nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not...
TRANSCRIPT

Nociceptor-derived brain-derived neurotrophic factor regulates
acute and inflammatory but not neuropathic pain
Jing Zhao,a,1 Anjan Seereeram,a,1 Mohammed A. Nassar,a Alessandra Levato,a Sophie Pezet,c
Gareth Hathaway,b Cruz Morenilla-Palao,a Caroline Stirling,a Maria Fitzgerald,b
Stephen B. McMahon,c Maribel Rios,d and John N. Wooda,*
London Pain ConsortiumaMolecular Nociception Group, Department of Biology, University College London, London WC1E 6BT, UKbAnatomy and Developmental Biology Department, University College London, London WC1E 6BT, UKcWolfson CARD, The Wolfson Wing, Hodgkin Building, Guy’s Campus, King’s College, London, SE1 1UL, UKdNeuroscience Department, Tufts University Medical School, Boston, MA 02111, USA
Received 10 August 2005; revised 10 November 2005; accepted 17 November 2005
Available online 18 January 2006
Conditional mouse knock-outs provide an informative approach to
drug target validation where no pharmacological blockers exist or
global knock-outs are lethal. Here, we used the Cre-loxP system to
delete BDNF in most nociceptive sensory neurons. Conditional null
animals were healthy with no sensory neuron loss. However, pain-
related behavior was substantially altered. Baseline thermal thresholds
were reduced. Carrageenan-induced thermal hyperalgesia was
inhibited. Formalin-induced pain behavior was attenuated in the
second phase, and this correlated with abolition of NMDA receptor
NR1 Ser896/897 phosphorylation and ERK1 and ERK2 activation in the
dorsal horn; AMPA receptor phosphorylation (GluR1/Ser831) was
unaffected. NGF-induced thermal hyperalgesia was halved, and
mechanical secondary hyperalgesia caused by intramuscular NGF
was abolished. By contrast, neuropathic pain behavior developed
normally. Nociceptor-derived BDNF thus plays an important role in
regulating inflammatory pain thresholds and secondary hyperalgesia,
but BDNF released only from nociceptors plays no role in the
development of neuropathic pain.
D 2005 Elsevier Inc. All right reserved.
Keywords: BDNF; DRG; Conditional knock-outs; Inflammatory pain;
Neuropathic pain; NMDA receptor; Phosphorylation; ERK1; ERK2
Introduction
Brain-derived neurotrophic factor (BDNF) is a member of the
neurotrophin family (Huang and Reichardt, 2003). First implicat-
ed in the survival and maintenance of the peripheral sensory
system during development (Jones et al., 1994; Liu et al., 1995),
BDNF also acts as a regulator of neuronal excitability and
modulator of synaptic plasticity in the central nervous system (Li
et al., 2005; McAllister et al., 1999; Rivera et al., 2004). Evidence
obtained using neutralizing TrkB receptor bodies suggests that
BDNF acts as a neuromodulator when released from small
diameter nociceptive neurons, playing an important role in pain
pathways (Kerr et al., 1999; Thompson et al., 1999). BDNF is
synthesized in the cell bodies of primary sensory neurons and
expressed by a sub-population of small-diameter sensory neurons
with unmyelinated axons that terminate in the superficial laminae
of the dorsal horn (Ernfors et al., 1990). BDNF undergoes
anterograde transport to the dorsal horn where it is associated with
synaptic vesicles of nociceptive neurons and may be released onto
first-order spinal neurons (Michael et al., 1997). BDNF expres-
sion levels in the nervous system are altered in a number of pain
models including peripheral inflammation (Cho et al., 1997a,b),
axotomy and nerve injury and neuropathic pain paradigms (Cho et
al., 1998; Ha et al., 2001; Zhang et al., 2000; Zhou et al., 2000).
TrkB, which is expressed by post-synaptic neurons of the dorsal
horn, is the high affinity receptor for BDNF and NT4. Noxious
stimulation increases the phosphorylation of TrkB in the rat spinal
dorsal horn consistent with the release of BDNF, and this is
associated with increased ERK kinase auto-phosphorylation in the
superficial dorsal horn (Pezet et al., 2002a,b). BDNF also appears
to enhance NMDA-receptor-mediated responses in the dorsal horn
(Kerr et al., 1999; Garraway et al., 2003, 2005). Acute or chronic
noxious stimuli increase the phosphorylation of various NMDA
1044-7431/$ - see front matter D 2005 Elsevier Inc. All right reserved.
doi:10.1016/j.mcn.2005.11.008
* Corresponding author. Molecular Nociception Group, Department of
Biology, University College London, London WC1E 6BT, UK.
E-mail address: [email protected] (J.N. Wood).1 The first two authors contributed equally to his work.
Available online on ScienceDirect (www.sciencedirect.com).
www.elsevier.com/locate/ymcne
Mol. Cell. Neurosci. 31 (2006) 539 – 548

receptor (NMDAR) subunits in the spinal cord in vivo (Guo et al.,
2002; Brenner et al., 2004). Exogenous BDNF has also been
shown to modulate NR1 phosphorylation (Slack and Thompson,
2002; Slack et al., 2004), although other studies have argued
against an effect of BDNF on NMDA receptor function
(Heppenstall and Lewin, 2001). A role for BDNF in neuropathic
pain has also been proposed. BDNF levels increase in uninjured
DRG neurons after neuropathic insults (Fukuoka et al., 2001).
Antibodies to TrkB or tyrosine kinase inhibitors and TrkB-
neutralizing receptor bodies have been shown to block neuro-
pathic pain (Yajima et al., 2002). On the other hand, a gene
therapy study suggested that exogenous BDNF can have analgesic
effects in neuropathic pain (Eaton et al., 2002). The role of BDNF
in neuropathic pain is thus contentious.
BDNF knock-out animals die during the second postnatal week
precluding a behavioral assessment of the role of BDNF in the
mature sensory system in vivo (Rios et al., 2001). Electrophys-
iological studies of ventral root potentials in p4–p7 BDNF null
mutant mice support a role for BDNF in modulating pain
pathways, consistent with the neutralizing receptor body experi-
ments (Heppenstall and Lewin, 2001; Kerr et al., 1999). However,
the neutralizing receptor bodies may fail to completely sequester
all BDNF and may also sequester NT4; the source of the BDNF
that may regulate synaptic input in the dorsal horn also remains
unknown. The development of a nociceptor-specific Cre strain
(Nav.1.8-Cre) allowed us to generate adult animals deficient in
BDNF in most nociceptive sensory neurons (Stirling et al., 2005).
The aims of this study were to determine the involvement of
nociceptor-derived BDNF in the establishment and maintenance of
a range of behavioral assays including responses to innocuous and
noxious thermal, chemical and mechanical stimuli under both
normal and inflammatory conditions (Kerr et al., 1999; Malcangio
and Lessmann, 2003; Pezet et al., 2002b). Here, we provide
evidence that BDNF released from nociceptors plays a major role
in regulating acute and inflammatory pain but not neuropathic
pain.
Results
Genotyping analysis of floxed BDNF mice and BDNF conditional
null mice
PCR and Southern blotting were used to detect the genotype of
floxed BDNF mice and BDNF conditional null mutants (BDNF
KO). PCR results showed that the floxed BDNF band (440 bp) and
Nav1.8 band (460 bp) were amplified from the tail DNA of floxed
BDNF mice and conditional null mutants. The Cre band (250 bp)
was amplified from the tail DNA of BDNF conditional null
mutants but not floxed BDNF mice (Fig. 1A). Southern blotting
demonstrates that the genotype of BDNF conditional null mutants
is heterozygous Nav1.8-Cre and the genotype of floxed BDNF
mice is homozygous Nav1.8 (Fig. 1B). Assessment of the genotype
ratios gave the expected ratios generated from the breeding
program. Furthermore, the weights of the two groups of females
used for behavioral studies (8–10 weeks post natal) were not
significantly different. Nav1.8-specific BDNF conditional null
mutants 16.88 T 0.88 g compared with wild-type (WT) littermates
17.72 T 0.42 g (P = 0.43; n = 6).
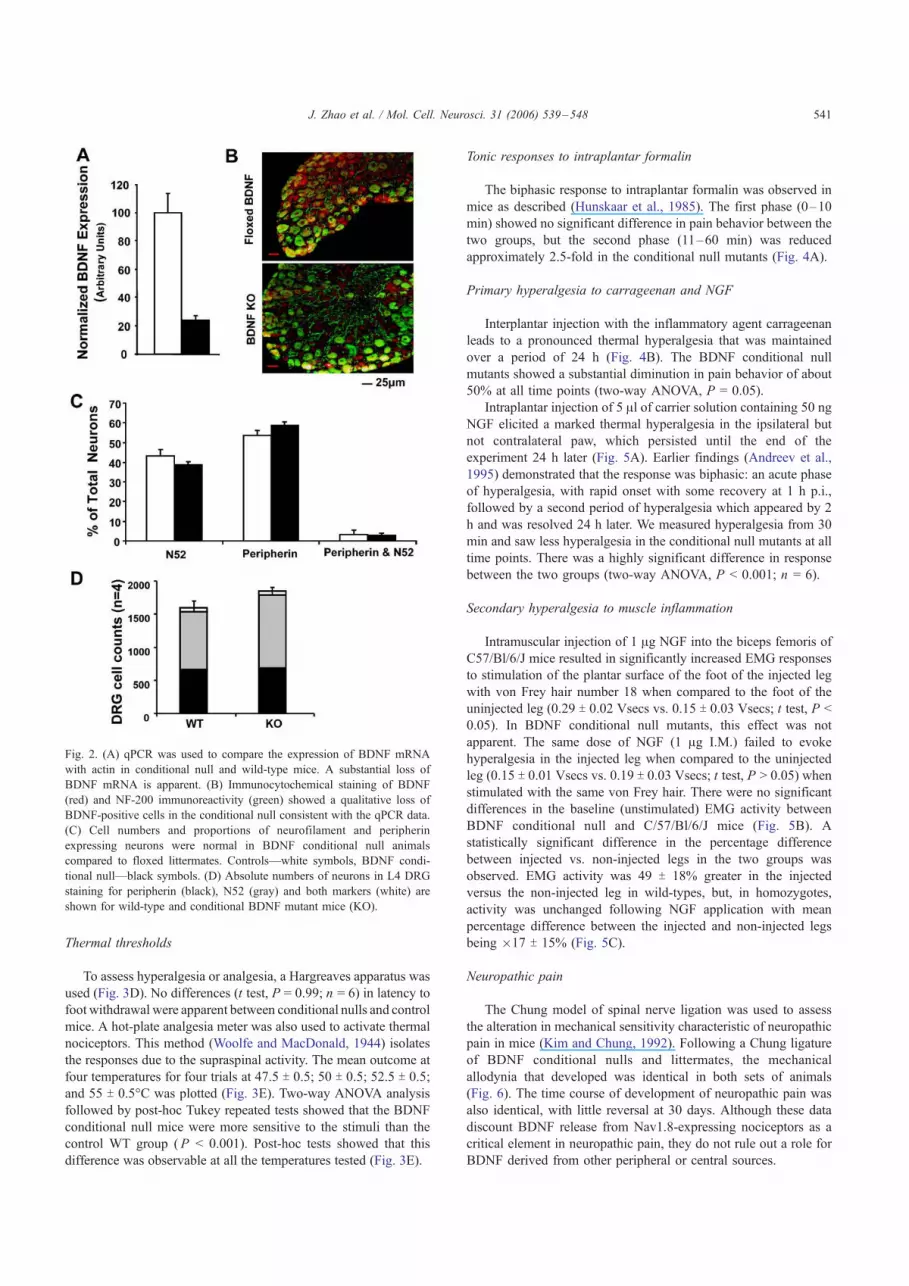
BDNF levels were compared in control and conditional null
mutants by immunocytochemistry and qPCR. Taking the
normalized expression of BDNF in WT mice as 100%, the
expression of BDNF mRNA in the null mutant is about 23% of
this value. This agrees with qualitative immunocytochemical
studies that show a reduction in BDNF-immunoreactive neurons
in sections of DRG taken from null mutants, compared with
controls (Figs. 2A and B).
BDNF null mutants have been shown to contain a normal
complement of DRG neurons (Heppenstall and Lewin, 2001). Not
surprisingly, the same result is true in the conditional null, where
the total number and proportion of neurofilament and peripherin-
positive neurons are identical in wild-type and null littermates
(Figs. 2C and D).
Behavioral studies
Motor coordination measured by means of the Rota-rod test did
not show any significant difference between nulls and littermates
(Fig. 3A). All animals (n = 12) in both groups were able to stay on
the rod, turning at a constant speed of 20 rpm for the maximum
time of 300 s. Subjecting the animals to the increasing speed test
also failed to highlight any differences in time spent on the rotor
rod by the two groups, BDNF null 140.1 T 19.6 s; control
littermates 157.0 T 14.5 s (t test, P = 0.43).
Mechanical pressure thresholds
The Randall – Sellito test, which measures nociceptive
responses to pressure, was applied to the tails of 9 animals of
each genotype (Randall and Sellito, 1957). The means obtained
were BDNF null 68.3 T 4.3 g; control 71.6 T 8.3 g. There is no
significant difference (t test, P = 0.74) in the responses of the
control vs. BDNF KO group (Fig. 3B). Punctate low threshold
mechanical sensitivity was measured using von Frey hairs (Fig.
3C). Again, there were no significant differences (t test, P = 0.59)
between wild-type (0.79 T 0.15 g; n = 9) and BDNF conditional
null mice (0.68 T 0.08 g; n = 7).
Fig. 1. Genotyping analysis of floxed BDNF mice and BDNF conditional
null mice. (A) PCR was used to detect the genotype for floxed BDNF mice
and BDNF conditional null mice. The tail DNA of littermate was amplified
with floxed BDNF primers, Nav1.8 primers and Cre primers. (B) Southern
blotting with BamHI and 3V arm external probe confirms heterozygous
Nav1.8Cre in BDNF conditional null mice and homozygous Nav1.8 in
floxed BDNF mice.
J. Zhao et al. / Mol. Cell. Neurosci. 31 (2006) 539–548540
Thermal thresholds
To assess hyperalgesia or analgesia, a Hargreaves apparatus was
used (Fig. 3D). No differences (t test, P = 0.99; n = 6) in latency to
foot withdrawal were apparent between conditional nulls and control
mice. A hot-plate analgesia meter was also used to activate thermal
nociceptors. This method (Woolfe and MacDonald, 1944) isolates
the responses due to the supraspinal activity. The mean outcome at
four temperatures for four trials at 47.5 T 0.5; 50 T 0.5; 52.5 T 0.5;
and 55 T 0.5-C was plotted (Fig. 3E). Two-way ANOVA analysis
followed by post-hoc Tukey repeated tests showed that the BDNF
conditional null mice were more sensitive to the stimuli than the
control WT group (P < 0.001). Post-hoc tests showed that this
difference was observable at all the temperatures tested (Fig. 3E).
Tonic responses to intraplantar formalin
The biphasic response to intraplantar formalin was observed in
mice as described (Hunskaar et al., 1985). The first phase (0–10
min) showed no significant difference in pain behavior between the
two groups, but the second phase (11–60 min) was reduced
approximately 2.5-fold in the conditional null mutants (Fig. 4A).
Primary hyperalgesia to carrageenan and NGF
Interplantar injection with the inflammatory agent carrageenan
leads to a pronounced thermal hyperalgesia that was maintained
over a period of 24 h (Fig. 4B). The BDNF conditional null
mutants showed a substantial diminution in pain behavior of about
50% at all time points (two-way ANOVA, P = 0.05).
Intraplantar injection of 5 Al of carrier solution containing 50 ngNGF elicited a marked thermal hyperalgesia in the ipsilateral but
not contralateral paw, which persisted until the end of the
experiment 24 h later (Fig. 5A). Earlier findings (Andreev et al.,
1995) demonstrated that the response was biphasic: an acute phase
of hyperalgesia, with rapid onset with some recovery at 1 h p.i.,
followed by a second period of hyperalgesia which appeared by 2
h and was resolved 24 h later. We measured hyperalgesia from 30
min and saw less hyperalgesia in the conditional null mutants at all
time points. There was a highly significant difference in response
between the two groups (two-way ANOVA, P < 0.001; n = 6).
Secondary hyperalgesia to muscle inflammation
Intramuscular injection of 1 Ag NGF into the biceps femoris of
C57/Bl/6/J mice resulted in significantly increased EMG responses
to stimulation of the plantar surface of the foot of the injected leg
with von Frey hair number 18 when compared to the foot of the
uninjected leg (0.29 T 0.02 Vsecs vs. 0.15 T 0.03 Vsecs; t test, P <
0.05). In BDNF conditional null mutants, this effect was not
apparent. The same dose of NGF (1 Ag I.M.) failed to evoke
hyperalgesia in the injected leg when compared to the uninjected
leg (0.15 T 0.01 Vsecs vs. 0.19 T 0.03 Vsecs; t test, P > 0.05) when
stimulated with the same von Frey hair. There were no significant
differences in the baseline (unstimulated) EMG activity between
BDNF conditional null and C/57/Bl/6/J mice (Fig. 5B). A
statistically significant difference in the percentage difference
between injected vs. non-injected legs in the two groups was
observed. EMG activity was 49 T 18% greater in the injected
versus the non-injected leg in wild-types, but, in homozygotes,
activity was unchanged following NGF application with mean
percentage difference between the injected and non-injected legs
being �17 T 15% (Fig. 5C).
Neuropathic pain
The Chung model of spinal nerve ligation was used to assess
the alteration in mechanical sensitivity characteristic of neuropathic
pain in mice (Kim and Chung, 1992). Following a Chung ligature
of BDNF conditional nulls and littermates, the mechanical
allodynia that developed was identical in both sets of animals
(Fig. 6). The time course of development of neuropathic pain was
also identical, with little reversal at 30 days. Although these data
discount BDNF release from Nav1.8-expressing nociceptors as a
critical element in neuropathic pain, they do not rule out a role for
BDNF derived from other peripheral or central sources.
Fig. 2. (A) qPCR was used to compare the expression of BDNF mRNA
with actin in conditional null and wild-type mice. A substantial loss of
BDNF mRNA is apparent. (B) Immunocytochemical staining of BDNF
(red) and NF-200 immunoreactivity (green) showed a qualitative loss of
BDNF-positive cells in the conditional null consistent with the qPCR data.
(C) Cell numbers and proportions of neurofilament and peripherin
expressing neurons were normal in BDNF conditional null animals
compared to floxed littermates. Controls—white symbols, BDNF condi-
tional null—black symbols. (D) Absolute numbers of neurons in L4 DRG
staining for peripherin (black), N52 (gray) and both markers (white) are
shown for wild-type and conditional BDNF mutant mice (KO).
J. Zhao et al. / Mol. Cell. Neurosci. 31 (2006) 539–548 541

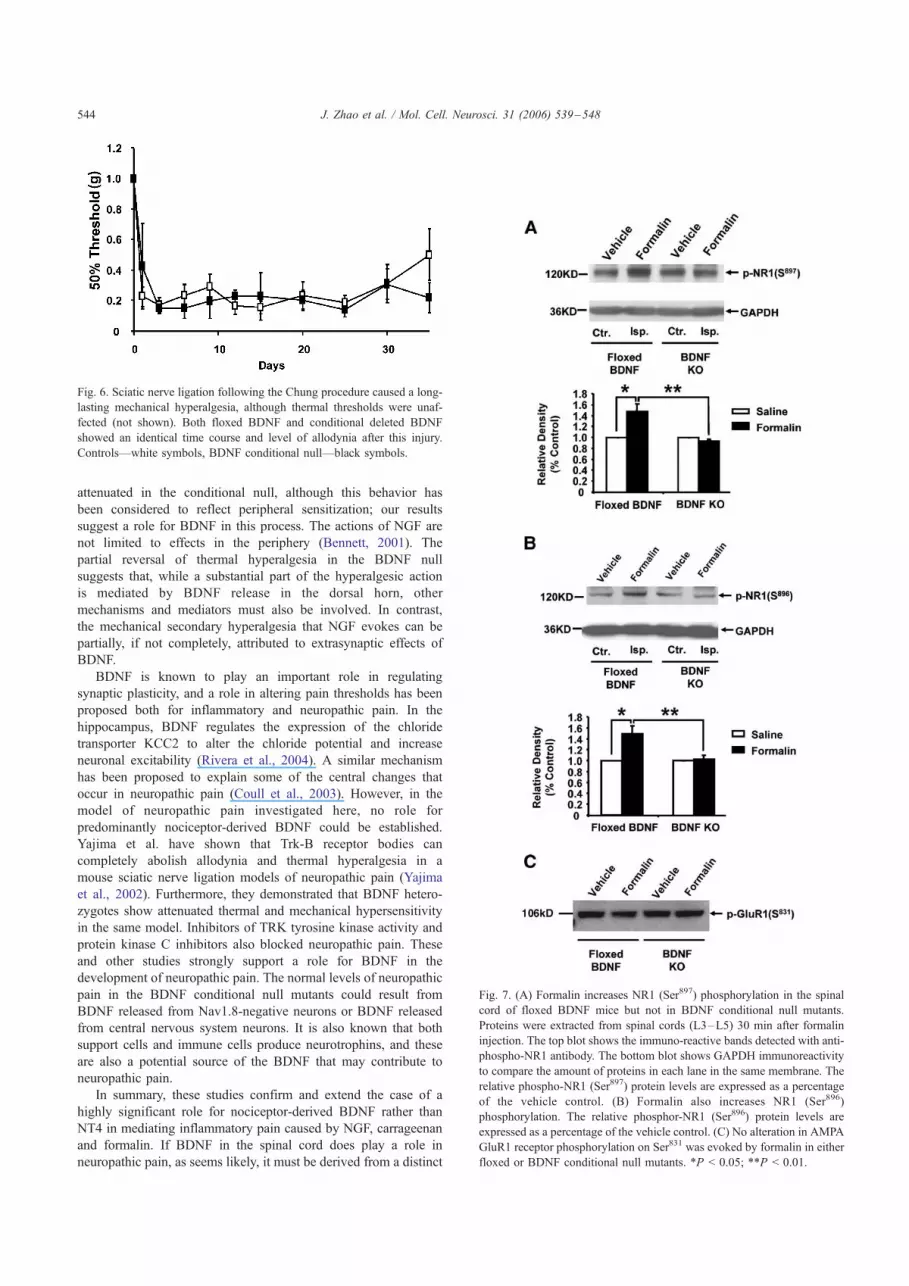
Biochemical events associated with inflammatory pain
We used antibodies directed against phosphorylated residues
in glutamate receptors to examine the biochemical correlates of
formalin-induced pain behavior. Formalin induces an increase of
NMDA receptor NR1 (Ser897, Ser896) phosphorylation in the
dorsal spinal cord of control floxed BDNF mice. Western blot
analysis showed that phospho-NR1 (Ser897 and Ser896) could be
detected in the contralateral spinal cord, but there was no
increase in phosphorylation in response to formalin. Formalin
induced a significant increase of phospho-NR1 (Ser897 and
Ser896) in the ipsilateral spinal cord (L3–L5) of control floxed
BDNF mice compared with the contralateral side (Figs. 7A and
B). Values of phospho-NR1 in formalin-stimulated cord com-
pared with control phospho-NR1 were 148 T 13.7% (Ser897; t
test, P < 0.05; n = 4) and 152 T 10.9% (Ser896; t test, P < 0.05;
n = 4). In null mutants, formalin failed to induce any increase of
phospho-NR1 Ser897 or Ser896 in the spinal cord compared with
saline control. Similarly, formalin did not induce GluR1 (Ser831)
phosphorylation in the spinal cord of floxed BDNF control mice
or BDNF conditional null mice (Fig. 7C). Thus, BDNF released
from primary afferents seems to be required for the phosphor-
ylation of NMDA NR1 subunits on Ser896 and 897, while GluR1
Ser831 is not regulated by BDNF released from Nav1.8-positive
primary afferent fibers.
Extracellular-signal-regulated kinase (ERK) is also known to
be activated by inflammatory stimuli such as formalin in a BDNF-
dependent manner (Pezet et al., 2002a,b). Injection of 20
Al formalin induced in floxed BDNF control mice a robust
induction of ERK phosphorylation in the spinal cord ipsilateral to
the injection of formalin. This activation was observed mostly in
superficial laminae (10–17 neurons per section in the superficial
laminae, 5–7 in deeper laminae) in lumbar levels L3 to L5 (Fig.
8). The contralateral site showed only few positive p-ERK
neurons in deep laminae of the cord (3–5 neurons per section).
In BDNF conditional null mice, the induction of ERK was
significantly reduced compared to floxed BDNF control mice in
all L3 to L5 levels (t test, P < 0.05; Fig. 8) in superficial layers of
the cord. In deep laminae of the cord in contrast, where it is
known that BDNF is not abundant, the induction of ERK was not
modified in BDNF conditional null mice, compared to floxed
BDNF controls.
Discussion
Target validation using null mutant mice has proved valuable
in assessing new approaches to analgesic drug development.
When gene deletion causes perinatal lethality, as with BDNF
null mutant mice, tissue-specific gene ablation may provide
important information about the possible relevance of a new
drug target. In addition, identifying the source of release for
neuromodulators such as BDNF provides additional insights into
physiological mechanisms. BDNF heterozygotes are viable and
have provided some insights to BDNF function, although the
absolute levels of BDNF in different tissues have not been
quantitated, while it has proved possible to record electrophys-
iologically from homozygous null mutants up to about day 7.
Fig. 3. (A) Rota-rod studies showed no motor deficits in conditional BDNF null animals, (B) acute mechanical pressure applied with the Randall–Sellito
apparatus also demonstrated identical behavior in nulls and wild-type mice. Responses to low-threshold mechanical stimulation by von Frey hairs are normal
(C) in the null mutant compared to littermate controls. (D) Hargreaves apparatus demonstrates identical latencies of response to thermal stimulation, while
supra-spinal reflexes to heat (E) are sensitized in the null mutant animals. Controls—white symbols, BDNF conditional null—black symbols. ***P < 0.001.
J. Zhao et al. / Mol. Cell. Neurosci. 31 (2006) 539–548542

However, behavioral studies of the role of BDNF in pain
pathways have relied on exogenous application of high doses of
BDNF or the effects of neutralizing receptor bodies that
sequester both NT4 and BDNF. Here, we have investigated
the effects of deleting BDNF expression in Nav1.8-positive,
mainly nociceptive sensory neurons, on pain behavior and
dorsal horn responses to noxious stimuli in an attempt to assess
the role of peripheral BDNF in regulating pain behavior. These
studies are unable to provide information about BDNF
expressed in Nav1.8-negative sensory neurons; the expression
of BDNF is, however, known to be plastic, altering in different
sensory neuron populations in different pain states (Obata et al.,
2003). Nevertheless, where clear pain behavioral deficits occur,
then these can be ascribed to the loss of BDNF in Nav1.8-
positive predominantly nociceptive neurons. The genetic back-
ground of the Cre mouse has been extensively studied and is
unlikely to produce a contribution to altered pain behavior
(Stirling et al., 2005).
Despite the presence of a normal complement of sensory
neurons, there was increased pain responsiveness to noxious
thermal stimulation in the hot-plate test. Mechanical thresholds
were unchanged. In global BDNF heterozygous animals, there is,
in contrast, no change in thermal and mechanical thresholds to
acute pain (MacQueen et al., 2001). The second phase of the
formalin response has, however, been shown to be attenuated in
BDNF heterozygous null mutants (MacQueen et al., 2001).
Results obtained with the conditional sensory neuron BDNF null
mice are consistent with these data. Here, we found that
activation of nociceptors by formalin caused a BDNF release-
dependent increase in NMDA receptor and ERK phosphorylation
that correlates with second phase pain behavior. This mechanism
may also contribute to carrageenan and NGF-induced hyper-
algesia which are both partially reversed in the absence of
predominantly nociceptor-derived BDNF. NGF has been shown
to be involved in nociception and pathological pain conditions
(Lewin et al., 1993; Apfel, 2000). Application of exogenous
NGF has been shown to reduce both thermal and mechanical
withdrawal thresholds (Lewin et al., 1993). Interestingly, thermal
responses at the earliest time points measured (30 min) were
Fig. 4. (A) Intraplantar formalin caused two phases of licking behavior,
the second of which is attenuated in the BDNF conditional null mouse.
(B) Intraplantar injection of carrageenan caused a long-term thermal
hyperalgesia that is attenuated in BDNF conditional nulls. Controls—
white symbols, BDNF conditional null—black symbols. **P < 0.01;
***P < 0.001.
Fig. 5. (A) Intraplantar NGF injections that cause thermal hyperalgesia
resulted in a diminished hyperalgesia in conditional BDNF nulls (open
triangles—saline-injected controls, white squares—NGF-injected control
mice, black squares—NGF-injected conditional BDNF nulls). (B) Sec-
ondary hyperalgesia quantitated electrophysiologically after intra-muscular
injections of NGF were also completely abolished in the conditional
BDNF null mouse. Controls—white symbols, BDNF conditional null—
black symbols. (C) Percentage differences in EMG activity between injected
and non-injected legs are significantly different between groups. Controls—
white symbols, BDNF conditional null—black symbols. *P < 0.05;
**P < 0.01.
J. Zhao et al. / Mol. Cell. Neurosci. 31 (2006) 539–548 543
attenuated in the conditional null, although this behavior has
been considered to reflect peripheral sensitization; our results
suggest a role for BDNF in this process. The actions of NGF are
not limited to effects in the periphery (Bennett, 2001). The
partial reversal of thermal hyperalgesia in the BDNF null
suggests that, while a substantial part of the hyperalgesic action
is mediated by BDNF release in the dorsal horn, other
mechanisms and mediators must also be involved. In contrast,
the mechanical secondary hyperalgesia that NGF evokes can be
partially, if not completely, attributed to extrasynaptic effects of
BDNF.
BDNF is known to play an important role in regulating
synaptic plasticity, and a role in altering pain thresholds has been
proposed both for inflammatory and neuropathic pain. In the
hippocampus, BDNF regulates the expression of the chloride
transporter KCC2 to alter the chloride potential and increase
neuronal excitability (Rivera et al., 2004). A similar mechanism
has been proposed to explain some of the central changes that
occur in neuropathic pain (Coull et al., 2003). However, in the
model of neuropathic pain investigated here, no role for
predominantly nociceptor-derived BDNF could be established.
Yajima et al. have shown that Trk-B receptor bodies can
completely abolish allodynia and thermal hyperalgesia in a
mouse sciatic nerve ligation models of neuropathic pain (Yajima
et al., 2002). Furthermore, they demonstrated that BDNF hetero-
zygotes show attenuated thermal and mechanical hypersensitivity
in the same model. Inhibitors of TRK tyrosine kinase activity and
protein kinase C inhibitors also blocked neuropathic pain. These
and other studies strongly support a role for BDNF in the
development of neuropathic pain. The normal levels of neuropathic
pain in the BDNF conditional null mutants could result from
BDNF released from Nav1.8-negative neurons or BDNF released
from central nervous system neurons. It is also known that both
support cells and immune cells produce neurotrophins, and these
are also a potential source of the BDNF that may contribute to
neuropathic pain.
In summary, these studies confirm and extend the case of a
highly significant role for nociceptor-derived BDNF rather than
NT4 in mediating inflammatory pain caused by NGF, carrageenan
and formalin. If BDNF in the spinal cord does play a role in
neuropathic pain, as seems likely, it must be derived from a distinct
Fig. 6. Sciatic nerve ligation following the Chung procedure caused a long-
lasting mechanical hyperalgesia, although thermal thresholds were unaf-
fected (not shown). Both floxed BDNF and conditional deleted BDNF
showed an identical time course and level of allodynia after this injury.
Controls—white symbols, BDNF conditional null—black symbols.
Fig. 7. (A) Formalin increases NR1 (Ser897) phosphorylation in the spinal
cord of floxed BDNF mice but not in BDNF conditional null mutants.
Proteins were extracted from spinal cords (L3–L5) 30 min after formalin
injection. The top blot shows the immuno-reactive bands detected with anti-
phospho-NR1 antibody. The bottom blot shows GAPDH immunoreactivity
to compare the amount of proteins in each lane in the same membrane. The
relative phospho-NR1 (Ser897) protein levels are expressed as a percentage
of the vehicle control. (B) Formalin also increases NR1 (Ser896)
phosphorylation. The relative phosphor-NR1 (Ser896) protein levels are
expressed as a percentage of the vehicle control. (C) No alteration in AMPA
GluR1 receptor phosphorylation on Ser831 was evoked by formalin in either
floxed or BDNF conditional null mutants. *P < 0.05; **P < 0.01.
J. Zhao et al. / Mol. Cell. Neurosci. 31 (2006) 539–548544

cellular source than Nav1.8-positive sensory neurons. These studies
support a therapeutic approach to inflammatory pain based on
down-regulating peripheral levels of BDNF in sensory neurons.
Experimental methods
Generation of Nav1.8-specific BDNF knock-out mice
Floxed mice containing loxP sites flanking BDNF exon 5 (Rios et
al., 2001) were crossed with the Nav1.8-Cre strain (Stirling et al., 2005)
to affect BDNF gene ablation in a defined subset of sensory neurons.
The study population contained the homozygous floxed BDNF gene and
one copy of the Nav1.8-Cre allele, while homozygous floxed BDNF
littermates were used as controls. Genotyping of all animals was done
with PCR as previously described (Stirling et al., 2005; Rios et al.,
2001). Mice were housed with a 12-h light:12-h (lights on at 07:00)
dark cycle and maintained under standard condition (21 T 1-C, food and
water ad libitum). Behavioral studies were carried out during the light
cycle. Experiments were carried out on female animals between 2 and 3
months old, drawn from different litters to avoid any ‘‘litter effect’’ and
tested blind. Real-time RT-PCR analysis of BDNF was performed using
an iCycler (Bio-Rad, CA). DRG RNA was extracted using TRIzolR
reagent (Gibco BRL), treated with RQ1 RNase-free DNase (Promega)
and equal amounts of total RNA were reversed transcribed using random
hexamers, and Superscripti II RT (Invitrogen) PCR reactions were
performed using PlatinumR SYBRR Green qPCR Supermix UDG
(Invitrogen) and BDNF gene-specific primers, forward primer: 5V-GCATCTGTTGGGGAGACAAG-3V; reversed primer: 5V-TGGTCAT-CACTCTTCTC ACCTG-3V. Reactions were performed in triplicate,
and threshold cycle values were normalized to h-Actin gene expression,
forward primer: 5V-TCTGTGTGGATCGGTGGCTC-3V; reversed primer:
5V-CTGCTTGCTGATCCACAT-CTG-3V. The specificity of the products
was determined by melting curve analysis, and their correct sizes were
analyzed by electrophoresis. The ratio of the relative concentration of
BDNF to h-Actin of each sample was calculated by using the 2DCT
formula.
Cell counts and immunocytochemistry
DRG 11 AM sections were blocked in 10% goats’ serum in PBS
overnight at 4-C then incubated in a mixture of 1:1000 NF200 antibodies
(mouse monoclonal, Sigma) and 1:1000 anti-peripherin antibody (rabbit
polyclonal, Chemicon) diluted in 10% goat serum in PBS overnight at 4-C.The slides were then washed in three changes of PBS (2 � 5 min, 1 � 30
min). They were then incubated with 1:600 FITC-conjugated goat anti-
Fig. 8. Activation of the MAPK ERK induced by intraplantar formalin is reduced in BDNF conditional null mice. (A–D) Representative immunostaining for p-
ERK in floxed control mice (A–B) or BDNF KO (C–D) 5 min after the injection of formalin in superficial (laminae I, II of Rexed, graph on the left) or (F) in
deep layers of the cord (laminae III–VI). The right hemicord on the picture is the cord ipsilateral to the injection of formalin. B and D are higher power
magnification of A and C, respectively. (E) Quantification of the mean number (TSEM) of positive p-ERK neurons in the spinal cord L3 to L5 ipsilateral to the
injection of formalin in superficial (graph on the left) or (F) in deep layers of the cord. Scale bar for figures A–D; A, C: 94 Hm; B, D: 34 Hm. *P < 0.05.
J. Zhao et al. / Mol. Cell. Neurosci. 31 (2006) 539–548 545

mouse IgG and 1:1000 Alexa-Fluor-conjugated goat anti-rabbit IgG for 3
h at room temperature. The sections underwent three more washes in PBS
(2 � 5 min, 1� overnight) and were then cover-slipped with PBS/Glycerol
(CITIFluor).
The number of neurons was counted in every 8th section throughout the
DRG. Thus, the DRG was sampled every 88 Al to avoid double counting of
the large diameter cells. Every visible cell was counted whether the
nucleolus was present or not. Each DRG was counted twice, and the results
were pooled. The percentages of NF200-positive, peripherin-positive and
double-stained cells were calculated for each section. Mean and SE of these
percentages were evaluated for wild-type and mutant groups. The raw data
from the cell counts passed the test for normality (Kolmogorov–Smirnov,
P > 0.200 for all groups), so significance was determined using a two-tailed
unpaired heteroscedastic t test.
Other primary antibodies used: monoclonal mouse anti-neurofilament-
H antibody, N52 (1:1000, Sigma), anti-BDNF rabbit antiserum (Abcom
ab6201, 1:400). The following secondary antibodies were used: anti-mouse
(goat) (Fab)2 FITC (1:1000, Jackson) and anti-rabbit Alexa Fluor 594 goat
IgG (1:1000, Molecular Laboratories).
Western immunoblot analysis
After 30 min of treatment with formalin (20 Al of 5% formalin plantar
surface injection in right paw, 20 Al of saline in left paw), floxed BDNF
mice and BDNF conditional null mice were killed. The dorsal half of the
L3–L5 spinal cord tissues was removed and homogenized (50 mM Tris–
Cl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% NP40, 0.5% Na-
deoxycholate, 0.1% SDS, 1 mM Na3VO4, 5 mM NaF, 1 mM PMSF, 1
U/ml aprotinin, 10 Ag/ml antipain, 1 Ag/ml leupeptin and 1 Ag/ml
pepstatin A). The homogenate was centrifuged at 14,000 rpm for 20 min
at 4-C. The supernatant was removed. The protein concentration was
determined with a BCA Kit. Proteins (40 Ag) were separated on a 7.5%
SDS-PAGE gel and blotted to nitrocellulose membrane with a Bio-Rad
Transfer Cell system. The blots were blocked with blocking buffer 1 (TBS
contains 0.1% Triton X-100 and 0.1% BSA) at room temperature for 1 h.
The blots were washed with TBST (TBS containing 0.1% Tween20) for
anti-phospho-NR1 Ser897 twice or PBST (PBS containing 0.1% Tween20)
for anti-phospho-NR1 Ser896 twice. After a rinse in water, the membrane
was incubated again with TBST–MLK (TBST containing 5% nonfat dry
milk, for anti-phospho-NR1 Ser897) or PBST–MLK (PBST containing 5%
nonfat dry milk, for anti-phospho-NR1 Ser896 and anti-phospho-GluR1
Ser831) at room temperature for 1 h. Blots were incubated in blocking
buffer 2 with the respective antibody overnight at 4-C. The membrane
was washed with TBST or PBST and incubated for 1 h with anti-goat IgG
horseradish peroxidase (1:2000) in TBST–MIL or PBST–MLK at room
temperature for 1 h. The membrane was then washed three times with
TBST or PBST, and immunoreactivity was finally detected by using ECL
kits (Amersham). Antibodies were used at dilutions suggested by the
makers instructions: anti-phospho-NR1 Ser897 (Upstate), anti-phospho-
NR1 Ser896 (Upstate), anti-phosphor-GluR1 Ser831 (Upstate), anti-
GAPDH (Chemicon), anti-rabbit IgG horseradish peroxidase (Amersham),
anti-mouse IgG horseradish peroxidase (Amersham).
Immunohistochemistry for p-ERK
Adult floxed BDNF mice (n = 4) or BDNF conditional mice (n = 4)
were anaesthetized with urethane (1.25 g/kg, i.p.) and received intraplantar
injections of formalin (Sigma, 20 Al injected in plantar surface of the left
hind paw). Five minutes after the injection, animals were transcardially
perfused with 30 ml heparinized saline (0.9% w/v NaCl) followed by 100
ml of 4% w/v paraformaldehyde in 0.1 M phosphate buffer pH 7.4 (PFA),
15% of a saturated solution of picric acid. The spinal cord was post-fixed in
the same fixative overnight and cryoprotected overnight in 20% w/v
sucrose in 0.1 MPB at 4-C. The spinal cords were embedded in OCT
embedding compound (BDH) on liquid nitrogen and cut serially (20 Amthickness) on a cryostat and collected onto superfrost slides (BDH). Every 6
sections were kept for p-ERK immunostaining. After several washes in
PBS, slides were incubated overnight in mouse anti-phosphorylated-ERK1/
ERK2 (recognizing sites Thr 202 and Tyr 204, New England Biolabs, UK,
1:200). After several washes in PBS, sections were incubated in goat anti-
rabbit Alexa Fluor 488 antibody (Molecular Probes, 1:1000) for 2 h.
Finally, slides were washed and mounted with Vectashield medium (Vector
laboratories). p-ERK staining was visualized using a fluorescent Leica
microscope. Images were taken using Hamamatsu Camera and software.
The same setup of acquisition was used to acquire the pictures of both
groups of mice. In each animal, the number of positive p-ERK neurons was
counted blind in 5 sections of the following lumbar spinal cord levels: L3,
L4 and L5.
Electrophysiology
Mice were injected with 1 Ag NGF unilaterally in the biceps femoris 24
h prior to EMG recording. Animals were subsequently anesthetized with
isoflurane (2%) and ventilated. Animals were mounted in a small animal
spinal frame and hindlimbs secured in a slight extension with the plantar
foot surface exposed for cutaneous stimulation. Bipolar EMG electrodes
(Ainsworks, London) were placed through a small incision into the belly of
the biceps femoris. Raw signals were amplified using a headstage amplifier
(NL100, Neurolog, Digitimer), preamplified and filtered (NL104, NL125)
and displayed on a digital storage oscilloscope (Hameg HM205). The
signal was fed to an analogue-to-digital signal converter for further analysis
using MacLab software (PowerLab 4S, AD Instruments, Castle Hill,
Australia). von Frey hairs (Stoelting U.S.A.) of graded intensity were
applied to the hindpaw and the EMG response recorded. The mechanical
withdrawal threshold was defined as the lowest number von Frey hair that
elicited an EMG response. Up to three vFh above threshold were
sequentially applied at 1-min intervals. Recordings were made from both
limbs, and the order in which this was done was randomized. Animals were
equilibrated for 30 min on the frame prior to recordings. The percentage
difference in EMG activity between injected and non-injected legs was
calculated, and comparisons between homozygote and wild-type mice
performed.
Behavioral analysis
Rota-rod test
Mice were acclimatized to the stationary rod (model 7650, Ugo Basile,
Italy) for 120 s (Crawley and Paylor, 1997), and 5 s later, the rod was
rotated at a constant speed of 20 rpm for 300 s. The time taken for the
animal to either fall off or rotate passively was recorded. Three further trials
(one trial per day) with the rod accelerating from 20 rpm to the maximum of
40 rpm within the 5-min test period were carried out. The time that each
mouse was able to stay on the rod without either falling or passively
rotating was recorded. The mean T SEM was determined and analyzed with
a Student’s t test.
Randall–Sellito test
Mice were passively restrained in a Perspex tube allowing the apparatus
to access the tail. After a 15-min acclimatization period, the area of the tail
approximately 1 cm from the base was subjected to an increasing pressure
and the force (grams) at which a struggling response was elicited noted.
This was repeated three times, and the mean T SEM calculated and assessed
by means of a Student’s t test (Randall and Sellito, 1957).
Formalin test
The two groups of animals (n = 6 each group) were singly housed in
Perspex boxes and given 30 min to habituate to the testing environment.
Animals were then injected subcutaneously with formalin (15 Al, 5%
dilution of stock formalin (40% w/v) in saline). Nociceptive behavior was
taken to be licking and biting the injected paw only. The time that these two
activities were displayed by the animal was recorded in 5-min bins until 60
min had passed. Differences between the two groups were assessed with
two-way ANOVA followed by post-hoc Tukey tests (McCall et al., 1996;
Hunskaar et al., 1985).
J. Zhao et al. / Mol. Cell. Neurosci. 31 (2006) 539–548546

Hot-plate test
The response to thermal stimuli was tested using a hot-plate
analgesia meter (Ugo Basile, Italy) (Crawley and Paylor, 1997). Animals
were habituated for the equipment for 15 min before the basal metal
plate was heated to the test temperature. The time taken for a response
was recorded. Lifting of any paw, biting and licking, rearing up on hind
legs, paw flinching or jumping was taken as the cut-off point with a
maximum of 60 s for temperatures under 52.5 T 0.5-C and 30 s above
53 T 0.5-C. One test was conducted per animal per day. Four
temperatures were assessed on subsequent days with each test group
having 12 animals. The means for each temperature T SEM were
determined and subjected to a two-way ANOVA test of significance
followed by post-hoc Tukey test.
von Frey test of mechanical thresholds
After acclimatization to the testing environment (30 min), mechan-
ical sensory thresholds were determined by paw withdrawal to
application of a series of von Frey filaments to the glabrous surface
of the hind paws. Calibrated von Frey filaments were applied five times
per paw with enough force to cause buckling of the filament. Eight
filaments were tested in ascending order from 0.219 g to 7.59 g. The
percentage response for each filament was determined by scoring the
positive responses (both supraspinal; biting, licking and simple hyper-
reflexia; paw lifting); i.e. number of trials accompanied by a response
divided by 5 and multiplied by 100. Left and right hind paws were
tested with 3-min breaks between subsequent tests. Each group
contained 12 animals, and the mean T SEM was assessed using two-
way ANOVA with post-hoc Tukey tests.
Hargreaves test of thermal nociceptive thresholds
The Hargreaves method (Hargreaves et al., 1988) was used to measure
thermal hyperalgesia using the plantar test (Hargreaves’ Basile Plantar test
model 7370; Ugo Basile, Comerio, Italy). Mice were habituated for 30 min
to the apparatus. The equipment was calibrated to give a mean paw
withdrawal latencies (PWL) of approximately 10 s by adjusting the infra-
red (IR) intensity. PWLs were taken three times for both hind paws with at
least 5-min intervals between each subsequent test. The mean of the three
measures represented the latency of paw withdrawal and was taken as a
measure of thermal pain responses. A cut-off point of 20 s was used to
prevent tissue damage.
Carrageenan and nerve growth factor (NGF) induced thermal hyperalgesia
Mice were injected either with 20 Al carrageenan (2%, w/v) or 50 ng
NGF (5 Al carrier volume) (human recombinant nerve growth factor 4.2
mg/ml in 20 mM succinate buffer diluted to 10 Ag/ml with saline, Sigma
Chemicals, UK) into the subcutaneous plantar surface of the left hind paw.
The injection was done under anesthesia induced by 4% halothane/oxygen
to ensure reproducible injections of a consistent position and depth. The
animals recovered within 3 min. The development of the thermal
hyperalgesia was tracked using the Hargreaves apparatus after the injection
up to a maximum of 24 h. Two-way ANOVA was carried out followed by
post-hoc Tukey analysis.
Sciatic nerve injury
Baseline thermal and mechanical thresholds were recorded from null
and wild-type littermates using Hargreaves test and von Frey hairs (using
the up–down method (Chaplan et al., 1994)) respectively. Animals were
anesthetized using halothane. A midline incision was made in the skin of
the back at the L4–S2 levels and the left paraspinal muscles separated from
the spinous processes, facet joints and transverse processes at the L4–S1
levels. The left L5 spinal nerve was tightly ligated using silk thread (Kim
and Chung, 1992). Mechanical thresholds were measured after injury, and
the results for the two groups were compared using a two-way repeated
measures analysis of variance test. Results of the 50% withdrawal threshold
were expressed as the relative change in the 50% threshold at each time
point after injection (test/baseline) for each group T SEM. The results for
the two groups were compared using a two-way repeated measures analysis
of variance test.
Statistical analysis
All data are presented as mean T SEM. Data were assessed for
normality, and normally distributed data sets were compared with two-way
analysis of variance (ANOVA) followed by post-hoc Tukey multiple
comparison tests. Non-normal data were assessed with Student’s unpaired t
test. P < 0.05 was regarded as significant. All calculations were done using
SigmaStat 2.01.
Acknowledgments
We acknowledge Sarah E. Slack for great help in detection of
phosphorylation of NMDA receptors in spinal cord. We also thank
the MRC, the BBSRC and the Wellcome Trust for funding this
work.
References
Andreev, N.Y., Dimitrieva, N., Koltzenburg, M., McMahon, S.B., 1995.
Peripheral administration of nerve growth factor in the adult rat
produces a thermal hyperalgesia that requires the presence of
sympathetic post-ganglionic neurones. Pain 63, 109–115.
Apfel, S.C., 2000. Neurotrophic factors and pain. Clin. J. Pain 16, S7–S11.
Bennett, D.L., 2001. Neurotrophic factors: important regulators of
nociceptive function. Neuroscientist 7, 13–17.
Brenner, G.J., Ji, R.R., Shaffer, S., Woolf, C.J., 2004. Peripheral noxious
stimulation induces phosphorylation of the NMDA receptor NR1
subunit at the PKC-dependent site, serine-896, in spinal cord dorsal
horn neurons. Eur. J. Neurosci. 20, 375–384.
Chaplan, S.R., Bach, F.W., Pogrel, J.W., Chung, J.M., Yaksh, T.L., 1994.
Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci.
Methods 53, 55–63.
Cho, H.J., Kim, J.K., Zhou, X.F., Rush, R.A., 1997a. Increased brain-
derived neurotrophic factor immunoreactivity in rat dorsal root ganglia
and spinal cord following peripheral inflammation. Brain Res. 764,
269–272.
Cho, H.J., Kim, S.Y., Park, M.J., Kim, D.S., Kim, J.K., Chu, M.Y., 1997b.
Expression of mRNA for brain-derived neurotrophic factor in the
dorsal root ganglion following peripheral inflammation. Brain Res.
749, 358–362.
Cho, H.J., Kim, J.K., Park, H.C., Kim, J.K., Kim, D.S., Ha, S.O., Hong,
H.S., 1998. Changes in brain-derived neurotrophic factor immunoreac-
tivity in rat dorsal root ganglia, spinal cord, and gracile nuclei following
cut or crush injuries. Exp. Neurol. 154, 224–230.
Coull, J.A., Boudreau, D., Bachand, K., Prescott, S.A., Nault, F., Sik, A.,
De Koninck, P., De Koninck, Y., 2003. Trans-synaptic shift in anion
gradient in spinal lamina I neurons as a mechanism of neuropathic pain.
Nature 424, 938–942.
Crawley, J.N., Paylor, R., 1997. A proposed test battery and constellations
of specific behavioral paradigms to investigate the behavioral pheno-
types of transgenic and knockout mice. Horm. Behav. 31, 197–211.
Eaton, M.J., Blits, B., Ruitenberg, M.J., Verhaagen, J., Oudega, M., 2002.
Amelioration of chronic neuropathic pain after partial nerve injury by
adeno-associated viral (AAV) vector-mediated over-expression of
BDNF in the rat spinal cord. Gene Ther. 9, 1387–1395.
Ernfors, P., Wetmore, C., Olson, L., Persson, H., 1990. Identification of
cells in rat brain and peripheral tissues expressing mRNA for members
of the nerve growth factor family. Neuron 5, 511–526.
Fukuoka, T., Kondo, E., Dai, Y., Hashimoto, N., Noguchi, K., 2001. Brain-
derived neurotrophic factor increases in the uninjured dorsal root
ganglion neurons in selective spinal nerve ligation model. J. Neurosci.
21, 4891–4900.
J. Zhao et al. / Mol. Cell. Neurosci. 31 (2006) 539–548 547

Garraway, S.M., Petruska, J.C., Mendell, L.M., 2003. BDNF sensitizes the
response of lamina II neurons to high threshold primary afferent inputs.
Eur. J. Neurosci. 18, 2467–2476.
Garraway, S.M., Anderson, A.J., Mendell, L.M., 2005. BDNF-induced
facilitation of afferent evoked responses in lamina II neurons is reduced
following neonatal spinal cord contusion injury. J. Neurophysiol. 94,
1798–1804.
Guo, W., Zou, S., Guan, Y., Ikeda, T., Tal, M., Dubner, R., Ren, K., 2002.
Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor
in the spinal cord during the development and maintenance of
inflammatory hyperalgesia. J. Neurosci. 22, 6208–6217.
Ha, S.O., Kim, J.K., Hong, H.S., Kim, D.S., Cho, H.J., 2001. Expression of
brain-derived neurotrophic factor in rat dorsal root ganglia, spinal cord
and gracile nuclei in experimental models of neuropathic pain.
Neuroscience 107, 301–309.
Hargreaves, K., Dubner, R., Brown, F., Flores, C., Joris, J., 1988. A new
and sensitive method for measuring thermal nociception in cutaneous
hyperalgesia. Pain 32, 77–88.
Heppenstall, P.A., Lewin, G.R., 2001. BDNF but not NT-4 is required
for normal flexion reflex plasticity and function. Proc. Natl. Acad. Sci.
U. S. A. 98, 8107–8112.
Huang, E.J., Reichardt, L.F., 2003. Trk receptors: roles in neuronal signal
transduction. Annu. Rev. Biochem. 72, 609–642.
Hunskaar, S., Fasmer, O.B., Hole, K., 1985. Formalin test in mice, a
useful technique for evaluating mild analgesics. J. Neurosci. Methods
14, 69–76.
Jones, K.R., Farinas, I., Backus, C., Reichardt, L.F., 1994. Targeted
disruption of the BDNF gene perturbs brain and sensory neuron
development but not motor neuron development. Cell 76, 989–999.
Kerr, B.J., Bradbury, E.J., Bennett, D.L., Trivedi, P.M., Dassan, P., French,
J., Shelton, D.B., McMahon, S.B., Thompson, S.W., 1999. Brain-
derived neurotrophic factor modulates nociceptive sensory inputs and
NMDA-evoked responses in the rat spinal cord. J. Neurosci. 19,
5138–5148.
Kim, S.H., Chung, J.M., 1992. An experimental model for peripheral
neuropathy produced by segmental spinal nerve ligation in the rat. Pain
50, 355–363.
Lewin, G.R., Ritter, A.M., Mendell, L.M., 1993. Nerve growth factor-
induced hyperalgesia in the neonatal and adult rat. J. Neurosci. 13,
2136–2148.
Li, Y., Jia, Y.C., Cui, K., Li, N., Zheng, Z.Y., Wang, Y.Z., Yuan, X.B., 2005.
Essential role of TRPC channels in the guidance of nerve growth cones
by brain-derived neurotrophic factor. Nature 434, 894–898.
Liu, X., Ernfors, P., Wu, H., Jaenisch, R., 1995. Sensory but not motor
neuron deficits in mice lacking NT4 and BDNF. Nature 375, 238–241.
MacQueen, G.M., Ramakrishnan, K., Croll, S.D., Siuciak, J.A., Yu, G.,
Young, L.T., Fahnestock, M., 2001. Performance of heterozygous
brain-derived neurotrophic factor knockout mice on behavioral
analogues of anxiety, nociception, and depression. Behav. Neurosci.
15, 1145–1153.
Malcangio, M., Lessmann, V., 2003. A common thread for pain and
memory synapses? Brain-derived neurotrophic factor and trkB recep-
tors. Trends Pharmacol. Sci. 24, 116–121.
McAllister, A.K., Katz, L.C., Lo, D.C., 1999. Neurotrophins and synaptic
plasticity. Annu. Rev. Neurosci. 22, 295–318.
McCall, W.D., Tanner, K.D., Levine, J.D., 1996. Formalin induces biphasic
activity in C-fibers in the rat. Neurosci. Lett. 208, 45–48.
Michael, G.J., Averill, S., Nitkunan, A., Rattray, M., Bennett, D.L., Yan, Q.,
Priestley, J.V., 1997. Nerve growth factor treatment increases brain-
derived neurotrophic factor selectively in TrkA-expressing dorsal root
ganglion cells and in their central terminations within the spinal cord.
J. Neurosci. 17, 8476–8490.
Obata, K., Yamanaka, H., Dai, Y., Tachibana, T., Fukuoka, T.,
Tokunaga, A., Yoshikawa, H., Noguchi, K., 2003. Differential
activation of extracellular signal-regulated protein kinase in primary
afferent neurons regulates brain-derived neurotrophic factor expres-
sion after peripheral inflammation and nerve injury. J. Neurosci. 23,
4117–4126.
Pezet, S., Malcangio, M., Lever, I.J., Perkinton, M.S., Thompson, S.W.,
Williams, R.J., McMahon, S.B., 2002a. Noxious stimulation induces
Trk receptor and downstream ERK phosphorylation in spinal dorsal
horn. Mol. Cell. Neurosci. 21, 684–695.
Pezet, S., Malcangio, M., McMahon, S.B., 2002b. BDNF: a neuromodulator
in nociceptive pathways? Brain Res. Brain Res. Rev. 40, 240–249.
Randall, L.O., Sellito, J.J., 1957. A method for measurement of
analgesic activity on inflamed tissue. Arch. Int. Pharmacodyn. Ther.
111, 409–419.
Rios, M., Fan, G., Fekete, C., Kelly, J., Bates, B., Kuehn, R., Lechan, R.M.,
Jaenisch, R., 2001. Conditional deletion of brain-derived neurotrophic
factor in the postnatal brain leads to obesity and hyperactivity. Mol.
Endocrinol. 15, 1748–1757.
Rivera, C., Voipio, J., Thomas-Crusells, J., Li, H., Emri, Z., Sipila, S.,
Payne, J.A., Minichiello, L., Saarma, M., Kaila, K., 2004. Mechanism
of activity-dependent downregulation of the neuron-specific K–Cl
cotransporter KCC2. J. Neurosci. 24, 4683–4691.
Slack, S.E., Thompson, S.W., 2002. Brain-derived neurotrophic factor
induces NMDA receptor 1 phosphorylation in rat spinal cord. Neuro-
Report 13, 1967–1970.
Slack, S.E., Pezet, S., McMahon, S.B., Thompson, S.W., Malcangio, M.,
2004. Brain-derived neurotrophic factor induces NMDA receptor
subunit one phosphorylation via ERK and PKC in the rat spinal cord.
Eur. J. Neurosci. 20, 1769–1778.
Stirling, L.C., Forlani, G., Baker, M.D., Wood, J.N., Matthews, E.A.,
Dickenson, A.H., Nassar, M.A., 2005. Nociceptor-specific gene
deletion using heterozygous Na(V)1.8-Cre recombinase mice. Pain
113, 27–36.
Thompson, S.W., Bennett, D.L., Kerr, B.J., Bradbury, E.J., McMahon, S.B.,
1999. Brain-derived neurotrophic factor is an endogenous modulator of
nociceptive responses in the spinal cord. Proc. Natl. Acad. Sci. U. S. A.
96, 7714–7718.
Woolfe, G., MacDonald, A.D., 1944. The evaluation of the analgesic action
of pethidine hydrochloride (Demerol). J. Pharmacol. Exp. Ther. 80,
300–307.
Yajima, Y., Narita, M., Narita, M., Matsumoto, N., Suzuki, T., 2002.
Involvement of a spinal brain-derived neurotrophic factor/full-length
TrkB pathway in the development of nerve injury-induced thermal
hyperalgesia in mice. Brain Res. 958, 338–346.
Zhang, J.Y., Luo, X.G., Xian, C.J., Liu, Z.H., Zhou, X.F., 2000.
Endogenous BDNF is required for myelination and regenera-
tion of injured sciatic nerve in rodents. Eur. J. Neurosci. 12,
4171–4180.
Zhou, X.F., Deng, Y.S., Xian, C.J., Zhong, J.H., 2000. Neurotrophins from
dorsal root ganglia trigger allodynia after spinal nerve injury in rats. Eur.
J. Neurosci. 12, 100–105.
J. Zhao et al. / Mol. Cell. Neurosci. 31 (2006) 539–548548