bunker, C. Segre, unpublished results.(1990) 1021.
[34] S. Mukerjee, J. McBreen, J. Electrochem. Soc. 146[3] T. Iwasita, F.C. Nart, W. Vielstich, Ber. Bunsenges. Phys.
(1999) 600.Chem. 94 (1990) 1030.[35] B.I. Dunlap, N. Rosch, Adv. Quantum Chem. 21 (1990)[4] P.A. Christensen, A. Hamnett, S.A. Weeks, J. Electroanal.
317.Chem. 250 (1988) 127.[36 ] G. te Velde, E.J. Baerends, Chem. Phys. 177 (1993) 399.[5] T. Iwasita, W. Vielstich, J. Electroanal. Chem. 250[37] M.-S. Liao, Y. Ishikawa, Int. J. Quantum Chem., submit-(1988) 451.
ted for publication.[6 ] J. Willsan, J. Heitbaum, Electrochim. Acta 31 (1986) 943.[38] H. Sellers, E.M. Pattrito, P. Paredes-Olivera, Surf. Sci. 356[7] B. Beden, F. Hahn, S. Juanto, C. Lamy, J.-M. Leger,
(1996) 222.J. Electroanal. Chem. 225 (1987) 215.[39] H. Sellers, E.M. Pattrito, P. Paredes-Olivera, Surf. Sci. 380[8] T. Iwasita, W. Vielstich, E. Santo, J. Electroanal. Chem.
(1997) 264.229 (1987) 367.[40] H. Sellers, E.M. Pattrito, P. Paredes-Olivera, Surf. Sci. 418[9] T. Iwasita, F.C. Nart, J. Electroanal. Chem. 317 (1991)
(1998) 376.291.[41] B.A. Sexton, A.E. Hughes, Surf. Sci. 140 (1984) 227.[10] E. Ticanell, J.G. Beery, M.T. Paffett, S. Gottesfeld,[42] E. Shustorovich, Adv. Catal. 37 (1990) 101.J. Electroanal. Chem. 258 (1989) 61.[43] C. Zheng, Y. Apeloig, R. Hoffmann, J. Am. Chem. Soc.[11] H.A. Gasteiger, N. Markovic, P.N. Ross Jr., E.J. Cairns,
J. Phys. Chem. 98 (1994) 617. 110 (1988) 749.

80 Y. Ishikawa et al. / Surface Science 463 (2000) 66–80
[44] G. Ertl, M. Neumann, K.M. Streit, Surf. Sci. 64 (1977) 393. [51] P. Paredes-Olivera, E.M. Pattrito, H. Sellers, Surf. Sci. 327(1995) 330.[45] C.E. Wartnaby, A. Stuck, Y.Y. Yeo, D.A. King, J. Phys.
Chem. 100 (1996) 12483. [52] W. Vielstich, P.A. Christensen, S.A. Weeks, A. Hamnett,J. Electroanal. Chem. 242 (1987) 327.[46 ] G.E. Gdowski, J.A. Fair, R.J. Madix, Surf. Sci. 127
(1983) 541. [53] W.F. Lin, M.S. Zei, M. Eiswirth, G. Ertl, T. Iwasita, W.Vielstich, J. Phys. Chem. B 103 (1999) 6968.[47] A.B. Anton, D.C. Cadogan, Surf. Sci. 239 (1990) L548.
[48] E. Shustorovich, H. Sellers, Surf. Sci. Rep. 31 (1998) 1. [54] A.A. Deckert, J.L. Brand, C.H. Mak, B.G. Koehler, S.M.George, J. Chem. Phys. 87 (1987) 1936.[49] D. Lide (Ed.), CRC Handbook of Chemistry and Physics,
75th ed. (1994), CRC Press, Boca Raton, FL, 1995, [55] B.A. Sexton, Surf. Sci. 102 (1981) 271.[56 ] T. Frelink, W. Visscher, J.A.R. van Veen, Electrochim.pp. 9–51.
[50] E. Shustorovich, A.T. Bell, Surf. Sci. 253 (1991) 386. Acta 39 (1994) 1871.