natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration
TRANSCRIPT

Q39
Q3
The American Journal of Pathology, Vol. -, No. -, - 2015
1234567891011121314151617181920212223242526272829303132333435363738394041424344454647484950515253545556575859606162
ajp.amjpathol.org
636465666768697071
7273747576Natural Killer Cells Limit Cardiac Inflammationand Fibrosis by Halting Eosinophil Infiltration
777879
SuFey Ong,* Davinna L. Ligons,y Jobert G. Barin,y Lei Wu,* Monica V. Talor,y Nicola Diny,* Jillian A. Fontes,*Elizabeth Gebremariam,y David A. Kass,z Noel R. Rose,*y and Daniela �Ciháková*y
80818283
From The W. Harry Feinstone Department of Molecular Microbiology and Immunology,* Johns Hopkins University, Bloomberg School of Public Health,Baltimore; and the Departments of Pathologyy and Cardiology,z Johns Hopkins University School of Medicine, Baltimore, Maryland
848586
Accepted for publication
2
C
P
h
87888990919293
November 18, 2014.
Address correspondence toDaniela �Ciháková, M.D., Ph.D.,Johns Hopkins University, RossResearch Bldg, Room 659, 720Rutland Ave, Baltimore,MD 21215. E-mail:[email protected].
opyright ª 2014 American Society for Inve
ublished by Elsevier Inc. All rights reserved
ttp://dx.doi.org/10.1016/j.ajpath.2014.11.023
949596979899100101102
F
Myocarditis is a leading cause of sudden cardiac failure in young adults. Natural killer (NK) cells, asubset of the innate lymphoid cell compartment, are protective in viral myocarditis. Herein, wedemonstrated that these protective qualities extend to suppressing autoimmune inflammation.Experimental autoimmune myocarditis (EAM) was initiated in BALB/c mice by immunization withmyocarditogenic peptide. During EAM, activated cardiac NK cells secreted interferon g, perforin, andgranzyme B, and expressed CD69, tumor necrosis factorerelated apoptosis-inducing ligand treatment,and CD27 on their cell surfaces. The depletion of NK cells during EAM with anti-asialo QGM1 antibodysignificantly increased myocarditis severity, and was accompanied by elevated fibrosis and a 10-foldincrease in the percentage of cardiac-infiltrating eosinophils. The resultant influx of eosinophils tothe heart was directly responsible for the increased disease severity in the absence of NK cells, becausetreatment with polyclonal antibody asialogangloside GM-1 did not augment myocarditis severity ineosinophil-deficient DdoubleGATA1 mice. We demonstrate that NK cells limit eosinophilic infiltrationboth indirectly, through altering eosinophil-related chemokine production by cardiac fibroblasts,and directly, by inducing eosinophil apoptosis in vitro. Altogether, we define a new pathway ofeosinophilic regulation through interactions with NK cells. (Am J Pathol 2015, -: 1e16; http://dx.doi.org/10.1016/j.ajpath.2014.11.023)
Supported by NIH/National Heart, Lung, and Blood Institute NationalResearch Service Awards F31 HL112665-01A1 (S.O.), R01HL118183(D.C.), and R01HL113008 (N.R.R.), the Michel Mirowski MD DiscoveryFoundation (D.C.), a W.W. Smith Charitable Trust Heart Research grantH1103 (D.C.), and the Children’s Cardiomyopathy Foundation Q1(D.C.).
Disclosures: None declared.Current address of D.L.L., Experimental Immunology Branch, Center for
Cancer Research, National Cancer Institute, National Institutes of Health,Bethesda, MD.
103104105106107108109110111112113114115116117118119120121122123
Myocarditis is a leading cause of sudden cardiac failure inindividuals <40 years, with 9% to 16% of cases progressingto inflammatory dilated cardiomyopathy.1e3 Necrotizingeosinophilic myocarditis, a subset of myocarditis, is charac-terized by extensive cardiac eosinophilic infiltration, pro-nounced cardiomyocyte death, and higher fatality rates.4e9
Correlations between eosinophil frequency and poor clinicaloutcomes have been reported in other chronic inflammatorydisease models, including asthma, inflammatory bowel dis-ease, and experimental autoimmune encephalomyelitis.10e12
Herein, we investigated the connection between eosinophilsand natural killer (NK) cells, highlighting a new pathwayresponsible for the control of eosinophilic accumulation insites of inflammation.
Our group and others have reported that NK cells, aninnate lymphoid cell subset, are protective in coxsackievirusB3 and murine cytomegalovirus animal models of myocar-ditis by limiting viral replication.13e15 Because myocarditis is
stigative Pathology.
.
LA 5.2.0 DTD � AJPA1936_proof �
also an autoimmune-mediated disease, it is unknown if NKcells can protect against disease through limiting viral repli-cation, as well as by reducing the autoimmune response.16,17
The data regarding NK cells and autoimmunity are extensive,but conflicting. NK cells accumulate in joints during rheu-matoid arthritis (RA), skin lesions during psoriasis, andbrain lesions during multiple sclerosis.18,19 Activated NKcells from the joints of RA patients induce differentiationof monocytes, signifying an active role in the immune
124
23 January 2015 � 9:00 pm � EO: AJP14_0604

Q4
Q5
Q6
7
8
9
10
Ong et al
125126127128129130131132133134135136137138139140141142143144145146147148149150151152153154155156157158159160161162163164165166167168169170171172173174175176177178179180181182183184185186
187188189190191192193194195196197198199200201202203204205206207208209210211212213214215216217218219220221222223224225226227228229230231232233234235236237238239240241242243244245246247
environment,20 and indicating that NK cells play a proin-flammatory role in autoimmunity.
This directly contradicts the observations that myocar-ditis, RA, Sjögren syndrome, and systemic lupus erythe-matosus patients have decreased NK cell numbers andcytotoxicity potential.21e25 A limited study of biopsyspecimens from myocarditis patients revealed a lack of NKcells in the cardiac tissue.26 Peripheral NK cells from RApatients failed to induce apoptosis in major histocompati-bility complex Iedeficient K562 cells versus healthy con-trols in vitro. Patients with multiple sclerosis in remissionhad higher frequencies of activated peripheral NK cells thanthose with active disease, supporting the notion that defectsin NK cells are associated with increased risk of autoim-munity.27 Altogether, it is unclear whether autoimmunediseases are exacerbated by deficiencies or excesses of NKcells, making animal studies necessary.
Herein, we investigated the role of NK cells in autoimmunemyocarditis using a mouse model of experimental autoim-mune myocarditis (EAM). EAM is induced by s.c. immuni-zation of myocarditogenic peptide in complete Freund’sadjuvant, the same antigen targeted by autoaggressive T cellsin CB3-induced myocarditis.28e31 Susceptible mice strainsdevelop myocarditis, followed by inflammatory dilated car-diomyopathy.29 EAM induces the immune response inde-pendent of persistent virus, allowing us to separateautoimmune- from virus-mediated disease. We report hereinthe ability of NK cells to control myocarditis in the absence ofa viral pathogen.
Materials and Methods
Mice
BALB/c, Rag1�/�, C.Cg-Gata1tm6Sho/J (DdoubleGATA1),CD3d-IL5Tg NJ.1636, Ccr3�/�, interferon g receptor 1(IFNgR1)�/�, and IFNg�/� mice were purchased from theJackson Laboratory and were bred and maintained in theconventional housing facilities at Johns Hopkins University(Baltimore, MD). All protocols have been reviewed andapproved by the Johns Hopkins Animal Care and UseCommittee.
Immunization with MHC614-629 and Assessment of EAM
Male 6- to 8-week-old BALB/c mice were injected s.c. with100 mg of myocarditogenic peptide of cardiac myosin heavychain (MyHC), MyHC614-629, emulsified in an equal volumeof complete Freund’s adjuvant (Sigma-Aldrich, St. Louis,MO), supplemented with 4 mg/mL of H37Ra extract (Difco,Lawrence, KS) on days 0 and 7, as previously described.29
Pertussis toxin (500 ng in 100 mL of phosphate-bufferedsaline (PBS; Sigma-Aldrich) was administrated i.p. on day0. Mice were sacrificed on day 21; hearts were collected andsections were stained with hematoxylin and eosin, as previ-ously described.32e35 The degree of myocardial infiltration
2FLA 5.2.0 DTD � AJPA1936_proof
and fibrosis was determined blindly by two individuals,and Qhistology was scored as follows: 0, no infiltration;1, �10%; 2, 11% to 30%; 3, 31% to 50%; 4, 51% to 90%;and 5, >90%.29
Assessment of Fibrosis
Mice were sacrificed on day 21. Hearts were collected andsections were stained with Masson’s trichrome, as previ-ously described.32e35 Images of Masson’s trichromeestained cardiac sections were uploaded into ImageJ software Q
(NIH, Bethesda, MD). The background space was deletedand the left ventricle, the region of interest, was selectedusing the freeform loop tool. Pixels within the selected areawere deconstructed into red (tissue) or blue (collagen)channels, and fibrosis was calculated as a percentage of blueversus total red plus blue pixels in the region of interest.
Echocardiography
An Acuson Sequoia 256 high-resolution microimagingsystem with a 13-MHz transducer was used (Visualsonic,Toronto, ON, Canada). In conscious mice, the heart wasimaged in the two-dimensional mode in the parasternalshort-axis view. From the M-mode, the left ventricular (LV)wall thickness and chamber dimensions were measured. TheM-mode cursor was positioned perpendicular to the intra-ventricular septum and the LV posterior wall, with three tofive readings taken for each measurement. The LV enddiastolic dimension, LV end systolic dimension, LV pos-terior wall thickness at end diastole, and the intraventricularseptal wall thickness at end diastole were measured from afrozen M-mode tracing. Fractional shortening, ejectionfraction, and relative wall thickness were calculated aspreviously described.32
Intracardiac and Splenic Flow Cytometry
The aorta was cannulated to perfuse hearts with 15 mL ofcold 1� PBS for 3 minutes to remove blood. To generatecardiac single-cell suspensions, hearts were bisected, placedin C-tubes, and dissociated on the GentleMACS system(Miltenyi Biotech, Bergisch Gladbach, Germany) underprogram Qheart_01. Cells were shaken with 10 mg of colla-genase II and 1.5 mg of DNase I (Worthington Biochemical,Lakewood, NJ) for 30 minutes at 37�C. Cells were disso-ciated again and rinsed twice with 1� PBS with 0.05%bovine serum albumin (BSA; Sigma-Aldrich) and 2 mmol/LEDTA (Corning Cellgro, Corning, NY). To generate asplenic single-cell suspension, spleens were dissociatedbetween two frosted glass slides and incubated with 2 mL ofACK Lysing Buffer Qfor 1 minute. The cells were rinsed with1� PBS and filtered through a 40-mm mesh. Cells (1 to 3 �106) were incubated with 1 mL of LIVE/DEAD Aqua(Invitrogen, Carlsbad, CA) for 30 minutes in 1� PBS tostain dead cells. Cells were then incubated with 2 mg of
ajp.amjpathol.org - The American Journal of Pathology
248
� 23 January 2015 � 9:00 pm � EO: AJP14_0604

Q11
Q12
Q13
14
15
16
17
18
NK Cells in Autoimmune Myocarditis
249250251252253254255256257258259260261262263264265266267268269270271272273274275276277278279280281282283284285286287288289290291292293294295296297298299300301302303304305306307308309310
311312313314315316317318319320321322323324325326327328329330331332333334335336337338339340341342343344345346347348349350351352353354355356357358359360361362363364365366367368369370371
aCD16/32 at 4�C for 10 minutes before the addition offluorescent antibodies (CD3, CD4, CD8, CD45, Ly6G,SiglecF, NKp46, DX5, CD11b, CD11c, and F4/80) (eBio-science, San Diego, CA). Samples were incubated withantibodies at 4�C for 10 to 20 minutes, washed in 1 mL of0.5% BSA in 1� PBS, and fixed in fixation and per-meabilization buffer (BD Bioscience, Franklin Lakes, NJ)for 30 minutes. For intracellular cytokine staining, suspen-sions were incubated for 4 to 6 hours with 20 ng/mL 4b-phorbol 12-myristate 13-acetate, 1 mg/mL ionomycin, andGolgistop (BD Bioscience). Cells were surface stained andthen permeabilized with 1� permeabilization buffer (BDBioscience) overnight at 4�C. Cells were then incubatedwith anti-cytokine antibodies (eBioscience) for 30 minutesat 4�C. Cells were washed in 1� permeabilization bufferand resuspended in 100 to 200 mL of buffer. Samples wereacquired on the LSR II quad-laser cytometer runningFACSDiva 6 (BD Immunocytometry, Franklin Lakes, NJ).Data were analyzed with FlowJo 7.6 (Treestar Software,Ashland, OR).
Depletion of NK Cells with Anti-Asialo GM1 Antibody
To deplete NK cells before immunization, 6-week-old maleBALB/c mice were injected i.p. with 1 mg of anti-asialo GM1(Wako Chemicals USA, Richmond, VA) antibody every day 6days before the first immunization (days -6 to 0).36,37 Tomaintain decreased levels of NK cells after the first immuni-zation (day 0), 1 mg of anti-asialo GM1 antibody was admin-istered every other day until day 20. Control mice received1 mg of rabbit IgG (Sigma-Aldrich) by the same schedule.
Isolation of Primary Adult Mouse Cardiac Fibroblasts
Primary adult mouse cardiac fibroblasts were isolated withminor modifications from protocols previously described.38
Hearts dissected from 6- to 8-week-old naïve BALB/c micewere perfused through the aorta with warmed 37�C calcium-free buffer, followed by collagenase type II (WorthingtonBiochemical, Lakewood, NJ) for 15 minutes. Tissue wasdissolved into a single-cell suspension and filtered through a70-mm mesh. Cells were seeded, and nonadherent cells werewashed off after 1 hour. Cells were either collected imme-diately in TRIzol reagent (Invitrogen) for ex vivo experi-ments or passaged twice before in vitro use in completeDulbecco’s modified Eagle’s medium with 20% fetal bovineserum (Hyclone Laboratories, Logan, UT), 1� penicillin/streptomycin, 25 mmol/L HEPES, 1� Anti-Anti, and 1�nonessential amino acids.
Isolation of Primary NK Cells
NK cells were negatively isolated from Rag1�/� BALB/cspleens by manual magnetic cell sorting using the MouseNK Isolation Kit II (Miltenyi Biotech) and cultured for24 hours with 10 ng/mL IL-12 and 5 ng/mL IL-15.
The American Journal of Pathology - ajp.amjpathol.orgFLA 5.2.0 DTD � AJPA1936_proof �
Isolation of Primary Eosinophils
Eosinophils were isolated from naïve CD3d-IL5Tg NJ.1636peripheral blood mononuclear cells using a Percoll (GE Life-sciences, Marlborough, MA) gradient and subsequent negativefluorescence-activated cell sorting for SSChiLy6G�DX5�
eosinophils.
Apoptosis Measurement
Cells were harvested from culture and rinsed twice with 1�PBS with 0.05% BSA (Sigma-Aldrich) and 2 mmol/L EDTA(Corning Cellgro). The cells were rinsed with 1� PBS andincubated with 1 mL of LIVE/DEAD Aqua (Invitrogen) for30 minutes in 1� PBS to stain dead cells. Cells were thenincubated with 2 mg of aCD16/32 at 4�C for 10 minutesbefore the addition of fluorescent antibodies (Ly6G, SiglecF,and NKp46) (eBioscience). Samples were incubated withantibodies at 4�C for 10 to 20 minutes and washed in 1 mL of0.5% BSA in 1� PBS. Cells were then resuspended in 1�Annexin Binding Buffer (eBioscience) and stained with 2 mLof annexin V. Cells were acquired after 15 minutes of incu-bation on ice on the LSRII Qflow cytometer.
mRNA
For real-time quantitative PCR (qPCR Q), cells or tissues werehomogenized in TRIzol reagent and chloroform extracted.Samples were DNase treated, and cDNA libraries weremade using iScript Reverse Transcriptase Supermix (Bio-Rad, Hercules, CA). mRNA was amplified using SYBRGreen (Applied Biosystems, Foster City, CA), and allvalues were calculated against HPRT QmRNA. Values werecontrolled against isotype control groups and shown as afunction of fold induction using the formula 2�D(DCT).
Murine primers Qwere as follows (forward and reverseprimers, respectively): Hprt, 50-TCCTCCTCAGACCGCT-TTT-30 and 50-TCTGCTGGAGTCCCCTTG-30; collagen(Col) 1a1, 50-AGCAGGTCCTTGGAAACCTT-30 and 50-AAGGAGTTTCATCTGGCCCT-30; Col1a2, 50-GTGAAC-GGGGCGAAGCTGGTT-30 and 50-GCGGCTCCTGGAA-GCCCATTTG-30; Col1a3, 50-AACCTGGTTTCTTCTCA-CCCTTC-30 and 50-ACTCATAGGACTGACCAAGGTGG-30; Il4, 50-AAGGCAACTTTCTTGATATT-30 and 50-GG-CCTTTCAGACTAATCTT-30; Il13, 50-TGAGGAGCT-GAGCAACATCACACA-30 and 50-TGCGGTTACAGAG-GCCATGCAATA3-3 Q0; eosinophil peroxidase, 50-AGATG-CAACAACAAGAAGCATCC-30 and 50-TGATTGGAGA-CATCCCGGAC-30; major basic protein 2, 50-TGAAA-CTTCTGACTCCAAAAGCC-30 and 50-CGGCATTAGCT-CTTCCCCT-30; Il1b, 50-CAACCAACAAGTGATATTC-TCCATG-30 and 50-GATCCACACTCTCCAGC-TGCA-30;transforming growth factor (Tgf) b1, 50-CTCCCGTGGC-TTCTAGTGC-30 and 50-GCCTTAGTTTGGACAGGATC-TG-30; chemokine ligand 11 (Ccl11), 50-GAATCACCAACA-ACAGATGCAC-30 and 50-TCCTGGACCCACTTCTTCTT-
3
372
23 January 2015 � 9:00 pm � EO: AJP14_0604

19
print&web4C/FPO
Ong et al
373374375376377378379380381382383384385386387388389390391392393394395396397398399400401402403404405406407408409410411412413414415416417418419420421422423424425426427428429430431432433434
435436437438439440441442443444
30; Ccl24, 50-TCTTAGGGCCCTTCTTGGTG-30 and 50-AA-TTCCAGAAAACCGAGTGG-30; and Cxcl9, 50-GTGGAG-ACCACCAGAGTTGG-30 and 50-TGCCACTAAGCTACA-GCCAC-30.
Enzyme-Linked Immunosorbent Assays
Samples were run using tissue homogenates in 1� PBS orcell culture supernatant using enzyme-linked immunosorbent
4FLA 5.2.0 DTD � AJPA1936_proof
assay kits for Ccl11 (RND Biosystems, Minneapolis, MN) ora multiplex LINCO kit (Millipore, Jaffery, NH).
Statistical Analysis
Multiple group comparisons were performed by ordinary one-way analysis of variance, followed by the Tukey-Kramer posttest (if parametric) orKruskal-Wallis, followed byDunn’s posttest (if nonparametric) (GraphPad Prism Q; GraphPad, San
ajp.amjpathol.org - The American Journal of Pathology
445446447448449450451452453454455456457458459460461462463464465466467468469470471472473474475476477478479480481482483484485486487488489490491492493494495496
� 23 January 2015 � 9:00 pm � EO: AJP14_0604

½F1�½F1�
½F2�½F2�
½F3�½F3�
20
21
22
NK Cells in Autoimmune Myocarditis
497498499500501502503504505506507508509510511512513514515516517518519520521522523524525526527528529530531532533534535536537538539540541542543544545546547548549550551552553554555556557558
559560561562563564565566567568569570571572573574575576577578579580581582583584585586587588589590591592593594595596597598599600601602603604605606607608609610
Diego, CA). All statistics of two groups were performedby Student’s t-test. P < 0.05 was considered statisticallysignificant.
Results
NK Cells Suppress Cardiac Inflammation and SevereMyocarditis in EAM
To determine whether NK cells modulate EAM, we depletedNK cells in vivo using 1 mg of anti-asialogangloside GM-1(ASGM-1) polyclonal antibody injected i.p. from day -6 to day21, as described in Materials and Methods (Figure 1A). Thedepletion caused myocarditis to increase severity as assessedby histology and total count of infiltrating CD45þ cells inthe heart at the peak of inflammation on day 21 (Figure 1,BeD). This was accompanied by increased circulating antieMyHCa614-629-specific IgG antibodies (Figure 1E). At thistime, ASGM-1 antibody reduced CD3�DX5þNKp46þ NKcells, but not CD3þDX5þ NK T cells (Figure 1, F and G) inthe spleen and heart during EAM. ASGM-1 had little effect onother cell types in naïve animals, because antibody treatmentdepleted CD3�DX5þNKp46þ NK, but not CD3þDX5þ
NKp46þ, NK T cells or Ly6GloSiglecFþ eosinophils(Supplemental Figure S1). Development of severemyocarditisafter depletion of NK cells suggests that NK cells protectfrom severe cardiac inflammation.
Absence of NK Cells Increases Collagen Deposition inthe Heart
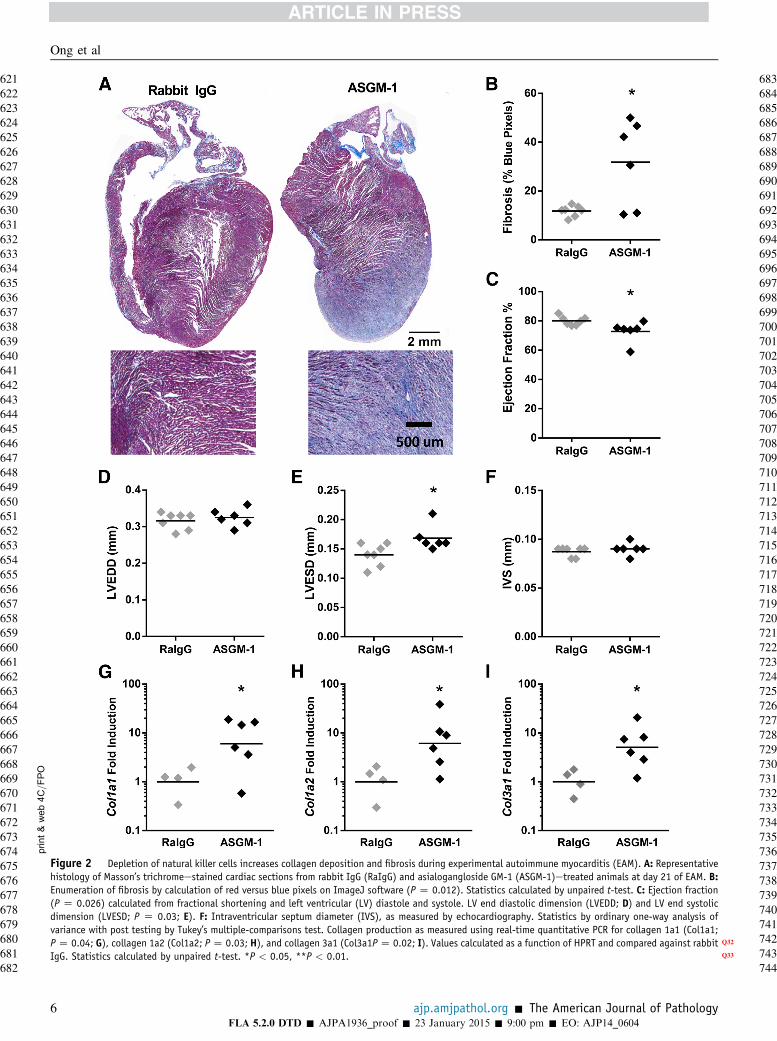
We explored whether NK cells inhibit fibrosis developmentand inflammation. To assess fibrosis quantitatively, imagesof cardiac histology slides stained by Masson’s trichromewere processed by ImageJ software to enumerate collagen-positive (blue) versus nonfibrotic (red) pixels. NK-depletedanimals had increased cardiac collagen deposition on day 21of EAM (Figure 2, A and B). This accompanied a decline incardiac function, as shown by decreased ejection fractionwith increased LV end systolic diameter and no changes inLV end diastolic diameter or intraventricular septal thick-ness (Figure 2, CeF). We quantified active collagen pro-duction in NK-depleted mice at day 21 and showed that NKdepletion led to increased mRNA levels of collagen 1 andcollagen 3 in the heart (Figure 2, GeI). Thus, the depletionof NK cells during EAM leads to increased cardiac fibrosisand a decline in cardiac function.
Figure 1 Depletion of natural killer (NK) cells increases cardiac inflammaA: Schematic of phosphate-buffered saline (PBS), rabbit IgG, and asialoganglossentative histology from rabbit IgG (RaIgG) and ASGM-1etreated animals (B) andIgG, and ASGM-1 antibody treated animals at day 21 of EAM (analysis of varianceand ASGM-1etreated animals at day 21 of EAM, as assessed by flow cytometry (P <
chain (MyHC)614-629 total IgG antibodies in the serum of PBS, isotype control, andPercentage of CD3�DX5þNKp46þ NK cells in the heart (P < 0.001) and spleen (Pand ASGM-1 monoclonal antibodyetreated wild-type (WT) mice at day 21 of EAM
The American Journal of Pathology - ajp.amjpathol.orgFLA 5.2.0 DTD � AJPA1936_proof �
Activated NK Cells Accumulate in the Heart during EAM
To examine NK cell kinetics during EAM, we quantifiedcardiac NK cells by flow cytometry (Figure 3A).CD3�DX5þNKp46þ NK cells increased from day 0 to day21 in absolute counts; however, their proportion out ofCD45þ cells was stable (Figure 3, B and C). To determinetheir phenotype, we profiled cytokine and receptor expressionfrom NK cells by flow cytometry on day 21 of EAM to see ifthey represented an activated population compared to those inthe periphery. Cardiac NK cells produced more Ifng thansplenic NK cells, but equivalent levels of Il-13 Q(Figure 3, Dand E). Greater proportions of cardiac NK cells up-regulatedperforin and were positive for lysosomal-associated mem-brane protein 1 (LAMP-1), a correlate for granzyme Bsecretion (Figure 3, F and G). In addition, cardiac NK cellsup-regulated activation markers CD27, CD69, and tumornecrosis factorerelated apoptosis-inducing ligand treatment(Figure 3, HeJ), with decreased NKG2D Q(Figure 3K). Thus,cardiac NK cells secrete Ifng and have increased expressionof multiple activation receptors compared to NK cells inperiphery during the course of myocarditis.
Activated Fibroblasts Are Not Targeted for CytotoxicKilling by NK Cells in Vitro
The depletion of NK cells led to accelerated fibrosis(Figure 2) in the heart; therefore, we hypothesized that NKcells lysed activated fibroblasts, as described in animalmodels of liver fibrosis.39,40 We explored this putativemechanism using a co-culture system in vitro with anoutcome focused on cell death. First, adult cardiac fibro-blasts were isolated from naïve BALB/c hearts and passagedtwice. The presence of contaminating macrophages wasruled out by qPCR and immunofluorescence (SupplementalFigure S2 Q). Second, NK cells were isolated from naïveor poly(I:C)-treated BALB/c spleens (SupplementalFigure S3A). Cardiac fibroblasts were activated withangiotensin II, illustrated by increased expression ofvimentin (Supplemental Figure S2B). Finally, we co-cultured cardiac fibroblasts and NK cells together for 48hours at a 1:1 ratio. Cardiac fibroblasts were stained greenwith calcein AM (live cells) and red with ethidium homo-dimer (dead cells) and examined by immunofluorescencemicroscopy (Supplemental Figure S3C). Although NK cellsinduced cell death of cardiac fibroblasts, no difference wasseen between untreated and activated cardiac fibroblasts
tion and the severity of experimental autoimmune myocarditis (EAM).ide GM-1 (ASGM-1) antibody treatment schedule throughout EAM. Repre-scores of hematoxylin and eosinestained cardiac sections from PBS, rabbitP < 0.01; C). D: Total CD45þ cells infiltrating the hearts of isotype control0.001). E: Levels of anti-myocarditogenic peptide of cardiac myosin heavy
ASGM-1etreated animals at day 21 of EAM (analysis of variance P < 0.001).< 0.002) (F) and CD3þDX5þ NK T cells out of CD45þ cells (G) of rabbit IgG, as assessed by flow cytometry. **P < 0.05, ***P < 0.001.
5
611612613614615616617618619620
23 January 2015 � 9:00 pm � EO: AJP14_0604
print&
web4C=FPO
Figure 2 Depletion of natural killer cells increases collagen deposition and fibrosis during experimental autoimmune myocarditis (EAM). A: Representativehistology of Masson’s trichromeestained cardiac sections from rabbit IgG (RaIgG) and asialogangloside GM-1 (ASGM-1)etreated animals at day 21 of EAM. B:Enumeration of fibrosis by calculation of red versus blue pixels on ImageJ software (P Z 0.012). Statistics calculated by unpaired t-test. C: Ejection fraction(P Z 0.026) calculated from fractional shortening and left ventricular (LV) diastole and systole. LV end diastolic dimension (LVEDD; D) and LV end systolicdimension (LVESD; P Z 0.03; E). F: Intraventricular septum diameter (IVS), as measured by echocardiography. Statistics by ordinary one-way analysis ofvariance with post testing by Tukey’s multiple-comparisons test. Collagen production as measured using real-time quantitative PCR for collagen 1a1 (Col1a1;PZ 0.04; G), collagen 1a2 (Col1a2; PZ 0.03; H), and collagen 3a1 (Col3a1PZ 0.02; I). Values calculated as a function of HPRT Q32and compared against rabbitIgG. Statistics calculated by unpaired t-test. *P Q33< 0.05, **P < 0.01.
Ong et al
6 ajp.amjpathol.org - The American Journal of Pathology
621622623624625626627628629630631632633634635636637638639640641642643644645646647648649650651652653654655656657658659660661662663664665666667668669670671672673674675676677678679680681682
683684685686687688689690691692693694695696697698699700701702703704705706707708709710711712713714715716717718719720721722723724725726727728729730731732733734735736737738739740741742743744
FLA 5.2.0 DTD � AJPA1936_proof � 23 January 2015 � 9:00 pm � EO: AJP14_0604

½F4�½F4�
print&web4C=FPO
Figure 3 Activated natural killer (NK) cells accumulate in the heart during experimental autoimmune myocarditis (EAM). A: Gating strategy for cardiacCD3�DX5þNKp46þ NK cells. Absolute numbers (analysis of variance P Z 0.002; B) and percentages (C) of CD3�DX5þNKp46þ NK cells on days 0, 14, and 21 ofEAM by flow cytometry. Significance by ordinary one-way analysis of variance with post testing by Tukey’s multiple-comparisons test. To determine NKphysiology, cardiac and splenic CD3�DX5þNKp46þ NK cells were stained at day 21 of EAM. Percentage of interferon g (IFNg; P < 0.001; D), Il-13 (E), andperforin (P < 0.001; F) positive cells were based on intracellular antibody staining after 4 to 6 hours of 4b-phorbol 12-myristate 13-acetate/ionomycin andGolgistop in vitro. G: Percentage of Q34LAMP-1þ, a marker for granzyme B release, staining (P < 0.001). Percentage of NK cells positive for activation-associatedmarkers CD27 (P < 0.001; H), CD69 (P < 0.001; I), tumor necrosis factorerelated apoptosis-inducing ligand treatment (TRAIL; P < 0.001; J), and NKG2D(P < 0.001; K). DeF: Significance calculated by unpaired t-test. **P < 0.01, ***P < 0.001. FSC Q35, ---; SSC, ---.
NK Cells in Autoimmune Myocarditis
745746747748749750751752753754755756757758759760761762763764765766767768769770771772773774775776777778779780781782783784785786787788789790791792793794795796797798799800801802803804805806
807808809810811812813814815816817818819820821822823824825826827828829830831832833834835836837838839840841842843844845846847848849850
(Supplemental Figure S3, D and E), indicating that NK-mediated killing of activated fibroblasts was not a likelymechanism.
851852853854855856857858859860861862863864865866867
NK Cells Prevent Eosinophils from Accumulating in theHeart during EAM
Given the lack of direct killing seen between NK cells andactivated fibroblasts, we hypothesized that NK cells altereddisease severity and fibrosis in EAM through alterationsin the hematopoietic populations infiltrating the heart.Therefore, we examined changes in the cardiac infiltrateduring EAM after the depletion of NK cells by flowcytometry. NK-depleted animals had increased proportionof SSChiCD45þ granulocytic cells (Figure 4A). We identi-fied that SSChiLy6GloSiglecFþ eosinophils were responsiblefor the increase in granulocytic cells. SSChiLy6GloSiglecFþ
eosinophils increased twofold on day 14 and 10-fold on day
The American Journal of Pathology - ajp.amjpathol.orgFLA 5.2.0 DTD � AJPA1936_proof �
21 out of CD45þ cells (Figure 4B). We found no changes inSSChiLy6Ghi neutrophils on day 14 or 21 (Figure 4C).
To determine whether the increased eosinophils in theheart seen in ASGM-1etreated mice during EAM werea cardiac-specific observation or a reflection of systemiceosinophilia in response to NK depletion, we analyzed theblood and spleens of mice depleted of NK cells during EAMat days 14 and 21. We found that, although there was a mildtrend toward increased eosinophils, it was not significant(Supplemental Figure S4), underlining that the mechanismof eosinophilic control by NK cells is specific for the cardiacenvironment during EAM.
We also explored whether there were shifts in any otherinfiltrating cardiac populations during EAM. SSCmidCD11bþ
myeloid cells, as a percentage of total CD45þ cells(Supplemental Figure S5A), remained unchanged, as wereCD11bþLy6Chi and CD11bþLy6Clo monocytes as a pro-portion of total SSCmidCD11bþ cells on day 21(Supplemental Figure S5, B and C). The proportions of total
7
868
23 January 2015 � 9:00 pm � EO: AJP14_0604

½F5�½F5�
23
½F6�½F6�
Figure 4 Depletion of natural killer (NK) cells increases eosinophilicinfiltration in the heart. A: Representative side-scatter histograms of iso-type and asialogangloside GM-1 (ASGM-1) antibody treated animals at day21. Percentage of Ly6GloSiglecFþ eosinophils from cardiac CD45þ cells onday 14 (P Z 0.043) and day 21 (P < 0.001) (B) and SiglecF�Ly6Ghi
neutrophils at days 14 and 21 (C). Significance calculated by unpaired t-test. *P < 0.05, ***P < 0.001. SSC, ---.
Ong et al
869870871872873874875876877878879880881882883884885886887888889890891892893894895896897898899900901902903904905906907908909910911912913914915916917918919920921922923924925926927928929930
931932933934935936937938939940941942943944945946947948949950951952953954955956957958959960961962963964965966967968969970971972973974975976977978979980981982983984985986987988989990991
infiltrating CD45þ cells of both CD11chi dendritic cells andFcεRIaþcKitþ mast cells were also unaltered (SupplementalFigure S5, D and E). Also comparable were proportions ofCD11b�B220þ B cells and CD3þ and CD3þCD4þ T cells(Supplemental Figure S5, FeH) of total CD45þ cells.Because EAM and eosinophil trafficking are influenced byCD4þ T-cell polarization,12,28,41 we examined if NK cellsaffected types 1, 2, and 17 helper T cell (Th1, Th2, andTh17, respectively) populations during EAM.12,28,37 Nochanges in IFNgþCD3þCD4þ Th1 cells in the heart afterNK depletion were found (Supplemental Figure S6A).However, Il-13þ Th2 and Il-17Aþ Th17 cells, as a fractionof total CD3þCD4þ cells, increased at day 21, but not day 14
8FLA 5.2.0 DTD � AJPA1936_proof
(Supplemental Figure S6, B and C). Thus, depletion of NKcells resulted in accumulation of eosinophils in the heart,reaching up to a 10-fold increase at the peak of inflammation,and was accompanied by a shift toward mixed Th2 and Th17milieu in the heart.
Eosinophils in the Heart of ASGM-1eTreated Mice Havean Activated Profile
To determine whether the increased flux of eosinophils tothe heart played a role in the increased disease severity, weexamined the phenotype of the heart-infiltrating eosinophilsfor their maturation and activation status. It has been shownthat mature eosinophils from the lungs of helminth-infectedor the blood of asthma-induced mice have increasedexpression of CD11b and SiglecF protein and decreasedexpression of eosinophil granuleeassociated genes dueto these granules being fully preloaded with protein.42,43
Indeed, paraffin-embedded cardiac tissue sections ofASGM-1etreated animals with EAM showed positivestaining for eosinophil granule major basic protein(Figure 5A), and cardiac eosinophils from ASGM-1etreatedhearts had up-regulated levels of SiglecF and CD11b proteincompared to spleen cells at day 21 (Figure 5, B and C).Consistent with this activated phenotype, cardiac eosino-phils down-regulated expression of eosinophil granuleeassociated eosinophil peroxidase and major basic protein 2due to their granules having already been formed during theimmature stages of development (Figure 5, D and E). Car-diac eosinophils also had increased expression of Il-1b,Ccl11, and Il-6, and showed no changes in Il-4 or Il-13expression (Figure 5, FeJ). In summary, eosinophils infil-trating the heart at day 21 of ASGM-1etreated micerepresent a distinct and activated population.
Eosinophils Are Necessary for Increased MyocarditisSeverity in the Absence of NK Cells
To establish if the greater EAM severity in NK-depletedanimals depended on the influx of activated and matureeosinophils, we depleted NK cells from eosinophil-deficientDdblGATA1 mice.44 These mice have a deletion in theGATA1 promoter, leading to a specific ablation in eosino-phils due to the inability of progenitors to differentiate in thebone marrow. In the absence of eosinophils, NK depletionhad no effect on cardiac inflammation, as assessed fromhistological readings by two independent investigators Qusinghematoxylin and eosinestained cardiac slides at day 21 ofEAM (Figure 6, A and B). In addition, we saw no increasesin fibrosis evaluated by Masson’s trichrome staining. Inaddition, cardiac function was preserved, as shown by nodifferences in echocardiographic parameters between NK-depleted and isotype DdblGATA1 groups at day 21(Figure 6, CeG). DdblGATA1 mice did not respond toASGM-1 treatment with the increased collagen 1 and 3mRNA seen in wild-type mice (Figure 6, HeJ). Therefore,
ajp.amjpathol.org - The American Journal of Pathology
992
� 23 January 2015 � 9:00 pm � EO: AJP14_0604

print&web4C=FPO
Figure 5 Activated eosinophils infiltrate the heart in the absence of natural killer cells. A: Immunofluorescence staining of paraffin-embedded cardiacsections from asialogangloside GM-1 (ASGM-1)etreated mice on day 21 of experimental autoimmune myocarditis (EAM). Sections stained with 1:500 rat anti-major basic protein (MBP) primary or rat IgG antibody, 1:200 anti-rat donkey phosphatidylethanolamine (PE)eTexas Red antibody, and DAPI. Relative meanfluorescence intensity of SiglecF (P < 0.001; B) and CD11b (P < 0.001; C) on Ly6GloSiglecFþ eosinophils from the heart and spleen of ASGM-1etreatedanimals at day 21 of EAM by flow cytometry. Levels of eosinophil peroxidase (Epx; PZ 0.05; D), major basic protein 2 (Prg2; PZ 0.05; E), Il1b (PZ 0.04; F),chemokine ligand (Ccl) 11 (PZ 0.03; G), Il6 (PZ 0.02; H), Il4 (I), and Il13 (J) mRNA from fluorescence-activated cell sorted Ly6GloSiglecFhi eosinophils fromASGM-1etreated mice at day 21 of EAM. Values shown as fold induction compared to spleen and controlled against Q36HPRT. Significance calculated by Student’st-test. *P < 0.05, ***P < 0.001. MFI, mean fluorescence intensity.
NK Cells in Autoimmune Myocarditis
The American Journal of Pathology - ajp.amjpathol.org 9
9939949959969979989991000100110021003100410051006100710081009101010111012101310141015101610171018101910201021102210231024102510261027102810291030103110321033103410351036103710381039104010411042104310441045104610471048104910501051105210531054
10551056105710581059106010611062106310641065106610671068106910701071107210731074107510761077107810791080108110821083108410851086108710881089109010911092109310941095109610971098109911001101110211031104110511061107110811091110111111121113111411151116
FLA 5.2.0 DTD � AJPA1936_proof � 23 January 2015 � 9:00 pm � EO: AJP14_0604

print&web4C/FPO
Ong et al
10 ajp.amjpathol.org - The American Journal of Pathology
11171118111911201121112211231124112511261127112811291130113111321133113411351136113711381139114011411142114311441145114611471148114911501151115211531154115511561157115811591160116111621163116411651166116711681169117011711172117311741175117611771178
11791180118111821183118411851186118711881189119011911192119311941195119611971198119912001201120212031204120512061207120812091210121112121213121412151216121712181219122012211222122312241225122612271228122912301231123212331234123512361237123812391240
FLA 5.2.0 DTD � AJPA1936_proof � 23 January 2015 � 9:00 pm � EO: AJP14_0604

½F7�½F7�
Q24
Q25
½F8�½F8�
NK Cells in Autoimmune Myocarditis
12411242124312441245124612471248124912501251125212531254125512561257125812591260126112621263126412651266126712681269127012711272127312741275127612771278127912801281128212831284128512861287128812891290129112921293129412951296129712981299130013011302
13031304130513061307130813091310131113121313131413151316131713181319132013211322132313241325132613271328132913301331133213331334133513361337133813391340134113421343134413451346134713481349135013511352135313541355
myocarditis severity and cardiac fibrosis in DdblGATA1mice are unaffected by NK cell depletion. This wouldindicate that NK cells protect the heart from eosinophilicaccumulation and the subsequent development of severemyocarditis.
Ccl11 Is Involved But Not Required for the Control ofEosinophilic Infiltration by NK Cells
NK cells are not major producers of eosinophil-associatedchemokines.45e47 However, we have determined that car-diac fibroblasts are an active source of cytokines and che-mokines and can control the types of immune cellsinfiltrating the heart during myocarditis.48 Eotaxins areprominent modulators of eosinophil trafficking and accu-mulation. In EAM, the depletion of NK cells increasedeotaxin expression in whole heart tissue (Figure 7A) andisolated cardiac fibroblasts on day 21 of EAM (Figure 7,AeC), as seen by qPCR. To examine if NK cells affectedeotaxin production in vitro, we co-cultured naïve adultcardiac fibroblasts and NK cells for 96 hours. NK cellssuppressed Il-4emediated Ccl11 (Figure 7D) and increasedIP-10 (Cxcl10), a negative eosinophil regulator, producedby cardiac fibroblasts (Supplemental Figure S7A). Down-regulation of monocyte chemoattractant protein-1 (Ccl2),macrophage inflammatory protein-1b (Ccl4), regulated onactivation normal T cell expressed and secreted (Ccl5), andKC (Cxcl1) (Supplemental Figure S7, BeE) also occurredin the presence of NK cells. In response to Il-4, cardiac fi-broblasts produced Il-5, but were unresponsive to NK cells(Supplemental Figure S7F).
To address whether NK suppression of eotaxins would besufficient to prevent eosinophil accumulation, we used micedeficient in Ccr3, the sole receptor of Ccl11 and Ccl24.49
Depletion of NK cells in Ccr3�/� mice still resulted in theinflux of eosinophils into the heart during EAM (Figure 7E),indicating that NK cells do not control eosinophils throughonly eotaxins and that other eosinophilic chemokines areable to drive eosinophil trafficking in the absence of NKcells. Thus, Ccl11 is involved, but not required, for thecontrol of eosinophilic infiltration in the heart by NK cells.
Ifng and Cxcl9 Are Not Required by NK Cells to ControlEosinophilic Infiltration
During EAM, IFNg was significantly up-regulated in car-diac NK cells accumulating in the heart. MIG (Cxcl9), amajor negative regulator of eosinophil trafficking, is directly
Figure 6 Eosinophil-deficient mice are phenotypically unresponsive to natuasialogangloside GM-1 (ASGM-1) antibody-treated (A) and scores of rabbit phosph(analysis of variance; B). Representative histology (C) and percentage of fibroexperimental autoimmune myocarditis (EAM) from rabbit IgG and ASGM-1etreatedF), and left ventricular end systolic dimension (LVESD; G) by echocardiography at dPCR for collagen 1a1 (Col1a1; H), collagen 1a2 (Col1a2; I), and collagen 3a1 (Col3of EAM. Values calculated as a function of HPRT levels and compared against rab
The American Journal of Pathology - ajp.amjpathol.orgFLA 5.2.0 DTD � AJPA1936_proof �
controlled by Ifng. We determined that in ASGM-1etreatedmice, expression of Cxcl9 was decreased compared to theisotype control groups at day 21 of EAM (SupplementalFigure S8A), and that similar to Cxcl10 (SupplementalFigure S7A), the presence of NK cells in culture with pri-mary cardiac fibroblasts significantly increased levels ofCxcl9 in the supernatant (Supplemental Figure S8B). Inaddition, our data indicate that Ifng produced by NK cells isabsolutely required for Cxcl9 production being instigated,because Ifng receptoredeficient (IFNgR1�/�) cardiac fi-broblasts were unresponsive to the presence of NK cells(Supplemental Figure S8C). These findings were consistentin vivo, because we observed significant decreases in theprotein levels of Cxcl9 in the hearts of IFNg�/� animals atday 14 of EAM (Supplemental Figure S8D). By usingIFNg�/� mice to model the effects of diminished Cxcl9 andthe absence of NK-sourced Ifng, we depleted NK cells inIFNg�/� mice and examined eosinophilic accumulation inthe heart on day 21. Similar to the Ccr3�/� mice, IFNg�/�
mice still continued to accumulate significantly more eo-sinophils in the heart (Supplemental Figure S8E). Inconclusion, both Cxcl9 and IFNg may contribute to the NK-mediated control of eosinophils in the heart, but they are notrequired.
NK Cells Directly Induce Apoptosis of Eosinophils
Finally, we examined whether NK cells could inhibiteosinophil infiltration through direct interactions betweenthe two cell types. Recent studies showed that human NKcells induce activation and apoptosis of eosinophils.50,51 Weexplored this as one avenue of NK-mediated control ofeosinophil accumulation in the heart during cardiacinflammation. NK cells and eosinophils were negativelysorted from naïve Rag1�/� spleens and Cd3deIL-5etransgenic (NJ.1638) blood, respectively, and co-culturedfor 3 hours at 5:1 and 10:1 ratios. Compared to eosino-phils alone, NK cells induced apoptosis, but not degranu-lation, as measured by side scatter, of eosinophils in a dose-dependent manner (Figure 8A). At this 3-hour time point,early apoptosis was measurable using annexin V, but nochanges were yet seen with a viability dye measuredthrough cell permeabilization (Figure 8, B and C). NK cellsdid not induce eosinophil activation either, as shown bychanges in levels of SiglecF and CD11b, unlike reports inhuman cells (Figure 8, D and E).51 In summary, we showthat, similarly to human NK cells, mice NK cells causeapoptosis of eosinophils.
ral killer depletion. Representative histology from rabbit IgG (RaIgG) andate-buffered saline, IgG, and ASGM-1 antibody-treated DdblGATA1 animalssis (D) by Masson’s trichrome staining of cardiac sections at day 21 ofmice. Ejection fraction (E), left ventricular end diastolic dimension (LVEDD;ay 21. Collagen 1 and 3 production as measured using real-time quantitativea1; J) mRNA in hearts of rabbit IgG and ASGM-1etreated animals at day 21
Q37bit IgG. Statistics calculated by unpaired t-test.
11
135613571358135913601361136213631364
23 January 2015 � 9:00 pm � EO: AJP14_0604

Q26
Q27
Figure 7 Chemokine ligand (Ccl) 11 is involved, but not required, for the suppression of eosinophils by natural killer (NK) cells. Ccl11 mRNA at day 21 ofexperimental autoimmune myocarditis (EAM) in whole heart (P Z 0.004; A) and isolated cardiac fibroblasts of rabbit IgG and asialogangloside GM-1(ASGM-1)etreated animals (B). C: CCL24 mRNA in isolated cardiac fibroblasts of rabbit IgG (RaIgG) and ASGM-1etreated animals at day 21 of EAM. D:Ccl11 in the supernatant after a 96-hour culture of wild-type cardiac fibroblasts with or without NK cells (P < 0.001). E: Percentage of Ly6GloSiglecFþ
eosinophils at day 21 of EAM in rabbit IgG and ASGM-1etreated Ccr3�/� animals. Significance by Student’s t-test. *P < 0.05, ***P < 0.001.
Ong et al
13651366136713681369137013711372137313741375137613771378137913801381138213831384138513861387138813891390139113921393139413951396139713981399140014011402140314041405140614071408140914101411141214131414141514161417141814191420142114221423142414251426
1427142814291430143114321433143414351436143714381439144014411442144314441445144614471448144914501451145214531454145514561457145814591460146114621463146414651466146714681469147014711472147314741475147614771478147914801481148214831484148514861487
Discussion
Previous studies have demonstrated that NK cells limitdisease severity in CB3- and MCMV-induced myocarditisby suppressing virus replication. In addition, we showedthat depletion of NK cells rendered previously resistantstrains of mice susceptible to viral myocarditis.13 Herein, wedemonstrate that the protective qualities of NK cells extendbeyond viral inhibition. Activated NK cells accumulated inthe heart throughout EAM and actively suppressedautoimmune-mediated inflammation, because the depletionof NK cells during EAM led to increased cardiac inflam-mation and fibrosis. This was caused primarily by a 10-foldincrease in the proportion of eosinophils in the cardiacinfiltrate, because eosinophil-deficient DdblGATA1 animalsdid not display increased myocarditis severity in response toNK cell depletion. Furthermore, this increase of eosinophilswas specific for the cardiac environment and was not foundin the periphery during EAM. On the basis of these data, weconclude that NK cells limited eosinophil accumulation inthe heart during EAM and in the absence of NK cells, in-creases in eosinophils drove the amplified disease severity.
12FLA 5.2.0 DTD � AJPA1936_proof
Similar to studies in RA patients, we showed that NK cellsoccupying local sites of autoimmune-mediated inflammationhave a distinct and activated phenotype compared to thosein the periphery.20,22 Cardiac NK cells up-regulated CD27,tumor necrosis factorerelated apoptosis-inducing ligandtreatment, and CD69 on their cell surfaces, but down-regulated NKG2D. The down-regulation of NKG2D is sur-prising because nonobese diabetes can be prevented withanti-NKG2D antibodies and NKG2Dþ cells are correlatedwith clinical Crohn’s and RA severity.52e54 However, totalNKG2D in these studies was targeted, including thoseexpressed on NK T and dg T cells.55 Therefore, the role ofNKG2D on NK cells is undetermined and its down-regulation in EAM indicates it may have a minimal role inautoimmunity.Our finding that eosinophils promote severe cardiac
inflammation is supported by other reports of eosinophilsdriving myocarditis severity. In CB3-induced myocarditis,infiltrating eosinophils significantly increased disease. SolubleST2, an Il-33 receptor decoy, prevented eosinophilia andreduced myocarditis without altering viral burden.56 Similarly,IFNg�/�Il17A�/� mice display massively inflamed hearts and
ajp.amjpathol.org - The American Journal of Pathology
1488
� 23 January 2015 � 9:00 pm � EO: AJP14_0604

print&web4C=FPO
Figure 8 Natural killer (NK) cells induce apoptosis in eosinophils (Eos)in vitro. A: Representative side scatter versus SiglecF and LIVE/DEADviability dye versus annexin V plots of Ly6GloSiglecFþ eosinophils after a 3-hours co-culture with primary naïve NK cells. Percentage of annexinVepositive (analysis of variance P < 0.001; B) and live/deadQ38 staine-positive Ly6GloSiglecFþ eosinophils (C). Average mean fluorescence in-tensity of activation markers SiglecF (D) and CD11b (E) of Ly6GloSiglecFhi
eosinophils. Statistics calculated by one-way analysis of variance with posttesting by Tukey’s test. *P < 0.05, ***P < 0.001. SSC, ---.
NK Cells in Autoimmune Myocarditis
14891490149114921493149414951496149714981499150015011502150315041505150615071508150915101511151215131514151515161517151815191520152115221523152415251526152715281529153015311532153315341535153615371538153915401541154215431544154515461547154815491550
1551155215531554155515561557155815591560156115621563156415651566156715681569157015711572157315741575157615771578157915801581158215831584158515861587158815891590159115921593159415951596159715981599160016011602160316041605160616071608160916101611
up to 50% fatality by day 21 of EAM, a phenotype that isreversed in the absence of eosinophils by crossing these ani-mals to dblGATA1 mice.57
Although the numbers of eosinophils infiltrating theheart after NK cell depletion (10% to 15%) during EAM donot approach clinical or animal models of eosinophilicmyocarditis (up to 50%), we clearly demonstrate that eventhis moderate increase has significant consequences ondisease outcome.56,57 Clinically, infiltrating eosinophils arefound in severe giant cell myocarditis and these patients,along with necrotizing eosinophilic myocarditis, have poorclinical prognosis.58,59 Other clinical entities in whichmoderate levels of eosinophils are found infiltrating themyocardium include hypersensitivity and drug reactions,parasitic infections, vasculitis and granulomatous diseases,
The American Journal of Pathology - ajp.amjpathol.orgFLA 5.2.0 DTD � AJPA1936_proof �
and idiopathic hypereosinophilic syndrome.60 Little is knownabout the cardiac eosinophil profile; however, in all cases,cardiac necrosis, thrombosis, and fibrosis are found.61
We found that eosinophils in the hearts of ASGM-1etreated animals displayed a mature, activated profile withfully formed granules containing cationic proteins. Cardiactissue stained positive for major basic protein by immuno-fluorescence. qPCR results showed the down-regulation ofeosinophil peroxidase and major basic protein mRNA ineosinophils isolated from the heart versus the spleen at day21 of EAM, indicating that the active transcription of theseproteins, characteristic of immature eosinophils, was nolonger taking place. Mature eosinophils contain fullyformed granules of these cationic proteins to be releasedinstantly on specific activation and, therefore, have no needfor further transcription of these genes.43 Furthermore, theirup-regulation of SiglecF and CD11b is consistent withactivated lung eosinophils from Nippostrongylus brasi-liensiseinfected mice.42,50 This up-regulation of activationmarkers cannot be attributed to the collagenase treatmentimplemented for cardiac dissociation because we havefound the treatment with collagenase does not alter cellphenotypes (data not shown). Cardiac eosinophils did notup-regulate Il-4 and Il-13, despite reports that eosinophilscan regulate muscle repair through Il-4 and Il-13.62 How-ever, cardiac eosinophils significantly up-regulated Il-6 andTgf-b, mirroring activated eosinophils in airway inflamma-tion and fibrosis models.50,63e65 These cytokines areinvolved in multiple fibrosis pathways and may be respon-sible for the eosinophil-mediated fibrosis seen in ASGM-1etreated wild-type mice.
An alternative hypothesis for the regulation of diseaseseverity by NK cells through eosinophils involves alter-ations in monocytes. Monocytes, as the majority of cardiacinfiltrating cells during EAM, can alter disease severity.33,48
We have shown that monocyte control of disease was due toalterations in Th17 cells.33,34,41,66 However, the increaseddisease in the absence of NK cells was not accompanied bychanges in SSCmidCD11bþ monocytes or the Ly6Chi in-flammatory monocyte subpopulation. Furthermore,although we did see increases in Th2 and Th17 populations,these shifts occurred after increases in eosinophilic infiltra-tion and are likely symptomatic, as opposed to causative, ofthese changes. Therefore, we do not believe that NK cellsprotect against autoimmune-mediated inflammation throughthe regulation of monocytic or T-cell populations.
To investigate how NK cells prevent eosinophils fromaccumulating in the heart during myocarditis, we turned ourattention to interactions between cardiac fibroblasts and NKcells. Studies have shown that tumor fibroblasts interfere withNK cell cytotoxicity, and synoviocytes in RA express NK-receptor ligands that result in NK cell activation.67,68 How-ever, we did not find that NK cells targeted activated cardiacfibroblasts for killing, unlike studies in liver fibrosis.39,40,69
Our group recently showed that Il-17Aestimulated cardiacfibroblasts produce key chemokines and cytokines that are
13
1612
23 January 2015 � 9:00 pm � EO: AJP14_0604

Q28
Ong et al
16131614161516161617161816191620162116221623162416251626162716281629163016311632163316341635163616371638163916401641164216431644164516461647164816491650165116521653165416551656165716581659166016611662166316641665166616671668166916701671167216731674
1675167616771678167916801681168216831684168516861687168816891690169116921693169416951696169716981699170017011702170317041705170617071708170917101711171217131714171517161717171817191720172117221723172417251726172717281729
critical downstream effectors in recruiting and differentiatingmyeloid cells.70,71 We suspect that cardiac fibroblasts secreteunique profiles of cytokines and chemokines when exposedto a different cytokine milieu or on interaction with immunecells in the heart. Indeed, we showed that NK cells controlledchemokine profiles secreted by cardiac fibroblasts duringEAM. On depletion of NK cells, cardiac fibroblastederivedchemokines generated a pro-eosinophilic environmentin vivo, in vitro, and ex vivo.
Of these chemokines, eotaxins and CXCL9, major con-trollers of eosinophilic trafficking in asthma and allergy, werethe most dysregulated.72e75 We were able to show thatCXCL9 was directly controlled by NK-derived IFNg and thatin the absence of IFNg, CXCL9 levels were diminished.However, most chemokines are notorious for functionalredundancy and it is difficult to obtain phenotypic evidencefor their role in disease.76,77 Ccl11 and CXCL9 are noexception to this rule. This was supported by the failure ofCcr3�/� and IFNg�/� mice to show any changes in eosin-ophilic accumulation in the absence of NK cells. Further-more, although this relationship between NK cells andresident cardiac fibroblasts was undeniably present in EAM,it may not be the predominant manner in which NK cellscontrol eosinophilic infiltration.
Therefore, we explored whether NK cells could directlyinhibit eosinophils, by either deactivation or killing. Reportsin human cells indicated that the co-culture of NK cells withnaïve eosinophils resulted in eosinophil activation anddegranulation.78,79 By annexin V staining, a marker of earlyapoptosis, we show that NK cells induce apoptosis of eo-sinophils in vitro. Delayed apoptosis of eosinophils has beenimplicated in multiple asthma models, and the induction ofapoptosis during pharmacological agents contributes to theclearance of disease.80e82 Therefore, the increased eosino-phil numbers seen in the absence of NK cells may be theresult of a deficiency in apoptotic signals during cardiacinflammation.
These data open the possibility for NK cells or theirproducts as a biological therapy for myocarditis. NK-relatedtherapies are an area of avid cancer research, and the sameresources could be used to treat autoimmune disorders.83e85
Developments in the treatment for clinical myocarditis areconstricted by the opposing needs in the viral and autoim-mune components of disease. Ideally, an intervention couldbe designed that would target both needs simultaneously.Our laboratory has now shown that NK cells are clearlyprotective in both viral and autoimmune-mediated drivenforms of myocarditis. In essence, NK cells or NK-derivedproducts concurrently could serve as an antiviral therapyand also as an immunosuppressant.
173017311732173317341735
Acknowledgment
We thank Djahida Bedja for technical assistance in per-forming echocardiography on mice.
14FLA 5.2.0 DTD � AJPA1936_proof
Supplemental Data
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2014.11.023.
References
1. Andreoletti L, Leveque N, Boulagnon C, Brasselet C, Fornes P: Viralcauses of human myocarditis. Arch Cardiovasc Dis 2009, 102:559e568
2. Cihakova D, Rose NR: Pathogenesis of myocarditis and dilatedcardiomyopathy. Adv Immunol 2008, 99:95e114
3. Rose NR, Cihakova D: Cardiomyopathies. Autoimmunity 2004, 37:347e350
4. Seshadri S, Narula J, Chopra P: Asymptomatic eosinophilicmyocarditis: 2 þ 2 Z 4 or 5! Int J Cardiol 1991, 31:348e349
5. Huston B, Froloff V, Mills K, McGee M: Death due to eosinophilicnecrotizing myocarditis despite steroid treatment. Am J Forensic MedPathol 2013, 34:95e97
6. Baandrup U: Eosinophilic myocarditis. Herz 2012, 37:849e8527. Al Ali AM, Straatman LP, Allard MF, Ignaszewski AP: Eosinophilic
myocarditis: case series and review of literature. Can J Cardiol 2006,22:1233e1237
8. Carniel E, Sinagra G, Bussani R, Di Lenarda A, Pinamonti B,Lardieri G, Silvestri F: Fatal myocarditis: morphologic and clinicalfeatures. Ital Heart J 2004, 5:702e706
9. Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M,Kuhl U, Levine GN, Narula J, Starling RC, Towbin J, Virmani R: Therole of endomyocardial biopsy in the management of cardiovasculardisease: a scientific statement from the American Heart Association,the American College of Cardiology, and the European Society ofCardiology. Endorsed by the Heart Failure Society of America andthe Heart Failure Association of the European Society of Cardiology.J Am Coll Cardiol 2007, 50:1914e1931
10. Wensky AK, Furtado GC, Marcondes MC, Chen S, Manfra D,Lira SA, Zagzag D, Lafaille JJ: IFN-gamma determines distinctclinical outcomes in autoimmune encephalomyelitis. J Immunol2005, 174:1416e1423
11. Al-Haddad S, Riddell RH: The role of eosinophils in inflammatorybowel disease. Gut 2005, 54:1674e1675
12. Kay AB: The role of eosinophils in the pathogenesis of asthma.Trends Mol Med 2005, 11:148e152
13. Fairweather D, Kaya Z, Shellam GR, Lawson CM, Rose NR: Frominfection to autoimmunity. J Autoimmun 2001, 16:175e186
14. Godeny EK, Gauntt CJ: Involvement of natural killer cells in cox-sackievirus B3-induced murine myocarditis. J Immunol 1986, 137:1695e1702
15. Loh J, Chu DT, O’Guin AK, Yokoyama WM, Virgin HW: Naturalkiller cells utilize both perforin and gamma interferon to regulatemurine cytomegalovirus infection in the spleen and liver. J Virol2005, 79:661e667
16. Johansson S, Berg L, Hall H, Hoglund P: NK cells: elusive players inautoimmunity. Trends Immunol 2005, 26:613e618
17. Perricone R, Perricone C, De Carolis C, Shoenfeld Y: NK cells inautoimmunity: a two-edg’d weapon of the immune system. Auto-immun Rev 2008, 7:384e390
18. Santiago-Schwarz F, Kay C, Panagiotopoulos C, Carsons SE:Rheumatoid arthritis serum or synovial fluid and interleukin 2abnormally expand natural killer-like cells that are potentstimulators of IgM rheumatoid factor. J Rheumatol 1992, 19:223e228
19. Pridgeon C, Lennon GP, Pazmany L, Thompson RN, Christmas SE,Moots RJ: Natural killer cells in the synovial fluid of rheumatoidarthritis patients exhibit a CD56bright,CD94bright,CD158negativephenotype. Rheumatology (Oxford) 2003, 42:870e878
ajp.amjpathol.org - The American Journal of Pathology
1736
� 23 January 2015 � 9:00 pm � EO: AJP14_0604

NK Cells in Autoimmune Myocarditis
17371738173917401741174217431744174517461747174817491750175117521753175417551756175717581759176017611762176317641765176617671768176917701771177217731774177517761777177817791780178117821783178417851786178717881789179017911792179317941795179617971798
1799180018011802180318041805180618071808180918101811181218131814181518161817181818191820182118221823182418251826182718281829183018311832183318341835183618371838183918401841184218431844184518461847184818491850185118521853185418551856185718581859
20. Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ,Callan MF: CD56bright NK cells are enriched at inflammatory sitesand can engage with monocytes in a reciprocal program of activation.J Immunol 2004, 173:6418e6426
21. Ren J, Feng Z, Lv Z, Chen X, Li J: Natural killer-22 cells in thesynovial fluid of patients with rheumatoid arthritis are an innatesource of interleukin 22 and tumor necrosis factor-alpha. J Rheumatol2011, 38:2112e2118
22. Dalbeth N, Callan MF: A subset of natural killer cells is greatlyexpanded within inflamed joints. Arthritis Rheum 2002, 46:1763e1772
23. Shi FD, Takeda K, Akira S, Sarvetnick N, Ljunggren HG: IL-18directs autoreactive T cells and promotes autodestruction in thecentral nervous system via induction of IFN-gamma by NK cells. JImmunol 2000, 165:3099e3104
24. Yang YZ, Jin PY, Wang QD: [Natural killer cell activity and in-duction of alpha and gamma interferon in patients with Coxsackie Bviral myocarditis] Chinese. Zhonghua Xin Xue Guan Bing Za Zhi1988, 16:337e339, 382
25. Kanda T, Ohshima S, Yuasa K, Watanabe T, Suzuki T, Murata K:Idiopathic myocarditis associated with T-cell subset changes anddepressed natural killer activity. Jpn Heart J 1990, 31:741e744
26. Chow LH, Ye Y, Linder J, McManus BM: Phenotypic analysis ofinfiltrating cells in human myocarditis: an immunohistochemicalstudy in paraffin-embedded tissue. Arch Pathol Lab Med 1989, 113:1357e1362
27. Takahashi K, Aranami T, Endoh M, Miyake S, Yamamura T: Theregulatory role of natural killer cells in multiple sclerosis. Brain 2004,127:1917e1927
28. Afanasyeva M, Georgakopoulos D, Rose NR: Autoimmunemyocarditis: cellular mediators of cardiac dysfunction. AutoimmunRev 2004, 3:476e486
29. Cihakova D, Sharma RB, Fairweather D, Afanasyeva M, Rose NR:Animal models for autoimmune myocarditis and autoimmunethyroiditis. Methods Mol Med 2004, 102:175e193
30. Neu N, Beisel KW, Traystman MD, Rose NR, Craig SW: Autoan-tibodies specific for the cardiac myosin isoform are found in micesusceptible to Coxsackievirus B3-induced myocarditis. J Immunol1987, 138:2488e2492
31. Rose NR: The significance of autoimmunity in myocarditis. ErnstSchering Res Found Workshop 2006, (55):141e154
32. Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D,Zheng D, Gabrielson K, Iwakura Y, Rose NR, Cihakova D: Inter-leukin-17A is dispensable for myocarditis but essential for the pro-gression to dilated cardiomyopathy. Circ Res 2010, 106:1646e1655
33. Barin JG, Baldeviano GC, Talor MV, Wu L, Ong S, Quader F,Chen P, Zheng D, Caturegli P, Rose NR, Cihakova D: Macrophagesparticipate in IL-17-mediated inflammation. Eur J Immunol 2012, 42:726e736
34. Barin JG, Talor MV, Baldeviano GC, Kimura M, Rose NR,Cihakova D: Mechanisms of IFNgamma regulation of autoimmunemyocarditis. Exp Mol Pathol 2010, 89:83e91
35. Cihakova D, Barin JG, Afanasyeva M, Kimura M, Fairweather D,Berg M, Talor MV, Baldeviano GC, Frisancho S, Gabrielson K,Bedja D, Rose NR: Interleukin-13 protects against experimentalautoimmune myocarditis by regulating macrophage differentiation.Am J Pathol 2008, 172:1195e1208
36. Kasai M, Yoneda T, Habu S, Maruyama Y, Okumura K, Tokunaga T:In vivo effect of anti-asialo GM1 antibody on natural killer activity.Nature 1981, 291:334e335
37. Habu S, Fukui H, Shimamura K, Kasai M, Nagai Y, Okumura K,Tamaoki N: In vivo effects of anti-asialo GM1, I: reduction of NKactivity and enhancement of transplanted tumor growth in nude mice.J Immunol 1981, 127:34e38
38. O’Connell TD, Rodrigo MC, Simpson PC: Isolation and culture ofadult mouse cardiac myocytes. Methods Mol Biol 2007, 357:271e296
The American Journal of Pathology - ajp.amjpathol.orgFLA 5.2.0 DTD � AJPA1936_proof �
39. Gonzalez A, Katz JD, Mattei MG, Kikutani H, Benoist C, Mathis D:Genetic control of diabetes progression. Immunity 1997, 7:873e883
40. Poirot L, Benoist C, Mathis D: Natural killer cells distinguishinnocuous and destructive forms of pancreatic islet autoimmunity.Proc Natl Acad Sci U S A 2004, 101:8102e8107
41. Barin JG, Cihakova D: Control of inflammatory heart disease byCD4þ T cells. Ann N Y Acad Sci 2013, 1285:80e96
42. Johansson MW: Activation states of blood eosinophils in asthma.Clin Exp Allergy 2014, 44:482e498
43. Voehringer D, van Rooijen N, Locksley RM: Eosinophils develop indistinct stages and are recruited to peripheral sites by alternativelyactivated macrophages. J Leukoc Biol 2007, 81:1434e1444
44. Yu C, Cantor AB, Yang H, Browne C, Wells RA, Fujiwara Y,Orkin SH: Targeted deletion of a high-affinity GATA-binding site inthe GATA-1 promoter leads to selective loss of the eosinophil lineagein vivo. J Exp Med 2002, 195:1387e1395
45. Lanier LL: Activating and inhibitory NK cell receptors. Adv ExpMed Biol 1998, 452:13e18
46. Lanier LL: NK cell recognition. Annu Rev Immunol 2005, 23:225e274
47. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S: Functions ofnatural killer cells. Nat Immunol 2008, 9:503e510
48. Wu LOS, Talor MV, Barin JG, Baldeviano GC, Kass DA, Bedja D,Zhang H, Sheikh A, Margolick JA, Iwakura Y, Rose NR,Cihakova D: Cardiac fibroblasts mediate IL-17A-driven inflammatorydilated cardiomyopathy. J Exp Med 2014, 211:1449e1464
49. Xanthou G, Duchesnes CE, Williams TJ, Pease JE: CCR3 func-tional responses are regulated by both CXCR3 and its ligandsCXCL9, CXCL10 and CXCL11. Eur J Immunol 2003, 33:2241e2250
50. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S,Wechsler ME, Israel E, Levy BD: Lipoxin A4 regulates natural killercell and type 2 innate lymphoid cell activation in asthma. Sci TranslMed 2013, 5:174ra126
51. Awad A, Yassine H, Barrier M, Vorng H, Marquillies P,Tsicopoulos A, Duez C: Natural killer cells induce eosinophil acti-vation and apoptosis. PLoS One 2014, 9:e94492
52. Sungur CM, Tang-Feldman YJ, Zamora AE, Alvarez M, Pomeroy C,Murphy WJ: Murine NK-cell licensing is reflective of donor MHC-Ifollowing allogeneic hematopoietic stem cell transplantation in mu-rine cytomegalovirus responses. Blood 2013, 122:1518e1521
53. Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T: Regulationof experimental autoimmune encephalomyelitis by natural killer (NK)cells. J Exp Med 1997, 186:1677e1687
54. Lassen MG, Lukens JR, Dolina JS, Brown MG, Hahn YS: Intra-hepatic IL-10 maintains NKG2AþLy49- liver NK cells in a func-tionally hyporesponsive state. J Immunol 2010, 184:2693e2701
55. Fathman JW, Bhattacharya D, Inlay MA, Seita J, Karsunky H,Weissman IL: Identification of the earliest natural killer cell-committed progenitor in murine bone marrow. Blood 2011, 118:5439e5447
56. Abston ED, Barin JG, Cihakova D, Bucek A, Coronado MJ,Brandt JE, Bedja D, Kim JB, Georgakopoulos D, Gabrielson KL,Mitzner W, Fairweather D: IL-33 independently induces eosinophilicpericarditis and cardiac dilation: ST2 improves cardiac function. CircHeart Fail 2012, 5:366e375
57. Barin JG, Baldeviano GC, Talor MV, Wu L, Ong S, Fairweather D,Bedja D, Stickel NR, Fontes JA, Cardamone AB, Zheng D,Gabrielson KL, Rose NR, Cihakova D: Fatal eosinophilic myocarditisdevelops in the absence of IFN-gamma and IL-17A. J Immunol 2013,191:4038e4047
58. Hyogo M, Kamitani T, Oguni A, Kawasaki S, Miyanaga H,Takahashi T, Kunishige H, Andachi H: Acute necrotizing eosino-philic myocarditis with giant cell infiltration after remission of idio-pathic thrombocytopenic purpura. Intern Med 1997, 36:894e897
59. Litovsky SH, Burke AP, Virmani R: Giant cell myocarditis: an entitydistinct from sarcoidosis characterized by multiphasic myocyte
15
1860
23 January 2015 � 9:00 pm � EO: AJP14_0604

Ong et al
18611862186318641865186618671868186918701871187218731874187518761877187818791880188118821883188418851886188718881889189018911892189318941895189618971898189919001901190219031904190519061907190819091910191119121913191419151916191719181919192019211922
19231924192519261927192819291930193119321933193419351936193719381939194019411942194319441945194619471948194919501951195219531954195519561957195819591960196119621963
destruction by cytotoxic T cells and histiocytic giant cells. ModPathol 1996, 9:1126e1134
60. Rezaizadeh H, Sanchez-Ross M, Kaluski E, Klapholz M, Haider B,Gerula C: Acute eosinophilic myocarditis: diagnosis and treatment.Acute Card Care 2010, 12:31e36
61. Weller PF, Bubley GJ: The idiopathic hypereosinophilic syndrome.Blood 1994, 83:2759e2779
62. Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC,Locksley RM, Rando TA, Chawla A: Type 2 innate signals stimulatefibro/adipogenic progenitors to facilitate muscle regeneration. Cell2013, 153:376e388
63. Louch WE, Sheehan KA, Wolska BM: Methods in cardiomyocyteisolation, culture, and gene transfer. J Mol Cell Cardiol 2011, 51:288e298
64. Lim YC, Luscinskas FW: Isolation and culture of murine heart andlung endothelial cells for in vitro model systems. Methods Mol Biol2006, 341:141e154
65. Afanasyeva M, Georgakopoulos D, Belardi DF, Ramsundar AC,Barin JG, Kass DA, Rose NR: Quantitative analysis of myocardialinflammation by flow cytometry in murine autoimmune myocar-ditis: correlation with cardiac function. Am J Pathol 2004, 164:807e815
66. Barin JG, Rose NR, Cihakova D: Macrophage diversity in cardiacinflammation: a review. Immunobiology 2012, 217:468e475
67. Nielsen N, Pascal V, Fasth AE, Sundstrom Y, Galsgaard ED,Ahern D, Andersen M, Baslund B, Bartels EM, Bliddal H,Feldmann M, Malmstrom V, Berg L, Spee P, Soderstrom K: Balancebetween activating NKG2D, DNAM-1, NKp44 and NKp46 andinhibitory CD94/NKG2A receptors determine NK degranulation to-wards RA synovial fibroblasts. Immunology 2014, 142:581e593
68. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C,Boitano M, Queirolo P, Vermi W, Facchetti F, Moretta A, Moretta L,Mingari MC, Vitale M: Melanoma-associated fibroblasts modulateNK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci US A 2009, 106:20847e20852
69. Shi FD, Wang HB, Li H, Hong S, Taniguchi M, Link H, Van Kaer L,Ljunggren HG: Natural killer cells determine the outcome of B cell-mediated autoimmunity. Nat Immunol 2000, 1:245e251
70. Wu L, Ong S, Talor MV, Barin JG, Baldeviano GC, Kass DA,Bedja D, Zhang H, Sheikh A, Margolick JB, Iwakura Y, Rose NR,Cihakova D: Cardiac fibroblasts mediate IL-17A-driven inflammatorydilated cardiomyopathy. J Exp Med 2014, 211:1449e1464
71. Fujiu K, Nagai R: Contributions of cardiomyocyte-cardiac fibroblast-immune cell interactions in heart failure development. Basic ResCardiol 2013, 108:357
16FLA 5.2.0 DTD � AJPA1936_proof
72. Zhang J, Wu Y, Weng Z, Zhou T, Feng T, Lin Y: Glycyrrhizinprotects brain against ischemia-reperfusion injury in mice throughHMGB1-TLR4-IL-17A signaling pathway. Brain Res 2014, 1582:176e186
73. Wang JY, Shyur SD, Wang WH, Liou YH, Lin CG, Wu YJ, Wu LS:The polymorphisms of interleukin 17A (IL17A) gene and its asso-ciation with pediatric asthma in Taiwanese population. Allergy 2009,64:1056e1060
74. Jin R, Guo S, Wu L, Fan X, Ma H, Lowrie DB, Zhang J: Il17Aautoantibody induced by recombinant protein Ag85A-Il17A. ApplBiochem Biotechnol 2013, 169:502e510
75. Wu X, Zeng Z, Xu L, Yu J, Cao Q, Chen M, Sung JJ, Hu P: Increasedexpression of IL17A in human gastric cancer and its potential roles ingastric carcinogenesis. Tumour Biol 2014, 35:5347e5356
76. Mantovani A: The chemokine system: redundancy for robust outputs.Immunol Today 1999, 20:254e257
77. Lukacs NW, Oliveira SH, Hogaboam CM: Chemokines and asthma:redundancy of function or a coordinated effort? J Clin Invest 1999,104:995e999
78. Fujisawa T, Velichko S, Thai P, Hung LY, Huang F, Wu R: Regu-lation of airway MUC5AC expression by IL-1beta and IL-17A: theNF-kappaB paradigm. J Immunol 2009, 183:6236e6243
79. Chen XQ, Yu YC, Deng HH, Sun JZ, Dai Z, Wu YW, Yang M:Plasma IL-17A is increased in new-onset SLE patients and associatedwith disease activity. J Clin Immunol 2010, 30:221e225
80. Fujisawa T, Chang MM, Velichko S, Thai P, Hung LY, Huang F,Phuong N, Chen Y, Wu R: NF-kappaB mediates IL-1beta- and IL-17A-induced MUC5B expression in airway epithelial cells. Am JRespir Cell Mol Biol 2011, 45:246e252
81. Zhao N, Hao J, Ni Y, Luo W, Liang R, Cao G, Zhao Y, Wang P,Zhao L, Tian Z, Flavell R, Hong Z, Han J, Yao Z, Wu Z, Yin Z:Vgamma4 gammadelta T cell-derived IL-17A negatively regulatesNKT cell function in Con A-induced fulminant hepatitis. J Immunol2011, 187:5007e5014
82. Kong Q, Xue Y, Wu W, Yang F, Liu Y, Gao M, Lai W, Pan X: IL-22exacerbates the severity of CVB3-induced acute viral myocarditis inIL-17A-deficient mice. Mol Med Rep 2013, 7:1329e1335
83. Ljunggren HG, Malmberg KJ: Prospects for the use of NK cells inimmunotherapy of human cancer. Nat Rev Immunol 2007, 7:329e339
84. Lee SK, Gasser S: The role of natural killer cells in cancer therapy.Front Biosci (Elite Ed) 2010, 2:380e391
85. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L: Targetingnatural killer cells natural killer T cells in cancer. Nat Rev Immunol2012, 12:239e252
ajp.amjpathol.org - The American Journal of Pathology
196419651966196719681969197019711972197319741975197619771978197919801981198219831984
� 23 January 2015 � 9:00 pm � EO: AJP14_0604

Q29
Q30
Q31
1985198619871988198919901991199219931994199519961997199819992000200120022003200420052006200720082009201020112012201320142015201620172018201920202021202220232024202520262027202820292030
2031203220332034203520362037203820392040204120422043204420452046204720482049205020512052205320542055205620572058205920602061206220632064206520662067206820692070207120722073207420752076
Supplemental Figure S1 The polyclonal antibody asialogangloside GM-1 (ASGM-1) selectively depletes natural killer (NK) cells from naïve animals.Percentage of CD3�DX5þNKp46þ NK cells (A), CD3þDX5þNKp46þ NK T cells (B), and Ly6GloSiglecFþ eosinophils (C) in the liver, heart, lung, and spleen ofnaïve wild-type (WT) mice after 3 days of rabbit IgG (RaIgG) or ASGM-1 treatment of 1 mg of antibody per day. All statistics calculated by unpaired t-tests.*P < 0.05, **P < 0.01, and ***P < 0.001.
Supplemental Figure S2 Cardiac fibroblasts can be isolated from adult mice. A: Immunofluorescence images of adult mouse cardiac fibroblasts (AMCFs)isolated and passaged twice and primary bone marrowederived macrophages (BMMs), fixed with 4% paraformaldehyde and stained with DAPI, 1:1000 anti-asmooth muscle rabbit IgG antibody and 1:200 anti-rabbit rat fluorescein isothiocyanateeconjugated antibody, and 1:500 anti-CD11b rat antibody and 1:100anti-rat Texas Redeconjugated antibody. B and C: Levels of CD11b Itgam mRNA as measured by real-time quantitative PCR relative to Hprt in untreated cardiacfibroblasts and BMMs (B) and untreated cardiac fibroblasts and naïve BALB/c heart homogenates (C). ND, no data.
Supplemental Figure S3 Activated fibroblasts are not targeted for cytotoxic killing by natural killer (NK) cells in vitro. A: Expression of NKG2D and CD69in splenic NK cells from naïve or poly(I:C)-treated mice after 24 hours. B: Levels of vimentin mRNA in cardiac fibroblasts after 1 mmol/L angiotensin II (AngII)treatment for 1 hour, as controlled by Hprt (P Z 0.02). C: Representative images and percentage of live cardiac fibroblasts after 48 hours of co-culture withnaïve and activated NK cells, as determined by red versus green cells. D and E: Quantification of naïve (D) and activated (E) NK cells in C. Statistics for Bcalculated by unpaired t-test and for D and E by one-way analysis of variance with Tukey’s post testing. *P < 0.05. AMCF, adult mouse cardiac fibroblast; NT,no treatment.
Supplemental Figure S4 Depletion of the polyclonal antibody asialogangloside GM-1 (ASGM-1) during experimental autoimmune myocarditis does notinduce peripheral eosinophilia. Percentage of Ly6G�SiglecFþ eosinophils in the blood (A) and spleen (B) in isotype control and natural killer (NK)celledepleted animals at day 14 and the spleen at day 21 (C) as a function of total CD45þ hematopoietic cells. Statistics calculated by unpaired t-test. RaIgG,rabbit IgG.
Supplemental Figure S5 Noneosinophil CD45þ populations are unchanged proportionally by natural killer cell depletion during experimental auto-immune myocarditis (EAM). A: SSCmidCD11bþ monocytes as a percentage of total CD45þ cardiac cells at day 14 of EAM in isotype and asialogangloside GM-1(ASGM-1)etreated wild-type (WT) animals. Ly6Chi inflammatory (B) and Ly6Clomid resident (C) subsets as a percentage of total SSCmidCD11bþ cardiacmonocytes. CD11cþ dendritic cells (D) and FCεR1aþcKitþ mast cells (E) as a percentage of total CD45þ cardiac cells at day 14 of EAM in isotype and ASGM-1etreated WT animals. Lymphocyte percentages of B220þ B cells (F), CD3þ T cells (G), and CD3þCD4þ helper T cells (H) as a percentage of total CD45þ cardiaccells at day 21 of EAM in isotype control and ASGM-1etreated animals. Significance determined by unpaired t-test. RaIgG, rabbit IgG.
Supplemental Figure S6 Depletion of natural killer cells increases type 2 helper T cell (Th2) and Th17 proportions at later stages of experimentalautoimmune myocarditis (EAM). T-cell subset examination by intracellular cytokine staining for interferon g (IFNg) (Th1; A), Il-13 (Th2; B), and Il-17A (Th17;C), as a proportion of cardiac CD3þCD4þ cells at days 14 and 21 of EAM after 4 to 6 hours of 4b-phorbol 12-myristate 13-acetate and ionomycin stimulationwith Golgistop. Significance calculated by unpaired t-test. **P < 0.01, ***P < 0.001. RaIgG, rabbit IgG.
Supplemental Figure S7 Natural killer (NK) cells alter levels of eosinophil-associated chemokines from cardiac fibroblasts in vitro. Cardiac fibroblastscultured with Il-4 or Il-4 and magnetically sorted NK cells (1:2 ratio) for 94 hours. Levels of Cxcl10 (P < 0.001; A), chemokine ligand (Ccl2; P < 0.001; B), Ccl4(P < 0.001; C), Ccl5 (P Z 0.002; D), Cxcl1 (P < 0.001; E), and Il-5 (P < 0.001; F) protein in supernatant were measured by enzyme-linked immunosorbentassay. Significance by ordinary one-way analysis of variance with post testing by Tukey’s multiple-comparisons test. ***P < 0.001.
Supplemental Figure S8 Cxcl9 and interferon g (IFNg) are not required by natural killer (NK) cells for control of eosinophil trafficking. A: Levels of Cxcl9mRNA by real-time quantitative PCR (qPCR) from rabbit IgG (RaIgG) and asialogangloside GM-1 (ASGM-1)etreated hearts at day 21 of experimental auto-immune myocarditis (EAM; P Z 0.004). Cxcl9 protein from supernatants of cardiac fibroblasts co-cultured with or without naïve NK cells for 96 hours(P < 0.001; B) and naïve NK co-cultures from wild-type (WT) or IFNg receptor 1 (IFNgR1)�/� cardiac fibroblasts for 96 hours (P < 0.001; C). D: Cxcl9 mRNA byqPCR from WT and IFNg�/� hearts at day 14 of EAM (P Z 0.050). E: Percentage of Ly6GloSiglecFþ eosinophils at day 14 of EAM in rabbit IgG and ASGM-1etreated IFNg�/� animals. Significance by Student’s t-test. *P < 0.05, **P < 0.01, and ***P < 0.001.
FLA 5.2.0 DTD � AJPA1936_proof � 23 January 2015 � 9:00 pm � EO: AJP14_0604