neuro-protective effects of growth hormone (gh) after hypoxia–ischemia injury in embryonic chicken...
TRANSCRIPT

General and Comparative Endocrinology 183 (2013) 17–31
Contents lists available at SciVerse ScienceDirect
General and Comparative Endocrinology
journal homepage: www.elsevier .com/locate /ygcen
Neuro-protective effects of growth hormone (GH) after hypoxia–ischemia injuryin embryonic chicken cerebellum
Clara Alba-Betancourt a, José Luis Luna-Acosta a, Candy Elizabeth Ramírez-Martínez a,Daniela Ávila-González a, Estefany Granados-Ávalos a, Martha Carranza a, Hilda Martínez-Coria b,Carlos Arámburo a, Maricela Luna a,⇑a Departamento de Neurobiología Celular y Molecular, Instituto de Neurobiología, Universidad Nacional Autónoma de México, Campus Juriquilla, Querétaro, Qro. 76230, Mexicob Department of Neurobiology and Behavior, University of California, Irvine, CA 92697-4545, USA
a r t i c l e i n f o
Article history:Received 19 August 2012Revised 27 November 2012Accepted 2 December 2012Available online 19 December 2012
Keywords:Growth hormone (GH)NeuroprotectionHypoxia and low glucose (HLG)Anti-apoptotic effectsPI3K/AktWortmannin
0016-6480/$ - see front matter � 2012 Elsevier Inc. Ahttp://dx.doi.org/10.1016/j.ygcen.2012.12.004
⇑ Corresponding author. Address: Instituto de NeurUniversidad Nacional Autónoma de México, Queréta+52 (442)238 1005.
E-mail address: [email protected] (M. Lun
a b s t r a c t
Neuroprotection is a mechanism within the central nervous system (CNS) that protects neurons fromdamage as a result of a severe insult. It is known that growth hormone (GH) is involved in cell survivaland may inhibit apoptosis in several cell types, including those of the CNS. Both GH and GH-receptor(GHR) genes are expressed in the cerebellum. Thus, we investigated the possible neuroprotective roleof GH in this organ, which is very sensitive to hypoxic/ischemic conditions. Endogenous GH levelsincreased in the brain and cerebellum (30% and 74%, respectively) of 15-day-old chicken embryosexposed to hypoxia during 24 h compared to normoxia. In primary embryonic cerebellar neuron culturestreated under hypoxia (0.5% O2) and low glucose (1 g/L) conditions (HLG) for 1 h, GH levels increased1.16-fold compared to the control. The addition of 1 nM recombinant chicken GH (rcGH) to cultures dur-ing HLG increased cell viability (1.7-fold) and the expression of Bcl-2 (1.67-fold); in contrast the caspase-3 activity and the proportion of apoptotic cells decreased (37% and 54.2%, respectively) compared to HLG.rcGH activated the PI3K/Akt pathway both under normoxic and HLG conditions, increasing the propor-tion of phosphorylated Akt (1.7- and 1.4-fold, respectively). These effects were abolished by wortmanninand by immunoneutralization, indicating that GH acts through this signaling pathway. Furthermore, the15-kDa GH variant (10 nM) significantly increased cell viability and decreased caspase-3 activity duringHLG condition. Thus GH may act as a paracrine/autocrine neuroprotective factor that preserves cellularviability and inhibits apoptotic cell death.
� 2012 Elsevier Inc. All rights reserved.
1. Introduction
Recent studies show that GH gene expression is not confined tothe pituitary gland, as it also occurs in neural [1,36,37,65], immune[54,66], reproductive [38,52,53], integumentary, muscular, skele-tal, and cardiovascular systems [63], where it appears to act as alocal growth or differentiation factor in the autocrine and/or para-crine regulation of cellular differentiation and proliferation[34,35,39,68]. Recent reports also show that GH might have someeffects on the central nervous system (CNS), since both GH andits receptor (GHR) are expressed and have been located in the brainof several species including humans [48,62], rats [13,50], turkeyand dove [64], chicken embryo [37], and chickens of different ages[1] as well as in different cell types (such as neurons, astrocytes,oligodendrocytes, microglia) of the CNS in rat [50]. This hormone
ll rights reserved.
obiología, Campus Juriquilla,ro, Qro. 76230, Mexico. Fax:
a).
acts not only on growth and development of the brain but it alsohas a role as a neuroprotective factor against a brain insult [74].The CNS is very sensitive to injurious influences such as the lackof nutrients and oxygen (commonly known as ischemia), and com-plex cerebral dysfunctions may result as a consequence of this in-sult [85]. Strokes can affect any part of the brain and occur due tothe interruption of blood flow to the affected area [23]. Ischemicstroke is the second most common cause of death in the worldand the leading cause of acquired disability in adults [67]. Mortal-ity from stroke is estimated to be 25%, which makes it the thirdhighest cause of death in industrialized countries [58]. Moreover,there is a high incidence of neonatal hypoxia, which represents amajor risk for disability manifested mainly by cognitive, behav-ioral, attentional, and socialization deficits as well as motor impair-ment [86].
In ischemia, there are at least three mechanisms that lead tocell death: excitotoxicity and ionic imbalance; oxidative andnitrosative stress; and apoptosis. These mechanisms are interre-lated; they show a similar temporal development and affect

18 C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31
neurons, glial, and vascular cells [51]. Delayed neuronal death oc-curs after ischemia, and the severity and duration of the ischemicinsult are of critical importance for the extent of brain damage[10,80]. Although there is still controversy about the mechanismof cell death after ischemia, apoptosis and necrosis could occur asa result of this insult, depending on its severity [9]. Sugawara andcolleagues [81] have demonstrated that both the intrinsic andextrinsic pathways of apoptosis are involved during the ischemicinjury process. Apoptosis, which has been reported as a feature ofpost-hypoxic brain damage in various experimental models [8], in-volves the systematic activation of a group of cysteine proteases,termed caspases [26], as well as a number of other componentssuch as the apoptosis-inducing factor (AIF) and Bcl-2 family pro-teins [55,57,82].
In order to reduce cell death resulting from an insult, the CNShas endogenous neuroprotective strategies such as the produc-tion of neurotrophic factors [82]. A number of growth factors(insulin-like growth factor-I, IGF-I; fibroblast growth factor,FGF; neural growth factor, NGF) and hormones (GH, prolactin,progesterone) are induced in the area of brain damage after hy-poxia; they have been shown to regulate cell death and havebeen implicated in the process of neural rescue after damage[30,44,90]. There is growing evidence that GH, like other neuro-trophins, might influence the functions of the CNS as a neuropro-tective factor in cerebral tissue after hypoxia–ischemia (HI)damage [78]. Previous studies showed that GH mRNA and pro-tein expression were induced in the rat hippocampus by chronicor short-term hypoxia exposure, although these increases wereclearly more prominent and sustained after chronic hypoxia[49]. Early reports demonstrated widespread expression of GHmRNA and GH protein within the normal rodent brain and theirup-regulation following brain injury, particularly in the hippo-campus and hypothalamus [22,74,89]. The protective effect ofGH appears to be at least partly mediated via a specific increasein post-injury neurogenesis [76]. In the infant brain, GH has beenreported to inhibit apoptosis following hypoxic–ischemic injury[78]. Moreover, there is evidence that GH activates the PI3k/Aktpathway, which is known to have anti-apoptotic effects due tothe phosphorylation of Akt and subsequent inactivation of cas-pase-9 and Bad [43]. GH has also been reported to have auto-crine/paracrine actions that regulate cell death occurring duringdevelopment in the retinal ganglion cells (RCGs) by exertinganti-apoptotic effects [69,70] that are mediated by Akt phosphor-ylation and by caspase-dependent and independent signalingpathways [39,71]. The balance of the pro- and anti-apoptoticBcl-2/Bax family proteins seems critical for the development ofneonatal hypoxia-induced brain injury [28,33]. Likewise, recentstudies link GH, IGF-I, and ghrelin with neuroprotective actionsdue to reduced expression of Bax and Bad [24,16,90], which areinvolved in the control of apoptosis.
Recently, we have shown that both the GH mRNA and the pro-tein it encodes are expressed in the chicken cerebellum, particu-larly in the Purkinje cells [1]. The hormone showed molecularheterogeneity and the most abundant isoform corresponded to a15 kDa GH variant. Also, the growth hormone receptor (GH) wasfound to co-localize in the same cells [1], suggesting that the lo-cally expressed GH may play a role through paracrine/autocrinemechanisms.
In this work, we employed chicken embryos (at 15ED and 18EDdevelopmental stages), and embryonic cerebellar cell cultures asmodels to investigate the possible anti-apoptotic actions of GH,and a 15-kDa variant of GH, in response to an ischemic insult, suchas the application of hypoxia and low glucose (HLG) conditions.Moreover, we examined if the PI3K/Akt signaling pathway andthe expression of Bcl-2 protein are involved in the anti-apoptoticeffects of GH in cerebellar neurons.
2. Materials and methods
2.1. Animals
Pathogen-free, fertilized eggs (Gallus gallus, White Leghorn)were obtained from Alpes (Tehuacán, México) and were incubatedat 38 �C in a humidified air chamber (IAMEX, Mexico). The eggswere rotated one-quarter of a revolution every 50 min duringincubation.
2.2. In vivo treatments
Fertilized chicken eggs were incubated under normal condi-tions. Aerogenic hypoxia was induced by wrapping half the egg-shell with a polyvinyl film to decrease air exchange and induce avery low oxygen exchange. Controls were maintained with a nor-mal air exchange (21% oxygen) [14]. The following experimentalgroups were established, each with its corresponding normoxiccontrol group: (a) embryos at embryonic day of development(ED) 15 and ED18, which were exposed to 24 h of hypoxia followedby 24 h of re-oxygenation and then sacrificed immediately (groupsA and B, HLG-24 h); (b) embryos of ED15 and ED18, which were ex-posed to 24 h of hypoxia, followed by re-oxygenation and sacrificeuntil one day after hatching (groups C and D, HLG-1ph). The em-bryos or chickens were weighed and then sacrificed by cervical dis-location, and the whole brain or cerebellum were obtained,immediately frozen, and kept in the ultra freezer at �70 �C untiluse.
2.3. Preparation of tissue extracts for ELISA and Western blot
Frozen tissues were homogenized (200 mg/ml) for 1 min with aPolytron in an EDTA-free, protease-inhibitor cocktail (Mini-Com-plete, Roche) containing 1 mM PMSF, pH 9.0, at 4 �C. The homoge-nates were agitated on a magnetic stirrer for 2 h at 4 �C, and thencentrifuged at 12,000 rpm at 4 �C for 15 min (Sorvall RMC14 centri-fuge). The supernatants were collected, and the protein content ofeach sample was determined by the Bradford micro-method (Bio-Rad, Hercules, CA, USA).
2.4. ELISA
The presence of GH-immunoreactivity (GH-IR) in the tissue orcell culture extracts was investigated using an indirect enzyme-linked immunosorbant assay (ELISA, [56]). Briefly, 96-well microti-ter plates (Immulon 2HB, Chantully, VA, USA) were coated over-night at 4 �C with 12 ng recombinant chicken GH (rcGH) in100 ll of 0.1 M carbonate buffer, pH 10.3. The plates were washedfive times with TPBS (0.01 M sodium phosphate, 0.15 mM NaCl,0.05% w/v Tween 20, pH 7.0) using an automatic microplate immu-nowasher (Biotrak II, Washer 2, Amersham Biosciences, Niskayuna,NY, USA). This wash procedure was performed after each incuba-tion step. Tissue extracts or serial dilutions of rcGH (1024–0.5 ng/ml) in TPBS containing 1% w/v non-fat dry milk were then incu-bated for 16 h with 100 ll primary antibody CAP-1 (at a final con-centration of 1:100,000) raised in rabbits against native pituitaryGH [2]. This antibody is specific for GH and has no cross-reactivity(<0.001%) with any other pituitary hormones [2]. The samples andstandards (100 ll) were then added to the coated wells and incu-bated for a further 2 h at room temperature. Horseradish peroxi-dase–anti-rabbit IgG conjugate (Bio-Rad) was then added (at adilution of 1:3000 in 5% [w/v] non-fat dry milk in 0.1 M TPBS, pH7.0) and incubated for 2 h at room temperature. Bound secondaryantibodies were then detected by reaction with 2,20amino-di-[3-ethylbenzothiazoline sulfate] substrate (Roche). The plates were

C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31 19
read 30 min later in an automatic ELISA Microplate Reader (Bio-Rad) at a wavelength of 405 nm. The assay has a sensitivity of2 ng/well (20 ng/ml), and the inter-assay and intra-assay coeffi-cients of variation are <4% [56].
2.5. Cerebellar primary cell cultures
Cerebellums from chicken embryos at ED 15 of developmentwere removed and treated in 0.5% trypsin solution (SIGMA–AL-DRICH, St. Louis, MO, USA) at 37 �C for 10 min with stirring. Trypsindigestion was halted by the addition of an equal volume of DMEM(GIBCO-BRL, Burlington, Ont., Canada) containing 10% fetal bovineserum (GIBCO). The digested tissue was then centrifuged at 1800gfor 5 min, and the pellet was resuspended in 2 ml plating medium(Neurobasal medium supplemented with 2% B27, 0.5 mM L-gluta-mine, and 1% penicillin–streptomycin; all from GIBCO). The tissuewas subsequently triturated 10 times with two fire-polished, sili-conized Pasteur pipettes of narrow bore size. After filteringthrough a 40 lm nylon mesh, 1 � 106 cells were plated on a 10-mm culture fluorodish (World Precision Instruments Inc., Sarasota,FL, USA) coated with 50 lg/ml poly-L-lysine and grown in the sameNeurobasal medium (NB, glucose: 6 g/L) with supplements at 37 �Cin a humidified atmosphere of 95% air and 5% CO2 for 72 h.
2.6. In vitro treatments
To induce hypoxia and low glucose conditions (HLG) the Neuro-basal-B27 culture medium was replaced with DMEM–Low Glucose1� (1 g/L, GIBCO-BRL, LG), and the cultures were then incubatedfor 1 h in a humidified, 37 �C hypoxic chamber (Napco E Series,Model 302 CO2 incubator, which was previously flushed with agas mixture of 5% CO2 and 95% N2 for 20 min resulting in a levelof 0.5% O2, that was maintained throughout the experiment andcontinuously monitored with an ambient oxygen sensor [BW Tech-nologies, Arlington, TX]). Replacing the LG medium with NB med-ium terminated HLG condition, and the cultures were thenincubated for an additional 24 h under normoxic conditions (N,95% air and 5% CO2).
ED15 cells were cultured as described, and then the mediumwas removed and replaced with defined medium (NB or LG) con-taining the treatments: recombinant chicken GH (rcGH, CyanamidAC 4797-100) at different concentrations (1, 10, and 100 nM);wortmannin (100 nM, Calbiochem, EMD Biosciences, Inc. La Jolla,CA); purified IgGs directed against rcGH (1:20 dilution, obtainedin our lab by immuno-affinity chromatography, following [3]), ora 15-kDa fragment obtained from the proteolytic cleavage of rcGHwith thrombin as described in [5] at 1 and 10 nM. Then cells wereincubated for 1 h in either normoxia or HLG conditions. All the sub-stances tested were added only once, at the beginning of the 1-hincubation.
The following parameters were measured: cell viability deter-mination in the cultures was carried out after the 24-h re-oxygen-ation period; caspase-3 and Bcl-2 studies were carried out incultures harvested after the 24-h re-oxygenation term; the TUNELassay was performed in cells cultured on a Fluorodish after the 24-h re-oxygenation period; p-Akt determination was carried out incultures harvested immediately after the 1-h treatments.
The effects of HLG incubation conditions upon these parametersin the cell cultures were initially compared against normoxic con-ditions. Then, the effects of the different treatments were evalu-ated against either the normoxic or the HLG controls.
2.7. Cell viability determination
Cell viability was determined by either of two methods: the try-pan blue exclusion assay [84], or the MTT assay. In short, for the
first assay, cells (3 � 106 cells per well) were seeded in 6-wellplates and were stabilized for 72 h at 37 �C with 5% CO2 in ahumidified chamber. After this stabilization time, cells were sub-jected to HLG or to HLG in the presence of 1 nM GH for 1 h andre-oxygenated for another 24 h. Then, cells were harvested in a1 ml suspension, and an aliquot of 10 ll was mixed with another10 ll 0.05% trypan blue solution, and analyzed in a Neubauerchamber under a microscope where at least 100 cells (in duplicate)were studied for viability. The mean percentage of living cells wascalculated. The MTT assay (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, Roche, Manheim, Germany) wasperformed according to the manufacturer’s instructions. Briefly,cells (1 � 105 cells per well) were seeded in 96-well microtiterplates (0.1 ml/well) and were stabilized for 72 h at 37 �C with 5%CO2 in a humidified chamber. After this stabilization time, cellswere subjected to HLG or to HLG in the presence of 1 nM GH for1 h and re-oxygenated for another 24 h. Then, 10 ll of MTT solu-tion (5 g/L) was added to each well, and the plates were incubatedfor an additional 4 h at 37 �C under normoxic conditions. To dis-solve formazan crystals formed in viable cells, 100 ll dimethylsulfoxide (DMSO) was added to each well before measuring theabsorbance at 540 nm (Microplate reader, Model 550, Bio-Rad,Hercules, CA, USA). All treatments [rcGH (1 nM), wortmannin(100 nM), purified IgGs against rcGH (1:20), and the 15-kDa frag-ment obtained from the proteolytic cleavage of rcGH [5] (1 and10 nM)] were used in the same microtiter plate for 1 h of eithernormoxic or hypoxic conditions. Each plate contained multiplewells of a given experimental condition and multiple control wells.This procedure was repeated for three to five plates for each condi-tion. A one-way ANOVA test was used to analyze the data.
2.8. Immunocytochemistry of cell cultures
Cerebellar cell cultures were fixed with 4% paraformaldehyde inPBS (pH 7.4) for 30 min, then washed with PBS (3 � 10 min) andincubated in PBS-5% non-fat dry milk (Bio-Rad) for 2 h. Doublestaining and confocal analysis were performed to determine GHco-localization with neuron-cell markers (neuronal specific nucle-ar protein, NeuN, and b-tubulin III). These cultures were incubatedovernight with a specific rabbit polyclonal antibody directedagainst recombinant chicken GH (C1, 1:200 dilution, [2]), andeither anti-mouse NeuN (1:200 dilution, Chemicon InternationalInc., CA, USA), or anti-mouse b-tubulin III (1:200 dilution, CovanceInc., Princeton, NJ, USA). After washing (3 � 10 min) in TPBS, sec-tions were incubated for 2 h at room temperature with the second-ary antibodies: goat anti-rabbit IgG-FITC (diluted 1:100, ZymedLaboratories, San Francisco, CA, USA), and goat anti-mouse IgG-TRITC (diluted 1:100). Alternatively, goat anti-rabbit IgG-Cy3 anti-bodies (diluted 1:2000, Zymed) were used in some experiments.The specificity of C1 anti-GH staining was determined by preab-sorbing this antibody with an excess of recombinant chicken GH(250 lg/ml; antibody final dilution was 1:500); also non-immunesera were employed as a negative control. All antibodies were di-luted in TPBS containing 1% non-fat dry milk (Bio-Rad). The cul-tures were analyzed for NeuN or b-tubulin III and cGH co-localization using a Carl Zeiss LSM 510 confocal microscope withlaser excitation wavelengths of 488 nm (FITC), 514 nm (TRITC),and 561 nm (Cy3). These cultures were also counterstained with300 nM 40,6-diamidino-2-phenylindole (DAPI) (Invitrogen) in TBSfor 45 min, rinsed 3 times with TBS, and total cells were countedemploying a Coherent-XR multiphotonic laser at 350 nm.
2.9. Evaluation of apoptosis
The enzymatic activity of caspase-3 in cerebellar cell culturelysates was determined using a caspase-3 colorimetric assay kit

20 C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31
(Assay Designs Inc., Ann Arbor, MI, USA). The samples (8 lg of pro-tein) of cell lysates from each treatment, standards, p-nitroaniline(pNA) standard, and blank controls were transferred in duplicate to96-well microplates. After a 3-h incubation at 37 �C the reactionwas stopped with 1 N HCl, and absorbance at 405 nm was readimmediately in a 3350-UV microplate reader (BioRad). The activityof caspase-3 in the samples was calculated as units per microgramprotein, then it was normalized and expressed as percent activityrelative to the HLG control (which was considered as 100%) [18].
Apoptosis was also measured with the TUNEL assay using theAPO-BrdU TUNEL Assay kit [Roche Diagnostics] to study theappearance of apoptotic bodies. After a 1-h exposure to HLG and24 h of re-oxygenation, the cultures were fixed with 4% parafor-maldehyde in PBS for 30 min and then washed with PBS. Thenthe cultures were treated with proteinase K (2.5 lg/ml) for10 min at room temperature and washed with PBS. Cultures werethen incubated with the reaction mixture from the assay kit (50 llper sample) for 1 h at 37 �C in a humid chamber. Cultures werewashed 3 � 5 min in PBS and analyzed with fluorescence micros-copy. TUNEL labeling was quantified using Image Pro Plus Soft-ware. The proportion of apoptosis was the number of TUNEL-positive cells detected in the field as a percentage of the total num-ber of cells counterstained with DAPI (40,6-diamidino-2-phenylin-dole dihydrochloride), a marker of the cell nuclei [66]. Thequantification was made in 10 fields for each culture dish. Statisti-cal analysis was performed by one-way ANOVA.
2.10. GH effect on the anti-apoptotic signaling pathway during HLG
Changes in phosphorylated Akt (p-Akt) or Bcl2 in response totreatments were evaluated by SDS–PAGE and Western blotting.Cell lysates were prepared in a protease-inhibitor cocktail (mini-complete, Roche), pH 9.0. The supernatants were collected, andtheir protein content was determined by the Bradford micro-meth-od (Bio-Rad). Samples (containing 40 lg protein) were analyzed byone-dimensional sodium dodecyl sulfate–polyacrylamide gel elec-trophoresis (SDS–PAGE) in 1.0 mm thick, 6 cm long, 12.5% gelsusing the buffer system of Laemmli [47] in a mini-Protean II cell(Bio-Rad) under reducing conditions (in the presence of 5% (w/v)2-mercaptoethanol). After electrophoresis, the gels were equili-brated in transfer buffer (25 mM Tris–HCl, 192 mM glycine, 20%(v/v) methanol, pH 8.3) for 30 min and the proteins were electro-transferred (at 200 mA for 60 min) to nitrocellulose membranes(Bio-Rad). After transfer, the membranes were washed with30 mM Tris, 500 mM NaCl, pH 7.5 (TBS) for 5 min, then blockedwith 5% (w/v) nonfat dry milk (Bio-Rad) in TBS, for 2 h at roomtemperature. The membranes were washed with TPBS containing1% nonfat dry milk for 15 min and were incubated overnight atroom temperature with either monoclonal p-Akt antibody (Abcam,Cambridge, MA, USA) or monoclonal Bcl-2 antibody (Invitrogen),both diluted 1:750 in TPBS. The membranes were then rinsed 3times (for 15 min) in TTBS, and antibody binding was visualizedusing anti-mouse IgG horseradish peroxidase conjugate (Bio-Rad), diluted 1:1000 in 1% (w/v) nonfat dry milk in TTBS for 2 h.Immunoreactive bands were developed by incubating the mem-branes in ECL chemiluminescent reagent (Amersham-Pharmacia,Buckinghamshire, UK) for 1 min and exposing them to Kodak Bio-max ML film. All blots measuring p-Akt and Bcl-2 were stripped offbound antibodies and re-probed to detect the total amounts of Akt(Santa Cruz Biotechnology, CA, USA, 1 ll of antibody in 2.0 ml ofTTBS containing 1% (w/v) nonfat dry milk) and Actin (Santa CruzBiotechnologies, 1 ll of antibody in 3.0 ml TTBS containing 1% non-fat dry milk), respectively. The p-Akt band intensity was normal-ized to Akt band intensity, and the Bcl-2 band intensity to actin.
2.11. Densitometric analysis
The relative proportions of immunoreactive p-Akt and Bcl2moieties were determined by densitometric analysis after digital-izing the luminograms in a Cannon scanner, by using IP LabGel2.2 software (Scanalytics, Fairfax, VA, USA).
2.12. Statistical analysis
Data are presented as mean ± SEM. Statistical analysis betweengroups was performed using one-way ANOVA followed by Tuckey’spost hoc test (GraphPad Software, Inc., La Jolla, CA, USA). Only re-sults with a value of p < 0.05 were considered significant.
3. Results
3.1. Effects of hypoxia on GH content in the brain, in the cerebellum,and in cerebellar cell cultures
In vivo experiments showed that local growth hormone levelsincreased both in whole brain and cerebellum when chicken em-bryos were exposed to hypoxia for 24 h (Fig. 1). Thus, the GH con-centration from neural origin increased by 29.2% (from172.3 ± 11.8 to 222.64 ± 13.23 ng GH/mg protein) in brains ofED15 (Fig. 1A), and significantly by 32.6% (from 176.46 ± 7.01 to234 ± 18.8) in brains of ED 18 (Fig. 1B) chicken embryos that hadbeen subjected to 24 h of hypoxia followed by 24 h of re-oxygena-tion just before sacrifice (H-24 h), as compared to controls undernormal oxygen incubation conditions (normoxia, N). Furthermore,there was still a significant increase in brain GH concentration inembryos exposed to 24 h hypoxia at ED15 (16%, from207.81 ± 6.5 to 240.8 ± 6.7 ng GH/mg protein) (Fig. 1C), or atED18 (44%, from 205.93 ± 8.9 to 296.6 ± 30.1) (Fig. 1D), respec-tively, but not sacrificed until 1-day post-hatching (H-1ph), com-pared with brains from normoxic controls. On the other hand,cerebellums obtained from ED15 chicken embryos exposed to24 h of hypoxia and sacrificed after 24 h of re-oxygenation, showeda 74% increase in the GH concentration compared with controls(from 675.1 ± 86.46 to 1177 ± 108.5) (Fig. 1E, H-24 h).
An analogous response was observed in vitro, when ED15 cere-bellar cell cultures were subjected to 1 h hypoxia and low glucosemedium, the GH concentration increased significantly (by 17%,from 361.26 ± 39.5 to 421.26 ± 33.4 ng GH/mg protein) when mea-sured after 24 h of re-oxygenation (Fig. 1F, HLG) (p < 0.05).
3.2. Immunocytochemical characterization of cerebellar cell cultures
Embryonic chicken cerebellar neuron culture conditionswere standardized, and the viability was evaluated with thetrypan blue exclusion method after 72 h of incubation. The re-sults indicated 98% viability, and the cell density and morphol-ogy were similar to previous observations where groups ofneurons with extensive neurites were reported [42]. To verifythat the cultured cells were of neuronal identity, they werestained for NeuN, a well-known neuron-specific antigen [59].Since GH expression has been described in the chicken cerebel-lum [1], the cells were also stained for GH. The cultured cellsformed a confluent monolayer (Fig. 2), and around 85% of thecells analyzed gave a positive signal against both GH (Fig. 2a)and NeuN (Fig. 2b). When the images were merged, co-localiza-tion of these two markers was observed (Fig. 2c). At highermagnification, the GH immunoreactive signal was found withinthe nucleus and also in some processes (Fig. 2d), whereas theNeuN signal, as expected, was observed mainly in the cell nuclei(Fig. 2e); after merging, these two markers co-localized in the

Fig. 1. GH concentration measured by ELISA and expressed in ng of GH per mg of protein. (A) and (B) 15- and 18-day-old chicken embryos (ED15 and ED18), respectively,were subjected to hypoxia for 24 h followed by 24 h of re-oxygenation (H-24 h); after this time, embryos were immediately sacrificed, and whole brains were obtained for GHdetermination. (C) and (D) Chicken embryos (ED15 and ED18) were exposed to hypoxia for 24 h and then maintained under normoxic conditions until 1 day after hatching(H-1ph), when the animals were sacrificed and the GH concentrations in whole brains were obtained. (E) Embryonic chicks (ED15) were subjected to hypoxia for 24 hfollowed by 24 h of re-oxygenation; after this time, embryos were immediately sacrificed and the cerebellum was collected for the GH measurements. (F) Primary cerebellarcell cultures from ED15 were subjected to hypoxia for 1 h followed by 24 h of re-oxygenation, and then the cells were harvested and maintained in an EDTA-free, protease-inhibitor cocktail (Mini-Complete, Roche) containing 1 mM PMSF, pH 9.0, and analyzed for GH concentration. Each bar represent mean ± SEM for: [(A) and (B) panels] n = 7(N) and 5 (H-24 h) independent embryos (3 three replicates for each); [(C) and (D) panels] n = 9 (N) and 5 (H-1ph) independent embryos (3 replicates for each); and [(E) and(D) panels] n = 5 independent embryos (3 replicates for each) in both conditions.
C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31 21
nucleus (Fig. 2f). Further characterization of the cell culturesshowed immunoreactivity for b-tubulin III, a specific markerfor the cytoskeleton in the cytoplasm and neuronal pro-cesses 2(Fig. 2h); the GH signal was more intense within and
around the nucleus, although it was also found in some pro-cesses (Fig. 2g). A significant fraction of these signals co-local-ized, mainly in the cytoplasmic region around the nucleus(Fig. 2i).

Fig. 2. GH-IR and NeuN-IR are present in chicken cerebellar cell cultures after 72 h of incubation with Neurobasal medium supplemented with B27. Confocal images at 40�magnification of double-stained ED15 cell cultures were analyzed to identify neurons containing NeuN (b, green, 1st Ab: monoclonal anti-NeuN antibody, 1:200; 2nd Ab: goatanti-mouse IgG-FITC 1:200) and GH (a, red, 1st Ab: anti-cGH 1:200; 2nd Ab: goat anti-rabbit IgG-TRITC 1:200). Panel c represents the overlap of GH and NeuNimmunoreactivity, indicating co-expression (yellow, arrows). Scale bars = 10 lm. Higher magnification (63�) is shown in d (cGH), e (NeuN), and f (merged). Immunoreactivesignal for NeuN is observed mainly in the nucleus (arrows), and it co-localizes with the GH signal. In panel g, GH immunoreactivity is also observed in the perikarya and insome cell processes; panel h clearly shows neural processes immunoreactive for b-tubulin III, and this signal also co-localized with GH, as shown in panel i. Magnification10�, bar: 50 lm (a–c); magnification 63�, bar: 10 lm (d–f); magnification 40�, bar: 10 lm (g and h). (For interpretation of the references to colour in this figure legend, thereader is referred to the web version of this article.)
22 C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31
3.3. Effects of hypoxia and low glucose incubation conditions (HLG)
The exposure of cerebellar cell cultures to hypoxia (0.5% O2) andlow glucose (1 g/L) conditions (HLG) for 1 h, followed by 24 h of re-oxygenation induced a significant decrease in the number of cells(Fig. 3Af–j) compared with normoxic conditions (Fig. 3Aa–e).Although less than half of the cells survived the harsh HLG incuba-tion conditions (Fig. 3Ai), most of the cells that did survive werestill able to express both GH, mainly in the nucleus and perikarya(Fig. 3Af, arrows); and NeuN, whose signal was also observed with-in the nucleus in HLG-treated cells (Fig. 3Ag). These signals co-localized when the confocal images were merged (Fig. 3Ah). Toquantify these data we counted the number of cells stained withDAPI per unit area (Fig. 3B, left panel). Compared to normoxic con-ditions (100%, corresponding to 211.6 ± 8.5 cells per field), the totalnumber of cells decreased significantly after HLG to 47 ± 2% (corre-sponding to 99.4 ± 5.9 cells per field). However, the addition of1 nM GH during these severe incubation conditions (HLG + GH)
promoted cell survival to 71 ± 3% (corresponding to 151.4 ± 8.4cells per field) (Fig. 3B, right panel). This meant GH promoted cellsurvival 1.5-fold in comparison with HLG.
3.4. GH effects on cell viability
We determined the viability of chicken cerebellar neurons atphysiological O2 and glucose concentrations (normoxia,91.3 ± 11%) and compared it with their viability in culture mediumunder HLG conditions after the cells were further incubated an-other 24 h under normoxic conditions, and with the cell viabilityafter addition of GH (1, 10 and 100 nM) treatments. As shown inFig. 4A cell viability, measured by trypan-blue exclusion, decreasedto 41 ± 8.04% under HLG conditions (Fig. 4A). The addition of 1 nMrcGH (HLG + GH1) promoted significant viability recovery (to70.3 ± 14.3%, 1.7-fold in comparison to HLG), although no furtherchanges were found with 10 and 100 nM rcGH (63.6 ± 12.4% and61 ± 10.6% viability, respectively). Under normoxic conditions, GH
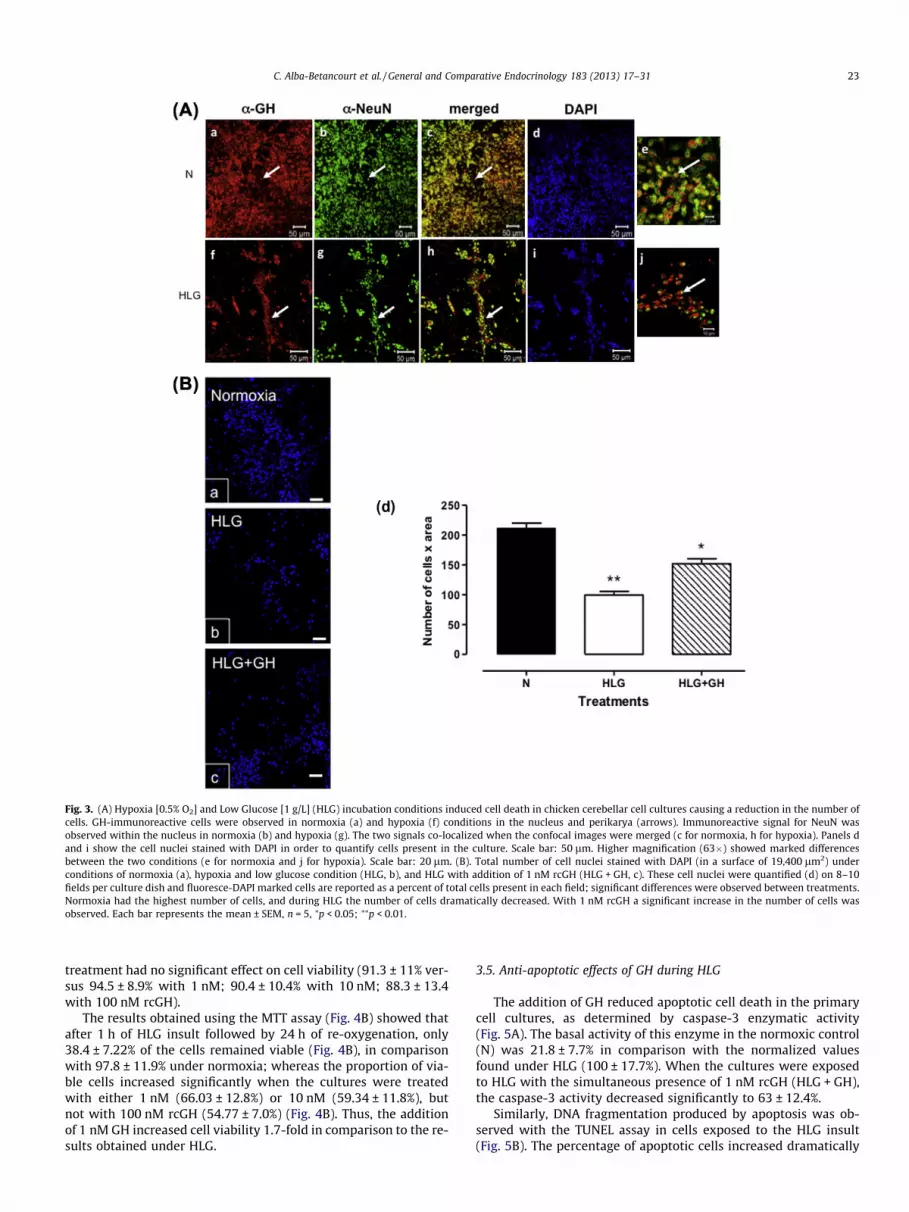
Fig. 3. (A) Hypoxia [0.5% O2] and Low Glucose [1 g/L] (HLG) incubation conditions induced cell death in chicken cerebellar cell cultures causing a reduction in the number ofcells. GH-immunoreactive cells were observed in normoxia (a) and hypoxia (f) conditions in the nucleus and perikarya (arrows). Immunoreactive signal for NeuN wasobserved within the nucleus in normoxia (b) and hypoxia (g). The two signals co-localized when the confocal images were merged (c for normoxia, h for hypoxia). Panels dand i show the cell nuclei stained with DAPI in order to quantify cells present in the culture. Scale bar: 50 lm. Higher magnification (63�) showed marked differencesbetween the two conditions (e for normoxia and j for hypoxia). Scale bar: 20 lm. (B). Total number of cell nuclei stained with DAPI (in a surface of 19,400 lm2) underconditions of normoxia (a), hypoxia and low glucose condition (HLG, b), and HLG with addition of 1 nM rcGH (HLG + GH, c). These cell nuclei were quantified (d) on 8–10fields per culture dish and fluoresce-DAPI marked cells are reported as a percent of total cells present in each field; significant differences were observed between treatments.Normoxia had the highest number of cells, and during HLG the number of cells dramatically decreased. With 1 nM rcGH a significant increase in the number of cells wasobserved. Each bar represents the mean ± SEM, n = 5, ⁄p < 0.05; ⁄⁄p < 0.01.
C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31 23
treatment had no significant effect on cell viability (91.3 ± 11% ver-sus 94.5 ± 8.9% with 1 nM; 90.4 ± 10.4% with 10 nM; 88.3 ± 13.4with 100 nM rcGH).
The results obtained using the MTT assay (Fig. 4B) showed thatafter 1 h of HLG insult followed by 24 h of re-oxygenation, only38.4 ± 7.22% of the cells remained viable (Fig. 4B), in comparisonwith 97.8 ± 11.9% under normoxia; whereas the proportion of via-ble cells increased significantly when the cultures were treatedwith either 1 nM (66.03 ± 12.8%) or 10 nM (59.34 ± 11.8%), butnot with 100 nM rcGH (54.77 ± 7.0%) (Fig. 4B). Thus, the additionof 1 nM GH increased cell viability 1.7-fold in comparison to the re-sults obtained under HLG.
3.5. Anti-apoptotic effects of GH during HLG
The addition of GH reduced apoptotic cell death in the primarycell cultures, as determined by caspase-3 enzymatic activity(Fig. 5A). The basal activity of this enzyme in the normoxic control(N) was 21.8 ± 7.7% in comparison with the normalized valuesfound under HLG (100 ± 17.7%). When the cultures were exposedto HLG with the simultaneous presence of 1 nM rcGH (HLG + GH),the caspase-3 activity decreased significantly to 63 ± 12.4%.
Similarly, DNA fragmentation produced by apoptosis was ob-served with the TUNEL assay in cells exposed to the HLG insult(Fig. 5B). The percentage of apoptotic cells increased dramatically

Fig. 4. Effect of recombinant growth hormone (rcGH) in embryonic ED15 primarycerebellar cultures treated during 1 h with hypoxia and low glucose (HLG) andfollowed by 24 h of reoxygenation. Cell viability was evaluated with the trypan-blue exclusion assay (A) and the MTT assay (B). The graph shows viable cells as apercent of total cells. Cells were subjected to HLG in the presence of 1 nM GH(H + GH1), 10 nM GH (H + GH10), and 100 nM GH (H + GH100). Controls (N) weremaintained under normoxic conditions (37 �C, 95% air, 5% CO2). Each bar representsthe mean ± SEM, n = 5 independent experiments with three replicates for eachsample. Groups with different letters are significantly different from each other byANOVA, p < 0.05.
24 C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31
(23.1-fold) in the HLG cultures (78.6 ± 13% Fig. 5C) compared tocell cultures under normoxia conditions (3.4 ± 1.6% Fig. 5C). Thepresence of 1 nM GH during this insult significantly decreased (toabout half) the number of apoptotic nuclei (36 ± 8%, Fig. 5C). Quan-tification (Fig. 5C) was performed within 8–10 fields per culturedish, and cells with TUNEL-positive nuclei (Fig. 5Bii, v and viii)are given as a percentage of the total cells present in each fieldquantified with DAPI (Fig. 5Bi, iv and Bvii, and overlapped imagesin Fig. 5Biii, vi and ix) using a confocal microscopy.
3.6. Effects of wortmannin on cell viability and apoptosis
To preliminary explore which mechanisms could be involved inthe anti-apoptotic influence of GH during HLG, cerebellar neuroncultures were also treated with wortmannin, a specific inhibitorof the PI3K-Akt pathway, and its effect examined upon cell viabilityand the appearance of apoptotic bodies (Fig. 6). As previously de-scribed, HLG conditions significantly decreased cell viability to52.9 ± 0.6% in comparison to normoxia (79.7 ± 2.2%), and additionof 1 nM GH significantly recovered viability to 69.1 ± 2.6%. Whencultures under normoxia were treated with 100 nM wortmannincell viability significantly decreased to 50.2 ± 1.0%. In the presenceof wortmannin, GH was unable to restore viability both undernormoxic (N + W + GH, 51.8 ± 0.6%) or hypoxic (HLG + W + GH,49.4 ± 3.9%) conditions (Fig. 6A). On the other hand, HLG condi-tions provoked a significant and dramatic increase of apoptoticcells (80.3 ± 1.4%) in comparison to normoxia (8.2 ± 0.8%), andtreatment with 1 nM GH significantly promoted a recovery byreducing the number of apoptotic bodies (40.0 ± 2.9%) in HLG
treated cultures. The addition of 100 nM wortmannin significantlyincreased apoptosis in cell cultures under normoxia (N + W,81.6 ± 2.2%). Again, in the presence of wortmannin GH was notcapable to prevent apoptosis, both under normoxia (N + W + GH,78.2 ± 2.7%) or hypoxia (HLG + W + GH, 80.6 ± 2.6%) conditions(Fig. 6B).
3.7. GH effect on activation of the PI3K/Akt pathway
The serine-threonine kinase, Akt, is involved in survival signal-ing pathways in many cell systems. In the present study, we exam-ined Akt phosphorylation at serine 473 (p-Akt) immediately after1 h of HLG insult. P-Akt and Akt were evident as bands of 60 kDain cell homogenates from cerebellar cultures in both normoxia(Fig. 7A; lane N) and HLG (Fig. 7B, lane H) conditions. To quantifythese results P-Akt was reported as a percentage of total Akt(Fig. 7C and 7D). P-Akt was constitutively expressed in normoxiaconditions (32.3 ± 12.7%) and showed a prominent increase(1.63-fold) after 1 h of HLG conditions (52.8 ± 4.9%). Treatmentwith 1 nM GH increased the proportion of p-Akt (1.7-fold) in nor-moxia (55.5 ± 12.7%, Fig. 7C), and also in HLG (1.4-fold,74.6 ± 15.1%, Fig. 7D) conditions, respectively. Cultured cells trea-ted with purified IgGs against GH (immunoneutralization) showedan important decrease (around two thirds) in the p-Akt levels to11.3 ± 5.1% for normoxia conditions (Fig. 7A and C, lane A) and to19.6 ± 3% for HLG (Fig. 7B and D, lane A). The stimulatory effectof 1 nM GH treatment upon p-Akt levels was also abolished whenco-incubated with the specific antibodies against GH (Fig 7, laneA + GH), decreasing to 10.8 ± 4.7% for normoxia (Fig. 7A and C)and 24.4 ± 6.4% for HLG conditions (Fig. 7B and D), respectively.These data indicated that the antibodies were able to neutralizethe effect of both local and externally added GH. When the cellswere treated with 100 nM wortmannin, very little p-Akt was de-tected in either condition or with simultaneous addition of GH(Fig. 7, lane W, W + GH; 11.5 ± 4.9% and 3.8 ± 1.1% respectivelyfor normoxia, and correspondingly 7.4 ± 3.7% and 5.0 ± 2.0% forHLG conditions), thus indicating that this specific inhibitor abol-ished the GH effect.
3.8. GH activation of Bcl-2
Another major pathway involved in the control of cell survivaldepends on members of the Bcl-2 family. To assess whether GHmay promote cell survival by regulating Bcl2 levels, we evaluatedthe effect of GH treatment on Bcl2 content in cerebellar cell cul-tures in normoxia (Fig. 8A, lane N) or HLG conditions (Fig. 8B, laneH). To quantify these results Bcl-2 was reported as a percentage ofBcl-2/Actin ratio (Fig. 8C and D). Basal expression of Bcl-2(49.6 ± 13.4%) was found in normoxia conditions (Fig. 8C) and itshowed a prominent increase (1.4-fold) after 1 h of HLG conditions(71.4 ± 16.9, Fig. 7D). Treatment with 1 nM GH significantly in-creased (1.6-fold) the proportion of Bcl-2 in normoxia to80.7 ± 24.1% (Fig. 8C, lane GH). A similar effect of GH was found un-der HLG conditions (Fig. 8D, lane GH) increasing Bcl-2 levels to119 ± 26.4% (1.6-fold), compared to their respective controls. Cul-tured cells treated with purified IgGs against GH (immunoneutral-ization) showed an important decrease in the Bcl-2 levels to22.7 ± 5.7% for normoxia conditions (Fig. 8C, lane A) and to13.8 ± 4.4% for HLG (Fig. 8D, lane A). Also, the effect observed with1 nM GH treatment on Bcl-2 levels was abolished when co-incu-bated with the specific antibodies against GH (Fig. 8, laneA + GH), decreasing to 15.1 ± 5.6 for normoxia (Fig. 8C) and14.8 ± 5 for HLG (Fig. 8D) conditions, respectively. These data indi-cated that the antibodies were able to neutralize the effect of bothlocal and externally added GH. On the other hand, the cells treatedwith wortmannin showed decreased levels of Bcl-2 in both

Fig. 5. Effect of added recombinant growth hormone upon apoptosis of embryonic chicken cerebellar primary cell cultures after 1 h of HLG treatment followed by a 24-hreoxygenation. (A) Caspase-3 activity was measured with a colorimetric assay kit, and the activity is reported as percent of activity after normalizing the activity obtainedunder HLG value as 100%. Addition of 1 nM rcGH during HLG decreased the caspase-3 activity. (B) Apoptotic bodies (arrows) were revealed by the TUNEL assay. Control cellcultures under normoxic conditions (N) displayed very low TUNEL reactivity (i–iii); cells subjected only to HLG without GH treatment showed the highest TUNEL signal (iv–vi), and with GH (HLG + GH) the TUNEL signal decreased significantly (vii–ix). (C) Quantization was performed on 8–10 fields per culture dish, and fluoresce-marked cells(green, ii, v, vii) are reported as a percent of total cells present in each field, observed with DAPI using a confocal microscope (i, iv, vii). Each bar represents the mean ± SEM,n = 5. Groups with different asterisks are significantly different from each other by one-way ANOVA and Tukey’s multiple-comparison post hoc tests, p < 0.05.
C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31 25
normoxia (Fig. 8C, lane W; 33.4 ± 5.1%) and HLG conditions(Fig. 8D, lane W; 20.3 ± 4.9%). When this inhibitor was adminis-tered together with GH, Bcl-2 levels decreased to 34.2 ± 7.3% innormoxia and to 8.8 ± 4.1% in HLG, as a shown in Fig. 8C and D.
3.8.1. 15-kDa GH effect during HLGPrevious data showed that the major GH isoform expressed in
the cerebellum during chicken development corresponded to a15-kDa GH variant [1]. We evaluated the effect of a 15-kDa frag-ment obtained by proteolytic (thrombin) cleavage of rcGH [5] oncellular viability and caspase-3 activity (Fig. 9). Cells were sub-jected to HLG or maintained in normoxia conditions in the absenceor presence of 1 or 10 nM 15-kDa GH fragment for 1 h and then re-oxygenated for 24 h. Cell viability (95.4 ± 21.94% under normoxia)decreased significantly to 30.7 ± 5.4% (3.1-fold) during HLG condi-tions; co-incubation of the HLG cultures with 1 and 10 nM 15 kDaGH, increased cell viability to 36.08 ± 14.14% (1.2-fold) and51.19 ± 10.7% (1.7-fold), respectively (Fig. 9A). On the other hand,the caspase-3 activity decreased from 100 ± 5.9% under HLG, to85.7 ± 6.6% (one seventh) and a significant 74.5 ± 5.0% (one fourth)with the addition of 1 and 10 nM 15 kDa GH, respectively (Fig. 9B).In comparison, basal activity of caspase-3 under normoxic condi-tions corresponded to 6.7 ± 1.38%.
4. Discussion
Previous studies have shown that GH may have a role as a neu-roprotective factor following an insult, acting through autocrine/
paracrine mechanisms [39,69,71]. Not only is there an increase inGH-like immunoreactivity on injured brain cells, but also whenexogenous GH is administered it is capable to enhance cell survivaland revert damage.
In this work we showed, with the in vivo study, an increase ofGH concentration in the whole brain of ED15 and ED18 chickenembryos after 24 h hypoxia followed by 24 h of re-oxygenationand sacrifice (groups A and B), and this increase was maintaineduntil one day posthatching (groups C and D). On the other hand,in the isolated cerebellum, this increase was more pronouncedthan in the whole brain, with 3.9- and 5.3-fold higher GH levelsin normoxia and HLG, respectively (Fig. 1E).
Similar results were observed by Scheepens et al. [73], whoshowed an increase in the expression of GH mRNA and protein inrat brain, after hypoxia damage [30,75]. Several studies have dem-onstrated that GH treatment not only provides some degree ofneuroprotection [30,73,75], but it also improves neurocognitiveoutcome following hypoxic-ischemic brain injury [90] and attenu-ates apoptosis. Furthermore, GH increases IGF-1 expression in ratcerebellum [24], suggesting that IGF-1 plays a role in mediatingthe protective effect of GH/GHR in hypoxic-ischemic brain injury[30,49,75].
In adult avian brains, GH-IR has been reported to be present inseveral hypothalamic nuclei, in the median eminence, and in somecircumventricular organs, as well as in the hippocampus of chick-ens, turkeys, and doves [64]. In late-stage embryos, GH-IR cells atED 14 were present in different areas of the brain, for example inthe molecular and pyramidal layers of the cerebral cortex, in thegray matter of the cerebellum, in the choroid plexus, in the pineal

Fig. 6. Effect of recombinant growth hormone (rcGH) and wortmannin upon cell viability and apoptosis in embryonic ED15 primary cerebellar cultures treated during 1 hwith hypoxia and low glucose (HLG) and followed by 24 h of re-oxygenation. (A) Cell viability was evaluated with the trypan-blue exclusion assay under normoxia condition(N), hypoxia and low glucose (HLG) or in presence of 1 nM GH (N + GH), (HLG + GH) respectively. Both groups were also incubated in presence of the PI3K/Akt pathwayinhibitor (100 nM) wortmannin, N + W or HLG + W and the combination of N + W + GH or HLG + W + GH. (B). The proportion of apoptotic cells was evaluated by TUNEL assay.Treatment groups were similar to the above mentioned, with the inclusion of and additional control (N + GH). Each bar represents the mean ± SEM, n = 3 independentexperiments with 5 replicates each. Groups with different letters are significantly different from each other by one-way ANOVA and Tukey’s multiple- comparison post hoctests, p < 0.001.
Fig. 7. GH activation of the PI3K/Akt signaling pathway. (A and B) Cell cultures were maintained either under normoxic or 1 h HLG conditions, and exposed to the followingtreatments: without (N and H, respectively) or with addition of 1 nM rcGH (lane GH); purified IgGs against GH (lane A); purified IgGs against GH + 1 nM rcGH (lane A + GH),100 nM wortmannin (lane W); or 100 nM wortmannin + 1 nM rcGH (lane W + GH). After treatment, cell homogenates were prepared and analyzed by Western Blot for Akt, p-Akt, and actin. Activation of the PI3K/Akt pathway was indicated by the phosphorylation of Akt (60 kDa). (C and D) Densitometric analysis: each bar represents the mean ofthe p-Akt/Akt ratio ± SEM, expressed in%; n = 5 independent experiments with three replicates for each sample. Groups with different letters are significantly different fromeach other by one-way ANOVA and Tukey’s multiple- comparison post hoc tests, p < 0.05.
26 C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31
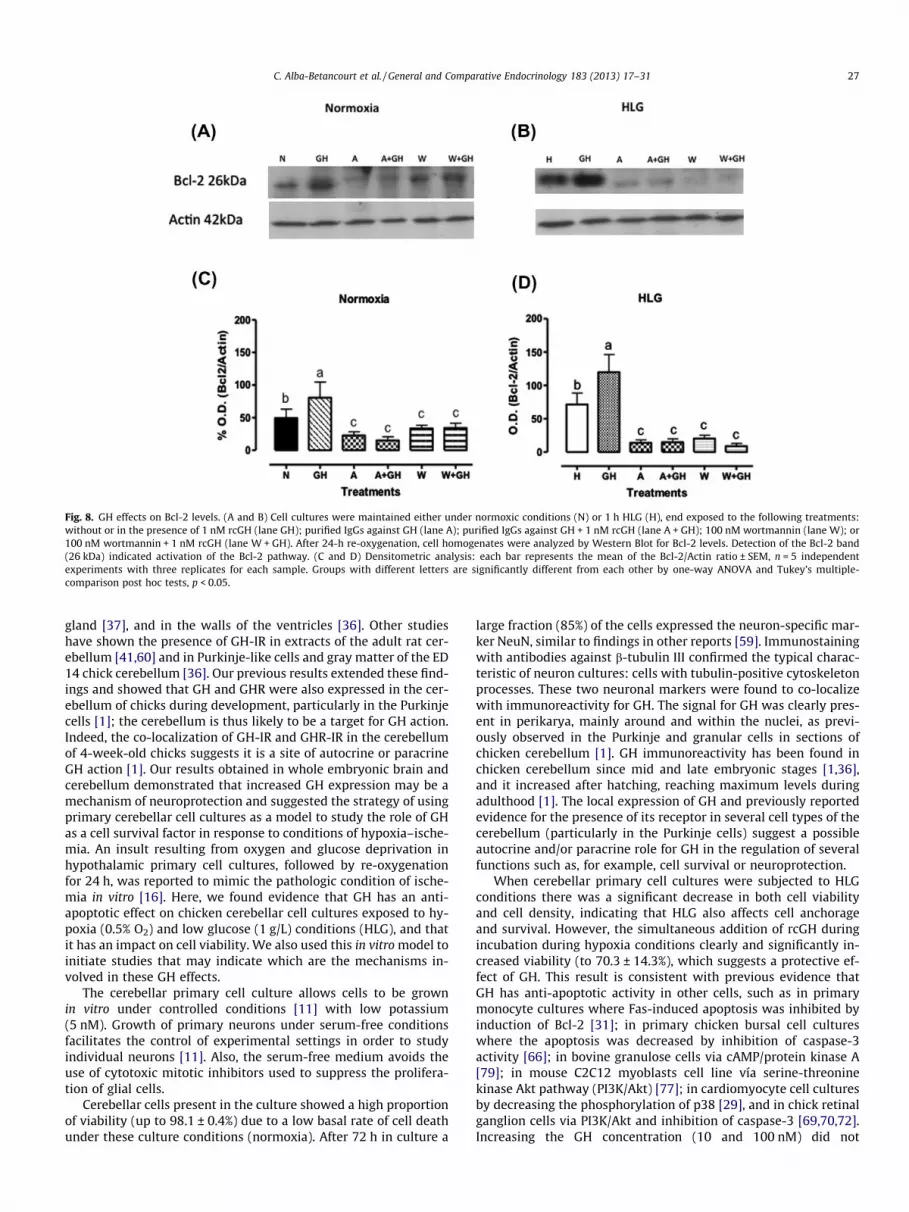
Fig. 8. GH effects on Bcl-2 levels. (A and B) Cell cultures were maintained either under normoxic conditions (N) or 1 h HLG (H), end exposed to the following treatments:without or in the presence of 1 nM rcGH (lane GH); purified IgGs against GH (lane A); purified IgGs against GH + 1 nM rcGH (lane A + GH); 100 nM wortmannin (lane W); or100 nM wortmannin + 1 nM rcGH (lane W + GH). After 24-h re-oxygenation, cell homogenates were analyzed by Western Blot for Bcl-2 levels. Detection of the Bcl-2 band(26 kDa) indicated activation of the Bcl-2 pathway. (C and D) Densitometric analysis: each bar represents the mean of the Bcl-2/Actin ratio ± SEM, n = 5 independentexperiments with three replicates for each sample. Groups with different letters are significantly different from each other by one-way ANOVA and Tukey’s multiple-comparison post hoc tests, p < 0.05.
C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31 27
gland [37], and in the walls of the ventricles [36]. Other studieshave shown the presence of GH-IR in extracts of the adult rat cer-ebellum [41,60] and in Purkinje-like cells and gray matter of the ED14 chick cerebellum [36]. Our previous results extended these find-ings and showed that GH and GHR were also expressed in the cer-ebellum of chicks during development, particularly in the Purkinjecells [1]; the cerebellum is thus likely to be a target for GH action.Indeed, the co-localization of GH-IR and GHR-IR in the cerebellumof 4-week-old chicks suggests it is a site of autocrine or paracrineGH action [1]. Our results obtained in whole embryonic brain andcerebellum demonstrated that increased GH expression may be amechanism of neuroprotection and suggested the strategy of usingprimary cerebellar cell cultures as a model to study the role of GHas a cell survival factor in response to conditions of hypoxia–ische-mia. An insult resulting from oxygen and glucose deprivation inhypothalamic primary cell cultures, followed by re-oxygenationfor 24 h, was reported to mimic the pathologic condition of ische-mia in vitro [16]. Here, we found evidence that GH has an anti-apoptotic effect on chicken cerebellar cell cultures exposed to hy-poxia (0.5% O2) and low glucose (1 g/L) conditions (HLG), and thatit has an impact on cell viability. We also used this in vitro model toinitiate studies that may indicate which are the mechanisms in-volved in these GH effects.
The cerebellar primary cell culture allows cells to be grownin vitro under controlled conditions [11] with low potassium(5 nM). Growth of primary neurons under serum-free conditionsfacilitates the control of experimental settings in order to studyindividual neurons [11]. Also, the serum-free medium avoids theuse of cytotoxic mitotic inhibitors used to suppress the prolifera-tion of glial cells.
Cerebellar cells present in the culture showed a high proportionof viability (up to 98.1 ± 0.4%) due to a low basal rate of cell deathunder these culture conditions (normoxia). After 72 h in culture a
large fraction (85%) of the cells expressed the neuron-specific mar-ker NeuN, similar to findings in other reports [59]. Immunostainingwith antibodies against b-tubulin III confirmed the typical charac-teristic of neuron cultures: cells with tubulin-positive cytoskeletonprocesses. These two neuronal markers were found to co-localizewith immunoreactivity for GH. The signal for GH was clearly pres-ent in perikarya, mainly around and within the nuclei, as previ-ously observed in the Purkinje and granular cells in sections ofchicken cerebellum [1]. GH immunoreactivity has been found inchicken cerebellum since mid and late embryonic stages [1,36],and it increased after hatching, reaching maximum levels duringadulthood [1]. The local expression of GH and previously reportedevidence for the presence of its receptor in several cell types of thecerebellum (particularly in the Purkinje cells) suggest a possibleautocrine and/or paracrine role for GH in the regulation of severalfunctions such as, for example, cell survival or neuroprotection.
When cerebellar primary cell cultures were subjected to HLGconditions there was a significant decrease in both cell viabilityand cell density, indicating that HLG also affects cell anchorageand survival. However, the simultaneous addition of rcGH duringincubation during hypoxia conditions clearly and significantly in-creased viability (to 70.3 ± 14.3%), which suggests a protective ef-fect of GH. This result is consistent with previous evidence thatGH has anti-apoptotic activity in other cells, such as in primarymonocyte cultures where Fas-induced apoptosis was inhibited byinduction of Bcl-2 [31]; in primary chicken bursal cell cultureswhere the apoptosis was decreased by inhibition of caspase-3activity [66]; in bovine granulose cells via cAMP/protein kinase A[79]; in mouse C2C12 myoblasts cell line vía serine-threoninekinase Akt pathway (PI3K/Akt) [77]; in cardiomyocyte cell culturesby decreasing the phosphorylation of p38 [29], and in chick retinalganglion cells via PI3K/Akt and inhibition of caspase-3 [69,70,72].Increasing the GH concentration (10 and 100 nM) did not

Fig. 9. Effect of a thrombin-produced 15-kDa rcGH peptide on cellular viability asmeasured by MTT (A), and on caspase-3 activity (B). Cells were subjected to HLG (H)or maintained under normoxia conditions with no addition (N), or with 1 nM(H + F1) or 10 nM (H + F10) 15-kDa rcGH fragment for 1 h, then re-oxygenated for24 h prior to measuring cell viability and caspase-3 activity. Each bar represents themean ± SEM, n = 5. Groups with different letters are significantly different fromeach other by one-way ANOVA and Tukey’s multiple-comparison post hoc tests,p < 0.05.
28 C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31
significantly increase the protective effect during HLG. These dataare similar to those previously reported with neuronal hybrid cells(ventral spinal cord 4.1) where cells treated with GH (6–600 nM)proliferated by 20% compared to the control, but the number of livecells began to decrease in a dose-dependent manner at a concen-tration of 600 nM or higher [55]. On the other hand, it was foundthat GH actions on cognitive processes are mediated by an in-verted-U dose–response curve. This means that while intermediatedoses enhance the cognitive processes, low doses of GH have no ef-fect and higher doses either have no effect or may even impair cog-nitive function [20]. These data can be explained by the finitenumber of receptors and the effect should be proportional to thefraction of the receptors occupied by the hormone and the maxi-mum effect should occur when all receptors are occupied. Largedoses of the ligand, however, might produce smaller effects thanthe lower dose [20]. This may be explained by the prevention ofGH receptor dimerization through Site 1 interactions at high GHconcentrations, leading to receptor saturation [27].
The early signaling events regulating the expression and activa-tion of apoptotic pathways that induce impaired function and celldeath are still not well understood. Hypoxia/ischemia induces Baxtranslocation from cytosol to mitochondria leading to cytochromec release from the mitochondrial intermembrane region andresulting in caspase-9 activation [61].
We found that the inhibition of HLG-induced apoptosis by GHtreatment is accompanied by a 37% decrease in caspase-3 activityrelative to HLG control levels; these data support the view that this
caspase is part of the network of pathways involved in neuron celldeath. Moreover, GH reduced by half the number of apoptotic cellsdetected with the TUNEL assay (from 78.6 ± 13% in the HLG controlto 36 ± 8%) with 1 nM GH, indicating a role of the hormone as ananti-apoptotic factor. These results are consistent with previous re-ports in explants of ED6 and ED8 chick embryo retinal or purifiedretinal ganglion cells incubated with 10�6 M GH, where GH in-creased cell survival and inhibited apoptosis by regulating thetranscription and protein expression of caspase-3, caspase-9, andthe presence of the TUNEL–positive nuclei in the RGCs [69–71].
The mechanisms by which GH may contribute to prevent cellsfrom entering a death program have not been completely defined,but it has been suggested that the phosphatidylinositol kinase(PI3K) pathway as well as activation of NfjB have been implicated[43]. Akt functions in a wortmannin-sensitive pathway involvingPI3K and is activated by various growth and survival factors[12,25]. We studied the effects of blocking the PI3K/Akt pathwaywith wortmannin both under normal and HLG conditions, and alsoin the presence of rcGH. Cell viability under normoxia decreasedone third when treated with wortmannin, resembling the effectobtained when the cultures were subjected to HLG. In contrast tothe result obtained when rcGH increased (30.6%) cell viability un-der HLG, when co-incubated with the specific inhibitor rcGH wasnot capable to exert its protective effect, neither under normoxiaor HLG conditions. Likewise, the number of apoptotic cells greatlyincreased (9.7-fold) when normoxic cultures were treated withwortmannin in comparison to the control, and were very similarto what happened when the cultures were subjected to HLG condi-tions. Although rcGH was capable to protect significantly by reduc-ing one half the number of apoptotic cells when added to culturesunder HLG conditions, it was unable to do so when co-incubatedwith wortmannin, both under normoxia or HLG, implicating thatthe PI3K/Akt pathway is involved in the neuroprotective actionsof GH in this model of ischemia.
It has been reported that GH and growth hormone (GH)-releas-ing peptide-6 (GHRP-6) increase phosphorylation of Akt in the rathypothalamus, hippocampus, and cerebellum [24], and also thatGH treatment reduces Akt levels, while raising phospho-Akt levels,in the embryonic neural retina [72]; therefore, we examinedwhether intracellular signaling mechanisms involving Akt couldbe activated by GH in the cerebellar cultured cells during HLG con-ditions. We observed that HLG induces activation of Akt (1.6-fold)compared to control (normoxia), and that 1 nM GH treatment in-creases this activation in both HLG and normoxia conditions, sug-gesting that GH may exert its neuroprotective and anti-apoptoticeffects through this signaling pathway. Akt prevents apoptosis byinactivating several targets, including BAD [19,21], caspase-9, andcaspase-3 [15]. Akt activation was also decreased 2.8-fold com-pared to control (normoxia) when cells were immunoneutralizedwith purified IgGs against GH, indicating that endogenous GH pres-ent in the cell culture may have a role in Akt activation. Similarly,when the cells were treated with specific IgG’s against GH duringHLG, the phosphorylation of Akt decreased 2.7-fold, suggestingthat the endogenous GH was blocked, and that this local GH mightbe activating the PI3K/Akt pathway, through a putative autocrineand/or paracrine action.
Since Akt is involved in the PI3K pathway, inhibition of phos-phoinositide 3-kinases with wortmannin resulted in the blockageof Akt phosphorylation, as we observed with the 100 nM wortman-nin treatment (W) during HLG (7-fold decrease), as well as with100 nM wortmannin plus 1 nM GH (W + GH), where Akt activationwas reduced (9.4-fold) compared to the HLG control.
Results suggest that the action of GH is putatively mediated byactivation of the PI3K/Akt pathway and expression of Bcl-2, result-ing in a decrease of apoptosis, as determined by analysis of bothcaspase-3 activity and DNA fragmentation observed with TUNEL.

C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31 29
Overall, data obtained in this study suggest that this pathway maybe involved in the neuroprotective actions of GH in the cerebellarcells exposed to HLG, since addition of wortmannin significantlydecreases cell viability, induces apoptosis, and blocks the recoveryeffects induced by GH, while it also abolishes the pAkt increase in-duced by both local and externally added GH. These results are ingood agreement with those found in embryonic retinal ganglioncells (RGC), where GH treatment stimulates pAkt levels and theaddition of wortmannin to RGC cultures simultaneously with GHsignificantly reduced the anti-apoptotic actions of GH [70–72].
Another major pathway in apoptosis involves the release ofcytochrome c from mitochondria into the cytoplasm, where it acti-vates caspases by interacting with cytosolic factors, includingApaf-1 [92] and caspase-9, both of which play essential roles inthe cytochrome c-dependent mitochondrial pathway of apoptosis[32,46,93,93]. Bcl-2, inhibits cytochrome c translocation, therebyblocking caspase activation and apoptosis [45,88]. Up-regulationof Bcl-2 expression has been identified as a critical step by whichgrowth factors promote cell survival [6,83] and neurons expressingBcl-2 protein rarely exhibit gross nuclear or biochemical evidenceof apoptosis [17]. It has been suggested that Akt activation attenu-ates Bcl-2 suppression by BAD after an insult, since Bcl-2 and Bcl-XL proteins are negatively regulated by BAD [87,91]. We observeda significant increase in Bcl-2 expression during HLG (1.4-fold)compared to control (normoxia); furthermore addition of 1 nMGH increased Bcl-2 expression 1.6-fold during normoxia, and 1.7-fold during HLG; this increase suggests that Bcl-2 could performan important role in regulating protective mechanisms in responseto ischemia after 24 h of re-oxygenation. These results are consis-tent with those observed in rats infused with GH (100 lg/d) andGHRP-6 (hexareline, 150 lg/d), which increased IGF-I levels inhypothalamus (300%), hippocampus (200%), and cerebellum(150%) in comparison to the vehicle. These results coincided withincreases in Akt activation and Bcl-2 levels in the same areas thatIGF-I was augmented, and showed that GH and GHRP-6 couldstimulate the intracellular pathways involved in cell survival in re-sponse to growth factors [24]. Furthermore, when local GH wasimmunoneutralized by the anti-cGH IgGs, the Bcl-2 expression de-creased 2.1-fold during normoxia and 5.1-fold during HLG, sug-gesting that the locally expressed GH may have a role in theactivation of Bcl-2 neuroprotective mechanisms. Similarly in theretinal ganglion cells, GH immunoneutralization induced the inhi-bition of Akt-phosphorylation and initiated apoptosis through acti-vation of caspase-3 and PARP-1 cleavage [70].
GH exists in a variety of molecular isoforms or variants [4], andwe demonstrated a pattern of GH heterogeneity in the chicken cer-ebellum that is different from that observed in the pituitary [1]. Ithas been shown that the submonomeric 15-kDa variant is the mostabundant GH isoform expressed in the chicken cerebellum, inaccordance with previous results showing that the 15-kDa GH isproduced in the chick embryo [1,4].
A 15 kDa rcGH fragment, obtained by controlled proteolysis, hasshown several bioactivities, such as: stimulation of 3H-thymidineincorporation into bovine brain endothelial cells in a dose-depen-dent manner, in an angiogenic bioassay; strong inhibition of50-monodeiodinase in embryonic chicken hepatocyte cultures;and stimulation of proliferation of bovine endothelial cellsin vitro [5]. We tested this purified 15-kDa rcGH fragment [5] inthe chicken cerebellar cell cultures during HLG; it had an effecton cellular viability analogous to that observed with the completercGH, although required a higher concentration (10 nM) to show astronger effect (1.6-fold). This result correlates with a dose-depen-dent effect of the purified 15-kDa GH fragment observed on theendothelial proliferation bioassay [5]. Since this GH variant in-creased cellular viability, we also tested its effect on caspase-3activity during HLG in order to determine its anti-apoptotic effects.
At a concentration of 10 nM, the 15-kDa GH fragment significantlydecreased caspase-3 activity (1.3-fold). In previous reports, ourgroup has shown that cGH can be enzymatically cleaved to pro-duce a 15 kDa fragment, corresponding to the N-terminus of themonomeric hormone, which was recognized marginally (0.5%)and with much less affinity by the conventional liver GH receptorbut it was, however, still able to exert clearly several bioactivities[5]. It has also been described that a 15 kDa variant is the mostabundant GH moiety in the embryonic neural retina and the vitre-ous humor [7], and that a retinal tissue extract is capable to metab-olize exogenous chicken pituitary GH and produce this 15-kDaisoform [40]. This 15 kDa variants is also present in the embryonicpituitary [4,7] where it reaches its maximum and then decreasesits relative proportion after hatching. Data available suggest thatthe 15 kDa fragment might exert its biological effects through aseparate receptor [5]. Further studies are needed to determine ifthe 15 kDa variant present in the cerebellum [1] is structurallysimilar to that used in this work, produced in vitro by controlledproteolysis. Also, since the 15 kDa variant is the most abundantGH moiety found in the cerebellum [1], a deeper characterizationof the GH receptor in this organ should be encouraged.
In summary, we showed in this work that in response to hypox-ia or ischemic insults, GH concentration increases both in thewhole brain and in cerebellum of live embryos, as well as inembryonic cerebellar cells cultured in vitro. On the other hand, incells treated under HLG conditions, GH increases cell viabilityand survival, exerts an anti-apoptotic effect that involves a de-crease in DNA fragmentation and a diminished activity of cas-pase-3. In this model of ischemia, GH is capable to activate anti-apoptotic signaling pathways (PI3K/Akt and Bcl-2), but its actionsare abolished by wortmannin. Also, a 15-kDa fragment of rcGHshowed analogous effects on cellular viability and in the inhibitionof apoptosis in the model studied. Future studies should determineand characterize how these effects may be mediated by autocrine/paracrine mechanisms.
Acknowledgments
C.A.B. and J.L.A. are enrolled in the Biomedical Sciences| Ph.D.Program, whereas C.E.R.M. and D.A.G. were enrolled in the Neuro-biology M.Sc. Program, both at UNAM. We thank Gerardo Courtois(Lab assistant) as well as E. Nydia Hernández from the MicroscopyUnit, and Ramón Martínez from the Informatics Unit for technicalsupport, Dr. Michael C. Jeziorski for help in the use of the hypoxicchamber, Dr. Luz Navarro for help in statistical analysis and Doro-thy D. Pless for critically editing the manuscript. Grants fromCONACyT (F1-60296, 118353, 178335) and PAPIIT-UNAM (IN-210209) of México supported this work. CAB, JLA, CERM and DAGreceived graduate fellowships from CONACYT (184939, 200220,225281 and 231731, respectively).
References
[1] C. Alba-Betancourt, J. Ávila-Mendoza, S.M. Ahumada-Solórzano, A.J. Rodríguez-Méndez, M. Carranza, S. Harvey, C. Arámburo, M. Luna, Expression, cellulardistribution, and heterogeneity of growth hormone in the chicken cerebellumduring development, Gen. Comp. Endocrinol. 170 (2011) 528–540.
[2] C. Arámburo, R. Sáchez-García, B. Fenton-Navarro, J.G. Perera-Marín, C.Valverde-Rodríguez, Desarrollo de un radioinmunoensayo homólogo yespecífico para la determinación de hormona del crecimiento de pollo (cGH),Veterinaria México 20 (1989) 397–405.
[3] C. Arámburo, S. Navarrete, J.L. Montiel, R. Sánchez, L.R. Berghman, Purificationand electrophoretic analysis of glycosylated chicken growth hormone (G-cGH): evidence of G-cGH isoforms, Gen. Comp. Endocrinol. 84 (1991) 135–146.
[4] C. Arámburo, M. Luna, M. Carranza, M. Reyes, H. Martínez-Coria, C.G. Scanes,Growth hormone size variants: changes in the pituitary during development ofthe chicken, Proc. Soc. Exp. Biol. Med. 223 (2000) 67–74.
[5] C. Arámburo, M. Carranza, M. Reyes, M. Luna, H. Martínez-Coria, L. Berúmen,C.G. Scanes, Characterization of a bioactive 15 kDa fragment produced by

30 C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31
proteolytic cleavage of chicken growth hormone, Endocrine 15 (2001) 231–240.
[6] N.L. Baker, V. Carlo Russo, O. Bernard, A.J. D’Ercole, G.A. Werther, Interactionsbetween bcl-2 and the IGF-I system control apoptosis in the developing mousebrain, Brain Res. Dev. Brain Res. 118 (1999) 109–118.
[7] M.L. Baudet, E.J. Sanders, S. Harvey, Retinal growth hormone in the chickembryo, Endocrinology 144 (2003) 5459–5468.
[8] E.J. Beilharz, C.E. Williams, M. Dragunow, E.S. Sirimanne, P.D. Gluckman,Mechanisms of delayed cell death following hypoxic–ischemic injury in theimmature rat: evidence for apoptosis during selective neuronal loss, Mol. BrainRes. 29 (1995) 1–14.
[9] E. Bonfoco, D. Krainc, M. Ankarchona, P. Nicotera, S.A. Lipton, Apoptosis andnecrosis: two distinct events induced respectively, by mild and intense insultswith N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures,Proc. Natl. Acad. Sci. USA 92 (1995) 162–166.
[10] C. Bossenmeyer, R. Chihab, S. Muller, H. Schroeder, J.L. Daval, Hypoxia/reoxygenation induces apoptosis through biphasic induction of proteinsynthesis in central neurons, Brain Res. 787 (1998) 107–116.
[11] G.J. Brewer, J.R. Torricelli, E.K. Evege, P.J. Price, Neurobasal 1% Medium/B27supplement: a new serum-free medium combination for survival of neurons,Focus 16 (1994) 6–9.
[12] B.M. Burgering, P.J. Coffer, Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction, Nature 376 (1995) 599–602.
[13] K.A. Burton, E.B. Kabigting, D.K. Clifton, R. Steiner, Growth hormone receptormessenger ribonucleic acid distribution in the adult male rat and itscolocalization in hypothalamic somatostatin neurons, Endocrinology 130(1992) 958–963.
[14] E.J. Camm, M.E. Gibbs, R. Harding, T. Mulder, S.M. Rees, Prenatal hypoxiaimpairs memory function but does not result in overt structural alterations inthe postnatal chick brain, Dev. Brain Res. 160 (2005) 9–18.
[15] M.H. Cardone, N. Roy, H.R. Stennicke, G.S. Salvesen, T.F. Franke, E. Stanbridge,S. Frisch, J.C. Reed, Regulation of cell death protease caspase-9 byphosphorylation, Science 282 (1998) 1318–1321.
[16] H. Chung, E. Kim, D.H. Lee, S. Seo, S. Ju, D. Lee, H. Kim, S. Park, Ghrelin inhibitsapoptosis in hypothalamic neuronal cells during oxygen–glucose deprivation,Endocrinology 148 (2007) 148–159.
[17] R.S. Clark, J. Chen, S.C. Watkins, P.M. Kochanek, M. Chen, R.A. Stetler, J.E.Loeffert, S.H. Graham, Apoptosis suppressor gene bcl-2 expression aftertraumatic brain injury in rats, J. Neurosci. 17 (1997) 9172–9182.
[18] G.M. Cohen, Caspases: the executioners of apoptosis, Biochem. J. 326 (1997) 1–16.
[19] S.R. Datta, H. Dudek, X. Tao, S. Masters, H. Fu, Y. Gotoh, M.E. Greenberg, Aktphosphorylation of BAD couples survival signals to the cell-intrinsic deathmachinery, Cell 91 (1997) 231–241.
[20] J.B. Deijen, L.I. Arwert, Cognitive status of adult growth hormone (GH)-deficient patients and GH-induced neuropsychological changes, in: F. Nyberg(Ed.), The Somatotrophic Axis in Brain Function, Elsevier Academic Press, SanDiego, California, USA, 2006, pp. 287–300.
[21] L. del Peso, M. Gonzalez-Garcia, C. Page, R. Herrera, G. Nunez, Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt, Science 278(1997) 687–689.
[22] C.P. Donahue, R.V. Jensen, T. Ochiishi, I. Eisenstein, M. Zhao, T. Shors, K.S. Kosik,Transcriptional profiling reveals regulated genes in the hippocampus duringmemory formation, Hippocampus 12 (2002) 821–833.
[23] I. Ferrer, A.M. Planas, Signaling of cell death and cell survival following focalcerebral ischemia: life and death struggle in the penumbra, J. Neuropathol.Exp. Neurol. 62 (2003) 329–339.
[24] L.M. Frago, C. Pañeda, A.L. Dickson, A.K. Hewson, J. Argente, J.A. Chowen,Growth hormone (GH) and GH-releasing peptide-6 increase brain insulin-likegrowth factor-I expression and activate intracellular signaling pathwaysinvolved in neuroprotection, Endocrinology 143 (2002) 4113–4122.
[25] T.F. Franke, S.I. Yang, T.O. Chan, K. Datta, A. Kazlauskas, D.K. Morrison, D.R.Kaplan, P.N. Tsichlis, The protein kinase encoded by the Akt proto-oncogene isa target of the PDGF-activated phosphatidylinositol 3-kinase, Cell 81 (1995)727–736.
[26] P. Fuentes-Prior, G.S. Salvesen, The protein structures that shape caspaseactivity, specificity, activation and inhibition, Biochem. J. 384 (2004) 201–232.
[27] G. Fuh, B.C. Cunningham, R. Fukunaga, S. Nagata, D.V. Goeddel, J.A. Wells,Rational design of potent antagonists to the human growth hormone receptor,Science 256 (1992) 1677–1680.
[28] M.E. Gibson, B.H. Han, J. Choi, C.M. Knudson, S.J. Korsmeyer, M. Parsadanian,D.M. Holtzman, BAX contributes to apoptotic-like death following neonatalhypoxia–ischemia: evidence for distinct apoptosis pathways, Mol. Med. 7(2001) 644–655.
[29] J.R. González-Juanatey, R. Piñeiro, M.J. Iglesias, O. Gualillo, P.A. Kelly, C.Diéguez, F. Lago, GH prevents apoptosis in cardiomyocytes cultured in vitrothrough a calcineurin-dependent mechanism, J. Endocrinol. 180 (2004) 325–335.
[30] K. Gustafson, H. Hagberg, B. Bengtsson, C. Brantsing, J. Isgaard, Possibleprotective role of growth hormone in hypoxia-ischemia in neonatal rats,Pediatr. Res. 43 (1999) 318–323.
[31] A. Haeffner, O. Déas, B. Mollereau, J. Estaquier, A. Mignon, N. Haeffner-Cavaillon, B. Charpentier, A. Senik, F. Hirsch, Growth hormone prevents humanmonocytic cells from Fas-mediated apoptosis by up-regulating Bcl-2expression, Eur. J. Immunol. 29 (1999) 334–344.
[32] R. Hakem, A. Hakem, G.S. Duncan, J.T. Henderson, M. Woo, M.S. Soengas, A.Elia, J.L. de la Pompa, D. Kagi, W. Khoo, J. Potter, R. Yoshida, S.A. Kaufman, S.W.Lowe, J.M. Penninger, T.W. Mak, Differential requirement for caspase-9 inapoptotic pathways in vivo, Cell 94 (1998) 339–352.
[33] U. Hallin, E. Kondo, Y. Ozaki, H. Hagberg, F. Shibasaki, K. Blomgren, Bcl-2phosphorylation in the BH4 domain precedes caspase-3 activation and celldeath after neonatal cerebral hypoxic–ischemic injury, Neurobiol. Dis. 21(2006) 478–486.
[34] S. Harvey, Extrapituitary growth hormone, Endocrine 38 (2010) 335–359.[35] S. Harvey, C.D. Johnson, P. Sharma, E.J. Sanders, K.L. Hull, Growth hormone: a
paracrine growth factor in embryonic development?, Comp Biochem. Physiol.C: Pharmacol. Toxicol. Endocrinol. 119 (1998) 305–315.
[36] S. Harvey, C.D. Johnson, E.J. Sanders, Growth hormone in neural tissues of thechick embryo, J. Endocrinol. 169 (2001) 487–498.
[37] S. Harvey, K. Hull, Neural growth hormone, J. Mol. Neurosci. 20 (2003) 1–14.[38] S. Harvey, M.L. Baudet, A. Murphy, M. Luna, K.L. Hull, C. Arámburo, Testicular
growth hormone (GH): GH expression in spermatogonia and primaryspermatocytes, Gen. Comp. Endocrinol. 139 (2004) 158–167.
[39] S. Harvey, M.L. Baudet, E.J. Sanders, Growth hormone and cell survival in theneural retina: caspase dependence and independence, NeuroReport 17 (2006)1715–1718.
[40] S. Harvey, B.T. Martin, M.L. Baudet, P. Davis, Y. Sauve, E.J. Sanders, Growthhormone in the visual system: comparative endocrinology, Gen. Comp.Endocrinol. 153 (2007) 124–131.
[41] S. Hojvat, G. Baker, L. Kirsteins, A.M. Lawrence, Growth hormone (GH)immunoreactivity in the rodent and primate CNS: distribution,characterization and presence post-hypophysectomy, Brain Res. 239 (1982)543–557.
[42] C.M. Jacobs, P. Aden, G.H. Mathisen, E. Khoung, M. Gaarder, E.M. Loberg, J.Lomo, J. Maehlen, R.E. Paulsen, Chicken cerebellar granule neurons rapidlydevelop excitotoxicity in culture, J. Neurosci. Methods 156 (2006) 129–135.
[43] S. Jeay, G.E. Sonenshein, M.C. Postel-Vinay, E. Baixeras, Growth hormoneprevents apoptosis through activation of nuclear factor-kB in interleukin-3-dependent Ba/F3 cell line, Mol. Endocrinol. 14 (2000) 650–661.
[44] W. Kiess, B. Gallaher, Hormonal control of programmed cell death/apoptosis,Eur. J. Endocrinol. 138 (1998) 482–491.
[45] R.M. Kluck, E. Bossy-Wetzel, D.R. Green, D.D. Newmeyer, The release ofcytochrome c from mitochondria: a primary site for Bcl-2 regulation ofapoptosis, Science 275 (1997) 1132–1136.
[46] K. Kuida, Caspase-9, Int. J. Biochem. Cell Biol. 32 (2000) 121–124.[47] U.K. Laemmli, Cleavage of structural proteins during assembly of the head of
bacteriophage T4, Nature 227 (1970) 680–685.[48] Z. Lai, M. Emtner, P. Roos, F. Nyberg, Characterization of putative growth
hormone receptors in human choroid plexus, Brain Res. 546 (1991) 222–226.[49] R.C. Li, S.Z. Guo, M. Raccurt, E. Moudilou, G. Morel, K.R. Brittian, D. Gozal,
Exogenous growth hormone attenuates cognitive deficits induced byintermittent hypoxia in rats, Neuroscience 196 (2011) 237–250.
[50] P.E. Lobie, J. Garcia-Aragon, D.T. Lincoln, R. Barnard, J.N. Wilcox, M.J. Walters,Localization and ontogeny of growth hormone receptor gene expression in thecentral nervous system, Brain Res. Dev. Brain Res. 74 (1993) 225–233.
[51] E.H. Lo, T. Dalkara, M.A. Moskowitz, Mechanisms, challenges and opportunitiesin stroke, Nat. Rev. Neurosci. 4 (2003) 399–415.
[52] M. Luna, L. Huerta, L. Berumen, H. Martínez-Coria, S. Harvey, C. Arámburo,Growth hormone in the male reproductive tract of the chicken: heterogeneityand changes during ontogeny and maturation, Gen. Comp. Endocrinol. 137(2004) 37–49.
[53] M. Luna, N. Barraza, L. Berumen, M. Carranza, E. Pedernera, S. Harvey, C.Arámburo, Heterogeneity of growth hormone immunoreactivity in lymphoidtissues and changes during ontogeny in domestic fowl, Gen. Comp. Endocrinol.144 (2005) 28–37.
[54] M. Luna, A.J. Rodríguez-Mendez, L. Berumen, M. Carranza, J. Riesgo-Escovar,M.L. Baudet, S. Harvey, C. Arámburo, Immune growth hormone (GH):localization of GH and GH mRNA in the bursa of Fabricius, Dev. Comp.Immunol. 32 (2008) 1313–1325.
[55] E. Lyuh, H.J. Kim, M. Kim, J.K. Lee, K.S. Park, K.Y. Yoo, K.W. Lee, Y.O. Ahn, Dose-specific or dose-dependent effect of growth hormone treatment on theproliferation and differentiation of cultured neuronal cells, Growth Horm. IGFRes. 17 (2007) 315–322.
[56] H. Martínez-Coria, J. López-Rosales, M. Carranza, L. Berumen, M. Luna, C.Arámburo, Differential secretion of chicken growth hormone variants afterGHRH stimulation, in vitro, Endocrine 17 (2002) 91–102.
[57] S.L. Mehta, N. Manhas, R. Raghubir, Molecular targets in cerebral ischemia fordeveloping novel therapeutics, Brain Res. Rev. 54 (2007) 34–66.
[58] A. Meisel, K. Prass, T. Wolf, U. Dirnagl, Stroke, in: M. Bähr (Ed.),Neuroprotection, Models, Mechanisms and Therapies, Wiley-VCH,Weinheim, Germany, 2004, pp. 1–30.
[59] R.J. Mullen, C.R. Buck, A.M. Smith, NeuN, a neuronal specific nuclear protein invertebrates, Development 116 (1992) 201–211.
[60] A. Mustafa, A. Adem, P. Roos, F. Nyberg, Sex differences in binding of humangrowth hormone to rat brain, Neurosci. Res. 19 (1994) 93–99.
[61] F.J. Northington, D.M. Ferriero, D.L. Flock, L.J. Martin, Delayedneurodegeneration in neonatal rat thalamus after hypoxia–ischemia isapoptosis, J. Neurosci. 21 (2001) 1931–1938.
[62] F. Nyberg, Growth hormone in the brain: characteristics of specific braintargets for the hormone and their functional significance, Front.Neuroendocrinol. 21 (2000) 330–348.

C. Alba-Betancourt et al. / General and Comparative Endocrinology 183 (2013) 17–31 31
[63] A. Palmetshofer, D. Zechner, T.A. Luger, A. Barta, Splicing variants of the humangrowth hormone mRNA: detection in pituitary, mononuclear cells and dermalfibroblast, Mol. Cell. Endocrinol. 113 (1995) 225–234.
[64] R. Ramesh, W.J. Kuenzel, J.D. Buntin, J.A. Proudman, Identification of growthhormone and prolactin containing neurons within the avian brain, Cell TissueRes. 299 (2000) 371–383.
[65] C.L. Render, K.L. Hull, S. Harvey, Neural expression of the pituitary GH gene, J.Endocrinol. 147 (1995) 413–422.
[66] A.J. Rodríguez-Méndez, J.L. Luna-Acosta, M. Carranza, S. Harvey, C. Arámburo,M. Luna, Growth hormone expression in stromal and non-stromal cells in thebursa of Fabricius during bursal development and involution: causalrelationships?, Gen Comp. Endocrinol. 167 (2010) 297–307.
[67] W. Rosamond, K. Flegal, G. Friday, K. Furie, A. Go, K. Greenlund, N. Haase, M.Ho, V. Howard, B. Kissela, S. Kittner, D. Lloyd-Jones, M. McDermott, J. Meigs, C.Moy, G. Nichol, C.J. O’Donnell, V. Roger, J. Rumsfeld, P. Sorlie, J. Steinberger, T.Thom, S. Wasserthiel-Smoller, Y. Hong, Heart disease and stroke statistics –2007 update: a report from the American Heart Association StatisticsCommittee and Stroke Statistics Subcommittee, Circulation 115 (2007) e69–e171.
[68] E.J. Sanders, S. Harvey, Growth hormone as an early embryonic growth anddifferentiation factor, Anat. Embryol. 209 (2004) 1–9.
[69] E.J. Sanders, E. Parker, C. Arámburo, S. Harvey, Retinal growth hormone is ananti-apoptotic factor in embryonic retinal ganglion cell differentiation, Exp.Eye Res. 81 (2005) 551–560.
[70] E.J. Sanders, E. Parker, S. Harvey, Retinal ganglion cell survival in development:mechanisms of retinal growth hormone action, Exp. Eye Res. 83 (2006) 1205–1214.
[71] E.J. Sanders, E. Parker, S. Harvey, Growth hormone-mediated survival ofembryonic retinal ganglion cells: signaling mechanisms, Gen. Comp.Endocrinol. 156 (2008) 613–621.
[72] E.J. Sanders, M.L. Baudet, E. Parker, S. Harvey, Signalling mechanismsmediating local GH action in the neural retina of the chick embryo, Gen.Comp. Endocrinol. 163 (2009) 63–69.
[73] A. Scheepens, E. Sirimanne, E. Beilharz, B.H. Breier, M.J. Waters, P.D. Gluckman,C.E. Williams, Alterations in the neural growth hormone axis followinghypoxic–ischemic brain injury, Brain Res. Mol. Brain Res. 68 (1999) 88–100.
[74] A. Scheepens, C.E. Williams, B.H. Breier, J. Guan, P.D. Gluckman, A role for thesomatotropic axis in neural development, injury and disease, J. Pediatr.Endocrinol. Metab. 13 (2000) 1483–1491.
[75] A. Scheepens, E.S. Sirimanne, B.H. Breier, R.G. Clark, P.D. Gluckman, C.E.Williams, Growth hormone as a neuronal rescue factor during recovery fromCNS injury, Neuroscience 104 (2001) 677–687.
[76] A. Scheepens, T.A. Moderscheim, P.D. Gluckman, The role of growth hormonein neural development, Horm. Res. 64 (2005) 66–72.
[77] H.B. Segard, S. Moulin, S. Boumard, C. Augier de Crémiers, P.A. Kelly, J. Finidori,Autocrine growth hormone production prevents apoptosis and inhibitsdifferentiation in C2C12 myoblasts, Cell. Signal. 15 (2003) 615–623.
[78] D.H. Shin, E. Lee, J.W. Kim, B.S. Kwon, M.K. Jung, Y.H. Jee, J. Kim, S. Bae, Y.P.Chang, Protective effect of growth hormone on neuronal apoptosis afterhypoxia–ischemia in the neonatal rat brain, Neurosci. Lett. 354 (2004) 64–68.
[79] A.V. Sirotkin, A.V. Makarevich, GH regulates secretory activity and apoptosis incultured bovine granulosa cells through the activation of the cAMP/proteinkinase A system, J. Endocrinol. 163 (1999) 317–327.
[80] T. Sugawara, M. Fujimura, Y. Morita-Fujimura, M. Kawase, P.H. Chan,Mitochondrial release of cytochrome c corresponds to the selectivevulnerability of hippocampal CA1 neurons in rats after transient globalcerebral ischemia, J. Neurosci. 19 (1999) 1–6.
[81] T. Sugawara, M. Fujimura, N. Noshita, G.W. Kim, A. Saito, T. Hayashi, P.Narasimhan, C.M. Maier, P.H. Chan, Neuronal death/survival signalingpathways in cerebral ischemia, NeuroRx 1 (2004) 17–25.
[82] A. Takeda, H. Onodera, A. Sugimoto, K. Kogui, M. Obinata, S. Shibahara,Coordinated expression of messenger RNAs for nerve growth factor, brain-derived neurotrophic factor and neurotrophin-3 in the rat hippocampusfollowing transient forebrain ischemia, Neuroscience 55 (1993) 23–31.
[83] M. Tamatani, S. Ogawa, G. Nuñez, M. Tokyama, Growth factors preventchanges in Bcl-2 and Bax expression and neuronal apoptosis induced by nitricoxide, Cell Death Differ. 5 (1998) 911–919.
[84] J.R. Tennant, Evaluation of the trypan blue technique for determination of cellviability, Transplantation 2 (1964) 685–694.
[85] R. Vannucci, Experimental biology of cerebral hypoxia-ischemia: relation toperinatal brain damage, Pediatr. Res. 27 (1990) 317–326.
[86] J.J. Volpe, Cerebellum of the premature infant: rapidly developing, vulnerable,clinically important, J. Child Neurol. 24 (2009) 1085–1104.
[87] E. Yang, J. Zha, J. Jockel, L.H. Boise, C.B. Thompson, S.J. Korsmeyer, BAD, aheterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes celldeath, Cell 80 (1995) 285–291.
[88] J. Yang, X. Liu, K. Bhalla, C.N. Kim, A.M. Ibrado, J. Cai, T.I. Peng, D.P. Jones, X.Wang, Prevention of apoptosis by Bcl-2: release of cytochrome c frommitochondria blocked, Science 275 (1997) 1129–1132.
[89] H. Yoshizato, T. Fujikawa, H. Soya, M. Tanaka, K. Nakashima, The growthhormone (GH) gene is expressed in the lateral hypothalamus: enhancement byGH-releasing hormone and repression by restraint stress, Endocrinology 139(1998) 2545–2551.
[90] H. Yoshizato, T. Fujikawa, M. Shibata, M. Tanaka, K. Nakashima, Stimulation ofgrowth hormone gene expression in the pituitary and brain by Panax ginsengC. A. Meyer, Endocr. J. 46 (1999) 85–88.
[91] J. Zha, H. Harada, E. Yang, J. Jockel, S.J. Korsmeyer, Serine phosphorylation ofdeath agonist BAD in response to survival factor results in binding to 14–3-3not BCL-X(L), Cell 87 (1996) 619–628.
[92] H. Zou, W.J. Henzel, X. Liu, A. Lutschg, X. Wang, Apaf-1, a human proteinhomologous to C. elegans CED-4, participates in cytochrome c-dependentactivation of caspase-3, Cell 90 (1997) 405–413.
[93] H. Zou, Y. Li, X. Liu, X. Wang, An APAF-1 cytochrome c multimeric complex is afunctional apoptosome that activates procaspase-9, J. Biol. Chem. 274 (1999)11549–11556.