molecular model and docking of brugia malayi
TRANSCRIPT

59
MAIN
©1996-2012 All Rights Reserved. Online Journal of Bioinformatics . You may not store these pages in any form except for your own personal use. All other usage or distribution is illegal under international copyright treaties. Permission to use any of these pages in any other way besides the before mentioned must be gained in writing from the publisher. This article is exclusively copyrighted in its entirety to OJB publications. This article may be copied once but may not be, reproduced or re-transmitted without the express permission of the editors. This journal satisfies the refereeing requirements (DEST) for the Higher Education Research Data Collection (Australia). Linking:To link to this page or any pages linking to this page you must link directly to this page only here rather than put up your own page.
OJBTM
Online Journal of Bioinformatics ©
Volume 13 (1):59-79 2012
Molecular model and docking of Brugia malayi glutathione reductase
Sudhanshu Shekhar Yadav1*, Vinay Kumar Singh2, Eva Liebau3, Sushma Rathaur1
1Department of Biochemistry, 2Centre for Bioinformatics, Faculty of Science, Banaras Hindu University, Varanasi 221005, U.P., India 3Westfalische Wilhelms-Uinversitat, Institute of Animal Physiology, Department of Molecular Physiology, Hindenburgplatz-55, Muenster, Germany
ABSTRACT
Yadav SS, Singh VK, Liebau E, Rathaur S., Molecular model and docking of Brugia
malayi glutathione reductase, Online J Bioinform, 13 (1):59-79, 2012. Brugia malayi Glutathione Reductase (BmGR), a redox enzyme that reduces glutathione disulfide (GSSG) to the sulfhydryl form GSH, plays an important role in filariasis and other diseases. BmGR was modeled using a 3DK9 template with 324 hydrogen Bonds, 18 helices, 32 strands and 47 turns and accepted at PMDB database (PM0077742). The model was used to dock and simulate the ant filarial drugs diethylcarbamazine citrate and albendazole, specific inhibitors of GR; 3,4 Dihydroxybenzyleamine and 1,3-bis(2-chloroethyl)-1-nitrosourea and substituted chalcones (SK series compounds). The active site analysis alignment of HsGR revealed ten stretches as active binding sites in BmGR. The amino acid residues SER19, GLU39, GLU40, THR41, THR46, TYR146, ASP162, ARG267, ARG272, ASP307 and THR315 were involved in hydrogen bonding, hydrophobic, polar, cation-pi and other interactions with NADPH. The binding of GSSG with protein especially with amino acids ALA142 and VAL143 and overlapped with NADPH binding site except SER19, ARG267, ARG272, ASP307 and THR315. In-silico investigation revealed that interaction of DEC with amino acids TYR181, ILE182 & ASP307 and SER19, THR46, ALA142, VAL143 & ALA318 interacting with ALB and specific inhibitors; DHBA showing interaction with amino acids GLU39, GLU40, THR41, THR46 & THR146 and SER19, GLY20 & THR46 with BCNU are involved in hydrogen, hydrophobic and polar level interactions. However, the interaction sites of antifilarials are similar to the binding sites of GSSG but positions are different and the binding sites of specific inhibitors are similar to the substrate i.e. active site

domain II of BmGR protein. While SK series compounds are interact with highest coordination of the major portion of the active binding site as well as NADPH binding site of the protein. Based on estimated free energy of binding, lowest inhibition constant (Ki) and lower frequency percentage ALB, BCNU and substituted chalcones were showing better interactions. Specific inhibitors are showing competitive type binding (stretch 1 & 2) with substrate while substituted chalcones non-competitive (stretch 1, 2, 3 & 6). However, Stretch 5 and 7 may be the activator sites of the protein, which are the binding sites of antifilarials. Amino acids SER19, CYS47, CYS52 and PHE165 may be better inhibitory sites for drug designing using BmGR as template. Among substituted chalcone SK-3 and SK-5 are showing lower estimated free energy of binding, lowest inhibition constant (Ki) and lower frequency percentage. These substituted chalcones may be good inhibitors of glutathione metabolism and have better antifilarial activity. The BmGR structural information and docking studies could aid in screening new antifilarials or selective inhibitors for chemotherapy against filariasis.
Key Words
DEC; Diethylcarbamazine citrate, ALB; Albendazole, DHBA; 3,4 Dihydroxybenzyleamine, BCNU; 1,3-bis(2-chloroethyl)-1-nitrosourea, Substituted chalcones, filariasis.
INTRODUCTION
Glutathione reductase (oxido-reductase) (GR), Glutathione peroxidase (GPx) and glutathione S-transferase (GST) play a major role in the cellular detoxification of oxidative damaging, and genotoxic chemicals. In parasites, their role can be elaborated by the fact that some of these organisms lack the cytochrome P450 system, a phase I detoxification enzyme (Precious and Barrett, 1989). Glutathione metabolism is the major phase II detoxification system in parasites and because of their detoxifying nature (Sheehan et al. 2001) and maintenance of cellular redox status (Lefeuvre et al., 2004), they are widely used in drug inhibition studies and have become plausible drug targets in various helminth diseases (Brophy and Barrett, 1999; Mannervik and Danielson, 1998; Ibarraa et al. 2003). The flavoenzyme glutathione reductase (GR) is a dimeric disulfide oxidoreductase that converts oxidized glutathione (GSSG) to two molecules of reduced glutathione (GSH) using an NADPH cofactor and an FAD prosthetic group. Glutathione plays a critical role in maintaining the cell’s reducing environment and defense against oxidative stress that appears to be associated with several neurodegenerative diseases (Berkholz et al. 2008; Halliwell, 1992; Bowling and Beal, 1995; Bains and Shaw, 1997). Human erythrocyte GR is a homodimer of 52 kD monomers, each with three domains: an NADPH-binding domain, an FAD-binding domain, and a dimerization domain. The NADPH and FAD-binding domains meet at the active site, in which both monomers participate (Karplus and Schulz, 1987; Karplus and Schulz, 1989; Sustmann et al. 1989). In our earlier reports we have shown the induction of filarial GST from Setaria cervi

by diethylcarbamazine (DEC, known antifilarials) (Gupta and Rathaur, 2005), whereas, our recent communication establishes the inhibition of S. cervi GST (ScGST) & GR (ScGR: data unpublished) activity by substituted chalcones, proposed them as an antifilarials (Awasthi et al. 2009). These studies were undertaken in vitro to observe the effect of afore-mentioned compounds on parasite survival. In order to understand the interaction of antifilarials/ compounds with nematode GR at atomic level, we have performed homology modeling of BmGR followed by its docking studies. The information of predicted binding map of BmGR from this study would aid in structural analysis, screening and designing new antifilarials.
MATERIALS AND METHODS
Protein comparative modeling
Glutathione reductase from Brugia malayi with Accession Number XP_001892443.1 was retrieved from NCBI Database (http://www.ncbi.nlm.nih.gov/). Protein sequence of Glutathione reductase was taken for homology modeling using Swiss-Model server (http://swissmodel.expasy.org/).
Structure refinement
Ramachandran Plot statistics to check the stereochemical property and G-factor were calculated using PDBSum database (http://www.ebi.ac.uk/pdbsum/). The structure was evaluated by ANOLEA force field, GROMOS96 and ERRAT (http://nihserver.mbi.ucla.edu/ERRATv2/). Refined and validated structure was submitted to protein model database [PMDB] (http://mi.caspur.it/PMDB/).
Ligand Designing and Retrieval
Reported inhibitory Drugs/ compounds were retrieved from PubChem Compound Database (http://www.ncbi.nlm.nih.gov/pccompound) and other inhibitors were designed using ChemSketch and geometry optimization were done using Discovery studio 3.1/Docking server (www.dockingserver.com). Docking server was used to calculate molecular weight, molecular formula, molar weight and MMFF94 energy calculation of all retrieved and designed inhibitors.
Active site identification and molecular docking
Active site of modelled protein glutathione reductase was analysed using Q-site finder (www.modelling.leeds.ac.uk/qsitefinder/). Docking simulation was performed using the LGA and Solis and Wets Local server method using docking server. Superimposition and evaluation
Discovery studio 3.1 was used for Pose validation and evaluation. 3-D Protein structure comparison and alignment were done using the combinatorial extension (CE) method (http://cl.sdsc.edu/). ClustalW (www.ebi.ac.uk/Tools/msa/clustalw2/) was used for sequence alignment and alignment was visualised by seaview tool (http://pbil.univ-lyon1.fr/software/seaview.html).
RESULTS AND DISCUSSION
Homology modeling, structural validation and refinement
Based on the input BmGR protein in Swiss model server, 3dk9 was selected as a template with sequence identity 56.93% and E-value: 0.00e-1. Quality QMEAN Z-Score was 0.05. The QMEAN4 score is a composite score consisting of a linear combination of 4 statistical potential terms (estimated model reliability between 0-1). The pseudo-energies of the contributing terms are given below together with their Z-scores with respect to the scores obtained for high-resolution experimental structures of similar size solved by X-ray crystallography (Table 1).
Table 1: QMEAN4 global scores
Scoring function term Raw score Z-score
C-beta interaction energy -166.73 0.29
All-atom pairwise energy -10224.40 -0.31
Solvation energy -43.62 0.16
Torsion angle energy -126.40 0.09
QMEAN4 score 0.763 0.05
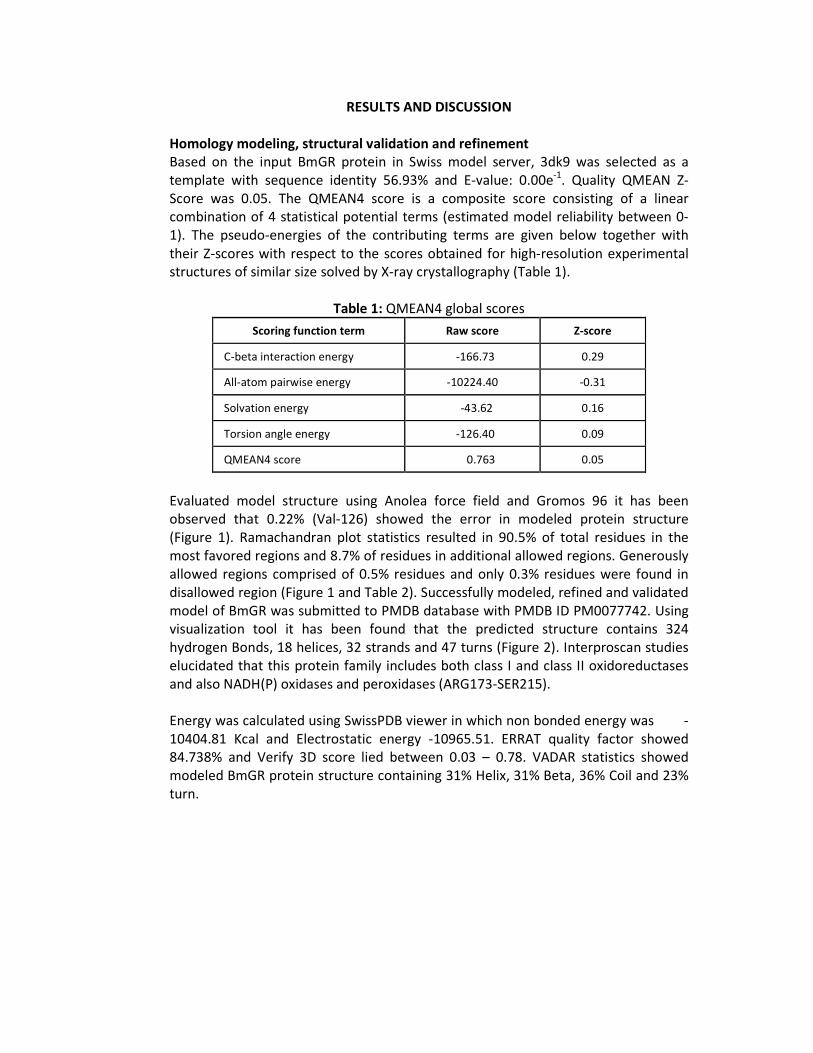
Evaluated model structure using Anolea force field and Gromos 96 it has been observed that 0.22% (Val-126) showed the error in modeled protein structure (Figure 1). Ramachandran plot statistics resulted in 90.5% of total residues in the most favored regions and 8.7% of residues in additional allowed regions. Generously allowed regions comprised of 0.5% residues and only 0.3% residues were found in disallowed region (Figure 1 and Table 2). Successfully modeled, refined and validated model of BmGR was submitted to PMDB database with PMDB ID PM0077742. Using visualization tool it has been found that the predicted structure contains 324 hydrogen Bonds, 18 helices, 32 strands and 47 turns (Figure 2). Interproscan studies elucidated that this protein family includes both class I and class II oxidoreductases and also NADH(P) oxidases and peroxidases (ARG173-SER215).
Energy was calculated using SwissPDB viewer in which non bonded energy was -10404.81 Kcal and Electrostatic energy -10965.51. ERRAT quality factor showed 84.738% and Verify 3D score lied between 0.03 – 0.78. VADAR statistics showed modeled BmGR protein structure containing 31% Helix, 31% Beta, 36% Coil and 23% turn.
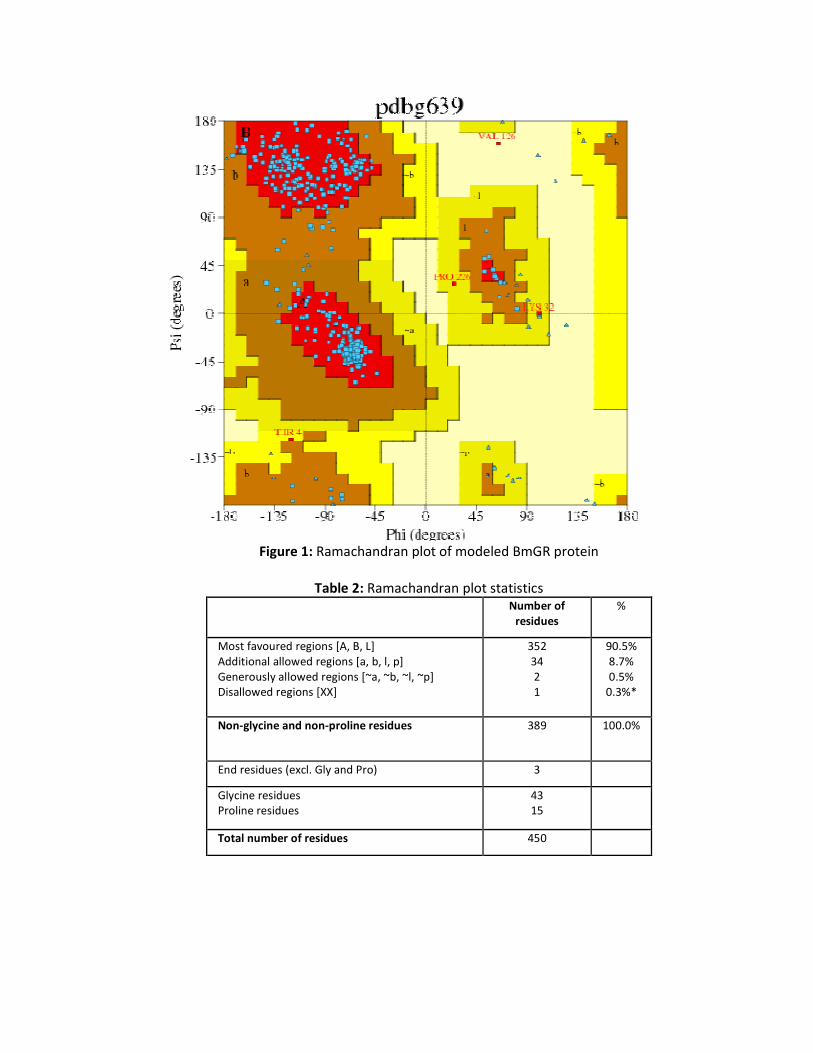
Figure 1: Ramachandran plot of modeled BmGR protein
Table 2: Ramachandran plot statistics Number of
residues %
Most favoured regions [A, B, L] Additional allowed regions [a, b, l, p] Generously allowed regions [~a, ~b, ~l, ~p] Disallowed regions [XX]
352 34 2 1
90.5% 8.7% 0.5%
0.3%*
Non-glycine and non-proline residues 389 100.0%
End residues (excl. Gly and Pro) 3
Glycine residues Proline residues
43 15
Total number of residues 450

Table 3: Calculation of G factor Parameter Score Average score
Dihedral angles:- Phi-psi distribution Chi1-chi2 distribution Chi1 only Chi3 & chi4 Omega
-0.15 0.16 0.04 0.47 -0.31
-0.06
Main-chain covalent forces:- Main-chain bond lengths Main-chain bond angles
0.45 0.20
0.31
OVERALL AVERAGE 0.09
G-factors provide a measure of how unusual, or out-of-the-ordinary, a property is. Values below -0.5* - unusual; Values below -1.0** - highly unusual
Figure 2: Schematic representation of secondary structure of BmGR, The α-helix has been represented by red, β-sheet by cyan and loops by green
Insilico characterization and functional identification
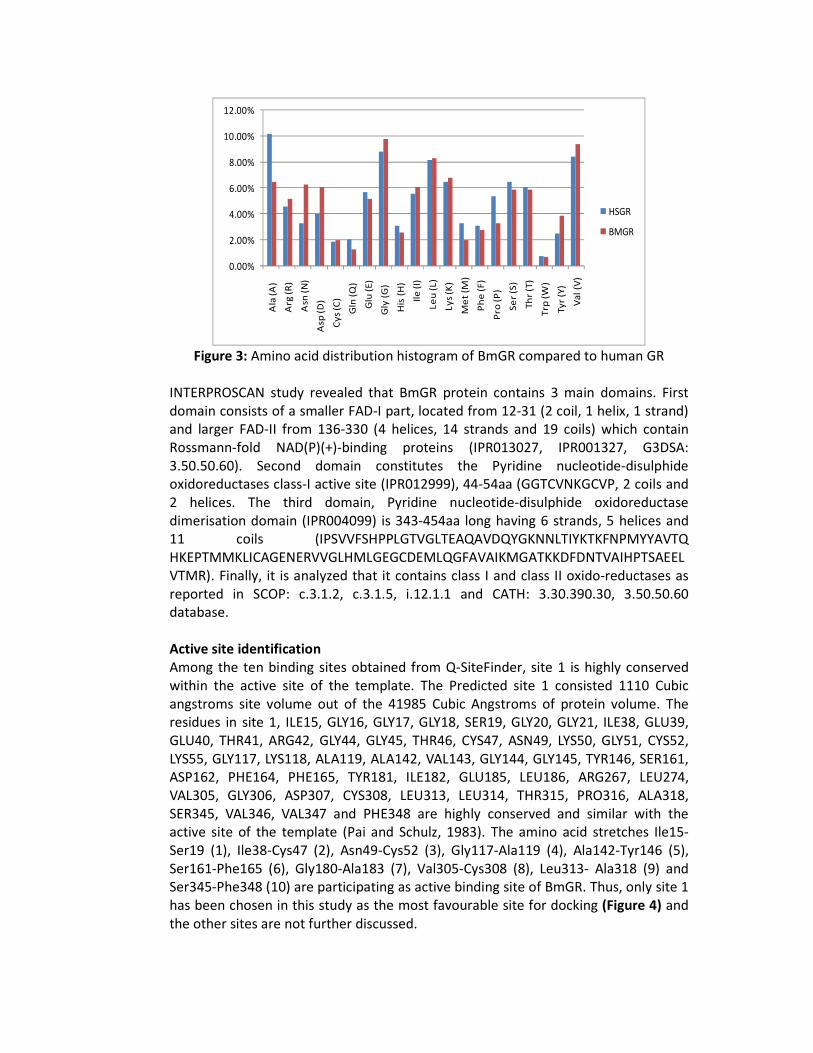
Comparative physico-chemical properties of both HsGR and BmGR showed that HsGR residues, Ala (10.20%), Gln (2.10%), Glu (5.70%), His (3.10%), Met (3.30%), Phe (3.10%), Pro (5.40%), Ser (6.50%) and Thr (6.10%) are present in higher number than BmGR. In case of BmGR, Arg (5.20%), Asn (6.30%), Asp (6.10%), Gly (9.8%), Tyr (3.90%) and Val (9.40%) were higher in comparison to HsGR (Figure 3).
0.00%
2.00%
4.00%
6.00%
8.00%
10.00%
12.00%
Ala
(A
)
Arg
(R
)
Asn
(N
)
Asp
(D)
Cy
s (C
)
Gln
(Q
)
Glu
(E
)
Gly
(G
)
His
(H
)
Ile
(I)
Le
u (
L)
Ly
s (K
)
Me
t (M
)
Ph
e (
F)
Pro
(P
)
Se
r (S
)
Th
r (T
)
Trp
(W
)
Tyr
(Y)
Va
l (V
)
HSGR
BMGR
Figure 3: Amino acid distribution histogram of BmGR compared to human GR
INTERPROSCAN study revealed that BmGR protein contains 3 main domains. First domain consists of a smaller FAD-I part, located from 12-31 (2 coil, 1 helix, 1 strand) and larger FAD-II from 136-330 (4 helices, 14 strands and 19 coils) which contain Rossmann-fold NAD(P)(+)-binding proteins (IPR013027, IPR001327, G3DSA: 3.50.50.60). Second domain constitutes the Pyridine nucleotide-disulphide oxidoreductases class-I active site (IPR012999), 44-54aa (GGTCVNKGCVP, 2 coils and 2 helices. The third domain, Pyridine nucleotide-disulphide oxidoreductase dimerisation domain (IPR004099) is 343-454aa long having 6 strands, 5 helices and 11 coils (IPSVVFSHPPLGTVGLTEAQAVDQYGKNNLTIYKTKFNPMYYAVTQ HKEPTMMKLICAGENERVVGLHMLGEGCDEMLQGFAVAIKMGATKKDFDNTVAIHPTSAEELVTMR). Finally, it is analyzed that it contains class I and class II oxido-reductases as reported in SCOP: c.3.1.2, c.3.1.5, i.12.1.1 and CATH: 3.30.390.30, 3.50.50.60 database. Active site identification
Among the ten binding sites obtained from Q-SiteFinder, site 1 is highly conserved within the active site of the template. The Predicted site 1 consisted 1110 Cubic angstroms site volume out of the 41985 Cubic Angstroms of protein volume. The residues in site 1, ILE15, GLY16, GLY17, GLY18, SER19, GLY20, GLY21, ILE38, GLU39, GLU40, THR41, ARG42, GLY44, GLY45, THR46, CYS47, ASN49, LYS50, GLY51, CYS52, LYS55, GLY117, LYS118, ALA119, ALA142, VAL143, GLY144, GLY145, TYR146, SER161, ASP162, PHE164, PHE165, TYR181, ILE182, GLU185, LEU186, ARG267, LEU274, VAL305, GLY306, ASP307, CYS308, LEU313, LEU314, THR315, PRO316, ALA318, SER345, VAL346, VAL347 and PHE348 are highly conserved and similar with the active site of the template (Pai and Schulz, 1983). The amino acid stretches Ile15-Ser19 (1), Ile38-Cys47 (2), Asn49-Cys52 (3), Gly117-Ala119 (4), Ala142-Tyr146 (5), Ser161-Phe165 (6), Gly180-Ala183 (7), Val305-Cys308 (8), Leu313- Ala318 (9) and Ser345-Phe348 (10) are participating as active binding site of BmGR. Thus, only site 1 has been chosen in this study as the most favourable site for docking (Figure 4) and the other sites are not further discussed.

Substrate binding site of template (3dk9) with FAD Substrate binding gives opportunity to check the specificity of binding residues in the modeled protein. Based on the interaction of 3dk9 with FAD complex, GLY27, GLY29, SER30, GLY31, GLU50, SER51, LYS53, GLY56, THR57, CYS58, GLY62, CYS63, LYS66, GLY128, HIS129, ALA130, ALA155, THR156, GLY157, TYR197, ARG291, ASN294, LEU298, GLY330, ASP331, LEU337, LEU338, THR339, PRO340, HIS467 and PRO468 residues were found on active binding site (Karplus et al., 1988; Savvides and Karplus, 1996; Gallwitz et al., 1999).
In case of BmGR, ILE15, GLY16, GLY17, GLY18, SER19, GLY20, GLY21, ILE38, GLU39, GLU40, THR41, ARG42, GLY44, GLY45, THR46, CYS47, ASN49, LYS50, GLY51, CYS52, LYS55, GLY117, LYS118, ALA119, ALA142, VAL143, GLY144, GLY145, TYR146, SER161, ASP162, PHE164, PHE165, TYR181, ILE182, GLU185, LEU186, ARG267, LEU274, VAL305, GLY306, ASP307, CYS308, LEU313, LEU314, THR315, PRO316, ALA318, SER345, VAL346, VAL347 and PHE348 residues were found to be potential active binding sites for FAD (Figure 4).
Figure 4: Predicted active sites determination in BmGR protein using Q-SiteFinder After structural alignment of BmGR with HsGR, it has been found that the amino acids GLY16, GLY18, SER19, GLY20, GLU39, GLU40, ARG42, GLY45, THR46, CYS47, GLY51, CYS52, LYS55, GLY117, LYS118, SER120, ALA142, VAL143, GLY144, TYR181, ARG267, ALA270, LEU276, GLY306, ASP307, LEU313, LEU314, THR315 and PRO316 of BmGR were similar to the active site amino acids of HsGR but may be located at different positions. However, some substitutions were found in BmGR at positions GLU39, ARG42, LYS118 and ALA270 in comparison to HsGR at positions SER51, LYS53, HIS129 and ASN294 respectively (Figure 5).
Structural alignment of target and template

The structural superimposition of Cα trace of the template (3DK9) and predicted structure of BmGR was performed using combinatorial extension of polypeptides (http://cl.sdsc.edu/). Structural alignment between target (BmGR) and template (3DK9) shows 0.2Å Root mean square deviation with 8.2 Z-Score having 58.1% sequence identity (Figure 6).
Figure 5: Alignment of BmGR with HsGR. (Red blocks showing conserved amino acids which are involved in the activity of the enzyme)
Figure 6: Superimposition of BmGR protein structure with template 3DK9. (Red circle showing dissimilarities between BmGR and 3DK9)

Ligands retrieval and preparation
The known ligands were successfully retrieved from Pubchem, reported synthesized substituted chalcones (SK series compounds) were designed using ChemSketch from ACDLAB12.0 and optimized using Discovery studio 3.1, Protein health module. All optimized compounds were submitted to docking server for the calculation of molecular weight, molecular formula, molar weight and MMFF94 energy as mentioned in Table 4 (Figure 7).
ClN
O
Cl
N
N
O
Cl
O
N OMe
O
N
OMe
O
N
N
N
N OMe
O
N
O
OMe
O
N
1 2
3 4
5
6
7
3-(4-chlorophenyl)-1-(4-pyrrole-1-yl-phenyl)prop-2-en-1-one 3-(4-chlorophenyl)-1-(4-pyrazole-1-yl-phenyl)prop-2-en-1-one
3-(4-chlorophenyl)-1-(4-piperidin-1-yl-phenyl)prop-2-en-1-one 3-(4-methoxyphenyl)-1- (4-pyrazole-1-yl-phenyl)prop-2-en-1-one
1-(4-benzotriazol-1-yl-phenyl)-3-(4-methoxyphenyl)prop-2-en-1-one
1-(4-morpholine-1-yl-phenyl)-3-(4-methoxyphenyl)prop-2-en-1-one
3-(4-methoxyphenyl)-1-(4-pyrrolidin-1-yl-phenyl)prop-2-en-1-one
Figure 7: Structure of SK series compounds (substituted chalcone): Chloro substituted; SK-1, 2, & 3 and methoxy substituted; 4, 5, 6 & 7) designed by ChemSketch
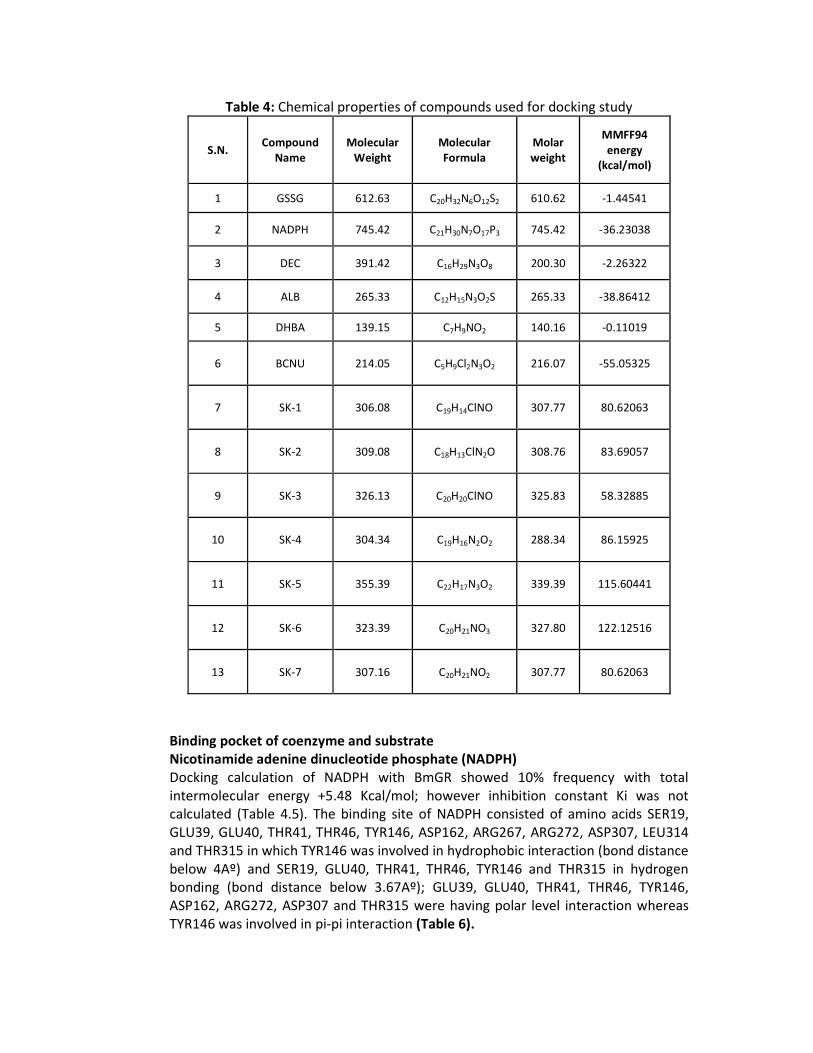
Table 4: Chemical properties of compounds used for docking study
S.N. Compound
Name Molecular
Weight Molecular
Formula Molar
weight
MMFF94
energy
(kcal/mol)
1 GSSG 612.63 C20H32N6O12S2 610.62 -1.44541
2 NADPH 745.42 C21H30N7O17P3 745.42 -36.23038
3 DEC 391.42 C16H29N3O8 200.30 -2.26322
4 ALB 265.33 C12H15N3O2S 265.33 -38.86412
5 DHBA 139.15 C7H9NO2 140.16 -0.11019
6 BCNU 214.05 C5H9Cl2N3O2 216.07 -55.05325
7 SK-1 306.08 C19H14ClNO 307.77 80.62063
8 SK-2 309.08 C18H13ClN2O 308.76 83.69057
9 SK-3 326.13 C20H20ClNO 325.83 58.32885
10 SK-4 304.34 C19H16N2O2 288.34 86.15925
11 SK-5 355.39 C22H17N3O2 339.39 115.60441
12 SK-6 323.39 C20H21NO3 327.80 122.12516
13 SK-7 307.16 C20H21NO2 307.77 80.62063
Binding pocket of coenzyme and substrate
Nicotinamide adenine dinucleotide phosphate (NADPH)
Docking calculation of NADPH with BmGR showed 10% frequency with total intermolecular energy +5.48 Kcal/mol; however inhibition constant Ki was not calculated (Table 4.5). The binding site of NADPH consisted of amino acids SER19, GLU39, GLU40, THR41, THR46, TYR146, ASP162, ARG267, ARG272, ASP307, LEU314 and THR315 in which TYR146 was involved in hydrophobic interaction (bond distance below 4Aº) and SER19, GLU40, THR41, THR46, TYR146 and THR315 in hydrogen bonding (bond distance below 3.67Aº); GLU39, GLU40, THR41, THR46, TYR146, ASP162, ARG272, ASP307 and THR315 were having polar level interaction whereas TYR146 was involved in pi-pi interaction (Table 6).
Oxidized Glutathione (GSSG)
Docking calculation of GSSG with BmGR also showed 10% frequency with total intermolecular energy -8.88 Kcal/mol and calculated inhibition constant (Ki) 28.2 mM (Table 4.5). The binding site of GSSG consisted of amino acids GLU39, GLU40, THR41, THR46, ALA142, VAL143, TYR146 and ASP162 in which VAL143 and TYR146 were involved in hydrophobic interaction (bond distance below 4Aº) and GLU39, GLU40, THR41, THR46, ALA142 & TYR146 in hydrogen bonding (bond distance below 3.67Aº); GLU39, GLU40, THR41, THR46, TYR146 and ASP162 as in NADPH have polar level interaction and TYR146 was involved in cation-pi interaction (Table 6).
Molecular Docking
A total of 13 compounds, in which two known anti-filarial drugs (DEC & ALB), two specific inhibitors (BCNU and DHBA) and seven SK series compounds (SK-1, SK-2, SK-3, SK-4, SK-5, SK-6 & SK-7) are used for docking calculation (Tomar et al., 2010). These calculations are based on Est. Free Energy of Binding, Est. Inhibition Constant, vdW + H bond + desolv Energy, Electrostatic Energy with Brugia malayi Glutathione Reductase (BmGR). Better interaction among compounds/drugs-protein is based on the docking intermolecular energy. Substrates (GSSG and NADPH) and specific inhibitors of GR (BCNU and DHBA) are also used for the identification of interacting binding sites in BmGR model to evaluate the comparative binding sites for drugs/ compounds (Table 5). It has been observed that ALB, BCNU, SK-5 and SK-3 showed lowest inhibition constant (Ki) and minimum vdW + Hbond + desolv energy with lowest electrostatic energies among drugs, inhibitors and substituted chalcones. Table 5: Docking calculation of compounds with BmGR
S.N.
Compound
Name Est. Free
Energy of
Binding
(kcal/mol)
Est. Inhibiton
Constant, Ki VdW + Hbond
+ desolv
Energy
(kcal/mol)
Electrostatic
Energy
(kcal/mol)
Total
Intermolec.
Energy
(kcal/mol)
Frequency
1 GSSG -2.11 28.20 mM -8.37 -0.51 -8.88 10
2 NADPH +13.96 - +4.61 +0.87 +5.48 10
3 DEC -4.84 281.79 µM -4.56 -0.94 -5.50 60
4 ALB -6.71 12.15 µM -7.74 -0.10 -7.85 50
5 DHBA -5.12 176.38 µM -4.34 -1.59 -5.93 100
6 BCNU -5.50 92.58 µM -6.99 +0.09 -6.90 20
7 SK-1 -9.30 152.45 nM -10.39 -0.07 -10.47 10
8 SK-2 -8.89 306.45 nM -10.03 -0.03 -10.06 30
9 SK-3 -10.13 37.82 nM -11.23 -0.05 -11.28 10
10 SK-4 -8.19 987.21 nM -9.38 +0.01 -9.37 10
11 SK-5 -10.30 28.18 nM -11.66 -0.03 -11.69 10
12 SK-6 -9.08 220.41 nM -10.24 -0.01 -10.26 10
13 SK-7 -8.41 688.90 nM -9.56 -0.00 -9.56 30

Binding pocket of Antifilarials
Diethylcarbamazine citrate (DEC)
Docking calculation of DEC with BmGR showed 60% frequency with total intermolecular energy -5.50 Kcal/mol and calculated inhibition constant (Ki) 282µM (Table 5). The binding site of DEC consisted of amino acids THR46, CYS52, LYS55, TYR181, ILE182, ASP307 and THR315 in which TYR181 & ILE182 are involved in hydrophobic interaction (bond distance below 4Aº) and TYR181 and ASP307 in hydrogen bonding (bond distance below 3.5Aº) whereas SER19, GLU39, GLU40 and THR46 are involved in other interaction (Table 6 & Figure 8A). DEC mainly involved in interaction with stretches 7 and 8 of BmGR protein. It has been observed that during the interaction of DEC with protein, only three amino acid residues are involved in coordination, which reflects that DEC compound is having partial interaction with lowest affinity.
Albendazole (ALB)
Docking calculation of ALB with BmGR showed 50% frequency with total intermolecular energy -7.85 Kcal/mol and calculated inhibition constant (Ki) 12.15µM (Table 5). The binding site of ALB consisted of amino acids SER19, GLU39, GLU40, THR46, ALA142, VAL143, ASP307, THR315 and ALA318 in which VAL143 and ALA318 are involved in hydrophobic interaction (bond distance below 3.45Aº). SER19 and THR46 are involved in polar level interaction (bond distance below 3.56Aº) and ALA142 involved in hydrogen bonding (bond distance below 2.94Aº). Whereas, SER19, GLU39, GLU40 and THR46 are involved in other interaction (Table 6 & Figure 8B). ALB mainly involved in interaction with stretches 1, 2, 5 and 9 of BmGR protein.
Binding pocket of Specific inhibitors
1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU)
Docking calculation of BCNU with BmGR showed 20% frequency with total intermolecular energy -6.90 Kcal/mol and calculated inhibition constant (Ki) 92.58 µM (Table 4.5). The binding site of DHBA consisted of amino acids SER19, GLY20, THR46, ALA142, VAL143, ASP307, THR315 and ALA318 in which SER19 was involved in polar level interaction, GLY20 and THR46 in hydrogen bonding and SER19, ALA142, VAL143 and ASP307 in halogen bonding. SER19 and THR46 were involved in other interaction (Figure 8C). BCNU mainly involved in interaction with stretches 1, 2, 5 and 8 of BmGR protein.
3, 4 Dihydroxybenzyleamine (DHBA) Docking calculation of DHBA with BmGR showed 100% frequency with total intermolecular energy -5.93 Kcal/mol and calculated inhibition constant (Ki) 176.38µM (Table 5). The binding site of DHBA consisted of amino acids GLU39, GLU40, THR41, THR46 and THR146 in which GLU39, GLU40, THR41, THR46 and TYR146 are involved in polar level interaction and GLU39, THR41 and THR46 in hydrogen bonding. GLU39, GLU40, THR41 & THR46 are involved in other interaction (Table 6 & Figure 8D). DHBA mainly involved in interaction with stretches 2 and 3 of BmGR protein.

Binding pocket of substituted chalcones (SK series compounds) SK-1 (3-(4-chlorophenyl)-1-(4-pyrrole-1-yl-phenyl) prop-2-en-1-one)
Docking calculation of SK-1 with BmGR showed 10% frequency with total intermolecular energy -10.47 Kcal/mol and calculated inhibition constant (Ki) 152.45nM (Table 5). The binding site of SK-1 consisted of amino acids THR46, CYS52, LYS55, PHE165, TYR181, ILE182, GLU185, LEU186, ASP307, THR315 and ALA318 in which CYS52, ILE182, LEU186 and ALA318 are involved in hydrophobic interaction and PHE165 in pi-pi interaction. THR46, CYS52, LYS55, TYR181, GLU185, ASP307 and THR315 are involved in other interaction (Table 6 & Figure 9A). SK-1 mainly involved in interaction with stretches 2, 3, 6, 7 and 9 of BmGR protein.
C-BCNU
A-DECB-ALB
D-DHBA
Figure 8: Binding pocket of Antifilarial drugs and specific inhibitors with BmGR
SK-2 (3-(4-chlorophenyl)-1-(4-pyrazole-1-yl-phenyl) prop-2-en-1-one)
Docking calculation of SK-2 with BmGR showed 30% frequency with total intermolecular energy -10.06 Kcal/mol and calculated inhibition constant (Ki) 306.45nM (Table 5). The binding site of SK-2 consists of amino acids SER19, THR46, LYS55, SER161, PHE165, TYR181, GLU185, LEU186, ASP307 and THR315 in which SER19 and TYR181 are involved in polar level interaction. SER19, THR46, LYS55, SER161, TYR181, GLU185, LEU186, ASP307 & THR315 are involved in other interaction (Table 6 & Figure 9B). SK-2 mainly involved in interaction with stretches 1, 2, 3, 6, 7, 8 and 9 of BmGR protein.
SK-3 (3-(4-chlorophenyl)-1-(4-piperidin-1-yl-phenyl) prop-2-en-1-one)
Docking calculation of SK-3 with BmGR showed 10% frequency with total intermolecular energy -11.28 Kcal/mol and calculated inhibition constant (Ki) 37.82nM (Table 5). The binding site of SK-3 consists of amino acids SER19, THR46,

CYS52, LYS55, PHE165, TYR181, ILE182, GLU185, LEU186, ASP307, THR315 and ALA318 in which TYR181 is involved in pi-pi interaction, THR315 in hydrogen bonding and CYS52, PHE165, ILE182, LEU186 and ALA 318 are involved in hydrophobic interaction. SER19, THR46, CYS52, LYS55, TYR181, ILE182, GLU185, ASP307 and THR315 are involved in other interaction (Table 6 & Figure 9C). SK-3 mainly involved in interaction with stretches 3, 6, 7, 8 and 9 of BmGR protein. SK-4 (3-(4-methoxyphenyl)-1-(4-pyrazole-1-yl-phenyl) prop-2-en-1-one)
Docking calculation of SK-4 with BmGR showed 10% frequency with total intermolecular energy -9.37 Kcal/mol and calculated inhibition constant (Ki) 987.21nM (Table 5). The binding site of SK-4 consists of amino acids SER19, THR46, GLS52, LYS55, PHE165, TYR181, ILE182, GLU185, ASP307, LEU314 THR315 and ALA318 in which PHE165 is involved in pi-pi interaction, GLU185 is involved in polar level interaction and CYS52, ILE182 and ALA318 are involved in hydrophobic interaction. SER19, THR46, LYS55, TYR181, ILE182, ILE182, GLU185, ASP307, and THR315 are involved in other interaction (Table 6 & Figure10A). SK-4 mainly involved in interaction with stretches 3, 6, 7 and 9 of BmGR protein.
SK-5 (1-(4-benzotriazol-1-yl-phenyl)-3-(4-methoxyphenyl)prop-2-en-1-one)
Docking calculation of SK-5 with BmGR showed 10% frequency with total intermolecular energy -11.69 Kcal/mol and calculated inhibition constant (Ki) 28.8nM (Table 5). The binding site of SK-5 consists of amino acids SER19, THR46, CYS47, CYS52, LYS55, PHE165, TYR181, ASP307 and THR315 in which SER19 is involved in hydrogen bonding, SER19 and TYR181 in polar level interaction and CYS47, CYS52 and PHE165 are involved in hydrophobic interaction. SER19, THR46, LYS55, TYR181, ASP307, and THR315 are involved in other interaction (Table 6 & Figure 10B). SK-5 mainly involved in interaction with stretches 1, 2, 3 and 6 of BmGR protein.
A- SK-1 B- SK-2
C- SK-3
Figure 9: Binding pocket of chloro substituted chalcone (SK-1, SK-2 & SK-3) with BmGR

SK-6 (1-(4-morpholine-1-yl-phenyl)-3-(4-methoxyphenyl)prop-2-en-1-one)
Docking calculation of SK-6 with BmGR showed 10% frequency with total intermolecular energy -10.26 Kcal/mol and calculated inhibition constant (Ki) 220.41nM (Table 5). The binding site of SK-6 consists of amino acids SER19, CYS47, CYS52, LYS55, SER161, PHE165, TYR181, GLU185, ASP307, THR315 and ALA318 in which CYS47, CYS52 and ALA318 are involved in hydrophobic interaction and THR315 in polar level interaction. SER19, CYS52, LYS55, SER161, PHE165, TYR181, GLU185, ASP307 and THR315 are involved in other interaction (Table 6 & Figure 10C). SK-6 mainly involved in interaction with stretches 2, 3 and 9 of BmGR protein. SK-7 (3-(4-methoxyphenyl)-1-(4-pyrrolidin-1-yl-phenyl) prop-2-en-1-one)
Docking calculation of SK-7 with BmGR showed 30% frequency with total intermolecular energy -9.56 Kcal/mol and calculated inhibition constant (Ki) 688.90nM (Table 5). The binding site of SK-7 consists of amino acids SER19, GLY20, GLU39, GLU40, THR46, CYS52, TYR181, ASP307, THR315 and ALA318 in which CYS52 is involved in hydrophobic interaction and GLY20 and THR315 in hydrogen bonding. SER19, GLU39, GLU40, THR46, CYS52, TYR181, ASP307, THR315 and ALA318 are involved in other interaction (Table 6 & Figure 10D). SK-7 mainly involved in interaction with stretches 1, 3 and 9 of BmGR protein.
E- SK-5
F- SK-6G- SK-7
D- SK-4
Figure 10: Binding pocket of methoxy substituted chalcone (SK-4, SK-5, SK-6, & SK-7) with BmGR
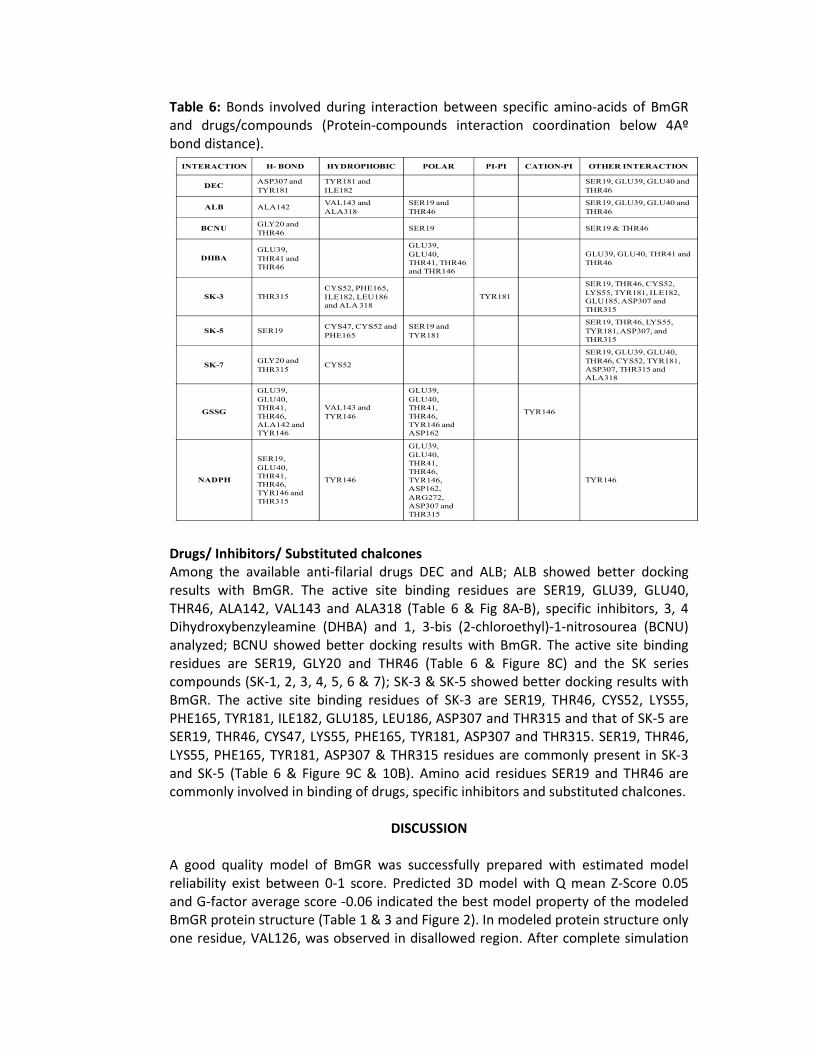
Table 6: Bonds involved during interaction between specific amino-acids of BmGR and drugs/compounds (Protein-compounds interaction coordination below 4Aº bond distance).
I&TERACTIO& H- BO&D HYDROPHOBIC POLAR PI-PI CATIO&-PI OTHER I&TERACTIO&
DECASP307 and
TYR181
TYR181 and
ILE182
SER19, GLU39, GLU40 and
THR46
ALB ALA142VAL143 and
ALA318
SER19 and
THR46
SER19, GLU39, GLU40 and
THR46
BC&UGLY20 and
THR46 SER19 SER19 & THR46
DHBA
GLU39,
THR41 and
THR46
GLU39,
GLU40,
THR41, THR46
and THR146
GLU39, GLU40, THR41 and
THR46
SK-3 THR315
CYS52, PHE165,
ILE182, LEU186
and ALA 318
TYR181
SER19, THR46, CYS52,
LYS55, TYR181, ILE182,
GLU185, ASP307 and
THR315
SK-5 SER19CYS47, CYS52 and
PHE165
SER19 and
TYR181
SER19, THR46, LYS55,
TYR181, ASP307, and
THR315
SK-7GLY20 and
THR315 CYS52
SER19, GLU39, GLU40,
THR46, CYS52, TYR181,
ASP307, THR315 and
ALA318
GSSG
GLU39,
GLU40,
THR41,
THR46,
ALA142 and
TYR146
VAL143 and
TYR146
GLU39,
GLU40,
THR41,
THR46,
TYR146 and
ASP162
TYR146
&ADPH
SER19,
GLU40,
THR41,
THR46,
TYR146 and
THR315
TYR146
GLU39,
GLU40,
THR41,
THR46,
TYR146,
ASP162,
ARG272,
ASP307 and
THR315
TYR146
Drugs/ Inhibitors/ Substituted chalcones
Among the available anti-filarial drugs DEC and ALB; ALB showed better docking results with BmGR. The active site binding residues are SER19, GLU39, GLU40, THR46, ALA142, VAL143 and ALA318 (Table 6 & Fig 8A-B), specific inhibitors, 3, 4 Dihydroxybenzyleamine (DHBA) and 1, 3-bis (2-chloroethyl)-1-nitrosourea (BCNU) analyzed; BCNU showed better docking results with BmGR. The active site binding residues are SER19, GLY20 and THR46 (Table 6 & Figure 8C) and the SK series compounds (SK-1, 2, 3, 4, 5, 6 & 7); SK-3 & SK-5 showed better docking results with BmGR. The active site binding residues of SK-3 are SER19, THR46, CYS52, LYS55, PHE165, TYR181, ILE182, GLU185, LEU186, ASP307 and THR315 and that of SK-5 are SER19, THR46, CYS47, LYS55, PHE165, TYR181, ASP307 and THR315. SER19, THR46, LYS55, PHE165, TYR181, ASP307 & THR315 residues are commonly present in SK-3 and SK-5 (Table 6 & Figure 9C & 10B). Amino acid residues SER19 and THR46 are commonly involved in binding of drugs, specific inhibitors and substituted chalcones.
DISCUSSION
A good quality model of BmGR was successfully prepared with estimated model reliability exist between 0-1 score. Predicted 3D model with Q mean Z-Score 0.05 and G-factor average score -0.06 indicated the best model property of the modeled BmGR protein structure (Table 1 & 3 and Figure 2). In modeled protein structure only one residue, VAL126, was observed in disallowed region. After complete simulation

of this protein we found that 90.5% residues in most favored region which reflect the predicted model quality is good (Table 2 and Figure 1). G-factor analysis indicate that out of the total 450 residues 43 glycine and 15 proline residues were present in the protein. As per the compositional study, the hydrophobic residues GLY, LEU and VAL were found to be more than 8.0%. These identified rich residues are assumed to play an important role in the structural stability and hydrophobic interaction with drugs/ compounds (Figure 3). Active site residues (GLY16, GLY18, SER19, GLY20, GLU39, GLU40, ARG42, GLY45, THR46, CYS47, GLY51, CYS52, LYS55, GLY117, LYS118, SER120, ALA142, VAL143, GLY144, TYR181, ARG267, ALA270, LEU276, GLY306, ASP307, LEU313, LEU314, THR315 and PRO316) of BmGR are involved in the interaction of compounds/ ligands (Figure 4). Active site residues (CYS47, CYS52 and TYR181) of BmGR are well conserved with human GR (CYS58, CYS63 & TYR197) which involved in formation of a stable charge-transfer complex (Berkholz et al. 2008). Based on earlier evidences, it has been found that a small NADH (P) binding domain within a larger FAD binding domain exists from ARG173-SER215. Similarly, Interproscan Accession IPR023753 includes both classes I and class II oxido-reductases. SCOP: c.3.1.2, c.3.1.5, i.12.1.1 and CATH: 3.30.390.30, 3.50.50.60 proves that BmGR protein having both class with parallel beta pleated sheet (beta-alpha-beta unit), FAD/NAD (P) binding domain consist of 3 layers, beta/beta/alpha; central parallel beta-sheet of 5 strands and top antiparallel beta-sheet of 3 strands. BmGR protein contains mainly three domains. Domain 1 having two sub domains; smaller (20 amino acids forms 2 coils, 1 helix, 1 strand) and larger (195 amino acids forms 4 helices, 14 strands and 19 coils) which contains FAD binding site. The larger domain also contains NADPH binding site. Domain 2 is a smaller domain (11 amino acids forms 2 coils and 2 helices) which is involved in substrate (GSSG) binding and domain 3 is the larger domain (112 amino acids forms 6 strands, 5 helices and 11 coils) which is mainly involved in dimerization of protein therefore known as dimerization domain. Docking studies revealed that the ALA142 and VAL143 are mainly involved in GSSG binding and SER19, ARG267, ARG272, ASP307, LEU314 and THR315 involved in NADPH binding with BmGR and rest residues were common in binding of both substrates. The N-terminal of BmGR structure is responsible for GSSG/GSH binding, middle part of protein was involved in NADPH/NADP+ binding and C terminal residues are involved in dimer formation. This is the first report defining the structural information of BmGR and direct molecular interaction of available antifilarials (DEC and ALB), specific inhibitors (BCNU and DHBA) and substituted chalcones. The observed difference in binding modes of various compounds with BmGR was firmly associated with their varied chemical structure. We observed a potential interaction and inhibition constant (Ki) of all SK series compounds, Ki in nM range. While specific inhibitor BCNU and antifilarial drug ALB, Ki was µM range with structural BmGR protein (Table 5). From

the overall docking evaluation, we could propose that the binding site of NADPH/ NADP+ with protein shared by the binding site of chalcones (SK series). Similarly, GSSG/GSH binding site was same as that for specific inhibitors (DHBA and BCNU). However, antifilarial drug DEC have different binding position than substrate. The combination of binding energy, hydrogen bonding interaction and inhibition constant (Ki) data suggests that SER19, THR46, CYS47, CYS52, LYS55, PHE165, TYR181, ASP307 & THR315 are more specific common sites for SK series compounds (Figure 9A-C & 10A-D and Table 6). The non binding interaction of drug and specific inhibitor is similar to the binding site of substrates i.e. active site domain II of BmGR protein. While, binding of SK series compounds is similar to the FAD binding site (domain I) as well as substrates (Domain II). Table 6 shows Drugs, Inhibitors and SK-series compounds binding interaction activity is better with the active binding site of BmGR. Docking of SK series compounds having better coordination with active site i.e. Pyridine nucleotide-disulphide oxidoreductases class-I active site (IPR012999), 44-54aa (GGTCVNKGCVP, 2 coils and 2 helices) as well as NADPH binding site. It may help in inhibition of redox enzyme. However, The docking of antifilarials with BmGR protein other than active site may help in the activation of GR activity at Ex-vivo and In-vitro level (data unpublished). We also proposed BmGR (a redox system enzyme) as template for drug target against filariasis. On the basis of unpublished result of Ex-vivo and In-vitro effect of these compounds on bovine filarial parasites, we can also propose one activation and two inhibitions stretches of the BmGR protein. Stretch 5 (ALA142 & VAL143) and 7 (TYR181 & ILE182) may be the activator sites of the GR protein. However, stretch 1 (SER19) & 2 (CYS47) and 3 (CYS52) & 6 (PHE165) are the inhibitory sites of the protein. Specific inhibitors are showing competitive type binding because the binding sites of substrates are same (stretch 1 & 2), while substituted chalcones are showing non competitive because binding sites are different from substrate (stretch 1, 2, 3 & 6). Amino acids SER19, CYS47, CYS52 and PHE165 may be better inhibitory sites for drug designing using BmGR as template. The results of one chloro (SK-3) and two methoxy-substituted chalcones (SK-5 and 7) have potent inhibitory effect on parasite motility, viability, GSH level and GST activity supporting the antifilarial efficacy of chalcones. The most significant effect on parasites was exerted by methoxy substituted chalcones, suggesting that these substituents can be used for further studies against filariasis (Awasthi et al., 2009; Yadav et al., 2010). Meanwhile, these compounds were also screened against other antioxidant enzymes i.e. Glutathione Reductase (GR) and Thioredoxin Reductase (TR) in Ex-vivo and In-vitro studies (Data not published). This study therefore concludes that out of the seven SK series compounds, SK3 and SK5 have shown better interaction with BmGR resulting in minimum energy, lowest inhibition constant (Ki) and lower frequency percentage with highest coordination of reported pyridine nucleotide-disulphide oxidoreductases class-I active site. Also, these compounds were effective against the adult filarial parasites, raising a

possibility to explore BmGR as a potential drug target. So, these compounds can be used as better candidates for new drugs/antifilarials against lymphatic filariasis. Acknowledgments The authors gratefully acknowledge the financial support from AVH fellowship Germany and UGC research fellowship BHU, Varanasi. The computational facilities provided by DBT funded SUB-DIC, Centre for Bioinformatics, School of Biotechnology, Faculty of Science, Banaras Hindu University, Varanasi is also thankfully acknowledged.
REFERENCES
Awasthi SK, Mishra N, Dixit SK, Singh A, Yadav M, Yadav SS, Rathaur S (2009) Antifilarial
activity of 1,3-diarylpropen-1-one: effect on glutathione-S-transferase, a phase II detoxification enzyme. Am. J. Trop. Med. Hyg. 80(5):764–768
Bains JS, Shaw CA (1997) Neurodegenerative disorders in humans, the role of glutathione in oxidative stress-mediated neuronal death. Brain Res Rev 25:335–358
Berkholz DS, Faber HR, Savvides SN, Karplus PA (2008) Catalytic cycle of human glutathione reductase near 1 A resolution. J. Mol. Biol. 382(2):371-384
Bowling AC, Beal MF (1995) Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sci. 56:1151–1171
Brophy PM, Barrett J (1999) Glutathione transferase in helminthes. Parasitology 100:345–349
Gallwitz H, Bonse S, Martinez-Cruz A, Schlichting I, Schumacher K, Krauth-Siegel RL (1999) Ajoene is an inhibitor and subversive substrate of human glutathione reductase and Trypanosoma cruzi trypanothione reductase: crystallographic, kinetic, and spectroscopic studies (PubMed PMID: 9986706). J. Med. Chem. 42(3):364-372
Gupta S, Rathaur S (2005) Filarial glutathione S-transferase, its induction by xenobiotics and potential as drug target. Acta Biochim Pol. 52:493–500
Halliwell B (1992) Reactive oxygen species and the central nervous system. J Neurochem 59:1609–1623
Ibarraa MPC, Grillob ML, Belloc M, Nucettellic TK, Atkinsa WM, Bammlerd (2003) Exploration of in vitro pro-drug activation and futile cycling by glutathione S-transferases: thiol ester hydrolysis and inhibitor maturation. Arch Biochem Biophys 414:303–311
Karplus PA, Krauth-Siegel RL, Schirmer RH, Schulz GE (1988) Inhibition of human glutathione reductase by the nitrosourea drugs 1,3-bis(2-chloroethyl)-1-nitrosourea and 1-(2-chloroethyl)-3-(2-hydroxyethyl)-1-nitrosourea, A crystallographic analysis. Eur. J. Biochem. 171(1-2):193-198
Karplus PA, Schulz GE (1987) Refined structure of glutathione reductase at 1.54 Å resolution (PubMed: 3656429). J. Mol. Biol. 195(3):701–729
Karplus PA, Schulz GE (1989) Substrate binding and catalysis by glutathione reductase as derived from refined enzyme: substrate crystal structures at 2 Å resolution (PubMed: 2585516). J. Mol. Biol. 210(1):163–180
Lefeuvre M, Bourd K, Loriot MA, Goldberg M, Beaune P, Perianin A, Stanislawski L (2004) Tegdma modulates glutathione transferase P1 activity in gingival fibroblasts. J. Dental Res. 83(12):914–919
Mannervik B, Danielson UH (1998) Glutathione transferases, structure and catalytic activity. CRC Crit Rev Biochem 23:283–337
Precious WY, Barrett J (1989) The possible absence of cytochrome, P-450 linked xenobiotic metabolism in helminthes. Biochim Biophys Acta 992:215–222
Savvides SN, Karplus PA (1996) Kinetics and crystallographic analysis of human glutathione

reductase in complex with a xanthene inhibitor (PMID: 8626496). J. Biol. Chem. 271(14):8101–8107
Sheehan D, Meade G, Foley VM, Dowd CA (2001) Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 360:1–16
Sustmann R, Sicking W, Schulz GE (1989) The Active Site of Glutathione Reductase: An Example of Near Transition-State Structures. Angew. Chem. Int. Ed. Eng. 28(8):1023–1025
Yadav M, Singh A, Rathaur S, Liebau E (2010) Structural modeling and simulation studies of Brugia malayi glutathione-S-transferase with compounds exhibiting antifilarial activity: Implications in drug targeting and designing. Journal of Molecular Graphics and Modelling 28:435–445