nucleotide-binding oligomerization domain containing 2
TRANSCRIPT

Nucleotide-binding Oligomerization Domain Containing 2 Characterization and Function during Mycobacterium tuberculosis Infection of Human Macrophages
DISSERTATION
Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University
By
Michelle Nichole Brooks
Graduate Program in Microbiology
The Ohio State University
2011
Dissertation Committee:
Professor Larry Schlesinger, M.D., Advisor
Professor John Gunn, Ph.D.
Professor Chad Rappleye, Ph.D.
Professor Mark Wewers, M.D.

Copyright by
Michelle Nichole Brooks
2011

ii
Abstract
Macrophages are important innate immune regulators in the recognition and clearance of
invading pathogens. Mycobacterium tuberculosis (M.tb), which causes tuberculosis, is a
host-adapted intracellular pathogen of macrophages. Intracellular pattern recognition
receptors in macrophages such as nucleotide-binding oligomerization domain (NOD)
proteins regulate pro-inflammatory cytokine production. NOD2-mediated signaling
pathways in response to M.tb have been studied primarily in mouse models and cell lines
but not in primary human macrophages. Work in this dissertation examined whether
NOD2 is expressed in human macrophages, controls the growth of M.tb, and functions in
inducible nitric oxide synthase (iNOS) and novel protein kinase c (PKC) signaling
pathways.
We examined NOD2 expression during monocyte differentiation and observed a marked
increase in NOD2 transcript and protein following 2-3 days in culture. Pre-treatment of
human monocyte-derived and alveolar macrophages with the NOD2 agonist muramyl
dipeptide enhanced production of TNF-α and IL-1β in response to M.tb and M. bovis
BCG (BCG) in a receptor interacting protein 2 (RIP2)-dependent fashion. The NOD2-
mediated cytokine response was significantly reduced following knockdown of NOD2
expression by using small interfering RNA (siRNA) in human macrophages. Finally,
NOD2 controlled the growth of both M.tb and BCG in human macrophages, whereas

iii
controlling only BCG growth in murine macrophages. Together, our results provide
evidence that NOD2 is an important intracellular receptor in regulating the host response
to M.tb and BCG infection in human macrophages.
Our observation of enhanced growth in NOD2 deficient cells at 24 hours (h) is of interest
because during this time M.tb is becoming acclimated to the macrophage. This led us to
examine the involvement of NOD2 in early innate immune responses. In order to study
new biological functions and molecular mechanisms of NOD2 during early M.tb
infection of human macrophages, we performed expression profiling of M.tb-infected
NOD2 deficient macrophages using a microarray. Notably, we identified that inducible
nitric oxide synthase (iNOS) expression is regulated by NOD2 during M.tb infection.
iNOS expression is enhanced at 24 and 48 h after M.tb and BCG incubation in human
macrophages. Our data show iNOS expression is decreased in NOD2 deficient cells
stimulated with M.tb and the M.tb-specific NOD2 agonist N-glycolylated muramyl
dipeptide (GMDP), indicating that NOD2 is involved in iNOS protein expression. These
data provide evidence for a novel pathway involving NOD2 that controls M.tb growth in
human macrophages through the regulation of iNOS expression.
We examined other potential signaling partners for NOD2 in human macrophages. Data
show that the NOD2 agonist, GMDP, stimulates phosphorylation and activation of PKCδ.
Corroborating these data, NOD2 deficient cells stimulated with PMA are significantly
reduced in PKCδ phosphorylation. PMA is a known stimulus for classical and novel

iv
PKC isoforms. Additionally, the kinase known to interact with NOD2, RIP2, is activated
by PMA. This activation is deficient upon knockdown of NOD2 in human macrophages.
Thus, our findings strongly support a role for NOD2 in human macrophages during M.tb
infection as well as in early innate immune responses that include iNOS and PKC
pathways.

v
Dedication
This document is dedicated to my family and friends who encouraged me throughout life
and molded me into the person I am today. I would like to specifically dedicate this to
my mother, Katherine L. Brooks, who struggled in life so that I may succeed. My father,
Michael S. Brooks, who taught me happiness and perseverance are the keys to life. My
fiancé, Samuel E. Landes, who I am eternally grateful to for providing a shoulder to lean
on during difficult times, listening to endless scientific presentations, and making my life
during graduate school filled with fun and laughter. Thank you!

vi
Acknowledgments
I am truly grateful to my advisor, Dr. Larry Schlesinger, M.D., for the
encouragement and expectation of scientific rigor, which led to much success during my
dissertation research. I am positive the skills obtained during my Ph.D. in his laboratory
will be invaluable to my career in science.
Foremost I would like to thank past and present laboratory members, who
everyday gave me scientific advice and encouragement. Specifically, I would like to give
thanks to Dr. Murugesan Rajaram, Ph.D., who undoubtedly guided the bulk of my work
in the laboratory and taught me how to execute experiments efficiently. Without him
instilling the excitement of obtaining the next result, none of my scientific success would
have been possible. I am also appreciative of Drs. Abul Azad, Ph.D. and Jordi Torrelles,
Ph.D., who helped me begin my training and taught me fundamental Schlesinger
laboratory techniques, while always taking time to answer my scientific questions.
I thank my graduate dissertation committee, John Gunn, Ph.D., Chad Rappleye,
Ph.D, Mark Wewers, M.D., for their contributions to my research. I am forever indebted
to them for shaping me into a true scientist who is confident and able to convey my
science. In addition, I would like to thank members of the CMIB for their support and
scientific advice for my dissertation research. Specifically, Drs. Amal Amer, M.D.,

vii
Ph.D, Joanne Turner, Ph.D., and Erin Rottinghaus, Ph.D., who devoted a great deal of
personal time to enhance my confidence and ability to communicate my science. I would
not be the scientist I am today without you.
Finally, I would like to acknowledge my funding sources including the
Microbiology graduate student fellowship, NSF Graduate Research Fellowship Program,
and the CMIB NIH/NIAID T32 training grant.

viii
Vita
August 14, 1984.............................................Born – Atlanta, Georgia
2006................................................................B.S.A. Biological Sciences,
University of Georgia
2006 to present ..............................................Graduate Research Fellow, Department of
Microbiology, The Ohio State University
2006 to 2007………………………………..Microbiology Graduate Student Fellowship
2007 to 2010………………………………..NSF Graduate Research Fellowship Program
2009………………………………………….1st place pre doctoral graduate poster
competition, DHLRI
2009………………………………………… Travel Award Winner 9th Annual OSUMC
Trainee Research Day poster competition
2010…………………………………………2nd place pre doctoral graduate poster
competition, DHLRI
2011…………………………………………Keystone Symposium Underrepresented
Minority Scholarship; TB Immunology and
Vaccine Design
2011 to present………………………………CMIB NIH/NIAID T32 training grant

ix
Publications
Brooks, M, M. Rajaram, A. Azad, A. Amer, M. Valdivia-Arenas, J. Park, G. Nuñez, and
L. Schlesinger. 2011. NOD2 controls the nature of the inflammatory response and
subsequent fate of Mycobacterium tuberculosis and M. bovis BCG in human
macrophages. Cellular Microbiology. 13(3):402-418
Carlson, T, M. Brooks, M. Rajaram, L. Henning, D. Myer, and L. Schlesinger. 2011.
Pulmonary innate immunity: soluble and cellular host defenses of the lung. In:
Regulation of Innate Immune Functions Book Chapter. Research Signpost, STM Books.
Rajaram, M, M. Brooks, J. Morris, J. Torrelles, A. Azad, L. Schlesinger. 2010.
Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-
Activated receptor γ linking mannose receptor recognition to regulation of immune
responses. Journal of Immunology. 185:929-942
Fields of Study
Major Field: Microbiology

x
Table of Contents
Abstract ............................................................................................................................... ii
Dedication ........................................................................................................................... v
Acknowledgments.............................................................................................................. vi
Vita................................................................................................................................... viii
List of Figures ................................................................................................................. xvii
CHAPTER 1 : Introduction ............................................................................................... 1
1.1 The immune system .................................................................................................. 1
1.2 Innate immunity ........................................................................................................ 1
1.2.1 PKC isoforms ..................................................................................................... 3
1.2.2 iNOS and NO production ................................................................................... 5
1.3 Adaptive immunity.................................................................................................... 6
1.4 Pulmonary innate immunity ...................................................................................... 8
1.5 Alveolar macrophage ................................................................................................ 9
1.6 Macrophage activation .............................................................................................. 9
1.7 Macrophage receptors ............................................................................................. 11

xi
1.8 Pattern recognition receptors................................................................................... 12
1.9 NOD-like receptors ................................................................................................. 14
1.9.1 Discovery.......................................................................................................... 14
1.9.2 NLR structure ................................................................................................... 15
1.9.3 LRR domain ..................................................................................................... 15
1.9.4 NOD domain..................................................................................................... 16
1.9.5 CARD domain .................................................................................................. 16
1.9.6 NOD agonists ................................................................................................... 18
1.9.7 Generation of NOD agonists ............................................................................ 18
1.10 NLRs and innate immunity ................................................................................... 19
1.11 NOD2 signaling cascade ....................................................................................... 19
1.12 NOD2 and TLR crosstalk...................................................................................... 22
1.13 NOD2 and microbial immunity............................................................................. 23
1.14 NOD2 and inflammatory diseases......................................................................... 24
1.15 NOD2: human versus mouse................................................................................. 25
1.16 Tuberculosis .......................................................................................................... 26
1.16.1 Drug therapy and vaccines ............................................................................. 27
1.16.2 Infection with M.tb ......................................................................................... 28
1.16.3 M.tb and alveolus interaction.......................................................................... 29

xii
1.16.4 M.tb and AM receptor interaction .................................................................. 30
1.16.5 M.tb and phagosome environment.................................................................. 31
1.16.6 M.tb secretion systems.................................................................................... 32
1.16.7 M.tb and entry into the cytosol ....................................................................... 35
1.17 M.tb and NOD2..................................................................................................... 36
1.18 Mouse and human differences in the immune system .......................................... 38
1.19 Summary ............................................................................................................... 39
1.20 Specific aims ......................................................................................................... 40
CHAPTER 2 : NOD2 controls the nature of the inflammatory response and subsequent
fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages............. 42
2.1 Introduction ............................................................................................................. 42
2.2 Materials and Methods ............................................................................................ 45
2.2.1 Buffers and reagents ......................................................................................... 45
2.2.2 Antibodies......................................................................................................... 46
2.2.3 Bacterial strains and culture ............................................................................. 47
2.2.4 Isolation and culture of human monocytes and macrophages.......................... 47
2.2.5 Mice and mouse macrophages.......................................................................... 48
2.2.6 RNA isolation................................................................................................... 48
2.2.7 Gene expression studies by qRT-PCR ............................................................. 48

xiii
2.2.8 Confocal microscopy........................................................................................ 49
2.2.9 Transfection of MDMs ..................................................................................... 49
2.2.10 Macrophage stimulation / infection, cell lysis, immunoprecipitation, and
western blotting ......................................................................................................... 50
2.2.11 ELISAs ........................................................................................................... 52
2.2.12 M.tb and BCG intracellular growth assays in macrophages........................... 52
2.2.13 M.tb and BCG cell association assay within macrophages ............................ 53
2.2.14 Cell enumeration of NOD2 siRNA transfected MDMs in monolayer culture53
2.2.15 NOD2 antibody peptide competition assay .................................................... 54
2.2.16 Statistics.......................................................................................................... 54
2.3 Results ..................................................................................................................... 55
2.3.1 NOD2 mRNA and protein expression increase as monocytes differentiate into
macrophages .............................................................................................................. 55
2.3.2 Activation of NOD2 enhances the M.tb- and BCG-mediated pro-inflammatory
cytokine response through the NF-κB pathway ........................................................ 58
2.3.3 siRNA-mediated knockdown of NOD2 activity in human macrophages leads to
loss of its activity....................................................................................................... 61
2.3.4 NOD2 regulates the macrophage TNF- and IL-1β cytokine response to M.tb
and BCG .................................................................................................................... 63

xiv
2.3.5 Expression of NOD2 in human AMs and its role in regulating the production of
TNF- and IL-1β in response to M.tb ....................................................................... 66
2.3.6 NOD2 controls the intracellular growth of M.tb and BCG in human
macrophages .............................................................................................................. 68
2.3.7 Effects of NOD2 on M.tb and BCG growth in mouse macrophages and on the
cytokine response to infection ................................................................................... 70
2.4 Discussion ............................................................................................................... 72
CHAPTER 3 : Critical role for NOD2 in Mycobacterium tuberculosis induced iNOS
expression in human macrophages ................................................................................... 81
3.1 Introduction ............................................................................................................. 81
3.2 Materials and Methods ............................................................................................ 84
3.2.1 Buffers and reagents ......................................................................................... 84
3.2.2 Antibodies......................................................................................................... 85
3.2.3 Bacterial strains and culture ............................................................................. 85
3.2.4 Isolation and culture of human macrophages ................................................... 86
3.2.5 RNA isolation................................................................................................... 86
3.2.6 Gene expression studies by qRT-PCR ............................................................. 86
3.2.7 Transfection of MDMs ..................................................................................... 87
3.2.8 Microarray analysis .......................................................................................... 87

xv
3.2.9 Macrophage stimulation / infection, cell lysis, and Western blotting .............. 88
3.2.10 DAF-FM diacetate staining ............................................................................ 89
3.2.11 Statistics.......................................................................................................... 90
3.3 Results ..................................................................................................................... 90
3.3.1 NOD2 regulates many genes during M.tb infection of human macrophages... 90
3.3.2 NOD2 agonists regulate iNOS expression and NO production in human
macrophages .............................................................................................................. 93
3.3.3 M.tb-mediated iNOS expression is NOD2-dependent ..................................... 96
3.4 Discussion ............................................................................................................... 99
CHAPTER 4 : The role of PKCδ in the NOD2 signaling pathway in human macrophages
......................................................................................................................................... 106
4.1 Introduction ........................................................................................................... 106
4.2 Materials and Methods .......................................................................................... 109
4.2.1 Buffers and reagents ....................................................................................... 109
4.2.2 Antibodies....................................................................................................... 110
4.2.3 Isolation and culture of human macrophages ................................................. 110
4.2.4 Transfection of MDMs ................................................................................... 111
4.2.5 Macrophage stimulation and preparation for ELISAs and Western blotting . 111
4.2.6 Kinase assays.................................................................................................. 112

xvi
4.2.7 ELISA............................................................................................................. 113
4.2.8 Statistics.......................................................................................................... 113
4.3 Results ................................................................................................................... 114
4.3.1 Activation of NOD2 enhances the PMA-mediated pro-inflammatory cytokine
response through its signaling cascade intermediates ............................................. 114
4.3.2 Phosphorylated PKCδ expression and activity is NOD2-dependent.............. 117
4.3.3 RIP2 activation is PKCδ-dependent ............................................................... 121
4.4 Discussion ............................................................................................................. 123
CHAPTER 5 : Synthesis................................................................................................ 128
Bibliography ................................................................................................................... 140

xvii
List of Figures
Figure 1.1: NOD1 and NOD2 structure and domain functions ....................................... 17
Figure 1.2: NOD2 signaling cascades.............................................................................. 21
Figure 2.1: NOD2 expression increases during monocyte differentiation to macrophages.
........................................................................................................................................... 57
Figure 2.2: The NOD2 ligand MDP enhances M.tb- and BCG-mediated cytokine
production in human macrophages, a process involving activation of RIP2 and IkBα.... 60
Figure 2.3: Loss of NOD2 activity in human macrophages by siRNA knockdown........ 62
Figure 2.4: NOD2 controls M.tb- and BCG-mediated cytokine production and RIP2
activation in human macrophages..................................................................................... 65
Figure 2.5: M.tb-mediated cytokine production in human AMs treated with MDP........ 67
Figure 2.6: NOD2 controls the intracellular growth of M.tb and BCG in human
macrophages. .................................................................................................................... 69
Figure 2.7: Effect of NOD2 on M.tb and BCG growth and cytokine response in mouse
WT and NOD2-/- BMMs. .................................................................................................. 71
Figure 3.1: NOD2 regulates gene expression in human macrophages during M.tb
infection. ........................................................................................................................... 92

xviii
Figure 3.2: NOD2 agonists regulate the mRNA and protein expression of iNOS and NO
production in human macrophages. .................................................................................. 95
Figure 3.3: M.tb-mediated induction of iNOS expression and NO production is NOD2-
dependent in human macrophages. ................................................................................... 98
Figure 4.1: The NOD2 signaling pathway is involved in PMA-stimulated cytokine
production. ...................................................................................................................... 116
Figure 4.2: NOD2 and PKCδ agonists stimulate PKCδ phosphorylation and activity in a
NOD2-dependent manner. .............................................................................................. 120
Figure 4.3: RIP2 phosphorylation mediated by PMA and GMDP is PKCδ-dependent.122
Figure 4.4: PKCδ is necessary for optimal NOD2 activity............................................ 127

xix
Abbreviations
ABC = ATP-binding cassette
AM = alveolar macrophage
APC = antigen presenting cell
BAL = bronchoalveolar lavage
BCG = Mycobacterium bovis BCG
BMM = bone marrow derived macrophage
BSA = bovine serum albumin
CARD = caspase activation and recruitment domain
CFP10 = culture filtrate proteins of 10 kDa
CR = complement receptor
CRD = carbohydrate recognition domain
DAF-FM = 4-amino-5-methylamino-2’,7’-difluorofluorescein diacetate
DAG = diacylglycerol
DAN = diaminonapthalene
DAP = diaminopimelic acid
DC-SIGN = dendritic cell-specific intercellular adhesion molecule-2 grabbing non-integrin
DUOX2 = dual oxidase 2

xx
EBD = effector binding domain
ELISA = enzyme-linked immunosorbent assay
ESAT6 = early secreted antigen target of 6 kDa
ESX-1 = early secretory antigenic target 6 system 1
GlcNAc = N-acetylglucosamine
GMDP = N-glycolyl-MDP
HIV = human immunodeficiency virus
IFN = interferon
Ig = immunoglobulin
IL = interleukin
iNOS = inducible nitric oxide synthase
IRG = immunity-related guanosine triphosphatase
JAK = janus kinases
LM = lipomannan
LPS = lipopolysaccharide LRD = ligand recognition domain LRR = leucine rich repeats ManLAM = mannose capped lipoarabinomannan
MAP = mitogen-activated protein
MDP = muramyl dipeptide
MDM = monocyte derived macrophage
MDR-TB = multi-drug resistant tuberculosis

xxi
MHC = major histocompatibility complex
MOI = multiplicity of infection
MR = mannose receptor
M.tb = Mycobacterium tuberculosis
MurNAc = N-acetylmuramic acid
NADPH = nicotinamide adenine dinucleotide phosphate hydrogen
NamH = N-acetyl muramic acid hydroxylase
NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells
NK = natural killer
NLR = nod-like receptor
NO = nitric oxide
NOD = nucleotide-binding oligomerization domain
PAMPs = pathogen associated molecular patterns
PBMC = peripheral blood mononuclear cell
PKC = protein kinase c
PPARγ = peroxisome proliferator activated receptor
PRK = PKC related kinases
PRR = pattern recognition receptor
PMA = phorbol 12-myristate 13-acetate
qRT-PCR = quantitative real-time PCR
R = resistance
RCN = relative copy number

xxii
RD1 = region of difference 1
RIP2 = receptor interacting protein 2
RNI = reactive nitrogen intermediates
ROI = reactive oxygen intermediates
SDS-PAGE = sodium dodecyl sulfate polyacrylamide gel electrophoresis
siRNA = small interfering RNA
SP-A = surfactant protein A
SP-D = surfactant protein D
SR = scavenger receptor
STAT = signal transducers and activators of transcription
TAK1 = TGF activated kinase 1
Tat = twin-arginase transport
TB = tuberculosis Th1 = type 1 helper T cells Th2 = type 2 helper T cells
TIR = toll IL-1 receptor
TLR = toll like receptor TNF- = Tumor necrosis factor alpha XDR-TB = extensively drug resistant WHO = World Health Organization

1
CHAPTER 1 : Introduction
1.1 The immune system
The immune system is highly complex, involving soluble components, cells, tissues, and
organs that are responsible for keeping the host healthy from disease. There are many
different cell types that make up host immunity including T cells, neutrophils,
eosinophils, basophils, mast cells, and antigen presenting cells (1). These cell types are
each unique in their function and work together to keep balance and order in the host.
The immune system is made of two arms, adaptive and innate, that work together in order
to keep the host healthy (2).
1.2 Innate immunity
The innate immune response is the first line of defense against invading pathogens, and is
also known as native immunity. This response occurs rapidly upon encountering an
infectious agent. There are four main principle components of innate immunity: physical
and chemical barriers, phagocytic cells and natural killer (NK) cells, blood proteins, and
cytokines (1). The major components that work in concert in order to protect the host
include: soluble and membrane bound molecules, cells, and processes. The soluble

2
components include complement, collectins, and acute phase proteins that are known to
decorate the surface of infectious agents and elicit a response. Membrane bound
components are those found on innate immune cells such as pattern recognition receptors
(PRR) (3). The cells of the innate immune response play a critical role in defending
against pathogens. These cells include mast cells, eosinophils, basophils, macrophages,
neutrophils, dendritic cells, and NK cells. The processes that occur include phagocytosis,
reactive oxygen/nitrogen intermediate killing, phagosome acidification, and antimicrobial
protein and peptide production (4). Macrophages are an important cell type in innate
immunity. These cells use three mechanisms in order to uptake small particles or
infectious agents. Pinocytosis is the uptake of substances that are < 1 μm, and is also
known as fluid phase endocytosis. Receptor-mediated endocytosis is the uptake of
specific substances that bind to surface receptors and this is mediated by clathrin.
Phagocytosis is the uptake of microbes that are 1 μm or larger, and this mechanism is
mediated by actin. Endocytosis uses clathrin-coated pits while phagocytosis uses
cytoskeletal components such as paxillin, talin, and vinculin (5).
Once engulfed inside the macrophage, the pathogen is in a phagosome. This is known as
an early phagosome characterized by Rab5 and the presence of cathepsin H. The
phagosome matures and acquires Rab7 and cathepsin S. This is known as the late
phagosome. Finally, the phagosome fuses with a lysosome that contains antimicrobial
proteins and peptides accompanied by a lower pH (6).

3
Upon phagocytosis the macrophage uses a mechanism called the respiratory burst as its
first line of defense, which causes the production of superoxide and hydrogen peroxide.
Upon ligation of phagocytic receptor(s), phospholipase C releases diacylglycerol (DAG)
and IP3. This is followed by calcium release from the endoplasmic reticulum (ER), which
in conjunction with DAG activates protein kinase C (PKC) isoforms. PKC then
phosphorylates the cytosolic component p47phox. This then activates the formation of the
NADPH oxidase, allowing its components to translocate to the phagosome or plasma
membrane, where the complex binds flavocytochrome subunits gp91 phox and p22 phox
(7). One of the end products of the respiratory burst, superoxide anion, can dismutate to
hydrogen peroxide, and can generate hydroxyl radicals and singlet oxygen. In
neutrophils, hydrogen peroxide can also be converted by myeloperoxidase into
hypochlorous acid and chloramines (4).
1.2.1 PKC isoforms
PKCs are a family of enzymes and were one of the first kinases identified. The major
function of these kinases is to phosphorylate hydroxyl groups on the serine and threonine
residues of other proteins in order for them to function properly. PKCs are most notably
known for their function in the respiratory burst but are also important in cytokine
signaling (8,9). PKCs are activated by phosphotidylserine or DAG in a calcium-
dependent manner as well as by tumor-promoting phorbol esters such as phorbol 12-
myristate 13-acetate (PMA) (10). There are 12 identified PKC isotypes in the
mammalian PKC family, and this contributes to the complexity of understanding PKC

4
function. There are four subfamilies of isoforms that are classified based on the
secondary messenger that is required for activation. Classical (conventional) PKCs have
been the most thoroughly studied and require DAG, calcium, and phospholipid for
activation. They include the isoforms PKCα, PKCβI, PKCβII, and PKCγ (11). PKCs that
require DAG but not calcium for activation are known as novel PKCs and include the
isoforms PKCδ, PKCθ, PKCε, and PKCη (12). Atypical PKCs require neither calcium
nor DAG for activation and include PKCι, PKCζ, PK-N1, PK-N2 (13). Both classical
and novel PKC subfamilies can be activated by phorbol esters in the presence of
phosphatidylserine. PMA is able to activate the enzymes by abolishing the requirement
of DAG and decreasing the concentration of calcium needed for activation (14). A final
group, PKC-related kinases (PRK), are similar to atypical PKCs; however, they were
discovered to bind and activate RhoA GTPase (15-18).
The novel PKCs are of interest here, specifically PKCδ. PKCδ is involved in B-cell
signaling, and the regulation of growth, apoptosis, and differentiation of a variety of cell
types (19-21). Phosphorylation of various tyrosine residues is necessary for activation of
the enzyme; one specific residue is Tyrosine 311 (22). Interactions between this kinase
include, but are not limited to, phosphoinositide-dependent kinase-1, STAT1, and the
insulin receptor (23-26). PKCθ functions to activate T-cells, and is necessary in the
activation of NF-κB and AP-1 through the T-cell receptor signaling complex (27,28).
This kinase has been shown to interact with a few proteins including, RAC-alpha
serine/threonine-protein kinase (AKT1) and Proto-oncogene tyrosine-protein kinase

5
(FYN) (29,30). Interestingly, PKCθ has been shown to phosphorylate CARD11
(CARMA1) and activation of NF-κB is mediated by these two proteins and B-cell
lymphoma/leukemia 10 (BCL10) in T cells (31).
1.2.2 iNOS and NO production
Reactive nitrogen species (RNS) are also prominent in macrophages. Inducible nitric
oxide synthase (iNOS) is regulated at the transcriptional level and is necessary in order
for nitric oxide (NO) to be produced (4). NO is produced on the cytoplasmic side of the
phagosome and can diffuse across membranes to reach intraphagosomal targets (32).
Upon encountering ROS in the luminal environment NO can undergo spontaneous or
catalytic conversion to RNS products that are important mediators in bacterial killing
such as nitric dioxide, peryoxynitrite, dinitrogen trioxide, and dinitrosyl iron complexes.
The products can cause protein inactivation and lipid conversion by oxidative damage,
causing impaired bacterial metabolism and inhibition of replication (33). Signal
transduction is one fundamental role RNS plays in biology and has been shown
experimentally during infections (34,35). It is now widely accepted that NO signaling is
important in the cardiovascular system as well as in vascular homeostasis (36). Another
role for NO is in controlling inflammation. While the mechanism is still unclear as to
how exogenous NO puts forth an anti-inflammatory effect, it is thought that it inhibits
NF-κB (37).

6
iNOS and NO production are dependent on many factors. iNOS mRNA is regulated at
multiple steps, including transcription, stability, and translation (38). The regulation of
iNOS protein may occur by calmodulin binding, dimer formation, substrate depletion (L-
arginine), substrate recycling, tetrahydrobiopterin availability, end product inhibition,
phosphorylation, and subcellular localization. Tetrahydrobiopterin plays a major role in
iNOS enzymatic activity; however, production can be influenced by cytokines and LPS
(38,39). Upon NO binding to the iron in heme, it acts as a feedback inhibitor of iNOS
(40,41). iNOS mRNA can be induced by Interferon-gamma (IFNγ) through the binding
of Interferon receptor 1 (IFNR1) and IFNR2 complex, which activates janus kinases
(JAK) and signal transducers and activators of transcription (STAT) pathways. The
transcription factor interferon response factor-1 (IRF1) is activated and iNOS mRNA is
produced (42). Production of iNOS mRNA is also induced by toll-like receptor (TLR)
ligands and use NF-κB transcription factors (43). While initial studies showed lower
levels of iNOS expression and NO production in human cells, the availability of better
techniques to study NO production has led to more convincing data for its presence in
human cells (44).
1.3 Adaptive immunity
When the innate immune response cannot control an infection, it also serves to give
warning that an infection is present and in turn activates the adaptive immune response.
While the innate immune system is notorious for its immediate response to an infectious
agent, the adaptive immune response is valued for its specificity and ability to recognize

7
a re-occurring previous antigen. Activation of this system is dependent on co-stimulatory
molecules on antigen presenting cells (APCs) and T lymphocytes (T cells). There are
two major types of adaptive immune responses, humoral and cell-mediated immunity.
Humoral immunity is the production of antibodies by B lymphocytes (B cells) and cell-
mediated is the interaction of T cells with macrophages. Extracellular microbes are
neutralized and targeted for elimination by antibodies, using mechanisms such as
phagocytosis or releasing inflammatory mediators from immune cells to remove them.
T cells are responsible for infectious agents that are able to subvert the innate immune
response and reside inside the macrophage where antibodies cannot reach. A major
function of T cells is to mediate the killing of infected cells in order to control the
infection. Activation of the adaptive immune response can elicit the production of
several cytokines including, IL-2, IL-4, and IFNγ. The T cell response to an infection is
dependent on the cytokine produced, e.g. TH1 (IL-12) or TH2 (IL-4) (1).
Other characteristics of adaptive immunity include specificity and diversity, memory,
specialization, self-limitation, and non-reactivity to self. Specificity is the ability of
individual lymphocytes to recognize specific epitopes of antigens. The lymphocyte
repertoire of an individual is great and this is due to the diversity of the structure of the
antigen-binding site. Memory is another key characteristic of adaptive immunity. The
second time an individual is exposed to an antigen, the response is quicker and greater
compared to the primary interaction. Specialization refers to the variety of responses
elicited, either humoral or cell-mediated, which allows the system to be more effective at

8
clearing an infection. The ability of the adaptive immune system to return to basal
conditions and only functioning when an antigen is present is called self-limitation.
Finally, nonreactivity to self may be one of the most important features because it
controls the system from reacting to an individual’s own antigenic substances (45).
1.4 Pulmonary innate immunity
While the lung is known for its role in respiration, it is also a major player in innate
immunity. The lung is constantly bombarded with particulates and microbes and serves
as a major interface between the host and the external environment. An intricate
pulmonary immune system is in place in order to protect the host, and this system
encompasses innate and adaptive responses (46).
Upon inhalation, a particulate or microbe (< 5 μm) is able to avoid ciliary beat, the cough
reflex, and mucus clearance to travel down the trachea and through the bronchi where it
eventually settles in the alveolus. The alveolus is where the protection of the host occurs.
Pulmonary innate immunity is controlled by cellular and soluble components. Airway
and alveolar epithelial cells, as well as leukocytes are cellular components, while the
antimicrobial products (e.g. collectins, defensins, lactoferrin, and cathelicidins) secreted
into the epithelial lining fluid compose the soluble components of pulmonary innate
immunity (47).

9
1.5 Alveolar macrophage
An important mediator of pulmonary immunity and one of the first lines of cellular
defense in the alveolus is the alveolar macrophage (AM). There are only one to two AMs
per alveolus which range in size from 9-40 μm in diameter (48). There are two major
ways AMs are generated. The majority of AMs are thought to originate from monocytes
circulating in the blood that eventually find their place in the alveolus, where they
differentiate into macrophages. The second way, considered more controversial, is that
the mononuclear phagocytes present in the lung replicate in the alveolus (48). In the
alveolus, AMs are continuously bathed in surfactant, which is an important immune
modulator that is produced by type II epithelial cells (49).
Two important functions of the AM are phagocytosis and inhibiting excessive pro-
inflammatory signaling. It is necessary for these cells to have enhanced phagocytosis in
order to clear the particles and microbes that are encountered by the alveolus. It is
important to maintain homeostasis within the alveolus, and this is achieved by a balanced
production of pro- and anti-inflammatory cytokines. AMs are unique in this ability to
regulate cytokine production and are labeled alternatively activated for this purpose
(48,50,51).
1.6 Macrophage activation
There are currently three types of activation states for macrophages based on the stimulus
encountered: type I, type II, and alternative activation (52,53). Type I, also known as

10
classically activated macrophages or M1 macrophages, are those that encounter IFNγ and
a stimulus that induces TNF-α, such as a microbe. Upon stimulation these cells produce
pro-inflammatory cytokines such as TNF-α, IL-12, and IL-6, along with oxidants,
increased MHCII expression, and a decrease in mannose receptor (MR) expression.
Macrophages that encounter immune complexes are known as type II macrophages.
These are similar to type I macrophages in that they produce pro-inflammatory cytokines,
oxidants, and increased MHCII expression; however, they also produce the anti-
inflammatory cytokine IL-10, and have less IL-12 production when compared to type I
macrophages (53).
Alternatively activated, also known as M2 macrophages, are the third type of activation
state. In vitro TH2 cytokines, IL-4 and IL-13, induce this macrophage type, which is
consistent with the phenotype of AMs. These cells are characterized by having
enhanced PRR expression, such as the MR, and decreased CD14 expression. They also
produce fewer amounts of pro-inflammatory cytokines and oxidants, but instead secrete
anti-inflammatory cytokines IL-10 and TGFβ more readily. Other markers of alternative
activation found through mouse studies include receptors (CD23 and CD163), metabolic
factors (L-arginase and lipoxygenase), and soluble proteins (FIZZI and Ym1/2) (52). Our
laboratory has identified several features of alternatively activated human macrophages
such as increased MR and TLR2 expression (54), decreased NADPH oxidase (55) and
TLR activities (54), and increased activity of the nuclear receptor PPARγ which is linked
to the MR (56).

11
1.7 Macrophage receptors
Phagocytosis is mediated by several different receptors. Opsonic phagocytosis occurs
through the complement receptors (CR), CR1 and CR3, and Fcγ receptors. CR1 can bind
mannose binding lectin (MBL), C1q, C3b, C4b, and requires additional signals to
mediate phagocytosis. CR3 (Mac 1 or CD11b/CD18) binds C3bi-opspnized bacteria
(57). The Fcγ receptors recognize particles opsonized with IgG antibodies (58). Other
receptors are involved in non-opsonic phagocytosis. The MR is a C-type lectin,
membrane bound Pattern Recognition Receptor (PRRs, see below) found on the cell
membrane as well as on endosomes within the cell. Its function is normally to bind
terminal mannose and fucose residues of host glycoproteins and glycolipids, which are
also common components of bacterial cell walls. The tail of the receptor is necessary in
order to induce phagocytosis (59). The scavenger receptor (SR) is known to phagocytose
low-density lipoproteins (LDL) circulating in the body that can no longer interact with
the LDL receptor, but has been shown to bind microbes as well (60).
There are several other membrane receptors that are not directly involved in
phagocytosis, but instead signal in order to activate the phagocyte and in some cases
regulate phagocytic receptors. These include dectin-1, TLRs, and G protein-coupled
receptors. Dectin-1 is involved in signaling after recognition of non-opsonic beta glucan
particles found in fungi (61). TLRs are trans-membrane PRRs that recognize various
pathogen-associated molecular patterns (PAMPS) and induce a signaling cascade that

12
typically results in the production of inflammatory cytokines (62). G protein-coupled
receptors recognize short peptides containing N-formylmethionyl residues and function
to stimulate the recruitment of lymphocytes to the site of infection. The macrophage also
has a secondary line of foreign molecule recognition with receptors located in the cytosol.
These PRRs are known as the Nucleotide-binding Oligomerization Domain (NOD)-Like
Receptors (NLRs) and function to induce an inflammatory response upon recognition of
their PAMP intracellularly (63).
1.8 Pattern recognition receptors
As noted above, PRRs are unique in that they recognize specific patterns or PAMPS.
These can range from bacterial peptidoglycan fragments, carbohydrates, lipids or flagella
to intracellular DNA or single stranded RNA. They play a major role in innate immunity
through functions such as phagocytosis and signaling an inflammatory response. PRRs
can have a transmembrane domain and be located on the cell surface or endosomes, the
cytosol, or secreted.
Two major membrane-bound PRRs are the MR and TLRs. The MR is 180 kDa protein
that, along with its transmembrane domain, has four other domains: an extracellular
amino terminal cysteine rich domain, a fibronectin type II repeat domain, eight tandem
cytsteine rich lectin-like carbohydrate recognition domains (CRD), and a cytoplasmic
carboxy-terminal domain (64). TLRs are an important class of PRRs. There are 10
identified mammalian TLRs that are distinctly membrane-associated type I receptors (65-

13
67). The exterior amino terminus contains variable arrangements of leucine-rich repeats
(LRRs) which serve to recognize PAMPs. The cytosolic carboxy terminus of TLRs is
highly homologous to the IL-1 receptor and contains a Toll-IL-1R (TIR) domain that
forms the scaffold for the assembly of signaling intermediates such as Myd88, IRAK1,
IRAK4, IRAKM and Mal/Tiram. TLR4 and TLR2 are the most thoroughly studied
TLRs. TLR4 has been found to recognize the endotoxin (lipopolysaccharide, LPS) of
Gram-negative organisms, while TLR2 recognizes Gram-positive PAMPS, such as
lipoteichoic acid. In general, TLR signaling drives NF-κB activation via phosphorylation
of IκBα (67-69), although several negative regulators such as IRAKM have been
identified (70).
Cytoplasmic PRRs do not contain a transmembrane domain and are associated with
cytoplasmic networks. NLRs are involved in processes such as apoptosis and pro-
inflammatory signaling. RNA helicases recognize single stranded or double stranded
RNA. The two recognized in mammalian cells are RIG-1 and MDA5, which activate
antiviral signaling (71). Secreted PRRs include MBL, serum amyloid protein, and C -
reactive protein (1,2).

14
1.9 NOD-like receptors
1.9.1 Discovery
NLRs were initially discovered in plants. Plants are able to recognize pathogens by
disease resistance (R) genes, and in turn induce a response that allows the infected area to
be isolated through cell wall remodeling, production of reactive oxygen species, cell
death, and production of anti-pathogenic molecules at the site of infection (72). There are
two classes of R genes, one that produces transmembrane proteins similar to TLRs and
another that produces cytosolic NOD proteins. These cytosolic NOD proteins generally
contain amino-terminal α-helix-rich or TIR domains, a central NOD, and carboxyl-
terminal LRRs. Since there are known similarities between plants and animals, genome
wide searches for homology between these plant R proteins and the mammalian system
were embarked upon by investigators. The first searches yielded apoptosis regulator,
Apaf-1 and its nematode homolog, CED-4, which revealed two proteins NOD1 (CARD4)
and NOD2 (CARD15) (73). Further studies have described 23 mammalian NLRs in the
innate immune recognition of intracellular pathogens (74). The current literature is
heavily focused on NOD1 and NOD2. They are mainly expressed in APCs and epithelial
cells. NOD1 protein is abundantly found in intestinal epithelial cells, while NOD2 protein
is low or undetectable in these cells. NOD2 protein expression has been found in Paneth
cells found at the base of intestinal crypts (75). NOD2 mRNA is present in monocytes
but protein expression is low; however, NOD2 protein expression is abundant in human
macrophages (76,77).

15
1.9.2 NLR structure
The structure of NOD1 and NOD2 are very similar to Apaf-1 and CED-4. They both
contain amino terminal caspase activating and recruitment domains (CARDs) linked to a
NOD domain; however, the mammalian NOD proteins contain LRRs in their carboxy
termini (73). Most mammalian NOD family members contain 3 distinct functional
domains, an amino terminal effector binding domain (EBD), a centrally located NOD,
and a carboxy-terminal ligand recognition domain (LRD). Depending on the downstream
signaling partner, the EBD is involved in homophilic and heterophilic interactions. The
NLR family can be divided into four subfamilies depending on the composition of their
N-terminal EBD. The differing N-terminal domains are as follows with subfamily name:
acidic transactivation domains (NLRAs), CARD (NLRCs), pyrin domains (NLRPs), and
baculovirus IPA repeat domains (NLRBs). NOD1 and NOD2 are part of the NLRC
subfamily and contain a LRR domain, NOD domain, and CARD domains (78).
1.9.3 LRR domain
The LRR domain is made up of leucine rich repeats that are needed for the sensing of
agonists. There are approximately 11 LRRs in a horseshoe shape made up of beta strands
that constitute the concave face of the LRRs. Functional analysis of NOD2 revealed
that the most C-terminal LRRs, 6-11, are critical for bacterial recognition (79). These
residues are confined to the β-strand/β-turn (xxLxLxx) motif (L = leucine, x = amino

16
acid) (80). There are six non-conserved amino acids in the corresponding regions of the
LRRs for each protein. These include G879, W907, V935, E959, K989, and S991. These
amino acids face outward and are thought to be important in recognition of the specific
agonist for each NOD protein (79). Currently, no one has been able to show direct
binding of NOD proteins with their specific agonist. Some speculate that there may be an
intermediate that has not been discovered that causes activation.
1.9.4 NOD domain
This region contains an ATP-binding cassette (ABC) that includes catalytic residues with
canonical binding site motifs for phosphate residues (Walker’s A box) and magnesium
ions (Walker’s B box). Similarities have been found between the AAA+ family of
ATPases oligomerization module and ABC region of NOD-LRR proteins (81). In this
region, there are also motifs involved in hydrolysis of nucleosides, ATP, deoxyATP
and/or GTP. Nucleotide hydrolysis is necessary for protein activation of NOD1 and
NOD2. Mutational analysis identified 21 critical residues in the NOD domain important
for signaling (79).
1.9.5 CARD domain
The CARD domain is necessary for interactions with the CARD domain of the
downstream kinase receptor interacting protein 2 (RIP2), which is necessary for the
signaling cascade to occur and NF-κB activation. The CARD effector domain is
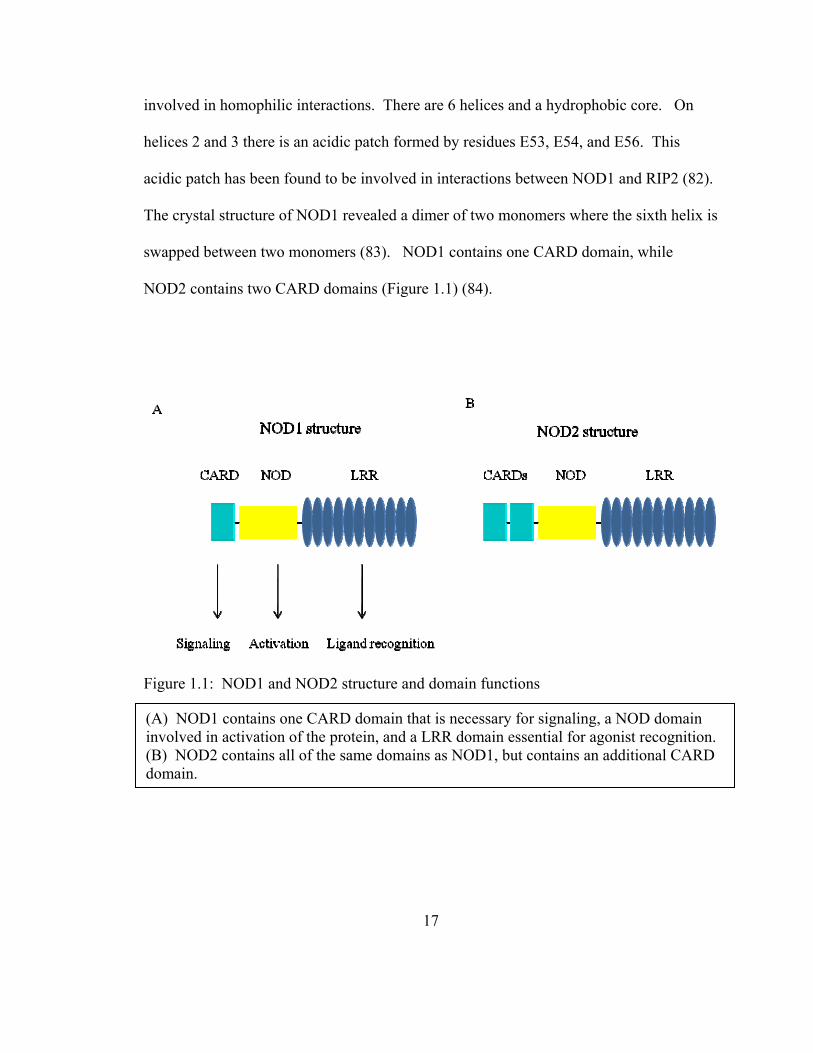
involved in homophilic interactions. There are 6 helices and a hydrophobic core. On
helices 2 and 3 there is an acidic patch formed by residues E53, E54, and E56. This
acidic patch has been found to be involved in interactions between NOD1 and RIP2 (82).
The crystal structure of NOD1 revealed a dimer of two monomers where the sixth helix is
swapped between two monomers (83). NOD1 contains one CARD domain, while
NOD2 contains two CARD domains (Figure 1.1) (84).
Figure 1.1: NOD1 and NOD2 structure and domain functions
(A) NOD1 contains one CARD domain that is necessary for signaling, a NOD domain involved in activation of the protein, and a LRR domain essential for agonist recognition. (B) NOD2 contains all of the same domains as NOD1, but contains an additional CARD domain.
17

18
1.9.6 NOD agonists
The NLRC proteins, NOD1 and NOD2, recognize specific muropeptides found in the
peptidoglycan layer of Gram-positive and Gram-negative bacteria. The repeating units of
N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) are arranged in
parallel by stem peptides made up of amino acids. Cleavage of these chains leads to the
agonists for NOD1 and NOD2. NOD2 recognizes muramyl dipeptide (MDP), which is
found in Gram-positive and Gram-negative bacteria, while NOD1 recognizes meso-
diaminopimelic acid (meso-DAP) only found in Gram-negative bacteria (85). Recently,
it was found that NOD2 recognizes an N-glycolylated form of MDP (GMDP) that can be
found in Mycobacterium tuberculosis (M.tb). The chemically synthesized GMDP is a
more potent stimulator of the immune response (86). Likewise the synthetic truncated
form of DAP, γ-D-glutamyl-meso-DAP, is sufficient to activate NOD1 (87,88).
1.9.7 Generation of NOD agonists
Even though the agonists are positioned in the peptidoglycan layer there are several ways
these muropeptides can be generated by the bacteria and host. As bacteria replicate, cell
division occurs, peptidoglycan remodeling occurs, and small fragments are released (89).
During peptidoglycan biosynthesis bacteria are also able to produce small molecules such
as MDP and iE-DAP (89-91). Lysozyme is secreted from the host in tears, saliva,
mucous, and is also found inside phagocytic lysosomes in order to breakdown the cell
wall of bacteria. This glycoside hydrolase catalyzes the hydrolysis of MurNAc and
GlcNAc thereby generates muropeptides, which include MDP and iE-DAP (92). N-

19
acetylmuramoyl-L-alanine amidases are found in bacteria and host cells. This amidase
cleaves the bond between MurNAc and the stem peptide in order to generate MDP.
There are proposed models for the agonists to get into the cytosol from inside the
phagosome in order to be recognized by NLRs. One mechanism is bacteria can divide
inside the phagosome and cause it to burst, in turn releasing the bacteria as well as any
peptidoglycan fragments that may have been generated by bacterial replication or host
response factors. The other method is through bacterial secretion systems (93).
1.10 NLRs and innate immunity
NLRs play a major role in innate immunity. They are a second line of recognition and
are responsible for inducing a pro-inflammatory response to bacteria that get inside the
macrophage. While there are 23 NLRs with a wide range of known functions including
cell signaling and cell death/survival pathways, the NLRC family’s established function
is in inducing an inflammatory response through NF-κB upon recognition of specific
muropeptides from bacteria.
1.11 NOD2 signaling cascade
The determining region of whether a NOD protein is active or inactive has been revealed
by mutational analysis to be the most carboxy-terminal region of the NOD domain also
known as the “switching region” (79). Upon recognition of MDP, the NOD domain of
NOD2 undergoes oligomerization, which allows the CARD domain to be exposed

20
leading to an electrostatic interaction with the CARD domain of the adapter protein RIP2
(85,88). This in turn causes autophosphorylation of RIP2 at serine 176 in cell lines and
murine macrophages (94). The NOD2-RIP2 complex then binds TGF activated kinase 1
(TAK1), which can then either directly activate by phosphorylation of the inhibitor of
NF-κB, IκBα, and or activate the MAPKs and thus indirectly allow for NF-κB and AP-1
activation leading to the production of pro-inflammatory cytokines (95,96). More
recently researchers discovered that NOD2 can directly bind and activate caspase-1 and
interact with the NALP1/NALP3 inflammasome, which causes the activation of caspase-
1, a necessary enzyme needed to cleave pro-IL-1β into its active secreted form (97)
(Figure 1.2). Also, a truncated form of NOD2 exists that is able to negatively regulate
NOD2 activity (98).
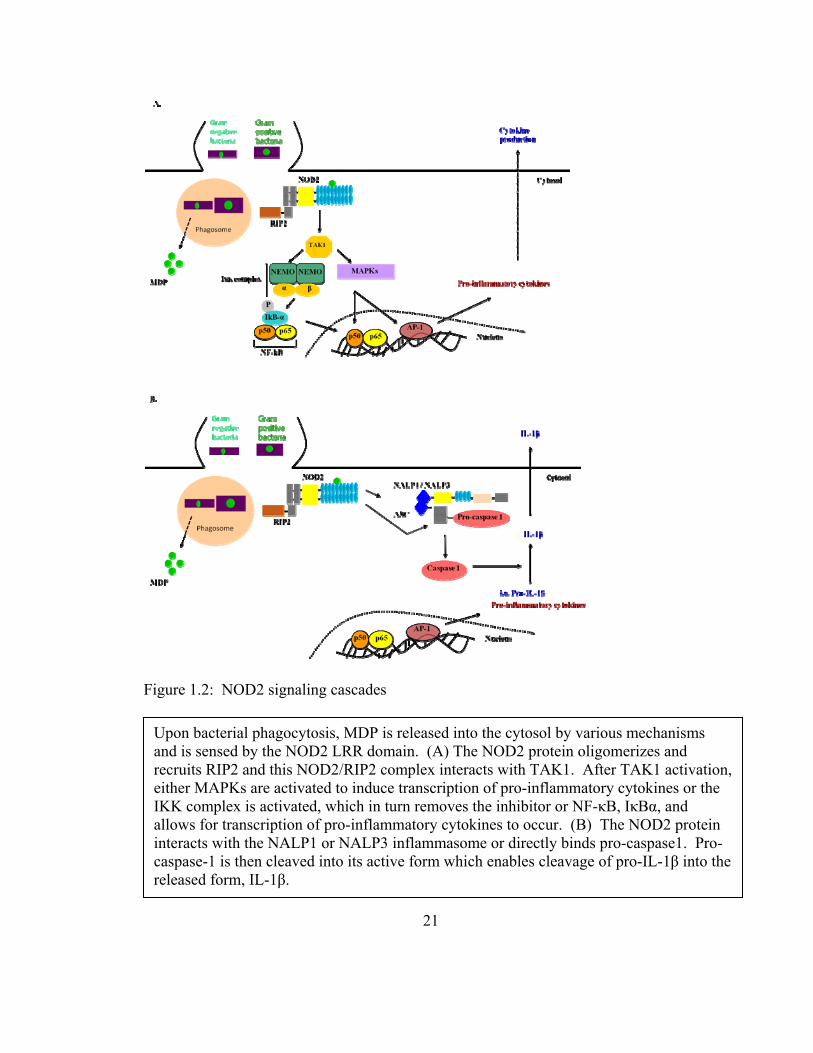
Figure 1.2: NOD2 signaling cascades
Upon bacterial phagocytosis, MDP is released into the cytosol by various mechanisms and is sensed by the NOD2 LRR domain. (A) The NOD2 protein oligomerizes and recruits RIP2 and this NOD2/RIP2 complex interacts with TAK1. After TAK1 activation, either MAPKs are activated to induce transcription of pro-inflammatory cytokines or the IKK complex is activated, which in turn removes the inhibitor or NF-κB, IκBα, and allows for transcription of pro-inflammatory cytokines to occur. (B) The NOD2 protein interacts with the NALP1 or NALP3 inflammasome or directly binds pro-caspase1. Pro-caspase-1 is then cleaved into its active form which enables cleavage of pro-IL-1β into the released form, IL-1β.
21

22
1.12 NOD2 and TLR crosstalk
There are many reports on the synergy between TLR agonists and NOD2 agonists in pro-
and anti-inflammatory cytokine release (99-106). Proper signaling through NF-κB by
NOD2 and TLRs involves the same mechanism of K63-linked ubiquitination of NEMO
(107). A biochemical mechanism for the synergy between TLRs and NOD2 has been
reported. Both signaling cascades utilize TAK1/TAB/Ubc13 to activate NF-κB, which
allows TLR and NOD2 signaling to synergistically induce cytokine release. However the
primary source of this activation is different, NOD2 uses RIP2 while TLRs utilize
TRAF6 (108).
On the level of transcription it has been shown that TLR activation and cytokines induce
NOD2 mRNA expression (109,110). MDP enhances MyD88 expression, which is a
necessary adapter protein in TLR signaling and may explain the high sensitivity of TLR
responses after peptidoglycan priming (111). The location of the NOD proteins may also
be a factor in crosstalk between TLRs and NOD2. While NOD2 is found in the cytosol,
studies have shown that it can be found localized at the cell-membrane and this
localization may be important in signaling (112-114). The proximity of NOD2 at the
membrane where TLRs can be found also suggests a mechanism for crosstalk.

23
1.13 NOD2 and microbial immunity
The biological function of NOD2 in innate and adaptive immune responses is poorly
understood; however, studies over the past decades on purified or synthetic MDP have
given some insight. In animal models the administration of MDP induces activity against
bacteria, fungi, viruses, protozoa, and tumor growth (115). The microbicidal activity of
MDP can be explained by the induction of TNF-α and IL-1β. This was thoroughly tested
by antibody neutralization studies (116-121). Further studies proved that MDP activates
NF-κB and that all MDP-inducible genes require NOD2 activity (122). MDP has been
described to be involved in the secretion of multiple immune mediators that are important
in pathogen clearance: pro-inflammatory cytokines (TNF-α, IL-1α, IL-1β), chemokines
(IL-8, CCL3, CCL4), and hematopoietic growth/survival factors (IL-6, G-CSF, GM-CSF)
(116-119,121-123). Adhesion molecules (ICAM1, CD44, TNFAIP6) known to be
important in the recruitment of inflammatory cells are also induced upon MDP
stimulation. NOD2 has also been shown to play a role in linking the innate and adaptive
immune response due to its adjuvant activity. MDP generates antigen specific T- and B-
cell responses, delayed-type hypersensitivity, and antibody production (115). This
activity is due to the ability of MDP to induce the expression of costimulatory molecules
(CD40, CD80, CD86) in monocytes and dendritic cells therefore differentiating naive T
cells into effector T cells (123-125).

24
Several microbial models have substantiated the importance of NLRs in the recognition
of pathogens including Shigella flexneri, Chlamydophila pneumonia, Pseudomonas
aeruginosa, Helicobacter pylori, Streptocooccus pneumoniae, Mycobacterium
tuberculosis, Bacillus antracis, Listeria monocytogenes, Legionella pneumophila, and
Staphylococcus aureus (77,97,126-137).
1.14 NOD2 and inflammatory diseases
Mutations in the NOD2 gene, CARD15, have been linked to many inflammatory diseases
including: Crohn’s disease, Blau syndrome, and early-onset sarcoidosis (138-141).
Mutations are found in the NOD domain for Blau syndrome and early-onset sarcoidosis,
while mutations in the LRR domain are linked to Crohn’s disease. Crohn’s disease
presents as inflammation of the gut along with granulomas, and is mediated by over
production of IL-1β, IL-6, IL-12, and TNF-α. There are two effective therapies; one is
neutralizing antibodies against TNF-α, IL-6, and IL-12 or NF-κB inhibitors and the
second is antibiotics (142). While debated, a correlative study reported that patients with
Crohn’s disease are also likely to be infected with Mycobacterium avium subspecies
paratuberculosis (143,144).
Studies have described mutations in the LRR domain as resulting in a ‘loss-of-function’
and ‘gain-of-function’. A ‘loss-of-function’ mutation results in a NOD2 protein that
cannot activate the immune response. One hypothesis for the ‘loss of function’
phenotype is that this leads to excessive inflammation to the bacterial burden due to the

25
lack of NOD2-induced defense mechanisms. Another hypothesis is that NOD2 is
involved in inhibiting the TLR2-mediated macrophage response and the lack of inhibition
leads to excessive activation of NF-κB and IL-12 expression (104). A ‘gain-of-function’
mutation results in a new or abnormal function. The ‘gain-of-function’ mutation
hypothesis is derived from ‘knockout-knockin’ CARD15 mouse studies which have a
homologous mutation found in human Crohn’s disease. Stimulation of these mouse
macrophages with MDP resulted in increased activation of NF-κB and caspase-1
mediated IL-1β processing and release (145). The excessive IL-1β production could lead
to enhanced inflammation in the intestines. This study is debated as recent studies using
human peripheral blood mononuclear cells (PBMCs) from patients with the homozygous
NOD2 mutation displayed decreased IL-1β production in response to MDP (146). These
differences reported are likely due to the difference in cell type, i.e. from mouse and
human.
1.15 NOD2: human versus mouse
Researchers discovered that the promoter regions of NOD2 in humans and mice are
different. Sequence alignment analysis revealed the canonical NF-κB binding site (-16 to
-25 bp) is not found in the mouse or rat NOD2 core promoter (147). Furthermore, the
NOD2 promoter (-500 to +300 bp) sequence alignment showed that there is only 49.7%
similarity to mouse. Hu et al found that the NOD2 proximal promoter between human
and chimpanzees is 98% identical (147). This indicates that human and chimpanzee
NOD2 genes are regulated by a highly similar transcriptional mechanism, while the lack

26
of similarity between human and mouse is due to divergent mechanisms in NOD2
regulation. These differences are important to remember when studying the function of
NOD2 in the mouse model.
1.16 Tuberculosis
Mycobacterium tuberculosis (M.tb) is the infectious agent that causes tuberculosis (TB).
One third of the world’s population is currently infected with this bacillus, however only
5-10% of the people will develop clinical disease during their lifetime. In 2008, the
World Health Organization (WHO) revealed that sub-Saharan Africa and the South-East
Asia region are where TB cases are most heavily reported (148). In 2009, 1.7 million
people died from TB with most deaths in the African region. This high incidence of TB
deaths is due to the enhanced number of human immunodeficiency virus (HIV) positive
people in Africa. HIV causes patients to be severely immuno-compromised (particularly
in CD4 T cell number and function), and thus the probability of developing TB is greater.
The WHO reported that TB is the leading cause of death among people who are HIV
positive in Africa, and since 1990 HIV has been the single largest contributor to the
increase in TB cases (148). The prevalence of TB combined with the increasing lack of
effective drug therapy give reason to study the interaction of M.tb with the host in more
depth. Understanding the molecular interactions of M.tb with host immune cells will
potentially yield greater insight into the development of new targets for drug therapy as
well as vaccine design.

27
1.16.1 Drug therapy and vaccines
The first TB vaccine was attempted by Robert Koch, who identified M.tb as the causative
agent of TB. His vaccine used sterile filtrates from M.tb culture but it was not protective
against TB; however it did open the door to further studies. Two French scientists,
Albert Calmette and Camille Guerin, used a strain of mycobacteria that infects cattle, M.
bovis made attenuated, by making serial passages for 13 years. This strain, M. bovis
BCG (BCG), became avirulent in mice and vaccination of infants resulted in 90%
reduction of mortality. Vaccination by BCG is effective against M.tb in children;
however, its efficacy in adults varies from 80% to 0% (149,150). While the vaccine is
not as effective in adults, it protects children, who are a highly susceptible group to
disseminated and meningeal TB (151). While the exact reason is unknown, host and
environmental factors are attributed to the lack of efficacy (152). The search continues
for a cost effective vaccine that is protective for adults in order to one day eradicate TB.
Researchers are attempting to synthesize different types of vaccines including DNA, sub-
unit, and living-attenuated vaccines. While there is currently no new TB vaccine in phase
III clinical trials, a non-profit organization, the Aeras Global TB Vaccine Foundation, has
a portfolio of promising vaccines. With its location in high incidence areas (South
Africa and India), the company is capable of pursuing phase III clinical trials (153).
Although there are drugs to treat TB, incomplete or erratic usage has led to the
development of drug resistant strains. Currently there are two categories of drug
resistance, multi-drug resistant (MDR-TB) and extensively drug resistant tuberculosis

28
(XDR-TB) strains. MDR-TB strains are resistant to the two major first-line drugs,
Isoniazid and Rifampin, while XDR-TB strains are resistant to the same drugs as well as
fluoroquinolones, and any second-line injectible drugs (154). With 68 countries
reporting in 2010 at least one case of XDR-TB, and the cost and long treatment regimen
of current available drugs, it is clear that TB is a major public health concern and new
drugs need to be identified to control the spread of this disease (155). As of 2010, there
are ten new compounds for the treatment of active TB progressing through the clinical
development pipeline. These drugs focus on novel regimens with 2-4 months of
treatment (156).
1.16.2 Infection with M.tb
Individuals with active pulmonary TB can expel bacilli on minute aerosol droplets by
coughing. A droplet containing as few as 1-10 bacilli is enough to cause an infection in a
healthy individual (157). These droplets are small enough to subvert upper airway
immune mechanisms such as ciliary beat and mucus clearance in order to reach the
alveolus. In the alveolus the bacilli encounter antimicrobial products as well as the major
cellular component of pulmonary innate immunity, the AM. The AM engulfs the
bacteria, but M.tb is able to subvert the immune response through many different
mechanisms (158).
The unique cell wall of M.tb, consisting of glycoproteins, glycolipids, lipoglycans and
long-chain fatty acids, enables the bacteria to survive in the AM, while at the same time

29
these factors can be processed for presentation to T-cells. Over 2-12 weeks following
primary infection the bacteria spread through the lymphatics and become systemic while
the immune system recruits cells, notably T-cells, to the infected macrophages forming a
granuloma. This cellular mass allows the healthy individual to contain the bacteria and
remain asymptomatic; the individual is termed as having a latent infection. Active
disease occurs when an individual becomes immunocompromised such as by old age or
acquiring another infection, particularly HIV. At this time, the bacteria are able to
replicate within the granuloma and induce a clinically evident disease state that the host
cannot control. Reactivation occurs in the lung 80% of the time [most notably seen in the
apex of the upper lobes of the lung (159)]; the remainder of cases are called
extrapulmonary TB and occur in the lymph nodes (scrofula), spine (Potts disease), or
systemic (miliary TB) among others.
1.16.3 M.tb and alveolus interaction
Upon entry into the alveolus, M.tb encounters surfactant, and two surfactant-associated
components that regulate the early interaction of M.tb in the lung, SP-A and SP-D. SPA
has been shown in vitro to enhance PRR activity, increase phagocytosis, alter production
of pro-inflammatory cytokines, and decrease RNI and ROIs in response to stimuli
(55,160). SPA enhances phagocytosis of M.tb in mouse and human macrophages, and
also has been shown to inhibit M.tb-mediated NO production from IFNγ stimulated
mouse macrophages (161-163). In contrast, SPD decreases phagocytosis of M.tb by
binding to the mannose caps of ManLAM and inhibiting the normal interaction with the

30
MR (164). In vitro studies have shown that SPD-opsonized M.tb that do enter the
macrophage have reduced intracellular growth that is due in part to the increased
phagosome-lysosome fusion (164,165).
1.16.4 M.tb and AM receptor interaction
M.tb interacts with a number of receptors on the AM surface as well as in the cytosol. In
vitro studies have shown that non-opsonic and serum opsonized bacteria coated with
complement C3 are taken up by the CR3 on macrophages (166). While CR3 plays a
clear role in phagocytosis, its role in pathogenesis is unclear. No affect was seen on the
production of ROIs, NO or bacterial survival in CR3 deficient murine BMMs infected
with M.tb (167) or in vivo in the mouse (168). Whether CR3 functions differently in the
mouse compared to man with regards to TB pathogenesis is unknown. The importance of
the MR during M.tb infection is better understood. M.tb is recognized by the MR through
its mannosylated lipoarabinomannan (ManLAM) found on the cell wall. Upon
phagocytosis by the MR, M.tb resides in a unique phagosome that does not fuse with the
lysosome. Scavenger receptor A (SR-A) and CD-14 have been shown to mediate
phagocytosis of unopsonized M.tb in different cell types (169,170). Avirulent and
attenuated mycobacterial strains have been shown to induce dectin-1/TLR2 TNF-α
production in murine macrophages (171). FcγRs mediate the phagocytosis of M.tb
opsonized with immunoglobulin in the immune host (172,173). The 19 kDa lipoprotein,
lipomannan (LM), and lower order PIMs found on the surface of the mycobacterial cell
wall have all been shown to interact with TLR2 (174-176).

31
Recent studies have provided data to support M.tb activating intracellular receptors, i.e.
NLRs. Masters et al published that M.tb inhibits inflammasome activation and IL-1β
production in primary murine macrophages; however, using a human monocytic cell line
researchers were able to show M.tb does activate the NLRP3/ASC inflammasome
(177,178). Although the ligand has not been determined, several reports indicate that
NOD1 and NOD2 recognize M.tb (127,135,179-181). Our recent data presented in this
dissertation provide evidence that NOD2 is important in controlling the growth of M.tb in
human macrophages and this is may be due to the role of NOD2 in inflammatory
cytokine production (77).
1.16.5 M.tb and phagosome environment
Once inside the macrophage M.tb resides in a unique phagosome. The environment in
the phagosome has a pH of 6.4, due to limited fusion of pre-formed lysosomes (182,183).
Phagosomes containing M.tb resemble early endosomes. The M.tb cell wall component,
ManLAM, is able to inhibit calcium increase in the cytosol that occurs normally during
phagocytosis and without this calcium PI3K does not become activated (184). M.tb also
inhibits sphingosine kinase, which inhibits calcium efflux (185). Due to the ability of
M.tb to inhibit calcium- and Rab5-dependent recruitment of PI3K, PI3P is not formed on
the membrane and there is a lack of Rab5 activity. This lack of activity inhibits full
maturation of the phagosome by the lack of recruitment of Rab5 effector proteins EEA1
and syntaxin-6 (186). Entry through the MR leads to an anti-inflammatory program

32
along with a unique phagosome that has reduced fusion with the lysosome (187,188).
This phagosome does not acidify normally in part because it does not acquire all subunits
of the vacuolar proton ATPase (189,190).
1.16.6 M.tb secretion systems
The complex, thick and relatively impermeable cell wall of mycobacteria suggests the
need for a secretion system, and in fact there are many secretion systems identified in
mycobacteria including early secretory antigenic target 6 system 1 (ESX-1) through
ESX-5. Many reports provide evidence for the release of proteins by M.tb into the
supernatants of in vitro broth cultures (191). Specifically, culture filtrate proteins of 10
kDa (CFP10) and early secreted antigen target of 6 kDa (ESAT6), which are products of
the ESX-1 secretion system, are particularly important. These two proteins are encoded
by genes found in the region of difference 1 (RD1) and are attributed to M.tb virulence
(192,193). During serial passage, the attenuated BCG lost 38 open reading frames,
including the RD1 (192,193).
Putative analysis of the clustering of genes that encode membrane associated proteins
with the genes that encode CFP10 and ESAT6 led to the idea and eventual confirmation
of the ESX-1 secretion system (194-196). Addition of the RD1 region back into BCG
restored the production of ESAT6. Also, disruption of genes in the ESX-1 locus,
Rv3870, Rv3871, and Rv3877 inhibited secretion of CFP10 and ESAT6. Further studies
indicate that there are 14 possible genes in the RD1 necessary for optimal function of

33
ESX-1, and a number of these proteins have been associated with known functional
domains. These include Rv3868, a putative cytoplasmic chaperone with an AAA+
ATPase domain, Rv3883c (MycP1), a subtilisin-like serine protease, and
Rv3870/Rv3871, which together form an FtsK/SpoIIIE-like ATPase. Rv3860, Rv3877,
Rv3881c, and Rv3882 are involved in ESAT6 secretion but have no known functions due
to lack of homology with described proteins. These proteins are thought to be located in
the cytoplasmic membrane. Rv3877 is predicted to be a multi-trans-membrane-spanning
protein and hypothetically could be the translocation pore in the cytosolic membrane
(191). The discovery of a second gene cluster, the Rv3614c-Rv3616c locus, involved in
ESAT6 secretion has given further complexity to the ESX-1 system (197,198). The lack
of similarity to type I- type VI secretion systems and the identification of additional ESX-
1 systems within mycobacteria and other Gram-positive species has led to the
characterization of a type VII secretion system (191).
While there is no structure determined yet for the type VII secretion system there are
protein-protein interaction studies that indicate the mechanism of secretion. In order for
secretion to occur, ESAT6 and CFP10 form a tight dimer held together by hydrophobic
interactions. These two proteins are dependent on each other for secretion (199-201).
Yeast two-hybrid screening showed Rv3871 interacts with Rv3870 along with CFP10.
One hypothesis is that Rv3871 is involved in recognition of the CFP10-ESAT6 substrate
pair and delivers it in an ATP-dependent manner to Rv3870 and thereby to the cell
membrane secretion machinery. This is mediated by an interaction of Rv3871 with the

34
carboxy-terminus of CFP10 (199). This carboxy-terminus is similar to the secretion
signal found in the type IV secretion system (202-204). EspA or Rv3616c and Rv3615c
are also substrates that are secreted similarly to CFP10 and ESAT6 (198). All of the
substrates of the ESX-1 system are dependent on each other for secretion. While the
mechanism is not yet understood, in the absence of EspA or Rv3615c, the CFP10/ESAT6
dimer is not secreted (197,198).
Although their roles in virulence are not as clear as the type VII system, mycobacteria
contain two other types of secretion systems, general secretion pathways (Sec) and the
twin-arginase transport (Tat) pathway. Mycobacteria contain a general secretion
pathway, like all bacteria, in order to secrete unfolded proteins across the cytosolic
membrane. SecA2 was identified in mycobacteria as the non-essential homologue of
SecA and its role in pathogenesis is unclear. However, SecA2 functions in L.
monocytogenes as an accessory protein to secrete enzymes that remodel the
peptidoglycan layer and these broken down products are shown to modify the innate
immune response to favor bacterial colonization (205). The mycobacterial SecA2 mutant
induces a more robust inflammatory immune response in infected macrophages
indicating a role for it in suppressing the immune response (206). The Tat pathway is a
Sec-independent pathway that has the ability to transport folded protein substrates across
the plasma membrane (207,208). In the mouse model, four M.tb phospholipase C
enzymes are secreted by Tat and are required for full M.tb virulence (207,209).

35
Secretion systems may play a key role in the ability of intracellular NLRs to sense
mycobacteria, since the bacteria reside in a distinctive phagosome separate from the
cystosol. Given that mycobacteria secrete proteins, the potential exists for these proteins
to get across the phagosomal membrane and into the cytosol of infected macrophages to
interact with NLRs. Secretion systems are reported to be important in enabling NLRs to
sense H. pylori and Legionella pneumophila (93,137,210,211).
1.16.7 M.tb and entry into the cytosol
It is widely accepted that M.tb resides in a unique phagosome, which allows for its
survival in antigen presenting cells along with the recruitment of CD4 T cells though
MHCII-based antigen presentation. However, the mechanisms underlying the ability of
virulent M.tb to use MHCI-based antigen presentation in order to recruit CD8 T cells
remains less clear.
While still debated, there is evidence that M.tb can escape from the phagosome and reside
in the cytosol. The first report, using electron microscopy (EM), found that at 18-24 h 60
– 100% of endocytosed M.tb H37Rv by rabbit alveolar macrophages resulted in bacteria in
the cytosol. While the less virulent strain, M.tb H37Ra was located in intact phagosomes
99% of the time (212). Another report found, also using EM, that M.tb H37Rv was
located in the cytosol or vesicles 50% of the time four days post infection. The authors
found the M.tb H37Ra was intermediate, while BCG was never found in the cytosol (213).
These results are debated due to the cell types as well as the harsh fixation techniques

36
used to prepare samples for EM. The cells in this study were J744.16, a mouse
macrophage cell line, and also human monocytes. It is important to note that monocytes
do not express the MR, and M.tb entry through this receptor causes a decrease in
phagosome-lysosome fusion (188,214).
Recently, another group reported visualizing M.tb in the cytosol using cryogenic-EM, a
more credible method for the preparation of samples when using EM. Cryogenic-EM is
the use of low temperatures in order to keep the sample in its native state. They also
used immunogold labeling, which allowed visualization of phagolysosomes. Using
monocyte-derived dendritic cells, the authors report visualizing M.tb and M. leprae in the
cytosol 48 to 96 hours post infection. While they did visualize BCG in a phagolysosome
7 days post infection, no bacteria were found in the cytosol. The data also indicate the
genes encoding CFP-10 and ESAT6 are necessary for mycobacterial entry into the
cytosol (215). The presence of M.tb in the cystosol has implication for recognition by
intracellular NLRs.
1.17 M.tb and NOD2
Recent studies provide evidence that NOD2 is involved in the pathogenesis of
mycobacterial infections. For example, a genome analysis of patients with leprosy found
a single nucleotide polymorphism in the NOD2 gene (216) which is linked to
Mycobacterium leprae susceptibility. Furthermore, African American patients with TB
have variants in their NOD2 gene rendering them more susceptible to M.tb infection

37
(217). In addition, polymorphisms in CARD15, which encodes NOD2, have been linked
to inflammatory diseases, including Crohn’s disease (138,218), whose cause is speculated
to be of bacterial origin (219). The association of Crohn’s disease with Mycobacterium
avium subspecies paratuberculosis continues to be reported (144).
The majority of studies to examine the role of NOD2 in the host response to M.tb
infection have been done in mice, murine macrophages, or in transfected cell lines
(127,135,179-181). There are limited reports using either normal PBMCs, PBMCs from
patients with the NOD2 mutation associated with Crohn’s disease, mixed
bronchoalveolar lavage (BAL) leukocytes, and none prior to our work using human
monocyte derived macrophages (MDMs) (127,220). A recent report indicates that NOD2
and TLR pathways are non-redundant in the recognition of M.tb, but can synergize to
induce a robust pro-inflammatory response (127). In addition, one study shows that RIP2
polyubiquitination occurs during M.tb incubation in a NOD2-dependent manner in
BMMs (180). Other studies in mouse macrophages have shown that NOD2 does not
have a significant role in controlling M.tb growth during early infection (179) but may
have during late infection (135). A decrease in pro-inflammatory cytokine production
was observed in mouse NOD2 knockout BMMs and naive murine AMs in response to
M.tb without affecting intracellular bacterial growth (135,179). Studies using human
macrophages report the same role for NOD2 in cytokine production in response to M.tb;
however in contrast, data indicate an important role for NOD2 in controlling the growth

38
of M.tb (77). This finding is yet another example of the difference between human and
mouse.
1.18 Mouse and human differences in the immune system
While the human and mouse genomes only differ in approximately 300 genes, they differ
greatly on development, activation, and response to challenge in the innate and adaptive
immune system (221). For example, there are differences in the bronchus-associated
lymphoid tissue that is present in the mouse but not human (222). Though it is not clear
the consequence, mice have more lymphocyte rich blood compared to humans who have
more neutrophils present (223). In the area of microbial defense, defensins are produced
by neutrophils in humans and by Paneth cells in mice. Humans produce 2 defensins from
Paneth cells, compared to mice, which produce over 20 defensins from this cell type
(224). NO production is easily detected in mouse cells whereas it is more difficult to
measure in human macrophages. It is debated whether or not the assays used to detect
NO are sensitive enough to measure the NO produced in human macrophages, but it is
clear that mouse macrophages produce a great deal of this antimicrobial mediator
compared to human macrophages (225). There are five structurally distinct homologues
of the receptor Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing (DC-
SIGN) in mice and only two found in humans (226). DC-SIGN is an adhesion, signaling,
and uptake receptor that functions to recognize mannose type carbohydrates found on
bacteria and fungi (227). Non-integrin FcR receptors are important links between the
innate and adaptive immune response and there are numerous differences between mice

39
and humans. Mice lack the FcαRI receptor, which is an important IgA receptor found in
humans, but they likely use other receptors for this function. FcγRIIA and FcγRIIC are
two IgG receptors expressed in humans and not mice (228,229). It has also been found
that depending on the mouse strain there are differences in the expression of IgG isotypes
as well as class switching. For example IL-4 in humans induces class switching of IgG4
and IgE, whereas in mice it induces switching to IgG1 and IgE (230). A few other
significant differences to be noted between mice and humans are in B cell development,
development and regulation of T cells, and IFN-α signaling (231-233). Finally, mice and
humans differ in immunity-related guanosine triphosphatases (IRGs), which are
important in innate immunity against pathogens and play a role in autophagy. There are
23 identified IRG genes in mice and only 3 in humans (234,235). The enhanced number
of IRG genes in mice could potentially lead to different functional outcomes in mice
when compared to humans.
1.19 Summary
The mammalian immune system, adaptive and innate, is uniquely designed to recognize
and respond to invading pathogens. The innate immune system plays an important role
in this function through the deployment of phagocytes that have extracellular and
intracellular PRRs.
One bacterial pathogen in particular that has found mechanisms to evade the innate
immune system is M.tb. The necessity to find better methods to control this pathogen is

40
at an all time high due to the emergence of MDR- and XDR-TB strains. In order to do
this, the mechanisms for how this bacterium is recognized inside of the macrophage is
important to understand because it will allow for the development of new ideas for the
generation of novel microbe- and host-directed therapeutic approaches. The intracellular
PRR, NOD2, has emerged as an important innate immune player in the recognition and
control of pathogens in the host and data suggest that NOD2 may be important in the
pathogenesis of M.tb.
1.20 Specific aims
It is important to study the interaction between M.tb and the immune system in order to
understand its pathogenesis and find new potential drug targets. Studying M.tb’s
interaction with the first major innate immune cell it comes in contact with in the
alveolus, the macrophage, is of particular relevance. The interaction of M.tb and
membrane receptors has been well studied; however, M.tb’s interaction with cytosolic
receptors has yet to be thoroughly studied in human macrophages. We hypothesize that
NOD2 is important in controlling the growth of M.tb in human macrophages by
regulating the nature of the inflammatory response in order to contain infection. We also
hypothesize that NOD2 plays a role in several novel early innate immune responses in
human macrophages and focus on its role in iNOS expression and PKCδ activation. The
specific aims of this dissertation are as follows:
(1) Characterize NOD2 expression in human macrophages and determine the role
of NOD2 during M.tb infection of human macrophages

41
(2) Determine if NOD2 is necessary for iNOS expression in human macrophages
in response to agonists as well as M.tb
(3) Determine NOD2 signaling partners in the innate immune response,
specifically the function of NOD2 in the novel PKC signaling cascade

42
CHAPTER 2 : NOD2 controls the nature of the inflammatory response and subsequent
fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages
2.1 Introduction
Tuberculosis (TB) is currently the leading cause of death in HIV-infected persons (236).
Multi-drug resistant Mycobacterium tuberculosis (M.tb) strains have limited our ability to
use the current antibiotic supply, and the development of new antibiotics requires a better
understanding of the biological mechanisms underlying M.tb growth and survival within
the host. Humans are the only known natural host for M.tb and alveolar macrophages
(AMs) in the lung microenvironment are the first immune cells that encounter inhaled
mycobacteria (237). Thus, knowledge about pathogenic events for M.tb in primary
human macrophages is particularly important.
Macrophages are equipped with an array of defense mechanisms to recognize and clear
invading pathogens. Several studies demonstrate that M.tb interacts with macrophage
membrane receptors such as the mannose receptor (MR), complement receptors, Toll-like
receptors (TLR) and scavenger receptors (237). Engagement of specific receptor

43
pathways during phagocytosis of mycobacteria regulates the early host response through
defined signaling pathways (158,175,238). Among these are the group of cytosolic
receptors called Nucleotide-binding Oligomerization Domain (NOD)-like receptors
(NLRs) (93) which function as intracellular pattern recognition receptors (PRRs) that
recognize pathogens or pathogen secreted molecules (239).
There are 23 NLRs which can be divided into four subfamilies depending on the
composition of their N-terminal effector domain. For example, those containing a
caspase recruitment domain (CARD) as their effector domain are known as the NLRC
subfamily and consist of proteins that initiate a pro-inflammatory response by signaling
through their CARD domain. NOD2, a member of this subfamily, is found in epithelial
cells and antigen presenting cells such as macrophages (76,240,241). NOD2 regulates the
production of inflammatory mediators in response to muramyl dipeptide (MDP) which is
found in Gram-negative and Gram-positive bacteria, and mycobacteria (86,88). Many
studies have found that NOD2 is able to recognize and generate a response to a variety of
microorganisms (97,126,127,136,242-248). NOD2 senses bacteria through its leucine
rich repeats (LRR), then oligomerizes through its NOD domain and is able to recruit
receptor interacting protein 2 (RIP2) through an electrostatic interaction of the CARD
domain in each protein. This leads to a signaling cascade in which activation and
translocation of nuclear factor-κB (NF-κB) into the nucleus occurs in order to transcribe
pro-inflammatory cytokines (108,147,241,249,250).

44
As reported earlier in the introduction section (See: 1.17 M.tb and NOD2), the literature
indicates that during mycobacterial infections NOD2 is an important player in the host
response. Two genomic analysis studies revealed an association of NOD2 with
susceptibility to mycobacterial infections. Patients with Mycobacterium leprae or
African American patients with TB had polymorphisms or variants in their NOD2 gene
(216,217). Patients with Crohn’s disease, an inflammatory granulomatous disease, are
reported to have polymorphisms in their NOD2 gene (CARD15) (138,218). While
debated, it is reported that the cause of Crohn’s disease is Mycobacterium avium
subspecies paratuberculosis (144).
The role of NOD2 in the host response to M.tb infection has mainly been studied in mice,
murine macrophages, or transfected cell lines, with limited reports using PBMCs from
healthy donors, PBMCs from patients with mutations in their NOD2 gene, and mixed
BAL leukocytes (127,127,135,179-181,220). NOD2 and TLR pathways are non-
redundant in the recognition of M.tb, but can synergize to induce a robust pro-
inflammatory response (127). In addition, RIP2 polyubiquitination occurs during M.tb
incubation with BMMs in a NOD2 dependent manner (180). While a decrease in pro-
inflammatory cytokine production in NOD2 knockout BMMs and naïve murine AMs in
response to M.tb was found, NOD2 did not control intracellular bacterial growth during
early in vivo infection in mice (179) but did during late infection (135).

45
Interest in the potential interactions between M.tb and cytosolic PRRs such as NOD2
continues to grow based on recent reports that M.tb secretes virulence factors and
immune activators or suppressors through a type VII secretion system (191,251,252).
Other reports provide evidence that M.tb may be able to escape the phagosome to the
cytosol under specific conditions (213,215).
Based on the known differences between human and mouse macrophages in receptor
expression, signaling pathways, and innate immune function (253-256), we decided to
study NOD2 in human macrophages and in response to M.tb by optimizing an antibody
to NOD2 and by accomplishing effective small interfering RNA (siRNA)-mediated
knockdown of NOD2 in these cells. We show that NOD2 expression increases during
monocyte differentiation. In contrast to previously published work using mouse
macrophages, we report that NOD2 is important in controlling both pro-inflammatory
cytokine production and the growth of M.tb in human macrophages; whereas NOD2
controls the growth of the related attenuated BCG strain in both human and mouse
macrophages.
2.2 Materials and Methods
2.2.1 Buffers and reagents
Dulbecco’s PBS with and without Ca2+ and Mg2+ ions, Iscoves, FBS, TRIzol, and RPMI
1640 medium with L-glutamine were purchased from Invitrogen (Invitrogen Life

46
Technologies). RHH medium [RPMI 1640 plus 10 mM HEPES (Invitrogen) plus 0.4%
human serum albumin (CSL Behring LLC)] was used for cell culture experiments. N-
Acetylmuramyl-L-alanyl-D-isoglutamine hydrate (MDP; Sigma-Aldrich) and H-Ala-D-γ-
Glu-diaminopimelic acid (iE-DAP; AnaSpec) were used for functional assays involving
ELISAs. 7H11 agar was prepared with Bacto Middlelebrook 7H11 agar, oleic acid-
albumin-dextrose-catalase enrichment medium, and glycerol (Difco Laboratories).
2.2.2 Antibodies
Unconjugated rabbit anti-human NOD2 was purchased from ProSci Inc. and another anti-
human NOD2 antibody was obtained from Dr. Nunez, University of Michigan, and
available by ebioscience. Syk (N-19), TLR2, actin, goat anti-rabbit IgG-HRP and
donkey anti-goat IgG-HRP antibodies were purchased from Santa Cruz Biotechnology.
The aforementioned antibodies were used for Western blot. The NOD2 antibody from
ProSci Inc., Alexa Fluor 488-conjugated goat anti-rabbit IgG (Molecular Probes and
Invitrogen) and normal rabbit IgG (Santa Cruz Biotechnology) were used for confocal
microscopy experiments. Monoclonal anti-human NOD2 (Cayman Chemical) and anti-
mouse IgG1 kappa (Abcam) were used for IP experiments. Antibodies specific for
phospho-RIP2 (Ser176) and phospho-IκB (Ser32) were purchased from Cell Signaling
Technology. The caspase-1 antibody was a kind gift from Dr. Mark Wewers, Ohio
State University.

2.2.3 Bacterial strains and culture
Lyophilized M.tb H37Rv (ATCC #25618) and BCG (ATCC #35734) were obtained from
American Tissue Culture Collection, reconstituted, and used as described (172). The
concentration of bacteria (1–2 x 108 bacteria /mL) and the degree of clumping ( 10%)
were determined by counting in a Petroff-Hausser chamber. Bacteria prepared in this
fashion are 90% viable by CFU assay.
2.2.4 Isolation and culture of human monocytes and macrophages
Blood obtained from healthy, Purified Protein Derivative negative human volunteers
using an approved protocol by The Ohio State University Institutional Review Board
(IRB) was processed as described (257). Briefly, PBMCs were isolated from heparinized
blood on a Ficoll-Paque cushion (Amersham Biosciences/GE Healthcare) and cultured in
Teflon wells (Savillex) for 1 (monocytes) through 5 (MDMs) days in the presence of
RPMI 1640 medium containing 20% autologous serum (2.0 x106 PBMCs/mL) at
37ºC/5% CO2 (258). Human AMs were isolated from BALs of healthy human donors
as previously described (161) using an approved protocol by The Ohio State University
IRB. Briefly, the BAL was centrifuged (200 x g, 4°C, 10 min), supernatant was removed,
and the pellet re-suspended in RPMI and washed two more times with RPMI. AMs were
plated in tissue culture plates with 10% human serum supplemented with penicillin
(1000U/mL), incubated in CO2 incubator at 37ºC for 2 h, washed with warm RPMI to
remove non-adherent cells and penicillin, and used for experiments.
47

48
2.2.5 Mice and mouse macrophages
Wild-type C57BL/6 and NOD2-/- mice were backcrossed to C57BL/6 background for 10
generations (242). BMMs were prepared from femurs of five to eight week-old mice as
previously described (137,259).
2.2.6 RNA isolation
One to 5 day-old PBMCs in Teflon wells were harvested and mononuclear phagocytes
adhered to a 6-well tissue culture plate with 10% autologous serum (8 x 106 PBMCs/mL).
After washing away lymphocytes, the monocytes or MDMs (8 x 105 cells) were lysed in
TRIzol and total RNA was isolated by using the Qiagen RNAeasy column method. The
Experion (Bio-Rad) was used to determine the quality and quantity of RNA samples.
2.2.7 Gene expression studies by qRT-PCR
RNA (100ng) was reverse transcribed to cDNA by reverse transcriptase enzyme,
(SuperScript II, Invitrogen) and qRT-PCR was performed using a human NOD2 or IL-1β
TaqMan gene expression kit (Applied Biosystems). NOD2 and IL-1β amplifications
were normalized to β actin as a housekeeping gene (∆Ct). Relative copy number (RCN)
and fold change were determined. RCN was calculated as follows: RCN = E-∆Ct x 100,
where E is the efficiency (2 = 100% efficiency) and Ct = Ct (target) – Ct (reference)
(260). Fold change was calculated from the RCN values compared to day 1 for NOD2

49
mRNA experiments, and compared to the siRNA resting group for IL-1β mRNA
experiments. Triplicate samples were analyzed in duplicate wells in each experiment.
2.2.8 Confocal microscopy
Untransfected MDMs or transfected (scramble siRNA or NOD2 siRNA) MDMs (2 x 105)
were adhered to a glass coverslip in a 24-well tissue culture plate for 2 h at 37°C and
washed to remove lymphocytes. The cells were washed, fixed with 2% paraformaldehyde
(10 min at room temperature), permeabilized by treatment with 100% methanol for 4
min, washed and then incubated overnight in 10% non-fat milk in PBS as blocking
reagent. The cells were then incubated with NOD2 (2.5µg/mL) or the appropriate isotype
(2.5µg/mL) control antibody for 1 h at room temperature, washed with blocking
reagent, and counterstained with an Alexa Fluor 488-conjugated secondary antibody
(1:500 dilution) for 1 h at room temperature. Nuclei were labeled with 0.1g/mL of the
DNA stain 4,6-diamidino-2-phenylindole (DAPI; Invitrogen/Molecular Probes) in PBS
for 5 min at room temperature. The cells were next washed with PBS and coverslips
were mounted on glass slides. Slides were viewed using an Olympus Flowview 1000
Laser Scanning Confocal microscope.
2.2.9 Transfection of MDMs
PBMCs were transfected with Accell scramble siRNA or NOD2 siRNA purchased from
Thermo Scientific Dharmacon RNAi technologies that targeted the sequence

50
CUUUAGGAUGUACAGUUA (368nM) using the Amaxa Nucleofector (Lonza Group
Ltd) Briefly, PBMCs (1× 107) were resuspended in 100 μl of nucleofector solution
followed by the addition of 5ug/mL of scramble siRNA or NOD2 siRNA, incubated at
room temperature for 5 min, and nucleofected according to the manufacturer’s
instructions. PBMCs were then seeded on 12-well plates containing 1.0 ml of RPMI
supplemented with 10% autologous serum and incubated for 2 h at 37°C and 5% CO2.
After 2 h, adhered transfected cells (MDMs) were washed and repleted with warm RPMI
containing 20% autologous serum for 72 h. Transfected cells were used for subsequent
experiments, such as CFU assays, cytokine assays, confocal microscopy, cell
enumeration assays, and Western blotting. Assessment of monolayer density in
experiments was performed as described below.
2.2.10 Macrophage stimulation / infection, cell lysis, immunoprecipitation, and western
blotting
Day 5 MDMs or MDMs transfected with scramble siRNA or NOD2 siRNA (2.0 x 105)
were incubated with M.tb or BCG (MOI 5:1; triplicate wells) in RHH for 30 min at 37°C
in 5% CO2 on a platform shaker for equal dispersion of bacteria followed by an additional
incubation for 90 min without shaking. The cells were then washed three times with
warm RPMI, repleted with RPMI containing 1% human autologous serum and incubated
for 24 h at which time supernatants were collected for ELISAs (see below). For
experiments using the NOD2 ligand MDP, 2.0 x 105 day 5 MDMs were adhered to a 24-
well plate for 2 h at 37°C in 5% CO2, washed to remove lymphocytes, and then incubated

51
with MDP (5ug/mL) for 90 min. Cells were then incubated with bacteria as described
above (2 h total), washed and repleted with MDP in 1% autologous serum for 24 h. Cell
culture supernatants were harvested and used for ELISAs. The monolayers were washed
once with PBS and lysed in TN-1 lysis buffer [50 mM Tris (pH 8.0), 10 mM EDTA, 10
mM Na4PO7, 10 mM NaF, 1% Triton X-100, 125 mM NaCl, 10 mM Na3VO4, 10
µg/ml aprotinin, and 10 µg/ml leupeptin)], incubated on ice for 10 min and then
centrifuged at 17,949 x g for 10 min at 4°C to remove cell debris (261). For infections,
MDM monolayers were incubated with M.tb or BCG in RHH for 15 min, 30 min, 1 h, 1.5
h, 2 h, and 3 h (MOI 5:1). At each time point, cells were washed with PBS and lysed as
above. For NOD2 knockdown assessment, lysates were collected as above at 72 h and 96
h post-knockdown. Protein concentrations of the cleared lysates were measured using the
BCA-protein assay kit (Pierce). Proteins were separated by SDS-PAGE and analyzed by
Western blot by probing with the primary and secondary antibody of interest and
development using ECL (Amersham Biosciences). For the IP experiment whole cell
lysates were collected from HEK293T (empty vector) and NOD2-expressing HEK293T
cells. Equal amounts of protein were incubated overnight with NOD2 antibody (Cayman
Chemicals) or control IgG1, kappa. Then cells were incubated for 2 h with Protein G
agarose beads (Invitrogen), washed, pellets boiled in Laemmli buffer and loaded on a
7.5% SDS-PAGE gel, and Western blot was performed using a NOD2 antibody (ProSci
Inc.). The protein band intensities were measured using the online ImageJ software
provided by the NIH. Background intensity was subtracted from each sample and then

52
normalized to β-4actin. Fold change was determined as follows: (treated sample band
intensity) / (untreated sample band intensity).
2.2.11 ELISAs
For MDM experiments, cell free culture supernatants were collected at 24 h and used to
measure TNF-α and IL-1β by ELISA (R&D Systems). For mouse macrophage studies,
WT and NOD2-/- macrophage monolayers were incubated with serum-opsonized
mycobacteria (M.tb MOI 1:1 and BCG MOI 2.5:1) in the same manner as described for
MDMs. Cell free culture supernatants were collected at 24 h, 72 h, and 168 h and
measured for IL-1β by ELISA (BD Biosciences).
2.2.12 M.tb and BCG intracellular growth assays in macrophages
A CFU assay using infected macrophage lysates was performed as described (262).
Briefly, scramble siRNA or NOD2 siRNA transfected MDM monolayers were incubated
with M.tb or BCG (MOI 5:1; triplicate wells) in RHH for 30 min at 37°C in 5% CO2 on a
platform shaker for equal dispersion of bacteria and for an additional 90 min stationary.
The cells were then washed three times with warm RPMI and either lysed or repleted
with RPMI containing 1% human autologous serum and incubated for 2 h or 24 h. The
supernatants and MDM monolayers were then processed for ELISA or enumeration of
CFUs, respectively (triplicate samples in each test group). The number of colonies was
counted after 4 weeks and data analyzed. For mouse macrophage experiments, WT and

53
NOD2-/- macrophages were incubated with serum-opsonized mycobacteria (M.tb MOI
1:1 and BCG MOI 2.5:1) in Iscoves plus 10% FBS using the same incubation protocol as
for MDMs and then lysed at 2 h, 24 h, 72 h, and 168 h.
2.2.13 M.tb and BCG cell association assay within macrophages
Scramble siRNA or NOD2 siRNA transfected MDM monolayers (2.0 x 105), or WT or
NOD2-/- mouse macrophages monolayers (2.5 x 105) were incubated with M.tb or BCG
(MOI 10:1; triplicate wells) in RHH for 30 min at 37°C in 5% CO2 on a platform shaker
for equal dispersion of bacteria and then for an additional 90 min without shaking. The
cells were then washed three times with warm RPMI and fixed with 10% formalin for 10
min at room temperature. Coverslips were then washed three times with PBS, dried and
cell-associated M.tb or BCG was stained with auramine-rhodamine (BD Biosciences).
Three hundred consecutive macrophages/coverslip/test group were counted using phase-
contrast and fluorescence microscopy (166).
2.2.14 Cell enumeration of NOD2 siRNA transfected MDMs in monolayer culture
Scramble siRNA and NOD2 siRNA transfected MDMs were plated in 24 well plates and
after 2 h, washed and repleted with 20% autologous serum for 72 h. Cells were then
washed one time with PBS and treated with 1% Cetavlon in 0.1M citric Acid with 0.05%
Napthol blue black (Sigma-Aldrich), pH 2.2, for 15 min at room temperature. Cell

54
lysates were then loaded on a hemacytometer and stained nuclei enumerated using phase
contrast microscopy (263).
2.2.15 NOD2 antibody peptide competition assay
The NOD2 antibody and the NOD2 peptide (ProSci Inc.) used to produce the antibody
were incubated in a ratio of 2:1 (peptide:antibody) at 37°C for 30 min in a 5% milk
solution in low retention tubes. Day 5 MDM lysates were run in parallel on a 7.5% SDS-
PAGE gel, transferred to a nitrocellulose membrane and the membrane cut so that each
lane could be probed with either the antibody-peptide mixture or antibody alone
overnight at 4°C. Western blotting was performed as described above. Membranes were
then developed using ECL.
2.2.16 Statistics
A paired one tailed student’s t test was used to analyze the differences between two
groups in figures showing one representative figure of three independent experiments.
An unpaired one tailed student’s t test was used to analyze differences between two
groups in figures showing cumulative data from three independent experiments.
Significance was P < 0.05.

55
2.3 Results
2.3.1 NOD2 mRNA and protein expression increase as monocytes differentiate into
macrophages
The expression of NOD2 in human macrophages has not been studied and there are few
reports on NOD2 mRNA expression in human monocytes (76,220). We began our
studies by determining the expression of NOD2 during monocyte differentiation to
MDMs in an established model by quantitative real-time PCR (qRT-PCR) and Western
blot (54). Results from qRT-PCR provide evidence that NOD2 transcript levels are
increased during the differentiation of monocytes to macrophages. While we saw
production of mRNA in day 1 monocytes, the results in Fig. 2.1A show a significant
increase in mRNA at day 3 of differentiation which remains elevated as the cells
differentiate further into mature macrophages. Next, we compared the mRNA levels with
the amount of protein produced during cell differentiation by Western blotting. Due to
the sensitivity and specificity issues in using antibodies specific for human NOD2, we
optimized an available commercial antibody (ProSci Inc.) with human macrophage
lysates. The specificity of this antibody was confirmed by a peptide competition assay
(Figure 2.1B), an immunoprecipitation (IP) experiment (Figure 2.1C), and in siRNA
experiments shown later. For IP experiments, proteins in cell lysates from HEK293T
cells expressing human NOD2 or HEK293T cells containing the empty vector were
immunoprecipitated with NOD2 antibody (Cayman Chemical) and then probed with
NOD2 antibody from ProSci Inc. As shown in Figure 2.1C, NOD2 antibody specifically
binds to the human NOD2 protein expressed in the HEK293 cell line. We were also able

56
to detect NOD2 in human lysates using the antibody available from eBioscience,
although with more background (data not shown). Thus, in all subsequent experiments
we used the antibody from ProSci Inc. In accordance with the mRNA studies, NOD2
protein levels in cell lysates increased markedly at day 3 of differentiation and remained
high on day 5 MDMs (Figure 2.1D upper panel). The densitometric analysis of Western
blots from three independent experiments shows that NOD2 protein expression levels
increased during cell differentiation (Figure 2.1D lower panel). Finally, our results were
confirmed using confocal microscopy where NOD2 protein was easily detected
throughout MDMs in a punctuate appearance (Figure 2.1E).
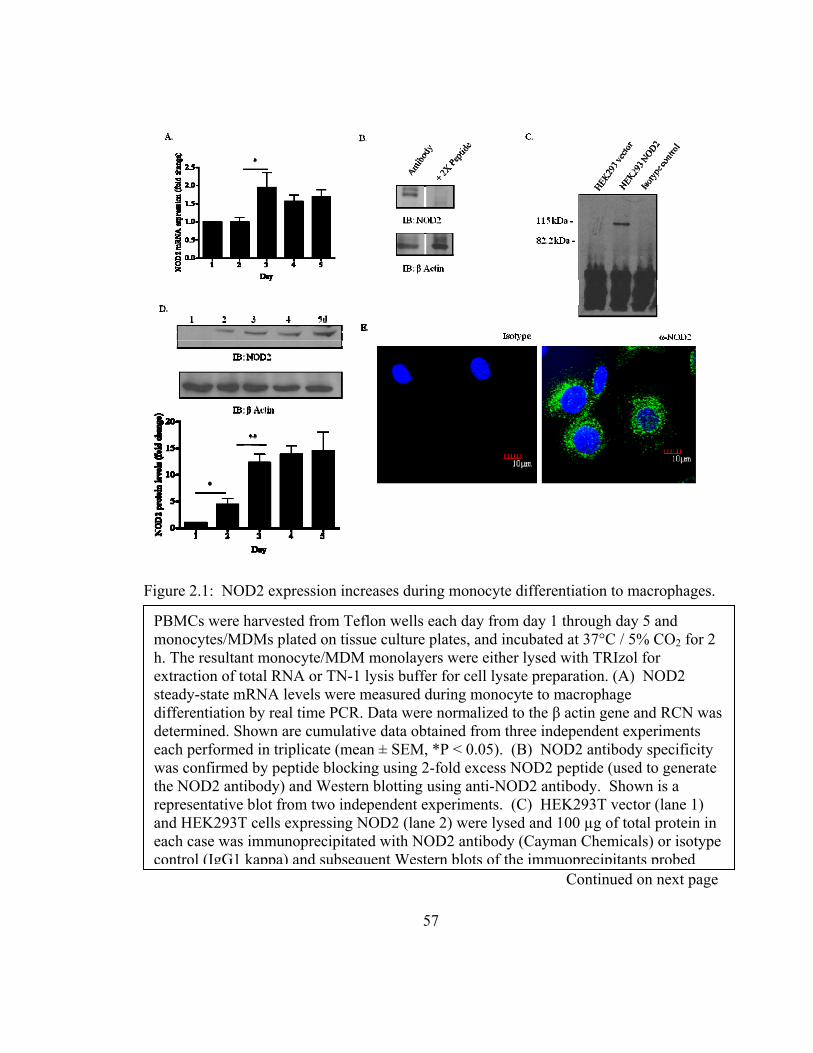
Figure 2.1: NOD2 expression increases during monocyte differentiation to macrophages.
Continued on next page
PBMCs were harvested from Teflon wells each day from day 1 through day 5 and monocytes/MDMs plated on tissue culture plates, and incubated at 37°C / 5% CO2 for 2 h. The resultant monocyte/MDM monolayers were either lysed with TRIzol for extraction of total RNA or TN-1 lysis buffer for cell lysate preparation. (A) NOD2 steady-state mRNA levels were measured during monocyte to macrophage differentiation by real time PCR. Data were normalized to the β actin gene and RCN was determined. Shown are cumulative data obtained from three independent experiments each performed in triplicate (mean ± SEM, *P < 0.05). (B) NOD2 antibody specificity was confirmed by peptide blocking using 2-fold excess NOD2 peptide (used to generate the NOD2 antibody) and Western blotting using anti-NOD2 antibody. Shown is a representative blot from two independent experiments. (C) HEK293T vector (lane 1) and HEK293T cells expressing NOD2 (lane 2) were lysed and 100 µg of total protein in each case was immunoprecipitated with NOD2 antibody (Cayman Chemicals) or isotype control (IgG1 kappa) and subsequent Western blots of the immuoprecipitants probed
57

58
with NOD2 antibody (ProSci Inc.). Shown is a representative Western blot from two independent experiments. (D) Monocyte/macrophage cell lysates were analyzed for NOD2 protein expression by Western blot using NOD2 antibody. The same membrane was re-probed with actin to verify equal loading of lysate proteins. Shown is a representative Western blot and the bar graph shows the cumulative densitometric analysis of Western blots from three different donors (mean ± SEM, *P < 0.05 ** P < 0.005). (E) Day 5 MDM monolayers on coverslips were fixed, permeabilized, and stained using either NOD2 antibody or isotype control antibody followed by Alexa flour 488 conjugated secondary antibody. Cells were then examined by confocal microscopy (Olympus FV1000). Shown is a representative section (63x mag) of a Z series image from one of three independent experiments performed on duplicate coverslips.
2.3.2 Activation of NOD2 enhances the M.tb- and BCG-mediated pro-inflammatory
cytokine response through the NF-κB pathway
We next investigated whether activation of NOD2 leads to an increase in the M.tb- and
BCG-mediated pro-inflammatory cytokine response in macrophages. Incubation of
macrophages with MDP, a NOD2 agonist, is known to induce the production of pro-
inflammatory cytokines such as IL-1β (122). A recent report indicates that in some TB
patients, NOD2 mRNA is enhanced in PBMCs; furthermore, patients on anti-tuberculosis
treatment have enhanced NOD2 levels in leukocytes (220). MDMs were pre-incubated
with the NOD2 ligand MDP for 90 min followed by incubation with M.tb or BCG as
described in the methods in the presence of MDP. Cell free culture supernatants were
analyzed for TNF-α and IL-1β secretion at 24 h. The data show an additive (TNF-α) or
synergistic (IL-1β) effect on cytokine production in cells pre-treated with MDP compared
to cells incubated with mycobacteria alone (Figure 2.2A and B). Thus, our results
indicate that pre-engagement of NOD2 by its ligand significantly enhances M.tb- or

59
BCG-mediated pro-inflammatory cytokine production. This effect is likely due to the
activation of NF-κB through the phosphorylation of IκBα and its subsequent degradation
(180,264).
To further explore the signaling pathway(s) involved, we examined whether M.tb- or
BCG-mediated signaling events include those known for the NOD2 pathway. NOD2
engagement leads to recruitment of a serine threonine kinase adapter protein called RIP2
by an electrostatic interaction through their CARD domains, which in turn causes
autophosphorylation of RIP2 at serine 176 (94) in cell lines and murine macrophages.
Activated RIP2 phosphorylates inhibitor of kBα (IkBα) resulting in the activation and
translocation of NF-κB. MDMs were incubated with M.tb or BCG for different time
points and the cell lysates were analyzed for activation of RIP2 and IkBα by Western
blotting of the phosphorylated proteins. The data show that both M.tb and BCG
phosphorylate RIP2 during incubation but with different kinetics and pattern. BCG- and
M.tb-mediated phosphorylation of RIP2 peaks at 30 min and 1 h, respectively, and the
phosphorylation decreased at 3 h during M.tb incubation (Figure 2.2C). M.tb and BCG
infection also leads to phosphorylation of serine 32 of IkBα (Figure 2.2D) which is
known to result in its degradation and subsequent activation of NF-B. We found that
phosphorylation occurs as early as 15 min for both mycobacteria and continues through 2
h for BCG but not M.tb. In addition, there is a decrease at 30 min which can be explained
by the degradation of IκBα and recruitment of newly re-synthesized pools of IκBα from
the cytoplasm, as previously described (264).
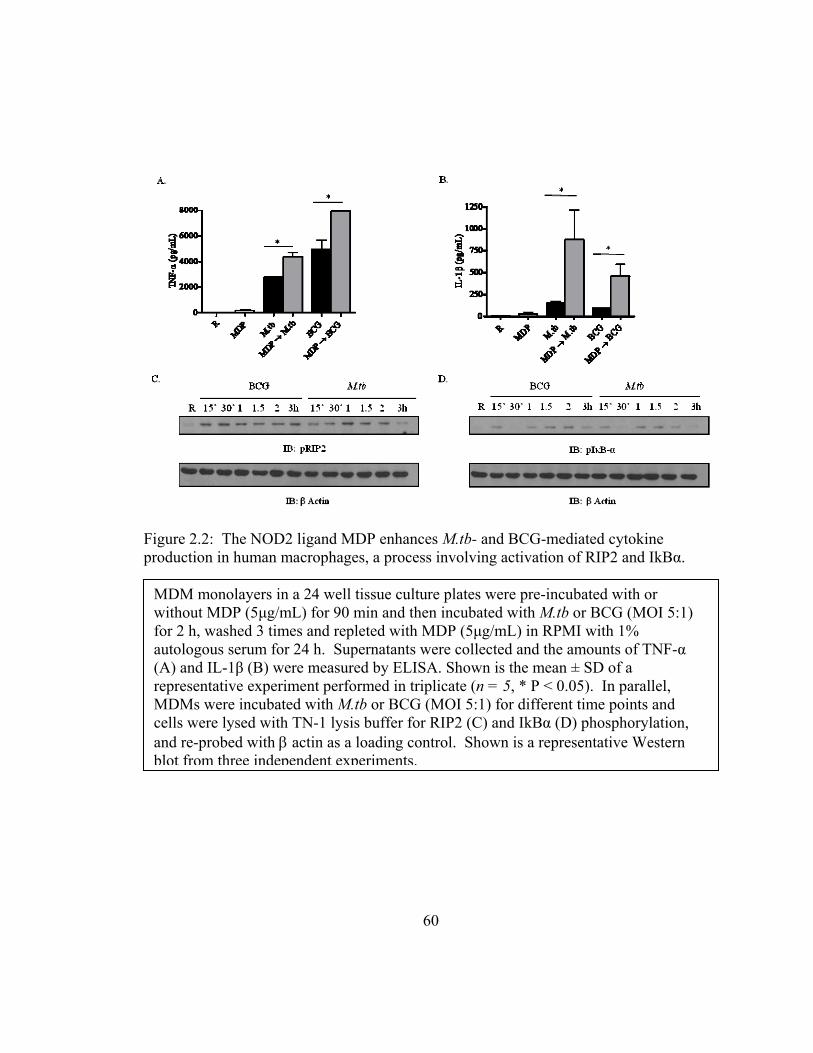
Figure 2.2: The NOD2 ligand MDP enhances M.tb- and BCG-mediated cytokine production in human macrophages, a process involving activation of RIP2 and IkBα.
MDM monolayers in a 24 well tissue culture plates were pre-incubated with or without MDP (5μg/mL) for 90 min and then incubated with M.tb or BCG (MOI 5:1) for 2 h, washed 3 times and repleted with MDP (5μg/mL) in RPMI with 1% autologous serum for 24 h. Supernatants were collected and the amounts of TNF-α (A) and IL-1β (B) were measured by ELISA. Shown is the mean ± SD of a representative experiment performed in triplicate (n = 5, * P < 0.05). In parallel, MDMs were incubated with M.tb or BCG (MOI 5:1) for different time points and cells were lysed with TN-1 lysis buffer for RIP2 (C) and IkBα (D) phosphorylation, and re-probed with actin as a loading control. Shown is a representative Western blot from three independent experiments.
60

61
2.3.3 siRNA-mediated knockdown of NOD2 activity in human macrophages leads to loss
of its activity
In order to more definitively examine the causal role of NOD2 in controlling the nature
of the inflammatory response and growth of mycobacteria in human macrophages, we
developed an effective and reliable protocol for knockdown of NOD2 in human
macrophages by using NOD2-specific siRNA and Amaxa nucleofector reagent. MDMs
were transfected with scramble siRNA or NOD2-specific siRNA under optimized
conditions, plated in tissue culture plates, and lysed after 72 h and 96 h. The cell lysates
were analyzed for NOD2 protein levels by Western blotting. Results shown in Figure
2.3A demonstrate the loss of NOD2 protein in MDMs transfected with NOD2-specific
siRNA at both time points. These results were confirmed by confocal microscopy
(Figure 2.3B). Cell enumeration of scramble siRNA- and NOD2 siRNA-treated MDMs
evaluated at 72 h post nucleofection by napthol blue black nuclear staining of cells
demonstrated no loss of cells on the monolayer in either experimental group (Figure
2.3C). There was also no difference in monolayer cell viability as seen by trypan blue
exclusion staining (95% viable in both groups, data not shown). An assay was performed
in order to verify that NOD2 protein knockdown led to loss of its function. Scramble
siRNA and NOD2 siRNA transfected macrophages were treated with MDP and the cell
free culture supernatants were analyzed for TNF-α production (Figure 2.3D). NOD2
knockdown led to a significant reduction in TNF-α production in response to MDP. We
also tested the specificity of NOD2 knockdown by stimulating the NOD2 siRNA
transfected MDMs with the NOD1 ligand H-Ala-D-γ-Glu-diaminopimelic acid (iE-DAP)
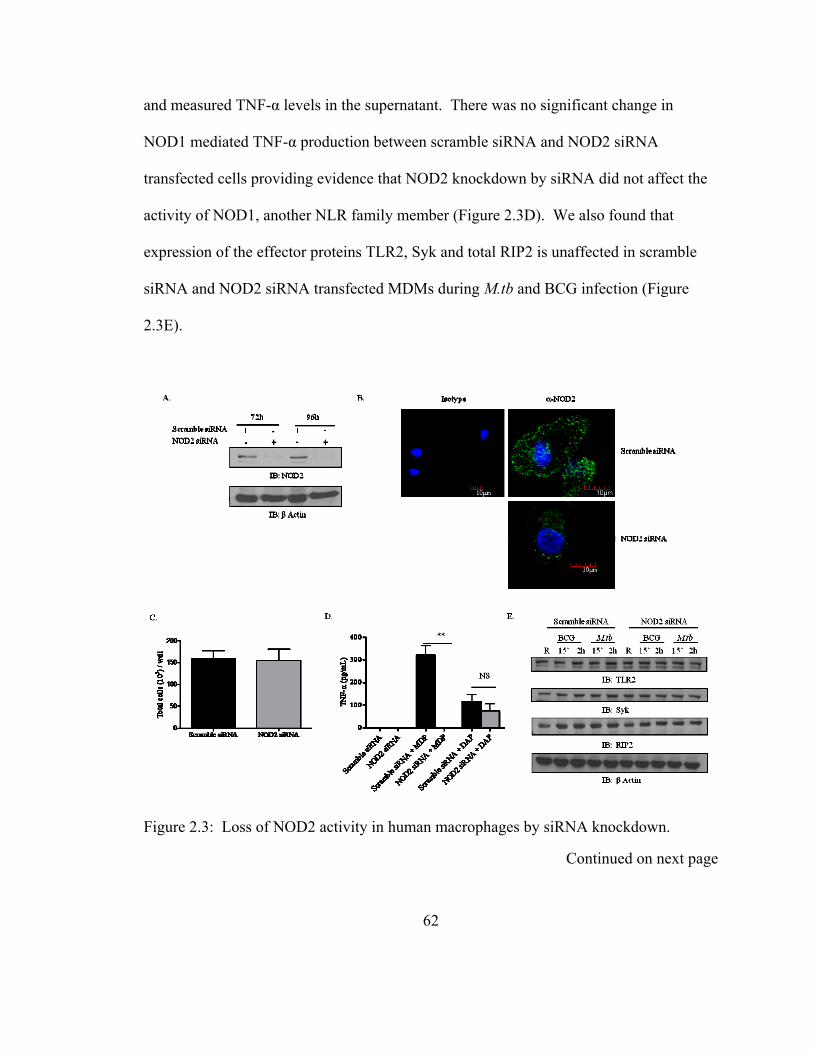
and measured TNF-α levels in the supernatant. There was no significant change in
NOD1 mediated TNF-α production between scramble siRNA and NOD2 siRNA
transfected cells providing evidence that NOD2 knockdown by siRNA did not affect the
activity of NOD1, another NLR family member (Figure 2.3D). We also found that
expression of the effector proteins TLR2, Syk and total RIP2 is unaffected in scramble
siRNA and NOD2 siRNA transfected MDMs during M.tb and BCG infection (Figure
2.3E).
Figure 2.3: Loss of NOD2 activity in human macrophages by siRNA knockdown.
Continued on next page
62

MDMs were transfected with either 368nM of scramble siRNA or NOD2 siRNA using the Amaxa nucleofector. (A) The transfected macrophages were plated in RPMI containing 20% autologous serum and incubated for the indicated time points (72 h and 96 h). At each time point macrophages were lysed and NOD2 or the loading control actin protein levels were examined by Western blot (representative blot from n = 3). (B) NOD2 or scramble siRNA trasfected MDM monolayers on coverslips were incubated for 72 h, washed, fixed, permeabilized, and stained using either NOD2 antibody or isotype control antibody followed by Alexa flour 488 conjugated secondary antibody. Cells were then examined by confocal microscopy. Shown is a representative micrograph from the Z series (63x mag for isotype; 126x mag for scramble/NOD2 siRNA, n = 3). (C) Cell numbers were assessed 72 h post transfection to ensure equal numbers of cells between scramble siRNA and NOD2 siRNA transfected cells. Nuclei in cell lysates were counted on a hemacytometer after nuclei were stained with Napthol blue black. Shown are cumulative data obtained from three independent experiments each performed in triplicate (mean ± SEM) (D) Scramble siRNA or NOD2 siRNA transfected MDM monolayers in culture for 72 h were treated with MDP (5μg/mL) or DAP (10μg/mL) for 24 h. Cell supernatants were analyzed for TNF-α by ELISA. Shown is a representative experiment performed in triplicate (mean ± SD, n =2, ** P < 0.005; N.S. P = 0.1519). (E) Scramble siRNA or NOD2 siRNA transfected MDM monolayers were incubated with M.tb or BCG (MOI 5:1) for the indicated time points (15 min and 2 h) and cells were lysed with TN-1 buffer. TLR2, Syk, and RIP2 were analyzed by Western blot. Shown is a representative experiment of two independent experiments.
2.3.4 NOD2 regulates the macrophage TNF- and IL-1β cytokine response to M.tb and
BCG
We next investigated whether NOD2 regulates the production of pro-inflammatory
cytokines during mycobacterial infection which can control the host defense response
through autocrine and paracrine functions (265). Results shown in Figure 2 had already
indicated a potential link between NOD2 and the cytokine response to mycobacteria.
Scramble and NOD2 siRNA transfected MDM monolayers were incubated for 72 h to
achieve the maximum NOD2 knockdown and subsequently incubated with M.tb or BCG
63

64
(MOI 5:1) for 24 h. The cell free culture supernatants were analyzed for cytokine levels
by ELISA. We found that NOD2 deficiency leads to a significant decrease in the
production of TNF-α and IL-1β in response to M.tb and BCG (Figure 2.4 A and B). Next,
we examined RIP2 activation in NOD2 knockdown MDMs in response to M.tb and BCG
infection. NOD2 deficient MDM monolayers were incubated with M.tb or BCG for short
time periods and cell lysates were analyzed for RIP2 phosphorylation by Western blot.
Our results show that NOD2 siRNA transfected MDMs have a partial but reproducible
decrease in RIP2 phosphorylation (Figure 2.4C: upper panel, Western blot; lower panel,
band densitometry), particularly at 2 h, compared to scramble siRNA transfected MDMs
in the case of both M.tb and BCG. The partial reduction of RIP2 phosphorylation may be
explained by the fact that the mycobacteria also produce NOD1 agonists that activate
RIP2 independently of NOD2. The smaller reduction of RIP2 phosphorylation in NOD2
knockdown cells incubated with M.tb compared to BCG may relate to the amount of the
NOD1 agonist DAP in the former (266).
Next we elected to study whether M.tb and BCG regulate IL-1β at the transcriptional
level or through caspase-1, the enzyme necessary for cleavage of pro IL-1β to the
secreted form. During BCG and M.tb infection, NOD2 knockdown macrophages
demonstrate either no effect or an increase in pro IL-1β transcription (Figure 2.4D). On
the other hand, since NOD2 knockdown leads to a significant reduction in active
(cleaved) caspase-1, NOD2 does appear to play a role in caspase-1 activation during M.tb
infection (Figure 2.4E). Similar results were found for caspase-1 using TCA
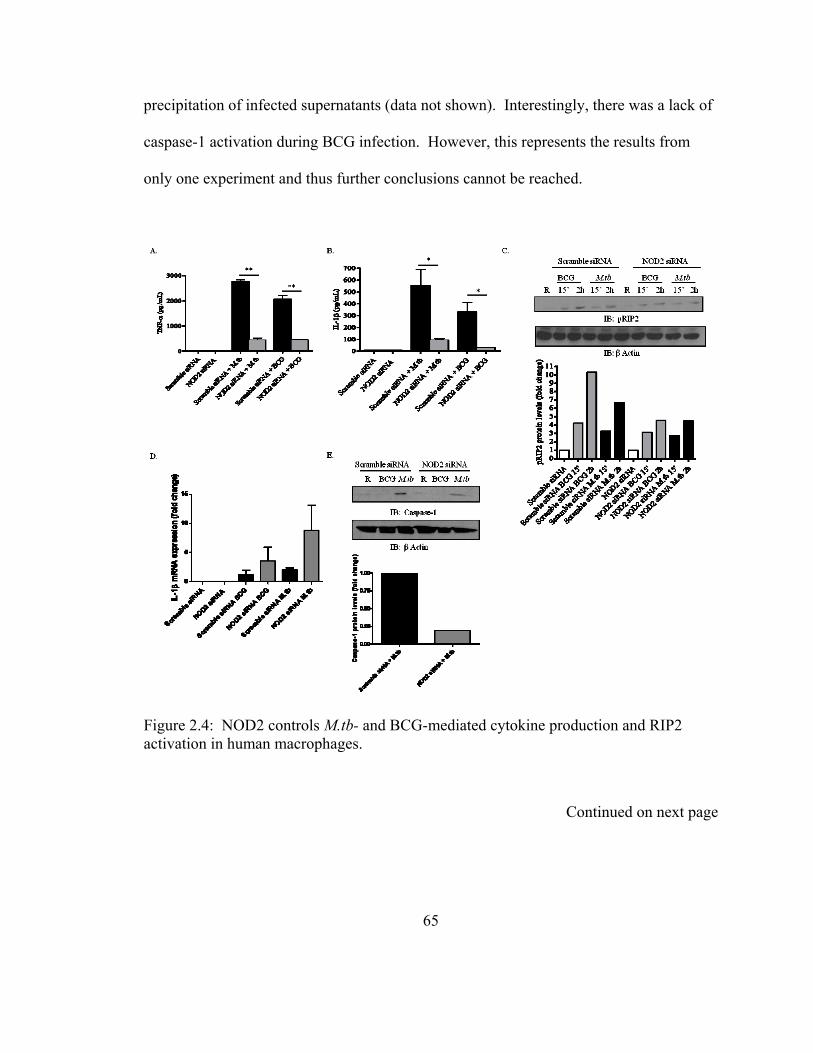
precipitation of infected supernatants (data not shown). Interestingly, there was a lack of
caspase-1 activation during BCG infection. However, this represents the results from
only one experiment and thus further conclusions cannot be reached.
Figure 2.4: NOD2 controls M.tb- and BCG-mediated cytokine production and RIP2 activation in human macrophages.
Continued on next page
65

Scramble or NOD2 siRNA transfected MDM monolayers were incubated with M.tb or BCG (MOI 5:1) for 2 h, washed 3 times with warm RPMI, and repleted in 1% autologous serum for 24 h. Cell culture supernatants were analyzed for TNF-α (A) and IL-1β (B) production by ELISA. The graphs shown are from a representative experiment of n = 3. *P < 0.05 or **P < 0.005 (C) Scramble or NOD2 transfected MDM monolayers were incubated with M.tb or BCG (MOI 5:1) for the indicated time points (15 min and 2 h) and cells were lysed with TN-1 buffer. NOD2-dependent RIP2 phosphorylation was analyzed by Western blot of cell lysates from scramble and NOD2 deficient MDMs (upper panel). Shown is a representative experiment of three independent experiments. Densitometry of the band intensities in this experiment is shown as a bar graph (lower panel). (D) IL-1β steady-state mRNA levels were measured during BCG and M.tb infection by real time PCR. Data were normalized to the β actin gene and RCN was determined. Shown is a representative experiment from two independent experiments each performed in triplicate. (E) Macrophage cell lysates were analyzed for caspase-1 protein expression by Western blot using caspase-1 antibody. The same membrane was re-probed with actin to verify equal loading of lysate proteins. Shown is a representative Western blot, and the bar graph below shows the densitometric analysis during M.tb infection (M.tb n = 2; BCG n = 1).
2.3.5 Expression of NOD2 in human AMs and its role in regulating the production of
TNF- and IL-1β in response to M.tb
A recent report showed the presence of NOD2 transcripts in human alveolar lavage cells,
but to date there is no evidence for the presence of NOD2 protein in AMs (220).
Therefore we examined whether NOD2 protein is expressed in human AMs. Cell lysates
were prepared from MDMs and AMs, the latter were isolated from healthy donors and
both cell types were analyzed for NOD2 expression by Western blot. Results shown in
Figure 2.5A demonstrate that AMs express a detectable amount of NOD2 protein but the
level is less than that in an equivalent number of MDMs. To further examine the
potential involvement of NOD2 in the production of inflammatory cytokines in response
66
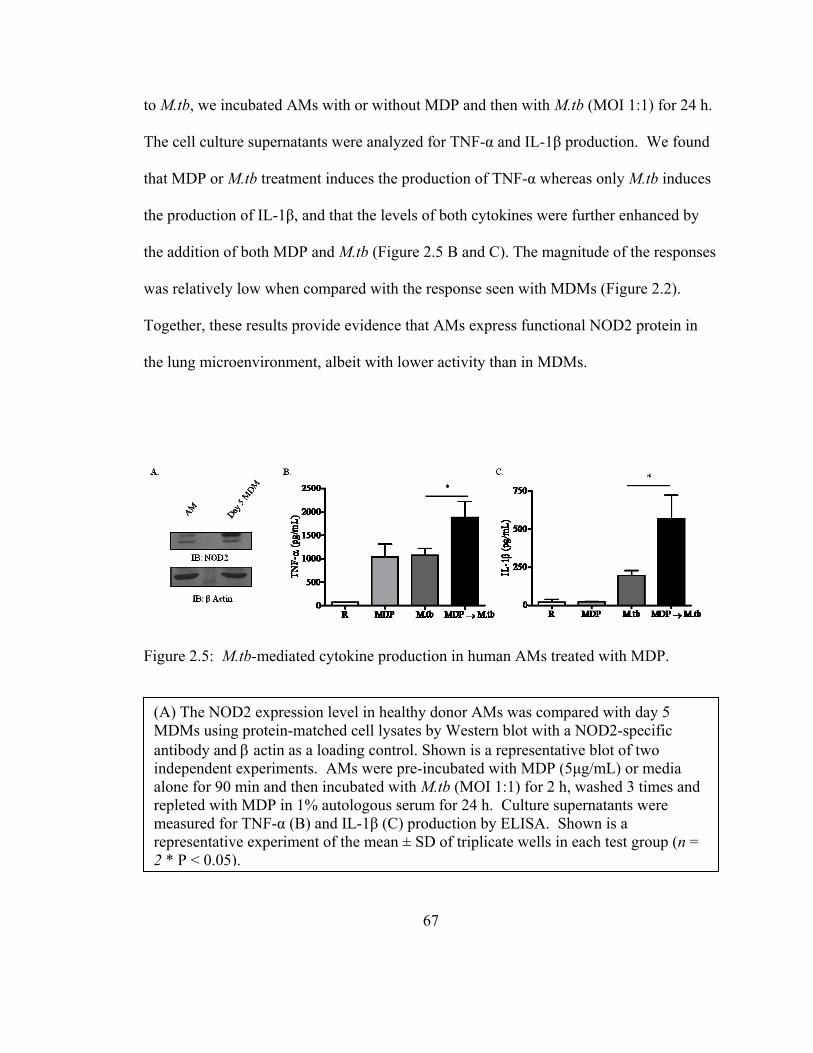
to M.tb, we incubated AMs with or without MDP and then with M.tb (MOI 1:1) for 24 h.
The cell culture supernatants were analyzed for TNF-α and IL-1β production. We found
that MDP or M.tb treatment induces the production of TNF-α whereas only M.tb induces
the production of IL-1β, and that the levels of both cytokines were further enhanced by
the addition of both MDP and M.tb (Figure 2.5 B and C). The magnitude of the responses
was relatively low when compared with the response seen with MDMs (Figure 2.2).
Together, these results provide evidence that AMs express functional NOD2 protein in
the lung microenvironment, albeit with lower activity than in MDMs.
Figure 2.5: M.tb-mediated cytokine production in human AMs treated with MDP.
(A) The NOD2 expression level in healthy donor AMs was compared with day 5 MDMs using protein-matched cell lysates by Western blot with a NOD2-specific antibody and actin as a loading control. Shown is a representative blot of two independent experiments. AMs were pre-incubated with MDP (5μg/mL) or media alone for 90 min and then incubated with M.tb (MOI 1:1) for 2 h, washed 3 times and repleted with MDP in 1% autologous serum for 24 h. Culture supernatants were measured for TNF-α (B) and IL-1β (C) production by ELISA. Shown is a representative experiment of the mean ± SD of triplicate wells in each test group (n = 2 * P < 0.05).
67

68
2.3.6 NOD2 controls the intracellular growth of M.tb and BCG in human macrophages
We next focused our studies on determining the role of NOD2 in controlling the growth
of mycobacteria in human macrophages. MDMs were transfected with scramble or
NOD2 siRNA and after 72 h of transfection, macrophages were incubated with M.tb or
BCG (MOI 5:1) for 24 h. Experiments were performed for 24 h due to the fact that
NOD2 protein levels in macrophages began to reappear after 120 h of knockdown.
Intracellular growth of M.tb and BCG was determined by a colony forming unit (CFU)
assay (262). The results in Figure 2.6A show that silencing of NOD2 in macrophages
significantly enhances the growth of M.tb (84.6 ± 8.0%, n = 3). A similar result was
observed for BCG growth in Figure 2.6B (108.4 ± 30.4%, n = 5). Western blotting
confirmed NOD2 knockdown in each experiment (Figure 2.6C). We next determined
whether the increase in mycobacterial growth at 24 h was the result of increased cell
association (both bacterial attachment and phagocytosis). We compared the association of
bacteria with scramble and NOD2 siRNA transfected MDMs by phase contrast and
fluorescence microscopy after 2 h infection. Our results show that bacterial association is
not altered by NOD2 siRNA transfection (Figure 2.6D and E). Thus, these results
indicate that NOD2 plays a role in regulating the early host defense response during
infection of both M.tb and BCG.
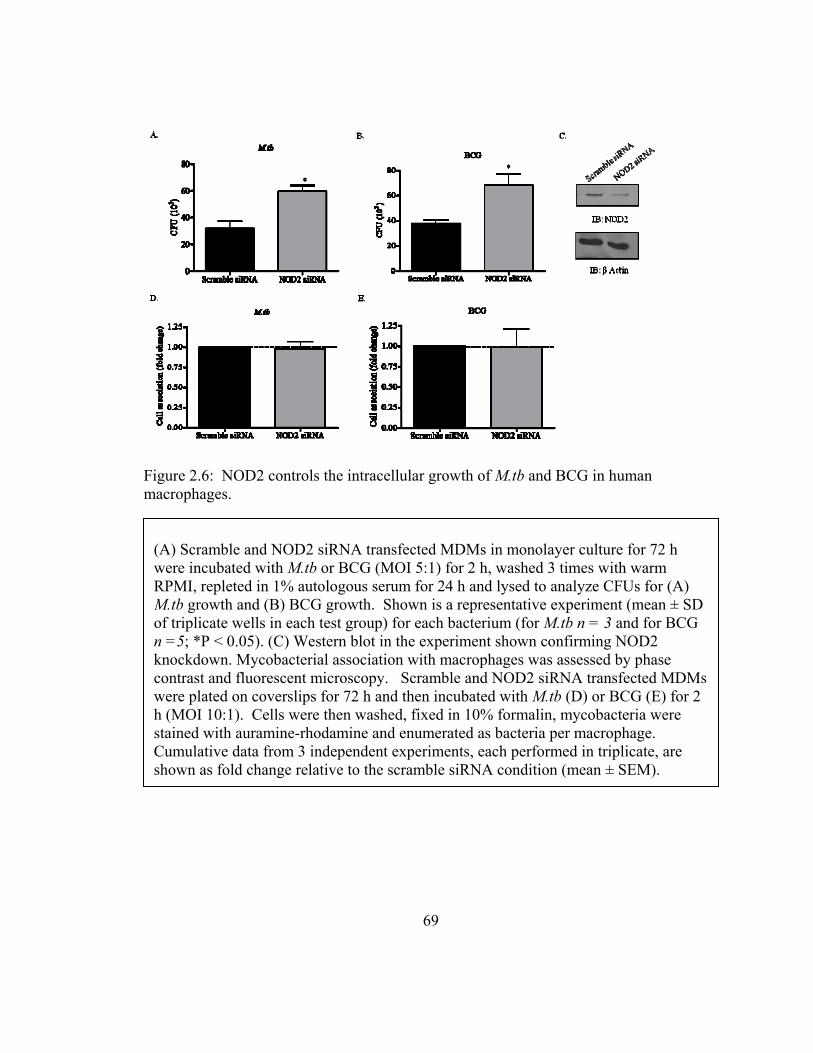
Figure 2.6: NOD2 controls the intracellular growth of M.tb and BCG in human macrophages.
(A) Scramble and NOD2 siRNA transfected MDMs in monolayer culture for 72 h were incubated with M.tb or BCG (MOI 5:1) for 2 h, washed 3 times with warm RPMI, repleted in 1% autologous serum for 24 h and lysed to analyze CFUs for (A) M.tb growth and (B) BCG growth. Shown is a representative experiment (mean ± SD of triplicate wells in each test group) for each bacterium (for M.tb n = 3 and for BCG n =5; *P < 0.05). (C) Western blot in the experiment shown confirming NOD2 knockdown. Mycobacterial association with macrophages was assessed by phase contrast and fluorescent microscopy. Scramble and NOD2 siRNA transfected MDMs were plated on coverslips for 72 h and then incubated with M.tb (D) or BCG (E) for 2 h (MOI 10:1). Cells were then washed, fixed in 10% formalin, mycobacteria were stained with auramine-rhodamine and enumerated as bacteria per macrophage. Cumulative data from 3 independent experiments, each performed in triplicate, are shown as fold change relative to the scramble siRNA condition (mean ± SEM).
69

70
2.3.7 Effects of NOD2 on M.tb and BCG growth in mouse macrophages and on the
cytokine response to infection
Previous studies have reported that NOD2 is not critical for controlling the growth of
M.tb and BCG in mouse macrophages (135,179). Based on our results in human
macrophages, we elected to examine the host response to these mycobacteria in mouse
macrophage experiments. We reproduced the published finding that NOD2 does not
control the growth of M.tb in mouse macrophages using WT and NOD2-/- BMMs (Figure
2.7A). In contrast to the published work with BCG that analyzed growth in naive murine
AMs at 48 h (135), our data show that NOD2 is important in controlling the growth of
BCG at the later time points of 72 h and 168 h in WT and NOD2-/- BMMs (Figure 2.7B).
We also examined the cell association of bacteria with WT and NOD2-/- BMMs by
microscopy and found no difference with M.tb and only a small difference with BCG
(Figure 2.7C and D). Finally, consistent with the literature, we found a reduction in the
pro-inflammatory cytokine IL-1β in response to M.tb and BCG at 24 h, 72 h, and 168 h in
NOD2-/- BMMs (Figure 2.7E).
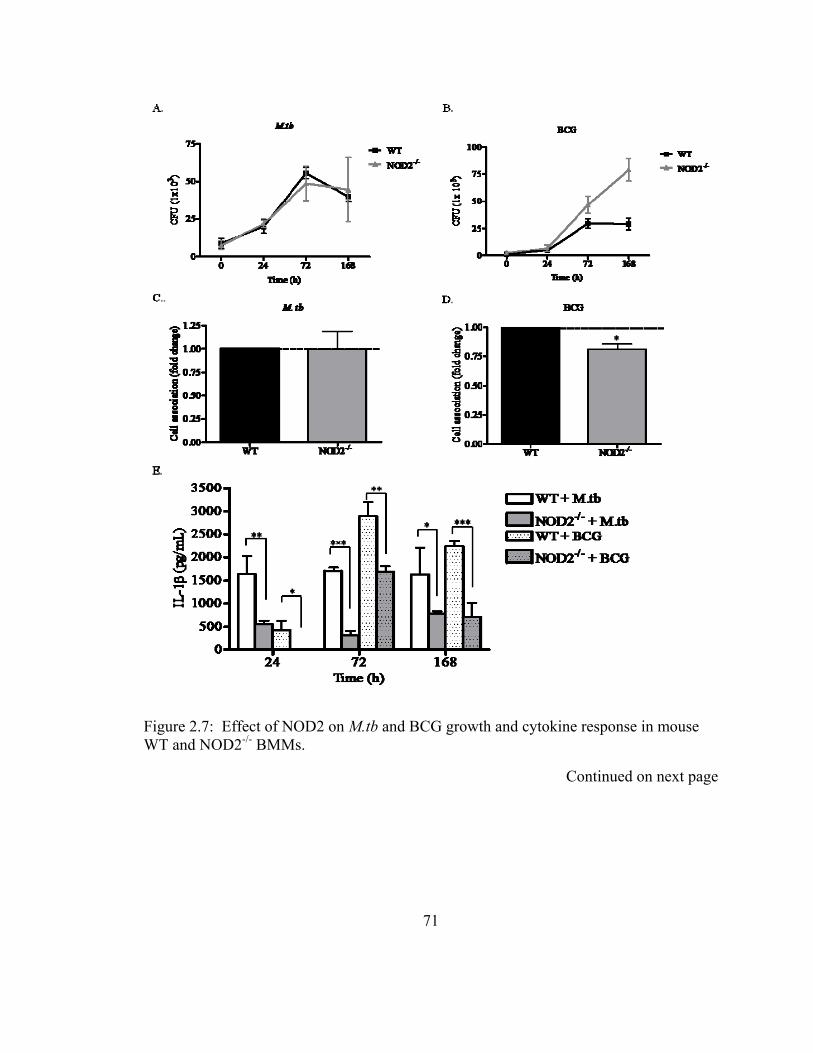
Figure 2.7: Effect of NOD2 on M.tb and BCG growth and cytokine response in mouse WT and NOD2-/- BMMs.
Continued on next page
71

BMMs from WT and NOD2-/- mice were plated in 24 well tissue culture plates and incubated with M.tb (1:1) or BCG (2.5:1) for 2 h, washed 3 times, and immediately lysed or incubated for different time points (2 h, 24 h, 72 h and 168 h) prior to lysis for assessment of CFUs for M.tb (A) and BCG (B). Shown is a representative experiment for each bacterium (mean ± variance of duplicate wells in each test group, n = 2). Cell association of M.tb (C) and BCG (D) with WT and NOD2-/- BMMs was analyzed at 2 h post-infection by microscopy. Cumulative data from 3 independent experiments, each performed in triplicate, are shown as fold change relative to WT condition (mean ± SEM, *P<0.05). The cytokine response IL-1β (E) was analyzed by ELISA in culture supernatants harvested from M.tb and BCG infected WT and NOD2-
/- BMMs. The data shown are from a representative experiment (mean ± SD, n = 2, with four test results in each group * P < 0.05; ** P < 0.005, ***P < 0.0005).
2.4 Discussion
Macrophages play a critical role in the recognition and removal of pathogens through
various PRRs, including intracellular receptors such as NLR family CARD-containing
domain 4 (NLRC4), Baculoviral IAP repeat-containing protein 1 (NAIP), NOD1, and
NOD2. Studies on host recognition of pathogens by human macrophages have
demonstrated the importance of transmembrane receptors; however the field is relatively
lacking in defining the role of intracellular receptors in these cells. NOD2 is an
intracellular PRR that senses bacteria through its LRR domain and induces a pro-
inflammatory response. While this inflammatory response can activate the macrophage,
it also allows for the recruitment and activation of other immune cells to the site of
infection. NOD2 has been shown to serve as a microbial sensor and activator of the pro-
inflammatory response to several pathogens, including M.tb (126,127,135,179,242-244).
In the case of H. pylori, it is thought that the bacterium is able to use a secretion system
to secrete peptidoglycan fragments that are then recognized by NOD2 in the cytosol (93).
72

73
Since M.tb is an intracellular pathogen, and known to secrete virulence factors, it is
necessary to understand the role of intracellular receptors and immune functions in
human macrophages with regards to this pathogen. Demonstrating a role for NOD2 can
lead to the development of new methods for targeting this host defense molecule/pathway
for therapy in the case of M.tb.
The presence of NOD2 mRNA has only been reported in monocytes (76). Thus, we
initiated studies of NOD2 mRNA transcript levels as monocytes differentiate into
macrophages using an established in vitro model. Our results indicate that NOD2 steady
state mRNA transcripts increase as monocytes differentiate into macrophages,
specifically on day three of differentiation. This result is similar to our previously
published data on a membrane PRR that recognizes M.tb, the MR, whose mRNA
transcript levels are also enhanced in macrophages when compared to monocytes. On the
other hand, TLR2 and TLR4 expression follows a different pattern on these cells (54).
Since mRNA levels do not always correlate with protein expression, we elected to
explore NOD2 protein expression in human macrophages compared to monocytes.
Studies in the field have been limited to assaying for mRNA levels in primary human
cells; whereas protein expression has been studied in cell lines by flag tagging NOD2
(114,267). Thus, a major contribution to the field by the current work is the optimization
of available antibodies for characterization of NOD2 protein expression in primary
human monocytes/macrophages by Western blot and confocal microscopy. Our findings

74
demonstrate that NOD2 protein is expressed in human macrophages and levels increase
during monocyte to macrophage differentiation, suggesting a particularly important role
for NOD2 in human macrophages. Our microscopy results show NOD2 protein in a
punctuate pattern in cells, suggesting the possibility that it resides within organelles or in
complexes present in the cytoplasm.
We also detected NOD2 protein in human AMs from healthy donors. An interesting
finding in our work is that AMs appear to express less of NOD2 protein than MDMs. It
is possible that reduced NOD2 levels in AMs represent another phenotypic marker of
alternatively activated macrophages of which AMs are a prototype and consequently
leads to less effective killing of M.tb on first contact during the innate immune response.
On the other hand, later during TB disease, NOD2 may play an important role in host
defense since NOD2 mRNA levels are increased in patients with TB disease (220).
NOD2 is engaged in response to pathogens which leads to the production of pro-
inflammatory cytokines. In cell lines expressing NOD2, incubation with M.tb enhances
NF-κB activation (86,127). We found that pre-stimulation of human macrophages (both
MDMs and AMs) with the NOD2 ligand MDP followed by M.tb or BCG incubation
caused a significant increase in TNF-α and IL-1β production. Although the addition of
MDP is not the paradigm for human infection, the result provided evidence for the
potential involvement of NOD2 in augmenting the cytokine response to M.tb and BCG.
This result raised the question as to what are the major downstream signaling kinases

75
activated in response to M.tb or BCG. RIP2 is a kinase that autophosphorylates at serine
176, and its kinase activity is necessary for NOD2-mediated pro-inflammatory cytokine
production (94). We found that M.tb and BCG both activate RIP2 by phosphorylation at
serine 176 in a NOD2-dependent manner; however the kinetics and pattern of activation
were different for these two bacteria. RIP2 phosphorylation peaked earlier for BCG (30
min) and continued throughout the time course; whereas RIP2 phosphorylation peaked at
1 h for M.tb and the level of phosphorylation decreased by 3 h. Consistent with this we
observed that BCG continues to activate downstream IκBα [thereby enabling the
transcriptional activity of NF-κB and the resultant up-regulation of expression of
macrophage pro-inflammatory response genes (264)] through 2 h whereas activation of
IκBα decreases for M.tb at this time point. Thus, it appears that M.tb but not BCG can
down-regulate RIP2 and IκBα activation perhaps as a mechanism to dampen the innate
immune response. One possibility for this down-regulation is that M.tb possesses
virulence factors that affect this pathway that are not present in BCG such as those
present in the Region of Difference-1 (RD1) which is responsible for the production of
several virulence-associated proteins in M.tb (191). For example, culture filtrate protein-
10 kDa (CFP-10) and early secreted antigenic target-6 kDa (ESAT-6) in M.tb but not in
BCG have been shown to inhibit lipopolysaccharide-induced NF-kB transactivation by
inhibiting reactive oxygen species production (251). Apart from RIP2, recent work from
our laboratory has identified a PPAR-mediated pathway for M.tb that is not operative for
BCG in human macrophages (56).

76
To more directly assess a causal role for NOD2 in regulating the inflammatory response
to mycobacteria in primary human macrophages, we developed a reliable knockdown
protocol for NOD2 that is specific and does not appreciably affect the viability of the
cells on the monolayer. To date, studies on the role of NOD2 in response to different
pathogens in primary human cells have relied on either expressing the NOD2 allele with
the Crohn’s disease mutation in cell lines (139) or by obtaining cells from individuals
homozygous for the NOD2 mutation, which was shown to render this protein reduced in
activity or non-functional (85). Obtaining cells from Crohn’s disease patients with this
mutation is difficult and the results may not be consistent among patients. Also, since
these patients have a mutation in this protein, their cells may be primed in other ways that
may alter the immunological response. The accomplished knockdown of NOD2 in
human macrophages is an important advance enabling the study of NOD2 more
comprehensively in primary monocytes/macrophages.
Our results are consistent with those found with mouse macrophages with regards to the
role of NOD2 in controlling the inflammatory response to mycobacteria. Gandotra et al
found that TNF-α and IL-12 production is decreased in NOD2 knockout BMMs in
response to M.tb (179) and Divingahi et al found that these cytokines are decreased in
NOD2 knockout naive alveolar mouse macrophages in response to BCG (135). We
observed that both TNF-α and IL-1β production are significantly reduced in NOD2-
deficient human macrophages and found a decrease in RIP2 phosphorylation in response
to M.tb and BCG infection. It is noteworthy that RIP2 activity is only partially reduced

77
rather than abolished by the knockdown of NOD2, which may be due to the activation of
RIP2 by other intracellular receptors. For example, NOD1 is involved in the activation
of RIP2 through the CARD-CARD interaction (94).
The role of NOD2 in IL-1β production during M.tb and BCG infection is of interest. A
recent publication reports that PBMCs from Crohn’s disease patients that are
homozygous for the 3020insC NOD2 mutation exhibit decreased production of IL-1β in
response to M.tb (268). The mechanism for the production of mature IL-1β in response
to M.tb involved the activation of ERK, p38 and RIP2 following recognition by
TLR2/TLR6 and NOD2 in PBMCs; however, the mechanism was not explored in human
macrophages. In PBMCs, NOD2 is reported to have a dual effect on pro IL-1β mRNA
transcription and secretion of mature IL-1β (269). Our preliminary observations propose
that during M.tb infection, NOD2 may play a small role in down-regulating pro IL-1β
transcription, suggesting that NOD2 may play a negative regulatory role (Figure 2.4D).
However, NOD2 is responsible for enhancing caspase-1 activation during M.tb infection,
since we observed a decrease in active caspase-1 expression in NOD2 deficient
macrophages (Figure 2.4E). In our experiments in which we added MDP to un-
transfected macrophages followed by M.tb infection, we observed an additive effect on
TNF-α secretion, but a synergistic effect on IL-1 secretion (Figure. 2.2 and 2.5). We
speculate that the differential response of TNF-α and IL-1β may be explained by NOD2
being relatively more important in the release of mature IL-1β due to a role in processing

78
pro IL-1β through caspase-1. The relationship between NOD2 and caspase-1 has been
reported (97,270).
Previous studies have reported that NOD2 knockout in mouse macrophages has no effect
on controlling M.tb growth (135,179). In contrast, we demonstrate here for the first time
that NOD2 plays a role in controlling intra-macrophage growth of M.tb and BCG in
human macrophages. The difference in the role of NOD2 in controlling mycobacterial
growth is validated by doing a direct side by side comparison of mycobacterial growth in
NOD2-deficient human macrophages and macrophages from NOD2 knockout mice. This
difference can be generally explained by the growing list of differences seen in the innate
immune response of human versus mouse macrophages (225,254,271-275). It is relevant
that the NOD2 knockout mouse does not show any signs of abnormalities in the intestinal
tract, which is inconsistent with the NOD2 mutations leading to Crohn’s disease and
inflammatory bowel disease in humans (276). In addition, differences in promoter
structure between humans and mice indicate divergent mechanisms in NOD2 regulation
(147).
There have been reports over time providing evidence that M.tb can translocate into the
cytosol of macrophages under defined conditions (212,213,215,277) and also that
mycobacterial phagosomes are porous (278). Nonetheless, the weight of the evidence to
date is that mycobacteria replicate in phagosomes in human and mouse macrophages. In
the context of the current work, we speculate that M.tb interacts with NOD2 by either

79
secreting virulence factors from the phagosome into the cytosol via a secretion system(s)
(181,191), or possibly translocates proteins across the phagosome through the action of a
yet to be defined flippase. Alternatively, a NOD2-RIP2 complex has been reported to
signal from the cell membrane along with the need for NOD2 to be present at the
membrane of intestinal epithelial cells for signaling to occur (114,279). These reports
raise the possibility that NOD2 may localize with the phagosome of M.tb leading to
engagement of this cytosolic PRR during residence of the bacterium within the
phagosome.
The mechanism(s) involved in the recognition of NOD2 by M.tb and BCG may differ
since BCG lacks the ESX-1 secretion system and is reported to be unable to stimulate
NOD2 dependent RIP2 polyubiquitination in murine macrophages (181). Nonetheless,
BCG does generate TNF-α in a NOD2-dependent manner [this report and (86)]. Of
relevance, mycobacteria contain the N-acetyl muramic acid hydroxylase, NamH, which
changes the acetyl group on MDP to a glycolyl group, causing it to be a more potent
stimulator of the immune response via NOD2 suggesting that NOD2 may be more tuned
at sensing mycobacterial infections in generating an immune response (86).
In summary, here we report that NOD2 is present and functional in human macrophages,
including AMs. We provide evidence that NOD2 plays a role in controlling the growth
of M.tb and BCG in human macrophages along with regulating the nature of the
inflammatory response. The role for NOD2 differs with respect to growth of M.tb and

80
BCG in human and mouse macrophages. Finally, the assays established and described in
this report now enable a more thorough evaluation of the role of NOD2 in regulating the
innate immune response of human macrophages to mycobacteria and other infectious
agents.

81
CHAPTER 3 : Critical role for NOD2 in Mycobacterium tuberculosis induced iNOS
expression in human macrophages
3.1 Introduction
Mycobacterium tuberculosis (M.tb) is the causative agent of tuberculosis (TB). TB
infects approximately one-third of the world’s populations, and was responsible for 1.7
million deaths in 2009. The high incidence of death is attributed to the increase in HIV
infections, drug resistant strain development, and length/cost of drug treatment (155). It
is imperative to understand the mechanism that underlies the survival of M.tb in humans
in order to effectively treat patients for this disease as well as create useful vaccines.
An important immune cell that is involved in recognizing and aiding in the clearance of
pathogens is the macrophage. Macrophages use a myriad of defense mechanisms in
order to protect the host including the production of reactive oxygen/nitrogen
intermediates (ROI/RNI), pro-inflammatory cytokines, and proteases, as well as
acidification of the phagosome. The type of response induced during the interaction of
M.tb with the macrophage is dependent on the receptor activated (186). For example,

82
entry through the mannose receptor leads to limited phagosome-lysosome fusion and an
anti-inflammatory response (238).
A group of intracellular receptors, Nucleotide-binding oligomerization domain (NOD)-
like receptors (NLR), have been described to recognize and induce a pro-inflammatory
response to specific portions of the peptidoglycan layer of Gram-negative and Gram-
positive bacteria cell walls (85). NOD2 is a NLR that was originally found to recognize
N-acetyl muramyl dipeptide (MDP) found in Gram- negative and positive bacteria. M.tb
produces N-glycolyl-MDP (GMDP), and this change in functional groups (acetyl to
glycolyl) enhances the NOD2 stimulating activity (86). As discussed in chapter 2, our
laboratory has recently provided evidence that in primary human macrophages NOD2
plays an important role in controlling the inflammatory response as well as the growth of
M.tb (77).
The role of NOD2 during early innate immune responses is of interest to our laboratory,
specifically the role of NOD2 in RNI production. Inducible nitric oxide synthase (iNOS
or NOS2) is responsible for the production of the RNI, nitric oxide (NO). NO is known
to play a role in signaling during inflammation, which suggests a potential role for it in
the NOD2 biochemical pathway (37,280). NOD1 is a close family member of NOD2,
and has been found to be involved in iNOS induction in rat vascular smooth muscle cells
and a murine macrophage cell line (281). Furthermore, interferon gamma (IFNγ)
activated mouse macrophages stimulated with MDP induces NO production (282). It has

83
also been shown that in mouse macrophages MDP is able to induce iNOS expression
(283). Little is known about the relationship of NOD2 and iNOS during M.tb infection.
A study using IFNγ-activated NOD2 deficient mouse macrophages showed a decrease in
NO production in response to M.tb (179). In the mouse model for TB NO plays an
important roles in controlling M.tb growth (284). However, it has been difficult to
duplicate these data using human macrophages, which may be due to differences in the
amount of NO produced by mouse macrophages and human macrophages as well as a
lack in sensitivity of assays (285). There are also differences in upstream pathways of
NO production that exist between human and mouse macrophages. L-arginine is
required for the production of NO in mouse macrophages, but not in human macrophages
(286,287). This is possibly due to the lack of tetrahydrobiopterin in human
macrophages, which is a necessary cofactor in the conversion of L-arginine to NO;
however it has been shown that addition of this cofactor does not induce NO production
(288). Recent studies indicate that, while low in comparison to mouse macrophages,
human monocytes produce NO in response to M.tb (289). Furthermore, clinical studies
indicate that patients with TB have increased iNOS expression in the inflammatory zone
of granulomas (290). More specifically, alveolar macrophages of patients with TB have
enhanced iNOS protein expression (291).
Our recent finding of the ability of NOD2 to control M.tb growth at 24 h is of interest,
and led us to believe that NOD2 is involved in early innate immune mechanisms. We
performed a microarray at this time point using NOD2-deficient human macrophages and

84
found inducible nitric oxide synthase (iNOS) to be down-regulated. Based on reports of
M.tb inducing iNOS and NO using in vitro cell models as well as clinical specimens, the
recent evidence that NOD2 plays a role in NO production during M.tb infection of IFNγ
activated mouse macrophages, and our microarray data, we decided to study the role of
NOD2 in the iNOS pathway during M.tb infection of human macrophages. Here we
show that NOD2 agonists increase the expression of iNOS mRNA and protein expression
as well as NO production. In coordination with previous reports we show that M.tb
induces iNOS expression and NO production, however, we show for the first time in
human macrophages that this phenomena is NOD2-dependent. Together these findings
provide evidence for a novel role for NOD2 and its importance in the macrophage
induction of NO in response to pathogens.
3.2 Materials and Methods
3.2.1 Buffers and reagents
Dulbecco’s PBS with and without Ca2+ and Mg2+ ions, Iscoves, FBS, TRIzol, and RPMI
1640 medium with L-glutamine containing phenol red or phenol red deficient were
purchased from Invitrogen (Invitrogen Life Technologies). RHH medium [RPMI 1640
plus 10 mM HEPES (Invitrogen) plus 0.4% human serum albumin (CSL Behring LLC)]
was used for cell culture experiments. N-Acetylmuramyl-L-alanyl-D-isoglutamine
hydrate (MDP; Sigma-Aldrich), N-Glycolyl-muramyldipeptide (GMDP; Invivogen), and
Interferon gamma (IFNγ; BD Biosciences) were used for assays involving iNOS

expression and NO production. 4-amino-5-methylamino-2’,7’-difluorofluorescein
diacetate (DAF-FM diacetate) was purchased from invitrogen for NO production assays.
7H11 agar was prepared with Bacto Middlelebrook 7H11 agar, oleic acid-albumin-
dextrose-catalase enrichment medium, and glycerol (Difco Laboratories).
3.2.2 Antibodies
Unconjugated rabbit anti-human NOD2 was purchased from ProSci and optimization for
human macrophages can be found in the previous report (77). actin, goat anti-rabbit
IgG-HRP and donkey anti-goat IgG-HRP antibodies were purchased from Santa Cruz
Biotechnology. Antibodies specific for iNOS were purchased from Cell Signaling
Technology. The aforementioned antibodies were used for Western blot.
3.2.3 Bacterial strains and culture
Lyophilized M.tb H37Rv (ATCC #25618) and BCG (ATCC #35734) was obtained from
the American Tissue Culture Collection, reconstituted, and used as described (172). The
concentration of bacteria (1–2 x 108 bacteria /mL) and the degree of clumping ( 10%)
were determined by counting in a Petroff-Hausser chamber. Bacteria prepared in this
fashion are 90% viable by CFU assay.
85

86
3.2.4 Isolation and culture of human macrophages
Blood obtained from healthy, Purified Protein Derivative negative human volunteers
using an approved protocol by The Ohio State University Institutional Review Board
(IRB) was processed as described (292). Briefly, PBMCs were isolated from heparinized
blood on a Ficoll-Paque cushion (Amersham Biosciences/GE Healthcare) and cultured in
Teflon wells (Savillex) for 1 (monocytes) through 5 (MDMs) days in the presence of
RPMI 1640 medium containing 20% autologous serum (2.0 x106 PBMCs/mL) at
37ºC/5% CO2 (258).
3.2.5 RNA isolation
Day 5 PBMCs in Teflon wells were harvested and mononuclear phagocytes adhered to a
12-well tissue culture plate with 10% autologous serum (4 x 106 PBMCs/mL). After
washing away lymphocytes, the MDMs (4 x 105 cells) were lysed in TRIzol and total
RNA was isolated by using the Qiagen RNAeasy column method. For microarray studies
RNA was not processed through the Qiagen RNAeasy column. The Experion (Bio-
Rad) was used to determine the quality and quantity of all RNA samples.
3.2.6 Gene expression studies by qRT-PCR
RNA (100ng) was reverse transcribed to cDNA by reverse transcriptase enzyme,
(SuperScript II, Invitrogen) and qRT-PCR was performed using a human NOS2 TaqMan
gene expression kit (Applied Biosystems). NOS2 amplification was normalized to β

87
actin as a housekeeping gene (∆Ct). Relative copy number (RCN) and fold change were
determined. RCN was calculated as follows: RCN = E-∆Ct x 100, where E is the
efficiency (2 = 100% efficiency) and Ct = Ct (target) – Ct (reference) (260). Fold change
was calculated from RCN compared to resting group for NOS2 mRNA experiments.
Triplicate samples were analyzed in duplicate wells in each experiment.
3.2.7 Transfection of MDMs
PBMCs were transfected with Accell scramble siRNA or NOD2 siRNA purchased from
Thermo Scientific Dharmacon RNAi technologies that targeted the sequence
CUUUAGGAUGUACAGUUA (368nM) using the Amaxa Nucleofector (Lonza Group
Ltd) Transfected cells were used for subsequent experiments, such as the DAF-FM
diacetate assay and Western blotting. See section 2.2.9 for more details of the transfection
protocol. Assessment of the NOD2 siRNA specificity and effects on monolayer
density/viability were performed and can be found in chapter 2 (77).
3.2.8 Microarray analysis
Scramble and NOD2 siRNA-transfected cells were incubated with M.tb for 24 h in 1%
autologous serum. Cells were then lysed with TRIzol and RNA was isolated as described
above. Subsequent labeling and hybridization to Affymetrix hgu133plus2 chips was
performed at The Ohio State University (OSU) Comprehensive Cancer Center microarray
facility. Analysis was performed at the OSU Comprehnsive Cancer Center Biomedical

88
Informatics core laboratory. The Venn diagram was generated using Bioinformatics
Organization’s Gene List Venn Diagram program
(http://www.bioinformatics.org/gvenn/).
3.2.9 Macrophage stimulation / infection, cell lysis, and Western blotting
Day 12 MDMs or day 5 MDMs transfected with scramble or NOD2 siRNA (2.0 x 105)
for 72 h were incubated with M.tb or BCG (MOI 5:1; triplicate wells) in RHH for 30 min
at 37°C in 5% CO2 on a platform shaker for equal dispersion of bacteria followed by an
additional incubation for 90 min without shaking. The cells were then washed three times
with warm RPMI, repleted with RPMI containing 1-2% human autologous serum and
incubated for 24 h. For experiments using the NOD2 agonists MDP, GMDP, or the
cytokine IFNγ, 2.0 x 105 day 5 MDMs were adhered to a 24-well plate for 2 h at 37°C in
5% CO2, washed to remove lymphocytes, and then incubated with MDP (5ug/mL),
GMDP (5ug/mL), or IFNγ (10ng/mL), for 24 h in RPMI containing 1 % autologous
serum. The monolayers were washed once with PBS and lysed in TN-1 lysis buffer [50
mM Tris (pH 8.0), 10 mM EDTA, 10 mM Na4PO7, 10 mM NaF, 1% Triton X-100, 125
mM NaCl, 10 mM Na3VO4, 10 µg/ml aprotinin, and 10 µg/ml leupeptin)], incubated on
ice for 10 min and then centrifuged at 17,949 x g for 10 min at 4°C to remove cell debris
(261). For NOD2 knockdown assessment, lysates were collected as above at 72 h or 96 h
post-knockdown. Protein concentrations of the cleared lysates were measured using the
BCA-protein assay kit (Pierce). Proteins were separated by SDS-PAGE and analyzed by
Western blot by probing with the primary and secondary antibody of interest and

89
development using ECL (Amersham Biosciences). The protein band intensities were
measured using the online ImageJ software provided by the NIH. Background intensity
was subtracted from each sample and then normalized to β-actin. Fold change was
determined as follows: (treated sample band intensity) / (untreated sample band
intensity).
3.2.10 DAF-FM diacetate staining
Untransfected MDMs or transfected (scramble or NOD2 siRNA) MDMs (2 x 105) were
adhered to glass coverslips in a 24-well tissue culture plate for 2 h at 37°C and washed to
remove lymphocytes. The cells were treated with GMDP, IFNγ, or M.tb for 24 h in no
phenol red RPMI containing 1% autologous serum. At 24 h cells were washed one time
with warm no phenol red RPMI and repleted with 5 μM/mL DAF-FM diacetate for 30
min at 37°C. Next cells were washed with warm no phenol red RPMI 3 times and left in
the third wash for 15 min at 37°C to allow for deesterification of the dye. Finally, cells
were fixed with 10% formalin (10 min at room temperature), washed with PBS, and
mounted on glass slides. Slides were viewed using an Olympus Flowview 1000 Laser
Scanning Confocal microscope at an excitation/emissions maxima of 495/515 nm. One
hundred and fifty to three hundred consecutive macrophages/test group were counted and
Mean fluorescence intensity (MFI) was calculated using Olympus FluoView FV1000
software.

90
3.2.11 Statistics
A paired one tailed student’s t test was used to analyze the differences between two
groups in figures showing one representative figure of three independent experiments.
An unpaired one tailed student’s t test was used to analyze differences between two
groups in figures showing cumulative data from three independent experiments.
Significance was P < 0.05.
3.3 Results
3.3.1 NOD2 regulates many genes during M.tb infection of human macrophages
As discussed in chapter 2, our laboratory has published that NOD2 plays an important
role in controlling the growth of M.tb in human macrophages at 24 h post infection.
Between initial infection and 24 h there is limited bacterial replication; the bacteria are
becoming acclimated to the phagosome of the macrophages. Thus, enhanced bacterial
viability at 24 h in NOD2-deficient macrophages suggests an alteration in primary host
defense mechanisms of the cell. In order to study this enhanced viability at 24 h, we used
our established method to knock-down NOD2 in human macrophages and performed a
microarray of scramble and NOD2 siRNA-transfected MDMs 24 h post infection. In
total, 1,583 transcripts were found to be significantly different with a fold difference of 2
or more, which is depicted by scatterplot (Figure 3.1A).
In order to quantify the overlap in the number of genes NOD2 regulates under resting
conditions and during M.tb infection, a Venn diagram was created from the 1,583

91
regulated transcripts (Figure 3.1B). Of these, 22% were regulated by NOD2 under
resting conditions, (scramble versus NOD2 siRNA-transfected MDMs) indicating that
NOD2 plays a role in regulating genes involved in homeostasis of macrophages. 54% of
the genes were regulated by NOD2 during M.tb incubation for 24 h (scramble versus
NOD2 siRNA-transfected MDMs incubated with M.tb) and 24% of the genes were
regulated under both conditions (resting and M.tb incubation combined). While it is of
interest that NOD2 regulates genes involved in normal macrophage homeostasis, we
focused on the role of NOD2 during M.tb infection due to the fact that approximately half
of the genes obtained in the microarray were regulated by NOD2 under this condition.
We first analyzed the data by assessing genes regulated in major macrophage signaling
pathways. We found genes affected by NOD2 deficiency during M.tb infection in the
iNOS, JAK/STAT, TLR, autophagy, TGF-β, and apoptosis signaling pathways (Figure
3.1C). While the percentage of genes in each pathway affected in the microarray may
differ depending on the number of genes involved, it is of interest to note that a large
number of genes were identified to be affected in the apoptosis signaling cascade.
When looking further at the fold change of up- or down-regulated genes in each cascade,
we found that iNOS was down-regulated 3.07-fold. We were able to validate that NOD2
deficiency in human macrophages led to a decrease in iNOS mRNA expression during
M.tb infection using qRT-PCR (Figure 3.1D). This was of interest to us because of
previous reports indicating that patients with TB have enhanced iNOS expression in
macrophages when compared to non-TB patients (291).
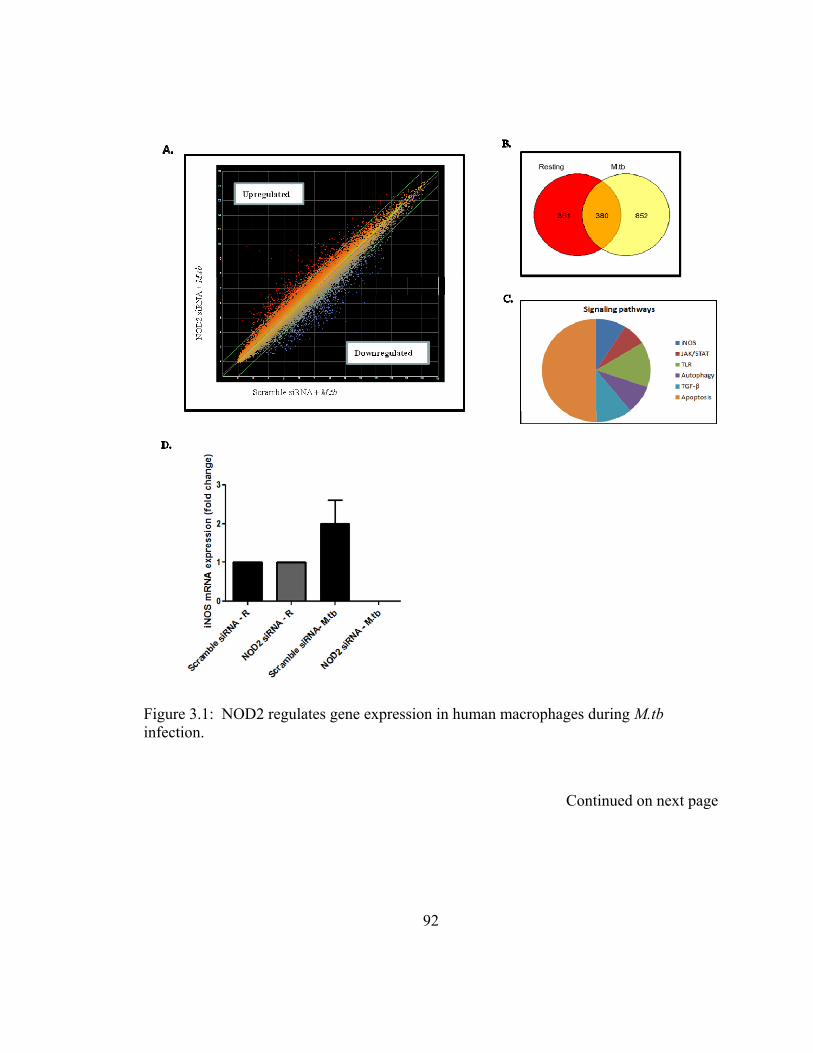
Figure 3.1: NOD2 regulates gene expression in human macrophages during M.tb infection.
Continued on next page
92

Scramble or NOD2 siRNA-transfected MDM monolayers were incubated with M.tb (MOI 5:1) for 2 h, washed 3 times with warm RPMI, and repleted in 1% autologous serum for 24 h. Cells were lysed with TRIzol to obtain total RNA . An Affymetrix Human Genome U133 plus 2.0 array was performed and processed. (A) Shown is a scatter plot (log base 2 scale) with NOD2 siRNA treated cells incubated with M.tb (y-axis) plotted against scramble siRNA treated cells incubated with M.tb (x-axis). (B) A Venn Diagram showing all significantly different genes that were up- or down-regulated twofold or more, with 380 genes common to both conditions (Resting = Scramble vs NOD2 siRNA treated cells and M.tb = Scramble vs NOD2 siRNA treated cells incubated with M.tb). (C) A pie chart showing the major signaling pathways analyzed, and the proportion of genes in each pathway that are regulated by NOD2 during M.tb infection of human macrophages. (D) Scramble or NOD2 siRNA transfected MDM monolayers were incubated with M.tb (MOI 5:1) for 24 h, and then monolayers were lysed with TRIzol for extraction of total RNA. Quantitative real time RT-PCR was used to determine iNOS mRNA levels. Shown is the mean ± SD of a representative graph of two independent experiments.
3.3.2 NOD2 agonists regulate iNOS expression and NO production in human
macrophages
We next determined whether NOD2 agonists were able to induce iNOS mRNA in human
macrophages by using quantitative real-time PCR (qRT-PCR). MDM monolayers were
incubated with MDP or GMDP for 24 h and then lysed in order to isolate RNA for qRT-
PCR analysis or cell lysates were collected for Western blotting analysis of iNOS.
Results from qRT-PCR provide evidence that MDP, the NOD2 agonist found in Gram
negative and Gram positive bacteria, induces iNOS mRNA expression (Figure 3.2A).
However the agonist for NOD2 generated from the cell wall of mycobacteria, GMDP,
induces iNOS mRNA at a significantly higher level. This is not surprising because it has
93

94
been shown that GMDP is a more potent stimulator of the immune response (86). Next,
we compared the iNOS mRNA levels with the amount of protein expressed during MDM
stimulation with NOD2 agonists by Western blotting. While there were no differences in
the amount of iNOS protein expression between agonists, we did observe that expression
is induced by NOD2 agonists (Figure 3.2B). IFNγ was used as a positive control for the
ability of MDMs to induce iNOS protein expression (294). We next investigated whether
the NOD2 agonist GMDP and IFNγ activation of iNOS is NOD2-dependent. Scramble
and NOD2 siRNA-transfected MDM monolayers were incubated for 72 h in order to
achieve the maximum NOD2 knockdown and then incubated with GMDP or IFNγ for 24
h. At this time cell lysates were collected and analyzed for iNOS expression. We found
that NOD2 deficiency leads to the loss of iNOS expression by GMDP and IFNγ (Figure
3.2C). This result indicates that iNOS expression is NOD2-dependent. Finally, we
sought to confirm whether the iNOS expression we observed in response to these
agonists in MDMs resulted in NO production. It was previously observed that IFNγ
activated mouse macrophages stimulated with MDP led to the production of NO, and this
was NOD2-dependent (179). In our MDM model, we used a membrane permeable, NO-
sensitive fluorescent dye, DAF-FM diacetate, in order to measure the production of NO
(293). Figure 2D shows that exposure of MDMs to GMDP for 24 h led to a 45% increase
in NO production. IFNγ stimulation for 24 h resulted in a 57% increase in NO
production, similar to previously reported data using primary human alveolar
macrophages (294). Two representative images from each test group are shown in figure
3.2E.
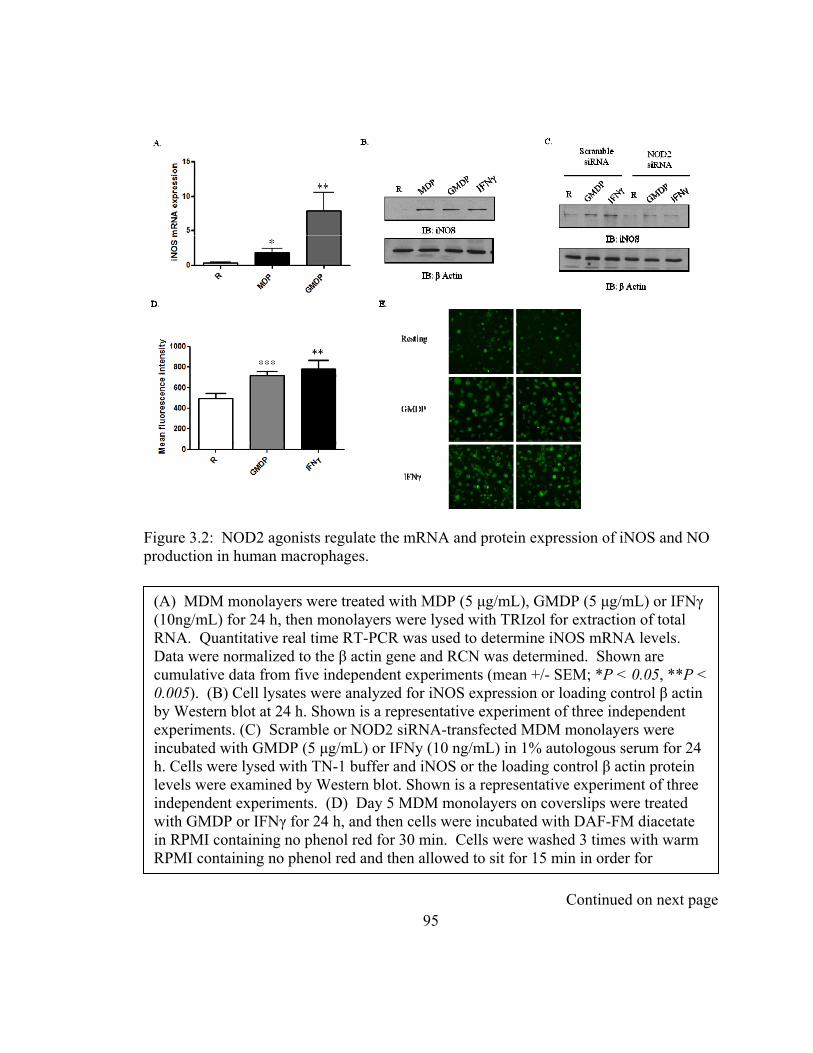
Figure 3.2: NOD2 agonists regulate the mRNA and protein expression of iNOS and NO production in human macrophages.
(A) MDM monolayers were treated with MDP (5 μg/mL), GMDP (5 μg/mL) or IFNγ (10ng/mL) for 24 h, then monolayers were lysed with TRIzol for extraction of total RNA. Quantitative real time RT-PCR was used to determine iNOS mRNA levels. Data were normalized to the β actin gene and RCN was determined. Shown are cumulative data from five independent experiments (mean +/- SEM; *P < 0.05, **P < 0.005). (B) Cell lysates were analyzed for iNOS expression or loading control β actin by Western blot at 24 h. Shown is a representative experiment of three independent experiments. (C) Scramble or NOD2 siRNA-transfected MDM monolayers were incubated with GMDP (5 μg/mL) or IFNy (10 ng/mL) in 1% autologous serum for 24 h. Cells were lysed with TN-1 buffer and iNOS or the loading control β actin protein levels were examined by Western blot. Shown is a representative experiment of three independent experiments. (D) Day 5 MDM monolayers on coverslips were treated with GMDP or IFNγ for 24 h, and then cells were incubated with DAF-FM diacetate in RPMI containing no phenol red for 30 min. Cells were washed 3 times with warm RPMI containing no phenol red and then allowed to sit for 15 min in order for
Continued on next page 95

deesterification to occur. Next, cells were fixed and mounted on slides. Cells were then examined by confocal microscopy with excitation/emission maxima of 495/515 nm (Olympus FV1000). Shown are cumulative data of the mean fluorescence intensities for each test group obtained from three independent experiments (mean +/- SEM; **P < 0.005, ***P < 0.0005). (E) Two representative micrographs (40x) are shown of DAF-FM diacetate staining for each test group of three independent experiments performed on duplicate coverslips.
3.3.3 M.tb-mediated iNOS expression is NOD2-dependent
It is known that the attenuated M.tb strain, H37Ra, enhances iNOS mRNA and protein
expression as well as NO production in human alveolar macrophages (295). The virulent
strain, Erdman, was shown to induce NO production in human monocytes and monocytic
cell lines (289). Corroborating these data, it was discovered that patients with TB have
higher levels of iNOS protein expression in alveolar macrophages (295). We next
examined whether the virulent strain of M.tb, H37RV, and the attenuated vaccine strain,
M. bovis BCG (BCG), caused increased iNOS expression in MDMs. Day 12 MDM
monolayers were incubated with M.tb or BCG (MOI 5:1) for 24, 48, 72, and 96 h at
which time cell lysates were analyzed for iNOS expression by Western blotting (Figure
3.3A). We found iNOS protein expression is enhanced at 24 and 48 h for both bacteria
and decreased at 72 h. A notable difference between the virulent and attenuated strain is
seen at 24 h, M.tb increases iNOS expression to a > level than BCG. We next confirmed
that M.tb-mediated induction of iNOS resulted in NO production in MDMs. MDM
monolayers were incubated with M.tb for 24 h and then exposed to DAF-FM diacetate in
order to quantify NO production. Our results show a 71% increase of NO production in
96

97
MDMs infected with M.tb (Figure 3.3B). Two representative images of each test group
are shown in figure 3.3C. The previous observation of a decrease in iNOS expression
upon stimulation of NOD2 deficient MDMs with the component of peptidoglycan that
can be found in M.tb, GMDP, led us to study whether NOD2 is responsible for the M.tb-
induced iNOS expression. MDMs were transfected with scramble or NOD2 siRNA, and
after 72 h of transfection MDMs were incubated with M.tb. At 24 h cells were lysed and
analyzed for iNOS expression by Western blotting (Figure 3.3D). Silencing of NOD2 in
MDMs and incubation with M.tb resulted in decreased iNOS protein expression. Thus,
our results indicate that in human macrophages, NOD2 is an important mediator of iNOS
expression during M.tb infection.
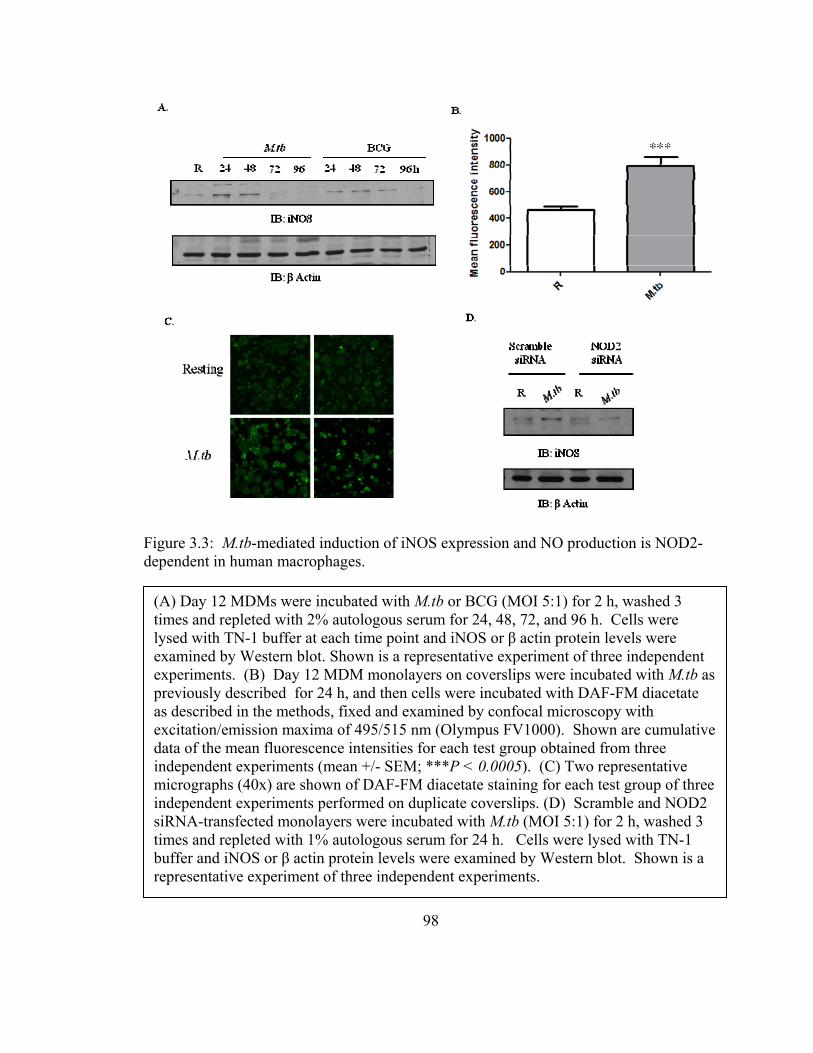
Figure 3.3: M.tb-mediated induction of iNOS expression and NO production is NOD2-dependent in human macrophages.
(A) Day 12 MDMs were incubated with M.tb or BCG (MOI 5:1) for 2 h, washed 3 times and repleted with 2% autologous serum for 24, 48, 72, and 96 h. Cells were lysed with TN-1 buffer at each time point and iNOS or β actin protein levels were examined by Western blot. Shown is a representative experiment of three independent experiments. (B) Day 12 MDM monolayers on coverslips were incubated with M.tb as previously described for 24 h, and then cells were incubated with DAF-FM diacetate as described in the methods, fixed and examined by confocal microscopy with excitation/emission maxima of 495/515 nm (Olympus FV1000). Shown are cumulative data of the mean fluorescence intensities for each test group obtained from three independent experiments (mean +/- SEM; ***P < 0.0005). (C) Two representative micrographs (40x) are shown of DAF-FM diacetate staining for each test group of three independent experiments performed on duplicate coverslips. (D) Scramble and NOD2 siRNA-transfected monolayers were incubated with M.tb (MOI 5:1) for 2 h, washed 3 times and repleted with 1% autologous serum for 24 h. Cells were lysed with TN-1 buffer and iNOS or β actin protein levels were examined by Western blot. Shown is a representative experiment of three independent experiments.
98

99
3.4 Discussion
While NOD2 is one of the most well characterized members of the NLR family, there are
limited reports of the functions of NOD2 in other innate immune responses besides
cytokine production. These recent reports indicate a role for NOD2 in autophagy and
MHC class I and class II presentation (296-298). Our own findings of the growth of M.tb
being controlled at an early time point, led us to question whether other early innate
immune responses were being affected by NOD2 (77). Since NOD2 plays such a major
role in bacterial recognition and clearance, we sought to study other roles of NOD2
through a microarray (299).
Our microarray results show that NOD2 regulates 1,583 transcripts in human
macrophages. While we did not pursue this avenue, our microarray results indicate that
NOD2 is important for normal macrophage homeostasis (independent of M.tb). The role
of NOD2 in homeostatic macrophage functions has not been studied and may be an
important aspect of the receptor to study. Over half of the genes are up- or down-
regulated during M.tb infection of NOD2 deficient macrophages, which suggests that
NOD2 plays a role in many more macrophage responses during infection than previously
reported. Our array analysis revealed that during M.tb infection of human macrophages
NOD2 plays a role in many signaling cascades including: iNOS, JAK/STAT, TLR,
autophagy, TGFβ, and apoptosis. Thus a major contribution to the field by this current
work is the identification of important genes involved in inflammation/immunity affected
by NOD2 deficiency in human macrophages.

100
We chose to study the role of NOD2 in the iNOS signaling pathway due to its relevance
to M.tb infection. In the mouse model, NOS2 is identified as a protective locus against
TB. Mice deficient in the NOS2 gene succumb to infection within 33-45 days compared
to WT mice which survive an average of 160 days. Large, granulomatous lesions were
found in the lung, liver, and spleens at 30 days post infection, while few were found in
WT mice at this time point (284). Similar results of mortality and tissue damage were
found using iNOS inhibitors, aminoguanidine and NG-monomethyl-L-arginine, in the in
vivo mouse model (300). The role of NO during M.tb pathogenesis in humans has been a
controversial topic in the past; however, there is now more evidence in the literature that
supports its impact. Initial reports testing human cells for the production of NO were
negative, due to the low amounts of NO produced by these cells and the usage of
insensitive assays. The discovery of a more sensitive assay using the fluorescent probe
diaminonapthalene (DAN), which detects as little as one pmol of nitrite, allowed
researchers to detect nitrite in human monocytes incubated with M.tb (301). Jagannath et
al reported using the DAN assay that M.tb is able to induce NO production in human
monocytes and this NO is bacteriostatic (289). Whether or not NO is bacteriostatic or
bactericidal is dependent on the specificity of the RNI and the strain of mycobacteria
(302-304). Although the mechanism has not been identified, the ability of M.tb to resist
the effects of RNI is due to a novel gene, noxR1 (305). It is interesting to note that iNOS
deficient mice treated with the same drugs that cure M.tb infected immunocompetent

101
mice are ineffective. This suggests that the effectiveness in vivo of tuberculocidal drugs
may be dependent on iNOS-derived NO (306).
The precise mechanism(s) underlying how NO antagonizes M.tb is unknown; however, it
may include disruption of bacterial DNA, proteins, signaling, or induction of apoptosis.
Since we recently found that NOD2 is an important modulator of the immune response to
M.tb and is necessary to control its growth in primary human macrophages, we decided to
study whether NOD2 plays a role in the expression of the necessary mediator of NO
production, iNOS. Our microarray results indicated that iNOS mRNA expression is
down-regulated during M.tb infection of NOD2 deficient human macrophages. We
verified this result using quantitative RT-PCR. Recently, it was found in mouse
macrophages that MDP induces iNOS protein expression in a RIP2-dependent manner
(283). Thus, we initiated our studies by investigating whether or not NOD2 agonists
induced iNOS mRNA expression in human macrophages. Our results indicate that the
agonist found in Gram negative and positive bacteria, MDP, is a relatively poor inducer
of iNOS mRNA at 24 h. However, the agonist produced by M.tb, GMDP, induces robust
mRNA expression of iNOS. This is not surprising because comparative studies of MDP
and GMDP indicate that GMDP is a more robust stimulator of the immune response (86).
mRNA expression does not always translate to protein expression in eukaryotic cells, so
we explored the ability of NOD2 agonists to induce iNOS protein expression in human
macrophages. Our findings demonstrate that both NOD2 agonists are effective at

102
inducing iNOS protein expression and this is at the same level as a known inducer of
iNOS in human alveolar macrophages, IFNγ (294). These NOD2 agonists are known to
induce the production of pro-inflammatory cytokines, TNF-α and IL-1β. As discussed in
chapter 2, our laboratory has shown that the M.tb-induced cytokine production is NOD2-
dependent in human macrophages (77). Alveolar macrophages from TB patients produce
TNF-α and IL-1β at a higher level than healthy patients and the usage of an inhibitor of
NO led to a reduction in TNF-α and IL-1β (307). It is known that iNOS expression can
be induced by pro-inflammatory cytokines (308,309). The lack of these cytokines being
produced is a possible reason why iNOS is not expressed when NOD2 is deficient.
In order to directly evaluate the role of NOD2 in regulating iNOS expression, we used a
well established method in our laboratory to knockdown NOD2 in human macrophages
(77). To date the role of NOD2 in iNOS and NO production has relied solely on the use
of primary mouse macrophages or the iNOS knockout mouse model (179,282,283). Our
results show that NOD2 is necessary to induce the protein expression of iNOS in human
macrophages. Since iNOS is a critical enzyme that catalyzes the production of NO, this
suggests that NOD2 plays a role in iNOS-induced NO production. NO is an effective
host-defense molecule against microbial pathogens. NO can damage DNA as well as
interact with accessory protein targets and result in enzymatic inactivation (310). Using a
compound that is non-fluorescent until it binds NO to form a fluorescent benzotrizole, we
found that the GMDP-induced iNOS expression consequently results in NO production.

103
Since GMDP is a portion of the cell wall of M.tb, it is possible that it mediates M.tb-
induced iNOS expression and NO production.
Our results are consistent with previous findings that M.tb enhances iNOS expression.
However, here we compare virulent M.tb to the attenuated vaccine strain, BCG. We
observed that M.tb and BCG induce iNOS at 24 h, but M.tb enhances iNOS expression at
a > level when compared to BCG. However, the levels decrease at 72 h for both strains.
In this study, we were able to confirm that the M.tb-induced iNOS expression results in
NO production using a different technique than previously published (DAF-FM
diacetate). This NO could potentially be bacteriostatic or bactericidal as previously
mentioned. Human alveolar macrophages isolated from patients with fibrosis and
challenged with BCG for 24 h displayed enhanced iNOS expression. This led to
peroxynitrate production and the subsequent killing of BCG (311).
For the first time we show that the induction of iNOS by M.tb in human macrophages is
NOD2-dependent. This cytosolic PRR is an important protein in the iNOS signaling
pathway, which leads to the production of NO. The role of a PRR in the induction of an
inflammatory mediator such as iNOS is not reported in human macrophages. In mouse
macrophages, iNOS expression is mediated by TLR4 in MyD88-dependent and
independent manners (312); however its expression in human macrophages has not been
reported. For example in mouse macrophages bacterial lipoprotein activation of TLR2
leads to NO-dependent killing of M.tb, but in human alveolar macrophages and

104
monocytes, the TLR2 activated antimicrobial pathway was found to be NO-independent
(313). This finding is important because it identifies a cytosolic PRR potentially
involved in the antimicrobial killing effects of NO. NOD2 is thought to be a secondary
line of defense in macrophages to fight bacteria that are able to avoid the initial host
defense mechanisms at the surface, but now these data suggest an even more important
role for this receptor in inducing a pathway that results in bacterial killing. This is of
great importance during M.tb infection, where the bacteria are able to modulate the host
immune response and reside in a unique phagosome. Clinical samples have high levels
of iNOS expression in the inflammatory zone surrounding tuberculous granulomas and
NOD2 could be the mechanism for this expression (290).
Patients with Crohn’s disease have inflamed mucosa. They are homozygous for the
3020insC NOD2 mutation (loss-of-function) and are reported to have enhanced levels of
iNOS expression and NO production in the active mucosa (314). This is somewhat
contradictory to the results in this manuscript, since NOD2 knockdown led to decreased
iNOS expression. However, the enhanced iNOS expression and NO production in
Crohn’s disease patients is likely due to other infiltrating cells such as neutrophils or
epithelial cells found in the area. It has been shown that elicited but not circulating
neutrophils produce larger amounts of NO (315,316). Furthermore, we show here that
NOD2 is important in iNOS expression but we did not study endothelial NOS, which is
expressed in cultured human bronchiolar epithelium and is also responsible for NO

105
production (317). These reasons could explain why patients with Crohn’s disease have
increased iNOS and NO levels.
In summary, we report results of a microarray indicating that NOD2 plays a role in many
signaling pathways important in inflammation/immunity. We confirm published work in
that human macrophages are able to produce NO, dependent on NOD2 agonists as well
as M.tb. Finally, we provide evidence that the cytosolic PRR, NOD2, is a mediator in the
production of iNOS in response to M.tb. These findings uncover a new mechanism for
how NOD2 plays a role during M.tb pathogenesis and point towards GMDP as being
clinically useful for controlling M.tb infection in an NO-dependent manner.

106
CHAPTER 4 : The role of PKCδ in the NOD2 signaling pathway in human macrophages
4.1 Introduction
Inflammation is a process that involves the production of cytokines that are important to
control infections and involves various regulators. The most notable transcription factor
that is involved in pro-inflammatory cytokine production is NF-κB. There are several
signaling cascades responsible for NF-κB activation, including Protein kinase C (PKC)
signaling (318). The PKC family of enzymes consists of several isoforms that are
important kinases in ensuring that other proteins function properly through the
phosphorylation of hydroxyl groups on serine and threonine residues. The PKC
subfamilies of isoforms are classified based on their activation stimulus and are called
classical, atypical, and novel. Classical PKCs require DAG, calcium, and phospholipid
for activation and include the isoforms PKCα, PKCβI, PKCβII, and PKCγ (11). Atypical
PKCs require neither calcium nor DAG for activation. They include the isoforms PKCι,
PKCζ, PK-N1, PK-N2 (13). The novel PKCs are activated by diacylglycerol and do not
require calcium. They include the isoforms PKCδ, PKCθ, PKCε, and PKCη (12). PKCδ
is of interest because it is expressed in many cell types including macrophages and is

107
thought to be important in microbial infections (319,320). During Listeria
monocytogenes infection PKCδ is necessary for the confinement of bacteria to the
phagosome (320). PKCδ is also necessary for the control of Raf and MAPK signaling in
response to the bacterium H. pylori (321). The role of PKCδ in intracellular events
during infection points to the possibility that it may be involved in intracellular signaling
pathways that involve cytosolic PRRs.
A more recently discovered intracellular group of receptors are the Nucleotide-binding
oligomerization domain (NOD)-like receptors (NLR). These receptors recognize specific
muropeptides from the peptidoglycan of Gram-negative and Gram-positive bacteria and
induce a pro-inflammatory cytokine response through activation of NF-κB and MAPK
(85). Of the 23 NLRs, NOD2 is one of the best characterized. It recognizes the
muropeptide, muramyl dipeptide (MDP), which is found in both Gram-negative and
Gram-positive bacteria, as well as a glycolyl-MDP (GMDP) found in mycobacteria (86).
While it is a well characterized NLR, there are still several fundamental questions that
need to be answered regarding the signaling pathways that it is involved in. For example,
it is established that MDP is an agonist of NOD2 and that NOD2 “senses” the
muropeptide, but the underlying biochemistry for this recognition is still not understood.
It is hypothesized that another molecule may be involved in the recognition of MDP
which ultimately binds to NOD2; however it is yet to be discovered (74,322).

108
It is important to study the signaling cascades involving NOD2 in order to have a clearer
understanding of how this PRR functions during bacterial infection. The current model
of NOD2 signaling includes recruitment of a key kinase, receptor interacting protein 2
(RIP2). This kinase has a CARD domain that interacts with the CARD domain of NOD2
by a homotypic interaction and induced proximity signaling (76). The interaction only
occurs upon activation and oligomerization of the NOD2 protein (85,88). The next
interaction that occurs is the binding of TGF activated kinase 1 (TAK1) by the NOD2-
RIP2 complex, which leads to subsequent NF-κB and AP-1 mediated activation and pro-
inflammatory cytokine production (95,96). It was previously thought using cell lines that
RIP2 kinase activity was not necessary for proper NOD2 signaling (107). However a
recent study using RIP2 knockout mice, RIP2 kinase-dead knockin mice, as well as RIP2
deficient primary mouse cells proved that RIP2 kinase activity is necessary for protein
stability and downstream signaling of NOD1 and NOD2 (94). However, the exact role of
RIP2 kinase activity in the activation of signaling molecules is still unknown.
A short isoform of NOD2 has been shown to inhibit the NOD2-RIP2 assembly complex
by interacting with these two proteins and interfering with the oligomerization of NOD2
(98). The protein Erbin, a member of the leucine rich repeat and PDZ domain (LAP)
family, also binds NOD2 through the CARD domain and inhibits NOD2-mediated NF-
κB activation (112,113). Another protein, CARD8, has also been shown to inhibit NOD2
signaling through a physical interaction with NOD2 (323). While these studies give
insight into possible mechanisms of proteins that interfere with NOD2 signaling, they are

109
all done in transfected cell lines and thus provide only indirect evidence for the signaling
pathways that may be occurring in primary cells.
Studies indicate that NOD2 may play a role in PKC signaling. In epithelial cells, NOD2
interacts with dual oxidase 2 (DUOX2) and co-localizes at the plasma membrane.
DUOX2 is involved in NOD2-dependent ROS production (324). PKC activation with
low concentrations of PMA causes DUOX2 to be highly induced (325). A more recent
study using mouse macrophages indicates that MDP stimulates PKCβII phosphorylation
in a RIP2-dependent manner (283). These studies along with the lack of full
understanding of NOD2 signaling in primary cells led us to study the role of PKCδ in the
NOD2 signaling cascade. PKCδ has been shown to play an important role in signal
transduction pathways active during infection. We show that NOD2 agonists activate
PKCδ kinase activity and PKCδ-induced cytokine production through a NOD2-
dependent mechanism. We also provide evidence to support that PKCδ is needed for
RIP2 phosphorylation. Together these findings indicate that PKCδ is a newly identified
important mediator in the NOD2 signaling cascade.
4.2 Materials and Methods
4.2.1 Buffers and reagents
Dulbecco’s PBS with and without Ca2+ and Mg2+ ions, and RPMI 1640 medium with L-
glutamine containing phenol red were purchased from Invitrogen (Invitrogen Life

110
Technologies). RHH medium [RPMI 1640 plus 10 mM HEPES (Invitrogen) plus 0.4%
human serum albumin (CSL Behring LLC)] was used for cell culture experiments.
Phorbol myristate acetate (PMA; Sigma-Aldrich), N-Acetylmuramyl-L-alanyl-D-
isoglutamine hydrate (MDP; Sigma-Aldrich), and N-Glycolyl-muramyldipeptide
(GMDP; Invivogen) were used for assays involving PKCδ expression and kinase activity.
4.2.2 Antibodies
Unconjugated rabbit anti-human NOD2 was purchased from ProSci and optimization for
human macrophages can be found in chapter 2 and our recently published work (77).
Total PKCδ, actin, goat anti-rabbit IgG-HRP and donkey anti-goat IgG-HRP antibodies
were purchased from Santa Cruz Biotechnology. Phospho-RIP2, Phospho-PKCδ,
Phospho-PKCθ, and Phospho-PKC/βII antibodies were purchased from Cell Signaling
Technology. The aforementioned antibodies were used for Western blot.
4.2.3 Isolation and culture of human macrophages
Blood obtained from healthy, Purified Protein Derivative negative human volunteers
using an approved protocol by The Ohio State University Institutional Review Board
(IRB) was processed as described (292). Briefly, PBMCs were isolated from heparinized
blood on a Ficoll-Paque cushion (Amersham Biosciences/GE Healthcare) and cultured in
Teflon wells (Savillex) for 1 (monocytes) through 5 (MDMs) days in the presence of
RPMI 1640 medium containing 20% autologous serum (2.0 x106 PBMCs/mL) at
37ºC/5% CO2 (258).

111
4.2.4 Transfection of MDMs
PBMCs were transfected with Accell scramble siRNA, NOD2 siRNA, or PKCδ siRNA
purchased from Thermo Scientific Dharmacon RNAi technologies that targeted for
NOD2 the sequence CUUUAGGAUGUACAGUUA (368nM) and for PKCδ the
sequence GGCUGAGUUCUGGCUGGACTT (250nM) using the Amaxa Nucleofector
(Lonza Group Ltd) Transfected cells were used for subsequent experiments, such as
ELISAs and Western blotting. See section 2.2.9 for more details on the transfection
protocol. Assessment of the NOD2 siRNA specificity and effects on monolayer
density/viability were performed and can be found in chapter 2 and our recently
published work (77). Assessment of the PKCδ siRNA specificity can be found in
previous reports (326). PKCδ siRNA did not affect monolayer density/viability.
4.2.5 Macrophage stimulation and preparation for ELISAs and Western blotting
Day 5 MDMs or day 5 MDMs transfected with scramble, NOD2 siRNA, or PKCδ siRNA
(supernatants, 2.0 x 105; lysates 4.0 x 105 ) for 72 h were incubated with NOD2 agonists
MDP (5ug/mL), GMDP (5ug/mL), or the phorbol diester PMA (0.123ug/mL in RHH for
various time points between 0.5 and 4 h at 37°C in 5% CO2. For day 5 MDM ELISA
experiments using the NOD2 agonist MDP for pre-stimulation, 2.0 x 105 day 5 MDMs
were adhered to a 24-well plate for 2 h at 37°C in 5% CO2, washed to remove
lymphocytes, and then incubated with MDP (5ug/mL) for 90 min. Cells were then

112
washed and incubated with PMA for 1 h, 2 h, 4 h, 6 h, or 24 h. Cell culture supernatants
were harvested and used for ELISAs (see below). For Western blotting experiments, the
monolayers were washed once with PBS at the time points indicated and lysed in TN-1
lysis buffer [50 mM Tris (pH 8.0), 10 mM EDTA, 10 mM Na4PO7, 10 mM NaF,
1% Triton X-100, 125 mM NaCl, 10 mM Na3VO4, 10 µg/ml aprotinin, and 10 µg/ml
leupeptin)], incubated on ice for 10 min and then centrifuged at 17,949 x g for 10 min at
4°C to remove cell debris (261). For NOD2 knockdown assessment, lysates were
collected as above at 72 h post-knockdown. Protein concentrations of the cleared lysates
were measured using the BCA-protein assay kit (Pierce). Proteins were separated by
SDS-PAGE and analyzed by Western blot by probing with the primary and secondary
antibody of interest and development using ECL (Amersham Biosciences). The protein
band intensities were measured using the online ImageJ software provided by the NIH.
Background intensity was subtracted from each sample and then normalized to β-actin.
Fold change was determined as follows: (treated sample band intensity) /
(untreated sample band intensity).
4.2.6 Kinase assays
MDMs were lysed in TN-1 lysis buffer and 250 μg of cell lysates were
immunoprecipitated overnight at 4°C, with 200 ng of anti-PKCδ or PKC (SC-937) or
IgG isotype control (SC-2027) antibodies followed by 1 h incubation with 25 μl of
protein G-agarose beads (Amersham Biosciences). Immunoprecipitates were rinsed four
times with 700 ul of TN-1 buffer followed by one rinse with PKCδ-kinase buffer (25 mM
HEPES pH 7.4, 10 mM MnCl2, 1 mM MgCl2, 1 mM DTT, 0.1 mM PMSF) and

113
subjected to in vitro kinase assays by incubating the immunoprecipitates for 1 h at 37°C
in the presence of 20 μl of kinase assay buffer containing 2 μCi of [γ-32P]-ATP (Perkin
Elmer, Boston, MA), 0.5 mM ATP, 200 μg/ml phosphatidylserine, 20 μg/ml
diacylglycerol, and 1 μg histone 2B (H2B) (Roche Applied Science, Mannheim,
Germany), the latter as exogenous substrate.
4.2.7 ELISA
For MDM experiments, cell free culture supernatants were collected at 1 h, 2 h, 4h, 6h,
and 24 h and used to measure TNF-α by ELISA (R&D Systems).
4.2.8 Statistics
A paired one tailed student’s t test was used to analyze the differences between two
groups in figures showing one representative figure of three independent experiments.
An unpaired one tailed student’s t test was used to analyze differences between two
groups in figures showing cumulative data from three independent experiments.
Significance was P < 0.05.

114
4.3 Results
4.3.1 Activation of NOD2 enhances the PMA-mediated pro-inflammatory cytokine
response through its signaling cascade intermediates
We began our studies by investigating whether activation of NOD2 leads to an increase
in the phorbol-myristate acetate (PMA)-mediated pro-inflammatory cytokine response in
macrophages. PMA is a phorbol ester that activates various PKC isoforms by mimicking
the structure of diacylglycerol (DAG). Incubation of macrophages with MDP, a NOD2
agonist, is known to induce the production of pro-inflammatory cytokines such as TNF-α
(85). MDMs were pre-incubated with the NOD2 agonist MDP for 90 min followed by
incubation with PMA. Cell free culture supernatants were analyzed for TNF-α secretion
at 1 h, 2 h, and 4 h. The data show a synergistic effect on cytokine production in cells
pre-treated with MDP compared to cells incubated with PMA alone (Figure 4.1A). Since
our agonist studies indicated a potential link between NOD2 and the cytokine response to
PMA, we next wanted to explore if the PMA-induced TNF-α production was dependent
on NOD2. Scramble and NOD2 siRNA transfected MDM monolayers were incubated
for 72 h to achieve the maximum NOD2 knockdown and subsequently incubated with
PMA for 3 h. The cell free culture supernatants were analyzed for cytokine levels by
ELISA. We found that NOD2 deficiency leads to a significant decrease in the production
of TNF-α in response to PMA (Figure 4.1B). Similar results were found at 6 h and 24 h
(data not shown). Thus, our results indicate that pre-engagement and hence activation of
NOD2 by its agonist significantly enhances PMA-mediated pro-inflammatory cytokine

115
production. Furthermore, we show that PMA-mediated cytokine production is NOD2-
dependent.
The role of NOD2 in PMA-mediated TNF-α production led us to study the downstream
signaling molecules during PMA stimulation of macrophages. RIP2 has been identified
as a signaling partner for NOD2 that is autophosphorylated upon interaction with the
CARD domain of NOD2 which leads to NF-κB-mediated pro-inflammatory cytokine
production (94). MDMs were incubated with PMA or MDP for 0.5 h, 1.5 h, and 3 h.
Cell lysates were analyzed for phosphorylation of RIP2 by Western blotting of the
phosphorylated protein. The data show that both PMA and MDP phosphorylate RIP2
during incubation but with different intensities. The NOD2 agonist MDP has a distinct
kinetic pattern in the activation of RIP2 peaking at 0.5 h and decreasing over time, while
PMA more robustly stimulates the phosphorylation of RIP2 and does not cause a wane in
expression (Figure 4.1C). In order to understand whether the enhanced expression of
RIP2 phosphorylation was NOD2-dependent or mediated by a different mechanism, we
used siRNA to knockdown NOD2 in macrophages. Scramble and NOD2 siRNA
transfected MDM monolayers were incubated with PMA for 3 h and cell lysates were
analyzed for phosphorylation of RIP2 by Western blotting. We observed that PMA-
mediated phosphorylation of RIP2 in day 5 MDMs was greatly enhanced when compared
to scramble siRNA transfected cells incubated with PMA (Figure 4.1C and D). Even
though the level of PMA-mediated RIP2 phosphorylation of scramble siRNA treated
cells was lower than day 5 MDMs, we did find that NOD2 deficiency led to a decrease in
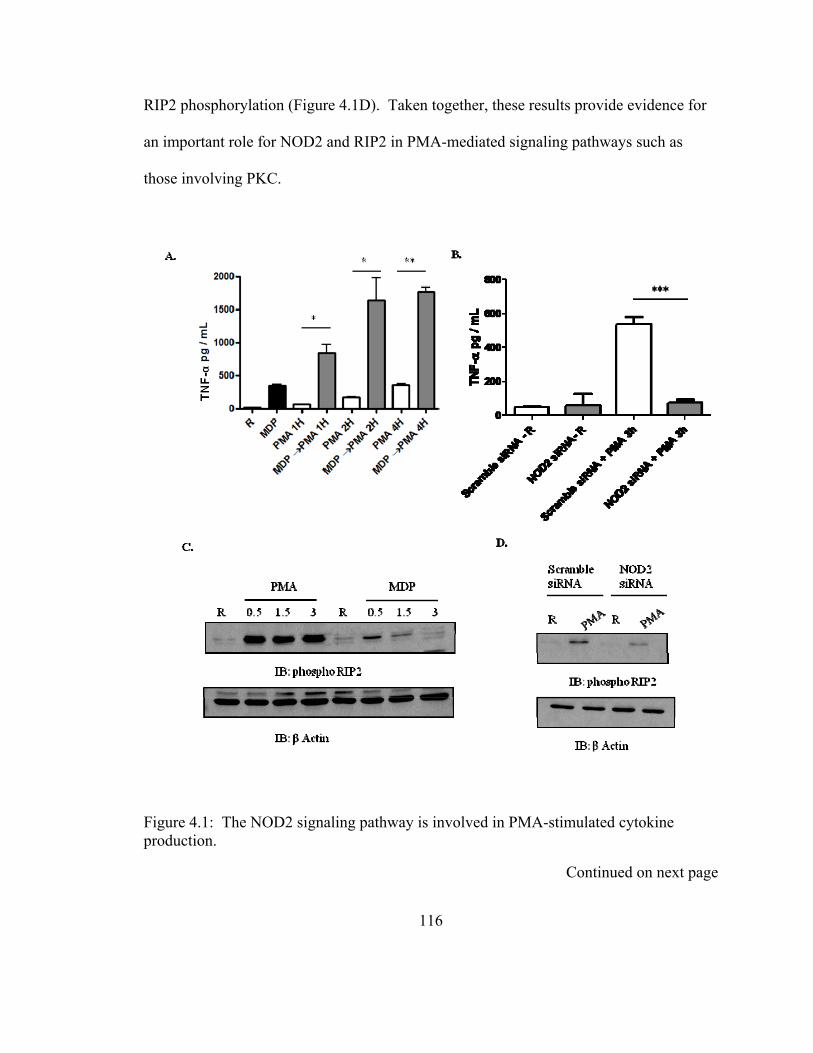
RIP2 phosphorylation (Figure 4.1D). Taken together, these results provide evidence for
an important role for NOD2 and RIP2 in PMA-mediated signaling pathways such as
those involving PKC.
Figure 4.1: The NOD2 signaling pathway is involved in PMA-stimulated cytokine production.
Continued on next page
116

(A) MDM monolayers in 24 well tissue culture plates were pre-incubated with or without MDP (5 μg/mL) for 90 min and then incubated with PMA (0.123 μg/mL) in RHH for the indicated time points. Supernatants were collected and the amount of TNF-α was measured by ELISA. Shown is the mean ± SD of a representative experiment performed in triplicate (n = 2, * P < 0.05; **P < 0.005). (B) Scramble or NOD2 siRNA-transfected MDM monolayers were incubated with PMA in RHH for 3 h. Supernatants were collected and the amount of TNF-α was measured by ELISA. Shown is the mean ± SD of a representative experiment performed in triplicate (n = 2, ***P < 0.0005). (C) Day 5 MDM monolayers were incubated with PMA or MDP for the indicated time points, and cells were lysed with TN-1 lysis buffer and probed for RIP2 phosphorylation, and then re-probed for actin as a loading control. Shown is a representative Western blot from two independent experiments. (D) Scramble or NOD2 siRNA-transfected MDM monolayers were incubated with PMA in RHH for 3 h and cells were lysed with TN-1 lysis buffer and probed for RIP2 phosphorylation, and then re-probed for actin as a loading control. Shown is a representative Western blot from three independent experiments.
4.3.2 Phosphorylated PKCδ expression and activity is NOD2-dependent
The ability of PMA to induce pro-inflammatory cytokine production and robustly
activate RIP2 in a NOD2-dependent manner led us to formally investigate whether PKC
isoforms were involved in this pathway. PMA is a known activator of several PKC
isoforms including PKCα, PKCβ, PKCδ, and PKCθ (14). We elected to examine
whether the NOD2 agonist, MDP, led to increased phosphorylation of PKC isoforms by
using a PKC phospho-array of isoforms, δ, θ, and, α / βII (Figure 4.2A, B, C). MDP
caused enhanced phosphorylation of PKC δ, PKCθ, and PKCα / βII. PKCθ is not well
characterized in macrophages (327,328). So, we focused our studies on PKCδ and PKCα
/ βII due to their presence in macrophages and roles in signaling during microbial entry of
host cells (320,329). We first compared the NOD2 agonists MDP and GMDP, the latter
117

118
is a NOD2 agonist from the cell wall of mycobacteria (see chapter 2), for their abilities to
induce PKCδ phosphorylation. MDMs were incubated with MDP or GMDP for 0.5 h,
1.5 h, and 3 h. Cell lysates were analyzed for phosphorylation of PKCδ by Western
blotting of the phosphorylated protein. The data show that GMDP leads to
phosphorylation of PKCδ at early time points (0.5 h and 1.5 h) and is lost at the later time
point (3 h), while MDP only slightly increased the phosphorylation of PKCδ at 0.5 h
(Figure 4.2D). Since GMDP was a more potent stimulator of PKCδ phosphorylation, we
proceeded in our studies using this agonist.
Detection of phosphorylation of PKC isoforms by Western blotting is not an accurate
indicator of the magnitude of its kinase activity because in order to be active the kinase
must be phosphorylated at multiple sites. In order to test for PKCδ kinase activity, we
performed a PKCδ kinase assay in immunoprecipitates using GMDP and PMA. The
results indicate that PKCδ activity is non-existent at 0.5 h and peaks at 1.5 h when
stimulated with GMDP (Figure 4.2E). This difference in kinetics of phosphorylation and
kinase activity of PKCδ is not surprising, as it may take time for all of the relevant sites
to be phosphorylated and the kinase to be fully functional. We also elected to study the
kinase activity of PKCα specifically during PMA and GMDP stimulation; however the
results were less consistent when compared to delta. Despite phosphorylation of
PKCα/βII at 1.5 h and 3 h (Figure 4.2C), the kinase assay shows PKCα is active only at
0.5 h (Figure 4.2F). Also, the kinetics of activation does not match RIP2. PKCα is
activated by GMDP at 0.5 h, which is at the same time that RIP2 is phosphorylated,

119
indicating that they would be working simultaneously. While this may occur it suggests
that other mechanisms are involved, making it more complicated to understand the
general role of PKCs in NOD2 signaling. Thus, we elected to move forward with
studying only PKCδ.
The ability of a NOD2 agonist to fully activate PKCδ led us to study whether NOD2 is
important in mediating PKCδ phosphorylation. Scramble and NOD2 siRNA transfected
MDM monolayers were incubated with PMA for 3 h and cell lysates were analyzed for
phosphorylation of PKCδ by Western blotting. Our results show decreased expression of
phosphorylated PKCδ in response to PMA in NOD2 deficient MDMs (Figure 4.2G). We
next wanted to determine whether the NOD2-dependent PKCδ phosphorylation was only
through the PMA-mediated pathway or also through the NOD2-mediated pathway.
Scramble and NOD2 siRNA transfected MDM monolayers were incubated with GMDP
for 3 h and cell lysates were analyzed for phosphorylation of PKCδ by Western blotting.
Our results show a marked decrease in phosphorylated PKCδ in NOD2 deficient MDMs
(Figure 4.2H). These results indicate that NOD2 is important in the classical PKCδ
signaling cascade (PMA) and the NOD2-signaling cascade (GMDP).
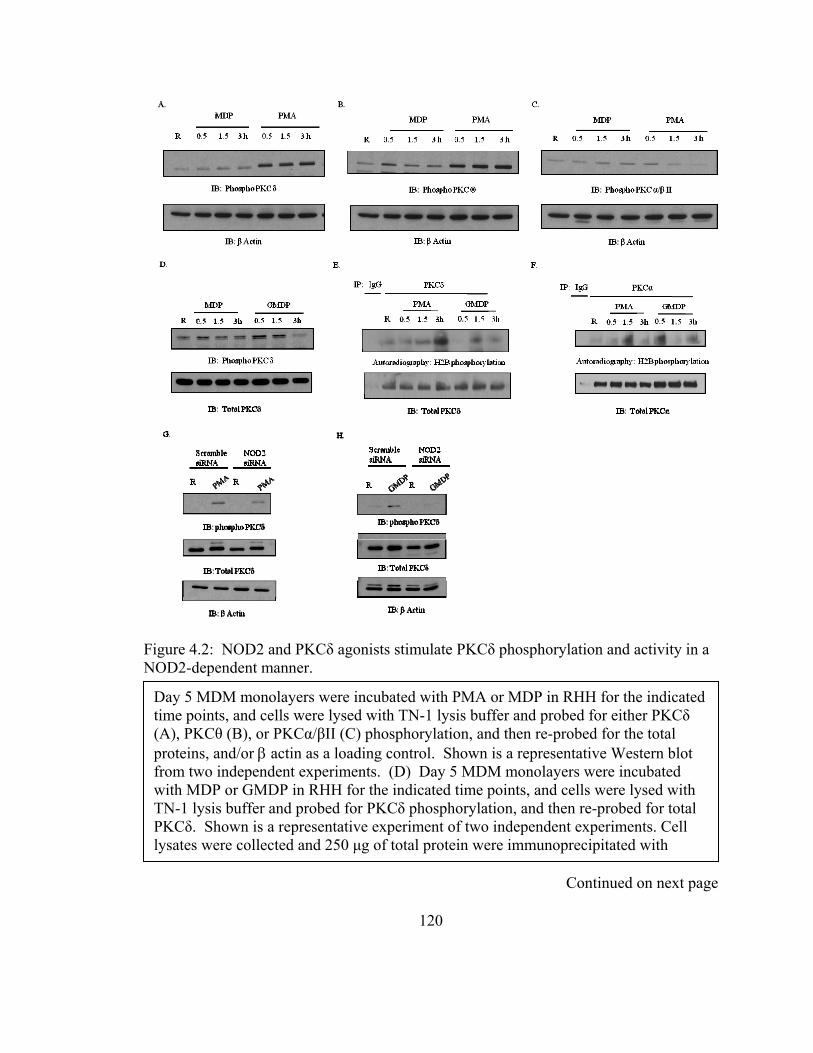
Figure 4.2: NOD2 and PKCδ agonists stimulate PKCδ phosphorylation and activity in a NOD2-dependent manner.
Day 5 MDM monolayers were incubated with PMA or MDP in RHH for the indicated time points, and cells were lysed with TN-1 lysis buffer and probed for either PKCδ (A), PKCθ (B), or PKCα/βII (C) phosphorylation, and then re-probed for the total proteins, and/or actin as a loading control. Shown is a representative Western blot from two independent experiments. (D) Day 5 MDM monolayers were incubated with MDP or GMDP in RHH for the indicated time points, and cells were lysed with TN-1 lysis buffer and probed for PKCδ phosphorylation, and then re-probed for total PKCδ. Shown is a representative experiment of two independent experiments. Cell lysates were collected and 250 μg of total protein were immunoprecipitated with
Continued on next page
120

PKCδ (E) or PKCα (F) antibody or isotype control. Immunoprecipitates were subjected to in vitro kinase assays. Shown are representative kinase assays of phosphorylation of recombinant H2B visualized by autoradiography from two independent experiments. Scramble or NOD2 siRNA-transfected MDM monolayers were incubated with PMA (G) or GMDP (H) in RHH for 3 h and cells were lysed with TN-1 lysis buffer and probed for PKCδ phosphorylation, and then re-probed for total PKCδ and actin as a loading control. Shown are representative Western blots from three independent experiments.
4.3.3 RIP2 activation is PKCδ-dependent
Our results indicate that NOD2 is necessary for the activation of PKCδ and this result
points to it being a potential player in the NOD2 signaling cascade. Therefore we next
sought to determine where PKCδ activation fits into the known NOD2 signaling cascade.
RIP2 is felt to be the next identified kinase to interact with NOD2, so we studied whether
PKCδ is necessary for RIP2 phosphorylation. Scramble and PKCδ siRNA transfected
MDM monolayers were incubated with PMA for 3 h and cell lysates were analyzed for
phosphorylation of RIP2 by Western blotting. Our results show decreased amounts of
phosphorylated RIP2 in PKCδ deficient MDMs (Figure 4.3A). We next wanted know if
this PKCδ dependent RIP2 phosphorylation was only through the PMA-induced pathway
or also through NOD2-induced pathway. Scramble and PKCδ siRNA transfected MDM
monolayers were incubated with GMDP for 1.5 h and cell lysates were analyzed for
phosphorylation of RIP2 by Western blotting. Our results show a small but consistent
decrease in amounts of phosphorylated RIP2 in PKCδ deficient MDMs (Figure 4.3B).
121
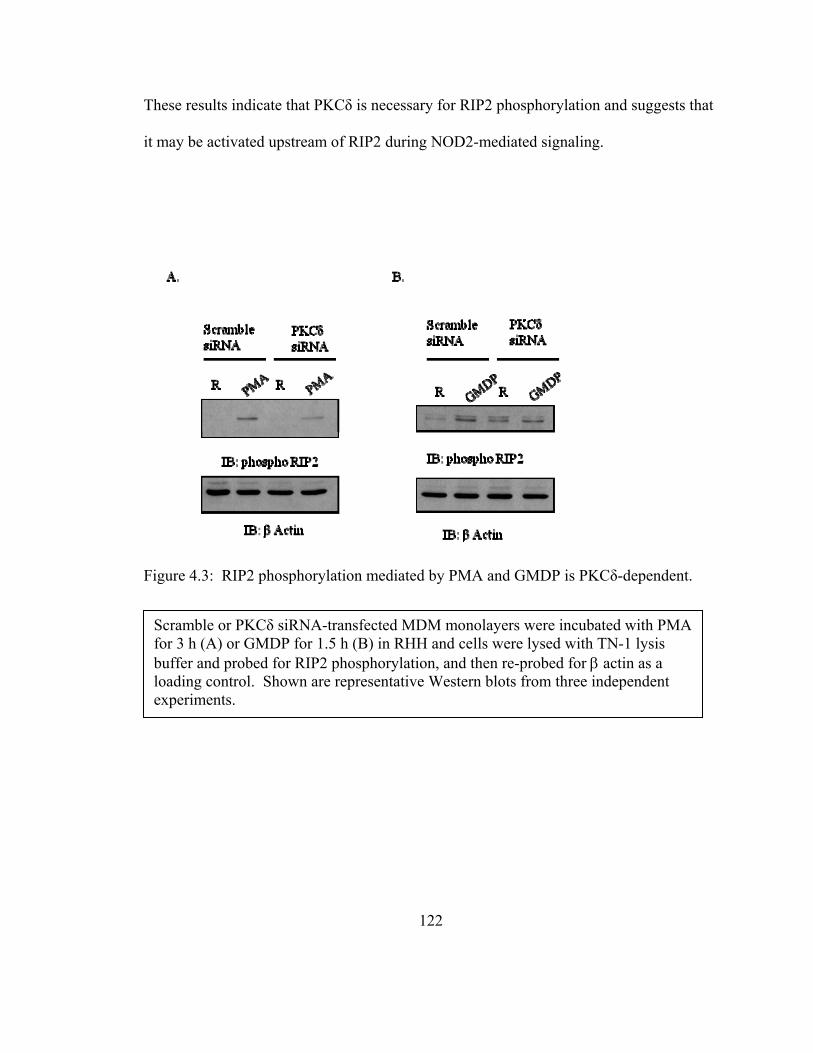
These results indicate that PKCδ is necessary for RIP2 phosphorylation and suggests that
it may be activated upstream of RIP2 during NOD2-mediated signaling.
Figure 4.3: RIP2 phosphorylation mediated by PMA and GMDP is PKCδ-dependent.
Scramble or PKCδ siRNA-transfected MDM monolayers were incubated with PMA for 3 h (A) or GMDP for 1.5 h (B) in RHH and cells were lysed with TN-1 lysis buffer and probed for RIP2 phosphorylation, and then re-probed for actin as a loading control. Shown are representative Western blots from three independent experiments.
122

123
4.4 Discussion
NOD2 is an important intracellular receptor in controlling bacterial infections (77,299).
There are many reports on the function of this receptor during bacterial infection, but
much less is known about the biochemical mechanisms (i.e. signaling pathways)
underlying its activation and ability to mediate its functions. Increased knowledge in this
area will not only expand our understanding of the role of NOD2 in host defense but may
also provide new targets for host-directed therapies that alter the activity of this receptor.
Currently there are reports of several proteins that potentially interact with NOD2 in in
vitro cell systems, and these reports point towards the ability of NOD2 to interact with
proteins other than its main regulator RIP2 (98,112,113,323).
The PKC enzyme family plays a role in pro-inflammatory cytokine production through
the transcription factor NF-κB (318). They also play a major role in signal transduction
cascades through their ability to phosphorylate hydroxyl groups of serine and threonine
residues. A member of the novel PKC family, PKCδ, is of interest in this chapter.
Diacylglycerol is necessary in order to activate this novel PKC, however, PMA is used
instead of diacylglycerol in vitro to mimic its actions (12). Our interest lies in PKCδ
because it is found in macrophages and recent findings provide evidence that PKCδ is
involved in microbial infections, including those due to Listeria monocytogenes and
Helicobacter pylori (319-321). PKCδ’s established role in signal transduction pathways
(including those that regulate ROS production) along with the discovery of its role during
microbial infection makes it plausible that it may intersect with pathways involving

124
intracellular receptors involved in microbial recognition such as NOD2. Thus, we sought
to study the role of PKCδ in the NOD2 signaling cascade in human macrophages
(283,325).
We initially found that using the NOD2 agonist MDP as an activator of NOD2 followed
by PMA stimulation caused a significant increase in TNF-α production at early time
points. We also found that PMA-induced cytokine production was NOD2-dependent.
PMA is a phorbol ester that mimics DAG, a lipid involved in second messenger
signaling. A major function of DAG is activation of PKCs. Upon activation, PKCs
translocate to the plasma membrane and perform their function of phosphorylating other
proteins that mediate the induction of cytokines. The substrates that PKCs activate are
dependent on the cell type. The recent finding that NOD2 co-localizes with an activator
of PKCs, DUOX2, at the plasma membrane indicates a role for PKCs in NOD2 signaling
(324).
In our model of human macrophages, we found that PMA led to the phosphorylation of
PKCs as expected, but were surprised to find that PMA also led to the phosphorylation of
the downstream signaling partner of NOD2, RIP2. The level of RIP2 phosphorylation
was in fact much higher than the known inducer of RIP2 phosphorylation, MDP. We
also found that this phosphorylation is partially NOD2-dependent. RIP2 is a serine
threonine kinase that is felt to act as a scaffolding protein more than a kinase during NF-
κB activation; however in NOD2 signaling the kinase activity is important for protein

125
stability (330). RIP2 has been shown to associate with TNFR1, TRAF1, TRAF5,
TRAF6, CIAP1, Cflip, TAK1, as well as caspase-1 (330-333). It is interesting to note
that PKCδ interacting protein kinase (DIK), also known as RIP4, shares 45% similarity to
the RIP2 kinase domain (334). RIP4 is a membrane-associated kinase with ankyrin
repeats that is known to bind PKCβ and contributes to activation of NF-κB (334,335).
This activation is dependent on the catalytic activity of RIP4. The ability of RIP2 to
interact with other proteins as well as the capability of a RIP family member to interact
with PKCs suggested to us that RIP2 may interact with PKCδ. Our findings along with
the literature provide evidence for the convergence of the NOD2 and PKC signaling
pathways. This discovery raises new possibilities for the role of NOD2 in a number of
macrophage host defense functions.
The ability of a NOD2 agonist to induce novel PKC isoforms is of interest. The
enhancement of PKCδ activity by NOD2 agonists indicates that NOD2 and PKCδ
signaling may be dependent on one another. Our studies show that PKCδ activity is
NOD2-dependent when stimulated with its classical agonist, PMA, or the NOD2 agonist
GMDP. The discovery that NOD2 is necessary for optimal PKCδ kinase activity is a
novel finding and may be an important mechanism for PKC-dependent cytokine
production. In order to evaluate the location of PKCδ in the NOD2 pathway, we used a
well-established method of knocking down PKCδ (326). We also show that the
downstream kinase of NOD2 is dependent on PKCδ, which is of interest. Our results
indicate that NOD2 is necessary for PKCδ activity and PKCδ is needed for RIP2

126
phosphorylation. We speculate in Figure 4.4 that NOD2, PKCδ, and RIP2 may be in
complex at the membrane that in turn binds TAK1 for proper NF-κB induced cytokine
production to occur. Upon PKC activation, RACK proteins translocate the kinase to the
membrane. It has been previously shown that NOD2 and RIP2 are at the plasma
membrane and signaling can occur here as well (112,114,279). In addition, we found that
PKCα is activated at an earlier time point which implies that this isoform and RIP2 are
activated simultaneously. While we did not pursue this area of interest, the data suggest
other possible signaling pathways may be involved. For example, NOD2 and PKCα may
be involved in earlier events, like p47phox activation for reactive oxygen species
production through the NADPH oxidase.
In summary, here we report a new addition to the NOD2 signaling pathway and provide
new insight into PKC-induced cytokine production. We show that NOD2 is important for
PKCδ activity as well as PMA-induced cytokine production. In addition, we show that
RIP2 phosphorylation is PKCδ-dependent. These findings divulge a new mechanism for
the NOD2 signaling cascade and for PKC-mediated cytokine production (Figure 4.4). It
will be interesting to further define the importance of PKCδ to the NOD2 signaling
pathway. These data provide useful knowledge for targeting NOD2 in host-directed
therapies to control bacterial infections.
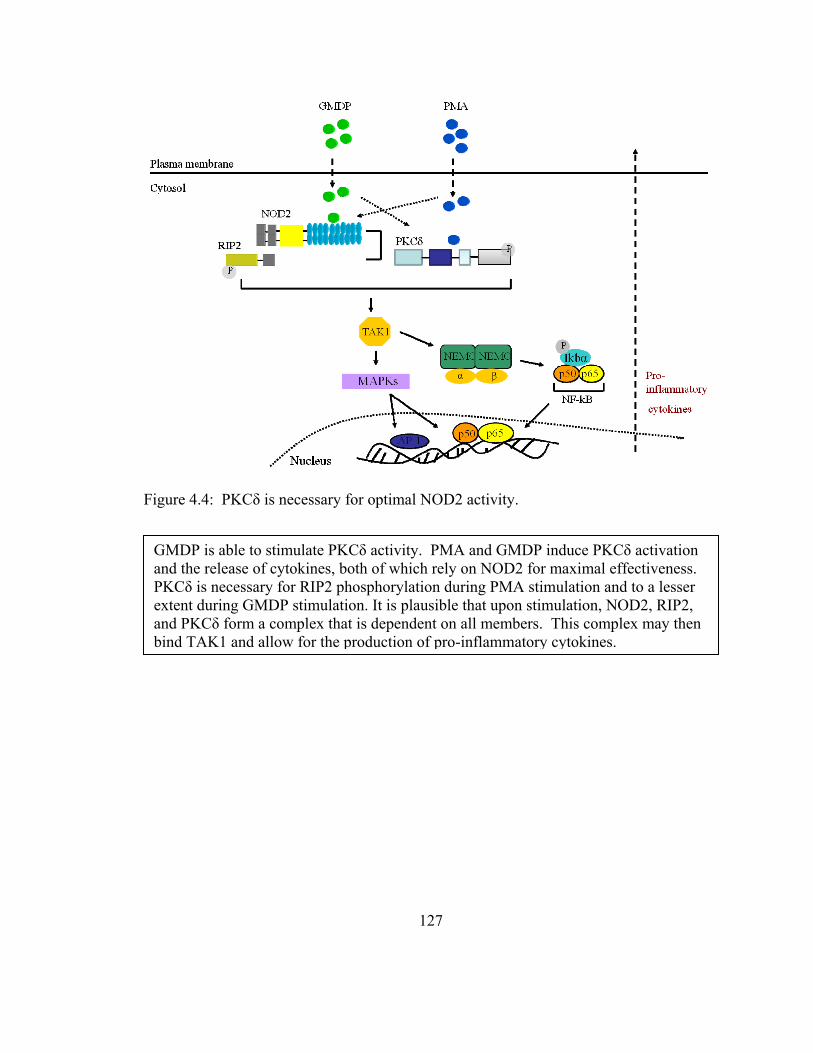
Figure 4.4: PKCδ is necessary for optimal NOD2 activity.
GMDP is able to stimulate PKCδ activity. PMA and GMDP induce PKCδ activation and the release of cytokines, both of which rely on NOD2 for maximal effectiveness. PKCδ is necessary for RIP2 phosphorylation during PMA stimulation and to a lesser extent during GMDP stimulation. It is plausible that upon stimulation, NOD2, RIP2, and PKCδ form a complex that is dependent on all members. This complex may then bind TAK1 and allow for the production of pro-inflammatory cytokines.
127

128
CHAPTER 5 : Synthesis
The human immune system is a well defined entity that humans depend on to protect
them against pathogens. Humans are equipped with a complex immune system that
encompasses an adaptive and innate immune response. While both components are
necessary in the clearance and eventual immunity against pathogens, the innate immune
system is the first line of defense against invading organisms. Epithelial cells,
neutrophils, monocytes, and macrophages are cell types that play key roles in protecting
the host. Important components of these cells involved in the recognition and clearance
of microorganisms are PRRs (1). The innate immune system is not always enough to
eliminate pathogens, wherein lies the role of the adaptive immune response. The
adaptive immune response protects the host by remembering previous invaders and
producing an enhanced immune response in order to control the infection (1,336).
Our laboratory is interested in the innate immune response to the pathogen M.tb. More
specifically we study the interaction of M.tb with human macrophages. M.tb infects one-
third of the world’s population and is a great public health concern. It is easily
transmissible by aerosol droplets with a low infectious dose of ten bacilli causing

129
infection (157). One of the first immune cells that M.tb comes in contact with in the
alveolus is the alveolar macrophage. While treatments are available, their expense and
long regimens make it difficult for under developed countries to control the spread of
disease. The recent increase in multi- and extensive-drug resistant strains makes it
difficult for even developed countries to control. It is important to gain more
understanding of the pathogenesis of M.tb in order to be successful in the development of
more effective drug therapies and possible vaccines. This study focuses on one of the
recently described PRRs found in macrophages, NOD2, and its role during M.tb
pathogenesis.
In order to study the interaction of NOD2 with M.tb, our laboratory uses the most
clinically relevant model for M.tb, human MDMs and alveolar macrophages. These two
cell types are critical during M.tb infection and are relevant since this bacterium only
naturally infects humans. While most of our studies use MDMs, this work and previous
work from our laboratory show that they have similarities to alveolar macrophages. For
example, they both express common PRRs such as the MR, TLRs, CRs, and NOD2
(54,77,258). Therefore MDMs are an excellent model to study the first interaction of
M.tb with the host and understand the early innate immune response.
Prior to our NOD2 studies, data were lacking regarding basal mRNA and protein
expression on mononuclear phagocytes during monocyte differentiation into
macrophages. Previous NOD2 studies were limited to cell lines, murine macrophages,

130
and some human monocyte and mixed BAL studies (127,127,135,179-181,220).
Importantly, there are significant differences between the mouse and human promoter of
NOD2 (147). Furthermore, the field was only able to identify NOD2 mRNA expression
in human PBMCs and not protein. Therefore, we began our research by examining
NOD2 expression during monocyte differentiation into macrophages using our MDM
model [Chapter 2]. Interestingly, we observed NOD2 mRNA and protein expression is
low during day one and two of differentiation and significantly increases at day three of
differentiation after which it remains relatively unchanged (Figure 2.1). These data
suggest that NOD2 plays a relatively more important role during an effective innate
immune response in macrophages, while monocytes more readily rely on other PRRs to
control infection. We also observed that NOD2 protein has a punctate rather than diffuse
appearance in the cytosol (Figure 2.1, 2.3). This appearance suggests that NOD2 may be
located either in organelles or as complexes in the cytoplasm. There also appeared to be
less NOD2 protein in human alveolar macrophages than in MDMs (Figure 2.5). Since
NOD2 is an important receptor in controlling early infection, this finding provides one
explanation for why alveolar macrophages are less effective at controlling initial M.tb
infection. However, we were able to show that pre-treatment of alveolar macrophages
with the agonist for NOD2, MDP, followed by infection leads to enhanced pro-
inflammatory cytokine production (Figure 2.5). Even though NOD2 expression is less in
HAMs when compared to MDMs (Figure 2.5), this result implies that this receptor can be
up-regulated, at least to some degree, in alveolar macrophages to potentially control
infection.

131
After determining NOD2 expression during the differentiation into macrophages, we
examined whether MDP pre-treatment followed by M.tb or BCG incubation leads to
enhanced cytokine production. We found that stimulating NOD2 with its agonist
followed by M.tb or BCG leads to increased pro-inflammatory cytokine production.
Additionally, these two bacteria are able to activate downstream mediators (RIP2 and
IκBα) in the NOD2 signaling cascade, albeit with different expression patterns (Figure
2.2). The differences in expression indicate that M.tb, which has a shorter and less robust
activation compared to BCG, is able to dampen the immune response. One possibility is
that M.tb utilizes secreted virulence proteins found in the RD1 to dampen NOD2-
mediated signaling. These proteins are absent in the attenuated BCG strain (192,193).
The enhanced pro-inflammatory cytokine production after NOD2 pre-engagement,
suggests that NOD2 is involved during mycobacterial infection of macrophages.
However, use of an agonist is not the most direct method to study a receptor’s activity.
Thus, we next used siRNA knockdown of NOD2 in human macrophages and observed
decreased RIP2 phosphorylation and pro-inflammatory cytokine production in response
to the two mycobacteria (Figure 2.4). In addition, we found enhanced M.tb and BCG
growth in NOD2 deficient macrophages (Fig. 2.6). This increase in growth is different
from previously published data (135,179), as well as our own data, using NOD2
knockout BMMs incubated with M.tb (Figure 2.7). In contrast, BCG showed enhanced
growth in NOD2 knockout BMMs suggesting that M.tb has acquired a mechanism for

132
evading the immune system of the mouse, while BCG has not. One possibility is that
under normal conditions M.tb utilizes virulence proteins, that BCG is lacking (such as
those in the RD1 in the genome), in order to inhibit proper NOD2 functioning. These
virulence proteins may be more effective in inhibiting NOD2 function in mice because
they target the promoter of NOD2, which has been found to be structurally different in
mice (147).
It is of interest that NOD2 plays a greater role in IL-1β production during M.tb infection,
with slight enhancement of pro-IL-1β transcription and a decrease in caspase-1
expression in NOD2 deficient MDMs (Figure 2.4). As one explanation, NOD2 may play
a relatively more important role in enabling the release of mature IL-1β through caspase-
1 activation in response to M.tb than in increasing TNF-α production, since NOD2 has
been shown to interact with inflammasomes (97,270).
Our studies of NOD2 involvement during M.tb infection lead to unanswered questions,
such as how does NOD2 recognize M.tb and what molecule(s) does NOD2 regulate in
order to affect viability of mycobacteria at 24 h? Several potential mechanisms exist for
NOD2 recognition of M.tb, including the ability of M.tb to escape the phagosome, the
bacteria’s ability to secrete virulence proteins, and the possibility that NOD2 signals from
the phagosomal membrane. Since our data show that NOD2 regulates cytokine
production and M.tb growth at 24 h, we reason that M.tb is not escaping the phagosome,
which has been shown to begin to occur in human dendritic cells at 48 h post infection

133
(215). In addition, our results with the attenuated vaccine strain, BCG, argue that
secreted virulence proteins are not the sole mechanism for NOD2-mediated recognition
of M.tb; however, we cannot rule out the possibility that M.tb down-regulates
downstream signaling molecules important to NOD2 signaling when compared to BCG.
Mycobacteria express a hydroxylase, NamH, which glycolylates MDP (GMDP) causing
it to induce a strong immune response (86). Our NOD2 confocal data support the
possibility that NOD2 localizes at the M.tb phagosome membrane. NOD2 signaling in
epithelial cells occurs at the plasma membrane and NOD2 must be localized there for this
signaling to occur (114,324). Therefore, we hypothesize that NOD2 localizes at the
mycobacterium-containing phagosome and is engaged by bacterial cell wall products
such as GMDP released during replication of the bacteria inside the phagosome. Such a
hypothesis would require that bacterial cell wall products be locally transported to the
cytosolic face of the phagosome, possibly through a secretion or transport system. In
order to confirm or refute this hypothesis, we would need to either isolate
mycobacterium-containing phagosomes to look for the presence of NOD2 by Western
blotting or perform immunofluorescence microscopy studies to see if NOD2 is located on
the M.tb-containing phagosome membrane.
While we observed that NOD2 regulates the viability of M.tb and BCG in human
macrophages, the mechanism(s) behind this role remains unknown. One possible
mechanism that our data support is through the production of pro-inflammatory cytokines
which can activate the macrophages through autocrine and paracrine means. We

134
performed experiments using MDP to “prime” the macrophage to induce early innate
immune responses such as ROIs / RNIs in a NOD2-dependent manner. We found a
consistent decrease in the growth of BCG and M.tb under these conditions, but the results
were variable (10-26% decrease for BCG and 4-29% for M.tb). Thus, the results are
generally consistent with NOD2 being involved in controlling the viability of these
mycobacteria, however it is possible that the experimental conditions were not optimized,
e.g. other parameters such as time and dose should be further tested. In order to
determine the role of NOD2 on other early innate immune responses during M.tb
infection, we would need to examine which processes are involved. Such studies would
broaden the roles of NOD2 in innate immunity beyond those that involve enhancement of
pro-inflammatory cytokine production.
Related to the discussion above, we became interested in what processes NOD2 regulates
during infection [Chapter 3]. We began these studies by performing a microarray of
NOD2 deficient macrophages infected with M.tb for 24 h. Interestingly, we observed
many of the genes in signaling pathways involved in host defense were regulated by
NOD2 in human macrophages including iNOS, autophagy, and apoptosis (Figure 3.1).
Although somewhat controversial, studies in the literature support involvement of iNOS,
and thus its product NO, during M.tb infection of human cells (290,291). Thus, we
decided to study this enzyme in the context of NOD2. NOD2 agonists have previously
been shown to induce iNOS protein expression in mouse macrophages (283), but no one
has studied the role of NOD2 in iNOS activity in human macrophages. We observed that

135
iNOS mRNA and protein are expressed in response to NOD2 agonists (Figure 3.2). It is
of interest that higher mRNA expression was observed in response to the mycobacterial
NOD2 agonist, GMDP, when compared to the Gram negative / Gram positive NOD2
agonist, MDP, but no differences were observed in iNOS protein expression. One
potential explanation for this result is that GMDP is able to induce iNOS protein
expression for a longer time period as a result of increased stability of transcript levels.
This idea was not studied in the context of the current work but if this was found to be the
case, the result would add to our understanding of iNOS transcriptional regulation.
After determining that NOD2 may be involved in iNOS expression through the usage of
NOD2 agonists, we more directly assessed the role of NOD2 using siRNA knockdown.
NOD2 deficient macrophages treated with GMDP or IFNγ resulted in decreased iNOS
expression (Figure 3.2). Thus, these data indicate that NOD2 is involved in iNOS
expression in human macrophages. In addition, we observed that NO was enhanced in
response to the NOD2 agonist GMDP (Figure 3.2). These results raise the possibility that
NOD2 is necessary for NO production in response to invading organisms. Ongoing
studies involve assessing NO production in NOD2 deficient cells stimulated with these
agonists.
We next explored whether M.tb induces iNOS expression and NO production in our
MDM model, since the literature is not in agreement on this subject matter (337). We
observed enhanced iNOS expression and NO production in response to M.tb (Figure 3.3).

136
In addition we found that M.tb-induced iNOS expression is NOD2-dependent (Figure
3.3). It is of interest that NOD2 regulates M.tb-induced iNOS expression and potentially
NO production. This links a cytosolic PRR with the induction of iNOS and the
antimicrobial killing effects of NO. It would be of interest to study which molecules in
the iNOS signaling pathway are regulated by NOD2 during M.tb infection. This
experiment would add to our understanding of the relationship between NOD2 and an
antimicrobial mediator, iNOS. It would also be interesting to identify if GMDP is the
main component of M.tb responsible for iNOS expression. To study this we are currently
using M.tb deficient in NamH, the gene necessary for the production of GMDP (86), to
analyze iNOS expression and NO production.
Since NOD2 is a relatively new PRR and its ability to recognize its agonist is still
unknown, we decided to study the signaling cascade to get a better understanding of how
NOD2 signals in response to pathogens [Chapter 4]. We began these studies by
stimulating NOD2 deficient cells with PMA and observed a decrease in cytokine
production. In addition, we found that PMA induces the NOD2 downstream signaling
kinase RIP2 in a NOD2-dependent manner (Figure 4.1). This is of interest because PMA
induces many PKC isoforms by mimicking DAG, and suggests that NOD2 and PKC
signal in concert to produce pro-inflammatory cytokines.
A PKC phospho-array identified PKCδ as a kinase phosphorylated by the NOD2
agonists, MDP and GMDP, and kinase assays confirmed that PKCδ is active (Figure 4.2).

137
In addition, we observed that PMA and GMDP-induced PKCδ phosphorylation is NOD2-
dependent (Figure 4.2). These data indicate that NOD2 is a necessary molecule in
optimally activating PKCδ.
To delineate PKCδ’s involvement in PMA-induced RIP2 phosphorylation and where
PKCδ belongs in the NOD2 signaling cascade, we incubated PKCδ deficient
macrophages with PMA or GMDP. We observed a decrease in RIP2 phosphorylation,
indicating that PKCδ must be activated prior to RIP2 (Figure 4.3). These data provide
new evidence for PKCδ being a necessary kinase in the NOD2 signaling pathway. We
hypothesize that PKCδ is in the NOD2-RIP2 complex, and therefore it would be of
interest to perform immunoprecipitations with NOD2 and PKCδ or RIP2 and PKCδ in
order to confirm. Additionally, we would like to know whether this complex binds to the
downstream molecule TAK1, which the NOD2-RIP2 complex is known to bind (95,96).
Finally we would like to understand the importance to PKCδ in the NOD2 signaling
cascade. Since PKCδ is known to phosphorylate hydroxyl groups on serine and
threonine residues, we hypothesize that it does this to RIP2 in order for RIP2 to be active.
Currently the literature suggests that RIP2 is autophosphorylated when recruited by its
CARD domain to NOD2 (338). More experiments in this area would add to our
understanding of the NOD2 signaling cascade that contributes to the innate immune
response to invading organisms.

138
New approaches to the treatment and prevention of inflammatory or infectious diseases
could involve the identification of novel activators or inhibitors of NOD2. Since NOD2
mutations (gain and loss of function) are associated with Blau syndrome and Crohn’s
disease, future treatment for these diseases could include NOD2 replacement or
modifying therapy (depending on the disease) (339). NOD2 polymorphisms in certain
populations are associated with susceptibility to TB and leprosy, so NOD2 replacement
or modifying therapy could also be given to high risk individuals as a preventative. Our
findings that NOD2 is important in controlling early M.tb infection of macrophages
suggest that activators such as MDP, could be given to individuals with primary infection
or potentially to those with reactivation disease. MDP has been used for decades as a key
component in complete Freund’s adjuvant to elicit adjuvant activity to injected antigens,
and the use of MDP in immunotherapy of infections and cancer is currently being studied
(340,341). However, since NOD2 gain of function mutations result in Blau syndrome,
MDP therapy should be used in moderation and studies should be done to test for the
correct dosage and length of administration.
In summary, work in this dissertation adds to our understanding of the complex role of
the cytosolic receptor, NOD2, in innate immunity. We focused on the role of NOD2
during M.tb infection. This PRR has been shown to regulate NF-κB and subsequent pro-
inflammatory cytokine secretion in response to specific bacterial products. In this
dissertation, we observed in human macrophages the role of NOD2 in cytokine secretion
and viability of M.tb, its importance in M.tb-induced iNOS expression, and its

139
relationship to a new molecule in the NOD2 signaling cascade. In addition, we
optimized antibodies for the detection of NOD2 in human cells, which led to the first
report of NOD2 protein expression in human macrophages. Furthermore, the
contribution of a new model, siRNA mediated knockdown of NOD2, to study NOD2 in
human cells is of great importance to the field. We would argue that previous studies
using mouse models have given misleading results on the role of NOD2 in TB. As a
result of work presented in this dissertation, we have elucidated the role of NOD2 using a
more clinically relevant model, which has the potential of identifying new drug targets to
treat or prevent the disease. Our studies on the relationship between NOD2 and iNOS
bring us a step closer to understanding the role of NOD2 in controlling the viability of
M.tb in human macrophages. Moreover, our reports on the involvement of PKCδ in the
NOD2 signaling cascade will be beneficial in future drug development studies. In all, our
data provide strong evidence for the role of NOD2 during M.tb infection of human
macrophages. Further studies will identify how NOD2 recognizes M.tb, the molecules
involved in controlling M.tb viability, whether NOD2 is directly involved in NO
production, and the importance of PKCδ in the NOD2 signaling cascade, thus further
increasing our knowledge of the role of NOD2 in innate immunity.

140
Bibliography
1. Janeway, C. A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu. Rev. Immunol. 20: 197-216
2. Medzhitov, R. 2007. Recognition of microorganisms and activation of the immune response. Nature 449: 819-826
3. Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21: 335-376
4. Flannagan, R. S., G. Cosio, and S. Grinstein. 2009. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 7: 355-366
5. Underhill, D. M., and A. Ozinsky. 2002. Phagocytosis of microbes: complexity in action. Annu. Rev. Immunol. 20: 825-852
6. Underhill, D. M. 2005. Phagosome maturation: steady as she goes. Immunity. 23: 343-344
7. Forman, H. J., and M. Torres. 2002. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am. J Respir. Crit Care Med 166: S4-S8
8. Kontny, E., M. Kurowska, K. Szczepanska, and W. Maslinski. 2000. Rottlerin, a PKC isozyme-selective inhibitor, affects signaling events and cytokine production in human monocytes. J Leukoc. Biol. 67: 249-258
9. Kontny, E., M. Ziolkowska, A. Ryzewska, and W. Maslinski. 1999. Protein kinase c-dependent pathway is critical for the production of pro-inflammatory cytokines (TNF-alpha, IL-1beta, IL-6). Cytokine 11: 839-848
10. Nishizuka, Y. 1984. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 308: 693-698

141
11. Takai, Y., A. Kishimoto, Y. Iwasa, Y. Kawahara, T. Mori, and Y. Nishizuka. 1979. Calcium-dependent activation of a multifunctional protein kinase by membrane phospholipids. J Biol. Chem 254: 3692-3695
12. Ono, Y., T. Fujii, K. Ogita, U. Kikkawa, K. Igarashi, and Y. Nishizuka. 1988. The structure, expression, and properties of additional members of the protein kinase C family. J Biol. Chem 263: 6927-6932
13. Ono, Y., T. Fujii, K. Igarashi, T. Kuno, C. Tanaka, U. Kikkawa, and Y. Nishizuka. 1989. Phorbol ester binding to protein kinase C requires a cysteine-rich zinc-finger-like sequence. Proc. Natl. Acad. Sci. U. S. A 86: 4868-4871
14. Castagna, M., Y. Takai, K. Kaibuchi, K. Sano, U. Kikkawa, and Y. Nishizuka. 1982. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol. Chem. 257: 7847-7851
15. Palmer, R. H., J. Ridden, and P. J. Parker. 1995. Cloning and expression patterns of two members of a novel protein-kinase-C-related kinase family. Eur. J Biochem. 227: 344-351
16. Palmer, R. H., L. V. Dekker, R. Woscholski, J. A. Le Good, R. Gigg, and P. J. Parker. 1995. Activation of PRK1 by phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate. A comparison with protein kinase C isotypes. J Biol. Chem 270: 22412-22416
17. Amano, M., H. Mukai, Y. Ono, K. Chihara, T. Matsui, Y. Hamajima, K. Okawa, A. Iwamatsu, and K. Kaibuchi. 1996. Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science 271: 648-650
18. Watanabe, G., Y. Saito, P. Madaule, T. Ishizaki, K. Fujisawa, N. Morii, H. Mukai, Y. Ono, A. Kakizuka, and S. Narumiya. 1996. Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science 271: 645-648
19. Mecklenbrauker, I., K. Saijo, N. Y. Zheng, M. Leitges, and A. Tarakhovsky. 2002. Protein kinase Cdelta controls self-antigen-induced B-cell tolerance. Nature 416: 860-865
20. Brodie, C., and P. M. Blumberg. 2003. Regulation of cell apoptosis by protein kinase c delta. Apoptosis. 8: 19-27
21. Ohba, M., K. Ishino, M. Kashiwagi, S. Kawabe, K. Chida, N. H. Huh, and T. Kuroki. 1998. Induction of differentiation in normal human keratinocytes by adenovirus-mediated introduction of the eta and delta isoforms of protein kinase C. Mol. Cell Biol. 18: 5199-5207

142
22. Blake, R. A., P. Garcia-Paramio, P. J. Parker, and S. A. Courtneidge. 1999. Src promotes PKCdelta degradation. Cell Growth Differ. 10: 231-241
23. Hodgkinson, C. P., E. M. Sale, and G. J. Sale. 2002. Characterization of PDK2 activity against protein kinase B gamma. Biochemistry 41: 10351-10359
24. Kristof, A. S., J. Marks-Konczalik, E. Billings, and J. Moss. 2003. Stimulation of signal transducer and activator of transcription-1 (STAT1)-dependent gene transcription by lipopolysaccharide and interferon-gamma is regulated by mammalian target of rapamycin. J Biol. Chem. 278: 33637-33644
25. Braiman, L., A. Alt, T. Kuroki, M. Ohba, A. Bak, T. Tennenbaum, and S. R. Sampson. 2001. Insulin induces specific interaction between insulin receptor and protein kinase C delta in primary cultured skeletal muscle. Mol. Endocrinol. 15: 565-574
26. Rosenzweig, T., L. Braiman, A. Bak, A. Alt, T. Kuroki, and S. R. Sampson. 2002. Differential effects of tumor necrosis factor-alpha on protein kinase C isoforms alpha and delta mediate inhibition of insulin receptor signaling. Diabetes 51: 1921-1930
27. Marsland, B. J., and M. Kopf. 2008. T-cell fate and function: PKC-theta and beyond. Trends Immunol. 29: 179-185
28. Sun, Z., C. W. Arendt, W. Ellmeier, E. M. Schaeffer, M. J. Sunshine, L. Gandhi, J. Annes, D. Petrzilka, A. Kupfer, P. L. Schwartzberg, and D. R. Littman. 2000. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature 404: 402-407
29. Ron, D., E. W. Napolitano, A. Voronova, N. J. Vasquez, D. N. Roberts, B. L. Calio, R. H. Caothien, S. M. Pettiford, S. Wellik, J. B. Mandac, and L. M. Kauvar. 1999. Direct interaction in T-cells between thetaPKC and the tyrosine kinase p59fyn. J Biol. Chem. 274: 19003-19010
30. Bauer, B., N. Krumbock, F. Fresser, F. Hochholdinger, M. Spitaler, A. Simm, F. Uberall, B. Schraven, and G. Baier. 2001. Complex formation and cooperation of protein kinase C theta and Akt1/protein kinase B alpha in the NF-kappa B transactivation cascade in Jurkat T cells. J Biol. Chem. 276: 31627-31634
31. Takeda, K., Y. Harada, R. Watanabe, Y. Inutake, S. Ogawa, K. Onuki, S. Kagaya, K. Tanabe, H. Kishimoto, and R. Abe. 2008. CD28 stimulation triggers NF-kappaB activation through the CARMA1-PKCtheta-Grb2/Gads axis. Int. Immunol. 20: 1507-1515

143
32. Webb, J. L., M. W. Harvey, D. W. Holden, and T. J. Evans. 2001. Macrophage nitric oxide synthase associates with cortical actin but is not recruited to phagosomes. Infect. Immun. 69: 6391-6400
33. Fang, F. C. 2004. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2: 820-832
34. Cooper, A. M., L. B. Adams, D. K. Dalton, R. Appelberg, and S. Ehlers. 2002. IFN-gamma and NO in mycobacterial disease: new jobs for old hands. Trends Microbiol. 10: 221-226
35. Alam, M. S., T. Akaike, S. Okamoto, T. Kubota, J. Yoshitake, T. Sawa, Y. Miyamoto, F. Tamura, and H. Maeda. 2002. Role of nitric oxide in host defense in murine salmonellosis as a function of its antibacterial and antiapoptotic activities. Infect. Immun. 70: 3130-3142
36. Li, H., and U. Forstermann. 2000. Nitric oxide in the pathogenesis of vascular disease. J Pathol. 190: 244-254
37. Laroux, F. S., D. J. Lefer, S. Kawachi, R. Scalia, A. S. Cockrell, L. Gray, H. H. Van der, J. M. Hoffman, and M. B. Grisham. 2000. Role of nitric oxide in the regulation of acute and chronic inflammation. Antioxid. Redox. Signal. 2: 391-396
38. Nathan, C., and Q.-W. Xie. 1994. Regulation of biosynthesis of nitric oxide. J. Biol. Chem. 269: 13725-13728
39. Rosenkranz-Weiss, P., W. C. Sessa, S. Milstien, S. Kaufman, C. A. Watson, and J. S. Pober. 1994. Regulation of nitric oxide synthesis by proinflammatory cytokines in human umbilical vein endothelial cells. Elevations in tetrahydrobiopterin levels enhance endothelial nitric oxide synthase specific activity. J Clin. Invest 93: 2236-2243
40. Assreuy, J., F. Q. Cunha, F. Y. Liew, and S. Moncada. 1993. Feedback inhibition of nitric oxide synthase activity by nitric oxide. Br. J Pharmacol. 108: 833-837
41. Rogers, N. E., and L. J. Ignarro. 1992. Constitutive nitric oxide synthase from cerebellum is reversibly inhibited by nitric oxide formed from L-arginine. Biochem. Biophys. Res. Commun. 189: 242-249
42. Martin, E., C. Nathan, and Q. Xie. 1994. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J. Exp. Med. 180: 977-984
43. Davis, R. L., A. C. Sanchez, D. J. Lindley, S. C. Williams, and P. J. Syapin. 2005. Effects of mechanistically distinct NF-kappaB inhibitors on glial inducible nitric-oxide synthase expression. Nitric. Oxide. 12: 200-209

144
44. Weinberg, J. B. 1998. Nitric oxide production and nitric oxide synthase type 2 expression by human mononuclear phagocytes: a review. Mol. Med. 4: 557-591
45. Abbas, A. K., and A. H. Lichtman. 2005. Cellular and Molecular Immunology.
46. Zhang, P., W. R. Summer, G. J. Bagby, and S. Nelson. 2000. Innate immunity and pulmonary host defense. Immunol. Rev. 173: 39-51
47. Zaas, A. K., and D. A. Schwartz. 2005. Innate immunity and the lung: defense at the interface between host and environment. Trends Cardiovasc. Med. 15: 195-202
48. Fels, A., and Z. A. Cohn. 1986. The alveolar macrophage. J. Appl. Physiol. 60: 353-369
49. Williams, M. C. 2003. Alveolar type I cells: molecular phenotype and development. Annu. Rev. Physiol 65: 669-695
50. Hoidal, J. R., D. Schmeling, and P. K. Peterson. 1981. Phagocytosis, bacterial killing, and metabolism by purified human lung phagocytes. J. Infect. Dis. 144: 61-71
51. Lohmann-Matthes, M. L., C. Steinmuller, and G. Franke-Ullmann. 1994. Pulmonary macrophages. Eur. Respir. J 7: 1678-1689
52. Gordon, S. 2003. Alternative activation of macrophages. Nat. Rev. Immunol. 3: 23-35
53. Mosser, D. M. 2003. The many faces of macrophage activation. J Leukoc. Biol 73: 209-212
54. Henning, L. N., A. K. Azad, K. V. Parsa, J. E. Crowther, S. Tridandapani, and L. S. Schlesinger. 2008. Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J. Immunol. 180: 7847-7858
55. Crowther, J. E., V. K. Kutala, P. Kuppusamy, J. S. Ferguson, A. A. Beharka, J. L. Zweier, F. X. McCormack, and L. S. Schlesinger. 2004. Pulmonary surfactant protein a inhibits macrophage reactive oxygen intermediate production in response to stimuli by reducing NADPH oxidase activity. J Immunol. 172: 6866-6874
56. Rajaram, M. V., M. N. Brooks, J. D. Morris, J. B. Torrelles, A. K. Azad, and L. S. Schlesinger. 2010. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J Immunol. 185: 929-942

145
57. Brown E.J. 1991. Complement Receptors and Phagocytosis. Curr. Opin. Immunol. 3: 76-82
58. Ravetch, J. V., and S. Bolland. 2001. IgG Fc receptors. Annu. Rev. Immunol. 19: 275-290
59. Stephenson, J. D., and V. L. Shepherd. 1987. Purification of the human alveolar macrophage mannose receptor. Biochem. Biophys. Res. Commun. 148: 883-889
60. Palecanda, A., and L. Kobzik. 2001. Receptors for unopsonized particles: the role of alveolar macrophage scavenger receptors. Curr. Mol. Med 1: 589-595
61. Taylor, P. R., G. D. Brown, D. M. Reid, J. A. Willment, L. Martinez-Pomares, S. Gordon, and S. Y. Wong. 2002. The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J Immunol. 169: 3876-3882
62. Underhill, D. M. 2003. Toll-like receptors: networking for success. Eur. J Immunol. 33: 1767-1775
63. Chen, S. K. a. N. n. 2009. NOD-Like Receptors: Role in Innate Immunity and Inflammatory Disease. The Annual Review of Pathology: Mechanisms of Disease
64. Stahl, P. D., and R. A. Ezekowitz. 1998. The mannose receptor is a pattern recognition receptor involved in host defense. Curr. Opin. Immunol. 10: 50-55
65. Gibson, G. a. W. 2010. Expression and Functions of Innate Pattern Recognition Receptors in T and B Cells. Immun. , Endoc. & Metab. Agents in Med. Chem 10: 11-20
66. Basu, S., and M. J. Fenton. 2004. Toll-like receptors: function and roles in lung disease. Am. J. Physiol Lung Cell Mol. Physiol 286: L887-L892
67. Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell 124: 783-801
68. Poltorak, A., X. He, I. Smirnova, M. Y. Liu, H. C. Van, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, M. Freudenberg, P. Ricciardi-Castagnoli, B. Layton, and B. Beutler. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282: 2085-2088
69. Underhill, D. M., A. Ozinsky, A. M. Hajjar, A. Stevens, C. B. Wilson, M. Bassetti, and A. Aderem. 1999. The toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401: 811-815

146
70. Kobayashi, K., L. D. Hernandez, J. E. Galan, C. A. Janeway, Jr., R. Medzhitov, and R. A. Flavell. 2002. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110: 191-202
71. Creagh, E. M., and L. A. O'Neill. 2006. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 27: 352-357
72. Hulbert, S. H., C. A. Webb, S. M. Smith, and Q. Sun. 2001. Resistance gene complexes: evolution and utilization. Annu. Rev. Phytopathol. 39: 285-312
73. Harton, J. A., M. W. Linhoff, J. Zhang, and J. P. Ting. 2002. Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J Immunol. 169: 4088-4093
74. Inohara, Chamaillard, C. McDonald, and G. Nunez. 2005. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu. Rev. Biochem. 74: 355-383
75. Ogura, Y., S. Lala, W. Xin, E. Smith, T. A. Dowds, F. F. Chen, E. Zimmermann, M. Tretiakova, J. H. Cho, J. Hart, J. K. Greenson, S. Keshav, and G. Nunez. 2003. Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut 52: 1591-1597
76. Ogura, Y., N. Inohara, A. Benito, F. F. Chen, S. Yamaoka, and G. Nunez. 2001. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem 276: 4812-4818
77. Brooks, M. N., M. V. Rajaram, A. K. Azad, A. O. Amer, M. A. Valdivia-Arenas, J. H. Park, G. Nunez, and L. S. Schlesinger. 2011. NOD2 controls the nature of the inflammatory response and subsequent fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages. Cell Microbiol. 13: 402-418
78. Ting, J. P., R. C. Lovering, E. S. Alnemri, J. Bertin, J. M. Boss, B. K. Davis, R. A. Flavell, S. E. Girardin, A. Godzik, J. A. Harton, H. M. Hoffman, J. P. Hugot, N. Inohara, A. MacKenzie, L. J. Maltais, G. Nunez, Y. Ogura, L. A. Otten, D. Philpott, J. C. Reed, W. Reith, S. Schreiber, V. Steimle, and P. A. Ward. 2008. The NLR gene family: a standard nomenclature. Immunity. 28: 285-287
79. Tanabe, T., M. Chamaillard, Y. Ogura, L. Zhu, S. Qiu, J. Masumoto, P. Ghosh, A. Moran, M. M. Predergast, G. Tromp, C. J. Williams, N. Inohara, and G. Nunez. 2004. Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J 23: 1587-1597

147
80. Kajava, A. V., and B. Kobe. 2002. Assessment of the ability to model proteins with leucine-rich repeats in light of the latest structural information. Protein Sci. 11: 1082-1090
81. Martinon, F., and J. Tschopp. 2004. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 117: 561-574
82. Manon, F., A. Favier, G. Nunez, J. P. Simorre, and S. Cusack. 2007. Solution structure of NOD1 CARD and mutational analysis of its interaction with the CARD of downstream kinase RICK. J Mol. Biol. 365: 160-174
83. Coussens, N. P., J. C. Mowers, C. McDonald, G. Nunez, and S. Ramaswamy. 2007. Crystal structure of the Nod1 caspase activation and recruitment domain. Biochem. Biophys. Res. Commun. 353: 1-5
84. Rosenstiel, P., A. Till, and S. Schreiber. 2007. NOD-like receptors and human diseases. Microbes. Infect. 9: 648-657
85. Inohara, N., Y. Ogura, A. Fontalba, O. Gutierrez, F. Pons, J. Crespo, K. Fukase, S. Inamura, S. Kusumoto, M. Hashimoto, S. J. Foster, A. P. Moran, J. L. Fernandez-Luna, and G. Nunez. 2003. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem 278: 5509-5512
86. Coulombe, F., M. Divangahi, F. Veyrier, L. L. de, J. L. Gleason, Y. Yang, M. A. Kelliher, A. K. Pandey, C. M. Sassetti, M. B. Reed, and M. A. Behr. 2009. Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J. Exp. Med. 206: 1709-1716
87. Chamaillard, M., M. Hashimoto, Y. Horie, J. Masumoto, S. Qiu, L. Saab, Y. Ogura, A. Kawasaki, K. Fukase, S. Kusumoto, M. A. Valvano, S. J. Foster, T. W. Mak, G. Nunez, and N. Inohara. 2003. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4: 702-707
88. Girardin, S. E., I. G. Boneca, J. Viala, M. Chamaillard, A. Labigne, G. Thomas, D. J. Philpott, and P. J. Sansonetti. 2003. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278: 8869-8872
89. Holtje, J. V. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62: 181-203

148
90. Sinha, R. K., and R. S. Rosenthal. 1980. Release of soluble peptidoglycan from growing conococci: demonstration of anhydro-muramyl-containing fragments. Infect. Immun. 29: 914-925
91. Cookson, B. T., A. N. Tyler, and W. E. Goldman. 1989. Primary structure of the peptidoglycan-derived tracheal cytotoxin of Bordetella pertussis. Biochemistry 28: 1744-1749
92. Araki, Y., S. Fukuoka, S. Oba, and E. Ito. 1971. Enzymatic deacetylation of N-acetylglucosamine residues in peptidoglycan from Bacillus cereus cell walls. Biochem. Biophys. Res. Commun. 45: 751-758
93. Franchi, L., J. H. Park, M. H. Shaw, N. Marina-Garcia, G. Chen, Y. G. Kim, and G. Nunez. 2008. Intracellular NOD-like receptors in innate immunity, infection and disease. Cell Microbiol. 10: 1-8
94. Nembrini, C., J. Kisielow, A. T. Shamshiev, L. Tortola, A. J. Coyle, M. Kopf, and B. J. Marsland. 2009. The kinase activity of Rip2 determines its stability and consequently Nod1- and Nod2-mediated immune responses. J Biol. Chem. 284: 19183-19188
95. Chen, C. M., Y. Gong, M. Zhang, and J. J. Chen. 2004. Reciprocal cross-talk between Nod2 and TAK1 signaling pathways. J. Biol. Chem. 279: 25876-25882
96. Kim, J. Y., E. Omori, K. Matsumoto, G. Nunez, and J. Ninomiya-Tsuji. 2008. TAK1 is a central mediator of NOD2 signaling in epidermal cells. J. Biol. Chem. 283: 137-144
97. Hsu, L. C., S. R. Ali, S. McGillivray, P. H. Tseng, S. Mariathasan, E. W. Humke, L. Eckmann, J. J. Powell, V. Nizet, V. M. Dixit, and M. Karin. 2008. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. U. S. A 105: 7803-7808
98. Rosenstiel, P., K. Huse, A. Till, J. Hampe, S. Hellmig, C. Sina, S. Billmann, K. O. von, G. H. Waetzig, M. Platzer, D. Seegert, and S. Schreiber. 2006. A short isoform of NOD2/CARD15, NOD2-S, is an endogenous inhibitor of NOD2/receptor-interacting protein kinase 2-induced signaling pathways. Proc. Natl. Acad. Sci. U. S. A 103: 3280-3285
99. Netea, M. G., R. Sutmuller, C. Hermann, C. A. Van der Graaf, J. W. Van der Meer, J. H. van Krieken, T. Hartung, G. Adema, and B. J. Kullberg. 2004. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol. 172: 3712-3718

149
100. Netea, M. G., G. Ferwerda, D. J. de Jong, T. Jansen, L. Jacobs, M. Kramer, T. H. Naber, J. P. Drenth, S. E. Girardin, B. J. Kullberg, G. J. Adema, and J. W. Van der Meer. 2005. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J Immunol. 174: 6518-6523
101. Uehara, A., Y. Sugawara, S. Kurata, Y. Fujimoto, K. Fukase, S. Kusumoto, Y. Satta, T. Sasano, S. Sugawara, and H. Takada. 2005. Chemically synthesized pathogen-associated molecular patterns increase the expression of peptidoglycan recognition proteins via toll-like receptors, NOD1 and NOD2 in human oral epithelial cells. Cell Microbiol. 7: 675-686
102. van Heel, D. A., S. Ghosh, K. A. Hunt, C. G. Mathew, A. Forbes, D. P. Jewell, and R. J. Playford. 2005. Synergy between TLR9 and NOD2 innate immune responses is lost in genetic Crohn's disease. Gut 54: 1553-1557
103. Watanabe, T., A. Kitani, P. J. Murray, and W. Strober. 2004. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat. Immunol. 5: 800-808
104. Watanabe, T., A. Kitani, P. J. Murray, Y. Wakatsuki, I. J. Fuss, and W. Strober. 2006. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 25: 473-485
105. Wolfert, M. A., T. F. Murray, G. J. Boons, and J. N. Moore. 2002. The origin of the synergistic effect of muramyl dipeptide with endotoxin and peptidoglycan. J Biol. Chem 277: 39179-39186
106. Yang, S., R. Tamai, S. Akashi, O. Takeuchi, S. Akira, S. Sugawara, and H. Takada. 2001. Synergistic effect of muramyldipeptide with lipopolysaccharide or lipoteichoic acid to induce inflammatory cytokines in human monocytic cells in culture. Infect. Immun. 69: 2045-2053
107. Abbott, D. W., A. Wilkins, J. M. Asara, and L. C. Cantley. 2004. The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr. Biol. 14: 2217-2227
108. Abbott, D. W., Y. Yang, J. E. Hutti, S. Madhavarapu, M. A. Kelliher, and L. C. Cantley. 2007. Coordinated regulation of Toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol. Cell Biol. 27: 6012-6025
109. Rosenstiel, P., M. Fantini, K. Brautigam, T. Kuhbacher, G. H. Waetzig, D. Seegert, and S. Schreiber. 2003. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology 124: 1001-1009

150
110. Takahashi, Y., K. Isuzugawa, Y. Murase, M. Imai, S. Yamamoto, M. Iizuka, S. Akira, G. M. Bahr, E. Momotani, M. Hori, H. Ozaki, and K. Imakawa. 2006. Up-regulation of NOD1 and NOD2 through TLR4 and TNF-alpha in LPS-treated murine macrophages. J Vet. Med. Sci. 68: 471-478
111. Takada, H., S. Yokoyama, and S. Yang. 2002. Enhancement of endotoxin activity by muramyldipeptide. Journal of Endotoxin Research 8: 337-342
112. McDonald, C., F. F. Chen, V. Ollendorff, Y. Ogura, S. Marchetto, P. Lecine, J. P. Borg, and G. Nunez. 2005. A role for Erbin in the regulation of Nod2-dependent NF-kappaB signaling. J Biol. Chem 280: 40301-40309
113. Kufer, T. A., E. Kremmer, D. J. Banks, and D. J. Philpott. 2006. Role for erbin in bacterial activation of Nod2. Infect. Immun. 74: 3115-3124
114. Barnich, N., J. E. Aguirre, H. C. Reinecker, R. Xavier, and D. K. Podolsky. 2005. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-{kappa}B activation in muramyl dipeptide recognition. J. Cell Biol. 170: 21-26
115. Takada, H., and S. Kotani. 1995. In The Theory of Pratical Application of Adjuvants. Wiley: Chichester, ed., pp. 171.
116. Inamura, N., S. Sone, A. Okubo, E. Kunishige, M. Nakanishi, and T. Ogura. 1989. Tumor cytotoxicity of human monocyte membrane-bound interleukin-1 alpha induced by synergistic actions of interferon-gamma and synthetic acyltripeptide, FK-565. Cancer Immunol. Immunother. 28: 164-170
117. Hasan, S. I., K. Ahmed, and J. L. Turk. 1990. Effect of anticancer drugs on the release of tumour necrosis factor in vitro. Cancer Immunol. Immunother. 30: 363-366
118. Maeda, M., T. Asano, and E. S. Kleinerman. 1993. Anti-(tumor necrosis factor) alters the response of human monocytes to liposomal muramyl tripeptide. Cancer Immunol. Immunother. 37: 203-208
119. Morikage, T., Y. Mizushima, and S. Yano. 1991. Prevention of bacterial infection and LPS-induced lethality by interleukin-1 alpha in mice. Tohoku J Exp. Med. 163: 47-57
120. Morikage, T., Y. Mizushima, and S. Yano. 1991. Prevention of bacterial infection and LPS-induced lethality by interleukin-1 alpha in mice. Tohoku J Exp. Med. 163: 47-57

151
121. Asano, T., A. McWatters, T. An, K. Matsushima, and E. S. Kleinerman. 1994. Liposomal muramyl tripeptide up-regulates interleukin-1 alpha, interleukin-1 beta, tumor necrosis factor-alpha, interleukin-6 and interleukin-8 gene expression in human monocytes. J Pharmacol. Exp. Ther. 268: 1032-1039
122. Li, J., T. Moran, E. Swanson, C. Julian, J. Harris, D. K. Bonen, M. Hedl, D. L. Nicolae, C. Abraham, and J. H. Cho. 2004. Regulation of IL-8 and IL-1beta expression in Crohn's disease associated NOD2/CARD15 mutations. Hum. Mol. Genet. 13: 1715-1725
123. Nau, G. J., J. F. Richmond, A. Schlesinger, E. G. Jennings, E. S. Lander, and R. A. Young. 2002. Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. U. S. A 99: 1503-1508
124. Todate, A., T. Suda, H. Kuwata, K. Chida, and H. Nakamura. 2001. Muramyl dipeptide-Lys stimulates the function of human dendritic cells. J Leukoc. Biol. 70: 723-729
125. Heinzelmann, M., H. C. Polk, Jr., A. Chernobelsky, T. P. Stites, and L. E. Gordon. 2000. Endotoxin and muramyl dipeptide modulate surface receptor expression on human mononuclear cells. Immunopharmacology 48: 117-128
126. Opitz, B., A. Puschel, B. Schmeck, A. C. Hocke, S. Rosseau, S. Hammerschmidt, R. R. Schumann, N. Suttorp, and S. Hippenstiel. 2004. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J. Biol. Chem. 279: 36426-36432
127. Ferwerda, G., S. E. Girardin, B. J. Kullberg, L. Le Bourhis, D. J. de Jong, D. M. Langenberg, R. van Crevel, G. J. Adema, T. H. Ottenhoff, J. W. Van der Meer, and M. G. Netea. 2005. NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS. Pathog. 1: 279-285
128. Girardin, S. E., R. Tournebize, M. Mavris, A. L. Page, X. Li, G. R. Stark, J. Bertin, P. S. DiStefano, M. Yaniv, P. J. Sansonetti, and D. J. Philpott. 2001. CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2: 736-742
129. Kim, J. G., S. J. Lee, and M. F. Kagnoff. 2004. Nod1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by toll-like receptors. Infect. Immun. 72: 1487-1495
130. Marriott, I., D. M. Rati, S. H. McCall, and S. L. Tranguch. 2005. Induction of Nod1 and Nod2 intracellular pattern recognition receptors in murine osteoblasts following bacterial challenge. Infect. Immun. 73: 2967-2973

152
131. Opitz, B., S. Forster, A. C. Hocke, M. Maass, B. Schmeck, S. Hippenstiel, N. Suttorp, and M. Krull. 2005. Nod1-mediated endothelial cell activation by Chlamydophila pneumoniae. Circ. Res. 96: 319-326
132. Travassos, L. H., L. A. Carneiro, S. E. Girardin, I. G. Boneca, R. Lemos, M. T. Bozza, R. C. Domingues, A. J. Coyle, J. Bertin, D. J. Philpott, and M. C. Plotkowski. 2005. Nod1 participates in the innate immune response to Pseudomonas aeruginosa. J Biol Chem 280: 36714-36718
133. Viala, J., C. Chaput, I. G. Boneca, A. Cardona, S. E. Girardin, A. P. Moran, R. Athman, S. Memet, M. R. Huerre, A. J. Coyle, P. S. DiStefano, P. J. Sansonetti, A. Labigne, J. Bertin, D. J. Philpott, and R. L. Ferrero. 2004. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 5: 1166-1174
134. Opitz, B., A. Puschel, W. Beermann, A. C. Hocke, S. Forster, B. Schmeck, L. van, V, T. Chakraborty, N. Suttorp, and S. Hippenstiel. 2006. Listeria monocytogenes activated p38 MAPK and induced IL-8 secretion in a nucleotide-binding oligomerization domain 1-dependent manner in endothelial cells. J. Immunol. 176: 484-490
135. Divangahi, M., S. Mostowy, F. Coulombe, R. Kozak, L. Guillot, F. Veyrier, K. S. Kobayashi, R. A. Flavell, P. Gros, and M. A. Behr. 2008. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J. Immunol. 181: 7157-7165
136. Deshmukh, H. S., J. B. Hamburger, S. H. Ahn, D. G. McCafferty, S. R. Yang, and V. G. Fowler, Jr. 2009. Critical role of NOD2 in regulating the immune response to Staphylococcus aureus. Infect. Immun. 77: 1376-1382
137. Amer, A., L. Franchi, T. D. Kanneganti, M. Body-Malapel, N. Ozoren, G. Brady, S. Meshinchi, R. Jagirdar, A. Gewirtz, S. Akira, and G. Nunez. 2006. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281: 35217-35223
138. Hugot, J. P., M. Chamaillard, H. Zouali, S. Lesage, J. P. Cezard, J. Belaiche, S. Almer, C. Tysk, C. A. O'Morain, M. Gassull, V. Binder, Y. Finkel, A. Cortot, R. Modigliani, P. Laurent-Puig, C. Gower-Rousseau, J. Macry, J. F. Colombel, M. Sahbatou, and G. Thomas. 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411: 599-603
139. Ogura, Y., D. K. Bonen, N. Inohara, D. L. Nicolae, F. F. Chen, R. Ramos, H. Britton, T. Moran, R. Karaliuskas, R. H. Duerr, J. P. Achkar, S. R. Brant, T. M. Bayless, B. S. Kirschner, S. B. Hanauer, G. Nunez, and J. H. Cho. 2001. A

153
frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411: 603-606
140. Miceli-Richard, C., S. Lesage, M. Rybojad, A. M. Prieur, S. Manouvrier-Hanu, R. Hafner, M. Chamaillard, H. Zouali, G. Thomas, and J. P. Hugot. 2001. CARD15 mutations in Blau syndrome. Nat. Genet. 29: 19-20
141. Kanazawa, N., I. Okafuji, N. Kambe, R. Nishikomori, M. Nakata-Hizume, S. Nagai, A. Fuji, T. Yuasa, A. Manki, Y. Sakurai, M. Nakajima, H. Kobayashi, I. Fujiwara, H. Tsutsumi, A. Utani, C. Nishigori, T. Heike, T. Nakahata, and Y. Miyachi. 2005. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood 105: 1195-1197
142. Fritz, J. H., R. L. Ferrero, D. J. Philpott, and S. E. Girardin. 2006. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 7: 1250-1257
143. Sanderson, J. D., M. T. Moss, M. L. V. Tizard, and J. Hermon-Taylor. 1992. Mycobacterium paratuberculosis DNA in Crohn's disease tissue. Gut 33: 890-896
144. Hermon-Taylor, J. 2009. Mycobacterium avium subspecies paratuberculosis, Crohn's disease and the Doomsday scenario. Gut Pathog. 1: 15
145. Maeda, S., L. C. Hsu, H. Liu, L. A. Bankston, M. Iimura, M. F. Kagnoff, L. Eckmann, and M. Karin. 2005. Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1beta processing. Science 307: 734-738
146. Netea, M. G., G. Ferwerda, D. J. de Jong, S. E. Girardin, B. J. Kullberg, and J. W. Van der Meer. 2005. NOD2 3020insC mutation and the pathogenesis of Crohn's disease: impaired IL-1beta production points to a loss-of-function phenotype. Neth. J Med. 63: 305-308
147. Hu, C., L. Sun, Y. Hu, D. Lu, H. Wang, and S. Tang. 2010. Functional characterization of the NF-kappaB binding site in the human NOD2 promoter. Cell Mol. Immunol. 7: 288-295
148. WHO. 2009. 2009 Update Tuberculosis Facts.
149. Fine, P. E. M. 1995. Variation in protection by BCG: Implications of and for heterologous immunity. Lancet 346: 1339-1345
150. Brewer, T. F. 2000. Preventing tuberculosis with bacillus Calmette-Guerin vaccine: a meta-analysis of the literature. Clin. Infect. Dis. 31 Suppl 3: S64-S67
151. Udani, P. M. 1994. BCG vaccination in India and tuberculosis in children: newer facets. Indian J Pediatr. 61: 451-462

154
152. Smith, D., E. Wiegeshaus, and V. Balasubramanian. 2000. An analysis of some hypotheses related to the Chingelput bacille Calmette-Guerin trial. Clin. Infect. Dis. 31 Suppl 3: S77-S80
153. Gupta, U. D., V. M. Katoch, and D. N. McMurray. 2007. Current status of TB vaccines. Vaccine 25: 3742-3751
154. Guy, and Mallampalli. 2008. Review: Managing TB in the 21st century: existing and novel drug therapies. Therapeutic Advances in Respiratory Disease 2: 401-408
155. WHO. 2010. Multidrug and extensively drug-resistant TB (M/XDR-TB): 2010 global report on surveillance and response. Zignol and M, eds. WHO Press, Geneva, Switzerland, pp. 1-72.
156. Ma, Z., C. Lienhardt, H. McIlleron, A. J. Nunn, and X. Wang. 2010. Global tuberculosis drug development pipeline: the need and the reality. Lancet 375: 2100-2109
157. Sundaramurthy, V., and J. Pieters. 2007. Interactions of pathogenic mycobacteria with host macrophages. Microbes. Infect. 9: 1671-1679
158. Torrelles, J. B., A. K. Azad, L. N. Henning, T. K. Carlson, and L. S. Schlesinger. 2008. Role of C-type lectins in mycobacterial infections. Curr. Drug Targets. 9: 102-112
159. Kaufmann, S. H. 2001. How can immunology contribute to the control of tuberculosis? Nature Reviews Immunology 1: 20-30
160. Wright, J. R. 2005. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 5: 58-68
161. Gaynor, C. D., F. X. McCormack, D. R. Voelker, S. E. McGowan, and L. S. Schlesinger. 1995. Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J. Immunol. 155: 5343-5351
162. Downing, J. F., R. Pasula, J. R. Wright, H. L. Twigg, III, and W. J. Martin, II. 1995. Surfactant protein A promotes attachment of Mycobacterium tuberculosis to alveolar macrophages during infection with human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 92: 4848-4852
163. Pasula, R., J. R. Wright, D. L. Kachel, and W. J. Martin, III. 1999. Surfactant protein A suppresses reactive nitrogen intermediates by alveolar macrophages in response to Mycobacterium tuberculosis. J. Clin. Invest. 103: 483-490

155
164. Ferguson, J. S., J. L. Martin, A. K. Azad, T. R. McCarthy, P. B. Kang, D. R. Voelker, E. C. Crouch, and L. S. Schlesinger. 2006. Surfactant protein D increases fusion of Mycobacterium tuberculosis-containing phagosomes with lysosomes in human macrophages. Infect. Immun. 74: 7005-7009
165. Ferguson, J. S., D. R. Voelker, F. X. McCormack, and L. S. Schlesinger. 1999. Surfactant protein D binds to Mycobacterium tuberculosis bacili and lipoarrabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J. Immunol. 163: 312-321
166. Schlesinger, L. S. 1993. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J. Immunol. 150: 2920-2930
167. Rooyakkers AW, and R. W. Stokes. 2005. Absence of complement receptor 3 results in reduced binding and ingestion of Mycobacterium tuberculosis but has no significant effect on the induction of reactive oxygen and nitrogen intermediates or on the survival of the bacteria in resident and interferon-gamma activated macrophages., pp. 57-67.
168. Hu, C., T. Mayadas-Norton, K. Tanaka, J. Chan, and P. Salgame. 2000. Mycobacterium tuberculosis infection in complement receptor 3-deficient mice. J. Immunol. 165: 2596-2602
169. Zimmerli, S., S. Edwards, and J. D. Ernst. 1996. Selective receptor blockade during phagocytosis does not alter the survival and growth of Mycobacterium tuberculosis in human macrophages. Am. J. Respir. Cell Mol. Biol. 15: 760-770
170. Peterson, P. K., G. Gekker, S. Hu, W. S. Sheng, W. R. Anderson, R. J. Ulevitch, P. S. Tobias, K. V. Gustafson, T. W. Molitor, and C. C. Chao. 1995. CD14 receptor-mediated uptake of nonopsonized Mycobacterium tuberculosis by human microglia. Infect. Immun. 63: 1598-1602
171. Yadav, M., and J. S. Schorey. 2006. The {beta}-glucan receptor Dectin-1 fucntions together with TLR2 to mediate macrophage activation by mycobacteria. Blood 108: 3168-3175
172. Schlesinger, L. S., C. G. Bellinger-Kawahara, N. R. Payne, and M. A. Horwitz. 1990. Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J. Immunol. 144: 2771-2780
173. Armstrong, J. A., and P. D. Hart. 1975. Phagosome-lysosome interactions in cultured macrophages infected with virulent tubercle bacilli: Reversal of the usual nonfusion pattern and observations on bacterial survial. J. Exp. Med. 142: 1-16

156
174. Lopez, M., L. M. Sly, Y. Luu, D. Young, H. Cooper, and N. E. Reiner. 2003. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J. Immunol. 170: 2409-2416
175. Dao, D. N., L. Kremer, Y. Guerardel, A. Molano, W. R. Jacobs, Jr., S. A. Porcelli, and V. Briken. 2004. Mycobacterium tuberculosis lipomannan induces apoptosis and interleukin-12 production in macrophages. Infect. Immun. 72: 2067-2074
176. Vergne, I., R. A. Fratti, P. J. Hill, J. Chua, J. Belisle, and V. Deretic. 2004. Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol. Biol. Cell 15: 751-760
177. Master, S. S., S. K. Rampini, A. S. Davis, C. Keller, S. Ehlers, B. Springer, G. S. Timmins, P. Sander, and V. Deretic. 2008. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host. Microbe 3: 224-232
178. Mishra, B. B., P. Moura-Alves, A. Sonawane, N. Hacohen, G. Griffiths, L. F. Moita, and E. Anes. 2010. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol. 12: 1046-1063
179. Gandotra, S., S. Jang, P. J. Murray, P. Salgame, and S. Ehrt. 2007. Nucleotide-binding oligomerization domain protein 2-deficient mice control infection with Mycobacterium tuberculosis. Infect. Immun. 75: 5127-5134
180. Yang, Y., C. Yin, A. Pandey, D. Abbott, C. Sassetti, and M. A. Kelliher. 2007. NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J. Biol. Chem. 282: 36223-36229
181. Pandey, A. K., Y. Yang, Z. Jiang, S. M. Fortune, F. Coulombe, M. A. Behr, K. A. Fitzgerald, C. M. Sassetti, and M. A. Kelliher. 2009. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS. Pathog. 5: e1000500
182. Deretic, V., S. Singh, S. Master, J. Harris, E. Roberts, G. Kyei, A. Davis, H. S. De, J. Naylor, H. H. Lee, and I. Vergne. 2006. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol. 8: 719-727
183. Russell, D. G. 2001. Mycobacterium tuberculosis: here today, and here tomorrow. Nature Reviews 2: 1-9

157
184. Vergne, I., J. Chua, and V. Deretic. 2003. Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. J. Exp. Med 198: 653-659
185. Malik, Z. A., C. R. Thompson, S. Hashimi, B. Porter, S. S. Iyer, and D. J. Kusner. 2003. Cutting edge: Mycobacterium tuberculosis blocks Ca2+ signaling and phagosome maturation in human macrophages via specific inhibition of sphingosine kinase. J Immunol. 170: 2811-2815
186. Schlesinger LS, Azad AK, Torrelles JB, E. Roberts, I. Vergne, and V. Deretic. 2008. Determinants of Phagocytosis, Phagosome Biogenesis and Autophagy for Mycobacterium tuberculosis. In Handbook of Tuberculosis. Immunology and Cell Biology. S. H. E. Kaufmann and W. J. Britton, eds. Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim, Germany, pp. 1.
187. Aderem, A., and D. M. Underhill. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17: 593-623
188. Kang, B. K., A. K. Azad, J. B. Torrelles, T. M. Kaufman, A. A. Beharka, E. Tibesar, L. E. Desjardin, and L. S. Schlesinger. 2005. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp. Med 202: 987-999
189. Singh, C. R., R. A. Moulton, L. Y. Armitige, A. Bidani, M. Snuggs, S. Dhandayuthapani, R. L. Hunter, and C. Jagannath. 2006. Processing and presentation of a mycobacterial antigen 85B epitope by murine macrophages is dependent on the phagosomal acquisition of vacuolar proton ATPase and in situ activation of cathepsin D. J Immunol. 177: 3250-3259
190. Sturgill-Koszycki, S., P. H. Schlesinger, P. Chakraborty, P. L. Haddix, H. L. Collins, A. K. Fok, R. D. Allen, S. L. Gluck, J. Heuser, and D. G. Russell. 1994. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263: 678-681
191. Abdallah, A. M., N. C. Gey van Pittius, P. A. Champion, J. Cox, J. Luirink, C. M. Vandenbroucke-Grauls, B. J. Appelmelk, and W. Bitter. 2007. Type VII secretion--mycobacteria show the way. Nat. Rev. Microbiol. 5: 883-891
192. Mahairas, G. G., P. J. Sabo, M. J. Hickey, D. C. Singh, and C. K. Stover. 1996. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M-bovis. J. Bacteriol. 178: 1274-1282
193. Gordon, S. V., R. Brosch, A. Billault, T. Garnier, K. Eiglmeier, and S. T. Cole. 1999. Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol. Microbiol. 32: 643-655

158
194. Tekaia, F., S. V. Gordon, T. Garnier, R. Brosch, B. G. Barrell, and S. T. Cole. 1999. Analysis of the proteome of Mycobacterium tuberculosis in silico. Tuber. Lung Dis. 79: 329-342
195. Gey van Pittius, N. C., J. Gamieldien, W. Hide, G. D. Brown, R. J. Siezen, and A. D. Beyers. 2001. The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G+C Gram-positive bacteria. Genome Biol. 2: Research0044
196. Pallen, M. J. 2002. The ESAT-6/WXG100 superfamily -- and a new Gram-positive secretion system? Trends Microbiol. 10: 209-212
197. MacGurn, J. A., S. Raghavan, S. A. Stanley, and J. S. Cox. 2005. A non-RD1 gene cluster is required for Snm secretion in Mycobacterium tuberculosis. Mol. Microbiol. 57: 1653-1663
198. Fortune, S. M., A. Jaeger, D. A. Sarracino, M. R. Chase, C. M. Sassetti, D. R. Sherman, B. R. Bloom, and E. J. Rubin. 2005. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc. Natl. Acad. Sci. U. S. A 102: 10676-10681
199. Stanley, S. A., S. Raghavan, W. W. Hwang, and J. S. Cox. 2003. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc. Natl. Acad. Sci. U. S. A 100: 13001-13006
200. Renshaw, P. S., K. L. Lightbody, V. Veverka, F. W. Muskett, G. Kelly, T. A. Frenkiel, S. V. Gordon, R. G. Hewinson, B. Burke, J. Norman, R. A. Williamson, and M. D. Carr. 2005. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J 24: 2491-2498
201. Brodin, P., M. I. de Jonge, L. Majlessi, C. Leclerc, M. Nilges, S. T. Cole, and R. Brosch. 2005. Functional analysis of early secreted antigenic target-6, the dominant T-cell antigen of Mycobacterium tuberculosis, reveals key residues involved in secretion, complex formation, virulence, and immunogenicity. J Biol. Chem 280: 33953-33959
202. Nagai, H., E. D. Cambronne, J. C. Kagan, J. C. Amor, R. A. Kahn, and C. R. Roy. 2005. A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc. Natl. Acad. Sci. U. S. A 102: 826-831
203. Vergunst, A. C., M. C. van Lier, A. den Dulk-Ras, T. A. Stuve, A. Ouwehand, and P. J. Hooykaas. 2005. Positive charge is an important feature of the C-terminal transport signal of the VirB/D4-translocated proteins of Agrobacterium. Proc. Natl. Acad. Sci. U. S. A 102: 832-837

159
204. Christie, P. J., K. Atmakuri, V. Krishnamoorthy, S. Jakubowski, and E. Cascales. 2005. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu. Rev. Microbiol. 59: 451-485
205. Lenz, L. L., S. Mohammadi, A. Geissler, and D. A. Portnoy. 2003. SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc. Natl. Acad. Sci. U. S. A 100: 12432-12437
206. Kurtz, S., K. P. McKinnon, M. S. Runge, J. P. Ting, and M. Braunstein. 2006. The SecA2 secretion factor of Mycobacterium tuberculosis promotes growth in macrophages and inhibits the host immune response. Infect. Immun. 74: 6855-6864
207. McDonough, J. A., K. E. Hacker, A. R. Flores, M. S. Pavelka, Jr., and M. Braunstein. 2005. The twin-arginine translocation pathway of Mycobacterium smegmatis is functional and required for the export of mycobacterial beta-lactamases. J Bacteriol. 187: 7667-7679
208. Posey, J. E., T. M. Shinnick, and F. D. Quinn. 2006. Characterization of the twin-arginine translocase secretion system of Mycobacterium smegmatis. J Bacteriol. 188: 1332-1340
209. Raynaud, C., C. Guilhot, J. Rauzier, Y. Bordat, V. Pelicic, R. Manganelli, I. Smith, B. Gicquel, and M. Jackson. 2002. Phospholipases C are involved in the virulence of Mycobacterium tuberculosis. Mol. Microbiol. 45: 203-217
210. Molofsky, A. B., B. G. Byrne, N. N. Whitfield, C. A. Madigan, E. T. Fuse, K. Tateda, and M. S. Swanson. 2006. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp. Med. 203: 1093-1104
211. Ren, T., D. S. Zamboni, C. R. Roy, W. F. Dietrich, and R. E. Vance. 2006. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2: e18
212. Myrvik, Q. N., E. S. Leake, and M. J. Wright. 1984. Disruption of phagosomal membranes of normal alveolar macrophages by the H37Rv strain of Mycobacterium tuberculosis. A correlate of virulence. Am. Rev. Respir. Dis. 129: 322-328
213. McDonough, K. A., Y. Kress, and B. R. Bloom. 1993. Pathogenesis of tuberculosis: Interaction of Mycobacterium tuberculosis with macrophages. Infect. Immun. 61: 2763-2773

160
214. Stahl, P. D. 1990. The macrophage mannose receptor: current status. Am. J. Respir. Cell Mol. Biol. 2: 317-318
215. van der Wel, N., D. Hava, D. Houben, D. Fluitsma, Z. M. van, J. Pierson, M. Brenner, and P. J. Peters. 2007. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129: 1287-1298
216. Zhang, F. R., W. Huang, S. M. Chen, L. D. Sun, H. Liu, Y. Li, Y. Cui, X. X. Yan, H. T. Yang, R. D. Yang, T. S. Chu, C. Zhang, L. Zhang, J. W. Han, G. Q. Yu, C. Quan, Y. X. Yu, Z. Zhang, B. Q. Shi, L. H. Zhang, H. Cheng, C. Y. Wang, Y. Lin, H. F. Zheng, X. A. Fu, X. B. Zuo, Q. Wang, H. Long, Y. P. Sun, Y. L. Cheng, H. Q. Tian, F. S. Zhou, H. X. Liu, W. S. Lu, S. M. He, W. L. Du, M. Shen, Q. Y. Jin, Y. Wang, H. Q. Low, T. Erwin, N. H. Yang, J. Y. Li, X. Zhao, Y. L. Jiao, L. G. Mao, G. Yin, Z. X. Jiang, X. D. Wang, J. P. Yu, Z. H. Hu, C. H. Gong, Y. Q. Liu, R. Y. Liu, D. M. Wang, D. Wei, J. X. Liu, W. K. Cao, H. Z. Cao, Y. P. Li, W. G. Yan, S. Y. Wei, K. J. Wang, M. L. Hibberd, S. Yang, X. J. Zhang, and J. J. Liu. 2009. Genomewide association study of leprosy. N. Engl. J Med. 361: 2609-2618
217. Austin, C. M., X. Ma, and E. A. Graviss. 2008. Common nonsynonymous polymorphisms in the NOD2 gene are associated with resistance or susceptibility to tuberculosis disease in African Americans. J Infect. Dis. 197: 1713-1716
218. Le, B. L., S. Benko, and S. E. Girardin. 2007. Nod1 and Nod2 in innate immunity and human inflammatory disorders. Biochem. Soc. Trans. 35: 1479-1484
219. Sartor, R. B. 2006. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 3: 390-407
220. Lala, S., K. Dheda, J. S. Chang, J. F. Huggett, L. U. Kim, M. A. Johnson, G. A. Rook, S. Keshav, and A. Zumla. 2007. The pathogen recognition sensor, NOD2, is variably expressed in patients with pulmonary tuberculosis. BMC. Infect. Dis. 7: 96
221. Waterston, R. H., K. Lindblad-Toh, E. Birney, J. Rogers, J. F. Abril, P. Agarwal, R. Agarwala, R. Ainscough, M. Alexandersson, P. An, S. E. Antonarakis, J. Attwood, R. Baertsch, J. Bailey, K. Barlow, S. Beck, E. Berry, B. Birren, T. Bloom, P. Bork, M. Botcherby, N. Bray, M. R. Brent, D. G. Brown, S. D. Brown, C. Bult, J. Burton, J. Butler, R. D. Campbell, P. Carninci, S. Cawley, F. Chiaromonte, A. T. Chinwalla, D. M. Church, M. Clamp, C. Clee, F. S. Collins, L. L. Cook, R. R. Copley, A. Coulson, O. Couronne, J. Cuff, V. Curwen, T. Cutts, M. Daly, R. David, J. Davies, K. D. Delehaunty, J. Deri, E. T. Dermitzakis, C. Dewey, N. J. Dickens, M. Diekhans, S. Dodge, I. Dubchak, D. M. Dunn, S. R. Eddy, L. Elnitski, R. D. Emes, P. Eswara, E. Eyras, A. Felsenfeld, G. A. Fewell, P. Flicek, K. Foley, W. N. Frankel, L. A. Fulton, R. S. Fulton, T. S. Furey, D.

161
Gage, R. A. Gibbs, G. Glusman, S. Gnerre, N. Goldman, L. Goodstadt, D. Grafham, T. A. Graves, E. D. Green, S. Gregory, R. Guigo, M. Guyer, R. C. Hardison, D. Haussler, Y. Hayashizaki, L. W. Hillier, A. Hinrichs, W. Hlavina, T. Holzer, F. Hsu, A. Hua, T. Hubbard, A. Hunt, I. Jackson, D. B. Jaffe, L. S. Johnson, M. Jones, T. A. Jones, A. Joy, M. Kamal, E. K. Karlsson, D. Karolchik, A. Kasprzyk, J. Kawai, E. Keibler, C. Kells, W. J. Kent, A. Kirby, D. L. Kolbe, I. Korf, R. S. Kucherlapati, E. J. Kulbokas, D. Kulp, T. Landers, J. P. Leger, S. Leonard, I. Letunic, R. Levine, J. Li, M. Li, C. Lloyd, S. Lucas, B. Ma, D. R. Maglott, E. R. Mardis, L. Matthews, E. Mauceli, J. H. Mayer, M. McCarthy, W. R. McCombie, S. McLaren, K. McLay, J. D. McPherson, J. Meldrim, B. Meredith, J. P. Mesirov, W. Miller, T. L. Miner, E. Mongin, K. T. Montgomery, M. Morgan, R. Mott, J. C. Mullikin, D. M. Muzny, W. E. Nash, J. O. Nelson, M. N. Nhan, R. Nicol, Z. Ning, C. Nusbaum, M. J. O'Connor, Y. Okazaki, K. Oliver, E. Overton-Larty, L. Pachter, G. Parra, K. H. Pepin, J. Peterson, P. Pevzner, R. Plumb, C. S. Pohl, A. Poliakov, T. C. Ponce, C. P. Ponting, S. Potter, M. Quail, A. Reymond, B. A. Roe, K. M. Roskin, E. M. Rubin, A. G. Rust, R. Santos, V. Sapojnikov, B. Schultz, J. Schultz, M. S. Schwartz, S. Schwartz, C. Scott, S. Seaman, S. Searle, T. Sharpe, A. Sheridan, R. Shownkeen, S. Sims, J. B. Singer, G. Slater, A. Smit, D. R. Smith, B. SPENCER, A. Stabenau, N. Stange-Thomann, C. Sugnet, M. Suyama, G. Tesler, J. Thompson, D. Torrents, E. Trevaskis, J. Tromp, C. Ucla, A. Ureta-Vidal, J. P. Vinson, A. C. Von Niederhausern, C. M. Wade, M. Wall, R. J. Weber, R. B. Weiss, M. C. Wendl, A. P. West, K. Wetterstrand, R. Wheeler, S. Whelan, J. Wierzbowski, D. Willey, S. Williams, R. K. Wilson, E. Winter, K. C. Worley, D. Wyman, S. Yang, S. P. Yang, E. M. Zdobnov, M. C. Zody, and E. S. Lander. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562
222. Pabst, R., and I. Gehrke. 1990. Is the bronchus-associated lymphoid tissue (BALT) an integral structure of the lung in normal mammals, including humans? Am. J Respir. Cell Mol. Biol. 3: 131-135
223. Doeing, D. C., J. L. Borowicz, and E. T. Crockett. 2003. Gender dimorphism in differential peripheral blood leukocyte counts in mice using cardiac, tail, foot, and saphenous vein puncture methods. BMC Clin. Pathol. 3: 3
224. Risso, A. 2000. Leukocyte antimicrobial peptides: multifunctional effector molecules of innate immunity. J Leukoc. Biol. 68: 785-792
225. Bogdan, C. 2001. Nitric oxide and the immune response. Nat. Immunol. 2: 907-916
226. Koppel, E. A., K. P. van Gisbergen, T. B. Geijtenbeek, and Y. Van Kooyk. 2005. Distinct functions of DC-SIGN and its homologues L-SIGN (DC-SIGNR) and mSIGNR1 in pathogen recognition and immune regulation. Cell Microbiol. 7: 157-165

162
227. Svajger, U., M. Anderluh, M. Jeras, and N. Obermajer. 2010. C-type lectin DC-SIGN: an adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell Signal. 22: 1397-1405
228. Monteiro, R. C., and J. G. van De Winkel. 2003. IgA Fc receptors. Annu. Rev. Immunol. 21: 177-204
229. Daeron, M. 1997. Fc receptor biology. Annu. Rev. Immunol. 15: 203-234
230. Snapper, C. M., and Finkelman F.D. 1999. Immunoglobulin class switching. In Fundamental Immunology. p. 831.
231. Minegishi, Y., J. Rohrer, E. Coustan-Smith, H. M. Lederman, R. Pappu, D. Campana, A. C. Chan, and M. E. Conley. 1999. An essential role for BLNK in human B cell development. Science 286: 1954-1957
232. Tokugawa, Y., M. Koyama, and J. Silver. 1997. A molecular basis for species differences in Thy-1 expression patterns. Mol. Immunol. 34: 1263-1272
233. Farrar, J. D., Smith J.D., Murphy T.L., Leung S., Stark G.R., and Murphy K.M. 2000. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse Stat2. Nature Immunology
234. Bekpen, C., J. P. Hunn, C. Rohde, I. Parvanova, L. Guethlein, D. M. Dunn, E. Glowalla, M. Leptin, and J. C. Howard. 2005. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol. 6: R92
235. Singh, S. B., A. S. Davis, G. A. Taylor, and V. Deretic. 2006. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313: 1438-1441
236. WHO. 2006. Clinical Aspects of HIV/AIDS: Pulmonary Manifestations.
237. Fenton, M. J., L. W. Riley, and L. S. Schlesinger. 2005. Receptor-Mediated Recognition of Mycobacterium tuberculosis by Host Cells. In Tuberculosis and the Tubercle Bacillus. S. T. Cole, K. D. Eisenach, D. N. McMurray, and W. R. Jacobs, Jr., eds. ASM Press, New York, pp. 405.
238. Kang, P. B., A. K. Azad, J. B. Torrelles, T. M. Kaufman, A. Beharka, E. Tibesar, L. E. Desjardin, and L. S. Schlesinger. 2005. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp. Med. 202: 987-999
239. Ting, J. P., J. A. Duncan, and Y. Lei. 2010. How the noninflammasome NLRs function in the innate immune system. Science 327: 286-290

163
240. Gutierrez, O., C. Pipaon, N. Inohara, A. Fontalba, Y. Ogura, F. Prosper, G. Nunez, and J. L. Fernandez-Luna. 2002. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem 277: 41701-41705
241. Inohara, N., and G. Nunez. 2003. NODs: intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 3: 371-382
242. Kobayashi, K. S., M. Chamaillard, Y. Ogura, O. Henegariu, N. Inohara, G. Nunez, and R. A. Flavell. 2005. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307: 731-734
243. Herskovits, A. A., V. Auerbuch, and D. A. Portnoy. 2007. Bacterial ligands generated in a phagosome are targets of the cytosolic innate immune system. PLoS. Pathog. 3: e51
244. Kapetanovic, R., M. A. Nahori, V. Balloy, C. Fitting, D. J. Philpott, J. M. Cavaillon, and M. dib-Conquy. 2007. Contribution of phagocytosis and intracellular sensing for cytokine production by Staphylococcus aureus-activated macrophages. Infect. Immun. 75: 830-837
245. Kim, Y. G., J. H. Park, M. H. Shaw, L. Franchi, N. Inohara, and G. Nunez. 2008. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity. 28: 246-257
246. Till, A., P. Rosenstiel, K. Brautigam, C. Sina, G. Jacobs, H. H. Oberg, D. Seegert, T. Chakraborty, and S. Schreiber. 2008. A role for membrane-bound CD147 in NOD2-mediated recognition of bacterial cytoinvasion. J Cell Sci. 121: 487-495
247. Hruz, P., A. S. Zinkernagel, G. Jenikova, G. J. Botwin, J. P. Hugot, M. Karin, V. Nizet, and L. Eckmann. 2009. NOD2 contributes to cutaneous defense against Staphylococcus aureus through alpha-toxin-dependent innate immune activation. Proc. Natl. Acad. Sci. U. S. A 106: 12873-12878
248. Loving, C. L., M. Osorio, Y. G. Kim, G. Nunez, M. A. Hughes, and T. J. Merkel. 2009. Nod1/Nod2-mediated recognition plays a critical role in induction of adaptive immunity to anthrax after aerosol exposure. Infect. Immun. 77: 4529-4537
249. Strober, W., P. J. Murray, A. Kitani, and T. Watanabe. 2006. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat. Rev. Immunol. 6: 9-20
250. Kufer, T. A. 2008. Signal transduction pathways used by NLR-type innate immune receptors. Mol. Biosyst. 4: 380-386

164
251. Ganguly, N., P. H. Giang, C. Gupta, S. K. Basu, I. Siddiqui, D. M. Salunke, and P. Sharma. 2008. Mycobacterium tuberculosis secretory proteins CFP-10, ESAT-6 and the CFP10:ESAT6 complex inhibit lipopolysaccharide-induced NF-kappaB transactivation by downregulation of reactive oxidative species (ROS) production. Immunol. Cell Biol. 86: 98-106
252. Simeone, R., D. Bottai, and R. Brosch. 2009. ESX/type VII secretion systems and their role in host-pathogen interaction. Curr. Opin. Microbiol. 12: 4-10
253. Schneemann, M., and G. Schoedon. 2002. Species differences in macrophage NO production are important. Nat. Immunol. 3: 102
254. Mestas, J., and C. C. Hughes. 2004. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 172: 2731-2738
255. Powlesland, A. S., E. M. Ward, S. K. Sadhu, Y. Guo, M. E. Taylor, and K. Drickamer. 2006. Widely divergent biochemical properties of the complete set of mouse DC-SIGN-related proteins. J. Biol. Chem. 281: 20440-20449
256. Schneemann, M., and G. Schoeden. 2007. Macrophage biology and immunology: man is not a mouse. J. Leukoc. Biol. 81: 579
257. Horwitz, M. A., and S. C. Silverstein. 1980. Legionnaries' disease bacterium (Legionella pneumophila) multiplies intracellularly in human monocytes. J. Clin. Invest. 66: 441-450
258. Beharka, A. A., C. D. Gaynor, B. K. Kang, D. R. Voelker, F. X. McCormack, and L. S. Schlesinger. 2002. Pulmonary surfactant protein A up-regulates activity of the mannose receptor, a pattern recognition receptor expressed on human macrophages. J. Immunol. 169: 3565-3573
259. Brieland, J., P. Freeman, R. Kunkel, C. Chrisp, M. Hurley, J. Fantone, and C. Engleberg. 1994. Replicative Legionella pneumophila lung infection in intratracheally inoculated A/J mice. A murine model of human Legionnaires' disease. Am. J Pathol. 145: 1537-1546
260. Gavrilin, M. A., I. J. Bouakl, N. L. Knatz, M. D. Duncan, M. W. Hall, J. S. Gunn, and M. D. Wewers. 2006. Internalization and phagosome escape required for Francisella to induce human monocyte IL-1beta processing and release. Proc. Natl. Acad. Sci U. S. A 103: 141-146
261. Rajaram, M. V., L. P. Ganesan, K. V. Parsa, J. P. Butchar, J. S. Gunn, and S. Tridandapani. 2006. Akt/Protein kinase B modulates macrophage inflammatory response to Francisella infection and confers a survival advantage in mice. J Immunol. 177: 6317-6324

165
262. Olakanmi, O., B. E. Britigan, and L. S. Schlesinger. 2000. Gallium disrupts iron metabolism of mycobacteria residing within human macrophages. Infect. Immun. 68: 5619-5627
263. Nakagawara, A., and C. F. Nathan. 1983. A simple method for counting adherent cells: application to cultured human monocytes, macrophages and multi-nucleated giant cells. J. Immunol. Methods 56: 261-268
264. Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 18: 621-663
265. Flesch, I. E. A., and S. H. E. Kaufmann. 1993. Role of cytokines in tuberculosis. Immunobiology 189: 316-339
266. Phiet, P. H., J. Wietzerbin, E. Zissman, J. F. Petit, and E. Lederer. 1976. Analysis of the cell wall of five strains of Myocbacterium tuberculosis BCG and of an attenuated human strain, W 115. Infect. Immun. 13: 677-681
267. Legrand-Poels, S., G. Kustermans, F. Bex, E. Kremmer, T. A. Kufer, and J. Piette. 2007. Modulation of Nod2-dependent NF-kappaB signaling by the actin cytoskeleton. J Cell Sci. 120: 1299-1310
268. Kleinnijenhuis, J., L. A. Joosten, D. Van, V, N. Savage, C. R. van, B. J. Kullberg, d. van, V, T. H. Ottenhoff, C. A. Dinarello, J. W. Van der Meer, and M. G. Netea. 2009. Transcriptional and inflammasome-mediated pathways for the induction of IL-1beta production by Mycobacterium tuberculosis. Eur. J Immunol. 39: 1914-1922
269. Ferwerda, G., M. Kramer, J. D. de, A. Piccini, L. A. Joosten, I. Devesaginer, S. E. Girardin, G. J. Adema, J. W. Van der Meer, B. J. Kullberg, A. Rubartelli, and M. G. Netea. 2008. Engagement of NOD2 has a dual effect on proIL-1beta mRNA transcription and secretion of bioactive IL-1beta. Eur. J Immunol. 38: 184-191
270. Pan, Q., J. Mathison, C. Fearns, V. V. Kravchenko, C. J. da Silva, H. M. Hoffman, K. S. Kobayashi, J. Bertin, E. P. Grant, A. J. Coyle, F. S. Sutterwala, Y. Ogura, R. A. Flavell, and R. J. Ulevitch. 2007. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc. Biol. 82: 177-183
271. Boot, R. G., A. P. Bussink, M. Verhoek, P. A. de Boer, A. F. Moorman, and J. M. Aerts. 2005. Marked differences in tissue-specific expression of chitinases in mouse and man. J Histochem. Cytochem. 53: 1283-1292
272. Reginato, A. M., and B. R. Olsen. 2007. Genetics and experimental models of crystal-induced arthritis. Lessons learned from mice and men: is it crystal clear? Curr. Opin. Rheumatol. 19: 134-145

166
273. Rigamonti, E., G. Chinetti-Gbaguidi, and B. Staels. 2008. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler. Thromb. Vasc. Biol. 28: 1050-1059
274. Amer, A. O. 2010. Modulation of caspases and their non-apoptotic functions by Legionella pneumophila. Cell Microbiol. 12: 140-147
275. Gavrilin, M. A., S. Mitra, S. Seshadri, J. Nateri, F. Berhe, M. W. Hall, and M. D. Wewers. 2009. Pyrin critical to macrophage IL-1beta response to Francisella challenge. J Immunol. 182: 7982-7989
276. Pauleau, A. L., and P. J. Murray. 2003. Role of nod2 in the response of macrophages to toll-like receptor agonists. Mol. Cell Biol. 23: 7531-7539
277. Leake, E. S., Q. N. Myrvik, and M. J. Wright. 1984. Phagosomal membranes of Mycobacterium bovis BCG-immune alveolar macrophages are resistant to disruption by Mycobacterium tuberculosis H37Rv. Infect. Immun. 45: 443-446
278. Teitelbaum, R., M. Cammer, M. L. Maitland, N. E. Freitag, J. Condeelis, and B. R. Bloom. 1999. Mycobacterial infection of macrophages results in membrane-permeable phagosomes. Proc. Natl. Acad. Sci USA 96: 15190-15195
279. Lecine, P., S. Esmiol, J. Y. Metais, C. Nicoletti, C. Nourry, C. McDonald, G. Nunez, J. P. Hugot, J. P. Borg, and V. Ollendorff. 2007. The NOD2-RICK complex signals from the plasma membrane. J Biol. Chem. 282: 15197-15207
280. Korhonen, R., A. Lahti, H. Kankaanranta, and E. Moilanen. 2005. Nitric oxide production and signaling in inflammation. Curr. Drug Targets. Inflamm. Allergy 4: 471-479
281. Moreno, L., S. K. McMaster, T. Gatheral, L. K. Bailey, L. S. Harrington, N. Cartwright, P. C. Armstrong, T. D. Warner, M. Paul-Clark, and J. A. Mitchell. 2010. Nucleotide oligomerization domain 1 is a dominant pathway for NOS2 induction in vascular smooth muscle cells: comparison with Toll-like receptor 4 responses in macrophages. Br. J Pharmacol. 160: 1997-2007
282. Totemeyer, S., M. Sheppard, A. Lloyd, D. Roper, C. Dowson, D. Underhill, P. Murray, D. Maskell, and C. Bryant. 2006. IFN-gamma enhances production of nitric oxide from macrophages via a mechanism that depends on nucleotide oligomerization domain-2. J Immunol. 176: 4804-4810
283. Bansal, K., and K. N. Balaji. 2011. Intracellular pathogen sensor NOD2 programs macrophages to trigger Notch1 activation. J Biol. Chem 286: 5823-5835

167
284. MacMicking, J. D., R. J. North, R. LaCourse, J. S. Mudgett, S. K. Shah, and C. F. Nathan. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA 94: 5243-5248
285. Nims, R. W., J. C. Cook, M. C. Krishna, D. Christodoulou, C. M. Poore, A. M. Miles, M. B. Grisham, and D. A. Wink. 1996. Colorimetric assays for nitric oxide and nitrogen oxide species formed from nitric oxide stock solutions and donor compounds. Methods Enzymol. 268: 93-105
286. Cameron, M. L., D. L. Granger, J. B. Weinberg, W. J. Kozumbo, and H. S. Koren. 1990. Human alveolar and peritoneal macrophages mediate fungistasis independently of L-arginine oxidation to nitrite or nitrate. Am. Rev. Respir. Dis. 142: 1313-1319
287. Weinberg, J. B., M. A. Misukonis, P. J. Shami, S. N. Mason, D. L. Sauls, W. A. Dittman, E. R. Wood, G. K. Smith, B. McDonald, K. E. Bachus, A. F. Haney, and D. L. Granger. 1995. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): Analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood 86: 1184-1195
288. Sakai, N., and S. Milstien. 1993. Availability of tetrahydrobiopterin is not a factor in the inability to detect nitric oxide production by human macrophages. Biochem. Biophys. Res. Commun. 193: 378-383
289. Jagannath, C., J. K. Actor, and R. L. Hunter, Jr. 1998. Induction of nitric oxide in human monocytes and monocyte cell lines by Mycobacterium tuberculosis. Nitric. Oxide. 2: 174-186
290. Choi, H. S., P. R. Rai, H. W. Chu, C. Cool, and E. D. Chan. 2002. Analysis of nitric oxide synthase and nitrotyrosine expression in human pulmonary tuberculosis. Am. J Respir. Crit Care Med. 166: 178-186
291. Nicholson, S., M. da Gloria, M. D. G. Bonecini-Almeida, J. R. L. E. Silva, C. Nathan, Q. W. Xie, R. Mumford, J. R. Weidner, J. Calaycay, J. Geng, N. Boechat, C. Linhares, W. Rom, and J. L. Ho. 1996. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J. Exp. Med. 183: 2293-2302
292. Schlesinger, L. S., and M. A. Horwitz. 1991. Phagocytosis of Mycobacterium leprae by human monocyte-derived macrophages is mediated by complement receptors CR1(CD35), CR3(CD11b/CD18), and CR4(CD11c/CD18) and interferon gamma activation inhibits complement receptor function and phagocytosis of this bacterium. J. Immunol. 147: 1983-1994

168
293. Sheng, J. Z., D. Wang, and A. P. Braun. 2005. DAF-FM (4-amino-5-methylamino-2',7'-difluorofluorescein) diacetate detects impairment of agonist-stimulated nitric oxide synthesis by elevated glucose in human vascular endothelial cells: reversal by vitamin C and L-sepiapterin. J Pharmacol. Exp. Ther. 315: 931-940
294. Howard, M., J. Roux, H. Lee, B. Miyazawa, J. W. Lee, B. Gartland, A. J. Howard, M. A. Matthay, M. Carles, and J. F. Pittet. 2010. Activation of the stress protein response inhibits the STAT1 signalling pathway and iNOS function in alveolar macrophages: role of Hsp90 and Hsp70. Thorax 65: 346-353
295. Rich, E. A., M. Torres, E. Sada, C. K. Finegan, B. D. Hamilton, and Z. Toossi. 1997. Mycobacterium tuberculosis (MTB)-stimulated production of nitric oxide by human alveolar macrophages and relationship of nitric oxide production to growth inhibition of MTB. Tuber. Lung Dis. 78: 247-255
296. Cooney, R., J. Baker, O. Brain, B. Danis, T. Pichulik, P. Allan, D. J. Ferguson, B. J. Campbell, D. Jewell, and A. Simmons. 2010. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16: 90-97
297. Travassos, L. H., L. A. Carneiro, M. Ramjeet, S. Hussey, Y. G. Kim, J. G. Magalhaes, L. Yuan, F. Soares, E. Chea, B. L. Le, I. G. Boneca, A. Allaoui, N. L. Jones, G. Nunez, S. E. Girardin, and D. J. Philpott. 2010. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11: 55-62
298. Homer, C. R., A. L. Richmond, N. A. Rebert, J. P. Achkar, and C. McDonald. 2010. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn's disease pathogenesis. Gastroenterology 139: 1630-41, 1641
299. Tattoli, I., L. H. Travassos, L. A. Carneiro, J. G. Magalhaes, and S. E. Girardin. 2007. The Nodosome: Nod1 and Nod2 control bacterial infections and inflammation. Semin. Immunopathol. 29: 289-301
300. Chan, J., K. Tanaka, D. Carroll, J. Flynn, and B. R. Bloom. 1995. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect. Immun. 63: 736-740
301. Misko, T. P., R. J. Schilling, D. Salvemini, W. M. Moore, and M. G. Currie. 1993. A fluorometric assay for the measurement of nitrite in biological samples. Anal. Biochem. 214: 11-16

169
302. Rhoades, E. R., and I. M. Orme. 1997. Susceptibility of a panel of virulent strains of Mycobacterium tuberculosis to reactive nitrogen intermediates. Infect. Immun. 65: 1189-1195
303. Yu, K., C. Mitchell, Y. Xing, R. S. Magliozzo, B. R. Bloom, and J. Chan. 1999. Toxicity of nitrogen oxides and related oxidants on mycobacteria: M. tuberculosis is resistant to peroxynitrite anion. Tuber. Lung Dis. 79: 191-198
304. Bryk, R., P. Griffin, and C. Nathan. 2000. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature 407: 211-215
305. Ehrt, S., M. U. Shiloh, J. Ruan, M. Choi, S. Gunzburg, C. Nathan, Q. Xie, and L. W. Riley. 1997. A novel antioxidant gene from Mycobacterium tuberculosis. J Exp. Med. 186: 1885-1896
306. Nathan, C., and M. U. Shiloh. 2000. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts andmicrobial pathogens. Proc. Natl. Acad. Sci USA 97: 8841-8848
307. Kuo, H. P., C. H. Wang, K. S. Huang, H. C. Lin, C. T. Yu, C. Y. Liu, and L. C. Lu. 2000. Nitric oxide modulates interleukin-1beta and tumor necrosis factor-alpha synthesis by alveolar macrophages in pulmonary tuberculosis. Am. J Respir. Crit Care Med. 161: 192-199
308. Balligand, J. L., D. Ungureanu-Longrois, W. W. Simmons, D. Pimental, T. A. Malinski, M. Kapturczak, Z. Taha, C. J. Lowenstein, A. J. Davidoff, R. A. Kelly, and . 1994. Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. Characterization and regulation of iNOS expression and detection of iNOS activity in single cardiac myocytes in vitro. J Biol. Chem 269: 27580-27588
309. Adams, V., B. Nehrhoff, U. Spate, A. Linke, P. C. Schulze, A. Baur, S. Gielen, R. Hambrecht, and G. Schuler. 2002. Induction of iNOS expression in skeletal muscle by IL-1beta and NFkappaB activation: an in vitro and in vivo study. Cardiovasc. Res. 54: 95-104
310. Chan, E. D., J. Chan, and N. W. Schluger. 2001. What is the role of nitric oxide in murine and human host defense against tuberculosis? Current knowledge. Am. J. Respir. Cell Mol. Biol. 25: 606-612
311. Nozaki, Y., Y. Hasegawa, S. Ichiyama, I. Nakashima, and K. Shimokata. 1997. Mechanism of nitric oxide-dependent killing of Mycobacterium bovis BCG in human alveolar macrophages. Infect. Immun. 65: 3644-3647
312. Toshchakov, V., B. W. Jones, P. Y. Perera, K. Thomas, M. J. Cody, S. Zhang, B. R. Williams, J. Major, T. A. Hamilton, M. J. Fenton, and S. N. Vogel. 2002.

170
TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 3: 392-398
313. Thoma-Uszynski, S., S. Stenger, O. Takeuchi, M. Ochoa, M. Engele, P. Sieling, P. Barnes, M. Rollinghoff, P. Bolcskei, M. Wagner, S. Akira, M. Norgard, J. Belisle, P. Godowski, B. Bloom, and R. Modlin. 2001. Induction of direct antimicrobial activity through mammalian Toll-like receptors. Science 291: 1544-1547
314. Kimura, H., S. Miura, T. Shigematsu, N. Ohkubo, Y. Tsuzuki, I. Kurose, H. Higuchi, Y. Akiba, R. Hokari, M. Hirokawa, H. Serizawa, and H. Ishii. 1997. Increased nitric oxide production and inducible nitric oxide synthase activity in colonic mucosa of patients with active ulcerative colitis and Crohn's disease. Dig. Dis. Sci. 42: 1047-1054
315. Grisham, M. B., K. Ware, H. E. Gilleland, Jr., L. B. Gilleland, C. L. Abell, and T. Yamada. 1992. Neutrophil-mediated nitrosamine formation: role of nitric oxide in rats. Gastroenterology 103: 1260-1266
316. Grisham, M. B., R. D. Specian, and T. E. Zimmerman. 1994. Effects of nitric oxide synthase inhibition on the pathophysiology observed in a model of chronic granulomatous colitis. J Pharmacol. Exp. Ther. 271: 1114-1121
317. Shaul, P. W., A. J. North, L. C. Wu, L. B. Wells, T. S. Brannon, K. S. Lau, T. Michel, L. R. Margraf, and R. A. Star. 1994. Endothelial nitric oxide synthase is expressed in cultured human bronchiolar epithelium. J Clin. Invest 94: 2231-2236
318. Shames, B. D., C. H. Selzman, E. J. Pulido, X. Meng, D. R. Meldrum, R. C. McIntyre, Jr., A. H. Harken, and A. Banerjee. 1999. LPS-Induced NF-kappaB activation and TNF-alpha release in human monocytes are protein tyrosine kinase dependent and protein kinase C independent. J Surg. Res. 83: 69-74
319. Wadsworth, S. J., and H. Goldfine. 2002. Mobilization of protein kinase C in macrophages induced by Listeria monocytogenes affects its internalization and escape from the phagosome. Infect. Immun. 70: 4650-4660
320. Schwegmann, A., R. Guler, A. J. Cutler, B. Arendse, W. G. Horsnell, A. Flemming, A. H. Kottmann, G. Ryan, W. Hide, M. Leitges, C. Seoighe, and F. Brombacher. 2007. Protein kinase C delta is essential for optimal macrophage-mediated phagosomal containment of Listeria monocytogenes. Proc. Natl. Acad. Sci. U. S. A 104: 16251-16256
321. Brandt, S., S. Wessler, R. Hartig, and S. Backert. 2009. Helicobacter pylori activates protein kinase C delta to control Raf in MAP kinase signalling: role in

171
AGS epithelial cell scattering and elongation. Cell Motil. Cytoskeleton 66: 874-892
322. Sumaroka, M. V., I. S. Litvinov, S. V. Khaidukov, T. N. Golovina, M. V. Kamraz, R. L. Komal'eva, T. M. Andronova, E. A. Makarov, V. A. Nesmeyanov, and V. T. Ivanov. 1991. Muramyl peptide-binding sites are located inside target cells. FEBS Lett. 295: 48-50
323. von, K. O., S. Lipinski, A. Till, S. J. Martin, W. Nietfeld, H. Lehrach, S. Schreiber, and P. Rosenstiel. 2010. Caspase recruitment domain-containing protein 8 (CARD8) negatively regulates NOD2-mediated signaling. J Biol. Chem 285: 19921-19926
324. Lipinski, S., A. Till, C. Sina, A. Arlt, H. Grasberger, S. Schreiber, and P. Rosenstiel. 2009. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J Cell Sci. 122: 3522-3530
325. Rigutto, S., C. Hoste, H. Grasberger, M. Milenkovic, D. Communi, J. E. Dumont, B. Corvilain, F. Miot, and D. De, X. 2009. Activation of dual oxidases Duox1 and Duox2: differential regulation mediated by camp-dependent protein kinase and protein kinase C-dependent phosphorylation. J Biol. Chem 284: 6725-6734
326. Voss, O. H., S. Kim, M. D. Wewers, and A. I. Doseff. 2005. Regulation of monocyte apoptosis by the protein kinase Cdelta-dependent phosphorylation of caspase-3. J Biol. Chem 280: 17371-17379
327. Monick, M. M., A. B. Carter, G. Gudmundsson, L. J. Geist, and G. W. Hunninghake. 1998. Changes in PKC isoforms in human alveolar macrophages compared with blood monocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 19: L389-L397
328. Meller, N., Y. Elitzur, and N. Isakov. 1999. Protein kinase C-theta (PKCtheta) distribution analysis in hematopoietic cells: proliferating T cells exhibit high proportions of PKCtheta in the particulate fraction. Cell Immunol. 193: 185-193
329. Mellor, H., and P. J. Parker. 1998. The extended protein kinase C superfamily. Biochem. J 332 ( Pt 2): 281-292
330. Thome, M., K. Hofmann, K. Burns, F. Martinon, J. L. Bodmer, C. Mattmann, and J. Tschopp. 1998. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr. Biol. 8: 885-888
331. McCarthy, J. V., J. Ni, and V. M. Dixit. 1998. RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J Biol. Chem 273: 16968-16975

172
332. Sarkar, A., M. Duncan, J. Hart, E. Hertlein, D. C. Guttridge, and M. D. Wewers. 2006. ASC directs NF-kappaB activation by regulating receptor interacting protein-2 (RIP2) caspase-1 interactions. J Immunol. 176: 4979-4986
333. Inohara, N., P. L. del, T. Koseki, S. Chen, and G. Nunez. 1998. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J Biol. Chem 273: 12296-12300
334. Meylan, E., F. Martinon, M. Thome, M. Gschwendt, and J. Tschopp. 2002. RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-kappa B and is processed during apoptosis. EMBO Rep. 3: 1201-1208
335. Chen, L., K. Haider, M. Ponda, A. Cariappa, D. Rowitch, and S. Pillai. 2001. Protein kinase C-associated kinase (PKK), a novel membrane-associated, ankyrin repeat-containing protein kinase. J Biol. Chem 276: 21737-21744
336. Dempsey, P. W., S. A. Vaidya, and G. Cheng. 2003. The art of war: Innate and adaptive immune responses. Cell Mol. Life Sci. 60: 2604-2621
337. Kobzik, L. 2009. Translating NO biology into clinical advances: still searching for the right dictionary? Am. J Respir. Cell Mol. Biol. 41: 9-13
338. Tigno-Aranjuez, J. T., J. M. Asara, and D. W. Abbott. 2010. Inhibition of RIP2's tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev. 24: 2666-2677
339. Kelsall, B. 2005. Getting to the guts of NOD2. Nat. Med. 11: 383-384
340. Dzierzbicka, K., A. Wardowska, and P. Trzonkowski. 2011. Recent developments in the synthesis and biological activity of muramylpeptides. Curr. Med. Chem 18: 2438-2451
341. Wang, L. Z., X. L. Li, X. Y. Pang, and L. R. Sun. 2011. Muramyl Dipeptide modulates differentiation, maturity of dendritic cells and anti-tumor effect of DC-mediated T cell in acute leukemia children. Hum. Vaccin. 7: