naringenin and 17β-estradiol coadministration prevents hormone-induced human cancer cell growth
TRANSCRIPT

Research Communication
Naringenin and 17b-Estradiol Coadministration PreventsHormone-Induced Human Cancer Cell Growth
Pamela Bulzomi1, Alessandro Bolli1,2, Paola Galluzzo1, Stefano Leone1, Filippo Acconcia1
and Maria Marino1,2*1Department of Biology, University Roma Tre, Viale G. Marconi, Roma, Italy2National Institute of Biostructures and Biosystems, Viale Medaglie d’Oro, Roma, Italy
Summary
Flavonoids have been described as health-promoting, disease-preventing dietary components. In vivo and in vitro experimentsalso support a protective effect of flavonoids to reduce the inci-dence of certain hormone-responsive cancers. In particular, ourprevious results indicate that the flavanone naringenin (Nar),decoupling estrogen receptor a (ERa) action mechanisms, drivescancer cells to apoptosis. Because these studies were conducted inthe absence of the endogenous hormone 17b-estradiol (E2), thephysiological relevance of these findings is not clear. We investi-gate whether the antiproliferative Nar effect persists in the pres-ence of physiological E2 concentration (i.e. 10 nM), using bothERa-transfected (HeLa cells) and ERa-containing (HepG2 cells)cancer cell lines. Ligand saturation experiments indicate that Nardecreases the binding of E2 to ERa without impairing the estro-gen response element (ERE)-driven reporter plasmid activity. Incontrast, Nar stimulation prevents E2-induced extracellular regu-lated kinases (ERK1/2) and AKT activation and still induces theactivation of p38, the proapoptotic member of mitogen-activatingprotein kinase (MAPK) family. As a consequence, Nar stimula-tion impedes the E2-induced transcription of cyclin D1 promoterand reverts the E2-induced cell proliferation, driving cancer cellto apoptosis. Thus, these results suggest that coexposure to thislow-affinity, low-potency ligand for ERa specifically antagonizesthe E2-induced ERa-dependent rapid signals by reducing theeffect of the endogenous hormone in promoting cellular prolifera-tion. As a whole, these data indicate that Nar is an excellentcandidate as a chemopreventive agent in E2-dependentcancers. � 2009 IUBMB
IUBMBLife, 62(1): 51–60, 2010
Keywords 17b-estradiol; estrogen receptor a; cell proliferation; nar-ingenin.
INTRODUCTION
Flavonoids, plant secondary metabolites, are defined chemi-
cally as substances composed of a common phenylchromanone
structure (C6-C3-C6) with one or more hydroxyl substituents
(1). Flavonoids are present in fruits, vegetables and beverages
derived from plants (e.g. tea, red wine, orange, and grapefruit
juices), and in many dietary supplements or herbal remedies
(1). Flavonoids have been described as health-promoting, dis-
ease-preventing dietary components; moreover, in vivo and
in vitro experiments support a protective effect of flavonoids to
reduce the incidence of certain hormone-responsive cancers (1–
5). In addition, they are extremely safe and associated with low
toxicity, making them good candidates as chemopreventive
agents.
The cancer-protective effects of flavonoids have been attrib-
uted to a wide variety of mechanisms (6). These include pro-
and/or antioxidant effects, and the modulation of kinase
activities as well as protein functions through competitive or
allosteric interactions. However, flavonoid-dependent kinases
modulation and antioxidant effects are only reported after
administration of high flavonoid concentration ([50 lM) (see
Ref. 6 and literature cited therein). Currently, the relative
importance of these pathways and their putative cross-talk
remain to be established. Furthermore, their clinical significance
at nutritionally relevant concentrations remains unsolved (7). Atconcentrations more physiologically achievable in the plasma
after the consumption of meals rich in flavonoids (i.e. 0.1–10lM), these compounds interact with estrogen receptors (ERaand ERb) and affect their resulting cellular responses (3, 8),thus leading to estrogenic or antiestrogenic effects. Because of
this ability to interfere with E2 action, flavonoids are actually
defined as dietary phytoestrogens (9).
We recently demonstrated that the flavanone naringenin
(Nar, 5,7,40-trihydroxyflavanone) hampers ERa-mediated rapid
activation of signaling kinases [i.e. extracellular regulated ki-
nases (ERK1/2) member of mitogen-regulated protein kinase
Address correspondence to: Maria Marino, Department of Biology,
University ‘‘Roma Tre’’, Viale G. Marconi, 446, I-00146 Roma, Italy.
Tel: 139 06 55176345. Fax: 139 06 55176321.
E-mail: m.marino@ uniroma3.it
Received 26 June 2009; accepted 8 October 2009
ISSN 1521-6543 print/ISSN 1521-6551 online
DOI: 10.1002/iub.279
IUBMB Life, 62(1): 51–60, January 2010

(MAPK) and phosphatidyl inositol 3 kinase (PI3K)/AKT] and
cyclin D1 transcription, important for cell cycle progression,
only when HeLa cells, devoid of any ER isoforms, were
endowed with human ERa (8, 10, 11). On the other hand, in
the presence of ERb, Nar does not impair the ERb-mediated
activities. Rather, Nar acts as an estrogen mimetic (8). These
results increase the possibility that Nar could reduce the effect
of the potent endogenous 17b-estradiol (E2) in promoting cellu-
lar proliferation when administrated in sufficient quantities, with
the net effect of antagonizing the ERa-dependent E2 effects.
Thus, the aim of the study was to evaluate the antagonistic
effect of Nar by investigating the effects of physiological E2
concentration (i.e. 10 nM) in the presence of different concen-
trations of Nar in ERa-expressing cells. In particular, the HeLa
cell line was chosen because it is devoid of endogenous expres-
sion of ERs but it can be rendered E2 sensitive after the tran-
sient transfection with ERa expression vector without any com-
plication because of the presence of ERb. Moreover, we previ-
ously reported that the full complement of coactivators,
corepressors, and signalling kinases necessary for the full ER
activity are present in this line (8, 12). In addition, the hepa-
toma cell line (HepG2), which endogenously express low levels
of ERa (13), was also used.
MATERIALS AND METHODS
Reagents
Naringenin, 17b-estradiol, gentamicin, penicillin and other
antibiotics, GenElute plasmid maxiprep kit, Dulbecco Modified
Eagle Medium (DMEM) and RPMI-1640 media without phenol
red, and charcoal-stripped fetal calf serum were purchased from
Sigma–Aldrich (St. Louis, MO). Lipofectamine reagent was
obtained from GIBCO-BRL Life-technology (Gaithersburg,
MD). The luciferase kit was obtained from Promega (Madison,
WI). Bradford protein assay was obtained from BIO-RAD Lab-
oratories (Hercules, CA). [6,7-3H]E2 (specific activity 5 44.8
Ci/mmol) was purchased from Perkin-Elmer Life Sciences
(Cambridge, UK). The human recombinant ERa was obtained
by PanVera (Madison, WI). The antiphospho-ERK1/2, anti-
AKT, anti-b-tubulin, anti-ERa, anticaspase-3, antipoly(ADP-
ribose)polymerase (PARP), and anti-ERK1/2 antibodies were
obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The
polyclonal antiphospho-AKT, anti-phospho-p38, and anti-p38
antibodies were purchased from New England Biolabs (Beverly,
MA). ECL, chemiluminescence reagent for Western blot was
obtained from Amersham Biosciences, (Little Chalfont, UK).
All the other products were from Sigma–Aldrich. Analytical or
reagent grade products were used without further purification.
Ligand Binding Analysis
The reversible binding of E2 to human recombinant ERawas studied by ligand saturation experiments. Recombinant
ERa (final concentration 1.0 3 10210 M) was incubated for 2 h
at 25 8C in the binding buffer (Tris–HCl 4.0 3 1022 M, EDTA
1.0 3 1023 M, DDT 1.0 3 1023 M, 1% (w/v) yeast extract
and 10% (v/v) glycerol, pH 7.4) with [3H]E2 (final concentra-
tion, ranging between 1.0 3 10210 and 4.0 3 1028 M). In par-
allel, recombinant ERa (final concentration, 1.0 3 10210 M)
was incubated for 2 h at 25 8C in the binding buffer with
[3H]E2 (final concentration, ranging between 1.0 3 10210 and
4.0 3 1028 M) in the presence of 1.0 3 1026 or 1.0 3 1025
M Nar. In all saturation ligand-binding experiments, the free
and ERa-bound radioligand were separated by vacuum filtration
through a 12-sample Millipore filter manifold (Bedford, MA),
holding glass microfibre filters (Whatman Ltd, UK) (14). Radio-
activity retained on each filter was counted in 5 mL of the scin-
tillation cocktail (Perkin Elmer, Cambridge, UK) with a
2100TR Tri-Carb liquid scintillation analyzer (Packard Instru-
ments CO., Meriden, CT).
The value of the apparent dissociation equilibrium constant
for [3H]E2 binding to ERa (Kd0), in the absence and presence of
1.0 3 1026 M and 1.0 3 1025 M Nar, was determined from
the dependence of the radioactivity retained on filters (i.e. R) on
the [3H]E2 concentration, according to Equation (1) (14):
Y ¼ R=Rtot ¼ ð13½L�Þ=ðK0d þ ½L�Þ (1)
where Rtot is the maximum asymptotic value of radioactivity
measured when the complete saturation of the receptor was
achieved and [L] denotes the free radioligand concentration (i.e.
[[3H]E2]). In the absence of Nar, Kd0 corresponds to the intrinsic
dissociation equilibrium constant for E2 binding to recombinant
ERa (i.e. KE2d ). According to the competitive inhibition mecha-
nism, the intrinsic equilibrium dissociation constant for Nar
binding to ERa (i.e. KNard ) was determined from the dependence
of K0d from the Nar concentration, according to Equation (2)
(14):
K0d ¼ ðKE2
d
�KNard Þ 3 ½Nar� þ KE2
d (2)
As expected from Eq. (2), for [Nar] 5 0, Kd0 corresponds to
KE2d .
Cell Culture
The ER devoid of human cervix epitheloid carcinoma cell
line (HeLa) and the ERa containing human hepatoma cell line
(HepG2) were routinely grown in air containing 5% CO2in
modified, phenol red-free, DMEM (HeLa cells) or RPMI-1640
medium (HepG2 cells) containing 10% (v/v) charcoal-stripped
fetal calf serum, L-glutamine (2.0 mM), gentamicin (10 mg/
mL), and penicillin (100 U/mL). Cells were passaged every
2 days (HeLa cells) or every 3 days (HepG2 cells).
Plasmids, Cell Transfection, and Luciferase Assay
The gene reporter plasmids complement 3-luciferase (pC3),
Cyclin D1-luciferase (pXP2-D1-2966-luciferase, pD1), and the
plasmids containing the vector expression for pCR3.1-b-galacto-
52 BULZOMI ET AL.

sidase and the wild-type human ERa pSG5-HE0 have been
described elsewhere (8). Furthermore, an empty vector,
pCMV5, was used as control. A luciferase dose response curve
showed that the maximum effect was obtained when 1.0 lg of
plasmids was transfected together with 1.0 lg of pCR3.1-b-ga-lactosidase to normalize for transfection efficiency (�50–60%).
Plasmids were purified for transfection using the GenElute plas-
mid maxiprep kit according to the manufacturer’s instructions.
HeLa cells were grown to �70% confluence and then trans-
fected using Lipofectamine Reagent according to the manufac-
turer’s instructions. Six hours after transfection, the medium
was changed and 24 h after the cells were stimulated for 24 h
with either Nar (1.0 3 1026 M) or E2 (1.0 3 1028 M) or with
different concentration (1.0 3 1028 to 1.0 3 1024 M) of Nar
in the presence of 1.0 3 1028 M E2. The cell lysis procedure
as well as the subsequent measurement of luciferase gene
expression was performed using the luciferase kit according to
the manufacturer’s instructions with a EC & G Berthold lumi-
nometer (Bad Wildbad, Germany).
Cell Viability and Cell Cycle
HeLa cells were grown to �70% confluence in six-well
plates, transfected with human pSG5-hERa or pCMV5 (empty
vector) and, after 24 h, stimulated with different concentration
(1.0 3 10210 to 1.0 3 1024 M) of Nar or E2 for 24 h or with
different concentration (1.0 3 10210 to 1.0 3 1024 M) of Nar
in the presence of 1.0 3 1028 M E2. After treatment, cells
were harvested with trypsin, centrifuged, stained with trypan
blue solution, and counted in a hemocytometer (improved Neu-
bauer chamber) in quadruplicate. For cell cycle analysis, 106
HeLa cells were transfected with either human pSG5-hERa or
pCMV5 expression vectors and, 24 h after, stimulated with Nar
1.0 3 1026 M in the presence or absence of E2 1.0 3 1028 M
for 24 h. After stimulation, cells were fixed with 1 mL ice-cold
70% ethanol and subsequently stained with 2 mg/mL DAPI/
PBS solution. The fluorescence of DNA was measured with
DAKO Galaxy flow-cytometer equipped with HBO mercury
lamp, and the percentage of cells present in sub-G1, G1, S and
G2/M phases was calculated using a FloMax� Software.
Western Blot
Cells were stimulated with either E2 (final concentration, 1.0
3 1028 M in ethanol/phosphate-buffered saline, PBS, 1:10, v/v)
or Nar (final concentration, 1.0 3 1026 M in DMSO/PBS 1:10,
v/v) or E2 1 Nar (final concentration, 1.0 3 1028 M and 1.0
3 1026 M, respectively) or vehicle (ethanol/PBS 1:10, v/v). In
some experiments, HepG2 cells were treated with E2 (1.0 31028 M) and different concentrations of Nar (1.0 3 1026 to 1.0
3 1024 M). After stimulation, cells were lysed and solubilized
in 0.125 M Tris, pH 6.8, containing 10% (w/v) SDS, 1.0 mM
phenylmethylsulfonyl fluoride, and 5.0 lg/mL leupeptin; then
the cell lysates were boiled for 2 min. Total proteins were quan-
tified using the Bradford protein assay. Solubilized proteins
(20 lg) were resolved by 7 or 10% SDS-PAGE at 100 V for 1
h at 24 8C and then electrophoretically transferred to nitrocellu-
lose for 45 min at 100 V and 4 8C. The nitrocellulose was
treated with 3% (w/v) BSA in 138.0 mM NaCl, 25.0 mM Tris,
pH 8.0, at 24 8C for 1 h and then probed overnight at 4 8C with
either anti-ERa or anticaspase-3 or anti-PARP or antiphospho-
ERK1/2 or antiphospho-AKT or antiphospho-p38 antibodies.
The nitrocellulose was stripped by Restore Western Blot Strip-
ping Buffer (Pierce Chemical Company, Rockford, IL) for 10
min at room temperature and then probed with either anti-
ERK1/2 or anti-AKT or anti-p38 and anti-b-tubulin antibodies.
Antibody reaction was visualized with chemiluminescence
Western blot detection reagent (Amersham Biosciences, Little
Chalfont, UK). Densitometric analyses were performed by
ImageJ software for Windows.
Statistical Analysis
A statistical analysis was performed by using Student0 t testwith the GraphPad INSTAT3 software system for Windows. In
all cases, P values\0.05 were considered significant.
Figure 1. Naringenin (Nar) effect on 17b-estradiol (E2) bindingto human recombinant ERa. Panel a: Dependence of the intrin-
sic molar fraction (Y) of [3H]E2-bound to ERa from [3H]E2
concentration in the absence (squares) and in the presence of 2
representative Nar concentrations (1.0 3 1026 M [diamonds]
and 1.0 3 1025 M [circles]). Data are the means 6 SD of five
different experiments. Panel b: Dependence of the apparent dis-
sociation equilibrium constant for [3H]E2 binding to ERa (Kd0)
from Nar concentrations (ranging from 1.0 3 1026 M to 1.0 31025 M). Data are means 6 SD of five different experiments.
For details see the text.
53E2 AND NARINGENIN ANTAGONIST EFFECTS ON PROLIFERATION
RESULTS
E2 and Nar Binding to ERa
To assess the Nar ability to compete with E2 for binding to
human recombinant ERa, E2 saturation experiments have been
performed in the absence and presence of 1.0 3 1026 and
1.0 3 1025 M Nar. Both in the absence and in the presence of
Nar, E2 binding to ERa follows a simple equilibrium as postu-
lated by Equation (1), the Hill coefficient being 1.0 6 0.1. In
the absence of Nar, E2 binding to ERa is characterized by an
intrinsic equilibrium dissociation constant (KE2d ) of (2.0 6 0.5)
3 10210 M. In the presence of unlabeled Nar, the apparent
equilibrium constant for E2 binding to ERa increased to Kd0 5
(5.2 6 0.6) 3 1029 M and (3.1 6 0.4) 3 1028 M in the pres-
ence of 1.0 3 1026 and 1.0 3 1025 M of Nar, respectively
(Fig. 1). The linear dependence of Kd0 on the Nar concentration
(Fig. 1b) indicates that a simple competition mechanism is oper-
ative (15, 16). Data reported in Figure 1b, analyzed according
to Eq. (2), allowed the determination of the intrinsic dissocia-
tion constant for Nar binding to ERa (KNard 5 1.4 6 0.3 3
1027 M). This confirms that Nar binds to ERa with an affinity
lower by about three orders of magnitude than that of E2. These
data indicate that Nar and E2 bind competitively to ERa; more-
over, in the presence of nutritionally relevant Nar concentra-
tions, the molar fraction of E2 bound to ERa decreases.
ERa Transcriptional Activities
The result of Nar binding to ERa prompted us to evaluate
the effect of co-stimulation of E2 and Nar on the ERa activities.
We first assessed the ERa-mediated direct gene transcription
(i.e. estrogen responsive element (ERE)-dependent) (13). HeLa
cells, transiently transfected with ERa or empty vector, and the
ERE-containing reporter plasmid (pC3) were incubated with
either E2 alone (1.0 3 1028 M) or Nar alone (1.0 3 1026 M)
or in the presence of E2 (1.0 3 1028 M) and different Nar
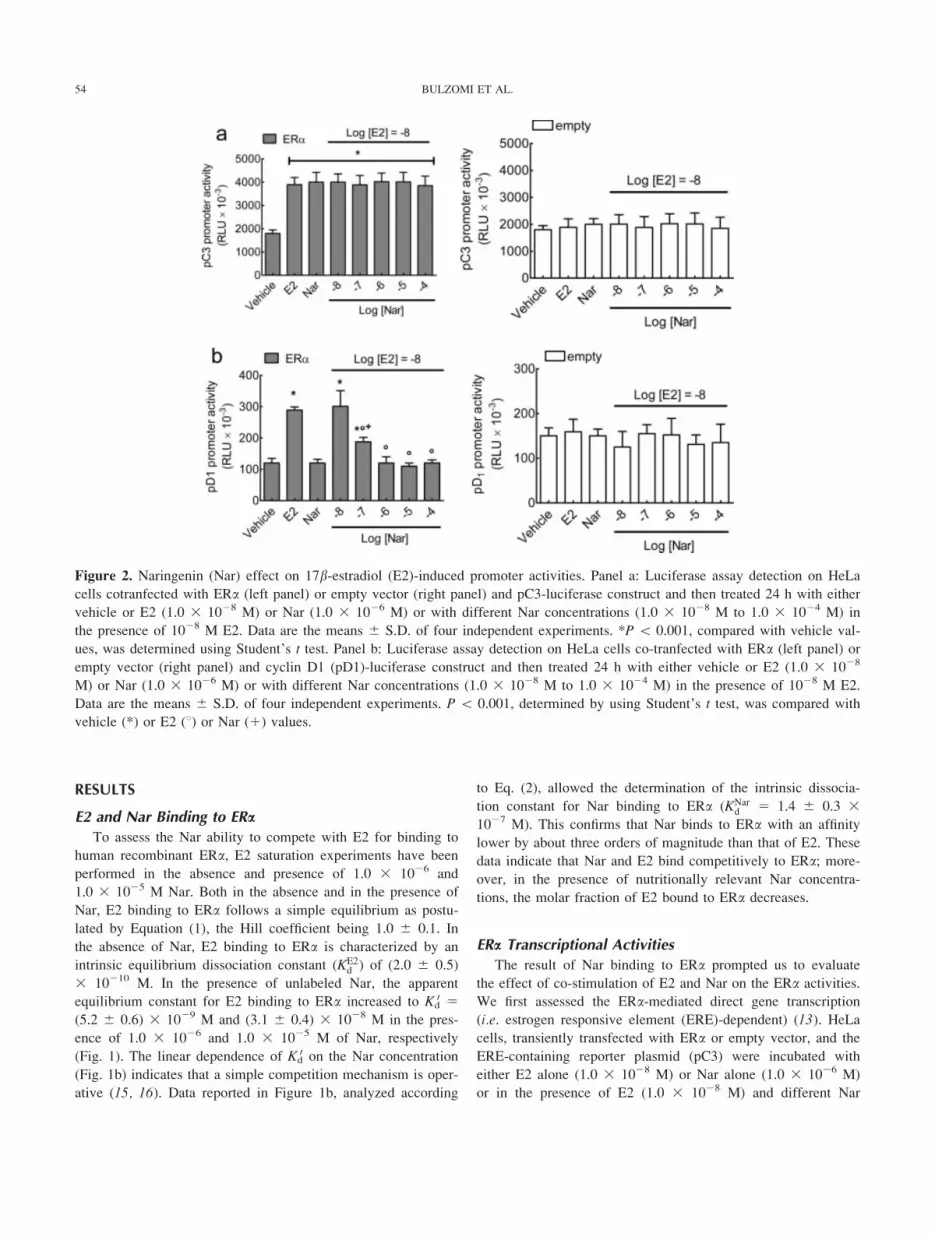
Figure 2. Naringenin (Nar) effect on 17b-estradiol (E2)-induced promoter activities. Panel a: Luciferase assay detection on HeLa
cells cotranfected with ERa (left panel) or empty vector (right panel) and pC3-luciferase construct and then treated 24 h with either
vehicle or E2 (1.0 3 1028 M) or Nar (1.0 3 1026 M) or with different Nar concentrations (1.0 3 1028 M to 1.0 3 1024 M) in
the presence of 1028 M E2. Data are the means 6 S.D. of four independent experiments. *P \ 0.001, compared with vehicle val-
ues, was determined using Student’s t test. Panel b: Luciferase assay detection on HeLa cells co-tranfected with ERa (left panel) or
empty vector (right panel) and cyclin D1 (pD1)-luciferase construct and then treated 24 h with either vehicle or E2 (1.0 3 1028
M) or Nar (1.0 3 1026 M) or with different Nar concentrations (1.0 3 1028 M to 1.0 3 1024 M) in the presence of 1028 M E2.
Data are the means 6 S.D. of four independent experiments. P \ 0.001, determined by using Student’s t test, was compared with
vehicle (*) or E2 (8) or Nar (1) values.
54 BULZOMI ET AL.

concentrations. Nar, alone or with E2, induced the ERE-contain-
ing promoter activity to a level comparable with that of E2
alone (Fig. 2a). No pC3 promoter activity was present when
HeLa cells, transiently transfected with the empty plasmid, were
stimulated with different ERa ligands (Fig. 2a), thus demon-
strating the ERa dependence of this effect. The indirect tran-
scriptional activity of ERa [i.e. through interaction with activa-
tor protein-1 (AP-1) or stimulating protein 1 (Sp1) transcription
factors] (13) was assessed by transfection with cyclin D1 (pD1)
promoter. In fact, cyclin D1 is a well-known E2-responsive
gene, even if ERE-like sequence in its promoter has not been
detected (17). As expected, cell treatment with E2 resulted in a
significant increase in cyclin D1 promoter activity (Fig. 2b)
comparable with those previously reported (13). Notably, 1.0 31027 M Nar reduced the E2 effect, and higher Nar concentra-
tions (i.e. 1.0 3 1026 to 1.0 3 1024 M) completely prevented
E2-induced pD1 promoter activity (Fig. 2b). To determine the
ER involvement in the ligand-induced cyclin D1 promoter ac-
tivity, experiments were performed also in HeLa cells trans-
fected with the empty plasmids (Fig. 2b). Results indicate that
no pD1 promoter activity was present when these cells were
stimulated with E2 or Nar (Fig. 2b).
ERa-Dependent Rapid Signals
The E2-induced cyclin D1 promoter activity requires rapid sig-
nal transduction pathways. In particular, the rapid (15 min) E2-
induced activation of ERK1/2 and PI3K/AKT cascades are funda-
mental for E2-induced pD1 promoter activity (13, 18). On the
other hand, Nar stimulation induces the rapid and persistent
(15 min to 24 h) activation of p38, another component of MAPK
family (8, 11). Thus, the ability of E2 to still induce rapid signal
kinase cascades even in the presence of 1.0 3 1026 M Nar was
evaluated in HeLa cells transfected with the empty vector or with
ERa expression vector. No kinase activation was detected in
HeLa cells devoid of ERa stimulated with E2 or Nar (data not
shown), whereas E2 ability to induce the rapid (15 min) ERK1/2
and AKT activation without any effect on the persistent (24 h)
p38 activation has been confirmed in ERa-containing HeLa cells
(Fig. 3). Remarkably, Nar stimulation prevents E2-induced
ERK1/2 and AKT activation and still induces the persistent p38
phosphorylation even in the presence of E2 (Fig. 3).
ERa-Dependent E2-Induced Cell Proliferation
Cyclin D1 represents the upstream sensor of E2-induced pro-
liferative signals, which, in turn, depends on the rapid activation
of upstream E2-induced kinase (12, 13). However, in the pres-
ence of ERa, Nar prevents cell proliferation inducing a proa-
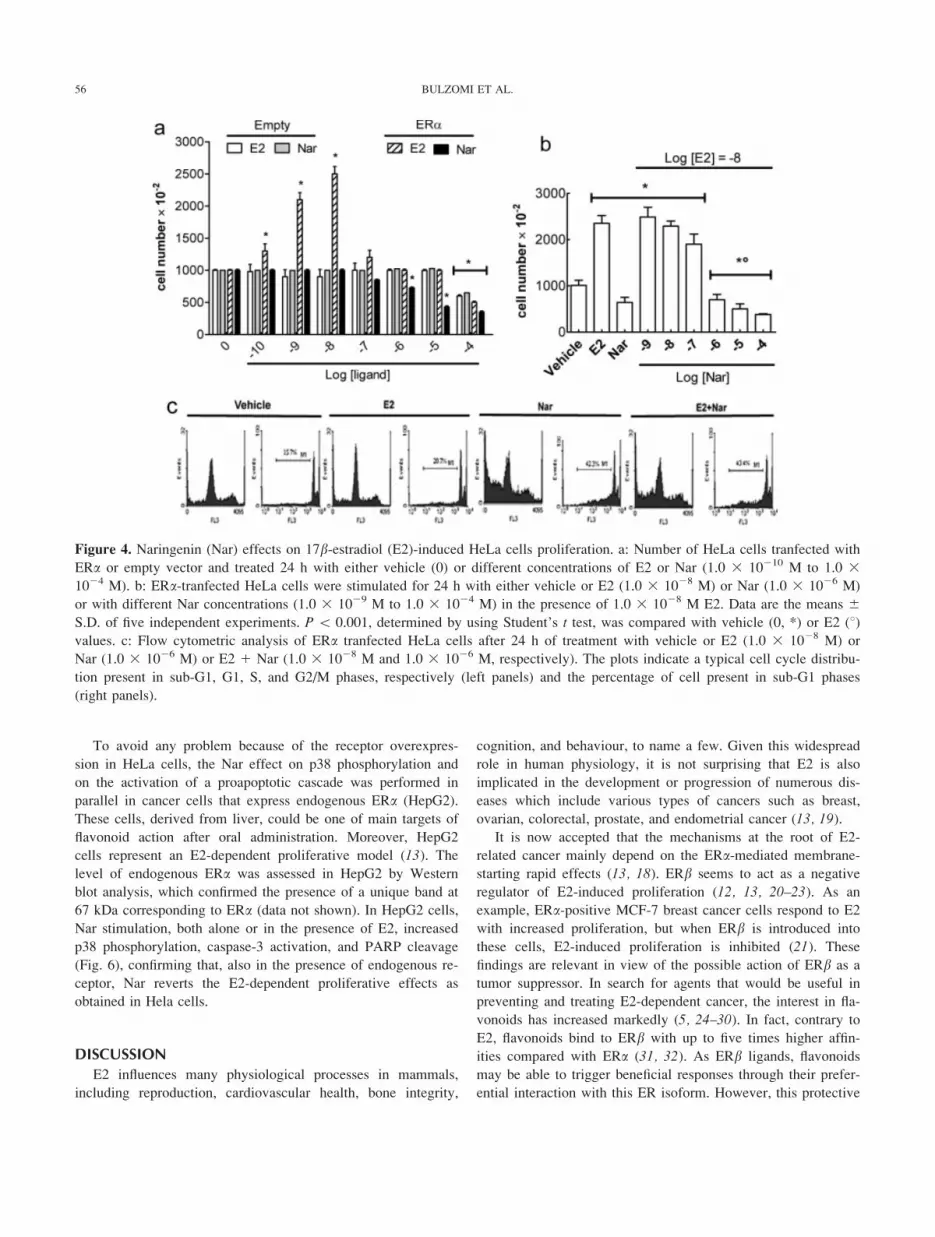
poptotic cascade (8, 11). Figure 4 confirms that 1.0 3 1026, 1.0
3 1025, and 1.0 3 1024 M Nar reduced cell number only in
ERa-containing HeLa cells, whereas physiological E2 concen-
trations (i.e. 1.0 3 1029, and 1.0 3 1028 M) doubled the cell
numbers in 24 h (Fig. 4a). Note that high Nar or E2 concentra-
tion (1.0 3 1024 M) reduced cell numbers also in empty vec-
tor-transfected HeLa cells, suggesting an ERa-independent cyto-toxic effects for both substances (Fig. 4a). Intriguingly, Nar
stimulation reverted the E2-induced effect on cell proliferation
significantly reducing the number of cells in a dose-dependent
manner (Fig. 4b). Furthermore, 1.0 3 1026 M Nar changed the
E2-induced distribution of cell population in the cell cycle
phases (Fig. 4c), decreasing the cells present in G1 phase and
increasing the number of cell present in sub-G1 phase of the
cell cycle as follows 15.0 6 1.3 % (Vehicle), 20.2 6 0.5%
(E2), 42.0 6 0.7 % (Nar), and 43.4 6 1.0 % (E21Nar) (Fig.
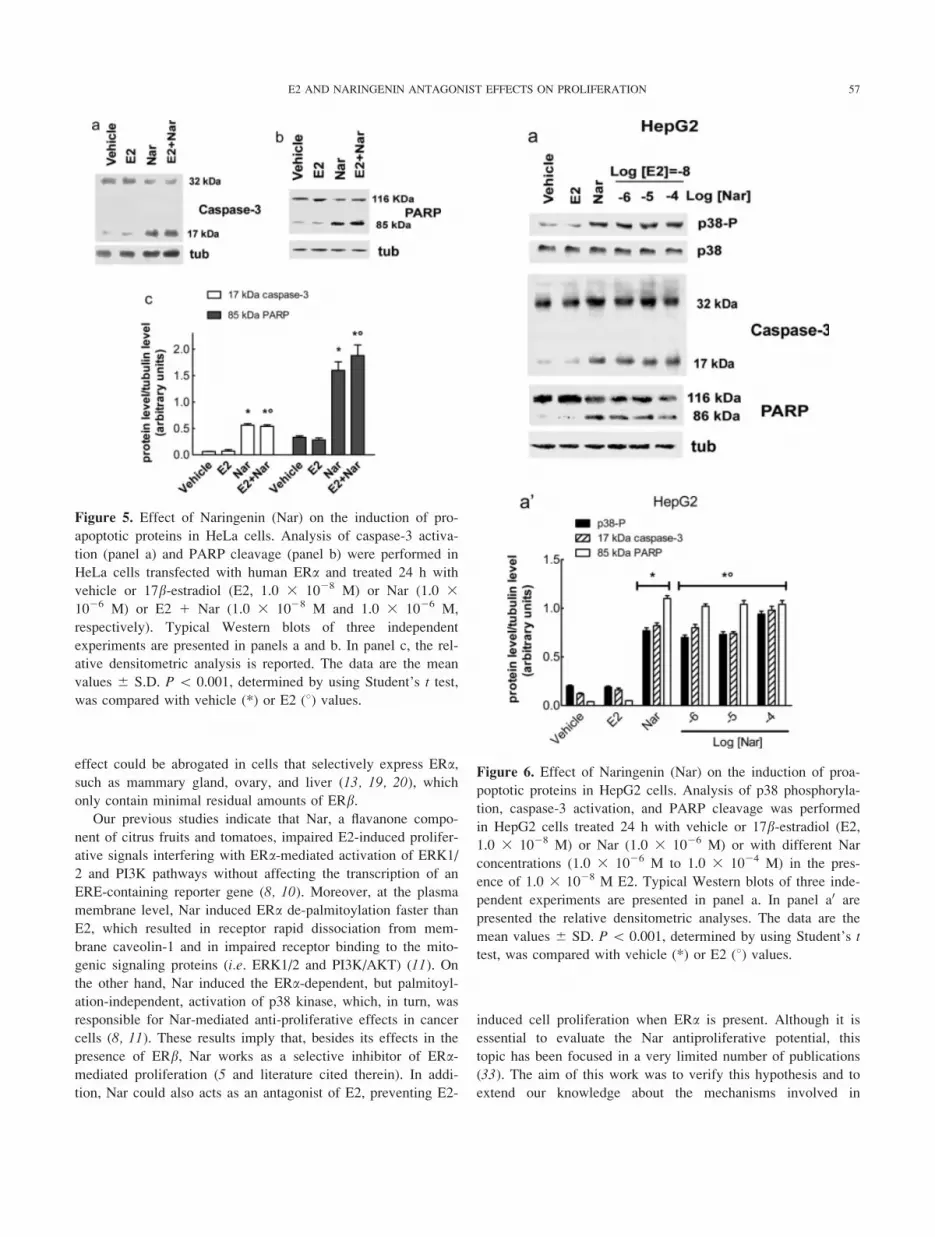
4b). In line with these results, Nar increased the level of the
active caspase-3 (i.e. 17 kDa band, Fig. 5a) as demonstrated by
the increased level of poly(ADP-ribose)polymerase (PARP)
cleavage, a caspase-3 substrate, even in the presence of 1.0 31028 M E2 (Fig. 5b), thus demonstrating the strong antagonistic
effects of this flavanone on E2-induced proliferation.
Figure 3. Naringenin (Nar) effects on 17b-estradiol (E2)-
induced rapid ERa activities. ERa-transfected HeLa cells were
treated with either vehicle or E2 (1.0 3 1028 M) or Nar (1.0 31026 M) or with a mixture of Nar (1.0 3 1026 M) 1 E2 (1.0
3 1028 M). After 15 min (left panel, ERK1/2 and AKT) or af-
ter 24 h (right panel, p38), the phosphorylation of the kinases
was evaluated. The amounts of protein were normalized by
comparison with un-phosphorylated ERK1/2 or AKT or p38
and tubulin antibodies. Upper panels show representative West-
ern blots, lower panel shows the densitometric analysis. Data
are the means 6 S.D. of four independent experiments. P \0.001, determined by using Student’s t test, was compared with
vehicle (*) or E2 (8) values.
55E2 AND NARINGENIN ANTAGONIST EFFECTS ON PROLIFERATION
To avoid any problem because of the receptor overexpres-
sion in HeLa cells, the Nar effect on p38 phosphorylation and
on the activation of a proapoptotic cascade was performed in
parallel in cancer cells that express endogenous ERa (HepG2).
These cells, derived from liver, could be one of main targets of
flavonoid action after oral administration. Moreover, HepG2
cells represent an E2-dependent proliferative model (13). The
level of endogenous ERa was assessed in HepG2 by Western
blot analysis, which confirmed the presence of a unique band at
67 kDa corresponding to ERa (data not shown). In HepG2 cells,
Nar stimulation, both alone or in the presence of E2, increased
p38 phosphorylation, caspase-3 activation, and PARP cleavage
(Fig. 6), confirming that, also in the presence of endogenous re-
ceptor, Nar reverts the E2-dependent proliferative effects as
obtained in Hela cells.
DISCUSSION
E2 influences many physiological processes in mammals,
including reproduction, cardiovascular health, bone integrity,
cognition, and behaviour, to name a few. Given this widespread
role in human physiology, it is not surprising that E2 is also
implicated in the development or progression of numerous dis-
eases which include various types of cancers such as breast,
ovarian, colorectal, prostate, and endometrial cancer (13, 19).
It is now accepted that the mechanisms at the root of E2-
related cancer mainly depend on the ERa-mediated membrane-
starting rapid effects (13, 18). ERb seems to act as a negative
regulator of E2-induced proliferation (12, 13, 20–23). As an
example, ERa-positive MCF-7 breast cancer cells respond to E2
with increased proliferation, but when ERb is introduced into
these cells, E2-induced proliferation is inhibited (21). These
findings are relevant in view of the possible action of ERb as a
tumor suppressor. In search for agents that would be useful in
preventing and treating E2-dependent cancer, the interest in fla-
vonoids has increased markedly (5, 24–30). In fact, contrary to
E2, flavonoids bind to ERb with up to five times higher affin-
ities compared with ERa (31, 32). As ERb ligands, flavonoids
may be able to trigger beneficial responses through their prefer-
ential interaction with this ER isoform. However, this protective
Figure 4. Naringenin (Nar) effects on 17b-estradiol (E2)-induced HeLa cells proliferation. a: Number of HeLa cells tranfected with
ERa or empty vector and treated 24 h with either vehicle (0) or different concentrations of E2 or Nar (1.0 3 10210 M to 1.0 31024 M). b: ERa-tranfected HeLa cells were stimulated for 24 h with either vehicle or E2 (1.0 3 1028 M) or Nar (1.0 3 1026 M)
or with different Nar concentrations (1.0 3 1029 M to 1.0 3 1024 M) in the presence of 1.0 3 1028 M E2. Data are the means 6S.D. of five independent experiments. P \ 0.001, determined by using Student’s t test, was compared with vehicle (0, *) or E2 (8)values. c: Flow cytometric analysis of ERa tranfected HeLa cells after 24 h of treatment with vehicle or E2 (1.0 3 1028 M) or
Nar (1.0 3 1026 M) or E2 1 Nar (1.0 3 1028 M and 1.0 3 1026 M, respectively). The plots indicate a typical cell cycle distribu-
tion present in sub-G1, G1, S, and G2/M phases, respectively (left panels) and the percentage of cell present in sub-G1 phases
(right panels).
56 BULZOMI ET AL.
effect could be abrogated in cells that selectively express ERa,such as mammary gland, ovary, and liver (13, 19, 20), which
only contain minimal residual amounts of ERb.Our previous studies indicate that Nar, a flavanone compo-
nent of citrus fruits and tomatoes, impaired E2-induced prolifer-
ative signals interfering with ERa-mediated activation of ERK1/
2 and PI3K pathways without affecting the transcription of an
ERE-containing reporter gene (8, 10). Moreover, at the plasma
membrane level, Nar induced ERa de-palmitoylation faster than
E2, which resulted in receptor rapid dissociation from mem-
brane caveolin-1 and in impaired receptor binding to the mito-
genic signaling proteins (i.e. ERK1/2 and PI3K/AKT) (11). On
the other hand, Nar induced the ERa-dependent, but palmitoyl-
ation-independent, activation of p38 kinase, which, in turn, was
responsible for Nar-mediated anti-proliferative effects in cancer
cells (8, 11). These results imply that, besides its effects in the
presence of ERb, Nar works as a selective inhibitor of ERa-mediated proliferation (5 and literature cited therein). In addi-
tion, Nar could also acts as an antagonist of E2, preventing E2-
induced cell proliferation when ERa is present. Although it is
essential to evaluate the Nar antiproliferative potential, this
topic has been focused in a very limited number of publications
(33). The aim of this work was to verify this hypothesis and to
extend our knowledge about the mechanisms involved in
Figure 5. Effect of Naringenin (Nar) on the induction of pro-
apoptotic proteins in HeLa cells. Analysis of caspase-3 activa-
tion (panel a) and PARP cleavage (panel b) were performed in
HeLa cells transfected with human ERa and treated 24 h with
vehicle or 17b-estradiol (E2, 1.0 3 1028 M) or Nar (1.0 31026 M) or E2 1 Nar (1.0 3 1028 M and 1.0 3 1026 M,
respectively). Typical Western blots of three independent
experiments are presented in panels a and b. In panel c, the rel-
ative densitometric analysis is reported. The data are the mean
values 6 S.D. P \ 0.001, determined by using Student’s t test,
was compared with vehicle (*) or E2 (8) values.
Figure 6. Effect of Naringenin (Nar) on the induction of proa-
poptotic proteins in HepG2 cells. Analysis of p38 phosphoryla-
tion, caspase-3 activation, and PARP cleavage was performed
in HepG2 cells treated 24 h with vehicle or 17b-estradiol (E2,1.0 3 1028 M) or Nar (1.0 3 1026 M) or with different Nar
concentrations (1.0 3 1026 M to 1.0 3 1024 M) in the pres-
ence of 1.0 3 1028 M E2. Typical Western blots of three inde-
pendent experiments are presented in panel a. In panel a0 are
presented the relative densitometric analyses. The data are the
mean values 6 SD. P\ 0.001, determined by using Student’s t
test, was compared with vehicle (*) or E2 (8) values.
57E2 AND NARINGENIN ANTAGONIST EFFECTS ON PROLIFERATION

Nar-mediated antiproliferative activity by investigating the
effect of this flavonoid on cancer cells growth in the presence
of an E2 background.
Current findings confirm that the Nar concentration required
to half-saturate ERa is about 1000-fold higher than that reported
for E2; however, in the presence of Nar, the Kd of E2 for its re-
ceptor linearly increased, suggesting a decreased affinity
between E2 and ERa. Remarkably, this decreased affinity
between E2:ERa did not impair the E2 ability, in the presence
of Nar, to trigger gene transcription through the direct binding
of ERa to ERE-containing reporter gene (i.e. pC3). This result
implies that the coactivator recruitment on the ligand-bound
ERa is not prevented by Nar as well as the arrangement of a
macromolecular complex, which provides the platform on which
the components of transcriptional machinery are assembled.
However, ligand bound to ERa could mediate gene transcription
even in a manner that does not require the ERa direct binding
to DNA. This is referred to as ‘‘indirect genomic mechanism’’
which requires the ERa interaction with specific transcription
factors such as Sp1 and AP-1. The ERa-Sp1 and ERa-AP-1complexes interact with response elements (GC-rich and TRE,
respectively) within target promoters. Genes activated by E2
through this genomic pathway include cathepsin D, c-fos, reti-
noic acid receptor a1, adenosine deaminase, IGF-binding pro-
tein 4, Bcl2, E2f1, thymidylate synthase, vascular endothelial
growth factor (Vegf), and cyclin D1 (34).
Intriguingly, in the presence of Nar, E2 lacks its ability to
activate cyclin D1 promoter, suggesting that E2-induced ERainteraction to Sp1 and AP-1 is impaired. Our previous data
indicate that E2 stimulation of ERa-containing HeLa cells
induced an increase in AP-1 binding to DNA, whereas Nar or
other flavonoids (i.e. quercetin) were unable to do this (10).
Although evidence indicate the ability of flavonoids to bind
both ER isoforms maintaining the ERs gene transcriptional
ability (31, 32, 35), current data indicate that Nar only allows
the E2-induced direct transcriptional activity of ERa, highlight-ing a role for Nar as an antagonist of E2-induced indirect
gene expression.
The physiological role played by rapid membrane-starting
pathways has been clarified at least for some E2 targets (19,
36–38). Among other cellular functions, the mechanisms by
which E2 exerts proliferative effects is assumed to be exclu-
sively mediated by rapid membrane-starting actions (13, 18). In
HepG2 cells, multiple and parallel membrane-starting pathways
are rapidly activated by the E2:ERa complex (13), and the
blockade of ERK1/2 and PI3K/AKT pathways completely pre-
vents the E2-induced DNA synthesis (13). ERK1/2/MAPK and
PI3K/AKT pathways, rapidly activated by E2:ERa complex,
also have a critical role in E2 action as a survival agent. In
fact, these pathways enhance the expression of the antiapoptotic
protein Bcl-2, block the activation of the p38/MAPK, reduce
the proapoptotic caspase-3 activation, and promote G1-to-S
phase transition through the enhancement of the cyclin D1
expression. Thus, in both ERa-transfected HeLa cells and in
HepG2 cells, the E2 inability to activate rapid signal transduc-
tion pathways, in the presence of Nar, was paralleled by the
block of E2-induced proliferation and by the induction of the
apoptotic cascade (i.e. caspase-3 activation and PARP cleav-
age).
As a whole, the assays with Nar against a background
level of E2 allowed us to assess the estrogenic versus anties-
trogenic activity of this flavanone. The results of this study
demonstrate that Nar treatment do not impair E2-induced
ERE-dependent ERa transcriptional activity, whereas Nar
reverts the proliferative effects of E2 impairing ERa-mediated
rapid signals and inducing different proapoptotic signal trans-
duction pathways. Moreover, the preventive effects elicited by
Nar on E2-dependent cancers may be enhanced, in some tis-
sues, through the induction of specific ERb-dependent proa-
poptotic signalling (8). In addition, these results increase the
list of Nar effects on human health adding up a possible ther-
apeutic benefit of regular consumption of these flavonoids,
which may counteract the E2 proliferative action. Collectively,
our data suggest that the regular consumption of Nar may
slow the rate at which E2-dependent cancer cell proliferate. In
addition, this study indicates that the studies, which only focus
on the transactivation capacity of various naturally derived es-
trogenic ligands, could be misleading in that they are actually
assaying just one of the diverse action mechanisms elicited by
the ERs.
Finally, a number of pleiotropic molecular effects of Nar
have been reported in cancer cells, which include the modula-
tion of cell signalling pathways, the regulation of the cell
cycle, the inhibition of glucose uptake, and antioxidant activ-
ities (1, 6). Some of these mechanisms may even occur inde-
pendently of ER binding (39, 40), but requires high plasma
Nar concentrations (i.e. 0.8 to 25 3 1025 M), which are diffi-
cult to obtain by the oral ingestion of food rich in this biofla-
vonoid. In the best case scenario, only 15% of ingested Nar
will get absorbed in the human gastrointestinal tract. A full
glass of orange juice would supply about enough Nar to
achieve a concentration of about 0.5 3 1026 M. In a study
conducted by Erlund et al. (41) in which five subjects drank
grapefruit juice containing approximately 200 mg Nar (similar
to the Nar content in one medium-sized grapefruit), the peak of
plasma naringenin concentrations ranged from 0.7 to 14.8
3 1026 M, demonstrating that Nar plasma concentration
depends on the ability of each individual to adsorb and metabo-
lize this compound (42). These concentrations could be
increased by the use of flavonoids as dietary supplements. A
huge number of plant extracts or mixtures containing varying
amounts of isolated flavonoids are commercially available on
the market as dietary supplements and healthy products. The
commercial success of these supplements is evident, even
though the health consequences of flavonoids exposure may be
not universally beneficial and, in certain physiological phases
of human life, could even increase the risk of diseases (43,
44). Thus, further investigations on the complex role of
58 BULZOMI ET AL.

nutritional molecules in human beings are warranted before
including flavonoids in specific nutritional recommendations.
ACKNOWLEDGEMENTS
Dr. Alessandro Bolli is supported by a grant from National
Institute of Biostructures and Biosystems (INBB). This work
was supported by grants from Ateneo Roma Tre and Italian
Health Ministry to M.M.
REFERENCES1. Galluzzo, P. and Marino, M. (2006) Nutritional flavonoids impact on
nuclear and extranuclear estrogen receptor activities. Genes Nutr. 1,
161–176.
2. Adlercreutz, H., Heinonen, S. M., and Penalvo-Garcia, J. (2004) Phy-
toestrogens, cancer and coronary heart disease. Biofactors 22, 229–236.
3. Birt, D. F., Hendrich, S., and Wang, W. (2001) Dietary agents in cancer
prevention: flavonoids and isoflavonoids. Pharmacol. Therap. 90, 157–
177.
4. Moutsatsou, P. (2007) The spectrum of phytoestrogens in nature: our
knowledge is expanding. Hormones 6, 173–193.5. Marino, M. and Bulzomi, P. (2009) Mechanisms at the root of flavonoid
action in cancer: a step toward solving the Rubik’s cube. In Flavonoids:
Biosynthesis, Biological Effects and Dietary Sources. (Keller, R. B.,
ed.). Nova Science Publisher, New York (In Press).
6. Virgili, F. and Marino, M. (2008) Regulation of cellular signals from
nutritional molecules: a specific role for phytochemicals, beyond antiox-
idant activity. Free Radic. Biol. Med. 45, 1205–1216.7. Manach, C., Manach, A., Morand, S. C., Remesy, C., and Jimenez, L.
(2004) Polyphenols: food sources and bioavailability. Am. J. Clin. Nutr.
79, 727–747.
8. Totta, P., Acconcia, F., Leone, S., Cardillo, I., and Marino, M. (2004)
Mechanisms of naringenin-induced apoptotic cascade in cancer cells:
involvement of estrogen receptor a and b signalling. IUBMB Life 56,
491–499.
9. Saarinen, N. M., Makela, S., Penttinen, P., Warri, A., Lorenzetti, S.,
Virgili, F., Mortensen, A., Sørensen, I. K., Bingham, C., Valsta, L. M.,
Vollmer, G., and Zierau, O. (2006) Tools to evaluate estrogenic potency
of dietary phytoestrogens: a consensus paper from the EU Thematic
Network ‘‘Phytohealth’’ (QLKI-2002–2453). Genes Nutr. 1, 143–158.10. Virgili, F., Acconcia, F., Ambra, R., Rinna, A., Totta, P., and Marino,
M. (2004) Nutritional flavonoids modulate estrogen receptor a signaling.
IUBMB Life 56, 145–151.
11. Galluzzo, P., Ascenzi, P., Bulzomi, P., and Marino, M. (2008) The nutri-
tional flavanone naringenin triggers antiestrogenic effects by regulating
estrogen receptor a-palmitoylation. Endocrinology 149, 2567–2575.
12. Acconcia, F., Totta, P., Ogawa, S., Cardillo, I., Inoue, S., Leone, S.,
Trentalance, A., Muramatsu, M., and Marino, M. (2005) Survival versus
apoptotic 17b-estradiol effect: role of ERa and ERb activated non-
genomic signaling. J. Cell. Physiol. 203, 193–201.
13. Ascenzi, P., Bocedi, A., and Marino, M. (2006) Structure-function rela-
tionship of estrogen receptor a and b: impact on human health. Mol.
Aspects Med. 27, 299–402.
14. Bolli, A., Galluzzo, P., Ascenzi, P., Del Pozzo, G., Manco, I., Vietri,
M. T., Mita, L., Altucci, L., Mita, D. G., and Marino, M. (2008) Lac-
case treatment impairs bisphenol A-induced cancer cell proliferation
affecting estrogen receptor a-dependent rapid signals. IUBMB Life 60,
843–852.
15. Antonini, E., and Brunori, M. (1971) Hemoglobin and Myoglobin in
their Reactions with Ligands. North Holland Publishing Co., Amsterdam
and London.
16. Cheng, Y.-C., and Prusoff, W. H. (1973) Relationship between the inhi-
bition constant (Ki) and the concentration of inhibitor which causes
50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22,
3099–3108.
17. Herbert, B., Truss, M., Beato, M., and Muller, R. (1994) Inducibile reg-
ulatory elements in the human cyclin D1 promoter. Oncogene 9, 1295–
1304.
18. Castoria, G., Migliaccio, A., Bilancio, A., Di Domenico, M., de Falco,
A., Lombardi, M., Fiorentino, R., Varricchio, L., Barone, M. V., and
Auricchio, F. (2001) PI3-kinase in concert with Src promotes the S-
phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 20, 6050–
6059.
19. Deroo, B. J., and Korach, K. S. (2006) Estrogen receptors and human
disease. J. Clin. Invest. 116, 561–570.
20. Matthews, J., and Gustafsson, J.-A. (2003) Estrogen signalling: a subtle
balance between ERa and ERb. Mol. Interv. 3, 281–292.
21. Koehler, K. F., Helguero, L. A., Haldosen, L. A., Warner, M., and Gus-
tafsson, J.-A. (2005) Reflections on the discovery and significance of
estrogen receptor b. Endocr. Rev. 26, 465–478.22. Korach, K. S., Emmen, J. M., Walker, V. R., Hewitt, S. C., Yates, M.,
Hall, J. M., Swope, D. L., Harrell, J. C., and Couse, J. F. (2003) Update
on animal models developed for analyses of estrogen receptor biological
activity. J. Steroid. Biochem. Mol. Biol. 86, 387–391.23. Strom, A., Hartman, J., Foster, J. S., Kietz, S., Wimalasena, J., and Gus-
tafsson, J.-A. (2004) Estrogen receptor b inhibits 17b-estradiol-stimu-
lated proliferation of the breast cancer cell line T47D. Proc. Natl. Acad.Sci. USA 101, 1566–1571.
24. Dixon, R. A. (2004) Phytoestrogens. Ann. Rev. Plant Biol. 55, 225–261.
25. Konstantakopoulos, N., Montgomery, K. G., Chamberlain, N., Quinn,
M. A., Baker, M. S., Rice, G. E., Georgiou, H. M., and Campbell, I. G.
(2006). Changes in gene expressions elicited by physiological concen-
trations of genistein on human endometrial cancer cells. Mol. Carcinog.
45, 752–763.
26. Martınez, C., Vicente, V., Yanez, J., Alcaraz, M., Castells, M. T., Can-
teras, M., Benavente-Garcıa, O., and Castillo, J. (2005) The effect of
the flavonoid diosmin, grape seed extract and red wine on the pulmo-
nary metastatic B16F10 melanoma. Histol. Histopathol. 20, 1121–1129.
27. McCarty, M. F., Barroso-Aranda, J., and Contreras, F. (2009) Genistein and
phycocyanobilin may prevent hepatic fibrosis by suppressing proliferation
and activation of hepatic stellate cells.Med. Hypotheses 72, 330–332.
28. McCarty, M. F. (2006) Isoflavones made simple – genistein’s agonist
activity for the b-type estrogen receptor mediates their health benefits.
Med. Hypotheses 66, 1093–114
29. Messina, M. and Wu, A. H. (2009) Perspectives on the soy-breast can-
cer relation. Am. J. Clin. Nutr. 89, 1673S–1679S.30. Rice, S. and Whitehead, S. A. (2006). Phytoestrogens and breast can-
cer-promoters or protectors? Endocr. Relat. Cancer 13, 995–1015.
31. Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Haggblad, J.,
Nilsson, S., and Gustafsson, J.-A. (1997) Comparison of the ligand
binding specificity and transcript tissue distribution of estrogen receptors
a and b. Endocrinology 138, 863–870.
32. Kuiper, G. G., Lemmen, J. G., Carlsson, B., Corton, J. C., Safe, S. H.,
van der Saag, P. T., van der Burg, B., and Gustafsson, J.-A. (1998)
Interaction of estrogenic chemicals and phytoestrogens with estrogen re-
ceptor b. Endocrinology 139, 4252–4263.33. Ruh, M. F., Zacharewski, T., Connor, K., Howell, J., Chen, I., and Safe,
S. H. (1995) Naringenin: a weakly estrogenic bioflavonoid that exhibits
antiestrogenic activity. Biochem. Pharmacol. 26, 1485–1493.34. Safe, S. H. and Kim, K. (2008) Non-classical genomic estrogen receptor
(ER)/specificity protein and ER/activating protein-1 signaling pathways.
J. Mol. Endocrinol. 41, 263–275.35. Harris, D. M., Besselink, E., Henning, S. M., Go, V. L. W., and Heber,
D. (2005) Phytoestrogens induce differential estrogen receptor a- or b-mediated responses in transfected breast cancer cells. Exp. Biol. Med.230, 558–568.
59E2 AND NARINGENIN ANTAGONIST EFFECTS ON PROLIFERATION

36. Farach-Carson, M. C. and Davis, P. J. (2003) Steroid hormone interac-
tions with target cells: cross talk between membrane and nuclear path-
ways. J. Pharmacol. Exper. Therap. 30, 839–845.
37. Kousteni, S., Chen, J. R., Bellido, T., Han, L., Ali, A. A., O’Brien,
C. A., Plotkin, L., Fu, Q., Mancino, A. T., Wen, Y., Vertino, A. M.,
Powers, C. C., Stewart, S. A., Ebert, R., Parfitt, A. M., Weinstein,
R. S., Jilka, R. L., and Manolagas, S. C. (2002) Reversal of bone
loss in mice by nongenotropic signaling of sex steroids. Science 298,
843–846.
38. Chambliss, K. L., Simon, L., Yuhanna, I. S., Mineo, C., and Shaul, P.
W. (2005) Dissecting the basis of nongenomic activation of eNOS by
estradiol: role of ERa domains with known nuclear functions. Mol.Endocrinol. 19, 277–289.
39. Sarkar, F. H., and Li, Y. (2002) Mechanisms of cancer chemoprevention
by soy isoflavone genistein. Cancer Metastasis Rev. 21, 265–280.
40. Magee, P. J. and Rowland, I. R. (2004) Phyto-oestrogens, their mecha-
nism of action: current evidence for a role in breast and prostate cancer.
Br. J. Nutr. 91, 513–531.
41. Erlund, I., Meririnne, E., Alfthan, G., and Aro, A. (2001) Plasma
kinetics and urinary excretion of the flavanones naringenin and hespere-
tin in humans after ingestion of orange juice and grapefruit juice. J.
Nutr. 131, 235–241.
42. Harmon, A. W. and Patel, Y. M. (2004) Naringenin inhibits glucose
uptake in MCF-7 breast cancer cells: a mechanism for impaired cellular
proliferation. Breast Cancer Res. Treat. 85, 103–110.
43. Katzenellenbogen, J. A. and Muthyala, R. S. (2003) Interactions of ex-
ogenous endocrine active substances with nuclear receptors. Pure Appl.Chem. 75, 1797–1817.
44. Whitten, P. L., Lewis, C., Russell, E., and Naftolin, F. (1995) Potential
adverse effects of phytoestrogens. J. Nutr. 125, 771S–776S.
60 BULZOMI ET AL.