novel amodiaquine congeners as potent antimalarial agents
TRANSCRIPT

EUROPEAN FEDERATION FOR MEDICINAL CHEMISTRY SOCIETÀ CHIMICA ITALIANA - DIVISIONE DI CHIMICA FARMACEUTICA
European School of Medicinal Chemistry (XXVIII Advanced Course of Medicinal Chemistry and
"E. Duranti" National Seminar for PhD Students)
UNIVERSITÀ DEGLI STUDI DI URBINO “CARLO BO”
PROCEEDINGS OF PhD STUDENT POSTER SESSION

2
Scientific Committee
Gloria CRISTALLI University of Camerino - Director Gabriele COSTANTINO University of Parma Carlo DE MICHELI University of Milan Romano DI FABIO GlaxoSmithKline - Verona Roberta FRUTTERO University of Turin Marco MACCHIA University of Pisa Stefano MORO University of Padua Maria PAPPALARDO University of Catania Giorgio TARZIA University of Urbino Mario VARASI Genextra S.p.A. - Milan
Organizing Committee
Lucia BEDINI Giuseppe DIAMANTINI Americo SALVATORI Gilberto SPADONI Istituto di Chimica Farmaceutica University of Urbino
The Organizers thank for their support:
GlaxoSmithKline, Verona (I)
Nikem Research, Milan (I)
Novartis, Cambridge (USA)
Siena Biotech, Siena (I)
Toscana Life Sciences Foundation, Siena (I)
Città di Urbino - Assessorato alla Cultura ed al Turismo
Università "Carlo Bo" di Urbino
Genextra S.p.A., Milan (I)

3
PROGRAM
SUNDAY, JULY 6
15.00 - 18.30 Registration
19.00 - 22.00 Poster Exhibition and Buffet Dinner
MONDAY, JULY 7
Opening of ESMEC 2008
Luisa MOSTI - President of the Medicinal Chemistry Division of the Italian Chemical Society
THERAPEUTIC APPROACHES FOR HUNTINGTON'S DISEASE
Chairperson: Gabriele COSTANTINO - University of Parma (I)
Introduction to the Session
Maria Pia ABBRACCHIO - University of Milan (I) General Features of Neurodegenerative Diseases
10.15 - 11.00 Robert SCHWARCZ - MPRC, University of Maryland, Baltimore (USA) The Molecular Basis of Huntington’s Disease: Renewed Focus on the Kynurenine Pathway of Tryptophan Degradation
11.00 - 11.30 Coffee Break
11.30 - 12.15 Eduardo Gonzales COUTO - Siena Biotech, Siena (I) Integrating Omics Data to Unraveling Pathologic Mechanism and Identify New Target in Huntington’s Disease
12.15 - 13.30 Poster Session 1 - Chairperson: Maria PAPPALARDO - University of Catania (I)
13.30 - 15.00 Lunch
Chairperson: Marco MACCHIA - University of Pisa (I)
15.00 - 15.45 Salvatore LA ROSA - Siena Biotech, Siena (I) New Trends in Huntington’s Disease Drug Discovery
15.45 - 16.30 Maria Pia ABBRACCHIO - University of Milan (I) Huntington's Disease and Other Brain Disorders
16.30 - 18.00 Workshop
20.30 Dinner
TUESDAY, JULY 8
Chairperson: Roberta FRUTTERO - University of Turin (I)
9.00 - 9.15 Introduction to the Session
9.15 - 10.00
10.00 - 10.45 Drug Metabolism: Reactions Performed by CYP450 and Beyonds. Comprehensive
Review of the Chemistry Behind Drug Metabolism and Enzymes Involved
10.45 - 11.15 Coffee Break
11.15 - 12.00 Angeliki KOUROUNAKIS - University of Athens (GR)
Adenosine A2A Receptors as a Target for Novel Neuroprotective Strategies in
The Value of Profiling and Understanding Metabolic Pathways in Discovery Upendra ARGIKAR - Novartis, Cambridge (USA)
Mahamud KAJBAF - GSK, Verona (I)
9.00 - 9.30
9.30 - 9.45
9.45 - 10.15
Gloria CRISTALLI - Director of the European School of Medicinal Chemistry
NEW PARADIGMS IN DRUG METABOLISM, EXCRETION, AND TOXICITY

4
Recent Developments in Retrometabolic Drug Design and Targeting Strategies
12.00 - 13.30 Poster Session 2 - Chairperson: Stefano MORO - University of Padua (I)
13.30 - 15.30 Lunch
Chairperson: Romano DI FABIO - GSK, Verona (I)
15.30 - 16.15 Predicting Metabolism in Silico: a Brief Overview
16.15 -17.00 Cinzia STELLA - University of Geneve (CH) Application of Metabonomics for the Study of Human Biocomplexity
20.30 Dinner
WEDNESDAY, JULY 9
SYNTHESIS AND REACTIVITY OF HETEROCYCLES
Chairperson: Carlo DE MICHELI - University of Milan (I)
Introduction to the Session
9.15 - 10.00 Albert PADWA - Emory University, Atlanta (USA) The Synthesis of Heterocycles Using Cascade Chemistry
Alberto BRANDI - University of Florence (I) Stereocontrolled Cycloaddition Processes en Route for Sugar Mimetics
10.45 - 11.15 Coffee Break
11.15 - 12.00 Peter MATYUS - Semmelweis University, Budapest (H) Synthesis of Aza-Heterocycles via C-C Bond Formation Reactions
12.00 - 12.45 Daniele ANDREOTTI - GSK, Verona (I) Synthesis of Heterocycles as Drug Intermediates
12.45 - 15.00 Lunch
15.00-15.45 Albert PADWA - Emory University, Atlanta (USA) Alkaloids - The Playground of Synthetic Heterocyclic Chemistry
15.45 - 17.45 Workshop
17.30 - 18.30 Meeting of Coordinators of Pharmaceutical Sciences Doctorates
17.30 - 18.30 Meeting of "Gruppo Giovani" of the Medicinal Chemistry Division of Italian Chemical Society
20.30 Dinner
THURSDAY, JULY 10
HOT TOPICS
Chairperson:
9.00 - 9.15 Introduction to the Session
9.15 - 10.00 Giovanni GAVIRAGHI - Siena Biotech, Siena (I) Target-based and Target Deconvolution Strategy in Drug Discovery
10.00 - 10.45 Daniele FANCELLI - Congenia S.r.l. - Genextra Group, Milan (I) Stem Cells and Opportunities for Drug Discovery
10.45 - 11.15 Coffee Break
11.15 - 12.00 Germano CARGANICO - Fondazione Toscana Life Sciences, Siena (I) Spin-off and Start-up: Challenges and Opportunities
12.00 - 12.45 Paolo PEVARELLO - Centro Nacional de Investigaciones Oncologicas, Madrid (E)
Franco LOMBARDO - Novartis, Cambridge (USA)
9.00 - 9.15
10.00 - 10.45
Mario VARASI - Genextra S.p.A., Milan (I)

5
Drugs Approved by EMEA and FDA in 2007 (To Market! To Market!)
12.45 - 14.45 Lunch
Chairperson: Giorgio TARZIA - University of Urbino (I)
14.45 - 18.00 Presentation and Discussion of Selected Posters
18.00 Concluding Remarks
and Training Committee of EFMC
Gloria CRISTALLI - University of Camerino (I) - ESMEC Director
20.30 Gala Dinner
FRIDAY, JULY 11
9.00 Leaving of participants
Peter MATYUS - Semmelweis University, Budapest (H) - Member of Education

6
CONTENTS
Jamila Isabella ALI SYNTHESIS AND BIOLOGICAL EVALUATION OF NEW iNOS INHIBITORS..................................................11
Gabriella AMATO DESIGN, SYNTHESIS AND PHARMACOLOGICAL RESULTS OF NEW POTENTIAL LARGE-CONDUCTANCE CALCIUM–ACTIVATED POTASSIUM-CHANNEL (BKCa) OPENERS..................................12
Francesca ANTONIETTI NOVEL SELECTIVE INHIBITORS OF N-ACYLETHANOLAMINE-HYDROLYZING ACID AMIDASE (NAAA) AS POTENTIAL ANTI-INFLAMMATORY AGENTS ..............................................................................14
Serena BASILI COMPUTATIONAL APPROACHES TO THE RATIONAL DESIGN OF NOVEL TOPOISOMERASE I POISONS AS POTENTIAL ANTICANCER DRUGS................................................................................................16
Asunción BURGUETE ANTICANCER, ANTI-INFLAMMATORY AND ANTIOXIDANT ACTIVITIES OF NEW QUINOXALINE AND QUINOXALINE DI-N-OXIDE DERIVATIVES...............................................................................................18
Mariangela CANTORE 6,7-DIMETHOXYTETRAHYDROISOQUINOLINE DERIVATIVES:POTENT P-GLYCOPROTEIN LIGANDS REVERSING MULTIDRUG RESISTANCE............................................................................................19
Rita CAPELA COUPLING ARTEMISININ TO A VINYL SULFONE SCAFFOLD TO IMPROVE ANTIMALARIAL ACTIVITY ...................................................................................................................................................................21
Antonia CAROLI APPLICATIONS OF 3-D QSAR METHODS TO DIFFERENT CLASS OF DRUGS...............................................23
Laura CARRO SYNTHESIS AND BINDING AFFINITY OF NEW QUINAZOLINONE DERIVATIVES AS POTENTIAL ATYPICAL ANTIPSYCHOTICS................................................................................................................................26
Manolo CASAGRANDE NOVEL AMODIAQUINE CONGENERS AS POTENT ANTIMALARIAL AGENTS ............................................28
Siew Lee CHEONG SYNTHESIS, CHARACTERIZATION AND EVALUATION OF NEW ADENOSINE RECEPTORS’ ANTAGONISTS ..........................................................................................................................................................30
Elena CICHERO DOCKING STUDIES AND QSAR ANALYSIS ON CB1 ANTAGONISTS: A COMPUTATIONAL APPROACH TOWARDS THE IDENTIFICATION OF NEW LIGANDS.................................................................32
Paolo COGHI NOVEL SYNTHETHIC APPROACH TO ANTIMALARIAL COMPOUNDS: 4-AMINOQUINOLINES THROUGH MICROWAVE-ASSISTED SNAR REACTIONS AND COMBINATORIAL APPROACH TO 2,4,6-TRISUBSTITUTED TRIAZINES................................................................................................................33
Catia CORNACCHIA L-DOPA-THIOL ANTIOXIDANT CODRUGS AS NEW ANTI-PARKINSON AGENTS WITH FREE RADICAL SCAVENGING PROPERTIES .................................................................................................................35
Delphine CRESSENDREACTIVITY OF SUBSTITUTED XANTHONES TOWARD PEROXYL, ABTS AND DPPH RADICALS ........37
Marco CROSETTI DESIGN AND SYNTHESIS OF A NEW SERIES OF DERIVATIVES ENDOWED WITH POTENTIAL INTEREST IN CANCER IMMUNOTHERAPY.........................................................................................................39

7
Fabio DEL BELLO 1,4-DIOXANE NUCLEUS AS A SUITABLE SUBSTRUCTURE FOR THE CHARACTERIZATION OF DIFFERENT RECEPTOR SYSTEMS ..................................................................................................................41 Marco ELEOPRA NOVEL BISPHOSPHONATES AS γδ-T LYMPHOCYTES ACTIVATORS............................................................43
Stella FIORINI SYNTHESIS AND BIOLOGICAL ACTIVITY OF HUMAN NEUROPEPTIDES ANALOGUES MODIFIED IN POSITION 2 .......................................................................................................................................44
Roberta FRASSON STRUCTURE AND FUNCTION OF NATURALLY OCCURRING VARIANTS OF HUMAN ALPHA-THROMBIN ..................................................................................................................................................45
Valentina GANDIN EFFECTS OF GOLD COMPOUNDS IN CISPLATIN-SENSITIVE AND-RESISTANT OVARIAN CANCER CELLS .....................................................................................................................................46
Michele GIAMPIERI STUDY ON DRUGS FOR CYSTIC FIBROSIS THERAPY ......................................................................................48
Francesca GORI SYNTHESIS AND SEMI-SYNTHESIS OF GLYCOSYLATED PROTEINS AS PHARMACEUTICAL AND DIAGNOSTIC TOOLS ......................................................................................................................................50
Amandine GUILLOT HIGHLY LIPOPHILIC COMPOUNDS: DETERMINATION OF LIPOPHILICITY BY RP-LC AND UPLC .........52
Enise Ece GURDAL COMFA STUDY ON SIGMA (σ) RECEPTOR LIGANDS........................................................................................54
Carmela INGLESE DESIGN AND BIOLOGICAL EVALUATION OF FLUORESCENT-, [3H]-, OR [11C]-SIGMA RECEPTOR LIGANDS AS NOVEL TOOLS IN CANCER DIAGNOSIS.................................................................56
Mario IPPOLITO MULTIVARIATE METHODS AND MOLECULAR MODELING TECHNIQUES IN THE STUDY OF ANTITUMOR AGENTS .......................................................................................................................................58
Dhuldeo Dnyandeo KACHARE DESIGN, SYNTHESIS, CHARACTERISATION AND BIOLOGICAL ACTIVITY OF LIGANDS FOR P2 RECEPTORS .................................................................................................................................................60
Meenakshisundaram KANDHAVELU TITLE OF THE RESEARCH: MOLECULAR CHARACTERIZATION OF THE DUALISTIC RECEPTOR GPR17.....................................................................................................................................................62
Ilaria LAZZARI A NEW ANTAGONIST OF Bv8-PROKINETICIN RECEPTORS FOR THE DEVELOPMENT OF NEW ANALGESICS AND ANTI-INFLAMMATORY DRUGS .........................................................................................64
Francesco LAZZARIN NEUROPEPTIDES IN THE REGULATION OF FEEDING......................................................................................66
Mariaelisa MANGANARO SYNTHESIS AND PHARMACOLOGICAL EVALUATION OF A NEW CLASS OF CARDIOPROTECTIVE BENZOPYRAN-BASED KATP OPENERS .........................................................................68
Erika MARTINA SYNTHESIS AND EVALUATION NOVEL CONJUGATES BASED ON THE COMBINED MITOXANTRONE–AMSACRINE PHARMACOPHORES ......................................................................................70
Andrea MILELLI DESIGN, SYNTHESIS AND BIOLOGICAL ACTIVITY OF NEW CAPROCTAMINE-BASED

8
COMPOUNDS AS ANTI-ALZHEIMER DRUGS......................................................................................................72
Beata MORAK-M ODAWSKA SYNTHESIS AND PROPERTIES OF NOVEL 10-SUBSTITUTED 2,7-DIAZAPHENOTHIAZINES..............................................73
Erika MORIZZO G PROTEIN-COUPLED RECEPTORS AS POTENTIAL DRUG TARGET: FROM RECEPTOR TOPOLOGY TO RATIONAL DRUG DESIGN, AN IN SILICO APPROACH .........................................................75
Marina MUSCARELLA 7-AZAINDOLO-FUSED HETEROCYCLES WITH POTENTIAL ANTITUMOR ACTIVITY ...............................77
Carmela NAPOLITANO DESIGN AND SYNTHESIS OF NOVEL ADENOSINE NUCLEOTIDE ANALOGUES: THE INTRODUCTION OF DIVERSITY INTO THE CARBOHYDRATE OR THE BASE SUBUNITS OF MODIFIED NUCLEOTIDES AS PROMISING STRATEGIES TO IDENTIFY SPECIFIC P2 RECEPTOR LIGANDS..........................................................................................................................................79
Thi Hanh Thuy NGUYENINVESTIGATION OF ANTIPRION ACTIVITY OF ACRIDINE DERIVATIVES ..................................................80
Roberto NUTI STRUCTURAL AND CONFORMATIONAL ASPECTS AFFECTING THE MOLECULAR RECOGNITION OF SUBSTRATES AND INHIBITORS BY INDOLEAMINE-2,3-DIOXYGENASE (IDO), A NOVEL TARGET FOR CANCER THERAPY ...........................................................................................82
Dóra ONDRÉ SYNTHESIS OF NEW STEROIDAL 2-OXAZOLIDONES, AS NOVEL POTENTIAL INHIBITORS OF 17 -HYDROXYLASE-C17,20-LYASE..................................................................................................................84
Dmitry OSOLODKIN MOLECULAR DESIGN OF NEW SELECTIVE AND NON-SELECTIVE INHIBITORS OF GLYCOGEN SYNTHASE KINASE 3...............................................................................................................................................85
Rossana PASCALE SYNTHESIS AND BIOLOGICAL EVALUATION OF N-(ARYLOXYALKYL)PHTHALIMIDES AND ISOLOGUES AS NOVEL -GLUCOSIDASE INHIBITORS ....................................................................................87
Francesco PISCITELLI DEVELOPMENT OF NEW INDOLYL ARYL SULFONES (IASs) AS POTENT ANTI-HIV AGENTS ................89
Anita PLAZINSKA BINDING OF FENOTEROL DERIVATIVES AND STEREOISOMERS OF FENOTEROL TO THE
2 ADRENERGIC RECEPTOR. A MOLECULAR MODELING STUDY................................................................91
Giovanni PROTA NEW LIGANDS FOR ESTROGEN RECEPTOR β....................................................................................................92
Stefano RIZZO DESIGN AND SYNTHESIS OF MULTI-TARGET-DIRECTED COMPOUNDS FOR THE TREATMENT OF ALZHEIMER’S DISEASE ...........................................................................................................94
Alessia ROMUSSI SYNTHESIS AND ENANTIOMER SEPARATION OF NEW PDE4 INHIBITORS AS POTENTIAL DRUGS IN ALZHEIMER DISEASE ..........................................................................................................................96
Simone RONSISVALLE (+)-MR200 DERIVATIVES. MODIFICATIONS ON THE AMINO AND CARBOXYLATE MOIETIES IN VITRO AND IN VIVO PHARMACOLOGICAL EVALUATION......................................................98
Sabrina RUGGIERI DESIGN, SYNTHESIS AND PRELIMINARY PHARMACOLOGICAL EVALUATION OF NEW POTENTIAL TOOLS FOR THE TREATMENT OF CENTRAL NERVOUS SYSTEM DISEASES .....................100

9
Giulia SAPONARO N6-SUBSTITUTED NECA DERIVATIVES AS USEFUL TEMPLATES FOR THE DEVELOPMENT OF A2B ADENOSINE RECEPTOR AGONISTS ......................................................................................................102
Stefania SARTINI DESIGN, SYNTHESIS AND BIOPHARMACOLOGICAL EVALUATION OF NOVEL ALDOSE REDUCTASE INHIBITORS: THREE DIFFERENT SCAFFOLDS AT COMPARISON .......................................105
Raquel SEIXAS A NOVEL SYNTHETHIC APPROACH OF NEW BENZO[b]ACRIDONES WITH POTENTIAL ANTIOXIDANT AND ANTITUMOUR ACTIVITY................................................................................................107
Hong-May SIM 4, 6-DIMETHOXYAURONES AS DUAL MODULATORS OF P-GLYCOPROTEIN (P-gp) AND BREAST CANCER RESISTANCE PROTEIN (BCRP) ..........................................................................................................109
Virginia SPANÒPYRROLO-FUSED HETEROCYCLES AS PHOTOCHEMOTHERAPEUTIC AGENTS......................................111
Khac-Minh THAI HERG-FREE: A COMPUTATIONAL APPROACH................................................................................................113
Raffaella TRISOLINI SYNTHESIS AND BIOLOGICAL EVALUATION OF CHIRAL 2-PHENOXY-3-PHENYLPROPANOIC ACID DERIVATES WITH PPAR / DUAL ACTIVITY .......................................................................................115
Michael VANNI SYNTHESIS OF NEW P-GLYCOPROTEIN INHIBITORS AND THEIR SCREENING: POTENTIAL TOOLS TO REDUCE THE MULTIDRUG RESISTANCE......................................................................................117
Patrizia VITA SUGAR-MODIFIED NUCLEOSIDES AND NUCLEOTIDES: SYNTHESIS, CONFORMATIONAL ANALYSIS AND BIOLOGICAL EVALUATION...................................................................................................119
Elisa VITTORINO SYNTHESIS OF 4’-THIONUCLEOSIDES BY 1,3-DIPOLAR CYCLOADDITIONS AS POTENTIAL ANTIVIRAL AGENTS..............................................................................................................................................121
Olga YUZLENKO SEARCH FOR SELECTIVE ADENOSINE A1 AND A2A RECEPTOR LIGANDS: SYNTHESIS, PHARMACOLOGY, 3D-QSAR STUDIES AND MOLECULAR MODELLING...................................................123
Elena ZANGONI Tc-99m LABELLING APPROACHES OF NEW OCTREOTIDE ANALOGUES FREE FROM DISULPHUR BRIDGE WITH HIGH AFFINITY TOWARDS SSTR EXPRESSING TUMOURS.........................126
Laura ZAPPALÀ PROAPOPTOTIC AND CYTOTOXIC EFFECTS OF MRJF4 ON HUMAN PROSTATE CANCER CELL......................................................................................................................................... 128
Teresa Fabiola MISCIOSCIA A NEW AUTOMATED STRATEGY TO JOIN LIGAND- AND STRUCTURE-BASED DRUG DESIGN .........130
Miriam SGOBBA VALIDATION OF THE BINDING SITE LOCATED INTO THE C-TERMINAL DOMAIN OF HSP90: A NEW OPPORTUNITY TO DESIGN ANTICANCER DRUGS.............................................. 132


11
SYNTHESIS AND BIOLOGICAL EVALUATION OF NEW iNOS INHIBITORS
Jamila Isabella ALI
Dipartimento di Scienze del Farmaco, Università degli Studi G.d’Annunzio, Chieti Dottorato di ricerca in Scienze del farmaco – XXI Ciclo
Nitric Oxide (NO) regulates numerous physiological processes, including neurotransmission, smooth muscle contractility, platelet reactivity and cytotoxic activity of immune cells. NO is crucial for many physiological functions and inappropriate release of this mediator has been linked to a number of pathologies [1]. NO is formed endogenously by a family of enzymes known as NO synthases (NOS). NOS converts L-arginine and O2 to L-citrulline and NO with concomitant oxidation of NADPH. Three NOS isoforms has been identified, that differ in cellular distribution, regulation and activity. Endotelial NOS (eNOS) regulates vascular tone and smooth muscle tension. Neuronal NOS (nNOS) produced NO functions as a diffusible neurotransmitter, whereas NO generated by inducible NOS (iNOS) generates cytotoxins with both protective and pathologic effects [2]. There are a number of pathological processes associated with an overproduction or underproduction of NO. For example, nNOS is implicated in stroke and migraine and iNOS is implicated in septic shock, arthritis and multiple sclerosis. The possibility of treating these and other conditions by inhibiting NOS has elicited intense efforts to identify or design selective NOS inhibitors. Three classes of NOS inhibitors are known, and among these there are the ligand-based inhibitors [3]. They could have peptidic or not peptidic structures, correlated to the natural substrate L-arginine. In this work the synthesis either of amidinic or peptidic compounds as potential iNOS inhibitors is described [4]. In the first case we selected as lead compound the N-(3-(Aminomethyl)benzyl)acetamidine (1400W), that was reported to be a slow, tight-binding, and highly selective inhibitor of iNOS in vitro and in vivo. We have modified this compound in three positions:
- the 3-aminomethyl group was removed or bulky groups were bonded to nitrogen; - substituents were introduced on benzyl carbon connected to the acetamidine function; - the amidinic group was incorporated in a imidazolic ring.
NH2N
H
NH
In the second case thymopentin (TP-5) was the lead compound; it is a synthetic pentapeptide (Arg-Lys-Asp-Val-Tyr) corresponding to the active site of human hormone thymopoietin. In a pilot study was assessed the efficacy of TP-5 in Sèzary syndrome, a cutaneos T-cell lymphoma marked by erythroderma, circulating atypical lymphoid cells, and extensive lymph node and visceral involvement [5]. Since the presence of the amino acid L-arginine in this pentapeptide, the involvement of NOS in this pathology has been thought. On this basis, small peptides structurally related to TP-5 were synthesized and tested as iNOS inhibitors. This work was performed in collaboration with the Department of Scienza e Tecnologia del Farmaco of the Turin University. The results of preliminary studies showed that all new compounds are able to inhibit iNOS and the majority of them are also selective for this isoform.
[1] Vallance P., Leiper J., Nat. Rev. Drug Disc., 2002, 1, 939.[2] Ignarro L.J., Cirino G., Casini A., Napoli C., J. Cardiovasc. Pharmacol., 1999, 34, 876.[3] Li H., Raman C.S., Martàsek P. Masters B.S.S., Poulos T.L., Biochem., 2001,40, 5399 [4] Ali I.J. et al., Atti del XXII Congresso Nazionale della Società Chimica Italiana, 2006, FAR-P-063 [5] Bernengo M.G., Appino A., Bertero M., Novelli M., Fierro M.T., Doveil G.C., Lisa F., Journal of National Cancer Institute, 1992, 17, 1341.
Deletion or substitution on NSubstitution
cyclization

12
DESIGN, SYNTHESIS AND PHARMACOLOGICAL RESULTS OF
NEW POTENTIAL LARGE-CONDUCTANCE CALCIUM–ACTIVATED
POTASSIUM-CHANNEL (BKCa) OPENERS.
Gabriella AMATO
Dip. Scienze Farmaceutiche, Università di Pisa Dottorato di Ricerca in “Scienza del Farmaco e delle Sostanze Bioattive”- XXI ciclo
Maxi-K+ channels, found in both excitable and non-excitable cells, are proteins which selectively allow K+ ion flux across the cell membrane. Once opened, a potent feed back control of the vascular smooth muscle tone is mediated by these channels, whose activation can be promoted by both a rise of the intracellular free calcium concentration as well as membrane depolarization. So, BK-openers are expected to have application for the therapy of cardiovascular diseases associated with diabetes and hypercholesterolemia, coronary disease and hypertension. But also they are crucial in modulating the bronco-tracheal, urethral, uterine tone or gastro-intestinal musculature. Only in the last decade, BK channels have been viewed as an explicit target of selective drugs. The first BK-activators resulted from the development of a series of arylimidazolones NS004 and NS1619 (A). The presence of the benzimidazolone nucleus does not appear as an obligatory structural requirement of a BK-activator but, up to now, these two compounds represent the reference models, which led to the design of several chemically heterogeneous BK-openers. In recent years our research program has concerned synthesis and pharmacological evaluation of new compounds as BK-openers. In particular 1,2,3-triazole derivatives (B) and substituted benzanilides and benzylanilides [1,2] (D), were tested and high pharmacological activity was discovered, in some cases higher than NS1619 (the derivatives C,E are the best compounds). On the basis of these results and the suggestions reported in the literature a pharmacophoric model was hypothesized, consisting in two suitable substituted phenyl rings bond to a linker of varied nature (F). The aim of this work was to investigate structural modifications of the best compounds (C,E).
R= CF3 NS1619 R= Cl NS004
More recently, for some BK-openers, the hypothesis of symmetrical pharmacophore emerged [3]. With reference to the symmetrical structures of the literature and to the large series of benzanilides (D) [1], which had shown interesting pharmacological results, I synthesized many substituted symmetrical and asymmetrical diarylureas (G), corresponding to the formal opening of the benzimidazolone heterocycle. The symmetrical compounds gave interesting pharmacological results. Moreover, I decided to transform the linker chain into a cyclic one to obtain substituted 2-arylamino-4H-3,1-benzoxazin-4-ones (H) and 3-substituted-2,4(1H,3H)-quinolindiones (I).
N
HN
O
OH
R
F3C
A
X
NN
N
Y
R1
R2 R3
R4
B
LINKER
GWE OH
R1R2F
N
HN
O
OH
R
F3C
A
XHN
X
O
OCH3
R1
R2
D
HN
O
NHR1
R2
G
N
O
O
NH
R1R2
H
NH
N
O
O
R1
R2
I
N
NN
HO
C
NH
O
OH
Cl
Cl
R2H3CO
E
XHN
X
O
OCH3
R1
R2
D

13
Then I considered the idea of simplifying the linear benzanilidic linker (E) to make it less flexible and stiff. The Base Shiff’s derivatives (L) presented an imino group between the two aromatic rings. The compounds (L) were reduced to give the corresponding products (M) with a more flexible spacer unit. These were, ultimately, subjected to an internal Mannich reaction to give the new molecules (N), with a cyclic linker fused with one of the aromatic rings, similar to the bezimidazolones (A).
Up to now, the highest vasorelaxing activity of 1-(2-hydroxybenzyl)-4-benzyl-1.2.3-triazole (C) on BK channels attested that it is the best compound synthesized in our lab. The evidence led us to investigate structural modifications to this compound. So, I proposed to change the heterocyclic 1,2,3-triazole linker with a 1,3,4-oxadiazole one. In addition, I decided to amplify the linker spacer. In fact the compounds (O) presented a methylene bridge too, while in the derivatives (P) two different bridges were introduced: on one side of the 1,3,4-oxadiazole ring a methylene group and on the other side an NH function, as a possible H-bond donor, which is important in channel interaction.
Finally, in a previous work, starting from the substituted benzanilides (D) and from the 1,2,3-triazoles (B), I decided to prepare new derivatives with a double spacer (Q,R). Unfortunately, the pharmacological results suggested that these modifications were ineffective. Placing our hope on increasing pharmacological results of these structures, the amidic spacer was translated from 4-position to 5-position and also a substituted aromatic ring was introduced in 4-position to increase the lipophilicity of the compounds (S,T). Thanks to these structural modifications we could evaluate the possible interaction between the aromatic rings and a hypothetical lipophic pocket of the potassium channels. Furthermore, some molecules (T) presented a methylene bridge that contributes to giving more flexibility to the structure.
XHN
X
O
OCH3
R1
R2
D
N
R1 R2
L
CH2
NH
R1 R2
MO
N
R1
R2
N
N
NN
HO
C
N
O
N
R1
R2
O
R1
O
N
N
HNR2
P
XHN
X
O
OCH3
R1
R2
D
X
NN
N
Y
R1
R2 R3
R4
B
N
NN
HO
C
R1N
NNR2
NH2
S
NN
N
H3C
HN
OX
R1
n
R2
Q
R1
N
NN
CH3
NH
O
NH R2
R
R1
NN
N
HN
R3
R2
O
T
[1] Calderone V., et al., IX. Eur. J. Med. Chem., 41,761-7, (2006). [2] Calderone V., et al., Eur. J. Med. Chem., 41,761-7, (2006). [3] Toshima T., Bioorganic & Med. Chem., 14, 8014-31, (2006).

14
NOVEL SELECTIVE INHIBITORS OF N-ACYLETHANOLAMINE-HYDROLYZING ACID
AMIDASE (NAAA) AS POTENTIAL ANTI-INFLAMMATORY AGENTS
Francesca ANTONIETTI
Istituto di Chimica farmaceutica, Facoltà di Farmacia, Università degli Studi di Urbino “Carlo Bo” Dottorato di Ricerca in Scienze Chimiche e Scienze Farmaceutiche (XXI ciclo)
N-Acylethanolamines (NAEs) are ethanolamides of long-chain fatty acids which represent a class of signalling lipids widely spread in the animal tissues. Among the polyunsaturated NAEs, N-arachidonoylethanolamide (anandamide, AEA, Figure 1) is the best known endogenous ligand of cannabinoid receptors (CB).1 The monounsaturated and saturated NAEs, although apparently inactive at CB receptors, show a variety of biological effects. N-oleoylethanolamine (OEA, Figure 1) is involved in feeding and body weight regulation2, whilst N-palmitoylethanolamine (PEA, Figure 1) has several pharmacological effects such as anti-inflammation,3 analgesia,4 anti-epilepsy and neuroprotection. A recent study showed that when applied as a drug, PEA can activate the nuclear receptor PPAR-α(Peroxisome Proliferator Activated Receptor-α) and such activation underlies its ability to inhibit the inflammatory response.3 However, the signalling functions of endogenous PEA are still under debate and the demonstration that PEA acts as an endogenous activator of PPAR-α requires further experimentation. The levels of PEA in the tissues depend on its biosynthesis and degradation. PEA is derived from membrane phospholipids by two enzymatic reactions, one of which involves cleavage of N-palmitoyl phosphatidilethanolamine (NPPE), catalyzed by a NAPE-specific phospholipase D (NAPE-PLD). In terms of hydrolysis, fatty acid amide hydrolase (FAAH)5 degrades PEA, along with other NAEs, into palmitic acid and ethanolamine. Recently, another amidase, called N-acylethanolamine-hydrolyzing acid amidase (NAAA), which preferentially hydrolyzes PEA, was molecularly cloned.6
OHNH
O
NH
O
OHNH
O
OH
PEA OEA AEA
Figure 1. Structures of endogenous bioactive N-acylethanolamines (NAEs)
In this study, we asked ourselves whether pharmacological inhibition of PEA hydrolysis by FAAH or NAAA could lead to an anti-inflammatory effect through increased signalling at PPAR-mediated by an elevation of endogenous levels of PEA. To investigate the role of PEA in inflammation, in vivo and in vitro inflammation models were used; data showed that endogenous PEA levels result in a decrease in inflammation. Administration of the selective FAAH inhibitor URB597,7 that did not inhibit NAAA activity at any concentration tested, was not able to restore PEA levels in inflammatory cells suggesting that NAAA rather than FAAH might be involved in mediating PEA degradation in these models. NAAA reveals no sequence homology with FAAH and belongs to the choloylglycine hydrolase family, which is included in the N-therminal nucleophile amino hydrolase superfamily. These are specialized in the cleavage of linear amide and have a cysteine, serine or threonine at the first position of their aminoacidic sequence, acting as the nucleophilic agent responsible for the catalytic attack.
Potent and selective NAAA inhibitors are not currently available. To discover such inhibitors we assumed that cysteine 131 is essential to the catalytic activity of NAAA, as suggested by Tsuboi et al.6
Screening a set of commercially available compounds known to target cysteine residues in proteins led to the identification of SD-41, that inhibited rat recombinant NAAA activity with an IC50 of 3.0±0.3 µM and native rat lung NAAA activity with an IC50 of 6.6±1.8 µM. The requirements for NAAA inhibition by analogues of SD-41 were investigated by a small series of compounds. An open analogue of SD-41 showed no activity suggesting a critical role of an intact β-lactone in NAAA inhibition. Indeed, the replacement of the carbamic fragment of SD-41 with an amide led to URB783, having an IC50 of 0.42±0.02 µM. Interestingly, its (R) enantiomer was significantly less effective (IC50 = 6.00±0.60 µM), suggesting that NAAA inhibition requires a specific recognition process, which may favour a covalent reaction between the active Cys131 and the lactone ring. This hypothesis is also supported by the lack of inhibition observed for compounds where the β-lactone ring was replaced with a cyclobutanone, a cyclobutane or a γ-lactone. On the other hand, racemic analogue of URB783, in which the carbamoyl group has been replaced by a dimethylene fragment retained some inhibitory activity (IC50 = 11.00±2.20

15
µM), even if its reduced potency, compared to the amide analogues, indicates a role of the carbamoyl fragment in the recognition step.
NAAA inhibition by URB783 is consistent with a non-competitive, partially reversible, and time-independent mechanism, which may be related to covalent binding between the inhibitor and the enzyme. In principle, the lactone ring of URB783 can react with the catalytic cysteine by a nucleophilic attack either at its carbonyl group, leading to enzyme acylation, or at its β-carbon, giving enzyme alkylation. The partial recovery of enzyme activity, upon extensive dialysis in the presence of dithiothreitol, is more consistent with the first pathway, suggesting the formation of a reversible thioester adduct. In order to confirm the correct mechanism of NAAA inhibition by β-lactones compounds, further experiments are needed.
URB783 resulted the most potent compound in this first series. It also showed to be selective for NAAA over cannabinoid/NAE (no inhibitory effect on FAAH, NAPE-PLD, monoacylglycerol lipase) or inflammation related enzymes (phospholipase A2, COX-2). Administration of URB783 restored cellular PEA levels in a dose-dependent manner both in vivo and in vitro. Furthermore, the normalization of PEA levels by URB783 decreased the inflammatory cells number in wild-type mice but not in mice lacking PPAR-α. Therefore, the novel NAAA inhibitor URB783 exerts anti-inflammatory effects by restoring cellular PEA which leads to an increase in the activity of the ligand-activated receptor PPAR-α.
In conclusion, our results show that an increase in PEA by blockade of its endogenous degradation leads to activation of PPAR-α which results in anti-inflammatory effect. The NAAA inhibitors developed allowed us to establish a role for endogenous PEA in the regulation of inflammation and represent a class of anti-inflammatory agents acting with an unprecedented mechanism.
References
(1) Devane, W. A.; Hanuš, L.; Breuer, A.; Pertwee, R. G.; Stevenson, L. A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and Structure of a Brain Constituent That Binds to the Cannabinoid Receptor. Science 1992, 258, 1946-1949.
(2) Fu, J.; Gaetani, S.; Oveisi, F.; LoVerme, J.; Serrano, A.; Rodríguez De Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; Piomelli, D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 2003, 425, 90-93.
(3) Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The Nuclear Receptor Peroxisome Proliferator-Activated Receptor-α Mediates the Anti-Inflammatory Actions of Palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15-19.
(4) Calignano, A.; La Rana, G.; Giuffrida, A.; Piomelli, D. Control of pain initiation by endogenous cannabinoids, Nature 1998, 394, 277-281.
(5) Cravatt, B. F.; Giang, D. K.; Mayfield, S. P.; Boger, D. L.; Lerner, R. A.; Gilula, N. B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384,83-87.
(6) Tsuboi, K.; Sun Y.-X.; Okamoto, Y.; Araki, N.; Tonai, T.; Ueda N. Molecular Characterization of N-Acylethanolamine-hydrolyzing Acid Amidase, a Novel Member of the Choloylglycine Hydrolase Family with Structural and Functional Similarity to Acid Ceramidase. J. Biol. Chem. 2005, 280,11082-11092.
(7) Kathuria, S.; Gaetani, S.; Fegley, D.; Valiño, F.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; La Rana, G.; Calignano, A.; Giustino, A.; Tattoli, M.; Palmery, M.; Cuomo, V.; Piomelli, D. Modulation of Anxiety Through Blockade of Anandamide Hydrolysis. Nat. Med. 2003, 9, 76-81.

16
COMPUTATIONAL APPROACHES TO THE RATIONAL DESIGN OF NOVEL
TOPOISOMERASE I POISONS AS POTENTIAL ANTICANCER DRUGS.
Serena BASILI
Università degli Studi di Padova – Dipartimento di Scienze Farmaceutiche Scuola di Dottorato in Scienze Molecolari – Scienze Farmaceutiche
Topoisomerase I (Top1) is an essential DNA –targeting enzyme which alters the supercoiling of DNA through a concerted process of breaking and rejoining of a DNA strand, thereby controlling the DNA topology required for replication and transcription1. Top1 mediates relaxation of supercoiled DNA by creating a transient single-strand break in the DNA duplex that originates from a transesterification reaction involving a nucleophilic attack by the active-site tyrosine (Tyr723) hydroxyl group on a DNA phosphodiester bond situated at the site of cleavage. The resulting 3’-phosphotyrosine enzyme-DNA complex (“covalent binary complex”) is then reversed, after DNA relaxation through strand passage, when the released 5’-OH of the nicked strand reattacks the phosphotyrosine intermediate in a second transesterification reaction. These events result in the relaxation of the DNA structure, which is required during transcription or replication. Top1 is a specific target for the pentacyclic alkaloid Camptothecin (see figure below) and its derivatives, known as Top1 poisons.
These molecules block DNA religation, thus converting Top1 into a DNA-damaging agent. In the presence of a Top1 poison a ternary complex between DNA, an intercalator and topoisomerase is formed. Such a ternary complex is more stable than the DNA-Top1 associate, which may lead to an enhanced lifetime of the initially cleaved DNA. As a consequence, the religation of the strands cannot take place, i.e. the strand breaks persist, so that the topoisomerase acts as an endogenous poison under these circumstances. Therefore, intercalators which form such stabilized ternary complexes with DNA and Top1 exhibit a high potential as DNA-targeting anticancer drugs. Camptothecin was early shown to be clinically problematic because, in addition to its negligible water solubility, its active “ring-closed” -hydroxylactone form is rapidly converted under physiological conditions to the “open” carboxylate form, which is inactive and readily binds to human serum albumin, making it inaccessible for cellular uptake2. To date, only two semisynthetic analogs of Camptothecin (Topotecan and Irinotecan) have been approved by FDA for the clinical treatment of the ovarian, small cell-lung and colorectal cancers. Solving crystal structures3 of Top1 in complex both with Camptothecin and Topotecan and with structurally different molecules (indolocarbazoles and indoloquinolines) has significantly increased the amount of structural information about the interaction between the Top1-DNA binary complex and the poison molecule. This encouraged the application of structure-based drug design to investigate and rationalize the activity of Top1 poisons and to rationally design new potential anticancer drugs. In order to describe the binding mode of different classes of poisons to Top1-DNA binary complex we used a classical molecular docking approach through molecular docking programs, including GLIDE, GOLD, MOE-Dock and FlexX. All of these software employ an approximate physical chemistry-based representation of protein-ligand interactions, obtaining charges from a molecular mechanics force-field. However, improving accuracy in docking can be attempted via the use of mixed quantum mechanical/molecular mechanics (QM/MM) methods to compute the ligand charge distribution. For this purpose, we used the quantum mechanics (QM)-polarized ligand docking (QPLD) protocol implemented

17
in the Schrodinger software suite4. The QPLD algorithm begins with a docking job that generates several geometrically unique protein-ligand complexes. A single-point energy calculation is then performed on each complex, treating the ligand with ab initio methods and deriving partial atomic charges using electrostatic potential fitting. The ligand is finally re-docked using each of the ligand charge sets calculated, and the QPLD algorithm returns the most energetically favorable pose. Here, we present the application of the QM-polarized ligand docking protocol to investigate the binding mode of a class of new 5-substituted Campthotecin derivatives.
BIBLIOGRAPHY:
1 Pommier Y., Nat. Rev. Canc. 2006, 6:789-801. 2 Esther, M., Laine, W., Tardy, C., Lansiaux, A., Iwao, M., Ishibashi, F., Bailly, C. and Gago, F.. J. Med. Chem. 2005, 48:3796-3808. 3 Staker, B.L., Feese, M.D., Cushman, M., Pommier, Y., Zembower, D., Stewart, L., Burgin, A.B. J. Med. Chem. 2005, 48:2336-2345. Staker, B.L., Hjerrild, K., Feese, M.D., Behnke, C:A:, Burgin, A:B: Jr, Stewart, L., PNAS 2002, 99:15387-15392. Ioanoviciu, A., Antony, S., Pommier, Y., Staker, B.L., Stewart, L., Cushman, M., J. Med. Chem 2005, 48:4803-4814. 4Schrdinger Suite 2007 QM-Polarized Ligand Docking protocol; Glide version 4.5, Schrdinger, LLC, New York, NY, 2005; Jaguar version 7.0, Schrdinger, LLC, New York, NY, 2005; QSite version 4.5, Schrdinger, LLC, New York, NY, 2005.

18
ANTICANCER, ANTI-INFLAMMATORY AND ANTIOXIDANT ACTIVITIES OF NEW
QUINOXALINE AND QUINOXALINE DI-N-OXIDE DERIVATIVES
Asunción BURGUETEa, Eleni PONTIKIb, Dimitra HADJIPAVLOU-LITINAb, Raquel VILLARa, Esther
VICENTEa, Beatriz SOLANOa, Saioa ANCIZUa, Mauricio CABRERAc, Laura CORCUERAc, Adela
LÓPEZ DE CERAINC, Silvia PÉREZ-SILANESA, Ignacio ALDANAa and Antonio MONGEa
aUnidad en Investigación y Desarrollo de Medicamentos, Centro de Investigación en Farmacobiología
Aplicada (CIFA), University of Navarra, C/ Irunlarrea s/n, 31080 Pamplona, Spain; bDepartment of
Pharmaceutical Chemistry, School of Pharmacy, Aristotle University of Thessaloniki, Thessaloniki
54124, Greece; cDepartamento de Toxicología, Centro de Investigación en Farmacobiología Aplicada
(CIFA), University of Navarra, C/ Irunlarrea s/n, 31080 Pamplona, Spain;
Quinoxaline derivatives display a broad spectrum of biological properties (antifungal, anticancer, antibacterial, antihelmintic, and antiviral). Oxidation of both nitrogens of the quinoxaline ring greatly increases some of these activities, such as hypoxia-selective anticancer activity1.
In our continuing efforts to identify new anticancer agents which can improve the current chemotherapeutic treatments, new series of quinoxaline di-N-oxide derivatives and some of their reduced analogues have been synthesized2 and tested for their in vitro anticancer activity. These compounds have also been tested in order to study their antioxidant activities, their role in inflammation, and their inhibition on lipoxygenase (LOX) since LOX inhibitors are able to induce the anti-carcinogenic enzymes and/or inhibit the pro-carcinogenic enzymes responsible for polyunsaturated fatty acid metabolism.
N+
N+
R7
R6
O
O O
R´
N
NR7
R6
O
R´
N+
N+
R7
R6
O
O NH
NR´
The tested compounds exhibit important scavenging activites. Two of them present higher in vivoanti-inflammatory activity than the reference drug, indomethacin. Furthermore, one of these compounds shows potent in vitro inhibition of LOX (IC50<1µM)2.
The in vitro anticancer activity of the compounds was evaluated against three cancer cell lines. Surprisingly, two of the reduced analogues are the most cytotoxic compounds in the colon cancer cell line HT-29 (IC50=19µM-24µM).
Acknowledgements: This work has been carried out thanks to the financial support of the FIS project (1051005, October 2005). A. Burguete. was awarded a PhD scholarship supported by the “Gobierno de Navarra”.
References1 Ganley, B.; Chowdhury, G.; Bhansali, J.; Daniels, J. S.; Gates, K. S. Bioorg. Med. Chem. 2001, 9, 2395-2401.2 Burguete, A.; Pontiki, E.; Hadjipavlou-Litina, D.; Villar, R.; Vicente, E.; Solano, B.; Ancizu, S.; Pérez-Silanes, S.; Aldana, I.; Monge, A. Bioorg. Med. Chem. Lett. 2007, 17, 6439–6443

19
6,7-DIMETHOXYTETRAHYDROISOQUINOLINE DERIVATIVES:POTENT P-GLYCOPROTEIN LIGANDS REVERSING MULTIDRUG RESISTANCE
Mariangela CANTORE
Dipartimento Farmacochimico, Universitá degli Studi di Bari, via Orabona, 4, 70125, Bari, Italy Dottorato in Scienze Farmaceutiche XXI ciclo
Multi Drug Resistance (MDR) is the major cause that limits the efficacy of chemotherapeutic treatment. Some tumours are intrinsically resistant to pharmacological therapy, while others, initially sensitive to chemotherapy, become resistant during the treatment. The most supported mechanism involved in MDR is the overexpression of several ATP-dependent efflux pumps, known as ATP Binding Cassette (ABC) transporters, in tumour cells.1 These transporters, localized in the cell membrane, bind ATP and employ its hydrolysis energy to extrude various molecules out of the cell and, in particular, chemotherapeutic drugs from tumour cells. The most involved ABC transporter in MDR is P-glycoprotein (P-gp, ABCB1subfamily).2 This pump, localized in several biological compartments, modulates the efflux of many structurally different drugs, regulates their intestinal absorption and their availability in Central Nervous System. At the present, the co-administration of an efflux P-gp inhibitor to chemotherapeutic agents represents the most probed strategy for reversing MDR. In the last years many P-gp modulators have been developed and, among them, last generation inhibitors such as Zosuquidar3, Elacridar4 and Tariquidar5 showed high potency for blocking P-gp. Although these compounds are evaluated in different phases of clinical trials, preliminary results are moderately satisfactory because they displays poor selectivity towards other ABC transporters involved in MDR such as BCRP and MRP-1. On the other hand, these compounds are complex molecules, and therefore Structure-Activity Relationship (SAR) studies are not so easy to carry out, and the role of each molecular portion is not still clarified. Therefore, to design small P-gp modulators and to develop corresponding SAR studies, represent the target of this research work. Our investigation started from PB28,6 a cyclohexylpiperazine sigma-2 receptor agonist, displaying a good P-gp modulating activity (EC50 = 0.55 µM) (Fig. 1).
Fig. 1 Advances in the design of potent P-gp inhibitors starting from PB28.
N
N
O
N
O
OX
R
N
O
OX
R
N
O
OX
N
O
OX
biphenyl series 2-naphthalenyl series
N
O
OO
PB28
1
X=CO,CH2
R = H, OH, OCH3
In order to improve P-gp modulating activity, the basic nucleus of PB28 has been replaced obtaining a set of compounds characterized by 6,7-dimethoxytetrahydroisoquinoline moiety, the same basic nucleus of Tariquidar and Elacridar, potent P-gp inhibitors. This modification led to identify compound 1 (Fig. 2), bearing a (E) double bond on the spacer and 5-methoxy substituent on tetraline ring. This compound displays similar potent P-gp inhibition activity of Elacridar (EC50 = 1.64 µM and 2.0 µM, respectively).

20
SAR studies demonstrated that the shifting or the absence of methoxy substituent, the shifting or the hydrogenation of double bond decreased P-gp inhibiting activity with respect to lead compound 1. Other details will be displayed in Poster section. This finding suggested that the conformational restriction of no basic moiety in this series could be a requirement for improving P-gp inhibition activity. Consequently, two different fragments have been employed: biphenyl and 2-naphthyl moieties.8,9 Several aspects have been investigated: a) the importance of the biphenyl linkage position (from 2 to 4 position); b) the influence of hydrogen bond donor or acceptor substituents (OH, OCH3); c) the influence of the basicity in each series, comparing the amines to the amides. Among tested ligands, the best result has been obtained for compound 2 (Fig. 2) displaying P-gp inhibitory activity in nanomolar range (EC50 = 0.050 µM).Compounds 1 and 2, were tested for determining the selectivity towards BCRP pump, another transporter involved in MDR. The results displayed that compound 1 inhibited BCRP pump in dose-dependent manner with a maximal effect at 100 µΜ. By contrast, compound 2 was unable to interact with BCRP pump (19% of effect at 100 µΜ). These preliminary results indicate that the fragment linked to 6,7-dimethoxytetrahydroisoquinoline nucleus could be considered a discriminative requirement for obtaining compounds with high P-gp inhibiting activity and, in the meantime, with good selectivity towards BCRP pump.
Fig. 2 The best P-gp inhibiting derivatives.
N
OH
O
ONO
O
O
compound 1
EC50 = 1.64 µµµµM
compound 2
EC50 = 0.050 µµµµM
References:
1) Altemberg, G. A. Curr. Med. Chem. 2004, 4, 53-62. 2) Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. J. Natl. Cancer Inst. 2000, 92, 1295-1302. 3) Morschhauser, F.; Zinzani, P. L.; Burgess, M.; Sloots, L.; Bouafia, F.; Dumontet, C. Leuk. Lymphoma2007, 48, 708-715. 4) Planting, A. S.; Sonneveld, P.; van der Gaast, A.; Sparreboom, A.; van der Burg, M. E.; Luyten, G. P.; de Leeuw, K.; de Boer-Dennert, M.; Wissel, P. S.; Jewell, R. C.; Paul, E. M.; Purvis, N. B.; Verweij, J. Cancer Chemother. Pharmacol. 2005, 55, 91-99. 5) Pusztai, L.; Wagner, P.; Ibrahim, N.; Rivera, E.; Theriault, R.; Booser, D.; Symmans, F. W.; Wong, F.; Blumenschein, G.; Fleming, D. R.; Rouzier, R.; Boniface, G.; Hortobagyi, G. N. Cancer 2005, 104, 682-691.6) Azzariti, A.; Colabufo, N. A.; Berardi, F.; Porcelli, L.; Niso, M.; Simone, M. G.; Perrone, R.; Paradiso, A. Mol. Cancer Ther. 2006, 5, 1807-1816. 7) Colabufo, N. A.; Berardi, F.; Cantore, M.; Perrone, M. G.; Contino, M.; Inglese, C.; Niso, M.; Perrone, R.; Azzariti, A.; Simone, G. M.; Porcelli, L.;Paradiso, A. Bioorg. Med. Chem. 2008, 16, 362-373.8) Colabufo, N. A.; Berardi, F.; Cantore, M.; Perrone, M. G.; Contino, M.; Inglese, C.; Niso, M.; Perrone, R.; Azzariti, A.; Simone, G. M.; Paradiso, A. Bioorg. Med. Chem. 2008, in press. 9) Multidrug Resistance: Biological and Pharmaceutical Advances in Antitumour Treatment. Review
Book ISBN 978-81-308-0258-9, 2008. Editor Colabufo Nicola Antonio.

21
COUPLING ARTEMISININ TO A VINYL SULFONE SCAFFOLD TO IMPROVE
ANTIMALARIAL ACTIVITY
Rita CAPELAa, Rudi OLIVEIRAa, Rui MOREIRAa, Philip ROSENTHALb, Jiri GUTb,Francisca LOPESa
a Medicinal Chemistry Group, iMed.UL, Faculty of Pharmacy, University of Lisbon, Portugal b Dep. Medicine, S. Francisco General Hospital, University of California, S. Francisco, USA
Malaria continues to be a potentially fatal threat to almost half of the world’s population and kills between 1 to 3 million people annually, mostly children under the age of five and pregnant women. Four Plasmodium species infect humans: P. falciparum, P. vivax, P. malariae and P. ovale. Almost all severe and fatal cases are caused by P. falciparum. The incidence of this disease is dramatically increasing because many P. falciparum strains became resistant to chloroquine and mefloquine, two drugs widely used in the antimalarial therapy. [1] World Health Organization (WHO) predicts that because of drug resistance and in the absence of new antimalarial strategies the population suffering from malaria will double by the year 2010. [2]
In the 60’s, the Chinese government launched a program to find new antimalarial drugs. Based in this search, in 1972, artemisinin, 1, was isolated from Artemisia annua, an herb used in cure for fever, nearly 2000 years ago. [3] The pharmacophore of artemisinin is the 1,2,4-trioxane scaffold. Despite the growing importance of artemisinin and derivatives, the exact mechanism of action is still unresolved and remains a matter of intense debate, but it appears that the formation of different C-centered radicals mediated by iron (II) is a key step. [4, 5] However, the therapeutic value of artemisinin is limited by its low solubility in both oil and water. In the search of more effective analogues for oral administration, a series of semisynthetic first-generation analogues such as artemether, 2, and arteether, 3, were prepared from dihydroartemisinin, 4. The need of more soluble drugs for i.v. administration, for the treatment of advanced cases of P. falciparum malaria, yield sodium artesunate, 5, and sodium artelinate, 6.Artemisinin and its derivatives are the most potent antimalarial drugs with no clinical resistance described. All this first-generation analogues shared poor bioavailability and pharmacokinetics, besides the fact that they have some neurotoxicity demonstrated in animal models. Much work has been invested in the development of the so-called second-generation artemisinins. [6]
O
O
O
O
CH3
CH3
CH3
O
H
O
O
O
O
CH3
CH3
CH3
OR
1 2, R= CH3; 3, R= C2H5; 4, R = H (α and β at C-11)
5, R= C(O)CH2CH2CO2Na; 6, R= CH2C6H4-4-CO2Na
There has been an increase interest for combination chemotherapy as a rational strategy to combat malaria.[7] It is anticipated that, in addition to a synergistic effect, combination therapy will delay the development of drug resistance in P. falciparum. WHO now recommends Artemisinin Combination Therapy (ACT) as part of the strategy for malaria control, as artemisinin shows fast antiparasitic action. [8,9] Very recently, an extension of this approach resulted in the synthesis of new potent antimalarial agents that contain two antimalarial pharmacophores, such as a 1,2,4-trioxane and aminoquinoline [10] or an aliphatic diamine (e.g. 7).[11] These can be appropriately called double drugs, because they combine two pharmacophores in a single molecule with the goal of creating a new chemical entity more effective than its individual components. In such double drugs, each pharmacophore should have independent mode of action to make the emergence of drug resistance less likely.

22
O
O
O
O
CH3
CH3
CH3
O
NN
R
7
Erythrocytic malaria parasites degrade haemoglobin to obtain aminoacids for protein synthesis. Falcipain-2 (FP-2) is a cysteine protease from the papain family that participates in the process of hemoglobin degradation, being a validated therapeutic target. Importantly, incubation of erythrocytic parasites with inhibitors of FP-2 blocks haemoglobin degradation and parasite development. Among the most potent FP-2 inhibitors are Michael acceptors. Vinyl sulfones (VS) and their analogues, such as sulfonamides and sulfonates, have been reported as a promising class of inhibitors for parasitic cysteine proteases. They act by irreversibly alkylating the active site cysteine residue via conjugate addition [12,13], and are now included in MMV’s portfolio [14].
This PhD project involves the synthesis of double drugs containing the artemisinin pharmacophore linked to a peptidyl vinylsulfone scaffold, 8. With the aim of designing effective inhibitors for FP-2, the chosen peptide sequence includes Gly-Phe and Phe-Phe, by analogy with some vinyl sulfones that were considered optimal for molecular recognition by this enzyme as well as by cruzain, a closely related cysteine protease from Trypanosoma cruzi. [12] The characterization of compounds 8 was made by usual techniques including COSY, HMQC and HMBC NMR. Preliminary results show that some compounds 8are active against P. falciparum W2 strain (chloroquine-resistant strain) at nM level, while inhibiting FP-2 in the µM range. The implications of these results for future lead optimization will be discussed.
C
O
O
O
O
CH3
CH3
CH3
O
H
O
NH
NH
O
SR
OOR1
3
R2
8
References
[1] Schlitzer, M. Arch. Pharm. Chem. Life. Sci. 2008, 341, 149-163. [2] O´Neill, P. M.; Posner, G. H. J. Med. Chem. 2004, 47, 12, 2945-2964. [3] Butler, A. R.; Wu, Y. Chem. Soc. Rev. 1992, 21, 85-90.[4] Tang, Y.; Dong, Y.; Vennerstrom, J. L. Med. Res. Rev. 2004, 24, 4, 425-448. [5] Krishna, S.; Uhlemann, A.; Haynes, R. K. Drug Resist. Updat. 2004, 7, 233-244. [6] Schlitzer, M. ChemMedChem. 2007, 2, 944-986. [7] Guerin, P. J.; Olliaro, P.; Nosten, F.; Druilhe, P.; Laxminarayan, R.; Binka, F.; Kilama, W. L.; Ford, N.; White, N. J. Lancet Infect. Dis. 2002, 2, 564 – 573. [8] WHO News Release 19-01-2006 [9] Nosten, F. ; Brasseur, P. Drugs, 2002, 62, 1315-1329. [10] Robert, A. ; Dechy-Cabaret, O. ; Cazelles, J. ; Meunier, B. Acc. Chem. Res. 2002, 35, 167 – 174 [11] Hindley, S.; Ward, S. A.; Storr, R. C.; Searle, N. L.; Bray, P. G.; Park, B. K.; Davies, J.; O’Neill, P. M. J. Med. Chem. 2002, 45, 1052 – 1063. [12] Powers, J. C.; Asgian, J. L.; Özlem, D. E.; James, K. E. Chem. Rev. 2002, 102, 4639-4750. [13] Lecaille, F.; Kaleta, J.; Brömme, D. Chem. Rev. 2002, 102, 4459-4488. [14] http://www.mmv.org/rubrique.php3?id_rubrique=38

23
APPLICATIONS OF 3-D QSAR METHODS TO DIFFERENT CLASS OF DRUGS
Antonia CAROLI
Dipartimento di Studi Farmaceutici, Università degli Studi di Roma “La Sapienza”. Dottorato di Ricerca in Scienze Farmaceutiche XXI Ciclo.
One important aim of drug design is to correlate the three-dimensional structure of drug molecules with their biological activities (IC50 or EC50), such a goal can be achieved with the construction of three-dimensional quantitative structure-activity relationships models (3-D QSAR). Using these models one should be able to design and predict the IC50 of new molecules to prioritize the synthesis of those predicted more active. The procedure entails the superimposition of a set of compounds whose activities have been measured, the computation of interaction energy fields for probes on a grid around each compound and partial least squares (PLS) statistical analyses to correlate fields with activity and to detect regions around the molecules where there are interactions that have an important impact on activity1. In 3-D QSAR methodologies the compounds are described by a large number of isolated grid-field variables. These grid-field values may represent total interaction energies, steric and electrostatic interactions, molecular electrostatic potential, hydrophobic interactions or a mixture of some of them represented graphically around molecules to help the interpretation of data results2. In the present work we developed 3-D QSAR studies using different approaches on five class of drugs which targets are listed below:
1. HDAC (Histone Deacetylase). 2. PRMT (Protein Arginine Methyltransferases). 3. Aromatase. 4. IFN (Interferon) 5. HSP90 (Heat Shock Protein 90)
The application of GRID/GOLPE procedure have been used to examine how steric, electrostatic, hydrophobic and hydrogen-bonding interactions could influence the inhibitory activity and to derive predictive 3-D QSAR models for designing and forecasting the activity of untested new compounds.
1. The reversible histone acetylation (HAT) and deacetylation (HDAC) are epigenetic phenomena that play critical roles in the modulation of chromatin topology and the regulation of gene expression3.Aberrant transcription due to altered expression or mutation of genes enzymes or their binding partners, has been clearly linked to carcinogenesis. In the present work the Molecular Modeling and 3-D QSAR studies have been performed on a series of 25 (aryloxopropenyl)pyrrolyl hydroxamates HDAC inhibitors with activity and selectivity against both maize HD1-B and HD1-A, two enzyme homologous of mammalian class I and class II HDACs, respectively. The studies have been accomplished by calculating alignment-independent descriptors (GRIND) using the ALMOND software4. Highly descriptive and predictive 3-D QSAR models were obtained using either class I or class II inhibitory activity displaying r2/q2 values of 0.96/0.81 and 0.98/0.85 for HD1-B and HD1-A, respectively. A deeper inspection revealed that in general a bent molecular shape structure is a prerequisite for HD1-A selective inhibitory activity while straight shape molecular skeleton leads to selective HD1-B compounds. The same conclusion could be achieved by molecular docking studies of the most selective inhibitors5.
2. Among post-translational covalent modifications is included also histone methylation. Histones can be methylated on lysine as well as arginine residue, preferentially on the amino-terminal tails of histones H3 and H4; this methylation is a stable epigenetic mark. The nine mammalian PRMTs identified share a highly conserved catalytic domain. Seven of them catalyze the transfer of a methyl group from S-adenosylmethionine (SAM) to guanidine nitrogen atoms of arginine residues. The PRMTs are over-expressed in prostate and breast cancer. The screening of the inhibition capabilities against both human PRMT1 and Aspergillus nidulans RmtA of dye-like small molecules from a focused library is reported as well as molecular modeling studies (homology modeling, molecular docking and 3-D QSAR) into the catalytic domain of the PRMT1 fungal homologue RmtA. The good correlation between computational and biological results proposes RmtA as a reliable tool for screening arginine methyltransferase inhibitors. In addition, the binding mode analyses of tested derivatives reveal the crucial role of two regions, the pocket formed by Ile12, His13, Met16 and Thr49 and the SAM cisteinic binding site. These regions should be taken into account in the design of novel PRMT inhibitors6.

24
3. Aromatase is a cytochrome P450 (CYP19) enzyme that catalyzes the conversion of androgens androstenedione and testosterone to the aromatic estrogenic steroids estrone and estradiol, respectively, through the aromatization of the A ring of the substrate. This enzyme is present in breast tissue and is an important pharmacological target in the anti-cancer therapy. Different series of imidazole- and triazole-structures previously reported as antifungal agents were included in a 3-D QSAR model using a flexible ligand-based approach. The data set includes 124 molecules and once aligned by SURFLEX software, the GRID/GOLPE procedure was applied. To this different probes were tried and DRY/OH pair was the combination that gave the best statistical results with q2, r2 and SDEP values of 0.70, 0.86 and 0.54, respectively. The final model showed good predictive capability using an external test set not previously reported in literature with a standard deviation error of prediction (SDEP) of only 0.997.
4. IFN inducer has yet been clinically employed for the treatment on hepatitis C. The present study is the application of 3-D QSAR methods and GRID/GOLPE combination to guide the design of new bioactive molecule IFN inducers for oral administration. Analyses were conducted on different published IFN-inducer series related to two different structural scaffolds: 8-hydroxy-adenines and 1H-imidazo-quinolines. The final model including 156 compounds displayed good statistical indexes displaying r2, q2 and cross-validated SDEP values of 0.73, 0.60 and 0.61 using OH probe and 0.89, 0.61 and 0.60 using the DRY probe respectively. An external test set formed of twenty compounds was used to evaluate the predictive capability of the models. To our knowledge this is the first 3-D QSAR application on different training set IFN-inducers8.
5. Hsp90, is one of the most abundant proteins in eukaryotic cells, comprising 1-2% of cellular proteins under non-stress conditions. It contributes to various cellular processes including signal transduction, protein folding, protein degradation and morphological evolution. Hsp90 works with other co-chaperones, playing an important role in the folding of newly synthesized proteins and stabilization and refolding of denatured proteins after stress9. A structure-based 3-D QSAR model was built using 26 inhibitors extracted from experimentally observed complexes with HSP90. The model display a high robustness (q2= 0.82, r2=0.95, SDEP=0.51) and good predictive capability using an external test set (SDEPext=0.48). From interpretation of grid maps was possible identify chemical groups that have an important impact for activity for building a general pharmacophore.
1. 2.
3. 4. 5.5.5.5.
References.
[1].A.R Ortiz; M. Pastor; A. Palomer; G. Cruciani; F. Gago; R.C. Wade; J. Med. Chem. 1997,40,1136-48 [2].G. Cruciani; S. Clementi and M. Pastor; Perspective in Drug Discovery and Design, 71-86, 1998. [3].A. Mai; S. Massa; D. Rotili; I. Cebara; S. Valente; R. Pezzi; S. Simeoni; R. Ragno; Med. Res. Reviews, 25, 3, 261-309, 2005. [4].M. Pastor; G. Cruciani; I. McLay; S. Pickett; S. Clementi; J. Med. Chem, 2000, 43, 3233-3243. [5].R. Ragno; S. Simeoni; D. Rotili; A. Caroli; G. Botta; G. Brosch; S. Massa; A. Mai; Eur. J. of Med. Chem., 2008, 43, 621-632.

25
[6].R. Ragno; S. Simeoni; S. Catellano; C. Vicidomini; A. Mai; A. Caroli; A. Tramontano; C. Bonaccini; P. Trojer; I. Bauer; G. Brosch; G. Sbardella; J. Med. Chem. 2007, 50, 1241-1253.[7].S. Castellano; G. Stefancich; R. Ragno; K. Schewe; M. Santoriello; A. Caroli; R.W. Hartman; G. Sbardella; Design, synthesis, 3-D QSAR and biological evaluation of novel selective CYP19 (Aromatase) inhibitors. Submitted.[8].R. Ragno; A. Caroli; S. Simeoni; I. Musmuca; 3-D QSAR studies on Interferon-inducers. A GRID/GOLPE approach on different series of compounds. Submitted.[9]. A.S. Sreedhar; E. Kalmar; P. Csermely; Y.F Schen; FEBS Letters 562 (2004) 11-15.

26
SYNTHESIS AND BINDING AFFINITY OF NEW QUINAZOLINONE DERIVATIVES
AS POTENTIAL ATYPICAL ANTIPSYCHOTICS
Laura CARRO
Organic Chemistry Department, Medicinal Chemistry Laboratory. Faculty of Pharmacy, University of Santiago de Compostela. 15782-Santiago de Compostela (Spain).
PhD Course: Organic Chemistry
Introduction
Schizophrenia is a complex disorder affecting approximately 1% of the population. For the treatment of this disease, classical (typical) neuroleptics such as haloperidol (Fig. 1) are currently used, but their use is associated with severe mechanism-related side effects, including induction of acute extrapyramidal symptoms (EPS), and they are ineffective against negative symptoms of schizophrenia.1 The clinical efficacy of classical antipsychotics in the treatment of schizophrenia and other psychotic disorders is directly related to their ability to block dopamine D2 receptors in the brain; however, it has been reported that dopamine receptor blockade in the striatum is closely associated with their extrapyramidal side effects.2
O
N ClF
OHN
NF
N O
O
N MeNH
NN
N
Me
Cl
Clozapine RisperidoneHaloperidol
Figure 1
The introduction of clozapine for treatment-resistant schizophrenia gave rise to a new group of atypical or non-classical antipsychotics which have no EPS and are effective against negative symptoms. These drugs exhibit potent antagonism at multiple receptor subtypes including serotonin and dopamine receptors, suggesting the implication of the serotoninergic system in this pathology.3 Meltzer et al.proposed that, in the efficacy of clozapine and other atypical antipsychotics such as risperidone or olanzapine, the most important factor is their relative affinities for D2 and 5-HT2A receptors: they proposed that the ratio between pKi for 5-HT2A and pKi for D2 may be used to discriminate atypical antipsychotics (ratio > 1.12) from classical antipsychotics (ratio < 1.09).4 Additionally, many of the atypical antipsychotic agents block not only 5-HT2A but other serotonin receptors, particularly 5-HT2C
receptors wich blockade, could be the responsible for reducing EPS and, consistently, a potential target in the treatment of psychotic illnesses.5
Over the last few years we have been working on modulation of the butyrophenone system with the aim of combining antagonism at 5-HT2 family and D2 receptors in a single molecule.6 As part of our ongoing work on the development of strategies for the preparation of new atypical antipsychotics, we explored the possibility of synthesizing analogues of aminobutyrophenones in which the benzene ring of the tetralone core was replaced by a pyrimidine, to form a tetrahydroquinazolinone system. The substitution of –CH= by –N= in aromatic rings has been one of the most successful applications of classical isosterism.7 In this communication we report the synthesis of the new quinazolinone derivatives 4a-d and 5a-d, and binding affinity on several dopamine and serotonin receptors.
Results and discussion
The synthesis of the cyclohexanedione derivative 2, a key intermediate in our synthetic proposals (Scheme 1), has been reported from 3,5-dimethoxybenzoic acid in a four step procedure.8 It is well established that formamide acetals react with active methylene ketones in a Vilsmeier–Haack-type reaction to produce enaminoketones which can subsequently yield, with the appropriate bifunctional nucleophile, a number of heterocycles such as pyrazoles, pyrimidines or isoxazoles, in a tandem Michael

27
addition-elimination/cyclodehydration process.9 Accordingly, synthon 2 was transformed by condensation with dimethylformamide dimethyl acetal (DMFDMA) into its enaminoketone with 95% yield. Then, cyclocondensation with a variety of amidine compunds in boiling AcOH or EtONa/EtOH gave tetrahydroquinazolinones 3a-d with good yields. Following our synthetic objective, the methyl ether of 3a-d was cleaved. The corresponding hydroxy compounds were obtained in moderate yields using a 1.0M sol. of BBr3 in CH2Cl2 at 0º for 24 h. Forcing conditions led to useless mixtures of compounds, and the use of other demethylating reagents did not improve the yield. The subsequent tosylation of the primary hydroxyl groups with p-toluenesulfonyl chloride in pyridine furnished the corresponding tosylates in good yields, which were converted into the desired amines 4a-d and 5a-d by nucleophilic displacement of the tosyl group by the corresponding substituted piperidines in bencene or acetonitrile in low yields.
N
O
N
N
R
O
F
N
O
N
N
R
NO
F
MeO
O
OHO
OMe
OMe
O
MeO
O
N
N
R
R = -H 3a
-SCH3 3b
-NHCH3 3c
-Ph 3d
12
4 a-d
5 a-d
Scheme 1
The binding affinities of the new aminomethylquinazolinones 4a-d and 5a-d at the serotonin 5-HT2A and 5-HT2C, and dopamine D2 human receptors have been determined and will be shown in the communication.
Acknowledgements
I would like to thank the Spanish Ministerio de Educación y Cultura for the financial support of this work (Ref SAF2005-08025-C03) and for a predoctoral fellowship.
References
1. Altar, A.; Martin, A. R.; Thurkauf, A. in ‘Burger's Medicinal Chemistry and Drug Discovery’, 6th edn.; Ed. D. J. Abraham, John Wiley & Sons, New Jersey, 2003, Vol. 6, p. 599.
2. Sawa, A.; Snyder, S. H. Science 2002, 296, 692. 3. Marino, M. J.; Knutsen, L. J. S.; Williams, M. J. Med. Chem. 2008, 51, 1077. 4. a) Meltzer, H. Y.; Matsubara, S.; Lee, J. C. Psychopharmacol. Bull. 1989, 25, 390; b) Roth, B. L.;
Tandra, S.; Burgess, L. H.; Sibley, D. R.; Meltzer, H. Y. Psychopharmacology 1995, 120, 365; c) Roth, B. L.; Meltzer, H. Y.; Khan, N. Adv. Pharmacol. 1998, 42, 482.
5. Roth, B. L.; Sheffler, D. J.; Kroeze, W. K. Nat. Rev. Drug Discov. 2004, 3, 353. 6. Dezi, C.; Brea, J.; Alvarado, M.; Raviña, E.; Masaguer, C. F.; Loza, M. I.; Sanz, F.; Pastor, M. J.
Med. Chem. 2007, 50, 3242. 7. a) Wermuth, C. G. in ‘The practice of medicinal chemistry’, 2nd edn., Ed. C. G. Wermuth,
Academic Press, Amsterdam, 2003, p. 193; b) Chen, X.; Wang, W. Annu. Rep. Med. Chem. 2003,38, 333; c) Kier, L. B.; Hall, L. H. Chem. Biodivers. 2004, 1, 138.
8. Pita, B.; Masaguer, C. F.; Raviña, E. Tetrahedron Lett. 2000, 41, 9835. 9. a) Stanovnik, B.; Svete, J. Chem. Rev. 2004, 104, 2433; b) Sekhar, B. C. J. Heterocyclic Chem.
2004, 41, 807; c) Molteni, V.; Hamilton, M. M.; Mao, L.; Crane, C. M.; Termin, A. P.; Wilson, D. M. Synthesis 2002, 1669.

28
NOVEL AMODIAQUINE CONGENERS AS POTENT ANTIMALARIAL AGENTS
Manolo CASAGRANDE
Istituto di Chimica Farmaceutica e Tossicologica “P. Pratesi”, Università degli Studi di Milano. Dottorato di Ricerca in Chimica del Farmaco XXI ciclo.
Novel, effective, safe and inexpensive antimalarial agents are required for the management of malaria in tropical and subtropical regions, where this disease afflicts approximately 500 million people annually.1,2
Presently the most promising and so far successful strategy in fighting malaria is a combination chemotherapy (ACT), in which an artemisinin derivative is used together with a conventional antimalarial drug to improve efficacy and delay onset of resistance.1
Despite the worldwide diffusion of resistance of P. falciparum to chloroquine (CQ), the 4-aminoquinoline derivatives continue to attract interest because the resistance seems to be compound-specific and not related to changes in the structure of the drug target. Indeed several CQ analogues, bearing different basic moieties, retain potent activity against CQ-resistant (CQ-R) strains of P. falciparum.Also amodiaquine (AQ) is active against many CQ-R strains of P. falciparum, but its clinical use has been severely restricted due to hepatotoxicity and agranulocytosis associated with long term prophylactic use. The situation changed in the recent years with the use of AQ in association with sulfadoxine/pyrimethamine (SP) or artesunate as first line treatment for uncomplicated P. falciparummalaria in different African countries.3
Since toxicity of AQ is related with the possibility to undergo in vivo oxidation to reactive quinoneimine derivatives, the structures of this drug was modified in order to prevent this kind of metabolic activation by the interchange of the positions of the basic head and the hydroxyl group, leading to isoquine (2).4
Moreover in most AQ-analogues the terminal diethylamino group, that characterizes both CQ and AQ, was replaced with a cyclic basic head or with a tert-butylamino group in order to prevent also the side chain metabolization, a process that produces N-dealkyl metabolites that are less potent against CQ-R strains. All these analogues resulted more resistant to metabolic oxidation and maintained the antimalarial efficacy.
NCl
HN
OH
N
1
Amodiaquine
NCl
HN
2
N
OH
Isoquine
Recently, our research group has synthesized new 4-aminoquinoline derivatives endowed with a remarkable in vitro activity against CQ-S and CQ-R strains of P. falciparum and orally active in murine models at doses comparable and lower than CQ and we have demonstrated that the presence of a bulky, strongly basic and lipophilic bicyclic moiety (such as the quinolizidine or the pyrrolizidine ring), which is supposed not to be easily metabolized, appears an interesting structural feature to overcome resistance.5-7
Pursuing our research, we have now explored the effect of replacing the diethylamino group of isoquine with the bulky (pyrrolizidin-7a-yl)alkylamino and (quinolizidin-1 -yl)methylamino moieties (compounds 3 d-f) that were present in our previous highly active CQ-analogues. We reasoned that the high lipophilicity of these substituents could improve the cellular permeation, while the presence of an additional protonable nitrogen atom should promote the endocellular accumulation of the drug.To further analyze the importance of the aromatic hydroxyl function, the hydroxyl group of compound 3f
was replaced by a chlorine atom (4f).In addition, to develop new classes of antimalarial agents, the possibility to replace the benzene ring of AQ and of its analogues with other aromatic nuclei, such as a pyrrole nucleus still linked to the quinoline moiety through the usual NH group (5 – 7), was also investigated. All the synthesized compounds were tested in vitro at the Department of Public Health-Microbiology-Virology of University of Milan against D-10 (CQ-S) and W-2 (CQ-R) strains of P.falciparum and exhibited from moderate to high activity against the CQ-S (D-10) strain with IC50 ranging from 8.5 to 222.2 nM. CQ IC50 was 27.7 nM.

29
NCl
HN
N
RR''
R'
R = OH : 3d-f
NCl
HNN
5a-d, f
R = Cl : 4f
NR''
R'
CH3
H3C
NCl
HNN
NR''
R'
CH3
R
R = H : 6a-c, f, g
R = Cl : 7a, b
NR''
R'N N HN NN
HN
HN N
NH H N
HN
H= ; ;;;; ;
a dcb fe g
Five of the new compounds exhibited also a strong activity against the CQ-R (W-2) strain, resulting from 10 to 28 times more active than CQ, with IC50 values as low as 14-59 nM compared to 481 nM of CQ. Moreover their resistance factor ranged from 1.2 to 1.7, resulting about ten times lower than that of CQ (17.7) and suggesting that these compounds were not (or only poorly) affected by the resistance mechanisms. Four additional compounds were in the order from 2- to 4.2 –fold more active than the reference drug. The most active compounds exhibit low toxicity against three different human or mouse cell lines with a therapeutic index, ranging between 555 and 34. In conclusion, the replacement of the phenolic ring, typical of AQ, tebuquine and isoquine, with a pyrrolic ring is associated with a good antimalarial activity. Therefore, these compounds represent a new class of antimalarial agents worthy of further investigation. Among the isoquine analogues, the most interesting compounds were 3f and 4f, both bearing a quinolizidinylmethylamino residue, which were 22 and 28 times more active than CQ against W-2 strain, respectively. On the basis of these results, the exchange of phenol ring of amodiaquine-like drugs with other aromatic moieties, as well as the replacement of the usual amino groups with pyrrolizidine and quinolizidine-derived basic heads, will be further investigated.
1. WHO (2001). "Antimalarial drug combination therapy. Report of a technical consultation." WHO, Geneva, CH WHO/CDS/RBM 35.
2. Snow, R. W., Guerra, C. A.; Noor, A.M.; Myint, H.Y.; Hay, S.I. Nature 2005, 434, 214. 3. Kiechel, J. R.; Pecoul, B. Med Trop (Mars), 2007 67,109. 4. O’Neill, P. M.; Mukhtar, A.; Stocks, P. A.; Randle, L. E.; Hindley, S.; Ward, S. A.; Storr, R. C.;
Bickley, J. F.; O’Neill, I. A.; Maggs, J. L.; Hughes, R. H.; Winstanley, P. A.; Bray, P. G.; Park, B. K. J. Med. Chem., 2003, 46, 4933.
5. Sparatore, A.; Basilico, N.; Parapini, S.; Romeo, S.; Novelli, F.; Sparatore, F.; Taramelli, D.. Bioorg Med Chem, 2005. 13, 5338.
6. Sparatore, A.; Romeo, S.; Basilico, N.; Parapini, S.; Taramelli, D.; Wittlin,S.; Brun, R.; Sparatore, F. 3rd Cost B22 Annual Congress “Drug discovery and development for parasitic diseases” Athens, 1-4 October 2006, Book of abstracts p. 133.
7. Lucantoni, L.; Sparatore, A.; Basilico, N.; Parapini, S.; Yardley, V.; Stewart, L.; Habluetzel, A.; Pasqualini, L.; Esposito, F.; Taramelli, D. 3rd Cost B22 Annual Congress “Drug discovery and development for parasitic diseases” Athens, 1-4 October 2006, Book of abstracts p. 137.

30
SYNTHESIS, CHARACTERIZATION AND EVALUATION OF
NEW ADENOSINE RECEPTORS’ ANTAGONISTS
Siew Lee, CHEONG∗; Giorgia, PASTORIN∗; Giampiero, SPALLUTO°
∗Department of Pharmacy, National University of Singapore ° Dipartimento di Scienze Farmaceutiche, Universita’ degli Studi di Trieste
Title of PhD course: PhD in Pharmacy
Introduction
There is growing evidence that adenosine receptors could be the promising therapeutic targets in a wide range of conditions. This has led to the development of adenosine antagonists, which have shown to represent potential therapeutic agents in various types of diseases, including Alzheimer’s disease, Parkinson’s disease, pulmonary disorders such as asthma1, as well as cancers. 2
Objectives
This project is aimed to synthesize and characterize a new series of adenosine receptors’ antagonists bearing the pyrazolo-triazolo-pyrimidine nucleus (Figure 1), in order to investigate the structural parameters at each position (2, N5 and N8) which are essential to have good affinity and selectivity at the adenosine receptors, especially at the human A3 (hA3) subtype.
R = CH3 R1 = H, PhCO, PhCH2CO R2 = Ph, 4-F-Ph, 4-Cl-Ph, 4-Br-Ph, 4-NO2-Ph
Figure 1: Chemical structure of new pyrazolo-triazolo-pyrimidine derivatives as adenosine receptor
antagonists.
Methodology
a) Synthesis of a new series of pyrazolo[4,3-e]1,2,4-triazolo-[1,5-c]-pyrimidine derivatives was based on a well established procedure3, which was further optimized to introduce suitable substituents at positions 2, N5 and N8 , followed by characterization of the synthesized compounds.
b) All the derivatives were then pharmacologically tested in binding assays: the affinity of a compound towards its receptor (expressed in Chinese Hamster Ovary cells (CHO) for hA1,hA2A, hA3 receptors) was evaluated through the measurement of displacement (Ki values) of selective radioligands, which were bound to the receptor expressed at the cell surface.4
c) The influence of structural parameters at position 2, N5 and N8 of the tricyclic nucleus on different receptor subtypes was then investigated based on the results obtained from the binding assays.
N N
N
N
N
N
HN
R
R2
R1
1
3
2
45
6
7
8

31
Results
The results from the assay showed that the series of synthesized pyrazolo-triazolo-pyrimidine derivatives has good affinity at hA3 receptors and selectivity towards the other subtypes, as indicated by the corresponding values of Ki in the nanomolar range. The finding confirmed the importance of small substituents (e.g. CH3) at the N8-position in affecting the affinity and selectivity at hA3 receptor. 5 It was also observed that concomitant introduction of benzoyl moiety (PhCO-) at position N5 has resulted in higher affinity at hA3 receptors and better selectivity against hA1 and hA2A receptors (S2, KihA3 = 5.0nM, hA1/hA3 = 124.4; hA2A/hA3 = 64.8) in comparison to either free NH2 (S1, KihA3 = 75nM, hA1/hA3 = 4.5; hA2A/hA3 = 1.6) or phenylacetyl (PhCH2CO-) group (S3, KihA3 = 60nM, hA1/hA3 = 5.1; hA2A/hA3 = 1.3). On top of that, the incorporation of 4-Br-Ph- and 4-F-Ph- groups at 2-position of the triazole ring, in combination with a free NH2 at N5
position, has also shown good affinity and selectivity at hA3 receptor subtype (S5, 4-Br-Ph-: KihA3 = 38.6nM, hA1/hA3 = 74.9, hA2A/hA3 = 38.9; S6, 4-F-Ph-: KihA3 = 31.4nM, hA1/hA3 = 32.2; hA2A/hA3 = 11.3). However, the introduction of substituent 4-NO2-Ph at position 2 has resulted in a relatively high nanomolar range in the binding assay towards A3 receptor (S7, KihA3
= 1,126nM), which indicates that the nitro group actually decreased the binding affinity of the compound towards hA3 receptor.
Compounds R R1 R2 hA1
(Ki nM)
(95% CI)
hA2A
(Ki nM)
(95% CI)
hA3
(Ki nM)
(95% CI) S1 CH3 H Ph 339.0
(319-359) 121.0
(100-147) 75.0
(63.1-90.4) S2 CH3 COPh Ph 622.0
(498-779) 324.0
(265-396) 5.00
(2.93-5.56) S3 CH3 COCH2Ph Ph 303.0
(246-372) 77.9
(68.9-88.2) 60.0
(50.6-72.2) S4 CH3 H 4-Cl-Ph 4,860
(3,360-7,010) 2,020
(1,060-3,840) 72.4
(71.3-73.6) S5 CH3 H 4-Br-Ph 2,890
(2,230-3,730) 1,500
(1,370-1,640) 38.6
(35.9-41.5) S6 CH3 H 4-F-Ph 1,010
(815-1,240) 355.0
(307-409) 31.4
(26.9-36.6) S7 CH3 H 4-NO2-Ph >100,000 8,530
(6,720-10,800) 1,126
(934-1,360)
Table 1: Binding affinities of pyrazolo-triazolo-pyrimidine derivatives from the binding assay.
Conclusion
In conclusion, this study has provided some information on the structural parameters at each position (2, N5 and N8) that are essential to have good affinity and selectivity at the adenosine receptors, particularly at the hA3 receptor subtypes, which implies the potential role of the new series of pyrazolo-triazolo-pyrimidine derivatives in the treatment of cancers in which the tumoral cells overexpress the hA3 receptors.
References
1. Sun, C.X. et al. J. Clin. Invest. 2006, 116, 2173-2182 2. Gessi, S. et al. Clin. Cancer Research 2004, 10, 5895-5901 3. Gatta, F. et al. Eur. J. Med. Chem. 1993, 28, 569-577 4. Klotz, K. N. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1998, 357, 1-9 5. Baraldi, P.G. et al. J. Med. Chem. 2000, 43, 4768-4780

32
DOCKING STUDIES AND QSAR ANALYSIS ON CB1 ANTAGONISTS:
A COMPUTATIONAL APPROACH TOWARDS THE IDENTIFICATION OF NEW LIGANDS
Elena CICHERO
Università degli Studi di Genova, Dipartimento Scienze Farmaceutiche, Viale Benedetto XV n°3, Genova 16132, Italy,
Scuola di Dottorato in Scienze e Tecnologie della Chimica e dei Materiali, Corso di Scienze Farmaceutiche Alimentari e Cosmetologiche
Rimonabant, 5-(4-Chlorophenyl)-1-(2,4-dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide, SR141716, Acomplia® is the most up-to-date cannabinoid receptor type 1 (CB1)antagonist, widely used as a tool to evaluate the mechanisms by which cannabinoid antagonists produce their pharmacological effects and to elucidate the physiological and the pathophysiological roles deriving from the inhibition of the CB1 receptors. Among the results of these studies, data obtained from different clinical trials clearly indicate that rimonabant may have benefits in the treatment of metabolic and cardiovascular disorders associated with overweight and obesity [1, 2]. In addition, other studies have pointed out its efficacy in reducing tobacco and drug dependence, thus suggesting that CB1 receptor antagonists might be of potential interest in these fields [3, 4]. However, its applications are actually limited by some adverse effects (mainly nausea and psychological disorders), which suggest the importance of finding new molecules provided with a higher safety profile. On these basis, a computational approach to support the synthesis of new CB1 antagonists has been taken. A data set of 78 antagonists belonging to five different chemical classes were selected from literature and docked into the human CB1 receptor model, built by homology modeling techniques. To gain further insights into the structure-activity relationships within the considered chemical classes, a pharmacophore model and a QSAR analysis were then developed, with a two steps approach. In a first step five alignments, one for each group of compounds, were generated. All of them were then merged to obtain a final pharmacophore model, representative of the whole dataset. On this pharmacophore a 3D-QSAR analysis, by means of the CoMFA methodology, was performed. The information thus derived has been used as a “filter” to identify potential CB1 ligands from an in-house library of diarylpyrazoles and other heterocyclic compounds, the most promising of which have been preliminary tested for their affinity towards the human CB1 receptor. Docking studies, pharmacophoric map and quantitative structure-activity relationships, plus some of the new molecules identified within this procedure, will be presented and discussed.
References
[1] Cooke D., Bloom S. The obesity pipeline: current strategies in the development of anti-obesity drugs. Nat. Rev. Drug. Discov. 2006, 5, 919-31. [2] Van Gaal L. F., Rissanen A. M., Scheen A. J., Ziegler O., Rossner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet 2005, 365, 1389-97.
[3] Boyd S. T., Fremming B. A., Rimonabant- a selective CB1 antagonist, Ann. Pharmacother. 2005, 39, 684-90. [4] Le Foll B., Goldberg S. R., Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence, J. Pharmacol. Exp. Ther. 2005, 312, 875-83 and literature cited therein.

33
NOVEL SYNTHETHIC APPROACH TO ANTIMALARIAL COMPOUNDS:
4-AMINOQUINOLINES THROUGH MICROWAVE-ASSISTED SNAR REACTIONS
AND COMBINATORIAL APPROACH TO 2,4,6-TRISUBSTITUTED TRIAZINES.
Paolo COGHI
Istituto di scienze e tecnologie molecolari C.N.R , Milano Dottorato in Chimica del Farmaco-XXI ciclo
Malaria nowadays remains one of the world’s greatest public health problems, especially in the developing countries. Despite numerous efforts expended to reduce its mortalityand morbidity,[1] malaria still threatens approximately 2 billion people worldwide, being responsible for almost two million deaths per year, mostly among young children in sub-Saharan Africa.[2] Among the four malaria species that infect humans, the parasite P. falciparum is universally considered the most aggressive. Particularly impressive is its ability in mutating forms in response to administered antiplasmodial treatment, rapidly giving rise to adaptation and resistance. 4-Aminoquinolines have recently been indicated to be an important class of chemotherapeutic agents for artemisinin based antimalarial combination therapy. A rapid, cheap, possibly clean and scalable route to 4-aminoquinolines endowed with multiple diversity is therefore badly needed. We here report microwave flash-heating chemistry to allow the efficient conversion of the available 4,7-dichloroquinoline into a library of aminoquinolines in high yields and purities, with no need for further purification steps and requiring very short reaction times. Some of the compounds in this library were active against chloroquine-sensitive and chloroquine-resistant parasite strains. [3]
N
Cl
Cl N
NR1R2
Cl
HNR1R2, (base)
(solvent), MWR1 = H, R2 = aliphatic
R1, R2 = N-substituted piperazine
R1 = H, R2 = aromatic
The combined use of conventional SNAr reaction and microwave irradiation is another approach developed, proved very valuable in producing derivatized triazines with tunable substituents. A library of 2,4,6-triamino-1,3,5-triazines was synthesized as both cycloguanil and chloroquine analogues, and were assessed in terms of in vitro antimalarial activity and toxicity. [4] [5]
N
N
N
Cl
Cl Cl
N
N
N
R1
R2 R3
i-iii
Scheme Reaction conditions: i) R1H, base, acetone, 0 °C, 3-4 h; ii) R2H, base,acetone, rt, 16 h; iii) R3H, base, DMSO, MW, 6 bar, 180 °C, 18 min. R1-3H =nucleophilic amines.
Some of the compounds in this library showed higher activity against CQ-resistant parasite strains and some exhibited CQ-sensitive activity in the same order as chloroquine itself.
References
[1] World Health Organization 2006: http://www.rollbackmalaria.org/malariaFAQ.html.

34
[2] C. A. Swales, P. L. Chiodini, B. A. J. Bannister, Infect. 2007,54, 107–110; b) D. A. van Schalkwyk, T. J. Egan, Drug Resist.Updates 2006, 9, 211–226.
[3] S. Melato, P. Coghi, N. Basilico, D. Prosperi, D. Monti, “Novel 4-Aminoquinolines through Microwave-Assisted SNAr Reactions: a Practical Route Antimalarial Agents”.Eur. J. Org. Chem. 2007, 6118–6123.
[4] S.Melato, D.Prosperi, P.Coghi, N.Basilico, and D.Monti. “A Combinatorial Approach to 2,4,6-Trisubstituted Triazines with Potent Antimalarial Activity: Combining Conventional Synthesis and Microwave-Assistance” ChemMedChem,2008, 1 – 4.
[5] Expert Meeting COST B22 "DRUG DISCOVERY & DEVELOPMENT FOR PARASITIC DISEASES", Modena, Italy, 19-20 February 2007.

35
L-DOPA-THIOL ANTIOXIDANT CODRUGS AS NEW ANTI-PARKINSON AGENTS WITH
FREE RADICAL SCAVENGING PROPERTIES
Catia CORNACCHIA
Dipartimento di Scienze Del Farmaco, Università degli Studi “G.D’Annunzio” di Chieti. Dottorato di ricerca in Scienze del Farmaco - XXI Ciclo
Parkinson’s disease (PD) is a neurodegenerative disease associated with degeneration of the pigmented neurons in the substantia nigra pars compacta (SNpc), resulting in decreased dopamine (DA) nigrostriatal system availability. Numerous drugs are available for treating PD, but none has surpassed the clinical efficacy of L-dopa (LD). Unfortunately LD seems to contribute to the progression of PD because of its pro-oxidant properties deriving from autoxidative metabolism [1]. During the last years, studies of the pathogenesis of PD have centered on the oxidative damage to the SNpc region of the midbrain. Biochemical changes occurring in PD [increased iron levels, inhibition of mitochondrial complex I activity and decreased reduced glutathione (GSH) levels] suggest that oxidative stress, free radical species production and an inadequate antioxidant defense system could contribuite to the nigrostriatal system degradation [2,3]. As a consequence, the inhibition of catecholamine autoxidation and the scavenging of ROS produced by such oxidation are important strategies for preventing or slowing the progression of aging and aged-related neurodegenerative disorders [4]. We have recently demonstrated that multifunctional codrugs, obtained by linking together LD with thiol-containing antioxidant agents, protect against the oxidative stress deriving from autoxidation and the MAO-mediated metabolism of DA [5,6]. In particular, we reported the two multifunctional codrugs 1-2 characterized by an amide linkage between LD and GSH possessing improved absorbition after oral administration with respect to LD alone and showing more efficacious central nervous system delivery than LD and GSH alone [6].
As a prosecution of our work in this field we report here preliminary data concerning the synthesis and activity of new codrugs 3-8 in which LD is linked together with thiol antioxidants such as N-acetyl-cysteine, methionine, N-acetyl-cysteinyl-cysteine and bucillamine, all possessing well documented free radical scavenging proprieties [7,8]. The obtained results emphasize the role of the selected thiol as antioxidants when combined with LD and suggest that conjugates 3-8 can represent useful dopaminergic agents able to permit a targeted delivery of the antioxidant directly to neurons in SNpc.
1 2
NH
HN
NH
AcHN
COOMe
OSH
O
O COOMeOH
OH NH
HN
HN
COOMe
OSH
O
COOMe
O
NHAc
HO
HO
AcHN
HN
O
SH
COOMe
OH
OH
AcHN
HN
O
COOMe
OH
OH
S
OH
OH
NH
O
COOMe
SH
AcHN
3 4 5
AcHN
OH
OH
NH
O
COOMe
S
NH
HN
O
SH
COOMe
OH
OH
O
HS
NH
HN
O
SH
COOMe
OH
OH
O
AcHN
HS
6 7 8

36
Codrugs 3-8 were synthesized employing solution phase procedures by elongation of the suitably protected aminoacid or dipeptide chain in the C- (compounds 3, 5, 7, 8) or N- (compounds 4, 6) direction with the LD derivative. All new compounds were also assayed “in vitro” to evaluate their chemical and enzymatic stability. Codrugs 3-8 possessed high stability in non-enzymatic Simulated Gastric Fluid (SGF) at pH 1.3 (t1/2>20 h); this stability implies that all compounds pass unhydrolyzed through the stomach after oral administration. At pH 7.4 they are stable enough (t1/2>7 h) to be absorbed intact from the intestine. In rat plasma amide bonds of the studied derivatives were cleaved and a rapid conversion to LD was observed. Hydrolysis in human plasma proceeds more slowly with formation of LD. The faster hydrolysis in rat than in human plasma may be ascribed to the different enzyme systems that are highly efficient in rat plasma. Neostriatal LD levels after administration of codrugs 3-8 were also evaluated and compared with those of the previous reported codrugs 1 and 2. In particular, compound 3 was able to induce a sustained release of both LD and DA in rat striatum with respect to equimolar dose of LD; the striatal levels of DA were still elevated after 12 hours. Studies are currently in progress in order to evaluate the antioxidant efficacy of the codrugs 3-8.Details of this study will be discussed in the poster session.
References:
[1] T. S. Smith, W. D. Parker, J. P. Bennet. Neuroreport. (1994), 5, 1009-1011. [2] M. Hsu, S. Bharath, K. Jyothi, S. Rajagopalan, J.K. Andersen. J. Neurochem. (2005), 92, 1091-1103. [3] S. J. Chinta, S. Rajagopalan, D. A. Butterfield, J. K. Andersen. Neurosci.Lett. (2006), 40, 137-141. [4] J. B. Schulz, J. Lindenau, J. Seyfried, J. Dichgans. Eur. J. Biochem. (2000), 267, 4904-4911. [5] A. Di Stefano, P. Sozio, A. Cocco, A. Iannitelli, E. Santucci, M. Costa, L. Pecci, C. Nasuti, F.
Cantalamessa, F. Pinnen. J. Med. Chem. (2006), 49, 1486-1493. [6] F. Pinnen, I. Cacciatore, C. Cornacchia, P. Sozio, A. Iannitelli, M. Costa, L. Pecci, C. Nasuti, F.
Cantalamessa, A. Di Stefano. J. Med. Chem. (2007), 50(10), 2506-2515. [7] G. Atmaca. Yonsei Med. J. (2004), 45(5), 776-784. [8] D. Mazor, L. Greenberg, D. Shamir, D. Meyerstein, N. Meyerstein. Biochem. Biophys. Res. Comm.
(2006), 349, 1171-1175.

• •
Unité de Pharmacochimie, Université de Genève, Université de Lausanne
Quai Ernest Ansermet, 30, CH-1211 Genève 4
Switzerland
Xanthones (dibenzo- -pyrones) of natural and synthetic origin have a great interest because of their
biological and pharmacological activities. They are systematic markers in chemotaxonomy and xanthone-
containing plant extracts are used in traditional medicine1. Furthermore they act as monoamine oxidase
(MAO)2 and acetylcholinesterase2 inhibitors and possess antioxidant activity3,4.
To further characterize their antioxidant properties, the reactivity of a series of hydroxy- and/or
methoxy-substituted xanthones (Table 1) and mangiferin (glycosyl-subsituted xanthone) toward peroxyl
(ROO•)5, 2,2-diphenylpicrylhydrazyl (DPPH•)6 and 2,2’-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid)
(ABTS•)7 radicals was examined. The results were compared to those obtained for a series of reference
phenols well known for their antioxidant activity, namely caffeic acid5, chlorogenic acid5, gallic acid6,
quercetin5, resveratrol5, -tocopherol8 and trolox5.
O
O R1
R2
R3
R4R5
R6
R7
R8
No R1 R2 R3 R4 R5 R6 R7 R8
1 H H H H H H H H
2 OH H H H H H H H
3 OMe H H H H H H H
4 H OH H H H H H H
5 H OMe H H H H H H
6 H H OMe H H H H H
7 H H H OH H H H H
8 H H H OMe H H H H
9 H H OMe OH H H H H
10 H H OH OMe H H H H
11 H H OMe OMe H H H H
12 OH H OMe H OH H H H
13 OH H OH H OH H H OH
14 OH H OH H H H OH OH
15 H OMe OMe OMe H H H H
16 H C-Glc OH H H OH OH H
Table 1: Xanthone derivatives. C-Glc stands for C-glycoside
The capacity of compounds to scavenge peroxyl radicals was evaluated by a recently developed
fluorimetric 96-well microplate assay5. Peroxyl radicals were generated by 2,2'-azobis-(2-methylpropionamidine) dihydrochloride (AAPH) and the decrease in catalytic activity of the enzyme
alkaline phosphatase (ALP) to hydrolyze 4-methylumbelliferyl phosphate (4-MUP) to the fluorescent
product 4-methylumbelliferone (4-MU) was monitored as marker of oxidative damage. The protection of
ALP from peroxyl radical-induced activity loss by test compounds allowed to assess their reactivity
toward ROO•. The capacity to preserve the catalytic effectiveness of the enzyme was expressed as pEC50
value, the negative logarithm of the antioxidant concentration that protects ALP to 50% from maximal
activity decrease.
The reactivity of compounds toward the persistent DPPH• and ABTS• radicals was measured by
spectroscopic 96-well microplate assays. ABTS• radicals were produced by reaction of ABTS with
potassium persulfate. In both assays, the radical scavenging effectiveness of compounds was assessed by
the decolorization (monitored at 515 nm for DPPH• and at 734 nm for ABTS•) of the radical in ethanol
solution. Results were expressed as EC50 value, corresponding to the effective concentration ratio of
37
Reactivity of substituted xanthones toward peroxyl, ABTS and DPPH radicals
Delphine CRESSEND, Marianne REIST, Julia DREYER, Pierre-Alain CARRUPT

antioxidant concentration to DPPH• or ABTS• concentration producing a 50% decrease in DPPH• or
ABTS• at steady state8.
As expected, unsubstituted xanthone (1), simply methoxy-substituted derivatives (3, 5, 6, 8, 11, 15)
and xanthone derivatives with hydroxyl substituents merely in position 1 (2) or 3 (10), showed no
reactivity toward any of the three radicals and were identified as devoid of antioxidant activity.
On the other hand, xanthone derivatives with a catechol moiety (norswertianin 14 and mangiferine
16) and/or with a hydroxyl substituent in position 2 (respectively 7) and/or 4 (respectively 5), as in
xanthone derivatives 4, 7, 9, 12, and 13, efficiently protected ALP from peroxyl radical-induced activity
loss. Their protective activity is comparable to the one of the measured reference polyphenols, all
classified as efficient antioxidants in the ALP test5 with pEC50 values above 5 and significantly higher
than the one of Trolox, classified as an intermediate antioxidant in this assay5 with a pEC50 value between
4 and 5.
All seven xanthones showing a good capacity to protect ALP form peroxyl radicals were also
reactive toward ABTS•, but the ranking of activities between the two tests was different. The EC50 values
of active xanthones were in the same range as the ones of the reference phenols tested.
Although all reference phenols showed to be active in the DPPH• assay, only three of the active
xanthones in the ALP assay were sufficiently efficient to reduce the stable DPPH• radical, namely
desmethylbellidifolin 13, with hydroxyl groups in positions 1, 3, 5, 8, and the catechols norswertianin 14
and mangiferine 16.
These results show that the reactivity of xanthones and of reference phenols toward the tested
radicals depends on the nature of the radical. Indeed, the ranking of reactivity of the tested compounds
toward the different radicals was poor. The three assays are thus complementary for the characterization
of antioxidant properties of compounds.
It can further be noted that all compounds showing antioxidant properties to protect ALP from
peroxyl radicals were identified as actives by the ABTS• assay whereas with the DPPH• assay only three
xanthones were classified as actives. Four xanthones with good antioxidant properties in the two other
tests were thus identified as inactive in the DPPH• assay. As the ABTS• test is simpler, easier to perform
and more cost-effective than the peroxyl radical/ALP assay, it seems to be the most convenient screening
assay between the tree performed to identify among a large series of new chemical entities the ones with
antioxidant properties. Further characterization of active hits should then be performed with additional
antioxidant tests.
References
1. Hostettmann K, Hostettmann M 1989. Xanthones. In: Methods In Plant Biochemistry, editor
Harborne YB. New York: Academic Press. p 493-508.
2. Urbain A, Marston A, Queiroz EF, Ndjoko K, Hostettmann K 2004. Xanthones from Gentiana campestris as new acetylcholinesterase inhibitors. Planta Med 70:1011-1014.
3. Salvi A, Brühlmann C, Migliavacca E, Carrupt PA, Hostettmann K, Testa B 2002. Protein
protection by antioxidants: development of a convenient assay and structure-activity
relationships of natural polyphenols. Helv Chim Acta 85:867-881.
4. Born M 1997. Redox studies of some natural and synthetic antioxidants. PhD Thesis, Lausanne:
University of Lausanne.
5. Bertolini F, Novaroli L, Carrupt PA, Reist M 2007. Novel screening assay for antioxidant
protection against peroxyl radical-induced loss of protein function. J Pharm Sci 96:2931-2944.
6. Brand-Williams W, Cuvelier ME, Berset C 1995. Use of a free radical method to evaluate
antioxidant activity. Food Sci Technol 28:25-30.
7. Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C 1999. Antioxidant activity
applying and improved abts radical cation decolorization assay. Free Rad Biol Med 26:1231-
1237.
8. Ancerewicz J, Migliavacca E, Carrupt PA, Testa B, Brée F, Zini R, Tillement JP, Labidalle S,
Guyot D, Chauvet-Monges AM, Crevat A, Le Ridant A 1998. Structure-property relationships of
trimetazidine derivatives and model compounds as potential antioxidants. Free Rad Biol Med25:113-120.
38

39
DESIGN AND SYNTHESIS OF A NEW SERIES OF DERIVATIVES ENDOWED WITH
POTENTIAL INTEREST IN CANCER IMMUNOTHERAPY
Marco CROSETTI
Dip. di Scienza e Tecnologia del Farmaco. Università degli Studi di Torino. Via P. Giuria 9, 10125 Torino. Italy
Host immunity has a critical role in the regulation of both tumor outgrowth and patient survival; however, the expansion of tumor-specific T cells is also limited by tumor growth. Recently, there has been a resurgence of interest in myeloid cells in association with their role in tumor progression and potential to limit therapeutic response.1
The accumulation of myeloid derived suppressor cells in the tumor microenvironment is one of the mechanism by which T cells are immunosuppressed. Thus the effector functions of tumor-antigen specific T lymphocytes are abolished and they are unable to kill cancer cells. Recents studies showed that nitric oxide-donating nonsteroidal anti-inflammatory drugs (NO-NSAIDs), obtained by the conjugation of an NSAID with an NO-donating moiety, could contribute significantly towards the development of effective chemoprevention strategies against cancer.2
The prototype of these products is an NO-aspirin in which the aspirin is linked through an ester bridge to a moiety containing an NO-donor function (Fig. 1).
Fig. 1. General structure
Following this design, in recent years a large number of derivatives with similar structure was synthesized and their in vitro metabolism and antitumor mechanism of action were studied.3, 4
NO-aspirins are not able to kill directly the tumor cells. However, by interfering with inhibitory enzymatic activities of myeloid cells, they increase the number and function of tumor-antigen-specific T lymphocytes, and enhance the therapeutic effectiveness of the antitumor immunity elicited by cancer vaccination.5
Starting from these considerations, our research group which for several years has been involved in designing of NO-aspirin analogues, decided to submit to a preliminary screening a large number of products previously synthetized. All these compounds are characterized by an NSAID moiety conjugated with a NO-donor portion with peculiar physical-chemical properties. The compounds were obtained through a versatile and simple synthesis. They were tested at the Department of Oncology and Surgical Sciences of the Istituto Oncologico Veneto through an in vitroassay which permits to evaluate the ability of our compounds to interfere with the immunosuppressive activity of myeloid cells and the possibility to create a favourable environment for lymphocytes activation.5
Preliminary results showed that most of these molecules possessed a micromolar antitumor activity; therefore we decided to realize a series of structural modulation in order to identify the essential structure endowed with antitumor activity which will be object of in vivo evaluations.
NO-DONORASPIRIN LINK

40
References
1Talmadge, J.E. “Pathways Mediating the Expansion and Immunosuppressive Activity of Myeloid-Derived Suppressor Cells and Their Relevance to Cancer Therapy” Clin Cancer Res 2007, 13, 5243-5248.2Rigas, B.; Kashfi, K. “Nitric-oxide-donating NSAIDs as agents for cancer prevention” TRENDS in Molecular Medicine 2004, 10, 324-330. 3Gao, J.; Kashfi, K.; Rigas, B. “In Vitro Metabolism of Nitric Oxide-Donating Aspirin: The Effect of Positional Isomerism” J. Pharm. Exp. Ther. 2005, 312, 989-997. 4Andrews, P.; Zhao, X.; Allen, J.; LI, F.; Chang, M. “A comparison of the effectiveness of selected non-steroidal anti-inflammatory drugs and their derivatives against cancer cells in vitro” Cancer Chemother. Pharmacol. 2008, 61, 203-214. 5De Santo, C.; Serafini, P.; Marigo, I.; Dolcetti, L.; Bolla, M.; Del Soldato, P.; Melani, C.; Guiducci, C.; Colombo, M. P.; Iezzi, M.; Musini, P.; Zanovello, P.; Bronte, V. “Nitroaspirin corrects immune dysfunction in tumor-bearing and promotes tumor eradication by cancer vaccination” PNAS 2005, 102,4185-4190.

41
1,4-DIOXANE NUCLEUS AS A SUITABLE SUBSTRUCTURE FOR THE
CHARACTERIZATION OF DIFFERENT RECEPTOR SYSTEMS
Fabio DEL BELLO
Dipartimento di Scienze Chimiche, Università di Camerino Dottorato di Ricerca in Scienze Farmaceutiche XXI ciclo
One of the strategies, which can lead to the discovery of novel biologically active compounds and over the last 20 years has emerged as a fruitful approach, is the identification and use of molecular scaffolds with versatile binding properties, such that they are able to provide potent and selective ligands for different specific targets through proper modification of functional groups. For such structures, able to recognize with high affinity only the members of a given family or interact with more unrelated targets, the term “privileged structure” has been proposed.1
In my research project I have demonstrated that the properly substituted 1,4-dioxane nucleus may be considered a privileged structure, as it has proven to be a suitable scaffold for: a) potent muscarinic acetylcholine receptor (mAChR) agonists and antagonists; b) α1D-adrenoreceptor (α D-AR) antagonists; c) potent 5-HT1A serotoninergic receptor full agonists.
On the basis of genetic and pharmacological characterizations, the muscarinic receptors, belonging to the G-protein-coupled receptor (GPCR) superfamily, are subdivided into five different subtypes (M1–M5). Each subtype has a unique distribution in central and peripheral nervous systems, and several are expressed both pre- and post-junctionally, the specific location varying between tissues. In the Central Nervous System, the muscarinic receptors are involved in vegetative, sensory cognitive, and motor functions. In the periphery, they mediate smooth muscle contraction, glandular secretion, and regulation of cardiac rate and force. Therefore, the potential therapeutic applications of ligands interacting with such receptors are numerous. However, the clinical use of muscarinic ligands is limited by various side effects due to the lack of a marked subtype-discrimination. Therefore, extensive efforts have been made to develop agonists and antagonists selective for each muscarinic receptor subtype. The 1,3-dioxolane nucleus has been used for many years as a scaffold for its ability to give potent muscarinic agonists or antagonists, depending on the size of the substituent in position 2. Indeed, the presence of a methyl group produces effective and potent muscarinic agonists (1), while the presence of bulky phenyl groups in the same position affords potent antagonists (2). Also the higher homologue of compound 1 and its corresponding 1,4-dioxane regioisomer have been reported to show a parasympathomimetic action.
O
OH3C
1
O
O
2
N(CH3)3 I N(CH3)3 I
Figure 1
Therefore, two series of 1,4-dioxane derivatives, whose general structure is reported in figure 2, have been designed, prepared and studied to obtain muscarinic agonists and antagonists.2
O
O
N(CH3)3
R'''R''R'
RI
a) agonist series: R = R' = R'' = R''' = H, or alkyl in different combinationsb) antagonist series: R = R' = R'' = R''' = H, or aryl in different combinations
Figure 2
Moreover, it is well known that muscarinic receptors display stereoselective requirements and the biological activity of muscarinic ligands is closely related to their stereochemistry. Therefore, the most interesting ligands within the two series have been resolved into their corresponding enantiomers.3
My research group has long been involved in designing new α1-AR antagonists structurally related to WB 4101 (3) (Figure 3), and in studying structure-affinity and structure-selectivity relationships to develop

42
high-affinity, site-selective ligands for each of the three α1-AR subtypes (α1a/A, α1b/B,α1d/D).4 I have expanded on the structure-activity relationship study by investigating the possibility that the quite planar 1,4-benzodioxane privileged structure of WB 4101 might be replaced by the less conformationally constrained 1,4-dioxane ring. The increased flexibility of 1,4-dioxane moiety might allow a better interaction with α1-AR subtypes or favour the interaction with only one of them, leading hopefully to selectivity.
ONH
O
O
OCH3
H3CO
3: WB 4101
Figure 3
Therefore, also on the basis that the stereochemically defined insertion of a phenyl ring in position 3 of WB 4101 (phendioxan),5 or in position 4 of its methylene bioisostere6 induced a significant modulation of α1-AR subtype selectivity, we designed the cis and trans 5- or 6-phenyl derivatives (Figure 4). Moreover, the observations that the enlargement of the 1,4-benzodioxane scaffold of WB 4101 by fusion with an additional benzene ring7 produced a significant α1a-AR subtype selectivity and its replacement with a 2,2-diphenyl-1,3-dioxolanyl structure, along with the contemporaneous removal of one or two methoxy groups in the 2,6-dimethoxyphenoxy moiety, afforded antagonists selective for the α1D-AR subtype,8
prompted us to design the 5- or 6-diphenyl derivatives (Figure 4).
O
O
NHO
R''
R'
O
O
NHO
R''
R'
R
RR = C6H5, or (C6H5)2R' = R'' = H, OCH3 in all combinations
Figure 4
All the novel compounds have been assayed at human cloned α1-ARs, expressed in Chinese hamster ovary (CHO), and native α1-ARs in rat vas deferens, thoracic aorta, and spleen. Moreover, it is well known that 5-HT1A serotoninergic receptor exhibits a high degree of homology to α1-ARs9 and benzodioxan derivatives and, particularly, WB 4101 and related compounds10 are effective ligands also for the 5-HT1A serotoninergic receptor. Therefore, all the compounds have been also evaluated on HeLa cells, expressing such a receptor. Finally, following our recent observation that α1D- and α1B-ARs are expressed in PC-3 prostate cancer cells and are involved in the modulation of apoptosis and cell proliferation,4 the cytotoxic effect of the new compounds and WB 4101 (1) on this cell line has been determined.
[1] Costantino, L.; Barlocco, D. Curr. Med. Chem. 2006, 13, 65-85. [2] Piergentili, A.; Quaglia, W.; Giannella, M.; Del Bello, F.; Bruni, B.; Buccioni, M.; Carrieri, A.; Ciattini, S. Bioorg. Med. Chem. 2007, 15, 886-96. [3] Piergentili, A.; Quaglia, W.; Giannella, M.; Del Bello, F.; Buccioni, M.; Nesi, M.; Matucci, R. Bioorg. Med. Chem. Lett. 2008, 18, 614-8. [4] Quaglia, W.; Santoni, G.; Pigini, M.; Piergentili, A.; Gentili, F.; Buccioni, M.; Mosca, M.; Lucciarini, R.; Amantini, C.; Nabissi, M. I.; Ballarini, P.; Poggesi, E.; Leonardi, A.; Giannella, M. J. Med. Chem. 2005, 48, 7750-7763. [5] Quaglia, W.; Pigini, M.; Giannella, M.; Melchiorre, C. J. Med. Chem. 1990, 33, 2946-2948. [6] Quaglia, W.; Pigini, M.; Piergentili, A.; Giannella, M.; Gentili, F.; Marucci, G.; Carrieri, A.; Carotti, A.; Poggesi, E.; Leonardi, A.; Melchiorre, C. J. Med. Chem. 2002, 45, 1633-1643. [7] Bolchi, C.; Catalano, P.; Fumagalli, L.; Gobbi, M.; Pallavicini M.; Pedretti A.; Villa L.; Vistoli G.; Valoti, E. Bioorg. Med. Chem. 2004, 12, 4937-4951. [8] Brasili, L.; Sorbi, C.; Franchini, S.; Manicardi M.; Angeli, P.; Marucci, G.; Leonardi, A.; Poggesi, E. J. Med. Chem. 2003, 46, 1504-1511. [9] Trumpp-Kallmeyer, S.; Hoflack, J.; Bruinvels, A.; Hibert, M. J. Med. Chem. 1992, 35, 3448-3462. [10] Quaglia, W.; Pigini, M.; Piergentili, A.; Giannella, M.; Marucci, G.; Poggesi, E.; Leonardi, A.; Melchiorre, C. J. Med. Chem. 1999, 42, 2961-2968.

43
NOVEL BISPHOSPHONATES AS γδγδγδγδ-T LYMPHOCYTES ACTIVATORS
Marco ELEOPRA
Dipartimento di Scienze Farmaceutiche, Università di Ferrara Dottorato in Scienze Farmaceutiche XXI Ciclo.
Geminal bisphosphonates (BPs) are analogues of endogenous pyrophosphate in which a carbon atom replaces the central atom of oxygen. Geminal bisphosphonates are drugs commonly used in bone resorption therapy, they are potent inhibitors of the enzyme farnesyl diphosphate synthase (FPPS) and recent clinical reports support the possibility of direct or indirect antitumuor effects of these compounds. Recent animal studies have shown that aminobisphosphonates, biphosphonates containing nitrogen in the side chain, have potent antiangiogenic activity that may contribute to these drugs antibone resorptive effect as well as provide additional mechanisms by which these drugs may have antimyeloma effects as well (1). A new potential antitumor mechanism for these compounds was recently reported for aminobisphosphonates (2). These drugs were shown to induce expansion of Vγ9δ2 T cells in peripheral blood mononuclear cell cultures and enhance cytotoxicity of malignant plasma cells in bone marrow cultures by these Vγ9Vδ2 T lymphocytes. Some of us (3) showed also that zoledronic acid was as able as the phosphoantigen isopentenylpyrophosphate (IPP) to induce expansion of Vγ9Vδ2 T cells upon culture in vitro in the presence of IL-2 indicating that in vivo treatment with zoledronic acid induces Vγ9Vδ2 T cells to mature toward an IFNγ-producing effector phenotype, which may induce more effective antitumor responses. Therefore, there is increasing evidence that bisphosphonates, especially the nitrogen-containing compounds (aminophosphonates) activating Vγ9Vδ2 T cell, can lead to direct and indirect effects that result not only in less bone loss but less tumor burden as well. As a consequence appears of big profit to have novel bisphosphonates that potently activate Vγ9Vδ2 T cells. Focusing our attention on bisphosphonates activating expansion of Vγ9Vδ2 T cells, here we report the discovery of a new lead compound endowed with a strong capacity to activate γδ-T lymphocytes, 1000 times more than zoledronic acid approximately. The compound shows a capacity comparable to the natural phosphoantigens (E)-4-Hydroxy-3-methyl-but-2-enyl-pyrophosphate to activate Vγ9Vδ2 T cells. The compounds shows also in vitro an unexpected high cytotoxic and apoptotic activity, and in vivo studies have demonstrated an interesting survival of mices receiving γδ T cells together the lead and rIL-2.
(1) Anderson, K.C.; Kyle, R.A. et al. Hematology. 2000; 147-165.(2) Kunzmann ,V; Bauer, E; Blood. 2000; 96, 384-392. (3) Dieli, F; Gebbia, N; et al. Blood. 2003, 102, 2310-2311.

44
SYNTHESIS AND BIOLOGICAL ACTIVITY OF HUMAN NEUROPEPTIDE
S ANALOGUES MODIFIED IN POSITION 2
Stella FIORINI
Department of Pharmaceutical Sciences and Biotechnology Center, University of Ferrara, Via Fossato di Mortara 17-19, 44100 Ferrara, Italy.
Dottorato in Scienze Farmaceutiche, XXI ciclo
Neuropeptide S (NPS)1 is one of the last endogenous peptide identified by the reverse pharmacology. The hNPS is a 20 residue peptide of the following primary sequence: SFRNGVGTGMKKTSFQRAKS which is highly conserved among species. After its pairing with NPS, the previously orphan GPCR was named NPSR. Cells stably expressing NPSR display a transient increase in the initial Ca2+ concentration levels in response to nanomole concentrations of NPS, thus suggesting that NPS behaves as an excitatory transmitter1. When injected supraspinally in rodents, NPS produces anxiolytic-like effects associated with stimulation of wakefulness. The biological originality of this peptide-receptor system relies on its combined effects on anxiety and arousal, a behavioral profile that differs from those of all the known anxiolytic and arousal promoting agents. The development of selective ligands for NPSR is of great importance to investigate the functions of this novel neuropeptide system. Previous structure-activity relationship (SAR)2 and conformational studies3,4 consistently demonstrated that the N-terminal part of the peptide is crucial for biological activity. In particular, based on findings obtained from Ala- and D-amino acid-scan studies, Phe2-Arg3-Asn4 were identified as the most important residues in hNPS sequence. Here, we present results obtained by replacing Phe2 of hNPS with coded amino acids and those obtained in a more focused second round of synthesis performed by replacing Phe2 with a series of non-coded Phe analogues. All the compounds were tested in a calcium mobilization assay performed on HEK293 cells stably expressing the mouse NPSR; in this assay NPS stimulated calcium mobilization in a concentration dependent manner with pEC50 and Emax values of 8.96 and 270% over the basal, respectively. Results obtained by testing hNPS analogues modified in position 2 indicated that: i) lipophilicity but not aromaticity is crucial, and a cyclic lipophilic side chain seems to be favored ii) both the size of the chemical moiety and its distance or position from the peptide backbone are important for biological activity; in particular the size can be enlarged up to a naphtyl or reduced down to isobutyl moieties, while the distance can be only increased by one carbon atom without major changes of biological activity iii) position 2 plays a role in both receptor binding and activation, as demonstrated by the reduction in efficacy displayed by [4,4’-Biphenyl-Ala2]hNPS (pEC50 and Emax values of 7.70 and 170% over the basal, respectively).
1. Xu, Y. L. et al., Neuron. 2004, 43, 487-497. 2. Roth, A. L. et al., J. Biol. Chem. 2006, 281, 20809-20816. 3. Tancredi, T. et al., J. Med. Chem. 2007, 50, 4501-4508. 4. Bernier, V. et al., J. Biol. Chem. 2006, 281, 24704-24712.

45
STRUCTURE AND FUNCTION OF NATURALLY OCCURRING VARIANTS
OF HUMAN ALPHA-THROMBIN
Roberta FRASSON
Pharmaceutical Science Department PhD Course in Molecular Sciences - Pharmaceutical Sciences Curriculum
ABSTRACT
THROMBIN is a multifunctional enzyme acting at the interface of coagulation, inflammation and cell growth. Thrombin is responsible of the final event of coagulation: the formation of fibrin plug, platelets activation and formation. Thrombin structure is similar to that of the other serine proteases of the chymotrypsin family. α-Thrombin consists of a light and a heavy chain covalently linked by a disulfide bond. The heavy chain is organized into two β-barrel domains with the catalytic site located at the interface between the two β-barrels. DISEASES related to malfunctioning of coagulative process represent the major cause of morbidity and mortality in the west world. Among the others, recessively inherited diseases are a minor cause of death, even if the phenomenon of immigration leads this problem also in our country, due to an higher rate of consanguineous marriages. PROTHROMBIN CONGENITAL DEFICIENCES are a rare cause of bleeding. 38 single-nucleotide mutations scattered along all the prothrombin gene were identified so far. For some of these mutations, the effects on the molecule’s functioning can be quite reasonably inferred. However, for many others the effect of mutations on thrombin activity is unpredictable and cannot be deduced merely from the analysis of the three-dimensional structure of the wild-type enzyme. With respect to this, little is known about the molecular and biochemical mechanisms specifying structure and function of thrombin natural variants. More than 170 crystallographic structures of wild-type or mutants of human alpha-thrombin have been reported in the Protein Data Bank. Nevertheless, none of them entails any of the naturally occurring thrombin variants identified so far. PRODUCTION OF ENOUGH QUANTITIES OF THROMBIN VARIANTS IS INSTRUMENTAL to conduct structure-function relationship studies on this enzyme, and to possibly envisage a therapeutic strategy for the treatment of these coagulation diseases. THE AIM OF THIS PROJECT regards the production as well as chemical, structural and functional characterization of recombinant thrombins based on some naturally occurring variants of the enzyme displaying a bleeding phenotype. The results of these studies will hopefully contribute to better understand the correlations existing between the changes in the chemical composition at the mutation sites and the structural and functional defects of thrombin, as well as a model of natural protein engineering on a key physiological enzyme. THE RESEARCH ACTIVITIES will be focused on the following issues:
1. Expression in E.coli of thrombin mutants corresponding to naturally occurring variants by recombinant DNA techniques.
2. Refolding and purification of thrombin mutants. 3. In vitro chemical, structural and functional characterization of thrombin mutants. 4. Crystallization and 3D structure determination of thrombin mutants by X-ray diffraction. 5. Analysis of functional data on the basis of the crystallographic structure.
THREE NATURAL VARIANTS have been selected. Two of them (i.e, Gly25Ser and des9Lys) contain mutations at protein sites connecting the heavy and the light chains of thrombin. In the third mutant, Arg67His, the mutation is in close proximity to the fibrinogen-binding site of thrombin. IN CONCLUSION, our results will contribute to provide novel insights into the correlations existing between the chemical composition of thrombin mutants and their altered structural and functional properties in vivo.

46
EFFECTS OF GOLD COMPOUNDS IN CISPLATIN-SENSITIVE
AND-RESISTANT OVARIAN CANCER CELLS
Valentina GANDIN
Dip. Scienze Farmaceutiche, Università di Padova, Via Marzolo 7, 35121 Padova, Italy Scuola di dottorato in scienze molecolari ad indirizzo Scienze Farmaceutiche.
Cisplatin is an effective antitumor agent essentially acting on DNA [1] and currently used as a first line chemotherapeutic agent for the treatment of testicular, ovarian, bladder and other carcinomas [2]. The development of resistance to cisplatin represents a serious clinical problem that has prompted a great deal of investigation concerning the mechanisms by which tumors cells become resistant to this chemotherapic agent [3]. So far, many tumor cell lines resistant to cisplatin have been isolated after prolonged exposure to low drug doses and they represent important tools for elucidating the factors underlying cisplatin resistance [4] that, however, has not been conclusively defined. In fact, there are several mechanisms responsible for cisplatin resistance, indicating the multifactorial nature of the problem [5]. These mechanisms include decreased cellular accumulation of cisplatin, increased intracellular thiols such as glutathione [6] and thioredoxin [7, 8], altered expression of regulatory genes, increased DNA repair/tolerance of platinum-DNA adducts [3]. There is a general agreement on deeming the decreased tendency of the cisplatin resistant cell to undergo apoptosis as a common basis where all the proposed resistance mechanisms converge. A substantial volume of experimental evidence points to the central role of mitochondria in apoptosis. Consequently, mitochondria represent an important target for anticancer drugs [9-10] and, in particular, the mitochondrial thiol-dependent redox systems formed by the glutathione and thioredoxin systems, can be altered after interaction with several antitumor agents [11,12]. The thioredoxin system, including NADPH, thioredoxin reductase and thioredoxin, participates in several cell processes including reduction of protein disulfides, removal of hydrogen peroxide through peroxiredoxins, formation of deoxyribonucleotides mediated by ribonucleotide reductase and regulation of transcription factors. The inhibition of both cytosolic and mitochondrial thioredoxin reductase can shift the redox balance toward a more oxidized state and hence alter the mitochondrial membrane permeability conditions with the consequent release of the segregated proapoptotic factors. Among the inhibitors of thioredoxin reductase, gold compounds are very effective and act at nanomolar level [13-14]. Auranofin is known to react with selenol-containing residues, and inhibits the thioredoxin reductase in near stoichiometric amounts with a formal Ki of 4 nM [14]. In the present work we have studied the effects of auranofin on cisplatin sensitive (2008) and resistant (C13*) human ovarian cancer cells [15]. Auranofin is more effective than cisplatin in decreasing cell viability and its effect is more marked in cisplatin-resistant than in sensitive cells. Furthermore, auranofin is able to permeate C13* cells more efficiently than 2008, determining a consistent release of cytochrome c both in resistant and in sensitive cells. Treatment with auranofin determines an increased production of reactive oxygen species that is larger in C13* cells compared to 2008. However, H2O2 production is counteracted in C13* cells by a large overexpression of thioredoxin reductase, while glutathione reductase is slightly incremented. In fact, Protein profile study performed by two-dimension liquid chromatography separation on the ProteomeLab PF 2D platform (Beckman Coulter Inc., Ca, U.S) and combined with differential expression analysis performed by DeltaVue™ software, confirms an enhanced expression of thioredoxin reductase in C13* cells.
Figure 1. Auranofin.
OS OAc
OAc
AcO
AcO
Au(C2H5)3P

47
Selenite is able to act as a substrate of thioredoxin reductases and to interact with glutathione. In addition, sodium selenite, in isolated mitochondria, determines a membrane permeability transition resulting from a thiol redox shift.[16] We have observed that, sodium selenite, in the nanomolar range, increases cell viability in both cell lines, while at higher concentrations decreases cell growth. Interestingly, cell pre-treatment with selenite dramatically increases the cytotoxic effect induced by auranofin in resistant, but not in sensitive cells. Moreover, selenite, at low doses, is able to increase thioredoxin reductases level, in both types of cell, but not glutathione peroxidase or glutathione reductase activities. In particular, TrxR2 (mitochondrial thioredoxin reductase) increases about four times with respect to the basal level.
1. G. Chu, J Biol Chem, 269, 1994, 787-790 2. P.J. O’Dwyer et al., In cisplatin. Chemistry and Biochemistry of Leading Anticancer Drug,
Lippert B. ed., 1999, 31-72 3. Z.H. Siddik et al., Oncogene 2, 2003, 7265-7279 4. L.R. Keòòand et al., J Inorg Chem 77, 1999, 111-115 5. M. Kartalou et al., Mutat Res. 478, 2001, 23-43 6. A.K. Godwin et al., Proc Natl Acad Sci. USA 89, 1992, 3070-3074 7. M. Yamada et al., Clin Cancer Res 2, 1996, 427-432 8. T. Sasada et al., Free Radic. Biol.Med. 27, 1999, 504-514 9. D.R. Green and G. Kroemer, Science 305 ,2004, 626-629 10. KM Debatin et al., Oncogene 21, 2002, 8786-8803 11. M.P. Rigobellon et al.,. Arch Biochem Biophys 441, 2005, 112-122 12. A.B. Witte et al. Free Radic Biol Med 39, 2005, 696-703 13. K. E. Hill et al., Biochem. Biophys Res Comm 234, 1997, 293-295. 14. M.P. Rigobello et al., Brit. J.Pharmacol. 136, 2002, 1162-1168. 15. Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello M.P., Free Radic Biol Med.
2007, 42, 872-81. 16. 1. Zheng Y., Zhong L., Shen X., J.Cell. Physiol., 2005, 205, 97-106.

48
STUDY ON DRUGS FOR CYSTIC FIBROSIS THERAPY
Michele GIAMPIERI
DISCIFAR, Università di Genova, Viale Benedetto XV, 3, 16132 – Genova, Italy Scuola di Dottorato in "Scienze e Tecnologie della Chimica e dei Materiali" – XXI ciclo
Cystic Fibrosis (CF) is an inherited disease caused by mutations that impair the function of the CFTR plasma membrane Cl- channel [1]. Until now, more than 1400 mutations have been discovered. One of the most severe and, unfortunately, common mutations is the deletion of a phenylalanine in position 508. This mutation is named ∆F508 and affects from 50 to 90% of CF patients (in relation to geographic setting). ∆F508 mutation causes both a mistrafficking of the protein (the protein remains entrapped in the endoplasmic reticulum [ER] and then is degraded by the quality control machinery of the cell) and a gating defect (decrease of the time spent by the channel in the open state on the plasma membrane). Other mutations, e. g. G551D and G1349D, are less frequent and cause only a gating defect, similar or even more severe than ∆F508 [2].
On the basis of the molecular defect, researchers have grouped CFTR mutations in five classes: lack of synthesis (I), maturation defect (II), gating defect (III), alteration of conductance (IV), shortage of synthesis (V) [3]. For instance, ∆F508 mutation belongs to class (II) and G551D and G1349D mutations belong to class (III).
In 2005, in order to find chemical compounds able to stimulate the activity of CFTR mutant proteins, we screened a library containing natural compounds and drugs that were already in use for the treatment of other human diseases. Indeed, 2000 compounds were tested on Fisher rat thyroid cells co-expressing ∆F508-CFTR (or other mutations) and a halide-sensitive yellow fluorescent protein (YFP). The YFP-based screening allowed the identification of the anti-hypertensive dihydropyridines (DHPs) nifedipine, felodipine, nicardipine, nimodipine and nitrendipine, as compounds being able to stimulate the activity of mutant CFTR: as such compounds act on CFTR mutants already in the plasma membrane (G551D and G1349D) or on ∆F508-CFTR saved to plasma membrane after incubation at low temperature (saved ∆F508-CFTR), we must define DHPs as “Potentiators” [4].
NH
COOR3
CH3
R2OOC
H3C
R1
1 : R1 = C(CH3)3, R2 = R3 = allyl
2 : R1 = CH(CH3)2, R2 = R3 = benzyl
3 : R1 = CH(CH3)2, R2 = benzyl, R3 = ethyl
From those results we could draw some SAR indications: 1) nitrogen in the position 1 must be unsubstituted; 2) methyl groups in position 2 and 6 are necessary; 3) substitutions with electron withdrawal groups at the position 4’ of the phenyl ring lead to a reduced activity; 4) substitutions at the position 2’ and 6’ by halogens give highly active compounds; 5) the phenyl ring in position 4 must be devoid of hydroxy groups [5].
Recently, we tested a panel of ca. 150 DHPs with the aim of finding DHPs active on mutant CFTR, but devoid of hypotensive activity. From that screening we found that DHPs 1 and 2 were active at µM

49
concentration on G551D and G1349D mutants and saved ∆F508-CFTR, but scarcely active on voltage-dependent calcium channels.
Following some data coming from molecular modeling showing the docking of DHPs in the first nucleotide binding domain of CFTR (NBD1), we hypothesized that it would be very interesting to obtain DHPs endowed with a strong asymmetry at ester level.
For this reason, asymmetrical DHPs (see compound 3 as an example) were synthesized. For instance, compound 3 was found very active on cited mutants, with no activity on calcium channels. The activity of 3 on CFTR mutants is about 60 nM. Other similar asymmetrical DHPs are in preparation. As the presence of two largely asymmetrical esters may be extremely important for a potent and stereo-selective activity, compound 3 and other similar structures were submitted to the enantiomeric separation. At present, pharmacological data on enantiomers are not available.
In the meantime, we began the synthesis of new compounds that may act as “Correctors” of ∆F508-CFTR mutant protein: such substances help the mutant CFTR to escape from the ER, allowing the mutant CFTR to reach the plasma membrane. To date, it is unknown if the binding site of such molecules is the CFTR itself or the chaperones leading the mutant protein to degradation [6]. Our research philosophy is based on the study of already known correctors (active but toxic) because some structural moiety is repeated in many correctors. Thus, we pointed our attention on the p-chloroanisole moiety with an ortho-substitution in respect to the methoxy group that, for instance, is present in the so-called corrector 4a [7].
N
S
S
NHN
H3CO
Cl
HNC
O
CH3
4a
Derivatives similar to corrector 4a, formed by a long molecule ending with a p-chloroanisole, are now under construction.
[1] F. Ratjen, G. Doring, Cystic Fibrosis, Lancet, 361, 681-689, 2003. [2] S. M. Rowe, S. Miller, E. J. Sorscher, Cystic fibrosis, N. Engl. J. Med., 352, 1992-2001, 2005. [3] M. Lim, P. L. Zeitlin, Therapeutic strategies to correct malfunction of CFTR, Paediatr. Respir. Rev.,2, 159-164, 2001.[4] N. Pedemonte, T. Diena, E. Caci, E. Nieddu, M. Mazzei, R. Ravazzolo, O. Zegarra-Moran, L.J.V. Galietta, Anti-hypertensive dihydropyridines as correctors of the CFTR channel gating defect caused by Cystic Fibrosis mutations, Mol. Pharmacology, 68, 1736-1746, 2005. [5] N. Pedemonte, D. Boido, O. Moran, M. Giampieri, M. Mazzei, R. Ravazzolo, L.J.V. Galietta, Structure-Activity Relationship of 1,4-Dihydropyridines as Potentiators of the CFTR Chloride Channel, Mol. Pharmacology, 72, 197-207, 2007. [6] M. D. Amaral, K Kunzelmann, Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis, Trends Pharmacol. Sci., 28, 334-341, 2007.[7] N. Pedemonte, G. L. Lukacs, K. Du, E. Caci, O. Zegarra-Moran, L.J.V. Galietta, A. S. Verkman, Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening, J. Clin. Investig., 115, 2564-2571, 2005.

50
SYNTHESIS AND SEMI-SYNTHESIS OF GLYCOSYLATED PROTEINS AS
PHARMACEUTICAL AND DIAGNOSTIC TOOLS
Francesca GORI
Laboratorio di Chimica e Biologia di Peptidi e Proteine, Dipartmento di Scienze Farmaceutiche, Università di Firenze
Dottorato in Chimica e Tecnologia del Farmaco
Several autoimmune diseases have been associated with post-translational modifications (PTMs), which can alter the function and immunogenicity of protein antigens (Ags). Glycosylation is the most important PTM of secreted proteins and plays a crucial role in several immune functions.1 Because of the heterogeneity of the potential specific sites of glycosylation present on each protein and of the diversity of enzymes involved in protein glycosylation, the study of the function of sugars is important and highly relevant. The role of autoantibodies (autoAbs) in autoimmune diseases has been reevaluated and it is accepted that a distinct pattern of Multiple Sclerosis (MS) pathology could involve an Ab-mediated demyelination.2
Some of the proposed biomarkers for MS, that may be objectively measured and evaluated as indicators for normal biological processes, are antibodies detectable as immunoassays. Previous studies showed that CSF114(Glc),3,4 a structured-based designed glycopeptide characterized by a β-D-glucopyranosyl moiety, is able to detect and isolate specific autoAbs present in the sera of a significant number of MS patients. Since this synthetic Ag could be considered as a mimetic of aberrantly glucosylated myelin protein(s) triggering autoimmunity in MS, we focused our interest on the role of myelin proteins. One of these is Myelin Oligodendrocyte Glycoprotein (MOG), a type I integral membrane protein specifically expressed on the outermost lamellae of myelin sheath and considered a putative autoantigen in MS.5 We decided to study the extracellular domain of MOG (MOGED) and to obtain this fragment properly glycosylated in order to characterize the molecular mechanisms of the form of MS in which the demyelinization is Ab mediated and to design new antigenic probes to detect auto-Abs as biomarkers. Therefore the aim of this research is to demonstrate a possible cross-reactivity of this glycoprotein, modified with a well defined glycosyl moiety, with the structure-based design glycopeptide CSF114(Glc). Production of glycoproteins with recombinant methods presents several difficulties. It’s well worth known that prokaryotes can be employed to express large quantities of recombinant proteins, but that they are not able to introduce specific PTMs on them. On the other hand mammalian cells can be used, but large scale preparation is very expensive and the final product is a heterogeneous mixture of glycoforms since protein glycosylation is not under direct genetic control. Thus, the preparation of specific glycoform may benefit from a chemical approach. During these last years several new interesting strategies have been developed to introduce noncanonical amino acids and biological probes into proteins. We are exploiting two different semi-synthetic strategies to get univocally glycosylated MOGED: the Cys-Ligation and the Expressed Protein Ligation (EPL).6,7
The former technique allows the selective reaction between a glucosyl iodoacetamide derivative and the cysteine free thiol of a protein (Scheme 1).
O
HO
HO
OH
HN
OH
O
I
O
HO
HO
OH
HN
OH
O
S-Cys-Protein
HS-Cys-Protein
Scheme 1. Cys-Ligation
We performed a site directed mutation on the plasmid pQE12rMOGED(His)6 to introduce a Cys residue at the position 31, native site of glycosylation, and to get the mutated rMOGEDN(31)C(His)6 protein by over expression in E. coli. After the purification and the characterization by LC-MS, the protein has been treated with the glycosyl derative and a tryptic digestion has been performed in order to investigate the result of the reaction.

51
The EPL is a protein engineering approach that allows synthetic and recombinant polypeptides to be chemoselectively and regioselectively joined together. This technique is an extension of the Native Chemical Ligation (NCL), based on the reaction between a peptide fragment containing a C-terminal thioester group and a second peptide containing a N-terminal Cys that generate an amide bond at the ligation junction after a spontaneous intramolecular S-N acyl shift. Studies performed on X-ray crystal structure of MOGED showed that this protein has a β-hairpin conformation, reminiscent of glycopeptide CSF114(Glc) one, in the segment 98-11.8 As a consequence it’s been decided to obtain MOGED synthetically glucosylated in position 104 in order to test it in immunological assays. We obtained the recombiant rMOGED(1-97) as C-terminal thioesther, using the IMPACTTM-TWIN (New England Biolabs) based on the protein splicing, and the peptide fragments [Gly103,Asn104(Glc)]MOGED(98-117) bearing a Cys residue at the N-terminus using the Solid Phase Peptide Syntehsis (SPPS). In this way the PTM has been introduced in the peptide sequence following the building-block approach: preformed glycosylated amino-acid building blocks are employed in the stepwise assembly of the peptide backbone.9,10
We are exploiting different conditions for the reaction between these two fragments. The obtained homogeneously glucosylated protein, carrying a glucosylated side-chain at natural glycosylation site, and the semi-synthetic protein with a well defined PTM will be tested by ELISA to study their ability to detect auto-Abs in MS patient sera, as compared to CSF114(Glc), and to characterize native MS autoantigen(s).
1 M.A. Daniels, et al., Nat. Immunol., 2002, 10, 903. 2 C. Lucchinetti, et al., Ann. Neurol., 2002, 47, 707-717. 3 A.M. Papini, P. Rovero, M. Chelli, & F. Lolli, PCT International Patent Application WO 03/000733. 4 F. Lolli, et al., Proc. Natl. Acad. Sci. USA, 2005, 102, 10273-10278. 5 T.G. Johns, C.C.A Bernard J., Neurochem., 1999, 72, 1-9. 6 D. Macmillan, R.M. Bill, K.A. Sage, D. Fern, S.L. Flitsch, Chemistry & Biology, 2001, 8, 133-145. 7 [a] T. Muir, Annu. Rev. Biochem., 2003, 72, 249-89; [b] R. Davies, M.P. Richter, A.G. Beck-Sickinger,
Eur. J. Biochem, 2204, 271, 663-677. 8 C. Breithaupt, A. Shubart, H. Zander, A. Skerra, R. Huber, C. Linington, U. Jacob, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 9446-9451. 9 M. Meldal, In: Neoglycoconjugates: Preparation and Application, ed. Y.C. Lee and R.T.Lee, Academic Press, Orlando, 1994, 145-198; [b] M. Meldal, K. Bock, Glycoconjugate J., 1994, 11, 59-63. 10 I. Paolini, F. Nuti, M. C. Pozo-Carrero, F. Barbetti, B. Kolesinska, Z. J. Kaminski, M. Chelli, A. M. Papini, Tetrahedron Letters, 2007, 48, 2901-2904

52
HIGHLY LIPOPHILIC COMPOUNDS: DETERMINATION OF LIPOPHILICITY BY RP-LC AND UPLC.
Amandine GUILLOT, Sophie MARTEL, Pierre-Alain CARRUPT
Unité de Pharmacochimie, Section des sciences pharmaceutiques, Université de Genève, Université de Lausanne, Quai Ernest-Ansermet 30,
CH-1211 Genève 4, Suisse
As inappropriate pharmacokinetic (PK) parameters has been recognized as the major factors leading to the withdrawal of new chemical entities (NCE) from drug development, important efforts were performed to predict PK behaviour as early as possible in the drug discovery process. Physicochemical properties successfully used as predictors of absorption, distribution, metabolism, excretion and toxicity (ADMET) required the development of HTS methods for their measurements in order to establish the NCE's physicochemical profile.
Among these physicochemical properties, lipophilicity, a key parameter in the prediction of pharmacokinetic processes of NCEs, was largely used in (quantitative) structure-property relationships, (Q)SPR. Many methods exist for the measurement or the determination of lipophilicity parameter. Due to their rapidity, their insensitivity to impurities and low sample consumption, chromatographic methods are more and more employed as an alternative to the time-consuming shake-flask. Reversed Phase-Liquid Chromatography (RP-LC) methods, based on correlation between log Poct and log kw (measured or extrapolated retention factors), were largely used for the determination of log Poct of neutral, acidic and basic compounds 1-3 presenting moderate lipophilicity. Furthermore, recently, the development of short columns packed with small particles (<2µm), used with very high pressure (UPLC), has demonstrated to be a promising approach for the high throughput determination of moderate lipophilicity with a gain in analysis time and chemical stability of stationary phases 4. However, these methods remain limited in term of lipophilicity range and currently no method exists for highly lipophilic compounds (log Poct > 5). So the lipophilicity of such compounds is often estimated by in silico methods with their well-known limitations and inaccuracies. In addition number of NCEs present high lipophilicity and then serious problems of bioaccumulation. Up to now, intestine and gill of fishes are the commonly used models for bioaccumulation estimation as they present similar permeability properties as mammalian intestine and blood-brain barrier respectively. Concurrently, a relation has been already established between log Poct and bioaccumulation (for moderate lipophilicity). Hence measuring lipophilicity (especially for these highly lipophilic compounds) instead of bioaccumulation is an approach of choice.
In this context, methods using RP-LC and UPLC techniques were developed and compared for the determination of lipophilicity indices of highly lipophilic compounds. Based on a series of reference compounds (neutral, acidic and basic compounds of low to moderate lipophilicity)5 and a series of selected rigid and highly lipophilic compounds, several experimental conditions were tested to optimize accuracy and analysis time and by this way enlarge the assessable lipophilicity range. Discovery® RP Amide C16 of 150 mm has already been used as stationary phase with RP-LC technique to evaluate log Poct of reference compounds 6. The use of shorter columns presents three main advantages: i) a significant reduction of the analysis time, ii) a diminution of organic modifier concentration even for lipophilic compounds, and iii) a broadening of log Poct range determination. Acetonitrile with its better elution capacity can be considered as the best organic modifier for lipophilicity measurements of highly lipophilic compounds. Hence to optimize lipophilicity range determination a Discovery® RP Amide C16 of 20 mm column was tested with acetonitrile as organic modifier. The
preliminary results have shown a good correlation between log kwnl(20) (log kw obtained by quadratic
extrapolation of a series of isocratic log k values against percentage of organic modifier used in the mobile phase) and log Poct or CLogP (version 4.0, Daylight Chemical Information System, Inc., Irvine, California, USA, http://www.daylight.com/, 2007) (for the highly lipophilic compounds). The diminution of columns length, recently employed to increase the throughput 1,7,8, also demonstrate the great advantage of reaching log Poct up to 8 with an analysis time which remained relatively moderate.
However to increase the throughput without compromising chromatographic performances, UPLC was also used. Two stationary phases were tested: an Hypersil Gold of 10 mm and an Acquity BEH Shield RP 18 of 30 mm. Two modes, isocratic and gradient, were also evaluated with both columns. Acetonitrile with its better elution capacity has been chosen in the mobile phase when isocratic mode was

53
applied. For technical reasons, methanol was preferred with gradient mode. Promising results were obtained with both stationary phases tested. Comparing the two modes, it has been shown that the correlation between log kw and log Poct (or CLogP) was enhanced using isocratic mode with Hypersil Gold, whereas Acquity Shield presents better results using the gradient mode. The two modes present their own advantages: isocratic mode allows the determination of a larger range of log Poct values compared to gradient mode. However, gradient mode increases the throughput since the analysis time is not compounds dependent and therefore become more relevant for the screening of a large number of lipophilic NCEs.
References
1. Lombardo F, Shalaeva MY, Tupper KA, Gao F, Abraham MH 2000. ElogPoct: a tool for lipophilicity determination in drug discovery. J Med Chem 43:2922-2928.
2. Nasal A, Siluk D, Kaliszan A 2003. Chromatographic retention parameters in medicinal chemistry and molecular pharmacology. Curr Med Chem 10:381-426.
3. Martel S, Guillarme D, Henchoz Y, Galland A, Veuthey JL, Rudaz S, Carrupt PA 2007. Chromatographic approaches for measuring log P. In Mannhold R, editor. Drug Properties: Measurement and Computation, Weinheim: Wiley-VCH. p 331-356.
4. Henchoz Y, Guillarme D, Rudaz S, Veuthey JL, Carrupt PA 2008. High-throughput log Pdetermination by ultraperformance liquid chromatography: a convenient tool for medicinal chemists. J Med Chem.
5. Pagliara A, Khamis E, Trinh A, Carrupt PA, Tsai RS, Testa B 1995. Structural properties governing retention mechanisms on RP-HPLC stationary phase used for lipophilicity measurements. J Liquid Chromatogr 18:1721-1745.
6. Stella C, Galland A, Liu X, Testa B, Rudaz S, Veuthey JL, Carrupt PA 2005. Novel RPLC stationary phases for lipophilicity measurement: solvatochromic analysis of retention mechanisms for neutral and basic compounds. J Sep Sci 28:2350-2362.
7. Lambert WJ 1993. Modeling oil-water partitioning and membrane permeation using reversed-phase chromatography. J Chromatogr 656:469-484.
8. Donovan SF, Pescatore MC 2002. Method for measuring the logarithm of the octanol-water partition coefficient by using short octadecyl-poly(vinyl alcohol) high-performance liquid chromatography columns. J Chromatogr A 952:47-61.

54
COMFA STUDY ON SIGMA (σσσσ) RECEPTOR LIGANDS
Enise Ece GURDAL
Yeditepe University, Faculty of Pharmacy, Department of Pharmaceutical Chemistry,
34755 Kayisdagi - Istanbul, TURKEY
INTRODUCTION
Sigma ( ) receptors are functional, membrane-bound proteins distributed throughout the brain and peripheral organs. 1 and 2 receptor types are clearly established, and further pharmacological differentiation may be possible. 1 receptors are implicated in central nervous system (CNS) disorders such as depression, schizophrenia and dementia. Further, 1 receptor agonists have value as neuroactive agents, while antagonists may help alleviate cocaine addiction. The significance of the receptor system to human health is augmented through the overexpression of sites by a number of cancers. Thus, receptor ligands might be used for the detection and treatment of malignant neoplastic diseases. 2
receptors may be of particular prognostic relevance, as the extent of their expression seems indicative of the proliferative status of tumors [1]. Structurally, ligands are represented by wide variety of chemical scaffolds, for example (+)-benzomorphans, phenylpiperidines, (+)-pentazocine and haloperidol [2]. Corbera et al. synthesized a series of novel tetrahydroindazole derivatives and tested for 1 and 2receptor binding [3].
Since sigma receptors are membrane-bound proteins, isolating and resolving their three-dimensional structure has proven to be difficult. QSAR (quantitative structure-activity relationship) methods assume that biological activity is correlated with chemical structure or properties and that as a consequence activity can be modeled as a function of computable physicochemical attributes. QSAR techniques are able to raise a predictive description of global structural requirements for interactions between substrates and receptor by using binding data.
In this study, we used apparent binding affinity (Ki) values of 27 sigma-1 receptor ligands to perform 2D and 3D-QSAR analysis. We carried out comparative molecular field analysis (CoMFA), a 3D-QSAR technique that is able to yield a predictive description of global structural requirements for interaction between substrates and protein.
METHODS
The QSAR functions in the Molecular Operating Environment (MOE) were used to compute theoretical molecular descriptors related to physicochemical properties. Correlation coefficient analysis, linear regression analysis and cluster analysis were done. We created a 3D database (Sybyl 6.8, Tripos Associates). Gasteiger-Hückel charges were calculated for each of the compounds. The structures were minimized by applying the MMFF94 force field. The CoMFA studies were performed with the QSAR module of SYBYL for combination of the two molecular fields - steric and electrostatic - which were sampled at each point of regularly spaced grid of 2 Å within automatically defined region. The steric and electrostatic fields were calculated using a sp3 –carbon with a +1 charge as probe. All regression analyses were done using the Partial-least squares (PLS) algorithms in SYBYL. Initial analyses were performed using full cross-validation (leave-one-out method). To minimize the influence of noisy columns, all cross-validated analyses were performed at a minimum (column filter) of 2.0 kcal/mol.
RESULTS
We have described a detailed QSAR study on tetrahydroindazole and tetrahydrocyclopenta[c]pyrazole series, in order to give better picture of action and to rationalize selection of substituents. After selection of molecular descriptors, a linear regression model can present the relationship between descriptors and binding data. The variability in the biological activity parameter `Ki` was best explained by SLogP (Log of the octanol/water partition coefficient (including implicit hydrogens) and ASA_H (Water accessible surface area of all hydrophobic atoms) with cross validated r2 of 0.80. The relative conrtibutions of the SLogP and ASA_H are 78.36 % and 21.63 %. The fact that contribution of the Log of the octanol/water

55
partition coefficient (SLogP) is more significant than Water accessible surface area of all hydrophobic atoms (ASA_H).
To derive 3D-QSAR models, their steric and electrostatic fields were correlated to their Ki values by CoMFA (using the QSAR option of SYBYL). After validation, a statistically significant CoMFA model was obtained (R2 = 0.759, q2 = 0.981).
In the CoMFA contour map, blue contoured areas show positive while red areas show negative charge regions. Green areas indicate regions where steric bulk increases while yellow indicates regions where steric bulk decreases binding affinity.
CONCLUSIONS
The 2D and 3D-QSAR models obtained in this study are useful for future predictions of sigma-1 ligands. Future studies might include synthesis and biological testing of the compounds to verify the predictions.
References
1. Lever, J.R., Gustafson, J.L., Xu, R., Allmon, R.L., Lever, S.Z., Synapse 59; 350-358 (2006). 2. Vangveravong, S., Xu, J., Zeng, C., Mach, R.H., Bioorg. Med. Chem. 14, 6988-6997 (2006). 3. Corbera, J. et al. ChemMedChem 1, 140-154 (2006).

56
DESIGN AND BIOLOGICAL EVALUATION OF FLUORESCENT-, [3H]-, OR [
11C]-SIGMA
RECEPTOR LIGANDS AS NOVEL TOOLS IN CANCER DIAGNOSIS
Carmela INGLESE
Dipartimento Farmacochimico, Università degli Studi di Bari, via Orabona, 4, 70125, Bari, Italy Dottorato in Scienze Farmaceutiche, XXI ciclo
Sigma receptors have been classified into sigma-1 and sigma-2 subtypes and they display a different
tissue distribution and a distinct physiological and pharmacological profile in the Central and Peripheral
Nervous System. In the Central Nervous System both receptors are involved in the modulation of
neurotransmitter release, in memory and in cognitive processes and in the regulation of movement,
whereas their role in the Peripheral Nervous System and their signal transduction remains to be clarified.
Sigma-1 and sigma-2 receptors have been detected in many tissues and they are highly expressed in
various tumor cell lines and tissues.1 These observations led to the development of sigma ligands as
molecular probes for diagnostic imaging and as a potential new strategy in tumor therapy. Our group in
the last decade carried out Structure Affinity Relationship (SAFIR) and Structure Activity Relationship
(SAR) studies on cyclohexylpiperazine derivatives developing a potent sigma-2 receptor ligand known as
PB28 (Fig. 1). This compound displayed high sigma-2 receptor affinity (Ki = 0.35 nM) and moderate
sigma-1 receptor affinity (Ki = 13.6 nM).2,3 Moreover, it displayed sigma-2 agonist and sigma-1
antagonist properties measured both in tumor cell lines overexpressing these receptors and in isolated
guinea pig ileum.4,5 Starting from PB28, many attempts have been carried out to obtain sigma-2 receptor
ligands suitable for: a) fluorescent probe in living cell studies;6b) tritium probe for binding receptor
assays;7c) [11C] carbon labeled probe for diagnostic imaging study by PET.8
Object a). In order to design a fluorescent sigma-2 receptor ligand, two strategies can be progressed: link
a fluorescent moiety by a spacer or design a fluorescent moiety that should be a structural feature of the
compound. Many failures have been reported for compounds linking fluorescent moiety because both
pharmacodynamic and pharmacokinetic properties of the starting compound are dramatically
compromised. For this purpose we chosen 2-naphthol as fluorescent moiety inserting it instead of 5-
methoxy-tetraline fragment of PB28. By SAFIR studies towards sigma-2 receptor and by evaluating the
fluorescent properties of resulting compounds, the best results have been obtained for compound I (Fig.
1).7 It displayed sigma receptor affinities (sigma-2 Ki = 26.4 nM; sigma-1 Ki = 6.78 nM) lower than PB28
but in acceptable sigma-2 affinity range (nanomolar). Moreover, it displayed an acceptable quantum yield
(Φ = 0.30). Compound I has been tested as fluorescent probe in saturation binding analyses towards
sigma-2 and sigma-1 receptors. The results displayed that it is an useful fluorescent probe to recognize
sigma-2 receptors in rat liver (Kd = 21.3 nM, Bmax = 470 fmol/mg of protein) whilst it failed to recognize
sigma-1 receptors in guinea-pig brain because it displayed high non-specific binding.7

57
Object b). In order to obtain a tritium
radiolabeled sigma-2 receptor ligand, the
radiosynthesis of PB28 has been designed
aiming to label the compound in the final step
of the reaction scheme. [3H]PB28 has been
obtained in quantitative yield (Specific
Activity = 8 mCi/mmol). The synthesized
radioligand has been employed in saturation
binding assay towards sigma-2 receptors in rat
liver displaying Kd = 0.13 nM and Bmax = 497
fmol/mg of protein.8 This result is consistent
to Ki value of unlabeled PB28 (Ki = 0.35 nM).
Moreover, [3H]PB28 has been employed in
competition binding assay to test DTG (Ki =
42 nM), Haloperidol (Ki = 5.85) and PB28 (Ki
= 0.57 nM). These findings were consistent to
the corresponding results obtained by
[3H]DTG, the reference radioligand for studing sigma-2 receptor ligands. Object c). In literature Kassiou
et al.9 prepared [11C]PB28 and tested it as probe in PET analysis. The results demonstrated that this
compound failed as PET probe because of high non-specific uptake in particular in Central Nervous
System. This failure could be explained considering that PB28 eluded P-glycoprotein (P-gp), the critical
efflux pump present both in the Brain Blood Barrier and in tumor cell membranes displaying Multi Drug
Resistance due to P-gp overexpression. Recently it was reported an animal model to recognize sigma-2
receptors in prostate carcinoma by PET analysis. PET radiotracer should display specific pharmacokinetic
and pharmacodynamic properties: to be a P-gp substrate in order to recognize only peripheral sigma-2
receptors and to show high sima-2 receptor affinity towards sigma-2 receptors. Considering these two
requirements, PB183, that is a P-gp substrate and displayed high sigma receptor affinities (sigma-2 Ki =
0.50 nM and sigma-1 Ki = 6.5 nM) has been selected.8 The results will be presented in the poster section.
References:
1) Vilner, B. J.; John, C. S.; Bowen, W. D. Cancer Res. 2005, 15, 408-413; 2) Berardi, F.; Colabufo, N. A.; Giudice, G.; Perrone, R.; Tortorella, V.; Govoni, S.; Lucchi, L. J. Med. Chem. 1996, 39, 176-182; 3) Perrone, R.; Berardi, F.; Colabufo, N. A.; Leopoldo, M.; Abate, C.; Tortorella, V. Med. Chem. Res. 2000,10, 201-207. 4) Colabufo, N. A.; Berardi, F.; Contino, M.; Perrone, R.; Tortorella, V. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2003, 368, 106-112; 5) Colabufo, N. A.; Berardi, F.; Contino, M.; Niso, M.; Abate, C.; Perrone, R.; Tortorella, V. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 370,106-113; 6) Ferorelli, S.; Abate, C.; Colabufo, N. A.; Niso, M.; Inglese, C.; Berardi, F.; Perrone, R. J.Med. Chem. 2007, 50, 4648-4655; 7) Colabufo, N. A.; Abate, C.; Contino, M.; Inglese, C.; Ferorelli, S.; Berardi, F.; Perrone, R. Bioorg. Med. Chem. Lett. 2008, 18, 1484-1488.; 8) Colabufo, N. A.; Abate, C.; Contino, M.; Inglese, C.; Niso, M.; Berardi, F.; Perrone, R. Bioorg. Med. Chem. Lett. 2008, 18, 1990-1993; 9) Kassiou, M.; Dannals, R. F.; Liu, X.; Wong, D. F.; Ravert, H. T.; Scheffel, U. A. Bioorg. Med. Chem. 2005, 13, 3623-3626.
N
O
NN
N
R2
R1
PB28compound I : R1 = OCH3; R2 = H
PB183 : R1 = H; R2 = OCH3
Fig. 1
O
N
N
O
N
NH
N
N
O
Scheme: radiosynthesis of [3H]PB28
**

58
MULTIVARIATE METHODS AND MOLECULAR MODELING TECHNIQUES
IN THE STUDY OF ANTITUMOR AGENTS
Mario IPPOLITO
Università di Palermo – Dipartimento Farmacochimico, Tossicologico e Biologico Via Archirafi, 32 – 90123 Palermo
Dottorato di Ricerca in Scienze Farmaceutiche – Ciclo 2005-2006 (XX)
The discovery of new compounds with anti-cancer activity is one of the hardest challenges of this era. In particular, beside the development of high-throughput biological assays, it emerged as necessary the use of computational techniques in order to analyze the entire amount of data deriving from in vitroscreening. Moreover, the application of molecular modeling techniques is also required to improve the understanding of the molecular features accounting for an optimal drug-receptor interaction.
In early 90’s the Developmental Therapeutics Program (DTP) of the National Cancer Institute (NCI) implemented a routinary protocol in order to evaluate the efficacy of many synthetic and natural compounds on a panel of 60 human tumor cell lines that have been grouped in subpanels including leukaemia, melanoma, and cancers of the lung, colon, breast, kidney, prostate, ovary, and central nervous system. The efficacy of each screened compound is reported as the “50% growth inhibition” (GI50) or the “Total growth inhibition” (TGI) concentrations. These measurements constitute a data vector of an array reflecting the pattern of cell killing. In order to detect privileged structures and analyze the pattern of differential growth inhibition among cell lines the COMPARE Algorithm [1] was firstly developed introducing a new methodological tool, the mean graph, which allowed the transformation of numerical cell line response data into graphic patterns. These patterns were further analyzed in order to rank a seed compound according to the similarity of the biological screening response, and the results are expressed qualitatively in terms of Pearson Correlation Coefficient. More recently [2] the 3D-Mind tool used the Self-Organizing Maps (SOM) in order to build two dimensional maps which allow to hypothesize a biochemical mechanism on the basis of the similarities in the pattern of cell killing.
Much part of in vitro screening data from the DTP forms the Anti-Cancer Agents Mechanism Database (ACAMD), a set of 122 compounds with a reasonably known mechanism of action for which are available drug screening data that measure their ability to inhibit the growth of a panel of 60 human tumour lines, explicitly designed as a training set for neural network and multivariate analysis. The compounds belonging to ACAMD are grouped according to their mechanism of action in these classes: Alkylating Agents, Antimytotic Agents, Topoisomerase I Inhibitors, Topoisomerase II Inhibitors, DNA/RNA Antimetabolites and DNA Antimetabolites. By using these data as source of information, a new method for the prediction of mechanisms of actions from the 3D structure of a drug candidate was built, through the combined use of Principal Components Analysis (PCA) and 3D-QSAR/QSPR, according to the figure; in this case the possible mechanism of action is predicted according to the structure of the tested compound, using the structure-property relationships.
Through the use of GI50 values as training data, it was possible to assign a correct mechanism of action with good statistic significance (more than 79%). Moreover, the analysis of molecular descriptors used in QSPR models is coherent with the information about structural features related to some anti-cancer activity. Thus, the application of statistics to anti-cancer data could also provide clues to the classification of drugs, the mechanism of which is unknown or controversial [3].
Among the different classes present in ACAMD, the Topoisomerase I Inhibitors are characterized by a well known molecular target which is highly interesting for the development of anti-cancer compounds. In fact, DNA Topoisomerases are enzymes partecipating to all metabolic processes associated with

59
separation of DNA strands, such as transcription, repair and replication [5]. In particular, Topoisomerase I (Topo1) is a monomeric enzyme requiring no cofactor to manage DNA superhelical tensions through the cleavage of one strand of duplex DNA. The inhibitors present in the NCI ACAMD belong to the family of Camptothecin (CMP), a natural alkaloid which has been shown to cause cell death through the cessation of DNA synthesis followed by a double-strand breakage when the replication fork encounters the camptothecin-trapped Topo1-DNA complex (termed as “ternary complex”). With the aim to investigate the inhibition of Topoisomerase I by camptothecin analogs included in the ACAMD, molecular docking experiments were performed [6]; four different structures of a ternary complex between Topo1, DNA, and four different inhibitors were used, in order to consider the different orientations of the active site residues. The obtained results allowed to analyze some conformations adopted by the inhibitors during active site binding, confirming the role of hydrogen bond and contributed to clarify the loss of activity due to single point mutations. In particular, poorly substituted camptothecins generally fit well into protein structure in which a simple camptothecin-like drug was crystallized (1K4T, 1T8I), whereas highly substituted compounds are better docked in those macromolecule crystallized with larger inhibitors. The analysis of the best docked conformations allowed to investigate the binding mode of compounds involved in this study, confirming the role of some aminoacids present in the active site as previously proposed. These results can also clarify and explain the role of some single point mutation in developing resistance to camptothecins, and are useful in order to understand the structural features required to improve the performance of camptothecin derivatives as Topoisomerase I inhibitors.
Considering the versatility of the techniques used, the studies were extended to other targets of anti-cancer therapeutic intervention. In particular the attention was focused on the cancer growth inhibition by acting on heat shock protein 90 (Hsp90) [7]. This protein chaperone is involved in the stabilization and conformational maturation of many signalling proteins that are deregulated in cancers; in particular it has recently shown that the Hsp90 inhibition results in the proteasome degradation of these client proteins, and leads to a potent antitumor activity. One of the factors involved in this process is the Hypoxia Inducible Factor 1 (HIF-1) [8] which activates the transcription of genes that are involved in crucial aspects of cancer biology, including angiogenesis, cell survival, glucose metabolism and invasion.
An initial molecular modeling approach to Hsp90 inhibition was carried out through the application of a particular molecular docking protocol, called Induced Fit, to the analysis of the binding modes of the Hsp90 inhibitors belonging to different classes (pyrazoles, purines, naphtols), whose crystallographic structure is available on the Protein Data Bank. This protocol allowed the generation of an accurate complex structure for a ligand known to be active but that cannot be docked in an existing (rigid) structure of the receptor, because it takes into account the active site flexibility, through the combined use of molecular dynamics and molecular docking. From the results it is possible to appreciate that several key residues involved in the drug-receptor interaction provide very different orientations, according to the three classes of compounds tested. Moreover, with the aim to evidence the common structural feature accounting for the receptor inhibition, the generation of pharmacophoric hypotheses was performed, one using the ligand conformations obtained from a previous molecular docking simulation, and one using vacuum-energy minimized conformation. This approach allowed to take into account the difference occurring between those conformation which fit into the active site and the global minima obtained from a conformational search. Further, the virtual screening protocol was extended to include a total of 130 derivatives belonging to eight structural classes; these experiments were carried out using a Hsp90 structure which emerged from previous molecular dynamics experiments to be suitable enough to dock differently shaped ligands. The most highly scored conformations were used for the calculation of a set of molecular descriptors; then, a protocol involving PCA and multivariate analysis was built in order to analyze, in terms of structural properties, the structural classes of Hsp90 inhibitors selected.
[1] Paull, K. D.; Shoemaker, R. H.; Hodes, L.; Monks, A.; Scudiero, D. A.; Rubinstein, L.; Plowman, J.; Boyd, M. R., J. Natl. Cancer. Inst. 1989, 81, 1088.
[2] Rabow, A. A.; Shoemaker, R. H.; Sausville, E. A.; Covell, D. G., J. Med. Chem. 2002, 45, 818. [3] Lauria, A.; Ippolito, M.; Almerico, A. M., EORTC 29th Pharmacology and Molecular
Mechanism Group Winter Meeeting, Palermo, January 30 - February 2, 2008. [4] Lauria, A.; Ippolito, M.; Almerico, A. M., J. Chem. Inf. and Modeling, submitted. [5] Pommier Y (1998) Biochimie 80:255. [6] Lauria, A.; Ippolito, M.; Almerico, A. M., J. Mol. Mod. 2007, 13, 393. [7] Kamal, A.; Boehm, M. F.; Burrows, F. J., Trends Mol. Med. 2004, 10, 283. [8] Minet, E.; Mottet, D.; Michel, G.; Roland, I.; Raes, M.; Remacle, J.; Michiels, C., Febs Lett.,
1999, 460, 251.

60
DESIGN, SYNTHESIS, CHARACTERISATION AND BIOLOGICAL ACTIVITY
OF LIGANDS FOR P2 RECEPTORS.
Dhuldeo Dnyandeo KACHARE
Dept. of Chemical Sciences, University of Camerino, Italy. Doctorate Course in Pharmaceutical Sciences- XXI cycle.
Purines and pyrimidines nucleosides and nucleotides are molecules involved in diverse physiological and pathophysiological processes through the activation of cell membrane receptors called purinergic receptors. The nucleotide ATP is abundant inside cells, where it provides the energy for many biological reactions, while in the extracellular space, although present in much lower amount, it modulates important physiological functions through the interaction with purinergic P2 receptors, classified as: ionotropic P2X receptors, which represent a class of ligand-gated ion channels and metabotropic P2Y receptors, which belong to the superfamily of heterotrimeric G-protein coupled receptors. The physiological functions modulated by P2 receptors include neurotransmission, muscle contractility, neural and endocrine secretion, immune cell regulation or cell proliferation. At sensory nerve terminals in the periphery, P2X3
and P2X2/3 receptors have been identified as the principal P2X purinoceptors present. Among P2X receptors, the P2X3 subtype showed ability to mediate pain sensation and there is strong evidence for a role of this subtype in neuropathic and chronic pain, in which plastic changes in the structure and function of P2X3 receptors are supposed to occur [1]. Hence, the availability of new selective P2X3 ligands can provide novel analgesics for the treatment of neuropathic pain, often associated with currently incurable diseases including cancer and neurodegenerative disorders. Xanthine (e.g. caffeine) and adenine derivatives are known to be purinergic P1 receptor antagonists [2, 3]. Since the phosphorylation of several xanthine derivatives led to the discovery of a new class of compounds, called “mininucleotides”, which showed high affinity at P2X receptors [4], a similar approach has been used on adenine analogues. In fact, the first part of this work has been focused on the design and characterization of novel P2 receptor ligands obtained through the phosphorylation of adenine derivatives bearing a hydroxybutyl chain in position 9. The choice of such substitution was due to the fact that the very flexible hydroxybutyl chain can mimic the nucleotide sugar portion of ATP and can maintain the same interaction with P2 receptors. Hence, a series of purine derivatives was synthesized by reacting selected purine analogues, substituted or not in 2 position, with 4-bromobutyl acetate and potassium carbonate in DMF, to give the desired purine derivatives containing a 4 carbon chain. Furthermore, different groups like halogens, alkynyl or aralkyl chains were introduced in position 2 by several reactions. Subsequent deprotection of the alkyl chain gave purine mininucleosides having a free hydroxyl group, which is essential for phosphorylation.
R = H, Cl, OCH2CH2Ph, NH2, I, n-Hexyne n = 1, 2, 3 Fig.1: Synthetic project-I ‘mininucleotides’
The newly synthesized mininucleosides were phosphorylated to obtain the corresponding mono, di- and tri-phosphates. More in detail, monophosphate derivatives were synthesized by reacting the corresponding mininucleosides with POCl3 in trimethylphosphate, while di- and tri-phosphates were prepared starting from the corresponding monophosphates. To this purpose, the tributylammonium salts of monophosphates were activated as phosphoroimidazolates by coupling with N,N’-carbonyl-diimidazole (CDI) and the latter was reacted with tri-n-butyl ammonium phosphate or bis-(tri-n-butyl ammonium) pyrophosphate to obtain the corresponding di- and tri-phosphates, respectively. The new mininucleotides (Fig.1) were tested to explore their potential agonist or antagonist activity on HEK cells expressing P2X3 receptors, in comparison with the full agonist alfa,beta-meATP by using

61
patch clamp technique. These studies demonstrated that certain “mininucleotides” behave as agonists of P2X3 receptors, thus producing rapid channel activation and desensitization. Recently several novel non-nucleotide small molecule P2X3 antagonists [5] have been reported. Among them, some substituted diammino-pyrimidine derivatives showed good potency in blocking P2X3
receptors (pIC50= 7.0) [6,7]. Hence, in the second part of this work, a new class of P2X3 ligands has been designed taking into account both the structure of the above mentioned non-nucleotide small molecule and the information obtained with molecular modeling techniques from superimposition of mininucleotide purine scaffold with the pyrimidine derivatives (Fig.2). In particular, aimed at mimicking the physical/chemical properties of diammino-pyrimidine derivatives to obtain antagonistic behavior, purine derivatives bearing different substituents in 9 position were synthesized and evaluated by using patch clamp technique to explore their antagonist activity, in comparison with the full agonist alfa,beta-meATP. Preliminary results showed that the new compounds seems to behave as P2X3 receptor antagonist.
Fig.2: Synthetic project-II. A. Designed structure. B. 3D alignment of one of designed purine
derivatives with one of pyrimidine derivatives. Non-polar hydrogen atoms are hidden.
References:
1. Burnstock, G. Purine-mediated signalling in pain and visceral perception. Trends Pharmacol. Sci. 2001, 22, (4), 182-8.
2. Camaioni, E.; Costanzi, S.; Vittori, S.; Volpini, R.; Klotz, K. N.; Cristalli, G. New substituted 9-alkylpurines as adenosine receptor ligands. Bioorg. Med. Chem. 1998, 6, (5), 523-33.
3. Volpini, R.; Costanzi, S.; Lambertucci, C.; Vittori, S.; Martini, C.; Trincavelli, M. L.; Klotz, K. N.; Cristalli, G. 2- and 8-alkynyl-9-ethyladenines: Synthesis and biological activity at human and rat adenosine receptors. Purinergic Signal. 2005, 1, (2), 173-181.
4. Fischer, B.; Yefidoff, R.; Major, D. T.; Rutman-Halili, I.; Shneyvays, V.; Zinman, T.; Jacobson, K. A.; Shainberg, A. Characterisation of “Mininucleotides” as P2X receptor agonists in rat cardiomyocyte cultures. An integrated synthetic, Biochemical, and Theoretical study. J. Med. Chem. 1999, 42, 2685-2696.
5. Donnelly-Roberts, D.; McGaraughty, S.; Shieh, C.C.; Honore, P.; Jarvis, M. F. Painful Puri-nergic Receptors. J. Pharmacol. Exp. Ther. 2008, 324, 409–415.
6. Gever, J. R.; Cockayne, D. A.; Dillon, M. P.; Burnstock, G.; Anthony, P. D.; Ford, W. Pharmacology of P2X channels. Eur. J. Physiol. 2006, 452, 513–537.
7. Carter, D.; Dillon, M. P.; Hawley, R. C.; Lin, C. J. J.; Parish, D. W.; Broka, C. A.; Jahangir, A. Diamminopyrimidines as P2X3 and P2X2/3 antagonists. PCT Int. Appl. WO2005095359, 2005,pp 1-144.

62
TITLE OF THE RESEARCH: MOLECULAR CHARACTERIZATION
OF THE DUALISTIC RECEPTOR GPR17
Meenakshisundaram KANDHAVELU
Department of Chemical Science, University of Camerino, Camerino, Italy. Pharmaceutical Sciences and Pharmaceutical Biotechnology
G-protein coupled receptors (GPCRs) are the largest family of receptor proteins in mammals and play important roles in many physiological and pathological processes including allergies, cardiovascular dysfunction, depression, obesity, cancer, pain, diabetes, and a variety of central nervous system disorders, and represent the most common target of drugs. The progress of human genome sequencing has revealed the existence of several hundred orphan GPCRs (1-3), whose deorphanization and identification is essential in order to clarify their physio-pathological role and identify new drug targets (4, 5). Recently, an orphan GPCR receptor phylogenetically and structurally related to known P2Y receptors (6) has been identified. This receptor, namely GPR17, is a dual uracil nucleotides/cysteinyl-leukotrines receptor, primarily expressed in the brain, and seems to play a key role in the progression of brain ischemic damage (7). This “dual” response is quite unusual, since all known G-protein-coupled receptors are activated by one single family of endogenous ligands. In order to design potent and specific agonists and antagonists for the GPR17 receptor, information about its structure, construction, and confirmation of the potential binding sites will be highly useful. Moreover, the availability of cell lines expressing human GPR17 (hGPR17) at high levels would be extremely useful for the functional characterization of the receptor and the screening of new potential anti-ischemic ligands. The first part of the research has been focused on the construction of a Flag tagged hGPR17 receptor. Flag tagged hGPR17 receptor vector construct has been expressed in COS7 cells and identified by both immuno-cytochemistry assay and western blotting analysis. This expression vector is utilized to purify the target hGPR17 receptor protein by chromatography, etc., The hGPR17 gene, cloned into pcDNA3.1/V5 His-Topo expression vector, has been transfected into mammalian cell lines (1321N1, HEK-293, COS-7 cells). In order to characterize this receptor, human astrocytoma 1321N1 has been chosen as the most appropriated cell line. In fact, this cell line expresses neither nucleotide (P2Y-like) receptors, nor CysLT receptors; therefore it is a useful tool to study the “dual” nature of the hGPR17. However, high levels of receptor expression are very difficult to obtain in these cells because they have a particular resistance to transfection. Therefore, different transfection methods such as Calcium phosphate, Lipofectamine, Fugene, Arrest-InTM, Microporation and Nucleofection have been used and expression of hGPR17 compared by quantitative real time PCR. The results showed different levels of transfection efficiency in the used techniques, with the Arrest-In lipo-polymeric formulation being the best method to promote an enriched hGPR17 gene delivery into human astrocytoma cell line. Furthermore, mutagenesis, useful to identify the ligand binding site including down and up-regulation of biochemical pathway, has been performed. Different techniques are available to introduce a mutation range from site-directed mutagenesis to random mutagenesis in decreasing order of specificity. Site-directed mutagenesis refers to the site-specific introduction of point mutations. In the GPR17, amino acid residues Arg 255, Lys 112 and Lys 116 were predicted by molecular modelling and docking experiments to be essential target in binding site for the receptor signal transduction (8-10). In order to investigate the importance of a single amino acid, a site directed mutagenesis at 255th position by changing Arginine into Isoleucine has been performed. For the identification of mutation, the plasmid has been isolated as well as sequenced by Primm Company. The results showed that 99% homology identities were common with the original gene sequence of GPR17 and also mutated site carrying Isoleucine instead of Arginine has been notified. The clones have been identified as positive site directed mutated clones by BLAST analysis. Moreover, mutation scanning for specific mutation at codon 255 of the gpr17 mutated gene using fluorescence monitoring during the melting experiment has been performed. Fluorescence melting peak analysis revealed a high melting transition with a melting temperature at 80 ˚C and at 82 ˚Ccorresponding to the mutated and wildtype gene sequences. The differences in Tm between the wildtype allele and mutant allele are correlated with the mismatch stability (GC: AT Arg to Ilu). In the last part of the work, tagging GPR17-GFP has been performed. Tagging of GPCRs with the green fluorescent protein (GFP) has enabled the direct visualization of real-time trafficking of GPCRs in living

63
cells. Hence, GPR17-GFP gene construct has been used to express tagged protein in different mammalian cells, allowing the investigation of the hGPR17 protein subcellular localization. Moreover, since hGPR17-GFP is comparable to the authentic protein and the possibility for the tag to affect normal folding of polypeptide chain during translation is minimized, hGPR17-GFP can be use to analyze protein-protein interaction by fluorescence resonance energy transfer (FRET) detection.
References:
1. Lee DK, Nguyen T, Lynch KR, Cheng R, Vanti WB, Arkhitko O, Lewis T, Evans JF, George SR, O'Dowd BF. Discovery and mapping of ten novel G protein-coupled reporter genes. Gene. 275: 83–91, 2001.
2. Joost P, Methner A. Phylogenetic analysis of 277 human G-protein-coupled receptors as a tool for the prediction of orphan receptor ligands. Genome Biol. 3: 1–16, 2002.
3. Wittenberger T, Schaller HC, Hellebrand S. An expressed sequence tag (EST) data mining strategy succeeding in the discovery of new G-protein coupled receptors. J. Mol. Biol. 307: 799–813, 2001.
4. Abbracchio MP, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Miras-Portugal MT, King BF, Gachet C, Jacobson KA, Weisman GA, Burnstock G. Characterization of the UDP-glucose receptor (re-named here the P2Y14 receptor) adds diversity to the P2Y receptor family. Trends Pharmacol. Sci. 24: 52–5, 2003.
5. Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Miras-Portugal MT, King BF, Gachet C, Jacobson KA, Weisman GA. The recently deorphanized GPR80 (GPR99) proposed to be the P2Y(15) receptor is not a genuine P2Y receptor. Trends Pharmacol. Sci. 26: 8–9, 2005.
6. Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. Update on the P2Y G Protein-Coupled Nucleotide receptors: From Molecular mechanisms and pathophysiology to Therapy. Pharmacol. Rev. 58: 1-53, 2006.
7. Ciana P, Fumagalli M, Trincavelli ML, Verderio C, Rosa P, Lecca D, Ferrario S, Parravicini C, Capra V, Gelosa P, Guerrini U, Belcredito S, Cimino M, Sironi L, Tremoli E, Rovati GE, Martini C, Abbracchio MP. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 25 (19): 4615-27, 2006.
8. Hoffmann C, Moro S, Nicholas RA, Harden TK, Jacobson, KA. The Role of Amino Acids in Extracellular Loops of the Human P2Y1 Receptor in Surface Expression and Activation Processes. J. Biol. Chem. 274: 14639-14647, 1999.
9. Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B, Mazarguil H, Vassart G, Parmentier M. and Costentin J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 377: 532-535, 1995.
10. Moro S, Hoffmann C, Jacobson KA. Role of the Extracellular Loops of G Protein-Coupled Receptors in Ligand Recognition: A Molecular Modeling Study of the Human P2Y1 Receptor. Biochemistry. 38: 3498-3507, 1999.

A NEW ANTAGONIST OF Bv8-PROKINETICIN RECEPTORS FOR THE DEVELOPMENT
OF NEW ANALGESICS AND ANTI-INFLAMMATORY DRUGS
Ilaria LAZZARI
Department of Pharmaceutical Sciences and Biotechnology Centre, University of Ferrara Dottorato in Scienze Farmaceutiche XXI Ciclo
An important consequence of tissue injury is the activation and sensitization of nociceptors. Recently it has been identified novel molecular entities which can lower pain threshold and modulate immune responses in laboratory animals and are over-expressed in inflammatory diseases. These molecules are small proteins named Bv8-Prokineticins1 (PK1 and PK2) that activate G-protein linked PK receptors (PKR1 and PKR2) in dorsal root ganglia neurons and in immune and inflammatory cells. PKR1 is widely distributed in the periphery, including the gastrointestinal system, lungs and heart; PKR2 is the dominant receptor in the adult brain, with particularly high expression in the hypothalamus, the olfactory ventricular region and the limbic system. It has been hypothesize that blocking PKR1 will be effective in ameliorating pain arising from tissue injury and additionally will accelerate recovery of normal sensitivity following injury. On the basis of a Janssen’s patent, we synthesized some 1,3,5-triazin-4,6-diones as potential prokineticin receptors antagonists: PC1, PC2 and PC3 (FIGURE 1).
N
N
N
OOO
HNNH2
N
N
N
OOO
HNHN N
H
N
PC2 PC3
N
N
N
OOO
HNHN NH2
NH
PC1
FIGURE 1
Starting from the commercially available di-Boc-S-methylisothiourea the fully protected compound 1 was obtained using classical Mitsunobu conditions. Treatment of compound 1 with TFA afforded the compound 2 in good yield and without further purification was used in the cyclisation reaction. Intermediate 3, with a second Mitsunobu reaction, allowed the insertion of the second benzyl moiety on the nitrogen N3 of the triazine (intermediate 4). The substitution of the thiomethyl function using 1,2-ethylendiammine gave the final compound PC2. Finally, the guanidine (PC1) and the imidazole (PC3) moieties instead of the primary amino function were obtained from PC2 using pyrazolo-1-carboxamidine hydrochloride and 2-thiomethyl-imidazole respectively. (SCHEME 1).
64

BocHN NBoc
SCH3
OH
O
BocN NBoc
SCH3
O
HN NH
SCH3
O
+a b
HN NH
SCH3
O
N
NH
N
S
OOO
c
N
N
N
S
OOOd
N
N
N
OOO
HNNH2
N
N
N
OOO
HNHN NH2
NH
N
N
N
S
OOO
e f
Conditions: a) THF, DEAD, PPh3, R.T. Y=78%; b) TFA, R.T. Y=91%; c) DCM, chlorocarbonyl isocianate, DIPEA,
Y=25% d) THF, DEAD, PPh3, R.T. Y=80%; e) Toluene, ethylendiamine, reflux Y=95%; f) CH3CN, DIPA, pyrazolo-1-carboxamidine or 2-thiomethyl-imidazole.
1 2
2 3 4
4 PC2 PC1
N
N
N
OOO
HNHN N
HN
PC3
SCHEME 1
PC1, PC2 and PC3 have been tested as PKRs antagonists at the Department of Human Physiology and Pharmacology, University of Rome. CHO cells stably transfected with the PKR1 or PKR2 receptors were utilized to evaluate the antagonistic effect of PC1 and analogues on protein Bv8 triggered cytoplasmatic Ca2+ signals. PC1, dose dependently, antagonized the 1 nM Bv8 induced intracellular Ca2+ mobilization in PKR1 and PKR2-transfected CHO cells (IC50 = 100 nM; 100% inhibition 300 nM). PC2 and PC3 slightly reduced Bv8 induced [Ca2+] at 100 nM.
1 L.Negri et al., Life Sciences 81 (2007) 1103-1116
65

NEUROPEPTIDES IN THE REGULATION OF FEEDING
Francesco LAZZARIN
Dipartimento di Scienze del Farmaco, Università degli Studi G. d’Annunzio, Chieti Dottorato di Ricerca in “Scienze del Farmaco” - XXI°Ciclo
INTRODUCTION
Feeding behavior is modulated by the interaction of central and peripheral signalling at the hypothalamic level, and aminergic neurotransmitters have long been implicated in feeding control [1]. Many endogenous peptides and hormones which are involved in feeding regulation have been shown to affect aminergic neurotransmitters in the hypothalamus, which could partially mediate their central nervous system activities. It has been described that cannabinoids can stimulate hunger in man, particularly for palatable foods [2]. Further studies confirmed that 9-tetrahydrocannabinol (THC) may cause overeating in laboratory animals and in humans [3, 4]. Anandamide (arachidonoylethanolamide) was the first endogenous fatty-acid ligand of cannabinoid receptors to be identified, and also induces overeating [5]. In the central nervous system this effect could in part be mediated by the activation of CB1 receptor, which mainly modulates neurotransmitter release at a presynaptic level [6-8].
Gut-derived hormones are powerful modulators of feeding both peripherally and at the central nervous system (CNS) level. Moreover gut peptides contribute to energy balance in humans by influencing feelings of hunger and satiety [9]. Glucagon-like peptide 1 (7-36) amide (GLP-1) is a gut-derived hormone which plays a satiety role after both peripheral and central administration [10, 11]. Exendin-4, isolated from Heloderma suspectum venom, shares 53% peptide sequence with GLP-1 and also inhibits food intake, possibly binding as an agonist to the same GLP-1 receptor [12].
AIMS
In order to further elucidate the mechanism of the central modulation of feeding, in the present study we have evaluated the role of anandamide, two synthetic CB1 receptor agonists [1a,2-(R)-5-(1,1-dimethylheptyl)-2-[5-hydroxy-2-(3-hydroxy-propyl)cyclohexyl]-phenol (CP 55,940) and R-(+)-(2,3-dihydro-5-methyl-3-[{4-morpholinyl}methyl]pyrol[1,2,3-de]-1,4-benz-ozoxazin-6-yl) (naphthalenyl)methanone monomethanesulfate (WIN 55,212-2), GLP-1 and Exendin-4 on dopamine (DA), norepinephrine (NE) and serotonin (5-HT) release from rat hypothalamic neuronal endings (synaptosomes) in vitro.
MATERIALS AND METHODS
Synaptosomes are prepared according to Gray and Whittaker, as previously described [13], suspended in Krebs-Ringer buffer and incubated at 37 °C for 15 min with either 3H-DA, 3H-NA or 3H-5-HT (uptake period). Then synaptosomes are layered onto 0.8 µm Millipore filters, placed into 37 °C water-jacketed superfusion chambers and perfused with Krebs-Ringer buffer (0.6 ml/min). After 30 min perfusion with buffer (equilibration period), perfusate is collected (1 min fractions for serotonin, and 2 min fractions for catecholamines release), and after the first 2 fractions (basal release), the peptide is added to the perfusion buffer for 10 min (stimulus), followed by 10 min with Krebs-Ringer buffer alone (return to basal). To evaluate the effects of the peptides on amine release during depolarization [3 min perfusion with K+(15 mM)], the peptide is added, after the equilibration period, both 20 min prior (pre-stimulus) and 3 min during K+(15 mM) perfusion (stimulus). Finally, β-emission from perfusate fractions, corresponding to each [3H]amine, is detected by liquid scintillation scanning.
ANALYSIS OF DATA
Amine release is calculated as either the means ± S.E.M. of the percentage of 3H-DA, 3H-NA or 3H-5-HT recovered in each fraction compared to total (fractions + filter) or the means ± S.E.M. of the area under
66

the time-response curve (AUC); each group represents the mean ± S.E.M. of 3-5 experiments performed in triplicate. Treatment and control group means are compared by the analysis of variance (ANOVA) followed by Newman-Keul’s multiple comparison test (GraphPad Prism 2.00 software).
RESULTS AND DISCUSSION
We have found that endocannabinoids did not modify basal amine release. On the other hand, anandamide, CP 55,940 and WIN 55,212-2 were able to inhibit depolarization-induced dopamine, norepinephrine and serotonin release in a dose-dependent manner. Glucagon-like peptide 1 (7-36) amide and exendin-4 did not modify dopamine and norephinephrine release. On the other hand glucagon-like peptide 1 (7-36) amide and exendin-4 stimulated basal serotonin release. In conclusion our study suggested that the central role of anandamide, in modulating feeding behavior, could in part be mediated by inhibition of hypothamic dopamine, norepinephrine and serotonin release and supported an involvement of CB1 receptor agonism in anadamide orexigenic activity in vivo [14]. Moreover our study indicated that the central anorexigenic role of GLP-1 and exendin-4 could in part be related to the stimulation of serotonin signalling in the hypothalamus [15].
REFERENCES
[1] Kalra, S.P., Dube, M.G., Pu, S., Xu, B., Horvath, T.L., Kalra, P.S., 1999. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr. Rev. 20, 68-100. [2] Greenberg L., Kuchnle J., Mendelson J. H., Bernstein J. G. Effects of marihuana use on body weight and caloric intake in humans. Psychopharmacol 1976; 49: 79–84. [3] Mattes R. D., Engelman K., Shaw L. M., Elsohly M. A. Cannabinoids and appetite stimulation. Pharmacol Biochem Behav 1994; 49: 187–195. [4] Williams C. M., Rogers P. J., Kirkham T. C. Hyperphagia in prefeed rats following oral D9-THC. Physiol Biol Behav 1998; 65: 343–346. [5] Williams C. M., Kirkham T. C. Anandamide induces overeating: meditation by central (CB1) receptors. Psychopharmacology 1999; 143: 315–317. [6] Christie M.J., Vaughan C.W., 2001. Cannabinoids act backwards. Nature 410, 527–530. [7] Gifford, A.N., Samiian, L., Gatley, S.J., Ashby, C.R., 1997. Examination of the effect of the cannabinoid receptor agonist, CP 55,940, on electrically evoked transmitter release from rat brain slices. Eur. J. Pharmacol. 324, 187-192. [8] Schlicker, E., Timm, J., Zentner, J., Gothert, M., 1997. Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Arch. Pharmacol. 356, 383-389. [9] Wynne, K., Stanley, S., Bloom, S., 2004. The gut and regulation of body weight. J. Clin. Endocrinol. Metab. 89, 2576-2582. [10] D’Alessio,D.A., Vogel, R., Prigeon, R.L., Laschansky, E., Koerker, D., Eng, J. Ensinck,J.W., 1996. Elimination of the action of Glucagon-like Peptide 1 causes an impairment of glucose tolerance after nutrient ingestion by healthy humans. J. Clin. Investigation 97, 133-138. [11] Tang-Christensen, M., Larsen, P.J., Goke, R., Fink-Jensen, A., Jessop, D.S., Moller, M., Sheikh, S.P., 1996. Central administration of GLP-1 (7-36) amide inhibits food and water intake in rats. Am. J. Physiol. 271, 848-856. [12] Rodriquez de Fonseca, F., Navarro, M., Alvarez, E., Roncero, I., Chowen, J.A., Maestre, O., Gomez, R., Munoz, R.M., Eng, J., Blazquez, E., 2000. Peripheral versus central effects of glucagon-like peptide-1 receptor agonists on satiety and body weight loss in Zucker obese rats. Metabolism 48, 709-717. [13] L. Brunetti, B. Michelotto, G. Orlando, M. Vacca. 1999. Leptin inhibits norepinephrine and dopamine release from hypothalamic neuronal endings. Eur. J. Pharmacol. 372, 237-240. [14] L. Brunetti, G. Orlando, C. Ferrante, A. Chiavaroli, S. Leone, F. Lazzarin, M. Vacca. Anandamide inhibits dopamine, Norepinephrine and serotonin release in the hypothalamus. 88th Annual meeting of the Endocrine Society, Boston, June 24-27, 2006, Abstract n° P3-159. [15] L. Brunetti, G. Orlando, S. Leone, L. Recinella, C. Ferrante, A. Chiavaroli, F. Lazzarin, M. Vacca. Effect of glucagon-like peptide 1 (7-36) amide (GLP-1) and exendin-4 on dopamine and norepinephrine release in the hypothalamus. 89th Annual meeting of the Endocrine Society, Toronto, June 2-5, 2007, Abstract n° P3-28.
67

SYNTHESIS AND PHARMACOLOGICAL EVALUATION OF A NEW CLASS OF
CARDIOPROTECTIVE BENZOPYRAN-BASED KATP OPENERS
Mariaelisa MANGANARO
Dipartimento di Scienze Farmaceutiche, Università di Pisa, Via Bonanno 6, 56126 Pisa Dottorato di ricerca in Scienza del Farmaco e delle Sostanze Bioattive- XXI Ciclo
ATP-sensitive potassium (KATP) channels link membrane potassium conductance to cellular metabolism by sensing intracellular nucleotide concentration [ATP]/[ADP]. Their functional role occurs in response to metabolic changes as in hypoxia, ischaemia or changes in glucose concentration. KATP are located in sarcolemmal and inner mitochondrial manner (sarc- and mito-KATP, respectively) in various tissues, such as pancreatic -cells, neurons, skeletal and smooth muscle. In the heart KATP channels have a protective function in response to hypoxia or ischaemia and several pharmacological evidences suggest their involvement in “ischaemic pre-conditioning” (IPC). The IPC is a biological phenomenon in which brief period of coronary occlusion trigger a “self-defence” mechanism which reduces the injury determined by a subsequent and more prolonged ischaemia and gives myocardiocytes an increased resistance against ischaemia-induced cell damage, resulting in a marked reduction of the infart size [1]. This phenomenon, not yet completely clarified, is triggered by several processes, both receptor-mediated (bradykinin, adenosine) and receptor-indipendent (involving NO and reactive oxygen species). Recent experimental data have indicated the endogenous activation of mito-KATP as the main effector in anti-ischemic cardiac protection [2]. Besides, drugs acting as activators of these ion channels are considered promising tools, which confer a “pharmacological pre-conditioning”, thus they represent an innovative rational therapy for myocardial ischaemia. KATP channel openers (KCOs) such as cromakalim, bimakalim and diazoxide, are chemically heterogeneous, but they have limited therapeutic usefulness because of many side-effects, such as hypotension, vasodilation, and hyperglicemia, due to their low selectivity towards the mito-KATP.
Benzopyran is the most investigated class of KCOs and a pharmacophoric model was elaborated in order to improve the cardioselectitvity: as regards substitution at C4 only few cases inserted the conformational restriction into a spiro-like structure, although X-ray analysis revealed that an orthogonally relationship with the plane of the benzopyran ring is necessary for good activity. Some new spiro-morpholone (A) and spiro-morpholine (B) benzopyran-based compounds were prepared in order to determine the effects of these structural modifications on both cardioprotective properties and hypotensive effects [3,4].
O
NY
OX
R
Small lipophylic groups required
Not essential for activity
Unbridged cycles, bridged cycles or acyclic substituents
Aromatic substituents increase cardio-selectivity
O=CH2>>>NH
H
O
A
R'
R'
R
EWG O
A:Y=CO; X= H, Br
B:Y= CH2 ; X= H, Br
4
Cardioselective benzopyran pharmacophore
68

R
O
N
O
O
XBr, H
CH2
C=S
C=O; C=S;
SO2;
Br Me OMe
CF3
NH2
NHMs NHAc
In order to extend the study of this new class of compounds, various substituents on the benzylic group linked to the nitrogen of the spiro-morpholone- and spiro-morpholine-ring were investigated. Further modifications regarded the spiro-heterocycle core and insertion of a thiocarbonyl and a sulphonyl group on the benzylic methylene directly linked to the nitrogen atom. Some of synthesized compounds, tested on Langerdoff perfused rat hearts, showed an appreciable anti-ischaemic cardioprotective effect leading to an improvemet of the functional post-ischaemic parameter and a clear reduction of the ischaemic injured areas. The vasorelaxing effects were also evaluated in vitro and in vivo and finally also the selectivity towards mito-KATP was evaluated.
REFERENCES:
[1] Testai, L.; Rapposelli S.; Calderone V. Cardiovasc. Haematol. Agants Med.Chem., 2007, 5, 79. [2] Garlid, K.D.; Dos Santos, P.; Xie, Z.J. ; Costa, A.D.; Paucek, P. Biochim. Biophys. Acta, 2003, 1606,1-21. [3] M. C. Breschi, V. Calderone, A.Martelli, F. Minutolo, S. Rapposelli, L. Testai, F. Tonelli, A. Balsamo J. Med. Chem. 2006, 49, 7600. [4] Balsamo A.; Calderone V.; Rapposelli S. PCT Int. Appl. 2008, WO2008007210.
69

SYNTHESIS AND EVALUATION NOVEL CONJUGATES BASED ON THE COMBINED
MITOXANTRONE–AMSACRINE PHARMACOPHORES
Erika MARTINA, Alessandra GIANONCELLI, Giuseppe ZAGOTTO
Università degli Studi di Padova – Dipartimento di Scienze Farmaceutiche, Via Marzolo 5 35131 Padova (Italy)
Scuola di Dottorato in Scienze Molecolari – Indirizzo Scienze Farmaceutiche
Abstract
Human toposiomerase are essential nuclear enzymes that regulates the topological state of the DNA. They are an established nuclear target for very effective anticancer drugs interfering with changes the DNA topological state such as anthracyclines, mitoxantrone and amsacrine. Changes of DNA topology are required for virtually all DNA-dependent events such as DNA replication, transcription, recombination, repair and nucleosome remodelling, chromosome condensation and segregation DNA and may cause the formation of tangling structures, such as supercoils or catenanes. The major role of topoisomerases is to prevent DNA tangling. Topoisomerases have been divided in two main groups with regard to their mechanism of interaction with DNA: type I topoisomerases act producing transient single-strand breaks in DNA whereas types II cut both the double-strand at the same time. Type II topoisomerases are more efficient in removing supercoils, but they need the energy from ATP hydrolysis while type I topoisomerase does not. Recently the interaction between the nuclear enzyme topoisomerase II, the DNA and two intercalating drugs, m-AMSA and mitoxantrone, attracted our interest. It is known that both the drugs interact with the nucleic acid intercalating between the base pairs. We decided to better determine the position of planar aromatic part relative to the base pairs and that of the side chains within the grooves.
N
NH
S
O
OO
O
O
OH
OH
NH
NH
NH
NH
OH
OH
m-AMSA
MITOXANTRONE
side chain interacting with the minor groove
side chains interacting with the major groove
Aromatic region which intercalates with the base pairs
To check the literature hypotheses we decided to synthesize new compounds sharing parts of the two molecules. The hybrid molecules should act as intercalators and interact with the topoisomerase II in a new way.
The structures of the new synthesized compounds are represented below:
70

O
O
NH
OS
O
O
R2
NH
NH
R1
R1
Where the mitoxantrone-like chains are made by an ethylamino spacer bearing a N-acetylamino, 2-hydroxy-amino, N-dimethylamino-methyl, , 2-hydroxy-ethoxy, piperazinyl, 3-morpholin-4-yl-methyl substituent. R2 is hydrogen or a methoxyl substituent.
NH
CH3
O
NCH
3
CH3
OOH
NH
OH
N
NH
N
O
R1 = N-acetylamino
N, N-dimethylaminomethyl
2-hydroxy-ethoxy
2-hydroxy-ethylamino
piperazinyl
morpholin-N-methyl
R1=
The compounds were synthesized, characterized and their citotoxicity and Top- II binding properties were tested in order to establish structure-activity relationships. This will help to better define the spatial properties of the topoisomerase II-DNA-dug cleavable-complex.
References:
1: Dal Ben D, Palumbo M, Zagotto G, Capranico G, Moro S., Curr Pharm Des. 2007;13(27):2766-80.
2: Sissi C, Moro S, Zagotto G, Ellis M, Krapcho AP, Menta E, Palumbo M. Anticancer Drug Des. 1999 14(3):265-74.
3: Capranico G, Guano F, Moro S, Zagotto G, Sissi C, Gatto B, Zunino F, Menta E, Palumbo M., J. Biol Chem. 1998 (21):12732-9.
4: Palumbo M, Gatto B, Moro S, Sissi C, Zagotto G. Biochim Biophys Acta. 2002, (2-3):145-54.
71

DESIGN, SYNTHESIS AND BIOLOGICAL ACTIVITY OF NEW CAPROCTAMINE-BASED
COMPOUNDS AS ANTI-ALZHEIMER DRUGS
Andrea MILELLI
Department of Pharmaceutical Sciences, Alma Mater Studiorum-Università di Bologna, Via Belmeloro, 6 - 40126 Bologna
Alzheimer’s disease (AD), the most common cause of senile dementia and a major cause of disability and death in the elderly, is a complex neurological affection that is clinically characterized by loss of memory and progressive deficits in different cognitive domains. AD is characterized by loss of neurons and synapses in the cerebral cortex and in certain subcortical regions. From a neuropathological point of view, the signs of AD are the presence of senile plaques, which are insoluble deposits of amyloid-beta ( A) protein derived from the cleavage of a precursor protein by enzymes such as - and -secretase, and neurofibrillary tangles. AD is also characterized by a pronounced degradation of cholinergic and other neurotransmitter systems, such as glutamatergic and serotoninergic ones. The primary approach used to control the progressive onset of the pathology is focus on increasing the levels of acetylcholine in the brain by using acetylcholinesterase inhibitors (AChEI) such as donepezil, galantamine and rivastigmine. Unfortunately, this kind of approach is just palliative and it does not take into account the multifactorial nature of AD. Nowadays, drug discovery in AD is focusing on the development of a single molecule able to interact with the multiple targets supposed to be responsible for the pathology; this strategy is known as “Multi-target-directed Ligands” (MTDLs)1.The discovery of Caproctamine2 (1) represents one of the first cases of a rationally designed AChEI characterized by a multimodal mechanism of action against AD. Structure activity relationships studies carried out on 13,4 provided derivatives endowed with multipotent biological profile. By exploiting “the frozen analogue approach” we subsequently designed and synthesized new 1-based compounds able to hit several targets involved in the AD pathogenesis.
NN
N
OCH3
O
N
O
OCH31
Such derivatives showed the ability to selectively inhibit Acetylcholinesterase, the AChE-induced and self-mediated A aggregation and -secretase.
(1) A. Cavallli et al., J. Med. Chem 2008, 51, 347 (2) C. Melchiorre et. al., J. Med. Chem. 1998, 41, 4186 (3) V. Tumiatti et al., J. Med. Chem. 2003, 46, 954 (4) V. Tumiatti et al., J. Med. Chem. 2004, 47, 6490
(This work was supported by MIUR, Roma, FIRB RBNE03FH5Y)
72

SYNTHESIS AND PROPERTIES OF NOVEL 10-SUBSTITUTED 2,7-DIAZAPHENOTHIAZINES
Beata MORAK-M ODAWSKA, Krystian PLUTA
Department of Organic Chemistry, The School of Pharmacy, The Medical University of Silesia, ul. Jagiellonska 4, Sosnowiec, Poland
Phenothiazines are the oldest and the largest group of neuroleptic drugs. These compounds are widely used in psychiatry, especially in the treatment of schizophrenia, maniac and delusion disorders. They exhibit valuable antiemetic, antihistaminic and antitussive properties. There have appeared numerous articles for last ten years on new biological properties of phenothiazines, among them anticancer, antibacterial, antiprotozoic, antiviral, antiprionic and multidrug resistance reversal activity [1,2]. Chemical modification of the phenothiazine structure was carried out by replacing the benzene ring with the pyridine ring and by introduction of new substituents in position 10.
Some modifications of phenothiazine structures were directed into azaphenothiazines, where the benzene ring was substituted with azine ring. We modified the phenothiazine structure with the pyridine ring to obtain novel tricyclic azaphenothiazine. 10H-2,7-Diazaphenothiazine 1 was obtained in cyclisation of 3,4-disubstituted pyridines. Further we transformed thiazine 1 into 10-substituted 2,7-diazapheno-thiazines 2-15 possessing alkyl, aryloalkyl, aryl, heteroaryl and dialkylaminoalkyl substituents [3,4].
2-15
N(CH2)2
CH3
1
S
NN
N
H
Ar Ar DMF DMF
N
Cl
NO2
+ +
N
SH
NO2
N
Cl
NH2
N
S
NH2
H
S
NN
N
R
7. -CH2COC6H5
NO2
N
O2N
N
N
O2N
H3C
NN
Cl
CH3
8.
9.
10.
11.
12. -(CH2)2N(C2H5)2
13. -(CH2)3N(CH3)2
14. -CH2CH(CH3)CH2N(CH3)2
15.
R
2. -CH3
3. -C2H5
4. -C4H9
5. -CH2CH=CH2
6. -CH2C6H5
RX
10H-2,7-Diazaphenothiazine 1 was also transformated into the amide 16, 18, 19, 22 and sulfonamide 20, 21 derivatives being the structural diaza-analogs of phenothiazine with potential anticancer and antibacterial activities.
or sulfonylation
+
N OO
CH2)n
NS
NN
(N
O
O
Br(CH2)n
16a, 16b
NS
NN
H
NaH, toluen
NH2NH2C2H5OH/HCl
(
NH2
CH2)n
NS
NN
17a, 17b
n = 3,41
H2)n
N
C
NS
NN
RH
(
��������
R 18 a,b -COCH3
19 a,b -COOC2H5
20 a,b -SO2CH3
21 a,b -SO2C6H5CH3
22 a,b -CONHCH2CH2Cl
18 - 22 a,b
73

The 2,7-diazaphenothiazine structure was confirmed by X-ray analysis of the pyridinyl derivative 9 which revealed the characteristic features of phenothiazines: the folded structure, the boat conformation of the thiazine ring and the quasi equatorial position of the pyridinyl substituent at the thiazine nitrogen atom [3].
9
NS
NN
N
O2N
The calculated (using program CLOGP, CambridgeSoft. Com.) and determined experimentally lipophilic parameters (using Reversed Phase - Thin Layer Chromatography) for 10-substituted 2,7-diazaphenothiazines were lower than the lipophilic parameters for neuroleptic phenothiazines [5,6]. The prediction of biological activity for 10-substituted 2,7-diazaphenothiazines using PASS (Prediction of Activity Spectra for Substance) program enabled to determine potential psychotropic, antimicrobial, anticancer and other activities [7]. 10H-2,7-diazaphenothiazine 1 exhibits in vitro promising anticancer activity with reference to 57 human cancer lines (5 types of leukemia, 9 types of non-small cell lung cancer, 7 types of colon cancer, 6 types of CNS cancer, 8 types of melanoma, 5 types of ovarian cancer, 8 types of renal cancer, 2 types of prostate cancer, 7 types of breast cancer), determined in National Cancer Institute in Bethesda in USA in Development Therapeutics Program 2005/2006 year. The best resalts are in the Table 1 [4].
Table 1.
Anticancer activity GI50 (x10-5 M) TGI50 (x10-5 M) Lung cancer HOP-62 1.43 5.74 Lung cancer HOP-92 0.85 7.66 Colon cancer COLO205 1.19 5.95 Colon cancer HCT-116 1.89 6.95 Renal cancer RXF393 1.56 4.23 Renal cancerA498 1.95 5.54 Leukemia HI-60(TB) 2.05 6.85
GI50 – growth inhibition of 50%, TGI – total growth inhibition
Some selected 10-substituted 2,7-diazaphenothiazines were estimated into examination of anticancer and immunosupressive activity.
References: 1. N. Motohashi, M. Kawase, K. Satoh, H. Sakagami, Curr. Drug Targets, 7, 1055 (2006). 2. L. Amaral, M. Viveiros, E. Kristiannsen, Trop. Med. Int. Health, 12, 1016 (2001). 3. B. Morak, K. Pluta, K. Suwi ska, Heterocyclic Commun., 8, 331 (2002). 4. B. Morak-M odawska, K. Pluta, Heterocycles 71, 1374 (2007). 5. B. Morak, M. Nowak, K. Pluta, J. Liq. Chromatogr. & Rel. Technol. 30, 1845 (2007). 6. B. Morak-M odawska, K. Pluta, J. Liq. Chromatogr. & Rel. Technol. 31, 611 (2008). 7. http://ibmc.msk.ru/PASS/
N4
C24
C25
C21
N5
O1
O2
N3
C3
C2
N1
C6
C5
C4
S1
C13
C12
N2
C16
C15
C14
C22
C23
74

G PROTEIN-COUPLED RECEPTORS AS POTENTIAL DRUG TARGET: FROM RECEPTOR
TOPOLOGY TO RATIONAL DRUG DESIGN, AN IN SILICO APPROACH
Erika MORIZZO
Università degli Studi di Padova – Dipartimento di Scienze Farmaceutiche Scuola di Dottorato in Scienze Molecolari – Indirizzo Scienze Farmaceutiche
G protein-coupled receptors (GPCRs) constitute a very large family of heptahelical, integral membrane proteins that mediate a wide variety of physiological processes, ranging from the transmission of the light and odorant signals to the mediation of neurotransmission and hormonal actions. GPCRs are dysfunctional or deregulated in several human diseases and are estimated to be the target of more than 40% of drugs used in clinical medicine today. The crystal structures of rhodopsin and the recent published crystal structures of human beta2-adrenergic receptor provide the information of the three-dimensional structure of GPCRs, which supports homology modeling studies and structure-based drug-design approaches.
Rhodopsin-based homology modeling represent a widely used approach to built GPCR three-dimensional models. Structural models can be used to describe the interatomic interactions between ligand and receptor and how the binding information is transmitted through the receptor. Both agonist and antagonist like states can be described by several different conformational receptor states depending on the nature of both ligand and receptor. Considering different complementarities, we might explore different conformations of the same pharmacological state. We investigated the molecular pharmacology of adenosine receptors and, in particular, the human A3
adenosine receptor (hA3AR) by using an interdisciplinary approach to speed up the discovery and structural refinement of new potent and selective hA3AR antagonists[1].Human A3 adenosine receptor belongs to adenosine receptors family of GPCRs, which consists of four distinct subtypes: A1, A2A, A2B, A3 that are ubiquitously expressed in the human body. The hA3AR, which is the most recently identified adenosine receptor, is implicated in a variety of important physiological processes. Activation of A3ARs increases the release of inflammatory mediators, such as histamine from rodent mast cells, and it inhibits the production of tumor necrosis factor- . The activation of the human A3AR seems to be involved in immunosuppression and in the response to ischemia of the brain and heart. Agonists or antagonists of A3ARs are potential therapeutic agents for the treatment of ischemic and inflammatory diseases[2].The first model of human A3AR has been built using a conventional rhodopsin-based homology modeling approach. The model has been used to probe atomic level specific interactions, detected using site-directed mutagenesis analysis. The rhodopsin-based model of the hA3AR in its resting state (antagonist-like state) has been revisited, taking into account a novel strategy to simulate the possible receptor reorganization induce by the
antagonist-binding. We called this new strategy ligand-based homology modeling (flow-chart on the left): it is an evolution of a conventional homology modeling algorithm: any selected atoms will be included in energy tests and in minimization stages of the modeling procedure. Ligand-based option is very useful when one wishes to build a homology model in the presence of a ligand docked to the primary template[3].Starting from the conventional rhodopsin-based homology model and
applying our ligand-based homology modeling implementation we can generate other antagonist-like conformational states of human A3AR in which the ligand recognition cavity is expanded.
75

Using different antagonist-like conformational states, we are able to rationalize the observed activities for all the compounds analyzed. Many severe analysis concerning false-positives and false-negatives situations are usually conducted. To strictly validate this methodology as novel tool to address the multi-conformational space of GPCRs, we have analyzed different classes of known human A3 antagonists in the corresponding putative ligand binding site: for example triazoloquinoxalin-1-one derivatives[4], arylpyrazolo-quinoline derivatives[5] and pyrazolo-triazolo-pyrimidines derivatives[6]. These studies led to the identification of groups for every class of antagonists that, introduced one by one in a suitable position, afford high human A3AR affinity and good selectivity. Starting from these binding requirements, we decided to perform an in silico molecular simplification approach to identify a suitable fragmentation route of the 4-amino-triazoloquinoxalin-1-one scaffold and explore which of the structural features were essential to guarantee an efficient ligand–receptor recognition. A schematic representation molecular simplification is shown in the following figure [7]:
With reference to different antagonist-like conformational states of the receptor, we are now investigating topological fluctuation of the binding pocket using molecular dynamic simulations in a POPC bilayer.
Bibliography:
1. Moro, S.; Bacilieri, M.; Deflorian, F.; Spalluto, G. New J. Chem. 2006, 30, 1-8. 2. Moro, S.; Gao, Z.G.; Jacobson, K.A.; Spalluto, G. Med Res Rev. 2006 Mar;26(2):131-59. 3. Moro, S.; Deflorian, F.; Bacilieri, M.; Spalluto, G. Curr. Pharm. Des. 2006, 12 (17), 2175-85. 4. Lenzi, O.; Colotta, V.; Catarzi, D.; Varano, F.; Filacchioni, G.; Martini, C.; Trincavelli, L.;
Ciampi, O.; Varani, K.; Marighetti, F.; Morizzo, E.; Moro, S. J. Med. Chem. 2006, 49(13):3916-3925.
5. Colotta, V., Catarzi, D., Varano, F., Capelli, F., Lenzi, O., Filacchioni, G., Martini, C.,Trincavelli, L., Ciampi, O., Pugliese, A.M., Pedata, F., Schiesaro, A., Morizzo, E., Moro, S. J. Med. Chem 2007, 50 (17):4061-74.
6. Bolcato, C., Cusan, C., Pastorin, G., Spalluto, P., Cacciari, B., Klotz, H.N., Morizzo, E., Moro, S. Purinergic Signalling 2008, 4:39-46.
7. Morizzo, E., Capelli, F., Lenzi, O., Catarzi, D., Varano, F., Filacchioni, G., Vincenzi, F., Varani, K., Borea, P.A., Colotta, V., Moro, S. J. Med. Chem. 2007, 50(26):6596-6606.
76

7-AZAINDOLO-FUSED HETEROCYCLES WITH POTENTIAL ANTITUMOR ACTIVITY
Marina MUSCARELLA
Università di Palermo – Dipartimento Farmacochimico, Tossicologico e Biologico Via Archirafi, 32 – 90123 Palermo
Dottorato di Ricerca in Scienze Farmaceutiche – Ciclo 2005-2006 (XX)
DNA represents one of the most important cellular targets for several chemotherapeutic drugs. Polycyclic nitrogen heterocycles can be good pharmacophores for classes of antineoplastic drugs because of their potential ability to bind to DNA by intercalating between the base pairs of the DNA duplex. Many 1,2,3-triazine derivatives are well known compounds endowed with a wide range of biological activities such as antineoplastic activity. Benzocondensation of 1,2,3-triazine system led to compounds that showed significant cytotoxicity towards M21 melanoma cells and TLX5 tumor [1,2]. When they are condensed with one or more heterocycles, as isoxazolo[5,4-d]1,2,3,-triazines[3], pyrido[3’,2’:4,5]thieno[3,2-d]-1,2,3-triazines, pyrido[3’,2’:4,5] dithieno[3,2-d]-1,2,3-triazines[4] and thieno[3,2-d]-1,2,3-triazines[5], they are useful as antiproliferative agents. Moreover indolo[1,2-c][1,2,3]triazines showed in vitro antitumor activity against leukemia-, lymphoma-, carcinoma-, and neuroblastoma-derived cell lines from micromolar to submicromolar concentrations (IC50 range 0.08-13 µM) [6]. Cinnoline derivatives also demonstrate a wide range of biological activities and particurarly antitumor activity[7,8]. Dibenzocinnolines are inhibitors of topoisomerases I and II and are effective as cytotoxic agents against cancer cells, including drug-resistant cancer cells[8,9]. Indole condensation with cinnolines led to derivatives that showed antiproliferative activity against leukaemia-, lymphoma-, and solid tumor-derived cell lines at micromolar concentrations.[11] In our attempts to search for novel antitumor compounds, we extended our interest to the 7-azaindole system and planned to synthesize 7-azaindole[3,2-c]cinnolines 5 and 7-azaindole[1,2-c][1,2,3] benzotriazines 9 with the aim of evaluating their antitumor activity.
N
Li
1
2
3
N
Me
CN
NH2 NN
H
NH2
4R
NN
N N
H
N
NN
NO
5
R
NN
H
NHAc
6R
NN
H
NHAcNO
7
NN
H
NH2
NO
8RRR
9
R
77

7-Azaindole intermediates 4 were obtained in good yields using an intramolecular Chichibabin-type reaction. Deprotonation of methylpyridine 1 with LDA, followed by reaction with substituted benzonitriles 3, gave an intermediate which reacted with a second equivalent of LDA to give derivatives 4. Diazotization reaction of amines 4, carried out at 0°C in acetic acid with stoichiometric amount of sodium nitrite, followed immediately by intramolecular cyclization at the 3 position of the 7-azaindole moiety gave, in high yields, the corresponding 7-azaindolocinnolines 5.
2-(2-Aminophenyl)-7-azaindoles 4 are also intermediates for the synthesis of the new ring system 7-azaindole[1,2-c][1,2,3]benzotriazine 9. Compounds 4 were acetylated to give the corresponding acetylamino derivatives 6 which were converted to 3-nitroso-7-azaindoles 7. Removal of the protecting group gave the amino compounds 8, which upon diazotization afforded the expected 7-azaindolo-benzo-1,2,3-triazines 9.All the synthesized derivatives were submitted to the National Cancer Institute, and five of them have been selected by the NCI for the evaluation of the antiproliferative properties.
[1] Faye, P.L. et al., Can. J. Chem., 1983, 61, 179. [2] Vaughan, K. et al., Can. J. Chem., 1985, 63, 2455. [3] Ryng, S. et al., Il Farmaco, 1997, 52, 105. [4] Quintela, J.M. et al. Eur. J. Med. Chem., 1998, 33, 887. [5] Paronikyan, E.G. et al., Pharm. Chem. J., 2006, 40, 293.[6] Cirrincione, G. et al., J. Med. Chem., 1999, 42, 2561. [7] Doria, G. et al., EP277791 1988 (Chem. Abstr. 1988, 109, P231043). [8] Lavoie, E.J. et al., WO 032631 2001 (Chem. Abstr. 2001, 134, P353314). [9] Yu, Y. et al., Bioorg. Med. Chem. 2003, 11, 1475. [10] Matsuna, K. et al., WO 9814431 1996 (Chem. Abstr. 1998, 128, P257447). [11] Barraja, P. et al., Bioorg. Med. Chem. 1999, 7, 1591.
78

DESIGN AND SYNTHESIS OF NOVEL ADENOSINE NUCLEOTIDE ANALOGUES: THE
INTRODUCTION OF DIVERSITY INTO THE CARBOHYDRATE OR THE BASE SUBUNITS
OF MODIFIED NUCLEOTIDES AS PROMISING STRATEGIES TO IDENTIFY SPECIFIC P2
RECEPTOR LIGANDS
Carmela NAPOLITANO
Department of Pharmaceutical Sciences, Univerity of Ferrara Via Fossato di Mortara 17-19, 44100 Ferrara (Italy)
Dottorato di Ricerca in Scienze Farmaceutiche – XXI° Ciclo
Extracellular nucleotides regulate a wide variety of functional responses in many cell types by stimulation of both G-protein coupled receptors (P2Y) and ATP-gated ion channel receptors (P2X).1 The binding motifs of these molecules are associated with a broad array of targets of therapeutic importance in biological systems.
Synthetic analogues of nucleotides have been the cornerstone of antiviral therapy over the past 30 years. Since the discovery of extracellular purine and pyrimidine nucleotides as endogenous cell function modulators, the interest in adenosine receptors has continued to grow as a promising target of immense therapeutic potential.2
The medicinal chemistry at P2 nucleotide receptors has been relatively slow to develop due to a variety of limitations in the study of nucleotide pharmacology. Nucleotides are subjected to metabolic processes, leading to both their degradation and production.3 As recently reported, 4 the isosteric substitution of the diphosphate moiety of natural substrates with phosphonoacetic acid ester and amide moieties lead to overcome the in vivo enzymatic hydrolysis: the introduction of both of those structural modifications reflects in a stronger interaction of the ligand with the active site.
Taking into account our previous results, we have explored the introduction of diversity into the carbohydrate or the base subunits of the modified nucleotides described above. In this preliminary communication, we describe the design and the preparation of novel analogues derived from adenosine nucleosides. These ligands would have the advantages of being selective for a single subclass, being relatively easy to synthesize, having a charge suitable for crossing cell membranes readily, being resistant to cleavage consequent to metabolism, and having activity that is greater than that of ATP itself. It is early recognized that introducing the modiifications discussed above represents a promising strategy to identify specific receptor ligands, enzyme inhibitors, or nucleoside function modifiers.
1 Ralevic, V.; Bursntock, G. Pharmacol. Reviews 1998, 50, 413. 2 Epple, R.; Kudirka, R.; Greenberg, W. A. J. Comb. Chem. 2003, 5, 292. 3 Jacobson, K. A.; Costanzi, S.; Ohno, M.; Joshi, B. V.; Besada, P.; Xu, B.; Tchilibon, S. Curr.Topics in Med. Chem. 2004, 4, 805. 4 M.-C. Bonache; Buzzoni, L.; Ciliberti, N.; Napolitano, C.; Manfredini, S. 8th International Symposium on Adenosine and Adenine Nucleotides, Ferrara (Italy), May 24-28, 2006.
79

INVESTIGATION OF ANTIPRION ACTIVITY OF ACRIDINE DERIVATIVES
Thi Hanh Thuy NGUYEN
Department of Pharmacy, National University of Singapore Medicinal Chemistry Program
Abstract
A library of acridine derivatives were synthesized and evaluated in vitro for their antiprion activity. Among compounds tested, compound 6-chloro-2-methoxy-N-(4-(4-methylpiperazin-1-yl)phenyl)acridin-9-amine (compound 17) was able to clear scrapie prion proteins in three different prion strains including two mouse strains (RML and 22L) and one human strain (Fukuoka-1) in N2a, a mouse neuroblastoma cell line and N2a#58 which overexpressed prion proteins. The effects on cellular prion proteins were investigated for these compounds but additional studies are required to confirm.
Introduction
Prion diseases or transmissible spongiform encephalopathies (TSEs) are a group of fatal neurodegenerative disorders that occur in humans and animals, e.g. scrapie, bovine spongiform encephalopathy, chronic wasting diseasein animals and Creutzfeldt-Jakob disease (CJD), kuru in humans. Currently, there is no therapeutic treatment and prophylactic tools.1 The causative agent of TSEs has been attributed to the conversion of the normal cellular prion protein (PrPC) to a scrapie form (PrPSc) whose biochemical properties are distinctly different.1 PrPSc forms aggregates in brain and it is believed that the conversion process is fatal.2
Quinacrine, a longstanding antimalarial drug, is a potentially useful antiprion agents (EC50 = 0.3 µMagainst the murine neuroblastoma ScN2a cell line)3. Unfortunately, its good in vitro activity did not translate to promising in vivo activity2 because of its high tissue binding. Based on these studies, we hypothesized that structural modification of the quinacrine template can give rise to compounds improved pharmacokinetic properties and potency
Library design and chemical synthesis
The 9-substituted arylamino-6-chloro-2-methoxyacridine compounds were synthesized by displacement of 6,9-dichloro-2-methoxyacridine and corresponding amines using catalytic HCl in ethanol (scheme 1) or via formation of phenoxy intermediates (scheme 1 and scheme 2).
NH+
Cl
OCH3
Cl
+ H2NR
NH+
Cl
OCH3
HNR
Scheme 1: Ethanol, cat. HCl, reflux, 24 hours.
NCl
OCH3
Cl
NCl
OCH3
OPh
a b
NCl
OCH3
NHR
Scheme 2: a)PhOH, 1h, 100oC b)RNH2, 4-5 h, 100oC
Some amines were synthesized by Hartwig-Buchwald amination followed by catalytic reduction (Scheme 3).
I
NO2 NO2
NHR3
NH2
NHR3
H2N
R3
a b
Scheme 3: a) Pd(OAc)2, BINAP, Cs2CO3, anhydrous toluene, 120oC b) Pd/C, H2
80

Biological assays
1) Screening for antiprion activity: The assay was based on the difference in proteinase K sensitivity of PrPC and PrPSc. The harvested proteins were subjected to digestive action of proteinase K. PrPC was readily digested by proteinse K and hence was not detected by western blotting. In contrast, PrPSc was resistant to enzymatic activity. It reacted with the prion antibody (anti-PrP mAb) which then bounb to the secondary antibody (antimouse IgG). The resulting complex was detected by chemiluminescence.
Figure 1: Antiprion activities of compound 17 against four cell lines
2) Evaluation of compounds’ effects on total PrPC and cell-surface PrP
C
To determine the effect on total prion expression, the prion protein was harvested from non-infectious N2a cells as described above without proteinase K digestion. The results showed that compounds did not inhibit the expression of PrPC.The cellular PrPC is a glycosylated cell-surface protein held in situ by a glycophosphatidylinositol (GPI) anchor4. To further probe the effect of the test compounds on cell-surface prion protein expression, N2a cells were incubated with fluorescent antibodies and the fluorescent signal was measured by flow cytometry. A reduction in the cell-surface prion protein expression would result in a (left) shift towards the origin.
Conclusion
We have shown that structural modification of the 9-substituted amino side chain of quinacrine resulted in several promising compounds with good antiprion potencies and selectivities on four prion infected murine cell models. This included a mouse adapted human prion strain (F3). Both side chains have not been previously associated with antiprion activity and are interesting leads for further modification. Further investigations would involve evaluating their activities in vivo, and to assess their permeability across the blood brain barrier.
References
1) ChemBioChem 2003, 4, 1268-1284. 2) Journal of Veterinary Pharmacology and Therapeutics 2003, 26, 315-326. 3) PNAS 2001, 98, 9836-9841. 4) Free Radical Biology and Medicine 1996, 20, 933-956.
45.7
32.5
18.4
Fluoresecence in log scale Figure 2: Retention of mAb-PrPC complexes on the cell surface. Overlapping of the control and test lines indicated that the compound did not affect cell-surface PrPC expression.
Cel
l num
ber
81

STRUCTURAL AND CONFORMATIONAL ASPECTS AFFECTING THE MOLECULAR
RECOGNITION OF SUBSTRATES AND INHIBITORS BY INDOLEAMINE-2,3-
DIOXYGENASE (IDO), A NOVEL TARGET FOR CANCER THERAPY.
Roberto NUTI
Dipartimento di Chimica e Tecnologia del Farmaco Università di Perugia, 06123 Perugia (Italy)Dottorato di Ricerca in Chimica e Tecnologia del Farmaco a Profilo Internazionale. Ciclo XXI°
Indoleamine 2,3-dioxigenase (IDO) is a heme-containing enzyme that catalyzes the first and rate-limiting step of the Kynurenine pathway along the major route of Tryptophan (L-Trp) catabolism.1 The reaction consists in the oxidative cleavage of the pyrrole ring of L-Trp and the production of N-formyl kynurenine. Since its discovery in 1963 by Hayashi,2 the only physiological role ascribed to IDO was the inhibition of the growth of various infections pathogens in vivo.3 The observation that the expression of IDO is induced by IFNγ3 and the discovery of the involvement of the enzyme in protecting the fetus from maternal immunity,4 have considerably increased the interest in the enzyme as an important key player in the control of the immune response. Furthermore, the involvement of IDO in the mechanisms of immune tolerance has led to associate the early observations of elevated expression of the enzyme in various human cancers to the participation of IDO in the tumor immuno-editing process.5 Accordingly, IDO promotes tumor outgrowth by counter-regulating inflammatory immune responses that would hamper tumor cell survival,6 and thereby making the host environment more hospitable to tumor survival and growth. A number of endogenous and non-endogenous compounds have been reported to inhibit IDO activity. Results from preclinical studies of small molecule inhibitors of the enzyme have shown the feasibility to block the IDO mediated tumor immuno-editing process and to enhance the efficacy of current chemotherapeutic agents.7 These data support the notion that IDO is a novel therapeutic target for the development of new cancer drugs. In my work of thesis, I have investigated substrate recognition and enhancer binding at IDO using extensive molecular docking experiments. Another part of the work consisted in studying the conformational transitions of IDO in response to substrate and enhancer binding through the application of coarse graining simulations.8 When compared to traditional molecular dynamic simulations, coarse graining simulations are not able to give information on the energy associated to the different conformational states of the protein. However, their main advantages are the low computational cost and the increased speed required to have a qualitative view of the conformational space of the protein in order to identify the molecular events leading the transitions between different conformational states. Since they do not utilize force-field potentials, coarse graining simulations also overcome issues related to parameterization of cofactors that usually affect the standard force-field based molecular dynamics simulations. The use of docking experiments on substrates and enhancers in the structure of human IDO aided to identify a putative binding site for the enhancers and to the construct of different complexes of the enzyme: IDO-Fe(III)/superoxide (complex A), IDO-Fe(III)/superoxide/L-Trp (complex B), IDO Fe(III)/superoxide/L-Trp/3-indole- ethanol (complex C). These complexes were used as input structures to perform a FIRST analysis in order to identify rigid and flexible regions of the protein and, then, a FRODA analysis to explore the conformational space of each complex. The results allowed to identify key structural and conformational elements involved in substrate recognition. In particular, different states of the enzyme are observed in the final conformations of complexes A-C that involve a diverse conformation of two key structural elements: an electrostatic gate composed by Arg231 and the 7-propionate moiety of heme, and the region 355-385. Each of these elements may adopt a closed and open conformation. While in the enzyme unbound state both elements are in the open conformation, in the substrate bound complex the electrostatic gate and region 355-385 are closed stabilizing the pose of the substrate within the catalytic site. The presence of the enhancer (IDOFe(III)/superoxide/L-Trp/3-indole- ethanol, complex D) induces the opening of region 355-385 while keeping closed the electrostatic gate over the substrate. Although we are aware of the current limitations suffered by the adopted computational methodologies, on the basis of these results we rise the hypothesis that the movements of the electrostatic gate and region 355-385 control the shift between the inactive and active states of the enzyme by regulating the access of
82

substrates inside the catalytic site and the exit of products. Along with this line, we propose that the mechanism of enhancement of the enzymatic activity by 3-indol-ethanol occurs increasing the breakdown rate of the ternary complex by opening the region 355-385 which favors the exit of products. In the last part of my work, the results of substrate recognition and coarse graining simulations have been instrumental to investigate the binding mode of the competitive inhibitors. At this aim, docking studies of inhibitors have been performed in different states and conformations of IDO. Six states were generated combining, the two different oxidative states of heme-iron, and the presence or not of superoxide/water molecules in the binding site. Three different rotamers of Arg231, at the binding site, were utilized to set up three IDO conformations. In this way, matching that, fifteen IDO models were generated and used for the docking of forty inhibitors of IDO. A Linear Regression Analysis (LRA) between the activity data and the binding energy of the inhibitors for every IDO models were performed to validate the models. The LRA point out that no statistically significant correlation exist between the ligands and the evaluate different states of enzyme. But If the inhibitors are considered separately, one groups the Brassinin derivates9 and the second one the Tryptophan derivates 7, a better results were noticed, with a statistical significant correlation between different IDO status and every of the two classes.These results can suggest a diverse binding mode for the competitive inhibitors of IDO in accord with differences in structure scaffolds. These information will be used to address the design of novel selective and potent IDO inhibitors and to set up structure-based virtual screening protocols.
Reference
1. Ruddick, J. P.; Evans, A. K.; Nutt, D. J.; Lightman, S. L.; Rook, G. A.; Lowry, C. A. Tryptophan metabolism in the central nervous system: medical implications. Expert Rev Mol Med 2006, 8, 1-27.
2. Higuchi, K.; Hayaishi, O. Enzymic formation of D-kynurenine from D-tryptophan. Arch Biochem Biophys 1967, 120, 397-403.
3. Taylor, M. W.; Feng, G. S. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. Faseb J 1991, 5, 2516-22.
4. Munn, D. H.; Zhou, M.; Attwood, J. T.; Bondarev, I.; Conway, S. J.; Marshall, B.; Brown, C.; Mellor, A. L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281, 1191-3.
5. Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B. J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 2003, 9, 1269-74.
6. Muller, A. J.; Prendergast, G. C. Indoleamine 2,3-dioxygenase in immune suppression and cancer. Curr Cancer Drug Targets 2007, 7, 31-40.
7. Muller, A. J.; Malachowski, W. P.; Prendergast, G. C. Indoleamine 2,3-dioxygenase in cancer: targeting pathological immune tolerance with small-molecule inhibitors. Expert Opin Ther Targets 2005, 9, 831-49.
8. Wells, S.; Menor, S.; Hespenheide, B.; Thorpe, M. F. Constrained geometric simulation of diffusive motion in proteins. Phys Biol 2005, 2, S127-36.
9. Gaspari, P.; Banerjee, T.; Malachowski, W. P.; Muller, A. J.; Prendergast, G. C.; DuHadaway, J.; Bennett, S.; Donovan, A. M. Structure-activity study of brassinin derivatives as indoleamine 2,3-dioxygenase inhibitors. J Med Chem 2006, 49, 684-92.
83

SYNTHESIS OF NEW STEROIDAL 2-OXAZOLIDONES, AS NOVEL POTENTIAL INHIBITORS OF 17 -HYDROXYLASE-C17,20-LYASE
Dóra ONDRÉ
Department of Organic Chemistry, University of Szeged, Dóm tér 8, H-6720 Szeged, Hungary
Prostatic cancer is the second leading cause of cancer-related mortality worldwide. 17 -hydroxylase-C17,20-lyase (P45017 ) is a key enzyme regulating the androgen biosynthetic pathway. Inhibition of this enzyme can block androgen synthesis at an early stage, and may therefore be useful in the treatment of prostatic carcinoma. A broad class of steroid derivatives substituted at C-17 with heterocyclic rings such as imidazole are also good inhibitors of this enzyme. We set out to synthesize a novel series of steroidal oxazolidinones, containing the heterocycle with two heteroatoms at position 17β.During the alkaline methanolysis of 3 -acetoxy-21-chloropregn-5-ene-20 -N-phenylurethane, and its 4-monosubstituted and 3,5-disubstituted phenyl derivatives (4),cyclization occurs, in the course of which 17 -[3-(N-phenyl)-2-oxazolidon-5-yl]androst-5-en-3 -ol and its substituted phenyl derivatives (5) are formed. The cyclization takes place with (N¯-5) neighboring group participation. The reaction of 3 -acetoxy-21-azidopregn-5-en-20 -ol (3d) with triphenylphosphine gave a phosphinimino derivative, which reacted in situ with carbon dioxide with the participation of the sterically favored 20 -OH to give the unsubstituted steroidal cyclic carbamate (8).Oppenauer oxidation of the 3 -hydroxy-exo-heterocyclic steroids yielded the corresponding 4-3-ketosteroids (7, 10). The inhibitory effects (IC50) of these compounds on rat testicular C17,20-lyase were investigated with an in vitro radioligand incubation technique. The N-unsubstituted 17 -(2-oxazolidon-5-yl)-androst-4-en-3-one derivative (10) was found to be a potent inhibitor.
AcO
C O
CH3
1AcO
C O
H2C
2
OAc
AcO
C
H2C
3
R
OHH
AcO4
C OCONHR
H2C Cl
R1O5 (R1 = H)
6 (R1 = Ac)
O
NOR
R1O
O
HNO
7
O
NOR
O
10
O
HNO
O
3 R
a OAc
b OH
c Cl
d N3
4, 5, 6, 7 R a phenyl b 4-fluorophenyl c 4-chlorophenyl d 4-bromophenyl e 4-methoxyphenyl f 3,5-dimethylphenyl
H
2'
4'
5'
8 (R1 = Ac)
9 (R1 = H)
References:Ondré, D.; Wölfling, J.; Iványi, Z.; Schneider, Gy.; Tóth, I.; Szécsi, M.; Julesz, J. Steroids (2008) submitted
84

MOLECULAR DESIGN OF NEW SELECTIVE AND NON-SELECTIVE INHIBITORS OF
GLYCOGEN SYNTHASE KINASE 3
Dmitry OSOLODKIN
Moscow State University, Department of Chemistry Ph.D. course: Organic & Medicinal Chemistry
The search for the kinase inhibitors is a very popular problem in the contemporary medicinal chemistry. There is about 600 different kinases identified in the human genome, all of them having the therapeutic potential. Search for the selective ATP-competitive kinase inhibitors is a difficult and fascinating problem because of the notable similarity between the ATP-binding sites of different kinases.
Glycogen synthase kinase 3 is a widely distributed serine/threonine kinase participating in several important pathways connected with such diseases as diabetes, Alzheimer's disease, Huntington's disease, inflammation, schizophrenia, mood disorders and cancer. Inhibitors of the kinase may be potentially used for the treatment of all these diseases except cancer, because in the oncogenic pathways the kinase is inhibited whereas in the other disease pathways it is activated. Nevertheless, GSK-3 inhibitors cannot cause cancer, because the initial activation of the tumorigenic process is necessary for the GSK-3 inhibition.
GSK-3 belongs to the CMGC family of kinases, which contains also MAP-kinases, Cyclin-dependent kinases (CDK) and CDK-like kinases. The selectivity of the GSK-3 inhibitors is usually measured against CDK-2, being the most close homolog of GSK-3. This kinase participates in the cell cycle, and its inhibition may be helpful in the cancer treatment, but the opposite action of GSK-3 inhibition in cancer should be unwanted. Thus, despite of potential interest for dual GSK/CDK inhibitors, selectivity is more actual problem.
The only approved drug targeting GSK-3 to date is the lithium ion. The mechanism of lithium inhibition is not ATP-competitive, but Mg-competitive. The binding of lithium to the kinase is poor (Ki 2 mM), so under therapeutic concentration (1 mM) only partial inhibition is achieved. Lithium is widely used for schizophrenia treatment, but it has unwanted side effects like possible teratogenicity.
ATP-competitive inhibitors of GSK-3 represent different structural classes. The most known classes are paullones, bisarylmaleimides, indirubins, pyrazolopyridines, pyrazolopyrimidines and pyrazolopyridazines; there are also examples of the kinase inhibitors from other structural classes. The activity of the inhibitors varies from micromolar to nanomolar range, and selectivity differs from poor to acceptable. There are about ten compounds currently on preclinical testing of about 900 compounds, structures of which were published in the available literature and patents.
There are also two classes of ATP-noncompetitve inhibitors discovered to date: manzamines (alkaloids from marine sponges) and thiadiazolidinones (TDZD). These compounds possess perfect selectivity, because they bind outside the conserved ATP-binding site, but their activity is in micromolar range. The new noncompetitive inhibitors are the most promising among many classes of GSK-3 inhibitors.
Three-dimensional structures of the complexes of GSK-3 with inhibitors and ATP analogues are available in the PDB. Good resolution (up to 1.8 Å) of the structures and diversity of conformations of the key residues together with different inhibitors allows extensive virtual screening against the ATP-competitive binding site and neighboring regions. Inhibitors with the features binding outside the ATP binding site are very promising because they should possess high selectivity due to differences between the kinase structures outside the ATP binding site. We have performed virtual screening of the ZINC databases and identified several classes of compounds which may be potential GSK-3 inhibitors. Also the binding mode of the known inhibitors was revised and revisited in some cases.
We have also performed the search for TDZD and manzamine binding site with the docking methods and identified two potential places which may accept the ligand: the axin binding site and the cavity near the primed phosphate binding site. The binding constants were calculated for both binding sites with the help of molecular dynamics and MM_PBSA method, and the best site was selected for virtual screening.
The 3D structure of the kinase can also be used for the de novo design of new potentially active compounds. We have used such method for the design of new derivatives of well-known scaffolds, such as paullone scaffold or indirubin scaffold. Then we selected the most synthetically accessible structures from the predicted database to perform further experimental investigation.
85

There were several QSAR and 3D-QSAR models built for different classes of compounds. Nevertheless, reasonable results of the 3D-QSAR modeling were obtained only for molecules without rotatable bonds, because the conformational space of conformationally flexible molecules was not investigated thoroughly and symmetry of the molecules was not taken into account. Our CoMFA and CoMSiA models lack these shortages and represent the correlation between the structure and the activity more strictly. CoMFA models were aligned with the crystal structure of the complexes where applicable, and there was an evidence that molecular fields correlate with the positions of the aminoacid residues crucial for the ligand binding. The CoMFA models can be used for the prediction of the activity of the new compounds.
We have also performed the 3D pharmacophoric search through the ZINC database. The most interesting compounds were selected for the further experimental investigation.
The problem of the kinase selectivity was tackled by different methods. The method of Molecular Field Topology Analysis (MFTA), developed in our laboratory, was used for the evaluation of selectivity inside the known classes of GSK-3 inhibitors. The results were checked by the CoMFA of the selectivity fields and docking into the X-ray structure of CDK-2.
86

SYNTHESIS AND BIOLOGICAL EVALUATION OF N-(ARYLOXYALKYL)PHTHALIMIDES
AND ISOLOGUES AS NOVEL -GLUCOSIDASE INHIBITORS
Rossana PASCALE
Dip. Farmaco-Chimico, Università degli Studi di Bari Dottorato di Ricerca in Scienze Farmaceutiche – XXI Ciclo
-Glucosidase is a hydrolytic enzyme catalyzing, in the small intestinal enterocytes, the cleavage of glycosidic bonds in carbohydrate digestive process. -Glucosidase inhibitors can retard mono- and disaccharide liberation from dietary complex carbohydrates and delay their absorption. The final result is a post-prandial hyperglycemia reduction.1
Therefore, molecules able to inhibit -glucosidase biological activity can be useful in the treatment of diabetes, obesity, and certain forms of hyperlipoproteinemia. The anti-glucosidase drugs acarbose (Precose®), miglitol (Glyset®) and voglibose (Basen®) are currently used in type II Diabetes Mellitusterapy.2
O
OH OH
OH
OH
OH OH
OH
NH
O
H3C
OH OH
O
O
OH OH
OH
O OH
N
OH
OH
OH
OH OH
OH
OH
NH
OH
OHOH
OH
OH
Acarbose Miglitol Voglibose
The low gastrointestinal tolerability (flatulence, abdominal discomfort, and sometimes diarrhoea) and the relatively high cost have substantially limited their use.3 As a consequence, recently the scientific interest towards new non-glycosidic inhibitors has grown.4, 5, 6
On the basis of pharmacological studies involving thalidomide, it was found that molecules with phthalimidic scaffold show significant in vitro anti-glucosidase activity.7
In order to investigate structure-activity relationships and to improve the druglikeness, about one hundred new phthalimide derivatives have been prepared (general structure 1) and tested on recombinant type I
-glucosidase from Saccharomyces cerevisiae.The intermediate chain length and patterns of substitution were investigated.
R1
= H, Cl R2 = H, Me, Ph
n = 0, 2− 6, 8−10 m = 0, 1
X = O, S, Se, NH, NCH3 Ar = substitued Ph, Naphthyl
N
O
O
(CH2)n CH (CH2)m
R1R1
R1
R1
X Ar
R2
1
87

Molecules with general formula 2 exhibited sub-micromolar IC50 values, being 100 times more potent than the well-known natural inhibitor 1-deoxynojirimycin (dNM) and 2–10 times more potent than phthalimide analogues reported in the literature.7
The synthetic strategies as well as biological results will be presented.
References
[1] Lebovitz, H.; Clin. Diabetes 1995, 13, 99–103. [2] Krentz, A. J.; Bailey, C. J.; Royal Society of Medicine Press 2001.
[3] Krentz, A. J.; Bailey, C. J.; Drugs 2005, 65, 385–411. [4] De Melo, E. B.; da Silveira Gomes, A.; Carvalho, I.; Tetrahedron 2006, 62, 10277–10302. [5] Hai-Wei X.; Gui-Fu D.; Gai-Zhi L. et al.; Bioorg. Med. Chem. 2007, 15, 4247–4255. [6] Gao H. and Kawabata J.; Bioorg. Med. Chem. 2008, 18, 812–815. [7] Takahashi, H.; Sou, S.; Yamasaki, R.; Sodeoka, M.; Hashimoto, Y.; Chem. Pharm. Bull. 2000,
48, 1494–1499.
N
O
O
(CH2)10 O R1
R2
R3
R4
2
R1 = H, Cl, NO2
R2 = H, Cl, NO2, CF3
R3 = H, Cl, NO2, CF3
R4 = H, Cl, NO2, CH3
88

DEVELOPMENT OF NEW INDOLYL ARYL SULFONES (IASs) AS POTENT
ANTI-HIV AGENTS
Francesco PISCITELLI
Istituto Pasteur – Fondazione Cenci Bolognetti,Dipartimento di Chimica e Tecnologia del Farmaco, Sapienza Università di Roma
PhD Course (XXI cycle) in Pasteurian Sciences
HIV (Human Immunedeficiency Virus) is the etiological agents of AIDS (Acquired Immuno Deficiency Syndrome), that caused 2.5 million death and 2.1 million newly infected people in 2007.
More than 20 antiretroviral drugs are available and fall into six class: nucleoside and nucleotide reverse transcriptase inhibitor (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitors (FIs), integrase inhibitors (IIs), and CCR5 antagonists. Three (or four) antiretroviral drugs are combined in the highly active antiretroviral therapy (HAART) that proved to be effective to reduce morbidity and mortality of HIV-infected people. However, HAART is unable to eradicate the viral infection and the needed long-term or permanent treatments favor the emergence of drug resistance, toxicity and unwanted side effects.
NNRTIs have received a great attention because of low toxicity and favorable farmacokinetics properties. Currently, four NNRTIs are approved for AIDS treatment (nevirapine (NVP), efavirenz (EFZ), delavirdine and etravirine), and more than 30 classes of structurally unrelated NNRTIs have been described.
IASs have showed a potent inhibitory activity against HIV-1 wild-type and other mutant strains. Figure 1 shows structure-activity relationships (SARs) for a good antiviral activity. The carboxamide function and chlorine atom at position 2 and 5 of the indole, respectively, are essential for the activity; reduction of sulfone group to sulfoxide or sulfur atom decreased the inhibitory activity; the presence of 3,5-dimethyl substitution at the aryl sulfonyl at position 3 of indole nucleus is required for a potent activity against mutant strains.
Molecular modeling study showed the interaction between IASs and HIV-1 reverse transcriptase (RT). The indole NH is essential for the H-bond with lys101; introduction of aminoacids at the carboxamide function increases inhibitor-RT interaction.
Figure 1. IASs SARs.
In the successive study we replaced the chlorine atom with a bromine atom or nitro group; furthermore we also evaluated the introduction of a second halogen atom at the other position of indole nucleus. Bromine and nitro derivatives retained the potent inhibitory activity, but in many instances the
89

toxicity was decreased. Among dihalo IASs, the introduction of fluorine atom at position 4 of indole nucleus increased the activity against HIV-1 wild-type and mutant strains. Compound 1 inhibited HIV-1 wild-type RT at subnanomolar concentration, and showed low toxicity (ED50 = 0.5 nM; TC50 > 20000 nM; SI(TC50/ED50) > 40000). Furthermore, 1 (Y181C: ED50 = 4 nM) was 2.5 times more active than EFV (Y181C: ED50 = 10 nM) against Y181C mutant strain and it was > 667-fold more active than NVP against K103N-Y181C double mutant strain.
We synthesized IASs derivatives with different aminoacids and different groups at the carboxamide function. The new compounds were synthesized (scheme 1) by reaction of appropriate ethyl indole-2-carboxylates with 1-(3,5-dimethylphenylthio)pyrrolidine-2,5-dione to obtain the corresponding 3-(3,5-dimethylphenylthio)-indole derivatives that was oxidized to sulfone by treatment with m-chloroperbenzoic acid (MPBCA). Hydrolysis of ester gave the corresponding carboxylic acids which were converted in the desidered compounds. In the case of ethyl 5-bromo-1H-indole-2-carboxylate, it was obtained by Fischer indole synthesis. 4-bromophenyl hydrazine hydrochloride was converted in the appropriate hydrazone that was cyclized by treatment with poliphosphoric acid (PPA). Ethyl 5-chloro-4-fluoro-1H-indole-2-carboxylate was initially synthesized by Fisher indole synthesis to obtained a mixture of 5-chloro-4-fluoro and 5-chloro-6-fluoro substituited indole that was separated by repeated column chromatography with overall yield of 5%. We improved the preparation of this indole protecting 3-fluoroaniline as N-pivalamide and then chlorinated with NCS to give N-(4-chloro-3-fluoro-2-methylphenyl)pivalamide (2). Acidic hydrolysis of N-pivalamide 2 afforded 4-chloro-3-fluoro-2-methylaniline (3) which was oxidized to the corresponding nitro derivative (4) with MCPBA. Reaction of 4 with diethyl oxalate in the presence of sodium ethoxide gave ethyl 3-(3-chloro-2-fluoro-6-nitrophenyl)-2-oxopropanoate (5). Finally, reduction of 5 with iron powder by heating at 60 °C in acetic acid and subsequent intramolecular cyclization of the intermediate amino derivative provided the desiderated indole with overall yield > 39%.
Scheme 1. Synthesis of IASs.
90

BINDING OF FENOTEROL DERIVATIVES AND STEREOISOMERS OF FENOTEROL TO
THE 2 ADRENERGIC RECEPTOR. A MOLECULAR MODELING STUDY.
Anita PLAZINSKA, Krzysztof JOZWIAK, Joanna KOZAK
Medical University of Lublin, Department of Analitycal Chemistry
Molecular generally higher affinities relative to their (R,S), (S,R) and (S,S) stereoisomers modeling is a
very attractive and a promising tool used by chemists to predict structure and properties of chiral
compounds, while some mathematical models are assumed. In particular, it can be applied to molecular
drug design, where enantioselectivity plays a crucial role. The binding of the stereoisomers of fenoterol
and fenoterol derivatives to the 2 adrenergic receptor has been analyzed. The binding of 26 agonists
(fenoterol and its derivatives) has been studied using comparative molecular field analysis (CoMFA). The
results indicate that the fenoterol derivatives interact with two separate binding sites and one steric
restricted site on the pseudo-receptor. A docking analysis has also been performed using the determined
binding sites and the template ligand TA2005. Both CoMFA and docking analysis demonstrate that (R,R)
stereoisomers have.
91

OH
R
NOH
XH
R'
R''
OH
OH
N
SOH
R
RR'
H
N
O
OH
NH
O
OH
R
R'
ON
OH
R
O
NNH2
R
NH
S
OH
R
R'
substituent(aromatic)
core structure
substituent
general estrogen ligand pharmacophore
AA'
oxime derivatives
central core
substituent
ERβ pharmacophore
A
A'
A'
A'
A'
X = O, NH, NMe, NEt
A'
A'
R, R' = H, OH, OMe
R, R' = H, OH, OMe
3
4
salicylaldoxime
amide thioamide
hydroxybenzosoxazoles
aminobenzosoxazoles hydroxybenzothioazoles
NEW LIGANDS FOR ESTROGEN RECEPTOR ββββ
Giovanni PROTA
Dipartimento di Scienze Farmaceutiche, Università di Pisa. Scuola di Dottorato in “Scienza del Farmaco e delle Sostanze Bioattive”- XXI Ciclo
The estrogen receptors (ERs), which are members of a superfamily of nuclear receptors, are able to mediate the estrogen action in many organs and tissues (reproductive, skeletal, cardiovascular and central nervous).
There are two subtypes of the estrogen receptor known as ERα and ERβ, that have different distributions in various estrogen target tissues and also have different functions. Selective estrogen receptor modulators (SERMs) are a special category of estrogens, that show an estradiol agonist-like action in some tissues, but antagonize estradiol in others. The typical estrogen ligand pharmacophoric model contains a generic and quite variable core structure bearing one phenolic ring (A, Figure 1) and a second aromatic substituent which can be differently substituted (R, Figure 1). In addition, this generalized ligand structure typically tolerates the presence of one or two additional substituents, one of which may be another aromatic group.
Despite the implication of ERα in the “typical” estrogenic action has been established, the role held by beta is not yet fully understood. Sometimes, ERβ seems to work against the activity of the other subtype ERα, the iso-form that induce cancer growth in some organs (uterus, breast glands, ovary). Moreover, it is now known that stimulation of ERβ by a β-selective agonist may lead to therapeutic action in some hormone dependent tumours. Thus, molecules possessive agonist activity and selectivity for ERβ(SERBAs) could be used in the treatment of these type of diseases.
The major differences between the non-selective general estrogen ligand pharmacophoric model and the ERβ pharmacophore are the lack of the third aromatic substituent and the presence of the phenolic A ring condensed to a central core, so that the ligand is smaller in volume (Figure 1).
In a previous investigation by our research group new potential selective estrogen receptor modulators (SERMs) with non-steroid structures were studied. It was shown that some oxime derivatives, comprised of a six-membered pseudocycle (A’) (see Figure 1) formed by an intramolecular H-bond, could isosterically replace one phenol ring (A) in ER ligands. Thus, a class of diarylsubstituted salicylaldoximes,1,2 that could efficiently bind the ERs, was developed.
Figure 1. Compound originated from the general estrogen ligands pharmacophore and from the ERβ pharmacophore.
92

In this series, the diaryl-salicylaldoxime bearing a single p-OH group in the 3-phenyl substituent (X = O, R’ = 4-OH, R = H, Figure 1) showed an improved ERβ affinity.2 Contemporarily, it was also found that the replacement of the pseudocyclic oxygen with a nitrogen atom, generated a new series of anthranylaldoximes, among which the N-methyl-substituted one (X = NMe, Figure 1) possessed the highest affinity for ERβ.3
My PhD thesis project was dedicated to the development of ERβ-selective ligands, possibly showing a prevalent agonist character. To this purpose, we have followed two different approaches.
In the first one, we have designed a new potential ERβ ligand, by combining the two structural features that had previously given the highest ERβ-selectivity in the diarylsubstituted compounds series, that are: 1) the N-Me-anthranyloxime central core; 2) the single p-OH group in the 3-phenyl substituent. These two features were introduced into a new N-Me-anthranyloxime possessing a 3-(p-OH-phenyl)-substituent (X = NMe, R’ =4-OH, R = H, Figure 1).4
The second approach that we followed was based upon the pharmacophore model for ERβ, that inspired the design and the synthesis of various classes of monoarylsubstituted molecules containg different central scaffolds, including salicylaldoximes,5 aminobenzoxazoles, hydroxybenzosoxazoles, and hydroxybenzothiazoles. In this approach, we have also developed phenol-substituted amides and thioamides that could be included in the same ERβ-pharmacophore model (Figure 1).
ERβ binding affinity properties of our molecules were determined by a radiometric competitive binding assay with [3H]-estradiol. Some of the most significant results we have obtained so far have shown that the monoarylsubstituted salicylaldoximes (Figure 1) possessing an additional phenol group were the most selective ERβ-agonist agents.5 In particular, the salicylaldoxime bearing a 3-Chloro and a 4-(p-OH-phenyl) substituents showed the highest of ERβ-selectivity together with a mostly agonist character.5 Studies are underway in order to complete the biological screening of all the compounds herein reported.
(1) Minutolo, F.; Bertini, S.; Papi, C.; Carlson, K. E.; Katzenellenbogen, J. A.; Macchia, M. J. Med. Chem. 2001, 44, 4288-4291. (2) Minutolo, F.; Antonello, M.; Bertini, S.; Rapposelli, S.; Rossello, A.; Sheng, S.; Carlson, K. E.; Katzenellenbogen, J. A.; Macchia, M.. Bioorg. Med. Chem. 2003, 11, 1247-1257. (3) Minutolo, F.; Antonello, M.; Bertini, S.; Ortore, G.; Placanica, G.; Rapposelli, S.; Sheng, S.; Carlson, K. E.; Katzenellenbogen, B. S.; Katzenellenbogen, J. A.; Macchia, M. J. Med. Chem. 2003, 46, 4032-4042.(4) Minutolo, F.; Tuccinardi, T.; Bestini, S.; Martinelli, A.; Ortore, G.; Placanica, G.; Prota, G.; Rapposelli, S.; Carlson, K. E. ; Katzenellenbogen J. A.; and Macchia M., J. M. C., 2006, 49, 5001-5012. (5) Minutolo, F.; Bellini, R.; Bertini, S.; Carboni, I.; Lapucci, A.; Pistolesi, L.; Prota, G.; Rapposelli, S.; Solati F.; Tuccinardi, T.; Martinelli, A.; Stossi, F., Carlson, K. E.; Katzenellenbogen, B. S.; Katzenellenbogen J. A. and Macchia M., J. M. C., 2008, 51, 1344–1351.
93

DESIGN AND SYNTHESIS OF MULTI-TARGET-DIRECTED COMPOUNDS FOR THE
TREATMENT OF ALZHEIMER’S DISEASE
Stefano RIZZO
Department of Medicinal Chemistry University of Bologna Doctorate School in Medicinal Chemistry
Alzheimer’s disease (AD) is a progressive brain disorder that seriously affects memory, thinking, and reasoning skills making the simplest daily activities tough to be carried out. Since the date of its discovery (1907) AD has always been a topical subject and a challenging matter to deal with for researchers worldwide. Through the decades, several steps have been taken towards the understanding of the pathology and nowadays two main hypotheses seem to explain its onset. The first one is called “cholinergic”, being related to those events occurring at the cholinergic synapses, such as a diminished synthesis of acetylcholine (ACh) due to a slower activity of choline acetyltransferase (ChAT), a deficit of neurotransmitter axonal transport and of choline reuptake, together with a numeric decrease of nicotinic and muscarinic M2 central presynaptic receptors. The second one is called “amyloid” and refers to an abnormal metabolism of APP protein, a transmembrane glycoprotein which is usually processed by the enzyme α-secretase to generate, in physiological conditions, small and soluble peptides. In pathological conditions, the sequential actions of β-secretase followed by γ-secretase generate two predominant Aβpeptides, either 40 or 42 amino acids in length, that are able to aggregate into fibrils via soluble oligomers resulting, as generally accepted, in neuronal toxicity. Other hypotheses involving the oxidative and the inflammatory stress, contribute to define the multifactor nature of the disease. Current treatment of AD focuses on symptomatic aspects of the pathology and is based on drugs increasing cholinergic neurotransmission by inhibiting acetylcholinesterase (AChE), like donepezil, rivastigmine or galantamine. On the other side, in recent times, the role of butyrylcholinesterase (BuChE) inhibition in AD has received increasing attention, both from the medicinal chemistry and the clinical points of view. Several lines of evidence indicate that BuChE might be a co-regulator of the activity of the neurotransmitter ACh. Remarkably, cortical levels of BuChE show a significant increase in AD. In addition, many drug discovery approaches are now targeting the slowing and/or blocking of the amyloid polymerization process. Thus, due to the complexity of AD and the involvement of different enzymes in its progression, the modulation of a single protein might not be sufficient to produce the desired efficacy. To this purpose, designing multi-target compounds is becoming a valid strategy and it could represent a more effective therapeutic approach. According to this new paradigm, we have synthesized a series of hybrid
O
O N
O
SKF-64346
O(CH2)7NCH2
CH3
R
general formula AChEI
OO(CH2)7NCH2
CH3
O
O(CH2)nNCH2
CH3
R
1
CH2 insertion
94

compounds(1) starting from 1, carrying a benzofuran motif, which is responsible for inhibiting Aaggregation,(2) linked by means of a heptyloxy spacer-chain to a benzylmethylamino moiety for the cholinesterases (AChE and BuChE) inhibition(3).SAR studies that we have performed so far have mainly focused on modifications of 1 affecting the furanic ring and the alkyloxy chain. In the first case a series of Friedel Crafts acylations have led to derivatives that modulate the A aggregatory activity. Furthermore, a methylene insertion at the indicated position was crucial to evaluate whether the coplanarity between the two aromatic rings had a role towards the biological activity. In the second case, we have tried to optimise the cholinesterase activity by lengthening or shortening the spacer-chain (n = 3-9 carbon atoms) and by moving the latter from the parato the metha position of the phenyl ring. These modifications turned out to have a positive effect on the anti-amyloid activity. Further biological investigation will try to elucidate the mechanism of action of these compounds. In 1996 Inestrosa and co-workers observed that AChE is prone to promote A aggregation and attributed a key role to its peripheral anionic site (PAS).(4) The PAS lies at the entrance of the enzyme’s gorge and is composed of five residues (Tyr 70, Asp 72, Tyr 121, Trp 279 and Tyr 334; Torpedo numbering). Associated with it are a number of surface loops, conferring a high degree of conformational flexibility to the area. Beside the amyloid deposition, the PAS is involved in the allosteric modulation of catalysis at the active centre, in cell adhesion and neurite outgrowth. Being aware of it all, we considered to synthesize a series of AChE dual binding inhibitors clearly inspired to the Tacrine’s structure. These compounds were designed with the aim to simultaneously interact with the central and the peripheral anionic sites so that they could show an A antiaggregant activity, in addition to the classic anticholinesterase one.
N
NH(CH2)7NHR
N
NH(CH2)nR
References
1) Rizzo, S.; Rivière, C,; Piazzi, L.; Bisi, A,; Gobbi, S.; Bartolini, M.; Andrisano, V.; Morroni, F.; Tarozzi, A.; Monti J.P.; Rampa, A. Benzofuran-Based Hybrid Compounds for the Inhibition of Cholinesterase Activity, Amyloid Aggregation, and A Neurotoxicity. J. Med. Chem. A.S.A.P.
2) Howlett, D. R.; Perry, A. E.; Godfrey, F.; Swatton, J. E.; Jennings, K.H.; Spitzfaden, C.; Wadsworth, H.; Wood S.J.; Markwell, R.E. Inhibition of fibril formation in -amyloid peptide by a novel series of benzofurans. Biochem. J. 1999, 340, 283-289.
3) Rampa A., Piazzi L., Belluti F., Gobbi S., Bisi A., Bartolini M., Andrisano V., Cavrini V., Cavalli A., Recanatini M., Valenti P. Acetylcholinesterase Inhibitors: SAR and kinetic studies on -[N-methyl-N-(3-alkylcarbamoyloxyphenyl)-methyl]-aminoalkoxyaryl derivatives. J. Med. Chem. 2001, 44, 3810-3820.
4) Inestrosa, N. C.; Alvarez, A.; Perez, C. A.; Moreno, R. D.; Vicente, M.; et al. Acetylcholinesterase accelerates assembly of amyloidbeta- peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881-891.
95

SYNTHESIS AND ENANTIOMER SEPARATION OF NEW PDE4 INHIBITORS AS
POTENTIAL DRUGS IN ALZHEIMER DISEASE
Alessia ROMUSSI
Dipartimento di Scienze Farmaceutiche - Università degli Studi di Genova Viale Benedetto XV, 3 16132 Genova
Scuola di dottorato in "Scienze e tecnologie della Chimica e dei Materiali" corso di "Scienze Farmaceutiche, Alimentari e Cosmetologiche" XXI Ciclo
Cyclic nucleotide phosphodiesterase type 4 (PDE4) is a family of isoenzymes playing an important role in the specific control of intracellular cAMP levels in many tissues, including brain.1 The selective inhibition of PDE4 provides potential, novel therapeutics for the treatment of inflammatory diseases, such as asthma, chronic obstructive pulmonary disease and psoriasis. Recently, a number of biological and pharmacological studies have shown PDE4 as critical modulators of memory processes and suggested PDE4 inhibitors as possible therapeutic agents in cognitive distresses. Rolipram, a well known PDE4 inhibitor, antagonizes the memory impairment caused by a deficit in postsynaptic cAMP/PKA/CREB (cAMP-Response-Element-Binding) pathway and counteracts the tendency of Abeta42 to suppress them, as recently confirmed by an experimental murine model of AD.2
The four PDE4 genes encodes different isoforms (A, B, C and D, classified in 21 subtypes) which are also named “long”, “short” or “super-short” on the base of its different primary structure.3 PDE4B and PDE4D (particularly PDE4D1-D5) are highly expressed in hippocampus and cortex regions4 but it is to ascertain what isoforms are over expressed in the different brain regions affected in AD patients. Rolipram indifferently inhibits all the PDE4 isoforms causing several side effects that dishearten its therapeutic use. In addition, the lack of selective inhibitors towards the different (long, short or super short) forms prevented, until now, a clear connection between different isoform, their brain localization and biological function. Our studies are focused on the search for selective PDE4 inhibitors, active on CNS, as potential therapeutics for Alzheimer’s disease. Several rolipram related 3-cyclopentyloxy-4-methoxybenzaldehyde derivatives (among which there were compounds 1a-c), previously synthesized by us,5 both inhibited the superoxide anion production and increased the cAMP level in human neutrophils TNFα stimulated; this activity was related to a good selective PDE4 inhibition. Compound 1c, showing the most interesting biological profile, was assumed as a lead to develop a new series of molecules possibly selective towards PDE4D isoform.
N
H
O
∗
OH
NR'R''
H3CO
O-NR'R'': a =
b = c =
N
N
ON1a-c
Preliminarily, a computational simulation of the binding of compound 1c into the catalytic unit of PDE4 enzyme was performed by a molecular docking procedure. Then, two little series of 1a-c acyl derivatives (compounds 2a-c and 3a-c) were designed to verify the role of the hydroxyl group in the propyl chain. The first series were synthesized as raceme form and they were submitted to human PDE4 in vitroenzymatic assay. In the meanwhile, we developed an analytical study for the enantiomers separation by chiral HPLC. Compounds 1a-c, 2a-c and 3a-c were screened on a chiral HPLC unit equipped with 12 chiral columns and a twelve positions valve which allows to analyze, at the same time, one compound on different columns with different eluants systems. The separation was optimized for all the compounds. In a second time, only the racemes resulted active as PDE4 inhibitors (1c, 2c and 3c) were resolved by semi-preparative chiral HPLC to afford, in very high e.e%, about 200 mg for each enantiomer, which also were tested on PDE4 assay. Many attempts to establish the enantiomers absolute configuration, by X-ray analysis of diastereoisomer derivatives, failed owing to the lack of suitable crystals.
96

On the basis of the biological results we planned further structural modifications of 1c to give a third series of active compounds in which morpholine has been replaced by other different amines. (compounds 4a-f). In addition, the propyl chain was chlorinated (compound 5) and its length was modified (compounds 4b, 7a,b and 10a,b). The reaction scheme, which will be discussed in session poster, is reported below:
H3CO
H
O
OBr
H3CO
H
O
HO
+N
H
OH N
H
N
H
+
N
H
HO
syn (E) 97% anti (Z) 3%
OO
O
∗
OH
NR'R''
1a-c, 4a-f
H3CO
O
H3CO
O
H3CO
O
H3CO
O
2a-c, 3a-c
O
R
N
H
O
∗
O
NR'R''
R
O
H
N
H3CO
O
O
O
Br
6
H
N
H3CO
O
O
O
NR'R"
7a,b
H
NO N
OCl
H3CO
O
5
H
N
H3CO
O
O OEt
OH
N
H3CO
O
O OH
O
H
N
H3CO
O
O NR'R''
O
8
10a,b
9
All the new compounds, in raceme form, were tested on human PDE4 and the most active one (4a) was screened on a panel of height different PDE4 isoforms (A4, B2, C2, D1, D2, D3, D4 and D5) considered as the major representatives. The same compound was also tested on PDE2, PDE3, PDE5 and PDE6 to verify its selectivity. Interestingly, the results of this extensive screening showed compound 4a as a selective inhibitor of the long forms PDE4D3, PDE4D4 and PDE4D5, even if less potent than the reference compound rolipram (see fig 1). No other PDE4D3-D5 selective inhibitors have been reported until now in the literature. The biological results suggested relevant SAR considerations that represent the rational base for the design of new, more potent PDE4 inhibitors selective toward the long isoforms D3, D4 and D5. Synthesis, analytical methods and complete biological data will be presented and discussed in session poster.
Figure 1.
0
10
20
30
40
50
60
70
80
90
100
PDE4A4 PDE4B2 PDE4C2 PDE4D1 PDE4D2 PDE4D3 PDE4D4 PDE4D5
% i
nib
izio
ne (
10
mic
roM
)
4a
Rolipram
1 Houslay, M. D. et al., Drug Discovery Today. 2005, 10, 1503-1519. 2 Bourtchouladze, R., Proc. Natl. Acad. Sci. USA 2003, 100, 10518-10522. 3 Houslay, M. D. Prog. Nucleic Acid Res. Mol. Biol. 2001, 69, 249-315. 4 H.T. Zhang et al., Psychopharmacology, 2005, 179, 613-619 5 Bruno, O. et al., Farmaco. 2004, 59(3), 223-235.
97

(+)-MR200 DERIVATIVES.
MODIFICATIONS ON THE AMINO AND CARBOXYLATE MOIETIES
IN VITRO AND IN VIVO PHARMACOLOGICAL EVALUATION
Simone RONSISVALLE
University of Catania, Dep. of Chemical and Pharmaceutical Sciences PhD in Pharmaceutical Sciences
Sigma receptors, represent a binding site constituted by a typical protein different from opioid, NMDA, dopaminergic, and other known neurotransmitter or hormone receptor families. Pharmacological data based on ligand binding studies, anatomical distribution, and biochemical features distinguish at least two subtypes of receptors. The sigma-1 subtype is better characterized at the functional and structural level and shows a very high affinity for the dextro isomers of cis-normetazocine derivatives such as (+)-pentazocine and (+)-SKF-10,047. The (+)-pentazocine represents a typical selective agonist that was used as tritiated ligand to label sigma-1 receptors. Other selective ligands are the putative antagonist NE-100 and AC915. Studies of anatomical distribution of sigma-1 subtype showed that in the central nervous system (CNS) the regions with high levels are the areas involved in motor, sensory, and endocrine functions, and memory. In peripheral tissues, they are present in placenta, liver, immune cells, and gastrointestinal tract. A protein of 223 amino acid corresponding to sigma-1 receptors has been purified and cloned, first from guinea pig liver and subsequently from human, rat, and mouse tissues. This protein shows no analogy with other mammalian proteins, but it has homology with a yeast protein (C8-C7
isomerase) involved in the ergosterol biosynthetic pathway. The high expression of sigma-1 receptors in steroid-producing tissues and in CNS suggests their possible role in functions of neuroendocrine and central neuroactive steroid system. Additional involvement of sigma-1 receptors has been postulated in psychosis with modulation of synthesis and release of neurotransmitters such as acetylcholine and dopamine. Moreover, they seem to be involved in modulation of the glutamatergic system with a neuroprotective effect and an improvement in learning and memory in animal models of amnesia. Pasternak et al. showed that sigma-1 receptors constitute a potent antiopioid system in which typical agonists such as (+)-pentazocine administered with analgesic opioid reduced notably the antinociceptive potency. Conversely, the coadministration of analgesic opioid with putative sigma-1 antagonist haloperidol increases opioid analgesia. The antiopioid modulator system involves all opioid receptors, but it seems that kappa opioid analgesia is particularly affected. The molecular mechanism involved in all the above-reported effects is not completely clear, but new interesting data provide evidence that modulation of intracellular Ca++ level by sigma-1 receptor/ankrin/IP3 receptor-complex is a possible key of action. Considering the sigma-2 subtype, few biochemical data are available. The reasons might be the absence of cloned protein and the small number of selective ligands with respect to sigma-1 or other receptor systems. The (+)-pentazocine showed a very low affinity for sigma-2 receptors. Conversely, analgesic ( )-pentazocine, antipsychotic haloperidol, guanidine DTG [N,N -(o-tolyl)gunidine], possess high affinity for both sigma subtypes. Selective sigma-2 subtype compounds, such as alkaloid ibogaine and phenylmorphans CB-64D and CB-184, have been reported, but unfortunately these compounds showed also activity and affinity for NMDA (ibogaine) and µ opioid (CB-64D, CB-184) receptors. Sigma-2subtypes have a different pattern of anatomical distributions in CNS and in peripheral tissues. A very high concentration of sigma-2 receptors in neuronal and non-neuronal tumor cell lines provides evidence of a possible role in cell proliferation and viability. In fact, putative sigma-2 agonists, such as ibogaine and CB-64D, induce apoptotic death by a new mechanism involving the increase of intracellular Ca++ level by release from endoplasmic reticulum and subsequently from mitochondrial stores. These effects are inhibited by putative sigma-2 antagonists BD1047 and BD1063. This physiological involvement of sigma-2 receptors led to the conclusion that potent and selective ligands could be diagnostic tools and drugs in anticancer therapy. Recently, we reported the synthesis and pharmacological evaluation of (+)-MR200 [(+)-methyl (1R,2S)-2-{[4-(4-chlorophenyl)-4-hydroxypiperidin-1-yl]methyl}-1-phenyl-cyclopropanecarboxylate] as a new selective ligand with high affinity for sigma receptors (1.5 and 21.9 nM for sigma-1 and sigma-2 respectively) and better selectivity with respect to dopaminergic, serotoninergic (5HT1A) and adrenergic (α1) receptors. Moreover, this compound increased opioid analgesia showing the same putative antagonist profile of haloperidol. Thus, (+)-MR200 could be a speculative tool to reduce adverse side effects related to opioid analgesia.
98

However, considering that only sigma1 is involved in modulation of opioid analgesia, the good binding affinity of this compound at the same time for sigma-2 could provide potential neurological side effects. To ameliorate the sigma-1/sigma-2 selectivity we have modified (+)-MR200, structurally related to haloperidol, as potent and selective sigma receptor ligands, onto amino and carboxylate moieties.
N
OHCl
O
OCH3
(+)-MR200 (+)-methyl (1R,2S)-2-{[4-(4-chlorophenyl)-4-hydroxypiperidin-1-yl]methyl}-1-phenylcyclopropanecarboxylate
Moreover, to evaluate the stereoselectivity, all single Z and E enantiomers were obtained with stereoselective reaction. The enantiomers (+)-Z-(1R,2S) and ( )-Z-(1S,2R) were obtained following modified procedures reported by Shuto et al. (1996). The (+)-E-(1R,2R) and ( )-E-(1S,2S) enantiomers were synthesized for the first time by us and absolute stereoconfiguration was assigned by X-ray crystallographic analysis of ester obtained from reaction of (+)-(1S)-10-camphorsulfonyl chloride and (+)-E-methyl 2-(hydroxymethyl)-1-phenylcyclopropanecarboxylate.
In vitro pharmacological agonist or antagonist activity of compounds with the better binding profile will be presented. In addition, for the most selective sigma-1 compounds, the relationship between sigma receptor and opioid analgesia will be also reported. For the selective sigma-2 compound [(–)-MRML16] the agonist/antagonist properties were evaluated in opportune cell lines through an in vitro citotoxicity test.
99

DESIGN, SYNTHESIS AND PRELIMINARY PHARMACOLOGICAL EVALUATION OF NEW
POTENTIAL TOOLS FOR THE TREATMENT OF CENTRAL NERVOUS SYSTEM DISEASES
Sabrina RUGGIERI
Department of Chemical Sciences , University of Camerino School of Advanced Studies in Pharmaceutical Sciences - XXI cycle
Several pathological conditions such as Parkinson’s disease (PD) and schizophrenia have been linked to a dysfunction of dopaminergic transmission1.Parkinson’s disease (PD) is a neurodegenerative disorder associated primarily with loss of dopamine (DA) neurons in the nigrostriatal system.2 Current therapy for PD is essentially symptomatic, and L-(-)-3-(3,4-Dihydroxyphenyl)alanine (L-DOPA), the immediate biological precursor of dopamine, is still considered the drug of choice in the treatment of Parkinson's disease. Substitution therapy with L-DOPA is, however, associated with a number of acute problems. The drug undergoes extensive decarboxylation to dopamine by amino acid decarboxylase (AADC) in the gastrointestinal tract before entering the systemic circulation and is converted by catechol-O-methyltransferase (COMT) into the inactive metabolite 3-O-methyldopa before crossing the blood-brain barrier.The peripheral conversion of L-DOPA to dopamine is responsible for the typical gastrointestinal (nausea, emesis) and cardiovascular (arrhythmia, hypotension) side effects. Although to minimize its peripheral conversion to dopamine L-DOPA is usually administrated in combination with peripheral inhibitors of AADC (carbidopa and benserazide), other central nervous side effects such as dyskinesia, on-off phenomenon and end-of-dose deterioration still remain. These effects might be reduced by attenuating peaks and rapid fluctuations of L-DOPA plasma levels3,4
The bioavailability, after oral administration, is only 33% being limited by metabolic instability, low water solubility (1,8 mg/ml) and low solubility in lipids (LogP = -2,38). In order to improve the bioavailability the prodrug approach appeared to be promising. Several L-DOPA prodrugs have been prepared over the past decades in an effort to overcome these problems.5
An ideal L-DOPA prodrug should be soluble in water and in lipids, completely absorbed by the gastrointestinal tract without any chemical degradation or metabolism, and thus deliver intact L-DOPA in the blood stream at a reproducible therapeutic level. Thus we designed a multi-protected L-DOPA prodrugs (1a-d) with the aim to increase the bioavailability after oral administration. Conjugation of L-DOPA with glycine will give a peptide that may be converted in carrier systems showing higher lipophylic profile, lower sensitivity to gastric and hematic chemical environment. At the same time the designed prodrugs should be able to release the drug by both spontaneous chemical or enzyme catalyzed hydrolysis. The new compounds have been synthesized and preliminarily evaluated for their water solubility, Log P, chemical stability and enzymatic stability. Schizophrenia is a CNS pathology first described over one century ago. Since that time many discoveries have been done, but still several pathophysiology details are unclear. Some neurochemical explanations have been proposed to rationalize an antipsychotic therapy, but the dopamine hypothesis, emerged forty years ago, still remain one of the main possibility. Since the discovery of chlorpromazine the antipsychotic drugs have been the treatment of choice for schizophrenia. Their usefulness appears to be accounted to the high affinity and antagonistic activity at D2 receptors. However, antipsychotic drugs are responsible of severe side effects mainly related to the dopamine receptor antagonistic activity.6,7 A newer approach seems to be represented by the use of D2-like receptor partial agonists namely drugs which have affinity, but limited intrinsic activity for the receptors.8 Drugs with such a mechanism of action could act as antagonists where the dopaminergic activity is oversized, and, at the same time, as agonists where the dopaminergic activity is reduced. In this manner, the negative side effects associated to the treatment with DA antagonists can be avoided. In most of DA D2-like agonist structures the pharmacophoric framework 2-(m-hydroxyphenyl)ethylamine or its bioisosters is easily recognizable. However molecules showing DA D2 partial agonist activity lacking of such structural requirement have been described.9 In the above structure-activity relationship
1a 1b 1c 1d
R1 H H COCH3 COC(CH3)3
HNN
COOCH3
RR
OR1
R1O
O
R CH3 CH2CH3 CH3 CH3
100

studies several phenolic and non-phenolic structures based on the new identified pharmacophoric moiety 2-(3-hydroxyphenoxy)ethylamine 2, are reported. The same authors discovered that conformational restrictions given by dioxane and dihydropyran rings 3 play a beneficial role on D2-like receptor affinity. On the other hand, conformational restrictions involving the benzylamine moiety have not been investigated yet. Thus, we have designed and synthesized a series of compounds with the general structure 4 embedding the reported pharmacophoric moiety in a partially rigid molecule.
NH
O
OH OH
O
NH
X
X= O; CH2
N
O
OH
R
R= H; CH3
2 3 4
Several studies report the versatility of the imidazoline ring in modulating the ligand profile, depending on the particular kind of substituent inserted in position 2 of imidazoline nucleus.10 Imidazoline compounds have not been yet explored as DA receptor ligands, but the well known prototype of I2
receptor ligands 2-(benzofuran-2-yl)-4,5-dihydro-1H-imidazole (2-BFI) 5 behaved also as DA indirect agonist and displayed a 47 µM binding to the DA D2 receptors.11 Therefore, with the aim to improve the DA D2-like receptor binding affinity, novel molecules 6a-i based on the 2-BFI scaffold were designed and synthesized. The substituents R
1-R
3 were chosen among those reported to be beneficial for the binding of DA D2-like receptor agonists. It can be observed that the 2-BFI molecule share a common part with the 3-hydroxyphenoxyethylamine 2. Being phenolic group fundamental for agonistic activity, one or two hydroxyl groups (R1, R2) have been introduced in the aromatic nucleus of 2-BFI.
[1] Seeman P., Bzowej N. H., Guan H. C., Bergeon C., Reynolds G. P., Bird E. D., Neuropsycopharmacology, 1987, 1, 5-15.
[2] Serra, P. A.; Esposito G.; Enrico P.; Mura M. A.; Migheli R.; Delogu M. R.; Miele M.; Desole M. S.; Grella G.; Miele E. Br. J. Pharmacol. 2000, 130, 937-945.
[3] Gundert-Remy, U.; Hildebrand, R.; Stiehl, A.; Weber, E.; Zurcher, G.; Da Prada, M. Eur. J. Clin. Pharmacol. 1983, 25, 69-72.
[4] Da Prada, M.; Keller, H. H.; Pieri, L.; Kettler, L.; Haefely, W.E. Experentia, 1984, 40, 1165-1172. [5] a) Garzon-Aburbeh, A.; Poupaert, J. H.; Claesen, M.; Dumont, P. J. Med. Chem. 1986, 29, 687-691.
b) Bodor, N.; Sloan, K. B.; Higuchi, T. J. Med. Chem. 1977, 20, 1435-1445. c) Ihara, M.; Nakajima, S.; Hisaka, A.; Tsuchiya, Y.; Sakuma, Y.; Suzuki, H.; Kitani, K.; Yano, M. J. Pharm. Sci. 1990,79, 703-708. d) Wang, H.; Lee, J.; Tsai, M.; Lu, H.; Hsu, W. Bioorg. Med. Chem. Lett. 1995, 5,2195-2198.
[6] J.A. Lieberman, CNS Drugs, 2004, 18, 4: 251-267. [7] A. Inoue, Y. Nakata, Jpn. J. Pharmacol. 2001, 86, 376-380; [8] C. A. Tamminga, J. Neural. Transm. 2002, 109, 411-420; [9] R. E. Mewshaw, R. L. Zhao, X. J. Shi, K. L. Marquis, , J. A. Brennan, H. Mazandarani, J. Coupet,
T. H. Andree, Bioorg. Med. Chem. Lett., 2002, 12, 271-274 and references within cited. [10] a) Gentili, F., Ghelfi, F., Giannella, M., Piergentili, A., Pigini, M., Quaglia, W., Vesprini, C.,
Crassous, P. A., Paris, H., Carrieri, A. J. Med. Chem. 2004, 47, 6160-6173; b) Gentili, F., Bousquet P., Brasili L., Dontenwill M., Feldman J., Ghelfi F., Giannella M., Piergentili A., Quaglia W., Pigini M. J. Med. Chem. 2003, 46, 2169-2176; c) Ferretti, G., Dukat M. Giannella M., Piergentili A., Quaglia W., Domaj M. I., Martin B. R., Glennon R. A. J. Med. Chem. 2002, 45, 4724-4731; d) Gentili, F., Pizzinat N., Ordener C., Marchal-Victorion S., Maurel A., Hofmann R., Renerd P., Delagrange P., Pigini M., Giannella M. J. Med. Chem. 2006, 49, 5578-5586.
[11] Sastre-Coll A., Esteban S., Miralles A., Zanetti R., Garcia-Sevilla J. A. Neuroscience lett. 2001,301, 29-32.
5 6a 6b 6c 6d 6e 6f 6g 6h 6i
R1 H H H H OH OH OH OH OH OH
R2 H OH OH OH H H H OH OH OH
O N
N
R3
R1
R2
R3 H H n-Pr CH2Ph H n-Pr CH2Ph H n-Pr CH2Ph
101

N6-SUBSTITUTED NECA DERIVATIVES AS USEFUL TEMPLATES FOR THE
DEVELOPMENT OF A2B ADENOSINE RECEPTOR AGONISTS
Pier Giovanni BARALDI,a Delia PRETI,a Mojgan Aghazadeh TABRIZI,a Francesca FRUTTAROLO,a
Giulia SAPONARO,a Stefania BARALDI,a Romeo ROMAGNOLI,a Allan R. MOORMAN,c Stefania GESSI,b Katia VARANIb and Pier Andrea BOREAb
aDipartimento di Scienze Farmaceutiche, Universita’ di Ferrara, 44100 Ferrara, Italy bDipartimento di Medicina Clinica e Sperimentale-sezione di Farmacologia, Universita’ di Ferrara,
44100 Ferrara, Italy cKing Pharmaceutical Research and Development, Inc., 4000 CentreGreen Way, Suite 300, Cary, NC
27513, USA
The identification of potent and selective A2B adenosine receptor (AR) agonists has been an ambitious goal for years in order to characterize the potential physiological role of A2B receptors. From a pharmacological point of view, the lack of highly selective agents has so far hampered the efforts to better characterize the adenosine A2B receptor subtype and consequently to fully define its therapeutic potential. 5’-N-Ethylcarboxamidoadenosine (NECA, Fig. 1) has been considered one of the most useful ligand at the A2B receptor subtype (EC50 = 160 nM), although it shows high affinity toward the other adenosine receptors (Ki from binding assays in the low nanomolar range). It has been reported that small modifications at the 6-position of NECA are able to produce significant changes in the selectivity pattern of the resulting compounds versus the four known adenosine receptors (A1, A2A, A2B and A3 AR). Considering its fundamental importance for the modulation of both affinity and selectivity, we decided to investigate the N6-positions, synthesising two new series of N6-functionalized 5’-N-ethylcarboxamidoadenosine analogues. According to the principles of bioisosterism, we replaced the (hetero)arylurea function at the 6 position of NECA of a reported series of A3 agonists1 (Fig. 2) with the isomeric (hetero)arylcarbonylhydrazino moiety and evaluated the effect on the binding and functional profile of the synthesized compounds. The new classes of 1-deoxy-1-[6-[((hetero)arylcarbonyl)-hydrazino]-9H-purin-9-yl]-N-ethyl- -D-ribofuranuronamide and 1-deoxy-1-[2-chloro-6-[((hetero)arylcarbonyl)-hydrazino]-9H-purin-9-yl]-N-ethyl- -D-ribofuranuronamide derivatives2 (Fig. 3) have been tested in binding assays at human (h) hA1,hA2A and hA3 AR, and in a functional assay at the hA2B subtype. This series of compounds are the first examples of both potent (EC50 in the nanomolar range) and selective A2B adenosine receptor agonists ever reported. For the design of new nucleoside A2B AR agonists, we considered the possibility to exploit some information deriving from the structural analysis of known A2B antagonists. Kim et al.3 reported that a (substituted)phenylcarbamoyl-methoxy-phenyl chain at the 8-position of a series of 1,3-dipropyl-xanthines (Fig. 4) selectively addresses the antagonists affinity to the adenosine A2B receptor. In light of this, and thanks to the identification of a versatile synthetic route which permitted us to functionalize the N6-position of the known non-selective adenosine agonists NECA and 2-Cl-NECA (2-chloro-5’-N-ethylcarboxamido-adenosine), we designed a series of adenosine analogues bearing a (substituted) phenylcarbamoyl- methoxy-phenyl chain at the N6-NECA position. Our attempt was to evaluate the possibility of identifying a molecular hybrid obtained by the molecular combination of the nucleoside core responsible for receptor activation with the structural element able to grant A2B selectivity to the series of known A2B AR antagonists. A new series of N6-[(hetero)aryl/(cyclo)alkyl-carbamoyl-methoxy-phenyl]-(2-chloro)-5 -N-ethylcarboxamido-adenosines4 (Fig. 5) has been synthesised and tested in binding assays at hA1, hA2A
and hA3 AR, and in a functional assay at the hA2B subtype. The examined compounds displayed high potency in activating A2B receptors with good selectivity versus A2A subtype. The introduction of an unsubstituted 4-[(phenylcarbamoyl)-methoxy]-phenyl chain led us to the recognition of a full agonist displaying the highest efficacy of the series (EC50 hA2B = 7.3 nM). These compounds represent the first report about adenosine-related structures capable of activating hA2B subtype in the low nanomolar range.
102

A3 Agonist
N
NN
HN
O
O
n-Pr
n-Pr
O
O
NH
N
NN
N
NH
O
OHOHHH
HN O
R
O
NH
O
R'
N
NN
N
HN
O
OHOHHH
HN O
NH
O
R'
R
N
NN
N
HN
O
OHOHHH
HN O
HN R'
R
O
N
NN
N
NH2
O
OHOHHH
HN O
Fig. 1
Fig. 2 Fig. 3
Fig. 5 Fig. 4
A2B Antagonist
103

1. Baraldi, P. G.; Cacciari, B.; Spalluto, G.; Ji, X.; Olah, M. E.; Stiles, G.; Dionisotti, S.; Zocchi, C.; Ongini, E.; Jacobson, K. A. J. Med. Chem. 1996, 39, 802.
2. Baraldi, P. G.; Preti, D.; Tabrizi, M. A.; Fruttarolo, F.; Romagnoli, R.; Carrion, M. D.; Lopez Cara, L. C.; Moorman, A. R.; Varani, K.; Borea, P. A. J. Med. Chem. 2007, 50, 374.
3. Kim, Y-C.; Ji, X.; Melman, N.; Linden, J.; Jacobson, K. A.; J. Med. Chem. 2000, 43, 1165. 4. Baraldi P. G., Preti, D.; Tabrizi, M. A.; Fruttarolo, F.; Saponaro G.; Baraldi S.; Romagnoli, R.;
Moorman, A. R.; Gessi S.; Varani, K.; Borea, P. A. Bioorg. Med. Chem., 2007, 15, 2514.
104

DESIGN, SYNTHESIS AND BIOPHARMACOLOGICAL EVALUATION OF NOVEL ALDOSE
REDUCTASE INHIBITORS: THREE DIFFERENT SCAFFOLDS AT COMPARISON
Stefania SARTINI
Dipartimento di Scienze Farmaceutiche, Università di Pisa Scuola di Dottorato in Scienza del Farmaco e delle Sostanze Bioattive- XXI Ciclo
Diabetes mellitus is a metabolic disorder characterized by high levels of blood glucose, resulting from defects in insulin production, insulin action, or both. It is recognized as a public health problem, as it affects a significant portion of the population worldwide and is rising to pandemic proportions.1 Diabetes can be successfully controlled by the administration of insulin and/or potent oral hypoglycemics, but it still remains the cause of significant morbidity and mortality, due to a progressive development of disabling complications.2-3 Experimental and clinical evidence demonstrates that the pathogenic mechanism leading to diabetic complications is causally linked to an increased activity of the enzyme Aldose Reductase (alditol:NADP+ oxidoreductase, EC 1.1.1.21, ALR2). ALR2 catalyzes the reduction of glucose to sorbitol in the so called polyol pathway, a metabolic pathway that becomes activated during diabetic conditions. Sorbitol is formed more rapidly than it is converted to fructose, so it accumulates increasing cellular osmolarity. Moreover, the activation of the polyol pathway causes an imbalance of cytosolic cofactor ratios contributing to the onset of hyperglycaemic oxidative stress. Osmotic stress, together with oxidative stress, trigger the activation of downstream mechanisms leading to the development of long-term diabetic complications, represented by a progressive impairment of nervous, renal, vascular and visual systems. Inhibition of ALR2 is therefore a useful therapeutic strategy to prevent the onset, or at least delay, the progression and the severity of diabetic complications. I focused my research interest in the aldose reductase inhibitors (ARIs) field with the aim to disclose novel drug candidate for the topical treatment of diabetic visual impairments, which are the leading cause of blindness in people aged from 20 to 75. Therefore, I developed three structurally different classes of ALR2 inhibitors, which proved to possess excellent in vitro and in vivo activities.4-6
Naphto[1,2-d]isothiazole Acetic Acid Derivatives
A number of acetic acid derivatives bearing a naphtoisothiazole heterocyclic core were synthesized and tested. The most active compounds of this series, 1 and 2, showed IC50 values of 0.14 and 0.55 µM,respectively. Derivative 1 proved also to prevent the development of nuclear cataract in severely galactosemic rats, after an eye-drops administration, but only after its conversion to the isopropyl ester prodrug. Moreover, crystallized in the ALR2 active site, compound 1 permitted to disclose a new subpocket, never seen before, which opens up to accommodate the naphthyl ring of the inhibitor.7,8
S
NO
O
O
COOH
COOH
1
S
NO
O
O
COOH
O
O
2
COOH
1,2,4-Oxadiazole Acetic Acid Derivatives
Another class of acetic acid derivatives which proved to inhibit ALR2 is represented by 1,2,4-oxadiazoles bearing an acetic acid function and a phenyl or benzyl moiety. The p-methoxyphenyl derivative, 3,showed the best inhibitory potency, with an IC50 value of 0.29 µM. In addition, administered as carboxylic acid in the pre-corneal region of galactosemic rats, proved to prevent the development of nuclear cataract. Actually, 3 provides the best data sets for an ocular uptake proving to be less ionized and more lipophylic than the other compounds of the whole series,
105

NO
N
COOH
H3CO
3
Pyrido[1,2-a]pyrimidin-4-one Derivatives
We then considered another aspect of diabetic hyperglycemia, represented by oxidative damages. In fact, under diabetic conditions, the activation of the polyol pathway causes an imbalance in cytosolic cofactor ratios contributing to the onset of hyperglycemic oxidative stress through the accumulation of reactive oxygen species (ROS). ROS, in turn, corroborate the development of long-term diabetic complications.9
Therefore, it should be useful to combine an ARI activity with an antioxidant one. It is well documented in the literature that such a class of molecules is represented by flavonoids. Therefore, we decided to develop pyrido[1,2-a]pyrimidin-4-one derivatives as bioisosters of flavonoids. In this class of ARIs, compounds that showed the best IC50 toward ALR2 proved to be cathecols 4, 5, 6 and 7. Furthermore, almost all the products exhibited significant antioxidant property, as evidenced by their ability in inhibiting the production of thiobarbituric acid reactive species, an index of peroxidation, at a concentration of 10 µM.
N
N
O
OH
OH
4
N
N
O
OH
OH
5
OH
N
N
O
OH
OH
6
OH
N
N
O
7
OH
OH
OH
1. King, H.; Aubert, R.E.; Herman, W.H. Diabetes Care 1998, 21, 1414-1431. 2. Kador, P.F. Med. Res. Rev. 1998, 8, 325-352. 3. Yabe-Nishimura, C. Pharmacol. Rev. 1998, 50, 21-33. 4. Da Settimo, F.; Primofiore, G.; La Motta, C.; Sartini, S.; Taliani, S.; Simorini, F.; Marini, A. M.;
Lavecchia, A.; Novellino, E.; Boldrini, E. J. Med. Chem. 2005, 48, 6897-6907. 5. La Motta, C.; Sartini, S.; Mugnaini, L.; Simorini, F.; Taliani, S.; Salerno, S.; Marini, A. M.; Da
Settimo, F.; Lavecchia, A.; Novellino, E.; Cantore, M.; Failli, P.; Ciuffi, M. J. Med. Chem. 2007,50, 4917-4927.
6. La Motta, C.; Sartini, S.; Salerno, S.; Simorini, F.; Taliani, S.; Marini, A. M.; Da Settimo, F.; Marinelli, L.; Limongelli, V.; Novellino, E. J. Med. Chem. Accepted for Publication
7. Steuber, H.; Zentgraf, M.; La Motta, C.; Sartini, S.; Heine, A.; Klebe, G. J. Mol. Biol. 2007, 369,186-197.
8. Zentgraf, M.; Steuber, H.; Koch, C.; La Motta, C.; Sartini, S.; Sotriffer, C. A.; Klebe, G. Angew. Chem. Int. Ed. 2007, 46, 3575-3578.
9. Williamson, J. R.; Chang, K.; Frangos, M.; Hasan, K. S.; Ido, Y.; Kawamura, T.; Nyengaard, J. R.; van der Enden, M.; Kilo, C.; Tilton, R. G. Diabetes 1993, 42, 801-813.
106

A NOVEL SYNTHETHIC APPROACH OF NEW BENZO[b]ACRIDONES WITH POTENTIAL
ANTIOXIDANT AND ANTITUMOUR ACTIVITY
Raquel SEIXAS
Department of Chemistry, University of Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal
PhD in Chemistry
Acridones are a group of naturally occurring nitrogen heterocyclic compounds found exclusively in plants belonging to the Rutaceae family. Both natural and synthetic acridone derivatives exhibit a variety of important biological activities. They are known to present important antiviral [1], antifungal, and antiparasitic, against leishmania and malaria, activities [2,3]. They are also very important anticancer compounds because they are able to inhibit Epstein-Barr virus early antigen (EBV-EA) activation [4]. Moreover they have shown a potent and selective inhibition of human immunodeficiency virus type 1 (HIV-1) replication in chronically HIV-1-infected cells [5].
There are only a few examples of benzo[b]acridones described in the literature, being the additional aromatic ring linearly fused to the acridone moiety. Most of the work that has been made with this kind of compounds is on the synthesis of benzo[b]acronycine derivatives. The benzo[b]acronycine was found to be more active than acronycine itself when tested in vitro against cancer cells proliferation and the cis-diacetyloxy derivative 1 had shown an impressive broad spectrum antitumour activity and it is currently in phase II clinical trials [6]. It is also worthy to mention other important potential applications of acridones, such as its use as fluorescent compounds, like labels for fluorescence detection of target biological materials, and as fluorescent anions receptors and sensors [7]. Taking into account the variety of potential applications of acridones, we started a programme to develop new synthetic methods for the synthesis of novel benzo[b]acridones aiming to find new potent antioxidant and antitumour agents. Acridones are usually prepared by Ullmann condensation of anilines with ortho-halogen-substituted benzoic acids followed by the acid-induced ring closure of the N-phenyl anthranilic acids; however harsh reaction conditions are generally required. Besides this classical synthesis other methods have been developed, such as: i) the intermolecular nucleophilic coupling of arynes with 2-aminobenzoates and subsequent intramolecular nucleophilic cyclisation, ii) the condensation of 3-amino-2-naphthalenecarboxylic acid with phloroglucinol and iii) the Friedel-Crafts reaction of 3,5-dimethoxyacetanilide with 2-methoxy-1-naphthoyl chloride followed by base-catalyzed cyclisation in order to prepare acronycine derivatives.
In this work we report a new method for the synthesis of novel benzo[b]acridones from Diels-Alder reaction of 3-formyl-4-quinolone, acting as dienophile, with the highly reactive dienes ortho-benzoquinodimethanes. Initial experiments considered the Diels-Alder reaction of 3-formyl-1-methyl-4-quinolone 3a with ortho-benzoquinodimethane 5b, formed in situ by thermal extrusion of sulfur dioxide from 1,3-dihydrobenzo[c]thiophene 2,2-dioxide 4b. 3-Formyl-1-methyl-4-quinolone 3a was easily prepared through a Vielsmeyer reaction of 2’-aminoacetophenone 2, followed by acidic hydrolysis and subsequent methylation of the amino group. This cycloaddition reaction afforded a diastereomeric mixture of benzo[b]1,6,6a,12a-tetrahydroacridone derivatives 6a and 7a and also the oxidised benzo[b]acridone 8a, in low yields even after an exhaustive study on the experimental conditions and using microwave irradiation. Since the obtained results were not satisfactory, it was decided to increase the reactivity of the C2=C3 double bound changing the N-protective group to a withdrawing group. After the introduction of an ethoxycarbonyl as protecting group, the Diels-Alder reaction of 1-ethoxycarbonyl-3-formyl-4-quinolone 3b with ortho-benzoquinodimethane 5b afforded the cis-benzo[b]tetrahydroacridone 7b, in good yield, traces of the trans-diastereomer 6b and also the corresponding oxidised benzo[b]acridone 8b. After the optimization of the reaction conditions we performed the reaction of 3b with substituted ortho-benzoquinodimethanes 5c-d, which gave the corresponding benzo[b]1,6,6a,12a-tetrahydroacridone 7c-d derivatives.
NO
CH3
OCH3 O
CH3OCO
OCOCH3
(1)
107

N
CHO
O
R1
SO2
R2
R3
R3
R2
R2
R3
R3
R2
N
OH
H
(7a-d)O
N
R1
(8a,b)
R1
N
OH
H
(6a,b)
R1 R2
R3
R2
R3
(5b-d)
(4b-d)
(3a,b)
a: R1= CH3; R2= H; R3= H
b: R1 = CO2Et; R2= H; R3= Hc: R1 = CO2Et; R2= OCH3; R
3= Hd: R1 = CO2Et; R2= OCH3, R
3= Br
N
O
CO2Et R2
R3
R2
R3
N
O
H R2
R3
R2
R3
N
CHO
Cl
(2)
NH2
O
N
O
H I
(9b-d) (10b-d)
(11)
Scheme 1
In order to achieve the corresponding benzo[b]acridones we also attempted the oxidation of benzo[b]1,6,6a,12a-tetrahydroacridones 7b-d. A first study was carried out using p-benzoquinone derivatives as oxidants, such as chloranil and 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ), however we only observed a small formation of the expected oxidised product 9b and a great decomposition. In a second approach we performed this transformation in refluxing dimethyl sulfoxide with a catalytic amount of iodine, a mixture used by our group in the cyclodehydrogenation of 2’-hydroxychalcones and similar compounds [8-10], and we obtained the expected 7-ethoxycarbonylbenzo[b]acridones 9b-d, the unprotected benzo[b]acridones 10b-d in good yields and also, in the case of the unsubstituted derivative a small amount of the undesirable iodine by-product 11. The experimental procedures and the structural characterisation of all synthesised compounds will be presented and discussed in this communication.
[1] N. Yamamoto, H. Furukawa, Y. Ito, S. Yoshida, K. Maeno, Y. Nishiyama, Antiviral Research, 1989,
12, 21-36. [2] K. M. Ahua, J. R. Ioset, A. Ransijn, J. Mauel, S. Mavi, K. Hostettmann, Phytochemistry, 2004, 65,
963-968. [3] R. W. Winter, J. X. Kelly, M. J. Smilkstein, R. Dodean, G. C. Bagby, R. K. Rathbun, J. I. Levin, D.
Hinrichs, M. K. Riscoe, Experimental Parasitology, 2006, 114, 47-56. [4] M. Itoigawa, C. Ito, T. Wu, F. Enjo, H. Tokuda, H. Nishin, H. Furukawa, Cancer Letters, 2003, 193,
133-138. [5] M. Fujiwara, M. Okamoto, M. Okamoto, M. Watanabe, H. Machida, S. Shigeta, K. Konno, T.
Yokota, M. Baba, Antiviral Research, 1999, 43, 189-199. [6] S. Michel, T. Gaslonde, F. Tillequin, European Journal of Medicinal Chemistry, 2004, 39, 649-655. [7] M. T. Blazquez, F. M. Muniz, S. Saez, L. M. Simon, A. Alonso, C. Raposo, A. Lithgow, V. Alcazar,
J. R. Moran, Heterocycles 2006, 69, 73-81. [8] A. M. S. Silva, D. C. G. A. Pinto, H. R. Tavares, J. A. S. Cavaleiro, M. L. Jimeno, J. Elguero,
European Journal of Organic Chemistry, 1998, 2031-2038. [9] A. I. R. N. A. Barros, A. M. S. Silva, Monatshefte Chemie, 2006, 137, 1505-1528. [10] A. I. R. N. A. Barros, A. M. S. Silva, Magnetic Resonance in Chemistry, 2006, 44, 1122-1127.
Acknowledgements: Thanks are due to my supervisor Professor Artur M.S. Silva, to Dr. Diana C .G. A. Pinto and Professor José A. S. Cavaleiro for the important contribution to this work, as well the University of Aveiro, FEDER and FCT for funding the Organic Chemistry Research Unit and project POCI/QUI/58835/2004 and also for a PhD (SFRH/BD/30734/2006) grant.
108

4, 6-DIMETHOXYAURONES AS DUAL MODULATORS OF P-GLYCOPROTEIN (P-gp) AND
BREAST CANCER RESISTANCE PROTEIN (BCRP)
Hong-May SIM
Department of Pharmacy, Faculty of Science, National University of Singapore PhD course in Pharmacy
Introduction
Multi-drug resistance (MDR) is a phenomenon involving resistance of tumors to chemically unrelated anticancer drugs. It is associated with the over-expression of the ATP-binding cassette (ABC) efflux proteins, mainly P-glycoprotein, P-gp and the Breast cancer resistance protein, BCRP1. Aurones is a sub-class of flavonoids that has been reported to modulate the activity of P-gp2.Modulatory activity of aurones on BCRP has not been extensively explored. Here we report the potential of aurones to function as modulators of P-gp and BCRP. 4, 6-dimethoxyaurones were synthesized and investigated as dual modulators of P-gp and BRCP. These compounds have the potential to improve chemoresponsiveness of drug resistant tumors.
Experimental
MDCKII/MDR1 MDCKII/WT MDA-MB-231/V MDA-MB-231/R
P-gp
actinBCRP
actin
Figure 1. Western blot analyses of P-gp and BCRP expression levels
4, 6-dimethoxyaurones were synthesized according to Scheme 1. Modulatory activity of the aurones on P-gp and BCRP were screened using the calcein-AM accumulation assay and the mitoxantrone (MX) accumulation assay respectively. Over-expression of P-gp and BCRP were confirmed in MDCKII/MDR1 and MDA-MB-231/R cell lines respectively by Western blot (Figure 1). Reversal of BCRP-mediated drug resistance by the aurones was shown via the MX cytotoxicity assay.
Scheme 1. Reagents and conditions: (a) Chloroacetic acid (1.1 eq), NaH, DMF, room temperature, 12 h. (b) Polyphosphoric acid, 80º C, 8 h. (c) R-substituted benzaldehyde, 50% KOH in MeOH/H2O, room temperature, 3 h.
OCH3
H3CO OH
OCH3
H3CO O
O
OOCH3
H3CO O
OOCH3
H3CO
a
b
c
COOH
R
O
OOCH3
H3CO
2'
3' 4'
A
B
C
R
4, 6- dimethoxyaurones
109

Results and Discussion
MX accumulation assay
At 5µM and 0.5µM, aurones showed good inhibitory activity on BCRP in the resistant MDA-MB-231/R cells. Most members showed comparable effects to fumitremorgin C (FTC), a specific inhibitor of BCRP at 5µM. Aurones with chloro, hydroxyl, methoxyl substituents and unsubstitution on ring B had higher MX accumulation with the 3 -OH analogue producing the greatest accumulation at 0.5 µM. Calcein-AM accumulation assay
At 10µM, aurones with 3 -substitution generally had better inhibitory activity on P-gp, accumulating more calcein-AM than the rest of the analogues. Activities of 3 -substituted aurones were comparable to verapamil, a known inhibitor of P-gp. MX Cytotoxicity Assay
Selected members of the aurones significantly reverse BCRP-mediated drug resistance. At 0.5µM, resistant MDA-MB-231/R cells treated with aurone (R= 3 -Cl) produced the greatest reversal index with a 24.6-fold difference in IC50 of MX (5.2-fold difference for FTC) compared to untreated cells. Cytotoxicity studies indicated that the aurones do not cause significant killing in parental MDA-MB-231/V and resistant MDA-MB-231/R cells.
Conclusion
Several aurones show potent inhibitory activities on both BCRP and P-gp. Functionalized aurones may yield novel and promising specific inhibitors for the reversal of BCRP-mediated drug transport. Non-toxic nature of the aurones favors their development as modulators of efflux proteins. Optimization of the aurone template is needed to improve modulatory activities against both BCRP and P-gp.
Acknowledgement
MDA-MB-231/V and MDA-MB-231/R cells were kindly provided by Dr Rachel Ee Pui-Lai (National University of Singapore) and Dr Douglas Ross (Greenebaum Cancer Center, University of Maryland, Baltimore, USA). MDCKII/WT and MDCKII/MDR1 cells were gifts from Dr Anton Berns of the Netherlands Cancer Institute, Antoni van Leeuwenhoek Hospital, Amsterdam, Netherlands.
References
1) Pérez-Tomás, R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem 2006, 13, 1859-76. 2) Boumendjel, A.; Beney, C.; Deka, N.; Mariotte, A.; Lawson, M.; Trompier, D.; Baubichon-Cortay, H.; Di Pietro, A. 4-Hydroxy-6-methoxyaurones with high-affinity binding to cytosolic domain of P-glycoprotein. Chem Pharm Bull (Tokyo) 2002, 50, 854-6.
110

PYRROLO-FUSED HETEROCYCLES AS PHOTOCHEMOTHERAPEUTIC AGENTS
Virginia SPANÒ
Università di Palermo – Dipartimento Farmacochimico, Tossicologico e Biologico Via Archirafi, 32 – 90123 Palermo
Dottorato di Ricerca in Scienze Farmaceutiche – Ciclo 2005-2006 (XX)
Psoralen 1 and Angelicin 2, linear and angular furocoumarins, are photoactivable drugs which upon UVA irradiation, intercalate into DNA and photobind with it.1 They are currently used in PUVA therapy for the treatment of various skin diseases such as psoriasis, vitiligo and tumors such as T-cell lymphoma when used in conjunction with long-wave (320-400 nm) ultraviolet light (UV-A). Recently the synthesis of their heteroanalogues were reported.2
O OO
O
O
O
1 Psoralen 2 Angelicin X-X= CH2-CH2 or CH=CH
6
XX
QW
Y
O
3 Y=NH, W = N-R, Q=C-Me
5 Y=S, W= N-R, Q=C-R 4 Y=NH, W = C-R, Q=N-R
XX
NR
For many years the research group I work with, has focused the attention on the synthesis of polycyclic systems containing the pyrrole mojety endowed with biological activity and more recently the interesting field of photochemotherapy has been investigated. With the aim of preparing and studying new photoreactive agents with antiproliferative activity and decreased toxic side effects, thus it was reported the synthesis of the new ring systems pyrrolo[2,3-h]quinolin-2-one 3, pyrrolo[3,4-h]quinolin-2-one 4 and thiopyrano[2,3-e]indol-2-one 5 whose derivatives showed higher cytotoxicity than 8-MOP (GI50 0.4-16.4 µM, 1.1-15.0 µM and 0.2-17.0 µM respectively).3
In this light considering the interesting results achieved in this field of research, it was planned for my project the synthesis of new ring systems of type 6 in which a six or a five membered ring was annelated to the isoindole moiety with the aim of evaluating their photochemotherapeutic activity. A first approach in this direction led us to investigate the synthesis and the biological activity of the new ring system pyrano[2,3-e]isoindol-2-one 10, where pyrrole replaces the furane ring of Angelicin. The starting ketons 7 were conveniently prepared according to procedures reported in literature.4 The first step of the synthetic pathway was the formylation of 7 with ethyl formate which led to the hydroxymethylene derivatives 8. Enaminoketones 9 (R4=Et) were successfully obtained by reacting these latter with diethylamine at room temperature, or directly by refluxing in toluene ketones 7 with an excess of t-butoxybis(dimethylamino)methane (TBDMAM) (R4=Me). Reaction of enaminoketones 9 with the proper dialkyl malonate, used as solvent, afforded the desired tricyclic derivatives 10 (R3=COOEt or COOMe, X-X= CH2-CH2) in moderate to good yields (30-73%). Alternatively, heating the hydroxymethylene derivatives 8 with (ethoxycarbonylmethylene)triphenilphosforane in TEG derivatives 10 (R3=H) where obtained. Full aromatization of adducts 10 was accomplished with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in refluxing anhydrous benzene (X-X= CH=CH). Having in hand the key intermediates 8 and 9, which are versatile synthons as they can react with a variety of dinucleophiles, we decided to study the synthesis of the new ring system pyrrolo[3,4-h]quinazolin-2-one, an heteroanalogue of the pyrroloquinolinone ring 4 i which an additional nitrogen atom replaces the -carbon of the pyridone ring. The quinazoline nucleus is of great interest because it is the scaffold of many antitumor drugs mainly acting as tyrosine kinase receptors (RTK); overexpression of these receptors is found in a number of cancers (e.g. breast, ovarian, colon, prostate).5 Leading examples are the anticancer agent Tarceva (OSI 774/CP358,774) which is in Phase III clinical trials and the clinically approved anticancer agent Iressa (ZD1839).6 Fused tricyclic pyrazolo- and pyrrolo-quinazoline showed GI50 values in the nanomolar range in the inhibition of (VEGFR).7 Additionally lipophilic
111

inhibitors of the dihydrofolate reductase (DHFR) containing the quinazoline ring are used for neoplastic diseases.8 Our synthetic approach for the synthesis of the mentioned ring system 11 consisted on the annelation of the pyrimidine ring on the isoindole moiety using hydroxymethylene isoindoles 8 asbuilding blocks. Thus reaction of 8 with urea, as dinucleophile, in refluxing anhydrous ethanol afforded the tricyclic derivatives 11 (X=CO) (30-62%). The same ring system 11 (X=CH) (30-65%) was obtained refluxing ketones 7 in formamide with tris-(formylamino)-methane whereas reaction of the enaminoketons 9 with guanidine nitrate afforded the 2-amino substituted derivatives 11 (X=C-NH2) (40-84%). We further investigated annelation on the isoidole moiety to achieve the new ring system isoxazolo[5,4-e]isoindole 12. The isoxazole nucleus is part of many drugs with antitumor activity. Among these diaryilisoxazoles showed strong growth inhibitory activities against human cancer cell lines. Moreover pteridine compounds containing the isoxazole moiety are active as dihydrofolate reductase (DHFR) inhibitors. These compounds showed growth in vitro inhibitory activities against breast (MCF7), lung (NCI-H460) and central nervous system (SF-268) cell lines.9
Ring closure on the hydroxymethylene building blocks 8 was easily achieved with hydroxylamine hydrochloride, in ethanol to give the desired derivatives 12 (40-72%). All derivatives of the new ring systems will be subjected to screenings to evaluate the photochemoterapeutic activity as well as the antiproliferative activity against a panel of human tumor cell lines.
8
9
NR
R1
R2
N O
NR
O
R1
R2
(R4)2NHC
NR
O
R1
R2
HOHC
NR
O
R1
R27 12
i
ii
viii
viivi iv,v
iii,v
ix
isoxazolo[5,4-e]isoindole
XX
NR
R1
R2
O
O
R3
10
pyrano[2,3-e]isoindol-2-one
NR
R1
R2
NX
N
11
pyrrolo[3,4-h]quinazoline
ii
(i) HCOOEt, benzene; (ii) HNEt2, benzene or TBDMAM, toluene; (iii) CH2(COOEt)2 or CH2(COOMe)2
(X-X= CH2-CH2); (iv) Ph3PCH2CO2Et, TEG (X-X= CH2-CH2); (v) DDQ, benzene (X-X= CH=CH); (vi)
CO(NH2)2, ethanol; (vii) (HCONH)3CH, formamide; (viii) H2NC(=NH)NH2.HNO3, ethanol; (ix) NH2OH.HCl, ethanol.
References: [1] Comber, M.F. et al. Synthesis 1992, 731; [2] Rodighiero, P. et al. J. Med. Chem. 1996, 39, 1293; [3] Barraja, P. et al. Bioorg. Med. Chem. Lett. 2003, 13, 2809; Barraja, P. et al. Bioorg. Med. Chem. Lett.2005, 15, 2291; Barraja, P. et al. Bioorg. Med. Chem., 2006, 14, 8712; [4] Spreitzer, H. et al. Chem. Ber.1990, 123, 413; Clezy, P.S. et al. Aus. J. Chem. 1977, 30, 1337; Gabbutt, C.D. et al. J. Chem. Soc. Perkins Trans. 1 2002, 2779; [5] Mtsuno, K. et al. J. Med. Chem. 2002, 45, 4513; Hennequin, L.F. et al. J. Med. Chem. 2002, 45, 1300; [6] Hunt, J.T. et al. J. Med. Chem. 2004, 47, 4054; Moyer, J.D. et al. Cancer Res. 1997, 57, 4838; [7] Palmer, B.D. et al. J. Med. Chem. 1997, 40, 1519; [8] Kuyper, L.F. et al. J. Med. Chem. 1996, 39, 892; [9] Sun, C.-M. et al. Bioorg. Med. Chem. Lett. 2007, 17, 1078; Chauhan, P.M.S. et al. Bioorg. Med. Chem. 2005, 13, 3513; Kaffy, J. et al. Bioorg. Med. Chem. 2006, 14, 4067.
112

HERG-FREE: A COMPUTATIONAL APPROACHK h a c � M i n h T H A I a n d G e r h a r d F . E C K E REmerging Field Pharmacoinformatics, Department of Medicinal Chemistry, University of Vienna,
Althanstrasse 14, A-1090 Vienna, Austria
IntroductionI n h i b i t i o n o f e t h e r � a � g o � g o � r e l a t e d � g e n e ( h E R G ) c h a n n e l s p r o l o n g s t h e v e n t r i c u l a r a c t i o n p o t e n t i a l w i t ht h e r i s k o f t o r s a d e d e p o i n t e s a r r h y t h m i a t h a t m a y r e s u l t i n s u d d e n c a r d i a c d e a t h . 1 T h e r e f o r e ,c o m p u t a t i o n a l a p p r o a c h e s f o r c l a s s i f i c a t i o n a n d p r e d i c t i o n o f h E R G a f f i n i t y i n a n e a r l y p h a s e o f t h e d r u gd i s c o v e r y a n d d e v e l o p m e n t p r o c e s s a r e o f i n c r e a s i n g i n t e r e s t . B o t h s t r u c t u r e � b a s e d a n d l i g a n d � b a s e da p p r o a c h e s h a v e b e e n u n d e r t a k e n t o s h e d m o r e l i g h t o n t h e m o l e c u l a r b a s i s o f d r u g � c h a n n e l i n t e r a c t i o n a sw e l l a s t o p r e d i c t h E R G a f f i n i t y a n d t o c l a s s i f y h E R G i n h i b i t o r s . 2 I n t h i s s t u d y w e r e p o r t b i n a r y Q S A Ra n d c o u n t e r � p r o p a g a t i o n n e u r a l n e t w o r k s ( C P G � N N ) a p p r o a c h e s f o r c l a s s i f i c a t i o n o f h E R G i n h i b i t o r s .T h e s e a p p r o a c h e s m i g h t p r o v i d e p o s s i b l e s t r a t e g i e s f o r i m p r o v i n g t h e p e r f o r m a n c e o f s e p a r a t i o n o f c l e a rn o � g o e s f r o m s a f e c o m p o u n d s .MethodsS t r a t e g i e s f o r b i n a r y Q S A R 3 a n d C P G � N N 4 a r e s h o w n i n F i g u r e 1 a n d 2 , r e s p e c t i v e l y .
Figure 1. Strategy for hERG binary QSAR
Figure 2. Flowchart for classification of hERG inhibitors by Counter-Propagation Neural Networks
Results and Discussion
Binary QSAR. B i n a r y Q S A R m o d e l s w i t h t h r e s h o l d v a l u e s a t I C 5 0 = 1 a n d 1 0 5 M , r e s p e c t i v e l y , w e r eg e n e r a t e d u s i n g t w o d i f f e r e n t s e t s o f d e s c r i p t o r s ; o n e s e t c o m p r i s i n g 3 2 P _ V S A d e s c r i p t o r s 3 a n d t h e o t h e r

o n e u t i l i z i n g a s e t o f d e s c r i p t o r s i d e n t i f i e d o u t o f a l a r g e s e t v i a a f e a t u r e s e l e c t i o n a l g o r i t h m . 4 F o r t h e f u l ld a t a s e t , t h e p o w e r f o r c l a s s i f i c a t i o n o f b l o c k e r s w a s 8 2 – 8 8 % , w h i c h m e e t s p r i o r c l a s s i f i c a t i o n m o d e l s . 5Table 1. S u m m a r y o f h E R G b i n a r y Q S A R m o d e l s b a s e d o n 1 1 r e l e v a n t d e s c r i p t o r sG H s c o r e o nI C 5 0 t h r e s h o l d( J M ) N u m b e r o f c o m p o u n d s( A c t i v e / I n a c t i v e ) T o t a l a c c u r a c y a c t i v e I n a c t i v e A c c u r a c y o n e x t e r n a l t e s t s e t( 5 8 c o m p o u n d s )1 2 8 7 ( 1 0 0 / 1 8 7 ) 0 . 8 7 0 . 8 0 0 . 9 0 0 . 8 41 0 2 8 7 ( 1 9 7 / 9 0 ) 0 . 8 2 0 . 8 7 0 . 6 9 0 . 7 8H i g h & w e a k 1 9 0 ( 1 0 0 / 9 0 ) 0 . 8 8 0 . 8 8 0 . 8 8 0 . 8 6
Counter-propagation neural networks. C P G u N N s w e r e u s e d t o d e v e l o p c o m p u t a t i o n a l m o d e l s f o rc l a s s i f i c a t i o n a n d p r e d i c t i o n o f h E R G p o t a s s i u m c h a n n e l b l o c k e r s . 6 T h r e e s e t s o f m o l e c u l a r d e s c r i p t o r sw e r e a p p l i e d t o c r e a t e C P G u N N m o d e l s i n c l u d i n g a s e t o f 3 2 P u V S A d e s c r i p t o r s , 3 a s e t o f 1 1 d e s c r i p t o r sr e t r i e v e d b y a f e a t u r e s e l e c t i o n m e t h o d f r o m 1 8 4 M O E d e s c r i p t o r s 4 a n d a s e t o f S I B A R d e s c r i p t o r s 7 . T h eC P G u N N w i t h a 3 u d i m e n s i o n a l o u t p u t l a y e r c o m b i n e d w i t h a s e t o f 1 1 h E R G r e l e v a n t d e s c r i p t o r s s h o w e db e s t p e r f o r m a n c e e s p e c i a l l y i n c l a s s i f y i n g c o m p o u n d s i n t h e m i d d l e a c t i v i t y c l a s s ( I C 5 0 = 1 u 1 0 � M ) . T h et o t a l a c c u r a c y v a l u e s o b t a i n e d f o r t r a i n i n g a n d t e s t s e t s a r e 0 . 9 3 u 0 . 9 5 a n d 0 . 8 3 u 0 . 8 5 , r e s p e c t i v e l y . I n e a c ha c t i v i t y c l a s s ( l o w , m e d i u m , h i g h ) , G H s c o r e s a r c h i v e d r a n g e f r o m 0 . 8 9 t o 0 . 9 7 f o r t h e t r a i n i n g s e t a n df r o m 0 . 7 4 t o 0 . 8 7 f o r t h e t e s t s e t . 8
Table 2. S u m m a r y o f h E R G c l a s s i f i c a t i o n p o w e r s b y C P G � N N a p p r o a c h b a s e d o n 1 1 r e l e v a n t d e s c r i p t o r sT o t a l a c c u r a c y C l a s s 1 � L o w h E R G a c t i v i t y( I C 5 0 > 1 0 � M ) C l a s s 2 � M i d d l e h E R G a c t i v i t y( I C 5 0 = 1 � 1 0 � M ) C l a s s 3 � H i g h h E R G a c t i v i t y( I C 5 0 < 1 � M )P a r a m e t e r T r a i n T e s t T r a i n T e s t T r a i n T e s t T r a i n T e s tA c c u r a c y 0 . 9 3 0 . 8 5 0 . 8 6 0 . 8 3 0 . 9 5 0 . 8 0 0 . 9 6 0 . 9 4P r e c i s i o n 0 . 9 2 0 . 8 3 0 . 9 3 0 . 9 4 0 . 9 2 0 . 7 9G H s c o r e 0 . 8 9 0 . 8 3 0 . 9 4 0 . 8 7 0 . 9 4 0 . 8 6Strategies to improve hERG classification by in silico approach.
W e c o u l d d e m o n s t r a t e t h a t b y u s i n gC P G u N N i t i s p o s s i b l e t o c l a s s i f y a l s o t h e c o m p o u n d s b e l o n g i n g t o t h e m i d d l e c l a s s ( I C 5 0 = 1 u 1 0 � M ) . Af u r t h e r i m p r o v e m e n t m i g h t b e a c h i e v e d w h e n a p p l y i n g a c o n s e n s u s s c o r i n g s c h e m e w h i c h c o m b i n e sm u l t i p l e m o d e l s a n d m u l t i p l e a p p r o a c h e s ( t r a d i t i o n a l Q S A R , s t r u c t u r e u b a s e d a p p r o a c h e s a n d m a c h i n el e a r n i n g m e t h o d s ) . 8 C o n s i d e r i n g t h e f a c t t h a t v e r y s m a l l a n d d i s c r e t e c h a n g e s i n t h e c h e m i c a l s t r u c t u r ec a n l e a d t o r e m a r k a b l y a l t e r e d h E R G a f f i n i t y , c l a s s i f i c a t i o n i n t o t h r e e c l a s s e s f o l l o w e d b y l o c a l i n s i l i c or e g r e s s i o n m o d e l s f o r e a c h c l a s s m i g h t e n h a n c e t h e p e r f o r m a n c e f o r p r e d i c t i n g h E R G a c t i v i t i e s .ConclusionsO u r b i n a r y Q S A R m o d e l s s h o w h i g h a c c u r a c i e s a n d a r e f a s t i n c a l c u l a t i o n . T h o s e m o d e l s m a y b e u s e i nl e a d o p t i m i z a t i o n p r o g r a m s f o r s h a p i n g c o m b i n a t o r i a l l i b r a r i e s i n o r d e r t o d e c r e a s e t h e r i s k f o r h E R Ga c t i v i t y . C P G u N N a p p r o a c h e s t o g e t h e r w i t h a 3 u d i m e n t i o n a l u o u t p u t l a y e r a c c u r a t e l y c l a s s i f i e d t h ec o m p o u n d s b e l o n g i n g t o t h e m i d d l e a c t i v i t y c l a s s ( I C 5 0 = 1 u 1 0 ¦ M ) . T h e C P G u N N w i t h S I B A Rd e s c r i p t o r s b a s e d o n t h e c a l c u l a t i o n o f 2 D a n d 3 D d e s c r i p t o r s a l s o p e r f o r m e d w e l l . T h o s e m o d e l s c a n b eu s e d a s c l a s s i f i c a t i o n s y s t e m f o r v i r t u a l s c r e e n i n g p r o t o c o l s , a l l o w i n g a n n o t a t i o n o f p o t e n t i a l l y t o x i cc o m p o u n d s i n a n e a r l y s t a g e o f t h e d r u g d i s c o v e r y p r o c e s s .U s i n g a r t i f i c i a l n e u r a l n e t w o r k s , o n e d i s a d v a n t a g e i s t h e d i f f i c u l t y t o r e c e i v e i n f o r m a t i o n o n t h e r e l a t i v ei m p o r t a n c e o f t h e d e s c r i p t o r s . H o w e v e r , o u r r e s u l t s s h o w t h a t a c o m b i n a t i o n o f C P G u N N a n d a f e a t u r es e l e c t i o n a l g o r i t h m l e a d s t o h i g h l y e n c o u r a g i n g r e s u l t s w h i c h m i g h t b e i m p l e m e n t e d a l s o f o r o t h e rp o l y s p e c i f i c p r o t e i n s s u c h a s t h e A B C t r a n s p o r t e r f a m i l y a n d t h e h e p a t i c c y t o c h r o m e P 4 5 0 f a m i l y .AcknowledgmentsT h i s w o r k w a s s u p p o r t e d b y t h e A u s t r i a n S c i e n c e F u n d ( g r a n t # L 3 4 4 � N 1 7 ) a n d t h e P h D p r o g r a m ‘ M o l e c u l a r D r u g T a r g e t s ’ o f t h eU n i v e r s i t y o f V i e n n a . K M T t h a n k s t h e A S E A � U n i n e t a n d t h e A u s t r i a n C o u n c i l f o r R e s e a r c h a n d T e c h n o l o g y D e v e l o p m e n t a n d t h eÖ A D f o r p r o v i d i n g a s c h o l a r s h i p . K M T i s g r a t e f u l f o r t h e f e l l o w s h i p s a w a r d t o p a r t i c i p a t e i n t h e 2 0 0 8 e d i t i o n o f E S M E C � U r b i n o .References1 . S a n g u i n e t t i , M . C . ; T r i s t a n i � F i r o u z i , M . Nature 2006, 440 , 4 6 3 .2 . a ) T h a i , K . � M . ; E c k e r , G . F . Curr. Med. Chem. 2007, 14 , 3 0 0 3 ; b ) R e c a n a t i n i , M . e t a l . Med. Res. Rev. 2005, 25 ,1 3 3 ; c ) A r o n o v , A . M . Curr. Opin. Drug Discov. Devel. 2008, 11 , 1 2 8 .3 . L a b u t e , P . J. Mol. Graph. Model. 2000 , 18 , 4 6 4 .4 . M O E C h e m i c a l C o m p u t i n g G r o u p I n c . , M o n t r e a l , H 3 A 2 R 7 C a n a d a . ( h t t p : / / w w w . c h e m c o m p . c o m )5 . T h a i , K . � M . ; E c k e r , G . F . Bioorg. Med. Chem. 2008, 16 , 4 1 0 7 .6 . S O N N I A . M o l e c u l a r N e t w o r k s G m b H , E r l a n g e n , G e r m a n y . ( h t t p : / / w w w . m o l � n e t . d e )7 . K l e i n C e t a l . J. Comp. Aided Mol. Des. 2002 , 16,
7 8 5 .8 . T h a i , K . � M . ; E c k e r , G . F . Chem. Biol. Drug Des. 2008 , s u b m i t t e d .

SYNTHESIS AND BIOLOGICAL EVALUATION OF CHIRAL
2-PHENOXY-3-PHENYLPROPANOIC ACID DERIVATES WITH PPAR / DUAL ACTIVITY
Raffaella TRISOLINI
Dipartimento Farmaco-Chimico, Università degli Studi di Bari Dottorato di ricerca in Scienze Farmaceutiche – XXI ciclo
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear receptor superfamily of ligand-activated transcription factors and play a central role in storage and catabolism of fatty acids. [1]Activation of PPARs leads to the formation of heterodimers with retinoid-X receptors (RXRs) forming a complex that interacts with specific DNA response elements (PPRE) within promoter regions of target genes. [2,3] When activated by agonist ligand binding, the heterodimer recruits transcription coactivators and the resulting complex regulates the transcription of genes involved in the lipid and carbohydrate metabolism control. There are three PPAR subtypes which are the products of distinct genes and are commonly designated PPAR , PPAR and PPAR . Each receptor has a distinct tissue expression profile, with PPAR the main subtype in the liver, PPAR the main subtype in adipose tissue and PPAR expressed in many tissues. All three PPARs are activated by naturally fatty acids and fatty acid metabolites, indicating that they function as the body’s fatty acid sensors. PPAR is the receptor for the fibrate drugs which are widely used to lower triglycerides and raise high-density lipoprotein cholesterol levels in the treatment and prevention of coronary artery disease. PPARplays a critical role in adipocyte differentiation and serves as the receptor for the glitazone class of insulin-sensiting drugs used in the treatment of type 2 diabetes. In contrast to PPAR and PPAR ,relatively little is known about the biology of PPAR , although recent findings suggest that this subtype also has a role in lipid homeostasis. Three-dimensional crystal structures reveal that the ligand-binding pockets of the PPARs are much larger and more accessible than those of other nuclear receptors, providing a molecular basis for the promiscuous ligand binding of these receptors. [1] Given the fundamental roles that PPARs play in energy balance, drugs that modulate PPAR activity are likely to be useful for treating a wide range of metabolic disorders, including atherosclerosis, dyslipidemia, obesity and type 2 diabetes. The combination therapy with drugs acting on both PPAR and PPAR isotypes may have synergistic and wider therapeutic effects improving both glucose and lipid metabolism and it represents a new exciting challenge for the therapy of patients suffering from Type 2 diabetes. This metabolic disorder is characterized by progressive insulin secretory dysfunction and insulin resistance at major target tissues such as skeletal muscle, liver and adipose tissue, and is usually associated with the so-called metabolic syndrome including dyslipidemia, hypertension, and obesity. Based on this hypothesis, the concept of identifying ligands that bind and activate both PPAR and PPAR represents a logical continuation in the field of PPAR research and many groups, in fact, have ongoing research programs to identify more potent and less toxic PPAR / dual agonists. [4] In the past, in order to obtain more information on the structural requirements allowing fibrates to interact with PPARs, we have synthesized and reported the effects on human PPARα and PPARγ of some chiral analogs of clofibric acid, the active metabolite of the hypolipidemic drug clofibrate. [4,5] The potency of these compounds in activating the receptor was increased by the presence on the phenoxy moiety of an electronwithdrawing group or by the enlargement of the same aromatic moiety. Moreover, these analogues displayed a high degree of stereoselectivity when a benzyl group was located in α to the carboxylic function with the (S)-isomers much more active than the (R)-isomers. From these studies we have identified two novel lead compounds, MS39 and LT175 (Figure 1), with mixed PPAR / agonist activity.
O COOH
X
O COOH
Cl
Clofibric Acid
X = Cl MS39
X = Ph LT175
Figure 1
115

With the aim of improving the potency and efficacy of these compounds, a new set of analogues of MS39 or LT175 was designed and synthesized. The main structural modifications were carried out by introduction of one or more substituents in different positions of MS39 phenoxy moiety while maintaining the chlorine atom in the para position (Figure 2a) and by introduction of different spacers between the aromatic groups of the LT175 diphenyl system (Figure 2b).The PPAR and PPAR activity of all derivatives was evaluated by the transactivation assay, a powerful and widely used assay that is generally accepted to correlate well with in vivo activity.
O COOH
R
R''
Cl R' [SPACER]
O COOH
R, R', R''= Halogens, Alkyl, Aryl
b)a)
Figure 2
The PPAR activity has not been evaluated given that 2-aryloxy-acetic acids with a bulky substituent situated alpha to the carboxylic group are expected to have very low or no activity on PPAR . [4,5] Considering the high degree of stereoselectivity shown from the previously reported derivatives of this series of chiral PPARs ligands, [4] the influence of the absolute configuration was also taken into account for the compounds esteemed to be more interesting. The results of the transactivation assay, revealing some interesting compounds with dual PPAR /agonist activity, will be reported in this work.
[1] Kliewer S.A., Xu E.H., Lambert M.H., Willson T.M. Recent Prog. Horm. Res.. 56, 2001, 239. [2] Kliewer, S. A.; Umesono, K.; Noonan, D. J.; Heyman, R. A.; Evans, R. M. Nature, 1992, 358, 771. [3] Mangelsdorf, D. J.; Evans, R. M. Cell, 1995, 83, 841. [4] Pinelli, A., Godio, C., Laghezza, A., Mitro, N., Fracchiolla, G., Tortorella, V., Lavecchia, A., Novellino, A., Fruchart, J.-C., Staels, B., Crestani, M., Loiodice, F. J. Med. Chem., 2005, 48, 5509. [5] G. Fracchiolla, A. Laghezza, L. Piemontese, G. Carbonara, A. Lavecchia, P. Tortorella, M. Crestani, E. Novellino, F. Loiodice, Chem. Med. Chem. 2007, 2, 641.
116

SYNTHESIS OF NEW P-GLYCOPROTEIN INHIBITORS AND THEIR SCREENING:
POTENTIAL TOOLS TO REDUCE THE MULTIDRUG RESISTANCE
Michael VANNI
Dipartimento di ScienzeFarmaceutiche, Università di PisaDottorato di ricerca in Scienza del Farmaco e delle Sostanze Bioattive-XXI Ciclo
One of the classical protection mechanisms of cancer cells involves an increased expression of drug efflux transport proteins like P-glycoprotein (P-gP). P-gP is an ABC drug efflux transporter which is physiologically express in many tissues such as the blood-brain-barrier (BBB) where it is involved in the protection of the CNS from potentially toxic agents. This protein is able to transport several and structurally different substrates, including many drugs used for a wild range of therapeutic applications. Therefore, the research of safe and effective P-gP inhibitors endowed of high selectivity and potency represent a great challenge in medicinal chemistry. In particular, the possibility to have selective molecules able to inhibit this protein could be useful to modulate the pharmacological behaviour of drugs including those used for chemo-therapy-resistant tumors. Up to now, many P-gP modulating agents, have been characterized including channel blockers, calmodulin antagonists, immunosuppressant and protein kinase inhibitors but all these compounds produced disappointing results in vivo because the use of high dose resulting in unacceptable toxicity. Some new drugs such as biricodar, tariquidar and elacridar have satisfactory inhibitory effects on P-gP. This latter displayed a high potency for P-gP transporter but it also inhibited breast cancer resistance protein (BCPR). On the basis of computational studies which suggested a general pharmacophore of a substrate/inhibitor of P-gP transporter, in which both a planar aromatic domain and a presence of a basic nitrogen atom within an extended side chain are described, we designed and synthesised new compounds of type A. These molecules possessed a benzylethereal side chain substituted by aliphatic spacer of 2, 3, or 4 methylenes linked to an arylpiperazine nucleus.
O
O
N
NR
n
R = 3-Cl-Ph 3-CF3-Ph
2-OCH3-Ph
4-F-Ph
2-Pyrimidil
n = 2-4
O
O
Nn
n = 2-4
O
O
N
NR
n
X
O
Ar
R = Me
n = 2-4
TETRAHYDROISOQUINOLINE SERIES
ARYLPIPERAZINE SERIES
N-METHYLPIPERAZINE SERIES
ARYLMETHYLOXY-and-
ARYLMETHYLAMINO-PHENYL
DERIVATIVES
Ar = 2Py; 3-Py; 4-Py 2-OCH3-Ph;3-OCH3-Ph;4-OCH3-Ph
2,3-OCH3-Ph;3,4-OCH3-Ph;3,5-OCH3-Ph X = O, NH
TYPE BTYPE A
All the synthesized compounds have been tested in vitro to evaluated the inhibitory activity on Caco2 cells in which P-gP is overexpressed and the best results have been obtained for compounds having a
117

four methylene chain. Unfortunately, as expected, some arylpiperazine derivatives displayed serotoninergic (5-HT1A) and dopaminergic (D2) receptor affinity. In order to improve P-gP affinity and to lift these subordinate activities some structural manipulations on these derivatives have been carried out. In particular we decided to bring the substitution of the aryl- or arylpiperazine- moieties with a methyl group or a tetrahydroisoquinoline nucleus, respectively. Besides, new ligand series of type B maintaing the arylmethyloxyphenyl- skeleton were prepared[1-3].Moreover the replacement of the oxygen atom with a nitrogen one led to arylmethylaminophenyl derivatives[2] which showed a higher potency than the one of oxygen-analogues[1].Since the limit of P-gP inhibitors such as elacridar is the poor selectivity toward other ABC transporters, in particular the BCRP pump, all the active compounds were tested for their ability to inhibit [3H]-mitoxantrone, a specific BCRP substrate. The results showed that only arylmethylaminophenyl derivatives displayed a good BCRP inhibition activity[1,2].These results led us to consider the 2-[(3-methoxyphenylethyl)phenoxy]- fragment, present in the more selective P-gP inhibitors, the pivotal molecular basis to design novel P-gP inhibitors.
REFERENCES
[1] Colabufo N. A.; Berardi F.; Perrone R.; Rapposelli S.; Digiacomo M.; Balsamo A. Arylmethyl oxyphenyl Derivatives: Small Molecules Displaying P-Glycoprotein Inhibition. J. Med. Chem. 2006,49, 6607–6613.
[2] Colabufo N. A.; Berardi F.; Perrone R.; Rapposelli S.; Digiacomo M.; Vanni M; Balsamo A. Synthesis and Biological Evaluation of (Hetero)Arylmethyloxy- and Arylmethylamine-phenyl Derivatives as Potent P-glycoprotein Modulatine Agents. J. Med. Chem. 2008, 51, 1415–1422.
[3] Balsamo A.; Berardi F.; Colabufo N. A.; Perrone R.; Rapposelli S.; “1-fenilalcossi-2- -feniletilderivati quali inibitori della glicoproteina-P (P-gP) utilizzabili in casi di farmacoresistenza” BREVETTO ITALIANO RM2006A000217
118

SUGAR-MODIFIED NUCLEOSIDES AND NUCLEOTIDES: SYNTHESIS,
CONFORMATIONAL ANALYSIS AND BIOLOGICAL EVALUATION
Patrizia VITA
Dipartimento di Scienze Chimiche, Università degli Studi di Camerino Dottorato di ricerca in Scienze Farmaceutiche, XXI ciclo
Nucleoside analogues are a pharmacologically diverse family that includes cytotoxic compounds, antiviral agents and immunosuppressive molecules. Considerable progress has been made in the search for novel nucleoside structures with anticancer and/or antiviral activity by modifications in the base or in the sugar moiety and several branched-chain sugar nucleosides have shown potent antitumor activity.1
After metabolic activation, these compounds act as antimetabolites inhibiting the DNA chain elongation or interfering with de novo synthesis of nucleosides and nucleotides through specific enzyme(s). One of these key enzymes is ribonucleotide reductase (RNR), a multisubunit radical-containing enzyme involved in the conversion of ribonucleoside diphosphates to their corresponding deoxyribonucleotides, which are building blocks for DNA replication and repair. Increased RNR activity has been associated with malignant transformation and tumor cell growth; it is therefore an excellent target for cancer chemotherapy. In recent years, several nucleoside RNR inhibitors have entered clinical trial or application such as arabinosylcytosine (ara-C), gemcitabine, cladribine, fludarabine and clofarabine. A previous structure-activity relationship study reported by us, identified the 3'-C-methyladenosine (3'-Me-Ado) as a mechanism-based RNR inhibitor endowed with a significant antitumor activity against a panel of human leukemia and carcinoma cell lines.2
To further investigate the structural determinants of 3'-Me-Ado required for the antitumor activity, in the first part of my PhD project, a number of 3'-C-methyl ribonucleosides with different nucleobases such as guanine, uracil, thymine, cytosine and the 3'-C-methyl derivatives of the antitumor agents 5-fluorouridine and 9-(β-D-arabinofuranosyl)adenine were synthesized and evaluated on human tumor cell lines.3
OHHO
HO
N
N
N
N
NH2
H3C
O OHHO
HO
B
H3C
O
HO
HO
N
N
N
N
NH2
H3C
O
OHB = guanineB = uracilB = thymineB = 5-F-uracilB = citosine3'-Me-Ado 3'-Me-ara-A
Among these, only 3'-C-methyluridine showed a cytotoxic effect against human leukemia K562, showing that the structure of 3'-Me-Ado is crucial for the antitumor activity of this type of ribose-modified nucleosides.3
However, on the basis that several adenosine derivatives monosubstituted at the N6-position demonstrated significant antitumor activity,4 a series of adenine 3'-C-methyl-ribonucleosides containing different substituent at N6-amino group of nucleobase such as a cycloalkyl, aryl or heteroaryl group were synthesized and in vitro tested as antitumor agents. Structure-activity relationships of the new compounds have been rationalized by docking studies utilizing the x-ray structure of the subunit Rnr1 of the RNR from Saccharomyces cerevisiae complexed with ADP.
OHHO
HO
N
N
N
N
NHR1
H3C
O
R
R = H, R1 =
Cycloalkyl Aryl Heteroaryl
R = Cl, R1 =
S
O
NH
N
119

Moreover, on the basis that the inhibition of RNR activity requires the intracellular conversion of nucleoside analogues into the corresponding 5'-diphosphates containing an unstable phosphate bridge, the methylenbisphosphonate derivative of 3'-Me-Ado was synthesized as a stable pyrophosphate-mimic. However, no cytoxicity was detected on human leukemia K562 cell line, probably due to inability of the nucleotide to across the cellular membrane and therefore to inhibit RNR. The ribose recognition domain appears to be of considerable importance also for activity at adenosine receptors.Adenosine affects a wide variety of physiological functions that are mediated by interaction with four receptor subtypes named A1, A2A, A2B and A3. The A1 adenosine receptors (A1ARs) are expressed in high density in the brain and in adipose tissue and in medium to low levels in many tissues as heart, lung and kidney. Thus, A1 agonists have many potential therapeutic applications, but their clinical use is hampered by cardiovascular side effects caused by strong hypotensive action and side effects at other organs.One way to circumvent the side effects of full agonists is the use of A1 selective partial agonists. In the search for potent and selective A1AR agonists we have previously investigated a series of 2'-C-methylribofuranosyl analogues of selective A1AR agonists and 2-chloro-2'-C-methyl-N6-cyclopentyl-adenosine (2'-MeCCPA) emerged as a potent and highly selective full agonist at rat, bovine and human A1 versus A2A, A2B and A3ARs.5
On the base of these findings, in the second part of my work, new purine nucleosides modified at C2'(5')-, and N6(C-2)-positions of ribose and purine moiety respectively, were designed as potential partial A1
agonists. 2'-C-methyl derivatives of N6-cyclopentyladenosine such as 2'-MeCPA, 2'-MeCCPA and of N6-[(R)-3-tetrahydrofuranyl]-analogue substituted at the 5'-position with a carbamoyl or thionocarbamoyl group were synthesized.
OHHO
R2O
N
N
N
N
NHR1
O
R
CH3
R = H or Cl, R1 = R2 = carbamoyl or thionocarbamoyl group
R = Cl, R1 = R2 = H (2'-Me-CCPA)
Oor
Evaluation of these nucleoside analogues for binding affinity at ARs and for relative intrinsic efficacy showed a different profiles at the receptors from bovine, porcine and human species. However, the tested nucleosides retained the A1 versus A2A, A2B, and A3 human ARs selectivity. Molecular modelling analysis explained the lower affinity of N6-tetrahydrofuranyl derivatives at human A1AR compared to that of the N6-cyclopentyl analogues. Moreover, 5'-methylthionocarbamoyl derivative of 2'-Me-CCPA proved to be partial agonist at porcine A1AR.6
References
(1) a) Niitsu, N.; Ishii, Y.; Matsuda, A.; Honma, Y. Cancer Res. 2001, 61, 178. b) Hattori, H.; Nozawa, E.; Iino, T.; Yoshimura, Y.; Shuto, S.; Shimamoto, Y.; Nomura, M.; Fukushima, M.; Tanaka, M.; Sasaki, T.; Matsuda, A. J. Med. Chem. 1998, 41, 2892
(2) Franchetti, P.; Cappellacci, L.; Pasqualini, M.; Petrelli, R.; Vita, P.; Jayaram, H. N.; Horvath, Z.; Szekeres, T.; Grifantini, M. J. Med. Chem. 2005, 48, 4983
(3) Cappellacci, L.; Franchetti, P.; Petrelli, R.; Riccioni, S.; Vita, P.; Jayaram, H. N.; Grifantini, M. Collect. Czech. Chem. Commun. 2006, 71, 1088
(4) Mlejnek, P. J. Cell. Biochem. 2001, 83, 678 (5) a) Franchetti, P.; Cappellacci, L.; Marchetti, S.; Trincavelli, L.; Martini, C.; Mazzoni, M. R.;
Lucacchini, A.; Grifantini, M. J. Med. Chem. 1998, 41, 1708. b) Cappellacci, L.; Franchetti, P.; Pasqualini, M.; Petrelli, R.; Vita, P.; Lavecchia, A.; Novellino, E.; Costa, B.; Martini, C.; Klotz, K.-N.; Grifantini, M. J. Med. Chem. 2005, 48, 1550
(6) Cappellacci, L.; Franchetti, P.; Vita, P.; Petrelli, R.; Lavecchia, A.; Costa, B.; Spinetti, F.; Martini, C.; Klotz, K.-N.; Grifantini, M. Bioorg. Med. Chem. 2008, 16, 336
120

SYNTHESIS OF 4’-THIONUCLEOSIDES BY 1,3-DIPOLAR CYCLOADDITIONS
AS POTENTIAL ANTIVIRAL AGENTS1
Elisa VITTORINO
Dipartimento di Scienze Chimiche, Università di Catania, viale A. Doria 6, 95125 Catania (Italy); PhD in Pharmaceutical Sciences-XXI cycle
In the past fifteen years, a wide variety of nucleoside analogues, modified in the carbohydrate moiety and/or in the nucleobase, have been prepared and tested for antiviral and antitumoural activities.2 One of the first modifications was the isosteric substitution of the oxygen atom with a sulfur atom in furanosyl nucleosides of adenine affording the corresponding 4’-thio-derivatives.3 These 4’-thionucleosides showed potent anti-herpes activities, and several other analogues, such as 4’-thiothymidine and 2’-deoxy-4’-thiocytidine, exhibited potent cytotoxicity. On a chemical level, it was envisaged that this modification would modulate stereoelectronic and steric effects in the tetrahydrothiophene moiety. Evidences were accumulating that the presence of a sulfur atom in the sugar ring stabilized the N-glycosidic bond with respect to phosphorolysis.4 Thus 4’-thioinosine was known to be resistant to phospholytic cleavage, which is a normal pathway in nucleoside catabolism. This is a major advantage of 4’-thionucleosides, since several ‘4’-oxy’ antivirals have a drawback with regard to their metabolic stability caused by nucleoside phosphorylases. In addition, the potent antiviral activity and cytotoxicity of 4’-thionucleosides suggest that they are well recognized as substrates by both viral and host cell kinases. Frequently, synthetic approaches to 4’-thionucleosides have made use of displacement reactions of a sugar leaving group with a sulfur-containing nucleophile, followed by ring closure or ring contraction,5
acetolysis of a sugar , -diethoxy episulfide, or ring closure of sugar dialkyl dithioacetal,6 since the initial stereochemistry of sugars directly controls that one of the final products through stereo-specific or -selective steps. Since tetrahydrothiophene syntheses starting from sugars have often met various difficulties, among which, for example, the frequent involvement of many steps, here, we planned to synthesize the tetrahydrothiophene moiety via a practical method involving the 1,3-dipolar cycloaddition of the simplest thiocarbonyl ylide 1,7 generated by CsF decomposition8 of the very easy to synthesize chloromethyltrimethylsilylmethyl sulfide,9 to a variety of appropriate alkenes bearing electron-withdrawing substituents, affording tetrahydrothiofene adducts 3 (Scheme 1). 4’-Thionucleosides 4 were then constructed by conventional synthetic methods, among which the coupling reaction of the corresponding sulfoxides with a persilylated nucleobase is largely applied in most approaches.10 Besides thymine and cytosine, uracil and its 5-fluoro derivative were also used as nucleobases for this thioglycosylation reaction based on the sila-Pummerer-type reaction.11
MeO2C
S3 (82%)
+
CO2MeMeO2C
S
MeSiCH2SCH2ClCsF
2
S
OHHO
B
4
1
CO2Me
Scheme 1
By following the same sequence of reactions by using (E)-3-benzoyloxypropenoyl-(1S)-camphorsultam 5with 1, tetrahydrothiophene rings 6 were obtained, from which the construction of the desired 4’-thionucleosides 8 bearing the nucleobase and the hydroxymethyl group in positions 3 and 4, was achieved. In addition, the tosyl derivative 7 was subjected to an other process including the elimination of the tosylate to produce the protected 4-hydroxymethyl thioglycal 9. The addition of phenylselenyl chloride and nucleobases, as silylated derivatives, to 9 provided adducts from which subsequently the phenylselenyl substituent was eliminated (Scheme 2). In this way, desired - and -thioapionucleosides
121
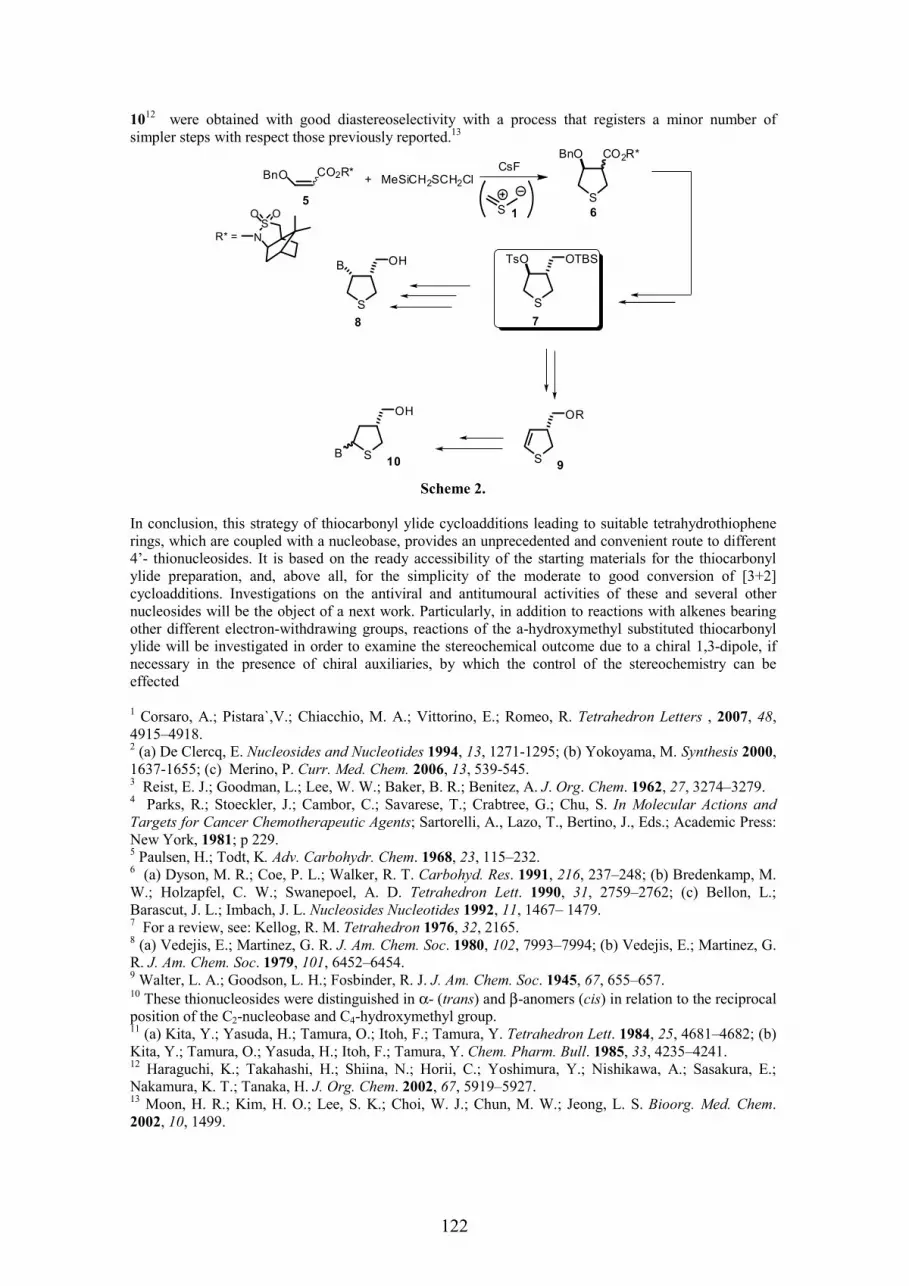
1012 were obtained with good diastereoselectivity with a process that registers a minor number of
simpler steps with respect those previously reported.13
BnO
S
+
S
N
O O
R* =
CO2R*BnO
S
65
MeSiCH2SCH2Cl
TsO
S
OTBS
S
OHB
78
CO2R*
S
OH
B10
1
S
OR
9
CsF
Scheme 2.
In conclusion, this strategy of thiocarbonyl ylide cycloadditions leading to suitable tetrahydrothiophene rings, which are coupled with a nucleobase, provides an unprecedented and convenient route to different 4’- thionucleosides. It is based on the ready accessibility of the starting materials for the thiocarbonyl ylide preparation, and, above all, for the simplicity of the moderate to good conversion of [3+2] cycloadditions. Investigations on the antiviral and antitumoural activities of these and several other nucleosides will be the object of a next work. Particularly, in addition to reactions with alkenes bearing other different electron-withdrawing groups, reactions of the a-hydroxymethyl substituted thiocarbonyl ylide will be investigated in order to examine the stereochemical outcome due to a chiral 1,3-dipole, if necessary in the presence of chiral auxiliaries, by which the control of the stereochemistry can be effected
1 Corsaro, A.; Pistara`,V.; Chiacchio, M. A.; Vittorino, E.; Romeo, R. Tetrahedron Letters , 2007, 48,4915–4918. 2 (a) De Clercq, E. Nucleosides and Nucleotides 1994, 13, 1271-1295; (b) Yokoyama, M. Synthesis 2000,1637-1655; (c) Merino, P. Curr. Med. Chem. 2006, 13, 539-545. 3 Reist, E. J.; Goodman, L.; Lee, W. W.; Baker, B. R.; Benitez, A. J. Org. Chem. 1962, 27, 3274–3279. 4 Parks, R.; Stoeckler, J.; Cambor, C.; Savarese, T.; Crabtree, G.; Chu, S. In Molecular Actions and Targets for Cancer Chemotherapeutic Agents; Sartorelli, A., Lazo, T., Bertino, J., Eds.; Academic Press: New York, 1981; p 229. 5 Paulsen, H.; Todt, K. Adv. Carbohydr. Chem. 1968, 23, 115–232. 6 (a) Dyson, M. R.; Coe, P. L.; Walker, R. T. Carbohyd. Res. 1991, 216, 237–248; (b) Bredenkamp, M. W.; Holzapfel, C. W.; Swanepoel, A. D. Tetrahedron Lett. 1990, 31, 2759–2762; (c) Bellon, L.; Barascut, J. L.; Imbach, J. L. Nucleosides Nucleotides 1992, 11, 1467– 1479. 7 For a review, see: Kellog, R. M. Tetrahedron 1976, 32, 2165. 8 (a) Vedejis, E.; Martinez, G. R. J. Am. Chem. Soc. 1980, 102, 7993–7994; (b) Vedejis, E.; Martinez, G. R. J. Am. Chem. Soc. 1979, 101, 6452–6454. 9 Walter, L. A.; Goodson, L. H.; Fosbinder, R. J. J. Am. Chem. Soc. 1945, 67, 655–657. 10 These thionucleosides were distinguished in α- (trans) and β-anomers (cis) in relation to the reciprocal position of the C2-nucleobase and C4-hydroxymethyl group. 11 (a) Kita, Y.; Yasuda, H.; Tamura, O.; Itoh, F.; Tamura, Y. Tetrahedron Lett. 1984, 25, 4681–4682; (b) Kita, Y.; Tamura, O.; Yasuda, H.; Itoh, F.; Tamura, Y. Chem. Pharm. Bull. 1985, 33, 4235–4241. 12 Haraguchi, K.; Takahashi, H.; Shiina, N.; Horii, C.; Yoshimura, Y.; Nishikawa, A.; Sasakura, E.; Nakamura, K. T.; Tanaka, H. J. Org. Chem. 2002, 67, 5919–5927. 13 Moon, H. R.; Kim, H. O.; Lee, S. K.; Choi, W. J.; Chun, M. W.; Jeong, L. S. Bioorg. Med. Chem.2002, 10, 1499.
122

SEARCH FOR SELECTIVE ADENOSINE A1 AND A2A RECEPTOR LIGANDS:
SYNTHESIS, PHARMACOLOGY, 3D-QSAR STUDIES AND MOLECULAR
MODELLING
Olga YUZLENKO, Katarzyna KIE -KONONOWICZ
Department of Technology and Biotechnology of Drugs, Medical College, Jagiellonian University, Medyczna Str. 9, 30-688 Kraków, Poland.
PhD work title: Search for selective A1 and A2A adenosine receptor ligands
The physiological effects of extracellular adenosine are mediated by four G protein-coupled receptors: A1, A2A, A2B and A3 adenosine receptors (ARs). Selective A1 AR antagonists have demonstrated promising therapeutic potential for the treatment of cognitive diseases, renal failure, Alzheimer’s disease and cardiac failure. Adenosine A2A receptor antagonists may be useful for the treatment of acute and chronic neurodegenerative disorders such as cerebral ischemia, Parkinson’s and Huntington’s diseases, as drugs controlling motor function and exhibiting neuroprotective properties. For adenosine A1 and A2A receptors xanthine derivatives have been designed as potential antagonists, synthesised and studied for their pharmacological activity. Tricyclic annelated xanthines exhibited high submicromolar affinity to the A1 and A2A ARs. The synthesis of tricyclic xanthine derivatives, the adenosine A1 and A2A receptors ligands was performed. The obtained derivatives possess main purine skeleton and annelated third ring with different heteroatoms. To study biological activity of the designed compounds, different substituents were introduced mainly at the nitrogen atom of the annelated ring. Investigated were compounds with aliphatic and aromatic substituents. The following groups of compounds were synthesized: 6-hydroxypyrimido[1,2-a]purinetriones (I), trimethylpyrimido[1,2-a]purinediones (II), [1,2,4]triazino[4,3-a]purinediones (III), [1,2,4]triazepino[4,3-a]purinediones (IV),[1,4]oxazino[4,3-a]purinediones (V). Among them there were molecules with high submicromolar affinity as well as selective compounds. The most favourable for both A1 and A2A ARs were compounds with annelated pyrimidine (trimethylpyrimido[1,2-a]purinediones) or triazepine ([1,2,4]triazepino[4,3-a]purinediones) rings. It was shown that the annelated ring substituents have a great impact not only on the ligands activity but also on ARs subtype selectivity.
N
N N
O
CH3
O
CH3
NN
OH R
O
R'
N
N N
O
CH3
O
CH3
NN
CH3
R
N
N N
O
O
CH3
CH3
N
N
N
R
R'
N
N N
O
O
CH3
CH3
N
NN
R
R'
N
N
O
CH3
O
CH3
N
N O
R
I II III
VIV
The obtained derivatives were tested in radioligand binding assays in vitro for A1, A2A and some for A3
adenosine receptors affinity. In vivo tests were performed in order to estimate anticonvulsant activity of the synthesized compounds. Trimethylpyrimido[1,2-a]- and [1,2,4]triazepino[4,3-a]purinediones were the most active groups with the derivatives which displayed submicromolar affinity to the studied adenosine receptor subtypes. The most preferable substituents in the third ring were long alkyl chains (pentyl or hexyl) as well as phenyl ring with a long linker. Most active compounds were: non-selective 1,3,8-trimethyl-9-pentylpyrimido[1,2-a]purinedione (Ki(A1AR)= 0.33 µM; Ki(A2AAR)= 0.36 µM) and A1
adenosine receptor ligand (Z)-3-ethyl-8,10-dimethyl-1-(3-pnenylpropyl)-4,5-dihydro-1H-[1,2,4]triazepino[4,3-a]purinedione, which showed 15-fold selectivity toward A2AAR (Ki(A1AR)= 0.29 µM; Ki(A2AAR)= 4.60 µM).
123

Crystallographic analysis was performed for selected ligands. This investigation revealed compounds’ spatial structure, which was used for further investigations in silico that included 3D-QSAR and docking studies. The molecules, including those previously obtained at the Department of Technology and Biotechnology of Drugs [1-4], were constructed by means of CAChe 7.6 software and optimized by semi-empirical and quantum chemistry methods giving the structures that were in accordance with the data from the X-Ray analysis. A library of all xanthine derivatives was created. Sixty-three active adenosine A1 receptor ligands and one hundred thirty nine active adenosine A2A receptor ligands were used for 3D-QSAR investigation to derive the equations with high predictive power for the affinity through the calculation of molecular descriptors. Two methods of validation were studied: subdividing the dataset into the training and test sets, as well as using the whole dataset and checking the predictive ability of the equations by means of cross-validated coefficients.
The reliable 3D-QSAR models able to rationalize the activity of A1 and A2A adenosine receptor for tricyclic xanthine ligands were developed. The analysis of the results depicts that for the A1 AR binding activity it is important for ligands to possess R1-propyl substituents along with the phenyl or benzyl substituents bearing halogen atom and phenethyl moiety. For A2A AR affinity it could be favourable to introduce phenethyl or phenyl substituent connected with the tricyclic ring by the alkoxy chain. The nature of R1 group may not significantly affect the A2A AR affinity. High predictive power of the equations implies that they can be used for further in silico design of the xanthine derivatives.
The molecular modelling studies of rat A1 and A2A adenosine receptors were performed [5]. The high-resolution crystal structures of bovine rhodopsin and recently discovered 2-adrenergic receptor were used for A1 and A2A adenosine receptors models building. The models were validated indirectly using MolProbity and Ramachandran plot programs, as well as structurally through the docking of the known ligands and investigation of mutagenesis data. The models were compared in docking studies for their ability to bind high affinity antagonists. It was concluded that for the docking experiments it is preferable to use the 2-adrenergic-based models especially for A2A AR, as they were able to better stabilize the ligands in their bindind cavities in comparison to those rhodopsine-based models. Moreover, this suggestion also arises form the higher homology between adenosine A1 and A2A receptors and 2-adrenergic receptor (35%) in comparison to bovine rhodopsin (21%).
The docking studies were performed using 2-adrenergic receptor-based A1 and A2A adenosine receptors models to explain the affinity of the synthesized ligands towards A1 and A2A ARs. The models were able to describe precisely the activity and selectivity of the ligands based on the active site cavity they occupied. The docking studies of the synthesized ligands confirmed the hypothesis of the presented 3D-QSAR investigations. For A2AAR subtype, the surface properties are of great importance, while for A1ARligands electrostatic properties are significant. A combined target-based and ligand-based approach can be used to improve the search for high affinity selective adenosine receptor ligands.
The 3D-QSAR equations developed in this research along with the analysis of the ligands docked into the active sites of the constructed receptors can be valuable for further discovery of active and selective A1
and A2A AR ligands.
This work was financially supported by the scholarships from: St. Vladimir the Baptist of Kiev Ruthenia Foundation, UNESCO Polish Committee, Academic Innovation for Ma opolska Program, Kasa Mianowskiego and International Visegrad Fund.
This research was supported by Polish Ministry of Science and Education Grant No. 2 P05F 014 28.
1. Drabczy ska, A.; Müller, C.E.; Lacher, S.K.; Schumacher, B.; Karolak-Wojciechowska, J.; Nasal, A.; Kawczak, P.; Yuzlenko, O.; Kie -Kononowicz. Synthesis and biological activity of tricyclic arylo-imidazo-, pyrimido-, and diazepinopurinediones. Bioorg. Med. Chem. 2006, 14, 7258-7281.
2. Drabczy ska, A.; Müller, C.E.; Karolak-Wojciechowska, J.; Schumacher, B.; Schiedel, A.; Yuzlenko, O.; Kie -Kononowicz. N9-Benzyl-substituted 1,3-dimethyl and 1,3-dipropyl-pyrimido[2,1-f]purinediones: Synthesis and structure-activity relationship at A1 and A2A adenosine receptors. Bioorg. Med. Chem. 2007, 15, 5003-5017.
124

3. Drabczy ska, A.; Müller, C.E.; Schiedel, A.; Schumacher, B.; Karolak-Wojciechowska, J.; Fruzi ski, A.; Zobnina, W.; Yuzlenko, O.; Kie -Kononowicz. Phenethyl-substituted pyrimido[2,1-f]purinediones and related compounds: structure activity relationships as adenosine A1 and A2A receptor ligands. Bioorg. Med. Chem. 2007, 15, 6956-6974.
4. Drabczy ska A.; Müller C.E.; Schiedel A.; Schumacher B.; Karolak-Wojciechowska J.; Yuzlenko O.; P kala E.; Kuder K.; Kie -Kononowicz K. Synthesis and biological activity of tricyclic cycloalkylimidazo-, pyrimido and diazepinopurinediones. Bioorg. Med. Chem. 2008, in preparation
5. O. Yuzlenko, K Kie -Kononowicz. Molecular modelling of A1 and A2A adenosine receptors: Comparison of rhodopsin- and 2-adrenergic-based homology models through the docking studies. J. Comput. Chem. 2008, XX, XXX-XXX
125

Tc-99m LABELLING APPROACHES OF NEW OCTREOTIDE ANALOGUES FREE FROM
DISULPHUR BRIDGE WITH HIGH AFFINITY TOWARDS SSTR EXPRESSING TUMOURS
Elena ZANGONI
Dept of Pharmaceutical Sciences, University of Padova Doctoral School in Molecular Sciences
Introduction. Somatostatin (SST) has many biological actions. Among them it appears to be an endogenous antiproliferative agent. SST analogues, by their wide spectrum of activities, could inhibit various tumours through multiple mechanisms. So far, five subtypes of somatostatin receptors (SSTR1-5) were cloned and functionally characterised. They belong to the guanine nucleotide-binding regulatory proteins (G-proteins)-linked receptor family. Since mRNAs of receptor subtypes are variably expressed in different cancers, a precise determination of SSTR profiles in tumour tissue before therapy with analogues is needed. The half-life of SST is very short; thus, its therapeutic use is impractical. Many SST analogues with more selective and prolonged activities were synthesis in the past, two or three of them being in use after labelling with appropriate radionuclides such as 111In, 123I, 188Re, or 90Y in cancer diagnosis or therapy. Radiolabeled SST analogues could allow treatment of SSTR-positive tumours by delivering a radioactive isotope to the cancer cell. The most used SST analogue, octreotide, has the highest affinity for SSTR2, lower affinities for SSTR3
and SSTR5, and no affinity for SSTR1 and SSTR4 [1].Basic knowledge of SSTR subtype profiles and of binding and internalisation of octreotide-analogue radiopharmaceuticals is important in evaluating the potential usefulness of receptor-mediated radiotherapy. Although SSTR specific analogues have been proposed, evaluation is complicated by profound disagreements among investigators relating to species specificity and expression systems in determining binding affinities to SSTR. Synthetic analogues such as BIM-23268D, L-362,855 (BIM-23208D), DC-23–99 (BIM-23052), and BIM-23059 have been proposed as SSTR5-selective agonists in different and sometimes conflicting reports. Recently, Ginj et coll describes the design, synthesis, and pre-clinical evaluation of new SST-based DOTA-coupled peptides. They designed and characterised two new DOTA-based peptides for diagnostic and therapeutic applications, [111In/90Y-DOTA]-NOC-ATE and [111In/90Y-DOTA]-BOC-ATE, which represented the first high SSTR2, SST3, and SST5 affinity SST-based radio-peptides with efficient internalisation into a SSTR3 expressing cell line. Our work should serve to resolve these questions and provide pre-clinical data defining appropriate agents for clinical trials starting from the synthesis of new SST analogues. The project will represent the background for the development of new pharmaceuticals with higher selectivity towards different SSTR subtypes and, thus, to obtain a favourable cellular targeting. SST analogues show, in most of the cases, the disulphide bridge of the parent somatostatin that can be subjected to the attacks of oxidizing and reducing agents. This prompted us to search a more stable tether bridging the active motif of somatostatin, identified as the Phe-d-Trp-Lys-Thr sequence. At the same time, the new side-chain to side-chain closure had to preserve the conformation of this sequence in octreotide. We previously synthesized the cyclic dicarba-analogue 1 that contained a -CH=CH- bridge[2]. More recently, we synthesized tree new unsaturated dicarba-analogues substituting respectively Thr10 with Phe and Tyr(Bzl) and Phe7 with 1-Nal starting from the parent linear hepta-peptides and accordingly to the solid phase peptide synthesis technique. The ring closure between the Hag1 and Hag7 were achieved by a
Figure 1: R:CH3-CH-(OH)-, R':C6H5CH2-; 2: R:C6H5CH2-, R':C6H5CH2- ; 3 R:C6H5CH2O-C6H4-CH2-, R':C6H5CH2- ; 4: R:CH3-CH-(OH)-, R':C10H7CH2-;
HO
HO CH3
HN
O
NH
O
R
NH
NH
NH2
HN
O
NH
R'
O
HN
O
NH
O
O
NHH
H
PN2S
126

Ring Closing Metathesis reaction using Grubbs catalyst. Peptide were cleaved from the resin and purified by RP-HPLC affording 1-4 (Fig 1). Results The absence of disulphur bridge allows the labelling with and 99mTc of the above derivatives without problems. Two already known labelling procedures were adopted [3,4]. The first approach consists in the modification of 4 derivative with PN2S (N-[N-(3,diphenylphosphinopropionyl) glycyl]cysteine) bifunctional ligand, followed by the labelling reaction by using the 99mTcO(gluconate)2
intermediate compound. The saline 99mTcO4- solution (40 µL, ∼37 MBq) was added to a peptide solution
in EtOH (1,2 mg/ml, 60 µl) also containing Nagluconate (0.01M in H2O, 1 µl) and SnCl2 (0.1M in HCl 0.1M, 2µl). The mixture was heated for 30 mins at 65°C, leading to a single labelled species, monitored by RP-HPLC, which remains stable for more than 24 h. The second approach was performed on 1 derivative, modified with the same PN2S bifunctional ligand, by using a (2+1) tricarbonyl approach developed by our group[3]. The reaction consists in the production of [99mTc(CO)3]
+ intermediate with kit Isolink™ and the addition of 30 µL of the labelled intermediate (∼37 MBq) to a solution formed by mixing a solution of the peptide in EtOH (6 mg/ml, 15 µl) and a solution of DTCM in EtOH (1 mg/ml, 5.2 µl). The final labelled species, monitored by RP-HPLC, was already observed at room temperature after few minutes. From developed studies, the best 1-(PN2S)/DTCM molar ratio for the preparation of the mixed complex is 5/1. Higest amounts of DTCM lead to tricarbonyl species with only DTCM, which reduce the peptide labelling yields.Biodistribution studies in vitro and in vivo are going to be developed for comparing the two different labelled species in the maintenance of the tumor receptor affinity.
References
[1] Patel Y.C., Front. Neuroendocrinology 1999, 20, 157.[2] Carotenuto, A.; D’Addona, D.; Rivalta, E.; Chelli, M.; Papini, A.M.; Rovero, P.; Ginanneschi, M.; Letters in Organic Chemistry, 2005, 2 (3), 274-279.- D’Addona D., Carotenuto A., Novellino E., Piccand V., Reubi J.C., Di Cianni A., Francesca G., Papini A.M., Ginanneschi M. J. Med. Chem., accepted. [3] Riondato M., Camporese D., Martin D., Suades J., Alvarez-Larena A., Mazzi U., Eur J Inorg. Chem., 2005, 20, 4048-4055 [4] Santimaria M., Blok D., Feitzma R.I.J., Mazzi U., Puwels E.K.J., J Nucl Med & Biol 1999, 26, 251-258.
127

PROAPOPTOTIC AND CYTOTOXIC EFFECTS OF MRJF4
ON HUMAN PROSTATE CANCER CELLS
Laura ZAPPALÀ
Department of Pharmaceutical Sciences-Pharmacology section, University of Catania, Viale A.Doria 6, 95125 Catania, Italy
PhD in Pharmaceut ica l Sciences XXI Cic lo
Prostate cancer is one of the most common causes of death in Western countries, and remains the second cause of cancer related death in men1. Initially cancer responds to hormonal therapy and after one or two years becomes resistant. The conversion of tumor cells from the androgen-sensitive into the androgen-refractory type, with consequent high risk of metastatic evolution, is a multiphasic process that remains a relevant clinical issue. The antiandrogenic drugs, in fact, induce cell death in androgen-sensitive cells, but are ineffective on androgen-insensitive cells. The development of several human cancer is linked to alteration of histone deacetilase (HDAC) activity that lead aberrant transcription of key genes regulating important cellular functions such as cell proliferation, cell-cycle regulation and apoptosis2. Actually new therapeutic strategies employ HDAC inhibitors alone or in combination with other drugs3. In this study, we sought to determine whether MRJF4, a new molecule obtained by fusion between an HDAC inhibitor and a sigma ligand, actively enter into the prostatic cancer cells that over-express sigma receptors4.Human prostate cancer cell lines, LNCaP and PC3, were exposed to graded concentrations of MRJF4. Cells were incubated for 96h and the cell viability was measured by MTT assay at different time. Apoptosis was evaluated by acridine orange(AO)/ethidium bromide (EB) nuclear staining to detect changes in cell morphology. The treatment with σ1 agonist and with σ2 antagonist show that the antiapoptotic effect of MRJF4 is determined by agonist activity σ2.
A. B.
MRJF4 treatment produced dose- and time-dependent cytotoxicity. Since the 24 hours the IC50 values are respectively 200-folds lower than 4-phenylbutyric acid (4-PB) and 20-folds lower than reduced
128

haloperidol alone and in association with 4- PB. Both cell lines when treated with MRJF4 showed some morphological features of apoptosis, such as chromatin condensation and cell shrinkage. Our results indicate that MRJF4 exerts pronounced proapoptotic effects in human prostatic cancer cells. These preliminary experimental data suggest a potential role of MRJF4 in the treatment of androgen-sensitive and androgen-refractory prostate cancer. Change in nuclear morphology were analyzed by double staining acridine orange/ethidium bromide. A e B LNCaP cells treated with MRFJ4 10 µM for 24h (A exitation AO; B exitation EB); C e D PC3cells treated with MRFJ4 10 µM for 24h (A exitation AO; B exitation EB). The arrows indicate nuclei of apoptotic cells characterized by marked chromatin condensation.
References
1. Li LC: Epigenetics of prostate cancer. Front Biosci. 2007 May 1;12:3377-97 2. So AI, Hurtado-Coll A. and Gleave, ME. World J. Urol. 2003; 21, 325-337. 3. Balakin KV, Ivanenkov YA, Kiselyov AS, Tkachenko SE. Anticancer Agents Med Chem. 2007;7(5):576-92. 4. Pan L, Lu J, Huang B. Cell Mol Immunol. 2007;4(5):337-343. 5. Colabufo N, Berardi F, Contino M, Ferorelli S, Niso M, Perrone R, Pagliarulo A, Saponaro P, Pagliarulo V. Cancer Lett. 2006; 237(1): 83-88.
129

A NEW AUTOMATED STRATEGY TO JOIN LIGAND- AND STRUCTURE-BASED DRUG DESIGN
Teresa Fabiola MISCIOSCIA
Dipartimento Farmaco-Chimico,Università di Bari, Via E Orabona 4, 70125 Bari
Dottorato di Ricerca in Scienze Farmaceutiche
Despite the efforts spent to develop newer docking algorithms, their potential to forecast
and properly sort all the calculated viable complexes still represent an unsolved
problem. Here, a novel strategy to join structure- and ligand-based drug design is
proposed. Developed entirely in-house, our automated computational package [1] called
@ゲic& (standing for aligning chemical ensembles) is able to generate molecular
superimpositions on the basis of a genetic algorithm which optimise the value of a
fitness function that computes at the same time the best regressions among binding
energy docking scores and affinities, and the minimal geometric deviations from
suitably defined crystal-based binding hypothesis. Molecular features of ligand binding
affinity associated to each molecular alignment were typified by adopting GRID/CPCA
approach, as implemented in GOLPE, while binding affinity of ligands in the prediction
set was estimated by adopting a novel multi-point equation.
@ゲic& was successfully used to model the well-known molecular series made of 88 (72
in training and 16 in test set, respectively) 3-amidinophenylalanine inhibitors [2] of
thrombin, trypsin and factor Xa whose x-ray crystals are available from the Protein Data
Bank with the code 1ETS, 1PPH and 1KYE, respectively.
In addition to remarkable statistics (0.728ø q2 ø0.777) whose robustness was proved by
a number of validation trials, @ゲic& resulted models provided with both easy
interpretability and impressive prediction.
Figure 1 Thrombin and trypsin surfaces and relevant amino acid residues along with the corresponding
docking solutions selected by @ゲic& are coloured in white and grey, respectively. Selected docking
poses of a highly thrombin selective inhibitor and the mutated S217E residues are shown.
Interestingly, the putative reason of molecular selectivity were also adequately
elucidated. And in fact, the probable existence of non-equivalent physico-chemical
130

interactions of differently oriented inhibitors into the active sites of two distinct proteins
(e.g. thrombin vs trypsin) was described through selectivity square-plots and easily
described by docking poses selected by @ゲic& for the same inhibitor into the two
different receptor pockets. (Figure 1).
References
1. Nicolotti, O.; Miscioscia, T. F.; Carotti, A.; Leonetti, F.; Carotti, A. J. Chem. Inf.
Model., in press.
2. Böhm, M.; Stürzebecher, J.; Klebe, G. J. Med. Chem., 1999, 42, 458-477.
131

VALIDATION OF THE BINDING SITE LOCATED INTO THE C-TERMINAL DOMAIN OF HSP90: A NEW OPPORTUNITY TO DESIGN ANTICANCER DRUGS
Miriam SGOBBA
Dipartimento di Scienze Farmaceutiche, Università di Modena e Reggio Emilia, via Campi 183, 41100 Modena, Italy
Scuola di dottorato in “Scienze e Tecnologie dei prodotti per la salute”, XXI ciclo Indirizzo Progettazione, sintesi e caratterizzazione
In the last decade, the chaperone Hsp90 has been recognized as a valuable and attractive target for anticancer drugs because of its central role in activation and stabilization of multiple oncogenic proteins. Examples of such proteins are steroid receptors, mutated p53, several protein kinases like ErbB2, Src, Abl, Raf, Akt and cyclin-dependent serine kinases. As a matter of fact, inhibition of Hsp90 prevents the association between the chaperone and these oncogenic proteins. In this way, by targeting a single specific protein, multiple signalling pathways important for growth and proliferation of cancer cells can be simultaneously inhibited.
Hsp90 is a homodimer composed by three domains, N-terminal (NT), middle (M) and C-terminal (CT), and its function is dependent on ATP binding and hydrolysis. The NT domain contains the principal ATP binding site which shows a Bergerat-fold present in different proteins such as bacterial DNA gyrase B, MutL, eukaryotic topoisomerase II and histidine-kinases. Several compounds, such as geldanamycin and radicicol, are known inhibitors of Hsp90 that compete with the nucleotide of the NT domain. In particular, two geldanamycin derivatives 17-AAG and 17-DMAG are currently in phase II and I clinical trials [1].
At the opposed terminus, the CT domain of Hsp90 has been recognized important for the dimerization of the chaperone and recent investigations showed that this domain contains a second ATP binding site. Moreover, compounds based on coumarin scaffolds, such as novobiocin, coumermycin A1 and their derivatives (A4), bind the CT domain by competing with the second ATP molecule thus inhibiting Hsp90 [2,3]. Structure-activity relationships and binding assays suggest that these new CT inhibitors have interesting antiproliferative activities against cancer cells. In the light of these findings, the design of small molecules that interfere with Hsp90 dimerization via CT domain provides a promising and new opportunity to develop anticancer agents.
Based on these premises, we started a research programme aimed at investigating the effects of small molecules on the dimer interface of Hsp90. In Figure 1 are shown the main steps of our strategy performed so far. For the first time, a homology model of the full-length human Hsp90 was obtained using the available X-ray structures of yeast chaperone as template. The structural information was used to study the dynamic behaviour of the chaperone and to infer the location of the binding site of the CT domain. For this purpose, a number of computational tools were applied to identify cavities inside the protein able to accommodate at least an ATP/inhibitor molecule and to suggest functional residues which have high probability to bind such molecules [4]. Further studies were developed to understand how the inhibitors bind to the CT domain using an integrated blind docking/molecular dynamics approach.
Finally, mutagenesis studies of the residues predicted to line the CT binding site are in progress in order to validate the proposed site.
132

Figure 1: computational strategy applied to infer the structure of human Hsp90 and identify the C-
terminal binding site. [1] Tian ZQ, Liu Y, Zhang D, Wang Z, Dong SD, Carreras CW, Zhou Y, Rastelli G, Santi DV, Myles DC. Bioorg Med Chem. 2004, 12, 5317-5329. [2] Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. J Biol Chem. 2000, 275, 37181-6. [3] Burlison JA, Blagg BS. Org. Lett. 2006, 8, 4855-4858. [4] Sgobba M, Degliesposti G, Ferrari AM, Rastelli G. Chem Biol Drug Des 2008, 71, 420-433.
Homology model of human Hsp90
MD simulation
Predicted binding site into the CT domain
Binding site predictions
Blind docking
NT domain
Middle domain
CT domain
133