new immortalized cell lines of patients with small supernumerary marker chromosome: towards the...
TRANSCRIPT

http://jhc.sagepub.com/Journal of Histochemistry & Cytochemistry
http://jhc.sagepub.com/content/55/6/651The online version of this article can be found at:
DOI: 10.1369/jhc.6A7161.2007
2007 55: 651J Histochem CytochemHasmik Mkrtchyan, Isolde Schreyer, Ferdinand von Eggeling, Anja Weise, Kristin Mrasek and Thomas Liehr
Schmidt, Berndt Schulze, Laura Rodríguez, Franz Binkert, Catharine Yardin, Nadezda Kosyakova, Marianne Volleth, Holger Tönnies, Joanna Pietrzak, Ewa Bocian, Kay MacDermont, Alma Kuechler, Britta Belitz, Udo Trautmann, Angela
Establishment of a Cell BankNew Immortalized Cell Lines of Patients With Small Supernumerary Marker Chromosome: Towards the
Published by:
http://www.sagepublications.com
On behalf of:
Official Journal of The Histochemical Society
can be found at:Journal of Histochemistry & CytochemistryAdditional services and information for
http://jhc.sagepub.com/cgi/alertsEmail Alerts:
http://jhc.sagepub.com/subscriptionsSubscriptions:
http://www.sagepub.com/journalsReprints.navReprints:
http://www.sagepub.com/journalsPermissions.navPermissions:
What is This?
- Jun 1, 2007Version of Record >>
by guest on June 7, 2014jhc.sagepub.comDownloaded from by guest on June 7, 2014jhc.sagepub.comDownloaded from

ARTICLE
New Immortalized Cell Lines of Patients With SmallSupernumerary Marker Chromosome: Towards theEstablishment of a Cell Bank
Holger Tonnies, Joanna Pietrzak, Ewa Bocian, Kay MacDermont, Alma Kuechler, Britta Belitz,Udo Trautmann, Angela Schmidt, Berndt Schulze, Laura Rodrıguez, Franz Binkert,Catharine Yardin, Nadezda Kosyakova, Marianne Volleth, Hasmik Mkrtchyan, Isolde Schreyer,Ferdinand von Eggeling, Anja Weise, Kristin Mrasek, and Thomas Liehr
Institute of Human Genetics, Charite Campus Virchow, Berlin, Germany (HT); Department of Medical Genetics, Institute ofMother and Child, Warsaw, Poland (JP,EB); The North West London Hospital, NHS Trust, Harrow, Middlesex, United Kingdom(KMacDermont); Institute of Human Genetics and Anthropology, Jena, Germany (AK,NK,HM,IS,FVE,AW,KMrasek,TL);Department of Pediatrics, University Clinic, Jena, Germany (AK); Practice for Human Genetics, Berlin, Germany (BB); Instituteof Human Genetics, Erlangen, Germany (UT); Practice for Human Genetics, Hannover, Germany (AS,BS); Estudio ColaborativoEspanol de Malformaciones Congenitas del Centro de Investigacion sobre Anomalıas Congenitas, Instituto de Salud Carlos III,Ministerio de Sanidad y Consumo, Madrid, Spain (LR); MCL Medical Laboratories, Niederwangen, Switzerland (FB); Serviced’Histologie Cytologie Cytogenetique Biologie Cell et de la Reproduction, Limoges, Cedex, France (CY); Research Centre forMedical Genetics, Moscow, Russia (NK); Institute of Human Genetics, Magdeburg, Germany (MV); and Department ofGenetics and Laboratory of Cytogenetics, State University, Jerewan, Armenia (HM)
SUMMARY Sixteen newly established cell lines with small supernumerary marker chro-mosomes (sSMC) derived from chromosomes 1, 2, 4, 6, 7, 8, 14, 15, 16, 18, 19, 21, and 22 arereported. Two sSMC are neocentric and derived from 15q24.1-qter and 2q35-q36, respec-tively. Two further cases each present with two sSMC of different chromosomal origin. sSMCwere characterized by multicolor fluorescence in situ hybridization for their chromosomalorigin and genetic content. Moreover, uniparental disomy of the sister chromosomes of thesSMC was excluded in all nine cases studied for that reason. The 16 cases provide informationto establish a refined genotype–phenotype correlation of sSMC and are available for futurestudies. (J Histochem Cytochem 55:651–660, 2007)
KEY WORDS
small supernumerary marker
chromosome
cell line
Epstein–Barr virus immortalization
genotype–phenotype correlation
cell bank
SMALL SUPERNUMERARY MARKER CHROMOSOMES (sSMC)are a diagnostic problem in clinical cytogenetics be-cause they are too small to be characterized for theirchromosomal origin by banding techniques alone; gen-erally, they are equal in size or smaller than a chro-mosome 20 of the same metaphase spread. Thus,molecular cytogenetic, i.e., fluorescence in situ hybrid-ization (FISH) approaches are needed for their classi-fication (Liehr et al. 2004). Recently, we collected allavailable reported sSMC cases and presented them onthe regularly updated sSMC homepage (Liehr 2006a).We concluded that sSMC are present in z2.7 million
people worldwide (prevalence: 0.044%) (Liehr et al.2004; Liehr 2006a).
For de novo sSMC, particularly those ascertainedprenatally, Paoloni-Giacobino et al. (1998) stated thatthey are not easy to correlate with a clinical outcome. Itis known that 32% of sSMC are derived from chro-mosome 15; 11% are i(12p) 5 Pallister-Killian, z10%are der(22)-, z7% are inv dup(22)-cat-eye-, and z6%are i(18p)-syndrome-associated sSMC (Liehr et al.2004). In general, the risk for an abnormal phenotypein prenatally ascertained de novo cases with sSMCis given as z13% (Warburton 1991). This has beenrefined to 7% (for sSMC from chromosome 13, 14,21, or 22) and 28% (for all non-acrocentric auto-somes) (Crolla 1998). Also, generally speaking, sSMCinherited from a normal sSMC carrier to its childrenare usually not correlated with clinical problems, eventhough exceptions are described (Liehr et al. 2004). The
Correspondence to: PD Dr. Thomas Liehr, Institut fur Human-genetik und Anthropologie, Kollegiengasse 10, D-07743 Jena, Ger-many. E-mail: [email protected]
Received for publication November 30, 2006; accepted February 8,2007 [DOI: 10.1369/jhc.6A7161.2007].
TheJournal
ofHistoch
emistry&
Cytoch
emistry
C The Histochemical Society, Inc. 0022-1554/07/$3.30 651
Volume 55(6): 651–660, 2007
Journal of Histochemistry & Cytochemistry
http://www.jhc.org
by guest on June 7, 2014jhc.sagepub.comDownloaded from

maternal line is predominantly involved in inheritance(Liehr 2006b).
It should be emphasized that a comprehensive markerchromosome characterization would be best for car-riers of sSMC; however, this is not always possibledue to lack of corresponding methods and/or sufficientprobe sample. After a complete molecular cytogeneticcharacterization, sSMC cell lines could also be avail-able for further research, e.g., depending on the modesof sSMC formation, karyotypic evolution of an sSMC,or changed expression profiles of cells due to presenceof sSMC.
We recently studied 19 of the z30 commerciallyavailable cell lines from the European Collection ofCell Cultures which, according to this company_s cata-logue, were positively karyotyped. Surprisingly, sixof nine cell lines with sSMC previously characterizedfor their chromosomal origin by others had to berevised, and no sSMC was detected in five others(Brecevic et al. 2006). Thus, there is an obvious need
for well-characterized sSMC cell lines. We present herethe beginning of a sSMC cell bank with 16 well-characterized sSMC cases.
Materials and Methods
(Molecular) Cytogenetics
Banding cytogenetics was done according to standard proce-dures (Seabright 1971). Multicolor FISH (mFISH) approachesapplied were also previously described in detail: centromere-specific mFISH (cenM-FISH) (Figure 1A) (Nietzel et al. 2001),multiplex FISH (M-FISH) using whole chromosome-paintingprobes (Speicher et al. 1996), subcentromere-specific mFISH(subcenM-FISH) (Figure 1B) (Starke et al. 2003), microdissec-tion and reverse painting (Starke et al. 2001), or multicolorbanding (MCB) (Liehr et al. 2002) was applied to differentextents in the 16 cases (see Table 1).
Analysis for Uniparental Disomy
Uniparental disomy (UPD) for the sister chromosomes of thesSMC (see Figure 2) were excluded either by microsatellite
Figure 1 (A) CenM-FISH results of the present case 05P0529: the derivative chromosome (arrowhead) is identified as a der(7). (B) SubcenM-FISH probe set for chromosome 7 revealed the presence of centromere near material 7p and 7q on the ring chromosome. Additionally,three different clones, one with a minute and two with ring chromosome shape, were identified; one ringtype was monocentric, the otherone dicentric.
TheJournal
ofHistoch
emistry&
Cytoch
emistry
652 Tonnies, Pietrzak, Bocian, MacDermont, Kuechler, Belitz, Trautmann, Schmidt, Schulze, Rodrıguez,Binkert, Yardin, Kosyakova, Volleth, Mkrtchyan, Schreyer, von Eggeling, Weise, Mrasek, Liehr
by guest on June 7, 2014jhc.sagepub.comDownloaded from
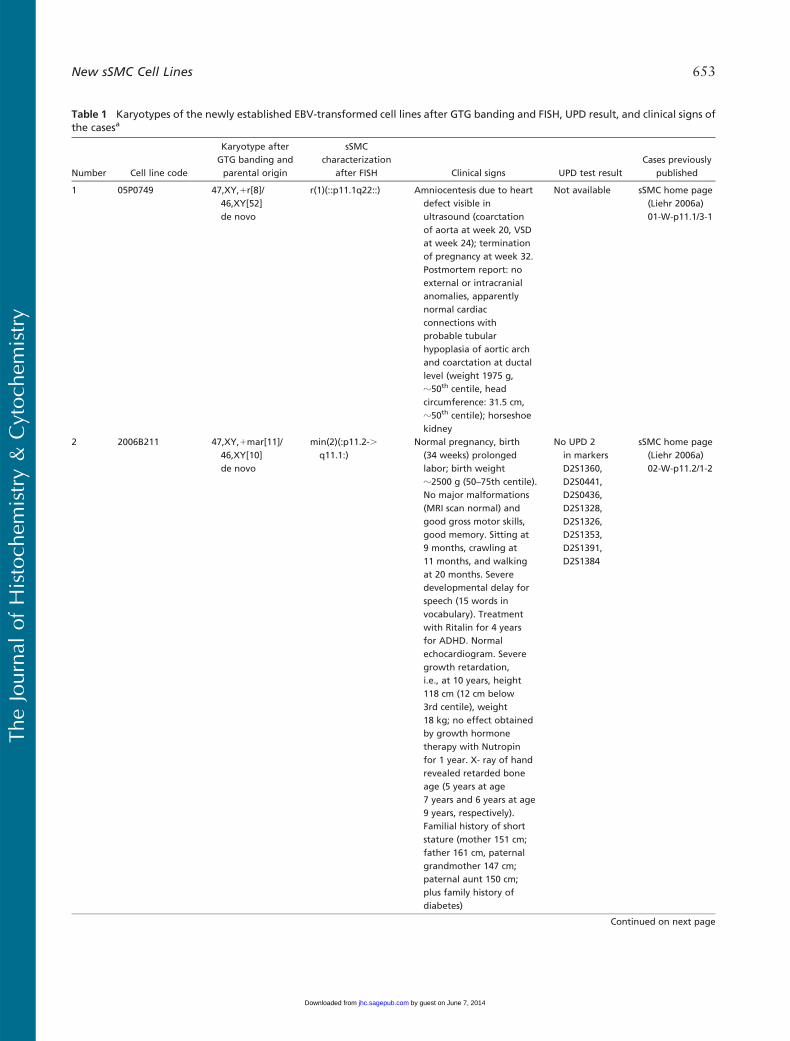
Table 1 Karyotypes of the newly established EBV-transformed cell lines after GTG banding and FISH, UPD result, and clinical signs ofthe casesa
Number Cell line code
Karyotype afterGTG banding andparental origin
sSMCcharacterization
after FISH Clinical signs UPD test resultCases previously
published
1 05P0749 47,XY,1r[8]/46,XY[52]de novo
r(1)(::p11.1q22::) Amniocentesis due to heartdefect visible inultrasound (coarctationof aorta at week 20, VSDat week 24); terminationof pregnancy at week 32.Postmortem report: noexternal or intracranialanomalies, apparentlynormal cardiacconnections withprobable tubularhypoplasia of aortic archand coarctation at ductallevel (weight 1975 g,z50th centile, headcircumference: 31.5 cm,z50th centile); horseshoekidney
Not available sSMC home page(Liehr 2006a)01-W-p11.1/3-1
2 2006B211 47,XY,1mar[11]/46,XY[10]de novo
min(2)(:p11.2-.q11.1:)
Normal pregnancy, birth(34 weeks) prolongedlabor; birth weightz2500 g (50–75th centile).No major malformations(MRI scan normal) andgood gross motor skills,good memory. Sitting at9 months, crawling at11 months, and walkingat 20 months. Severedevelopmental delay forspeech (15 words invocabulary). Treatmentwith Ritalin for 4 yearsfor ADHD. Normalechocardiogram. Severegrowth retardation,i.e., at 10 years, height118 cm (12 cm below3rd centile), weight18 kg; no effect obtainedby growth hormonetherapy with Nutropinfor 1 year. X- ray of handrevealed retarded boneage (5 years at age7 years and 6 years at age9 years, respectively).Familial history of shortstature (mother 151 cm;father 161 cm, paternalgrandmother 147 cm;paternal aunt 150 cm;plus family history ofdiabetes)
No UPD 2in markersD2S1360,D2S0441,D2S0436,D2S1328,D2S1326,D2S1353,D2S1391,D2S1384
sSMC home page(Liehr 2006a)02-W-p11.2/1-2
Continued on next page
TheJournal
ofHistoch
emistry&
Cytoch
emistry
New sSMC Cell Lines 653
by guest on June 7, 2014jhc.sagepub.comDownloaded from
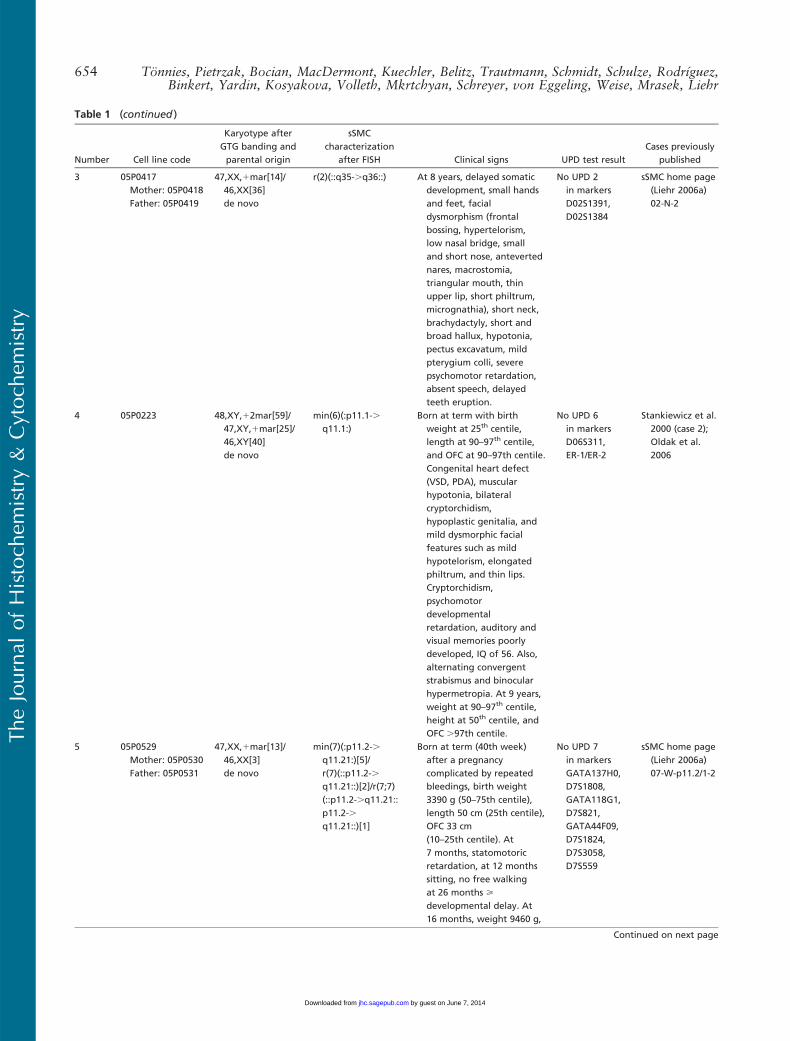
Number Cell line code
Karyotype afterGTG banding andparental origin
sSMCcharacterization
after FISH Clinical signs UPD test resultCases previously
published
3 05P0417Mother: 05P0418Father: 05P0419
47,XX,1mar[14]/46,XX[36]de novo
r(2)(::q35-.q36::) At 8 years, delayed somaticdevelopment, small handsand feet, facialdysmorphism (frontalbossing, hypertelorism,low nasal bridge, smalland short nose, antevertednares, macrostomia,triangular mouth, thinupper lip, short philtrum,micrognathia), short neck,brachydactyly, short andbroad hallux, hypotonia,pectus excavatum, mildpterygium colli, severepsychomotor retardation,absent speech, delayedteeth eruption.
No UPD 2in markersD02S1391,D02S1384
sSMC home page(Liehr 2006a)02-N-2
4 05P0223 48,XY,12mar[59]/47,XY,1mar[25]/46,XY[40]de novo
min(6)(:p11.1-.q11.1:)
Born at term with birthweight at 25th centile,length at 90–97th centile,and OFC at 90–97th centile.Congenital heart defect(VSD, PDA), muscularhypotonia, bilateralcryptorchidism,hypoplastic genitalia, andmild dysmorphic facialfeatures such as mildhypotelorism, elongatedphiltrum, and thin lips.Cryptorchidism,psychomotordevelopmentalretardation, auditory andvisual memories poorlydeveloped, IQ of 56. Also,alternating convergentstrabismus and binocularhypermetropia. At 9 years,weight at 90–97th centile,height at 50th centile, andOFC .97th centile.
No UPD 6in markersD06S311,ER-1/ER-2
Stankiewicz et al.2000 (case 2);Oldak et al.2006
5 05P0529Mother: 05P0530Father: 05P0531
47,XX,1mar[13]/46,XX[3]de novo
min(7)(:p11.2-.q11.21:)[5]/r(7)(::p11.2-.q11.21::)[2]/r(7;7)(::p11.2-.q11.21::p11.2-.q11.21::)[1]
Born at term (40th week)after a pregnancycomplicated by repeatedbleedings, birth weight3390 g (50–75th centile),length 50 cm (25th centile),OFC 33 cm(10–25th centile). At7 months, statomotoricretardation, at 12 monthssitting, no free walkingat 26 months >developmental delay. At16 months, weight 9460 g,
No UPD 7in markersGATA137H0,D7S1808,GATA118G1,D7S821,GATA44F09,D7S1824,D7S3058,D7S559
sSMC home page(Liehr 2006a)07-W-p11.2/1-2
Continued on next page
Table 1 (continued )
TheJournal
ofHistoch
emistry&
Cytoch
emistry
654 Tonnies, Pietrzak, Bocian, MacDermont, Kuechler, Belitz, Trautmann, Schmidt, Schulze, Rodrıguez,Binkert, Yardin, Kosyakova, Volleth, Mkrtchyan, Schreyer, von Eggeling, Weise, Mrasek, Liehr
by guest on June 7, 2014jhc.sagepub.comDownloaded from
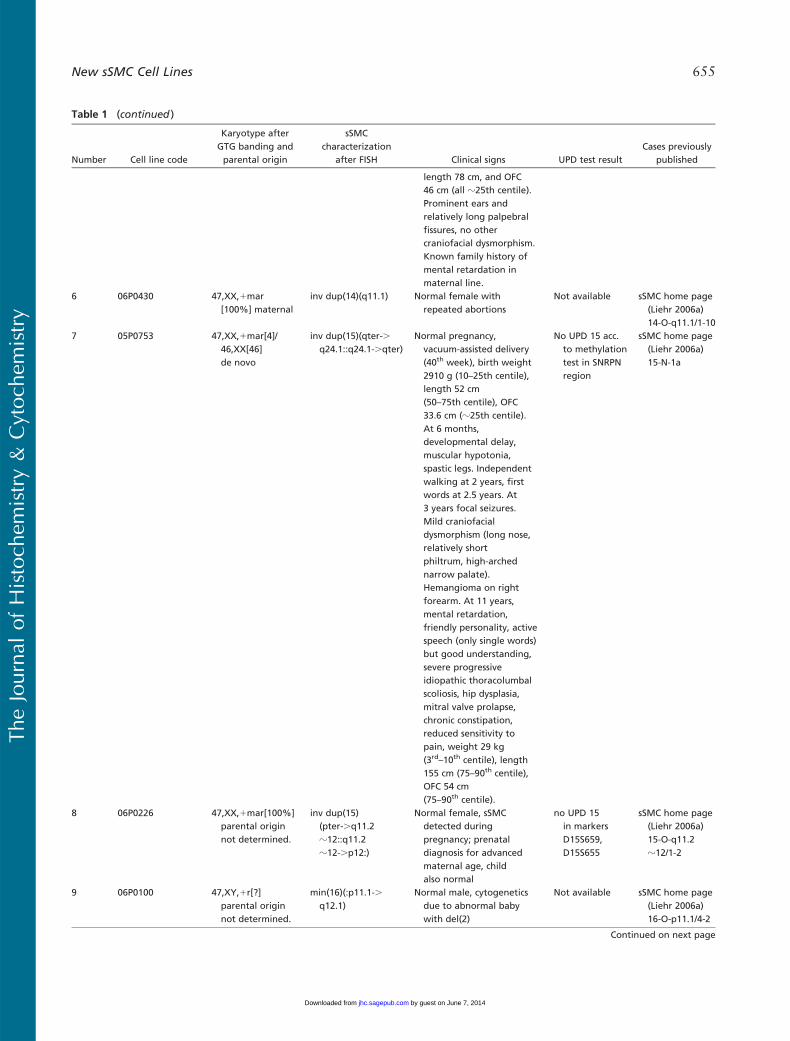
Number Cell line code
Karyotype afterGTG banding andparental origin
sSMCcharacterization
after FISH Clinical signs UPD test resultCases previously
published
length 78 cm, and OFC46 cm (all z25th centile).Prominent ears andrelatively long palpebralfissures, no othercraniofacial dysmorphism.Known family history ofmental retardation inmaternal line.
6 06P0430 47,XX,1mar[100%] maternal
inv dup(14)(q11.1) Normal female withrepeated abortions
Not available sSMC home page(Liehr 2006a)14-O-q11.1/1-10
7 05P0753 47,XX,1mar[4]/46,XX[46]de novo
inv dup(15)(qter-.q24.1::q24.1-.qter)
Normal pregnancy,vacuum-assisted delivery(40th week), birth weight2910 g (10–25th centile),length 52 cm(50–75th centile), OFC33.6 cm (z25th centile).At 6 months,developmental delay,muscular hypotonia,spastic legs. Independentwalking at 2 years, firstwords at 2.5 years. At3 years focal seizures.Mild craniofacialdysmorphism (long nose,relatively shortphiltrum, high-archednarrow palate).Hemangioma on rightforearm. At 11 years,mental retardation,friendly personality, activespeech (only single words)but good understanding,severe progressiveidiopathic thoracolumbalscoliosis, hip dysplasia,mitral valve prolapse,chronic constipation,reduced sensitivity topain, weight 29 kg(3rd–10th centile), length155 cm (75–90th centile),OFC 54 cm(75–90th centile).
No UPD 15 acc.to methylationtest in SNRPNregion
sSMC home page(Liehr 2006a)15-N-1a
8 06P0226 47,XX,1mar[100%]parental originnot determined.
inv dup(15)(pter-.q11.2z12::q11.2z12-.p12:)
Normal female, sSMCdetected duringpregnancy; prenataldiagnosis for advancedmaternal age, childalso normal
no UPD 15in markersD15S659,D15S655
sSMC home page(Liehr 2006a)15-O-q11.2z12/1-2
9 06P0100 47,XY,1r[?]parental originnot determined.
min(16)(:p11.1-.q12.1)
Normal male, cytogeneticsdue to abnormal babywith del(2)
Not available sSMC home page(Liehr 2006a)16-O-p11.1/4-2
Continued on next page
Table 1 (continued )
TheJournal
ofHistoch
emistry&
Cytoch
emistry
New sSMC Cell Lines 655
by guest on June 7, 2014jhc.sagepub.comDownloaded from
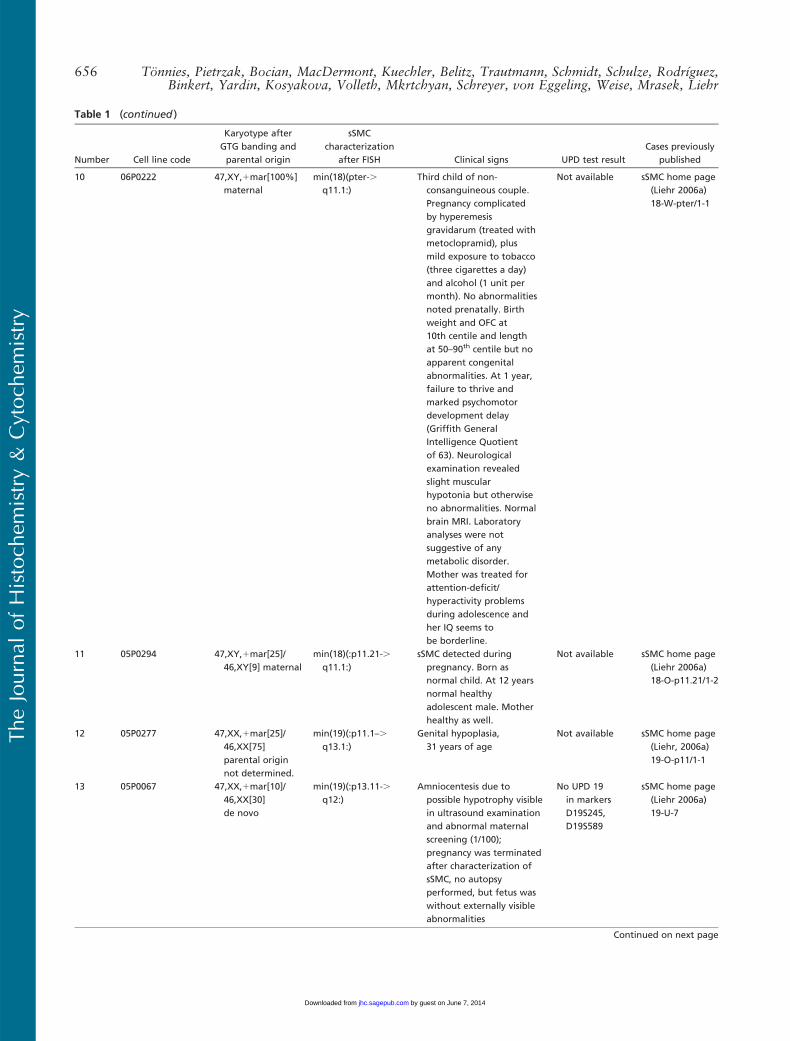
Number Cell line code
Karyotype afterGTG banding andparental origin
sSMCcharacterization
after FISH Clinical signs UPD test resultCases previously
published
10 06P0222 47,XY,1mar[100%]maternal
min(18)(pter-.q11.1:)
Third child of non-consanguineous couple.Pregnancy complicatedby hyperemesisgravidarum (treated withmetoclopramid), plusmild exposure to tobacco(three cigarettes a day)and alcohol (1 unit permonth). No abnormalitiesnoted prenatally. Birthweight and OFC at10th centile and lengthat 50–90th centile but noapparent congenitalabnormalities. At 1 year,failure to thrive andmarked psychomotordevelopment delay(Griffith GeneralIntelligence Quotientof 63). Neurologicalexamination revealedslight muscularhypotonia but otherwiseno abnormalities. Normalbrain MRI. Laboratoryanalyses were notsuggestive of anymetabolic disorder.Mother was treated forattention-deficit/hyperactivity problemsduring adolescence andher IQ seems tobe borderline.
Not available sSMC home page(Liehr 2006a)18-W-pter/1-1
11 05P0294 47,XY,1mar[25]/46,XY[9] maternal
min(18)(:p11.21-.q11.1:)
sSMC detected duringpregnancy. Born asnormal child. At 12 yearsnormal healthyadolescent male. Motherhealthy as well.
Not available sSMC home page(Liehr 2006a)18-O-p11.21/1-2
12 05P0277 47,XX,1mar[25]/46,XX[75]parental originnot determined.
min(19)(:p11.1–.q13.1:)
Genital hypoplasia,31 years of age
Not available sSMC home page(Liehr, 2006a)19-O-p11/1-1
13 05P0067 47,XX,1mar[10]/46,XX[30]de novo
min(19)(:p13.11-.q12:)
Amniocentesis due topossible hypotrophy visiblein ultrasound examinationand abnormal maternalscreening (1/100);pregnancy was terminatedafter characterization ofsSMC, no autopsyperformed, but fetus waswithout externally visibleabnormalities
No UPD 19in markersD19S245,D19S589
sSMC home page(Liehr 2006a)19-U-7
Continued on next page
Table 1 (continued )
TheJournal
ofHistoch
emistry&
Cytoch
emistry
656 Tonnies, Pietrzak, Bocian, MacDermont, Kuechler, Belitz, Trautmann, Schmidt, Schulze, Rodrıguez,Binkert, Yardin, Kosyakova, Volleth, Mkrtchyan, Schreyer, von Eggeling, Weise, Mrasek, Liehr
by guest on June 7, 2014jhc.sagepub.comDownloaded from
analysis (Starke et al. 2003) or by methylation test in SNRPNregion (Nietzel et al. 2003). Informative microsatellitemarkers are listed in Table 1.
Epstein–Barr Virus Transformation of PeripheralBlood B-lymphocytes
Epstein–Barr virus (EBV) was done as previously describedusing 2–10 ml of heparinized peripheral blood (Pattengaleet al. 1973; Neitzel 1986; Xie et al. 2005). This study wasapproved by the Ethical Commission of the Friedrich SchillerUniversity, Jena, Germany (internal code 1457-12/04).
Results
To date we have established for our sSMC cell bank16 new sSMC cell lines (see Table 1) by EBV transfor-mation of peripheral blood B-lymphocytes. sSMC werecharacterized by molecular cytogenetics for their chro-mosomal origin and genetic content (Table 1). Anexample for the applied FISH methods is given for case05P0529 in Figure 3.
Cell lines presented here contain sSMC derived fromchromosomes 1 (05P0749), 2 (2006B211, 05P0417),4 (05P0163), 6 (05P0223), 7 (05P0529), 8 (05P0208),14 (06P0430), 15 (05P0753, 06P0226), 16 (06P0100), 18(06P0222, 05P0294, 05P0163), 19 (05P0277, 05P0067),21 (05P0208), and 22 (05P0209). Two neocentric sSMCcell lines derived from 2q35-q36 and 15q24.1-qter, re-spectively, were established (05P0417, 05P0753). Intwo further cases, two sSMC of different chromosomalorigin each were present (05P0163, 05P0208).
Parental DNA was available for UPD analysis in9/16 cases, and a UPD was excluded in all (2006B211,05P0417, 05P0223, 05P0529, 05P0753, 06P0226,05P0067, 05P0163, 05P0208; Table 1).
Discussion
As mentioned above, there is a lack of sSMC cell lines;thus, 16 new EBV-transformed sSMC cell lines are pre-sented here (see Table 1). EBV transformation was chosenfor immortalization because it is a well-established and,
Number Cell line code
Karyotype afterGTG banding andparental origin
sSMCcharacterization
after FISH Clinical signs UPD test resultCases previously
published
14 05P0209 47,XX,1mar[100%]de novo
inv dup(22)(q11.21) At 2 years, motorretardation anddysmorphic featurestypical for cat eyesyndrome
Not available sSMC home page(Liehr 2006a)22-Wces-5-93
15 05P0163 48,XX,1mar1,1mar2[101]/47,XX,1mar1[38]/47,XX,1mar2[29]/46,XX[5]de novo
min(4)(:p11.1-.q12:); r(18)::(p11.21-.q11.1::p11.21-.q11.1::)[2]/inv dup(18)(:p11.1-.q11.1::q11.1-.p11.1:)[4]/min(18)(:p11.21-.q11.1::q11.1-.p11.1:)[4]
At 3 years, moderatepsychomotor retardation,short stature withshortening of upper andlower limbs, taperingfingers and facialdysmorphism (flat face,telecanthus, broad nose,prominent forehead)
No UPD 4 and18 in markersD4S2366,D4S1627,D4S2367,D4S2361,D18S858
sSMC home page(Liehr 2006a)mult 2-18
16 05P0208 47,XY,1mar1[37]/48,XY,1mar1,1mar2[52]/49,XY,1mar2x2[7]/46,XY[4] de novo
r(8)(:p12-.q11.1::q11.1-.p21.1:),min(8)(:p11.22-.q11.21::q11.21-.p21.1::p21.1-.p11.22:) [70%],min(21)(:p11.1-.q21.3:) [z30%]
Intrauterine and postnatalgrowth retardation, at6 years microcephaly,brachycephaly, hypotonia,psychomotor retardation,partial agenesis of corpuscallosum, abnormaldermatoglyphic patternsand facial dysmorphism(upslanting palpebralfissures, antevertednares, relatively largeears, prominent lower lip,long philtrum)
No UPD 21in markersD21S1432 Noinformativemarker forchromosome 8
sSMC home page(Liehr 2006a)mult 2-21
aPrevious references of the cases are listed together with the cell line codes.EBV, Epstein–Barr virus; sSMC, small supernumerary marker chromosome; FISH, fluorescence in situ hybridization; UPD, uniparental disomy; VSD, ventral sepaldefect; PDA, patent ductus arteriosus; ADHD, attencion-deficit hyperactivity disorder; OFC, occipitofrontal circumference.
Table 1 (continued )
TheJournal
ofHistoch
emistry&
Cytoch
emistry
New sSMC Cell Lines 657
by guest on June 7, 2014jhc.sagepub.comDownloaded from
in our hands, easily performed approach. It has beentaken into account that the mechanism of EBV transfor-mation is still not completely understood (Yamashitaet al. 2006), and problems were also reported concern-ing chromosomal (Okubo et al. 2001), genetic, andproteomic effects of the procedure (Toda et al. 2000).However, for the 16 reported immortalized cell lines,future studies will now be possible without the needfor taking further blood samples. However, for case05P0749, this would not be possible due to terminationof pregnancy.
For all 16 cell lines, clinical data could be collected,and the mosaic state of the sSMC could be determined,as well as the chromosomal origin and genetic contentpresent on the sSMC (see Table 1). Six sSMC carrierswere clinically normal, and five of those six had a eu-chromatic imbalance due to sSMC presence. Thus, those
cases could not only provide the genotype–phenotypecorrelation of sSMC (see below and Figure 3) but arealso important for future studies on the question of ifand how these genetically relevant regions are silenced,e.g., by heterochromatization.
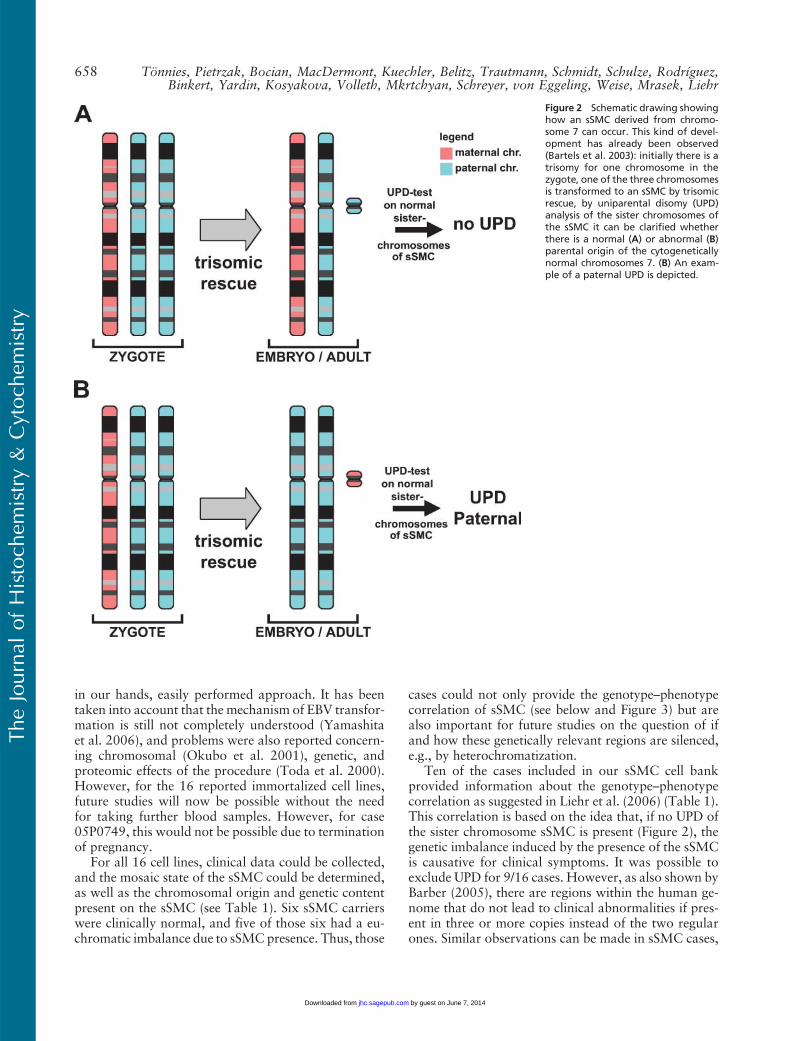
Ten of the cases included in our sSMC cell bankprovided information about the genotype–phenotypecorrelation as suggested in Liehr et al. (2006) (Table 1).This correlation is based on the idea that, if no UPD ofthe sister chromosome sSMC is present (Figure 2), thegenetic imbalance induced by the presence of the sSMCis causative for clinical symptoms. It was possible toexclude UPD for 9/16 cases. However, as also shown byBarber (2005), there are regions within the human ge-nome that do not lead to clinical abnormalities if pres-ent in three or more copies instead of the two regularones. Similar observations can be made in sSMC cases,
Figure 2 Schematic drawing showinghow an sSMC derived from chromo-some 7 can occur. This kind of devel-opment has already been observed(Bartels et al. 2003): initially there is atrisomy for one chromosome in thezygote, one of the three chromosomesis transformed to an sSMC by trisomicrescue, by uniparental disomy (UPD)analysis of the sister chromosomes ofthe sSMC it can be clarified whetherthere is a normal (A) or abnormal (B)parental origin of the cytogeneticallynormal chromosomes 7. (B) An exam-ple of a paternal UPD is depicted.
TheJournal
ofHistoch
emistry&
Cytoch
emistry
658 Tonnies, Pietrzak, Bocian, MacDermont, Kuechler, Belitz, Trautmann, Schmidt, Schulze, Rodrıguez,Binkert, Yardin, Kosyakova, Volleth, Mkrtchyan, Schreyer, von Eggeling, Weise, Mrasek, Liehr
by guest on June 7, 2014jhc.sagepub.comDownloaded from
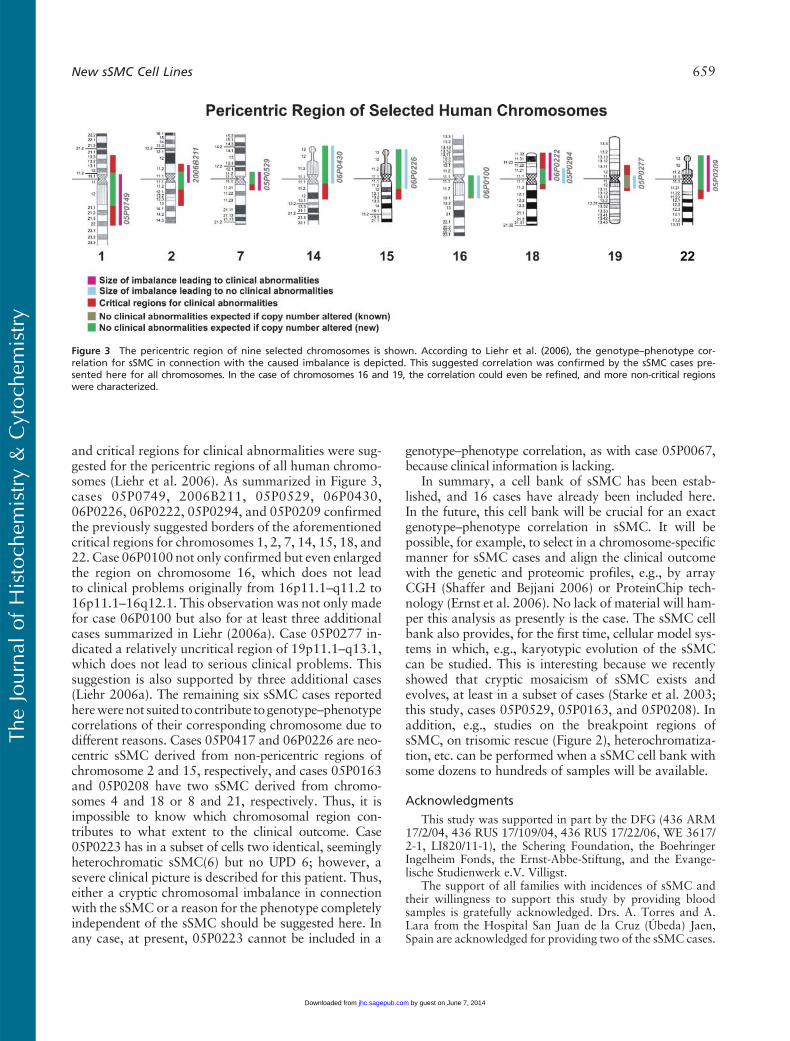
and critical regions for clinical abnormalities were sug-gested for the pericentric regions of all human chromo-somes (Liehr et al. 2006). As summarized in Figure 3,cases 05P0749, 2006B211, 05P0529, 06P0430,06P0226, 06P0222, 05P0294, and 05P0209 confirmedthe previously suggested borders of the aforementionedcritical regions for chromosomes 1, 2, 7, 14, 15, 18, and22. Case 06P0100 not only confirmed but even enlargedthe region on chromosome 16, which does not leadto clinical problems originally from 16p11.1–q11.2 to16p11.1–16q12.1. This observation was not only madefor case 06P0100 but also for at least three additionalcases summarized in Liehr (2006a). Case 05P0277 in-dicated a relatively uncritical region of 19p11.1–q13.1,which does not lead to serious clinical problems. Thissuggestion is also supported by three additional cases(Liehr 2006a). The remaining six sSMC cases reportedherewerenot suited to contribute togenotype–phenotypecorrelations of their corresponding chromosome due todifferent reasons. Cases 05P0417 and 06P0226 are neo-centric sSMC derived from non-pericentric regions ofchromosome 2 and 15, respectively, and cases 05P0163and 05P0208 have two sSMC derived from chromo-somes 4 and 18 or 8 and 21, respectively. Thus, it isimpossible to know which chromosomal region con-tributes to what extent to the clinical outcome. Case05P0223 has in a subset of cells two identical, seeminglyheterochromatic sSMC(6) but no UPD 6; however, asevere clinical picture is described for this patient. Thus,either a cryptic chromosomal imbalance in connectionwith the sSMC or a reason for the phenotype completelyindependent of the sSMC should be suggested here. Inany case, at present, 05P0223 cannot be included in a
genotype–phenotype correlation, as with case 05P0067,because clinical information is lacking.
In summary, a cell bank of sSMC has been estab-lished, and 16 cases have already been included here.In the future, this cell bank will be crucial for an exactgenotype–phenotype correlation in sSMC. It will bepossible, for example, to select in a chromosome-specificmanner for sSMC cases and align the clinical outcomewith the genetic and proteomic profiles, e.g., by arrayCGH (Shaffer and Bejjani 2006) or ProteinChip tech-nology (Ernst et al. 2006). No lack of material will ham-per this analysis as presently is the case. The sSMC cellbank also provides, for the first time, cellular model sys-tems in which, e.g., karyotypic evolution of the sSMCcan be studied. This is interesting because we recentlyshowed that cryptic mosaicism of sSMC exists andevolves, at least in a subset of cases (Starke et al. 2003;this study, cases 05P0529, 05P0163, and 05P0208). Inaddition, e.g., studies on the breakpoint regions ofsSMC, on trisomic rescue (Figure 2), heterochromatiza-tion, etc. can be performed when a sSMC cell bank withsome dozens to hundreds of samples will be available.
Acknowledgments
This study was supported in part by the DFG (436 ARM17/2/04, 436 RUS 17/109/04, 436 RUS 17/22/06, WE 3617/2-1, LI820/11-1), the Schering Foundation, the BoehringerIngelheim Fonds, the Ernst-Abbe-Stiftung, and the Evange-lische Studienwerk e.V. Villigst.
The support of all families with incidences of sSMC andtheir willingness to support this study by providing bloodsamples is gratefully acknowledged. Drs. A. Torres and A.Lara from the Hospital San Juan de la Cruz (Ubeda) Jaen,Spain are acknowledged for providing two of the sSMC cases.
Figure 3 The pericentric region of nine selected chromosomes is shown. According to Liehr et al. (2006), the genotype–phenotype cor-relation for sSMC in connection with the caused imbalance is depicted. This suggested correlation was confirmed by the sSMC cases pre-sented here for all chromosomes. In the case of chromosomes 16 and 19, the correlation could even be refined, and more non-critical regionswere characterized.
TheJournal
ofHistoch
emistry&
Cytoch
emistry
New sSMC Cell Lines 659
by guest on June 7, 2014jhc.sagepub.comDownloaded from

Literature Cited
Barber JC (2005) Directly transmitted unbalanced chromosome ab-normalities and euchromatic variants. J Med Genet 42:609–629
Bartels I, Schlueter G, Liehr T, von Eggeling F, Starke H, Glaubitz R,Burfeind P (2003) Supernumerary small marker chromosome(SMC) and uniparental disomy 22 in a child with confined placen-tal mosaicism of trisomy 22: trisomy rescue due to marker chro-mosome formation. Cytogenet Genome Res 101:103–105
Brecevic L, Michel S, Starke H, Muller K, Kosyakova N, Mrasek K,Weise A, et al. (2006) Multicolor FISH used for the characteri-zation of small supernumerary marker chromosomes (sSMC) incommercially available immortalized cell lines. Cytogenet GenomeRes 114:319–324
Crolla JA (1998) FISH and molecular studies of autosomal super-numerary marker chromosomes excluding those derived fromchromosome 15. II. Review of the literature. Am J Med Genet 75:367–381
Ernst G, Melle C, Schimmel B, Bleul A, von Eggeling F (2006) Proteo-histography—direct analysis of tissue with high sensitivity andhigh spatial resolution using ProteinChip technology. J HistochemCytochem 54:13–17
Liehr T (2006a) sSMC homepage. http://markerchromosomes.ag.vu orLiehr T (2006b) Familial small supernumerary marker chromosomes
are predominantly inherited via the maternal line. Genet Med 8:459–462
Liehr T, Claussen U, Starke H (2004) Small supernumerary markerchromosomes (sSMC) in humans. Cytogenet Genome Res 107:55–67
Liehr T, Heller A, Starke H, Rubtsov N, Trifonov V,Mrasek K,WeiseA, et al. (2002) Microdissection based high resolution multicolorbanding for all 24 human chromosomes. Int J Mol Med 9:335–339
Liehr T, Mrasek K, Weise A, Dufke A, Rodriguez L, MartinezGuardia N, Sanchis A, et al. (2006) Small supernumerary markerchromosomes—progress towards a genotype-phenotype correla-tion. Cytogenet Genome Res 112:23–34
Neitzel H (1986) A routine method for the establishment of perma-nent growing lymphoblastoid cell lines. Hum Genet 73:320–326
Nietzel A, Albrecht B, Starke H, Heller A, Gillessen-Kaesbach G,Claussen U, Liehr T (2003) Partial hexasomy 15pterY15q13 in-cluding SNRPN and D15S10: first molecular cytogenetically provencase report. J Med Genet 40:e28
Nietzel A, Rocchi M, Starke H, Heller A, Fiedler W, Wlodarska I,Loncarevic IF, et al. (2001) A new multicolor-FISH approach forthe characterization of marker chromosomes: centromere-specificmulticolor-FISH (cenM-FISH). Hum Genet 108:199–204
Okubo M, Tsurukubo Y, Higaki T, Kawabe T, Goto M, Murase T,Ide T, et al. (2001) Clonal chromosomal aberrations accompa-nied by strong telomerase activity in immortalization of human
B-lymphoblastoid cell lines transformed by Epstein-Barr virus.Cancer Genet Cytogenet 129:30–34
Oldak M, Waligora J, Gieruszczak-Bialek D, Skorka A, Bocian E,Brycz-Witkowska J, Stankiewicz P, et al. (2006) Congenitalanomalies and developmental delay in a boy with double chromo-some 6 derived supernumerary marker. Genet Counsel 19:27–32
Paoloni-Giacobino A, Morris MA, Dahoun SP (1998) Prenatal super-numerary r(16) chromosome characterized by multiprobe FISHwith normal pregnancy outcome. Prenat Diagn 18:751–752
Pattengale PK, Smith RW, Gerber P (1973) Selective transformationof B lymphocytes by E.B. virus. Lancet 2:93–94
Seabright M (1971) A rapid banding technique for human chromo-somes. Lancet 2:971–972
Shaffer LG, Bejjani BA (2006) Medical applications of array CGHand the transformation of clinical cytogenetics. Cytogenet Ge-nome Res 115:303–309
Speicher MR, Gwyn Ballard S, Ward DC (1996) Karyotyping humanchromosomes by combinatorial multi-fluor FISH. Nat Genet12:368–375
Stankiewicz P, Bocian E, Jakubow-Durska K, Obersztyn E, Lato E,Starke H, Mroczek K, et al. (2000) Identification of supernumer-ary marker chromosomes derived from chromosomes 5, 6, 19, and20 using FISH. J Med Genet 37:114–120
Starke H, Nietzel A, Weise A, Heller A, Mrasek K, Belitz B, KelbovaC, et al. (2003) Small supernumerary marker chromosomes(SMCs): genotype-phenotype correlation and classification. HumGenet 114:51–67
Starke H, RaidaM, Trifonov V, Clement JH, Loncarevic IF, Heller A,Bleck C, et al. (2001) Molecular cytogenetic characterization of anacquired minute supernumerary marker chromosome as the soleabnormality in a case clinically diagnosed as atypical Philadelphia-negative chronic myelogenous leukaemia. Br J Haematol 113:435–438
Toda T, Sugimoto M, Omori A, Matsuzaki T, Furuichi Y, Kimura N(2000) Proteomic analysis of Epstein-Barr virus-transformedhuman B-lymphoblastoid cell lines before and after immortaliza-tion. Electrophoresis 21:1814–1822
Warburton D (1991) De novo balanced chromosome rearrange-ments and extra marker chromosomes identified at prenatal diag-nosis: clinical significance and distribution of breakpoints. Am JHum Genet 49:995–1013
Xie J, Techritz S, Haebel S, Horn A, Neitzel H, Klose J, Schuelke M(2005) A two-dimensional electrophoretic map of human mito-chondrial proteins from immortalized lymphoblastoid cell lines: aprerequisite to study mitochondrial disorders in patients. Proteo-mics 5:2981–2999
Yamashita Y, Tsurumi T, Mori N, Kiyono T (2006) Immortalizationof Epstein-Barr virus-negative human B lymphocytes with minimalchromosomal instability. Pathol Int 56:659–667
TheJournal
ofHistoch
emistry&
Cytoch
emistry
660 Tonnies, Pietrzak, Bocian, MacDermont, Kuechler, Belitz, Trautmann, Schmidt, Schulze, Rodrıguez,Binkert, Yardin, Kosyakova, Volleth, Mkrtchyan, Schreyer, von Eggeling, Weise, Mrasek, Liehr
by guest on June 7, 2014jhc.sagepub.comDownloaded from