nitric oxide/cgmp protects endothelial cells from hypoxia-mediated leakiness
TRANSCRIPT

ARTICLE IN PRESS
European Journal of Cell Biology 87 (2008) 147–161
0171-9335/$ - se
doi:10.1016/j.ej
Abbreviations
monophosphate
endothelial cells
altitude pulmon
quinoxalin-1-on�CorrespondE-mail addr
www.elsevier.de/ejcb
Nitric oxide/cGMP protects endothelial cells from hypoxia-mediated
leakiness
Gopi Krishna Kollurua, K.P. Tamilarasana, Arun Stephen Rajkumarb, S. Geetha Priyaa,Megha Rajarama, Niyas K. Saleema, Syamantak Majumdera, B.M. Jaffar Alib,G. Illavazaganc, Suvro Chatterjeea,�
aVascular Biology Laboratory, AU-KBC Research Centre, MIT Campus, Anna University, Chennai 600 044, Tamil Nadu, IndiabOptical Nano-Manipulation Group, AU-KBC Research Centre, Anna University, Chennai, Tamil Nadu, IndiacDefence Institute of Physiology and Allied Sciences, DIPAS, Delhi, India
Received 14 July 2007; received in revised form 6 October 2007; accepted 8 October 2007
Abstract
Leakiness of the endothelial bed is attributed to the over-perfusion of the pulmonary bed, which leads to highaltitude pulmonary edema (HAPE). Inhalation of nitric oxide has been successfully employed to treat HAPE patients.We hypothesize that nitric oxide intervenes in the permeability of the pulmonary macrovascular endothelial bed torectify the leaky bed under hypoxia. Our present work explores the underlying mechanism of ‘hypoxia-mediated’endothelial malfunction by using human umbilical cord-derived immortalized endothelial cells, ECV-304, and bovinepulmonary artery primary endothelial cells. The leakiness of the endothelial monolayer was increased by two-foldunder hypoxia in comparison to cells under normoxia, while optical tweezers-based tethering assays reported a highermembrane tension of endothelial cells under hypoxia. Phalloidin staining demonstrated depolymerization of F-actinstress fibers and highly polarized F-actin patterns in endothelial cells under hypoxia. Nitric oxide, 8-Br-cGMP andsildenafil citrate (phosphodiesterase type 5 inhibitor) led to recovery from hypoxia-induced leakiness of the endothelialmonolayers. Results of the present study also suggest that ‘hypoxia-induced’ cytoskeletal rearrangements andmembrane leakiness are associated with the low nitric oxide availability under hypoxia. We conclude that nitric oxide-based recovery of hypoxia-induced leakiness of endothelial cells is a cyclic guanosine monophosphate (cGMP)-dependent phenomenon.r 2007 Elsevier GmbH. All rights reserved.
Keywords: Hypoxia; Endothelial cells; Nitric oxide; Actin filaments; cGMP; Sildenafil
e front matter r 2007 Elsevier GmbH. All rights reserved.
cb.2007.10.001
: ARDS, acute respiratory distress syndrome; BPAEC, bovine pulmonary aortic endothelial cells; cGMP, cyclic guanosine
; DAF-2DA, diaminofluorescein diacetate; DEAN, diethylamine NONOate; DMEM, Dulbecco’s modified Eagle’s medium; EC,
; ECV 304, human umbilical vein endothelial cells; eNOS, endothelial nitric oxide synthase; FBS, fetal bovine serum; HAPE, high
ary edema; L-NAME, nitro-L-arginine-methyl ester; NO, nitric oxide; NOS, nitric oxide synthase; ODQ, 1H-[1, 2, 4]oxadiazolo[4, 3-a]e; PBS, phosphate-buffered saline; sGC, soluble guanylyl cyclase; SNP, sodium nitroprusside.
ing author. Tel.: +9144 2223 4885x48; fax: +91 44 2223 1034.
ess: [email protected] (S. Chatterjee).

ARTICLE IN PRESSG.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161148
Introduction
Nitric oxide (NO), an endogenously produced auta-coid, modulates a variety of biological functionsincluding plasma membrane permeability and paracel-lular routes (Ignarro, 2002a, b; Moncada, 1997, 1999;Sessa, 2004). NO protects the lungs from increasedpermeability during injury in a number of species (Posset al., 1995; Garat et al., 1997; Searles et al., 2004).Benzing et al. (1998) demonstrated that inhaled NOlowers capillary pressure in patients with adult respira-tory distress syndrome. It has been suggested that NOserves a potential protective role in pulmonary liquidbalance by reducing microvascular pressure, therebyreducing fluid filtration (Mundy and Dorrington, 2000).
High altitude pulmonary edema (HAPE) is a hydro-static edema in the presence of normal left atrialpressure with non-inflammatory high permeabilityleakage of the alveolocapillary barrier and mild alveolarhemorrhage (Sartori et al., 2007). Uneven hypoxicpulmonary vasoconstriction has been proposed toexpose parts of the pulmonary capillary bed to highpressure and vascular injury in HAPE (Hultgren, 1978;Maggiorini et al., 2001). Autopsy findings in patientsdying of HAPE also demonstrated a protein-richalveolar fluid (Arias-Stella and Kruger, 1963). In therat model, inhaled NO improves survival from HAPE(Omura et al., 2000). Several studies have suggested thatinhaled NO improves oxygenation in pulmonary edema(Anand et al., 1998; Kinsella et al., 1997; Perrin et al.,2006; Roberts et al., 1997; Scherrer et al., 1996; Wesselet al., 1997). Work of Mundy and Dorrington (2000)showed that inhibition of NO synthesis augmentspulmonary edema in isolated perfused rabbit lung.Scientists from the Defence Research and DevelopmentOrganization (DRDO), India, successfully used gaseousNO to treat soldiers who suffered from HAPEsymptoms in the Himalayas (Himashree et al., 2003).However, the cellular and molecular basis of NO-basedrecovery from HAPE is not completely understood.
Bronchoalveolar lavage studies in subjects withHAPE show a significant elevation of protein content,suggesting that the vascular barrier to protein move-ment has been decreased (Maggiorini et al., 2001). Workof Kayyali et al. (2002) showed that hypoxia activatesMAPK-activated protein kinase MK2 in connectionwith HSP27 phosphorylation and reorganization of theactin cytoskeleton. Although these observations par-tially explain the hypoxia ‘downstream’ effects, therelationship between hypoxia-mediated reorganizationof the actin network and ‘‘membrane leakiness’’ isnot explored yet. In the present work, we show thatdelivery of NO recovers endothelial cells (ECs) fromhypoxia-induced leakiness by reorganizing the actin-based cytoskeleton and by reducing hypoxia-inducedmembrane tension. Hypoxia-mediated alterations in
ECs were prevented by treatment with sildenafil andcyclic guanosine monophosphate (cGMP) analogsunder hypoxic conditions, indicating a major roleof the NO/cGMP pathway in the hypoxia-inducedalteration of endothelial morphology, permeability andcytoskeletal pattern.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM) waspurchased from Hi-Media, Mumbai, India. Fetal bovineserum (FBS) was from Invitrogen Life technologies.Phalloidin-Alexa Fluor 568 (phalloidin) and diamino-fluorescein diacetate (DAF-2DA) from Molecularprobe, Oregon, USA. Sodium nitroprusside (SNP),propidium iodide, cytochalasin D, and L-NAME werepurchased from Sigma Chemical Co (St. Louis, MO).Polystyrene microspheres were purchased from Poly-sciences, Inc. (Warrington, PA). Diethylamine NON-Oate (DEAN) and guanosine-3050-monophosphate8-bromo-sodium salt (8-Br-cGMP) were obtained fromEMD Biosciences, Inc., California, USA. Dr. VijayShah, GI Research Unit, Mayo Clinic, Rochester kindlyprovided the eNOS-GFP plasmid. All other chemicalswere of reagent grade and were obtained commercially.
Cell culture
Human umbilical cord-derived endothelial cells (ECV304) with additional features of T24 cells were culturedin DMEM supplemented with 10% FBS (v/v) and 1%(w/v) penicillin and streptomycin.
Bovine pulmonary aortic endothelial cells (BPAEC)were isolated from the pulmonary artery of freshly killedanimals. At the slaughterhouse, large blood vessels(pulmonary artery) were collected aseptically and placedin phosphate-buffered saline (PBS) (pH 7.4). In thelaboratory, vessels were washed with PBS. Connectivetissue and fat were removed aseptically. Next, ECs wereharvested as described elsewhere (Ryan, 1984). Isolatedcells were confirmed as ECs by using antibodies againstendothelial markers, eNOS and factor VIII, respec-tively. The cells were used for experiments till passage 6.
Transfection
ECV 304 cells were grown in a 12-well plate for atleast 18–24 h prior to transfection. The cells were thentransfected with plasmid vector encoding eNOS-GFPusing the calcium phosphate method as describedelsewhere (Chatterjee et al., 2002).

ARTICLE IN PRESSG.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161 149
In vitro hypoxia model
ECs in the absence or presence of added compoundswere placed into a hypoxia chamber with an inlet andoutlet for purging the required percentage of O2. As acontrol, the cells were incubated under normoxicconditions for the same length of time. For all thehypoxia experiments, a gas mixture (10% O2+90% N2)was purged in the hypoxia chamber for 2 h. The dosageof hypoxia was standardized by varying the O2
concentrations and duration of treatments.
Trypan blue permeability assay
We measured the trans-cellular permeability of ECplasma membranes by using a modified trypan blue-based permeability assay (Grankvist et al., 1979). ECoverexpressing eNOS-GFP were grown on cover glassesand incubated under normoxia and hypoxia for 30min,1 and 2 h at 37 1C/5% CO2. Next, the cells weremounted in a live-cell chamber. Phase-contrast imageswith 8 s time-lapse were taken at 40� magnificationafter addition of trypan blue (0.004%). The rate ofinclusion of trypan blue dye was calculated from thenuclear color index (color intensity).
Permeability assay
In this manuscript, ‘‘leakiness’’ denotes to net ECmonolayer permeability, which is comprised of para-cellular and trans-cellular vascular solute fluxes. Effectsof NO, L-NAME, sildenafil citrate, and 8-Br-cGMP onthe permeability of endothelial monolayers undernormoxia and hypoxia, respectively, were studied. Inbrief, a half million EC were seeded on the collagen-coated polycarbonate membrane in the permeabilitychamber and incubated for 24 h at 37 1C/5% CO2. Thepermeability chamber with EC in the absence orpresence of added compounds was kept under hypoxiaand normoxia, respectively, for 2 h and incubated at37 1C/5% CO2. Trypan blue (0.004%) was added in theupper half of the permeability chamber, and incubatedfor 1 h at 37 1C/5% CO2. The solution from the lowerchamber was collected to measure the optical density at580 nm using a Varian Cary 4000 UV–vis spectro-photometer.
Fluorescence microscopy
EC were cultured on cover glasses in 12-well plates tillthey reach 40% confluence before starting the experi-ments. The cells were then incubated under hypoxia andnormoxia with and without 500 mM SNP in 37 1C/5%CO2 for 30min, 1 and 2 h, respectively. The cover slipswere washed gently with PBS, and cells were fixed in 2%
formaldehyde (freshly prepared from paraformalde-hyde) for 7min, permeabilized with 0.1% TritonX-100 for 2min and incubated with phalloidin (0.5 mMfinal concentration) for 1 h. Specimens were viewedunder a NIKON TE2000-U fluorescence microscopeat 560 nm emission. Photographs were taken with anAndor CCD camera.
Single cell migration assay
Cell migration was assessed using the wound healingmethod. One million ECV 304 cells in 2ml DMEM/10%FBS were seeded in a 35-mm dish. Twenty-four hourslater, when the cells reached confluence, a linear woundwas created by scratching the monolayer with a 1mmwide sterile plastic scraper. Cells were washed with PBS,treated with and without SNP (500 mM) and incubatedfor a fixed time period (2 h unless otherwise stated)under normoxia and hypoxia. Bright-field images weretaken with 4� and 40� magnifications under aninverted bright-field microscope. The rate of woundhealing was quantified from the images using ScionImage, Release Alpha 4.0 3.2 and Adobe Photoshopversion 6.0.
Measurement of membrane tension by optical
tweezers
The optical tweezers set-up was built around aninverted microscope (TE-2000U, Nikon Corporation,Japan). A 1.5W 1064 nm Nd:YAG laser (Laser Quan-tum, UK) focused through an oil-immersion objective(Plan Apo, N.A. 1.4) was used to form an optical trap.The motion of trapped particles was tracked by back-scattering using a 635 nm diode laser (Coherent, USA).Stiffness of the trap (was around 0.008 pN/nm) cali-brated by power spectrum analysis. However, foraccuracy of measurement, the trap stiffness wascalculated each time before the start of an individualexperiment.
ECV304 ECs treated under normoxia and hypoxiawere used for optical tweezers experiments. Prior toexperiments, the medium was replaced with PBScontaining 2-mm uncoated polystyrene spheres. Usingthe optical trap, membrane tethers were drawn bypressing the bead against the cell surface for 5 s andpulling out at the rate of 0.5 mm/s and held static at alength of 5 mm. After equilibrium for 20 s, the tetherforce, F0 (Dai and Sheetz, 1998) and time series of beadfluctuations were measured. The effective stiffness, keff,of the membrane-trap system was computed from powerspectral analysis of the time series. keff is a linearcombination of the trap stiffness ktrap, and membranetension Tm given by 1/Tm ¼ 1/ktrap+1/keff (Evans et al.,2005). From this relation, the membrane tension was
ARTICLE IN PRESSG.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161150
calculated. All membrane tension experiments werecarried out at a height of 5 mm above the cover slip.The necessary corrections due to the height of the beadfrom the surface were made.
NO-imaging with DAF-2DA
eNOS-transfected cells cultured on cover glasses in12-well plates were incubated under hypoxia andnormoxia at 37 1C/5% CO2 for 2 h. Cells were washedtwice with PBS and then loaded with 200 ml DAF-2DA(10 mM) and incubated for 5min. After another incuba-tion of 5min with 1 mM calcium ionophore, cells werestimulated with 0.5mM calcium chloride and effectswere observed under a NIKON TE-2000 invertedfluorescence microscope at 515 nm. Images were takenwith an Andor CCD camera. Fluorescence intensity ofthe cells was calculated by using the image analysismodule of Adobe Photoshop 7.0.
Statistical analysis
All experiments were performed in triplicate unlessotherwise specified. Data are presented as mean+SE.Data was analyzed using t-test, one-way and two-wayANOVA as appropriate. P-values p0.05 were selectedas the criterion for a statistically significant difference.
Results
Hypoxia attenuated NO production from eNOS-
transfected ECs
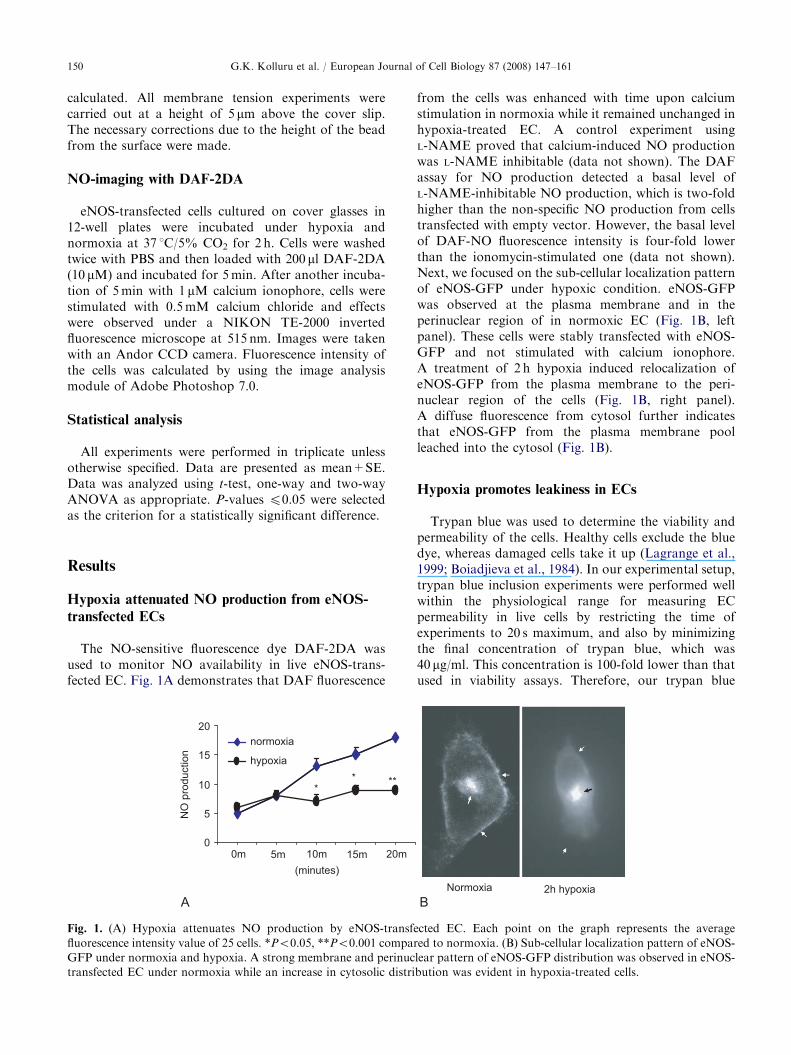
The NO-sensitive fluorescence dye DAF-2DA wasused to monitor NO availability in live eNOS-trans-fected EC. Fig. 1A demonstrates that DAF fluorescence
0
5
10
15
20
(minutes)
0m 5m 10m 15m 20m
NO
pro
duction
normoxia
hypoxia
**
**
A
Fig. 1. (A) Hypoxia attenuates NO production by eNOS-transfe
fluorescence intensity value of 25 cells. *Po0.05, **Po0.001 compar
GFP under normoxia and hypoxia. A strong membrane and perinuc
transfected EC under normoxia while an increase in cytosolic distri
from the cells was enhanced with time upon calciumstimulation in normoxia while it remained unchanged inhypoxia-treated EC. A control experiment usingL-NAME proved that calcium-induced NO productionwas L-NAME inhibitable (data not shown). The DAFassay for NO production detected a basal level ofL-NAME-inhibitable NO production, which is two-foldhigher than the non-specific NO production from cellstransfected with empty vector. However, the basal levelof DAF-NO fluorescence intensity is four-fold lowerthan the ionomycin-stimulated one (data not shown).Next, we focused on the sub-cellular localization patternof eNOS-GFP under hypoxic condition. eNOS-GFPwas observed at the plasma membrane and in theperinuclear region of in normoxic EC (Fig. 1B, leftpanel). These cells were stably transfected with eNOS-GFP and not stimulated with calcium ionophore.A treatment of 2 h hypoxia induced relocalization ofeNOS-GFP from the plasma membrane to the peri-nuclear region of the cells (Fig. 1B, right panel).A diffuse fluorescence from cytosol further indicatesthat eNOS-GFP from the plasma membrane poolleached into the cytosol (Fig. 1B).
Hypoxia promotes leakiness in ECs
Trypan blue was used to determine the viability andpermeability of the cells. Healthy cells exclude the bluedye, whereas damaged cells take it up (Lagrange et al.,1999; Boiadjieva et al., 1984). In our experimental setup,trypan blue inclusion experiments were performed wellwithin the physiological range for measuring ECpermeability in live cells by restricting the time ofexperiments to 20 s maximum, and also by minimizingthe final concentration of trypan blue, which was40 mg/ml. This concentration is 100-fold lower than thatused in viability assays. Therefore, our trypan blue
Normoxia
B2h hypoxia
cted EC. Each point on the graph represents the average
ed to normoxia. (B) Sub-cellular localization pattern of eNOS-
lear pattern of eNOS-GFP distribution was observed in eNOS-
bution was evident in hypoxia-treated cells.
ARTICLE IN PRESS
858 16 24
90
95
100
105
110
115
Time (sec)
Re
lative
in
cre
ase
in n
ucle
ar
co
lor
ind
ex
Normoxia
Hypoxia
Hypoxia + SNP
* *
@@
A
0
50
100
150
200
250
Control Control+SNP Hypoxia Hypoxia+SNP
Pe
rce
nta
ge
ch
an
ge
in
pe
rme
ab
ility
of E
C m
on
ola
ye
r
**
#
*
B
90
95
100
105
110
8 16 24
Time (sec)
Ra
te o
f try
pa
n b
lue
inclu
sio
n
(nu
cle
ar co
lor in
de
x)
Hyp eNOS-ve
Hyp eNOS+ve
**
C
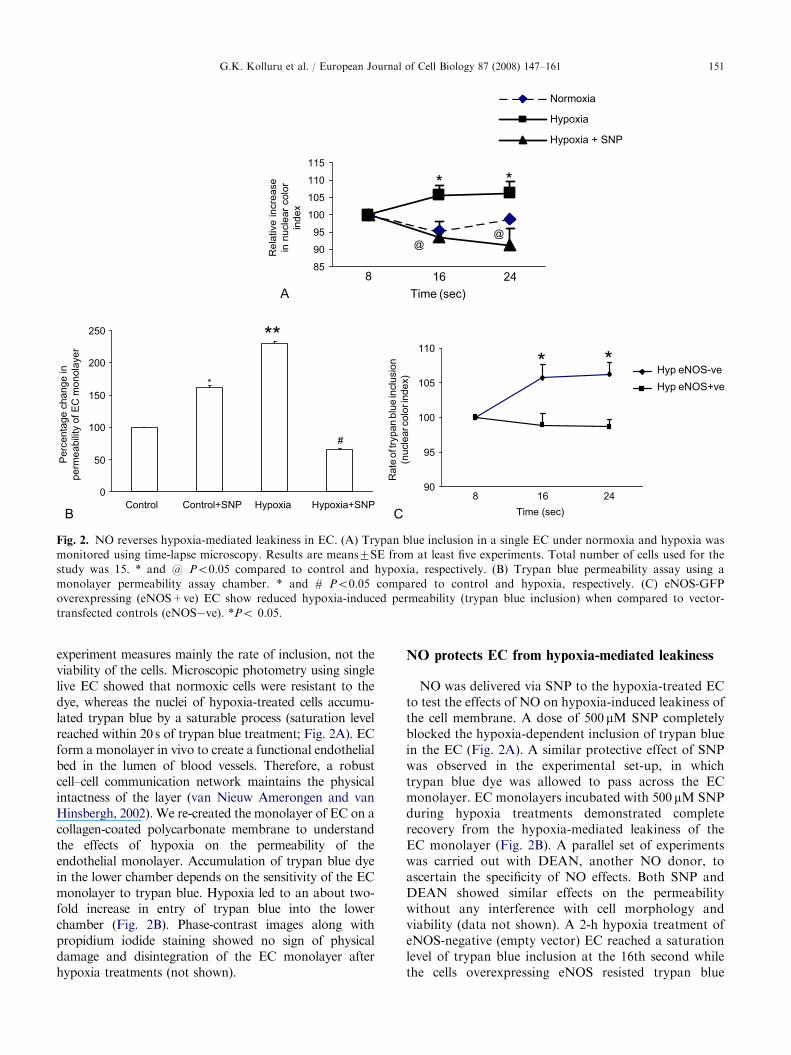
Fig. 2. NO reverses hypoxia-mediated leakiness in EC. (A) Trypan blue inclusion in a single EC under normoxia and hypoxia was
monitored using time-lapse microscopy. Results are means7SE from at least five experiments. Total number of cells used for the
study was 15. * and @ Po0.05 compared to control and hypoxia, respectively. (B) Trypan blue permeability assay using a
monolayer permeability assay chamber. * and # Po0.05 compared to control and hypoxia, respectively. (C) eNOS-GFP
overexpressing (eNOS+ve) EC show reduced hypoxia-induced permeability (trypan blue inclusion) when compared to vector-
transfected controls (eNOS�ve). *Po 0.05.
G.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161 151
experiment measures mainly the rate of inclusion, not theviability of the cells. Microscopic photometry using singlelive EC showed that normoxic cells were resistant to thedye, whereas the nuclei of hypoxia-treated cells accumu-lated trypan blue by a saturable process (saturation levelreached within 20 s of trypan blue treatment; Fig. 2A). ECform a monolayer in vivo to create a functional endothelialbed in the lumen of blood vessels. Therefore, a robustcell–cell communication network maintains the physicalintactness of the layer (van Nieuw Amerongen and vanHinsbergh, 2002). We re-created the monolayer of EC on acollagen-coated polycarbonate membrane to understandthe effects of hypoxia on the permeability of theendothelial monolayer. Accumulation of trypan blue dyein the lower chamber depends on the sensitivity of the ECmonolayer to trypan blue. Hypoxia led to an about two-fold increase in entry of trypan blue into the lowerchamber (Fig. 2B). Phase-contrast images along withpropidium iodide staining showed no sign of physicaldamage and disintegration of the EC monolayer afterhypoxia treatments (not shown).
NO protects EC from hypoxia-mediated leakiness
NO was delivered via SNP to the hypoxia-treated ECto test the effects of NO on hypoxia-induced leakiness ofthe cell membrane. A dose of 500 mM SNP completelyblocked the hypoxia-dependent inclusion of trypan bluein the EC (Fig. 2A). A similar protective effect of SNPwas observed in the experimental set-up, in whichtrypan blue dye was allowed to pass across the ECmonolayer. EC monolayers incubated with 500 mM SNPduring hypoxia treatments demonstrated completerecovery from the hypoxia-mediated leakiness of theEC monolayer (Fig. 2B). A parallel set of experimentswas carried out with DEAN, another NO donor, toascertain the specificity of NO effects. Both SNP andDEAN showed similar effects on the permeabilitywithout any interference with cell morphology andviability (data not shown). A 2-h hypoxia treatment ofeNOS-negative (empty vector) EC reached a saturationlevel of trypan blue inclusion at the 16th second whilethe cells overexpressing eNOS resisted trypan blue
ARTICLE IN PRESSG.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161152
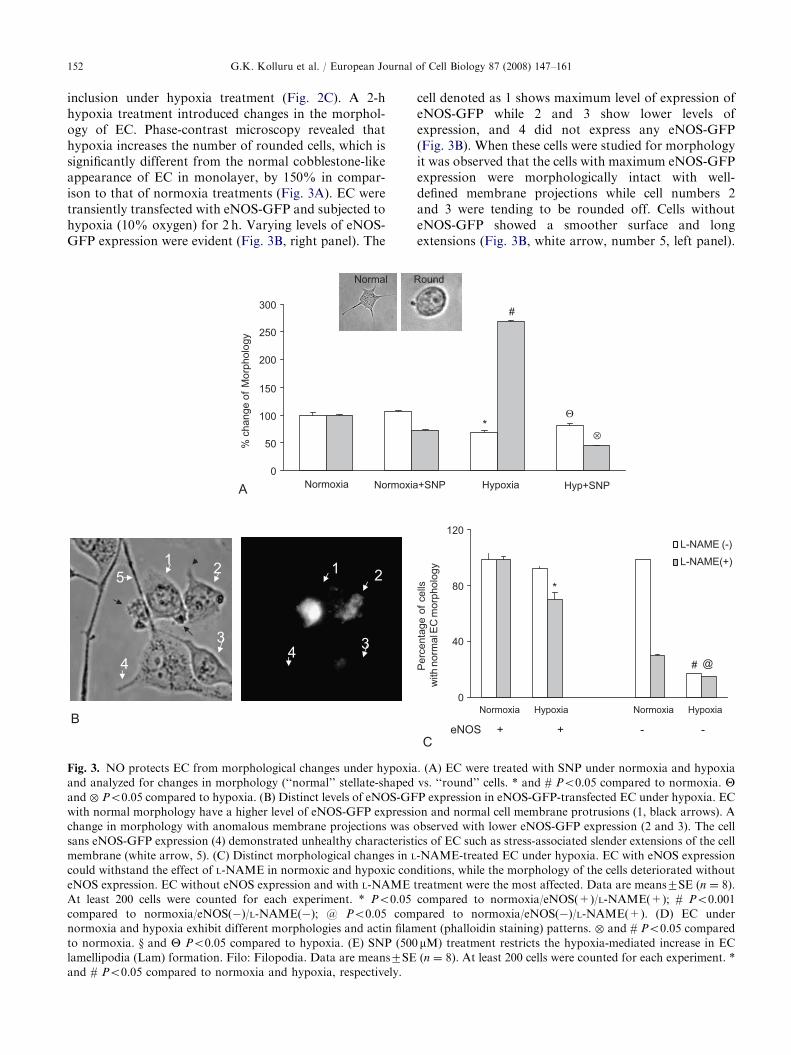
inclusion under hypoxia treatment (Fig. 2C). A 2-hhypoxia treatment introduced changes in the morphol-ogy of EC. Phase-contrast microscopy revealed thathypoxia increases the number of rounded cells, which issignificantly different from the normal cobblestone-likeappearance of EC in monolayer, by 150% in compar-ison to that of normoxia treatments (Fig. 3A). EC weretransiently transfected with eNOS-GFP and subjected tohypoxia (10% oxygen) for 2 h. Varying levels of eNOS-GFP expression were evident (Fig. 3B, right panel). The
0
50
100
150
200
250
300
Normoxia Normoxia
% c
ha
ng
e o
f M
orp
ho
log
y
Normal
A
12 1 2
3
4
5
34
B
Fig. 3. NO protects EC from morphological changes under hypoxia
and analyzed for changes in morphology (‘‘normal’’ stellate-shaped
and � Po0.05 compared to hypoxia. (B) Distinct levels of eNOS-GF
with normal morphology have a higher level of eNOS-GFP expressi
change in morphology with anomalous membrane projections was o
sans eNOS-GFP expression (4) demonstrated unhealthy characterist
membrane (white arrow, 5). (C) Distinct morphological changes in L
could withstand the effect of L-NAME in normoxic and hypoxic con
eNOS expression. EC without eNOS expression and with L-NAME t
At least 200 cells were counted for each experiment. * Po0.05
compared to normoxia/eNOS(�)/L-NAME(�); @ Po0.05 com
normoxia and hypoxia exhibit different morphologies and actin filam
to normoxia. y and Y Po0.05 compared to hypoxia. (E) SNP (500
lamellipodia (Lam) formation. Filo: Filopodia. Data are means7SE
and # Po0.05 compared to normoxia and hypoxia, respectively.
cell denoted as 1 shows maximum level of expression ofeNOS-GFP while 2 and 3 show lower levels ofexpression, and 4 did not express any eNOS-GFP(Fig. 3B). When these cells were studied for morphologyit was observed that the cells with maximum eNOS-GFPexpression were morphologically intact with well-defined membrane projections while cell numbers 2and 3 were tending to be rounded off. Cells withouteNOS-GFP showed a smoother surface and longextensions (Fig. 3B, white arrow, number 5, left panel).
+SNP Hypoxia Hyp+SNP
Round
*
#
Θ
0
40
80
120
Normoxia Hypoxia Normoxia Hypoxia
Perc
enta
ge o
f cells
with n
orm
al E
C m
orp
holo
gy
L-NAME (-)
L-NAME(+)
eNOS + + - -
*
# @
C
⊗
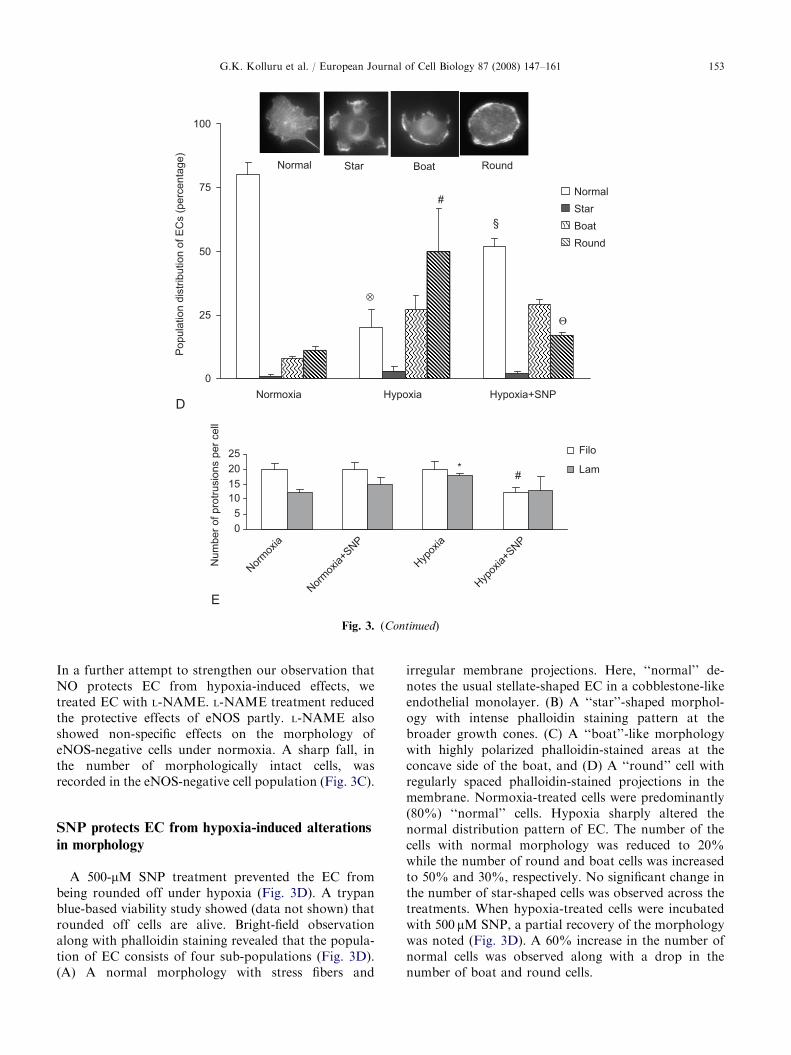
. (A) EC were treated with SNP under normoxia and hypoxia
vs. ‘‘round’’ cells. * and # Po0.05 compared to normoxia. YP expression in eNOS-GFP-transfected EC under hypoxia. EC
on and normal cell membrane protrusions (1, black arrows). A
bserved with lower eNOS-GFP expression (2 and 3). The cell
ics of EC such as stress-associated slender extensions of the cell
-NAME-treated EC under hypoxia. EC with eNOS expression
ditions, while the morphology of the cells deteriorated without
reatment were the most affected. Data are means7SE (n ¼ 8).
compared to normoxia/eNOS(+)/L-NAME(+); # Po0.001
pared to normoxia/eNOS(�)/L-NAME(+). (D) EC under
ent (phalloidin staining) patterns. � and # Po0.05 compared
mM) treatment restricts the hypoxia-mediated increase in EC
(n ¼ 8). At least 200 cells were counted for each experiment. *
ARTICLE IN PRESS
0
25
50
75
100
Normoxia Hypoxia Hypoxia+SNP
Po
pu
latio
n d
istr
ibu
tio
n o
f E
Cs (
pe
rce
nta
ge
)
#
Θ
§
⊗
D
0
5
10
15
20
25
Nor
mox
ia
Nor
mox
ia+S
NP
Hyp
oxia
Hyp
oxia+S
NP
Nu
mb
er
of p
rotr
usio
ns p
er
ce
ll
Filo
Lam*#
E
Normal Boat RoundStar
Normal
Star
Boat
Round
Fig. 3. (Continued)
G.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161 153
In a further attempt to strengthen our observation thatNO protects EC from hypoxia-induced effects, wetreated EC with L-NAME. L-NAME treatment reducedthe protective effects of eNOS partly. L-NAME alsoshowed non-specific effects on the morphology ofeNOS-negative cells under normoxia. A sharp fall, inthe number of morphologically intact cells, wasrecorded in the eNOS-negative cell population (Fig. 3C).
SNP protects EC from hypoxia-induced alterations
in morphology
A 500-mM SNP treatment prevented the EC frombeing rounded off under hypoxia (Fig. 3D). A trypanblue-based viability study showed (data not shown) thatrounded off cells are alive. Bright-field observationalong with phalloidin staining revealed that the popula-tion of EC consists of four sub-populations (Fig. 3D).(A) A normal morphology with stress fibers and
irregular membrane projections. Here, ‘‘normal’’ de-notes the usual stellate-shaped EC in a cobblestone-likeendothelial monolayer. (B) A ‘‘star’’-shaped morphol-ogy with intense phalloidin staining pattern at thebroader growth cones. (C) A ‘‘boat’’-like morphologywith highly polarized phalloidin-stained areas at theconcave side of the boat, and (D) A ‘‘round’’ cell withregularly spaced phalloidin-stained projections in themembrane. Normoxia-treated cells were predominantly(80%) ‘‘normal’’ cells. Hypoxia sharply altered thenormal distribution pattern of EC. The number of thecells with normal morphology was reduced to 20%while the number of round and boat cells was increasedto 50% and 30%, respectively. No significant change inthe number of star-shaped cells was observed across thetreatments. When hypoxia-treated cells were incubatedwith 500 mM SNP, a partial recovery of the morphologywas noted (Fig. 3D). A 60% increase in the number ofnormal cells was observed along with a drop in thenumber of boat and round cells.
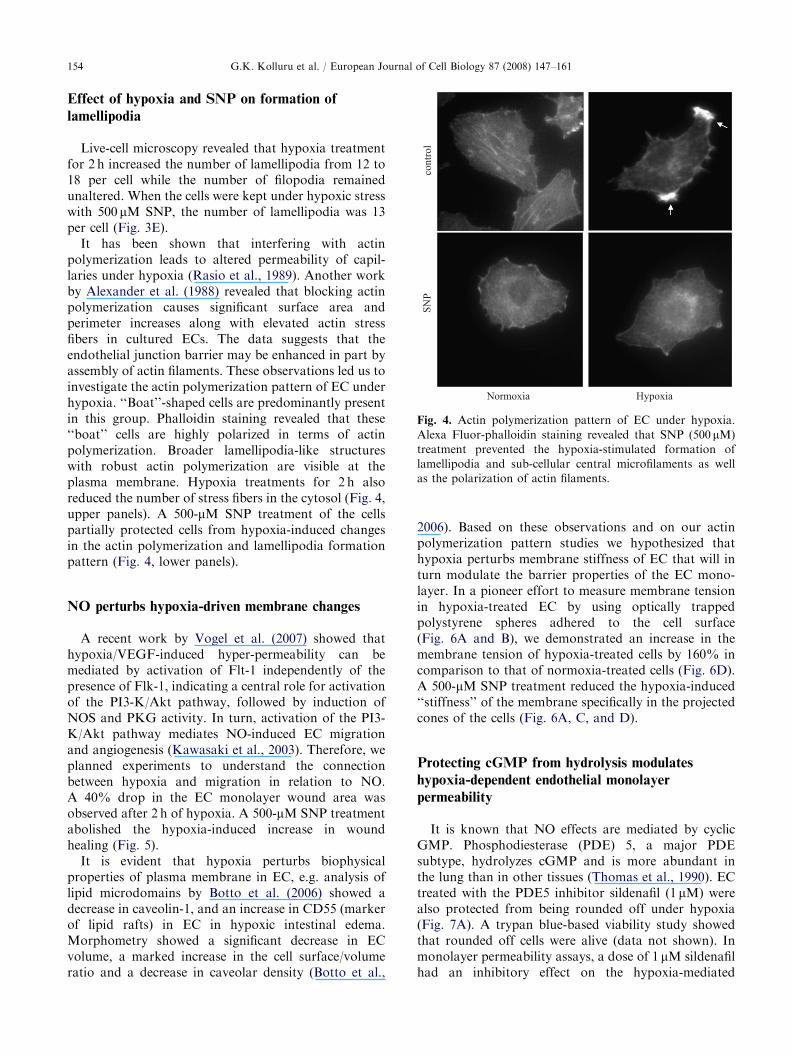
ARTICLE IN PRESS
control
SNP
Normoxia Hypoxia
Fig. 4. Actin polymerization pattern of EC under hypoxia.
Alexa Fluor-phalloidin staining revealed that SNP (500 mM)
treatment prevented the hypoxia-stimulated formation of
lamellipodia and sub-cellular central microfilaments as well
as the polarization of actin filaments.
G.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161154
Effect of hypoxia and SNP on formation of
lamellipodia
Live-cell microscopy revealed that hypoxia treatmentfor 2 h increased the number of lamellipodia from 12 to18 per cell while the number of filopodia remainedunaltered. When the cells were kept under hypoxic stresswith 500 mM SNP, the number of lamellipodia was 13per cell (Fig. 3E).
It has been shown that interfering with actinpolymerization leads to altered permeability of capil-laries under hypoxia (Rasio et al., 1989). Another workby Alexander et al. (1988) revealed that blocking actinpolymerization causes significant surface area andperimeter increases along with elevated actin stressfibers in cultured ECs. The data suggests that theendothelial junction barrier may be enhanced in part byassembly of actin filaments. These observations led us toinvestigate the actin polymerization pattern of EC underhypoxia. ‘‘Boat’’-shaped cells are predominantly presentin this group. Phalloidin staining revealed that these‘‘boat’’ cells are highly polarized in terms of actinpolymerization. Broader lamellipodia-like structureswith robust actin polymerization are visible at theplasma membrane. Hypoxia treatments for 2 h alsoreduced the number of stress fibers in the cytosol (Fig. 4,upper panels). A 500-mM SNP treatment of the cellspartially protected cells from hypoxia-induced changesin the actin polymerization and lamellipodia formationpattern (Fig. 4, lower panels).
NO perturbs hypoxia-driven membrane changes
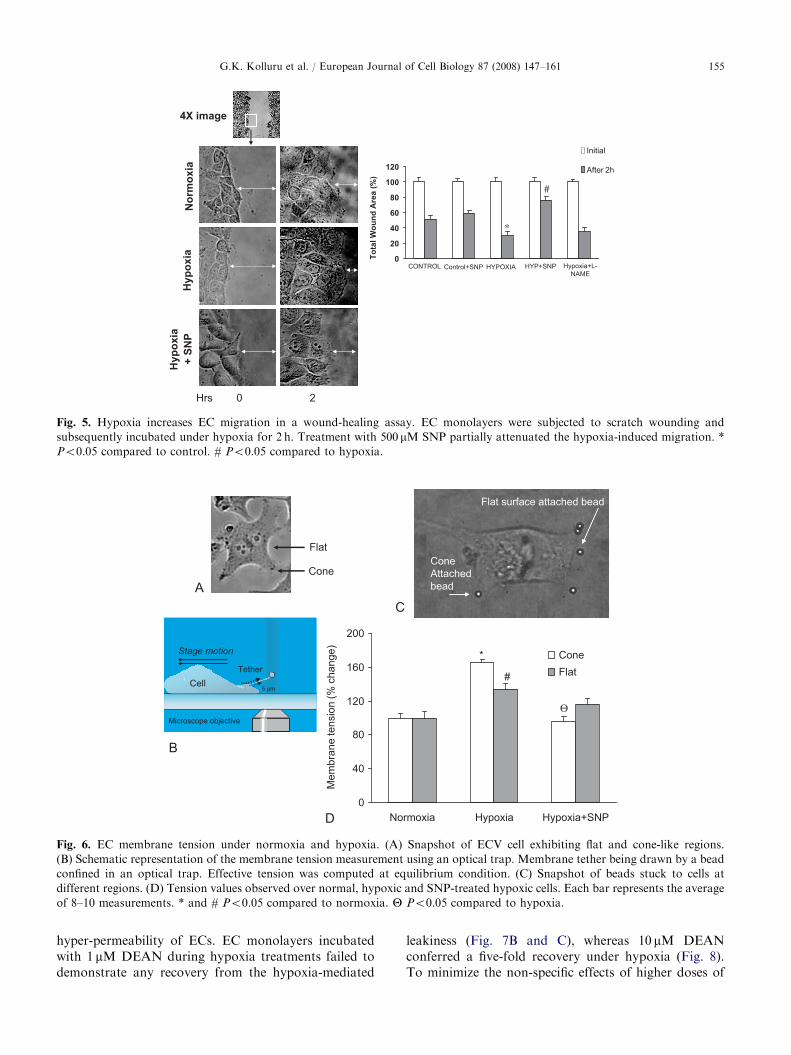
A recent work by Vogel et al. (2007) showed thathypoxia/VEGF-induced hyper-permeability can bemediated by activation of Flt-1 independently of thepresence of Flk-1, indicating a central role for activationof the PI3-K/Akt pathway, followed by induction ofNOS and PKG activity. In turn, activation of the PI3-K/Akt pathway mediates NO-induced EC migrationand angiogenesis (Kawasaki et al., 2003). Therefore, weplanned experiments to understand the connectionbetween hypoxia and migration in relation to NO.A 40% drop in the EC monolayer wound area wasobserved after 2 h of hypoxia. A 500-mM SNP treatmentabolished the hypoxia-induced increase in woundhealing (Fig. 5).
It is evident that hypoxia perturbs biophysicalproperties of plasma membrane in EC, e.g. analysis oflipid microdomains by Botto et al. (2006) showed adecrease in caveolin-1, and an increase in CD55 (markerof lipid rafts) in EC in hypoxic intestinal edema.Morphometry showed a significant decrease in ECvolume, a marked increase in the cell surface/volumeratio and a decrease in caveolar density (Botto et al.,
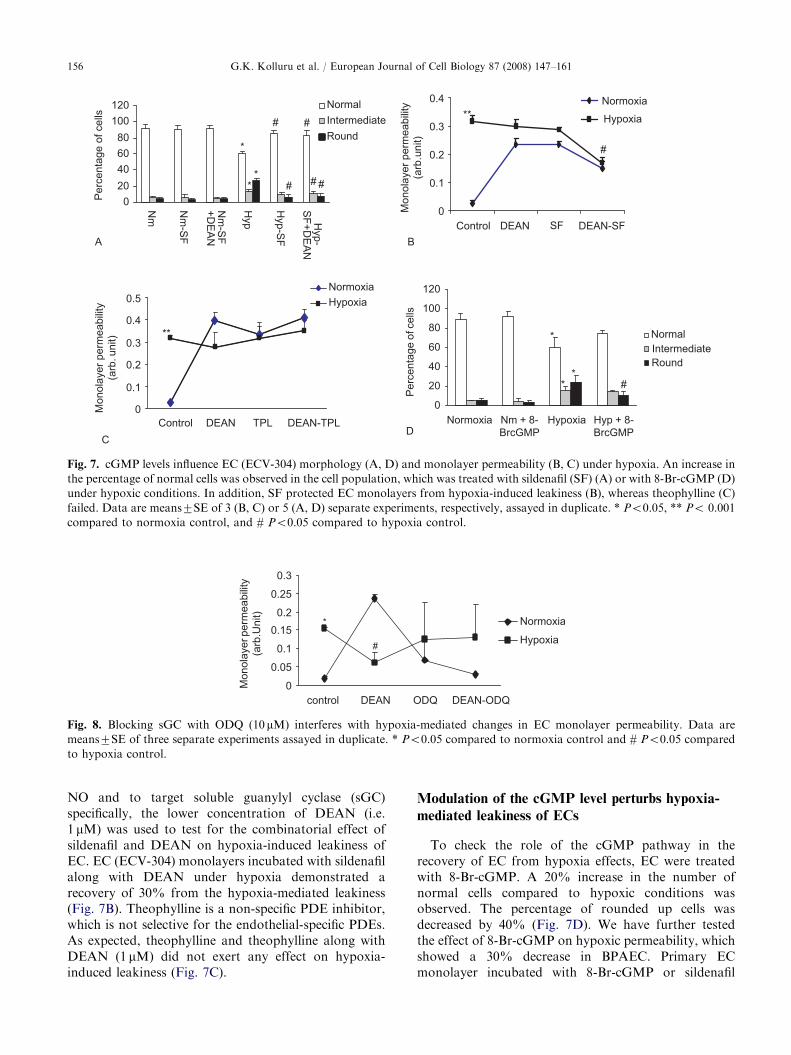
2006). Based on these observations and on our actinpolymerization pattern studies we hypothesized thathypoxia perturbs membrane stiffness of EC that will inturn modulate the barrier properties of the EC mono-layer. In a pioneer effort to measure membrane tensionin hypoxia-treated EC by using optically trappedpolystyrene spheres adhered to the cell surface(Fig. 6A and B), we demonstrated an increase in themembrane tension of hypoxia-treated cells by 160% incomparison to that of normoxia-treated cells (Fig. 6D).A 500-mM SNP treatment reduced the hypoxia-induced‘‘stiffness’’ of the membrane specifically in the projectedcones of the cells (Fig. 6A, C, and D).
Protecting cGMP from hydrolysis modulates
hypoxia-dependent endothelial monolayer
permeability
It is known that NO effects are mediated by cyclicGMP. Phosphodiesterase (PDE) 5, a major PDEsubtype, hydrolyzes cGMP and is more abundant inthe lung than in other tissues (Thomas et al., 1990). ECtreated with the PDE5 inhibitor sildenafil (1 mM) werealso protected from being rounded off under hypoxia(Fig. 7A). A trypan blue-based viability study showedthat rounded off cells were alive (data not shown). Inmonolayer permeability assays, a dose of 1 mM sildenafilhad an inhibitory effect on the hypoxia-mediated
ARTICLE IN PRESS
Hrs 0
No
rmo
xia
Hy
po
xia
Hy
po
xia
+ S
NP
4X image
0
20
40
60
80
100
120
CONTROL Control+SNP HYPOXIA HYP+SNP Hypoxia+L-
NAME
To
tal W
ou
nd
Are
a (
%)
Initial
After 2h
*
#
2
Fig. 5. Hypoxia increases EC migration in a wound-healing assay. EC monolayers were subjected to scratch wounding and
subsequently incubated under hypoxia for 2 h. Treatment with 500 mM SNP partially attenuated the hypoxia-induced migration. *
Po0.05 compared to control. # Po0.05 compared to hypoxia.
0
40
80
120
160
200
Normoxia Hypoxia Hypoxia+SNP
Mem
bra
ne tensio
n (
% c
hange)
Cone
FlatTether
5 µm
Stage motion
Microscope objective
Cell
Cone
Attached
bead
Flat surface attached bead
Cone
Flat
A
B
C
D
*
#
Θ
Fig. 6. EC membrane tension under normoxia and hypoxia. (A) Snapshot of ECV cell exhibiting flat and cone-like regions.
(B) Schematic representation of the membrane tension measurement using an optical trap. Membrane tether being drawn by a bead
confined in an optical trap. Effective tension was computed at equilibrium condition. (C) Snapshot of beads stuck to cells at
different regions. (D) Tension values observed over normal, hypoxic and SNP-treated hypoxic cells. Each bar represents the average
of 8–10 measurements. * and # Po0.05 compared to normoxia. Y Po0.05 compared to hypoxia.
G.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161 155
hyper-permeability of ECs. EC monolayers incubatedwith 1 mM DEAN during hypoxia treatments failed todemonstrate any recovery from the hypoxia-mediated
leakiness (Fig. 7B and C), whereas 10 mM DEANconferred a five-fold recovery under hypoxia (Fig. 8).To minimize the non-specific effects of higher doses of
ARTICLE IN PRESS
0
Control DEAN SF DEAN-SF
0.1
0.2
0.3
0.4
Monola
yer p
erm
eabili
ty
(arb
. unit)
**
#
B
0
0.1
0.2
0.3
0.4
0.5
Control DEAN TPL DEAN-TPL
Monola
yer
perm
eabili
ty
(arb
. unit)
Normoxia
Hypoxia
Normoxia
Hypoxia
**
C
0
20
40
60
80
100
120
Normoxia Nm + 8-
BrcGMP
Hypoxia Hyp + 8-
BrcGMP
Perc
enta
ge o
f cells
Normal
Round
*
**
#
D
A
0
20
40
60
80
100
120
Nm
Nm
-SF
Nm
-SF
+D
EA
N
Hyp
Hyp-S
F
Hyp-
SF
+D
EA
N
Perc
enta
ge o
f cells
Normal
Intermediate
Intermediate
Round*
**
#
#
#
##
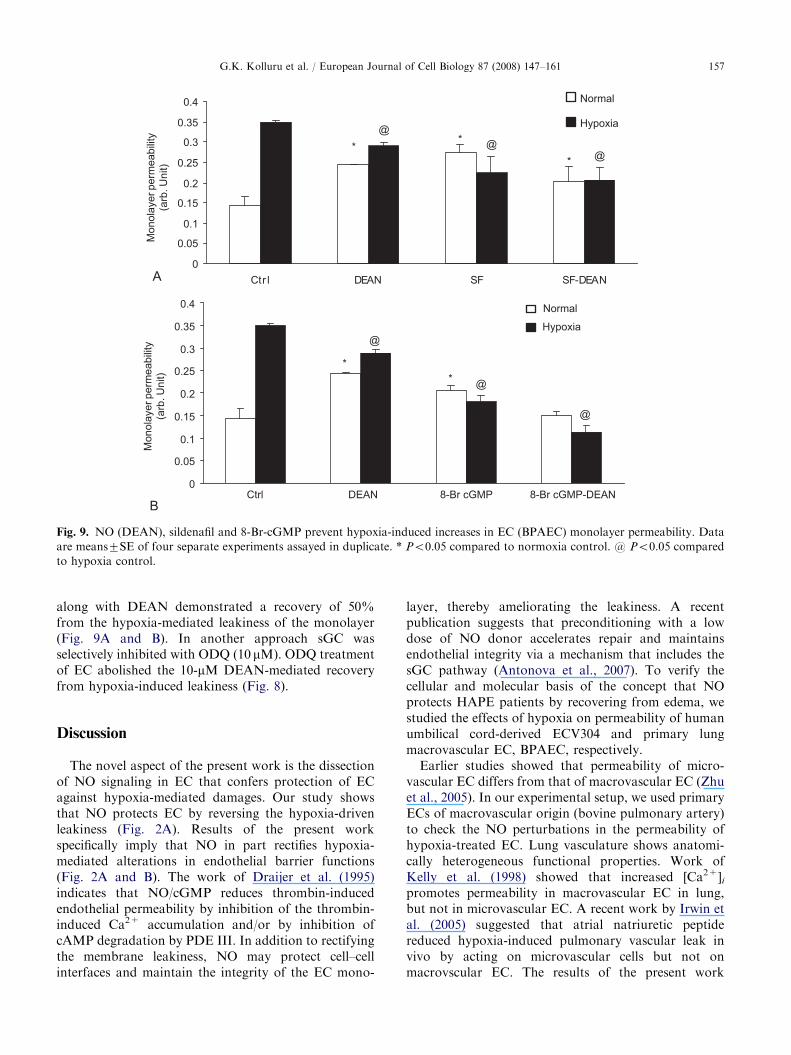
Fig. 7. cGMP levels influence EC (ECV-304) morphology (A, D) and monolayer permeability (B, C) under hypoxia. An increase in
the percentage of normal cells was observed in the cell population, which was treated with sildenafil (SF) (A) or with 8-Br-cGMP (D)
under hypoxic conditions. In addition, SF protected EC monolayers from hypoxia-induced leakiness (B), whereas theophylline (C)
failed. Data are means7SE of 3 (B, C) or 5 (A, D) separate experiments, respectively, assayed in duplicate. * Po0.05, ** Po 0.001
compared to normoxia control, and # Po0.05 compared to hypoxia control.
0
0.05
0.1
0.15
0.2
0.25
0.3
control DEAN ODQ DEAN-ODQ
Monola
yer perm
eabili
ty
(arb
.Unit)
Normoxia
Hypoxia
*
#
Fig. 8. Blocking sGC with ODQ (10 mM) interferes with hypoxia-mediated changes in EC monolayer permeability. Data are
means7SE of three separate experiments assayed in duplicate. * Po0.05 compared to normoxia control and # Po0.05 compared
to hypoxia control.
G.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161156
NO and to target soluble guanylyl cyclase (sGC)specifically, the lower concentration of DEAN (i.e.1 mM) was used to test for the combinatorial effect ofsildenafil and DEAN on hypoxia-induced leakiness ofEC. EC (ECV-304) monolayers incubated with sildenafilalong with DEAN under hypoxia demonstrated arecovery of 30% from the hypoxia-mediated leakiness(Fig. 7B). Theophylline is a non-specific PDE inhibitor,which is not selective for the endothelial-specific PDEs.As expected, theophylline and theophylline along withDEAN (1 mM) did not exert any effect on hypoxia-induced leakiness (Fig. 7C).
Modulation of the cGMP level perturbs hypoxia-
mediated leakiness of ECs
To check the role of the cGMP pathway in therecovery of EC from hypoxia effects, EC were treatedwith 8-Br-cGMP. A 20% increase in the number ofnormal cells compared to hypoxic conditions wasobserved. The percentage of rounded up cells wasdecreased by 40% (Fig. 7D). We have further testedthe effect of 8-Br-cGMP on hypoxic permeability, whichshowed a 30% decrease in BPAEC. Primary ECmonolayer incubated with 8-Br-cGMP or sildenafil
ARTICLE IN PRESS
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
Ctr l DEAN SF SF-DEAN
Monola
yer perm
eabili
ty
(arb
. U
nit)
Monola
yer perm
eabili
ty
(arb
. U
nit)
Normal
Hypoxia
Normal
Hypoxia
*
@
@@
*
*
A
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
Ctrl DEAN 8-Br cGMP 8-Br cGMP-DEAN
*
@
@
@
*
B
Fig. 9. NO (DEAN), sildenafil and 8-Br-cGMP prevent hypoxia-induced increases in EC (BPAEC) monolayer permeability. Data
are means7SE of four separate experiments assayed in duplicate. * Po0.05 compared to normoxia control. @ Po0.05 compared
to hypoxia control.
G.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161 157
along with DEAN demonstrated a recovery of 50%from the hypoxia-mediated leakiness of the monolayer(Fig. 9A and B). In another approach sGC wasselectively inhibited with ODQ (10 mM). ODQ treatmentof EC abolished the 10-mM DEAN-mediated recoveryfrom hypoxia-induced leakiness (Fig. 8).
Discussion
The novel aspect of the present work is the dissectionof NO signaling in EC that confers protection of ECagainst hypoxia-mediated damages. Our study showsthat NO protects EC by reversing the hypoxia-drivenleakiness (Fig. 2A). Results of the present workspecifically imply that NO in part rectifies hypoxia-mediated alterations in endothelial barrier functions(Fig. 2A and B). The work of Draijer et al. (1995)indicates that NO/cGMP reduces thrombin-inducedendothelial permeability by inhibition of the thrombin-induced Ca2+ accumulation and/or by inhibition ofcAMP degradation by PDE III. In addition to rectifyingthe membrane leakiness, NO may protect cell–cellinterfaces and maintain the integrity of the EC mono-
layer, thereby ameliorating the leakiness. A recentpublication suggests that preconditioning with a lowdose of NO donor accelerates repair and maintainsendothelial integrity via a mechanism that includes thesGC pathway (Antonova et al., 2007). To verify thecellular and molecular basis of the concept that NOprotects HAPE patients by recovering from edema, westudied the effects of hypoxia on permeability of humanumbilical cord-derived ECV304 and primary lungmacrovascular EC, BPAEC, respectively.
Earlier studies showed that permeability of micro-vascular EC differs from that of macrovascular EC (Zhuet al., 2005). In our experimental setup, we used primaryECs of macrovascular origin (bovine pulmonary artery)to check the NO perturbations in the permeability ofhypoxia-treated EC. Lung vasculature shows anatomi-cally heterogeneous functional properties. Work ofKelly et al. (1998) showed that increased [Ca2+]ipromotes permeability in macrovascular EC in lung,but not in microvascular EC. A recent work by Irwin etal. (2005) suggested that atrial natriuretic peptidereduced hypoxia-induced pulmonary vascular leak invivo by acting on microvascular cells but not onmacrovscular EC. The results of the present work

ARTICLE IN PRESSG.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161158
further indicate that NO may target specifically macro-vascular EC to modulate permeability under hypoxicconditions.
Basal vascular endothelial permeability is normallykept low in part by the restrictiveness of inter-endothelial junctions. We observed an increase inmonolayer permeability under hypoxia that could bethe result of trans-cellular hyper-permeability andincreasing inter-endothelial gaps (Fig. 2A). Endothelialpermeability is believed to be associated with morphol-ogy changes of the cells, such as we observed when theendothelial monolayer was subjected to hypoxia(Fig. 3A). Tiruppathi et al. (1992) demonstrated thata-thrombin widens the inter-endothelial junctions be-cause of ‘‘rounding up’’ of the cells and therebyincreases the permeability. Hinder et al. (1999) haveused an animal model of sepsis to demonstrate that NOinhalation may protect against pulmonary edemaassociated with endotoxemia and NOS inhibition. Ithas also been shown that pretreatment of cells withdifferent NO donors increased endothelial cGMPcontent and blocked hydrogen peroxide-related effectson permeability (Suttorp et al., 1996). The work ofMarumo et al. (1999) demonstrated that blocking ofNOS enhanced VEGF-induced hyper-permeability,while activation of NOS counteracts the VEGF effects.There is evidence that even basal release of NO helps tomaintain normal pulmonary vascular fluid balance(Arkowitz et al., 1997). We critically examined theprotective role of NO on permeability and morphologyof EC (Fig. 3B and C). We also found that supplemen-ted NO protects EC from hypoxia-mediated damages(Fig. 2A), while NO induces permeability in EC undernormoxia (Fig. 2B). It is evident that hypoxia promotessuperoxide production in EC that contributes to theenhanced permeability of EC membrane (Gertzberget al., 2004). NO is known for its high affinity forsuperoxide radicals (Inoue et al., 2000). Therefore,supplemented NO interacts with superoxide to formperoxynitirite, thereby neutralizing the permeability-enhancing effects of superoxide and protecting the ECfrom abnormal barrier functions. The present studyreveals that the NO/cGMP pathway perturbs endothe-lial barrier functions (Figs. 7B, 8, and 9A, B) and alsoindicates that eNOS-derived NO possibly tunes theintegrity of inter-endothelial junctions, and thus servesto maintain the low basal permeability of continuousendothelia (Predescu et al., 2005).
The normal function of the endothelium is highlydependent on the endothelial cytoskeleton arrange-ments. By using a simulation method known asdissipative particle dynamics, in which several atomsunited into a single particle. Groot and Rabone (2001)explained that stress and membrane damage are twointer-dependent phenomena. When we measured mem-brane tension of normoxia- and hypoxia-treated cells by
using optical tweezers, hypoxia-treated cells showedregion-specific variation in the level of tension. Themigration cones with denser actin polymerizationdeveloped a 20% higher tension than apparently flatterareas (Fig. 6), and hypoxic conditions with NO donorscould protect cells from hypoxia-mediated elevation ofmembrane tension (Fig. 6). NO has been implicated inthe migration of EC. It is evident from the work ofGoligorsky et al. (1999) that NO modulates focaladhesions in ECs. The results of our experiments showthat hypoxia-associated changes in actin polymerizationpattern, permeability and migration of EC are NOdependent (Figs. 2B, 3C, E, 4, and 5). This observationis further supported by earlier work of Baldwin et al.(1998), which confirms that inhibition of NO synthesisincreases venular permeability and alters the endothelialactin cytoskeleton. Work of Dai and Sheetz (1995, 1998)furnishes evidence in support to our result that hypoxia-induced polarization of actin in EC overstretches thesurface of hypoxia-induced cones and incites higherleakiness in those regions (Fig. 6A and D). We speculatethat a drastic rearrangement of cytoskeleton proteinsunder hypoxia rafts the actin monomers to polymerizein a polarized fashion (Fig. 4). We assume that restrictedstiffening of the EC membrane promotes eNOS loosen-ing from the membrane followed by reduced NOproduction under hypoxia (Fig. 1B). Our assumptionis further substantiated by a recent publication indicat-ing that hypoxia produced enlarged EC, with dysfunc-tional Golgi and loss of eNOS from the plasmamembrane (Mukhopadhyay et al., 2007).
PDE plays a crucial role in cGMP-dependent signal-ing by hydrolyzing the cGMP. PDE 5 is largelyresponsible for cGMP metabolism in the lung endothe-lium (Matthay, 2002). Sildenafil, a PDE inhibitor, helpsin keeping cGMP levels high by inhibiting PDE 5. Arecent publication suggests that pulmonary microvas-cular ECs do not express PDE 5 in culture, and possiblyin vivo as well (Zhu et al., 2005). In our experiments,sildenafil led to partial recovery of both types of EC(Fig. 7A and B). The sildenafil effect was also shown tobe potentiated by NO donors (Figs. 7A, B and 9A),whereas theophylline, a non-specific PDE inhibitor,failed to confer any protection against hypoxia-mediated damages (Fig. 7C). Sildenafil is known topromote vasodilatory effects in acute hypoxia-inducedpulmonary hypertension in eNOS KO mice (Prestonet al., 2004). Earlier work demonstrated that chronicadministration of DA-8159, an inhibitor of PDE5, couldattenuate the development of endothelial dysfunction indiabetes, and its effect is associated with an improve-ment in endothelial function (Ahn et al., 2005). Suttorpet al. (1993) suggested that PDE inhibition is a powerfulapproach to block the H2O2-induced increase inendothelial permeability. This concept appears espe-cially valuable when endothelial PDE isoenzyme pattern

ARTICLE IN PRESSG.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161 159
and PDE inhibitor profile are matched optimally, as ourresult shows that inhibition of endothelial-specific PDE5is more effective than inhibition of PDE4, a non-endothelial subtype (Fig. 7C). The importance of thecGMP-dependent pathway in maintaining the barrierfunction of the endothelium was further verified when ahigher dose (10 mM) of DEAN failed to restore thebarrier functions under hypoxia (Fig. 8). Our studydemonstrates that cGMP protects endothelial barrierfunctions under hypoxia. Occurrence of pathologicedema in ARDS, pulmonary hypertension, HAPE,and high altitude cerebral edema is attributed to theleaky endothelial barrier. Dissection of the NO-cGMPpathway, which improves membrane barrier functionsin EC, may help identifying a candidate drug from thegenre of pharmacological agents such as PDE inhibitorsor cGMP analogs. Our findings may be also helpful infurther dissecting the mechanism of NO-mediated actinpolymerization and polarization in EC under hypoxia.Since vascular permeability in the lung is tightlycontrolled by an array of physiological variables suchas lymph flow, lymph protein concentration, hetero-geneity of endothelial behavior, level of histamine andprostaglandins (Brigham et al., 1974; Brigham andOwen, 1975) presently this work does not offer a cure ofhypoxia-associated lung diseases. Instead, the workverifies the concept of NO-based recovery of HAPE atthe cellular level, and also prepares the springboard toexplore the possibilities of NO-based therapy ofhypoxia-associated lung diseases in animal models byconsidering the unique biochemical and biophysicalmilieu of the lung.
Acknowledgments
This study was financially supported by a taskforcegrant (Grant# 97(SC-AU)/DIPAS/2005) from DefenceInstitute of Physiology and Allied Sciences (DIPAS),Defence Research and Development Organization(DRDO), Government of India to S. Chatterjee.
References
Ahn, G.J., Yu, J.Y., Choi, S.M., Kang, K.K., Ahn, B.O.,
Kwon, J.W., Kang, S.K., Lee, B.C., Hwang, W.S., 2005.
Chronic administration of phosphodiesterase 5 inhibitor
improves erectile and endothelial function in a rat model of
diabetes. Int. J. Androl. 28, 260–266.
Alexander, J.S., Hechtman, H.B., Shepro, D., 1988. Phalloidin
enhances endothelial barrier function and reduces inflam-
matory permeability in vitro. Microvasc. Res. 35, 308–315.
Anand, I.S., Prasad, B.A., Chugh, S.S., Rao, K.R., Cornfield,
D.N., Milla, C.E., Singh, N., Singh, S., Selvamurthy, W.,
1998. Effects of inhaled nitric oxide and oxygen in high-
altitude pulmonary edema. Circulation 98, 2441–2445.
Antonova, G., Snead, C., Antonov, A., Dimitropoulou, C.,
Venema, R.C., Catravas, J.D., 2007. Nitric oxide precon-
ditioning regulates endothelial monolayer integrity via the
heat shock protein 90-soluble guanylate cyclase pathway.
Am. J. Physiol. Heart Circ. Physiol. 292, H893–H903.
Arias-Stella, J., Kruger, H., 1963. Pathology of high altitude
pulmonary edema. Arch. Pathol. 70, 43–53.
Arkowitz, M., Wispe, J.R., Garcia, V.F., Szabo, C., 1997.
Selective inhibition of the inducible isoform of nitric oxide
synthase prevents pulmonary transvascular flux during
acute endotoxemia. J. Pediatr. Surg. 31, 1009–1015.
Baldwin, A.L., Thurston, G., Al Naemi, H., 1998. Inhibition
of nitric oxide synthesis increases venular permeability and
alters endothelial actin cytoskeleton. Am. J. Physiol. Heart
Circ. Physiol. 274, H1776–H1784.
Benzing, A., Mols, G., Guttmann, J., Kaltofen, H., Geiger, K.,
1998. Effect of different doses of inhaled nitric oxide
on pulmonary capillary pressure and on longitudinal
distribution of pulmonary vascular resistance in ARDS.
Br. J. Anaesth. 80, 440–446.
Boiadjieva, S., Hallberg, C., Hogstrom, M., Busch, C., 1984.
Exclusion of trypan blue from microcarriers by endothelial
cells: an in vitro barrier function test. Methods Lab. Invest.
50, 239–246.
Botto, L., Beretta, E., Daffara, R., Miserocchi, G., Palestini,
P., 2006. Biochemical and morphological changes in
endothelial cells in response to hypoxic interstitial edema.
Respir. Res. 7, 7.
Brigham, K.L., Owen, P.J., 1975. Increased sheep lung
vascular permeability caused by histamine. Circ. Res. 37,
647–657.
Brigham, K.L., Woolverton, W.C., Blake, L.H., Staub, N.C.,
1974. Increased sheep lung vascular permeability caused by
Pseudomonas bacteremia. J. Clin. Invest. 54, 792–804.
Chatterjee, S., Cao, S., Shah, V., 2002. Inhibition of GTP-
dependent vesicle trafficking impairs internalization of
plasmalemmal eNOS and cellular nitric oxide production.
J. Cell Sci. 116, 3645–3655.
Dai, J., Sheetz, M.P., 1995. Mechanical properties of neuronal
growth cone membranes studied by tether formation with
laser optical tweezers. Biophys. J. 68, 988–996.
Dai, J., Sheetz, M.P., 1998. Cell membrane mechanics.
Methods Cell Biol. 55, 157–171.
Draijer, R., Atsma, D.E., van der Laarse, A., van Hinsbergh,
V.W., 1995. cGMP and nitric oxide modulate thrombin-
induced endothelial permeability. Regulation via different
pathways in human aortic and umbilical vein endothelial
cells. Circ. Res. 76, 199–208.
Evans, E., Heinrich, V., Leung, A., Kinoshita, K., 2005. Nano-
to microscale dynamics of P-selectin detachment from
leukocyte interfaces. I. Membrane separation from the
cytoskeleton. Biophys. J. 88, 2288–2298.
Garat, C., Jayr, C., Eddahibi, S., Laffon, M., Meigan, M.,
Adnot, S., 1997. Effects of inhaled nitric oxide or inhibition
of endogenous nitric oxide formation on hyperoxic lung
injury. Am. J. Respir. Crit. Care Med. 155, 1957–1964.
Gertzberg, N., Neumann, P., Rizzo, V., Johnson, A., 2004.
NAD(P)H oxidase mediates the endothelial barrier dys-
function induced by TNF-alpha. Am. J. Physiol. Lung Cell
Mol. Physiol. 286, L37–L48.

ARTICLE IN PRESSG.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161160
Goligorsky, M.S., Abedi, H., Noiri, E., Takhtajan, A., Lense,
S., Victor, R., Zachary, I., 1999. Nitric oxide modulation of
focal adhesions in endothelial cells. Am. J. Physiol. Cell
Physiol. 276, C1271–C1281.
Grankvist, K., Lernmark, A., Taljedal, I.B., 1979. Trypan blue
as a marker of plasma membrane permeability in alloxan-
treated mouse islet cells. J. Endocrinol. Invest. 2, 139–145.
Groot, R.D., Rabone, K.L., 2001. Mesoscopic simulation of
cell membrane damage, morphology change and rupture by
nonionic surfactants. Biophys. J. 81, 725–736.
Himashree, G., Dass, D., Banerjee, P.K., Selvamurthy, W.,
2003. Nitric oxide and the respiratory system. Curr. Sci. 85,
607–614.
Hinder, F., Stubbe, H.D., Van Aken, H., Waurick, R., Booke,
M., Meyer, J., 1999. Role of nitric oxide in sepsis-associated
pulmonary edema. Am. J. Respir. Crit. Care Med. 159,
252–257.
Hultgren, J.N., 1978. High altitude pulmonary edema. In:
Staub, N.C. (Ed.), Lung Water and Solute Exchange.
Marcel Dekker, Inc., New York, pp. 437–469.
Ignarro, L.J., 2002a. Nitric oxide as a unique signaling
molecule in the vascular system: a historical overview.
J. Physiol. Pharmacol. 53, 503–514.
Ignarro, L.J., 2002b. Wei Lun Visiting Professorial Lecture:
nitric oxide in the regulation of vascular function: an
historical overview. J. Cardiac Surg. 17, 301–306.
Inoue, M., Sato, E.F., Park, A.M., Nishikawa, M., Kasahara,
E., Miyoshi, M., Ochi, A., Utsumi, K., 2000. Cross-talk
between NO and oxyradicals, a supersystem that regulates
energy metabolism and survival of animals. Free Radic.
Res. 33, 757–770.
Irwin, D.C., Tissot van Patot, M.C., Tucker, A., Bowen, R.,
2005. Direct ANP inhibition of hypoxia-induced inflam-
matory pathways in pulmonary microvascular and macro-
vascular endothelial monolayers. Am. J. Physiol. Lung Cell
Mol. Physiol. 288, L849–L859.
Kawasaki, K., Smith, R.S., Hsieh, C.M., Sun, J., Chao, J.,
Liao, J.K., 2003. Activation of the phosphatidylinositol
3-kinase/protein kinase Akt pathway mediates nitric oxide-
induced endothelial cell migration and angiogenesis. Mol.
Cell. Biol. 23, 5726–5737.
Kayyali, U.S., Pennella, C.M., Trujillo, C., Villa, O., Gaestel,
M., Hassoun, P.M., 2002. Cytoskeletal changes in hypoxic
pulmonary endothelial cells are dependent on MAPK-
activated protein kinase MK2. J. Biol. Chem. 277,
42596–42602.
Kelly, J.J., Moore, T.M., Babal, P., Diwan, A.H., Stevens, T.,
Thompson, W.J., 1998. Pulmonary microvascular and
macrovascular endothelial cells: differential regulation of
Ca2+ and permeability. Am. J. Physiol. Lung Cell Mol.
Physiol. 274, L810–L819.
Kinsella, J.P., Truog, W.E., Walsh, W.F., Goldberg, R.N.,
Bancalari, E., Mayock, D.E., Redding, G.J., deLemos,
R.A., Sardesai, S., McCurnin, D.C., Moreland, S.G.,
Cutter, G.R., Abman, S.H., 1997. Randomized, multicenter
trial of inhaled nitric oxide and high-frequency oscillatory
ventilation in severe, persistent pulmonary hypertension of
the newborn. J. Pediatr. 131, 55–62.
Lagrange, P., Romero, I.A., Minn, A., Revest, P.A., 1999.
Transendothelial permeability changes induced by free
radicals in an in vitro model of the blood-brain barrier.
Free Radic. Biol. Med. 27, 667–672.
Maggiorini, M., Melot, C., Pierre, S., Pfeiffer, F., Greve, I.,
Sartori, C., Lepori, M., Hauser, M., Scherrer, U., Naeije,
R., 2001. High-altitude pulmonary edema is initially caused
by an increase in capillary pressure. Circulation 4,
2078–2083.
Marumo, T., Noll, T., Schini-Kerth, V.B., Harley, E.A.,
Duhault, J., Piper, H.M., Busse, R., 1999. Significance of
nitric oxide and peroxynitrite in permeability changes of the
retinal microvascular endothelial cell monolayer induced by
vascular endothelial growth factor. J. Vasc. Res. 36,
510–515.
Matthay, M.A., 2002. Alveolar fluid clearance in patients
with ARDS: does it make a difference? Chest 122,
340S–343S.
Moncada, S., 1997. Nitric oxide in the vasculature: physiology
and pathophysiology. Ann. NY Acad. Sci. 811, 60–67
discussion 67–69.
Moncada, S., 1999. Nitric oxide: discovery and impact on
clinical medicine. J. R. Soc. Med. 92, 164–169.
Mukhopadhyay, S., Xu, F., Sehgal, P.B., 2007. Aberrant
cytoplasmic sequestration of eNOS in endothelial cells after
monocrotaline, hypoxia, and senescence: live-cell caveolar
and cytoplasmic NO imaging. Am. J. Physiol. Heart Circ.
Physiol. 292, H1373–H1389.
Mundy, A.L., Dorrington, K.L., 2000. Inhibition of nitric
oxide synthesis augments pulmonary oedema in isolated
perfused rabbit lung. Br. J. Anaesth. 85, 570–576.
Omura, A., Roy, R., Jennings, T., 2000. Inhaled nitric oxide
improves survival in the rat model of high-altitude
pulmonary edema. Wild. Environ. Med. 11, 251–256.
Perrin, G., Roch, A., Michelet, P., Reynaud-Gaubert, M.,
Thomas, P., Doddoli, C., Auffray, J.P., 2006. Inhaled
nitric oxide does not prevent pulmonary edema after
lung transplantation measured by lung water content:
a randomized clinical study. Chest 129, 1024–1030.
Poss, W.B., Timmons, O.D., Farrukh, I.S., Hoidal, J.R.,
Michael, J.R., 1995. Inhaled nitric oxide prevents the
increase in pulmonary vascular permeability caused by
hydrogen peroxide. J. Appl. Physiol. 79, 886–891.
Predescu, D., Predescu, S., Shimizu, J., Miyawaki-Shimizu, K.,
Malik, A.B., 2005. Constitutive eNOS-derived nitric oxide
is a determinant of endothelial junctional integrity. Am. J.
Physiol. Lung Cell Mol. Physiol. 289, L371–L381.
Preston, I.R., Hill, N.S., Gambardella, L.S., Warburton, R.R.,
Klinger, J.R., 2004. Synergistic effects of ANP and
sildenafil on cGMP levels and amelioration of acute
hypoxic pulmonary hypertension. Exp. Biol. Med. 229,
920–925.
Rasio, E.A., Bendayan, M., Goresky, C.A., Alexander, J.S.,
Shepro, D., 1989. Effect of phalloidin on structure and
permeability of rete capillaries in the normal and hypoxic
state. Circ. Res. 65, 591–599.
Roberts Jr., J.D., Fineman, J.R., Morin III, F.C., Shaul, P.W.,
Rimar, S., Schreiber, M.D., Polin, R.A., Zwass, M.S.,
Zayek, M.M., Gross, I., Heymann, M.A., Zapol, W.M.,
1997. Inhaled nitric oxide and persistent pulmonary
hypertension of the newborn. The Inhaled Nitric Oxide
Study Group. N. Engl. J. Med. 336, 605–610.

ARTICLE IN PRESSG.K. Kolluru et al. / European Journal of Cell Biology 87 (2008) 147–161 161
Ryan, U.S., 1984. Isolation and culture of pulmonary
endothelial cells. Environ. Health Perspect. 56, 103–114.
Sartori, C., Allemann, Y., Scherrer, U., 2007. Pathogenesis of
pulmonary edema: learning from high-altitude pulmonary
edema. Respir. Physiol. Neurobiol. [E-pub ahead of print,
doi:10.1016/j.resp.2007.04.006].
Scherrer, U., Vollenweider, L., Delabays, A., Savcic, M.,
Eichenberger, U., Kleger, G.R., Fikrle, A., Ballmer, P.E.,
Nicod, P., Bartsch, P., 1996. Inhaled nitric oxide for
high-altitude pulmonary edema. N. Engl. J. Med. 334,
624–629.
Searles, C.D., Ide, L., Davis, M.E., Cai, H., Weber, M., 2004.
Actin cytoskeleton organization and posttranscriptional
regulation of endothelial nitric oxide synthase during cell
growth. Circ. Res. 95, 488–495.
Sessa, W.C., 2004. eNOS at a glance. J. Cell Sci. 117,
2427–2429.
Suttorp, N., Weber, U., Welsch, T., Schudt, C., 1993. Role of
phosphodiesterases in the regulation of endothelial perme-
ability in vitro. J. Clin. Invest. 91, 1421–1428.
Suttorp, N., Hippensteil, S., Fuhrmann, M., Krull, M.,
Podzuweit, T., 1996. Role of nitric oxide and phosphodies-
terase isoenzyme II for reduction of endothelial hyperper-
meability. Am. J. Physiol. Cell Physiol. 39, C778–C785.
Thomas, M.K., Francis, S.H., Corbin, J.D., 1990. Character-
ization of a purified bovine lung cGMP-binding cGMP
phosphodiesterase. J. Biol. Chem. 265, 14964–14970.
Tiruppathi, C., Malik, A.B., Vecchio, P.J.D., Keese, C.R.,
Giaever, I., 1992. Electrical method for detection of endothe-
lial cell shape change in real time: assessment of endothelial
barrier function. Proc. Natl. Acad. Sci. USA 89, 7919–7923.
Van Nieuw Amerongen, G.P., van Hinsbergh, V.W., 2002.
Targets for pharmacological intervention of endothelial
hyperpermeability and barrier function. Vasc. Pharmacol.
39, 257–272.
Vogel, C., Bauer, A., Wiesnet, M., Preissner, K.T., Schaper,
W., Marti, H.H., Fischer, S., 2007. Flt-1, but not Flk-1
mediates hyperpermeability through activation of the
PI3-K/Akt pathway. J. Cell. Physiol. 212, 236–243.
Wessel, D.L., Adatia, I., Van Marter, L.J., Thompson, J.E.,
Kane, J.W., Stark, A.R., Kourembanas, S., 1997. Improved
oxygenation in a randomized trial of inhaled nitric oxide
for persistent pulmonary hypertension of the newborn.
Pediatrics 100, E7.
Zhu, B., Strada, S., Stevens, T., 2005. Cyclic GMP-specific
phosphodiesterase 5 regulates growth and apoptosis in
pulmonary endothelial cells. Am. J. Physiol. Lung Cell
Mol. Physiol. 289, L196–L206.