neodymium-doped alkaline -earth oxide catalysts for propane oxidative dehydrogenation. part ii....
TRANSCRIPT

Applied Catalysis A: General 359 (2009) 55–61
Neodymium-doped alkaline-earth oxide catalysts for propane oxidativedehydrogenation. Part II. Catalytic properties
Bistra Savova a, Diana Filkova a,*, Dorel Crisan b, Maria Crisan b, Malina Raileanu b,Nicolae Dragan b, Lachezar Petrov c, Jacques C. Vedrine d
a Institute of Catalysis, Bulgarian Academy of Sciences, Acad. G. Bonchev Str., Bl. 11, 1113 Sofia, Bulgariab Ilie Murgulescu Institute of Physical Chemistry, Roumanian Academy, 202 Splaiul Independentei, 060021 Bucharest, Romaniac SABIC Chair in Catalysis, King Abdulaziz University, Jeddah, Saudi Arabiad Laboratoire de Reactivite de Surface, Universite P. et M. Curie-Paris 06, 4 Place Jussieu, F-75252 Paris, France
A R T I C L E I N F O
Article history:
Received 11 August 2008
Received in revised form 12 February 2009
Accepted 16 February 2009
Available online 27 February 2009
Keywords:
Propane ODH
Oxide catalysts
Lattice defects
Olefin selectivity
A B S T R A C T
Two series of alkaline-earth oxides of Mg, Ca, and Sr, doped with 5 mol% Nd2O3 and synthesised by two
different procedures: (i) evaporation of nitrate aqueous solutions followed by nitrate decomposition and
(ii) sol–gel technique, have been studied for propane oxidative dehydrogenation reaction. Solid solutions
of both Nd in alkaline-earth oxides and of alkaline-earth elements in Nd2O3 were shown to be formed as
a result of both preparation procedures. It was observed that Nd increased strongly propane conversion,
while propene and ethene selectivity generally followed the catalyst basicity measured by CO2-TPD. The
sol–gel preparation led to materials exhibiting enhanced alkene selectivity, higher basicity and higher
content of structural defects. The selectivity was observed to increase with propane conversion at
increased contact time at difference with what was usually reported for redox-type catalysts. The
relationship between catalyst preparation method, solid solution formation, lattice structural defects,
surface basicity, and catalytic performance has been discussed.
� 2009 Elsevier B.V. All rights reserved.
Contents lists available at ScienceDirect
Applied Catalysis A: General
journa l homepage: www.e lsev ier .com/ locate /apcata
1. Introduction
Oiland naturalgasaretheworldpremiersourcesofenergyandarefundamental to almost every important function of modern life.However, their limited natural reserves require wise and effectiveutilization. One route for achieving this goal is to upgrade lightalkanes C1–C4 [1] which are formed during the oil processing andwhicharepresentinvariableamountsinnaturalgas,dependingonitsorigin field. Creating effective catalytic processes for alkane utiliza-tion is a real challenge for scientists working in catalysis field and forcontemporary petrochemical and oil processing industry [2–4].
Common feature of light alkanes is their chemical inertness andthe higher reactivity of the reaction products, which easily undergosecondary reactions, thus diminishing the selectivity of theprocess. The activation of alkanes by dehydrogenation has beendeveloped industrially but it is not an effective process becausedehydrogenation is endothermic and high energy consuming. Thedehydrogenation catalysts have short lifetime due to fastdeactivation by coking and have to be regenerated periodically.
* Corresponding author. Tel.: +359 2 9792531; fax: +359 2 9712967.
E-mail addresses: [email protected] (D. Filkova), [email protected] (D. Crisan),
[email protected] (L. Petrov), [email protected] (J.C. Vedrine).
0926-860X/$ – see front matter � 2009 Elsevier B.V. All rights reserved.
doi:10.1016/j.apcata.2009.02.019
Regeneration process releases carbon oxides and other harmfulemissions. These disadvantages can be avoided by oxidativedehydrogenation (ODH), which is an exothermal process andpermits to avoid thermodynamically limited conversion at lowtemperatures. The lifetime of catalysts used in this reaction is alsomuch longer than for dehydrogenation catalysts.
Therefore, one can expect that the reaction of ODH is apromising way for alkanes transformation to olefins due tosubstantial advantages it offers compared to the ordinarydehydrogenation [5–9]. However, in spite of huge research effortsdevoted to light alkanes’ selective oxidation, since the pioneerwork by Centi and Trifiro [10], the only successful processindustrially realised was n-butane to maleic anhydride on V–P–O catalyst. In his review article devoted to ODH of ethane andpropane Cavani et al. [11] stated that the potential advantagesoffered by this process at the moment are not enough to convincecompanies to abandon well established technologies, unless betteryield than that currently achieved can be obtained.
There are three main classes of catalysts studied for selectiveoxidation of light alkanes:
� Catalysts based on reducible metal oxides, typically transitionmetal oxides. A redox Mars and van Krevelen mechanism hasbeen shown to operate.

Table 1Catalyst samples under study, their specific surface area measured by BET method
and catalytic performance at 550 8C.
Samples Spec. surf.
area, m2 g�1
Propane
conversion, %
Selectivity, %
Propene Ethene
MgNd–Ev 11 38 9 6
CaNd–Ev 2 29 16 16
SrNd–Ev 0.5 16 24 22
MgNd–SG 60 32 33 25
CaNd–SG 13 17 31 22
SrNd–SG 1 5 56 16
MgO 26 10 64 16
CaO 0.6 11 48 26
SrCO3 1.2 7 62 17
Nd2O3 6 27 20 28
VMgO (Mg/V = 0.94) 8 8 57 13
B. Savova et al. / Applied Catalysis A: General 359 (2009) 55–6156
� Catalysts based on non-reducible or hardly reducible metaloxides. In such a case it has been shown that the reaction isinitiated on the catalyst surface and is then transferred to the gasphase (heterogeneous–homogeneous radical-type mechanism).� Noble metal-based catalysts usually considered as total oxida-
tion catalysts, except in specific conditions, e.g. at very high flowrate.
Non-redox catalysts based on doped or modified alkaline-earthoxides have been studied for methane oxidative coupling and morerecently for propane ODH [12–14]. For the last reaction vanadiumoxide-based catalysts have been widely studied [15–20]. The highperformance of such catalysts arises from the specific activity ofthe V–O bond in the C–H bond activation of alkanes, which isconsidered the rate-limiting step of the reaction [21]. Once formedon the catalyst surface, the alkyl species may form either olefins oroxygenates, or transform further to carbon oxides. The overoxidation may be suppressed by increasing catalyst surfacebasicity which is expected to enhance the desorption of theproduced alkenes [22,23].
In this paper we report experimental data on basic alkaline-earth oxides (AEOs) used for propane ODH. Nd2O3 is chosen as apromoter as it has shown high dehydrogenation ability in methaneoxidative coupling (the most inactive light alkane) [24–26]. Theobjective of this work is to study both the influence of the surfacebasicity and of the method of preparation on the catalyticperformance of two series of doped AEO catalysts of the samecomposition.
2. Experimental
2.1. Catalysts preparation and characterization
Six samples of 5 mol% Nd2O3–AEO (AEO = MgO, CaO, and SrO)were prepared by two methods, namely by evaporation of nitratesolutions and subsequent decomposition of nitrate mixtures(designated Ev samples) or by sol–gel technique (designated SGsamples). Pure MgO, CaO, SrCO3, and Nd2O3 were prepared byevaporation and decomposition, too, and studied for comparison.VMgO catalyst (V:Mg atomic ratio 1:1), used as a standard, wasprepared by evaporation and decomposition, starting fromammonium metavanadate and magnesium nitrate. The procedurewas described in Part I of this work [27].
The samples characterization by ICP chemical analysis, BETspecific surface area measurement, XRD, CO2-TPD, FTIR, and XPSspectroscopy was described in Part I of this work [27].
2.2. Catalytic activity measurements
A fixed bed flow reactor was used to study the propane ODHreaction in the range 350–600 8C. The reaction temperature wasmeasured by thermocouple placed in a coaxial well of 5 mm ofdiameter, positioned in the middle of the reactor. The inner reactordiameter was of 11 mm, so that the loaded catalyst made a layer ofabout 2 mm thick. The free volume of the quartz reactor after thecatalyst bed was filled with quartz particles to minimize thehomogeneous reactions. The propane (99.95%, Messer Chimco GasLtd.)–air (99.99%, Messer Chimco Gas Ltd.)–nitrogen (99.99%,Messer Chimco Gas Ltd.) mixture entered the reactor upstreamthrough two coaxial tubes and mixed just before the catalyst bed toavoid any reaction in the heated pre-catalyst zone. Matheson massflow controllers were used to fix the gas flows. All the testing setwas placed in a thermostat. The lines to the gas chromatographswere heated at about 100 8C to prevent water condensation.
The feed gas composition was C3H8/O2/N2 = 20/10/70 vol.% attotal gas flow 30–90 cm3 min�1. Catalyst fractions of 0.2–
0.315 mm particle size and of 30–110 mg weight were loadedinto the reactor. The contact time varied between 0.017 and 0.14 s.The catalyst samples in the reactor were kept under air flow at600 8C for 1 h before the catalytic test.
The gas chromatographic analysis was performed on-line bythree GC columns, namely Porapak Q and Molecular sieve 5A withtwo thermal conductivity detectors, and modified g-Al2O3 with aflame ionisation detector. CO2 and water were separated onPorapak Q column; methane, ethane, ethene, propane, propene,and C4 hydrocarbons were separated on a modified g-Al2O3;hydrogen, oxygen, and CO were analyzed on Molecular sieve 5A.Carbon, oxygen, and hydrogen balances were calculated for eachexperiment.
3. Results
3.1. Catalyst samples under study and their characterization
The studied samples, their specific surface area and catalyticresults at 550 8C are shown in Table 1. As discussed in Part I, theXRD analyses revealed presence of AEO and Nd2O3, SrCO3 (about62 wt.%) in SrNd–SG sample, CaCO3 and Ca(OH)2 (about 5 wt.%) inCaNd–SG. Solid solutions of Nd in AEO and of alkaline-earthelements (AEEs) in Nd2O3 were observed in all samples. Based onpowder XRD peak profile analysis, lattice constants, unit cellvolume, average grain dimension, lattice micro-strain, unit cellmass modification, and scattering factor of the shape parameters ofthe profiles have been estimated [27].
3.2. Catalytic activity measurements
The experiments with the reactor filled with only quartzparticles showed that the homogeneous reaction occurred at verylow extent in the temperature range under study and reachedabout 5% conversion at 600 8C.
The balances calculated for carbon and oxygen equaled100 � 5%. The catalytic reaction gave together with propene andethene a number of side products such as carbon oxides, methane,and smaller quantities of ethane, C4 hydrocarbons, water andhydrogen. The main products were propene, ethene and COx. Themain carbon oxide was CO2, the presence of CO being only detectedfor MgNd–Ev, MgNd–SG and Nd2O3.
Catalytic results for pure oxides studied for comparison, arepresented in Figs. 1 and 2. However, as we were not successful inobtaining pure SrO by evaporation technique, SrCO3 sample wassynthesised and studied instead. Actually, SrO had too high basicityand strong adsorption capacity for CO2, so the surface of Sr-containing samples was covered by carbonate, as shown by XPS
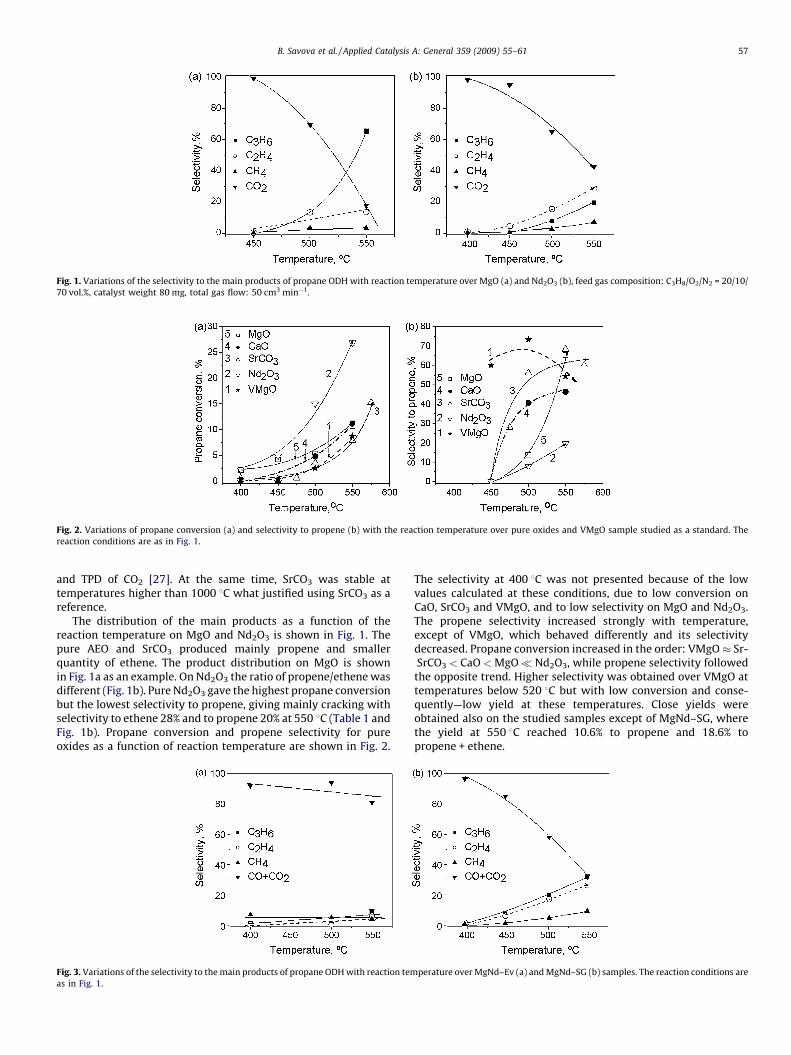
Fig. 1. Variations of the selectivity to the main products of propane ODH with reaction temperature over MgO (a) and Nd2O3 (b), feed gas composition: C3H8/O2/N2 = 20/10/
70 vol.%, catalyst weight 80 mg, total gas flow: 50 cm3 min�1.
Fig. 2. Variations of propane conversion (a) and selectivity to propene (b) with the reaction temperature over pure oxides and VMgO sample studied as a standard. The
reaction conditions are as in Fig. 1.
B. Savova et al. / Applied Catalysis A: General 359 (2009) 55–61 57
and TPD of CO2 [27]. At the same time, SrCO3 was stable attemperatures higher than 1000 8C what justified using SrCO3 as areference.
The distribution of the main products as a function of thereaction temperature on MgO and Nd2O3 is shown in Fig. 1. Thepure AEO and SrCO3 produced mainly propene and smallerquantity of ethene. The product distribution on MgO is shownin Fig. 1a as an example. On Nd2O3 the ratio of propene/ethene wasdifferent (Fig. 1b). Pure Nd2O3 gave the highest propane conversionbut the lowest selectivity to propene, giving mainly cracking withselectivity to ethene 28% and to propene 20% at 550 8C (Table 1 andFig. 1b). Propane conversion and propene selectivity for pureoxides as a function of reaction temperature are shown in Fig. 2.
Fig. 3. Variations of the selectivity to the main products of propane ODH with reaction tem
as in Fig. 1.
The selectivity at 400 8C was not presented because of the lowvalues calculated at these conditions, due to low conversion onCaO, SrCO3 and VMgO, and to low selectivity on MgO and Nd2O3.The propene selectivity increased strongly with temperature,except of VMgO, which behaved differently and its selectivitydecreased. Propane conversion increased in the order: VMgO � Sr-SrCO3 < CaO < MgO� Nd2O3, while propene selectivity followedthe opposite trend. Higher selectivity was obtained over VMgO attemperatures below 520 8C but with low conversion and conse-quently—low yield at these temperatures. Close yields wereobtained also on the studied samples except of MgNd–SG, wherethe yield at 550 8C reached 10.6% to propene and 18.6% topropene + ethene.
perature over MgNd–Ev (a) and MgNd–SG (b) samples. The reaction conditions are
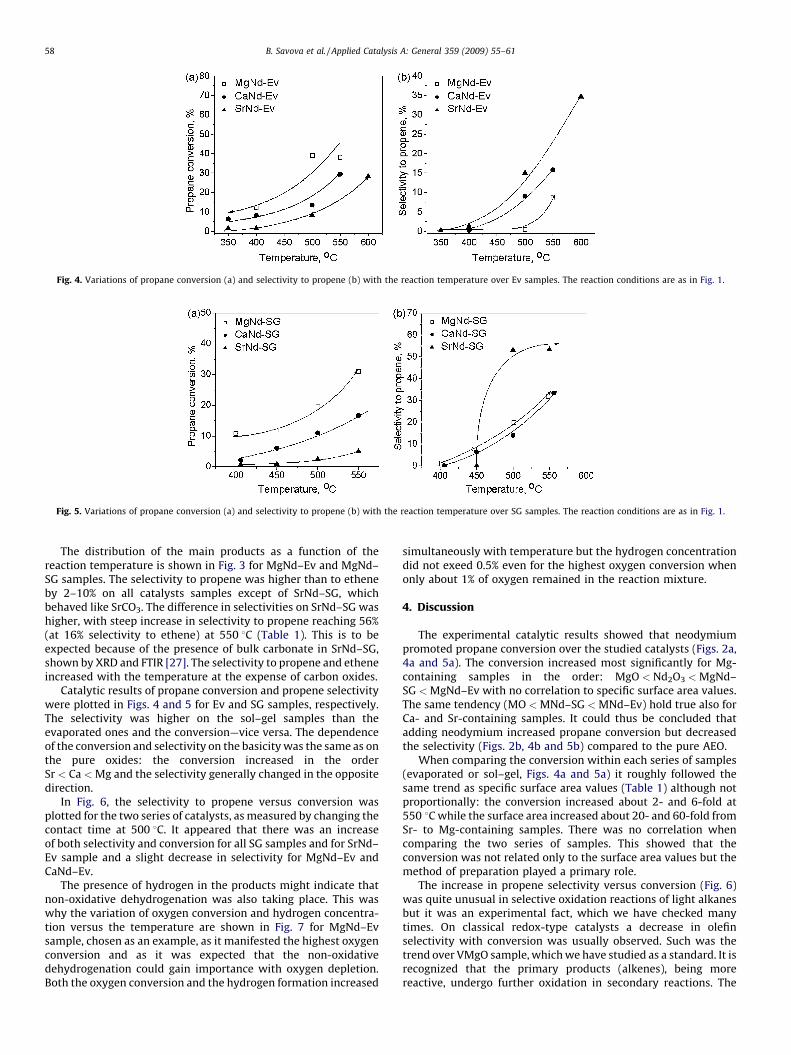
Fig. 4. Variations of propane conversion (a) and selectivity to propene (b) with the reaction temperature over Ev samples. The reaction conditions are as in Fig. 1.
Fig. 5. Variations of propane conversion (a) and selectivity to propene (b) with the reaction temperature over SG samples. The reaction conditions are as in Fig. 1.
B. Savova et al. / Applied Catalysis A: General 359 (2009) 55–6158
The distribution of the main products as a function of thereaction temperature is shown in Fig. 3 for MgNd–Ev and MgNd–SG samples. The selectivity to propene was higher than to etheneby 2–10% on all catalysts samples except of SrNd–SG, whichbehaved like SrCO3. The difference in selectivities on SrNd–SG washigher, with steep increase in selectivity to propene reaching 56%(at 16% selectivity to ethene) at 550 8C (Table 1). This is to beexpected because of the presence of bulk carbonate in SrNd–SG,shown by XRD and FTIR [27]. The selectivity to propene and etheneincreased with the temperature at the expense of carbon oxides.
Catalytic results of propane conversion and propene selectivitywere plotted in Figs. 4 and 5 for Ev and SG samples, respectively.The selectivity was higher on the sol–gel samples than theevaporated ones and the conversion—vice versa. The dependenceof the conversion and selectivity on the basicity was the same as onthe pure oxides: the conversion increased in the orderSr < Ca < Mg and the selectivity generally changed in the oppositedirection.
In Fig. 6, the selectivity to propene versus conversion wasplotted for the two series of catalysts, as measured by changing thecontact time at 500 8C. It appeared that there was an increaseof both selectivity and conversion for all SG samples and for SrNd–Ev sample and a slight decrease in selectivity for MgNd–Ev andCaNd–Ev.
The presence of hydrogen in the products might indicate thatnon-oxidative dehydrogenation was also taking place. This waswhy the variation of oxygen conversion and hydrogen concentra-tion versus the temperature are shown in Fig. 7 for MgNd–Evsample, chosen as an example, as it manifested the highest oxygenconversion and as it was expected that the non-oxidativedehydrogenation could gain importance with oxygen depletion.Both the oxygen conversion and the hydrogen formation increased
simultaneously with temperature but the hydrogen concentrationdid not exeed 0.5% even for the highest oxygen conversion whenonly about 1% of oxygen remained in the reaction mixture.
4. Discussion
The experimental catalytic results showed that neodymiumpromoted propane conversion over the studied catalysts (Figs. 2a,4a and 5a). The conversion increased most significantly for Mg-containing samples in the order: MgO < Nd2O3 < MgNd–SG < MgNd–Ev with no correlation to specific surface area values.The same tendency (MO < MNd–SG < MNd–Ev) hold true also forCa- and Sr-containing samples. It could thus be concluded thatadding neodymium increased propane conversion but decreasedthe selectivity (Figs. 2b, 4b and 5b) compared to the pure AEO.
When comparing the conversion within each series of samples(evaporated or sol–gel, Figs. 4a and 5a) it roughly followed thesame trend as specific surface area values (Table 1) although notproportionally: the conversion increased about 2- and 6-fold at550 8C while the surface area increased about 20- and 60-fold fromSr- to Mg-containing samples. There was no correlation whencomparing the two series of samples. This showed that theconversion was not related only to the surface area values but themethod of preparation played a primary role.
The increase in propene selectivity versus conversion (Fig. 6)was quite unusual in selective oxidation reactions of light alkanesbut it was an experimental fact, which we have checked manytimes. On classical redox-type catalysts a decrease in olefinselectivity with conversion was usually observed. Such was thetrend over VMgO sample, which we have studied as a standard. It isrecognized that the primary products (alkenes), being morereactive, undergo further oxidation in secondary reactions. The
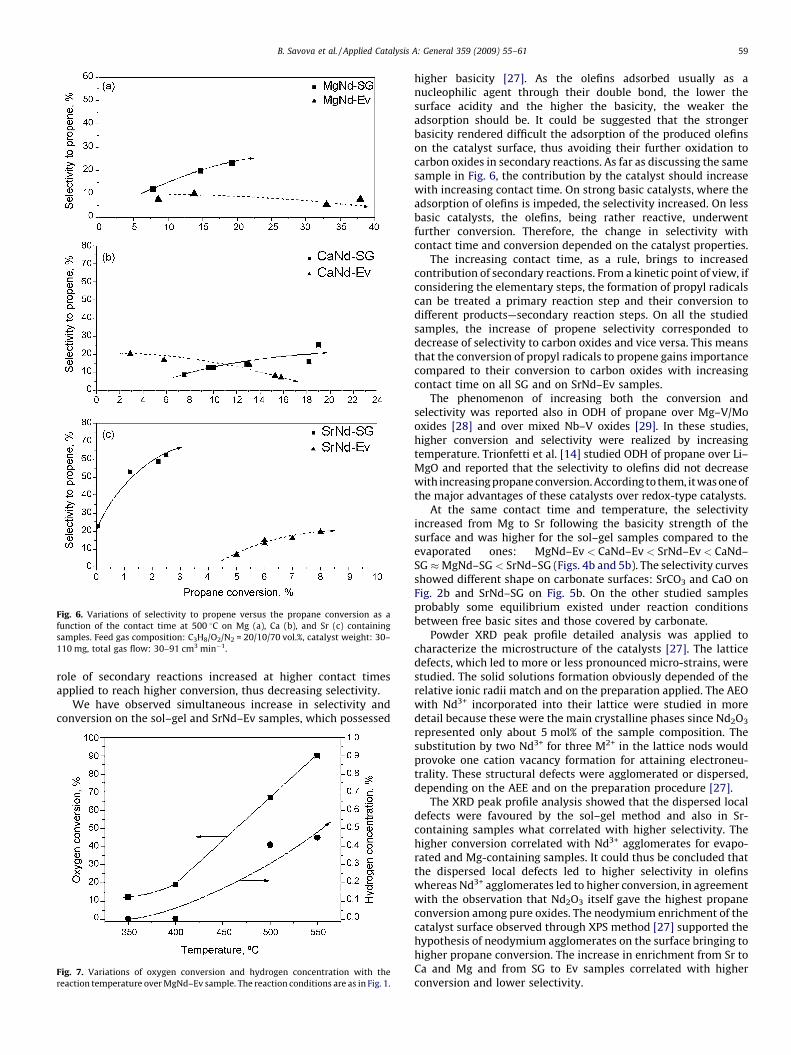
Fig. 6. Variations of selectivity to propene versus the propane conversion as a
function of the contact time at 500 8C on Mg (a), Ca (b), and Sr (c) containing
samples. Feed gas composition: C3H8/O2/N2 = 20/10/70 vol.%, catalyst weight: 30–
110 mg, total gas flow: 30–91 cm3 min�1.
B. Savova et al. / Applied Catalysis A: General 359 (2009) 55–61 59
role of secondary reactions increased at higher contact timesapplied to reach higher conversion, thus decreasing selectivity.
We have observed simultaneous increase in selectivity andconversion on the sol–gel and SrNd–Ev samples, which possessed
Fig. 7. Variations of oxygen conversion and hydrogen concentration with the
reaction temperature over MgNd–Ev sample. The reaction conditions are as in Fig. 1.
higher basicity [27]. As the olefins adsorbed usually as anucleophilic agent through their double bond, the lower thesurface acidity and the higher the basicity, the weaker theadsorption should be. It could be suggested that the strongerbasicity rendered difficult the adsorption of the produced olefinson the catalyst surface, thus avoiding their further oxidation tocarbon oxides in secondary reactions. As far as discussing the samesample in Fig. 6, the contribution by the catalyst should increasewith increasing contact time. On strong basic catalysts, where theadsorption of olefins is impeded, the selectivity increased. On lessbasic catalysts, the olefins, being rather reactive, underwentfurther conversion. Therefore, the change in selectivity withcontact time and conversion depended on the catalyst properties.
The increasing contact time, as a rule, brings to increasedcontribution of secondary reactions. From a kinetic point of view, ifconsidering the elementary steps, the formation of propyl radicalscan be treated a primary reaction step and their conversion todifferent products—secondary reaction steps. On all the studiedsamples, the increase of propene selectivity corresponded todecrease of selectivity to carbon oxides and vice versa. This meansthat the conversion of propyl radicals to propene gains importancecompared to their conversion to carbon oxides with increasingcontact time on all SG and on SrNd–Ev samples.
The phenomenon of increasing both the conversion andselectivity was reported also in ODH of propane over Mg–V/Mooxides [28] and over mixed Nb–V oxides [29]. In these studies,higher conversion and selectivity were realized by increasingtemperature. Trionfetti et al. [14] studied ODH of propane over Li–MgO and reported that the selectivity to olefins did not decreasewith increasing propane conversion. According to them, it was one ofthe major advantages of these catalysts over redox-type catalysts.
At the same contact time and temperature, the selectivityincreased from Mg to Sr following the basicity strength of thesurface and was higher for the sol–gel samples compared to theevaporated ones: MgNd–Ev < CaNd–Ev < SrNd–Ev < CaNd–SG �MgNd–SG < SrNd–SG (Figs. 4b and 5b). The selectivity curvesshowed different shape on carbonate surfaces: SrCO3 and CaO onFig. 2b and SrNd–SG on Fig. 5b. On the other studied samplesprobably some equilibrium existed under reaction conditionsbetween free basic sites and those covered by carbonate.
Powder XRD peak profile detailed analysis was applied tocharacterize the microstructure of the catalysts [27]. The latticedefects, which led to more or less pronounced micro-strains, werestudied. The solid solutions formation obviously depended of therelative ionic radii match and on the preparation applied. The AEOwith Nd3+ incorporated into their lattice were studied in moredetail because these were the main crystalline phases since Nd2O3
represented only about 5 mol% of the sample composition. Thesubstitution by two Nd3+ for three M2+ in the lattice nods wouldprovoke one cation vacancy formation for attaining electroneu-trality. These structural defects were agglomerated or dispersed,depending on the AEE and on the preparation procedure [27].
The XRD peak profile analysis showed that the dispersed localdefects were favoured by the sol–gel method and also in Sr-containing samples what correlated with higher selectivity. Thehigher conversion correlated with Nd3+ agglomerates for evapo-rated and Mg-containing samples. It could thus be concluded thatthe dispersed local defects led to higher selectivity in olefinswhereas Nd3+ agglomerates led to higher conversion, in agreementwith the observation that Nd2O3 itself gave the highest propaneconversion among pure oxides. The neodymium enrichment of thecatalyst surface observed through XPS method [27] supported thehypothesis of neodymium agglomerates on the surface bringing tohigher propane conversion. The increase in enrichment from Sr toCa and Mg and from SG to Ev samples correlated with higherconversion and lower selectivity.

B. Savova et al. / Applied Catalysis A: General 359 (2009) 55–6160
The literature data on the mechanism of propene formationover non-reducible catalysts is scarce as mostly catalysts contain-ing vanadium or other transition elements have been studied. It isgenerally accepted that on reducible metal oxides the reactionproceeds through Mars–van Krevelen redox mechanism. In most ofthe cases oxygen-containing compounds are formed either in smallquantity as by-products or as the main products. The non-redox-type catalysts do not give rise to oxygenates as they cannotintroduce oxygen into the hydrocarbon molecule but they may beeffective for hydrogen abstraction and generation of alkyl radicals.The processes with high probability proceed via the formation andfurther transformations of free radicals. This means that multiplereactions run—both homogeneous and heterogeneous [30,31]. Inthe case of propane oxidation, the activation of propane withgeneration of propyl radicals may occur either on the surface or bypyrolysis; this has been observed when operating at temperatureshigher than 450 8C [32–34].
For pure AEO and AEO–Nd2O3 samples, the reaction appeared tobe heterogeneously initiated on the catalyst surface with alkylradical fragments released into the gas phase and a significantcontribution of gas-phase reactions. The mechanism of ODH ofpropane on non-reducible catalysts was studied by Trionfetti et al.[35] who reported that ODH of propane followed a mixedheterogeneous–homogeneous radical chemistry over Li–MgOcatalysts.
In the presence of oxygen, peroxide species (CnH2n+1O2�)
formed which led mainly to oxygenates and carbon oxides. Thesespecies were stable at lower temperatures but at high tempera-tures the equilibrium:
CnH2nþ1� þ O2 $ CnH2nþ1O2
�
was strongly in favour of the hydrocarbon [31]. Thus, the relativecontribution of the reactions of alkyl radical fragments leading toolefins (either by direct abstraction of H from CnH2n+1
� by O2, or bydecomposition of alkylhydroperoxide species, both accompaniedwith HO2
� releasing) increased. The total homogeneous oxidationvia formation of alkylperoxyradicals was considered unlikely athigh temperatures [36]. By isotopic tracing experiments, Ekstrom[37] and Nelson and Cant [38] have demonstrated competition athigh temperature between ethane and methane to be oxidized oncatalyst surface sites and not in the gas phase. Probably the highcontribution of homogeneous reactions could explain the increasein the selectivity to olefins with increasing temperature on thepromoted AEO under study.
The presence of low amounts of hydrogen in the products(Fig. 7) might indicate that non-oxidative dehydrogenation wasalso taking place at very low extent. Hydrogen formation has beendiscussed by Leveles et al. [13] during dehydrogenation of propaneon lithium promoted magnesia catalyst. These authors consideredthat hydrogen was obtained by chain propagation reactions in thegas phase:
C3H7� ! C3H6þH�
H� þ C3H8 ! H2þC3H7�
Hydrogen formation during dehydrogenation of C2–C4 alkanesin the presence of oxygen has been discussed by Sinev et al. [39].The catalysts under consideration were V- and Sb-based complexoxides supported on alumina. The authors suggested developmentof homogeneous chain process what led to hydrogen along witholefin formation. On our catalyst samples, hydrogen formationprobably occurred through the gas-phase radical reactions alongwith olefin production regardless of oxygen presence.
According to the model proposed by Leveles et al. [13] and byTrionfetti et al. [35], propane molecule was activated byelectrophilic oxygen O� by abstracting hydrogen which was therate-determining step. Iso- or n-propyl radicals were formed and
released into the gas phase where they underwent chainpropagation reactions. According to the authors iso-propyl radicalsled to propene through b-scission of C–H bond and n-propylpreferentially followed a C–C cleavage in b-position leading toethene and methyl radical:
i-C3H7� ! C3H6þH�
n-C3H7� ! C2H4þCH3
�
The reaction of ODH of propane was supposed to take placethrough Eley–Rideal mechanism on non-reducible catalysts. Theactive centre was regenerated without lattice oxygen removal withgas phase O2 participation according to [13,40]:
O2þOHS� ! Os� þHO2
�
HO2� þ OHS
� ! Os� þH2O2
H2O2 ! 2OH�
OH� þ OHS� ! OS
� þH2O�
Obviously, some oxygen species on the catalyst surface activatethe hydrocarbon molecule. Such species may be either oxygen ionO2�, or electrophilic oxygen species (O�, O2
2�, O2�) which have
been studied as active centres mainly for methane activation andmay appear as a result of gas-phase oxygen trapping into surfacevacancies. A dynamic equilibrium between the gas-phase oxygenin the reaction mixture and the lattice oxygen has been reported inthe literature [12,41]:
ð1=2ÞO2ðgÞ $ Oa $ Ol $ O� $ O2�
It was shown that on lanthana catalysts, either promoted (1%Sr/La2O3) or not, lattice oxygen did not interact with methanewhen oxygen was not present in the gas phase, which supportedthe hypothesis of regeneration of active site with gas-phase oxygenparticipation [41].
It can thus be suggested that the reaction of propane ODH startsat the catalyst surface by propane activation through hydrogenabstraction and radicals release in the gas phase. Once released, theradicals form a pool that set up a complex oxidative reactionnetwork.
5. Conclusions
Basic catalysts of MgO, CaO and SrO doped with 5 mol% Nd2O3
have been synthesised by two procedures, namely (i) evaporationof nitrate solutions and subsequent decomposition of the nitratesand (ii) sol–gel method. Adding neodymium increased the propaneconversion but decreased the selectivity to propene and ethenecompared to the pure AEO. The selectivity to olefins depended onthe synthesis procedure and increased from Mg to Sr, i.e. followingthe basicity strength of the catalyst surface. An increase of bothselectivity and conversion for the SG and SrNd–Ev samples wasobserved with increasing contact time. Compared with the well-known VMgO catalyst, which behaved classically, this findingopened a way to increase propene formation by controllingcatalyst preparation and properties. On the studied catalysts thecatalytic reaction most probably proceeded according to a complexheterogeneous–homogeneous scheme through Eley–Ridealmechanism and the active centres were regenerated throughgas-phase O2 participation without lattice oxygen removal.
The application of sol–gel method led to improved selectivity toolefins compared to the procedure of evaporation and decom-position. Moreover, the sol–gel samples presented higher basicityas shown by CO2-TPD. This phenomenon could be explained by abetter incorporation of Nd into the AEO lattice, what createdcationic vacancies for attaining electroneutrality and renderednearby oxide anions coordinatively unsaturated and more basic.

B. Savova et al. / Applied Catalysis A: General 359 (2009) 55–61 61
Thus created structural defects were agglomerated or dispersed,depending on the preparation procedure and the cation radiimatch. It could be concluded that the dispersed local defects(favoured by the sol–gel method) led to higher selectivity whereasNd3+ agglomerates led to higher conversion in agreement with theobservation that Nd2O3 itself gave the highest propane conversion.
Acknowledgements
The financial support by the French Ministries of Foreign Affairsand of National High Education and Research via EGIDE and by theMinistries of Education and Science of Bulgaria and Romaniathrough the National Science Funds within the programmes Rilaand Brancusi is gratefully acknowledged.
References
[1] D. Sanfilippo, I. Miraca, M. Di Girolamo, in: E.G. Derouane, et al. (Eds.), NATOScience Series, vol. 191, Springer, Dordrecht, 2005, p. 217.
[2] J.-P. Lange, in: E.G. Derouane, et al. (Eds.), NATO Science Series, vol. 191, Springer,Dordrecht, 2005, p. 51.
[3] J. Gascon, C. Tellez, J. Herguido, M. Menendez, in: E.G. Derouane, et al. (Eds.), NATOScience Series, vol. 191, Springer, Dordrecht, 2005, p. 149.
[4] J.R. Rostrup-Nielsen, in: Derouane, et al. (Eds.), NATO Science Series, vol. 191,Springer, Dordrecht, 2005, p. 3.
[5] F. Cavani, F. Trifiro, Catal. Today 24 (1995) 307.[6] E.A. Mamedov, V. Cortes Corberan, Appl. Catal. A 127 (1995) 1.[7] M.M. Bettahar, G. Costentin, L. Savary, J.C. Lavalley, Appl. Catal. A 145 (1996) 1.[8] S. Albonetti, F. Cavani, F. Trifiro, Catal. Rev. Sci. Eng. 38 (1996) 413.[9] T. Blasco, J.M. Lopez-Nieto, Appl. Catal. A 157 (1997) 117.
[10] G. Centi, F. Trifiro, Catal. Today 3 (1988) 151.[11] F. Cavani, N. Ballarini, A. Cericola, Catal. Today 127 (2007) 113.[12] Z. Kalenik, E.E. Wolf, Catal. Lett. 9 (1991) 441.[13] L. Leveles, K. Seshan, J.A. Lercher, L. Lefferts, J. Catal. 218 (2003) 296.[14] C. Trionfetti, I.V. Babich, K. Seshan, L. Lefferts, Top. Catal. 39 (2006) 191.
[15] M. Chaar, D. Patel, H.H. Kung, J. Catal. 109 (1988) 463.[16] D. Siew Hew Sam, V. Soenen, J.C. Volta, J. Catal. 123 (1990) 417.[17] A. Corma, J.M. Lopez-Nieto, N. Paredes, J. Catal. 144 (1993) 425.[18] R. Solsona, A. Dejoz, M.I. Vazquez, P. Marquez, J.M. Lopez-Nieto, Appl. Catal. A 205
(2001) 99.[19] A. Klisinska, K. Samson, I. Gressel, B. Grzybowska, Appl. Catal. A 309 (2006) 10.[20] A. Klisinska, S. Loridant, B. Grzybowska, J. Stoch, I. Gressel, Appl. Catal. A 309
(2006) 17.[21] G. Centi, F. Cavani, F. Trifiro, Selective Oxidation by Heterogeneous Catalysis,
Kluwer-Academic Publ./Plenum Press, New York, 2001.[22] J.C. Vedrine, Top. Catal. 21 (2002) 97.[23] E.K. Novakova, J.C. Vedrine, in: J.L.G. Fierro (Ed.), Metal Oxides: Chemistry
and Applications, CRC Press, Taylor & Francis Group, BocaRaton, FL, USA, 2006,p. 413.
[24] M.Yu. Sinev, V.Yu. Bychkov, Yu.P. Tulenin, O.V. Kalashnikova, B.V. Rozentuller, P.A.Shiryaev, Catal. Today 13 (1992) 585.
[25] D. Filkova, M. Sinev, Yu. Tyulenin, L. Petrov, Catal. Lett. 13 (1992) 323.[26] D. Filkova, G. Gayko, D. Wolf, L. Petrov, M. Baerns, Appl. Catal. A 159 (1997) 33.[27] B. Savova, D. Filkova, D. Crisan, M. Crisan, M. R?ileanu, N. Dr?gan, A. Galtayries,
J.C. Vedrine, Appl. Catal. A: Gen. 359 (2009) 47.[28] J.D. Pless, B.B. Bardin, H.-S. Kim, D. Ko, M.T. Smith, R.R. Hammond, P.C. Stair, K.R.
Poeppelmeier, J. Catal. 223 (2004) 419.[29] M. Sarzi-Amade, S. Morselli, P. Moggi, A. Maione, P. Ruiz, M. Devillers, Appl. Catal.
A 284 (2005) 11.[30] E.V. Kondratenko, M.Yu. Sinev, Appl. Catal. A 325 (2007) 353.[31] F. Cavani, F. Trifiro, Catal. Today 51 (1999) 561.[32] J. Nilsson, A. Landa-Canovas, S. Hansen, A. Andersson, Catal. Today 33 (1997) 97.[33] R. Burch, E.M. Crabb, Appl. Catal. 100 (1993) 111.[34] K.T. Nguyen, H.H. Kung, J. Catal. 122 (1990) 415.[35] C. Trionfetti, S. Crapanzano, I.V. Babich, K. Seshan, L. Lefferts, Catal. Today, in Press,
doi:10.1016/j.cattod.2008.06.018.[36] J.H. Lunsford, in: Proceedings of the 10th ICC, L. Guczi, F. Solymoshi, P. Teteniy
(Eds.), Budapest, Hungary, 19–24 July 1992, p. 103.[37] A. Ekstrom, in: E.E. Wolf (Ed.), Methane Conversion by Oxidative Processes, Van
Nostrand Reinhold Catalysis Series, New York, 1992, p. 99.[38] P.F. Nelson, N.W. Cant, J. Phys. Chem. 94 (1990) 3756.[39] M.Yu. Sinev, Z.T. Fattakhova, Yu.P. Tulenin, P.S. Stennikov, V.P. Vislovskii, Catal.
Today 81 (2003) 107.[40] M.Y. Sinev, V.Y. Bychkov, Kinet. Katal. 34 (1993) 309.[41] J.T. Gleaves, J.R. Erbner, T.C. Kuechler, Catal. Rev. Sci. Eng. 30 (1988) 49.