native chemical ligation of polypeptides
TRANSCRIPT

UNIT 18.4Native Chemical Ligation of Polypeptides
The total synthesis and semisynthesis of proteins allows the site-specific incorporation
of unnatural amino acids, post-translational modifications, and biophysical/biochemical
probes into the target molecule. Among the various chemical and enzymatic approaches
available for the synthesis/semisynthesis of proteins, the native chemical ligation tech-
nique has proven especially useful and will be the exclusive focus of this unit. Native
chemical ligation allows native backbone proteins to be assembled from fully unprotected
polypeptide building blocks. To facilitate the ligation reaction, the α-carboxylate group
of the N-terminal polypeptide fragment must be mildly activated as an aryl thioester, while
the C-terminal polypeptide fragment must contain an amino-terminal cysteine residue.
The reaction is normally carried out in aqueous buffers at around neutral pH and can be
performed with complete regioselectivity in the presence of all the functionalities com-
monly found in proteins. Even the presence of additional cysteine sulfhydryl groups in
one or both peptide fragments does not affect the reaction regioselectivity. The unit first
discusses how to choose the ligation site(s) in the target protein and then outlines how to
obtain the necessary polypeptide building blocks using either chemical synthesis or
recombinant DNA expression (see Strategic Planning). Next, the synthesis of a protein
by native chemical ligation of two polypeptide fragments is described (see Basic Protocol
1). The synthesis of a protein from three polypeptide fragments using a sequential native
chemical ligation strategy is also described (see Basic Protocol 2). The support protocols
describe how to obtain the necessary polypeptide fragments using either chemical
synthesis or recombinant DNA expression.
NOTE: All the operations involving volatile chemicals should be performed in a well
ventilated fume hood. See APPENDIX 2A for general safety guidelines.
STRATEGIC PLANNING
The first step in synthesizing a protein by native chemical ligation is to choose the ligation
site(s), thereby defining the polypeptide fragments to be used in the ligation reactions.
The position of the ligation site depends upon several factors: the primary sequence of
the protein; the secondary and tertiary structures of the protein (if known); and the type
of strategy being used to generate the fragments, i.e., chemical synthesis or recombinant
DNA expression. Ideally the target protein should be divided up into synthetically
accessible fragments the size of which depends upon whether chemical synthesis or
biosynthesis is being used for their generation; optimized solid-phase peptide synthesis
(SPPS; also see UNIT 18.1) allows polypeptides of up to ∼50 residues to be efficiently
prepared, whereas recombinant DNA expression permits the preparation of much larger
fragments. For example, a hypothetical 100-amino-acid protein could be assembled either
from two ∼50-residue synthetic peptides or from a 20-residue synthetic fragment and an
80-residue recombinant fragment. Note that in the latter strategy (i.e., semisynthesis) it
is important to design the synthesis in such a way that the chemically synthesized fragment
encompasses the region of the protein to be engineered. Where possible, naturally
occurring X-Cys motifs in the native sequence should be chosen as the ligation sites,
where X can be any residue except Asp, Asn, Glu, Gln, and Pro. In the absence of an
endogenous Cys at the appropriate position in the primary sequence, it will be necessary
to mutate a native residue in order to facilitate ligation. The effect of this Cys mutation
on the structure and function of the protein can usually be minimized by following a few
simple rules: (1) If the tertiary structure of the protein is known, choose a region remote
from the active site of the protein, preferably in a flexible surface loop; (2) choose the
mutation to be as conservative as possible, e.g., Ser→Cys or Ala→Cys; (3) try to avoid
Supplement 15
Contributed by Julio A. Camarero and Tom W. MuirCurrent Protocols in Protein Science (1999) 18.4.1-18.4.21
Copyright © 1999 by John Wiley & Sons, Inc.
18.4.1
Preparation andHandling ofPeptides
mutating residues that are conserved across a gene family; and (4) take advantage of any
known mutational data on the system since the effect on structure/function of mutating a
particular residue may already be known.
Chemical synthesis and recombinant DNA expression each allow the generation of
polypeptides containing an amino-terminal cysteine residue (C-terminal fragment) and
the generation of polypeptides containing an α-thioester functionality (N-terminal frag-
ment). For polypeptides of ∼50 residues or less, chemical synthesis is usually the method
of choice since it is relatively quick and efficient and allows the incorporation of unnatural
amino acids. Amino-terminal cysteine–containing peptides can be chemically synthesized
via established t-butyloxycarbonyl (Boc) or 9-fluorenyloxycarbonyl (Fmoc) SPPS pro-
tocols using commercially available resins (e.g., from NovaBiochem, Bachem, Peptides
International, Applied Biosystems, or Neosystems) and amino acid derivatives (see Table
18.4.1). The synthesis of peptide α-thioesters can currently only be achieved using Boc
chemistry, due to the sensitivity of the thioester functionality to the base-deprotection
conditions associated with the Fmoc-SPPS strategy. Note, the resin required for the
synthesis of peptide α-thioesters is not commercial available, but can be prepared in the
laboratory in a single day.
For polypeptide fragments larger than ∼50 amino acids in length, a biosynthetic strategy
should be employed. It must be noted that this assumes the gene has been cloned.
Polypeptides containing amino-terminal Cys residues can be obtained using a mutagene-
sis/proteolysis strategy. This involves constructing an expression vector in which a DNA
sequence encoding the peptide Ile-Glu-Gly-Arg-Cys is inserted between a upstream
affinity-purification handle (e.g., GST or His tag) and the appropriate gene fragment of
interest (for cloning strategies see UNIT 6.6). Following expression and affinity purification
(UNIT 6.6), the fusion protein is treated with Factor Xa to give the desired recombinant
Cys-polypeptide. Polypeptides containing α-thioester functionalities can be obtained by
expressing the corresponding polypeptide sequence using the IMPACT expression system
(New England Biolabs). This results in the generation of a fusion protein in which the
polypeptide of interest is attached through its C-terminus to an intein-CBD, where the
intein is an engineered protein-splicing element and CBD (chitin binding domain) is an
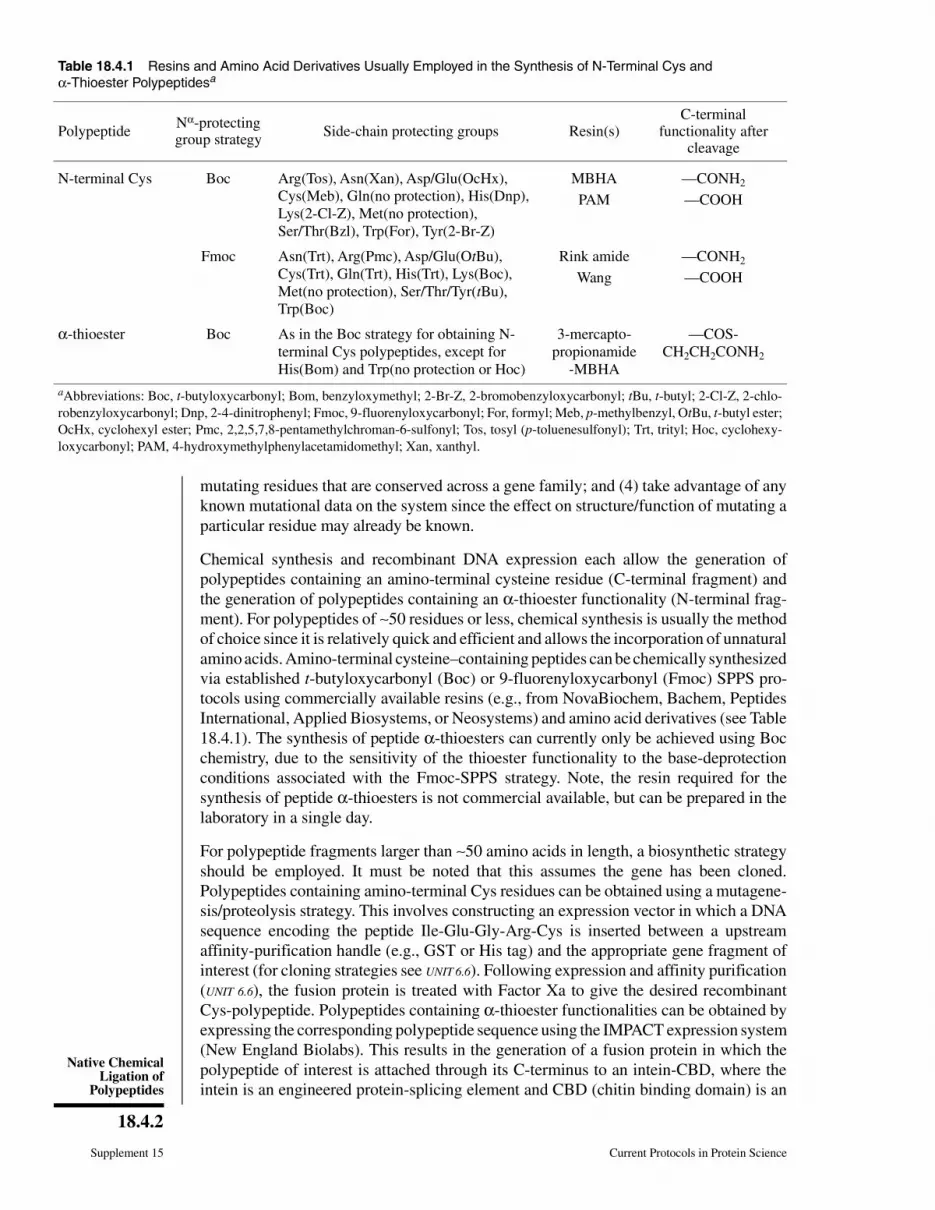
Table 18.4.1 Resins and Amino Acid Derivatives Usually Employed in the Synthesis of N-Terminal Cys and
α-Thioester Polypeptidesa
PolypeptideNα-protectinggroup strategy
Side-chain protecting groups Resin(s)C-terminal
functionality aftercleavage
N-terminal Cys Boc Arg(Tos), Asn(Xan), Asp/Glu(OcHx),
Cys(Meb), Gln(no protection), His(Dnp),
Lys(2-Cl-Z), Met(no protection),
Ser/Thr(Bzl), Trp(For), Tyr(2-Br-Z)
MBHA —CONH2
PAM —COOH
Fmoc Asn(Trt), Arg(Pmc), Asp/Glu(OtBu),
Cys(Trt), Gln(Trt), His(Trt), Lys(Boc),
Met(no protection), Ser/Thr/Tyr(tBu),
Trp(Boc)
Rink amide —CONH2
Wang —COOH
α-thioester Boc As in the Boc strategy for obtaining N-
terminal Cys polypeptides, except for
His(Bom) and Trp(no protection or Hoc)
3-mercapto-
propionamide
-MBHA
—COS-
CH2CH2CONH2
aAbbreviations: Boc, t-butyloxycarbonyl; Bom, benzyloxymethyl; 2-Br-Z, 2-bromobenzyloxycarbonyl; tBu, t-butyl; 2-Cl-Z, 2-chlo-
robenzyloxycarbonyl; Dnp, 2-4-dinitrophenyl; Fmoc, 9-fluorenyloxycarbonyl; For, formyl; Meb, p-methylbenzyl, OtBu, t-butyl ester;
OcHx, cyclohexyl ester; Pmc, 2,2,5,7,8-pentamethylchroman-6-sulfonyl; Tos, tosyl (p-toluenesulfonyl); Trt, trityl; Hoc, cyclohexy-
loxycarbonyl; PAM, 4-hydroxymethylphenylacetamidomethyl; Xan, xanthyl.
Supplement 15 Current Protocols in Protein Science
18.4.2
Native ChemicalLigation of
Polypeptides

affinity handle. The desired recombinant α-thioester polypeptide can be obtained by
simply treating the immobilized fusion protein with an appropriate thiol; the intein-CBD
construct remains attached to the affinity matrix.
BASIC
PROTOCOL 1
NATIVE CHEMICAL LIGATION OF TWO POLYPEPTIDES
This protocol describes the chemical ligation of two polypeptide segments to afford a
target protein containing both fragments linked through a native peptide bond. The
reaction is initiated by dissolving both peptides in aqueous buffer at pH 7.5 in the presence
of thiol cofactors. The reaction usually takes between 1 and 3 days to go to completion,
at which point the product is purified using reversed-phase high-performance liquid
chromatography (RP-HPLC) and subsequently folded. A schematic of the chemistry
behind the procedure is presented in Figure 18.4.1.
Materials
6 M guanidine⋅HCl buffer (see recipe)
Nitrogen source (ultrapure)
Benzyl mercaptan (Aldrich)
Thiophenol (Aldrich)
Polypeptide with N-terminal Cys residue (see Support Protocols 1 and 2)
Polypeptide with α-thioester functionality (see Support Protocols 3 and 4)
1 M NaOH
Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, 99% pure; Strem Chemicals)
OH
SHCO2–
NH
NH2H2N+
NH3
+
O
SRH3N
+H2N
HS
CO2–CONH
OH
SHCO2–
NH
NH2H2N+
NH3
+
Polypeptide 2 Polypeptide 1
OH
SHCO2–
NH
NH2H2N+
NH3
+
O
SH3N
+H2N
CONH
SHCO2–
NH
NH2H2N+
NH3
+
Polypeptide 1
reversible transthioesterification
spontaneous S to N acyl transfer
Polypeptide 2
OH
SHCO2–
NH
NH2H2N+
NH3
+
O
NHH3N
+SH
CO2–CONH
OH
SHCO2–
NH
NH2H2N+
NH3
+
Polypeptide 2Polypeptide 1
Polypeptide 2
OH
CO2–
Figure 18.4.1 The mechanism of native chemical ligation. The initial step is a reversible
transthioesterification reaction involving the thiol group of the N-terminal Cys-polypeptide (C-termi-
nal fragment) and the α-thioester moiety of the N-terminal polypeptide fragment. This intermediate
undergoes a spontaneous rearrangement to form a natural peptide bond at the ligation site.
continued
Current Protocols in Protein Science Supplement 15
18.4.3
Preparation andHandling ofPeptides

Buffers for analytical C18 reversed-phase HPLC
Buffer A: H2O containing 0.1% trifluoroacetic acid (TFA)
Buffer B: 1 part H2O/9 parts acetonitrile/0.1% TFA
Appropriate final buffer in which target protein will be dissolved (e.g., forlong-term storage, activity studies, or structural studies), containing 6 Mguanidine⋅HCl and 5 mM DTT
Appropriate final buffer in which target protein will be dissolved, containing 4 M,2 M, and 0 M guanidine⋅HCl (APPENDIX 3A) and 1 mM DTT
Dialysis membrane with MWCO substantially smaller than target protein
Centricon filter (Amicon; optional)
Tabletop centrifuge (Sorvall RT-7 or equivalent)
Additional reagents and equipment for analytical and semipreparative orpreparative C18 reversed-phase HPLC, electrospray ionization (ESI) ormatrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) massspectrometry (MS; UNITS 16.1 & 16.2), dialysis (UNIT 4.4 & APPENDIX 3B), andspectrophotometric determination of protein concentration (UNIT 3.1)
Prepare the ligation buffer
1. Degas the 6 M guanidine⋅HCl buffer by purging with ultrapure nitrogen for at least
20 min.
2. Add benzyl mercaptan and thiophenol, each at a concentration of 0.5% (v/v), to the
degassed buffer. Mix by vortexing to produce the ligation buffer.
IMPORTANT NOTE: This ligation buffer should be prepared just prior to use and should
not be stored for long periods of time. Thiols are, in general, toxic and have an unpleasant
odor; therefore thiol-containing solutions should be treated with 5% (v/v) commercial
bleach in 1:1 (v/v) methanol/water before being disposed of.
Since benzyl mercaptan and thiophenol are barely soluble in aqueous buffers, vortexing is
required to form a homogeneous thiol suspension in the ligation buffer.
Ligate the N-terminal and C-terminal polypeptide fragments
3. Dissolve equimolar amounts of the polypeptides to be ligated (i.e., one with an
N-terminal Cys residue and the other with an α-thioester functionality) in the ligation
buffer to a final concentration of at least 0.5 mM with respect to each peptide.
Reactions can be run in volumes of 50 to 100 µl for analytical purposes and up to 0.5 to 5
ml for preparative purposes.
Native chemical ligation reactions are usually carried out in the presence of chaotropic
agents (guanidium⋅HCl or urea) in order to maximize the concentration of the two reactants
and to increase the availability of reacting moieties by alleviating any steric hindrance that
might be associated with folded polypeptides. The use of chemical denaturants, of course,
presumes that the final product can be properly folded in vitro. In cases where the fragments
being ligated must be kept in a folded state, the ligation reaction can be carried out in the
absence of chemical denaturants.
4. Check the pH of the ligation mixture by spotting a 2- to 3-µl aliquot onto a strip of
pH paper. If it is above or below 7.0, adjust the pH by titrating in small increments
with 1 M HCl or NaOH, respectively. Repeat this process until a pH of ∼7.5 is reached.
Dissolving lyophilized peptides at high concentrations can cause the pH of the ligation
mixture to drop; this is particularly so when HPLC-purified materials are being used (i.e.,
from acidic conditions). It is extremely important to ensure that the pH of the reaction
mixture is kept between 7 and 8, since the efficiency of the native chemical ligation is
strongly dependent on the pH. As the pH drops below pH 7.0, the reaction slows down, and
as it increases above pH 8.0 the chemoselectivity of the process is lost.
Supplement 15 Current Protocols in Protein Science
18.4.4
Native ChemicalLigation of
Polypeptides

5. Keep the reaction at room temperature with gentle stirring for 24 hr.
6. After 24 hr, take a 5-µl aliquot of the reaction mixture (allowing the rest of the mixture
to continue reacting as in step 5) and dilute the aliquot in a polypropylene microcen-
trifuge tube with 50 µl of 6 M guanidine⋅HCl buffer in the presence of one crystal of
tris-(2-carboxyethyl)phosphine hydrochloride (TCEP, used as a reducing agent).
Shake the sample until the TCEP is completely dissolved, wait 10 min, then
microcentrifuge 5 min at 10,000 rpm.
7. Analyze 25 µl of the supernatant by analytical C18 reversed-phase HPLC, initially
using a linear gradient from 0% buffer B to 73% buffer B over 60 min at a flow rate
of 1 ml/min and refining as necessary based on the elution times of the reactants and
product. Monitor the run via UV detection at 214 nm (and 280 nm if aromatic residues
are present in the target protein). Manually collect every peak (except the solvent
peak) into a separate polypropylene microcentrifuge tube and analyze each by
ESI-MS (UNITS 16.1 & 16.2), which will allow the retention times of the reactants and
the product to be determined
Occasionally the peak corresponding to the ligation product is buried beneath the thiophe-
nol or benzyl mercaptan peaks. This can usually be resolved by optimizing the HPLC
gradient or, alternatively, by lyophilizing the reduced sample a few times and then
reanalyzing it by HPLC.
8. After 36 hr take another 5-µl aliquot of the reaction mixture and analyze it by HPLC
as in step 7. If significant amounts of both reactants still remain and the product peak
area is still increasing, then leave the reaction for another 12 hr and repeat the analysis.
If, on the contrary, both reactants have been consumed or if the product peak area has
not increased appreciably, then terminate the reaction as in step 9.
Ligation reactions normally take 24 to 36 hr to go to completion, although on rare
occasions longer periods (3 to 4 days) or shorter periods (a few hours) may be required.
9. Dilute the crude reaction mixture into 5 to 10 vol of 6 M guanidine⋅HCl buffer and
reduce by adding 15 molar equivalents of TCEP (i.e., based on the quantities of
reactants used; 15 times the number of moles of either one of the polypeptides added).
Shake the solution until all the TCEP is dissolved and wait for 20 min.
10. Centrifuge the sample 20 min at 3000 rpm in a tabletop centrifuge (Sorvall RT-7 or
equivalent), room temperature. Retain the supernatant.
A white cloudiness often appears in the reaction medium during the course of the reaction.
This is due to the slow formation of insoluble benzyl mercaptan and thiophenol disulfides.
The reactants and ligation product will generally remain in solution, allowing the unwanted
disulfides to be removed by centrifugation.
The crude ligation mixture can be stored at −80°C for 2 to 3 days, although it is advisable
to purify it as soon as possible.
Purify the target protein
11. Purify the crude reaction mixture by semipreparative or preparative reversed-phase
HPLC using an optimized gradient system. Analyze each HPLC fraction by mass
spectrometry (UNITS 16.1 & 16.2) and analytical reversed-phase HPLC in order to identify
those fractions containing the target protein. Pool the fractions of highest purity
(>95%) and lyophilize.
Reversed-phase HPLC purification will denature the majority of proteins, meaning that a
subsequent refolding step is required. In cases where the ligation reaction has been carried
out under more physiological conditions (i.e., without chemical denaturants in the ligation
buffer), the final purification step can be performed using, e.g., gel-filtration chromatog-
raphy (UNIT 8.3) or ion-exchange chromatography (UNIT 8.4) under conditions compatible
with protein folding.
Current Protocols in Protein Science Supplement 15
18.4.5
Preparation andHandling ofPeptides

Fold the purified protein
12. Dissolve the lyophilized target protein to a final concentration of ≤100 µM in the
appropriate freshly degassed buffer (e.g., for long-term storage, activity studies, or
structural studies), containing 6 M guanidine⋅HCl and 5 mM DTT.
Although the present protocol involves the stepwise dialysis from 6 M guanidium⋅HCl, it
should be stressed that this approach may not work for all proteins and that a certain
amount of optimization will be required for every system. It should also be noted that for
disulfide-containing proteins, an additional oxidation step will have to be performed.
In general, the lower the final concentration of protein the better, since this will tend to
minimize aggregation during the refolding step.
13. Dialyze the solution at 4°C (UNIT 4.4 & APPENDIX 3B) against the same buffer as in step
12, this time containing 1 mM DTT and decreasing amounts of guanidine⋅HCl (i.e.,
first against 4 M guanidine⋅HCl, then against 2 M guanidine⋅HCl, then against final
storage buffer). In each step perform dialysis for at least 12 hr and change the buffer
2 to 3 times during this period.
The MWCO of the dialysis membrane should be chosen to be substantially smaller than
the purified protein.
14. Remove sample from dialysis bag, centrifuge to remove any precipitate, and quantify
the supernatant by UV spectrophotometry at 280 nm (UNIT 3.1). If necessary concen-
trate the protein solution using a Centricon filter (UNIT 4.4; Amicon), then divide into
aliquots and freeze at −70°C.
BASIC
PROTOCOL 2
SEQUENTIAL CHEMICAL LIGATION OF THREE POLYPEPTIDES
If the region of interest in the protein is >50 residues from the N- or C-termini, then its
chemical modification will be extremely difficult using a two-fragment ligation strategy,
since this would require the chemical synthesis of a peptide >50 residues in length. This
problem can be overcome by assembling the target protein from three polypeptide
fragments using a sequential native ligation strategy. The basic method for sequential
native ligation uses a N-terminal Cys polypeptide (C-terminal fragment), an α-thioester
polypeptide (N-terminal fragment), and an Nα(methylsulfony)ethyloxycarbonyl-Cys,
α-thioester polypeptide (central fragment). The procedure starts by ligating the middle
fragment and the C-terminal fragment at pH 7.5 in the presence of thiol cofactors. The
Nα(methylsulfony)ethyloxycarbonyl (Msc) group is then removed and the resulting
polypeptide (central plus C-terminal fragment) is ligated with the N-terminal fragment
to give the target protein, which is purified and refolded. The general approach is depicted
in Figure 18.4.2.
Materials
Three polypeptide fragments to be ligated:
Polypeptide with N-terminal Cys residue (see Support Protocols 1 and 2)
Polypeptide with Nα(Msc)-Cys, α-thioester functionality (see Support Protocol 5)
Polypeptide with α-thioester functionality (see Support Protocols 3 and 4)
1 M HCl
Additional reagents and equipment for native chemical ligation of twopolypeptides (see Basic Protocol 1)
1. Prepare the ligation buffer (see Basic Protocol 1, steps 1 and 2). Ligate the central
[i.e., Nα(Msc)-Cys, α-thioester] and C-terminal (N-terminal Cys) polypeptide frag-
ments [see Basic Protocol 1, steps 3 to 8, but use the Nα(Msc)-Cys, α-thioester
polypeptide in the reaction mix in place of the α-thioester polypeptide].
Supplement 15 Current Protocols in Protein Science
18.4.6
Native ChemicalLigation of
Polypeptides

2. When the ligation reaction is complete, remove the Nα-Msc protecting group by
raising the pH of the crude ligation mixture to 13 with 1 M NaOH solution (see Basic
Protocol 1, step 4). After 1 min, lower the pH to 5.0 to 7.0 with 1 M HCl (use ∼1.1
to 1.2 times the volume of 1 M NaOH required to raise the pH to 13).
Usually, for a 1-ml ligation mixture, the pH can be raised by adding 75 µl of 1 M NaOH,
then dropped by adding 95 µl of 1 M HCl. It is recommended that the pH be dropped by
adding the acid all at once, instead of by titration, for reasons of speed.
The presence of thiols improves the yield in the deprotection step, since they trap the still
reactive methyl ethylenyl sulfone side product.
3. Analyze the crude reaction mixture (see Basic Protocol 1, steps 6 and 7) and examine
to see if the Msc deprotection is complete. Purify the polypeptide fragment (see Basic
Protocol 1, steps 9 to 11), which will consist of the central fragment ligated to the
C-terminal fragment.
4. Starting with the lyophilized (central plus C-terminal) polypeptide fragment prepared
in steps 1 to 3 above and the N-terminal polypeptide fragment (containing an
α-thioester functionality), perform the ligation reaction to generate the target protein
(see Basic Protocol 1, steps 3 to 10).
5. Purify and refold the target protein (see Basic Protocol 1, steps 11 to 15).
peptide 2
HS
Msc-HN-Cys CO-SRMsc-HN-Cys
HS
H2N-Cys peptide 1+
peptide 3 CO-SR
CO2–
peptide 2Msc-HN-Cys CO-NH- Cys peptide 1 CO2–
HS
aqueous buffer, pH 7.5
0.5%thiophenol
benzyl mercaptan
HS
ligation 1
HS
peptide 2 CO-NH- CysH2N-HN-Cys peptide 1 CO2
–
HS
removal of Msc group
aqueous buffer, pH 7.5
0.5%thiophenol
benzyl mercaptan
peptide 3 CO-NH-CysH3N-Cys +
HS
peptide 2 CO-NH-Cys
HS
peptide 1 CO2–
H3N+
ligation 2
pH 13
peptide 3
peptide 2
Figure 18.4.2 The principle of sequential native chemical ligation. The key to this approach is the
reversible protection of the α-amino group of the central peptide fragment, thereby preventing
self-reaction with the α-thioester moiety present in the same molecule. This can be accomplished
with the base-labile Msc [Nα(methylsulfony)ethyloxycarbonyl] group, which can be easily removed
by brief treatment with base after the first ligation step. The newly deprotected fragment is now
ready for the next ligation step.
Current Protocols in Protein Science Supplement 15
18.4.7
Preparation andHandling ofPeptides

SUPPORT
PROTOCOL 1
CHEMICAL SYNTHESIS OF N-TERMINAL CYS-POLYPEPTIDES
The chemical synthesis of N-terminal Cys-polypeptides can be easily carried out using
standard SPPS with Boc or Fmoc Nα-protected amino acids and the appropriate resins
(see Table 18.4.1 and UNIT 18.1). In both cases, the synthesis can be performed on automated
solid-phase peptide synthesizers, which are available in the core facilities of many
institutions.
NOTE: Extreme care must be taken to avoid exposing N-terminal Cys-polypeptides to
even trace amounts of carbonyl-containing compounds (e.g., acetone or formaldehyde).
These chemicals react rapidly with the α-amino and thiol groups of the N-terminal Cys
to give a very stable thiazolidine derivative which prevents the peptide from participating
in subsequent native chemical ligation reactions. For the same reason, the groups
benzyloxymethyl (Bom) and t-butyloxymethyl (Bum), used to protect the side-chain of
His in Boc and Fmoc SPPS, respectively, should be avoided during the synthesis of these
polypeptides since they release formaldehyde during the cleavage/deprotection step.
SUPPORT
PROTOCOL 2
BIOSYNTHESIS OF N-TERMINAL CYS POLYPEPTIDES
Biosynthesis represents a complementary approach to chemical synthesis (see Support
Protocol 1) for obtaining N-terminal-Cys polypeptides, and is especially useful when the
target polypeptide fragment is somewhat greater than 50 residues in length (i.e., too large
to be chemically synthesized). Assuming that the cDNA for the gene is available,
N-terminal Cys polypeptides can be obtained using a mutagenesis/proteolysis strategy
(Erlandson et al., 1996). This involves constructing an expression vector in which a DNA
sequence encoding the peptide Ile-Glu-Gly-Arg-Cys is inserted between a upstream
affinity-purification handle (e.g., MBP, GST, or His tag) and the appropriate gene
fragment of interest (for cloning and mutagenesis see UNIT 6.6). Following expression and
affinity purification, the fusion protein is treated with Factor Xa to give the desired
recombinant Cys-polypeptide, which can then be further purified if necessary. These
recombinant Cys-polypeptides can be used in native chemical ligation reactions as per
their synthetic counterparts.
SUPPORT
PROTOCOL 3
CHEMICAL SYNTHESIS OF α-THIOESTER POLYPEPTIDES
This protocol describes the chemical synthesis of a 3-mercaptopropionamide α-thioester
polypeptide on a 3-mercaptopropionamide-MBHA resin. This resin can be easily pre-
pared from commercially available MBHA resin using a three-step solid-phase procedure
(Fig. 18.4.3). The solid-phase synthesis of the polypeptide is achieved using Boc amino
acid derivatives employing in situ neutralization/HBTU [2-(1H-benzotriazolyl)-1,1,3,3-
tetramethyluronium hexafluorophosphate] activation protocols for Boc-SPPS (Schnölzer
et al., 1992). The corresponding α-thioester polypeptide, suitable for native chemical
ligation, is obtained after cleavage with hydrogen fluoride and purification.
Materials
Methylbenzhydrylamine (MBHA) resin (Peptides International)
Dimethylformamide (DMF, spectrophotometric grade; Fisher)
5% (v/v) diisopropylethylamine (DIEA, peptide synthesis grade; Perkin-ElmerApplied Biosystems) in DMF (store up to 1 month at room temperature)
97% 3-bromopropionic acid
Dichloromethane (DCM, spectrophotometric grade; Fisher)
99% diisopropyl carbodiimide (DIPC, 99%; Aldrich)
Diisopropylethylamine (DIEA, peptide synthesis grade; Perkin-Elmer AppliedBiosystems)
continued
Supplement 15 Current Protocols in Protein Science
18.4.8
Native ChemicalLigation of
Polypeptides

Ac2O/DIEA/DMF solution (see recipe)
AcSH/DIEA/DMF solution (see recipe)
BME/DIEA/DMF solution (see recipe)
Boc–amino acyl–N-hydroxysuccinimide ester (Boc-AA-OSu; where AA is thefirst amino acid to be incorporated into the polypeptide; Bachem)
Trifluoroacetic acid (TFA, BioGrade)
Ninhydrin test reagents: monitor 1, monitor 2, and monitor 3 (Perkin-ElmerApplied Biosystems)
HF/p-cresol solution (see recipe)
Diethyl ether, cold
10% to 50% acetonitrile in H2O containing 0.1% TFA
15-ml manual peptide synthesis vessel (Peptides International)
Black rubber tubing (1/4 in. i.d. × 5/8 in. o.d. × 3/16 in. wall thickness; Fisher),resistant to acids and organic solvents
2-liter side-arm flasks with rubber stoppers and glass tubing to fit
Pasteur pipet containing glass wool for filtration
2-ml polypropylene column (Microcolumn X from Isolab) with Teflon stopcock
13 × 100–mm glass test tube
110°C heating block
60% ethanol
HF cleavage apparatus (Peptides International)
Additional material and equipment for solid-phase peptide synthesis (Schnölzer etal., 1992; UNIT 18.1), preparative C18 reversed-phase HPLC (see Basic Protocol1, step 11), and ESI-MS (Chapter 16)
Prepare the MBHA resin
1. Build a manual solid-phase peptide synthesis system (Fig. 18.4.4) consisting of a
15-ml manual peptide synthesis vessel attached by 12 in. of black rubber tubing
through a rubber stopper to a 2-liter side-arm flask, which is in turn connected to a
vacuum source.
MBHA NH2 NH BrCO2H, DIPC
DCM
OCH3-COSH, DIEA
DMFNH S
O
CH3
Br
NH SH
O
NH S-AA-Boc
OBoc-AA-OSu
NSOH,DIEA,DMF
DMF
O
MBHA MBHA
MBHA MBHA
Figure 18.4.3 Scheme showing the preparation of the Boc-aminoacyl-3-mercaptopropionamide-MBHA resin using solid-
phase transformations. Abbreviations: AA, aminoacyl; Boc, t-butyloxycarbonyl; CH3-COSH, thioacetic acid; DCM, dichlo-
romethane; DIEA, diisopropylethylamine; DIPC, diisopropyl carbodiimide; DMF, dimethylformamide; MBHA,
methylbenzhydrylamine; OSu, hydroxysuccinimide ester.
Current Protocols in Protein Science Supplement 15
18.4.9
Preparation andHandling ofPeptides

2. Place 1 g of MBHA resin (1.0 mmol/g) into the peptide synthesis vessel, add enough
DMF to cover the dry resin (∼5 ml), and wait 30 min.
This preswelling step is crucial for the success of any reaction carried out on a solid
support, since it renders the functional groups on the polymer available for reaction.
3. Drain the resin. Cover the resin with 5% DIEA/DMF, leave for 1 min, then drain the
resin. Repeat this process two more times, then perform three 20-sec flow washes
with DMF—i.e., wash the resin with a continuous flow of DMF for 20 sec while
keeping a constant volume of solvent above the resin bed (usually 3 to 4 mm), then
drain the resin.
A continuous flow wash is an extremely efficient way of exchanging solvent in a swollen
polymer—much more so than the commonly used bulk wash (i.e., by adding solvent, mixing,
and draining).
Prepare the symmetrical anhydride of 3-bromopropionic acid
4. Dissolve 1.222 g 3-bromopropionic acid (8 mmol) in a minimal volume of DCM (∼1
to 2 ml), then add 630 µl of 99% DIPC (4 mmol). Shake the solution vigorously and
wait 10 min.
A white precipitate corresponding to the diisopropyl urea will appear during the activation
reaction. If no such precipitate forms, the reaction has not taken place and should be
repeated.
It is best to prepare the symmetrical anhydride fresh for each coupling reaction.
5. Filter the solution through a Pasteur pipet containing glass wool.
It is very important to remove the diisopropyl urea by filtration before adding the symmet-
rical anhydride solution to the resin. The authors have found that if this step is not carried
out the acylation reaction does not go to completion.
Couple the symmetrical anhydride of 3-bromopropionic acid to the MBHA resin
6. Add the filtered solution from step 5 to the MBHA resin in the peptide synthesis
vessel. Without applying any suction, add 800 µl DIEA (4.5 mmol) and the minimal
amount of DMF (1 to 2 ml) required to give a good slurry.
7. Leave the coupling reaction for 30 min with occasional stirring using a glass stirring
rod.
rubber tubing resistantto acids and organic solvents
waste container
solid-phase peptidereaction vessel
vacuum
resin
stopcock
Figure 18.4.4 Suggested apparatus for the manual solid-phase synthesis of thioester peptides.
Supplement 15 Current Protocols in Protein Science
18.4.10
Native ChemicalLigation of
Polypeptides

8. Drain the resin and wash thoroughly with three 20-sec DMF flow washes as in step
3.
9. Repeat the coupling process (steps 4 to 8) two more times.
10. Add 5 ml Ac2O/DIEA/DMF solution to the resin and wait 10 min. Drain the resin
and wash three times with DMF as in step 3.
This acetylation step ensures that the small amount of unreacted amine groups remaining
on the MBHA resin cannot participate in any subsequent acylation steps.
Prepare the 3-mercaptopropionamide-MBHA resin
11. Add 5 ml AcSH/DIEA/DMF solution to the resin and wait 20 min. Drain the resin
and wash three times with DMF as in step 3. Repeat this entire process (treatment
with AcSH/DIEA/DMF solution, along with the washings) two more times.
12. Add 5 ml BME/DIEA/DMF solution to the resin and wait 20 min. Drain the resin
and wash three times with DMF as in step 3. Repeat this entire process (treatment
with BME/DIEA/DMF solution, along with the washings) two more times.
The 3-mercaptopropionamide-MBHA resin which is the product of this step is susceptible
to oxidation upon long-term storage. The authors therefore recommend that it be immedi-
ately acylated with the appropriate Boc amino acid derivative.
Couple the first Boc amino acid to the 3-mercaptopropionamide-MBHA resin
13. Dissolve 3 molar equivalents of the appropriate Boc-AA-OSu (where AA is the first
amino acid to be incorporated in the synthesis) in ∼6 ml DMF, then add this solution
to the 3-mercaptopropionamide-MBHA resin. Add 714 µl DIEA (4 molar equiva-
lents, 4 mmol) and leave the coupling reaction for 3 to 4 hr with occasional stirring.
Most of the Boc amino acid derivatives are commercially available as N-hydroxysuccin-
imide esters. If the required Boc amino acid derivative is not commercially available it can
be readily prepared manually (Bodanszky and Bodanszky, 1994).
14. Drain the resin and wash three times with DMF as in step 3. Add 5 ml
Ac2O/DIEA/DMF solution to the resin and wait 10 min, then wash the resin three
times with dichloromethane (DCM) using 20-sec flow washes as described in step
3. Dry the resin under vacuum and store at −20°C.
Determine the final substitution of the Boc-amino acyl-3-mercaptopropionamide-
MBHA resin
15. Place ∼5 mg of the dry Boc–amino acyl–3-mercaptopropionamide–MBHA resin
from step 14 in a 2-ml polypropylene Microcolumn X column with a Teflon stopcock,
connected to a vacuum source.
16. Add 1 ml TFA to the resin and wait 2 min. Drain the column, then flow wash first
with DMF and then with DCM. Drain again and dry under vacuum.
17. Weigh an exact amount of the dried resin (∼3 to 5 mg) and place in a 13 × 100–mm
glass test tube.
18. Add 2 drops of ninhydrin test monitor 1 reagent, 4 drops of ninhydrin test monitor 2
reagent, and 2 drops of ninhydrin test monitor 3 reagent, then incubate 5 min at 110°C
in a heating block.
19. Dilute the blue solution with 60% ethanol to 25 ml and mix well. Measure the
absorbance at 570 nm using 1-cm path-length cuvette. Calculate the substitution of
the thioester resin (in mmol/g) as 1.67 × (A570/amount of resin in mg).
Current Protocols in Protein Science Supplement 15
18.4.11
Preparation andHandling ofPeptides

For a resin with an initial loading of ∼1 mmol/g the final substitution for the corresponding
acylated thioester resin is usually ∼0.2 mmol/g.
Perform solid-phase synthesis of the α-thioester polypeptide
20. Synthesize the polypeptide sequence using the in situ neutralization/HBTU activation
protocols for Boc-SPPS (Schnölzer et al., 1992; UNIT 18.1).
Solid-phase deprotection of the 2,6-dinitrophenyl (Dnp) and formyl (For) protecting groups
(commonly used for the protection of the side chains of His and Trp residues, respectively)
results in premature cleavage of α-thioester polypeptides from the resin. Therefore, these
residues should either be deprotected after the ligation reaction is complete or incorporated
as Boc-Trp-OH (no side-chain protection) or Boc-Trp(Hoc)-OH and Boc-His(Bom)-OH
during SPPS .
21. Once the synthesis is complete, flow wash with DMF and DCM as in step 14. Dry
the resin under vacuum and store at −20°C.
Cleave and purify the α-thioester polypeptide
22. Using 200 mg of peptide α-thioester resin from step 21, cleave the peptide-resin by
treating with 5 ml of HF:p-cresol solution for 1 hr at 4°C in an HF cleavage apparatus.
CAUTION: Anhydrous HF is a highly toxic and corrosive gas and should only be
manipulated in a fume hood using the commercially available specialized apparatus.
23. Remove the HF under vacuum and resuspend both peptide and resin in ∼40 ml cold
diethyl ether with gentle stirring for 10 min. Filter the suspension on a glass Buchner
funnel under vacuum, without letting air pass through the filter, as this could oxidize
the cleaved peptide.
24. Wash the material in the filter (containing the cleaved peptide and the resin) three
times, each time with 10 ml cold diethyl ether. Wash an additional three times, each
time with 10 ml dichloromethane, then wash with 10 ml cold diethyl ether again.
Discard the wash liquid.
These washes will remove most of the scavenger byproducts.
25. Add 10 ml freshly degassed 50% acetonitrile in water containing 0.1% TFA to the
filter, wait 10 min (to dissolve the cleaved peptide), then filter. Repeat this process
three more times, then recover and lyophilize the filtrates.
26. Analyze a 50-µl aliquot by reversed-phase HPLC and ESI-MS (Chapter 16; also see
Basic Protocol 1, step 7).
27. Purify the lyophilized propionamide α-thioester peptide by preparative C18 RP-
HPLC (see Basic Protocol 1, step 11).
SUPPORT
PROTOCOL 4
BACTERIAL EXPRESSION OF α-THIOESTER POLYPEPTIDES
This protocol describes the preparation of α-thioester polypeptides using bacterial ex-
pression in E. coli. The desired polypeptide-encoding gene fragment is first cloned into
a commercially available pTYB vector (Fig. 18.4.5). Following E. coli transformation,
soluble expression of the polypeptide-intein-CBD (chitin binding domain) fusion protein
is induced and the cells are harvested and lysed. The lysate is then loaded onto a chitin
column and the fusion protein affinity purified. Finally, the target α-thioester polypeptide
is cleaved from the column and eluted using a buffer containing ethanethiol.
NOTE: Initial studies using an MBP-intein-CBD system indicate that the majority of
amino acid residues, when located immediately before the intein N-terminal cysteine,
allow both purification of fusion proteins and efficient cleavage with thiols. However, Pro,
Supplement 15 Current Protocols in Protein Science
18.4.12
Native ChemicalLigation of
Polypeptides
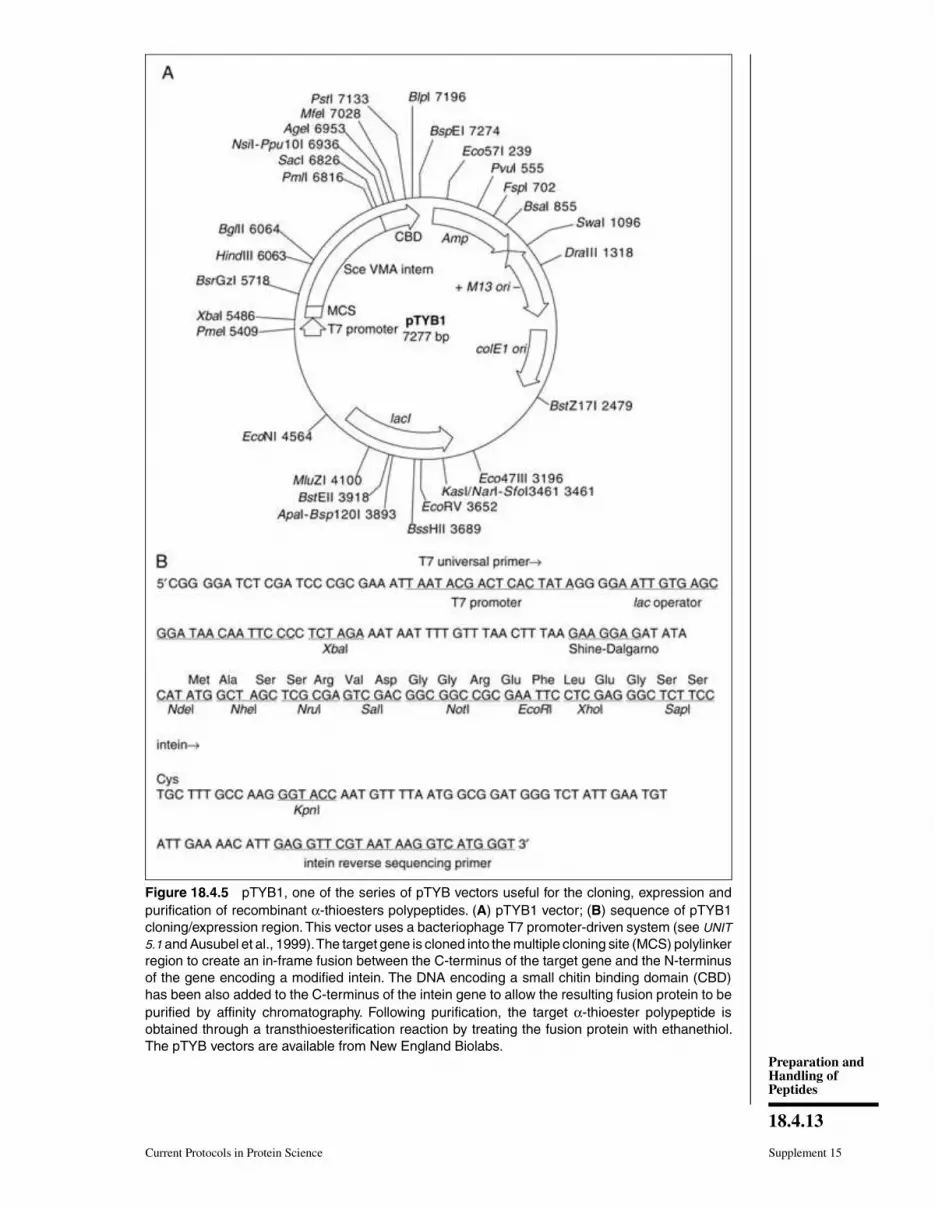
Figure 18.4.5 pTYB1, one of the series of pTYB vectors useful for the cloning, expression and
purification of recombinant α-thioesters polypeptides. (A) pTYB1 vector; (B) sequence of pTYB1
cloning/expression region. This vector uses a bacteriophage T7 promoter-driven system (see UNIT
5.1 and Ausubel et al., 1999). The target gene is cloned into the multiple cloning site (MCS) polylinker
region to create an in-frame fusion between the C-terminus of the target gene and the N-terminus
of the gene encoding a modified intein. The DNA encoding a small chitin binding domain (CBD)
has been also added to the C-terminus of the intein gene to allow the resulting fusion protein to be
purified by affinity chromatography. Following purification, the target α-thioester polypeptide is
obtained through a transthioesterification reaction by treating the fusion protein with ethanethiol.
The pTYB vectors are available from New England Biolabs.
Current Protocols in Protein Science Supplement 15
18.4.13
Preparation andHandling ofPeptides

Cys, and Asn residues were found to inhibit the in vitro cleavage with thiols. Note that
partial cleavage during bacterial expression can sometimes be observed in certain systems,
resulting in a decrease in the yield of fusion precursors. For example, in the MBP system,
Asp, Arg, His, and Glu residues gave rise to in vivo cleavage (>50%).
Materials
Gene construct encoding target polypeptide
pTYB1 vector (New England Biolabs)
E. coli BL21 (or any other suitable E. coli strain)
LB medium containing 100 µg/ml ampicillin (APPENDIX 4A)
1 M isopropyl-1-thio-β-D-galactopyranoside (IPTG), filter sterilized
Chitin beads slurry: 50% (w/v) suspension of chitin beads (New England Biolabs)in 40% ethanol
Column buffer (see recipe)
Lysis buffer (see recipe)
Cleavage buffer (see recipe)
Refrigerated centrifuge with Beckman JA-10 rotor and centrifuge buckets to hold1 liter of culture
1.5 × 10–cm glass or polypropylene column
Refrigerated centrifuge with Beckman JA-17 rotor and appropriate centrifuge tubes
Shaker
Additional reagents and equipment for introducing plasmid vectors into bacterialcells (APPENDIX 4D), growth of bacteria in liquid medium (APPENDIX 4A), lysis ofbacterial cells using a French press (UNIT 6.3), ESI-MS (UNITS 16.1 & 16.2)
Express the fusion protein
1. Insert the gene construct encoding the target polypeptide into the pTYB1 vector (see
manufacturer’s instruction for vector and Ausubel et al., 1999) so as to express the
target protein as an intein-CBD fusion. Introduce the vector into the E. coli BL21
cells (see APPENDIX 4D and Ausubel et al., 1999). Inoculate 15 ml LB/ampicillin with
E. coli BL21 containing a pTYB vector expressing the desired protein fusion. Grow
overnight with shaking at 37°C (APPENDIX 4A).
2. Inoculate 1000 ml LB/ampicillin with 10 ml of the overnight culture and grow with
shaking at 37°C to an OD600 of 0.5 to 0.6 (APPENDIX 4A).
3. Add 1 ml of 1 M IPTG (final concentration of 1 mM) to the culture and continue
incubation for 1 to 6 hr at 37°C.
The optimal induction conditions (i.e., incubation time, temperature, and final IPTG
concentration) for soluble expression will depend on the in vivo properties of the overex-
pressed protein, and should be optimized for every system.
4. Centrifuge the cell culture 10 min at 8700 × g (7000 rpm in Beckman JA-10 rotor),
4°C. Discard supernatant. Proceed to step 5 immediately or freeze pellet at −70°C
indefinitely.
If extract preparation is to be be carried out immediately, the chitin column may be prepared
during the centrifugation step.
Prepare the affinity column
5. Add ∼10 ml of chitin bead slurry to a 1.5 × 10–cm column and allow liquid to drain
just to the top of the packed resin bed (∼5 ml).
6. Wash the column with 100 ml column buffer at a flow rate of 1 ml/min.
Supplement 15 Current Protocols in Protein Science
18.4.14
Native ChemicalLigation of
Polypeptides

Prepare the cell extract
7. If cell pellet was frozen, (step 4), thaw on ice. Resuspend pellet in 30 ml ice-cold
lysis buffer. Lyse cells using a French press (UNIT 6.3).
IMPORTANT NOTE: From this step on, all procedures should be carried out at 4°C.
8. Centrifuge lysate for 30 min (or until supernatant is clear) at 25,000 × g (14,000 rpm
in Beckman JA-17 rotor), 4°C. Decant supernatant into a clean container on ice and
discard pellet.
The supernatant can be frozen and stored at −70°C indefinitely before continuing with the
procedure.
In order to monitor the expression, extraction, and purification steps, it is convenient to
take small aliquots at every step and analyze them by SDS-PAGE (UNIT 10.1).
Purify the fusion protein
9. If extract is frozen, thaw on ice. Load onto the chitin column (at a rate no faster than
0.5 ml/min). Collect flowthrough and reapply to column, then repeat this process one
more time.
Chitin beads have a capacity of ∼2 mg of CBD-tagged protein per ml packed beads.
Therefore, the amount of extract that can be loaded on the column will depend on the
amount of soluble fusion protein in the extract.
10. Wash the column with 100 ml column buffer at a flow rate of 1 ml/min. Discard
flowthrough. Be sure that all traces of crude extract have been washed off the sides
of the column.
Obtain the α-thioester polypeptide
11. Add 5 ml cleavage buffer to the column and gently shake the reaction slurry overnight
at room temperature.
12. Drain the beads, then wash with 15 ml cleavage buffer (three times, each time with
5 ml). Pool all of the fractions.
IMPORTANT NOTE: Thioesters are susceptible to hydrolysis under alkaline conditions;
consequently purification and storage of α-thioester polypeptides should be performed at
pH 6.0 or below.
Analyze and purify the α-thioester polypeptide
13. Analyze a 50-µl aliquot by reversed-phase analytical HPLC and ESI-MS (see UNITS
16.1 & 16.2; also see Basic Protocol 1, step 7).
14. Purify the target α-thioester polypeptide by reversed-phase preparative HPLC see
Basic Protocol 1, step 11).
SUPPORT
PROTOCOL 5
CHEMICAL SYNTHESIS OF Nα(Msc)-CYS, α-THIOESTER POLYPEPTIDES
This protocol describes how to introduce the Nα(methylsulfony)ethyloxycarbonyl (Msc)
protecting group onto the α-amino group of an N-terminal Cys polypeptide synthesized
on a 3-mercaptopropionamide-MBHA resin (see Support Protocol 3). The resulting
Nα(Msc)-Cys, α-thioester polypeptides are used in sequential native chemical ligation
reactions (see Basic Protocol 2).
NOTE: Boc-SPPS of α-thioester polypeptides requires the use of the Bom side-chain
protecting group for His. The Bom group releases fomaldehyde during its deprotection
with HF; however the formation of the thiazolidine adduct with the N-terminal Cys cannot
take place due to the presence of the NαMsc group.
Current Protocols in Protein Science Supplement 15
18.4.15
Preparation andHandling ofPeptides

Additional Materials (also see Support Protocol 3)
Fully protected Boc-polypeptide-3-mercaptopropionamide-MBHA resin (seeSupport Protocol 3, step 21), dried
(Methylsulfonyl)-ethyl 4-nitrophenyl carbonate (Msc-ONp; Fluka)
Prepare the Boc-polypeptide-3-mercaptopropionamide-MBHA resin
1. Place ∼0.5 g of fully protected Boc-polypeptide-3-mercaptopropionamide-MBHA
resin in a peptide synthesis vessel attached to a vacuum source and swell with DMF
(see Support Protocol 3, steps 1 and 2).
2. Deprotect the Boc group by adding ∼5 ml of TFA to the resin, waiting 1 min, then
draining the resin. Repeat this process one additional time, then drain the resin and
perform three 20-sec flow washes with DMF (see Support Protocol 3, step 3).
3. Neutralize the polypeptide-3-mercaptopropionamide-MBHA resin by treating with
5% DIAE/DMF and flow washing with DMF (see Support Protocol 3, step 3).
Introduce the Msc group on the α-amino group of the polypeptide-3-mercapto-
propionamide-MBHA resin
4. Dissolve 560 mg (2 mmol) of Msc-ONp in 4 to 5 ml DMF and add this mixture to
the peptide thioester resin, then add 340 µl (2 mmol) of DIEA. Leave the coupling
reaction for 3 hr, with occasional stirring using a glass stirring rod, then drain the
resin and perform three 20-sec flow washes with DMF.
5. Place ∼1 mg of resin from step 4, in a column (see Support Protocol 3, step 15). Flow
wash first with DMF and then with DCM, drain again, and dry under vacuum. Place
the aliquot in a glass test tube and perform the ninhydrin test (see Support Protocol
3, step 18). If the color of the solution is strongly blue, repeat the coupling reaction
(step 4) and test again; if not, continue with step 6, below.
Sometimes the ninhydrin test can give false positives (i.e., strong blue coloration) even
although the acylation reaction is complete. This is especially true when the α-amino group
is protected with a base-labile group (i.e., Fmoc or Msc). Therefore, if the ninhydrin test
is still positive after the third coupling, proceed to step 6.
6. Perform three 20-sec flow washes with DMF and then with DCM. Dry the resin under
vacuum and store at −20°C until further use.
7. Cleave and purify 200 mg of peptide-thioester resin (see Support Protocol 3, steps
22 to 26).
REAGENTS AND SOLUTIONS
Use Milli-Q-purified water or equivalent for the preparation of all buffers. For common stock solutions,see APPENDIX 2E; for suppliers, see SUPPLIERS APPENDIX.
Ac2O/DIEA/DMF solution
15 parts (v/v) 99% acetic anhydride (Ac2O)
15 parts (v/v) diisopropylethylamine (DIEA, peptide synthesis grade; Perkin-El-
mer Applied Biosystems)
70 parts (v/v) dimethylformamide (DMF, spectrophotometric grade; Fisher)
Prepare fresh
AcSH/DIEA/DMF solution
1 part (v/v) 96% thiolacetic acid (AcSH; Fluka)
1 part (v/v) diisopropylethylamine (DIEA, peptide synthesis grade; Perkin-Elmer
Applied Biosystems)
8 parts (v/v) dimethylformamide (DMF, spectrophotometric grade; Fisher)
Prepare fresh
Supplement 15 Current Protocols in Protein Science
18.4.16
Native ChemicalLigation of
Polypeptides

BME/DIEA/DMF solution
1 part (v/v) 98% 2-mercaptoethanol (BME)
1 part (v/v) diisopropylethylamine (DIEA, peptide synthesis grade; Perkin-Elmer
Applied Biosystems)
8 parts (v/v) dimethylformamide (DMF, spectrophotometric grade; Fisher)
Prepare fresh
Cleavage buffer
0.1 mM EDTA
200 mM sodium phosphate, pH 6.0
250 mM NaCl
3% (v/v) ethanethiol
0.1% (v/v) Triton X-100
Adjust pH to 6.0 with 1 M NaOH or HCl
Store up to 6 months at 4°C
Column buffer
0.1 mM EDTA
20 mM sodium phosphate, pH 7.2
250 mM NaCl
0.1% (v/v) Triton X-100
Adjust pH to 7.2 with 1 M NaOH or HCl
Store up to 6 months at 4°C
Guanidine⋅HCl buffer, 6 M
6 M guanidine⋅HCl (APPENDIX 3A)
0.1 M sodium phosphate
1 mM EDTA
Adjust pH to 7.5 with 1 M NaOH
Store up to 6 months at room temperature
HF/p-cresol solution
96 parts (v/v) hydrogen fluoride (HF; anhydrous)
4 parts (v/v) p-cresol
Prepare fresh
CAUTION: Anhydrous HF is a highly toxic and corrosive gas and should be manipulated
only in a fume hood, using the commercially available specialized apparatus (HF cleavage
apparatus from Peptides International).
Lysis buffer
0.1 mM EDTA
1 mM PMSF
25 mM HEPES, pH 8.0
250 mM NaCl
5% (v/v) glycerol
Adjust pH to 8.0 with 1 M NaOH
Store up to 6 months at 4°C
COMMENTARY
Background InformationThe introduction of solid-phase peptide-
synthesis (SPPS) by Bruce Merrifield revolu-
tionized the chemical synthesis of peptides
(Merrifield, 1963; also see UNIT 18.1). Despite
the enormous impact of SPPS in the generation
and study of small bioactive peptides, it is now
clear that the combination of incomplete acy-
lation/deprotection reactions and other well
documented side reactions places an intrinsic
limit on the size of peptides accessible by effi-
cient stepwise SPPS. Thus, polypeptides of up
Current Protocols in Protein Science Supplement 15
18.4.17
Preparation andHandling ofPeptides

to ∼50 residues in length can be prepared by
stepwise SPPS with reasonable confidence, but
beyond this, the chances of success fall off
precipitously. In order to overcome this size
limitation, recent years have seen renewed in-
terest in the use of convergent synthetic strate-
gies—i.e., the synthesis of large polypeptides
from smaller peptide building blocks which are
themselves accessible via the SPPS approach.
It should be noted that fragment condensation
has a long and illustrious history in the peptide
chemistry field; however, there have always
been serious problems associated with the ma-
nipulation of the fully protected peptides in
these classical convergent strategies. Although
the use of minimal protection strategies repre-
sented a step in the right direction (see Lloyd-
Williams et al., 1993 for an extensive review),
it was not until the early 1990s that a truly
practical way of performing fragment conden-
sations was developed—i.e., chemical ligation
(for reviews see Muir, 1995; Wallace, 1995).
The original chemical ligation strategies were
all based on the premise that an unnatural moi-
ety could be used to covalently join two fully
unprotected (and hence water-soluble)
polypeptides, each bearing unique and mutu-
ally reactive groups. A number of different
chemistries have been developed for this pur-
pose, all of which give rise to an unnatural
covalent structure at the ligation site (Muir,
1995; Wallace, 1995).
In 1994, a second-generation ligation chem-
istry was introduced, known as “native chemi-
cal ligation,” which allows the preparation of
proteins with native backbone structures from
fully unprotected peptide building blocks
(Dawson et al., 1994). This important extension
of the chemical ligation concept makes use of
the mild acylating power of the α-thioester
functionality. The principle of native chemical
ligation is depicted in Figure 18.4.1. The first
step involves the chemoselective reaction
which occurs between the free thiol group of
an unprotected N-terminal Cys-polypeptide
and a second, unprotected polypeptide contain-
ing an α-thioester group. This transthioesteri-
fication reaction gives rise to a thioester-linked
intermediate which spontaneously rearranges
to form a native peptide bond at the ligation
site. The target full-length polypeptide is thus
obtained without any further manipulation. Na-
tive chemical ligation reactions are performed
in aqueous buffers at pH 7 to 7.5 in the presence
of thiol cofactors. At this pH, the regioselectiv-
ity of the reaction is such that the reaction can
to be performed in the presence of all the
functionalities commonly found in proteins.
Even the presence of additional Cys residues in
one or both fragments does not affect the re-
gioselectivity of the ligation (Hackeng et al.,
1997). Small proteins or protein domains ∼100
to 120 amino acids in length can be reliably
generated from two peptide building blocks in
a single chemical ligation step (for a few exam-
ples see Dawson et al., 1994, 1997; Lu et al.,
1996; Hackeng et al., 1997; Camarero et al.,
1998).
The total chemical synthesis of larger pro-
tein targets (>100 residues) via the ligation of
just two fragments becomes more and more
problematic as the size increases. This is due to
the difficulties associated with the direct step-
wise SPPS of polypeptide segments bigger than
50 residues. This difficulty can be overcome by
performing multiple ligation reactions using
three or more synthetic peptides (e.g., Canne et
al., 1995). Native chemical ligation has been
extended to allow multiple ligation steps to be
performed sequentially in a controlled and di-
rected way (Muir et al., 1997; Camarero et al.,
1998). The general approach is depicted in
Figure 18.4.2. Key to this strategy is the tem-
porary protection of the α-amino group of the
central peptide with the base-labile 2-(methyl-
sulfonyl)ethyloxycarbonyl (Msc) group
(Tesser and Balvert-Geers, 1975). The presence
of this protecting group prevents the N-terminal
Cys from reacting in an intramolecular or in-
termolecular fashion with the α-thioester func-
tionality present in the same polypeptide frag-
ment. Once the first ligation reaction is fin-
ished, the Nα-Msc group can be efficiently
removed, allowing the next ligation step to be
performed (Fig. 18.4.2).
The native chemical ligation approach can
also be used in the semisynthesis of proteins
(Muir et al., 1998; Severinov and Muir, 1998;
Erlandson et al., 1996; Evans et al., 1998) from
recombinant and synthetic polypeptide frag-
ments. As described above, native chemical
ligation of two polypeptides requires that one
of the fragments possess an N-terminal Cys and
that the other contain an α-thioester moiety.
Polypeptides containing an N-terminal cyste-
ine residue for use in litigation can be obtained
using standard recombinant DNA expression
methods (see Chapter 5). Importantly, biosyn-
thetic methods are also now available for the
generation of α-thioester polypeptides (Muir et
al., 1998; Severinov and Muir, 1998; Evans et
al., 1998). This is made possible using the
IMPACT expression system, commercially
available from New England Biolabs (Chong
Supplement 15 Current Protocols in Protein Science
18.4.18
Native ChemicalLigation of
Polypeptides

et al., 1997). This system utilizes a protein
splicing element, an intein from the Sac-
charomyces cerevisiae VMA1 gene, in con-
junction with a chitin binding domain (CBD)
which allows purification by affinity chroma-
tography (Fig. 18.4.6). The natural intein has
been modified in the expression system
(Asn454→Ala) in order to block the normal
protein splicing reaction in midstream. This
results in the formation of a thioester linkage
between the polypeptide of interest and the
intein (Fig. 18.4.6). Cleavage of the thioester
can thus be induced by treatment of the ex-
pressed fusion protein with the appropriate
thiol to give the target α-thioester polypeptide
(Muir et al., 1998; Severinov and Muir, 1998;
Evans et al., 1998).
Critical Parameters andTroubleshooting
As mentioned in Basic Protocol 1, pH is the
most critical parameter in the native chemical
ligation of two fully unprotected peptides in
aqueous buffers. The efficiency of the ligation
reaction is strongly dependent on the pH; above
pH 8.0 the reaction loses its regioselectivity and
below pH 6.0 the reaction is usually very slow.
N-extein
A HS
NH intein
HS
NH2
NHC-extein
O
O
N→S acyl transfer
N-exteinHS
NH2
NH
O
Ointein C-exteinH2N
S
transthioesterification
S
NH2
NH
O
Ointein C-exteinH2N
HS
O
S→N acyl transfer
and succinimide formation
intein
SH
HN
O
N-extein
intein
NH
O
OinteinH2N
HS +
O
O
recombinant protein intein*CO-NH-Cys
clone protein gene in IMPACT system
express in E. coli
HS
recombinant protein intein*CO-NH-Cys
HS
recombinant protein CO-S
recombinant protein CO-SCH2CH3
intein*H2N-Cys
i) 3% ethanethiol at pH 6
overnight
ii) filtration
transthioesterification
N→S Acyl transfer
Affinity purification
on chitin beads
B
CBD
CBD
N-extein
CBD
C-extein
Figure 18.4.6 Principles of the biosynthetic preparation of α-thioester polypeptides by recombinant techniques. (A)
Scheme representing the proposed mechanism of protein splicing involving the intein from Saccharomyces cerevisiae VMA1
gene (Xu and Perler, 1996). (B) Expression, purification, and cleavage of polypeptide-intein*-CBD fusion protein (where the
asterisk refers to the mutation Asn454→Ala in the intein element and CBD refers to the chitin binding domain) with an
appropriate thiol to give the α-thioester polypeptide.
Current Protocols in Protein Science Supplement 15
18.4.19
Preparation andHandling ofPeptides

Another crucial factor is the concentration
of the two reactants. The bimolecular nature of
native chemical ligation (and ligations in gen-
eral) means that the concentration of both reac-
tants should be as high as possible for efficient
reaction. Generally the use of chemical dena-
turants (GdmCl or urea) will allow high con-
centrations of both reactants to be achieved.
Furthermore, the use of denaturing conditions
helps to alleviate potential steric problems that
may be associated with the use of folded
polypeptides.
Another important parameter for the success
of the ligation reaction is the availability of the
thiol and/or α-amino groups of the N-terminal
Cys polypeptide during the ligation reaction. If
one or both groups are chemically blocked, the
ligation will not take place. It is well known
that N-terminal Cys peptides can react rapidly
with carbonyl-containing compounds (e.g.,
acetone and formaldehyde) to give the corre-
sponding N-terminal thiazolidine adducts,
which are totally unreactive in the native liga-
tion process. It is thus crucial to avoid the use
of these substances while handling all peptides
and buffers. Note that the authors have found
acetone, a commonly used solvent for washing
glassware in many laboratories, to be particu-
larly problematic in this regard.
Anticipated ResultsNative chemical ligation has been applied to
the synthesis and semisynthesis of a large num-
ber of proteins and protein domains. These
studies indicate that very high yields (80% or
better) are typically obtained for the ligation
step. Following purification, the total yield usu-
ally drops to 50% to 60%. In sequential liga-
tions where the Nα-Msc group has to be depro-
tected in situ after the first ligation step, the
typical total yield after purification is ∼30% to
40%. It is also important to note that the pro-
tein-folding step may further decrease the final
yield of product.
Time ConsiderationsIn Basic Protocols 1 and 2, the chemical
ligation step typically requires 2 days, although
in some cases the ligation reaction can proceed
very rapidly (in a few hours) or somewhat more
slowly (in 4 days). The purification step (RP-
HPLC or other liquid chromatographies) can
be performed in half a day. Processing of the
purified samples (lyophilization or concentra-
tion) can take 1 to 2 days (depending on the
volume). Finally, folding of the protein (if nec-
essary) can be achieved in 3 days.
The thioester resin can be prepared in 1 day.
The time required for the chemical synthesis of
an α-thioester polypeptide (see Support Proto-
col 3) will depend on its size. Typically, for
peptides ∼50 residues in length, the solid-phase
chain assembly can be carried out in 3 days
manually or in 1 day using an automated syn-
thesizer. The deprotection-cleavage and purifi-
cation can be performed in 1 day, and lyophili-
zation of peptide fractions can take 1 to 2 days
(depending on the volume).
Bacterial expression of a peptide-intein-
CBD fusion protein and preparation of the
crude cell extract requires 2 days for the system
described here (see Support Protocol 4). Col-
umn preparation requires ∼2 to 3 hr. Loading
and washing the column can be done in 3 hr.
The cleavage of the α-thioester polypeptide
from the affinity column requires 10 to 15 hr
(overnight). Purification and processing of the
polypeptide fractions requires 1 to 2 days.
Literature CitedAusubel, F.A., Brent, R., Kingston, R.E., Moore,
D.D., Seidman, J.G., Smith, J.A., and Struhl, K.(eds.). 1999. Current Protocols in Molecular Bi-ology. John Wiley & Sons, New York.
Bodanszky, M. and Bodanszky, A. 1994. The Prac-tice of Peptide Synthesis, 2nd ed. Springer-Ver-lag, Berlin.
Camarero, J.A., Cotton, G.J., Adeva, A., and Muir,T.W. 1998. Chemical ligation of unprotectedpeptides directly from a solid support. J. Pept.
Res. 51:303-316.
Canne, L.E., Ferre-D’Amare, A.R., Burley, S.K.,and Kent, S.B.H. 1995. Total chemical synthesisof a unique transcription factor-related protein:cMyc-Max. J. Am. Chem. Soc. 117:2998-3007.
Chong, S., Mersha, F.B., Comb, D.G., Scott, M.E.,Landry, D., Vence, L.M., Perler, F.B., Benner, J.,Kucera, R.B., Hirvonen, C.A., Pelletier, J.J.,Paulus, H., and Xu, M.Q. 1997. Single-columnpurification of free recombinant proteins using aself-cleavable affinity tag derived from a proteinsplicing element. Gene 192:271-281.
Dawson, P.E., Muir, T.W., Clark-Lewis, I., and Kent,S.B.H. 1994. Synthesis of proteins by nativechemical ligation. Science 266:776-779.
Dawson, P.E., Churchill, M.J., Ghadiri, M.R., andKent, S.B.H. 1997. Modulation in native chemi-cal ligation through the use of thiol additives. J.
Am. Chem. Soc. 119:4325-4329.
Erlandson, D.A., Chytill, M., and Verdine, G.L.1996. The leucine zipper domain controls theorientation of AP-1 in the NFAT AP-1 DNAcomplex. Chem. Biol. 3(12):981-991.
Evans, T.C., Benner, J.J., and Xu, M.-Q. 1998. Semi-synthesis of cytotoxic proteins using a modifiedprotein splicing element. Protein Sci. 7:2256-2264.
Supplement 15 Current Protocols in Protein Science
18.4.20
Native ChemicalLigation of
Polypeptides

Hackeng, T.M., Mounier, C.M., Bon, C., Dawson,P.E., Griffin, J.H., and Kent, S.B. 1997. Totalchemical synthesis of enzymatically active hu-man type II secretory phospholipase A2. Proc.Natl. Acad. Sci. U.S.A. 94:7845-7850.
LLoyd-Williams, P., Albericio, F., and Giralt, E.1993. Convergent solid-phase peptide synthesis.Tetrahedron 49:11065-11133.
Lu, W., Qasim, M.A., and Kent, S.B.H. 1996. Com-parative total synthesis of turkey ovomucoidthird domain by both stepwise solid phase syn-thesis and native chemical ligation. J. Am. Chem.Soc. 118:8518-8523.
Merrifield, R.B. 1963. Solid phase peptide synthe-sis. I. The synthesis of a tetrapeptide. J. Am.
Chem. Soc. 85:2149-2154.
Muir, T.W. 1995. A chemical approach to the con-struction of multimeric protein assemblies.Structure 3:649-652.
Muir, T.W., Dawson, P.E., and Kent, S.B.H. 1997.Protein synthesis by chemical ligation of unpro-tected peptides in aqueous solution. MethodsEnzymol. 289:266-298.
Muir, T.W., Sondhi, D., and Cole, P.A. 1998. Ex-pressed protein ligation: A general method forprotein engineering. Proc. Natl. Acad. Sci.U.S.A. 95:6705-6710.
Schnölzer, M., Alewood, P., Jones, A., Alewood, D.,and Kent, S.B.H. 1992. In situ neutralization inBoc-chemistry solid phase peptide synthesis:Rapid, high yield assembly of difficult se-quences. Int. J. Pept. Protein Res. 40:180-193.
Severinov, K. and Muir, T.W. 1998. Expressed pro-tein ligation: A novel method for studying pro-tein-protein interactions in transcription. J. Biol.Chem. 273:16205-16209.
Tesser, G.I. and Balvert-Geers, I.C. 1975. Themethylsulfonylethyloxycarbonyl group: A newand versatile amino protective function. Int. J.Pept. Prot. Res. 7:295-305.
Wallace, C.J.A. 1995. Peptide ligation and semisyn-thesis. Curr. Opin. Biotechnol. 6:403-410.
Xu, M.-Q. and Perler, F.B. 1996. The mechanism ofprotein splicing and its modulation by mutation.EMBO J. 15:5146-5153.
Contributed by Julio A. Camarero and Tom W. MuirThe Rockefeller UniversityNew York, New York
Current Protocols in Protein Science Supplement 15
18.4.21
Preparation andHandling ofPeptides